

Abstract
Numerous studies have shown that cg05575921 methylation decreases in response to smoking. However, secondary to methodological issues, the magnitude and dose dependency of that response is as of yet unclear. This lack of certainty is a barrier to the use of DNA methylation clinically to assess and monitor smoking status. To better define this relationship, we conducted a joint analysis of methylation sensitive PCR digital (MSdPCR) assessments of cg05575921 methylation in whole blood and/or saliva DNA to smoking using samples from 421 smokers and 423 biochemically confirmed non-smokers from 4 previously published studies. We found that cg05575921 methylation manifested a curvilinear dose dependent decrease in response to increasing cigarette consumption. In whole blood DNA, the Receiver Operating Characteristic (ROC) Area Under the Curve (AUC) of cg05575921 methylation for predicting daily smoking status was 0.98. In saliva DNA, the gross AUC was 0.91 with correction for cellular heterogeneity improving the AUC to 0.94. Methylation status was significantly associated with the Fagerstrom Test for Nicotine Dependence score, but with significant sampling heterogeneity. We conclude that MSdPCR assessments of cg05575921 methylation are a potentially powerful, clinically implementable tool for the assessment and management of smoking.
Subject terms: Diagnostic markers, DNA methylation
Introduction
Despite sustained efforts, smoking continues to be one of the largest preventable cause of premature death in the world1. A variety of pharmacological and behavioral treatments for smoking exist2. But in actual clinical practice, these measures have unacceptably high failure rates2. Although there are a number of reasons for these high failure rates, one of the potentially more addressable reasons may be weaknesses in current biomarker approaches for quantifying smoking and monitoring smoking cessation.
Currently, two biomarkers are used clinically to guide the assessment and treatment of smoking3. The first is carbon monoxide (CO). CO measurements are easy to perform. However, the short half-life of CO prevents it from being used to assess daily levels of cigarette consumption and makes it insensitive to low intensity or sporadic smoking3,4. Cotinine assessments are more sensitive and can be used to more effectively gauge the level of cigarette consumption3,5. But the use of nicotine replacement therapy and the use of non-combustible products has rendered this valuable test less informative. Conceivably, a test that could be conducted in standard clinical settings and that could quantitate cigarette consumption even in the face of other forms of nicotine consumption could find clinical utility.
DNA methylation testing may be that new acceptable form of testing. Since 2008, scores of studies have examined the relationship of smoking to DNA methylation status6. In general, these studies have shown that smoking is associated with broad changes across the epigenome with the majority of these changes at least partially reverting in response to smoking cessation7–9. Using this methylation information, several groups have attempted to construct methylation indices for the prediction of smoking5,10,11. In general, these indices have worked well but have two substantial failings. First of all, many of these indices were constructed using samples from subjects whose smoking status was not biochemically verified. This is a concern because we have repeatedly shown that substantial numbers of subjects participating in research studies (4–10%) who deny any lifetime use of nicotine products have significant serum levels of cotinine12–14. Second, these indices rely on array hybridization techniques, which are costly, relatively imprecise, time consuming and reference dependent that do not produce reliable beta values for clinical interpretation15–17.
Precision Epigenetics methods targeting highly predictive loci may be a better approach. In particular, measurements of cg05575921, a CpG residue contained within an intronic transcription enhancer in the aryl hydrocarbon receptor repressor (AHRR) gene, may have considerable potential for clinical use18,19. Genome wide studies of smoking consistently show that it is the locus most highly associated with smoking in individuals of European, African and Asian ancestries with the difference in blood DNA methylation between smokers and non-smokers (i.e. ∆β) typically being between 15 and 25%19–21. Most critically, as opposed to other single and multipoint methods for assessing methylation status, ethnic specific variation does not affect the methylation set point of cg0557592122.
In 2018, we introduced a methylation sensitive digital PCR (MSdPCR) assay for assessing methylation status at cg05575921. The assay, which is configured to run on the FDA approved Bio-Rad QX200 platform, has inter-assay variation of 0.7% and can be completed in approximately 4 to 5 h23. Most importantly, unlike quantitative PCR tests whose determinations are dependent upon reference standards, digital PCR tests are reference/calibration-free methods with clearly established metrics for precision23–25. This allows the results from different runs or labs to be compared without need to control for batch variation. As such, if cg05575921 methylation can be used to predict smoking, this MSdPCR test theoretically has many of the characteristics of an ideal assay for Precision Epigenetic approaches for assessing smoking.
Over the past 3 years, we have used this assay to assess cg05575921 methylation in the DNA prepared from the whole blood (WB) of four populations of subjects who were clinically characterized for smoking status and with the non-use status of all control subjects being verified using both serum cotinine and exhaled carbon monoxide assessments26–29. In an attempt to provide a more robust understanding of the relationship of WB cg05575921 methylation to smoking status, we now jointly analyze those results. In addition, we introduce the methylation assessments of DNA from the paired saliva samples for many of those samples and conduct parallel analyses of those results.
Methods
The data for these analyses are derived from subjects included in four previously published studies of smoking or alcohol use26–29. In each of these studies, methylation status at cg05575921 status in WB or saliva DNA was determined using MSdPCR and the smoking status of each non-smoking control was verified using serum cotinine assessments and carbon monoxide assessments as previously described26,29. All procedures were performed in accordance with the Declaration of Helsinki with the individual institutional approvals noted separately for each cohort being noted in the supplementary methods section with each subject providing written informed consent.
The Nicotine Cessation (NC) cohort subjects were collected as part of a project whose goal was to help define the reversion curve of cg05575921 in response to smoking cessation26. In brief, subjects from an Iowa community substance use treatment who stated an interest in smoking cessation, reported smoking at least 8 cigarettes per day and had an expired carbon monoxide level of 8 ppm or greater, were eligible for inclusion for this longitudinal study of smoking cessation. After consent, adult subjects were interviewed with a modified version of the Semi-Structured Assessment for the Genetic of Alcoholism, Ver 2 (SSAGA-II) and the Substance Use Questionnaire30,31. Then, both WB and saliva specimens were collected to provide biomaterials for serum and DNA preparation. A total of 114 subjects enrolled in the study and completed the intake visit.
The Alcohol Cessation (ALC2A) subjects were collected for a project whose goal was to develop a blood-based test for heavy alcohol consumption27. In this protocol, two distinct groups of subjects were collected. The first group consisted of self-reported smokers who were admitted to one of three Iowa alcohol treatment centers for the treatment of severe alcohol use disorder in the context of current intoxication. The second group of subjects (i.e. controls) collected under this award were individuals solicited from the University of Iowa community who denied any history of substance abuse and reported at least one year of abstinence from alcohol. After informed consent was received, both sets of subjects (127 smokers and 144 controls) were then were interviewed with a modified version of the SSAGA-II and the Substance Use Questionnaire, with both saliva and blood collected for biomaterial preparation30,31.
The Smoke Free World (SFW) cohort was collected as part of a project to understand whether DNA methylation could differentiate those who use combustible forms of tobacco from those who use other nicotine containing products29. After providing informed consent, adult subjects were administered a 320 item REDCap® interview that assessed nicotine product use history, then phlebotomized and saliva sampled to provide biomaterials for the current study29,32. Only the data from the smoking subjects and the lifetime non-smoking controls were included in this study.
The Smoking Computerized Tomography (SCT) cohort was collected as part of a project whose purpose was to understand the relationship of sildenafil on pulmonary28. In this protocol, subjects who smoked at least 10 cigarettes per day and had a 5-pack year history of smoking were invited to complete a REDCap® administered prescreening interview to assess smoking history and study eligibility. After receiving informed consent, subjects were phlebotomized to provide blood for DNA for epigenetic analysis and serum.
Serum cotinine values presented herein for the SFW cohort were determined as previously described using enzyme-linked immunoassay reagents from AbNova (Taiwan) and a Molecular Devices Emax spectrophotometer (Sunnydale). Fagerstrom Test for Nicotine Dependence (FTND) scores for three of the cohorts (NC, AlcC and SCT) was assessed and scored as previously described33. Exhaled carbon monoxide levels were assessed using a Smokelyzer® according to the manufacturer’s protocol (CoVita, USA). All control subjects included in this study had serum cotinine values < 2 ng/ml and exhaled carbon monoxide levels of < 8 parts per million.
DNA from WB and saliva was prepared as previously described26. DNA methylation at cg05575921 and DMR11 for both WB and saliva DNA was determined as previously described. In brief, 1 µg of DNA of either whole blood or saliva DNA was bisulfite converted using a EpiTect® Fast DNA kit from Qiagen (Germany) according to manufacturer’s directions. An aliquot of each bisulfite-converted sample was pre-amplified, diluted 1:3000 with molecular grade water, and partitioned into ~ 1.5 nl droplets using an automated droplet generator. DNA amplicons contained within these droplets were then PCR amplified using proprietary primer probe sets (Smoke Signature® or DMR11) for each locus from Behavioral Diagnostics (Coralville, IA) and universal digital PCR reagents from Bio-Rad (Carslbad, CA). The number of droplets containing amplicons with at least one “C” allele (methylated CpG residue), one “T” allele (unmethylated CpG residue) or neither allele was then determined using Bio-Rad QX-200 droplet reader. Percent methylation was calculated using Quantisoft software by fitting the observed ratios to a Poisson distribution. Please also note that because MSdPCR is a reference free method, correction for batch variation is not necessary24,34.
The relative contribution of leukocyte DNA (X) to the total DNA sample was determined using the following equation: where DMR11obs is the methylation status at DMR11 in the saliva sample, and 0.01 and 0.99 are the fractional methylation values of DMR11 in leukocytes and buccal cells, respectively35.
Data Analyses: In general, statistical analyses were conducted with R statistical software (v 1.3.959), and JMP Version 10 (SAS Institute, Cary, NC). Linear regression was used to examine the relationships between cg05575921 methylation, demographic variables, FTND, and self-reported cigarette consumption. All reported R2 values are adjusted for the sample size and number of features. The significance of differences in group means was assessed using Welch’s Two Sample t-tests36. Smoothing splines (λ = 100,000) were fit in JMP to examine the nonlinear relationship of cg05575921 methylation to self-reported cigarette consumption37.
Models for predicting smoking status were generated separately using whole blood (WB, Model 1) and saliva (Model 2) DNA data. The first goal of each model building exercise was to examine the extent to which abstaining from smoking was predicted in a similar manner among the four studies. The second goal was to combine the data from all the studies and estimate general models for predicting smoking abstinence. Cox testing was performed to compare linear regression models using the coxtest function in the lmtest R package38.
For all analysis, logistic regression was used with the binary response being abstinence (0 = smoker, 1 = control)39. A complication was that NC and SCT cohorts had only smokers, and logistic regression models could not be estimated. Therefore, we trained regression models separately on ALC2A and SFW and tested them on all the data sets individually. Predictive performance was evaluated with the Brier score, as this can be computed regardless if a data set has only smokers, or a mix of smokers and controls40. The Brier score is defined as , where is the predicted probability for the th participant and is the observed value (0 or 1)40,41.
Two training models were considered, one for WB cg05575921(Model 1) and the other for saliva cg05575921 (Model 2). Model 1 had the single predictor of WB cg05575921 methylation (%). Model 2 had uncorrected saliva cg05575921 methylation (%), DMR11 methylation (%), and their interaction (product term).Testing consisted of using the estimated coefficients of a training model to compute the Brier score for ALC2A, SFW, NC, SCT (note that the training and test sets were sometimes identical). An additional complication was that the SCT cohort did not have saliva cg05575921 and thus, was excluded from the Model 2 testing. Confidence intervals were computed using the non-parametric bootstrap42.
Ethics approval and consent to participate
All procedures were performed in accordance with the Declaration of Helsinki. Institutional Review Board (IRB) approvals were provided by the University of Iowa IRB (IRB 201905678 and IRB201706713) and the Western Institutional Review Board (WIRB 20160135 and WIRB20162083). All subjects in each of the cohorts provided written informed consent for the procedures.
Results
Clinical demographics
The key demographic and tobacco use characteristics of the subjects from each of the four cohorts are given in Table 1. In total, the four cohorts contain 421 smoking and 423 non-smoking subjects (N = 843) with 603 subjects having both whole blood and saliva DNA methylation data.
Table 1.
Key clinical characteristics of subjects in each cohort.
| Cohort | Group | Gender | N | Age | Cigs/Day | Cg05575921 |
|---|---|---|---|---|---|---|
| ALC2A | Smoker | Male | 84 | 41 ± 11.7 | 19.4 ± 13.8 | 48.9% ± 18.0 |
| Smoker | Female | 43 | 40.8 ± 11.3 | 14.2 ± 9 | 49.6% ± 21.0 | |
| SFW | Smoker | Male | 45 | 42.8 ± 14.9 | 16.3 ± 9.6 | 53.7% ± 17.8 |
| Smoker | Female | 67 | 40.1 ± 11.5 | 14.5 ± 7.9 | 55.2% ± 20.1 | |
| SCT | Smoker | Male | 35 | 42.5 ± 10.9 | 19.9 ± 10.3 | 47.0% ± 17.6 |
| Smoker | Female | 32 | 45.1 ± 8.9 | 14.7 ± 4.2 | 57.3% ± 19.0 | |
| NC | Smoker | Male | 70 | 42.3 ± 11.7 | 14.1 ± 8.3 | 51.4% ± 14.2 |
| Smoker | Female | 45 | 41.5 ± 13.5 | 12.7 ± 7.5 | 52.0% ± 15.8 | |
| Total | Male | 234 | 41.9 ± 12.1 | 17.3 ± 11.3 | 50.2% ± 16.8 | |
| Female | 187 | 41.5 ± 11.6 | 14.1 ± 7.6 | 53.6% ± 19.2 | ||
| ALC2A | Control | Male | 68 | 40.2 ± 14.6 | – | 85.6% ± 3.4 |
| Control | Female | 86 | 42.2 ± 14.8 | – | 86.6% ± 2.3 | |
| SFW | Control | Male | 80 | 30.3 ± 12.8 | – | 86.0% ± 3.3 |
| Control | Female | 189 | 30.4 ± 11.4 | – | 87.1% ± 2.5 | |
| Total | Male | 148 | 34.9 ± 14.5 | – | 85.8% ± 3.3 | |
| Female | 275 | 34.1 ± 13.6 | – | 87.0% ± 2.5 |
Consistent with the overall distribution in the population, the majority of the smoking subjects were male (55%, 234 of 421). In contrast, the majority of the non-smoking controls were female (65%, 275 of 423).
Smokers in each of the four cohorts tended to be in their early to mid 40’s with an average age of 41.9 ± 12.1 and 41.5 ± 11.6 for males and females, respectively. The control subjects tended to be younger with both the male (34.9 ± 14.5 vs 41.9 ± 12.1, p < 0.0001) and the female (34.1 ± 14.6 vs 41.5 ± 11.6) subjects being significantly younger than their corresponding smoking counterparts.
The smoking subjects reported smoking as little as one cigarette and as much as 60 cigarettes per day. But overall, they reported consuming approximately ¾ of a pack daily over the past month with males smoking significantly more than females (17.3 ± 11.3 vs 14.1 ± 12.1, p < 0.001).
There was no significant difference between the WB cg05575921 levels of male and female smokers (50.3% ± 16.8 vs 53.6% ± 19.2, p < 0.685). Although the range of values was much more restricted (see Fig. 1), female non-smokers had a slight, but significantly higher levels of cg05575921 methylation than male non-smokers (87.0% vs 85.8%, p < 0.0001).
Figure 1.

Box plots of the distribution of cg05575921 values in WB samples from case (n = 421) and control (n = 423) subjects as a function of gender (M = male, F = female).
Consistent with the localities in which the subjects were ascertained, the majority of subjects in the study were White, with 84% for smokers and 88% for controls. However, there was significant representation of other ethnicities with 11% of the smoking subjects being African American and 3% for controls. Asians constituted 1% of smokers and 6% of controls (“other” ethnic categories constituted 4% of smokers and of controls).
Other biomarker status
We next examined the relationship of WB cg05575921 methylation to serum cotinine (COT) and exhaled carbon monoxide (CO) levels. Because the ALC2A, NC and SCT cohorts were ascertained as part of smoking cessation protocols with a number of the ALC2A subjects on the nicotine patch, the serum cotinine levels of these subjects may not represent steady state levels of cotinine. However, the SFW subjects were not ascertained as part of a treatment program. In these SFW subjects, serum cotinine values were highly correlated with cg05575921 methylation (Supplemental Fig. 1; r = − 0.82, p < 2.2e−16). Similarly, CO was also highly correlated (Supplemental Fig. 2; r = − 0.74, p < 2.2e−16).
Consumption and dependence variables
Next, we examined the ability of WB cg05575921 methylation to predict average daily cigarette consumption within the past month. Using a smoothing spline, a significant curvilinear dose response is seen (Fig. 2; p < 0.0001, R2 = 0.6446). A sharp drop in methylation with increasing levels of daily cigarette consumption, with a more gradual decline in methylation seen after ~ 20 cigarettes per day. We also examined the relationship of pack year history to cg05575921 methylation status. Using a smoothing spline, a significant non-linear dose response is seen (Fig. 3; p < 0.0001, R2 = 0.5365).
Figure 2.
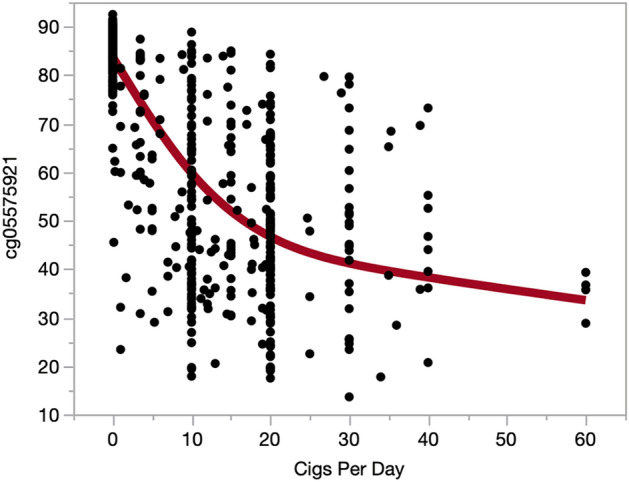
The relationship of WB cg05575921 methylation to self-reported daily cigarette consumption. The line in red represent a smoothing spline fit with a lambda of 100,000 (p < 0.0001, R2 = 0.6446).
Figure 3.
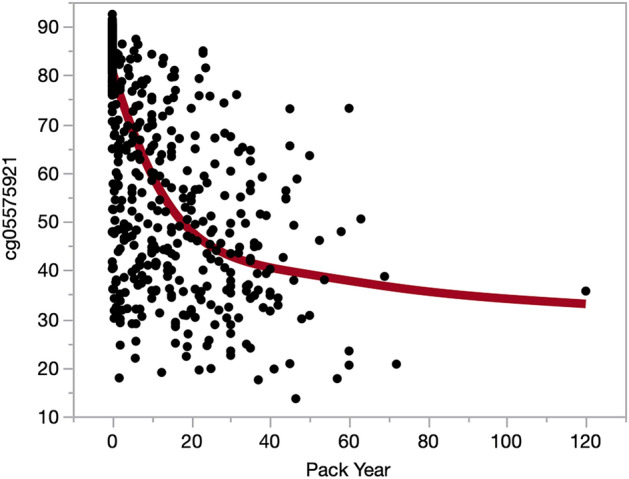
The relationship of WB cg05575921 methylation to lifetime cigarette consumption expressed as pack years. The line in red represent a smoothing spline fit with a lambda of 100,000 (p < 0.0001, R2 = 0.5365).
In pooled case subjects, FTND was negatively correlated with both WB cg05575921 methylation (p = 2.64e−05, R2 = 0.0612), and average self-reported cigarettes consumed per month (p = 0.006, R2 = 0.023). Age, sex and ethnicity were not significantly associated with FTND. Cox Testing demonstrated a significant effect of group status on the ability of cg05575921 to predict FTND (p < 0.0001). In the ALC2A and NC case subjects, FTND did not have a significant relationship with WB cg05575921 methylation, cigarettes consumed per month or key demographics. But in the SCT case subjects, significant relationships between FTND and WB cg05575921 methylation (p = 1.28e−04, R2 = 0.1966), self-reported average cigarettes consumed per month (p = 1.84e−05. R2 = 0.2392), and male gender (p = 0.0425, R2 = 0.04811) were observed. The SFW case subjects did not have FTND data available.
Prediction capacity
For the final set of analyses of the WB data, logistic regression was used to examine the ability of WB cg05575921 methylation to predict current smoking status. Table 2 shows the Brier score results of the training and testing sets for Model 1. The upper portion shows the Brier score and 95% CI when training on either ALC2A or SFW (left) and testing on the four data sets (top). The bold entries indicate that the testing and training data sets were identical. The first row shows that when the model is trained on ALC2A, the Brier score is slightly larger when testing on SFW and SCT than when testing on ALC2A itself, but slightly smaller when testing on NC. The second row shows that when the model is trained on SFW, the Brier score is larger when testing on SCT than testing on SFW itself, but slightly smaller when testing on ALC2A and NC. The lower portion of Table 3 shows the difference between the Brier score trained and tested on the same data set (bold entries in the upper table) and tested on a different data set. For ALC2A, all the 95% CIs for the difference contain 0. For SFW, the only difference for which the CI does not contain 0 is SFW-SFW vs SFW-ALC2A, indicating a smaller Brier score for the latter.
Table 2.
Model 1 (WB DNA) Brier scores and Brier scores differences for the test and training data sets.
| Train | Test | |||
|---|---|---|---|---|
| ALC2A | SFW | NC | SCT | |
| Brier score | ||||
| ALC2A | 0.0421 (0.0410, 0.0478) | 0.0493 (0.0468, 0.0580) | 0.0340 (0.0211, 0.0528) | 0.0705 (0.0509, 0.1000) |
| SFW | 0.0423 (0.0410, 0.0470) | 0.0480 (0.0468, 0.0536) | 0.0372 (0.0239, 0.0533) | 0.0740 (0.0536, 0.1021) |
| Difference | ||||
| ALC2A | – | − 0.0072 (− 0.0139, − 0.0023) | 0.0081 (− 0.0056, 0.0195) | − 0.0284 (− 0.0496, − 0.0127) |
| SFW | 0.0057 (0.0004, 0.0116) | – | 0.0108 (− 0.0058, 0.0249) | − 0.0260 (− 0.0540, − 0.0051) |
*Bolded numerical entries are the Brier scores for which the training and test data are identical.
**95% CI intervals for Brier Scores and differences are given in parentheses.
Table 3.
The ethnicity of smoking and non-smoking subjects.
| Smoker | Control | |
|---|---|---|
| Caucasian | 351 (83%) | 371 (88%) |
| African American | 48 (11%) | 11 (3%) |
| Asian | 5 (1%) | 24 (4%) |
| Other | 17 (4%) | 17 (4%) |
| Total | 421 | 423 |
Table 4 provides the parameters for the final Model 1. The area under the curve (AUC) is 0.984 (95% CI 0.975–0.991). Figure 4 illustrates the relationship between WB cg05575921 methylation level with the probability that they are a smoker in this cohort of subjects of whom almost exactly 50% reported smoking. Using the base cg05575921 model, there is a steady increase in probability of smoking as DNAm decreases from 90 to 83% (i.e. < 10% probability with 85% DNAm). Next, a sharp ~ 10% increase in probability is observed per 1% decrease in DNAm between 81 and 79% methylation (i.e., 56% probability with 79% DNAm). Then, the rate of probability levels out to 99% probability when DNAm is less than 68%.
Table 4.
Model 1 (WB DNA) Parameter estimates and calibration statistics with 95% CI bounds for the pooled data (top), ALC2A (middle), and SFW (bottom).
| Model | Effect/Statistic | Estimate | Lwr | Upr |
|---|---|---|---|---|
|
Model 1 Pooled (N = 779) |
Intercept | − 33.683 | − 40.3087 | − 27.0573 |
| WB cg05575921 | 0.4235 | 0.3434 | 0.5036 | |
| Brier Score | 0.0433 | 0.033 | 0.0537 | |
| AUC | 0.984 | 0.9753 | 0.991 | |
|
Model 1 ALC2A (N = 260) |
Intercept | − 34.3824 | − 50.3077 | − 23.4395 |
| WB cg05575921 | 0.4370 | 0.3045 | 0.6319 | |
| Brier Score | 0.0405 | 0.0221 | 0.0590 | |
| AUC | 0.9815 | 0.9610 | 0.9953 | |
|
Model 1 SFW (N = 351) |
Intercept | − 29.9781 | − 41.4922 | − 22.5292 |
| WB cg05575921 | 0.3838 | 0.2940 | 0.5165 | |
| Brier Score | 0.0466 | 0.0304 | 0.0629 | |
| AUC | 0.9699 | 0.9439 | 0.9883 |
Figure 4.

Predicted probability (95% confidence ribbon) of abstaining from smoking as a function of whole blood cg05575921 methylation percentage.
Saliva DNA analyses
As a first step, we analyzed the relationship of cg05575921 DNAm in WB DNA to that in saliva DNA in 629 paired samples (266 cases, 363 controls). Figure 5 illustrates the distribution of uncorrected cg05575921 methylation as a function of case or control and stratified by gender. As compared to WB cg05575921 methylation, the range and distribution of saliva DNA cg05575921 methylation values is visibly larger and broader in the control subjects. Figure 6 illustrates the uncorrected correlation of the paired samples. In general, the cg05575921 DNAm values between the two tissues were highly correlated in the pooled cohorts (p < 2.2e−16, r = 0.9) and the individual cohorts: SFW (p < 2.2e−16, r = 0.85), ALC2A (p < 2.2e−16, r = 0.95), and NC (p < 2.2e−16, r = 0.83). Please note that the SCT cohort did not have saliva DNA specimens.
Figure 5.

Box plots of the distribution of cg05575921 values in saliva samples from case (n = 266) and control (n = 363) subjects as a function of gender (M = male, F = female).
Figure 6.

The relationship of WB cg05575921 methylation to saliva cg05575921 methylation. The salmon colored line represents the best fit line for the smoking subjects while the blue line represents the best fit line for the non-smoking subjects.
Next, we examined the ability of uncorrected cg05575921 methylation to predict average daily cigarette consumption within the past month and pack year consumption. Using a smooth cubic spline, a similar dose response is observed in uncorrected saliva values to both daily cigarette (Supplement Fig. 3; p < 0.0001, R2 = 0.5340) and pack year consumption (p < 0.0001, R2 = 0.4352) to that observed in WB methylation.
The imperfect correlation of WB cg05575921 methylation in WB to that of saliva is due in part to the cellular heterogeneity found in saliva. To better understand this heterogeneity, we used the DMR11 assay to impute the ratio of buccal and leukocytes in each saliva sample (Fig. 7). The average contribution of leukocyte DNA to the total human DNA in saliva was 75.9% ± 16.7% (n = 603).
Figure 7.
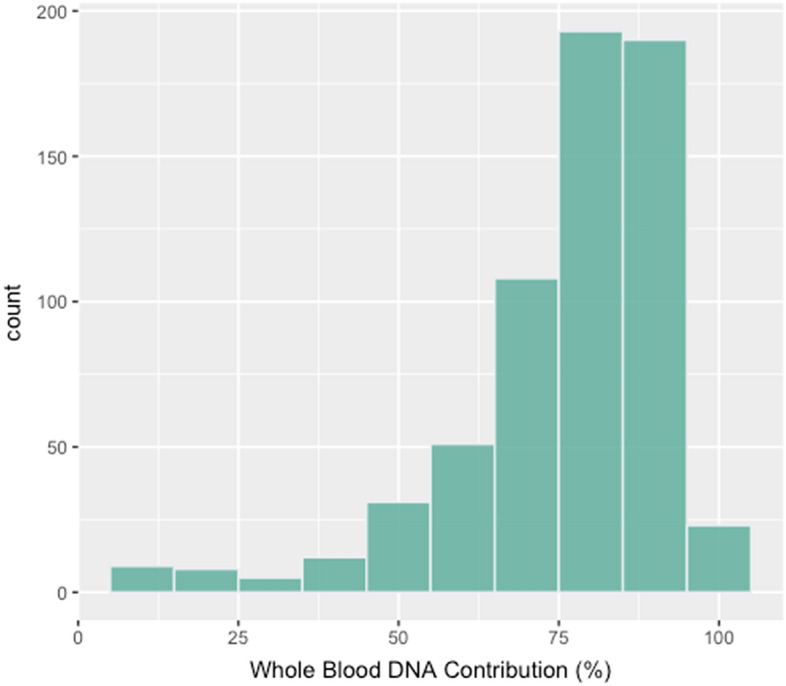
The WB contribution to the total human DNA component of saliva DNA as determined using the DMR11 assay (n = 601).
Prediction capacity
In our final set of analyses, we attempted to predict smoking status using methylation data only from the saliva samples. Table 5 shows the Brier score results for Model 2. The values are larger than for Model 1 indicating generally lower prediction accuracy for saliva assessments. The first row shows that training on ALC2A and testing on SFW produces a larger Brier score than testing on ALC2A itself, but a smaller value when testing on NC. The lower portion shows that these differences are not statistically significant (0 is not in the CI). In contrast, training on SFW and testing on ALC2A or NC was not statistically different than testing on SFW itself. The AUC using saliva DNA was 0.939 (95% CI 0.915 to 0.962) was lower than that for WB DNA.
Table 5.
Model 2 (Saliva DNA) Brier scores and Brier scores differences for the test and training data sets.
| Train | Test | |||
|---|---|---|---|---|
| ALC2A | SFW | NC | SCT | |
| Brier score | ||||
| ALC2A | 0.0638 (0.0608, 0.0739) | 0.1031 (0.0704, 0.1652) | 0.0424 (0.0239, 0.1120) | – |
| SFW | 0.0713 (0.0637, 0.0811) | 0.0665 (0.0641, 0.0715) | 0.0944 (0.0641, 0.1448) | – |
| Difference | ||||
| ALC2A | – | − 0.0393 (− 0.0999, − 0.0044) | 0.0214 (− 0.0492, 0.0414) | – |
| SFW | − 0.0048 (− 0.0157, 0.0039) | – | − 0.0280 (− 0.0771, 0.0035) | – |
*Bolded numerical entries are the Brier scores for which the training and test data are identical.
**95% CI intervals for Brier Scores and differences are given in parentheses.
Discussion
In this communication, we present the analyses of data from four prior studies in order provide a more complete picture of the ability of a MSdPCR assay to assess cigarette consumption and classify subjects as smokers or non-smokers using DNA from whole blood or saliva. These findings show that the assay is a robust method for assessing smoking status and intensity. However, before considering these findings, certain limitations should be noted. First, although the assay is commercially available, the data analyzed are only from our laboratory group. Second, even though there is considerable representation of non-White ethnicities, all of the subjects are from the Midwest region of the United States which has excellent air quality. If environmental influences such as air pollution substantially affect the methylation set point, the application of this assay to regions of the world with significant exposure may be affected. Third, this manuscript does not address the issue of classifying former smokers. This limitation is discussed in depth below.
Perhaps the interesting finding of the study is that methylation at this locus can be used “straight up”, without factoring in age or sex, to predict smoking status. This appears to be a fortunate finding because many other CpG loci experience “epigenetic drift”, a phenomenon that has been harvested to construct indices of epigenetic aging43–45. Establishing this lack of variability of the set point at this locus point is critical to the field because smoking is a major driver of accelerated aging with cg05575921 methylation being associated with methylation status at least 38% of 513 markers in one popular EA index46. Indeed, if EA indices are to be used in guiding healthcare decisions, it is essential that they be interpretable in a clinically meaningful and reliable manner. In this regard, estimates of smoking status can be derived from some EA indices47. But to the best of our knowledge, the inter-assay variability of these EA assessments has not been published nor are the methods through they impute smoking independently verified. In contrast, the inter-assay variability of this reference free MSdPCR method used herein is established (0.8%) with the current data giving a clear basis for interpreting results (e.g. cigarettes per day)23–25.
Establishing the methylation set point in non-smokers was not a trivial undertaking. Our efforts have had to address a number of problems including two key barriers in defining the set point of cg05575921 methylation in non-smoking individuals. The first barrier was the problem of subjects presenting themselves as non-smokers when they are in fact smoking. In our prior efforts, we have noted that between 3 and 5% of individuals presenting themselves as lifetime non-users of any tobacco or cannabis product have significant serum levels of cotinine or cannabinoids26,30. The rate of misrepresentation is even higher in some of our longitudinal studies with the rate of former smokers misrepresenting their past smoking being completely unknown13. The second challenge to establishing a set point is variability in air quality. Studies from Taiwan have shown that air quality can have a small, but significant effect on cg05574921 levels48. However, the levels of air pollution in Taiwan are relatively low and fortunately, the level of PAH in the Midwestern United States are relatively low49. But in other parts of the world, in particular those which rely on wood or coal fired power production, the exposure could be considerable50. Occupational exposure may magnify this problem. We note prior publications noting considerable levels of PAH exposure in steel workers51. Together with other problems, these issues have made establishing the non-smoking threshold of cg05575921 methylation a challenging proposition.
To address these challenges and establish the methylation set points of cg05575921 methylation, we have surveyed populations with low likelihood of significant underreporting to get a better understanding of the assay set point in various groups. The most notable effort prior to this communication has been the Healthy Iowan Study22. In that study, on the presumption that most high school sophomores will not yet have started smoking, we enrolled 15 year-old high school students and followed them for two years, drawing their blood and measuring their CO levels at three points of time. In the 364 subjects who denied any type of smoking and had negative cotinine and cannabinoid levels, the average level of cg05575921 methylation at age 15 using this assay was 86.8% ± 3.222. That is very similar to the 86.6% ± 2.9 found in these 423 biochemically screened adults and strongly argues against any significant age dependent changes in cg05575921 methylation in adult subjects.
For those conducting clinical studies, the most interesting results in these analyses may those for the saliva specimens. In brief, we show that consistent with prior analyses by ourselves and others, approximately 76% of the human portion of saliva DNA is derived from white blood cells and that the use of the DMR11 assay improves prediction26,52. When the National Institutes of Health (NIH) funded the commercialization projects supporting the development of this assay were initiated, the initial, explicit purpose was to develop a blood-based assay compatible with existing sample flow in hospital laboratories. Since that time, the COVID19 pandemic and the rise of telemedicine (TM) approaches for provision of patient care have led to the need for assay systems that do not require blood draws and are not temperature sensitive. As a result, subsequent NIH commercial awards (R43AA027423; Philibert PI) were made to develop an assay such as DMR11 and the previously described DMR16 assay, which are capable of correcting for the heterogeneity of saliva26,35,53 and can use DNA from commercially available sampling kits, such as those provided by DNA Genotek (www.dnagenotek.com)54. Using these types of kits, providers such as Intelelabs (www.intelelabs.com) are already using TM to offer genetic testing. The current data strongly suggests that epigenetic testing for cg05575921 could also be conducted using these kits via standard TM approaches. If so, this would add yet another method through which clinicians could assess and monitor smoking cessation therapy.
Although it is clear that for classification purposes, cg05575921 information is sufficient, one question that these data do not address is whether the addition of genetic information may aid in understanding the relationship of smoking intensity to cg05575921. This is an important question. Nicotine, though addictive, has very few other adverse effects at pharmacologically relevant doses55. Instead, it is the other constituents in tobacco smoke, such as polyaromatic hydrocarbons, that cause smoking associated diseases56. Nevertheless, even though it is clear that we can precisely measure cg05575921 methylation levels and the basal set point appears to be reliably determined, the relationship between increasing cigarette consumption and decreasing cg05575921 levels is only modestly strong (R2 ~ 0.62). There are several reasons for this less than complete accounting for the variance in the relationship. First, subjects do not always accurately recall their cigarette consumption. Second, the binning of responses into easy to express units (e.g. “half-pack”) may contribute to the error. Finally, subjects vary in the number and size of puffs that they extract from each cigarette57. But another factor that may affect the dose response relationship is that genetic variation in the cytochromes regulated in part by AHRR may alter the magnitude of response58. If so, by defining cytochrome variation, it may be possible to devise new methods to induce the detoxification of PAH that can be used for preventing cancer in those who cannot avoid PAH exposure.
Finally, as noted in the limitations section, this analysis does not address the use of cg05575921 methylation to classify former smokers. Several studies have shown that cg05575921 methylation undergoes a slow reversion to the mean as a function of initial smoking intensity7,8,28,59. In brief, those studies show that the longer and more intensively one smokes, the longer it takes to revert back to baseline after quitting. But the number of subjects and the time periods examined in our studies and those of others are limited. As a consequence, we do not believe that there is a sufficient evidence base to guide prediction of when a former smoker will cross the 80% threshold associated with non-smoking status.
Conclusions
In summary, in this joint analysis of samples of subjects from four rigorous studies, we show that cg05575921 methylation levels can be used to accurately classify subjects with respect to smoking and infer daily cigarette consumption using DNA from either whole blood or saliva. Critically, unlike methylation array approaches for assessing smoking, the MSdPCR method used in these assessments can be rapidly performed by any of the thousands of laboratories with digital PCR machines with the results being expressed as β or % methylation values that are directly interpretable. The resulting methylation levels will be of potentially informative for clinicians assessing patients for need for low dose CT lung screening60 or for guiding smoking cessation therapy28. Efforts to make this assay more available to clinicians through independent regional facilities are in progress.
Supplementary Information
Acknowledgements
Not Applicable.
Author contributions
K.D. and S.M. conducted the methylation assessments. K.D., J.L. and J.M. conducted the analyses. R.P., K.D. and J.L. wrote the first draft of the manuscript. E.H., A.A., R.R. and R.P. obtained the funding for the project. K.D., E.H., A.A., R.P., R.R., S.M. and J.L. contributed the writing of the final manuscript. All authors read and approved the final draft.
Funding
The collection and analyses of these data were supported by grants from the National Institutes of Health; 5R01HL130883 (Eric Hoffman, PI), R44CA213507 (Robert Philibert, PI) and R44AA022041 (Robert Philibert, PI) as well as the Smoke Free World Foundation.
Data availability
The data used in the analyses can be obtained upon reasonable request to the principle investigators for each study cohort.
Competing interests
Dr. Philibert is the Chief Executive Officer of Behavioral Diagnostics. The use of cg05575921 to assess smoking status is covered by existing and pending patents including US Patents 8,637,652 and 9,273,358. The use of DMR11 to impute cell heterogeneity is covered by pending patents assigned to Behavioral Diagnostics. Ms. Dawes, Mr. Mills, Drs. Andersen, Reimer, Hoffman and Long have no conflicts to report. Ms. Miller is an employee of Behavioral Diagnostics.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-01088-7.
References
- 1.World Health Organization. Tobacco (2020).
- 2.US Department of Health and Human Services. Smoking Cessation: A Report of the Surgeon General (2020).
- 3.Florescu A, Ferrence R, Einarson T, Selby P, Soldin O, Koren G. Methods for quantification of exposure to cigarette smoking and environmental tobacco smoke: Focus on developmental toxicology. Ther. Drug Monit. 2009;31(1):14–30. doi: 10.1097/FTD.1090b1013e3181957a3181953b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandberg A, Sköld CM, Grunewald J, Eklund A, Wheelock ÅM. Assessing recent smoking status by measuring exhaled carbon monoxide levels. PLoS ONE. 2011;6(12):e28864. doi: 10.1371/journal.pone.0028864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Florath I, Saum K-U, Brenner H. Self-reported smoking, serum cotinine, and blood DNA methylation. Environ. Res. 2016;146:395–403. doi: 10.1016/j.envres.2016.01.026. [DOI] [PubMed] [Google Scholar]
- 6.Kaur G, Begum R, Thota S, Batra S. A systematic review of smoking-related epigenetic alterations. Arch. Toxicol. 2019;93(10):2715–2740. doi: 10.1007/s00204-019-02562-y. [DOI] [PubMed] [Google Scholar]
- 7.Wilson R, Wahl S, Pfeiffer L, Ward-Caviness CK, Kunze S, Kretschmer A, Reischl E, Peters A, Gieger C, Waldenberger M. The dynamics of smoking-related disturbed methylation: A two time-point study of methylation change in smokers, non-smokers and former smokers. BMC Genomics. 2017;18(1):805. doi: 10.1186/s12864-017-4198-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guida F, Campanella G, Castagné R, Chadeau-Hyam M, Vermeulen RCH, Vineis P, Georgiadis P, Kyrtopoulos SA, Lund E, Sandanger TM, et al. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum. Mol. Genet. 2015;24(8):2349–2359. doi: 10.1093/hmg/ddu751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCartney DL, Stevenson AJ, Hillary RF, Walker RM, Bermingham ML, Morris SW, Clarke T-K, Campbell A, Murray AD, Whalley HC, et al. Epigenetic signatures of starting and stopping smoking. EBioMedicine. 2018;37:214–220. doi: 10.1016/j.ebiom.2018.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bollepalli S, Korhonen T, Kaprio J, Anders S, Ollikainen M. EpiSmokEr: A robust classifier to determine smoking status from DNA methylation data. Epigenomics. 2019;11(13):1469–1486. doi: 10.2217/epi-2019-0206. [DOI] [PubMed] [Google Scholar]
- 11.Sugden K, Hannon EJ, Arseneault L, Belsky DW, Broadbent JM, Corcoran DL, Hancox RJ, Houts RM, Moffitt TE, Poulton R. Establishing a generalized polyepigenetic biomarker for tobacco smoking. Transl. Psychiatry. 2019;9(1):1–12. doi: 10.1038/s41398-019-0430-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Philibert, R. et al. A quantitative epigenetic approach for the assessment of cigarette consumption. Front. Psychol.6 (2015). [DOI] [PMC free article] [PubMed]
- 13.Andersen AM, Philibert RA, Gibbons FX, Simons RL, Long J. Accuracy and utility of an epigenetic biomarker for smoking in populations with varying rates of false self-report. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017;174(6):641–650. doi: 10.1002/ajmg.b.32555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Philibert RA, Dogan MV, Mills JA, Long JD. AHRR methylation is a significant predictor of mortality risk in Framingham heart study. J. Insur. Med. 2019;48:90–102. doi: 10.17849/insm-48-1-1-1.1. [DOI] [PubMed] [Google Scholar]
- 15.The Blueprint consortium. Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat. Biotechnol.34, 726 (2016). [DOI] [PubMed]
- 16.Dedeurwaerder S, Defrance M, Bizet M, Calonne E, Bontempi G, Fuks F. A comprehensive overview of Infinium HumanMethylation450 data processing. Brief. Bioinform. 2013;15(6):929–941. doi: 10.1093/bib/bbt054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kruppa J, Sieg M, Richter G, Pohrt A. Estimands in epigenome-wide association studies. Clin. Epigenetics. 2021;13(1):98. doi: 10.1186/s13148-021-01083-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andersen AM, Dogan MV, Beach SRH, Philibert RA. Current and future prospects for epigenetic biomarkers of substance use disorders. Genes. 2015;6(4):991–1022. doi: 10.3390/genes6040991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeilinger S, Kuhnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, Weidinger S, Lattka E, Adamski J, Peters A, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE. 2013;8(5):e63812. doi: 10.1371/journal.pone.0063812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dogan MV, Shields B, Cutrona C, Gao L, Gibbons FX, Simons R, Monick M, Brody GH, Tan K, Beach SR, et al. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genomics. 2014;15:151. doi: 10.1186/1471-2164-15-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dogan MV, Xiang J, Beach SR, Cutrona C, Gibbons FX, Simons RL, Brody GH, Stapleton JT, Philibert RA. Ethnicity and smoking-associated DNA methylation changes at HIV co-receptor GPR15. Front. Psych. 2015;6:132. doi: 10.3389/fpsyt.2015.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dawes K, Andersen A, Papworth E, Hundley B, Hutchens N, El Manawy H, Becker A, Sampson L, Philibert W, Gibbons FX. Refinement of cg05575921 demethylation response in nascent smoking. Clin. Epigenetics. 2020;12(1):1–11. doi: 10.1186/s13148-020-00882-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Philibert, R. et al. Dose response and prediction characteristics of a methylation sensitive digital PCR assay for cigarette consumption in adults. Front. Genet.9, 137 (2018). [DOI] [PMC free article] [PubMed]
- 24.Pinheiro, L. & Emslie, K. R. Basic concepts and validation of digital PCR measurements. In Digital PCR: Methods and Protocols (eds Karlin-Neumann, G. & Bizouarn F.) 11–24 (Springer New York, 2018). [DOI] [PubMed]
- 25.White RA, Blainey PC, Fan HC, Quake SR. Digital PCR provides sensitive and absolute calibration for high throughput sequencing. BMC Genomics. 2009;10(1):116. doi: 10.1186/1471-2164-10-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Philibert R, Dogan M, Beach SRH, Mills JA, Long JD. AHRR methylation predicts smoking status and smoking intensity in both saliva and blood DNA. Am. J. Genet. 2019;183(1):51–60. doi: 10.1002/ajmg.b.32760. [DOI] [PubMed] [Google Scholar]
- 27.Philibert R, Miller S, Noel A, Dawes K, Papworth E, Black DW, Beach SRH, Long JD, Mills JA, Dogan M. A four marker digital PCR toolkit for detecting heavy alcohol consumption and the effectiveness of its treatment. J. Insur. Med. 2019;48(1):90–102. doi: 10.17849/insm-48-1-1-1.1. [DOI] [PubMed] [Google Scholar]
- 28.Philibert R, Mills JA, Long JD, Salisbury SE, Comellas A, Gerke A, Dawes K, Vander Weg M, Hoffman EA. The reversion of cg05575921 methylation in smoking cessation: A potential tool for incentivizing healthy aging. Genes. 2020;11(12):1415. doi: 10.3390/genes11121415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andersen A, et al. DNA methylation differentiates smoking from vaping and non-combustible tobacco use. Epigenetics. 2021 doi: 10.1080/15592294.2021.1890875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Philibert R, Penaluna B, White T, Shires S, Gunter T, Liesveld J, Erwin C, Hollenbeck N, Osborn T. A pilot examination of the genome-wide DNA methylation signatures of subjects entering and exiting short-term alcohol dependence treatment programs. Epigenetics. 2014;9(9):1212–1219. doi: 10.4161/epi.32252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bucholz KK, Cadoret R, Cloninger CR, Dinwiddie SH, Hesselbrock VM, Nurnberger JI, Jr, Reich T, Schmidt I, Schuckit MA. A new, semi-structured psychiatric interview for use in genetic linkage studies: a report on the reliability of the SSAGA. J. Stud. Alcohol. 1994;55(2):149–158. doi: 10.15288/jsa.1994.55.149. [DOI] [PubMed] [Google Scholar]
- 32.Harris PA, Taylor R, Minor BL, Elliott V, Fernandez M, O'Neal L, McLeod L, Delacqua G, Delacqua F, Kirby J. The REDCap consortium: Building an international community of software platform partners. J. Biomed. Inform. 2019;95:103. doi: 10.1016/j.jbi.2019.103208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heatherton TF, Kozlowski LT, Frecker RC, Fagerstrom KO. The Fagerstrom test for nicotine dependence: A revision of the fagerstrom tolerance questionnaire. Br. J. Addict. 1991;86(9):1119–1127. doi: 10.1111/j.1360-0443.1991.tb01879.x. [DOI] [PubMed] [Google Scholar]
- 34.Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011;83(22):8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Philibert R. Compositions and methods for applying epigenetic analysis to different tissues. In Edited by United States Patent and Trade Office (Behavioral Diagnostics LLC; Pending).
- 36.Welch BL. The significance of the difference between two means when the population variances are unequal. Biometrika. 1938;29(3/4):350–362. doi: 10.2307/2332010. [DOI] [Google Scholar]
- 37.Rice J, Rosenblatt M. Smoothing splines: Regression, derivatives and deconvolution. Ann. Stat. 1983;11(1):141–156. doi: 10.1214/aos/1176346065. [DOI] [Google Scholar]
- 38.Package ‘lmtest’ [http://mirrors.nics.utk.edu/cran/web/packages/lmtest/lmtest.pdf].
- 39.Hardin JW, Hilbe JM. Generalized Estimating Equations. Chapman and Hall; 2003. [Google Scholar]
- 40.Steyerberg EW, Vickers AJ, Cook NR, Gerds T, Gonen M, Obuchowski N, Pencina MJ, Kattan MW. Assessing the performance of prediction models: A framework for some traditional and novel measures. Epidemiology. 2010;21(1):128. doi: 10.1097/EDE.0b013e3181c30fb2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brier GW. Verification of forecasts expressed in terms of probability. Mon. Weather Rev. 1950;78(1):1–3. doi: 10.1175/1520-0493(1950)078<0001:VOFEIT>2.0.CO;2. [DOI] [Google Scholar]
- 42.Iba K, Shinozaki T, Maruo K, Noma H. Re-evaluation of the comparative effectiveness of bootstrap-based optimism correction methods in the development of multivariable clinical prediction models. BMC Med. Res. Methodol. 2021;21(1):9. doi: 10.1186/s12874-020-01201-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suñer D, Cigudosa JC, Urioste M, Benitez J, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U.S.A. 2005;102(30):10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bocklandt S, Lin W, Sehl ME, Sánchez FJ, Sinsheimer JS, Horvath S, Vilain E. Epigenetic predictor of age. PLoS ONE. 2011;6(6):821. doi: 10.1371/journal.pone.0014821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan J-B, Gao Y, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell. 2013;49(2):359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mills JA, Beach SR, Dogan M, Simons RL, Gibbons FX, Long JD, Philibert R. A direct comparison of the relationship of epigenetic aging and epigenetic substance consumption markers to mortality in the framingham heart study. Genes. 2019;10(1):51. doi: 10.3390/genes10010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu AT, Quach A, Wilson JG, Reiner AP, Aviv A, Raj K, Hou L, Baccarelli AA, Li Y, Stewart JD. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY) 2019;11(2):303. doi: 10.18632/aging.101684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tantoh DM, Wu M-C, Chuang C-C, Chen P-H, Tyan YS, Nfor ON, Lu W-Y, Liaw Y-P. AHRR cg05575921 methylation in relation to smoking and PM 2.5 exposure among Taiwanese men and women. Clin. Epigenet. 2020;12(1):1–9. doi: 10.1186/s13148-020-00908-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang J, Wang P, Li J, Mendola P, Sherman S, Ying Q. Estimating population exposure to ambient polycyclic aromatic hydrocarbon in the United States—Part II: Source apportionment and cancer risk assessment. Environ. Int. 2016;97:163–170. doi: 10.1016/j.envint.2016.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xia Z, Duan X, Tao S, Qiu W, Liu D, Wang Y, Wei S, Wang B, Jiang Q, Lu B, et al. Pollution level, inhalation exposure and lung cancer risk of ambient atmospheric polycyclic aromatic hydrocarbons (PAHs) in Taiyuan, China. Environ. Pollut. 2013;173:150–156. doi: 10.1016/j.envpol.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 51.Singh A, Kamal R, Ahamed I, Wagh M, Bihari V, Sathian B, Kesavachandran CN. PAH exposure-associated lung cancer: An updated meta-analysis. Occup. Med. 2018;68(4):255–261. doi: 10.1093/occmed/kqy049. [DOI] [PubMed] [Google Scholar]
- 52.Smith AK, Kilaru V, Klengel T, Mercer KB, Bradley B, Conneely KN, Ressler KJ, Binder EB. DNA extracted from saliva for methylation studies of psychiatric traits: Evidence tissue specificity and relatedness to brain. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2015;168(1):36–44. doi: 10.1002/ajmg.b.32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dawes K, Andersen A, Vercande K, Papworth E, Philibert W, Beach SR, Gibbons FX, Gerrard M, Philibert R. Saliva DNA methylation detects nascent smoking in adolescents. J. Child Adol. Psychopharm. 2019;29(7):535–544. doi: 10.1089/cap.2018.0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iwasiow, R., Desbois, A. & Birnboim, H. Long-term stability of DNA from saliva samples stored in the Oragene® self-collection kit†. In (Ed Genotek, D.) (2011).
- 55.Lindson, N. et al. Different doses, durations and modes of delivery of nicotine replacement therapy for smoking cessation. Cochrane Database Syst. Rev. 4(4), CD013308 (2019). [DOI] [PMC free article] [PubMed]
- 56.Hecht SS. Lung carcinogenesis by tobacco smoke. Int. J. Cancer. 2012;131(12):2724–2732. doi: 10.1002/ijc.27816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zacny JP, Stitzer ML, Brown FJ, Yingling JE, Griffiths RR. Human cigarette smoking: Effects of puff and inhalation parameters on smoke exposure. J. Pharmacol. Exp. Ther. 1987;240(2):554–564. [PubMed] [Google Scholar]
- 58.Moorthy B, Chu C, Carlin DJ. Polycyclic aromatic hydrocarbons: From metabolism to lung cancer. Toxicol. Sci. 2015;145(1):5–15. doi: 10.1093/toxsci/kfv040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Philibert R, Hollenbeck N, Andersen E, McElroy S, Wilson S, Vercande K, Beach SR, Osborn T, Gerrard M, Gibbons FX, et al. Reversion of AHRR demethylation is a quantitative biomarker of smoking cessation. Front Psychiatry. 2016;7:55. doi: 10.3389/fpsyt.2016.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bojesen SE, Timpson N, Relton C, Davey Smith G, Nordestgaard BG. AHRR (cg05575921) hypomethylation marks smoking behaviour, morbidity and mortality. Thorax. 2017;72:646–653. doi: 10.1136/thoraxjnl-2016-208789. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used in the analyses can be obtained upon reasonable request to the principle investigators for each study cohort.