

Graphical abstract
Keywords: Artificial intelligence, Functional genomics, Genomics, Proteomics, Epigenomics, Transcriptomics, Epitranscriptomics, Metabolomics, Machine learning, Deep learning
Abstract
We review the current applications of artificial intelligence (AI) in functional genomics. The recent explosion of AI follows the remarkable achievements made possible by “deep learning”, along with a burst of “big data” that can meet its hunger. Biology is about to overthrow astronomy as the paradigmatic representative of big data producer. This has been made possible by huge advancements in the field of high throughput technologies, applied to determine how the individual components of a biological system work together to accomplish different processes. The disciplines contributing to this bulk of data are collectively known as functional genomics. They consist in studies of: i) the information contained in the DNA (genomics); ii) the modifications that DNA can reversibly undergo (epigenomics); iii) the RNA transcripts originated by a genome (transcriptomics); iv) the ensemble of chemical modifications decorating different types of RNA transcripts (epitranscriptomics); v) the products of protein-coding transcripts (proteomics); and vi) the small molecules produced from cell metabolism (metabolomics) present in an organism or system at a given time, in physiological or pathological conditions. After reviewing main applications of AI in functional genomics, we discuss important accompanying issues, including ethical, legal and economic issues and the importance of explainability.
1. Introduction
In the last decades, the fast development of high-throughput technologies in biological sciences has led to the production of large amounts of data. These data are aimed at the quantification and characterization of selected ensembles of biological molecules, such as DNA, RNA, proteins and metabolites, with the ultimate goal to understand how these molecules contribute to determine the structure, function and dynamics of a living system, such as a cell, tissue or organism. Disciplines that aim at collecting and analysing large sets of biological data are generally indicated as “omics” by derivation of the word genome, used to indicate the whole amount of DNA present in each cell of an organism, with an extra flavour of openness to big challenges [1]. The different disciplines that contribute to generate this massive volume of biological data are named after the main target of investigation, be it the DNA information content of an organism or system (genomics), the modulations that DNA can reversibly undergo (epigenomics), the RNA transcripts originated by a genome (transcriptomics), the set and dynamics of RNA modifications (epitranscriptomics), the translational products of protein-coding transcripts (proteomics) or the metabolites (metabolomics) that can be present in a given organism or system, at a given time and condition, in physiological and pathological states. All these disciplines are independent fields of study, but the knowledge and data that they produce converge into the ambitious goal of functional genomics, a field of research aimed at characterizing the action and interaction of all main actors (DNA, RNA, proteins and metabolites, along with their modifications) that link a set of observable characteristics of a cell or individual (that is, the phenotype) to the functional interplay between the underlying genetic characteristics (the genotype) and the environmental conditions.
Omics data can get easily too bulky and complex to be investigated through visual analysis or statistical correlations. This has encouraged the use of the so-called Machine Intelligence or Artificial Intelligence (AI) [2], able not only to manage amounts of data that are intractable for human minds, but also to extract information that go beyond our current understanding of the system under investigation and, importantly, to improve automatically through experience gained on training data.
Within AI, independence from the need of being explicitly programmed to perform a given task is the distinguishing feature of Machine Learning (ML) algorithms, including Linear Regression, Clustering and Bayesian Networks (Table 1). The first applications of ML methods in Biology date back to the early 1980s [3]. More recently, ML programs have been applied in all research areas related to functional genomics, such as genomics [4], [5], [6], transcriptomics [7], proteomics [8], [9] and metabolomics [10], [11].
Table 1.
A summary of commonly used machine learning methods, including a brief description of their distinctive features and indication of core applications.
| Learning Type | Method | Description and Most Relevant Features | Main Applications | References |
|---|---|---|---|---|
| LR | Linear Regression is a supervised learning method to investigate the linear relationship between a dependent and one or more independent variables. LR was the oldest and the most widely used type of regression. To overcome the limit of linear assumption many regression techniques have been developed, varying in the type of cost function used: Non-Linear Regression, Polynomial Regression, Logistic Regression (Sigmoid function), Poisson Regression and many others. | Classification. Functional causal modelling. Metabolomics. Genotype-phenotype associations. |
2001 [49] 2005 [50] 2006 [51] 2008 [52] 2012 [53] 2014 [54] |
|
| SVM | Support Vector Machines are supervised learning methods for binary classification. SVMs represent data as points in space and construct a hyperplane or set of hyperplanes in a high-dimensional space to separate the points and predict the belonging to a category [21]. SVMs can perform linear classification and non-linear classification using kernel methods, a class of algorithms for high-dimensional pattern analysis. | Cancer genomics classification. Outliers detection. Discovery of new biomarkers and new drug targets. |
2003 [55] 2007 [56] 2008 [57] 2011 [58] 2013 [59] 2014 [60] |
|
| Supervised | RDF | Random Decision Forests are learning methods that train and average predictions provided by many Decision Trees (DTs). DTs are ML approaches in which predictions are represented by a series of decisions to predict the target value of a variable starting from features observations [61]. Target variable can take continuous (Regression Trees) or discrete values (Classification Trees). DTs are often unstable methods, but have the big advantage to be easily interpretable. | Genome-Wide Association (GWA). Epistasis detection. Pathway analysis. Visualization of decision processes. |
2003 [62] 2004 [63] 2006 [64] 2009 [65] 2012 [66] 2015[67] |
| Naive Bayes | Bayes Classifiers are ML methods that use the Bayes’ theorem for the classification process. A strong assumption for Naive Bayes is mutual feature-independence. These classifiers are very fast and, despite their simplicity, they are efficient in many complex tasks, also with small training data sets. | Short-sequences classification. Multi-class prediction. DNA barcoding. Biomarker selection. |
2001 [68] 2002 [69] 2006 [70] 2009 [71] |
|
| k-NN | The k-Nearest Neighbours is an instance-based learning algorithm used for classification or regression. The algorithm assigns weights to neighbour contribution. The nearest neighbours contribute more to the computed average than distant ones. | Cancer genomics classification. Gene expression analysis. |
2005 [72] 2006 [73] 2010 [74] |
|
| PCA | Principal Component Analysis is a statistical procedure for the reduction of the dimensionality of variable space. PCA consists in a linear coordinate transformation that projects variables from an high-dimensional space to a low-dimensional space trying to maintain the variance as much as possible[19]. One of the main limits of this method is that it can capture only linear correlations between variables. To overcome this disadvantage, Sparse PCA and Nonlinear PCA have been recently introduced. | Dimensionality reduction. Cancer classification. SNPs tagging. Visualization of genetic distances. Proteomic analysis. |
2004 [75] 2007 [76] 2009 [77] 2011 [78] 2013 [79] 2014 [80] |
|
| Unsupervised | DBNs | A Dynamic Bayesian Network is a Bayesian Network (a probabilistic graphical model that uses Bayesian inference for probability computations) with a temporal extension able to model stochastic processes over time [20]. The advantage of this kind of architectures is that they can model very complex time series and relationships between multiple time series. | Gene regulation analysis. Epigenetic data integration. Protein sequencing. |
2007 [81] 2010 [82] 2012 [83] 2014 [84] 2016 [85] |
| LDA | Linear Discriminant Analysis is a linear dimensionality reduction technique for the projection of a dataset on a lower-dimensional space. LDA is very similar to PCA, but in addition to maximizing data variance, LDA is also interested in finding axes that minimize variance. | Data pre-processing. Motifs identification. Cancer genomics classification. |
2000 [86] 2008 [87] 2009 [88] |
|
| k-Means | k-Means Clustering is a vector-quantization method for the partition of observations into k clusters. At each step the algorithm re-updates centroids as cluster barycenters and re-assigns each data point to the nearest centroid. k-Means is at the same time a simple and efficient algorithm for clustering problems. | Genome clustering. Gene expression pattern recognition. Image segmentation. |
2005 [89] 2007 [90] 2015 [91] 2016 [92] |
|
Among ML methods, the most promising to address omics-data complexity are the ones collectively known as Deep Learning (DL) methods. These methods process information by performing mathematical operations (named neurons, in analogy to the “computational elements” in the brain) arranged in multiple layers (thus deep) connected to one other (thus referred to as a neural network) (Table 2). Although the first neural network models were implemented more than 60 years ago, at the beginning they constituted a fascinating but unsustainable resource, due to prohibitive monetary and computational costs. The Perceptron, the first neural network architecture introduced to the scientific community in 1958 by Frank Rosenblatt [12], had limited learning ability. Moreover, despite being the size of a large room, the computer that was running the Perceptron algorithm had quite limited processing power. In general, effective application of DL methods has become possible only in the last decade thanks to steep increase in processor performance that reached their high computational demand, especially following the repurposing of gaming-aimed Graphical Processing Units (GPUs) [13]. In the same years, tremendous decrease of sequencing costs favoured availability of a flood of large genome-scale datasets and made functional genomics a fertile ground for DL applications [14], [15], [16].
Table 2.
A summary of most relevant deep learning architectures, including a brief description of their distinctive features and indication of core applications.
| Learning Type | Method | Description and Most Relevant Features | Main Applications | References |
|---|---|---|---|---|
| DNNs | Deep Neural Networks are neural networks with many hidden layers of artificial neurons. The output of a layer represents the input of the following layer [23]. This kind of architecture allows to capture non-linear relationships and provides complex representations of input data. | Cancer genomics. Protein sequence classification. Phenotype from genotype prediction. |
2017 [93] 2018 [94] 2018 [95] 2019 [96] 2020 [7] |
|
| MLP | The Single Layer Perceptron (SLP) is a ML algorithm for linear binary classification. Neurons of the layer learn optimal weights for input signals one at a time and generate two linearly separable classes [12]. The Multilayer Perceptron (MLP) contains many SLPs organized into three or more layers with feed-forward connections. Unlike SLP, MLP can perform also non-linear classifications. | Protein structure prediction. Molecular Classification. Cancer genomics. |
1982 [3] 2006 [97] 2009 [98] 2010 [99] 2013 [100] |
|
| CNNs | Convolutional Neural Networks are hierarchical architectures inspired by biological processes governing the organization of animals’ visual cortex. This architecture uses combination of convolution and pooling layers and can detect complex local and global patterns [27]. They work by scanning multidimensional arrays such as 2D images or weight matrices of DNA motifs. To be efficient, CNNs require many layers and large labelled datasets. | Modelling regulatory elements. Detection of DNA accessibility. Finding Binding-sites sequences. |
2016 [101] 2016 [102] 2016 [103] 2017 [104] 2018 [105] 2018 [106] 2019 [107] |
|
| Supervised | RNNs | Recurrent Neural Networks are deep architectures able to capture temporal dynamic behaviours. They are suitable for processing time series or sequential data and in general to predict outputs depending on previous states. RNNs hidden layers retain information from previous layers and feed to the next layer, providing the architecture with a sort of memory [25]. | Transcription factor binding sites prediction. Mutation and variants identification. Protein homology detection. |
2017 [108] 2017 [109] 2018 [110] 2019 [111] 2019 [107] |
| LSTM | Long Short Term Memory is a particular type of RNN architecture with feedback connections. It is able to retain information over a long time and to learn long-term dependencies [112]. LSTMs can overcome the vanishing gradient problem, typical of traditional RNNs. They are useful to make predictions, especially when dealing with time series with lags, even wide, between events. | Splicing prediction. Gene expression regulation. Detection of genomic long-term correlations. |
2015 [113] 2015 [114] 2016 [115] 2017 [116] 2019 [117] 2020 [118] |
|
| DBMs | Deep Boltzman Machines are a kind of RNNs based on a stochastic maximum likelihood algorithm. The network contains undirected connections between all layers and has the ability to learn internal representations that become increasingly complex and abstract [26]. The main disadvantage of this kind of networks is the slow speed. | Protein function prediction. SNPs pattern recognition. Cancer genomics. |
2004 [119] 2017 [120] 2017 [121] 2017 [122] 2018 [123] |
|
| DBN | Deep Belief Networks can be viewed as a composition of Restricted Boltzmann Machines (two-layers generative stochastic neural networks) where each layer learns the entire input [24]. Unlike DBMs, only the first two layers of DBNs have undirected connections. A disadvantage of this kind of networks is the high computational cost of the training process since layers must be trained one at a time. | Enhancers prediction. Gene expression pattern recognition. Cancer classification. Drug discovery. |
2014 [124] 2016 [125] 2016 [126] 2017 [127] 2017 [128] 2017 [129] |
|
| AEs | AutoEncoders are neural networks trained to reconstruct the input. The output layer of an AE has the same number of neurons as the input layer, while one or more hidden layers have a lower dimensionality, in order to force the AE to compress data and to extract important features neglecting unimportant ones [28], [29]. Many variants can make representation very robust and precise: Variational AEs, Spares AEs, Denoising AEs, Contractive AEs, Convolutional AEs. | Dimensionality Reduction. Mutation and variants identification. Methylation analysis. Drug discovery. |
2014 [130] 2016 [103] 2017 [131] 2018 [132] 2019 [133] 2020 [134] |
|
| Unsupervised | GANs | Generative Adversarial Networks are architectures made of two neural networks, one against the other [30]. The first neural network, called the generator, generates new instances, the second neural network, the discriminator, evaluates if the generated instances can belong to the training data-set or not. This way GANs are able to generate new data that are indistinguishable from the observed ones. | Data denoising. Data augmentation. Missing data imputation. Genome editing. |
2017 [135] 2018 [136] 2018 [137] 2019 [138] 2019 [139] 2020 [140] |
DL methods in recent years have opened up interesting and exciting perspectives in core areas of research (e.g., image analysis, language analysis and also omics sciences) [17], [18], having many important advantages over traditional ML techniques such as Principal Component Analysis (PCA) [19], Bayesian Methods (BMs) [20], Support Vector Machines (SVMs) [21], Random Forests (RFs) and Decision Trees (DTs) [22] (Table 1). The main advantage of DL over ML methods is the end-to-end learning, that is the possibility of obtaining classification or prediction results directly from the raw data. While not saving the process from possible sources of bias (e.g., input data selection for the network training phase), end-to-end learning benefits from avoiding the potential bias introduced by manual intervention in the various data processing stages. Also, DL methods ease the integration of different input data types (textual, numeric, images, audio files). Finally, DL architectures have a much higher capability of abstraction compared to traditional ML techniques.
Modern DL architectures, such as Deep Neural Networks (DNNs) [23], Deep Belief Networks (DBNs) [24], Recurrent Neural Networks (RNNs) [25], Deep Boltzmann Machines (DBMs) [26], Convolutional Neural Networks (CNNs) [27], AutoEncoders (AEs) [28], [29] and Generative Adversarial Networks (GANs) [30] (Table 2), have moved a long way from the Rosenblatt’s Perceptron in terms of both efficiency and performance. Yet, progress came at the cost of decreased transparency and loss of the ability to trace associative feature extraction and classification processes. This loss of explainability stems from the increased architecture complexity, moving from the single layer of neurons of the Perceptron to the many layers of hidden neurons intervening between the input and output layers of advanced DL models. Of note, loss of explainability entails new risks of obtaining variously biased results and, therefore, it is currently one of the most active research areas in AI (see Sections 5, 6). Explainability is indeed a major issue for the exploitation of DL potential, especially in the biomedical research and healthcare domains, where features selected by the learning system towards the output decision need to be made understandable in human terms. In fact, the ability of DL architectures to extract much more elaborate features than visual deduction and infer associations based on very high abstraction levels facilitates new investigative strategies. However, it also raises major ethical and legal issues due to the cryptic rationale supporting the machine decisions, that is given as a black-box impeding to evaluate the process and to clear possible sources of errors or biases (see Section 7). Finally, more and more sophisticated DL architectures are subject to increased training complexity, due to the exploding number of model configuration parameters (e.g., the weights - or contribution to the prediction - of each node in an artificial neural network) that need to be estimated from the training data. Moreover, DL architectures need a careful and long tuning of configuration hyper-parameters (e.g. the learning rate for training a neural network) that are external to the model and whose value cannot be estimated from the data, but that can strongly impact training speed and model performance. The reader is referred to excellent recent reviews [4], [31], [32] for a detailed discussion on parameter and hyper-parameter setting in biological applications of DL architectures. Taken together, these considerations make DL a powerful tool to be handled with care.
Here we review the main applications of AI methods in functional genomics and the interlaced fields of genomics, epigenomics, transcriptomics, epitranscriptomics, proteomics and metabolomics. In particular, we focus on recent years applications following the raising of big data production in functional genomics and the natural crossing of this discipline with the flourishing field of AI (Fig. 1). In this framework, we also discuss important aspects of data management, such as data integration, cleaning, balancing and imputation of missing data. Furthermore, we address legal, ethical and economic issues related to the application of AI methods in the functional genomics domain. Finally, we endeavour to provide a glimpse of possible future scenarios.
Alan Turing - Mathematician and philosopher.
It seems probable that once the machine thinking method had started, it would not take long to outstrip our feeble powers… They would be able to converse with each other to sharpen their wits. At some stage, therefore, we should have to expect the machines to take control.
Fig. 1.

A timeline of momentous events in functional genomics and artificial intelligence from their foundation until the time they crossed their paths.
2. Functional genomics
Functional genomics is the science that studies, on a genome-wide scale, the relationships among the components of a biological system - genes, transcripts, proteins, metabolites, etc. - and how these components work together to produce a given phenotype. The term ”functional genomics” takes root in the scientific community at the time of the rising of the first genome sequencing projects. These projects are ultimately aimed at determining the complete genome sequence of a given organism and to annotate functionally relevant features therein, such as protein-coding and non-coding genes as well as DNA regulatory regions. The landmark such endeavour is the Human Genome Project (HGP),1 a worldwide collaborative project launched in 1990 and officially completed in 2003 (International Human Genome Sequencing Consortium [33]). However, the first completely sequenced genome from a eukaryote, that of the budding yeast Saccharomyces cerevisiae, was released already in 1996 [34] and provided material to start exploring the complex relationships between genes and gene products at the genome scale. Indeed, a tentative definition of functional genomics was first published in 1997 by Hieter and Boguski [35], that at the beginning of their paper state: “An informal poll of colleagues indicates that the term [functional genomics] is widely used, but has many different interpretations. There is even some sentiment that the term is unnecessary and that it does nothing more than refer to biological research as a whole.” Nevertheless, in the same paper, they also recognize that “[…] the concept of functional genomics has arrived and it is stimulating the creation of new ideas and approaches to understanding biological mechanisms in the context of knowledge of whole genome structure.”. Functional genomics is eventually defined by these authors as a “new phase of genome analysis”, following the conclusion of the “structural genomics” phase (i.e., construction of a physical map and sequencing of the genome). This “new phase” consisted in developing and applying genome-wide experimental approaches and computational techniques to infer gene functions.
The impressive advances occurred since the beginning of this century in massively parallel sequencing technologies and related protocols have changed the face of functional genomics. Today, we can claim that the term is not open to different interpretations any more: it refers to a discipline integrating a large variety of “omics” data and relying on a plethora of high-throughput experimental methodologies and computational approaches to understand the behaviour of biological systems, being the system a cell, tissue or entire organism, in either healthy or pathological conditions.
Specifically, the data used in functional genomics analyses are produced in the context and with the technologies of “omics” disciplines, including genomics, epigenomics, transcriptomics, epitranscriptomics, proteomics and metabolomics (Fig. 2).
Elaine Rich – Computer scientist.
Artificial Intelligence is the study of how to make computers do things which, at the moment, people do better.
Fig. 2.

The many facets of functional genomics: contributing “omics” disciplines, target biological features and core high-throughput technologies for data production. Abbreviations: 4sU-Seq: 4-thiouridine (4sU)-labeled RNA Sequencing; BS-Seq: Bisulfite sequencing; ChIP-Seq: Chromatin ImmunoPrecipitation followed by sequencing; DNase-Seq: DNase I hypersensitive sites sequencing; GRO-seq: Global Run On Sequencing; HPLC: high-performance liquid chromatography; HTS: High- Throughput Sequencing; LC-MS: Liquid Chromatography coupled with Mass Spectrometry; LC-MS/MS: Liquid Chromatography coupled with tandem Mass Spectrometry; MALDI-TOF: Matrix-assisted laser desorption/ionization (MALDI) Time Of Flight; MALDI-TOF/TOF: MALDI coupled with tandem Time Of Flight; MS: Mass Spectrometry; MS/MS: tandem Mass Spectrometry; NET-Seq: Nascent RNA Transcript Sequencing; RNA-Seq: RNA Sequencing; SAGE: serial analysis of gene expression; TLC: thin-layer chromatography..
3. AI applications in functional genomics
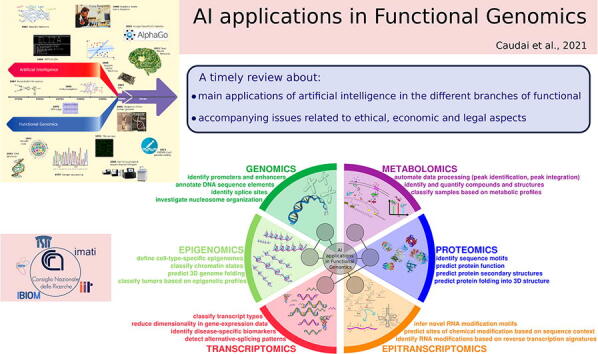
In the last decades, ML has been widely used in many areas of “omics” sciences, especially those characterized by the production of large amounts of data and/or complex mechanisms governed by the synergic participation of different factors. Important applications include: prediction of DNA regulatory regions; discovery of cell morphology and spatial organization; identification of associations between phenotypes and genotypes; classification of DNA methylation and histone modifications; biomarkers discovery; transcriptional enhancers detection; cancer diagnosis and analysis of evolutionary mechanisms [36], [37], [38], [39], [40], [41], [42] (see Fig. 3).
Fig. 3.

AI applications in functional genomics.
Since the 1980s we have witnessed the first attempts to apply supervised training techniques to “omics” sciences. In 1982, Stormo et al. used the Perceptron algorithm to distinguish E. coli translational initiation sites from all other sites in a library of over 78.000 nucleotides of mRNA sequence [3]. In 1993, Rost and Sander implemented a neural network to predict the protein secondary structure [43]. DL techniques began to be massively used in functional genomics only in the second decade of the 2000s, due to the improvement of PC performance and the collapse of genome sequencing costs [44], [45], [46].
In 2015, two important deep architectures have been implemented and applied to functional genomics, producing results of great scientific impact. DeepBind [47] is a fully automatic stand-alone software for the prediction of sequence specificities of DNA and RNA binding proteins. DeepSEA (deep learning-based sequence analyser) [48] predicts chromatin effects of sequence alterations with single-nucleotide resolution, by learning regulatory sequences from large-scale chromatin-profiling data. Both methods, based on deep architectures, have overcome many challenges such as the processing of millions of sequences, the generalization between data from different technologies, the tolerance of noise and missing data and the end-to-end and totally automatic learning, without the need for hand-tuning. These approaches outperformed other state-of-the-art methods and encouraged many scientists to follow similar exciting paths.
In the next sections, we analyse in detail some of the highest impact applications of ML and DL in the main disciplines converging into functional genomics.
Yoshua Bengio – Computer scientist.
I don’t think that any of the human faculties is something inherently inaccessible to computers. I would say that some aspects of humanity are less accessible and creativity of the kind that we appreciate is probably one that is going to be something that’s going to take more time to reach. But maybe even more difficult for computers, but also quite important, will be to understand not just human emotions, but also something a little bit more abstract, which is our sense of what’s right and what’s wrong.
3.1. Genomics
The concept of “genome” was first proposed in 1920 by Hans Winkler, then professor of Botany at the University of Hamburg, referring to “the haploid number of chromosomes” located in the nucleus [141]. In the current era of biological research, with the technological progress in sequencing and the discovery of the DNA complexity, this concept has been extended to the whole set of DNA sequences in a cell or organism (i.e., accounting for the number of copies of the basic set of chromosomes, or ploidy, and including the DNA material from extranuclear organelles such as the mitochondria).
Genomics can be defined as the “science of genomes”. The term was coined in 1986 by Thomas Roderick to describe the nascent discipline of sequencing, mapping, annotating and analysing genomes [35]. The first complete genome sequence of a eukaryotic organelle (the human mitochondrion, 16.6 kb in length) was determined in 1981 [142]; the first free living organism (H. influenzae, 1.8 Mb) was sequenced in 1995 [143]; and the first eukaryotic genome (S. cerevisiae, 12.1 Mb) was completed in 1996 [34]. Sequencing of the human genome (3 Gb), a milestone in the genomics field, took 13 years and was completed in 2003 [33].
Starting from early low-throughput methods (the first generation of sequencing technologies) [144], [145], [146], in a few decades the field was first revolutionized by high parallelization of sequencing reactions, reaching the production of millions of short reads in few hours of run (the second generation) [147], [148]; more recently, it further evolved towards single molecule sequencing and very long sequencing reads (the third generation) [149], [150], [151].
Today, thanks to the advent of high-throughput sequencing techniques, hundreds of thousands of genomes from different kingdoms have been fully sequenced, including over 15.000 eukaryotic genomes (Genome NCBI),2 and the term genomics has now expanded to include the investigation of DNA structure, function, evolution, and editing.
As pointed out by Libbrecht and Stafford Noble [152], ML has been widely used in genomics to annotate sequence elements, identify splice sites, find promoters and enhancers, etc. A large amount of genome sequences have been used to train ML models to recognize specific functional elements. In 1990, an important paper by Bucher [153] was published, where an Optimized-Weight-Matrix algorithm has been applied to hundreds of unrelated promoter sequences to identify promoter elements.
Of note, this ML application, as others in the following years, has been made possible by establishment of databases such as the Eukaryotic Promoter Database (EPD)3 or the European Nucleotide Archive (ENA).4 In 2002, SVM and NB prediction methods [69] have been applied for splice site prediction and showed improvements and advantages over traditional relevant features selection methods. In 2006, Segal et al. proposed an important combined experimental and computational approach [154] to investigate the nucleosome organization. In the proposed pipeline, nucleosome-bound sequences from yeast were isolated at high resolution and used to construct a probabilistic nucleosome-DNA interaction model for linking nucleosome positions to specific chromosome functions and predicting the genome-wide organization of nucleosomes. In 2007, Heintzman et al. mapped five histone modifications and four transcription factors on 30 Mb of the human genome using a clustering ML approach [90]. In 2012, Hoffman et al. applied an unsupervised Dynamic BN method [83] to analyse different types of omics data (such as histone modification marks and binding sites for modifiers of chromatin structure), all of which derived from a human chronic myeloid leukemia cell line, to analyse the entire genome at 1-bp resolution, despite the presence of noise and missing data (See Sections 4.2, 4.1).
DL methods have only been applied in the genomic field in more recent years. An open-source package based on CNNs named Basset [101] for the annotation and interpretation of the non-coding genome was proposed in 2016. The following year, Killoran et al. proposed a GAN to generate DNA sequences with specific properties [135]. More recently, Avsec et al. [155] introduced a DL approach to unravel the influence of motif spacing between neighbour transcription-factor binding sites on transcription factor cooperativity.
Unsupervised approaches, such as GANs and AEs (see Table 1) have a great ability to extract very representative features and learn complex representations of the input data without any type of supervision and addressing. Moreover, they can efficiently denoise and reduce dimensionality without loss of information.
In recent years, representation models used for Natural Language Processing (NLP) have been applied to biological sequence data processing [156], [157]. In a sense biological sequences may be considered as sentences of a language. A widely used method in NLP is LSTM [112], based on RNN architecture, which is suitable for extracting semantic and contextual information from long sequences. In 2013, Mikolov et al. proposed Word2Vec [158], an unsupervised word embedding method to perform low-dimensional vector representation of natural language words. This method is capable of capturing the context of a word in a document, underline relationships between words, and capture semantic and syntactic similarities. In 2015, Vaswani et al. [159] proposed Transformer, a new architecture based on attention mechanism. Transformers are designed to handle sequential data, like LSTM and RNN, and in this sense they are suitable for text translation and interpretation; however, they neither use recurrence nor process inputs in their order. Transformers use a random initialization and are based on dynamic word embeddings (unlike other NLP architectures that use static word embedding). In 2018, Devlin et al., introduced a new NLP method named BERT (Bidirectional Encoder Representations from Transformers) [160] where the authors applied the bidirectional training of Transformer, which was highly performing in capturing semantic meaning and context words.
Very recent papers reported interesting applications of unsupervised word embedding methods on biological sequences. Woloszynek et al. [161] used Word2Vec to embed nucleotide sequences, in particular k-mers obtained from 16S rRNA amplicon surveys, and managed to extract relevant features related to sequence context, taxonomy and classification. Ostrovsky-Berman et al. presented Immune2vect [162], an adaptation of Word2Vec for B-cell receptor sequencing data, where they embedded immune sequencing data in low-dimensional vector-representations to extract relevant features such as n-gram properties and classify immunoglobulin heavy-chain variable (IGHV) genes. Recently Le et al. [163] presented a new technique made up of a BERT and a CNN for DNA enhancer prediction. This approach turned out to be more efficient than Word2Vec in capturing the hidden information in DNA sequences because the word embedding generated with BERT is dynamic, and nucleotides can be represented in different positions and assume different vector values. This is an advantage over static word embeddings, where the same vectors are obtained for the same words regardless of their context, because it provides more detailed and accurate representations.
A major goal in genomics is the identification of genetic variants that underpin human traits, particularly diseases. HTS technologies greatly accelerated our ability to identify gene mutations responsible for human disorders that are caused by variation of large effect in a single gene (e.g., Huntington disease, Duchenne muscular dystrophy). Additionally, thousands of genome-wide association studies (GWAS) have produced long lists of genetic variants associated with common diseases (e.g., asthma, diabetes, heart disease), which are often due to weak contribution of multiple genes and environmental factors. Nevertheless, our understanding of the genetic determinants of these complex diseases still remains limited. This is partly due to unexpected phenotypic readouts originating from functional interactions between two or more genes, as in the case of a genetic mutation whose presence can mask the effects of an allele at another locus (a.k.a. epistasis). Systematic genetics screens conducted in model organisms have fostered a better understanding of the interplay between genotype and phenotype, and provide a framework for the development of personalized genetics in humans by mapping phenotypes between organisms [164]. The most comprehensive analyses have been conducted in the budding yeast Saccharomyces cerevisiae [165], [166], which has led to quantitative phenotypic measurements for tens of million pairs of mutations in yeast. These massive screening efforts have been leveraged together with ML and DL methods for multiple scopes, including: automatic prediction of growth impact of selected genetic interactions in yeast metabolic network, based on both regression and a genetic algorithm that improved prediction accuracy [167]; association of genetic interactions to functional impact, based on RF regression [168]; and costruction of an interpretable or ‘visible’ NN, named DCell, which simulates a basic eukaryotic cell growth [169] and predicts response to genetic perturbation in terms of cellular fitness.
The advent of powerful genome editing technologies, such as CRISPR-Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats-CRISPR-associated protein 9) has enabled scalable manipulation of DNA to functionally characterize genes and gene regulatory elements in a number of different organisms and in human cell model systems. A key point for successful application of CRISPR-Cas9 is the proper design of short RNAs (broadly referred to as gRNAs, acronym to guide RNAs), which provide scaffold to and guide the enzymatic complex to target sites for editing, based on sequence complementarity of 17–20 nucleotides at the 5’-end of the gRNA. In particular, in the gRNA design process, it is crucial to optimize the engineered sequence towards specific interaction with the editing target (on-target activity) while minimizing unintended interactions with other genomic sites (off-target activity), which may arise from sequence similarity with the genuine target. Various ML methods and DL methods have been developed to optimize gRNA design and predict both on-target and off-target activity, including: CRISTA [170], an RF-based regression model that scores the propensity of a genomic site to be cleaved by a given gRNA; DeepCRISPR [171], a computational platform that uses data augmentation technique to expand the training dataset of experimentally validated gRNA sequences and feeds two CNNs (one for on- and one for off-target activity prediction), with gRNA representations produced by pre-trained autoencoders; CROTON [172], an end-to-end framework based on deep multi-task CNNs and neural architecture search to predicting CRISPR-Cas9 editing outcomes; and the complementary tools CRISPR-ONT and CRISPR-OFFT [173], attention-based CNNs trained to predict gRNA on- and off-target activities, respectively.
Combining efficient gene perturbation provided by CRISPR-Cas9 technology with manifold transcriptional phenotyping provided by single-cell RNA sequencing (scRNA-seq) offers an unprecedented opportunity to explore genetic interactions in mammalian cells at large scale. This experimental framework was recently explored by Norman et al. [174], who applied recommender system ML for dimensionality reduction of the high-dimensional map of transcriptional states (phenotypes) associated to gene perturbation, to allow visual analysis and predict genetic interactions.
Several groups have explored ML and DL approaches both to identify disease-associated genetic interactions and to predict the genetic risk of complex diseases in populations from genome-wide maps of genetic variation, such as the occurrence of single-nucleotide polymorphisms (SNPs) or small nucleotide insertions or deletions (indels) in human genomes. In 2014, Kircher et al. proposed CADD (Combined Annotation-Dependent Depletion) [60], an SVM approach for the classification of functional, deleterious and pathogenic variants, which was trained with millions of both high-frequency human derived alleles and simulated variants. The method outperformed existing methods at distinguishing various pathogenic variants that underlie diseases from nearby benign variants. In 2016, Quang and Xie implemented DANN [115], a method for annotating the pathogenicity of genetic variants developed by using the same feature set and training data as CADD, but based a DNN, more suitable than SVMs to capture non-linear relationships between features. In the same year, Ionita-Laza et al. [175], proposed an alternative method based on unsupervised spectral approach (Eigen) that scores genetic variants for disease-association. In 2018, Zhou et al. [106] proposed ExPecto, an end-to-end framework based on a CNN, which was trained on multiple omics data obtained from 200 human tissues and cell types, to predict cell-type-specific effects of genetic sequence variation on gene expression and disease risk. Finally, concerning the analysis of raw sequencing data to identify the presence of genetic variation, in 2018 the genomics team at Google Brain published a deep learning architecture, named DeepVariant, based on a CNN trained to call SNPs and indels variants from piles of aligned sequencing reads [176]. This method won the highest performance award for SNPs in US Food and Drug Administration-sponsored variant calling Truth Challenge in May 2016.
3.1.1. Cancer genomics
In the last decades, the rise of NGS techniques has revolutionized the medical approach to cancer [177]. Genomics has become increasingly important in clinical study, prevention, treatment and monitoring practices. Cancer genomics studies differences in DNA sequences and gene expression between tumour and normal cells, with the aim to understand the dynamics underlying the formation and spread of tumours at the genetic, metabolic, systemic and environmental level. The Cancer Genome Atlas [178] project collected multi-level NGS data for 33 different types of common tumours, an enormous data resource made available to study tumour-specific as well as recurrent cancer mechanisms. The availability and integration of large quantities of genomic, proteomic and epigenomic information has allowed increasingly comprehensive representations of complex dynamics, such as cancer formation[179], to be obtained. Indeed, integration of multiple omics data can help overcome possible noise and/or bias of single data layers, thus improving the relevance of extracted representative features. In this framework, data integration has been an active field of research for ML and DL techniques applied to omics data, especially cancer genomics [180], [181] (see Section 4.3 for a more detailed discussion on data integration). In particular, the introduction of autoencoders, such as denoising autoencoders, has allowed robust representations of heterogeneous data to be provided, and extraction of highly representative and predictive features to be more easily performed [182], [183], [184]. Indeed, AI applications to cancer genomics can provide useful information for a rapid growth of precision medicine and for disease prevention and monitoring.
ML applications to mutation detection and interpretation can help in identifying cancer-predisposing genes such as BRCA1/2 and in predicting cancer risk [185], [186]. AI performances in cancer genomics are very promising. As an example, AI results in the diagnosis of melanoma and breast cancer are very reliable and often surpass expert evaluation [187], [188]. Many ML techniques have been applied to cancer detection and classification, and especially to biomarker identification. In 2003, Vlahou et al. obtained good ovarian cancer classification results by applying a Decision Tree [62]. In the early 2010s, two groups, Abeel et al. [189] and Chen et al. [190], applied SVM for cancer biomarkers identification. In recent years, deep architectures have been applied to variant calling and mutation detection. In 2016, Yuan et al. proposed DeepGene, a DNN cancer classifier, [191] and in 2018 Qi et al. used a MVP for prioritizing pathogenic missense variants [192]. In the same year, Malta et al. proposed a one-class LR for the extraction of transcriptomic and epigenetic features associated with dedifferentiated oncogenic states [193]. Survival models, such as SurvivalNet [194], a DL approach for the screening of large cancer genomic datasets, can be useful for prognosis accuracy improvement and prediction of cancer outcomes.
A recent and promising field of application for AI methods in cancer genomics concerns the computational investigation of synthetic lethal interactions in cancer cell lines to guide anti-cancer drug design. Synthetic lethality refers to a type of genetic interaction where the simultaneous perturbation of two genes leads to cell death or severe impairment of cell viability, while a perturbation of either gene alone does not. Concomitant availability of thorough maps of genetic interactions obtained in model organisms [166], catalogues of cancer genomics data [178], powerful tools for genome editing (e.g. CRISPR-Cas9 editing system) and single cell high-throughput sequencing technologies opened the way to systematic phenotypic discovery at single cell resolution, which is utterly important to tackle tumour cell heterogeneity. In 2017, Way et al. [195] developed an ML approach based on ensemble logistic regression, which was trained on both mutation and transcriptomic profiles of glioblastoma from The Cancer Genome Atlas [178], to predict genes that may exhibit synthetic lethality in cancer cells lacking the neurofibromin 1 tumour suppressor gene. In 2019, Das et al. implemented DiscoverSL [196], a multiparameter RF classifier trained on multi-omic cancer data from The Cancer Genome Atlas [178] to predict and visualize synthetic lethality in cancers. In 2020, Wan et al. developed EXP2SL [197], a semi-supervised NN-based method, which was trained on a large collection of cancer cell line expression signatures from the LINCS1000 Program [198], to predict cancer cell-line specific synthetic lethal interactions.
Other important applications of AI in cancer genomics concern identification of regulatory variants in noncoding domains [199], bioactivity prediction [200], anticancer drug prioritization [201] and sensitivity prediction [202], [203]. All of these applications represent important steps towards personalized medicine, increasing accurate and less invasive prevention, treatment and monitoring paths based on the specific characteristics of the patients and the environment in which they live [204].
Erik Brynjolfsson – Director of Stanford Human-Centered AI.
We can virtually eliminate global poverty massively reduce disease and provide better education to almost everyone on the planet. That said, AI and ML can also be used to increasingly concentrate wealth and power, leaving many people behind, and to create even more horrifying weapons… The right question is not ‘What will happen?’ but ‘What will we choose to do?’ We need to work aggressively to make sure technology matches our values.
3.2. Epigenomics
Epigenomics is a discipline that studies epigenetic processes at the genome scale. These processes include the regulatory mechanisms of gene activity and inheritance that are dictated by genome architecture and independent of changes in the DNA sequence. The term epigenetics, coined in 1942 by British biologist Conrad Waddington, indicates a regulatory layer of gene expression mainly mediated by small chemical compounds (such as Methyl-, Acetyl- or Phosphate-groups) that can be reversibly attached to DNA (e.g., DNA methylation) or chromatin proteins (e.g., methylation, acetylation, phosphorylation and other chemical modifications occurring at the tails of histone proteins). These epigenetic marks are dynamically orchestrated (i.e., layered, interpreted or removed) from the so-called “writer”, “reader” and “eraser” proteins. They cause DNA modulation both in terms of spatial organization and capacity to interact with the gene regulatory machinery, ultimately resulting in switching on or off the expression of the affected genes. In addition to DNA methylation and histone modifications, chromatin remodelling complexes in concert with other DNA binding proteins (such as enhancer-binding proteins and mediators of long-range chromatin looping) provide further epigenetic mechanisms that collectively define the three-dimensional (3D) organization of the genome. This, in turn, defines chromatin regions of active (i.e., transcriptionally competent) or repressed (i.e., inaccessible to transcriptional machinery) states. Epigenomics aims at systematically charting ensembles of epigenetic marks and landscapes of active and repressed genomic regions (i.e., the epigenome) in different cell types and states, to characterize the functional effect on gene expression. In fact, each cell type has a unique epigenome that allows a specific differentiation and reflects a specific state for the cell [205]. Identification of chromatin states, local density of epigenetic marks, long-range chromatin contacts and histone modification patterns has proven relevant for studying and interpreting regulatory regions, cell specific activity and disease-associated patterns. To this end, many ML and DL techniques have been applied to define cell type-specific profiles of DNA methylation (or methylomes) and histone modifications, classify chromatin regions into active and repressed states and, more recently, classify tumour types based on high-throughput methylome data and predict 3D genome folding [206], [207], [208].
In 2015, Ernst and Kellis developed ChromImpute [67], an ML approach based on regression trees to make large-scale prediction of epigenomic marks (such as DNA methylation and histone marks) and chromatin states (such as DNA accessibility). The authors demonstrated the performance of their inference method on a large compendium of publicly available epigenomic maps, achieving strong agreement between experimentally observed and computationally imputed signals. In 2016, Zhang and co-workers developed IDEAS [85], an integrative epigenome annotation system based on quantitative Hidden Markov Models (HMMs) for the characterization of epigenetic dynamics and the detection of regulatory regions. The proposed method is able to handle multiple genomes and to compare inferred epigenomic events at base resolution across different cell types, to identify recurrent as well as cell specific patterns. Wang et al. [209] developed a stacked denoising autoencoder architecture, named DeepMethyl, that uses both DNA sequence features and 3D genome structure to predict DNA methylation status of CpG sites. Recently, Kelley and co-workers proposed Basenji [105], a CNN approach to predict cell-type-specific epigenetic and transcriptional profiles using only DNA sequence as input.
Epigenomics data are often affected by noise and biases (see Section 4.2), and ML and DL methods have been widely used in recent years for data quality enhancement. In 2017, Koh et al. [210] used a CNN to denoise and improve data quality of histone ChIP-seq (chromatin immune-precipitation sequencing) data. In 2019, Hiranuma et al. proposed AIControl [211], a regression algorithm for genome-wide detection of binding-enriched regions, which integrates many publicly available control datasets to improve background subtraction and signal discrimination. The advantage of data integration exploited by AIControl is the ability to subtract different kind of biases affecting ChIP-seq data, thus providing an effective method to remove background signals from experiments lacking control samples. Most recently, Lal et al. introduced AtacWorks [212], a DL-based toolkit, which trains a residual NN model consisting of multiple stacked residual blocks, to denoise low-coverage or low-quality single-cell sequencing data obtained by ATAC-seq (Assay for Transposase-Accessible Chromatin using Sequencing), a high-throughput technique that captures genome-wide open chromatin sites as a proxy for active regulatory regions.
Several ML approaches have been applied to modelling chromatin structure from experimental data obtained by chromosome conformation capture (3C) and its derived technologies (such as 4C, 5C and Hi-C) [213], [214]. In 2012, Ernst and Kellis presented ChromHMM [215], an automated method based on a multivariate HMM for the inference of chromatin states starting from sets of aligned reads for each chromatin modification mark under investigation. In 2014, Gusmao et al. [84] proposed an HMM for the detection of transcription factor binding sites and open chromatin regions integrating structural information such as DNase I hypersensitivity and histone modifications. Chrom3D [216] and ChromStruct [217], [218] use Monte Carlo optimization with loss-score function minimization for the estimation of the chromatin structure starting for Hi-C data. Many of the computational frameworks for the 3D-modelling of chromatin also provide visualization tools [219], in order to allow chromatin structural patterns to be visually interpreted and relationships between chromatin states, genomic positions and pathological modifications to be more easily understood. In 2020, Fudenberg et al. developed Akita [220], a CNN that predicts local 3D genome structures in terms of locus-specific contact frequencies. The Akita algorithm, which was trained on a collection of high-resolution Hi-C maps, takes a genomic region of one million base pairs as input and predicts contact frequencies between any pair of 2,048 bp long windows of DNA sequence within this region. In the same year, Schwessinger et al. developed DeepC [221], a DNN that leverages transfer learning approach and tissue-specific Hi-C data, to train models that predict genome folding in megabase-sized DNA windows. These trained models are then exploited to predict both chromatin domain boundaries at high-resolution and sequence determinants of genome folding, which allows DeepC to also predict the impact of genetic variants of different size (e.g., from large structural variations to SNPs) on 3D-structure.
Ginni Rometty – CEO of IBM.
Some people call this artificial intelligence, but the reality is this technology will enhance us. So instead of artificial intelligence, I think we’ll augment our intelligence.
3.3. Transcriptomics
The transcriptome is the complete set of transcribed genes present within a cell at a given point of time. The first use and definition of the word “transcriptome” date back to 1997 in a work by Velculescu et al. [222], where the authors analysed and characterized the genes expressed in yeast, the only eukaryote for which the entire genome sequence was available at the time [34]. Transcripts were quantified using one of the earliest sequencing-based transcriptomic methods to be developed, namely the serial analysis of gene expression (SAGE) [223]. Velculescu et al. [222] define the transcriptome as “the identity of each expressed gene and its level of expression for a defined population of cells”. The term came later to be used in a broader meaning, and can now be applied to a defined population of cells, a tissue, an organ or an entire organism. It encompasses the whole transcript content, comprising both protein- and non-protein-coding transcribed genes, from the most commonly known infrastructural RNAs (transfer and ribosomal RNAs) and messenger RNAs (involved in protein translation) to the most recently identified small and long non-coding RNAs (defined by a heuristic length cut off of 200 bases [224]), circular RNAs [225], Piwi-interacting RNAs [226], and many other novel non-coding RNA (ncRNA) types. In fact, consortia-based projects for the systematic annotation and characterization of functional elements, such as the ENCyclopedia Of DNA Elements - ENCODE (www.encodeproject.org) [227], [228], detected an unexpected pervasive transcription across genomes, with about 80% of mammalian genomic DNA being actively transcribed, the vast majority of this classified as ncRNA. Compared to the genome, the transcriptome is intrinsically variable and dynamic, making its definition and analysis considerably more complicated.
Transcriptomics is the study of the transcriptome in given physiological or pathological conditions of interest, aimed at capturing the dynamic link between the genome of an organism and its phenotypical characteristics. Ideally, it tries to identify all RNA types and sequences present in a given cell at a given time; to determine the transcriptional structure of genes in terms of start sites, 5’ and 3’ ends, exons, introns and splicing patterns; to detect gene expression levels and unravel possible regulation mechanisms at the whole-genome scale using high-throughput techniques. Instead of focusing on the function of individual genes or transcripts, transcriptomics has the ambition to characterize the whole transcriptome and its changes across a variety of cells, developmental stages, in different biological and environmental conditions. Since the late 1990s, transcriptomics research has been repeatedly revolutionized by the new technological innovations in the field, re-specifying at each step what was possible to investigate. The development of microarrays [229], [230] and, later, NGS technologies [231], [232] have been two key moments in this process. Microarrays allow quantification of a set of already known and preselected RNA sequences since their output signals rely on hybridization of the target molecules with ad hoc designed probes being anchored on the array. NGS technologies applied to RNA sequencing (RNA-seq) [233], [234] are able to capture transcribed molecules independent of prior knowledge since they reconstruct the sequence of assayed RNA molecules as part of the detection step (such in the sequencing-by-synthesis approach, where the target sequence is revealed by synthesis of the complementary strand accompanied by a detection system of the nucleotides inserted during synthesis). As a result of increased throughput, higher accuracy, and lower cost of these specialized NGS technologies, the last two decades have witnessed an exponential growth in transcriptomics studies, which have provided valuable resources for extensive investigations of transcriptional and post-transcriptional regulation [235].
3.3.1. Protein-coding and non protein-coding transcript classification
One core goal of functional genomics is the classification of transcriptome elements, such as the annotation of transcripts as mRNAs (i.e., protein-coding) or ncRNAs, or the prediction of coding potential for each of the multiple transcript products (i.e., isoforms) originating from the same gene locus due to alternative-splicing (AS) events. Many in silico (bioinformatics) methods have attempted to solve this task, but it can be surprisingly difficult in practice. In fact, the proposed solutions often results in manually curated and time-consuming workflows with a number of limitations. The ENCODE and GENCODE projects [227], [228], [236] played a crucial role in this context. In the vast majority of cases, the characterization of novel transcripts is based on the comparison with current sets of genome annotations available from public databases, such as transcript and protein sequences collected from different organisms, known protein domains and structures, integrated with multi-omics experimental data. The more the supporting evidences, the higher the confidence to call the transcript under investigation as being or being not protein-coding [237], [238], [239].
Classification of transcript type provides one application where AI can be crucial. Indeed, this is a typical ML task for which several methods and tools, based on both supervised and unsupervised learning, have been made available. For example, SVM methods were successfully applied to assign coding potential to transcripts according to selected sequences and structure features. In particular, diverse classification algorithms variably integrated relevant characteristics such as: the length of an open reading frame (ORF), which is the specific mRNA sub-sequence dictating the series of amino acids to produce a protein; the corresponding amino acid composition; the predicted protein secondary structure; the predicted proportion of protein residues exposed to the solvent; the existence of corresponding homologous in other organisms; and the synonymous versus non-synonymous substitution rates [240], [241], [242], [243].
Furthermore, classifiers based on ML algorithms were also proposed to distinguish long ncRNAs (lncRNAs) from protein-coding transcripts. For example, Pian et al. [244] used an RF method with some new specific features. Since protein-coding transcripts seem to have longer ORFs compared to lncRNAs, the authors selected the following two specific features for better discrimination: [i.] the longer ORF length (MaxORF) obtained in the three possible lecture schemes (i.e., starting in silico translation of each triplet of nucleotides into the corresponding amino acid at position 1, 2 or 3 of the given transcript); and [ii.] the normalized MaxORF value, obtained by taking into account the total length of the transcript. Similarly, other algorithms that extract a selection of features from sequences and feed it into traditional ML algorithms to assess coding potential have been developed and are available [245], [246].
Even though the integration of additional information not intrinsically derived from transcript sequences may improve the transcript classification, it can also introduce dependence on reliable annotation and be limited by current scientific knowledge, which is biased towards mainstream topics or species (e.g., less available for lncRNAs compared to mRNAs, or for non-model compared to model organisms). Furthermore, manual feature selection made as in traditional ML can introduce biases in the classification, because they are designed and picked by hand. Conversely, DL methods using neural networks can de novo discover complex and hidden biological rules in transcript raw data, thus becoming a more powerful tool to investigate the transcriptomes of the myriad of species made available by current high-throughput sequencing technologies [247], [248], [249], [250].
3.3.2. Gene-expression data analysis
Gene expression is the dynamic process that converts the information encoded within the genome into final functional gene products, giving rise to a range of proteins or ncRNAs. Identifying the molecular mechanisms that control differential gene expression is a major goal of both basic and applied biological research. Gene-expression data from microarrays or RNA-seq platforms have been widely used to distinguish tissues, biological vs. physiological conditions, disease phenotypes, and identify valuable disease biomarkers. A typical problem with high-throughput technologies is the disproportion of dimensions between samples and variables in the dataset. In fact, the high-dimensional number of assayed variables, such as the expression levels of tens of thousands of genes or transcripts, typically far outnumbers that of available samples under investigation (e.g., biological replicates, individuals with a disease). Moreover, these high-dimensional datasets are often sparse and noisy (see Sections 4.1, 4.2 for a more detailed discussion). Practically, the increase in sparsity hampers the collection of data with statistical power, making it extremely difficult to gain biological insights from these data using traditional analytical approaches. This phenomenon is called the “curse of dimensionality”.
Specialized ML algorithms can be powerful tools to address such issues and other serious challenges. Unsupervised learning approaches such as clustering and PCA have been extensively used to find inherent patterns within the data without reference to prior knowledge, for example, to identify gene signatures in gene-expression profiles that might otherwise be overlooked. Global gene-expression correlations (or meta-analyses) are even possible, by comparing numerous genome-wide studies.
Talavera et al. [251] performed a meta-analysis of about 1,500 yeast microarray datasets containing several stress-related experiments. They used an agglomerative clustering algorithm to identify groups (blocks) of transcripts showing high correlation of RNA levels across multiple conditions. Subsequent functional enrichment analyses of the obtained transcriptional blocks, performed using yeast genome annotations of biological processes based on Gene Ontology subsets (also known as GO slims), showed that those groups of consistently up- or down-regulated genes were indeed associated with biological processes linked to responses to different external stimuli (e.g., oxidative stress, osmotic stress, DNA damage stimulus, glucose limitation). This strategy highlights how functional information at the transcript block level, rather than at the single-gene level, in differential expression analyses can effectively help to make hypotheses and model molecular biological mechanisms of the system under investigation.
Microarrays or RNA-seq data can also be used by AI approaches as training sets to effectively learn how to discriminate distinct clinical groups and correctly assign patients to them [252]. In a very recent work [253], the authors analysed about 200 soft-tissue sarcoma samples from The Cancer Genome Atlas project [178] to gain novel insights into the many subtypes differing in prognosis and treatment, which unfortunately have considerable morphological overlap among each other and make differential diagnosis really difficult. To this end, the authors applied different ML algorithms: PCA for dimensionality reduction; a DNN to investigate the overlap of gene-expression patterns of the soft-tissue sarcomas with gene-expression patterns of healthy human tissues; an RF approach to identify novel diagnostic markers. Finally, tumor subtype-specific prognostic genes were identified and tested as predictive of the metastasis-free interval using k-NN analysis.
Very interestingly, in the latest phase of the ENCODE project [254], hierarchical clustering was used to define core gene sets that correspond to major cell types in 53 primary cells from different locations in the human body. Clustering of these primary cells revealed that most cells in the human body share a few broad transcriptional programs, which define five major cell types: epithelial, endothelial, mesenchymal, neural, and blood cells. Based on gene-expression profiles, this new set of cell types redefined the basic histological paradigm by which tissues have been traditionally classified.
Due to the raise of technologies able to profile molecules within an individual cell, such as scRNA-seq, the task of dimensionality reduction to allow visualization and analysis of high-dimensional datasets has becoming increasingly demanding. Consequently, non-linear methods, such as t-distributed Stochastic Neighbour Embedding (t-SNE) [255] and Uniform Manifold Approximation and Projection (UMAP) [256], gained momentum when dealing with large and heterogeneous samples over conventional linear methods such as PCA [257].
The scarce amount of RNA material inherent to scRNA-seq experiments is reflected in the very noisy and incomplete nature of output data. In particular, one major problem related to scRNA-seq experiments is the high percentage of zero-valued observations (a.k.a. dropouts), which stimulated the development of several ML- and DL-based approaches for data imputation (See Section 4.1 for a more detailed discussion on data imputation). In 2018, Li et al. proposed ScImpute [258], an iterative LASSO regression for the imputation of dropout values in scRNA-seq data. Gong et al. developed DrImpute [259], a clustering-based approach that uses a consensus strategy to impute missing values for a given target gene in scRNA-seq data, based on gene expression values of other cells belonging to the same cluster. Arisdakessian et al. implemented DeepImpute [260], a DNN architecture embedding a divide-and-conquer approach to extract relevant patterns useful for imputation of missing expression values for target genes. Specifically, given a set of target genes with dropouts in a scRNA-seq data, DeepImpute builds multiple sub-neural networks, each aimed at learning the relationship between the input genes (predictor genes) and a subset of target genes with dropouts (with zero-values of gene expression to be imputed), thus reducing the complexity by learning smaller problems. Ghahramani et al. [136] applied a GAN to integrate and denoise different scRNA-seq datasets derived from diverse laboratories and experimental protocols, and perform dimensionality reduction. In 2019, Grønbech et al. used an unsupervised DL approach based on Variational AEs [133] to estimate gene expression levels directly from raw scRNA-seq data.
3.3.3. Alternative-splicing code detection
Eukaryotic mRNA AS constitutes an important source of protein diversity [261]. It has been reported that most (i.e., 95%) of multi-exon human genes can undergo AS events [262], [263]. Aberrant AS has been shown to be associated with many diseases [264], [265], [266], [267], [268], [269], [270], [271], [272]. In addition to providing information on RNA abundance, RNA-seq data can be used to infer AS patterns and identify differential AS events linked to different sample conditions, such as treatment vs. control, disease vs. normal, diverse developmental stages, etc. The seminal work on developing DL methods to decipher the splicing code was done by Leung and colleagues [271]. The authors have predicted splicing patterns from mouse RNA-Seq data by using a DNN, with millions of variables representing both the genomic features and the tissue context, which outperformed previous attempts that were based on shallower architectures.
3.3.4. Alternative polyadenylation event detection
Several tools have been introduced in the literature to predict polyadenylation sites (PASs) from human genomic sequences. DNAFSMiner [273] predicts PASs from sequences using k-mer features in an SVM model. Dragon PolyA Spotter [274] also predicts PASs from sequences using both an ANN and an RF. POLYAH [275] discriminates real PASs from other hexamer signals using a linear discriminant function. This algorithm focuses only on the canonical PAS (i.e., the AATAAA sequence motif) in the analysis, although alternative PASs (variants of the AATAAA sequence) may influence the site discrimination. Polyadq [276] uses a quadratic discriminant function to predict real PAS regions. This tool considers two PAS signals in the analysis.
However, the biology underlying alternative polyadenylation is more complicated, and the choice for the polyadenylation machinery to recognize a given PAS depends not only on the PAS itself but also on downstream U/GU-rich elements (AUEs and DAEs). Polyasvm [277] predicts polyA sites from sequences using an SVM model. PolyAR [278] also predicts polyA sites from sequences using a linear discriminant function. Both of these tools use hand-picked sequence features. In order to overcome this limit, DL models such as DeepPolyA [279], DeeReCT-PolyA [280], and Conv-Net [281] have been recently introduced to predict PASs and recognize relatively dominant gene PASs (i.e., most frequently used PASs in a given gene). Of note, all of these models use CNNs to extract features from input genomic sequences. Although the secondary structure near a PAS is also crucial for the PAS to be selected for the polyadenylation process [282], [283], [284], none of these tools consider RNA secondary structures in their prediction procedures.
Geoffrey Hinton - Cognitive psychologist and computer scientist.
I have always been convinced that the only way to get artificial intelligence to work is to do the computation in a way similar to the human brain. That is the goal I have been pursuing. We are making progress, though we still have lots to learn about how the brain actually works.
3.4. Epitranscriptomics
Among the diverse regulatory mechanisms of molecular biology, it has been emerging that all classes of cellular RNA are subject to co- and post-transcriptional modification. The transcriptome modification status is dynamic, revealing a novel and finer layer of complexity in gene expression regulation. Similar to epigenomics, this regulatory mechanism seems orchestrated by writer, reader, and eraser RNA-binding proteins, which can rapidly alter transcript expression levels upon environmental and developmental changes. Taken together, the multitude of RNA modifications, including both non-substitutional chemical modifications and editing events, constitute the “epitranscriptome” [285]. Early reports about RNA modifications deriving from studies on abundant non-coding RNAs such as transfer RNAs and ribosomal RNAs in prokaryotes and simple eukaryotes date back to decades ago [286], [287]. However, only recently, technical advances and refined computational approaches have revealed thousands of novel modification sites within different species of cellular RNA, including mRNAs and lncRNAs. Currently, over 150 distinct post-transcriptional modifications are known to occur on diverse RNA types [288], [289], and the number of discovered epitranscriptomic marks is ever-growing. Nevertheless, knowledge about the function and specific location of RNA modifications remains scarce thus far. Accordingly, epitranscriptomics is the research field devoted to identifying the full spectra of RNA modifications and characterizing them in both protein-coding and non-protein-coding RNA, where they seem to have roles beyond simply fine-tuning of RNA structure and function, as numerous studies on various disease syndromes have highlighted.
In 2012, two independent groups [290], [291] achieved a transcriptome-wide mapping of a specific type of modification (i.e., methylation on the sixth position of the purine ring in RNA adenine, or m6A). These results demonstrated the feasibility of identifying RNA modifications across the entire transcriptome and established the field of epitranscriptomics [292]. Availability of large collections of experimentally identified m6A modification sites stimulated the development of many supervised learning algorithms for the prediction of transcriptome modification sites. Among others, the top performing was SRAMP [293], a predictor of m6A modification sites based on multiple RF classifiers. In 2019, Chen et al. [294] developed WHISTLE, an ML approach that outperformed other algorithms by integrating multiple genomic features (e.g., gene expression profiles, RNA methylation profiles, and protein–protein interaction networks) to predict m6A modification sites rather than rely only on transcript sequences. The next year, Dao and co-workers [295] established iRNA-m6A, an SVM-based classifier for the identification of m6A sites in multiple tissues of human, mouse and rat. The classifier worked on a set of optimal features selected from three kinds of sequence encoding features (i.e., physical–chemical property matrix, mono-nucleotide binary encoding and nucleotide chemical property) computed from the input RNA sequences. Most recently, Zhang et al. introduced DNN-m6A [296], a DNN-based method outperforming preexisting methods in the same task (i.e., prediction of m6A modification sites in RNA sequences of different mammalian tissues).
As mentioned above, transcripts can either be edited (i.e., with base replacement), or covalently linked to small molecules. The former case (i.e., introduction of base changes) can be detected directly by using RNA-seq techniques due to the mismatches that will emerge when the sequencing reads are mapped back to the reference genome. The latter case (i.e., covalent link to small molecules) is more complicated to detect because conventional NGS approaches would erase information about the chemical modification during the sample preparation, specifically during the reverse transcription step. In this mandatory step of NGS protocols, an enzyme called reverse transcriptase (RT) converts RNA into complementary DNA (cDNA) by reading the trascript as a template and inserting base by base the complementary DNA nucleotide in the growing cDNA strand. Consequently, modifications that do not affect Watson–Crick base pairing during cDNA synthesis will be canceled out.
Experimental assays dedicated to the detection of non-mutational RNA modifications have been developed, such as immunoprecipitation with ad hoc antibodies. Importantly, these methods can be applied to a limited number of RNA modifications since they rely on availability of effective antibodies. Other methods exploit the natural consequence of a handful of RNA modifications to induce the RT to arrest during cDNA synthesis, or to make errors (i.e., incorporate non-complementary nucleotides) into the nascent cDNA. In both cases, disturbance in the RT processing will become visible in so-called RT-signatures, that are typical for a given RNA modification and will become visible by mapping the set of sequencing reads spanning the modified RNA position under investigation back to the reference genome. These RT-signatures include accumulation of sequencing reads with identical ends, which match the modified RNA position that caused the RT to stall, or in variable patterns of mismatches, which arise from misreading of the modified RNA residue by the RT. Most recently, Werner and co-workers [297] used an RF approach to predict RNA modifications based on RT-signatures. Their results show strong variability in the success rates depending not only on the type of RNA modification but also on the specific RT enzyme used in the cDNA synthesis step.
The prediction of transcriptome-wide modification sites from transcript sequences is a prototypical supervised learning task. However, for most RNA modifications the number of known positive cases (i.e., experimentally identified sites of modification on transcripts) is too scarce for training robust predictive models. Recently, Salekin and co-workers [298] proposed a GAN-based approach to overcome this problem by successfully mimicking the underlying data distribution and achieving RNA modification site prediction by unsupervised feature learning from input RNA sequences.
Eliezer Yudkowsky - Computer scientist and writer.
By far, the greatest danger of Artificial Intelligence is that people conclude too early that they understand it.
3.5. Proteomics
The proteome is the whole set of proteins expressed and modified after expression by a biological system, being this a cell, a tissue, an organ or an organism. It changes from one system to another (e.g., from cell to cell). It also changes over time in the same system, reflecting the underlying transcriptome and the complex regulatory systems that control protein expression levels, movements within the cell, post-translational modifications, and participation in metabolic pathways. Proteomics is the discipline that studies proteomes using large-scale approaches. The term “proteomics” was first used by Marc Wilkins in 1996 to indicate the “PROTein complement of a genOME” [299], though the highly dynamic nature of proteomes makes proteomics more complex than genomics [300].
Proteomics uses a variety of techniques to explore the overall protein content of a system at a given time, as well as analyze protein function, regulation, post-translational modifications, fluctuations in expression levels, movements and interactions. In particular, conventional (e.g., chromatography-based techniques, Western blotting, X-ray crystallography), advanced (e.g., protein microarrays, gel-based approaches), quantitative (e.g., isotopic protein labeling), and high throughput (e.g., mass spectrometry-based approaches) techniques are available to investigate proteomes [301]. Mass spectrometry (MS) is the leading high-throughput technique for the study of protein mixtures. It is used to determine the molecular weight of proteins through the measure of molecular mass-to-charge ratio (m/z). In MS, molecules are transformed to gas-phase ions, then a mass analyser is used to separate ions in electric or magnetic fields by their m/z values, and finally the amount of each ion species is measured [302]. In tandem MS (MS/MS) [303] two or more mass analysers are coupled together to increase the ability of identifying and separating distinct ions with similar molecular weights. Depending on the molecule at hand, different approaches to ion separation are adopted: liquid chromatography followed by mass spectrometry (LC-MS) and tandem LC-MS (LC-MS/MS) are analytical chemistry techniques in which LC is used to separate mixtures with multiple molecular species, and MS or tandem MS is used to detect individual ion species [304]. Both matrix-assisted laser desorption ionization-time of flight mass spectrometer (MALDI-TOF) [305] and tandem MALDI-TOF (MALDI-TOF/TOF) [306] couple a ionisation technique (MALDI), that creates ions from large molecules with minimal fragmentation, to a mass spectrometer (TOF), which measures the time ions take to reach the detector when accelerated through the same potential of an electric field.
Proteomics encompasses several levels of investigation, such as: protein primary structure (e.g., detection of homology and protein families, motif recognition, multiple sequence alignment, sequence classification); secondary structure (SS) (e.g., identification of local sub-structures); 3D structure (e.g., folding prediction, structure comparison, domain classification, identification of 3D patterns, analysis of chemical and topological features); protein function and functional interactors (e.g., function classification, prediction of active sites and critical residues, prediction of binding sites, analysis of substrates and modulators, analysis of structure–function relationships, drug design). In particular, study of the SS of proteins usually refers to Q3 or Q8 accuracy, respectively defined as the percent of residues for which 3 general states (helix, strand, and coil) or a finer dictionary of 8 such states [307] are well predicted.
In 2003, Kim and Park proposed the SVMpsi method [55], based on SVMs, to maximize the Q3 measure. In 2005, Garrow et al. introduced TMB-Hunt [72], a program that uses a k-NN algorithm for classifying protein sequences as trans-membrane beta-barrel (TMB) or non-TMB. DL-based methods for the study of SS are more recent, such as DeepCNF [103], which used CNNs to detect complex relationship between sequence and structure of proteins and SSREDNs [108], based on a Deep Recurrent Encoder-Decoder Networks architecture to capture complex nonlinear relationship between protein features and SS and also interactions among continuous residues. In the same years, Sønderby and Winther [113] implemented an LSTM, and Fang and co-workers [308] a Deep Inception-Inside-Inception Neural Network for the SS prediction starting from amino-acid sequences. ML methods have been also used for overall tertiary structure [58], [309], torsion angles [57], [310], [311] and loop prediction [312], [313]. Notably, in the 2018 edition of the Critical Assessment of Protein Structure Prediction (CASP), a NN-based software developed by the AI research team at Google DeepMind, named AlphaFold, outperformed all other participating methods in accurately predicting both overall folding and distance between pairs of residues [314]. In the 2020 edition, a new version of the AlphaFold method was presented, which used a completely different model [315], and provided unprecedented results that have resonated throughout the whole community of life science researchers. In particular, the latest AlphaFold architecture includes a new self-supervised learning module (the Evoformer module) based on two transformers (a two tower architecture) for the embedding of the following two pieces of core information that the system tries to collect from public databases, starting from a given input amino acid sequence: 1. a Multiple Sequence Alignment (MSA) and 2. a list of potentially similar structures (or templates). In the Evoformer module, the two tranformer-based representations of sequence and structure information communicate with each-other throughout the neural network for many learning cycles (48 cycles) until they reach solid representations that will be passed on to the last module (the Structure module). Finally, the NN of the Structure module outputs the predicted protein structure by mapping the abstract representations received from the Evoformer module to actual 3D atom coordinates (see Supplementary material in [315] for more details). Of note, the AlphaFold source code has been made freely available to the scientific community right after publication, and its outstanding ability to predict protein structures has been already leveraged to create a free database of structures that covers all 20,000 human proteins plus the full proteomes of several other biologically significant organisms (https://alphafold.ebi.ac.uk/).
Protein function prediction basically consists in the classification of protein functions associated with various structural characteristics. Experimental function annotations is expensive and time-consuming, and information concerning domains, motifs, families, interactions and linkages is large and complex. For these reasons, it is impossible to analyse this information without computational approaches. In 2017, Liu proposed an efficient RNN approach [109] for the classification of protein functions directly from the primary sequences. DeepFunc [96] and DeepPred [316] are two recent DL approaches for the prediction of protein function. Their promising results demonstrate that DL have significant potential in protein function prediction because of the complexity of the task and the size and variety of the datasets [317], [318].
Protein physical interactions, including protein–protein interactions, protein-drug interactions, and binding of proteins to DNA or RNA, are core determinants of cell function, and, effective tools for their systematic investigation would be desirable to gain a solid understanding of cell biology and disease mechanisms. Despite technological advances, experimental investigation of protein–protein interactions is still expensive, laborious, and limited in scale, thus precluding unbiased and systematic efforts. In the last years, accumulating wealth of sequence and structure data has promoted the use of computational approaches to address large-scale investigation of protein–protein interactions.Back in 2004, Dohkan et al. [119] proposed an SVM approach to predict protein–protein interactions based on several protein features, such as annotated functional domains. In 2016, An et al. [319] proposed RVM-BiGP, an ML method for predicting protein–protein interactions from protein sequence, based on RVM classifier combined with Bi-gram probabilities, for protein sequence feature representation, and PCA, for dimensionality reduction. In 2018, Huang et al.[320] proposed a DL method based on a NN and an autoencoder-like architecture to complete sparse and disconnected protein–protein interaction networks via prediction of missing interactions.
Among protein-DNA interactions, transcription factors that bind to regulatory regions in DNA play a central role in regulating various cellular processes by setting cell specific transcriptional programs, and modulating gene expression in response to internal and external stimuli. In 2017, Qin et al. developed TFImpute [321], a DNN that can predict cell-specific binding patterns by learning from experimental data concerning different transcription factors and cell lines. In 2018, Shen et al. proposed KEGRU [110], a DL model to predict TF binding sites based on a Bidirectional Gated Recurrent Unit network combined with k-mer embedding of DNA sequences.
Recently, Rives et al. [322] trained a transformer on 86 billion amino acids across 250 million protein sequences. This unsupervised pre-trained model provides a multiscale representation of protein structures, containing information on secondary and tertiary chains organization, homology, contacts, and mutational effects. The learned representations could also be used for promising application such as generating new sequences and designing functional proteins.
Elon Musk - Co-founder of OpenAI.
AI doesn’t have to be evil to destroy humanity. If AI has a goal and humanity just happens in the way, it will destroy humanity as a matter of course without even thinking about it, no hard feelings.
3.6. Metabolomics
Metabolomics is a discipline aimed at studying the comprehensive profile of metabolites in a cell, a tissue or a whole organism. Metabolites are small molecules that are transformed during metabolic processes, and the whole set produced by a specific cell (the metabolome) provides a functional readout of the cellular biochemical activity [323]. This new discipline emerged at the beginning of this century and rapidly grew thanks to the improvements in instrument technology. Metabolomics studies can be focused on a specific set of metabolites and the particular pathways they take part in (referred to as targeted metabolomics), or be aimed at global metabolite profiling [324] (referred to as untargeted or shotgun or discovery metabolomics). Targeted metabolomics experiments can be performed using mass spectrometry (MS) and nuclear magnetic resonance (NMR), whereas LC-MS is the technique of choice for untargeted metabolomics [323]. In metabolomics, ML and DL techniques have mainly been applied to data pre-processing (such as peak identification and peak integration), and compound identification and quantification [325]. In 2008, Yuan and co-workers used an LDA for the exploration of metabolomics data [87]. In the same year, Cavill et al. implemented a genetic algorithm [326] to analyse NMR spectra of rats' urine to classify liver and kidney toxicity. In 2012, Hao et al. proposed BATMAN [327], a Bayesian automated metabolite analyser for NMR spectra. The process of manual peak fitting, alignment and binning can be time-consuming and can introduce artefacts or errors, so the adoption of ML in this field is preferable to classical approaches. BAYESIL [328] is a fully-automatic and publicly-accessible system to automate compound identification and quantification from NMR spectra of complex mixtures, including biological samples. In particular, the algorithm is a spectral deconvolution system that views spectral matching as a Monte Carlo inference problem within a probabilistic graphical model, which rapidly approximates the input NMR spectrum with the most probable metabolic profile. After several spectral processing steps, BAYESIL couples the given spectrum against a reference compound library containing the signatures of more than 60 metabolites. The deconvolution process is able to capture both the identity and quantity of metabolites present in a complex mixture under examination, such as a person’s biofluid (specifically, serum or cerebrospinal fluid). Alakwaa and co-workers [329] applied feed-forward networks, a DL framework, to predict estrogen receptor status from breast cancer metabolomics data. The authors benchmarked their DL approach against six other ML-based methods, all of which were trained on a cohort of 271 breast cancer tissues (i.e., 204 estrogen receptor positive and 67 estrogen receptor negative) assayed by gas chromatography followed by time-of-flight mass spectrometry, and found that the DL approach is a better classifier for the given task. The biological interpretation of the DL hidden layers revealed significant pathways, such as central carbon metabolism in breast cancer and glutathione metabolism, and allowed the biosynthetic enzymes involved in the metabolomics pathways to be mapped.
Hector Klie - CEO of DeepCast.ai and Professor of Computational and Applied Mathematics at Rice University.
Ultimately, the physics we know needs to rely on data to unmask the physics that we do not yet know.
3.7. Modelling the system: Functional genomics, AI and Systems Biology
Two main approaches to leverage experimental results and enrich our understanding of biological processes are currently adopted: data-driven and model-based. Nowadays, the data-driven approach is mainly used in the domain of DL, which relies on black-box systems for automated decision making. Typically, ML and DL models map the features of a system into a class or a score without exposing the guiding reasons or explaining the structure and the dynamics of the underlying system. This is one of the key hurdles against an extensive use of AI for understanding biology. Indeed, for example, in clinical decisions people tend to pose little trust in results whose predictive mechanism is not known. The general aspects regarding this lack of interpretability and explainability are briefly treated in Section 5.
The model-based approach, conversely, is the traditional domain of systems biology, whose aim is to decipher the complexity of biological systems and understand their structure, components, relationships and dynamics based on biological, genetic, or chemical perturbation and monitoring of the effects on the system [330]. In this framework, to understand the system structure and operation mode, system components and their properties must be identified, and attempts must be made to infer how these interact and evolve dynamically to generate observable biological behavior [331]. In particular, dynamics is typically modelled by a set of ordinary differential equations that describe how chemical and molecular species in the system evolve over time. It is important to point out that the actual predictive value of the results strongly depends on accurate and effective estimation of model parameters, and the differential models depend on many unknown parameters (e.g., rate constants and initial concentrations), which are classically inferred from relatively few experimental measurements. For this limitation, it has been possible to successfully model only some relatively simple biological systems (e.g., lactose- and galactose-utilization systems in bacteria, such as Escherichia coli [332] and Streptococcus [333]), whilst modelling more complex systems still remains prohibitive.
Given the increasing success of AI techniques for large-scale biological data generation and analysis, experimental design and model validation, researchers are inquiring how data-driven approaches can be integrated into model-based strategies to solve the problem of parameter estimation and hidden dynamics inference, to help elucidate biological system structure, mechanisms, and dynamics. This possibility has been discussed during the meeting reported by Wang et al. [334], where convergence of data-driven and theoretical approaches was considered to be an important step to complete the cycle data-model-data, which is typical of experimental sciences like physics and biology. A promising approach to foster this convergence is to use existing theoretical models to constrain the AI results. Nowadays, availability of high-throughput data, and tools to handle it, permit efficient model validation and/or refinement. Along this line, some recent solutions proposed in the literature deserve attention, as they let model-based and data-driven approaches effectively converge towards a deep understanding of biological processes. Costello and Garcia Martin [335] predict the dynamics of the limonene and isopentenol pathway by solving a problem of system identification where the most appropriate model is selected by an ML strategy trained by time-series proteomics data. Yazdani et al. [336] leverage the principle of physics-informed neural networks [337], which further deviates from being purely data driven, in that a mathematical model (with parameters to be identified) is used as a strong constraint in training the network. On this basis, the authors developed a systems-biology-informed DL method capable of estimating model parameters as well as inferring hidden system dynamics. This approach was successfully tested on yeast glycolysis, cell apoptosis and ultradian endocrine models. Fortelny and Bock[338] also use knowledge from systems biology to constrain their results, thus following the same principle of physics-informed neural networks. The authors map a biological network into a neural network where each node represents a molecule and each edge represents an interaction, whose existence and strength, when known, are derived from a mechanistic model. New interactions can then be discovered from experimental evidence and used to refine the structure of the network. The review paper by Muzio et al. [339] explains the structure of graph convolutional networks and graph neural networks, and lists a series of applications where these networks are successfully used to analyze biological networks, including protein function prediction, protein–protein interactions and in silico drug discovery and development. Other review papers, e.g. by Eraslan et al. [15], Zampieri et al. [10], Antonakoudis et al. [340] and Gilpin et al. [341], raise the issue of the competitive vs. collaborative nature of data-driven and model-based approaches, stressing the importance of using appropriately constrained mathematical models to help AI tools produce new knowledge of biological mechanisms.
Klaus Schwab - Founder and CEO of the World Economic Forum.
We must address, individually and collectively, moral and ethical issues raised by cutting-edge research in artificial intelligence and biotechnology, which will enable significant life extension, designer babies, and memory extraction.
4. Data management issues for AI applications in functional genomics
Most ML or DL algorithms take their input in the form of a matrix, where to each column corresponds a sample and to each row a variable (i.e., feature) describing the samples. The nature of these matrices depends on both the context and the specific application. In functional genomics, and in bioinformatics in general, such a representation is advantageous since most data naturally come in this form. For example, RNA-seq data are usually arranged as matrices containing the quantification of gene or transcript abundance (rows) across a set of samples representing different conditions (columns). As a consequence of this mutual suitability of matrices for ML and bioinformatics, programming languages such as R and python, which use data matrices (data frames) as core data structure, have become popular tools.
In many real bioinformatics applications, however, data matrices may be incomplete and/or contain errors. Different protocols, experimental conditions and machineries may cause biases and artefacts. Moreover, some data point may be missing for some of the samples. Yet, the need of implementing a holistic approach to the understanding of every genomic element function requires considering data of different nature. In this scenario, data imputation, denoising and integration should be part of the design of ML and AI for functional genomics.
Vivienne Ming - Theoretical neuroscientist.
AI might be a powerful technology, but things won’t get better simply by adding AI.
4.1. Data imputation
As mentioned above, it is not uncommon to experience the nuisance of dealing with incomplete data. The reasons of these missing values are multifold, including: unavailability, measurement failures, and integration of databases with different schemas. According to the probability of data to be missing, Little and Rubin [342] have defined three classes: missing completely at random (MCAR), missing at random (MAR), and not missing at random (NMAR). The first class, MCAR, describes the case in which all the measures have the same probability to be missed. This is likely, for example, in microarray data where reading failures can happen everywhere. If the probability of missing data is evenly distributed only within a group, then the data are MAR. The last class covers the cases in which neither MCAR nor MAR apply.
In any case, before feeding ML or AI algorithms, the question about which strategy is more suitable for dealing with missing data needs to be answered. As explained by Cismondi et al. [343], there are two main alternatives: 1) remove either variables or samples with missing values, 2) impute missing data. The first is widely discouraged since it begets biases [344]. The second approach, i.e., data imputation, consists in filling the gaps of the data matrix by forecasting the most appropriate value for each missing measurement. Several strategies have been proposed in over thirty years of research on this topic, as witnessed by the wide literature [345], [346].
A common factor of most proposed approaches is that the concept of “appropriate value” coincides with accurately approximate the missing value. This concept has experimentally been challenged by Desouto et al. [347], who show that clustering and classification tasks do not benefit from complex imputation strategies. Conversely, simple methods, such as replacing missing values with average values, perform similarly well. The authors move the attention to the ability of imputation methods to preserve significance. This point of view implies that data imputation cannot be used as a black-box, but the method should be chosen in accordance with the specific task. In his seminal book [348], Stef Van Buuren provides an overview of imputation techniques by subdividing them in three classes: statistical imputation, that leverages on univariate (e.g., the mean) or multivariate (e.g., the k-NN [349]) statistics; multiple imputation, that creates complete datasets and then merges them by minimizing a given objective function; model based imputation, that utilizes ML approaches (e.g., clustering [350]). Due to their simplicity and broad applicability, methods of the first class are more common. However, in the absence of guidelines, the data analysts must rely only on their experience to choose the more appropriate imputation technique. To address this issue, in [351], the authors provide a broad experimentation of 6 univariate and multivariate statistical methods applied to different types of missing data (i.e., MAR, NMAR, MCAR). Their experiments revealed that, in general, multivariate methods are more accurate in predicting missing values.
A linked problem to the above is that of missing columns in multi-omics data integration. Here, we deal with multiple data tables, each describing a disjoint set of features of the same cohort of samples. To integrate this data, some algorithms, such as the similarity network fusion [352], require an exact correspondence between cohorts of samples. Consequently, the presence of just one missing column in the table is enough to compromise applicability of the method. Imputing an entire column is much more challenging than imputing spotted values, and statistical methods might not be adequate to the task. To face this problem, some specialized methods have been proposed. Voillet et al. [353] propose an approach where multiple imputation is mixed with PCA. In short, they generate a set of plausible imputations by “borrowing” missing columns from other samples, then they perform separate PCA analysis and choose the option that best fits the consensus PCA. Other alternatives are mainly based on PCA [354], [355].
Garry Kasparov - Chess Grand-master.
Deep Blue was intelligent the way your programmable alarm clock is intelligent. Not that losing to a 10 million alarm clock made me feel any better.
4.2. Data denoising
Omics data often consist of quantification of the abundance of certain features in a cell or in a bulk of cells. Abundance values are subject to several sources of bias, including both biological variability and technical artefacts. Moreover, measured features typically far outnumber available samples. In some cases, technology advances and availability of technical replicates can help to estimate and correct measurement errors, but cannot help against biological variability [356], which has generally a larger impact [357].
As the old adage in computer science “garbage in, garbage out” reminds, clean data is an essential prerequisite for ML and AI programs in general, which rely on this data to learn. Data denoising is the task of both correcting artefacts (i.e., data normalization) and removing incorrect or irrelevant measures (i.e., feature selection).
There are two classic approaches to denoising [358], differing in the use or lack of a model (which is often empirical). An example of the first class is the normalization of RNA-seq data [359], where the expression profile modelled as a negative binomial distribution. As for the second class, most denoising methods not using a model are based on setting thresholds [360]. Rather than apply value correction, these latter methods filter out weak signals and not significant features. Both approaches have their pros and cons. Model-based denoising can correct artefacts, therefore allowing heterogeneous datasets to be merged. This happens, for example, when normalizing gene expression data obtained by microarray technology [361]. The problem in this case is that both applicability and effectiveness of the method depend on the degree of adherence of the model to the given data set. Model-free approaches, on the other hand, are always applicable. However, thresholds are scale-dependent and an inadequate choice can cause the removal of relevant features which accidentally fails to meet the cut-off value. Multi-scale filters, such as that proposed by Nounou et al. [362], can mitigate this problem.
A recent approach to data denoising uses DL. The idea, exposed by Eraslan et al. [15], is to embed the computation of features into the ML model. Denoising autoencoders [363] (and in particular those based on deep networks [364]) are one of the most common solutions along this line. Briefly, these networks differ from the standard autoencoders in that they are trained to reconstruct the input from a corrupted version of it. Autoencoders (either standard or denoising) have the additional advantage of being modular, which allows them to be combined with other techniques. For example, denoising autoencoders can be stacked to build more complex networks [29] or combined with a Bayesian model to denoise single-cell sequencing data [365].
Gray Scott - Futurist and philosopher.
The real question is, when will we draft an artificial intelligence bill of rights? What will that consist of? And who will get to decide that?
4.3. Data integration
Using a metaphor, we could think of the different lines of omics data within a cell (genomics, proteomics, transcriptomics, etc.) as elements in a symphony orchestra, where the displayed cell phenotype is the result of the synchronous playing of all of the elements. Listening to a single element (i.e., analysing a single omics data track) can provide an idea of the melody, but most nuances would be missed. The rationale behind data integration in functional genomics is that only the comprehensive analysis of all omics events and their interactions can provide a complete view of the cell state. A well-known example supporting this view is provided by the relationship between DNA methylation and gene expression [366], [367].
In general, there can be three types of data integration [368]: horizontal integration, which involves disjoint data sets of the same omics type; vertical integration, which combines different types of omics data in the same cohort of samples; and parallel integration, which mixes the two cases above. The first type of integration is particularly useful to combine data sets coming from different sources, with the goal of reducing the disequilibrium between the number of samples and the number of genomic features. The second type of approach is mostly used to characterize the features that induce an observed phenotype. Finally, the third approach is best suitable in the presence of heterogeneous data.
From a mathematical point of view, the input of integration algorithms consists of a set of matrices (one for each type of omics data) where each row corresponds to a gene and columns are the samples. The goal here is that of producing a new matrix in which multi-level relationships are highlighted. In [369], the authors provide a taxonomy of integration algorithms. A major distinctive feature is whether they return their output in the form of a network or not. In the first case, the output is a square matrix, which can be conveniently interpreted as an adjacency matrix where each row/column corresponds to a gene and the values represent the strength of each relationship. The advantage of these algorithms is that they allow the use of pathways [370] and community detection algorithms [371] for subsequent analyses. The second approach returns a matrix whose rows represent the genomic features. The advantage of this class of algorithms stands in the possibility of using differential analysis or clustering for downstream analyses. On the other hand, these methods can impose restrictions on either the sample space or the feature space. For example, the gene-wise weights schema proposed in [372] (where each gene is assigned a score computed as a linear combination of the corresponding profiles) requires the input matrices to be in the same feature space (i.e., genes). This constraint can be bypassed in certain cases by using gene target prediction methods such as TargetScan [373].
Huang et al. [374] subdivide the integration algorithms according to an orthogonal taxon that discriminates based on whether phenotype labels of samples (e.g., disease or normal) are used or not. The latter category comprises unsupervised matrix factorization methods, such as Bayesian methods and network-based methods. Unsupervised matrix factorization consists in projecting the omics spaces into a lower dimensional space [375], [376]. These methods combine data integration and feature selection, and are particularly useful for clustering applications (see [377] for an extensive review). Bayesian methods [378], [376] leverage on a priori assumptions on data distribution and on the relationships among datasets. Thanks to Bayes’ rule, these methods can easily estimate the posterior probabilities of certain patterns to belong to a specific phenotype. Supervised methods have the same goals as Bayesian methods, namely the identification of complex interactions and/or profiles. However, they make explicit use of labels in the training set to learn the model. This opens to ML and AI approaches. For example, in [379] the authors make use of autoencoders to integrate three omics data types of liver cancer and learn patterns leading to different survival profiles. We expect that supervised integration will experience great advances thanks to ML and AI [380].
Yann LeCun - Computer scientist.
Our intelligence is what makes us human, and AI is an extension of that quality.
5. Explainability and interpretability of AI in functional genomics
Israelsen and Ahmed [381] define trust as the psychological process in which a subject decides to create a relationship of dependence on a trustee after examining its characteristics. This definition does not pose any limit on the nature of the trustee, which can be anything, including software. Besides people’s natural inclination, trust depends on two fundamental aspects: the reputation of the trustee and the cost derived from its failure. Bioinformatics applications are often characterized by high failure costs. For example, genomic research projects leverage on AI for finding loci of interest for a given genotype. Failure of the AI model in this case can cause the failure of the entire project. Not surprisingly, the AI community has started to invest substantial effort to develop techniques to enhance algorithm reliability so as to improve their reputation and, in turn, trust.
Both interpretability and explainability of AI algorithms go in the direction of enforcing trust [382]. Although a common formal definition of these concepts is not established, interpretability concerns how the AI model comes to its conclusions, whereas explainability is focused on why the model arrives to certain conclusions in a given case.
According to these informal definitions [383], interpretability has the ultimate goal of verifying that model accuracy derives from a correct representation of the problem and not from artefacts present in the training data. In fact, an AI system may achieve a high testing accuracy not on the basis of a “real understanding” but because it was able to find unknown hidden relationships among training and testing data. In the absence of such relationships, however, the accuracy of these classifiers can significantly drop, thereby making them unsuitable for real bioinformatics tasks [384].
Explainability is mainly linked to the principle of the right to explanation, namely, the right of an individual to be explained the reasons why an algorithm has taken a decision that affects his/her life. This principle is receiving more and more attention in many legislations. In the United States, for example, it is already recognized at least for certain business sectors (in particular finance) while in the European Union, thanks to the General Data Protection regulation (GDPR), it has been extended to any field.
Besides granting personal rights, explainability can be useful to perform ex-post analyses of the strategies used by an AI system to make certain choices and derive new knowledge from them. A famous example in this direction was the Go match where AlphaGo [385] won against a world pluri-champion using a move that had never been seen before. Ex-post analyses revealed the winning strategy adopted by the network.
Broadly speaking, explainability and interpretability can be achieved by exploiting two major strategies [386]: transparency orpost hocanalysis. The idea of transparency is that the explanation of the model is the model itself. This is the case of simple classifiers such as trees, rule-based classifiers and linear classifiers, which can easily be interpreted directly. In decision trees, the explanation of the reasons that led to a certain decision can easily be obtained by inspecting the path from which the decision was originated. The advantage of models of this class stands in their natural interpretability and explainability. On the other hand, these simple models are not capable of learning non-linear relationships and, consequently, they may not be suitable for complex applications.
The vast majority of AI systems are too complex to be directly understood. In this case they must be treated as black-boxes and explanation must be derived via post hoc analyses. These can be performed based on two main strategies: use of model-agnostic (i.e., methods that leverage only on the input and output) or model-dependent methods. The advantage of the former strategy is that it is always applicable, whereas the latter can take advantage of peculiar characteristics of the AI system of interest. In some cases, the explanation is achieved by deriving a transparent model that mimics the original one. For example, in [387] the authors derived a rule-based classifier from an SVM, while in [388] the authors made something conceptually similar with a neural network. However, this is not generally possible and explanation is often provided in the form of lower dimensional visualization of as a set of examples.
Kathy Baxter - Architect of Ethical AI Practice at Salesforce.
Unfortunately, we have biases that live in our data, and if we don’t acknowledge that and if we don’t take specific actions to address it then we’re just going to continue to perpetuate them or even make them worse.
6. Software and data sharing issues
Readers coming from the computer science field are often not used to sharing software and data. In spite of the fact that communities like Kaggle (www.kaggle.com) are providing the AI community with valuable datasets useful for algorithm design, for many applications the burden of data collection remains under the responsibility of developers. AI applications in bioinformatics are different in this respect, since both algorithm design and production of new knowledge can take advantage of the large amount of publicly available software and data. However, the nature of both this data and that of AI applications in healthcare (which, in the end, is one of the main final goals of all the bioinformatics subfields, including functional genomics), raise issues on the pros and cons of data sharing, as well as on the possible conflicts between economic and ethical aspects.
Amit Ray - Spiritual master and writer.
As more and more artificial intelligence is entering into the world, more and more emotional intelligence must enter into leadership.
6.1. Data sharing and privacy
Straight from the definition of functional genomics, a new paradigm of research that steps away from the inspection of a few target genes and considers the genome as a whole arises. As a direct consequence, we have now the opportunity of “recycling” datasets acquired using omics technologies for new research projects.
Recycling (and sharing) data has both ethical and economic advantages. From the ethical point of view, the circulation of data among laboratories reduces the need for animal experimentation without compromising the battle for health. When experiments are sources of distress or pain, reuse assumes a particular significance. As far as economic advantages are concerned, the cost of storing, protecting and sharing data is much lower than that of producing new data, considering high-throughput technologies cost decrease. It may be claimed that sharing is democratic as well, because it grants data access to small laboratories whose limited budget does not allow them to produce their own data. A further advantage of sharing is that it contributes to the creation of large data collections, which are required to both train AI algorithms and increase their accuracy [389].
In this framework, it is not surprising that stakeholders (starting from funders and publishers) are giving great impulse to data sharing (see Table 3 for a description of the most common data repositories for AI applications). The other side of the coin is that leaks due to malicious or incautious use of genomic data [390] may have severe privacy implications, and even lead to discrimination phenomena [391].
Table 3.
A list of AI-related useful resources publicly available from the word wide web, including some great lectures and courses as well as popular frameworks, cloud services, platforms, software libraries and data repositories.
| Source Type | Source Availability | Description | Hyperlink/Reference |
|---|---|---|---|
| MIT Open Learning Library | 6.036 Introduction to Machine Learning. | shorturl.at/aeCDL | |
| MIT Open Learning Library | 6.S191 Introduction to Deep Learning. | shorturl.at/sJWY3 | |
| UC Berkeley | CS182 Designing, Visualizing and Understanding Deep Neural Networks | shorturl.at/iCHJ3 | |
| Teaching Aid | The Royal Institution | Artificial Intelligence, the History and Future by Chris Bishop. | shorturl.at/mvOUZ |
| MIT Deep Learning Series | Deep Learning State of the Art 2020 by Lex Fridman | shorturl.at/jC246 | |
| A.I. Wiki | A Beginner’s Guide to Important Topics in AI, Machine Learning, and Deep Learning. | shorturl.at/hpuwH | |
| NVIDIA Developer Blog | Deep Learning in a Nutshell: History and Training. | shorturl.at/uDJMT | |
| Udacity AI | The school of Artificial Intelligence | udacity.com/school-of-ai | |
| DataCamp ML | Machine Learning Courses | shorturl.at/isSX0 | |
| Online Courses | Coursera | Artificial Intelligence Certifications | shorturl.at/ejzNV |
| Udemy | Artificial Intelligence Courses | shorturl.at/vV046 | |
| Google Developers | Machine Learning Crash Course | shorturl.at/bkrIV | |
| Google AI | Learn with Google AI | ai.google/education | |
| Microsoft Azure | Cloud computing platform created by Microsoft. | azure.microsoft.com [401] | |
| Cloud Computing Services | Amazon Web Service | On-demand cloud computing web services providing a variety of basic abstract technical infrastructure and distributed computing building blocks and tools. | aws.amazon.com [402] |
| Google Cloud Platform | Cloud computing services running on the same infrastructure used by Google | cloud.google.com [403] | |
| PyTorch | open-source machine learning library based on the Torch library | pytorch.org [404] | |
| Keras | Open-source software library with a Python interface for artificial neural networks. | keras.io [405] | |
| Scikit-learn | open-source machine learning library for Python, built upon the SciPy library (Scientific Python). | scikit-learn.org | |
| Neural Network Frameworks | Tensor Flow | Free and open-source software library for machine learning with a particular focus on training and inference of deep neural networks. | tensorflow.org [399], [406] |
| fast.ai | open-source deep learning library providing high-level components to quickly and easily provide state-of-the-art results. Based on Pytorch, provides online courses, documentation and community. | fast.ai | |
| Google Colab | Infrastructure optimized for ML/DL model implementation/ running; TensorFlow engine; Jupyter Notebooks environment; google account requested. | colab.research.google.com | |
| Tensor Flow Hub | Repository of hundreds well documented ML models from different domains, available as Jupyter notebooks, linked to and ready to be ran in Google Colab. | tfhub.dev | |
| Kaggle | Repository of community published data and code; Jupyter Notebooks environment. | kaggle.com | |
| Repositories | Scientific Data Collection | Multi-Omics Data Sharing: a compendium of multi-omics datasets ready for reuse. | shorturl.at/knLRZ |
| Open AI Microscope | Collection of visualizations of every significant layer and neuron of eight important and largely used vision models. | microscope.openai.com | |
| ImageNet | Image database for AI applications. | image-net.org | |
| Lucid | Collection of infrastructures and tools for research in neural network interpretability. | github.com/tensorflow/lucid | |
| Papers With Code | A free and open resource of ML papers, including code and evaluation tables, organized by topics. | paperswithcode.com | |
To preserve privacy, large collaborative sharing projects have invested in security by applying sophisticated anonymization algorithms and defining strict data access protocols. Public data are only available in the form of quantification of genomic features, while raw reads are restricted to qualified institutions. All these precautions, however, are not necessarily able to meet patients security and expectations in terms of privacy [392]. Anonymity, for example, is challenged by linking strategies [390], which make re-identification progressively easier with the increase of dataset dimensionality. As discussed in [393], and experimentally shown in [394], under certain conditions, re-identification can successfully disclose the majority of identities in large datasets. On the side of user rights, the basic principle of withdrawal, which grants the right to discontinue the participation into a research study, with consequent deletion of all personal data (both in raw and aggregated form), can be denied in large-scale international sharing projects due to the impracticability of keeping track of data. A thornier problem is the potential privacy violation that can derive from the advances of AI algorithms. Further progress may enable AI methods to derive new and more refined genotyping information, which had not been taken into account at the time the informed consent was signed [395].
At first sight, it would seem that we are called to decide whether to sacrifice privacy on the altar of AI driven healthcare or vice versa. In reality, there is a huge ongoing effort to find solutions that balance both needs [396]. De-identification can be enforced by selective data suppression. The k-anonymity [397], for example, is a method to selectively remove or generalize data until no attribute combination is shared by less than k records. Differential privacy [398] introduces controlled noise in the data to maximize the probability for the output of a given query of a database of n records, to be similar to that of the database with records. Due to the mathematical guarantees offered by this method, specialized AI algorithms have been developed. For example, Abadi et al. [399] introduce a framework for DL with differential privacy. Other privacy preserving solutions include learning from encrypted data or using generative neural network models to simulate realistic data [400].
Tristan Harris - Co-founder and CEO of Apture.
Humane technology starts with an honest appraisal of human nature. We need to do the uncomfortable thing of looking more closely at ourselves.
6.2. Open-source software: Liability and reliability
As for data, sharing software has fostered the development of functional genomics. Integrated software suites and standardized file formats have made the creation of analysis pipelines less stressful, while web applications have granted access to large-scale experimentation to laboratories with limited computational resources as well. Indeed, software sharing has also positive economic effects.
Nowadays, several research projects rely on the abundance of publicly available tools for large-scale screening, with the aim of narrowing down expensive and time-consuming in vitro experiments only to the most promising genomic features. In these cases, however, the project success strictly depends on available software reliability.
Most bioinformatics software is released under the GPL (General Public Licence) or the MIT licence, both of which offer the great opportunity to reuse code, thereby supporting fast development of new tools. At the same time, both have a disclaimer that heavily limits warranty and liability5, thus leaving all the responsibility of software failures to the end user. Although acceptable in general, this absence of guarantees calls for caution, for example in mission critical applications where programming protocols such as the defensive programming [407] should be used to enhance software quality.
The above observations raise the natural question about whether we can trust bioinformatics tools or not. As discussed by Lawlor and Walsh [408], several critical issues need to be addressed to improve the reliability of bioinformatics software. However, a careful choice allows us to give a positive answer to this question. For example, in [409] the authors make an ex-post assessment of three common alignment tools (bwa, bowtie and bowtie2). In the same work, the authors face the problem of the assessment in the absence of a golden standard providing useful suggestions for developers.
In general, the bioinformatics community is becoming aware of the need for reliable tools, as witnessed by both the request of publishers for accurate tests before publishing software and the number of proposals of best practices that are being discussed [410], [411]. The adoption of these practices is expected to become popular in the near future, with a consequent increase of tools reliability. In a recent work, Mahmud and co-workers [32] discuss the exploitation of a set of open-source DL tools and open access data, and compare these tools from qualitative, quantitative, and benchmarking points of view. Table 3 summarizes some useful sources of general-purpose software, cloud computing services and popular frameworks to support AI applications; such frameworks are appreciated also in the bioinformatics community.
Stephen Hawkins - Theoretical physicist and cosmologist.
Success in creating AI would be the biggest event in human history. Unfortunately, it might also be the last, unless we learn how to avoid the risks.
7. Legal, ethical and economic issues
AI applications in functional genomics, and in healthcare in particular, are often devoted to identify specific patterns that are subsequently used by decision makers for diagnosis and therapy. Classifiers, however, are not infallible and can commit two types of errors: assigning non-member elements to a class (false positives) or failing to recognize that some elements belong to a class (false negatives). In case the class is “diseased”, false positives may subject people to unnecessary distress, as well as increase costs, by promoting unnecessary and often expensive screenings [412]; on the other hand, false negatives would result in delays before the correct diagnosis is formulated, and have a dramatic impact in pathologies like cancer, where timing of diagnosis strongly affects the long term outcome of therapies. Unsurprisingly, misclassification is the major source of economic and legal issues. Due to the different scenarios caused by the two types of errors, some authors have proposed a cost-based assessment of classification performance [413]. However, the application of methods of this type is complicated. In fact, although the cost of false positives can be reasonably estimated based on screening costs, quantifying false negatives is definitely more problematic [414] since it would imply assigning a cost value to human life.
While pursuing error free methods, two strategies can be adopted to contrast misclassification: 1) adopt guidelines that involve the re-examination by human experts for the most complex cases; and 2) develop ad hoc AI methods trained to recognize classification errors. As an example of the latter case, Aboutalib et al. [415] proposed a deep network designed to reanalyse samples that were classified as positive with another method. The goal in this case was that of finding false positives.
Another important issue concerning the use of AI in either functional genomics or precision medicine is that of data fairness. Newspapers are plenty of stories telling about discriminatory behaviour of AI systems. This behaviour usually comes from the fact that algorithms are trained on biased or unbalanced data. Learning from population biased data, for example, could lead to identify ancestry-specific variants and biomarkers. If this can be useful to identify rare variants (see for example [416], where a population of Sardinian subjects was used to find new loci associated with the levels of blood lipids and inflammatory markers), it also involves the potential risk of increasing disparity among underrepresented and overrepresented populations [417]. In order to mitigate this risk, local initiatives of large-scale data collection (see for example the Japanese version of The Cancer Genome Atlas [418]) should be internationally supported and integrated with existing data banks.
Alan Kay - Computer scientist.
Some people worry that artificial intelligence will make us feel inferior, but then, anybody in his right mind should have an inferiority complex every time he looks at a flower.
8. Conclusion
The outbreak of AI has affected virtually all the fields of research, especially those dealing with big data, such as functional genomics. Among the different branches of functional genomics, next- and third-generation sequencing technologies have produced a vast amount of data in the last years. Uncovering the relationships between variants and diseases, epigenetic mutations and gene expression, binding site positions and regulatory processes has become more and more attractive, largely because of the development and availability of AI instruments. In particular, deep architectures can reach high levels of abstraction and ability to hierarchically organize large amounts of data of different nature but highly interconnected, making them more interpretable.
On the cons side, deep AI architectures make us unable to explain how correct results on critical tasks were eventually achieved. The relevance of identifying a cancer-related gene expression profile is undoubted, although obscurity of the underlying feature selection process may undermine the practical value of the finding. Lack of explainability of some DL paths also makes it difficult to choose the best architecture to use for a given task, which is why the sharing and free availability of software, resources and databases are crucial for the rise of AI applications in functional genomics.
In view of the crucial applications often addressed by biology and, in particular, by functional genomics, it would be preferable to deal with AI tools able to help a mechanistic understanding of biological processes. In other words, it is important to enable systems biology to draw advantages from AI results in functional genomics. This entails the ability to help validating, or even building, theoretical models aimed at having a predictive value on the static and/or dynamic behavior of biological systems of various complexities.
Interpretability, in the sense described above, can surely help AI to be more easily accepted in practical applications such as medicine. In our opinion, increase in the amount and variety of reliable massive data, and its integration with theoretical modelling will contribute to increase the trust of humans in AI-based predictions and decisions in the future. As far as the future of AI in systems biology is concerned, two different scenarios have been proposed, encompassing either competition or collaboration between data- and model-driven approaches. We believe that collaboration and integration between the two approaches would be most helpful to reach an understanding of system relationships and mechanisms. In fact, on the one hand, model-based approaches can provide knowledge-based constraints; on the other hand, AI results can help to establish parameters of systems biology models.
One of the most impressive achievements of AI methods in functional genomics so far is undoubtedly the revolution introduced by the DeepMind AlphaFold method in the field of protein structure prediction, to the point that this went from being one of the biggest challenge in biology to be considered as a solved problem [419]. The ground-breaking success of the AlphaFold software largely rely on transformers, which outperform state-of-the-arts deep architectures in sequence representations and context interpretation. In fact, their introduction has stimulated many applications in the field of biology, since unsupervised models based on the attention mechanism, pretrained on large datasets, performs effectively in homology detection, residue-residue interaction representation, secondary structure prediction and generative biology [156], [157], in addition to protein tertiary structure predictions [315].
Functional genomics, as well as all the fields of medicine, biology and other sciences where both individual and collective rights are involved, is a complex field of research. Those who want to use AI in this field will find that it is difficult to navigate, not only for the great variety and large amount of available data, not just to understand what question to ask and which instrument to use, but also because this field is highly sensitive to legal, ethical and moral aspects. This review is intended to help those willing to approach the application of AI methods to functional genomics to identify the multiple facets of the topic and find some useful tools for orientation. AI opens up many possibilities, which we must not refuse for fear of not understanding all the steps. The road that follows the development of AI is just beginning to unfold. It brings many promises and many potential dangers with it. The path is likely to be long and irreversible. It will change our lives and we need to change our minds as soon as possible in order to adapt, accept and manage the resulting changes in the best possible way, to ensure that they bring as many benefits as possible and as few negative consequences as possible.

Funding
This work has been supported by the Italian Ministry of Education, University and Research (MIUR), with the following grants: (1) Progetto Bandiera e di interesse – Collezione Nazionale di Composti Chimici e Centro Screening (CNCCS), FOE CNR 2020 to Veronica Morea and Andrea Ilari, and (2) Research Projects of National Relevance (PRIN 2017, Grant No.: 2017483NH8_005) to Veronica Morea.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This research has been conceived and developed within the activities of the working group ‘AI for Functional Genomics’ (AI4FG), which was launched in the framework of the CNR Observatory on Artificial Intelligence, an interdepartmental initiative established by the National Research Council of Italy in 2019.
Footnotes
shorturl.at/rAJT5.
The MIT licence states: “THE SOFTWARE IS PROVIDED “AS IS”, WITHOUT WARRANTY OF ANY KIND”
Contributor Information
Claudia Caudai, Email: claudia.caudai@cnr.it.
Teresa Colombo, Email: teresa.colombo@cnr.it.
References
- 1.McKusick V.A., Ruddle F.H. Editorial: A new discipline, a new name, a new journal. Genomics. 1987;1:1–2. [Google Scholar]
- 2.McCarthy, J., Minsky, M., Rochester, N. & Shannon, C.E.A proposal for the Dartmouth summer research project on artificial intelligence, August 31, 1955. AI Magazine 27, 12–14 (2006).
- 3.Stormo G.D., Schneider T.D., Gold L., Ehrenfeucht A. Use of the ‘perceptron’ algorithm to distinguish translational initiation sites in e. coli. Nucleic Acids Research. 1982;10:2997–3011. doi: 10.1093/nar/10.9.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Angermueller C., Pärnamaa T., Parts L., Stegle O. Deep learning for computational biology. Molecular Systems Biology. 2016;12 doi: 10.15252/msb.20156651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Ridder D., de Ridder J., Reinders M.J.T. Pattern recognition in bioinformatics. Briefings in Bioinformatics. 2013;14:633–647. doi: 10.1093/bib/bbt020. [DOI] [PubMed] [Google Scholar]
- 6.Camacho D.M., Collins K.M., Powers R.K., Costello J.C., Collins J.J. Next-generation machine learning for biological networks. Cell. 2018;173:1581–1592. doi: 10.1016/j.cell.2018.05.015. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Z. Deep learning in omics: a survey and guideline. Briefings in Functional Genomics. 2019;18:41–57. doi: 10.1093/bfgp/ely030. [DOI] [PubMed] [Google Scholar]
- 8.Park Y., Kellis M. Deep learning for regulatory genomics. Nature Biotechnology. 2015;33:825–826. doi: 10.1038/nbt.3313. [DOI] [PubMed] [Google Scholar]
- 9.Ragoza M., Hochuli J., Idrobo E., Sunseri J., Koes D.R. Protein-ligand scoring with convolutional neural networks. Journal of chemical information and modeling. 2017;57:942–957. doi: 10.1021/acs.jcim.6b00740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zampieri G., Vijayakumar S., Yaneske E., Angione C. Machine and deep learning meet genome-scale metabolic modeling. PLOS Computational Biology. 2019;15 doi: 10.1371/journal.pcbi.1007084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grapov D., Fahrmann J., Wanichthanarak K., Khoomrung S. Rise of deep learning for genomic, proteomic, and metabolomic data integration in precision medicine. OMICS. 2018;22:630–636. doi: 10.1089/omi.2018.0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenblatt F. The perceptron: A probabilistic model for information storage and organization in the brain. Psychological Review. 1958;65:386–408. doi: 10.1037/h0042519. [DOI] [PubMed] [Google Scholar]
- 13.Dettmers, T. Deep learning in a nutshell: History and training. https://devblogs.nvidia.com/parallelforall/deeplearning-nutshell-history-training/ (2015).
- 14.Zou J. A primer on deep learning in genomics. Nature Genetics. 2018;51:12–18. doi: 10.1038/s41588-018-0295-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eraslan G., Avsec Ž., Gagneur J., Theis F.J. Deep learning: new computational modelling techniques for genomics. Nature Reviews Genetics. 2019;20:389–403. doi: 10.1038/s41576-019-0122-6. [DOI] [PubMed] [Google Scholar]
- 16.Esteva A. A guide to deep learning in healthcare. Nature Medicine. 2019;25:24–29. doi: 10.1038/s41591-018-0316-z. [DOI] [PubMed] [Google Scholar]
- 17.LeCun Y., Bengio Y., Hinton G. Deep learning. Nature. 2015;521:436–444. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- 18.Marx V. The big challenges of big data. Nature. 2013;498:255–260. doi: 10.1038/498255a. [DOI] [PubMed] [Google Scholar]
- 19.Kramer M.A. Nonlinear principal component analysis using autoassociative neural networks. AIChE Journal. 1991;37:233–243. [Google Scholar]
- 20.Weiss R. Bayesian methods for data analysis. American journal of ophthalmology. 2010;149(2):187–188.e1. doi: 10.1016/j.ajo.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cortes C., Vapnik V.N. Support-vector networks. CiteSeerX. 1995;20:273–297. [Google Scholar]
- 22.Rokach L., Maimon O. Series in Machine Perception and Artificial Intelligence. 2014. Data mining with decision trees - theory and applications. (2nd Ed.) [Google Scholar]
- 23.Schmidhuber J. Deep learning in neural networks: An overview. Neural Networks. 2015;61:85–117. doi: 10.1016/j.neunet.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Hinton, G.E., Osindero, S. & W., T.Y.A fast learning algorithm for deep belief nets. Neural Computtion 18, 1527–1554 (2006). [DOI] [PubMed]
- 25.Williams R.J., Zipser D. A learning algorithm for continually running fully recurrent neural networks. Neural Computation. 1989;1:270–280. [Google Scholar]
- 26.Salakhutdinov R., Hinton G.E. Deep Boltzmann machines. Proc. Int. Conf. Artif. Intell. Stat. 2009;1 [Google Scholar]
- 27.LeCun Y., Buttou L., Bengio Y., Haffner P. Gradient-based learning applied to document recognition. Proceedings IEEE. 1998;86:2278–2324. [Google Scholar]
- 28.Hinton G.E., Salakhutdinov R.R. Reducing the dimensionality of data with neural networks. Science. 2006;313:504–507. doi: 10.1126/science.1127647. [DOI] [PubMed] [Google Scholar]
- 29.Vincent P., Larochelle H., Lajoie I., Bengio Y., Manzagol P.-A. Stacked denoising autoencoders: Learning useful representations in a deep network with a local denoising criterion. Journal of Machine Learning Research. 2010;11:3371–3408. [Google Scholar]
- 30.Goodfellow I. In: Ghahramani Z., Welling M., Cortes C., Lawrence N., Weinberger K.Q., editors. vol. 27. (Curran Associates Inc; 2014. Generative adversarial nets. (Advances in Neural Information Processing Systems). [Google Scholar]
- 31.Li Y. Deep learning in bioinformatics: Introduction, application, and perspective in the big data era. Methods (San Diego, Calif.) 2019;166:4–21. doi: 10.1016/j.ymeth.2019.04.008. [DOI] [PubMed] [Google Scholar]
- 32.Mahmud M., Kaiser M.S., McGinnity T.M., Hussain A. Deep learning in mining biological data. Cognitive computation. 2021;1–33 doi: 10.1007/s12559-020-09773-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Consortium, I.H.G.S. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004). [DOI] [PubMed]
- 34.Goffeau A. Life with 6000 Genes. Science. 1996;274:546–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 35.Hieter P., Boguski M. Functional genomics: it’s all how you read it. Science. 1997;278(5338):601–602. doi: 10.1126/science.278.5338.601. [DOI] [PubMed] [Google Scholar]
- 36.Ravì D. Deep learning for health informatics. IEEE Journal of Biomedical and Health Informatics. 2016;21:4–21. doi: 10.1109/JBHI.2016.2636665. [DOI] [PubMed] [Google Scholar]
- 37.Ching T. Opportunities and obstacles for deep learning in biology and medicine. Journal of the Royal Society Interface. 2018;15 doi: 10.1098/rsif.2017.0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao C. Deep learning and its applications in biomedicine. Genomics, Proteomics & Bioinformatics. 2018;16:17–32. doi: 10.1016/j.gpb.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yue, T. & Wang, H. Deep learning for genomics: A concise overview. https://arxiv.org/abs/1802.00810 (2018).
- 40.Wainberg M., Merico D., Delong A., Frey B.J. Deep learning in biomedicine. Nature Biotechnology. 2018;36:829–838. doi: 10.1038/nbt.4233. [DOI] [PubMed] [Google Scholar]
- 41.Min S., Lee B., Yoon S. Deep learning in bioinformatics. Briefings in Bioinformatics. 2016;18:851–869. doi: 10.1093/bib/bbw068. [DOI] [PubMed] [Google Scholar]
- 42.Miotto R., Wang F., Wang S., Jiang X., Dudley J.T. Deep learning for healthcare: review, opportunities and challenges. Briefings in Bioinformatics. 2018;19:1236–1246. doi: 10.1093/bib/bbx044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rost B., Sander C. Secondary structure prediction of all-helical proteins in two states. Protein Eng. 1993;6:831–836. doi: 10.1093/protein/6.8.831. [DOI] [PubMed] [Google Scholar]
- 44.Eickholt J., Cheng J. Predicting protein residue-residue contacts using deep networks and boosting. Bioinformatics. 2012;28(23):3066–3072. doi: 10.1093/bioinformatics/bts598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Asgari E., Mofrad M., Kobeissy F. Continuous distributed representation of biological sequences for deep proteomics and genomics. PLoS ONE. 2015;10 doi: 10.1371/journal.pone.0141287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spencer M., Eickholt J., Cheng J. A deep learning network approach to ab initio protein secondary structure prediction. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2015;12:103–112. doi: 10.1109/TCBB.2014.2343960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alipanahi B., Delong A., Weirauch M.T., Frey B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nature Biotechnology. 2015;33:831–838. doi: 10.1038/nbt.3300. [DOI] [PubMed] [Google Scholar]
- 48.Zhou J., Troyanskaya O.G. Predicting effects of noncoding variants with deep learning–based sequence model. Nature Methods. 2015;12:931–934. doi: 10.1038/nmeth.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pan X. Multiple linear regression for protein secondary structure prediction. Proteins. 2001;43(3):256–259. doi: 10.1002/prot.1036. [DOI] [PubMed] [Google Scholar]
- 50.Wagner M., Adamczak R., Porollo A.A., Meller J. Linear regression models for solvent accessibility prediction in proteins. Journal of computational biology: a journal of computational molecular cell biology. 2005;12(3):355–369. doi: 10.1089/cmb.2005.12.355. [DOI] [PubMed] [Google Scholar]
- 51.Anderson C.A., McRae A.F., Visscher P.M. A simple linear regression method for quantitative trait loci linkage analysis with censored observations. Genetics. 2006;173(3):1735–1745. doi: 10.1534/genetics.106.055921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu H., Yang L., Freitas M.A. A robust linear regression based algorithm for automated evaluation of peptide identifications from shotgun proteomics by use of reversed-phase liquid chromatography retention time. BMC Bioinformatics. 2008;9:347. doi: 10.1186/1471-2105-9-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ogutu J.O., Schulz-Streeck T., Piepho H.-P. Genomic selection using regularized linear regression models: ridge regression, lasso, elastic net and their extensions. BMC Proceedings. 2012;6:S10. doi: 10.1186/1753-6561-6-S2-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xi B., Gu H., Baniasadi H., Raftery D. Statistical analysis and modeling of mass spectrometry-based metabolomics data. Methods in Molecular Biology. 2014;333–353 doi: 10.1007/978-1-4939-1258-2_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim H., Park H. Protein secondary structure prediction based on an improved support vector machines approach. Protein Eng. 2003;16:553–560. doi: 10.1093/protein/gzg072. [DOI] [PubMed] [Google Scholar]
- 56.Liao C., Li S. ISBRA. 2007. A support vector machine ensemble for cancer classification using gene expression data. [Google Scholar]
- 57.Wu S., Zhang Y. Anglor: A composite machine-learning algorithm for protein backbone torsion angle prediction. PLOS ONE. 2008 doi: 10.1371/journal.pone.0003400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu S., Szilagyi A., Zhang Y. Improving protein structure prediction using multiple sequence-based contact predictions. Structure. 2011;19:1182–1191. doi: 10.1016/j.str.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang F., Kaufman H.L., Deng Y., Drabier R. Recursive SVM biomarker selection for early detection of breast cancer in peripheral blood. BMC Medical Genomics. 2013;6:S4. doi: 10.1186/1755-8794-6-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kircher M. A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rokach L., Maimon O. Series in Machine Perception and Artificial Intelligence. 2007. Data mining with decision trees - theory and applications. [Google Scholar]
- 62.Vlahou A., Schorge J.O., Gregory B.W., Coleman R.L. Diagnosis of ovarian cancer using decision tree classification of mass spectral data. J Biomed Biotechnol. 2003;2003:308–314. doi: 10.1155/S1110724303210032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qi Y., Klein-Seetharaman J., Bar-Joseph Z. Pacific Symposium on Biocomputing. Pacific Symposium on Biocomputing. 2004. Random forest similarity for protein-protein interaction prediction from multiple sources; pp. 531–542. [PubMed] [Google Scholar]
- 64.Blockeel, H., Schietgat, L., Struyf, J., Dzeroski, S. & Clare, A. Decision trees for hierarchical multilabel classification: A case study in functional genomics. In PKDD (2006).
- 65.Jiang R., Tang W., Wu X. & Fu, W.A random forest approach to the detection of epistatic interactions in case-control studies. BMC Bioinformatics. 2009;10:S65. doi: 10.1186/1471-2105-10-S1-S65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen X.D., Ishwaran H. Random forests for genomic data analysis. Genomics. 2012;99(6):323–329. doi: 10.1016/j.ygeno.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ernst J., Kellis M. Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nature Biotechnology. 2015;33:364–376. doi: 10.1038/nbt.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sandberg R. Capturing whole-genome characteristics in short sequences using a naïve bayesian classifier. Genome research. 2001;11(8):1404–1409. doi: 10.1101/gr.186401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Degroeve S., De Baets B., Van de Peer Y., Rouzé P. Feature subset selection for splice site prediction. Boinformatics. 2002;18:75–83. doi: 10.1093/bioinformatics/18.suppl_2.s75. [DOI] [PubMed] [Google Scholar]
- 70.Nielsen R., Matz M.V. Statistical approaches for DNA barcoding. Systematic biology. 2006;55(1):162–169. doi: 10.1080/10635150500431239. [DOI] [PubMed] [Google Scholar]
- 71.Silla C.N., Freitas A.A. 2009 Ninth IEEE International Conference on Data Mining. 2009. A global-model naive Bayes approach to the hierarchical prediction of protein functions; pp. 992–997. [Google Scholar]
- 72.Garrow A.G., Agnew A., Westhead D.R. Tmb-hunt: a web server to screen sequence sets for transmembrane beta-barrel proteins. Nucleic Acids Res. 2005;33:W188–W192. doi: 10.1093/nar/gki384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yao Z., Ruzzo W.L. A regression-based k nearest neighbor algorithm for gene function prediction from heterogeneous data. BMC Bioinformatics. 2006;7:S11. doi: 10.1186/1471-2105-7-S1-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Parry R.M. k-nearest neighbor models for microarray gene expression analysis and clinical outcome prediction. The Pharmacogenomics Journal. 2010;10:292–309. doi: 10.1038/tpj.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin Z., Altman R.B. Finding haplotype tagging snps by use of principal components analysis. American journal of human genetics. 2004;75(5):850–861. doi: 10.1086/425587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alexe G., Dalgin G.S., Ganesan S., DeLisi C., Bhanot G. Analysis of breast cancer progression using principal component analysis and clustering. Journal of Biosciences. 2007;32:1027–1039. doi: 10.1007/s12038-007-0102-4. [DOI] [PubMed] [Google Scholar]
- 77.Maisuradze G.G., Liwo A., Scheraga H.A. Principal component analysis for protein folding dynamics. Journal of molecular biology. 2009;385(1):312–329. doi: 10.1016/j.jmb.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.h. Taguchi, Y. & Okamoto, A. Principal component analysis for bacterial proteomic analysis. 2011 IEEE International Conference on Bioinformatics and Biomedicine Workshops (BIBMW) 961–963 (2011).
- 79.Worley B., Powers R. Multivariate analysis in metabolomics. Current. Metabolomics. 2013;1:92–107. doi: 10.2174/2213235X11301010092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hsu Y.-L., Huang P.-Y., Chen D.-T. Sparse principal component analysis in cancer research. Translational cancer research. 2014;3(3):182–190. doi: 10.3978/j.issn.2218-676X.2014.05.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Day N., Hemmaplardh A., Thurman R.E., Stamatoyannopoulos J.A., Noble W.S. Unsupervised segmentation of continuous genomic data. Boinformatics. 2007;23:1424–1426. doi: 10.1093/bioinformatics/btm096. [DOI] [PubMed] [Google Scholar]
- 82.ChenXiaoyu, HoffmanMichael, M., BilmesJeff, A., HesselberthJay, R. & NobleWilliam, S.A dynamic bayesian network for identifying protein-binding footprints from single molecule-based sequencing data. Bioinformatics (2010). [DOI] [PMC free article] [PubMed]
- 83.Hoffman M.M., Buske O.J., Wang J., Weng Z., Bilmes J.A. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nature Methods. 2012;9:473–476. doi: 10.1038/nmeth.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gusmao E.G., Dieterich C., Zenke M., Costa I.G. Detection of active transcription factor binding sites with the combination of dnase hypersensitivity and histone modifications. Bioinformatics. 2014;30:3143–3151. doi: 10.1093/bioinformatics/btu519. [DOI] [PubMed] [Google Scholar]
- 85.Zhang Y., An L., Yue F., Hardison R.C. Jointly characterizing epigenetic dynamics across multiple human cell types. Nucleic Acids Research. 2016;44:6721–6731. doi: 10.1093/nar/gkw278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang M.Q. Discriminant analysis and its application in dna sequence motif recognition. Briefings in bioinformatics. 2000;1(4):331–342. doi: 10.1093/bib/1.4.331. [DOI] [PubMed] [Google Scholar]
- 87.Yuan, Y., Liang, Y., Yi, L., Xu, Q. & Kvalheim, O.M. Uncorrelated linear discriminant analysis (ULDA): A powerful tool for exploration of metabolomics data. Chemometrics & Intelligent Laboratory Systems 93, 70–79 (208).
- 88.Huang D., Quan Y., He M., sen Zhou B. Comparison of linear discriminant analysis methods for the classification of cancer based on gene expression data. Journal of Experimental & Clinical Cancer Research: CR. 2009;28:149. doi: 10.1186/1756-9966-28-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pollard, K.S. & Van Der Laan, M.J. Cluster analysis of genomic data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor, 209–228 (Springer, 2005).
- 90.Heintzman N.D., Stuart R.K., Hon G., Fu Y. & el al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature Genetics. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 91.Handhayani T., Hiryanto L. Intelligent kernel k-means for clustering gene expression. Procedia Computer Science. 2015;59:171–177. [Google Scholar]
- 92.Oyelade J. Clustering algorithms: Their application to gene expression data. Bioinformatics and Biology Insights. 2016;10:237–253. doi: 10.4137/BBI.S38316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Angermueller C., Lee H.J., Reik W., Stegle O. DeepCpG: accurate prediction of single-cell dna methylation states using deep learning. Genome Biology. 2017;18 doi: 10.1186/s13059-017-1189-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sundaram L. Predicting the clinical impact of human mutation with deep neural networks. Nature Genetics. 2018;50:1161–1170. doi: 10.1038/s41588-018-0167-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Date Y., Kikuchi J. Application of a deep neural network to metabolomics studies and its performance in determining important variables. Analytical Chemistry. 2018;90:1805–1810. doi: 10.1021/acs.analchem.7b03795. [DOI] [PubMed] [Google Scholar]
- 96.Zhang, F. et al. Deepfunc: A deep learning framework for accurate prediction of protein functions from protein sequences and interactions. 2019, doi: 10.1002/pmic.201900019. [DOI] [PubMed]
- 97.Wang Z. Optimized multilayer perceptrons for molecular classification and diagnosis using genomic data. Bioinformatics. 2006;22(6):755–761. doi: 10.1093/bioinformatics/btk036. [DOI] [PubMed] [Google Scholar]
- 98.Kushwaha S.K., Shakya M. 2009 International Conference on Advances in Recent Technologies in Communication and Computing. 2009. Multi-layer perceptron architecture for tertiary structure prediction of helical content of proteins from peptide sequences; pp. 465–467. [Google Scholar]
- 99.Mojarad, S.A., Dlay, S.S., lok Woo, W. & Sherbet, G.V. Breast cancer prediction and cross validation using multilayer perceptron neural networks. 2010 7th International Symposium on Communication Systems, Networks & Digital Signal Processing (CSNDSP 2010) 760–764 (2010).
- 100.Oh S.-H. Protein disorder prediction using multilayer perceptrons. International Journal of Contents. 2013;9:11–15. [Google Scholar]
- 101.Kelley D.R., Snoek J., Rinn J.L. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Research. 2016;26:990–999. doi: 10.1101/gr.200535.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cheng S. Mirtdl: A deep learning approach for mirna target prediction. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2016;13:1161–1169. doi: 10.1109/TCBB.2015.2510002. [DOI] [PubMed] [Google Scholar]
- 103.Wang S., Peng J., Ma J., Xu J. Protein secondary structure prediction using deep convolutional neural fields. Scientific Reports. 2016;6 doi: 10.1038/srep18962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Min X., Zeng W., Chen S., Chen N., Jiang R. Predicting enhancers with deep convolutional neural networks. BMC Bioinformatics. 2016;18 doi: 10.1186/s12859-017-1878-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kelley, D.R. et al. Sequential regulatory activity prediction across chromosomes with convolutional neural networks. https://genome.cshlp.org/content/early/2018/03/27/gr.227819.117 (2018). [DOI] [PMC free article] [PubMed]
- 106.Zhou J. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nature Genetics. 2018;50:1171–1179. doi: 10.1038/s41588-018-0160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Quang D., Xie X. Factornet: A deep learning framework for predicting cell type specific transcription factor binding from nucleotide-resolution sequential data. Methods. 2019;166:40–47. doi: 10.1016/j.ymeth.2019.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang Y., Mao H., Yi Z. Protein secondary structure prediction by using deep learning method. Knowledge-Based Systems. 2017;118:115–123. [Google Scholar]
- 109.Liu, X. Deep recurrent neural network for protein function prediction from sequence. https://arxiv.org/abs/1701.08318 (2017).
- 110.Shen Z., Bao W., Huang D.-S. Recurrent neural network for predicting transcription factor binding sites. Scientific Reports. 2018;8 doi: 10.1038/s41598-018-33321-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu Q. Detection of dna base modifications by deep recurrent neural network on oxford nanopore sequencing data. Nature Communications. 2019;10 doi: 10.1038/s41467-019-10168-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hochreiter S., Schmidhuber J. Long short-term memory. Neural Computation. 1997;9:1735–1780. doi: 10.1162/neco.1997.9.8.1735. [DOI] [PubMed] [Google Scholar]
- 113.Sønderby, S.K. & Winther, O. Protein secondary structure prediction with long short term memory networks. https://arxiv.org/abs/1412.7828 (2015).
- 114.Sønderby S.K., Sønderby C.K., Nielsen H.B., Winther O. Convolutional LSTM networks for subcellular localization of proteins. AlCoB. 2015 [Google Scholar]
- 115.Quang D., Xie X. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of dna sequences. Nucleic Acids Research. 2016;44 doi: 10.1093/nar/gkw226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tran N.H., Zhang X., Xin L., Shan B., Li M. Proceedings of the National Academy of Sciences of the United States of America. 2017. De novo peptide sequencing by deep learning. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tavakoli, N. Modeling genome data using bidirectional LSTM. 2019 IEEE 43rd Annual Computer Software and Applications Conference (COMPSAC) 2, 183–188 (2019).
- 118.Lee D., Zhang J., Liu J., Gerstein M.B. Epigenome-based splicing prediction using a recurrent neural network. bioRxiv. 2020 doi: 10.1371/journal.pcbi.1008006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dohkan S., Koike A., Takagi T. Proceedings. Fourth IEEE Symposium on Bioinformatics and Bioengineering. 2004. Prediction of protein-protein interactions using support vector machines; pp. 576–583. [Google Scholar]
- 120.Zou X., Wang G., Yu G. Protein function prediction using deep restricted boltzmann machines. BioMed Research International. 2017;2017 doi: 10.1155/2017/1729301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hess M., Lenz S., Blätte T.J., Bullinger L., Binder H. Partitioned learning of deep Boltzmann machines for SNP data. Bioinformatics. 2017;33:3173–3180. doi: 10.1093/bioinformatics/btx408. [DOI] [PubMed] [Google Scholar]
- 122.Li H., Hou B., Lyu Q., Cheng J. Deep learning methods for protein torsion angle prediction. BMC Bioinformatics. 2017;18 doi: 10.1186/s12859-017-1834-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nivaashini M., Soundariya R. Deep Boltzmann machine based breast cancer risk detection for healthcare systems. Int. J. Pure Appl. Math. 2018;119:581–590. [Google Scholar]
- 124.Ibrahim R., Yousri N.A., Ismail M.A., El-Makky N.M. 36th Annual International Conference of the IEEE Engineering in Medicine and Biology Society. IEEE Engineering in Medicine and Biology Societ, IEEE; Chicago, IL: 2014. Multi-level gene/mirna feature selection using deep belief nets and active learning; pp. 3957–3960. [DOI] [PubMed] [Google Scholar]
- 125.Sun W., Zheng B., Qian W. SPIE Medical Imaging. 2016. Computer aided lung cancer diagnosis with deep learning algorithms. [Google Scholar]
- 126.Abdel-ZaherAhmed M., EldeibAyman M. Breast cancer classification using deep belief networks. Expert Systems With Applications. 2016 [Google Scholar]
- 127.Rachmatia, H., Kusuma, W. & Hasibuan, L. Prediction of maize phenotype based on whole-genome single nucleotide polymorphisms using deep belief networks. In Journal of Physics: Conference Series, vol. 835 - 1, 012003 (IOP Publishing, 2017).
- 128.Bu H., Gan Y., Wang Y., Zhou S., Guan J. A new method for enhancer prediction based on deep belief network. BMC Bioinformatics. 2017;18 doi: 10.1186/s12859-017-1828-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Karabulut E.M., Ibrikci T. Discriminative deep belief networks for microarray based cancer classification. Biomedical Research-tokyo. 2017;28:1016–1024. [Google Scholar]
- 130.Chicco D., Sadowski P., Baldi P. BCB ’14: Proceedings of the 5th ACM Conference on Bioinformatics, Computational Biology, and Health Informatics. 2014. Deep autoencoder neural networks for gene ontology annotation predictions; pp. 533–540. [Google Scholar]
- 131.Tan J. Unsupervised extraction of stable expression signatures from public compendia with an ensemble of neural networks. Cell Systems. 2017;5:63–71. doi: 10.1016/j.cels.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang D., Gu J. Vasc: Dimension reduction and visualization of single-cell rna-seq data by deep variational autoencoder. Genomics, Proteomics & Bioinformatics. 2018;16:320–331. doi: 10.1016/j.gpb.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Heje Grønbech, C. et al. scvae: Variational auto-encoders for single-cell gene expression data. https://www.biorxiv.org/content/10.1101/318295v3 (2019). [DOI] [PubMed]
- 134.Levy J.J. Methylnet: an automated and modular deep learning approach for dna methylation analysis. BMC Bioinformatics. 2020;21 doi: 10.1186/s12859-020-3443-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Killoran, N., Lee, L.J., Delong, A., Duvenaud, D. & Frey, B.J. Generating and designing dna with deep generative models. https://arxiv.org/abs/1712.06148 (2017).
- 136.Ghahramani, A., Watt, F.M. & Luscombe, N.M. Generative adversarial networks simulate gene expression and predict perturbations in single cells.
- 137.Yang Q. Low-dose ct image denoising using a generative adversarial network with wasserstein distance and perceptual loss. IEEE Transactions on Medical Imaging. 2018;37:1348–1357. doi: 10.1109/TMI.2018.2827462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gupta A., Zou J. Feedback GAN for DNA optimizes protein functions. Nature Machine Intelligence. 2019;1:105–111. [Google Scholar]
- 139.Liu Q., Lv H., Jiang R. hicGAN infers super resolution Hi-C data with generative adversarial networks. Bioinformatics. 2019;35:i99–i107. doi: 10.1093/bioinformatics/btz317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Marouf M.M. Realistic in silico generation and augmentation of single-cell rna-seq data using generative adversarial networks. Nature Communications. 2020;11 doi: 10.1038/s41467-019-14018-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Noguera-Solano R., Ruiz-Gutiérrez R., Rodriguez-Caso J.M. Genome: twisting stories with dna. Endeavour. 2013;37(4):213–219. doi: 10.1016/j.endeavour.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 142.Anderson S. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 143.Fleischmann R. Whole-genome random sequencing and assembly of haemophilus influenzae rd. Science. 1995;269(5223):496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 144.Sanger F., Nicklen S., Coulson A. Dna sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 1977;74(12):5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sanger F. Nucleotide sequence of bacteriophage phi x174 dna. Nature. 1977;265:687–695. doi: 10.1038/265687a0. [DOI] [PubMed] [Google Scholar]
- 146.Heather J., Chain B. The sequence of sequencers: The history of sequencing dna. Genomics. 2016;107:1–8. doi: 10.1016/j.ygeno.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Margulies M. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bentley D.R. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Braslavsky I., Hébert B., Kartalov E., Quake S. Sequence information can be obtained from single dna molecules. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:3960–3964. doi: 10.1073/pnas.0230489100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Haque, F., Li, J., chen Wu, H., Liang, X. & Guo, P. Solid-state and biological nanopore for real-time sensing of single chemical and sequencing of dna. Nano today 8 1, 56–74 (2013). [DOI] [PMC free article] [PubMed]
- 151.Bleidorn C. Third generation sequencing: technology and its potential impact on evolutionary biodiversity research. Systematics and Biodiversity. 2016;14:1–8. [Google Scholar]
- 152.Libbrecht M.W., Stafford Noble W. Machine learning applications in genetics and genomics. Nature Reviews Genetics. 2015;16:321–332. doi: 10.1038/nrg3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bucher P. Weight matrix descriptions of four eukaryotic RNA polymerase II promoter elements derived from 502 unrelated promoter sequences. Journal of Molecular Biology. 1990;212:563–578. doi: 10.1016/0022-2836(90)90223-9. [DOI] [PubMed] [Google Scholar]
- 154.Segal E., Fondufe-Mittendorf Y., Chen L. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Avsec Ž. Base-resolution models of transcription factor binding reveal soft motif syntax. Nature Genetics. 2021;53:354–366. doi: 10.1038/s41588-021-00782-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wu S.T. Deep learning in clinical natural language processing: a methodical review. Journal of the American Medical Informatics Association: JAMIA. 2020 doi: 10.1093/jamia/ocz200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Song B. Pretraining model for biological sequence data. Briefings in Functional Genomics. 2021 doi: 10.1093/bfgp/elab025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mikolov T., Chen K., Corrado G., Dean J. ICLR. 2013. Efficient estimation of word representations in vector space. [Google Scholar]
- 159.Vaswani A. Attention is all you need. ArXivabs/1706.03762. 2017 [Google Scholar]
- 160.Devlin J., Chang M.-W., Lee K., Toutanova K. NAACL. 2019. Pre-training of deep bidirectional transformers for language understanding. [Google Scholar]
- 161.Woloszynek S., Zhao Z., Chen J., Rosen G. 16s rRNA sequence embeddings: Meaningful numeric feature representations of nucleotide sequences that are convenient for downstream analyses. PLoS Computational Biology. 2019;15 doi: 10.1371/journal.pcbi.1006721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ostrovsky-Berman M., Frankel B., Polak P., Yaari G. Immune2vec: Embedding b/t cell receptor sequences in , using natural language processing. Frontiers in immunology. 2021;12 doi: 10.3389/fimmu.2021.680687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Le, N.Q.K., Ho, Q.-T., Nguyen, T.-T.-D. & Ou, Y.-Y.A transformer architecture based on bert and 2d convolutional neural network to identify dna enhancers from sequence information. Briefings in bioinformatics 22, 2021, doi: 10.1093/bib/bbab005. [DOI] [PubMed]
- 164.Lehner B. Genotype to phenotype: lessons from model organisms for human genetics. Nature reviews. Genetics. 2013;14:168–178. doi: 10.1038/nrg3404. [DOI] [PubMed] [Google Scholar]
- 165.Costanzo, M. et al. The genetic landscape of a cell. Science (New York, N.Y.) 327, 425–431, 2010, doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed]
- 166.Costanzo, M. et al. A global genetic interaction network maps a wiring diagram of cellular function. Science (New York, N.Y.) 353, 2016, doi: 10.1126/science.aaf1420. [DOI] [PMC free article] [PubMed]
- 167.Szappanos B. An integrated approach to characterize genetic interaction networks in yeast metabolism. Nat Genet. 2011;43:656–662. doi: 10.1038/ng.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yu M.K. Translation of genotype to phenotype by a hierarchy of cell subsystems. Cell systems. 2016;2:77–88. doi: 10.1016/j.cels.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ma J. Using deep learning to model the hierarchical structure and function of a cell. Nature Methods. 2018;15:290–298. doi: 10.1038/nmeth.4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Abadi S., Yan W.X., Amar D., Mayrose I. A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput Biol. 2017;13 doi: 10.1371/journal.pcbi.1005807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chuai G. Deepcrispr: optimized crispr guide rna design by deep learning. Genome biology. 2018;19:80. doi: 10.1186/s13059-018-1459-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Li V.R., Zhang Z., Troyanskaya O.G. CROTON: an automated and variant-aware deep learning framework for predicting CRISPR/Cas9 editing outcomes. Bioinformatics. 2021;37:i342–i348. doi: 10.1093/bioinformatics/btab268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhang G., Zeng T., Dai Z., Dai X. Prediction of CRISPR/Cas9 single guide RNA cleavage efficiency and specificity by attention-based convolutional neural networks. Comput Struct Biotechnol J. 2021;19:1445–1457. doi: 10.1016/j.csbj.2021.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Norman T.M. Exploring genetic interaction manifolds constructed from rich single-cell phenotypes. Science. 2019;365:786–793. doi: 10.1126/science.aax4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ionita-Laza I., McCallum K., Xu B., Buxbaum J.D. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nature Genetics. 2016;48:214–220. doi: 10.1038/ng.3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Poplin R. A universal SNP and small-indel variant caller using deep neural networks. Nature Biotechnology. 2018;36:983–987. doi: 10.1038/nbt.4235. [DOI] [PubMed] [Google Scholar]
- 177.Berger M.F., Mardis E.R. The emerging clinical relevance of genomics in cancer medicine. Nature Reviews Clinical Oncology. 2018;15:353–365. doi: 10.1038/s41571-018-0002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.NIH . 2020. The Cancer Genome Atlas - Cancer Genome - TCGA. [DOI] [Google Scholar]
- 179.Sánchez-Vega F., Mina M., Armenia J. Oncogenic signaling pathways in the cancer genome atlas. Cell. 2018;173:321–337. doi: 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Swan A.L., Mobasheri A., Allaway D., Liddell S., Bacardit J. Application of machine learning to proteomics data: Classification and biomarker identification in postgenomics biology. OMICS. 2013;17:595–610. doi: 10.1089/omi.2013.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Huang S. Applications of support vector machine (svm) learning in cancer genomics. Cancer Genomics and Proteomics. 2018;15:41–51. doi: 10.21873/cgp.20063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chaudhary K., Poirion O.B., Lu L., Garmire L.X. Deep learning-based multi-omics integration robustly predicts survival in liver cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2018;24:1248–1259. doi: 10.1158/1078-0432.CCR-17-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wang J. Denoising autoencoder, a deep learning algorithm, aids the identification of a novel molecular signature of lung adenocarcinoma. Genomics, Proteomics & Bioinformatics. 2020;18:468–480. doi: 10.1016/j.gpb.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Chai H. Integrating multi-omics data through deep learning for accurate cancer prognosis prediction. Computers in biology and medicine. 2021;134 doi: 10.1016/j.compbiomed.2021.104481. [DOI] [PubMed] [Google Scholar]
- 185.Li H., Giger M.L., Huynh B.Q., Antropova N. Deep learning in breast cancer risk assessment: evaluation of convolutional neural networks on a clinical dataset of full-field digital mammograms. Journal of Medical Imaging. 2017;4 doi: 10.1117/1.JMI.4.4.041304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Doncescu A., Tauzain B., Kabbaj N. 2009 International Conference on Advanced Information Networking and Applications Workshops. 2009. Machine learning applied to BRCA1 hereditary breast cancer data; pp. 942–947. [Google Scholar]
- 187.Bejnordi B.E., Veta M., van Diest P.J. Diagnostic assessment of deep learning algorithms for detection of lymph node metastases in women with breast cancer. JAMA. 2017;318:2199–2210. doi: 10.1001/jama.2017.14585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Haenssle H.A. Man against machine: diagnostic performance of a deep learning convolutional neural network for dermoscopic melanoma recognition in comparison to 58 dermatologists. Annals of Oncology. 2018;29:1836–1842. doi: 10.1093/annonc/mdy166. [DOI] [PubMed] [Google Scholar]
- 189.Abeel T., Helleputte T., de Peer Y.V., Dupont P., Saeys Y. Robust biomarker identification for cancer diagnosis with ensemble feature selection methods. Bioinformatics. 2010;26(3):392–398. doi: 10.1093/bioinformatics/btp630. [DOI] [PubMed] [Google Scholar]
- 190.Chen L., Xuan J., Riggins R.B., Clarke R., Wang Y.J. Identifying cancer biomarkers by network-constrained support vector machines. BMC Systems Biology. 2011;5:161. doi: 10.1186/1752-0509-5-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Yuan Y. Deepgene: an advanced cancer type classifier based on deep learning and somatic point mutations. BMC Bioinformatics. 2016;17 doi: 10.1186/s12859-016-1334-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Qi H. Mvp: predicting pathogenicity of missense variants by deep learning. bioRxiv. 2018 [Google Scholar]
- 193.Malta T.M. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell. 2018;173:338–354.e15. doi: 10.1016/j.cell.2018.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Yousefi S. Predicting clinical outcomes from large scale cancer genomic profiles with deep survival models. Scientific Reports. 2017;7 doi: 10.1038/s41598-017-11817-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Way G.P. A machine learning classifier trained on cancer transcriptomes detects nf1 inactivation signal in glioblastoma. BMC genomics. 2017;18:127. doi: 10.1186/s12864-017-3519-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Das S., Deng X., Camphausen K., Shankavaram U. Discoversl: an r package for multi-omic data driven prediction of synthetic lethality in cancers. Bioinformatics (Oxford, England) 2019;35:701–702. doi: 10.1093/bioinformatics/bty673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wan F. Exp2sl: A machine learning framework for cell-line-specific synthetic lethality prediction. Frontiers in pharmacology. 2020;11:112. doi: 10.3389/fphar.2020.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Stathias, V. et al. Lincs data portal 2.0: next generation access point for perturbation-response signatures. Nucleic acids research 48, D431–D439, doi: 10.1093/nar/gkz1023 (2020). [DOI] [PMC free article] [PubMed]
- 199.Kalinin A.A. Deep learning in pharmacogenomics: from gene regulation to patient stratification. Future Medicine. 2018;19 doi: 10.2217/pgs-2018-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Chen H., Engkvist O., Wang Y., Olivecrona M., Blaschke T. The rise of deep learning in drug discovery. Drug discovery today. 2018;23(6):1241–1250. doi: 10.1016/j.drudis.2018.01.039. [DOI] [PubMed] [Google Scholar]
- 201.Gupta S. Prioritization of anticancer drugs against a cancer using genomic features of cancer cells: A step towards personalized medicine. Scientific Reports. 2016;6 doi: 10.1038/srep23857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hejase H., Chan C.Y. Improving drug sensitivity prediction using different types of data. CPT: Pharmacometrics & Systems Pharmacology. 2015;4 doi: 10.1002/psp4.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zitnik M., Agrawal M., Leskovec J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics. 2018;34:i457–i466. doi: 10.1093/bioinformatics/bty294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Xu J. Translating cancer genomics into precision medicine with artificial intelligence: applications, challenges and future perspectives. Human Genetics. 2019;138:109–124. doi: 10.1007/s00439-019-01970-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Stueve T.R., Marconett C.N., Zhou B., Borok Z., Laird-Offringa I.A. The importance of detailed epigenomic profiling of different cell types within organs. Epigenomics. 2016;8:817–829. doi: 10.2217/epi-2016-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Holder L.B., Haque M.M., Skinner M.K. Machine learning for epigenetics and future medical applications. Epigenetics. 2017;12:505–514. doi: 10.1080/15592294.2017.1329068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Perez E., Capper D. Invited review: Dna methylation-based classification of paediatric brain tumours. Neuropathology and applied neurobiology. 2020;46:28–47. doi: 10.1111/nan.12598. [DOI] [PubMed] [Google Scholar]
- 208.Belokopytova P., Fishman V. Predicting genome architecture: Challenges and solutions. Frontiers in genetics. 2020;11 doi: 10.3389/fgene.2020.617202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Wang Y. Predicting DNA methylation state of CpG dinucleotide using genome topological features and deep networks. Scientific Reports. 2016;6 doi: 10.1038/srep19598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Koh P.W., Pierson E., Kundaje A. Denoising genome-wide histone chip-seq with convolutional neural networks. Bioinformatics. 2017;33:i225–i233. doi: 10.1093/bioinformatics/btx243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hiranuma N., Lundberg S.M., Lee S.-I. AIControl: replacing matched control experiments with machine learning improves ChIP-seq peak identification. Nucleic Acids Research. 2019;47:e58. doi: 10.1093/nar/gkz156. https://academic.oup.com/nar/article-pdf/47/10/e58/28761870/gkz156.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lal A. Deep learning-based enhancement of epigenomics data with atacworks. Nature Communications. 2021;12:1507. doi: 10.1038/s41467-021-21765-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Dekker J., Marti-Renom M.A., Mirny L.A. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nature reviews. Genetics. 2013;14:390–403. doi: 10.1038/nrg3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Lin D., Bonora G., Yardimci G.G., Noble W.S. Wiley interdisciplinary reviews. Systems biology and medicine 11. 2019. Computational methods for analyzing and modeling genome structure and organization. e1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Ernst J., Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nature Methods. 2012;9:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Paulsen J. Chrom3d: three-dimensional genome modeling from hi-c and nuclear lamin-genome contacts. Genome Biology. 2017;18 doi: 10.1186/s13059-016-1146-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Caudai C., Salerno E., Zoppè M., Tonazzini A. Estimation of the spatial chromatin structure based on a multiresolution bead-chain model. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019;16:550–559. doi: 10.1109/TCBB.2018.2791439. [DOI] [PubMed] [Google Scholar]
- 218.Caudai C., Salerno E., Zoppè M., Merelli I., Tonazzini A. Chromstruct 4: A python code to estimate the chromatin structure from hi-c data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019;16:1867–1878. doi: 10.1109/TCBB.2018.2838669. [DOI] [PubMed] [Google Scholar]
- 219.Serra F. Automatic analysis and 3d-modelling of hi-c data using tadbit reveals structural features of the fly chromatin colors. PLOS Computational Biology. 2017;13 doi: 10.1371/journal.pcbi.1005665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Fudenberg G., Kelley D.R., Pollard K.S. Predicting 3d genome folding from dna sequence with akita. Nature methods. 2020;17:1111–1117. doi: 10.1038/s41592-020-0958-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Schwessinger R. Deepc: predicting 3d genome folding using megabase-scale transfer learning. Nature methods. 2020;17:1118–1124. doi: 10.1038/s41592-020-0960-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Velculescu V.E. Characterization of the Yeast Transcriptome. Cell. 1997;88:243–251. doi: 10.1016/S0092-8674(00)81845-0. [DOI] [PubMed] [Google Scholar]
- 223.Velculescu V.E., Zhang L., Vogelstein B., Kinzler K.W. Serial Analysis of Gene Expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- 224.Nagano T., Fraser P. No-Nonsense Functions for Long Noncoding RNAs. Cell. 2011;145:178–181. doi: 10.1016/j.cell.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 225.Kristensen L.S. The biogenesis, biology and characterization of circular RNAs. Nature Reviews Genetics. 2019;20:675–691. doi: 10.1038/s41576-019-0158-7. [DOI] [PubMed] [Google Scholar]
- 226.Ozata, D.M., Gainetdinov, I., Zoch, A., OĆarroll, D. & Zamore, P.D. PIWI-interacting RNAs: small RNAs with big functions. Nature Reviews Genetics 20, 89–108, doi: 10.1038/s41576-018-0073-3 (2019). [DOI] [PubMed]
- 227.Djebali S. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Dunham I. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012 doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Chang T.W. Binding of cells to matrixes of distinct antibodies coated on solid surface. Journal of Immunological Methods. 1983;65:217–223. doi: 10.1016/0022-1759(83)90318-6. [DOI] [PubMed] [Google Scholar]
- 230.Schena M., Shalon D., Davis R.W., Brown P.O. Quantitative monitoring of gene expression patterns with a complementary dna microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 231.Morozova O., Marra M.A. Applications of next-generation sequencing technologies in functional genomics. 2008 doi: 10.1016/j.ygeno.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 232.Buermans H.P., den Dunnen J.T. Next generation sequencing technology: Advances and applications. 2014 doi: 10.1016/j.bbadis.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 233.Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics. 2010;10:57–63. doi: 10.1038/nrg2484.RNA-Seq. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Stark R., Grzelak M., Hadfield J. RNA sequencing: the teenage years. 2019 doi: 10.1038/s41576-019-0150-2. [DOI] [PubMed] [Google Scholar]
- 235.Lowe R., Shirley N., Bleackley M., Dolan S., Shafee T. Transcriptomics technologies. PLOS Computational Biology. 2017;13 doi: 10.1371/journal.pcbi.1005457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Frankish A. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Research. 2019;47:D766–D773. doi: 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Fickett J.W., Tung C.-S. Assessment of protein coding measures. Nucleic Acids Research. 1992;20:6441–6450. doi: 10.1093/nar/20.24.6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Frith M.C. Discrimination of Non-Protein-Coding Transcripts from Protein-Coding mRNA. RNA Biology. 2006;3:40–48. doi: 10.4161/rna.3.1.2789. [DOI] [PubMed] [Google Scholar]
- 239.Leoni G., Le Pera L., Ferrè F., Raimondo D., Tramontano A. Coding potential of the products of alternative splicing in human. Genome biology. 2011;12:R9. doi: 10.1186/gb-2011-12-1-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Liu J., Gough J., Rost B. Distinguishing protein-coding from non-coding RNAs through support vector machines. PLoS Genetics. 2006 doi: 10.1371/journal.pgen.0020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Kong L. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Research. 2007 doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Li A., Zhang J., Zhou Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinformatics. 2014 doi: 10.1186/1471-2105-15-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Schneider, e. a., Hugo W. A Support Vector Machine based method to distinguish long non-coding RNAs from protein coding transcripts. BMC Genomics 18, 804, doi: 10.1186/s12864-017-4178-4 (2017). [DOI] [PMC free article] [PubMed]
- 244.Pian C. LncRNApred: Classification of Long Non-Coding RNAs and Protein-Coding Transcripts by the Ensemble Algorithm with a New Hybrid Feature. PLOS ONE. 2016;11 doi: 10.1371/journal.pone.0154567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Wang L. CPAT: Coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Research. 2013 doi: 10.1093/nar/gkt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Kang Y.J. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Research. 2017 doi: 10.1093/nar/gkx428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Baek J., Lee B., Kwon S., Yoon S. LncRNAnet: Long non-coding RNA identification using deep learning. Bioinformatics. 2018 doi: 10.1093/bioinformatics/bty418. [DOI] [PubMed] [Google Scholar]
- 248.Hill S.T. A deep recurrent neural network discovers complex biological rules to decipher RNA protein-coding potential. Nucleic Acids Research. 2018;46:8105–8113. doi: 10.1093/nar/gky567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Amin N., McGrath A., Chen Y.P.P. Evaluation of deep learning in non-coding RNA classification. 2019 doi: 10.1038/s42256-019-0051-2. [DOI] [Google Scholar]
- 250.Camargo A.P., Sourkov V., Pereira G.A.G., Carazzolle M.F. RNAsamba: neural network-based assessment of the protein-coding potential of RNA sequences. NAR Genomics and Bioinformatics. 2020;2 doi: 10.1093/nargab/lqz024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Talavera D. Archetypal transcriptional blocks underpin yeast gene regulation in response to changes in growth conditions. Scientific Reports. 2018;8:7949. doi: 10.1038/s41598-018-26170-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Myszczynska M.A. Applications of machine learning to diagnosis and treatment of neurodegenerative diseases. Nature Reviews Neurology. 2020;16:440–456. doi: 10.1038/s41582-020-0377-8. [DOI] [PubMed] [Google Scholar]
- 253.van IJzendoorn, D.G. et al. Machine learning analysis of gene expression data reveals novel diagnostic and prognostic biomarkers and identifies therapeutic targets for soft tissue sarcomas. PLoS Computational Biology doi: 10.1371/journal.pcbi.1006826 (2019). [DOI] [PMC free article] [PubMed]
- 254.Breschi, A. et al. A limited set of transcriptional programs define major histological types and provide the molecular basis for a cellular taxonomy of the human body. bioRxiv 2019, doi: 10.1101/857169.
- 255.van der Maaten L., Hinton G. Visualizing data using t-SNE. Journal of Machine Learning Research. 2008;9:2579–2605. [Google Scholar]
- 256.McInnes, L., Healy, J. & Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction (2020). 1802.03426.
- 257.Yang Y. Dimensionality reduction by umap reinforces sample heterogeneity analysis in bulk transcriptomic data. Cell reports. 2021;36 doi: 10.1016/j.celrep.2021.109442. [DOI] [PubMed] [Google Scholar]
- 258.Li W.V., Li J.J. An accurate and robust imputation method scimpute for single-cell rna-seq data. Nature Communications. 2018;9 doi: 10.1038/s41467-018-03405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Gong W., Kwak I.-Y., Pota P., Koyano-Nakagawa N., Garry D.J. Drimpute: imputing dropout events in single cell rna sequencing data. BMC Bioinformatics. 2018;19 doi: 10.1186/s12859-018-2226-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Arisdakessian C.G., Poirion O.B., Yunits B., Zhu X., Garmire L.X. Deepimpute: an accurate, fast and scalable deep neural network method to impute single-cell rna-seq data. bioRxiv. 2018 doi: 10.1186/s13059-019-1837-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Maniatis T., Tasic B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature. 2002;418:236–243. doi: 10.1038/418236a. [DOI] [PubMed] [Google Scholar]
- 262.Wang E. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Mollet I. Unconstrained mining of transcript data reveals increased alternative splicing complexity in the human transcriptome. Nucleic Acids Research. 2010;38:4740–4754. doi: 10.1093/nar/gkq197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Garcia-Blanco M., Baraniak A.P., Lasda E.L. Alternative splicing in disease and therapy. Nature Biotechnology. 2004;22:535–546. doi: 10.1038/nbt964. [DOI] [PubMed] [Google Scholar]
- 265.Kahles A. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer cell. 2018;34(2):211–224.e6. doi: 10.1016/j.ccell.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Salovska B. Isoform-resolved correlation analysis between mRNA abundance regulation and protein level degradation. Molecular Systems Biology. 2020;16 doi: 10.15252/msb.20199170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Bretschneider H., Gandhi S., Deshwar A.G., Zuberi K., Frey B.J. COSSMO: Predicting competitive alternative splice site selection using deep learning. Bioinformatics. 2018 doi: 10.1093/bioinformatics/bty244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Jha A., Gazzara M.R., Barash Y. Integrative deep models for alternative splicing. Bioinformatics. 2017 doi: 10.1093/bioinformatics/btx268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Shen S. MATS: A Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Research. 2012 doi: 10.1093/nar/gkr1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Zhang Z. Deep-learning augmented RNA-seq analysis of transcript splicing. Nature Methods. 2019 doi: 10.1038/s41592-019-0351-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Leung M.K., Xiong H.Y., Lee L.J., Frey B.J. Deep learning of the tissue-regulated splicing code. Bioinformatics. 2014 doi: 10.1093/bioinformatics/btu277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Jaganathan K. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019;176:535–548.e24. doi: 10.1016/j.cell.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 273.Liu H., Han H., Li J., Wong L. DNAFSMiner: a web-based software toolbox to recognize two types of functional sites in DNA sequences. Bioinformatics. 2005;21(5):671–673. doi: 10.1093/bioinformatics/bth437. [DOI] [PubMed] [Google Scholar]
- 274.Kalkatawi M. Dragon polya spotter: predictor of poly(a) motifs within human genomic dna sequences. Bioinformatics. 2013;29 11:1484. doi: 10.1093/bioinformatics/btt161. [DOI] [PubMed] [Google Scholar]
- 275.Salamov A., Solovyev V. Recognition of 3’-processing sites of human mrna precursors. Computer applications in the biosciences: CABIOS. 1997;13(1):23–28. doi: 10.1093/bioinformatics/13.1.23. [DOI] [PubMed] [Google Scholar]
- 276.Tabaska J.E., Zhang M. Detection of polyadenylation signals in human dna sequences. Gene. 1999;231(1–2):77–86. doi: 10.1016/s0378-1119(99)00104-3. [DOI] [PubMed] [Google Scholar]
- 277.Cheng Y., Miura R., Tian B. Prediction of mRNA polyadenylation sites by support vector machine. Bioinformatics. 2006;22(19):2320–2325. doi: 10.1093/bioinformatics/btl394. [DOI] [PubMed] [Google Scholar]
- 278.Akhtar N. MicroRNA-27b regulates the expression of matrix metalloproteinase 13 in human osteoarthritis chondrocytes. Arthritis and rheumatism. 2010;62(5):1361–1371. doi: 10.1002/art.27329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Gao J., Yang Y., Zhou Y. Grid-based prediction of torsion angle probabilities of protein backbone and its application to discrimination of protein intrinsic disorder regions and selection of model structures. BMC Bioinformatics. 2018;19:29. doi: 10.1186/s12859-018-2031-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Xia Z. Deerect-polya: a robust and generic deep learning method for pas identification. Bioinformatics. 2019;35:2371–2379. doi: 10.1093/bioinformatics/bty991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Leung M., Delong A., Frey B. Inference of the human polyadenylation code. Bioinformatics. 2017;34:2889–2898. doi: 10.1093/bioinformatics/bty211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Bar-Shira A., Panet A., Honigman A. An RNA secondary structure juxtaposes two remote genetic signals for human t-cell leukemia virus type I RNA 3’-end processing. Journal of Virology. 1991;65(10):5165–5173. doi: 10.1128/jvi.65.10.5165-5173.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Brown P.H., Tiley L., Cullen B. Effect of RNA secondary structure on polyadenylation site selection. Genes & development. 1991;5(7):1277–1284. doi: 10.1101/gad.5.7.1277. [DOI] [PubMed] [Google Scholar]
- 284.Wu C., Alwine J. Secondary structure as a functional feature in the downstream region of mammalian polyadenylation signals. Molecular and Cellular Biology. 2004;24:2789–2796. doi: 10.1128/MCB.24.7.2789-2796.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Saletore Y. The birth of the Epitranscriptome: deciphering the function of RNA modifications. Genome biology. 2012;13:175. doi: 10.1186/gb-2012-13-10-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Agris P.F. The importance of being modified: roles of modified nucleosides and Mg2+ in RNA structure and function. Progress in Nucleic Acid Research and Molecular Biology. 1996;53:79–129. doi: 10.1016/s0079-6603(08)60143-9. [DOI] [PubMed] [Google Scholar]
- 287.Marbaniang C.N., Vogel J. Emerging roles of rna modifications in bacteria. Current Opinion in Microbiology. 2016;30:50–57. doi: 10.1016/j.mib.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 288.Machnicka M.A. Modomics: a database of rna modification pathways–2013 update. Nucleic Acids Research. 2013;41:D262–D267. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Mathlin J., Le Pera L., Colombo T. A census and categorization method of epitranscriptomic marks. International Journal of Molecular Sciences. 2020;21 doi: 10.3390/ijms21134684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Dominissini D. Topology of the human and mouse m6a rna methylomes revealed by m6a-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 291.Meyer K.D. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Frye, M., Jaffrey, S.R., Pan, T., Rechavi, G. & Suzuki, T. RNA modifications: What have we learned and where are we headed?, 2016, doi: 10.1038/nrg.2016.47. [DOI] [PubMed]
- 293.Zhou Y., Zeng P., Li Y.-H., Zhang Z., Cui Q. Sramp: prediction of mammalian n6-methyladenosine (m6a) sites based on sequence-derived features. Nucleic Acids Research. 2016;44 doi: 10.1093/nar/gkw104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Chen K. Whistle: a high-accuracy map of the human n6-methyladenosine (m6a) epitranscriptome predicted using a machine learning approach. Nucleic Acids Research. 2019;47 doi: 10.1093/nar/gkz074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Dao F.-Y. Computational identification of n6-methyladenosine sites in multiple tissues of mammals. Computational and Structural Biotechnology Journal. 2020;18:1084–1091. doi: 10.1016/j.csbj.2020.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Zhang L., Qin X., Liu M., Xu Z., Liu G. DNN-m6A: A cross-species method for identifying RNA N6-Methyladenosine sites based on deep neural network with multi-information fusion. Genes. 2021;12 doi: 10.3390/genes12030354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Werner S. Machine learning of reverse transcription signatures of variegated polymerases allows mapping and discrimination of methylated purines in limited transcriptomes. Nucleic Acids Research. 2020;48:3734–3746. doi: 10.1093/nar/gkaa113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Salekin S. Predicting sites of epitranscriptome modifications using unsupervised representation learning based on generative adversarial networks. Frontiers in physics. 2020;8 doi: 10.3389/fphy.2020.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Wilkins M. Progress with proteome projects: why all proteins expressed by a genome should be identified and how to do it. Biotechnology & Genetic Engineering Reviews. 1996;13:19–50. doi: 10.1080/02648725.1996.10647923. [DOI] [PubMed] [Google Scholar]
- 300.Lander E. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 301.Aslam B., Basit M., Nisar M.A., Khurshid M., Rasool M. Proteomics: Technologies and their applications. Journal of Chromatographic Science. 2017;55(2):182–196. doi: 10.1093/chromsci/bmw167. [DOI] [PubMed] [Google Scholar]
- 302.Yates, J.A century of mass spectrometry: from atoms to proteomes. Nature Methods 8, 633–637 (2011).
- 303.van Agthoven M.A., Lam Y.P.Y., O’Connor P., Rolando C., Delsuc M. Two-dimensional mass spectrometry: new perspectives for tandem mass spectrometry. European Biophysics Journal. 2019;48:213–229. doi: 10.1007/s00249-019-01348-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Zhang Z., Wu S., Stenoien D., Pasa-Tolic L. High-throughput proteomics. Annual review of analytical chemistry. 2014;7:427–454. doi: 10.1146/annurev-anchem-071213-020216. [DOI] [PubMed] [Google Scholar]
- 305.Hillenkamp F., Karas M., Beavis R., Chait B. Matrix-assisted laser desorption/ionization mass spectrometry of biopolymers. Analytical chemistry. 1991;63(24):1193A–1203A. doi: 10.1021/ac00024a002. [DOI] [PubMed] [Google Scholar]
- 306.Gogichaeva N.V., Williams T., Alterman M. Maldi tof/tof tandem mass spectrometry as a new tool for amino acid analysis. Journal of the American Society for Mass Spectrometry. 2007;18:279–284. doi: 10.1016/j.jasms.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 307.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 308.Fang C., Shang Y., Xu D. MUFOLD-SS: New deep inception-inside-inception networks for protein secondary structure prediction. Proteins. 2018;86:592–598. doi: 10.1002/prot.25487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Bonnel N., Marteau P.F. Lna: fast protein structural comparison using a laplacian characterization of tertiary structure. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012;9:1451–1458. doi: 10.1109/TCBB.2012.64. [DOI] [PubMed] [Google Scholar]
- 310.Faraggi E., Zhang T., Yang Y., Kurgan L., Zhou Y. Spine x: improving protein secondary structure prediction by multistep learning coupled with prediction of solvent accessible surface area and backbone torsion angles. Journal of Computational Chemistry. 2012;33:259–267. doi: 10.1002/jcc.21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Fang C., Shang Y., Xu D. Prediction of protein backbone torsion angles using deep residual inception neural networks. 2018 doi: 10.1109/TCBB.2018.2814586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Jacobson M.P. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 313.Nguyen S.P., Li Z., Xu D., Shang Y. New deep learning methods for protein loop modeling. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2017;16:596–606. doi: 10.1109/TCBB.2017.2784434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Senior A.W. Improved protein structure prediction using potentials from deep learning. Nature. 2020;577:706–710. doi: 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- 315.Jumper J. Highly accurate protein structure prediction with alphafold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Rifaioglu A.S., Doğan T., Martin M.J., Cetin-Atalay R., Atalay V. Deepred: Automated protein function prediction with multi-task feed-forward deep neural networks. Scientific Reports. 2019;9 doi: 10.1038/s41598-019-43708-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Kelchtermans P. Machine learning applications in proteomics research: How the past can boost the future. Proteomics. 2013;14:353–366. doi: 10.1002/pmic.201300289. [DOI] [PubMed] [Google Scholar]
- 318.Sonsare P.M., Gunavathi C. Investigation of machine learning techniques on proteomics: A comprehensive survey. Progress in Biophysics and Molecular biology. 2019;149:54–69. doi: 10.1016/j.pbiomolbio.2019.09.004. [DOI] [PubMed] [Google Scholar]
- 319.An J.-Y. Improving protein-protein interactions prediction accuracy using protein evolutionary information and relevance vector machine model. Protein science: a publication of the Protein Society. 2016;25:1825–1833. doi: 10.1002/pro.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Huang L., Liao L., Wu C.H. Completing sparse and disconnected protein-protein network by deep learning. BMC bioinformatics. 2018;19:103. doi: 10.1186/s12859-018-2112-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Qin Q., Feng J. Imputation for transcription factor binding predictions based on deep learning. PLoS computational biology. 2017;13 doi: 10.1371/journal.pcbi.1005403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Rives A. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proceedings of the National Academy of Sciences of the United States of America. 2021;118 doi: 10.1073/pnas.2016239118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Patti G., Yanes Ó., Siuzdak G. Innovation: Metabolomics: the apogee of the omics trilogy. Nature Reviews Molecular Cell Biology. 2012;13:263–269. doi: 10.1038/nrm3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Zamboni N., Saghatelian A., Patti G. Defining the metabolome: size, flux, and regulation. Molecular cell. 2015;58(4):699–706. doi: 10.1016/j.molcel.2015.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Pomyen Y. Deep metabolome: Applications of deep learning in metabolomics. Computational and Structural Biotechnology Journal. 2020;18:2818–2825. doi: 10.1016/j.csbj.2020.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Cavill R. Genetic algorithms for simultaneous variable and sample selection in metabonomics. Bioinformatics. 2009;25:112–118. doi: 10.1093/bioinformatics/btn586. [DOI] [PubMed] [Google Scholar]
- 327.Hao J., Astle W., De Iorio M., Ebbels T. Batman-an R package for the automated quantification of metabolites from nuclear magnetic resonance spectra using a bayesian model. Bioinformatics. 2012;28:2088–2090. doi: 10.1093/bioinformatics/bts308. [DOI] [PubMed] [Google Scholar]
- 328.Ravanbakhsh S. Accurate, fully-automated nmr spectral profiling for metabolomics. PLOS ONE. 2008;10 doi: 10.1371/journal.pone.0124219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Alakwaa F.M., Chaudhary K., Garmire L.X. Deep learning accurately predicts estrogen receptor status in breast cancer metabolomics data. Journal of Proteome Research. 2018;17:337–347. doi: 10.1021/acs.jproteome.7b00595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Ideker T., Galitski T., Hood L. A new approach to decoding life: systems biology. Annual Review of Genomics and Human Genetics. 2001;2:343–372. doi: 10.1146/annurev.genom.2.1.343. [DOI] [PubMed] [Google Scholar]
- 331.Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–1664. doi: 10.1126/science.1069492. [DOI] [PubMed] [Google Scholar]
- 332.Khodayari A., Maranas C.D. A genome-scale escherichia coli kinetic metabolic model k-ecoli457 satisfying flux data for multiple mutant strains. Nature Communications. 2016;7:13806. doi: 10.1038/ncomms13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Zeng L., Das S., Burne R.A. Utilization of lactose and galactose by streptococcus mutans: Transport, toxicity, and carbon catabolite repression. Journal of Bacteriology. 2010;192:2434–2444. doi: 10.1128/JB.01624-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Wang Y., Zhang X.-S., Chen L. Integrating data- and model-driven strategies in systems biology. BMC Systems Biology. 2018;12:38. doi: 10.1186/s12918-018-0562-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Costello Z., Martin Garcia. H.A machine learning approach to predict metabolic pathway dynamics from time-series multiomics data. Npj System Biology and Applications. 2018;4 doi: 10.1038/s41540-018-0054-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Yazdani A., Lu L., Raissi M., Karniadakis G.E. Systems biology informed deep learning for inferring parameters and hidden dynamics. PLOS Computational Biology. 2020;16 doi: 10.1371/journal.pcbi.1007575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Raissi M., Perdikaris P., Karniadakis E. Physics-informed neural networks: A deep learning framework for solving forward and inverse problems involving nonlinear partial differential equations. Journal of Computational Physics. 2019;378:686–707. doi: 10.1016/j.jcp.2018.10.045. [DOI] [Google Scholar]
- 338.Fortelny N., Bock C. Knowledge-primed neural networks enable biologically interpretable deep learning on single-cell sequencing data. Genome Biology. 2020;21:190. doi: 10.1186/s13059-020-02100-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Muzio G., O’Bray L., Borgwardt K. Biological network analysis with deep learning. Briefings in Bioinformatics. 2021;22:1515–1530. doi: 10.1093/bib/bbaa257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Antonakoudis A., Barbosa R., Kotidis P., Kontoravdi C. The era of big data: Genome-scale modelling meets machine learning. Comput Struct Biotechnol J. 2020;18:3287–3300. doi: 10.1016/j.csbj.2020.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Gilpin W., Huang Y., Forger D.B. Learning dynamics from large biological data sets: Machine learning meets systems biology. Current Opinion in Systems Biology. 2020;22:1–7. [Google Scholar]
- 342.Little R., Rubin D. John Wiley & Sons; 1987. Statistical Analysis with Missing Data. [Google Scholar]
- 343.Cismondi F. Missing data in medical databases: Impute, delete or classify? Artificial Intelligence in Medicine. 2013;58:63–72. doi: 10.1016/j.artmed.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 344.Heitjan D.F. Annotation: what can be done about missing data? approaches to imputation. American Journal of Public Health. 1997;87:548–550. doi: 10.2105/ajph.87.4.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Chen J., Shao J. Nearest neighbor imputation for survey data. Journal of Official Statistics. 2000;16:113. [Google Scholar]
- 346.Kim, J., Tae, D. & Seok, J.A survey of missing data imputation using generative adversarial networks. In 2020 International Conference on Artificial Intelligence in Information and Communication (ICAIIC), 454–456 (IEEE, 2020).
- 347.De Souto M.C., Jaskowiak P.A., Costa I.G. Impact of missing data imputation methods on gene expression clustering and classification. BMC Bioinformatics. 2015;16:64. doi: 10.1186/s12859-015-0494-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Van Buuren S. CRC Press; 2018. Flexible imputation of missing data. [Google Scholar]
- 349.Malarvizhi M.R., Thanamani A.S. K-nearest neighbor in missing data imputation. International Journal of Engineering Research and Development. 2012;5:5–7. [Google Scholar]
- 350.Gautam, C. & Ravi, V. Evolving clustering based data imputation. In 2014 International Conference on Circuits, Power and Computing Technologies [ICCPCT-2014], 1763–1769 (IEEE, 2014).
- 351.Petrazzini B.O., Naya H., Lopez-Bello F., Vazquez G., Spangenberg L. Evaluation of different approaches for missing data imputation on features associated to genomic data. BioData Mining. 2021;14:1–13. doi: 10.1186/s13040-021-00274-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Wang B. Similarity network fusion for aggregating data types on a genomic scale. Nature methods. 2014;11:333. doi: 10.1038/nmeth.2810. [DOI] [PubMed] [Google Scholar]
- 353.Voillet, V., Besse, P., Liaubet, L., San Cristobal, M. & González, I. Handling missing rows in multi-omics data integration: multiple imputation in multiple factor analysis framework. BMC Bioinformatics 17, 1–16 (2016). [DOI] [PMC free article] [PubMed]
- 354.Husson F., Josse J. Handling missing values in multiple factor analysis. Food Quality and Preference. 2013;30:77–85. [Google Scholar]
- 355.Josse J., Husson F. missmda: a package for handling missing values in multivariate data analysis. Journal of Statistical Software. 2016;70:1–31. [Google Scholar]
- 356.Hansen K.D., Wu Z., Irizarry R.A., Leek J.T. Sequencing technology does not eliminate biological variability. Nature Biotechnology. 2011;29:572–573. doi: 10.1038/nbt.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.McIntyre L.M. Rna-seq: technical variability and sampling. BMC Genomics. 2011;12:293. doi: 10.1186/1471-2164-12-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Nounou M.N., Nounou H.N., Mansouri M. Model-based and model-free filtering of genomic data. Network Modeling Analysis in Health Informatics and Bioinformatics. 2013;2:109–121. [Google Scholar]
- 359.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biology. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Van Hulse J., Khoshgoftaar T.M., Napolitano A., Wald R. Threshold-based feature selection techniques for high-dimensional bioinformatics data. Network Modeling Analysis in Health Informatics and Bioinformatics. 2012;1:47–61. [Google Scholar]
- 361.Lazar C. Batch effect removal methods for microarray gene expression data integration: a survey. Briefings in Bioinformatics. 2013;14:469–490. doi: 10.1093/bib/bbs037. [DOI] [PubMed] [Google Scholar]
- 362.Nounou, M., Nounou, H., Meskin, N. & Datta, A. Wavelet-based multiscale filtering of genomic data. In 2012 IEEE/ACM International Conference on Advances in Social Networks Analysis and Mining, 804–809 (IEEE, 2012).
- 363.Vincent P., Larochelle H., Bengio Y., Manzagol P.-A. Proceedings of the 25th international conference on Machine learning. 2008. Extracting and composing robust features with denoising autoencoders; pp. 1096–1103. [Google Scholar]
- 364.Saad O.M., Chen Y. Deep denoising autoencoder for seismic random noise attenuation. Geophysics. 2020;85:V367–V376. [Google Scholar]
- 365.Wang J. Data denoising with transfer learning in single-cell transcriptomics. Nature Methods. 2019;16:875–878. doi: 10.1038/s41592-019-0537-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Razin A., Cedar H. Dna methylation and gene expression. Microbiology and Molecular Biology Reviews. 1991;55:451–458. doi: 10.1128/mr.55.3.451-458.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Bell J.T. Dna methylation patterns associate with genetic and gene expression variation in hapmap cell lines. Genome Biology. 2011;12:R10. doi: 10.1186/gb-2011-12-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Richardson S., Tseng G.C., Sun W. Statistical methods in integrative genomics. Annual Review of Statistics and its Application. 2016;3:181–209. doi: 10.1146/annurev-statistics-041715-033506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Bersanelli M. Methods for the integration of multi-omics data: mathematical aspects. BMC Bioinformatics. 2016;17:S15. doi: 10.1186/s12859-015-0857-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Gui H., Li M., Sham P.C., Cherny S.S. Comparisons of seven algorithms for pathway analysis using the wtccc crohn’s disease dataset. BMC Research Notes. 2011;4:386. doi: 10.1186/1756-0500-4-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Pellegrini M., Baglioni M., Geraci F. Protein complex prediction for large protein protein interaction networks with the core&peel method. BMC Bioinformatics. 2016;17:37–58. doi: 10.1186/s12859-016-1191-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Louhimo R., Hautaniemi S. CNAmet: an R package for integrating copy number, methylation and expression data. Bioinformatics. 2011;27:887–888. doi: 10.1093/bioinformatics/btr019. [DOI] [PubMed] [Google Scholar]
- 373.Agarwal V., Bell G.W., Nam J.-W., Bartel D.P. Predicting effective microRNA target sites in mammalian mRNAs. elife. 2015;4 doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Huang S., Chaudhary K., Garmire L.X. More is better: recent progress in multi-omics data integration methods. Frontiers in genetics. 2017;8:84. doi: 10.3389/fgene.2017.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Shen R., Olshen A.B., Ladanyi M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics. 2009;25:2906–2912. doi: 10.1093/bioinformatics/btp543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Zhang S., Li Q., Liu J., Zhou X.J. A novel computational framework for simultaneous integration of multiple types of genomic data to identify microRNA-gene regulatory modules. Bioinformatics. 2011;27:i401–i409. doi: 10.1093/bioinformatics/btr206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Wu C. A selective review of multi-level omics data integration using variable selection. High-Throughput. 2019;8:4. doi: 10.3390/ht8010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Zhao B., Rubinstein B.I., Gemmell J., Han J. VLDB. 2012. A bayesian approach to discovering truth from conflicting sources for data integration. [Google Scholar]
- 379.Chaudhary K., Poirion O.B., Lu L., Garmire L.X. Deep learning–based multi-omics integration robustly predicts survival in liver cancer. Clinical Cancer Research. 2018;24:1248–1259. doi: 10.1158/1078-0432.CCR-17-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Hamamoto R., Komatsu M., Takasawa K., Asada K., Kaneko S. Epigenetics analysis and integrated analysis of multiomics data, including epigenetic data, using artificial intelligence in the era of precision medicine. Biomolecules. 2020;10:62. doi: 10.3390/biom10010062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Israelsen B.W., Ahmed N.R. dave...i can assure you...that it’s going to be all right...a definition, case for, and survey of algorithmic assurances in human-autonomy trust relationships. ACM Computing Surveys (CSUR) 2019;51:1–37. [Google Scholar]
- 382.Samek, W. & Müller, K.-R. Towards explainable artificial intelligence. In Explainable AI: interpreting, explaining and visualizing deep learning, 5–22 (Springer, 2019).
- 383.Montavon G., Samek W., Müller K.-R. Methods for interpreting and understanding deep neural networks. Digital Signal Processing. 2018;73:1–15. [Google Scholar]
- 384.Lapuschkin S. Unmasking clever hans predictors and assessing what machines really learn. Nature Communications. 2019;10:1–8. doi: 10.1038/s41467-019-08987-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Silver D. Mastering the game of Go with deep neural networks and tree search. Nature. 2016;529:484–489. doi: 10.1038/nature16961. [DOI] [PubMed] [Google Scholar]
- 386.Došilović, F.K., Brčić, M. & Hlupić, N. Explainable artificial intelligence: A survey. In 2018 41st International convention on information and communication technology, electronics and microelectronics (MIPRO), 0210–0215 (IEEE, 2018).
- 387.Martens D., Baesens B., Van Gestel T., Vanthienen J. Comprehensible credit scoring models using rule extraction from support vector machines. European Journal of Operational Research. 2007;183:1466–1476. [Google Scholar]
- 388.Zhou Z.-H., Jiang Y., Chen S.-F. Extracting symbolic rules from trained neural network ensembles. AI Communications. 2003;16:3–15. [Google Scholar]
- 389.Halevy A., Norvig P., Pereira F. The unreasonable effectiveness of data. IEEE Intelligent Systems. 2009;24:8–12. [Google Scholar]
- 390.Harmanci A., Gerstein M. Analysis of sensitive information leakage in functional genomics signal profiles through genomic deletions. Nature Communications. 2018;9:1–10. doi: 10.1038/s41467-018-04875-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Joly Y., Feze I.N., Song L., Knoppers B.M. Comparative approaches to genetic discrimination: chasing shadows? Trends in Genetics. 2017;33:299–302. doi: 10.1016/j.tig.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 392.Kaye J. The tension between data sharing and the protection of privacy in genomics research. Annual Review of Genomics and Human Genetics. 2012;13:415–431. doi: 10.1146/annurev-genom-082410-101454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.de Montjoye Y.-A., Farzanehfar A., Hendrickx J., Rocher L. Solving artificial intelligence’s privacy problem. Field Actions Science Reports. 2017;80–83 [Google Scholar]
- 394.Sweeney, L., Abu, A. & Winn, J. Identifying participants in the personal genome project by name (a re-identification experiment). arXiv preprint arXiv:1304.7605 (2013).
- 395.Greenbaum D., Sboner A., Mu X.J., Gerstein M. Genomics and privacy: implications of the new reality of closed data for the field. PLoS Computational Biology. 2011;7 doi: 10.1371/journal.pcbi.1002278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Azencott C.-A. Machine learning and genomics: precision medicine versus patient privacy. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences. 2018;376:20170350. doi: 10.1098/rsta.2017.0350. [DOI] [PubMed] [Google Scholar]
- 397.Sweeney L. k-anonymity: A model for protecting privacy. International Journal of Uncertainty, Fuzziness and Knowledge-Based Systems. 2002;10:557–570. [Google Scholar]
- 398.Nissim K. Differential privacy: A primer for a non-technical audience. Privacy Law Scholars Conf. 2017;3 [Google Scholar]
- 399.Abadi M. Proceedings of the 2016 ACM SIGSAC Conference on Computer and Communications Security. 2016. Deep learning with differential privacy; pp. 308–318. [Google Scholar]
- 400.Beaulieu-Jones B.K. Privacy-preserving generative deep neural networks support clinical data sharing. Circulation: Cardiovascular Quality and Outcomes. 2019;12 doi: 10.1161/CIRCOUTCOMES.118.005122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Salvaris M., Dean D., Tok W.H. Deep learning with azure. Apress. 2018 [Google Scholar]
- 402.Jackovich J., Richards R. (Packt Publishing; 2018. Machine Learning with AWS: Explore the power of cloud services for your machine learning and artificial intelligence projects. [Google Scholar]
- 403.Ciaburro G., Ayyadevara V.K., Perrier A. (Packt Publishing Ltd; 2018. Hands-on machine learning on google cloud platform: Implementing smart and efficient analytics using cloud ml engine. [Google Scholar]
- 404.Paszke A. NeurIPS. 2019. Pytorch: An imperative style, high-performance deep learning library. [Google Scholar]
- 405.Chollet, F. keras. URL:https://github.com/fchollet/keras (2015).
- 406.Rampasek L., Goldenberg A. Tensorflow: Biology’s gateway to deep learning? Cell Systems. 2016;2(1):12–14. doi: 10.1016/j.cels.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 407.Yang J., Lodgher A. 2019 International Conference on Security and Management. 2019. Fundamental defensive programming practices with secure coding modules. [Google Scholar]
- 408.Lawlor B., Walsh P. Engineering bioinformatics: building reliability, performance and productivity into bioinformatics software. Bioengineered. 2015;6:193–203. doi: 10.1080/21655979.2015.1050162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Giannoulatou E., Park S.-H., Humphreys D.T., Ho J.W. Verification and validation of bioinformatics software without a gold standard: a case study of bwa and bowtie. BMC Bioinformatics. 2014;15:S15. doi: 10.1186/1471-2105-15-S16-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Leprevost, F. d. V., Barbosa, V.C., Francisco, E.L., Perez-Riverol, Y. & Carvalho, P.C. On best practices in the development of bioinformatics software. Frontiers in Genetics 5, 199 (2014). [DOI] [PMC free article] [PubMed]
- 411.Seemann, T. Ten recommendations for creating usable bioinformatics command line software. GigaScience 2, 2047–217X (2013). [DOI] [PMC free article] [PubMed]
- 412.Wolff J., Pauling J., Keck A., Baumbach J. The economic impact of artificial intelligence in health care: Systematic review. Journal of Medical Internet Research. 2020;22 doi: 10.2196/16866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Landgrebe, T., Paclík, P., Tax, D.M., Verzakov, S. & Duin, R.P. Cost-based classifier evaluation for imbalanced problems. In Joint IAPR International Workshops on Statistical Techniques in Pattern Recognition (SPR) and Structural and Syntactic Pattern Recognition (SSPR), 762–770 (Springer, 2004).
- 414.Rao R.R., Makkithaya K. Learning from a class imbalanced public health dataset: A cost-based comparison of classifier performance. International Journal of Electrical and Computer Engineering. 2017;7:2215. [Google Scholar]
- 415.Aboutalib S.S. Deep learning to distinguish recalled but benign mammography images in breast cancer screening. Clinical Cancer Research. 2018;24:5902–5909. doi: 10.1158/1078-0432.CCR-18-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Sidore C. Genome sequencing elucidates sardinian genetic architecture and augments association analyses for lipid and blood inflammatory markers. Nature Genetics. 2015;47:1272. doi: 10.1038/ng.3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Martin A.R. Clinical use of current polygenic risk scores may exacerbate health disparities. Nature Genetics. 2019;51:584–591. doi: 10.1038/s41588-019-0379-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Nagashima T. Japanese version of the cancer genome atlas, jcga, established using fresh frozen tumors obtained from 5143 cancer patients. Cancer Science. 2020;111:687. doi: 10.1111/cas.14290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Callaway E. ‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures. Nature. 2020;588:203–204. doi: 10.1038/d41586-020-03348-4. [DOI] [PubMed] [Google Scholar]