

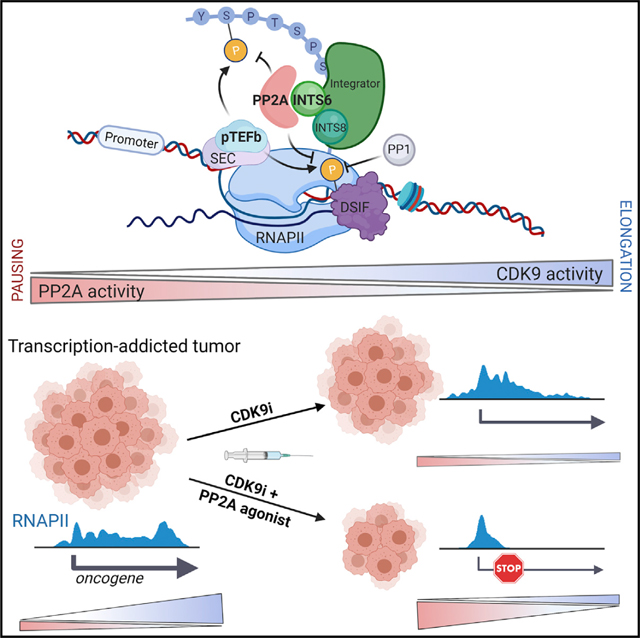
SUMMARY
Gene expression by RNA polymerase II (RNAPII) is tightly controlled by cyclin-dependent kinases (CDKs) at discrete checkpoints during the transcription cycle. The pausing checkpoint following transcription initiation is primarily controlled by CDK9. We discovered that CDK9-mediated, RNAPII-driven transcription is functionally opposed by a protein phosphatase 2A (PP2A) complex that is recruited to transcription sites by the Integrator complex subunit INTS6. PP2A dynamically antagonizes phosphorylation of key CDK9 substrates including DSIF and RNAPII-CTD. Loss of INTS6 results in resistance to tumor cell death mediated by CDK9 inhibition, decreased turnover of CDK9 phospho-substrates, and amplification of acute oncogenic transcriptional responses. Pharmacological PP2A activation synergizes with CDK9 inhibition to kill both leukemic and solid tumor cells, providing therapeutic benefit in vivo. These data demonstrate that fine control of gene expression relies on the balance between kinase and phosphatase activity throughout the transcription cycle, a process dysregulated in cancer that can be exploited therapeutically.
In brief
Interplay between PP2A and CDK9 provides a control point for gene expression that can be exploited to inhibit tumorigenesis.
Graphical Abstract
INTRODUCTION
Spatiotemporal control of gene expression is essential for appropriate organismal development and to direct functional responses to a range of intrinsic and extrinsic cellular cues (Roeder, 2019). This process is tightly regulated and highly dynamic, directed by the activity of RNA polymerase II (RNAPII) (Chen et al., 2018; Roeder, 2019) and the concerted action of cyclin-dependent kinases (CDKs) and their cognate cyclins in a process termed the transcription cycle (Malumbres, 2014). Following transcription initiation, RNAPII arrests shortly after transcribing the first 60–100 base pairs (bp) of most genes and acquires a “paused” conformation, stabilized by the DRB sensitivity inducing factor (DSIF) and the negative elongation factor (NELF) (Chen et al., 2018; Core and Adelman, 2019; Vos et al., 2018b). Paused RNAPII is still engaged in transcription, yet unable to elongate the nascent transcript owing to reduced mobility and impaired binding of NTPs. Dynamic phosphorylation of the RNAPII C-terminal domain (CTD), NELF, and DSIF by the positive transcription elongation factor (P-TEFb), comprised of CDK9 and cyclin T (Chen et al., 2018; Core and Adelman, 2019), facilitates the transition of the paused polymerase into active elongation through allosteric changes and modulation of RNAPII-associated factors (Chen et al., 2018; Core and Adelman, 2019; Vos et al., 2018a). P-TEFb activity is mediated by its recruitment to the transcriptional pause site by transcription factors such as c-MYC (Eberhardy and Farnham, 2001), the bromodomain-containing protein BRD4 (Hargreaves et al., 2009; Jang et al., 2005), and the super elongation complex (SEC) (Lin et al., 2010; Luo et al., 2012).
Small molecule CDK9 inhibitors (CDK9i) (e.g., flavopiridol) abrogate productive transcriptional elongation leading to accumulation of hypo-phosphorylated RNAPII at the transcriptional pause site (Chao and Price, 2001; Martin et al., 2020). The observation that transcriptional processes are fundamentally dysregulated in cancer and that P-TEFb interacts with oncogenic transcription factors such as c-MYC and MLL-fusion proteins (Bradner et al., 2017) has prompted the development and application of CDK9 inhibitors in the oncology setting (Fujinaga, 2020).
Despite extensive literature on the various RNAPII-CTD kinases including the “transcriptional kinases” (CDK7–CDK9 and CDK11–CDK13), less is known about the phosphatases that target RNAPII-CTD and oppose P-TEFb kinase activity (Jeronimo et al., 2016). A dedicated set of phosphatases have been demonstrated to de-phosphorylate RNAPII-CTD in mammals and lower organisms, which is particularly important for RNAPII recycling (Mayfield et al., 2016). Recently, CDK9-mediated phosphorylation of SPT5, a component of DSIF, was shown to be opposed by PP1 and PP4 during the transition from elongation to termination and at promoter proximal regions, respectively (Parua et al., 2018; Parua et al., 2020). This provides evidence for functional interplay between kinases and phosphatases to dynamically regulate the core transcriptional machinery.
Here, we used genome-wide CRISPR-screens to identify genes that regulate CDK9i-induced cell death and override the block in productive elongation. Deletion of INTS6, a component of the Integrator complex, suppressed CDK9i-induced cell death and allowed for transcriptional elongation in transformed and untransformed cell lines and across species. The 14-subunit Integrator complex associates with the RNAPII-CTD and cleaves nascent RNA species such as U small nuclear RNAs (snRNAs) and enhancer RNAs (eRNAs) (Lai et al., 2015; Rienzo and Casamassimi, 2016). Recently, Integrator was reported to regulate RNAPII pausing and elongation through its RNA-directed catalytic subunits INTS11/INTS9 (Elrod et al., 2019; Gardini et al., 2014). Here, we uncovered that INTS6, INTS8, and the PP2A phosphatase (Int-PP2A) form a distinct functional module of Integrator that is recruited to actively transcribed genes to oppose CDK9 activity by controlling the phosphorylation turnover of key CDK9 substrates, including the RNAPII-CTD and DSIF. These discoveries provided a molecular rationale to combine CDK9i with small molecule activators of PP2A (SMAPs), which led to enhanced transcriptional pausing and synergistic therapeutic efficacy in MLL-rearranged AML and solid tumors. We reveal how RNAPII-driven transcription is fine-tuned through the PP2A-Integrator-CDK9 axis, and the opposing enzymatic activities of CDK9 and PP2A present a viable therapeutic target for solid and hematopoietic malignancies.
RESULTS
Loss of the Integrator component INTS6 confers resistance to CDK9 inhibition
To identify proteins that functionally antagonize CDK9 activity on both the phenotypic and transcriptional level, we performed a series of genome-wide CRISPR-screens on MLL-rearranged leukemia cell lines THP-1 and MV4;11 treated with CDK9 inhibitors (CDK9i) using the following readouts: (1) long-term cell survival, and (2) nascent RNA transcription (Figure 1A). Cell survival screens performed in THP-1 cells treated with the CDK9i AZ5576, A159, or Dinaciclib (Wu et al., 2020) revealed selective enrichment of single guide RNAs (sgRNAs) targeting the Integrator complex subunit INTS6 (Figure 1B, left). This was reproduced in analogous screens in MV4;11 cells (Figures 1B, right, and S1A; Table S1), confirming that deletion of INTS6 conferred resistance to CDK9i.
Figure 1. Loss of INTS6 confers resistance to CDK9 inhibition.

(A) CRISPR-Cas9 genome-wide screens: Cas9-THP-1 and MV4;11 cells were transduced with a genome-wide sgRNA library and cultured with DMSO or CDK9i for 21 days (survival) or 16 h (nascent RNA).
(B) Enriched sgRNAs (CDK9i versus untreated; Tend) in THP-1-Cas9 survival screens using different CDK9 inhibitors (left) and sgRNA libraries (right), adjusted p value <0.1 for >3 sgRNAs.
(C) Enriched sgRNAs for THP-1-Cas9 nascent RNA screens (significance relative to T0).
(D) Enrichment of INTS6 targeting sgRNAs in CDK9i-treated THP-1-Cas9 cells.
(E) Comparison of enriched sgRNAs for nascent RNA and survival screens.
(F) Competitive proliferation assays for (H) and (L); cells expressing Cas9 and GFP/BFP-sgRNAs were mixed 1:1 and cultured with DMSO or under selective pressure.
(G and H) Western blot (G) and competitive proliferation assay (H) for THP-1-Cas9 cells expressing indicated sgRNAs and treated as indicated.
(I) Annexin-V analysis of THP-1-Cas9 cells expressing indicated sgRNAs.
(J) Schematic of analog-sensitive (AS) mutant CDK9 (CDK9AS/AS) bound by the inhibitory ATP analog 1-NA-PP1.
(K) Competitive proliferation assay for THP1 CDK9AS/AS cells expressing CFP-Cas9/sgSCR or mCherry(CH)-Cas9/sgINTS6 treated with 1-NA-PP1.
(L) Competitive proliferation assay for THP-1-Cas9 cells expressing indicated sgRNAs treated with indicated CDK inhibitors.
(M) Competitive proliferation assay for HeLa cells expressing CFP-Cas9/sgSCR or CH-Cas9/sgINTS6 treated with CDK9i.
(N) Western blot of HeLa cells expressing sgSCR or sgINTS6 (mixed) treated with CDK9i.
(O and P) Competitive proliferation assay for (O) MM1.S and (P) HS5 cells expressing CFP-Cas9/sgSCR or CH-Cas9/sgINTS6 treated with CDK9i.
(Q) Annexin-V analysis of D. melanogaster S2-Cas9 expressing SCR or IntS6 sgRNAs.
Blue dots (C and E) represent nominal p value <0.01. (G)–(I), (K)–(N), and (Q) are representative of 3 experiments. (O) and (P) are representative of 2 experiments. (H), (I), (K)–(M), and (Q) were analyzed by 2-way ANOVA, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To directly assess antagonism to CDK9 inhibition on a transcriptional level, we used a flow cytometry-based readout of nascent RNA production using click chemistry-based fluorescent 5-ethynyl uridine (EU) labeling (Figure S1B). As expected, CDK9i treatment of THP-1 cells resulted in a time- and dose-dependent reduction of EU incorporation (Figure S1C). Using genome-wide CRISPR-KO libraries, we screened for THP-1 cells that retained EU incorporation, and therefore transcription, in the presence of CDK9i (Figure S1D). sgRNAs targeting INTS6 were significantly enriched (Figures 1C, 1D, and S1E), revealing INTS6 as the only gene consistently identified across all screens (Figure 1E; Table S1). In contrast, the majority of Integrator subunits did not score significantly, although enrichment of sgRNAs targeting INTS8 and INTS12 was observed in individual phenotypic and transcription screens (Figure S1F), suggesting a possible role in regulating the CDK9i response.
In competitive proliferation assays, INTS6 deletion conferred a strong survival advantage to THP-1 and MV4;11 cells treated with AZ5576 (CDK9i) compared to scrambled sgRNA-expressing control cells, with no competitive advantage to DMSO-treated cells (Figures 1F–1H and S1G). INTS6 deletion desensitized cells to CDK9i-induced apoptosis in a time- and dose-dependent manner (Figures 1I and S1H), and conferred a survival advantage to targeted degradation of CDK9 using a proteolysis targeting chimera (22–533) (Figures S1I and S1J). Finally, INTS6 loss conferred resistance to CDK9 inhibition using the ATP analog 1-NA-PP1 in THP-1 cells engineered to express analog-sensitive CDK9 alleles (CDK9AS/AS) (Figures 1J, 1K, and S1K), demonstrating that the resistance observed is not the result of off-target effects of the small molecule CDK9i (Gressel et al., 2017).
Disruption of the Integrator complex through deletion of INTS3 and INTS11 did not confer resistance to CDK9 inhibition (Figures S1L and S1M). INTS6 loss did not provide a competitive advantage for small molecule inhibition of other transcriptional CDKs (t-CDKs) CDK7, CDK11, or CDK12/13 and did not confer resistance to the chemotherapeutic agent cytarabine (Figures 1L and S1N), highlighting the specificity of the INTS6 deletion phenotype. Importantly, ectopic expression of INTS6 re-sensitized cells with INTS6 loss to CDK9i (Figures S1O–S1Q), and INTS6 deletion allowed for efficient EU incorporation in the presence of CDK9i compared to control cells (Figure S1R). Moreover, INTS6 loss was associated with acquired resistance to CDK9i in MV4;11 cells cultured under selective pressure of increasing CDK9i doses for 11 weeks (Figure S1S).
INTS6 deletion also provided competitive advantage to CDK9i in HeLa, MM1.S, and two non-transformed cell lines (HS-5 and BJ-T) (Figures 1M–1P and S1T). Analogous results were obtained following deletion of IntS6 in D. melanogaster-derived S2 cells, a phenotype that was rescued by ectopic expression of full-length IntS6 protein (Figures 1Q, S1U, and S1V). These data show that experimental or acquired loss of INTS6 confers a selective resistance to CDK9i across species and cell lines, which is independent from disruption of the Integrator complex, off-target effects, or general antagonism of t-CDKs.
INTS6 bridges the interaction between Integrator and PP2A
To determine how INTS6 regulates the transcriptional and biological responses to CDK9i, liquid chromatography-tandem mass spectrometry (LC-MS/MS) was performed to investigate the INTS6/Integrator interactome. Immunoprecipitation (IP) of INTS6 from THP-1 and MV4;11 whole cell lysates revealed co-purification of the other 13 Integrator complex members, RNAPII core subunits, and enrichment of PP2A phosphatase complex subunits PPP2R1A/PPP2R1B and PP2A-C/PPP2CB (Figures 2A, S2A, and S2B). Affinity purification of PP2A-C from HeLa nuclear extracts identified Integrator and RNAPII as the most abundant interactors of nuclear PP2A, with the reciprocal IP for INTS6 identifying all Integrator complex subunits, RNAPII, PPP2R1A, and PP2A-C (Figure 2B). Similar results were obtained in epitope tagged IntS6 IP-MS experiments in D. melanogaster S2 cells, attesting to the evolutionary conservation of the Integrator, PP2A, and RNAPII association (Figure S2C). Altogether, these data reveal an interaction network between Integrator, RNAPII and PP2A as well as the catalytic subunit of the PP1 phosphatase (Figures 2A–2C). The interaction between endogenous PP2A and INTS6 was validated by coimmunoprecipitation (coIP) in THP-1 cells through endogenous V5-tagged PPP2R1A and in HeLa cells through endogenous INTS6 (Figures 2D and 2E).
Figure 2. INTS6 bridges the interaction between Integrator and PP2A.

(A) Log10 iBAQ protein scores for INTS6 IP mass spectrometry (MS) experiments in THP-1 versus MV4;11 cells (filtered for isotype-control).
(B) Log10 iBAQ proteins scores for INTS6 versus PP2A-C IP MS experiments from HeLa nuclear extract.
(C) Protein-protein interaction network of INTS6, RNAPII, and PP2A interaction partners identified in THP-1, MV4;11, and HeLa INTS6 IPs.
(D and E) CoIP western blot of (D) endogenous V5-tagged PPP2R1A IP in THP-1 cells and (E) endogenous PP2A-C, INTS3, and INTS11 IPs in HeLa cell nuclear extract.
(F) Log10 iBAQ proteins scores for PP2A-C IP MS experiments from shLUC versus shINTS6 HeLa cell chromatin extract.
(G and H) PP2A-C coIP western blot in (G) shLUC-, shINTS5-, shINTS6-, shINTS8-, and shINTS12-infected HeLa cell nuclear extract, and (H) THP-1-Cas9 sgSCR/INTS6-KO cells.
(I) Relative expression of misprocessed UsnRNA from shLUC-, shINTS6-, shINTS8-, or shINTS2-infected HeLa cells, normalized to 18S ribosomal RNA. GUSB is used as control mRNA.
(J) Glycerol gradient fractions of nuclear extracts from shLUC-, shINTS6-, shINTS8-, or shINTS2-infected HeLa cells. IP MS experiments are representative of 3 (THP-1) or 2 (HeLa, MV4;11) experiments. Western blots are representative of 3 experiments. qPCR was analyzed by one-way ANOVA with Tukey’s HSD post hoc test, ****p < 0.0001.
See also Figure S2.
Although most PP2A is cytoplasmic, sub-cellular fractionation of HeLa cells indicated that a substantial fraction of the phosphatase complex is found in the nuclear compartment and the insoluble chromatin fraction, where the Integrator complex is also detected (Figure S2D). Short hairpin RNA (shRNA)-mediated depletion of INTS6 in HeLa cells abrogated the interaction between PP2A-C and components of Integrator complex and RNAPII compared to shLuciferase (LUC) control cells (Figure 2F). Depletion of INTS6 and Integrator subunits 5, 8, and 12 revealed that the PP2A-C interaction was unperturbed by depletion of either INTS5 or INTS12, but was greatly diminished following loss of INTS6 and INTS8 (Figures 2G and S2E), suggesting that INTS8 also contributes to PP2A recruitment to Integrator. Loss of the Integrator-PP2A interaction was similarly observed in THP-1 cells on deletion of INTS6 in coIP experiments between INTS11 and PP2A-C (Figures 2H and S2F). INTS6 or INTS8 loss did not impact the catalytic activity or structural integrity of the Integrator complex because their depletion did not increase misprocessed UsnRNAs or perturb migration of the Integrator complex on a glycerol gradient, in contrast to the depletion of the core-subunit INTS2 (Figures 2I, 2J, S2G, and S2H). Interestingly, although INTS8 was required for INTS6 recruitment, deletion of INTS6 did not impact INTS8 incorporation into the Integrator complex (Figures S2H–S2J). These data demonstrate that INTS6 and INTS8 are part of an Integrator module responsible for recruitment of PP2A that is functionally and structurally distinct from the catalytic INTS4/9/11 module.
INTS6 mediates the recruitment of PP2A to the transcription pause-release checkpoint
To investigate co-localization of Integrator, PP2A, and CDK9 complexes across the genome, chromatin immunoprecipitation sequencing (ChIP-seq) was performed in THP-1 cells for CDK9, BRD4, RNAPII, INTS6, INTS11, and PPP2R1A. Similar to BRD4, RNAPII, and CDK9, Integrator subunits INTS6 and INTS11 and the PP2A subunit PPP2R1A localized to regions at or around transcription start sites (TSS) (Figure 3A). CDK9i treatment resulted in retention of RNAPII at TSS-proximal regions, consistent with the induction of transcriptional pausing concomitant with an increase in chromatin-bound CDK9 (Figures 3A, 3B, and S3A). CDK9i-induced CDK9 recruitment was also observed in INTS6 KO cells, albeit to a lesser extent, suggesting that resistance mediated by INTS6 loss does not arise from increased CDK9 recruitment to chromatin (Figure S3B). BRD4 and INTS11 also accumulated at the TSS following CDK9i treatment, consistent with the physical interaction between either BRD4 (Jang et al., 2005) or Integrator with RNAPII (Baillat et al., 2005). Notably, INTS6 followed an opposite trend, with slightly decreased recruitment after exposure to CDK9i (Figures 3A, 3B, and S3A).
Figure 3. INTS6-dependent dynamic recruitment of PP2A at actively transcribed genes.

(A and B) (A) Average TSS ChIP-seq profiles, metagene occupancy heatmaps and (B) representative ChIP-seq signal (FUT4 locus) for indicated proteins in THP-1 cells treated with CDK9i for 2 h.
(C) Representative RNAPII and PPP2R1A ChIP signal (BTG2 locus) in THP-1 cells treated with LPS for 3 h.
(D and E) Average gene profiles for (D) RNAPII and (E) PPP2R1A ChIP-seq at LPS-induced genes (n = 35) in THP-1 cells treated as per (C).
(F) PPP2R1A ChIP-seq coverage at LPS-induced genes under indicated conditions.
(G) Representative ChIP-seq signal (CTGF locus) for indicated proteins in HeLa cells treated with EGF (0.1 μg/mL; 15 min). INTS11 ChIP-seq tracks from Gardini et al. (2014).
(H and I) Average ChIP-seq profiles for (H) RNAPII and (I). PPP2R1A at EGF-induced genes (n = 50) in HeLa cells treated as per (G).
(J and K) (J) Average PPP2R1A ChIP-seq profile and (K) RNAPII-normalized PPP2R1A ChIP-seq coverage at PP2A-occupied genes (n = 1398, defined in S3F) in HeLa cells treated with 10 μM SMAP for 2 h.
(L) Average ChIP-seq profile for PP2A-C at EGF-induced genes in HeLa cells treated with EGF.
(M and N) (M) Average PPP2R1A ChIP-seq profile and N. RNAPII-normalized PPP2R1A ChIP-seq coverage at PP2A-occupied genes in shLUC- and shINTS6-infected HeLa cells.
(O) Representative PPP2R1A ChIP-seq signal (CITED4 locus) in shLUC- and shINTS6-infected HeLa.
(F), (K), and (N) were analyzed by unpaired, two-sided Student’s t test, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To assess the dynamic recruitment of PP2A in response to a transcriptional stimulus, ChIP-seq assays were performed using THP-1 cells treated with lipopolysaccharide (LPS). LPS treatment enhanced RNAPII recruitment, but also stimulated recruitment of PPP2R1A at the TSS and along the entire gene body of LPS-responsive genes (Figures 3C–3F and S3C). To further assess PP2A recruitment to chromatin in response to physiological stimulus, epidermal growth factor (EGF) was used to stimulate transcription of immediate early genes in HeLa cells (Gardini et al., 2014). A dramatic recruitment of RNAPII, PPP2R1A, PP2A-C, and INTS11 along the entire gene body and the 3′ end of EGF-responsive genes was observed following treatment with EGF (Figures 3G–3I, S3D, and S3E). In both THP-1 and HeLa cells, PP2A recruitment peaked immediately 3′ of the pausing site (Figures S3F and S3G). Peak-centric analysis revealed that conserved PPP2R1A peaks overlapped almost exclusively with RNAPII peaks (>98%) and marked genes with higher levels of nascent transcription (Figure S3H). These data indicate that PP2A co-localizes with RNAPII across the genome and suggests a correlation between PP2A recruitment and transcriptional output, in particular at regions encoding transcriptional regulators including RNAPII-associated DNA-binding transcription factors (Figure S3I).
To further establish the ability of PP2A to bind a broad set of expressed genes, we treated HeLa cells with a SMAP that stabilizes heterotrimeric PP2A complexes and activates catalytic PP2A-C in dimeric holoenzymes (Kauko et al., 2018; Leonard et al., 2020; Westermarck and Neel, 2020). Spike-in normalized ChIP-seq assays showed significant increase of PPP2R1A binding across the previously defined PPP2R1A-occupied genes in all replicates (Figures 3J and 3K), whereas total RNAPII levels remained unchanged (Figures S3J–S3L). Moreover, ChIP-seq profiles for the catalytic subunit (PP2A-C) demonstrated a distribution mirroring PPP2R1A across EGF-responsive genes and robust correlation across all RNAPII-transcribed genes (Figures 3L and S3M).
ChIP-seq assays in HeLa cells expressing shRNAs targeting INTS6 demonstrated that INTS6 depletion resulted in a reduction in PPP2R1A binding across actively transcribed genes, which was phenocopied by depletion of INTS8 (Figures 3M–3O and S3N–S3P). Total protein levels of PPP2R1A and PP2A-C were unchanged by INTS6 or INTS8 depletion (Figure 2G). Taken together, these findings demonstrate that PP2A co-occupies sites across the genome with P-TEFb and Integrator, and its dynamic recruitment relies on INTS6 and INTS8.
INTS6/PP2A dynamically controls CDK9 substrates including RNAPII CTD phosphorylation levels
To investigate whether the functional antagonism between CDK9 and INTS6/PP2A was reflected at the phosphorylation level, reverse phase protein arrays (RPPA) and global phospho-proteomics MS assays were performed (Figure 4A). RPPA analysis of RNAPII CTD phosphorylation (phospho-Ser2 [pS2], phosphoSer5 [pS5], and phospho-Ser7 [pS7]) demonstrated that no significant loss of phosphorylation was observed for any of the phospho-CTD sites in THP-1 cells with stable deletion of INTS6 (sgINTS6-KO) following CDK9i treatment, in contrast to sgSCR control cells (Figure 4B). Concordantly, total phospho-proteome analysis identified a large number of CDK9i-responsive phospho-peptides in sgSCR THP-1 cells that were resistant to CDK9i treatment in sgINTS6-KO cells (Figures 4C, S4A, and S4B) including key CDK9 substrates such as SUPT5H (DSIF, SPT5) (Figures 4D and S4C), LEO1, and degenerate heptad repeats in the RNAPII CTD (POLR2A) (Figures 4E and S4D). Gene Ontology (GO) analysis of INTS6-dependent CDK9i targets revealed a strong enrichment for mRNA processing and RNA metabolic processes, suggesting that INTS6/PP2A oppose CDK9 kinase activity on multiple levels (Figure S4E).
Figure 4. Loss of INTS6/PP2A results in decreased turnover of CDK9 substrates.

(A) Phosphorylation studies in THP-1-Cas9 sgSCR/sgINTS6-KO cells.
(B) RNAPII CTD reverse phase protein array (RPPA): log fold change in relative fluorescence intensity for indicated antibodies in CDK9i-treated versus untreated THP-1-Cas9 sgSCR/sgINTS6-KO cells.
(C) Heatmap of phospho-peptide Z scores in THP-1-Cas9 sgSCR/sgINTS6-KO cells treated for 2 h, p value <0.05. Differentially phosphorylated.
(D and E) (D) SUPT5H and (E) POL2RA peptides in THP-1-Cas9 sgSCR/sgINTS6-KO cells treated for 2 h.
(F) Western blot of THP-1-Cas9 sgSCR/sgINTS6-KO cells treated as indicated for 2 h.
(G) Western blot of THP-1 CDK9AS/AS sgSCR/sgINTS6 cells treated with 1-NA-PP1 for 2 h.
(H) In vitro recombinant kinase/phosphatase assay; recombinant RNAPII CTD peptide was incubated with ATP, CDK9/cyclinT1, and/or PP2A as indicated for 30 min.
(I) ADPGlo assay (left) and quantitation of ATP to ADP conversion by recombinant CDK9/cyclin T1 incubated with recombinant RNAPII CTD peptide, PP2A, and/or CDK9i as indicated for 30 min.
(J) Western blot of THP-1 cells treated as indicated (15 min pre-treatment with Phendione; CDK9i, 2 h). 3 (RPPA) and 10 (phospho-peptide MS) biological replicates were analyzed. Western blots are representative of 3 experiments.
(I) represents the mean ± SEM of 3 experiments. For (F) and (G), TUBULIN (#) is representative of individual blots for phospho-CTD and phospho-SPT5 sites. RPPA was analyzed using Welch unpaired t test and ADPGlo assay was analyzed using an unpaired student’s test, *p < 0.05, **p < 0.01.
Western blot analysis of phospho-residues of the RNAPII-CTD and phospho-DSIF (SPT5) confirmed that THP-1 sgINTS6-KO cells were refractory to the CDK9i-mediated loss of phosphorylation observed in control cells (Figure 4F), with this phenotype recapitulated in non-transformed BJ-T fibroblasts (Figure S4F). Moreover, INTS6 deletion abrogated the reduction in DSIF and RNAPII-CTD phosphorylation that was observed on inhibition of AS-CDK9 using 1-NA-PP1 in THP-1 CDK9AS/AS cells (Figure 4G). Importantly, INTS6 loss did not affect the molecular consequences of CDK12/13 inhibition with THZ-531, with a similar reduction of RNAPII CTD phosphorylation observed in both control and sgINTS6-KO cells (Figure S4G). IntS6 deletion in D. melanogaster S2 cells also conferred resistance to CDK9i-mediated loss of RNAPII-CTD pS2 levels, indicating that this role is evolutionarily conserved (Figure S4H).
To determine if PP2A can directly dephosphorylate RNAPII CTD residues that are phosphorylated by P-TEFb, in vitro kinase/phosphatase assays were performed using purified recombinant proteins (Figure S4I). Incubation of a recombinant RNAPII-CTD peptide with an active P-TEFb complex resulted in hyper-phosphorylation of the CTD peptide, as evidenced by pS2 and pan-phospho CTD western blot analysis (Figure 4H). Addition of recombinant PP2A-C greatly reduced the CDK9-mediated CTD phosphorylation, which was partially rescued through PP2A inhibition by okadaic acid, but not by simultaneous CDK9 inhibition with AZ5576 (Figure 4H). Assessment of CDK9 kinase activity in vitro by measuring ATP to ADP conversion demonstrated that CDK9 kinase activity was increased in the presence of its substrate, could be inhibited by addition of a CDK9 inhibitor, but was unaffected by addition of PP2A (Figure 4I). PP2A was still capable of dephosphorylating a CTD peptide pre-phosphorylated by CDK9, indicating that PP2A can act directly on the CTD (Figure S4J).
Consistent with the phenotype observed in sgINTS6-KO cells, PP2A inhibition with phendione (Yue et al., 2020) rescued the loss of RNAPII-CTD and SPT5 phosphorylation observed on CDK9 inhibition in THP-1 cells treated with CDK9i (Figure 4J) as well as THP-1 CDK9AS/AS cells treated with 1-NA-PP1 (Figure S4K). Rescue of CDK9-dependent phospho-marks was also observed on co-treatment with CDK9i and dual PP1/PP2A inhibitors calyculin A and okadaic acid, suggesting a complex interplay between these two phosphatases and CDK9 (Figure S4L). These data highlight an evolutionarily conserved kinase/phosphatase antagonism between CDK9 and INTS6/PP2A that regulates the critical transcription regulatory factors RNAPII and SPT5.
Loss of INTS6/PP2A overrides a CDK9i-induced block in transcription elongation
To assess the impact of INTS6/PP2A loss on CDK9i-induced suppression of nascent transcription, 4-thiouridine metabolic labeling followed by RNA sequencing (4sU-seq) was performed in sgINTS6-KO and sgSCR THP-1 cells (Figure 5A). Spike-in normalized 4sU-seq analysis revealed that the global CDK9i-dependent decrease of transcription seen in sgSCR THP-1 cells was significantly rescued across all expressed genes in THP-1 sgINTS6-KO cells, even after 6 h of sustained CDK9 inhibition (Figures 5B and S5A). Similarly, nascent transcription in THP-1 cells was significantly rescued by INTS6 loss at highly expressed genes (top 1,000), and the most CDK9i-sensitive genes, which are enriched for mRNA metabolic processes and immune effector processes, respectively (Figures 5C, 5D, and S5A; Table S2). A significant rescue of nascent transcription in sgINTS6-KO cells compared to sgSCR was also observed at key loci, including at the IL6R and MYC genes (Figures 5E, S5B, and S5C). ChIP-seq assays further demonstrated that compared to sgSCR control cells, sgINTS6-KO THP-1 cells exhibited reduced accumulation of total RNAPII at TSS proximal regions as well as increased maintenance of RNAPII-CTD pS2 throughout the gene body and at the transcription end site (TES) following CDK9i treatment, even relative to total RNAPII levels (Figures 5F and S5D–S5I). This phenotype was conserved across highly expressed genes and those most sensitive to CDK9 inhibition, whereas no difference was observed for genes that are least sensitive to CDK9i-mediated transcriptional inhibition that are enriched for cellular catabolic processes (Figure S5H; Table S2)). Calculation of the pausing index for all transcribed genes showed that INTS6 loss reduced CDK9i-induced pausing in comparison to control sgSCR THP-1 cells, suggesting effective RNAPII pause-release even when CDK9 activity is impaired (Figure 5G). This phenotype was conserved in D. melanogaster S2 cells, with reduced RNAPII pausing observed in CDK9i-treated sgIntS6 S2 cells compared to sgSCR control cells (Figure S5J).
Figure 5. INTS6/PP2A loss overrides CDK9i-induced transcriptional pausing.

(A) 4sU labeling and analysis of nascent transcription in THP-1-Cas9 sgSCR/sgINTS6-KO cells.
(B–D) Log fold change in 4sU-seq signal (CPM; CDK9i versus untreated) in THP-1-Cas9 sgSCR/sgINTS6-KO cells for (B) all expressed, (C) highly expressed (top 1,000; untreated sgSCR cells), and (D) the most and least sensitive genes to CDK9i.
(E) 4sU-seq signal (IL6R locus) under indicated conditions.
(F) RNAPII pS2 ChIP-seq signal (IL6R locus) in THP-1-Cas9 sgSCR/sgINTS6-KO cells treated for 2 h.
(G) Pausing index in THP-1-Cas9 sgSCR/sgINTS6-KO cells treated for 2 h.
(H) RNAPII and RNAPII pS2 ChIP-seq signal (DDIT4 locus) in shLUC- and shINTS6-infected HeLa cells treated with CDK9i and EGF.
(I) Average RNAPII pS2 ChIP-seq profile under the same conditions at EGF-response genes (n = 50).
(J) Fast-GRO signal (FOSB locus) in shLUC or shINTS6 infected HeLa cells treated with CDK9i and EGF.
(K) Average Fast-GRO signal across EGF response genes.
(L) Fast-GRO pausing index for CDK9i-treated shLUC- and shINTS6-infected HeLa cells at highest-expressed genes.
(B)–(D) were analyzed by unpaired, two-sided Student’s t test, ****p < 0.0001.
See also Figure S5.
To investigate whether INTS6 loss can rescue CDK9i-mediated suppression of acute EGF-induced transcriptional responses, INTS6 was depleted in HeLa cells that were pre-treated with CDK9i prior to stimulation with EGF (Figure S5K). Accumulation of total- and pS2-RNAPII in response to EGF stimulation at EGF-responsive genes was retained in INTS6-depleted cells in contrast to shLUC control cells treated with AZ5576 and the less specific CDK9 inhibitor flavopiridol (Figures 5H, 5I, and S5L–S5Q). In agreement with a role for INTS8 in PP2A recruitment, its depletion phenocopied INTS6 loss and conferred resistance to CDK9i-induced pausing (Figures S5O–S5Q). FastGRO assays (Barbieri et al., 2020) measuring nascent RNA production also showed that INTS6 depletion counteracted CDK9i-induced transcriptional pausing at all EGF-stimulated genes, whereas a broader analysis of the most highly expressed genes demonstrated that INTS6 depletion decreased the pausing index at nearly all active loci (Figures 5J–5L). To determine whether PP2A depletion can also drive escape from CDK9i-induced pausing, analogous assays were also performed in HeLa cells with shRNA-mediated knockdown of PPP2R1A (Figure S5R). Depletion of PPP2R1A resulted in sustained EGF-induced pS2 RNAPII coverage across EGF-stimulated genes in the presence of the CDK9i, particularly along the first half of the gene body (Figure S5S). These data demonstrate that the loss of INTS6/PP2A complex decreases RNAPII susceptibility to CDK9i-induced pausing under steady-state conditions and in response to acute-transcriptional cues.
The INTS6/PP2A axis fine-tunes acute transcriptional responses
We next investigated the importance of the Int-PP2A complex in controlling RNAPII-driven transcription under steady-state conditions and in response to acute pro-inflammatory and mitogenic stimuli. ChIP-seq experiments performed in THP-1 cells following acute INTS6 depletion (60 h) demonstrated increased RNAPII occupancy across the TSS region and gene body, indicating that steady-state RNAPII-driven transcription is modulated by INTS6 loss in the absence of pharmacological CDK9 inhibition (Figures S6A–S6C). FastGRO analyses of INTS6-depleted HeLa cells showed increased elongation along the body of the 3,000 highest expressed genes, even in the absence of CDK9i (Figure S6D).
In the context of the acute EGF-response in HeLa cells, INTS6 depletion increased RNAPII pS2 levels across the gene body and 3′ end of EGF-responsive genes compared to control cells, with moderate changes observed at TSS-proximal regions (Figures 6A and 6B). In support of the notion that INTS6 and PP2A functionally interact to regulate transcriptional elongation in this context, these findings were phenocopied by PPP2R1A depletion (Figures 6C and 6D). In contrast, no modulation of pS2 RNAPII levels was observed in INTS12-depleted cells. Moreover, perturbation of the INTS6/PP2A axis resulted in a significant increase of EGF-induced nascent RNA transcription as measured by transient-transcriptome sequencing (Figure S6E). Concordantly, 3′ mRNA-seq analysis of EGF responses revealed that INTS6-depletion amplified activation of EGF-responsive genes, an effect also observed in the presence of CDK9i (Figures 6E–6G and S6F). Similarly, the transcriptional response to LPS was significantly amplified in sgINTS6-KO THP-1 cells compared to sgSCR control cells across all time points, which included key inflammatory LPS-target genes, such as CXCL1, IL1B, IL23A, and SERPINE2 (Figures 6H–6L, S6G, and S6H). Collectively, these data provide evidence that PP2A recruitment via INTS6 regulates steady-state transcription and is required to fine-tune acute transcriptional responses to pro-inflammatory and mitogenic stimuli.
Figure 6. The INTS6/PP2A axis fine-tunes acute transcriptional responses.

(A and B) (A) RNAPII and RNAPII pS2 ChIP-seq signal (CTGF locus), and (B) average RNAPII pS2 ChIP-seq profile at EGF-response genes (n = 50) in shLUC-, shINTS6-, and shINTS12-infected HeLa cells after EGF treatment, with quantification of the total RNAPII-normalized pS2 ChIP-seq coverage.
(C and D) (C) RNAPII and RNAPII pS2 ChIP-seq signal (CTGF locus), and (D) average RNAPII pS2 ChIP-seq profile at EGF-response genes in shLUC- and shPPP2R1A-infected HeLa cells after EGF treatment, with quantification of the total RNAPII-normalized pS2 ChIP-seq coverage.
(E) Acute EGF stimulation of shLUC- and shINTS6-infected HeLa cells.
(F) Log fold change (CPM) in EGF-treated versus untreated shLUC- and shINTS6-infected HeLa cells.
(G) Log fold change difference between shLUC- and shINTS6-infected HeLa cells (EGF versus untreated).
(H) Acute LPS stimulation of THP-1-Cas9 sgSCR/sgINTS6-KO cells.
(I) GSEA profile of THP-1-Cas sgSCR cells treated with LPS for 2 h.
(J) Log fold change (CPM) in LPS-treated versus untreated THP-1-Cas9 sgSCR and sgINTS6-KO cells.
(K) Log fold change difference between THP-1-Cas9 sgINTS6-KO and sgSCR cells (LPS versus untreated).
(L) LPS-target gene expression in THP-1-Cas9 sgSCR and sgINTS6-KO cells (LPS versus untreated).
(B), (D), (F), and (J) were analyzed by unpaired, two-sided Student’s t test, **p < 0.01, ***p < 0.001, ****p < 0.0001.
See also Figure S6.
To investigate whether the Int-PP2A complex may be dysregulated in cancer, INTS6 expression and copy number data from The Cancer Genome Atlas (TCGA) were analyzed. This revealed that INTS6 deletion is a common occurrence in a wide variety of cancer types, which strongly correlates with INTS6 mRNA expression and overall poor-prognosis (Figures S6I–S6K). Analysis of gene-signatures associated with INTS6 deletion revealed that INTS6 loss was associated with the inductionof E2F cell cycle and MYC signatures with concurrent downregulation of genes located proximal to the INTS6 genomic locus on the long arm of chromosome 13 at position 14 (13q14) (Figures S6L–S6N). These data suggest that loss or disruption of the Int-PP2A complex may impact cancer etiology and pathogenesis.
Therapeutic synergy between CDK9 inhibition and PP2A activation in cancer
Targeting of CDK9 can be therapeutically efficacious in pre-clinical models of leukemia and lymphoma (Baker et al., 2016; Gregory et al., 2015), and CDK9i have progressed to clinical trials for both hematological and solid malignancies (Chou et al., 2020). Recently SMAPs have been developed that are therapeutically effective in combination with MEK inhibitors in KRAS mutant lung cancer models (Kauko et al., 2018). The observation that SMAPs potentiate recruitment of PP2A to chromatin at actively transcribed genes (Figures 3J and 3K) provided a strong rationale to target the PP2A-Integrator-CDK9 axis in cancer via concurrent CDK9 inhibition and SMAP-mediated PP2A activation. Drug-synergy studies in THP-1 and MV4;11 cells revealed that concurrent treatment with the SMAP DBK-1154 (Kastrinsky et al., 2015; Sangodkar et al., 2017) greatly enhanced CDK9i-induced cell death, with no single agent SMAP activity observed (Figures 7A, S7A, and S7B). Importantly, no synergy was observed in sgINTS6-KO THP-1 cells highlighting that an intact Int-PP2A complex is required for effective combination therapy (Figure 7A). Synergy between CDK9i and SMAPs was also observed to varying degrees across a panel of solid and hematopoietic cancer cell lines, including multiple myeloma, non-MLL-driven leukemia and colorectal cancer cells (Figures S7C–S7E). The synergy between CDK9i and SMAP was also reflected on the transcriptional level with enhanced RNAPII pausing observed in THP-1 cells treated with both compounds, compared to single agent CDK9i (Figures 7B and 7C). The minimal impact of single agent SMAP treatment on RNAPII pausing (Figures 7B and 7C) suggests that PP2A activity is not rate-limiting for maintenance of this transcriptional checkpoint under basal conditions. In HT-29 cells, dual CDK9i and SMAP treatment significantly suppressed global mRNA transcription compared to both single agents alone, with combination treatment resulting in increased downregulation of key genes associated with oncogenesis in colorectal cancer (Figures S7F and S7G).
Figure 7. Therapeutic and molecular synergy between PP2A agonist and CDK9i.

(A) Annexin-V analysis of THP-1-Cas9 sgSCR and sgINTS6-KO cells treated with CDK9i, DBK-1154, or combination. Orange lines indicate single agent CDK9i activity and maximal synergistic activity with DBK-1154.
(B) RNAPII ChIP-seq pausing index in THP-1 cells treated with CDK9i (170 nM, 2 h) with/without 15 min pre-treatment with DBK-1154 (10 μM).
(C) Representative RNAPII ChIP-seq signal (IL6R) locus under the same conditions as (B).
(D) Schematic of in vivo cross-sectional analysis and survival experiments.
(E) Quantitation of mCherry-positive AML blasts in bone marrow and spleen post-therapy.
(F) Kaplan-Meier survival curves at the conclusion of 4 rounds of therapy for MV4–11 and A431 tumor models. 1 mouse in the MV4–11 cohort (combination group) and 1 mouse in the A431 cohort (AZD′4573 group) were censored due to acute toxicity.
(G) Schematic of Integrator/PP2A/CDK9 axis.
(A) represents the mean ± SEM of 3 experiments, (E) represents the mean ± SD of 3 mice/group, and (F) represents 8 (MV4–11) or 7 (A431) mice per group. (A) was analyzed by 2-way ANOVA, (E) was analyzed by Student’s t-test, and (F) was analyzed by log-rank (Mantel-Cox) test, *p < 0.05, **p < 0.01, ***p < 0.001.
See also Figure S7.
The therapeutic efficacy of combined CDK9 inhibition with PP2A agonism in vivo was assessed in xenograft models of leukemic and solid cancer xenografts. NOD-scid IL2Rgnull (NSG) mice bearing mCherry/Luciferase-tagged MV4–11 leukemias or A431 subcutaneous tumors were treated with the in vivo-optimized CDK9 inhibitor AZD′4573 (Cidado et al., 2020) and DBK-1154 (Figure 7D). Cross-sectional analysis of bone marrow and spleen showed marked reduction in the number of mCherry-positive MV4;11 cells at 96 h and overall spleen size at 24 h post-therapy relative to single agent and vehicle controls (Figures 7E, S7H, and S7I). This suggests that AML blasts rapidly undergo apoptosis in response to combination therapy, concomitant with the induction of normal erythroblastic and myeloblastic maturation that are maintained at endpoint (Figures S7J and S7K). Importantly, this translated into significantly prolonged survival in leukemic mice treated with the combination regimen compared to vehicle or either single agent (Figure 7F). Similarly, combination therapy in the A431 solid tumor model demonstrated reduced tumor growth rates and significantly reduced mean tumor volume relative to the vehicle group, resulting in enhanced overall survival (Figures 7F, S7L, and S7M). Collectively, these data demonstrate that concomitant CDK9 inhibition and PP2A agonism results in enhanced anti-cancer effects in pre-clinical models of solid and hematopoietic malignancies.
DISCUSSION
RNAPII-driven transcription is a highly conserved process comprising distinct checkpoints, each controlled by dedicated transcriptional CDKs (t-CDKs) and their cognate cyclins (Adelman and Lis, 2012; Mayer et al., 2017). How phosphatases balance t-CDK-mediated RNAPII phosphorylation to control transcription cycle checkpoints in mammals is poorly understood (Mayfield et al., 2016). This study details the discovery of an auxiliary phosphatase module of the RNAPII-associated Integrator complex containing PP2A, INTS6, and INTS8 (Int-PP2A), which is actively recruited to chromatin in response to cellular cues to functionally antagonize CDK9 (Figure 7G). Loss of IntPP2A alters the phosphorylation dynamics throughout the transcription cycle, such that limited CDK9 activity is required for effective RNAPII-driven transcription, conferring molecular and biological resistance to CDK9 inhibition. Although INTS6 loss is tolerated under steady-state conditions, acute depletion of INTS6 or INTS8 globally disrupts RNAPII transcription. Importantly, the absence of Int-PP2A antagonism of CDK9 results in unopposed activation of acute transcriptional responses to cellular stress, growth signaling, or inflammatory cues. We posit that antagonism of CDK9 kinase activity at the pause-release checkpoint and throughout the rest of the transcription cycle is critical to fine-tune transcriptional output, and our findings establish a therapeutically targetable role for Int-PP2A in the dynamic control of RNAPII transcription in both normal and cancer settings.
The Integrator-PP2A-CDK9 axis may have co-evolved with pause-release in early metazoans, as organisms required a finer control of transcription in order to respond to increasingly diverse developmental, environmental, and immunological cues (Adelman and Lis, 2012; Baillat et al., 2005; Lai et al., 2015; Luo et al., 2012). Concordantly, the Integrator complex first arises in metazoans and has a multifaceted role in transcription. The INTS11/INTS9 catalytic core coordinates processing of short noncoding RNAs and the regulation of productive RNAPII elongation by affecting SEC recruitment and the cleavage of nascent transcripts generated by non-productive RNAPII elongation (Baillat et al., 2005; Beckedorff et al., 2020; Elrod et al., 2019; Gardini et al., 2014; Lai et al., 2015; Rienzo and Casamassimi, 2016; Rubtsova et al., 2019; Tatomer et al., 2019). Another proposed Integrator module (with INTS13) regulates transcription independent from the core complex and modulates enhancer activity (Barbieri et al., 2018). The Int-PP2A module and its role in RNAPII-mediated transcription were recently detailed by two other research groups. Zheng et al. (2020) resolved the structure of the Integrator-PP2A complex, termed INTAC, by cryo-electron microscopy (cryo-EM). Consistent with our results, this work confirmed the modular nature of the Integrator complex, which comprises a phosphatase module consisting of INTS6, PPP2R1A (PP2A-A), and PP2A-C (PP2CA) (Zheng et al., 2020) and demonstrates that INTS6 directly engages the PP2A holoenzyme in the absence of canonical beta-subunits. The observation that INTS8 stabilizes the phosphatase module by interacting with both INTS6 and PP2A (Zheng et al., 2020) is also in agreement with our data. Our observation that impaired assembly of Int-PP2A resulted in phenotypic resistance to CDK9i and a broad rescue of RNAPII CTD and SPT5 phosphorylation suggests that this complex antagonizes CDK9 throughout the transcription cycle and may extend to other distinct CDK9 substrates such as XRN2 (Laitem et al., 2015; Sansó et al., 2016). This is consistent with the model put forward by Zheng et al. (2020) that suggests a widespread rescue of RNAPII phosphorylation at multiple sites in the CTD (S2, S5 and S7). In contrast, Huang et al. (2020) suggest that INTS8 loss primarily affects RNAPII pause-release through modulation of Spt5 S666 and RNAPII CTD phosphorylation at S5 and S7. These discrepancies likely arise from the difficulty in pinpointing direct substrates of PP2A in vivo or may be due to unrecognized differences between the roles played by INTS6 and INTS8. Consistent with a role beyond pause-release, we observed that the recruitment of PP2A extends into the gene body and 3′ end of genes, mirroring RNAPII. Collectively, data from this study and complementary data from recently published work (Huang et al., 2020; Zheng et al., 2020) further demonstrate that the transcriptional role of Integrator is defined by the intricate interplay between the core complex, peripheral modules, and binding partners such as PP2A. Future studies will be required to address the precise dynamics of the Int-PP2A assembly and the role of INTS6 in driving substrate selectivity.
In addition to PP2A, PP1 and PP4 may play a critical role in mediating the dephosphorylation of CDK9 substrates at the elongation-termination transition and early stages of transcription, respectively (Cortazar et al., 2019; Eaton et al., 2020; Parua et al., 2018, 2020). In agreement with these findings, we also found PP1 immunoprecipitated with INTS6, and loss of RNAPII and DSIF phosphorylation by CDK9i was more potently antagonized when both PP2A and PP1 were inhibited. Interestingly, yeast PP1 plays an important role in regulating PP2A signaling by directly activating PP2A during mitosis (Grallert et al., 2015). This raises the intriguing possibility that in addition to its role in directly regulating SPT5 phosphorylation, PP1 may also indirectly regulate the turnover of CDK9 substrates by modulating the activity of PP2A complexes, such as Int-PP2A, during the RNAPII transcription cycle (Parua et al., 2018). Although the recruitment of PP2A is mediated by the Integrator complex, the recruitment of complexes comprising CDK9 to co-occupied loci remains to be investigated further but may rely on its interaction with the Pol2-associated-factor 1 (PAF1) containing complex (Chen et al., 2015; Yu et al., 2015; Xu et al., 2017).
INTS6 underwent a gene duplication event in mammals with the highly homologous INTS6-like (INTS6L/DDX26B) protein and the ancestral INTS6 protein proposed to locate to mitochondria (Rienzo and Casamassimi, 2016). In agreement with the disparate roles for INTS6 and INTS6L, our screens revealed no enrichment for loss of INTS6L. There is growing evidence that the loss of INTS6 may play an important role in cancer. Indeed, INTS6 was originally identified and named for being frequently deleted in cancer 1 (DICE1), with loss or downregulation of INTS6 commonly reported in prostate cancer, hepatocellular carcinoma, and hematopoietic malignancies (Filleur et al., 2009; Li et al., 2015; Röpke et al., 2005). The tumor suppressive role of INTS6 may be attributed to its proposed role in the sensor of the ssRNA (SOSS) complex, which controls the double stranded DNA damage response (Skaar et al., 2009). However, we propose that INTS6 loss may also promote oncogenic transcription by facilitating pause-release, as is observed in transcriptionally addicted MLL-rearranged leukemias and MYC-driven lymphomas (Bradner et al., 2017; Luo et al., 2012). Indeed, analysis of TCGA datasets revealed that INTS6 is frequently deleted in human cancers and correlated with poor prognosis in a subset of these malignancies. Reduction of PP2A activity through epigenetic silencing and/or somatic mutations also occurs in several tumors, including AML (Perrotti and Neviani, 2013; Shih et al., 2011). Moreover, point mutations in the PP2A scaffold subunit that recur in several gynecological malignancies may affect its binding to interaction partners such as the Integrator complex (Haesen et al., 2016; Shih et al., 2011). Notably, a recent phosphoproteome screen for phosphosites regulated by PP2A and its oncogenic inhibitory proteins CIP2A, PME-1 and SET identified a number of targets directly involved in RNAPII-mediated transcription and RNA processing, providing further putative links between PP2A, transcription, and cancer (Kauko et al., 2020). Taken together, dysregulation of the IntPP2A complex may contribute to dysregulated transcriptional states in cancer and affect disease initiation or progression, although the putative roles of INTS6 and PP2A as bona fide tumor suppressors remain to be formally demonstrated.
Using pre-clinical models of solid and hematopoietic malignancies, we demonstrated that concurrent PP2A activation and CDK9 inhibition resulted in enforced transcriptional pausing and synergistic cell death in vitro, and greatly reduced tumor burden and prolonged survival in vivo. Tricyclic sulfonamide activators of PP2A such as DBK-1154 and analogs such as DT-061 (SMAPs), can bind to and activate catalytic PP2A-C in dimeric holoenzymes devoid of B-subunits (Clark and Ohlmeyer, 2019; Kastrinsky et al., 2015; Sangodkar et al., 2017). At the molecular level, we found that SMAP treatment increased the recruitment of PP2A to chromatin, suggesting that SMAP binding to PP2A holoenzymes can facilitate formation of Int-PP2A complexes, similar to what has been observed with canonical B55 and B56 subunits (Leonard et al., 2020; Morita et al., 2020; Westermarck and Neel, 2020). The molecular mechanism by which SMAPs stabilize non-canonical PP2A trimers, such as the Int-PP2A module, is under investigation and may lead to development of a new generation of SMAPs that selectively stabilize Int-PP2A complexes for the treatment of transcriptionally addicted cancers. Taken together, these findings highlight the importance of molecular antagonism of CDKs by phosphatases in the transcription cycle and demonstrate the therapeutic benefit of dual CDK9/PP2A targeting in cancer, providing the basis for future pre-clinical and clinical studies.
Limitations of the study
There are several limitations to the current studies that require further investigation and refined in vitro and in vivo experimental model systems. In the current study, INTS6 is constitutively depleted that precludes the identification of RNAPII-related transcriptional defects that would be observed within hours of acute depletion of INTS6 using degron-systems. Similarly, acute degradation studies of PP2A components, in particular when engineered to be selective for those isoforms expressed in the nucleus, would enable the fine-dissection of PP2A’s role in RNAPII transcription in an acute setting in a manner that does not rely on small molecule inhibitors that require further optimization to ensure exclusive targeting of transcriptionally engaged PP2A-Integrator complexes. Finally, the role of the PP2A-Integrator complex in normal physiology and disruption in oncogenesis remain to be investigated in greater detail.
STAR⋆METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ricky Johnstone (ricky.johnstone@petermac.org).
Materials availability
This study did not generate any new unique reagents.
Data and code availability
Bioinformatics and proteomics datasets generated during this study are available from the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/; GSE163803, GSE163803, GSE163804, GSE163805, GSE145379) and the Proteomics Identifications Database (PRIDE, https://www.ebi.ac.uk/pride/archive/; PXD022962, PXD025060) repositories respectively.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
THP-1 (male cell line, ATCC® TIB-202™, RRID CVCL_0006), MV4;11 (male cell line, ATCC® CRL-9591™, RRID CVCL_0064), HT-29 (female cell line, ATCC® HTB-38™, RRID CVCL_0320), MM1.S (female cell line, ATCC® CRL-2974™, RRID CVCL_8792), JJN-3 (female cell line, DSMZ ACC 541, RRID CVCL_2078), OPM2 (female cell line, DSMZ ACC 50, RRID CVCL_1625), MOLM-13 (male cell line, DSMZ ACC 554, RRID CVCL_2119), HL-60 (female cell line, ATCC® CCL-240™, CVCL_A794) and OCI-AML3 (male cell line, DSMZ ACC 582 CVCL_1844) cells were cultured at 37°C and 5 percent carbon dioxide (CO2) in Roswell Park Memorial Institute medium (RMPI; ThermoFisher Scientific 11875093) supplemented with 10%–20% fetal bovine serum (FBS; Peter MacCallum Cancer Centre), 100U/mL penicillin, 100 μg/mL streptomycin (ThermoFisher Scientific 10378016) and 2mM GlutaMAX™ (ThermoFisher Scientific 35050061). THP-1 and MV4;11 cells were authenticated using short-tandem-repeat (STR) profiling. Henrietta Lacks (HeLa, female cell line - ATCC® CCL-2™, CVCL_0030) cells were cultured at 37°C and 5 percent carbon dioxide (CO2) in Dulbeccos Modified Eagle’s medium (DMEM: ThermoFisher Scientific 11995073) supplemented with 10% super calf serum (GEMcell) and 2mM L-glutamine (Corning). hTERT-immortalized BJ foreskin fibroblasts (BJ-T, male cell line; ATCC® CRL-2522™, RRID CVCL_6573), HS5 bone marrow stromal cells (male cell line, ATCC® CRL-11882™, CVCL_3720), A431 cells (female cell line, ATCC® CRL-1555™, RRID CVCL_0037) and human embryonic kidney (HEK) 293T cells (female cell line, ATCC® CRL-3216™, CVCL_0063); used for generation of lentivirus and retrovirus) were cultured at 37°C and CO2 in DMEM supplemented with 10% FBS, 100U/mL penicillin; 100 μg/mL streptomycin and 2mM GlutaMax™. Drosophila melanogaster (D.melanogaster) Schneider 2 cells (S2, male cell line - ATCC® CRL-9591 ™. RRID CVCL_Z232) were cultured in Scheinder’s Drosophila medium (ThermoFisher Scientific 21720) supplemented with 10% HI-FBS, 100U/mL penicillin; 100 μg/mL streptomycin and 2mM GlutaMax™ at room temperature and atmospheric CO2.. A431 and HT-29 cells were a gift from the Translational Research Laboratory (TRL) at the Peter MacCallum Cancer Centre.
In vivo animal studies
All in vivo experiments performed in this study were performed using procedures approved by the Peter MacCallum Cancer Centre (PMCC) Animal Ethics Committee under ethics approval numbers E555 and E592. NOD-scid IL2Rgnull (NSG) mice utilized were bred in-house (PMCC) and had not undergone any previous procedures. For disseminated MV4–11 therapy experiments, 4–6-week-old female NSG mice were IV-injected with 5 × 106 MV4;11 cells expressing the MSCV-Luc2-mCherry vector (Bots et al., 2014). For long-term survival experiments, treatment commenced when peripheral blood was detected to be 1% mCherry positive as determined by flow cytometry (day 18). Mice were randomly assigned to experimental groups (vehicle control, AZD’4573 only, DBK-1154 and combination therapy). Mice were administered 15 mg/kg AZD’4573 twice daily via intraperitoneal injection (IP; 100 mL) two hours apart, with 40 mg/kg DBK-1154 (100 mL) administered via oral gavage two hours prior to, and two hours post, the AZD’4573 injections. This regimen was implemented in a 2 days on, 5 days off manner and repeated for four consecutive cycles of therapy. For Cross-sectional analysis, therapy commenced 30 days post inoculation, and mice were treated as per the above regimen. Mice were culled 24 hours and 96 hours post the conclusion of 1 cycle of therapy. At endpoint in all experiments, blast cells were harvested from femur and tibial/fibial bone and spleen, and assessed for mCherry percentage via flow cytometry using a BD FACSymphony™. For A431 long-term survival experiments, 3 × 106 A431 cells in a 1:1 matrigel:PBS solution (In Vitro Technologies, 354230) were subcutaneously transplanted into the right flanks of 7-week old female NGS mice. Treatment commenced at 11 days post-transplant (mean tumor volume: 165.5mm3) using the same dosing regimen as described for MV4–11 transplanted mice. Tumor volume and body weight was monitored 3 times per week and mice were culled when tumor volume measured 1200mm3 or if mice exhibited a loss of 20 percent of their original body weight. All treatments were well tolerated with 1 mouse treated with AZD’4753 culled due to > 20 percent body weight loss. Kaplan-Meier survival analyses were performed using the Mantel Cox log-rank test and Bonferroni-corrected threshold for multiple pairwise comparisons. Tumor growth inhibition (TGI) for each mouse was measured as the change in tumor volume between the day one and day 15 of treatment. The mean change in tumor volume was calculated for control (ΔC) and treatment (ΔTi) groups and percentage TGI for each treatment group was calculated as 100 × (1 – (ΔTi / ΔC)), which was analyzed using unpaired Student’s t tests.
METHOD DETAILS
Reagents
AZ’5576 (CDK9i) and AZD’4573 were provided by AstraZeneca, and reconstituted in dimethyl sulfoxide (DMSO) for in vitro use. For in vivo use, AZD’4573 was reconstituted in a 10:30:70 ratio of N,N-dimethylacetamide (DMA), PEG-400, and 1% Tween-80 in H2O. Fresh AZD’4573 stocks were prepared weekly. The small molecule activator of PP2A (SMAP) DBK-1154 was provided by Michael Ohlmeyer (Icahn School of Medicine, Mount Sinai Hospital) and was reconstituted in DMSO (in vitro) or prepared daily in 10% DMA, 10% Kolliphor-HS-15 and 80% H2O (in vivo). The CDK9 degrader, 22–533, and THZ1 were provided by Nathaneal Gray and were reconstituted in DMSO. 1-NA-PP1 was purchased from Cayman Chemical (10954) and reconstituted in DMSO. LPS was purchased from Sigma Aldrich (L2630) and reconstituted in PBS. Phendione (1,10-Phenanthroline-5,6-dione) was purchased from Sigma Aldrich (496383) and reconstituted in DMSO. Calyculin A (ab141784) and Okadaic Acid (ab120375) were purchased from Abcam and reconstituted in DMSO. Cytarabine was obtained from the Peter MacCallum Cancer Centre pharmacy. EGF was purchased from GIBCO (PHG0311) and reconstituted in PBS. Flavopiridol was purchased from Sigma-Aldrich (F3055) and reconstituted in DMSO.
Click-EU labeling/nascent RNA
Cells treated with transcriptional inhibitors were labeled with 1mM ethynyl-uridine (EU, ThermoFisher Scientific E10345) for 1 hour prior to fixation/permeabilization (0.5% paraformaldehyde, 0.2% Tween20, 0.1% BSA, 0.1% Azide). Cells were washed once with PBS and incubated at room temperature for 30 minutes with 86.5 μL Click-iT reaction buffer, 4 μL CuSO4 buffer, 0.125 μL Alexa Fluor-azide-647 and 10.3 μL Click-iT reaction buffer additive from the Click-iT® RNA Imaging kit (ThermoFisher Scientific, C10329). Cells were washed twice with 200 μL Click-iT kit rinse buffer, and once each with PBS plus 0.1% Tween 20 and fixation/permeabilization buffer prior to analysis using the BDFortessa flow cytometer.
Genome-wide CRISPR-Cas9 screens
THP-1 and MV4;11 cells were engineered to stably express humanized S.pyogenes Cas9 endonuclease by lentiviral transduction with the FUCas9Cherry vector (Addgene 70182) and subsequent FACS-selection for mCherry-positive cells (THP-1-Cas9, MV4;11-Cas9). For THP-1-Cas9 survival screens, cells were transduced with the GeCKOV2 (125000 sgRNAs) (Sanjana et al., 2014) or Brunello (75000 sgRNAs) (Doench et al., 2016) genome-wide sgRNA libraries at a MOI of 0.3 and a fold representation of 500 for each individual sgRNA. For nascent RNA screens THP-1-Cas9 cells were transduced with the Brunello sgRNA library. Transduced cells were selected with puromycin (1 μg/mL) for 7 days, at which time cells were split into relevant treatment conditions (DMSO, CDK9i) and cell pellets were collected and snap-frozen for T0 reference controls. For survival screens, puromycin-selected cells were cultured in AZ5576 (150nM), A159 (50nM, GeCKOV2 only) or Dinaciclib (10nM, GeCKOV2 only) for 21 days to select for CDK9i-resistant populations and cell pellets were collected from DMSO- and CDK9i-resistant populations (Tend). For nascent RNA screens THP-1-Cas9 cells were transduced with the Brunello sgRNA library and puromycin-selected cells were cultured for 16 hours with DMSO or AZ5576 (170nM). Cells were labeled for 1 hour with EU and Click-iT RNA detection was performed as described previously prior to FACS-selection of EU-positive cells using a BDFACSAria sorter. For MV4;11-Cas9 survival screens, cells were transduced with the Brunello sgRNA library and puromycin-selected cells were cultured for 21 days with DMSO or 350nM AZ5576. Genomic DNA was isolated from T0, Tend and EU-positive cells using the DNeasy Blood and Tissue Kit (QIAGEN, 69504) as described. Cells were resuspended in 200 μL PBS and mixed with 20 μL Proteinase K (QIAGEN, 69504) and 200 μL QIAGEN AL buffer prior to incubation at 56°C for 10 minutes. Lysates were mixed with 200 μL 100% ethanol, loaded onto QIAGEN DNeasy columns and centrifuged at 6000 g for 1 minute. Columns were washed using QIAGEN AW1 buffer (500 μL, 6000 g, 1 min) and QIAGEN AW2 buffer (500 μL, 6000 g, 3 min) and DNA was eluted using QIAGEN AE buffer. Sequencing libraries were generated by PCR amplification as previously described (Doench et al., 2016; Joung et al., 2017; Kearney et al., 2018). For survival screens 20 PCR reactions were performed for each sample; individual 50 μL PCR reactions consisted of 195ng DNA, 0.75 μL ExTaq DNA Polymerase (Takara RR001C), 0.5 μL 100 μM P5 primer (Integrated DNA Technologies) and 10 μL of 5 μM uniquely barcoded P7 primer (Integrated DNA Technologies). For nascent RNA screens 1 PCR reaction was performed for each sample using all isolated genomic DNA. PCR cycling conditions were as follows: 95°C (1 min); 95°C (30 s), 53°C (30 s), 72°C (30 s) for 28 cycles; 72°C (10 minutes). 75 base-pair single-end reads were sequenced using the Illumina NextSeq500. Sequencing files were demultiplexed (Bcl2fastq, v2.17.1.14), reads were trimmed to 20 base-pair sequences using cutadapt (v2.1) and MAGeCK (v0.5.9) was used to count reads and perform sgRNA enrichment analysis. The R package ggplot2 (v3.3.0) was employed for data visualization of screens. Venn diagrams were generated using the online web tool BioVenn (Hulsen et al., 2008); represented genes had an adjusted p value of < 0.1 for > 3 sgRNAs in at least one replicate screens. Only genes with an adjusted p value less than for INTS6, INTS8 or INTS12 were included.
Stable expression of INTS6 in THP-1 cells
Human full-length INTS6 ORF in pCMV6-entry vector was purchased from Origene (RC208036). The INTS6 ORF was PCR amplified with 5′ Xho I and 3′ EcoRI restriction site overhangs and the coding sequence for a C-terminal V5 epitope tag was added in frame to the 3′ end of the INTS6 ORF (Table S3 for oligonucleotide sequences). The INTS6 PCR product and MSCVpuro vector (Addgene 68469) were digested with 1 μL XhoI (NEB R0146) and 1 μL EcoRI-HF (NEB R3101) in CutSmart™ buffer (NEB7204S) at 37°C overnight and purified digests were ligated with 1 μL T7 DNA Ligase in T7 DNA Ligase Reaction Buffer overnight at 16°C (NEB M0318). Ligation reactions were transformed into One Shot™ Stbl3™ chemically-competent E.coli cells (42°C heat-shock, 90 s; Invitrogen C737303) and positive clones were selected by Sanger sequencing. THP-1-Cas9 cells expressing FgH1t-UTG-sgSCR-GFP or FgH1tUTG-sgINTS6-GFP with stable knockout of INTS6 were engineered to express empty vector MSCVpuro or MSCVpuro-V5-INTS6 by retroviral transduction.
V5-PPP2R1A knock-in THP-1 cells
5 × 105 THP-1 cells were resuspended in 20μL nucleofection solution (16.4μL SG nucleofector solution + 3.6μL supplement 1) from the SG Cell Line 4D-Nucleofector™ X kit (Lonza, V4XC-3032). 300pmol PPP2R1A sgRNA (Synthego, Key Resources Table) was incubated with 100pmol Alt-R® S.p. HiFi Cas9 Nuclease V3 (Integrated DNA Technologies, 1081061) at room temperature for 20 minutes prior to the addition of 566ng mCherry-P2A-V5-PPP2R1A donor template DNA (Integrated DNA Technologies, Key Resources Table) on ice (6μL total volume). THP-1 cells in SG buffer were mixed with the sgRNA/Cas9/template complex and transferred to a 16-well nucleocuvette strip. Nucleofection was performed using the FF100 program of the Amaxa 4D Nucleofector X Unit (Lonza, AAF-1002X) and cells were incubated at 37°C for 10 minutes post-nucleofection before addition of culture media. Following expansion, mCherry cells were FACS-selected using a BD FACSAria™ Fusion sorter.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-Actin, mouse monoclonal | Sigma-Aldrich | Cat#A2228 |
| Anti-AKT (phospho Ser-473), rabbit polyclonal | Cell Signaling Technologies | Cat#9271 |
| Anti-Asunder (INTS13), rabbit polyclonal | Bethyl Laboratories | Cat#A303-575A |
| Anti-CDK9, rabbit polyclonal | Cell Signaling Technologies | Cat#2316 |
| Anti-DICE1 (INTS6), clone H-6, mouse monoclonal | Santa Cruz | Cat#sc-376524 |
| Anti-DSIF | BD Biosciences | Cat#611106 |
| Anti-GAPDH, rabbit monoclonal | Cell Signaling Technologies | Cat#2118 |
| Anti-GST, rabbit polyclonal | Upstate | Cat#06-332 |
| Anti- Drosophila H2Av, spike-in antibody | Active Motif | Cat#61686 |
| Anti-Histone H3, rabbit polyclonal | abcam | Cat#ab1791 |
| Anti-INTS1, clone 4.47, mouse monoclonal | Millipore, Sigma | Cat#MABS1984 |
| Anti-INTS3, rabbit polyclonal | Proteintech | Cat#16620-1-AP |
| Anti-INTS8, rabbit polyclonal | Sigma | Cat# HPA057299 |
| Anti-INTS11, rabbit polyclonal | Bethyl Laboratories | Cat#A301-274A |
| Anti-Mouse IgG-AlexaFluor-647 | Invitrogen, ThermoFisher Scientific | Cat#Z25008 |
| Anti-Mouse IgG-HRP, horse polyclonal | Cell Signaling Technologies | Cat#7076 |
| Anti-Mouse IgG-HRP, rabbit polyclonal | Agilent, Dako | Cat#16102 |
| Anti-PP2A, C subunit, clone 1D6, mouse monoclonal | Millipore, Sigma | Cat#05-421 |
| Anti-PPP2R1A, rabbit polyclonal | Bethyl Laboratories | Cat#A300-962A |
| Anti-Rabbit IgG-AlexaFluor-647 | Invitrogen, ThermoFisher Scientific | Cat#Z25308 |
| Anti-Rabbit IgG-HRP, goat polyclonal | Cell Signaling Technologies | Cat#7074 |
| Anti-Rabbit IgG-HRP, swine polyclonal | Agilent, Dako | Cat#P039901 |
| Anti-Rat IgG-AlexaFluor-647 | Invitrogen, ThermoFisher Scientific | Cat#A21247 |
| Anti-Rat IgG-HRP, rabbit polyclonal | Agilent, Dako | Cat#P0161202 |
| Anti-RNA Polymerase II, raised against the N terminus of subunit B1, rabbit polyclonal | Barbieri et al., 2018; Alessandro Gardini, Wistar Institute, Philadelphia, PA 19104, USA | N/A |
| Anti-RNA Polymerase II, clone CTD 4H8, mouse monoclonal | Millipore, Sigma | Cat#05-623 |
| Anti-RNA Polymerase II CTD repeat YSPTSPS, clone 8WG16, mouse monoclonal | Abcam | Cat#ab817 |
| Anti-RNA Polymerase II subunit B1 (phospho-CTD Ser-2), clone 3E10, rat monoclonal | Millipore, Sigma | Cat#04-1571-I |
| Anti-RNA Polymerase II subunit B1 (phospho-CTD Ser-5), clone 3E8, rat monoclonal | Active Motif | Cat#61085 |
| Anti-RNA Polymerase II subunit B1 (phospho-CTD Ser-7), clone 4E12, rat monoclonal | Millipore, Sigma | Cat#04-1570-I |
| Anti-RNA Polymerase II subunit B1 (phospho-CTD Thr-4), clone 6D7, rat monoclonal | Active Motif | Cat#61361 |
| Anti-RNA Polymerase II subunit B1 (phospho-CTD Tyr-1), clone 3E12, rat monoclonal | Active Motif | Cat#61383 |
| Anti-SBP-Tag, clone SB19-C4, mouse monoclonal | Santa Cruz | Cat#sc-101595 |
| Anti-SPT5 (phospho Thr-806) | Sansó et al., 2016; Robert P. Fisher, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA | N/A |
| Anti-α-Tubulin, clone B-5-1-2, mouse monoclonal | Merck, Sigma Aldrich | Cat#T5168 |
| Anti-V5-Tag, clone D3H8Q, rabbit monoclonal | Cell Signaling Technology | Cat#13202 |
| IRDye 800CW Anti-Mouse IgG, goat polyclonal | LI-COR | Cat#926-32210 |
| IRDye 680RD Anti-Rabbit IgG, goat polyclonal | LI-COR | Cat#926-68071 |
| IRDye 680RD Anti-Rat IgG, goat polyclonal | LI-COR | Cat#926-68076 |
| Purified Rabbit IgG | Bethyl Laboratories | Cat#P120-101 |
| Purified Mouse IgG2b, clone MPC-11 | BD Biosciences | Cat#557351 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| DH5-α chemically competent Escherichia coli | Invitrogen, Thermo Fisher Scientific | Cat#18265017 |
| One Shot Stbl3 chemically competent Escherichia coli | Invitrogen, Thermo Fisher Scientific | Cat#C737303 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| AZ5576, inhibitor of CDK9 | Hashiguchi et al., 2019; AstraZeneca | N/A |
| AZD’4573, inhibitor of CDK9 | Cidado et al., 2020; AstraZeneca | N/A |
| DBK-1154, agonist of PP2A | Merisaari et al., 2020; Michael Ohlmeyer, Icahn School of Medicine at Mount Sinai, New York, NY10029, USA | N/A |
| 22-533, PROTAC of CDK9 | Nathaneal Gray, Dana Farber Cancer Institute, Boston, MA 02215, USA | N/A |
| THZ1, inhibitor of CDK7 and CDK12 | Kwiatkowski et al., 2014; Nathaneal Gray, Dana Farber Cancer Institute, Boston, MA 02215, USA | N/A |
| 1-NA-PP1, inhibitory ATP analog | Cayman Chemical, Sapphire Bioscience | Cat#10954 |
| OTS-964 | MedChem Express | Cat#HY-12467 |
| Lipopolysaccharide (LPS) [for 3′RNA seq] | Merck, Sigma Aldrich | Cat#L2630 |
| Lipopolysaccharide (LPS) solution [for ChIP-seq] | eBioscience, Invitrogen | Cat#00497693 |
| 1,10-Phenanthroline (Phendione), inhibitor of PP2A | Merck, Sigma Aldrich | Cat#496383 |
| Calyculin A, inhibitor of PP2A/PP1 | Abcam | Cat#ab141784 |
| Okadaic Acid, inhibitor of PP2A/PP1 | Abcam | Cat#ab120375 |
| Cytarabine | Peter MacCallum Cancer Centre Pharmacy | N/A |
| Epidermal Growth Factor (EGF) Recombinant Human Protein | GIBCO | Cat#PHG0311 |
| Flavopiridol hydrochloride hydrate | Sigma-Aldrich | F3055 |
| 4-thiouridine (4sU) | Merck, Sigma Aldrich | Cat#T4509 |
| EZ-Link HPDP Biotin | ThermoFisher Scientific | Cat#21341 |
| Puromycin dihydrochloride from Streptomyces alboniger | Merck, Sigma Aldrich | Cat#P8833 |
| Hygromycin | ThermoFisher Scientific | Cat#10687010 |
| Doxycycline hyclate | Merck, Sigma Aldrich | Cat#D9891 |
| Dimethyl sulfoxide (DMSO) | Merck, Sigma Aldrich | Cat#D8418 |
| Dimethylformamide | Merck, Sigma Aldrich | Cat#227056 |
| Formaldehyde Solution (37%) | Merck, Sigma Aldrich | Cat#252549 |
| Paraformaldehyde | Merck, Sigma Aldrich | Cat#16005 |
| Sequa-brene | Merck, Sigma Aldrich | Cat#S2667-1VL |
| RNA Polymerase II CTD repeat YSPTSPS, recombinant protein | Abcam | Cat#ab81888 |
| 6X-His-CDK9/Cyclin T1, recombinant protein complex | Merck | Cat#140685 |
| GST-PP2A-alpha, recombinant protein | Merck, Sigma Aldrich | Cat#SRP5336 |
| Alt-R S.p. HiFi Cas9 Nuclease V3, recombinant protein | Integrated DNA Technologies | Cat#1081601 |
| Roswell Park Memorial Institute medium (RPMI) | GIBCO, ThermoFisher Scientific | Cat#11875093 |
| Dulbecco’s Modified Eagle’s medium (DMEM) | GIBCO, ThermoFisher Scientific | Cat#11995073 |
| Scheinder’s Drosophila medium | ThermoFisher Scientific | Cat#21720 |
| Penicillin / Streptomycin | ThermoFisher Scientific | Cat#10378016 |
| GlutaMAX | ThermoFisher Scientific | Cat#35050061 |
| Kolliphor ELP | Merck, Sigma Aldrich | Cat#30906 |
| Poly(ethylene glycol)-400 (PEG-400) | Merck, Sigma Aldrich | Cat#202398 |
| N,N-Dimethylacetamide (DMA) | Merck, Sigma Aldrich | Cat#185884 |
| XhoI restriction enzyme | New England Biolabs | Cat#R0146 |
| EcoRI-HF restriction enzyme | New England Biolabs | Cat#R3101 |
| KpnI-HF restriction enzyme | New England Biolabs | Cat#R3142 |
| BspQ1 | New England Biolabs | Cat#R0712 |
| CutSmart restriction digestion buffer | New England Biolabs | Cat#B7204 |
| BsmBI restriction enzyme | New England Biolabs | Cat#R0739 |
| Buffer 3.1 restriction digestion buffer | New England Biolabs | Cat#B7203S |
| T7 DNA Ligase kit | New England Biolabs | Cat#M0318 |
| ExTaq DNA Polymerase | Takara | Cat#RR001C |
| SensiFast SYBR Hi-Rox kit | Bioline | Cat#BIO-92005 |
| CLB1 RPPA lysis buffer | Zeptosens, Bayer | N/A |
| CSBL1 RPPA dilution buffer | Zeptosens, Bayer | N/A |
| BB1 RPPA blocking buffer | Zeptosens, Bayer | N/A |
| Sodium Bicarbonate | Merck, Sigma Aldrich | Cat#S7277 |
| IGEPAL CA-630 | Merck, Sigma Aldrich | I8896 |
| cOmplete Protease Inhibitors | Merck, Roche | Cat#1183580001 |
| Phosphatase Inhibitors | Pierce, ThermoFisher Scientific | Cat#A32957 |
| Trypsin | Promega | Cat#V5280 |
| Lysyl Endopeptidase | Wako | Cat#125-05061 |
| Acetonitrile | Merck, Sigma Aldrich | Cat#34851 |
| Benzonase | Merck, Sigma Aldrich | Cat#70746-5 |
| Annexin-V APC | BD Biosciences | Cat#550475 |
| TRIzol | Ambion, ThermoFisher Scientific | Cat#15596026 |
| Odyssey Blocking Buffer | LI-COR | Cat#927-50000 |
| Enhanced Chemiluminescence Plus Reagent (ECL) | Amersham, GE Healthcare | Cat#RPN2132 |
| Dynabeads Protein A | Invitrogen, ThermoFisher Scientific | Cat#10002D |
| Dynabeads Protein G | Invitrogen, ThermoFisher Scientific | Cat#10003D |
| Strepavidin Agarose | Pierce, ThermoFisher Scientific | Cat#20353 |
| Effectene Transfection Reagent | QIAGEN | Cat#301425 |
| 4’,6-diamidino-2-phenylindole (DAPI) | Invitrogen, ThermoFisher Scientific | Cat#1306 |
| Matrigel | In Vitro Technologies | Cat#354230 |
| U.S. Origin SuperCalf Serum | GemCell | Cat#100-510 |
|
| ||
| Critical commercial assays | ||
|
| ||
| DNesy Blood and Tissue Kit | QIAGEN | Cat#69504 |
| Direct-zol RNA mini-prep kit | Zymo Research | Cat#R2051 |
| RNeasy MinElute Cleanup kit | QIAGEN | Cat#74204 |
| Zymogen ChIP DNA Clean and Concentrator Kit | Zymo Research | Cat#D5205 |
| μMACS Streptavidin Kit | Miltenyi Biotec | Cat#130-074-101 |
| Tapestation High Sensitivity RNA kit | Agilent | Cat#067-5576 |
| BCA Protein Assay Kit | Pierce, ThermoFisher Scientific | Cat#23227 |
| Click-iT RNA Alexa Fluor Imaging Kit | Invitrogen, ThermoFisher Scientific | Cat#C10330 |
| SG Cell Line 4D-Nucleofector Kit | Lonza | Cat#V4XC-3032 |
| SG Cell Line 4D-Nucleofector Kit | Lonza | Cat#V4XC-1012 |
| P3 Primary Cell Line 4D-Nucleofector Kit | Lonza | Cat#V4XC-3012 |
| NEBNext Multiplex Oligos for Illumina (Index Primers) | New England Biolabs | Cat#E7335 |
| NEBNext Ultra II DNA Library Prep Kit | New England Biolabs | Cat#E7645 |
| QuantSeq 3′-mRNA Seq Library Prep Kit | Lexogen | Cat#015 |
| NEBNext Ultra II Directional RNA Library Prep Kit | New England Biolabs | Cat#E7760S |
| Revertaid first strand cDNA synthesis kit | Thermo Scientific | Cat#K1622 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat#G7570 |
| ADP-Glo Kinase Assay | Promega | Cat#V6930 |
|
| ||
| Deposited data | ||
|
| ||
| 4sU-seq data | This paper | GEO: GSE163803 |
| 3′-mRNA-seq data | This paper | GEO: GSE163802, GSE145379 |
| ChIP-seq data | This paper | GEO: GSE163804, GSE145379 |
| TT-seq data | This paper | GEO: GSE145379 |
| Fast-GRO data | This paper | GEO: GSE145379 |
| Proteomics data | This paper | PXD022962, PXD025060 |
| HeLa INTS11 ChIP-seq data | Gardini et al., 2014 | GEO: GSE58255 |
| HeLa NELF ChIP-seq data | Lai et al., 2015 | GEO: GSE68401 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Human: THP-1 | ATCC | Cat#TIB-202 |
| Human: MV4;11 | ATCC | Cat#CRL-9591 |
| Human: HeLa | ATCC | Cat#CCL-2 |
| Human: BJ | ATCC | Cat#CRL-2522 |
| Human: MM1.S | ATCC | Cat#CRL-2974 |
| Human: HS5 | ATCC | Cat#CRL-11882 |
| Human: JJN3 | DSMZ | Cat#ACC 541 |
| Human: A431 | ATCC | Cat#CRL-1555 |
| Human: HT-29 | ATCC | Cat#HTB-38 |
| Human: OPM2 | DSMZ | Cat#ACC 50 |
| Human: MOLM-13 | DSMZ | Cat#ACC 554 |
| Human: HL-60 | ATCC | Cat#CCL-240 |
| Human: OCI-AML3 | DSMZ | Cat#ACC 582 |
| Drosophila melanogaster: S2 | ATCC | Cat#CRL-1963 |
| Human: HEK293T | ATCC | Cat#CRL-11268 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mice: NOD-scid IL2Rgnull (NSG) mice | Peter MacCallum Cancer Center | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Scrambled sgRNA (sgSCR) for Human cells GCCTAGTCTCGGTAAGAGTG | Doench et al., 2016 | Non-Targeting Control |
| sgRNA A targeting Human INTS6 GGCTGTTACCTTGATAGCAT | Sanjana et al., 2014 | HGLibA_23426 |
| sgRNA B targeting Human INTS6 AGAGCCGCCCTATGCTATCA | Sanjana et al., 2014 | HGLibB_23394 |
| sgRNA A targeting Human INTS3 CCTAGCATGTCGTCTAACCC | Doench et al., 2016 | ID_48444 |
| sgRNA B targeting Human INTS3 GGAGATGGACAACCATATGT | Doench et al., 2016 | ID_48445 |
| sgRNA A targeting Human CPSF3L (INTS11) AAGATCGCCGTAGACAAGAA | Doench et al., 2016 | ID_41187 |
| sgRNA targeting Human PPP2R1A GGACGGAGCCAAGAUGGCGG | This paper | N/A |
| sgRNA targeting human CDK9 CACCGGCTCGCAGAAGTCGAACACC | Gressel et al., 2017 | N/A |
| DNA donor template for CDK9AS/AS mutation AAAGTGTGTTGGGTGT GGTTTTCTTGACTTTTTCTTCTTT CTATTCCTGCCTCAGCTTCCCC CTATAACCGCTGCAAGGGTAGT ATATACCTGGTCGCGGACTTCT GCGAGCATGACCTTGCTGGG CTGTTGAGCAATGTTTTGGTCA AGTTCACGCTGTCTGAGATCAA GAGGGTGATGCAGATGCTG CTTAACGGCCT | Gressel et al., 2017 | N/A |
| DNA donor template for mCherry-P2A-V5-PPP2R1A CCAGCGGAACCACGGCCTGGTAA CCCAAAACCTGCACACCCTCCAGCT CCCCACAGGACGTCACGTATTACCA CCGACGCACGCGCAGAAGCCTTCC CGGGGACTCAAGAAAGGGCAGGC TTAGCCTCCTCCCCATGTCGCCC CTCATTGGCTAGAAACTACTGCT CGTCTCGGTCGTTGTTAGCAGCG ACCAGGGCGGGTACAATCTTGGT CGCTAGGACACGGCTAACTTCCG CTTTCTTCCCCCTCTCCTAGGCTC AAACTAGTCAAATCTTGTTCACT CGACCAATGGCAAATCGGAA GTGGGCGGGACTTCACAAGTCC GGACCAAAGAAACGCGAGCTTA GCCCTGGGTAGCGCGGCCA ATGGCCGTGGAGCAGCCCCT GTAAACTGGCTCGGGCGCC CCCACGCCCGCCCTTCCTTC TTCTCCCAGCATTGCCCCC CCCACGTTTCAGCACAGC GCTGGCCGCAGTCTGACAG GAAAGGGACGGAGCCAAGA TGGTGAGCAAGGGCGAGGAG GATAACATGGCCATCATCAAG GAGTTCATGCGCTTCAAGGT GCACATGGAGGGCTCCGTGA ACGGCCACGAGTTCGAGATC GAGGGCGAGGGCGAGGGCC GCCCCTACGAGGGCACCCA GACCGCCAAGCTGAAGGTGAC CAAGGGTGGCCCCCTGCCCTT CGCCTGGGACATCCTGTCCCC TCAGTTCATGTACGGCTCCAAG GCCTACGTGAAGCACCCCGCC GACATCCCCGACTACTTGAAGC TGTCCTTCCCCGAGGGCTTCA AGTGGGAGCGCGTGATGAACTTC GAGGACGGCGGCGTGGTGACC GTGACCCAGGACTCCTCCCTGCA GGACGGCGAGTTCATCTACAA GGTGAAGCTGCGCGGCACCAAC TTCCCCTCCGACGGCCCCGTA ATGCAGAAGAAGACCATGGGCT GGGAGGCCTCCTCCGAGCGGAT GTACCCCGAGGACGGCGCCC TGAAGGGCGAGATCAAGCAGAG GCTGAAGCTGAAGGACGGCGG CCACTACGACGCTGAGGTCAAGA CCACCTACAAGGCCAAGAAGCC CGTGCAGCTGCCCGGCGCCTA CAACGTCAACATCAAGTTGGA CATCACCTCCCACAACGAGGA CTACACCATCGTGGAACAGT ACGAACGCGCCGAGGGCCGCC ACTCCACCGGCGGCATGGACG AGCTGTACAAGGCCACGAACTT CTCTCTGTTAAAGCAAGCAGGAG ACGTGGAAGAAAACCCCGGTCC TGGTAAGCCTATCCCTAACCCTC TCCTCGGTCTCGATTCTACGGG AGGATCAGGAGGAGGAGCGGC GGCCGACGGCGACGACTCGC TGTACCCCATCGCGGTGCTCA TAGACGAACTCCGCAATGAGG ACGTTCAGGTCCGGAGGCTAC GGGGGACTTGGGGAAGACGCG GAGGGGTACCTGGGGGCACG GGCGGCCCTCGCGGAGAAGA CTCAGCGTTCGCTGGGAGTGG CGGAAGGGGGCGACGGCCAATC AGCGTGCGTCTCTTATCTCCCCG GTTGCCCGGACTCCTTGAGACG GCGCTCCCGATTGGGTGTCGGC CCAGTGGAGGGCGGGGGCCAG CGCTAGCCTCGAGGGTCCCGGG CCTGCCCTGTGCGCGCGGCGG TCCGCGGTCCTGGGAGGTTGTG GCCAGGGCTGGGGTCTGCGGA CTGGGTCTGGGAGAGAGGAGG ACTCCGTGATTGGCGGCGGCC TCTGAATGGCCTCTTGGGGATG TGGGGCGCGCATGACTTG CTCCAAG | This paper | N/A |
| Please see Table S3 for additional oligonucleotide sequences | ||
|
| ||
| Recombinant DNA | ||
|
| ||
| FUCas9Cherry Cas9 expression vector | Aubrey et al., 2015 | Addgene Cat#70182 |
| FgH1tUTG-GFP sgRNA expression vector | Aubrey et al., 2015 | Addgene Cat#70183 |
| Human CRISPR Knockout Pooled Library (Brunello) | Doench et al., 2016 | Addgene Cat#73179 |
| Human CRISPR Knockout Pooled Library (GeCKO v2) | Sanjana et al., 2014 | Addgene Cat#1000000048 |
| Human full-length INTS6 ORF in pCMV6 | Origene | Cat#RC208036 |
| MSCV Puro expression vector | Akama-Garren et al., 2016 | Addgene Cat#68469 |
| pAc-sgRNA-Cas9 expression vector | Bassett et al., 2014 | Addgene Cat#49330 |
| pMK33-SBP expression vector | Yang and Veraksa, 2017 | N/A |
| D. melanogaster IntS6 coding sequence | FlyBase.org | Clone DmeI/SD04165 FBcl0286688 |
| pLKO.1 shRNA expression vector | Moffat et al., 2006 | Addgene Cat#10879 |
| VSV.G vector | Reya et al., 2003 | Addgene Cat#14888 |
| pMDLg/pRRE vector | Dull et al., 1998 | Addgene Cat#12251 |
| pRSV-REV vector | Dull et al., 1998 | Addgene Cat#12253 |
|
| ||
| Software and algorithms | ||
|
| ||
| Bcl2fastq2, v2.17.1.14 | https://support.illumina.com/downloads/bcl2fastq-conversionsoftware-v2-20.html | N/A |
| FASTQC, v0.11.6 | bioinformatics.babraham.ac.uk/projects/fastqc/ | N/A |
| HISAT2, v2.1.0 | Kim et al., 2015 | N/A |
| Subread, v2.0.0 | Liao et al., 2013 | http://subread.sourceforge.net/ |
| Bowtie2, v2.3.4.1 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/ |
| Samtools, v1.9 | Li et al., 2009 | http://www.htslib.org/ |
| Deeptools, v3.0.0 | Ramírez et al., 2014 | https://deeptools.readthedocs.io/en/dev elop / |
| MAGeCK, v0.5.9 | Li et al., 2014 | https://sourceforge.net/p/mageck/wiki/Home/ |
| Cutadapt, v2.1 | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| BWA | Li, 2013 | http://bio-bwa.sourceforge.net/ |
| MACS2 | Zhang et al., 2008 | https://github.com/macs3project/MACS/ |
| STAR, v2.5 | Dobin et al., 2013 | https://github.com/alexdobin/STAR/ |
| RSEM | Li and Dewey, 2011 | https://github.com/deweylab/RSEM/ |
| Valr, v0.6.1 | Riemondy et al., 2017 | https://rnabioco.github.io/valr/ |
| seqMINER, v1.3.4 | Ye et al., 2014 | https://sourceforge.net/projects/seqminer/ |
Generation of THP-1 CDK9AS/AS cells
sgRNAs targeting CDK9 (300pmol; Key Resources Table) were incubated for 20 minutes at room temperature with 100pmol Alt-R® SpCas9 nuclease (Integrated DNA Technologies, 1074182) prior to the addition of 100pmol of the CDK9 Phe-103-Ala dsDNA HDR template (Key Resources Table; final volume - 5 μL). The ribonucleoprotein complex was added to 5×106 THP-1 cells resuspended in 20 μL Nucleofector solution (16.4 μL SG nucleofector solution + 3.6 μL supplement 1) from the SG Cell Line 4D-Nucleofector™ Kit (Lonza, V4XC-3032). Cells were transferred to a 16-well Nucleocuvette™ strip and electroporated using the Amaxa 4D-Nucleofector™ X Unit (program FF-100, AAF-1002X). Electroporated cells were expanded in THP-1 culture media prior to isolation of single cell clones in 96-well culture plates using the Becton Dickinson Fusion FACS sorter. To screen CDK9AS/AS mutant clones, genomic DNA was isolated using the QIAGEN DNeasy Blood and Tissue Kit (QIAGEN, 69504) as described above and subjected to qPCR using the SensiFast SYBR Hi-Rox kit (Bioline BIO-92005; 100ng DNA, 1× 2 μM each of forward and reverse oligonucleotide primers (Table S3)). Mutant status was confirmed using Sanger sequencing of the CDK9 locus at the Australian Genome Research Facility (AGRF).
Generation of resistant MV4;11 cells
Wild-type MV4;11 AML cells were cultured continuously in the presence of AZ5576 (100nM, increasing to 350nM) or the vehicle control (DMSO) for 11 weeks until resistant clones expanded. Protein lysates were isolated and assessed by SDS-PAGE and Immunoblotting as described below.
Generation of D.melanogaster S2-Cas9 cells
Control (SCR) and IntS6 targeting sgRNAs (Sigma Aldrich, Key Resources Table) with BspQ1 overhangs were ligated using T7 DNA ligase as described above (NEB M0318) into pAc-sgRNA-Cas9 vector (Addgene 49330) (Bassett et al., 2014) digested overnight at 50°C with 1 μL BspQ1 (NEB R0712) in CutSmart™ buffer. D.melanogaster S2 cells were transfected with pAc-sgSCR-Cas9 or pAc-sgIntS6-Cas9 by Effectene transfection (QIAGEN 301425) and positive clones were selected with puromycin.
Expression of Ints6-SBP in D. melanogaster S2 cells
The D. melanogaster IntS6 coding sequence (FlyBase.org, Clone DmeI\SD04165. FBcl0286688) was PCR amplified with 5′ XhoI and 3′ KpnI restriction site overhangs (Table S3). The IntS6 PCR product and pMK33-SBP C-terminal vector (Yang and Veraksa, 2017) were digested with 1 μL each of XhoI and KpnI-HF (NEB R3142) in CutSmart™ buffer at 37°C overnight and purified digests were ligated overnight at 16°C using T7 DNA Ligase as described above. Ligation reactions were transformed into DH5-α chemically-competent cells (42°C heat-shock, 45 s; Invitrogen 18265017) and ampicillin-resistant clones were screened by sanger sequencing. D.melanogaster S2 clones expressing SCR sgRNA or with stable knockout of Ints6 (sgIntS6-5, clone D and clone E) were engineered to express pMK33-IntS6-SBP or pMK33-SBP by Effectene transfection and positive clones were selected using hygromycin (Yang and Veraksa, 2017) (300 μg/mL; ThermoFisher Scientific 10687010). Expression of IntS6-SBP was confirmed by western blot (SBP-Tag antibody, Key Resources Table).
Competitive Proliferation and Cell Death Assays
2 independent INTS6/3/11 sgRNAs or a control Scrambled (SCR) sgRNA (Sigma Aldrich, Key Resources Table and Table S3) with BsmBI compatible overhangs were ligated as described above into FgH1tUTG-GFP vectors (Addgene 70183) digested at 55°C overnight with 1 μL BsmBI (NEB R0739) in NEBuffer 3.1 (NEB B7203S). THP-1-Cas9 and MV4;11-Cas9 cells were engineered to stably express FgH1tUTG-sgINTS6/3/11-GFP or FgH1t-UTG-sgSCR-GFP vectors by lentiviral transduction. 6 μg FgH1tUTG-sgRNA DNA was incubated with 3 μg pMDLg/pRRE (Addgene 12251), 1.5 μg pRSV-REV (Addgene 12253), 1.8 μg VSV.G (Addgene 14888) DNA plus 62 μg polyethyleneimine (Sigma 913375) at room temperature for 20 minutes prior to addition to 5 × 106 HEK293T cells to produce lentiviral particles. Viral-supernatant was collected from HEK293T cells at 48 hours post-transfection and added to THP1-Cas9 and MV4;11-Cas9 cells supplemented with 4 μg/mL Sequa-brene (Sigma Aldrich S2667). Transduced cells were isolated by sorting for BFP- and GFP-expression using a BD FACSAria™ Fusion sorter. Cells were incubated with doxycycline (1 μg/mL; Sigma Aldrich D9891) for 7 days to induce sgRNA expression and depletion of INTS6 protein was confirmed by SDS-PAGE and western blotting as described below. For THP-1/MV4;11 competitive proliferation assays cells expressing FgH1tUTG-sgINTS6-GFP or FgH1t-UTG-sgSCR-GFP were mixed 1:1 with THP-1/MV4;11-Cas9 cells expressing FgH1t-UTG-sgSCR-BFP, incubated with doxycycline (1 μg/mL) and cultured in the presence of DMSO, CDK9i or the CDK9-degrader at indicated concentrations. The relative proportions of GFP- and BFP-positive cells was measured following mixing (T0) and at 4, 7, 11 and 14 days post-assay initiation using a BD LSRFortessa™ flow cytometer. For HeLa, MM1.S, BJ-T and HS5 competition assays, cells stably expressing either the FUCa-s9Cherry or FUCas9CFP vector were electroporated with Alt-R® S.p. HiFi Cas9 Nuclease V3 and INTS6-A targeting sgRNA or sgSCR (Synthego, Key Resources Table) respectively. 100pmol Alt-R® S.p. HiFi Cas9 Nuclease V3 and 300pmol synthetic guide RNA were pre-incubated with sterile, nuclease-free water (snRNP reaction; 5 μL volume) at room-temperature for 20 minutes. 0.5 ×106 cells were resuspended in 16.4 μL electroporation buffer plus 3.6 μL supplement (see below), mixed with the snRNP reaction, transferred to a 16-well Nucleocuvette™ strip and electroporated using the Amaxa 4D-Nucleofector™ X Unit (AAF-1002X). HeLa cells were prepared using SE buffer (Lonza V4XC-1012) and the CN-114 program, MM1.S cells were prepared using SG buffer (Lonza, V4XC-3032) and the FF-100 program, BJ-T and HS5 primary cells were prepared using P3 Primary Cell buffer (Lonza V4XP-3032) and the DS-138 program. At five days post-electroporation mCherry and CFP Cells were mixed 1:1 and incubated with increasing concentrations of AZ5576 for 96 hours. Cell populations were assessed by flow-cytometry using a BD LSRFortessa™ and SDS-PAGE/immunoblotting (HeLa). For cell death analysis, THP-1-Cas9 cells expressing FgH1tUTG-sgINTS6-A-GFP, FgH1tUTG-sgINTS6-B-GFP or FgH1t-UTG-sgSCR-GFP or D.melanogaster S2 cells expressing pAc-sgSCR-Cas9 or pAc-sgInts6-Cas9 were incubated with increasing doses of AZ5576, cytarabine, THZ1 or DBK-1154 for 72 hours. Cells were resuspended in Annexin binding buffer (10mM HEPES, 130mM NaCl, 5mM CaCl2) and stained with Annexin-V APC (BD Biosciences 550475; 1:100 dilution) for 15 minutes prior to flow cytometric analysis using a BD LSRFortessa™. The R package synergyfinder (v1.8.0) (He et al., 2018) was employed for Zero Interaction Potency (ZIP) synergy score computing between DBK-1154 and AZ5576. For solid cancer cell line (A431 and HT-29) combination assays, 5000 cells were seeded in triplicate in a flat-bottom 96-well plate and incubated for 24 hours prior to treatment with escalating doses of AZ5576 and/or DBK-1154. At endpoint (72 hours post-drug treatment) cells were incubated with a 1:2 dilution of the Promega CellTiter-Glo® Luminescent cell viability reagent (Promega G7570) for 10 minutes with gentle agitation. Luminescence was measured using a Biotek Cytation 3 Cell Imaging Multi-Mode reader and data (expressed as cell death percentage) was normalized to cells treated with DMSO. For hematological cancer cell lines (MM1.S, OPM2, JJN3, MOLM-13, HL-60, OCI-AML3) a D300e Digital Dispenser (Tecan Technologies) was used to seed 20000 cells per well in duplicate into flat-bottom 96-well plates and cells were incubated with escalating doses of of AZ5576 and/or DBK-1154. At endpoint (72 hours post-drug treatment) cells were washed once with PBS and stained with 4’,6-diamidino-2-phenylindole (DAPI, 5 μg/mL; Invitrogen™ D1306).
Infection of HeLa cells with shRNA
shRNA vectors pLKO.1-shINTS6, pLKO.1-shINTS8, pLKO.1-shINTS12, and pLKO.1-shPPP2R1A were obtained from the Molecular Screening Facility at the Wistar Institute. shINTS2 and shINTS5 were designed with the Broad Institute algorithm (https://portals.broadinstitute.org/gpp/public/) and subsequently cloned into pLKO.1 (Addgene #10879). For cloning, the pLKO.1 vector was gel purified after digestion with EcoRI and AgeI. shRNA oligos were annealed through incubation of 5 μL of 20 μM forward oligo, 5 μL of 20 μM reverse oligo, 5 μL 10X NEB buffer 2, and 35 μL nuclease-free water at 95°C for 4 minutes and cooled to room temperature over several hours. Annealed oligos were ligated into the digested pLKO.1 vector by combining 2 μL annealed oligo, 20ng digested pLKO.1, 2 μL 10X NEB T4 DNA ligase buffer, and 1 μL NEB T4 DNA ligase (NEB M0202), and brought to a total of 20 μL with nuclease-free water, and incubated at 16°C for 18 hours. Sequences of all shRNAs are listed in the Table S3. For infection, lentiviral particles were produced in HEK293T cells with pLKO.1-shRNA vectors as described above, with the following changes: viral supernatant was collected at 48 and 72 hours after transfection, passed through a 0.45 μM sterile filter, and viral particles were concentrated by ultracentrifugation at 150000 g for 90 minutes and resuspended in PBS at a volume of 10 μL per collection per plate. Concentrated virus was added to HeLa cell growing media (1 μL concentrated virus per 1mL media), and cells were incubated overnight in growing media plus virus and polybrene (Sigma-Aldrich, TR-1003). The following day, the virus-containing media was removed and replaced with growing media, and cells were selected with puromycin (Invivogen) at 2 μg/mL beginning 24 hours after infection. In all shRNA experiments, cells were treated and harvested within 72 hours of infection.
SDS-PAGE and Immunoblotting
For whole cell THP-1 lysates, cells were washed once with cold PBS and lysed with laemmli lysis buffer (60mM Tris-HCl pH 6.8, 10% (v/v) glycerol, 2% (w/v) SDS) for 10 minutes at 95°C and protein concentration was determined using the Pierce™ BCA Protein Assay Kit (ThermoFisher Scientific 23227). Protein samples were separated by SDS-PAGE using 4%–20% Mini-PROTEAN® TGX™ gels (BioRad, #4561094) in 25mM Tris, 192mM Glycine, 0.1% (w/v) SDS and wet-transferred to Immobulin-P (Merck Millipore, IPVH00010) or Immobulin-FL membranes (Merck Millipore, IPFL00010) in 25mM Tris, 192mM Glycine, 5% v/v methanol (4°C, 200mA, 2 hours). Membranes were blocked at room-temperature in Tris-buffered-saline (TBS) supplemented with 0.1% (v/v) Tween20 (TBST; Sigma Aldrich, P1379) and 5% skim-milk powder (local supermarket) or Odyssey® blocking buffer (LI-COR, 927–50000) prior to incubation at 4°C overnight with primary antibodies (Key Resources Table). Membranes were washed 3 times in TBST and incubated for 1 hour at room temperature with HRP-conjugated or LI-COR secondary antibodies (Key Resources Table). Membranes were washed 3 times in TBST and incubated with Amersham ECL PLUS™ (GE Healthcare, RPN2132) prior to exposure to Super RX film (Fujifilm, 03G01), or images were acquired using the Odyssey CLx machine (LI-COR, 83μm, medium setting).
For whole cell HeLa lysates, cells were harvested and washed three times in 1X PBS and lysed in ChIP lysis buffer (150 mM NaCl, 1% Triton X-100, 0.7% SDS, 500 mM DTT, 10 mM Tris-HCl, 5 mM EDTA) supplemented with 1 μg/mL aprotinin, 1 μg/mL leupeptin (Sigma) and 1 μg/mL pepstatin (BMB). Protein samples were loaded into Bolt 4%–12% Bis-Tris Plus gels (Invitrogen) and separated through gel electrophoresis (SDS-PAGE) in Bolt MES running buffer (Invitrogen). Separated proteins were wet-transferred to Immun-Blot PVDF membranes (BioRad) for antibody probing. Membranes were incubated with 10% BSA in TBST for 30 minutes at room temperature, then incubated for 2 hours at room temperature or overnight at 4°C with the suitable antibodies diluted in 5% BSA in 1X TBST. After incubation with the primary antibody, the membranes were washed with TBST, and incubated with a 1:10000 dilution of HRP-linked anti-mouse or anti-rabbit secondary antibody (Cell Signaling) for one hour at room temperature. HRP-linked antibodies were visualized using Clarity Western ECL substrate (Biorad) and imaged with Fujifilm LAS-3000 Imager (Fujifilm).
Co-immunoprecipitation experiments
For Co-IP experiments in THP-1 cells this protocol adapted from Gregersen et al. (2019) was used. 10 × 106 cells were used per IP and all lysis and wash buffers were supplemented with Roche cOmplete™ protease inhibitors (Merck, 11873580001) and Pierce phosphatase inhibitors (ThermoFisher Scientific, A32957). Cells were washed twice with cold PBS and resuspended in 2 pellet volumes of hypotonic lysis buffer (10mM HEPES pH 7.5, 10mM KCl, 1.5mM MgCl2) prior to incubation on ice for 40 minutes. Nuclei were pelleted at 4°C (3900rpm, 15 minutes) and the hypotonic cytoplasmic fraction was removed and discarded. Nuclei were resuspended in 2 original pellet volumes of nucleoplasmic lysis buffer (20mM HEPES pH 7.9, 1.5mM MgCl2, 150mM potassium acetate, 10% (v/v) glycerol, 0.05% (v/v) IGEPAL® CA-630) and incubated on ice for 20 minutes. Chromatin was pelleted at 4°C (20kg, 20 minutes) and the nucleoplasmic fraction was removed and stored on ice. Chromatin was incubated with 2 original pellet volumes of low salt chromatin digestion buffer (20mM HEPES pH 7.9, 1.5mM MgCl2, 10% (v/v) glycerol, 150mM NaCl, 0.1% (v/v) IGEPAL® CA-630, 250 U/mL benzonase (Merck, 70746–4)) on ice for 1 hour prior to centrifugation at 4°C (20kg, 20 minutes). The low salt chromatin fraction was removed and stored on ice. The remaining undigested chromatin was incubated with 2 original pellet volumes of high salt chromatin digestion buffer (20mM HEPES pH 7.9, 3mM EDTA, 1.5mM MgCl2, 10% (v/v) glycerol, 500mM NaCl, 0.1% (v/v) IGEPAL® CA-630) on ice for 20 minutes. 6 original pellet volumes of salt dilution buffer (20mM HEPES pH 7.9, 3mM EDTA, 1.5mM MgCl2, 10% (v/v) glycerol, 0.1% (v/v) IGEPAL® CA-630) was added prior to centrifugation at 4°C (20kg, 20 minutes). The nucleoplasmic, low salt chromatin and diluted high salt chromatin fractions were pooled and input samples taken. Pooled fractions were tumbled at 4°C for 1 hour with 5 μg of antibody (Key Resources Table) and 25 μL Dynabeads™ Protein A or Protein G (ThermoFisher Scientific, 10001D and 10003D respectively). Beads were washed 5 times (20mM Tris-HCl pH 7.5, 150mM NaCl, 1.5mM MgCl2, 3mM EDTA, 10% (v/v) glycerol, 0.1% (v/v) IGEPAL® CA-630) and incubated for 10 minutes at 95°C in 2X laemmli lysis buffer (120mM Tris-HCl pH 6.8, 4% (w/v) SDS, 1% β-mercaptoethanol, 0.02% (w/v) bromophenol blue) to elute immunoprecipitated proteins. Eluted fractions were analyzed by SDS-PAGE and immunoblotting as described previously.
For Co-IP experiments in HeLa, cells were washed twice with ice cold PBS before resuspension in buffer A (10mM Tris pH 7.9, 1.5mM MgCl2, 10mM KCl, 0.5mM DTT, 1 mg/ml each of protease inhibitors aprotinin, leupeptin, and pepstatin), and incubated at 4°C for 10 minutes. After spinning down at 2000rpm for 10 minutes, the pellet was resuspended in buffer A and subject to dounce homogenization (with B pestle), and spun down again. The supernatant was kept as cytoplasmic extract. The pellet was resuspended in buffer C (20mM Tris pH 8.0, 1.5mM MgCl2, 0.42M NaCl, 25% glycerol, 0.2mM EDTA, 0.5mM DTT, protease inhibitors) and dounce homogenized (with B pestle), followed by incubation at 4°C for 30 minutes. The resulting lysate was spun down at 12,000rpm for 30 minutes. The supernatant was kept as nuclear extract. The chromatin pellet was resuspended in Nuclease Incubation buffer (150mM HEPES pH 7.9, 1.5mM MgCl2, 150mM potassium acetate, 10% glycerol, 0.5mM DTT, and protease inhibitors) with Benzonase nuclease (Sigma-Aldrich), and incubated for 45 minutes at 4°C. The supernatant was kept as chromatin extract following centrifugation at 12,000rpm for 30 minutes. All extracts were dialyzed overnight in BC80 (20mM Tris pH 8.0, 80mM KCl, 0.2mM EDTA, 10% glycerol, 1mM B-mercaptoethanol, 0.2mM phenylmethylsulfonyl fluoride (PMSF)), cleared, and stored at −80°C. Prior to IP, 500ug of nuclear or chromatin extract was diluted in co-IP buffer (20mM Tris pH 7.9, 100mM NaCl, 0.1% NP-40, protease inhibitors). Each IP was incubated for 2 hours at 4°C with 2 μg antibody (Key Resources Table) and 25 μL of Dynabeads™ Protein A or Protein G. Beads were washed three times with co-IP buffer, followed by a final wash with 0.05% NP-40 in PBS. Elution was performed by agitation in 0.1M glycine pH 3.0 for one minute, and 1M Tris base pH 11.0 was added to neutralize the pH of the eluate. IP elutions were analyzed by SDS-PAGE and immunoblotting as described previously.
Immunoprecipitation-mass spectrometry (IP-MS)
For THP-1 and MV4;11 IP-MS experiments 50–80 × 106 cells were used per IP. Dynabeads™ Protein A or Protein G (Invitrogen 10002D and 10003D) were washed 5 times with 5% (w/v) BSA/PBS and tumbled at 4°C with relevant antibodies (Key Resources Table) for 1 hour. Cells were washed twice with cold PBS and incubated on 4°C roller for 10 minutes in nuclear extraction buffer (10mM HEPES, 10mM KCl, 1.5mM MgCl2, 0.05% (v/v) IGEPAL® CA-630, protease and phosphatase inhibitors) prior to centrifugation at 4°C (2000 g, 5 minutes). This step was repeated once before isolated nuclei were incubated in 1mL mass spectrometry buffer (20mM Tris-HCl pH 8, 150mM NaCl, 2mM EDTA, 0.5% (v/v) IGEPAL® CA-630, protease and phosphatase inhibitors) on ice for 10 minutes prior to sonication using the Covaris S220 Focused Ultrasonicator (8 minutes per sample). Sonicated lysates were cleared at 4°C (20000 g, 10 minutes) prior to tumbling at 4°C with 50 μL antibody-bound Dynabeads™ Protein A or Protein G for 1 hour. For IP-MS experiments for SBP or SPB-Ints6 in D.melanogaster S2 cells expressing pAc-sgSCR-Cas9 and pMK33-SBP or pMK33-IntS6-SBP, 100 × 106 cells were used per IP using 50 μL Pierce™ Strepavidin Agarose slurry (ThermoFisher Scientific, 20353) under the same conditions. Beads were washed 5 times (20mM Tris-HCl pH 8, 150mM NaCl, 2mM EDTA, 0.1% (v/v) IGEPAL® CA-630) and 2 times in 100mM ammonium bicarbonate (AMBIC). Beads were resuspended in 50 μmL 100mM AMBIC and incubated overnight at 37°C with 400ng Trypsin (Promega V5280). Beads were supplemented with an additional 400ng Trypsin and incubated for a further 4 hours at 37°C. The peptide-containing supernatant was isolated from the beads and acidified through the addition of 10% formic acid. Peptides were isolated using C18 ultra micro-spin columns (Pierce #87782) which had been pre-conditioned (100% acetonitrile followed by 2 rounds of 80% acetonitrile) and equilibrated (0.5% formic acid). Columns were washed twice (0.1% formic acid) and peptides were eluted using 40μL 80% acetronitrile diluted in 0.1% formic acid. Excess acetonitrile was evaporated using a vacuum centrifuge and samples were analyzed at the Bio21 Mass Spectrometry and Proteomics facility (Parkville, VIC 3052, Australia). LC MS/MS was carried out on a QExactive plus Orbitrap mass spectrometer (Thermo Scientific) with a nanoESI interface in conjunction with an Ultimate 3000 RSLC nanoHPLC (Dionex Ultimate 3000). The LC system was equipped with an Acclaim Pepmap nano-trap column (Dinoex-C18, 100 Å, 75 μm x 2 cm) and an Acclaim Pepmap RSLC analytical column (Dinoex-C18, 100 Å, 75 μm x 50 cm). The tryptic peptides were injected to the enrichment column at an isocratic flow of 5 μL/min of 2% v/v CH3CN containing 0.1% v/v formic acid for 5 min applied before the enrichment column was switched in-line with the analytical column. The eluents were 0.1% v/v formic acid (solvent A) and 100% v/v CH3CN in 0.1% v/v formic acid (solvent B). The flow gradient was (i) 0–6min at 3% B, (ii) 6–40 min, 3%–25% B (iii) 40–48 min, 25%–45% B (iv) 48–50 min, 40%–80% B (v) 50–53 min, 85%–85% B (vi) 53–54 min 85%–3% and equilibrated at 3% B for 10 minutes before the next sample injection. The QExactive plus mass spectrometer was operated in the data-dependent mode, whereby full MS1 spectra were acquired in positive mode, 70 000 resolution, AGC target of 3e6 and maximum IT time of 50ms. Fifteen of the most intense peptide ions with charge states R ≥ 2 and intensity threshold of 1.7e4 were isolated for MSMS. The isolation window was set at 1.2 m/z and precursors fragmented using normalized collision energy of 30, 17 500 resolution, AGC target of 1e5 and maximum IT time of 100ms. Dynamic exclusion was set to be 30sec. Raw files were analyzed using MaxQuant (version 1.5.8.3). The database search was performed using the Uniprot Homo sapiens or Drosophila melanogaster databases plus common contaminants with strict trypsin specificity allowing up to 3 missed cleavages. The minimum peptide length was 7 amino acids. Carbamidomethylation of cysteine was a fixed modification while N-acetylation of proteins N-termini and oxidation of methionine and phosphorylation of Serine/Threonine/Tyrosine were set as variable modifications. The iBAQ quantification option was selected. During the MaxQuant main search, precursor ion mass error tolerance was set to 4.5 ppm and fragment ions were allowed a mass deviation of 20 ppm. PSM and protein identifications were filtered using a target-decoy approach at a false discovery rate (FDR) of 1% with the match between runs option enabled. The RStudio package ggplot2 (v3.3.0) was employed for data visualization: a value of 10 was added to all raw iBAQ scores to allow log10 transformation. The average value of log10(iBAQ) of each technical replicate for each condition in THP1 and MV4;11 was calculated and subsequently filtered according the following criteria: log10(isotype) < 2 & log10(IP) > 2. Interaction networks were visualized in Rstudio (v3.5.1) using the packages concaveman (v1.0.0), dplyr (v0.8.1), ggraph (v2.0.3) and ggforce (v0.3.1).
For HeLa cell IP-MS experiments, IPs were performed as described previously, but with the following modifications: for each IP, 1–2mg of nuclear or chromatin extract was incubated with 4 μg antibody (Key Resources Table) and 50 μL Dynabeads™ Protein A or Protein G. Eluates were prepared for SDS-PAGE and run on a Novex WedgeWell 10% Tris-Glycine Gel (Invitrogen) with Tris-Glycine-SDS buffer (Bio-Rad), at 110V for 10 minutes. The gel was stained with Colloidal Blue staining kit (Invitrogen), and further processed at the proteomics facility at the Wistar Institute. Briefly, the gel lanes were excised, digested with trypsin, and analyzed by LC-MS/MS on a Q-Exactive Plus mass spectrometer. MS/MS spectra generated from the LC-MS/MS runs were searched with full tryptic specificity against the UniProt human database (https://www.uniprot.org; 10/01/2018) using the MaxQuant 1.6.2.3 program. False discovery rates for protein and peptide identifications were set at 1%. The RStudio package ggplot2 (v3.3.0) was used for data visualization: a value of 10 was added to all raw iBAQ scores to allow log10 transformation.
qRT-PCR
HeLa cells were lysed in (ThermoFisher Scientific 15596026) followed by RNA extraction using Direct-zol RNA MiniPrep kit (Zymo Research R2051). TRIzol® samples were mixed with 1 volume 100% ethanol, loaded into Zymo-Spin II columns and centrifuged at 11000 g for 30 s. Columns were washed with 400 μL RNA Wash Buffer (11000 g, 30 s) and incubated with 5 μL DNase I prepared in 75 μL DNA Digestion buffer for 15 minutes at room temperature. Columns were washed with 400 μL RNA PreWash Buffer (11000 g, 30 s) and 700 μL RNA Wash Buffer (11000 g, 30 s) and RNA was eluted using 50 μL nuclease-free water (11000 g, 30 s). Reverse transcription of template RNA into cDNA was performed using the Revertaid first strand cDNA synthesis kit (Thermo Scientific K1691) as follows: random hexamer primer (1 μL), template RNA (900ng), RT Reaction buffer (4 μL of 5X), dNTPs (2 μL of 10mM), and RevertAid RT (1 μL of 200U/μL), plus nuclease-free water to a total of 20 μL, was incubated for 5 minutes at 25°C, 60 minutes at 42°C, and 5 minutes at 70°C. Real-time quantitative PCR was performed with 50ng cDNA, 0.4mM primer, 10 μL iQ SYBR Green Supermix (BioRAD), in a final volume of 20 μL. We used the CFX96 real-time system (BioRAD), and thermal cycling parameters were as follows: 3 minutes at 95°C, followed by 40 cycles of [10 s at 95°C, 20 s at 63°C, 30 s at 72°C]. All samples were run in triplicate. 18S rRNA used to normalize. Primer sequences are reported in Table S3.
Glycerol gradient ultracentrifugation
Glycerol gradients were prepared fresh with 1X HEMG buffer (50mM HEPES (pH 7.9), 0.2mM EDTA, 30mM MgCl2, 200mM KCl, 0.5mM DTT, 1 mg/ml each of protease inhibitors aprotinin, leupeptin, and pepstatin) and glycerol from 11% to 50% in increments of 3%. Gradients were assembled on ice by carefully layering 320uL of 1X HEMG-glycerol in descending order of glycerol concentration, starting with 50%, into an ultracentrifuge tube (Beckman Coulter 344057). Prior to centrifugation, 500ug of nuclear extract (brought to 300uL and cleared) was layered on top. Gradients were ultracentrifuged in a swinging-bucket rotor at 48,000rpm (272,798 rcf.) for 16 hours (accel = 8, decel = 8). After centrifugation, gradient fractions were collected by piercing a hole in the bottom of the tube and collecting fractions of 320uL each. Protein was precipitated from each fraction with 1/10th volume trichloroacetic acid (TCA, Sigma-Aldrich), incubated overnight at −20°C, washed twice with 1 volume of cold acetone (Sigma-Aldrich), and resuspended in 70uL of 1X BOLT laemmli sample buffer (Invitrogen) for SDS-PAGE analysis.
Reverse Phase Protein Array
THP-1-Cas9 cells expressing FgH1t-UTG-sgSCR-GFP or FgH1tUTG-sgINTS6-A-GFP with stable knockout of INTS6 were treated with DMSO or AZ5576 for 2 or 6 hours. 3 biological replicates were assessed for each treatment condition. Treated cells were lysed with CLB1 buffer (Zeptosens, Bayer) and quantified using a Pierce™ BCA Protein Assay Kit (ThermoFisher Scientific 23227). Samples were diluted (100%, 63%, 40%, 25%) with 9:1 CSBL1:CBL1 buffer (Zeptosens, Bayer) using a Sciclone Caplier ALH3000 liquid handling root (Perkin Elmer). Diluted samples were spotted onto ZeptoChips (Zeptosens; 2 technical replicates) using a Nano-plotter-NP2 non-contact microarray system (GeSim). Chips were blocked with BB1 buffer (Zeptosens, 1 hour) prior to incubation with primary antibodies (1:500 dilution for 2 hours, Key Resources Table) and Alexa Fluor® conjugated secondary antibodies (1:1000 dilution for 4 hours, anti-rabbit Alexa Fluor®−647, ThermoFisher Z25308; anti-mouse Alexa Fluor®−647, ThermoFisher Z25008; anti-rat Alexa Fluor®−647, ThermoFisher A21247). Alexa Fluor® signal intensity was measured using a ZeptoREADER and relative fluorescence intensity (RFI) values were calculated using ZeptoVIEW software (Zeptosens, version 3.1). RFI values for each sample were normalized to secondary-antibody only controls.
Phospho-peptide mass spectrometry
THP-1-Cas9 cells expressing FgH1t-UTG-sgSCR-GFP or FgH1tUTG-sgINTS6-A-GFP with stable knockout of INTS6 were treated with DMSO or AZ5576 for 2 hours. 4 biological replicates were used for each condition. Cells were pelleted in Eppendorf® protein LoBind tubes (Sigma Aldrich Z666505) washed twice with ice cold PBS and snap-frozen. Thawed cell pellets were incubated at 95°C for 10 minutes in SDS-lysis buffer (5% SDS, 10mM TCEP, 40mM 2-CAA, 10mM Tris-HCl pH 7.5) prior to DNA hydrolysis using 1% TFA and sample neutralisation through the addition of 3M Tris-HCl pH 7.5 to a final concentration of 160mM. Samples were incubated with PureCUBE carboxy agarose magnetic beads (Cube Biotech 50201) and 70% acetonitrile at room temperature for 20 minutes prior to 2 washes with 70% ethanol and 1 wash with 100% acetonitrile. Beads were lyophilised to remove residual acetonitrile and were then incubated with lysis buffer (50 μL per sample; 10% trifluoroethanol, 100mM ammonium bicarbonate), Trypsin (Promega V5280) and Lysyl Endopeptidase (Wako 125–05061) at 1:25 ratio of enzyme:substrate. Beads were incubated for 2 minutes at room temperature in an ultrasonic water bath (Unisonics Australia) prior to incubation at 37°C for 4 hours with agitation (1200rpm). The lysis supernatant was isolated and the beads were washed with 1 volume of ultrapure H2O. The lysis supernatant and wash supernatant were pooled and TFA was added to a final concentration of 1%. Samples were centrifuged (20000 g) and the supernatant was isolated. TFA-supplemented acetonitrile was added to the supernatant for a final concentration of 80% acetonitrile and 0.1% TFA prior to incubation at room temperature with gentle shaking for 30 minutes with PureCUBE Fe(III)-NTA beads (Cube Biotech 31501-Fe). Beads were washed 3 times with 80% acetonitrile / 0.1% TFA and transferred to C8 stage tips pre-wetted with 100% acetonitrile. Isolated phospho-peptides were eluted using 50% acetonitrile / 2.5% ammonium hydroxide pH 10 (2 × 20 μL) and collected into tubes containing 30 μL 10% TFA. Samples were lyophilized and peptides were resuspended in 50 μL 5% formic acid prior to being transferred to C18 stage tips pre-wetted with 10% isopropyl alcohol, 60% acetonitrile and 5% formic acid. C18 stage tips were centrifuged (500 g, 1 minute) prior to washing twice with 5% formic acid. Phospho-peptides were eluted in 60% acetonitrile and 5% formic acid (50 μL) and were lyophilised to dryness prior to storage at minus 80°C. For analysis, phospho-peptides were resuspended in 2% acetonitrile and 1% formic acid and separated by reverse phase liquid chromatography using an ThermoFisher Scientific Easy-nLC 1200 UHPLC system (1.6 μm C18 packed emitter tip column, 250mm x 75 mm, Ion Opticks) with a linear 120 minute gradient at 400nL/min flow rate from 98% solvent A (0.1% formic acid / ultrapure H2O) to 37% solvent B (0.1% formic acid / acetonitrile). The UHPLC was coupled online to a Q-Exactive HF Orbitrap mass spectrometer equipped with a nanoelectrospray ionization source (ThermoFisher Scientific) and a column oven at 50°C (Sonation, Germany). The Q-Exactive HF was operated in a data-dependent mode, switching automatically between one full-scan and subsequent MS/MS scans of the ten most abundant peaks. The instrument was controlled using Exactive series version 2.9 and Xcalibur 4.1.31.9. Full-scans (m/z 350–1,850) were acquired with a resolution of 60,000 at 200 m/z. The 10 most intense ions were sequentially isolated with a target value of 10,000 ions and an isolation width of 1.4 m/z and fragmented using HCD with NCE of 27. Maximum ion accumulation times were set to 100ms for full MS scan and 110ms for MS/MS. Raw files were analyzed using MaxQuant (version 1.5.8.3). The database search was performed using the Uniprot Homo sapiens database plus common contaminants with strict trypsin specificity allowing up to 3 missed cleavages. The minimum peptide length was 7 amino acids. Carbamidomethylation of cysteine was a fixed modification while N-acetylation of proteins N-termini, oxidation of methionine and phosphorylation of Serine/Threonine/Tyrosine were set as variable modifications. During the MaxQuant main search, precursor ion mass error tolerance was set to 4.5 ppm and fragment ions were allowed a mass deviation of 20 ppm. PSM and protein identifications were filtered using a target-decoy approach at a false discovery rate (FDR) of 1% with the match between runs option enabled.
Further analysis was performed using a custom pipeline developed in R, which utilizes the MaxQuant output file evidence.txt. A feature was defined as the combination of peptide sequence, charge and modification. Features not found in at least 80% of the replicates in one group were removed. To correct for injection volume variability, feature intensities were normalized by converting to base 2 logarithms and then multiplying each value by the ratio of maximum median intensity of all replicates over median replicate intensity. Missing values were imputed using a random normal distribution of values with the mean set at mean of the real distribution of values minus 2.2 s.d., and a s.d. of 0.15 times the s.d. of the distribution of the measured intensities. The probability of differential peptide expression between groups was calculated using the Limma R package (version 3.34.9). Probability values were corrected for multiple testing using Benjamini–Hochberg method. The R package pheatmap (v1.0.12) was used to compute heatmaps of phospho-sites with the indicated significance cutoff. The values in the heatmaps represent the median of z-norm values of biological replicates for each condition. Filtering conditions were applied to phospho-sites in addition to significance cutoffs. To be plotted, z-norm values of phospho-sites were to meet the following criteria: SCR_CDK9i – SCR_UT < 0.3 & SCR_UT – INTS6_KO_UT < 0.3 & SCR_CDK9i ≤ INTS6_KO_UT & SCR_CDK9i ≤ INTS6_KO_CDK9i. Kmean cluster line plots and phospho-peptides scatterplots were generated by using the R package ggplot2 (v3.3.0). To be plotted, z-norm values of phospho-sites in kmean cluster and in single site plots were to meet the following criteria: SCR_CDK9i < SCR_UT & SCR_CDK9i ≤ INTS6_KO_UT & SCR_CDK9i ≤ INTS6_KO_CDK9i. In the phospho-peptides scatterplots, each bar represents the median of z-norm values of different biological replicates for the same condition. Gene ontology (GO) analysis was performed using Metascape (release 3.5) (Zhou et al., 2019).
Recombinant protein kinase / phosphatase assay
Reactions were performed in kinase reaction buffer (25mM Tris-HCl pH 7.4, 2mM dithiothreitol (DTT), 10mM MgCl2) supplemented with 200mM Mg+-ATP (Sigma Aldrich A9187) in 40 μL final reaction volumes. 0.5 μg recombinant human RNA polymerase II CTD repeat YSPTSPS peptide (Abcam ab81888) was incubated with/without 0.4 μg recombinant human 6X-His-CDK9/Cyclin T1 (Merck, 140685) and/or 0.4 μg recombinant human GST-tagged PP2A- α (Sigma, SRP5336) in the presence or absence of AZ’5576 or okadaic acid (Abcam ab120375) for 45 minutes at 30°C (300rpm). Reactions were stopped through the addition of 20 μL 2X laemmli lysis buffer (120mM Tris-HCl pH 6.8, 20% (v/v) glycerol, 4% (w/v) SDS) and incubation at 95°C for 10 minutes. Reactions were analyzed by SDS-PAGE and immunoblotting as described previously. To measure ATP conversion, reactions were performed as described above. ADP generation was analyzed using the ADP-Glo™ Kinase Assay (Promega, V6930): for each condition, 5 μL kinase reaction was aliquoted in triplicate in 384 well white-walled plates and incubated at room temperature with 5 μL ADP-Glo™ reagent (40 minutes) and 10 μL Kinase Detection Reagent (40 minutes). The ADP-Glo™ luminescence signal was measured using a Biotek Cytation 5 Cell Imaging Multi-Mode reader.
3′RNA Quant-Seq
For LPS experiments THP-1-Cas9 cells expressing FgH1t-UTG-sgSCR-GFP or FgH1tUTG-sgINTS6-A-GFP with stable knockout of INTS6 were treated with PBS or LPS (1μg/mL) for 1, 2, 4 and 6 hours (2 × 106 cells per condition in duplicate). For HT-29 experiments, HT-29 cells were seeded in 6 well plates 24 hours prior to treatment with DMSO, AZ-5576 (200nM) and/or DBK-1154 (5 μM) for 2 hours (1 × 106 cells per condition in triplicate). Cells were lysed with 600μL TRIzol® (ThermoFisher Scientific 15596026) and total RNA was isolated using the Direct-zol RNA mini-prep kit (Zymo Research R2051) as described above. Sequencing libraries were prepared using the QuantSeq 3®-mRNA Seq Library Prep Kit for Illumina (Lexogen) from 500ng total RNA and 75 base-pair single-end reads were sequenced on the Illumina NextSeq 500. Sequencing files were demultiplexed (Bcl2fastq, v2.17.1.14) and QC was performed on FASTQ files using FASTQC (v0.11.6). Sequencing reads were trimmed (cutadapt v2.1) and aligned to the Hg19 human reference genome using HISAT2 (v2.1.0). Read counting across genomic features was performed using FeatureCounts (Subread, v2.0.0) and differential gene expression analysis was performed using Voom-Limma in R (v3.42.2). Gene set enrichment analyses were performed using the Broad Institute GSEA software (Subramanian et al., 2005).
HeLa cells infected with shLUC or shINTS6 were treated with PBS or EGF (0.1μg/mL) for 15, 55, and 180 minutes. Cells were lysed in Tri-reagent and total RNA was isolated using the Zymo Research Direct-zol RNA mini-prep kit as described above. For spike-in, 200ng of RNA from D.melanogaster S2 cells was added to 1μg of total RNA for each time point. Sequencing libraries were prepared at the Wistar Genomics Facility using the QuantSeq 3′-mRNA Seq Library Prep Kit for Illumina (Lexogen) and 75 base-pair single-end reads were sequenced on the Illumina NextSeq 500. Reads were aligned to hg19 and dm5 reference genomes using STAR v2.5 (Dobin et al., 2013) in 2-pass mode with the following parameters: –quantMode TranscriptomeSAM –outFilterMultimapNmax 10 – outFilterMismatchNmax 10 –outFilterMismatchNoverLmax 0.3 –alignIntronMin 21 –alignIntronMax 0 –alignMatesGapMax 0 –alignS-JoverhangMin 5 –runThreadN 12 –twopassMode Basic –twopass1readsN 60000000 –sjdbOverhang 100. BAM files were filtered based on alignment quality (q = 10) using Samtools v0.1.19 (Li et al., 2009). FeatureCounts (Liao et al., 2014) was used to count reads mapping to each gene, and differential gene expression analysis was performed using Voom-Limma in RStudio (v3.42.2) with normalizationFactors calculated from the D.melanogaster spike-in. Data was visualized using ggplot2 (v3.3.1).
Chromatin Immunoprecipitation Sequencing
For THP-1 parental, THP-1-Cas9 FgH1t-UTG-sgSCR-GFP or FgH1tUTG-sgINTS6-A-GFP, 20 – 50 × 106 THP-1 cells were incubated with DMSO, AZ5576 or DBK-1154 for indicated time-points. Cells were washed once with cold PBS prior to cross-linking at room temperature for 10 minutes with 1/10th volume fresh formaldehyde solution (11% formaldehyde, 0.05mM EGTA, 1mM EDTA, 100mM NaCl, 50mM HEPES-KOH pH7.5). To quench the cross-linking reaction, 1/20th volume of 2.5M glycine was added and cells incubated at room temperature for a further 5 minutes. Cells were washed with ice-cold PBS and nuclei were isolated by 3 successive 10 minute incubations ice with cold nuclear extraction buffer (20mM Tris-HCl pH 8, 10mM NaCl, 0.5% IGEPAL® CA-630, 2mM EDTA). Cell nuclei were resuspended in sonication buffer (20mM Tris-HCl pH 7.5, 150mM NaCl, 2mM EDTA, 0.3% SDS, 1% IGEPAL® CA-630) and sonicated for 18 minutes using the Covaris S2 instrument (20% Duty Cycle, 1000 cycles/burst, 10 Intensity). Fragmented chromatin was diluted 1:1 with dilution buffer (20mM Tris-HCl pH 8, 150mM NaCl, 2mM EDTA, 1% Triton X-100). For each immunoprecipitation, diluted chromatin was incubated overnight at 4°C with Protein A and Protein G Dynabeads (ThermoFisher Scientific 10002D, 10004D; 25μL each) and indicated antibodies (Key Resources Table). Post-incubation, Dynabeads were washed once each with dilution buffer, ChIP wash buffer 1 (20mM Tris-HCl pH 8, 500mM NaCl, 2mM EDTA, 0.1% SDS, 1% Triton X-100) and ChIP wash buffer 2 (20mM Tris-HCl pH 8, 250mM LiCl, 2mM EDTA, 0.5% deoxycholate, 0.5% IGEPAL® CA-630) and washed twice with TE buffer (10mM Tris-HCl pH 7.5, 1mM EDTA). Washed beads were incubated with shaking at 55°C for 1 hours in reverse crosslinking buffer (200mM NaCl, 100mM NaHCO3, 1% SDS, 300μg/mL Proteinase-K) followed by incubation of the supernatant overnight at 65°C. DNA was isolated using the Zymogen ChIP DNA Clean and Concentrator Kit (Zymo Research D5205). 5 volumes ChIP binding buffer was added to the reverse-cross-linked DNA sample prior to loading on the Zymo-Spin™ column and centrifugation at 10000 g for 30 s. Columns were washed twice with 200 μL Wash Buffer (10000 g, 30 s) and DNA was eluted using 6 μL Elution Buffer (10000 g, 30 s). Sequencing libraries were prepared using the NEBNext Ultra II DNA Library Prep Kit (NEB E7645) and 200 – 500 base-pair size selection was performed using a Pippin Prep system (Sage Science). 75 base-pair single end reads or 40 base-pair paired end reads were sequenced on the Illumina NextSeq500. Sequencing files were demultiplexed (Bcl2fastq, v2.17.1.14) and QC was performed on FASTQ files using FASTQC (v0.11.6). Sequenced reads were aligned to the Hg19 human or dm3 D. melanogaster reference genomes using Bowtie2 (v2.3.4.1) and generated SAM files were converted to BAM files using Samtools (v.1.9) using the view function. Samtools was used to further sort, index and remove potential PCR duplicates (rmdup) from BAM files which were converted to BigWig files using Deeptools (v3.0.0; bamCoverage,–nomalizeUsing CPM–smoothLength 150–binSize 50 -e 200, scaleFactor 1). BigWig files were visualized using the Integrative Genomics Viewer (IGV; Broad Institute). Average read density across defined genomic intervals was computed using the Deeptools computeMatrix function and the resulting matrices were used to generate chromatin occupancy heatmaps using the plotHeatmap function. Pausing indices for Hg19 and dm3 were calculated as a ratio TSS read density (+/− 250bp) over gene body density (+500 bp from TSS and −250 bp from TES). In more detail, subread (v2.0.0; featureCounts -O -M -T 16 -F SAF) was used to count reads within TSS and gene body intervals generated using bedtools (v2.27.1) slop function that included Hg19 or dm3 chromosome sizes (downloaded from UCSC). Read density was calculated as RPKM in Rstudio (v3.6.1) using limma (v3.40.6) and edgeR (v3.26.8) packages and visualized using ggplot2 (v3.3.1).
For wild-type HeLa, cells were serum-starved (with 0.3% SCS) for 72 hours prior to induction of gene expression with 0.1μg/mL recombinant EGF (GIBCO) or PBS for 15 minutes. For knockdowns, HeLa cells infected with indicated shRNAs were placed under serum-starvation (0.3% SCS) the night before harvesting. Cells were treated for 2 hours with 10μM SMAP, 2 hours with 300nM AZ5576, 1 hour with 2μM Flavopiridol, or time-matched DMSO control, and subsequently treated with PBS or 0.1μg/mL EGF for 15 minutes. ChIP-seq was performed as previously described (Lai et al., 2015), with some modifications. To allow for accurate normalization between treatment conditions, tandem ChIP-seq was performed with a D.melanogaster- specific antibody similar to the protocol described by Egan et al. (2016). For each replicate, 10–20×106 HeLa cells were harvested and cross-linked with 1% formaldehyde for 5 minutes at room temperature and washed twice with PBS. The chromatin pellet was resuspended in ChIP lysis buffer (150mM NaCl, 1% Triton X-100, 0.7% SDS, 500mM DTT, 10mM Tris-HCl, 5mM EDTA), and supplemented at 5% with chromatin from 0.5–1×106 formaldehyde-crosslinked D.melanogaster S2 cells. The chromatin was sheared to an average length of 200–400 bp using a Covaris S220 Ultrasonicator. The chromatin lysate was diluted with SDS-free ChIP lysis buffer. For ChIP-seq, the following were added to the sheared chromatin and incubated at 4°C overnight: 10μg of human antibody, 0.5μg Drosophila spike-in antibody, and Protein A or Protein G Dynabeads (Invitrogen). After incubation, beads were washed twice with each of the following buffers: Mixed Micelle Buffer (150 mM NaCl, 1% Triton X-100, 0.2% SDS, 20 mM Tris-HCl, 5 mM EDTA, 65% sucrose), Buffer 500 (500 mM NaCl, 1% Triton X-100, 0.1% Na-deoxycholate, 25 mM HEPES, 10 mM Tris-HCl, 1 mM EDTA), LiCl/detergent wash (250 mM LiCl, 0.5% Na-deoxycholate, 0.5% NP-40, 10 mM Tris-HCl, 1 mM EDTA), followed by a final wash with 1X TE. Finally, beads were resuspended in 1X TE with 1% SDS and incubated at 65°C for 10 min to elute. Elution was repeated twice, and the samples were incubated overnight at 65°C to reverse cross-link, alongside the untreated input (5% of the starting material). After treatment with 0.5 mg/ml proteinase K for 3 hours, DNA was purified with Zymo ChIP DNA Clean Concentrator kit (Zymo Research D5205) as described above and quantified by QUBIT. Barcoded libraries were made with NEB Ultra II DNA Library Prep Kit for Illumina, and sequenced on Illumina NextSeq 500, producing 75 base pair single-end reads or 40 base pair paired-end reads. Sequences were aligned to the human reference hg19 and D.melanogaster dm5 genome using Burrows Wheeler Alignment tool (BWA), with the MEM algorithm (Li, 2013). Using Samtools, aligned reads were filtered based on mapping quality (MAPQ > 10) and PCR duplicates removed (rmdup). Data was visualized on the UCSC Genome Browser (http://genome.ucsc.edu/) with Bigwig files generated with deeptools (v2.4.2): bamCoverage–binSize 10–normalizeTo1× 3137161264–extendReads 150–ignoreForNormalization chrX. For D.melanogaster spike-in normalization, the number of hg19 filtered reads was divided by the lowest number of dm5 filtered reads for each set of experiments, resulting in a downsampling factor for each sample. Normalized BAM files were generated using samtools view –s with the downsampling factor, and normalized Bigwigs were generated from the normalized BAM files using bamCoverage–binSize 10–extendReads 150. Peaks were called using MACS2 (2.1.2) with the broad peak option (Zhang et al., 2008): -f BAM–broad–broad-cutoff 0.1 for single-end libraries, or -f BAMPE–broad–broad-cutoff 0.1 for paired-end libraries. Genomic intervals were generated using valr (v0.6.1) (Riemondy et al., 2017). To identify genes that overlap peaks, valr bed_intersect was used with hg19 known genes, filtered for RNA Polymerase II processed-transcripts (n = 20618). Only genes with peaks identified in multiple replicates were included for further analysis. Average read density across defined genomic intervals was computed using seqMINER 1.3.3 package (Ye et al., 2011) with the following parameters: left and right extensions = 5.0 kb; internal bins = 160; flanking region bins = 20. Read density matrices were exported to generate average profiles. Where indicated, total RNAPII-normalization was performed as follows: for average profiles, read density of RNAPII pS2 was normalized by TSS read-density of total RNAPII in the matched condition, and for boxplot quantifications of RNAPII pS2 and PPP2R1A, ChIP-seq coverage was normalized by per-gene coverage of RNAPII in the matched condition. Coverage of ChIP-seq datasets across genomic intervals was calculated using valr bed_coverage, an R-based implementation of BEDtools (Quinlan AR and Hall IM, 2010). Highest expressed genesets were identified by genes with largest coverage values in two replicates of HeLa shLUC control. Pausing index was calculated as a ratio of coverage at the TSS (−150bp/+50bp) and coverage of the gene body (+50 bp from TSS through the TES). Heatmaps were generated from the normalized bigwig files using deeptools computeMatrix (scale-regions–missingDataAsZero -m 20000 -bs 50 -a 5000 -b 5000) with or without k-means clustering, and visualized with plotHeatmap. All other data were visualized in RStudio with ggplot2 (v3.3.1) and ggpubr (v0.4.0).
4sU Nascent RNA sequencing
20 × 106 THP-1-Cas9 cells expressing FgH1t-UTG-sgSCR-GFP or FgH1tUTG-sgINTS6-A-GFP with stable knockout of INTS6 were treated with DMSO (6 hours) or AZ5576 for 2 or 6 hours. Cells were labeled with 500 μM 4-thiouridine (4sU; Sigma Aldrich T4509) for the final 30 minutes of the treatment time and were lysed in TRIzol. D.melanogaster S2 cells (20 × 106) were also labeled with 4sU as above. Lysates were mixed with 1/5th volume of chloroform and the aqueous phase was collected after centrifugation at 4°C (13000 g, 15 minutes). Total RNA was precipitated in isopropanol (room temperature for 10 minutes, 4°C for 10 minutes at 13000 g) and washed once with 70 percent ethanol (4°C for 15 minutes at 13000 g). RNA was resuspended in ultrapure nuclease-free H2O and denatured at 65°C for 10 minutes. 80 μg human RNA (with 5% S2 RNA spike-in) was incubated for 1.5 hours with constant rotation at room temperature with 10mM Tris pH7.4, 1mM EDTA, 200 μg/mL dimethylformamide (Sigma Aldrich, 227056) and 300 μg EZ-Link™ HPDP Biotin (ThermoFisher Scientific 21341). The ‘biotin-labeling reaction’ was supplemented with 1 volume chloroform and vortexed well prior to centrifugation at 4°C (20000 g, 5 minutes). The aqueous phase was isolated and chloroform isolation was repeated twice, prior to the addition of 5M NaCl to a final concentration of 0.5M and 1 volume of isopropanol. Precipitated RNA was centrifuged at 4°C (20000 g, 20 minutes), washed with 75% ethanol (4°C, 20000 g, 20 minutes) and resuspended in nuclease-free H2O (1 μg/1 μg RNA input) prior to incubation at 65°C for 10 minutes. Biotinylated RNA was incubated with constant rotation for 15 minutes at room temperature with μMACs streptavidin beads and was isolated using μMACs columns (μMACs Strepavidin Kit, Miltenyi Biotec 130–074-101) that had been previously equilibrated with wash buffer (100mM Tris-HCl pH 7.4, 10mM EDTA, 1M NaCl, 0.1% Tween-20). Columns were washed with wash buffer pre-warmed to 65°C (5 × 0.9mL washes) and room temperature wash buffer (5 × 0.9mL washes) and RNA was eluted using DTT (100mM, 2 × 100 μL) and collected into 700 μL RLT buffer from the RNeasy MinElute Cleanup Kit (QIAGEN 74204). RLT samples were mixed with 500 μL 100% ethanol, loaded into RNeasy MinElute columns and centrifuged at 8000 g for 15 s. Columns were washed with 500 μL RPE buffer (8000 g, 15 s) and 500 μL 80% ethanol (8000 g, 2 minutes) and excess ethanol was removed through centrifugation at 20000 g for 5 minutes. RNA was eluted using 12 μL RNase-free water (20000 g, 1 minute). RNA was quantified using the Agilent Tapestation High Sensitivity RNA ScreenTape kit (Agilent 067–5576) as described. 1 μL High Sensitivity RNA Sample buffer (Agilent 5067–5580) was added to 2 μL RNA sample, mixed at 2000rpm for 1 minute, incubated at 72°C for 3 minutes and then incubated on ice for 2 minutes prior to loading into the High Sensitivity RNA ScreenTape (Agilent 5067–5579) using the Agilent 2200 TapeStation instrument. RNA sequencing libraries were prepared using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina and 75 base-pair single-end reads were sequenced using the Illumina NEXTseq 500. Sequencing files were demultiplexed (Bcl2fastq, v2.17.1.14) and QC was performed on FASTQ files using FASTQC (v0.11.6). Sequenced reads were aligned to a combined Hg19/dm3 reference genome using Bowtie (v2.3.4.1) and BAM files were generated and processed as described for ChIP-seq. Normalization to the D.melanogaster S2 RNA spike-in was performed similarly to that described previously (Orlando et al., 2014), with reads mapping to Hg19 human and dm3 D.melanogaster genomes calculated using FeatureCounts (Subread v2.0.0) and a scaleFactor determined by calculating the proportion of reads mapping to the dm3 genome relative to the combined Hg19/dm3 genome. BigWig files were generated using Deeptools (v3.0.0) bamCoverage function using the appropriate scale Factor (–normalizeUsing CPM–smoothLength 150–binSize 50 –e 200 scaleFactor #). Scaled BigWig files were visualized in IGV and the bigwigCompare function (Deeptools). Data was visualized in RStudio using ggplot2 (v3.3.1), ggrepel (v0.8.2) and ggfortify (v0.4.10) packages.
Transient Transcriptome Sequencing
HeLa cells infected with indicated shRNAs were placed under serum-starvation (0.3% SCS) the night before harvesting. Cells were treated with EGF (0.1 μg/mL) and concurrently labeled with 500 μM 4-thiouridine (4sU; Sigma Aldrich T4509) for 15 minutes. Cells were lysed in QIAzol lysis reagent (QIAgen, #79306), mixed with 1/5th volume of chloroform, and the aqueous phase collected after centrifugation at 4°C (13000 g, 15 minutes). Total RNA was precipitated in isopropanol (room temperature for 10 minutes), centrifuged at 4°C for 10 minutes at 13000 g, and washed twice with 75 percent ethanol. Pellet was air-dried and then re-suspended in ultrapure nuclease-free H2O on ice for 10 minutes, then 60°C for 5 minutes. 300 μg human RNA (with 5% S2 4sU-labeled RNA spike-in) was fragmented with the Bioruptor Plus for one cycle of 30 s/30 s ON/OFF at high settings. Fragmentation was checked using the Agilent RNA ScreenTape Assay: 5 μL RNA Sample buffer (Agilent 5067–5577) was added to 1 μL RNA sample, mixed at 2000rpm for 1 minute, incubated at 72°C for 3 minutes and then incubated on ice for 2 minutes prior to loading into the RNA Screen-Tape (Agilent 5067–5578) using the Agilent 2200 TapeStation instrument. 150 μg fragmented RNA was incubated at 65°C for 10 minutes, then on ice for 5 minutes before adding biotin-labeling reagents (100mM Tris pH 7.5, 10mM EDTA, 200 μL dimethylformamide (DMF), 200 μg EZ-link™ HPDP Biotin (ThermoFisher Scientific 21341)) and incubating in the dark at 24°C and 800rpm for 2 hours. Chloroform was added to the reaction and mixed well. After centrifugation, the upper aqueous phase was collected and the RNA was precipitated by the addition of 1/10th volume of 5M NaCl and 1 volume of isopropanol, centrifuged at 4°C (20000 g, 30 minutes), and washed with ice cold 75% ethanol. RNA was resuspended in ultrapure nuclease-free H2O, incubated on ice for 10 minutes, then 65°C for 5 minutes. Biotin-labeled RNA was enriched using Streptavidin beads (Invitrogen). Beads were equilibrated with wash buffer (100mM Tris pH 7.5, 10mM EDTA, 1M NaCl, 0.1% (v/v) Tween-20) before incubation with biotinylated RNA at 4°C and 800rpm for 15 minutes. Beads were washed 3x with 65°C wash buffer and 3x with room temperature wash buffer. RNA was eluted twice by resuspending the beads in 100mM DTT buffer, and incubating for 5 minutes. RNA was purified using the RNA Clean and Concentrator kit (Zymo research R1015) as follows: 2 volumes of RNA Binding Buffer was added to each sample and mixed, followed by addition of an equal volume of ethanol, and transferred to the Zymo-Spin IC Column in a collection tube and centrifuged (all centrifugations performed at 16000 g for 30 s). The column was washed with 400 μL of RNA Wash Buffer before the addition of DNase I Reaction Mix (5 μL of 1 U/μL DNase I + 35 μL of DNA Digestion Buffer) and incubation at room temperature for 15 minutes. Following DNase treatment, the column was washed with 400 μL RNA Prep Buffer, 700 μL of RNA Wash Buffer, then 400 μL of RNA Wash Buffer, and eluted in 15 μL of DNase/RNase-Free water. Sequencing libraries were prepared with the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina and 75bp single-end or 40bp paired-end reads were sequenced using the Illumina NEXTseq 500. Reads were aligned to hg19 and dm5 reference genomes using STAR v2.5 (Dobin et al., 2013) in 2-pass mode with the same parameters described above for HeLa 3′-Quantseq. BAM files were filtered based on alignment quality (q = 10) using Samtools v0.1.19 (Li et al., 2009). Feature-Counts (Liao et al., 2014) was used to count reads mapping to each gene. Normalization to the D.melanogaster S2 RNA spike-in, and subsequent normalized BAM and BigWig file generation was performed as described above for HeLa ChIP-seq. Average read density across defined genomic intervals was computed on the normalized BAM files using seqMINER 1.3.3 package (Ye et al., 2011) with the following parameters: left and right extensions = 5.0 kb; internal bins = 160; flanking region bins = 20. Read density matrices, featureCounts, were exported for plotting in RStudio with ggplot2 (v3.3.1).
FastGRO
FastGRO was performed as described (Barbieri et al., 2020). HeLa cells infected with shLUC or shINTS6 were treated, 72 hours after infection, with DMSO or 300nM CDK9i AZ5576 for 2 hours, then subsequently treated for 15 minutes with 0.1 μg/mL EGF. Per condition, 20×106 cells were harvested and washed twice with ice-cold PBS before adding swelling buffer (10 mM Tris-HCL pH 7.5, 2mM MgCl2, 3 mM CaCl2, 2U/ml Superase-in (Invitrogen)). Cells were swollen for 5 min on ice, washed with swelling buffer with 10% glycerol, and then lysed in lysis buffer (10 mM Tris-HCL pH 7.5, 2mM MgCl2, 3mM CaCl2, 10% glycerol, 1mL Igepal (NP-40), 2 U/ml Superase-in) to isolate nuclei. Nuclei were washed twice with lysis buffer and resuspended in freezing buffer (40% glycerol, 5mM MgCl2, 0.1mM 0.5M EDTA, 50mM Tris-HCL pH 8.3) to a concentration of 2×107 nuclei per 100 μL. Nuclei were frozen on dry ice and stored at −80°C. For the run-on reaction, nuclei were thawed on ice, an equal volume of pre-warmed nuclear run-on reaction buffer (10mM Tris-HCl pH 8, 5mM MgCl2, 300mM KCl, 1mM DTT, 500 μM ATP, 500 μM GTP, 500 μM 4-thio-UTP, 2 μM CTP, 200 U/ml Superase-in, 1% Sarkosyl (N-Laurylsarcosine sodium salt solution) was added. This was incubated for 7 min at 30°C for the nuclear run-on. Nuclear run-on RNA was extracted with TRIzol LS reagent (Invitrogen 10296010) as follows: 3 volumes of TRIzol LS Reagent was added to 1 volume of sample, mixed, and incubated for 5 minutes at room temperature. Then 0.8 volume of chloroform was added, mixed, incubated for 3 minutes at room temperature, and centrifuged for 15 minutes at 12000 g at 4°C. The colorless upper aqueous phase containing the RNA was transferred into a new tube, and 2 volumes of isopropanol were added, mixed, incubated for 10 minutes at room temperature, centrifuged for 10 minutes at 12000 g at 4°C, and the supernatant discarded. The pellet was resuspended in 4 volumes of 75% ethanol, vortexed, centrifuged for 5 minutes at 7500 g at 4°C, and supernatant removed again. The RNA pellet was air-dried for 10 minutes at room temperature. RNA was then resuspended in RNase-free water and the concentration was determined with Qubit High Sensitivity Assay kit (Invitrogen). Spike-in D.melanogaster RNA was added at 10%, and RNA was fragmented with a Bioruptor UCD-200 for 1–5 cycles of 30 s ON/30 s OFF, high settings. Fragmentation efficiency was analyzed by running fragmented and unfragmented RNA using the Agilent High Sensitivity RNA ScreenTape Assay as described above. Fragmented RNA was incubated in Biotinylation Solution (100 mM Tris pH 7.5, 10 mM EDTA pH 8.0, 40% dimethylformamide, 200 g/mL EZ-link HPDP Biotin (Thermo Scientific) for 2h in the dark at 25°C and 800 rpm. After ethanol precipitation, the biotinylated RNA was resuspended in water and separated with M280 Streptavidin Dynabeads (Invitrogen). 100 μl/sample of beads were washed twice with 2 volumes of freshly prepared wash buffer (100 mM Tris pH 7.5, 10mM EDTA pH 8.0, 1M NaCl, 0.1% (v/v) Tween-20) and re-suspended in 1 volume of wash buffer and added to the biotinylated-RNA. After 15 minutes in rotation at 4°C, beads were washed three times with wash buffer pre-warmed at 65°C and three times with room temperature wash buffer. 4-S-UTP containing RNA was eluted in 100mM DTT buffer and purified with RNA Clean and Concentrator kit (Zymo Research) as described above with in-column DNase reaction to eliminate traces of genomic DNA. The eluted RNA was quantified with Qubit High Sensitivity Assay kit (Invitrogen) and used to produce barcoded RNA sequencing libraries using the NEBNext Ultra II Directional RNA Library Prep kit (New England Biolabs). Libraries were sequenced on Illumina NextSeq 500. Reads were aligned to hg19 and dm5 reference genomes using STAR v2.5 (Dobin et al., 2013) in 2-pass mode with the same parameters described above for HeLa 3′-Quantseq. BAM files were filtered based on alignment quality (q = 10) using Samtools v0.1.19 (Li et al., 2009). The latest annotations were obtained from Ensembl to build reference indexes for the STAR alignments. FeatureCounts (Liao et al., 2013) was used to count reads mapping to each gene. Normalization to the D.melanogaster S2 RNA spike-in, and subsequent normalized BAM and BigWig file generation, was performed as described above for HeLa ChIP-seq. Average read density across defined genomic intervals was computed on the normalized BAM files using seqMINER 1.3.3 package (Ye et al., 2011) with the following parameters: left and right extensions = 5.0 kb; internal bins = 160; flanking region bins = 20. Read density matrices and featureCounts outputs were exported for plotting in RStudio with ggplot2 (v3.3.1).
TCGA analysis
RNA-seq by Expectation Maximization (RSEM) scaled transcripts per million for TCGA samples were downloaded from the GDAC Firehose website (Li and Dewey, 2011) and copy number status for INTS6 were downloaded via the cBioPortal website (Gao et al., 2013). TCGA samples were stratified into copy number groups (change of 2,1, 0, −1, −2 from diploid) and PFI survival endpoints were fit to Kaplan-Meier and Cox regression models using the survival R package (version 3.1–8) (Therneau et al., 2018). Copy number and gene expression data from the Cancer Cell Line Encyclopedia were downloaded from the DepMap portal (20Q3 release) (Ghandi et al., 2019). Differential expression between wild-type samples and homo-deleted samples for each gene using the limma-trend method with the limma R package (version 3.42.2) (Phipson et al., 2016).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data is presented as the mean ± standard deviation (SD) or standard error of the mean (SEM) as indicated in the figure legends. The number of replicates for each experiment (number of independent experiments for in vitro cell-based assays, number of mice for in vivo animal studies) is indicated in the figure legends. To determine statistical significance 2-way ANOVA, Student’s t-Test and Log-rank (Mantel-Cox) tests were performed using GraphPad Prism 8 software (La Jolla, CA) and unpaired, two-sided tests were performed using RStudio (v3.5.1). p value of < 0.05 (*) was deemed statistically significant.
Supplementary Material
Highlights.
Loss of the INTS6 subunit of Integrator confers resistance to CDK9 inhibition
INTS6 recruits PP2A to chromatin and forms a submodule of Integrator (Int-PP2A)
Int-PP2A opposes CDK9 at the phosphorylation level to fine-tune transcription
PP2A activators synergize therapeutically with CDK9 inhibitors in cancer
ACKNOWLEDGMENTS
We thank members of the Johnstone and Gardini laboratories, the Translational Research Laboratory, the Victorian Centre for Functional Genomics (VCFG), Molecular Genomics Core and the Flow Cytometry Core Facilities at the Peter MacCallum Cancer Center (PMCC), the Proteomics Facility at the Walter and Eliza Hall Institute, and the Genomics Core and the Proteomics Core at The Wistar Institute (P30-CA010815) for their technical expertise and for providing reagents. We thank Prof. Robert Fisher for providing phosphoSpt5 antibodies. Illustrations were made with BioRender.com. R.W.J. was supported by the Cancer Council Victoria, National Health and Medical Research Council of Australia (NHMRC) and The Kids’ Cancer Project. The Gardini laboratory (A.G.) was supported the American Cancer Society (RSG-18-157-01-DMC), the G. Harold & Leila Y. Mathers Foundation (A.G.), the NIH (R01 HL141326), the Emerson Collective Cancer Research Fund, and the Ovarian Cancer Research Alliance. S.J.V. was supported by a Rubicon fellowship (NWO, 019.161LW.017), NHMRC EL1 fellowship (GNT1178339), and The Kids’ Cancer Project. S.A.W. was supported by a training grant from NIH (T32-GM071339). J.R.D. was supported by an Early Career Seed Grant from the Victorian Cancer Agency (VCA). S.J.H. was supported by a Postdoctoral Fellowship from the Cancer Council of Victoria (CCV). K.F.H. was supported by a NHMRC Senior Research Fellowship (APP1078220). J.W. and K.P. were supported by Academy of Finland (294850) and Finnish Cancer Foundation. K.P. was supported by Finnish Cultural Foundation. The VCFG ACRF Translational Reverse Phase Protein Array platform (K.J.S.) is funded by the Australian Cancer Research Foundation (ACRF), University of Melbourne Collaborative Research Infrastructure Program, and the PMCC Foundation. The PMCC Foundation and ACRF provide generous support for equipment and core facilities.
Footnotes
DECLARATION OF INTERESTS
The Johnstone laboratory receives funding support from Roche, BMS, Astra Zeneca, and MecRx. R.W.J. is a shareholder in MecRx and receives consultancy payments.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.04.022.
REFERENCES
- Adelman K, and Lis JT (2012). Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat. Rev. Genet 13, 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akama-Garren EH, Joshi NS, Tammela T, Chang GP, Wagner BL, Lee DY, Rideout WM 3rd, Papagiannakopoulos T, Xue W, and Jacks T. (2016). A Modular Assembly Platform for Rapid Generation of DNA Constructs. Sci. Rep 6, 16836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit I, Citri A, Shay T, Lu Y, Katz M, Zhang F, Tarcic G, Siwak D, Lahad J, Jacob-Hirsch J, et al. (2007). A module of negative feedback regulators defines growth factor signaling. Nat. Genet 39, 503–512. [DOI] [PubMed] [Google Scholar]
- Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O’Connor L, Milla L, Wilcox S, Tai L, Strasser A, and Herold MJ (2015). An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep. 10, 1422–1432. [DOI] [PubMed] [Google Scholar]
- Avni D, Yang H, Martelli F, Hofmann F, ElShamy WM, Ganesan S, Scully R, and Livingston DM (2003). Active localization of the retinoblastoma protein in chromatin and its response to S phase DNA damage. Mol. Cell 12, 735–746. [DOI] [PubMed] [Google Scholar]
- Bacon CW, and D’Orso I. (2019). CDK9: a signaling hub for transcriptional control. Transcription 10, 57–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillat D, Hakimi MA, Näär AM, Shilatifard A, Cooch N, and Shiekhattar R. (2005). Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase II. Cell 123, 265–276. [DOI] [PubMed] [Google Scholar]
- Baker A, Gregory GP, Verbrugge I, Kats L, Hilton JJ, Vidacs E, Lee EM, Lock RB, Zuber J, Shortt J, and Johnstone RW (2016). The CDK9 Inhibitor Dinaciclib Exerts Potent Apoptotic and Antitumor Effects in Preclinical Models of MLL-Rearranged Acute Myeloid Leukemia. Cancer Res. 76, 1158–1169. [DOI] [PubMed] [Google Scholar]
- Barbieri E, Trizzino M, Welsh SA, Owens TA, Calabretta B, Carroll M, Sarma K, and Gardini A. (2018). Targeted Enhancer Activation by a Subunit of the Integrator Complex. Mol. Cell 71, 103–116.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri E, Hill C, Quesnel-Vallières M, Zucco AJ, Barash Y, and Gardini A. (2020). Rapid and Scalable Profiling of Nascent RNA with fastGRO. Cell Rep. 33, 108373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AR, Tibbit C, Ponting CP, and Liu JL (2014). Mutagenesis and homologous recombination in Drosophila cell lines using CRISPR/Cas9. Biol. Open 3, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckedorff F, Blumenthal E, daSilva LF, Aoi Y, Cingaram PR, Yue J, Zhang A, Dokaneheifard S, Valencia MG, Gaidosh G, et al. (2020). The Human Integrator Complex Facilitates Transcriptional Elongation by Endonucleolytic Cleavage of Nascent Transcripts. Cell Rep. 32, 107917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bots M, Verbrugge I, Martin BP, Salmon JM, Ghisi M, Baker A, Stanley K, Shortt J, Ossenkoppele GJ, Zuber J, et al. (2014). Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood 123, 1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JE, Hnisz D, and Young RA (2017). Transcriptional Addiction in Cancer. Cell 168, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao SH, and Price DH (2001). Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J. Biol. Chem 276, 31793–31799. [DOI] [PubMed] [Google Scholar]
- Chen FX, Woodfin AR, Gardini A, Rickels RA, Marshall SA, Smith ER, Shiekhattar R, and Shilatifard A. (2015). PAF1, a Molecular Regulator of Promoter-Proximal Pausing by RNA Polymerase II. Cell 162, 1003–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FX, Smith ER, and Shilatifard A. (2018). Born to run: control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol 19, 464–478. [DOI] [PubMed] [Google Scholar]
- Chou J, Quigley DA, Robinson TM, Feng FY, and Ashworth A. (2020). Transcription-Associated Cyclin-Dependent Kinases as Targets and Biomarkers for Cancer Therapy. Cancer Discov. 10, 351–370. [DOI] [PubMed] [Google Scholar]
- Cidado J, Boiko S, Proia T, Ferguson D, Criscione SW, San Martin M, Pop-Damkov P, Su N, Roamio Franklin VN, Sekhar Reddy Chilamakuri C, et al. (2020). AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clin. Cancer Res 26, 922–934. [DOI] [PubMed] [Google Scholar]
- Clark AR, and Ohlmeyer M. (2019). Protein phosphatase 2A as a therapeutic target in inflammation and neurodegeneration. Pharmacol. Ther 201, 181–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core L, and Adelman K. (2019). Promoter-proximal pausing of RNA polymerase II: a nexus of gene regulation. Genes Dev. 33, 960–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortazar MA, Sheridan RM, Erickson B, Fong N, Glover-Cutter K, Brannan K, and Bentley DL (2019). Control of RNA Pol II Speed by PNUTS-PP1 and Spt5 Dephosphorylation Facilitates Termination by a “Sitting Duck Torpedo” Mechanism. Mol. Cell 76, 896–908.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, and Naldini L. (1998). A third-generation lentivirus vector with a conditional packaging system. J. Virol 72, 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton JD, Francis L, Davidson L, and West S. (2020). A unified allosteric/torpedo mechanism for transcriptional termination on human protein-coding genes. Genes Dev. 34, 132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardy SR, and Farnham PJ (2001). c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J. Biol. Chem 276, 48562–48571. [DOI] [PubMed] [Google Scholar]
- Egan B, Yuan CC, Craske ML, Labhart P, Guler GD, Arnott D, Maile TM, Busby J, Henry C, Kelly TK, et al. (2016). An Alternative Approach to ChIP-Seq Normalization Enables Detection of Genome-Wide Changes in Histone H3 Lysine 27 Trimethylation upon EZH2 Inhibition. PLoS ONE 11, e0166438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichhorn PJ, Creyghton MP, and Bernards R. (2009). Protein phosphatase 2A regulatory subunits and cancer. Biochim. Biophys. Acta 1795, 1–15. [DOI] [PubMed] [Google Scholar]
- Elrod ND, Henriques T, Huang KL, Tatomer DC, Wilusz JE, Wagner EJ, and Adelman K. (2019). The Integrator Complex Attenuates Promoter-Proximal Transcription at Protein-Coding Genes. Mol. Cell 76, 738–752.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filleur S, Hirsch J, Wille A, Schön M, Sell C, Shearer MH, Nelius T, and Wieland I. (2009). INTS6/DICE1 inhibits growth of human androgen-independent prostate cancer cells by altering the cell cycle profile and Wnt signaling. Cancer Cell Int. 9, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowle H, Zhao Z, and Graña X. (2019). PP2A holoenzymes, substrate specificity driving cellular functions and deregulation in cancer. Adv. Cancer Res 144, 55–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga K. (2020). P-TEFb as A Promising Therapeutic Target. Molecules 25, 8385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardini A, Baillat D, Cesaroni M, Hu D, Marinis JM, Wagner EJ, Lazar MA, Shilatifard A, and Shiekhattar R. (2014). Integrator regulates transcriptional initiation and pause release following activation. Mol. Cell 56, 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019). Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grallert A, Boke E, Hagting A, Hodgson B, Connolly Y, Griffiths JR, Smith DL, Pines J, and Hagan IM (2015). A PP1-PP2A phosphatase relay controls mitotic progression. Nature 517, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregersen LH, Mitter R, Ugalde AP, Nojima T, Proudfoot NJ, Agami R, Stewart A, and Svejstrup JQ (2019). SCAF4 and SCAF8, mRNA Anti-Terminator Proteins. Cell 177, 1797–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory GP, Hogg SJ, Kats LM, Vidacs E, Baker AJ, Gilan O, Lefebure M, Martin BP, Dawson MA, Johnstone RW, and Shortt J. (2015). CDK9 inhibition by dinaciclib potently suppresses Mcl-1 to induce durable apoptotic responses in aggressive MYC-driven B-cell lymphoma in vivo. Leukemia 29, 1437–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gressel S, Schwalb B, Decker TM, Qin W, Leonhardt H, Eick D, and Cramer P. (2017). CDK9-dependent RNA polymerase II pausing controls transcription initiation. eLife 6, e29736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, and Young RA (2007). A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haesen D, Abbasi Asbagh L, Derua R, Hubert A, Schrauwen S, Hoorne Y, Amant F, Waelkens E, Sablina A, and Janssens V. (2016). Recurrent PPP2R1A Mutations in Uterine Cancer Act through a Dominant-Negative Mechanism to Promote Malignant Cell Growth. Cancer Res. 76, 5719–5731. [DOI] [PubMed] [Google Scholar]
- Hargreaves DC, Horng T, and Medzhitov R. (2009). Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138, 129–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlen KM, and Churchman LS (2017). The code and beyond: transcription regulation by the RNA polymerase II carboxy-terminal domain. Nat. Rev. Mol. Cell Biol 18, 263–273. [DOI] [PubMed] [Google Scholar]
- Hashiguchi T, Bruss N, Best S, Lam V, Danilova O, Paiva CJ, Wolf J, Gilbert EW, Okada CY, Kaur P, et al. (2019). Cyclin-Dependent Kinase-9 Is a Therapeutic Target in MYC-Expressing Diffuse Large B-Cell Lymphoma. Mol. Cancer Ther 18, 1520–1532. [DOI] [PubMed] [Google Scholar]
- He L, Kulesskiy E, Saarela J, Turunen L, Wennerberg K, Aittokallio T, and Tang J. (2018). Methods for High-throughput Drug Combination Screening and Synergy Scoring. Methods Mol. Biol. 1711, 351–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KL, Jee D, Stein CB, Elrod ND, Henriques T, Mascibroda LG, Baillat D, Russell WK, Adelman K, and Wagner EJ (2020). Integrator Recruits Protein Phosphatase 2A to Prevent Pause Release and Facilitate Transcription Termination. Mol. Cell 80, 345–358.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsen T, de Vlieg J, and Alkema W. (2008). BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, and Ozato K. (2005). The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 19, 523–534. [DOI] [PubMed] [Google Scholar]
- Jeronimo C, Collin P, and Robert F. (2016). The RNA Polymerase II CTD: The Increasing Complexity of a Low-Complexity Protein Domain. J. Mol. Biol 428, 2607–2622. [DOI] [PubMed] [Google Scholar]
- Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE, and Zhang F. (2017). Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc 12, 828–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastrinsky DB, Sangodkar J, Zaware N, Izadmehr S, Dhawan NS, Narla G, and Ohlmeyer M. (2015). Reengineered tricyclic anti-cancer agents. Bioorg. Med. Chem 23, 6528–6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauko O, O’Connor CM, Kulesskiy E, Sangodkar J, Aakula A, Izadmehr S, Yetukuri L, Yadav B, Padzik A, Laajala TD, et al. (2018). PP2A inhibition is a druggable MEK inhibitor resistance mechanism in KRAS-mutant lung cancer cells. Sci. Transl. Med 10, eaaq1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauko O, Imanishi SY, Kulesskiy E, Yetukuri L, Laajala TD, Sharma M, Pavic K, Aakula A, Rupp C, Jumppanen M, et al. (2020). Phosphoproteome and drug-response effects mediated by the three protein phosphatase 2A inhibitor proteins CIP2A, SET, and PME-1. J. Biol. Chem 295, 4194–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney CJ, Vervoort SJ, Hogg SJ, Ramsbottom KM, Freeman AJ, Lalaoui N, Pijpers L, Michie J, Brown KK, Knight DA, et al. (2018). Tumor immune evasion arises through loss of TNF sensitivity. Sci. Immunol 3, eaar3451. [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, et al. (2014). Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F, Gardini A, Zhang A, and Shiekhattar R. (2015). Integrator mediates the biogenesis of enhancer RNAs. Nature 525, 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laitem C, Zaborowska J, Isa NF, Kufs J, Dienstbier M, and Murphy S. (2015). CDK9 inhibitors define elongation checkpoints at both ends of RNA polymerase II-transcribed genes. Nat. Struct. Mol. Biol 22, 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard D, Huang W, Izadmehr S, O’Connor CM, Wiredja DD, Wang Z, Zaware N, Chen Y, Schlatzer DM, Kiselar J, et al. (2020). Selective PP2A Enhancement through Biased Heterotrimer Stabilization. Cell 181, 688–701.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, arXiv:1303.3997. [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhai X, Wang H, Qian X, Miao H, and Zhu X. (2015). Bioinformatics analysis of gene expression profiles in childhood B-precursor acute lymphoblastic leukemia. Hematology 20, 377–383. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W. (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, and Shilatifard A. (2010). AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol. Cell 37, 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Lin C, and Shilatifard A. (2012). The super elongation complex (SEC) family in transcriptional control. Nat. Rev. Mol. Cell Biol 13, 543–547. [DOI] [PubMed] [Google Scholar]
- Malumbres M. (2014). Cyclin-dependent kinases. Genome Biol. 15, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J 10.14806/ej.14817.14801.14200. [DOI] [Google Scholar]
- Martin RD, Hébert TE, and Tanny JC (2020). Therapeutic Targeting of the General RNA Polymerase II Transcription Machinery. Int. J. Mol. Sci 21, 3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, Landry HM, and Churchman LS (2017). Pause & go: from the discovery of RNA polymerase pausing to its functional implications. Curr. Opin. Cell Biol 46, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield JE, Burkholder NT, and Zhang YJ (2016). Dephosphorylating eukaryotic RNA polymerase II. Biochim. Biophys. Acta 1864, 372–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merisaari J, Denisova OV, Doroszko M, Le Joncour V, Johansson P, Leenders WPJ, Kastrinsky DB, Zaware N, Narla G, Laakkonen P, et al. (2020). Monotherapy efficacy of BBB-permeable small molecule activators of PP2A in glioblastoma. bioRxiv. 10.1101/777276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298. [DOI] [PubMed] [Google Scholar]
- Morita K, He S, Nowak RP, Wang J, Zimmerman MW, Fu C, Durbin AD, Martel MW, Prutsch N, Gray NS, et al. (2020). Allosteric Activators of Protein Phosphatase 2A Display Broad Antitumor Activity Mediated by Dephosphorylation of MYBL2. Cell 181, 702–715.e20. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, and Guenther MG (2014). Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep. 9, 1163–1170. [DOI] [PubMed] [Google Scholar]
- Parua PK, Booth GT, Sansó M, Benjamin B, Tanny JC, Lis JT, and Fisher RP (2018). A Cdk9-PP1 switch regulates the elongation-termination transition of RNA polymerase II. Nature 558, 460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parua PK, Kalan S, Benjamin B, Sansó M, and Fisher RP (2020). Distinct Cdk9-phosphatase switches act at the beginning and end of elongation by RNA polymerase II. Nat. Commun 11, 4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotti D, and Neviani P. (2013). Protein phosphatase 2A: a target for anti-cancer therapy. Lancet Oncol. 14, e229–e238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipson B, Lee S, Majewski IJ, Alexander WS, and Smyth GK (2016). Robust Hyperparameter Estimation Protects against Hypervariable Genes and Improves Power to Detect Differential Expression. Ann. Appl. Stat 10, 946–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T. (2014). Deep-Tools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, and Weissman IL (2003). A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 423, 409–414. [DOI] [PubMed] [Google Scholar]
- Riemondy KA, Sheridan RM, Gillen A, Yu Y, Bennett CG, and Hesselberth JR (2017). valr: Reproducible genome interval analysis in R. F1000Res. 6, 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rienzo M, and Casamassimi A. (2016). Integrator complex and transcription regulation: Recent findings and pathophysiology. Biochim. Biophys. Acta 1859, 1269–1280. [DOI] [PubMed] [Google Scholar]
- Roeder RG (2019). 50+ years of eukaryotic transcription: an expanding universe of factors and mechanisms. Nat. Struct. Mol. Biol 26, 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röpke A, Buhtz P, Böhm M, Seger J, Wieland I, Allhoff EP, and Wieacker PF (2005). Promoter CpG hypermethylation and downregulation of DICE1 expression in prostate cancer. Oncogene 24, 6667–6675. [DOI] [PubMed] [Google Scholar]
- Rubtsova MP, Vasilkova DP, Moshareva MA, Malyavko AN, Meerson MB, Zatsepin TS, Naraykina YV, Beletsky AV, Ravin NV, and Dontsova OA (2019). Integrator is a key component of human telomerase RNA biogenesis. Sci. Rep 9, 1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangodkar J, Perl A, Tohme R, Kiselar J, Kastrinsky DB, Zaware N, Izadmehr S, Mazhar S, Wiredja DD, O’Connor CM, et al. (2017). Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J. Clin. Invest 127, 2081–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F. (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansó M, Levin RS, Lipp JJ, Wang VY, Greifenberg AK, Quezada EM, Ali A, Ghosh A, Larochelle S, Rana TM, et al. (2016). P-TEFb regulation of transcription termination factor Xrn2 revealed by a chemical genetic screen for Cdk9 substrates. Genes Dev. 30, 117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen I, Zhou X, Chernobrovkin A, Puerta-Cavanzo N, Kanno T, Salignon J, Stoehr A, Lin XX, Baskaner B, Brandenburg S, et al. (2020). DAF-16/FOXO requires Protein Phosphatase 4 to initiate transcription of stress resistance and longevity promoting genes. Nat. Commun 11, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih IeM., Panuganti PK, Kuo KT, Mao TL, Kuhn E, Jones S, Velculescu VE, Kurman RJ, and Wang TL (2011). Somatic mutations of PPP2R1A in ovarian and uterine carcinomas. Am. J. Pathol 178, 1442–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, et al. (2016). Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 529, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, Richard DJ, Saraf A, Toschi A, Bolderson E, Florens L, Washburn MP, Khanna KK, and Pagano M. (2009). INTS3 controls the hSSB1-mediated DNA damage response. J. Cell Biol 187, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatomer DC, Elrod ND, Liang D, Xiao MS, Jiang JZ, Jonathan M, Huang KL, Wagner EJ, Cherry S, and Wilusz JE (2019). The Integrator complex cleaves nascent mRNAs to attenuate transcription. Genes Dev. 33, 1525–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therneau TM, Lumley T, Atkinson E, and Crowson C. (2018). A Package for Survival Analysis in S. version 2.38. https://cran.r-project.org/web/packages/survival/index.html. [Google Scholar]
- Vos SM, Farnung L, Boehning M, Wigge C, Linden A, Urlaub H, and Cramer P. (2018a). Structure of activated transcription complex Pol II-DSIF-PAF-SPT6. Nature 560, 607–612. [DOI] [PubMed] [Google Scholar]
- Vos SM, Farnung L, Urlaub H, and Cramer P. (2018b). Structure of paused transcription complex Pol II-DSIF-NELF. Nature 560, 601–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermarck J, and Neel BG (2020). Piecing Together a Broken Tumor Suppressor Phosphatase for Cancer Therapy. Cell 181, 514–517. [DOI] [PubMed] [Google Scholar]
- Wu T, Qin Z, Tian Y, Wang J, Xu C, Li Z, and Bian J. (2020). Recent Developments in the Biology and Medicinal Chemistry of CDK9 Inhibitors: An Update. J. Med. Chem 63, 13228–13257. [DOI] [PubMed] [Google Scholar]
- Xu Y, Bernecky C, Lee CT, Maier KC, Schwalb B, Tegunov D, Plitzko JM, Urlaub H, and Cramer P. (2017). Architecture of the RNA polymerase II-Paf1C-TFIIS transcription elongation complex. Nat. Commun 8, 15741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, and Veraksa A. (2017). Single-Step Affinity Purification of ERK Signaling Complexes Using the Streptavidin-Binding Peptide (SBP) Tag. Methods Mol. Biol 1487, 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye T, Krebs AR, Choukrallah MA, Keime C, Plewniak F, Davidson I, and Tora L. (2011). seqMINER: an integrated ChIP-seq data interpretation platform. Nucleic Acids Res. 39, e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye T, Ravens S, Krebs AR, and Tora L. (2014). Interpreting and visualizing ChIP-seq data with the seqMINER software. Methods Mol. Biol 1150, 141–152. [DOI] [PubMed] [Google Scholar]
- Yu M, Yang W, Ni T, Tang Z, Nakadai T, Zhu J, and Roeder RG (2015). RNA polymerase II-associated factor 1 regulates the release and phosphorylation of paused RNA polymerase II. Science 350, 1383–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue J, Vendramin R, Liu F, Lopez O, Valencia MG, Gomes Dos Santos H, Gaidosh G, Beckedorff F, Blumenthal E, Speroni L, et al. (2020). Targeted chemotherapy overcomes drug resistance in melanoma. Genes Dev. 34, 637–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Qi Y, Hu S, Cao X, Xu C, Yin Z, Chen X, Li Y, Liu W, Li J, et al. (2020). Identification of Integrator-PP2A complex (INTAC), an RNA polymerase II phosphatase. Science 370, eabb5872. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Bioinformatics and proteomics datasets generated during this study are available from the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/; GSE163803, GSE163803, GSE163804, GSE163805, GSE145379) and the Proteomics Identifications Database (PRIDE, https://www.ebi.ac.uk/pride/archive/; PXD022962, PXD025060) repositories respectively.