

P-selectin expressed on the surface of activated platelets and endothelium binds PSGL-1 expressed on leukocytes. Wong et al describe a novel pegylated P-selectin inhibitor, P-G6, that blocks platelet-leukocyte and endothelium-leukocyte interactions. PG6 reduces thrombosis in a murine model of thrombosis without disrupting hemostasis, positioning it as a promising candidate for clinical studies for prevention of venous thrombosis.
Key Points
P-G6 inhibits P-selectin binding to PSGL-1 and reduces platelet-leukocyte aggregation in vitro and in vivo.
P-G6 inhibits venous thrombosis in a preclinical model of venous thromboembolism without increasing bleeding time.
Visual Abstract

Abstract
Events mediated by the P-selectin/PSGL-1 pathway play a critical role in the initiation and propagation of venous thrombosis by facilitating the accumulation of leukocytes and platelets within the growing thrombus. Activated platelets and endothelium express P-selectin, which binds P-selectin glycoprotein ligand-1 (PSGL-1) that is expressed on the surface of all leukocytes. We developed a pegylated glycomimetic of the N terminus of PSGL-1, PEG40-GSnP-6 (P-G6), which proved to be a highly potent P-selectin inhibitor with a favorable pharmacokinetic profile for clinical translation. P-G6 inhibits human and mouse platelet-monocyte and platelet-neutrophil aggregation in vitro and blocks microcirculatory platelet-leukocyte interactions in vivo. Administration of P-G6 reduces thrombus formation in a nonocclusive model of deep vein thrombosis with a commensurate reduction in leukocyte accumulation, but without disruption of hemostasis. P-G6 potently inhibits the P-selectin/PSGL-1 pathway and represents a promising drug candidate for the prevention of venous thrombosis without increased bleeding risk.
Introduction
As many as 1 million people in the United States are affected each year by venous thromboembolism (VTE), with significant associated morbidity and mortality.1,2 Pulmonary embolus and deep venous thrombosis constitute the majority of venous thrombotic events and necessitate systemic anticoagulation to prevent clot propagation and fatal pulmonary embolus.3 However, all anticoagulants increase the risk of bleeding, with hemorrhagic complications occurring in up to 10% of individuals during the initial course of treatment depending on the drug regimen, indication, and duration of therapy.4-6 Despite the initial promise that direct thrombin and factor Xa inhibitors would display superior safety profiles compared with warfarin, the risk of bleeding in “real-world” settings remains in the range of 5% to 10% or higher.6,7 As such, there is an urgent need for safer therapies.
It is now well recognized that the pathophysiology of VTE extends beyond Virchow’s triad of venous stasis, hypercoagulability, and endothelial dysfunction.8 Increased rates of VTE in patients with chronic inflammatory conditions, such as inflammatory bowel disease or rheumatoid arthritis, highlight that a systemic inflammatory state is an important risk factor for VTE.9,10 Mechanistic studies have confirmed that neutrophils, monocytes, and platelet-leukocyte aggregates are contained within venous thrombi, as well as revealed their critical role in venous thrombus initiation and propagation.11 Numerous studies have demonstrated that monocyte- and neutrophil-derived tissue factor (TF) is a decisive initial trigger for fibrin deposition12,13 and revealed a role for neutrophil extracellular traps in promoting thrombus propagation.14-18 In principle, targeting leukocyte-driven events, rather than the coagulation cascade alone, may provide a means to prevent or treat venous thrombosis while decreasing bleeding risk.
P-selectin is expressed on the surface of activated platelets and endothelium and binds its cognate ligand, P-selectin glycoprotein ligand-1 (PSGL-1), which is expressed on leukocytes and supports platelet-leukocyte aggregation and leukocyte rolling.19-24 Transgenic mouse studies have demonstrated that P-selectin mediates leukocyte recruitment, TF release, and fibrin deposition during the course of venous thrombosis.11,12,25,26 Significantly, in a variety of animal models, inhibitors of the P-selectin/PSGL-1 pathway, including monoclonal antibodies,27-29 recombinant PSGL-1–immunoglobulin G fusion (rPSGL-Ig),30-32 small molecule antagonists,33-36 and oligonucleotide aptamers,37 attenuated the VTE phenotype by reducing thrombus accumulation, accelerating clot resolution, and decreasing the local inflammatory response and late vein wall fibrosis.
Although supporting the potential clinical significance of the P-selectin/PSGL-1 pathway in VTE, these particular strategies have faced a number of barriers with regard to translation to the clinic. All therapeutic antibodies are susceptible to an antidrug immune response that may promote the loss of drug efficacy, particularly among patients requiring repeated dosing and long-term therapy.38 Evidence of anti-drug antibody production to chimeric and humanized monoclonal antibodies underscores the continued limitations that exist when attempting to reduce antibody immunogenicity.39 Recombinant protein production possesses its own inherent challenges, including imprecise posttranslation modifications in nonhuman cell lines with a failure to preserve structural fidelity under sensitive culture conditions.40 As an example, translational studies with rPSGL-Ig were only possible following the discovery that transfection of Chinese hamster ovarian cell lines with core-2 β1,6-GlcNAc transferase was required to preserve high-affinity bonding.41-43 Moreover, protein modifications can induce hypersensitivity, alter pharmacokinetics, and induce immunogenicity.44,45 As such, the production of recombinant proteins, especially therapeutic glycoproteins bearing complex glycans, requires precise control of an inherently complicated and sensitive biologic process, which is especially difficult to achieve on a commercial scale.46
Although synthetic small molecule inhibitors for P-selectin remain an attractive target, developing high-affinity synthetic antagonists continues to pose a significant challenge.47 First-generation P-selectin inhibitors were designed to mimic the tetrasaccharide sialyl LewisX moiety of PSGL-1 but failed to account for the crucial contributions of multiple clustered tyrosine sulfates on the N terminus of PSGL-1.36,48-53 High-affinity binding of P-selectin to PSGL-1 requires stereospecific interactions with clustered tyrosine sulfates and the sialyl LewisX–containing hexasaccharide epitope.53-55 P-selectin antagonists exhibit relatively low micromolar affinity for P-selectin (GMI-1070: 50% inhibitory concentration [IC50], 423 μM; PSI-697: IC50, 150 μM; PSI-421: IC50, 225 μM)48,49 compared with the nanomolar affinity observed for native P-selectin/PSGL-1 interactions (equilibrium dissociation constant, Kd, 70-300 nM).56,57 As a consequence, the lack of clinical efficacy of these compounds has been attributed, at least in part, to their significantly diminished binding affinity. For example, despite demonstrated ex vivo efficacy, PSI-697 failed to inhibit platelet-monocyte aggregation in a phase 1 clinical trial, whereas gram-scale dosing was required for GMI-1070 in clinical studies of sickle cell disease.58,59 Notably, crizanlizumab, the only approved P-selectin inhibitor, as an antibody, possesses a Kd in the low nanomolar range.60
There is a need to develop P-selectin inhibitors that exhibit high affinity and specificity. We recently reported a rationally designed novel glycosulfopeptide mimic of N-terminal PSGL-1, GSnP-6, as the first synthetic high-affinity (Kd; 22 nM) and specific P-selectin antagonist for human therapy. GSnP-6 reduces leukocyte rolling and platelet-leukocyte aggregation in vitro and in vivo.61 In this article, we demonstrate the efficacy of a pegylated GSnP-6 (P-G6) in a preclinical model of nonocclusive venous thrombosis and confirm that this lead drug candidate does not disrupt hemostasis.
Methods
General
All commercially available reagents and solvents were used without further purification. N,N-dimethylformamide (227056) and N,N′-diisopropylethylamine (387649) were purchased from Sigma-Aldrich; the PEGylation reagent, 40 kDa methoxypoly(ethylene glycol)-succinimidyl valerate (mPEG-SVA), was purchased from Advanced BioChemicals (#MOP2506; PDI 1.06). Reverse-phase high performance liquid chromatography (RP-HPLC) was performed using the Waters 2767 Gradient Purification System with a Waters 2489 UV/Vis Detection Module and a Waters 2545 Binary Gradient Module, equipped with a C18 100-Å column (preparative; 250 × 30 mm) or a C18 100-Å Column (analytical; 50 × 4.6 mm; both from Phenomenex). A Bruker AutoFlex II Matrix-Assisted LASER Desorption Ionization–Time of Flight Mass Spectrometer was used to analyze samples cocrystallized with Super-DHB (2,3-dihydroxybenzoic acid) matrix. Murine and human protocols were approved by the Institutional Animal Care and Use Committee and Institutional Review Board of Beth Israel Deaconess Medical Center.
Synthesis of P-G6
GSnP-6 was synthesized, as previously described.61 PEGylation of GSnP-6 was performed by adding mPEG-SVA (1 eq.) to GSnP-6 (2 eq.) in N,N-dimethylformamide (1.25 mM) at 0°C. Subsequently, N,N′-diisopropylethylamine was slowly added and shaken for 22 hours at 25°C to a final concentration of GSnP-6 of 20.1 μL/mg. The reaction was quenched using acetic acid, and the crude material was purified using RP-HPLC (preparative) to afford the final product, P-G6, in 62% yield (Figure 1), which was confirmed by matrix-assisted laser desorption/ionization time of flight (supplemental Figure 1; available on the Blood Web site). Purity was assessed using RP-HPLC (analytical). The RP-HPLC gradient used for preparative purposes included a solvent (A) consisting of water and 0.1% trifluoroacetic acid (TFA) and a solvent (B) consisting of acetonitrile: 0 to 10 minutes, 25% B; 10 to 40 minutes, 25% to 80% B; 40 to 41 minutes, 80% to 98% B; 41 to 50 minutes, 98% B; 50 to 51 minutes, 98% to 25% B; 51 to 60 minutes, 25% B (P-G6, retention time [Rt] = 20.43 minutes at a flow rate of 75 mL/min). The RP-HPLC gradient used for analytical purposes included a solvent (A) consisting of water and 0.1% TFA and a solvent (B) consisting of acetonitrile: 0 to 2 minutes, 5% B; 2 to 6 minutes, 5% to 98% B; 6 to 20 minutes, 98% B; 20 to 21 minutes, 98% to 5% B; 21 to 22 minutes, 5% B (P-G6, Rt = 4.35 minutes at a flow rate of 2.5 mL/min) (supplemental Figure 2).
Figure 1.

Synthesis of PEG40-GSnP-6 (P-G6). P-selectin antagonist GSnP-6 is modeled after the N terminus of human PSGL-1, including sialyl LewisX (N-acetylneuraminic acid)–containing hexasaccharide and sulfopeptide epitopes responsible for high-affinity binding to P-selectin. A linear 40-kDa mPEG-SVA was conjugated to the ε-amino residue of the N terminal lysine of GSnP-6 affording P-G6. The glycan short form (A) and full chemical structure (B) of P-G6 are shown.
Pharmacokinetic profile of P-G6
P-G6 was administered IV to C57BL/6 mice at a dose of 8 μmol/kg; blood was collected into lithium heparin tubes and centrifuged at 1500g for 10 minutes at room temperature. P-G6 was extracted from 20 μL of plasma by protein precipitation using 80 μL of cold methanol and centrifuged at 17000g for 20 min at 4°C. The supernatant (80 μL) was dried in vacuo and reconstituted in nanopure water (32 μL), and an aliquot (2 μL) was injected for preconcentration at a flow rate of 5 μL/min into an UltiMate 3000 RSLCnano ultrahigh-performance liquid chromatography (LC) system (Dionex) coupled to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific). A binary gradient was applied to an Accucore 150 amide hydrophilic interaction LC analytical column (150 mm × 75 μm ID, 2.6 μm; Thermo Fisher Scientific) maintained at 40°C using a solvent (A) consisting of 5 mM ammonium formate in water (pH 8.5) and a solvent (B) consisting of 5 mM ammonium formate in 80% (v/v) acetonitrile and 20% (v/v) water (pH 8.5) at a flow rate of 0.4 μL/min: 0 to 3 minutes, 99% B; 3 to 14 minutes, 99% to 89% (B); 14 to 26 minutes, 89% to 45% (B); 26 to 33 minutes, 45% (B); 33 to 34 minutes, 45% to 99% (B); 34 to 44 minutes, 99% (B). An offset voltage (30 V) was applied in the ion source after ultrahigh-performance LC separation, and mass spectrometry (MS) and tandem mass spectrometry (MS/MS) spectra were acquired in the negative mode with the following settings: radio frequency (RF) lens, 24%; MS automatic gain control, 5 × 105; MS maximum injection time, 50 ms; MS resolution, 120 000; MS/MS automatic gain control, 2 × 105; MS/MS maximum injection time, 200 ms; MS/MS resolution, 30000; precursor ion isolation width, 1.6 Da; and higher-energy collisional dissociation normalized collision energy, 20, 30, 40. Following in-source fragmentation, m/z 1201.11 (z = 3) was used as the quantifying peptide. Peak areas from the extracted chromatograms were used for quantification. An external standard curve was generated by spiking plasma with various concentrations of P-G6, followed by protein precipitation, as described. A linear regression curve was fitted to the data and used to determine concentrations of P-G6 in collected plasma samples. Compound half-life was determined using a noncompartmental analysis.
Flow cytometry
Flow cytometry was used to quantify binding inhibition of P-selectin Fc chimeras to human and mouse leukocytes.61 Whole mouse or human blood was collected into citrate-coated tubes. Leukocytes were isolated by centrifugation and incubated with increasing concentrations of GSnP-6 or P-G6 (0-100 μM) and Fc chimeras of human or mouse P-selectin (3 μg/mL; R&D Systems), followed by phycoerythrin (PE)-conjugated anti-Fc (1:100; #H10104; Life Technologies). The interaction of P-selectin with mouse and human leukocytes was analyzed by flow cytometry (BD LSR II) and quantified (FlowJo) as the percentage of inhibition. Inhibition experiments were conducted in triplicate, and representative curves are shown.
Assessment of platelet-leukocyte aggregation in vitro
Platelet-leukocyte aggregates were quantified in whole blood using dual-label flow cytometry. Anticoagulated human or mouse blood was incubated with 120 µM P-G6 at room temperature and stimulated with thrombin receptor-activating peptide (human PAR1-activating peptide 40 µM, mouse PAR4-activating peptide 200 µM) to induce platelet P-selectin expression. Anti–P-selectin blocking antibodies were included as a positive control (5 µg/mL human KPL1, 5 µg/mL mouse RB40.34).61 Two-color flow cytometry was used to quantify platelet-leukocyte aggregates by incubating human samples with anti-CD42a–PE and anti-CD45–APC and by incubating mouse samples with anti-CD41–PE and anti-CD45–APC. CD45+ monocyte and neutrophil populations were distinguished through characteristic side scatter and quantified as the percentage of positive platelets in saline control– or P-G6 (120 µM)–treated samples.
Murine model of nonocclusive venous thrombosis
Drug efficacy was evaluated in a preclinical mouse model in which nonocclusive venous thrombosis was induced by electrolytic injury of the inferior vena cava, as detailed elsewhere.62 P-G6 was administered intravenously (8 μmol/kg) to male C57BL/6 mice (8-12 weeks of age) immediately prior to electrolytic injury and 2 hours after injury. Enoxaparin was administered subcutaneously (6 mg/kg) 4 hours prior and 24 hours after electrolytic injury as a clinically relevant control. C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME) were anesthetized with 2% isoflurane, and the inferior vena cava was approached via a midline laparotomy. Venous side branches were ligated or cauterized, whereas posterior branches were left patent. A 25-gauge stainless steel needle, attached to a silver-coated copper wire, was inserted into the exposed caudal vena cava and positioned against the anterior wall (anode). A second wire was implanted subcutaneously to complete the circuit (cathode), and a 250 microamp current was applied for 15 minutes. Subsequently, the needle was removed, and a cotton swab was held in gentle contact with the puncture site to prevent bleeding. The vena cava and associated thrombus, immediately below the renal veins to just above the bifurcation, were excised 48 hours after injury for determination of wet thrombus weight and histological examination. Specimens were fixed overnight in 10% neutral buffered formalin and processed for paraffin embedding; 5-µm sections were stained with hematoxylin and eosin or antibodies specific to neutrophils (Ly-6G; BD Biosciences) and macrophages (CD68; Abcam, Cambridge, MA). The circumference of the vein wall was imaged at 20× magnification and leukocyte infiltration was quantified in 3 representative animals per group, with 5 to 10 sections per tissue sample.
Tail vein transection bleeding time
Hemostasis was assessed using a tail vein transection model to determine bleeding time.63 Mice were anesthetized with ketamine and xylazine by intraperitoneal injection and placed on a warming mat at 37°C. Sterile saline, enoxaparin (6 mg/kg), or P-G6 in 125 μL of sterile saline was injected into the penile vein. Five minutes after administration of test compound, the lateral tail vein was transected with a number 11 scalpel blade at a tail width of 2.3 mm and immediately submerged in 37°C phosphate-buffered saline. The bleeding time was determined at that point when bleeding had ceased for 30 seconds. Animals were excluded if arterial bleeding was present.
Intravital microscopy
Recombinant tumor necrosis factor-α (TNF-α; 0.33 μg; R&D Systems) was administered by intrascrotal injection 3 hours prior to imaging. Surgical preparation of the mouse cremaster was performed, as previously described.64 Mice were anesthetized with an intraperitoneal injection of ketamine HCl (125 mg/kg) and xylazine (12.5 mg/kg) and placed on a 37°C surgical blanket. The jugular vein was cannulated with PE10 tubing to allow the introduction of reagents, including P-G6 (8 μmol/kg), anti-platelet DyLight 649–anti-CD42b (#X649; 0.1 μg/g; Emfret), and Alexa Fluor 488 anti–Gr-1 (#50-167-80, 0.05 μg/g; Thermo Fisher Scientific), diluted to a final volume of 200 μL. The cremaster muscle was exteriorized, immobilized, and superfused with thermocontrolled bicarbonate-buffered saline. Images were obtained using an Olympus AX microscope with a 60× water immersion objective and recorded with a Hamamatsu C9300-201/GenIII videoscope with coordinated image acquisition and analysis using SlideBook software (Intelligent Imaging Innovations). For each treatment condition, platelet accumulation was characterized as median integrated fluorescence normalized to vessel area and plotted during a 3-minute interval from 7 to 9 postcapillary venules. The platelet signal was quantified as area under the curve (AUC) for each individual capture and normalized to vessel surface area. Similar levels of adherent leukocytes were observed among all groups.
Statistical analysis
Descriptive data are presented as mean ± standard error of the mean (SEM), unless otherwise stated. Group comparisons were conducted using 1-way analysis of variance (ANOVA) with Tukey’s multiple-comparison test or Welch’s ANOVA with Dunnett’s multiple-comparison test, as appropriate for variance of data. Statistical analysis was performed using GraphPad Prism version 8.0.0 for Windows (GraphPad Software, San Diego, CA).
Results
Pharmacokinetics of P-G6
We developed an LC-MS/MS method to determine the plasma concentration of P-G6. Specifically, low-flow hydrophilic interaction LC LC–Orbitrap Fusion Lumos mass spectrometry was used to quantify P-G6 in plasma. We developed a method to distinguish the analyte of interest from other matrix components and to identify an abundant precursor-product ion pair to reliably monitor. Optimal analytical performance was achieved using in-source fragmentation to de-PEGylate P-G6 prior to detection (Figure 2A). We did not detect competing or interfering ions that coeluted with GSnP-6 in plasma controls or plasma isolated from mice injected with saline vehicle, validating the utility of GSnP-6 as a quantifying peptide (supplemental Figure 3). The full peptide sequence, site-specific sulfation of phenylalanine, site-specific O-glycosylation of threonine, and O-glycan composition of GSnP-6 were confirmed via stepped higher-energy collisional dissociation (Figure 2B-C). Lability was minimized by setting the RF lens to <30%, maintaining buffered solvents at pH 8.5, and operating in negative ionization mode. A standard curve of P-G6 in pooled plasma was generated with excellent linearity over 4 orders of magnitude (Figure 2A). A single IV dose of P-G6 was administered to adult mice (4 per time point). Time-dependent changes in the plasma concentration of P-G6 were consistent with a calculated terminal half-life of 15.65 ± 3.55 hours (Figure 2D), which is substantially longer than the reported half-life of several minutes that is associated with structurally similar non-PEGylated glycosulfopeptide mimics of PSGL-1.54,65
Figure 2.

Quantification of P-G6 in plasma. (A-C) LC-MS/MS was used to generate a standard curve of P-G6 concentration in blood plasma. (A) LC-MS/MS parameters for in-source fragmentation and rigor of the calibration curve. (B) Generation of the quantifying peptide. (C) A stepped higher-energy collisional dissociation approach effectively fragmented P-G6 in a single MS/MS spectrum. Fragment ions: blue, b-ions; red, y-ions; green, glycan loss. Modified amino acid are in bold type. (D) Plasma concentration of P-G6 over time after IV administration of a single weight-based (8 µmol/kg) dose.
P-G6 inhibits leukocyte/P-selectin binding in a dose-dependent manner
Flow cytometry was used to assess the inhibition of P-selectin binding to murine and human leukocytes by GSnP-6 and P-G6. A recombinant mouse P-selectin Fc chimera (3 μg/mL) was incubated with murine leukocytes along with GSnP-6 or P-G6 (0-120 μM). In a similar manner, a recombinant human P-selectin Fc chimera (3 μg/mL) was incubated with peripheral blood leukocytes from healthy adult volunteers. The binding of P-selectin to leukocytes was measured using a fluorescent labeled anti-Fc antibody, quantified as mean fluorescent intensity, and presented as the percentage inhibition of binding. P-G6 inhibited P-selectin binding to mouse and human neutrophils and monocytes in a dose-dependent manner similar to that observed for GSnP-6 (Figure 3).
Figure 3.

GSnP-6 and P-G6 inhibit P-selectin binding to leukocytes. GSnP-6 and P-G6 (0-100 μM) were incubated with mouse (A-B) or human (C-D) neutrophils and monocytes. Flow cytometry was used to evaluate the percentage of binding inhibition of species-appropriate P-selectin chimera to neutrophils or monocytes produced by GSnP-6 and P-G6 compared with phosphate-buffered saline control. GSnP-6 and P-G6 inhibit P-selectin leukocyte interactions in a dose-dependent manner. Data are mean ± SEM, n = 3 per agent per study. PMN, polymorphonuclear cells.
P-G6 inhibits platelet-leukocyte aggregation in vitro and in vivo
Platelet-leukocyte aggregation is dependent on PSGL-1/P-selectin binding.66 In vitro platelet-monocyte and platelet-neutrophil aggregation was evaluated by flow cytometry. Platelet activation was induced by exposure to a PAR4-activating peptide in mouse blood or a PAR1-activating peptide in human blood in the presence of P-G6 (120 μM). Murine platelet-monocyte and platelet-neutrophil aggregates were reduced by 45% and 38%, respectively, and human platelet-monocyte and platelet-neutrophil aggregates were reduced by 29% and 42%, respectively (Figure 4A-B; supplemental Figure 4). Positive controls (anti-mouse and anti-human P-selectin) validate inhibition of P-selectin/PSGL-1 as a feasible therapeutic strategy to reduce platelet-leukocyte aggregation (supplemental Figure 4). Platelet-leukocyte adhesion in vivo was examined using fluorescently labeled platelets following TNF-α induction of venular inflammation. P-G6 or saline was administered IV (8 μmol/kg) prior to cremaster preparation and platelet microaggregate formation, and platelet-neutrophil binding was monitored (n = 3-8 mice per group, 7-9 vessels per mouse). P-G6 significantly reduced platelet aggregation as quantified by median integrated fluorescence or normalized fluorescence signal AUC (Figure 4C-F).
Figure 4.

P-G6 inhibition of platelet-leukocyte aggregation in vitro and in vivo. (A-B) Inhibition of platelet-leukocyte aggregation in vitro. Anticoagulated mouse or human blood was dosed with 120 µM P-G6 or saline control. Platelet-leukocyte aggregation was induced by adding species-specific PAR peptide, and samples were analyzed using flow cytometry to quantify platelet-positive monocytes or neutrophils. P-G6 significantly reduced platelet-leukocyte aggregation in mouse (A) and human (B) blood. Unstimulated control blood is included for reference. (C-F) Inhibition of platelet-leukocyte aggregation in vivo. Intravital microscopy was used to characterize TNF-α–induced venular inflammation. Immediately prior to cremaster exteriorization, mice were infused with P-G6 or saline vehicle control and antiplatelet CD42b-DyLight 649 (red)/antineutrophil Alexa Fluor 488 (green). Platelet fluorescent signal was normalized to vessel area and reported as median integrated fluorescence over time (C) and AUC (D). P-G6 significantly reduced platelet accumulation to adherent neutrophils (E) compared with mice administered saline vehicle (F). Data are mean ± SEM. ***P < .001, *P < .05, Student t test. PMN, polymorphonuclear cells.
P-G6 inhibits venous thrombosis without disruption of hemostasis
Administration of P-G6 led to a significant decrease in thrombus formation after electrolytic injury of the murine vena cava, an effect that was equivalent to that observed for enoxaparin (Figure 5A-B). Treatment with P-G6 or enoxaparin led to a significant reduction in infiltrating Ly6G+ neutrophils and CD68+ macrophages (Figure 5C-F). Tail vein transection bleeding time was prolonged after treatment with enoxaparin but was unaffected by administration of P-G6 (Figure 6).
Figure 5.

Treatment with P-G6 reduces venous thrombus formation. A nonocclusive thrombus was induced be electrolytic injury of the inferior vena cava, and vessel thrombus weight was measured 48 hours after injury to determine treatment efficacy. (A) Prophylactic administration of P-G6 (8 µmol/kg IV) and low molecular weight heparin (LMWH; 6 mg/kg, subcutaneously) resulted in a significant reduction in thrombus weight compared with mice administered saline vehicle (8 mice per group). (B) Representative images of excised infrarenal vena cava 48 hours after electrolytic injury. (C-F) Neutrophil and macrophage infiltration within the vein wall 48 hours after thrombus induction. Quantification of Ly6G+ neutrophils (C,E) and CD68+ macrophages (D,F) reveals significantly less wall inflammation compared with mice administered saline vehicle. Arrows indicate positive immunostaining scale bar displayed in saline group applies to all images. Data are mean ± SEM. ***P < .001, ANOVA with Tukey’s multiple-comparison test. L, lumen.
Figure 6.
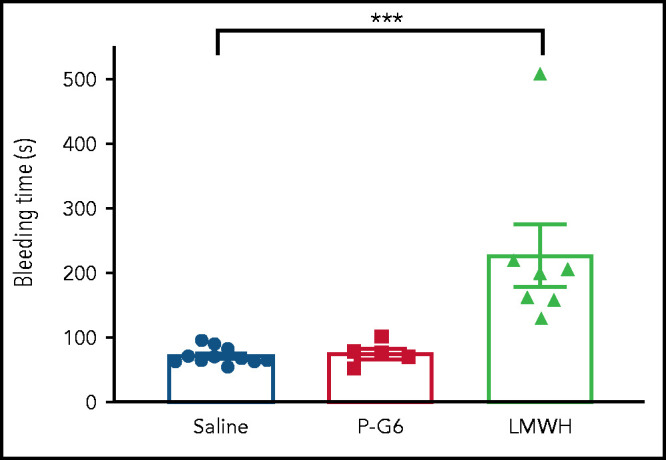
Treatment with P-G6 does not affect hemostasis. The effect of P-G6 on hemostasis was assessed using a tail vein bleeding assay. Mice were subjected to IV administration of saline vehicle, P-G6 (8 μmol/kg), or low molecular weight heparin (LMWH; 6 mg/kg) 5 minutes prior to transection of the lateral tail vein. LMWH resulted in a significant increase in bleeding time compared with mice receiving saline vehicle, whereas no increase in bleeding time was observed after administration of P-G6. Data are mean ± SEM. Group comparisons were conducted using Welch’s ANOVA with Dunnett’s multiple-comparison test. ***P < .001.
Discussion
Events mediated by the PSGL-1/P-selectin pathway are central to the pathogenesis of VTE. P-selectin plays a critical role in thrombus initiation, propagation, and the evolution of an attendant inflammatory response, which leads to the release of TF, fibrin deposition, and leukocyte recruitment.11,12,24-26 Moreover, clinical studies have shown that elevated plasma P-selectin levels are predictive of VTE in high-risk populations67,68; plasma P-selectin can also serve as a diagnostic marker of deep venous thrombosis when evaluated in combination with a clinical score, such as Wells criteria.69,70 As such, inhibition of the P-selectin/PSGL-1 pathway represents an attractive approach to reduce the risk of venous thrombosis. Nonetheless, P-selectin inhibitors have yet to be translated for prevention or treatment of VTE.
Blocking antibodies were the first P-selectin–targeted therapies to show efficacy in animal studies of VTE. Wakefield et al noted a synergistic effect in TNF-α and P-selectin antibody blockade with decreased leukocyte extravasation following vena cava ligation in a rat.29 Although the degree of thrombus accumulation was not affected in this model, subsequent studies in primates confirmed a reduction in thrombus formation and leukocyte extravasation.27 Notably, outcomes in a baboon model varied with underlying venous pathology. For instance, P-selectin antibody blockade led to a sixfold reduction in thrombus accumulation in segments with pure venous stasis, whereas thrombus persisted in regions associated with underlying endothelial damage caused by an occlusive balloon. Early studies of rPSGL-Ig also demonstrated reduced thrombosis in feline and primate occlusive models of VTE,28,30,31 with the treatment effect most marked in regions without underlying endothelial trauma.30
The contextual framework and model-dependent treatment effects highlight the inherent variability of existing animal models of VTE.71 Depending on the thrombotic stimulus and degree of flow restriction, models differ in the degree of hypoxia, endothelial activation and injury,72 initial source of TF,11,73 thrombus kinetics,62 and blood flow.74 Further, only baboon and feline models possess venous valves, considered the initial site of VTE in humans.75 As such, defining therapeutic potential in existing animal models of VTE is complicated by a lack of clarity of essential outcomes and translatable pathology. It is noteworthy that clinical trials of P-selectin–targeted therapies in VTE have lagged behind its evaluation in other diseases, including acute coronary syndrome, reperfusion injury, and sickle cell vaso-occlusive crisis. To date, the anti-PSGL-1 SelK2 monoclonal antibody is the only therapy targeting the P-selectin/PSGL-1 pathway in ongoing clinical trials. The modest affinity and variable efficacy of many of the previously reported P-selectin inhibitors, combined with the aforementioned challenges associated with recombinant glycoproteins and therapeutic antibodies, have delayed the introduction of P-selectin–targeted therapy for VTE.
Among the synthetic inhibitors of P-selectin, PSI-697 and second-generation PSI-421 are best characterized in models of VTE. PSI-697 reduced thrombus weight in mice and decreased wall stiffness, neointima formation, and the extravasation of inflammatory cells in rats.33,34 Notably, PSI-697 reduced thrombus accumulation in P-selectin- and E-selectin double-deficient mice, suggesting a potential lack of specificity for selectin signaling. In primate models, PSI-697 and PSI-421 enhanced recanalization of thrombosed veins and decreased radiologic evidence of inflammation.35,36 However, PSI-697 failed to inhibit platelet-monocyte aggregation in humans, presumably because of its modest affinity and specificity for P-selectin.58 The potential for P-selectin inhibition with oligonucleotide aptamers was demonstrated in a baboon model showing increased venous recanalization and preserved valve competency, but further studies are lacking.37 Moreover, translational challenges persist for aptamer therapy, including rapid renal clearance, enzymatic degradation, endosomal sequestration, and uncertain toxicology.76
In our studies, treatment with P-G6 results in a 60% reduction in thrombus weight after 48 hours. Notably, this effect is equivalent to that obtained with enoxaparin, yet it is not associated with increased bleeding time. Cellular infiltration of the venous wall is an important measure of the inflammatory component of thrombosis across preclinical models.11,27-29,32 P-G6 reduces neutrophil and monocyte extravasation at a magnitude similar to enoxaparin treatment. The electrolytic-induced vena cava injury model used in these studies creates a consistent partially occlusive thrombus with continuous exposure to venous flow and reliable drug exposure levels. As such, this is the preferred small animal model for comparing antithrombotic agents.74
Enoxaparin has an established history of clinical effectiveness in the treatment of VTE and is the first-line therapy for cancer-associated VTE and postoperative prophylaxis.3 Thus, the equivalent antithrombotic and anti-inflammatory effects of P-G6, without disruption of hemostasis, is a significant finding given the morbidity associated with anticoagulants, even at reduced dosing regimens. The 15-hour half-life (t1/2) of P-G6 is similar to that of many direct oral anticoagulants, as well as fondaparinux. However, it is significantly shorter and, therefore, potentially advantageous compared with other biologics that inhibit the P-selectin/PSGL-1 pathway whose systemic clearance may require several weeks (t1/2 for crizanlizumab, 10.6 days; t1/2 for rPSGL-Ig, 4 days).43,60 Other reported glycopeptide mimics of PSGL-1 possessed a half-life of several minutes, which limits clinical application.65 P-selectin/PSGL-1 interactions mediate leukocyte adhesion; as a consequence, the potential of adverse effects associated with the blockade of this pathway due to suppression of an innate immune response has been suggested, and further safety studies of inhibitors of this pathway are warranted. However, over a 1-year period, rates of urinary tract and upper respiratory infection, as well as pneumonia, were equivalent among patients with sickle cell disease treated with low-dose or high-dose crizanlizumab compared with those receiving placebo.77
The addition of a PEG moiety is a common strategy to increase drug exposure levels and extend circulating t1/2, but the PEGylation of a negatively charged glycopeptide bearing a complex oligosaccharide has yet to be reported.78 Few, if any, established protocols exist for the synthesis or purification of such compounds.79 In particular, we determined that RP-HPLC offered several advantages over anion-exchange chromatography for the purification of PEGylated glycopeptides, including the ability to visualize the progression of the reaction profile, separate GSnP-6 from P-G6 for reuse in the reaction scheme, and eliminate the presence of excess salt. PEGylation can also complicate MS-based peptide analysis. PEGylated peptides exhibit more complexity than traditional drugs of low molecular weight, including broad spectra, over a wide m/z range and high molecular weight ions that are outside of the detectable m/z range. To quantify P-G6 concentrations, we established an LC-MS/MS protocol that relied upon in-source fragmentation. In general, MS coupled to nanoflow ultrahigh-performance LC has proven quite versatile in determining the pharmacokinetic profile of a wide range of therapeutics because of its wide dynamic range, high sensitivity, analyte specificity, minimal sample preparation, and high-throughput capabilities.80-83
These studies have several limitations. First, we used an electrolytic vena cava injury model of venous thrombosis. Although this model reproduces endothelial activation, characteristic of the initiating event of human VTE, it does not recapitulate the role of venous stasis in thrombus initiation. Additionally, although our results reflect thrombus accumulation over 48 hours, this investigation did not evaluate thrombus resolution, venous fibrosis, or valve function. Ongoing studies are evaluating the ability of P-G6 to alter chronic inflammatory responses after venous thrombosis. Future work will include studies in a venous stasis model, as well as large animal models of venous thrombosis, to assess venous recanalization and valve competency.37 Inhibition was observed for human and mouse platelet and leukocyte binding assays. It is well documented that plasma P-selectin levels vary with disease and among individuals.67 For example, in a post hoc analysis of the SELECT-CABG trial, initial plasma levels of soluble P-selectin were predictive of a therapeutic effect.84 As such, plasma P-selectin may serve as an important biomarker of the relative effectiveness of P-G6 inhibition in at-risk individuals. Finally, although tail vein bleeding time is a standard preclinical method to assess hemostasis, this model is inexact in predicting clinical bleeding risk.
In summary, blockade of the P-selectin/PSGL-1 pathway by P-G6 inhibited murine and human leukocyte/P-selectin binding in a dose-dependent manner and reduced platelet-leukocyte aggregation in vitro and in vivo. P-G6 inhibited venous thrombosis in a preclinical model of VTE without impairing hemostasis. These findings support the development of P-G6 for the prevention of VTE.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The visual abstract was created using BioRender.com, RCSB PDB,85 and Mol*86 using PDB-1G1S.87
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute fellowship T32 HL007734 (D.J.W.), National Institutes of Health, National Heart, Lung, and Blood Institute grant R01 HL128237 (E.L.C.), and National Institutes of Health, National Institute of General Medical Sciences grant U01 GM116196 (E.L.C., R.D.C.).
Footnotes
Data sharing requests should be sent to Elliot L. Chaikof (echaikof@bidmc.harvard.edu).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: D.J.W., C.A.H., L.S., R.D.C., and E.L.C. designed and analyzed the experiments; D.D.P. performed pharmacokinetics studies; S.S.P., A.R.M., P.E., B.D., W.J.W., and M.H. performed synthesis, glycosylation, and pegylation reactions; J.C. conducted flow cytometry experiments; E.D. and L.L. performed in vivo studies and immunohistochemistry analysis; and D.J.W., C.A.H, D.D.P., S.S.P., and E.L.C. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Elliot L. Chaikof, Department of Surgery, Beth Israel Deaconess Medical Center, 110 Francis St, Suite 9F, Boston, MA 02115; e-mail: echaikof@bidmc.harvard.edu.
REFERENCES
- 1.Benjamin EJ, Muntner P, Alonso A, et al. ; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee . Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. 2019;139(10):e56-e528. [DOI] [PubMed] [Google Scholar]
- 2.Beckman MG, Hooper WC, Critchley SE, Ortel TL. Venous thromboembolism: a public health concern. Am J Prev Med. 2010;38(4 suppl):S495-S501. [DOI] [PubMed] [Google Scholar]
- 3.Kearon C, Akl EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: Chest guideline and expert panel report [published correction appears in Chest. 2016;150(4):988]. Chest. 2016;149(2):315-352. [DOI] [PubMed] [Google Scholar]
- 4.Carrier M, Abou-Nassar K, Mallick R, et al. ; AVERT Investigators . Apixaban to prevent venous thromboembolism in patients with cancer. N Engl J Med. 2019;380(8):711-719. [DOI] [PubMed] [Google Scholar]
- 5.Coulis AA, Mackey WC. A review of the efficacy and safety profiles of the novel oral anticoagulants in the treatment and prevention of venous thromboembolism. Clin Ther. 2018;40(12):2140-2167. [DOI] [PubMed] [Google Scholar]
- 6.Aryal MR, Gosain R, Donato A, et al. Systematic review and meta-analysis of the efficacy and safety of apixaban compared to rivaroxaban in acute VTE in the real world. Blood Adv. 2019;3(15):2381-2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ay C, Beyer-Westendorf J, Pabinger I. Treatment of cancer-associated venous thromboembolism in the age of direct oral anticoagulants. Ann Oncol. 2019;30(6):897-907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riva N, Donadini MP, Ageno W. Epidemiology and pathophysiology of venous thromboembolism: similarities with atherothrombosis and the role of inflammation. Thromb Haemost. 2015;113(6):1176-1183. [DOI] [PubMed] [Google Scholar]
- 9.Bacani AK, Gabriel SE, Crowson CS, Heit JA, Matteson EL. Noncardiac vascular disease in rheumatoid arthritis: increase in venous thromboembolic events? Arthritis Rheum. 2012;64(1):53-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grainge MJ, West J, Card TR. Venous thromboembolism during active disease and remission in inflammatory bowel disease: a cohort study. Lancet. 2010;375(9715):657-663. [DOI] [PubMed] [Google Scholar]
- 11.von Brühl ML, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(4):819-835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falati S, Liu Q, Gross P, et al. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J Exp Med. 2003;197(11):1585-1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darbousset R, Thomas GM, Mezouar S, et al. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood. 2012;120(10):2133-2143. [DOI] [PubMed] [Google Scholar]
- 14.Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463-469. [DOI] [PubMed] [Google Scholar]
- 15.Ammollo CT, Semeraro F, Xu J, Esmon NL, Esmon CT. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J Thromb Haemost. 2011;9(9):1795-1803. [DOI] [PubMed] [Google Scholar]
- 16.Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107(36):15880-15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ward CM, Tetaz TJ, Andrews RK, Berndt MC. Binding of the von Willebrand factor A1 domain to histone. Thromb Res. 1997;86(6):469-477. [DOI] [PubMed] [Google Scholar]
- 18.Massberg S, Grahl L, von Bruehl ML, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010;16(8):887-896. [DOI] [PubMed] [Google Scholar]
- 19.Larsen E, Celi A, Gilbert GE, et al. PADGEM protein: a receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell. 1989;59(2):305-312. [DOI] [PubMed] [Google Scholar]
- 20.McEver RP, Beckstead JH, Moore KL, Marshall-Carlson L, Bainton DF. GMP-140, a platelet α-granule membrane protein, is also synthesized by vascular endothelial cells and is localized in Weibel-Palade bodies. J Clin Invest. 1989;84(1):92-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doré M, Korthuis RJ, Granger DN, Entman ML, Smith CW. P-selectin mediates spontaneous leukocyte rolling in vivo. Blood. 1993;82(4):1308-1316. [PubMed] [Google Scholar]
- 22.Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood. 2011;118(26):6743-6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayadas TN, Johnson RC, Rayburn H, Hynes RO, Wagner DD. Leukocyte rolling and extravasation are severely compromised in P selectin-deficient mice. Cell. 1993;74(3):541-554. [DOI] [PubMed] [Google Scholar]
- 24.Palabrica T, Lobb R, Furie BC, et al. Leukocyte accumulation promoting fibrin deposition is mediated in vivo by P-selectin on adherent platelets. Nature. 1992;359(6398):848-851. [DOI] [PubMed] [Google Scholar]
- 25.Sullivan VV, Hawley AE, Farris DM, et al. Decrease in fibrin content of venous thrombi in selectin-deficient mice. J Surg Res. 2003;109(1):1-7. [DOI] [PubMed] [Google Scholar]
- 26.Myers D Jr, Farris D, Hawley A, et al. Selectins influence thrombosis in a mouse model of experimental deep venous thrombosis. J Surg Res. 2002;108(2):212-221. [DOI] [PubMed] [Google Scholar]
- 27.Downing LJ, Wakefield TW, Strieter RM, et al. Anti-P-selectin antibody decreases inflammation and thrombus formation in venous thrombosis. J Vasc Surg. 1997;25(5):816-827; discussion 828. [DOI] [PubMed] [Google Scholar]
- 28.Myers DD, Hawley AE, Farris DM, et al. P-selectin and leukocyte microparticles are associated with venous thrombogenesis. J Vasc Surg. 2003;38(5):1075-1089. [DOI] [PubMed] [Google Scholar]
- 29.Wakefield TW, Strieter RM, Downing LJ, et al. P-selectin and TNF inhibition reduce venous thrombosis inflammation. J Surg Res. 1996;64(1):26-31. [DOI] [PubMed] [Google Scholar]
- 30.Eppihimer MJ, Schaub RG. P-Selectin-dependent inhibition of thrombosis during venous stasis. Arterioscler Thromb Vasc Biol. 2000;20(11):2483-2488. [DOI] [PubMed] [Google Scholar]
- 31.Wakefield TW, Strieter RM, Schaub R, et al. Venous thrombosis prophylaxis by inflammatory inhibition without anticoagulation therapy. J Vasc Surg. 2000;31(2):309-324. [DOI] [PubMed] [Google Scholar]
- 32.Thanaporn P, Myers DD, Wrobleski SK, et al. P-selectin inhibition decreases post-thrombotic vein wall fibrosis in a rat model. Surgery. 2003;134(2):365-371. [DOI] [PubMed] [Google Scholar]
- 33.Myers DD Jr, Rectenwald JE, Bedard PW, et al. Decreased venous thrombosis with an oral inhibitor of P selectin. J Vasc Surg. 2005;42(2):329-336. [DOI] [PubMed] [Google Scholar]
- 34.Myers DD Jr, Henke PK, Bedard PW, et al. Treatment with an oral small molecule inhibitor of P selectin (PSI-697) decreases vein wall injury in a rat stenosis model of venous thrombosis. J Vasc Surg. 2006;44(3):625-632. [DOI] [PubMed] [Google Scholar]
- 35.Myers DD Jr, Wrobleski SK, Longo C, et al. Resolution of venous thrombosis using a novel oral small-molecule inhibitor of P-selectin (PSI-697) without anticoagulation. Thromb Haemost. 2007;97(3):400-407. [PubMed] [Google Scholar]
- 36.Meier TR, Myers DD Jr, Wrobleski SK, et al. Prophylactic P-selectin inhibition with PSI-421 promotes resolution of venous thrombosis without anticoagulation. Thromb Haemost. 2008;99(2):343-351. [DOI] [PubMed] [Google Scholar]
- 37.Diaz JA, Wrobleski SK, Alvarado CM, et al. P-selectin inhibition therapeutically promotes thrombus resolution and prevents vein wall fibrosis better than enoxaparin and an inhibitor to von Willebrand factor. Arterioscler Thromb Vasc Biol. 2015;35(4):829-837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prado MS, Bendtzen K, Andrade LEC. Biological anti-TNF drugs: immunogenicity underlying treatment failure and adverse events. Expert Opin Drug Metab Toxicol. 2017;13(9):985-995. [DOI] [PubMed] [Google Scholar]
- 39.Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010;2(3):256-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Solá RJ, Griebenow K. Glycosylation of therapeutic proteins: an effective strategy to optimize efficacy. BioDrugs. 2010;24(1):9-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mertens P, Maes A, Nuyts J, et al. ; PSALM investigators . Recombinant P-selectin glycoprotein ligand-immunoglobulin, a P-selectin antagonist, as an adjunct to thrombolysis in acute myocardial infarction. The P-Selectin Antagonist Limiting Myonecrosis (PSALM) trial. Am Heart J. 2006;152(1):125.e1-125.e8. [DOI] [PubMed] [Google Scholar]
- 42.Busuttil RW, Lipshutz GS, Kupiec-Weglinski JW, et al. rPSGL-Ig for improvement of early liver allograft function: a double-blind, placebo-controlled, single-center phase II study. Am J Transplant. 2011;11(4):786-797. [DOI] [PubMed] [Google Scholar]
- 43.Osama Gaber AO, Mulgaonkar S, Kahan BD, et al. YSPSL (rPSGL-Ig) for improvement of early renal allograft function: a double-blind, placebo-controlled, multi-center phase IIa study. Clin Transplant. 2011;25(4):523-533. [DOI] [PubMed] [Google Scholar]
- 44.Chung CH, Mirakhur B, Chan E, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-αetuxi1,3-galactose. N Engl J Med. 2008;358(11):1109-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov. 2009;8(3):226-234. [DOI] [PubMed] [Google Scholar]
- 46.Hossler P, Khattak SF, Li ZJ. Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology. 2009;19(9):936-949. [DOI] [PubMed] [Google Scholar]
- 47.Patel MS, Miranda-Nieves D, Chen J, Haller CA, Chaikof EL. Targeting P-selectin glycoprotein ligand-1/P-selectin interactions as a novel therapy for metabolic syndrome. Transl Res. 2017;183:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang J, Patton JT, Sarkar A, Ernst B, Magnani JL, Frenette PS. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood. 2010;116(10):1779-1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang A, Moretto A, Janz K, et al. Discovery of 2-[1-(4-chlorophenyl)cyclopropyl]-3-hydroxy-8-(trifluoromethyl)quinoline-4-carboxylic acid (PSI-421), a P-selectin inhibitor with improved pharmacokinetic properties and oral efficacy in models of vascular injury. J Med Chem. 2010;53(16):6003-6017. [DOI] [PubMed] [Google Scholar]
- 50.Kaila N, Janz K, DeBernardo S, et al. Synthesis and biological evaluation of quinoline salicylic acids as P-selectin antagonists. J Med Chem. 2007;50(1):21-39. [DOI] [PubMed] [Google Scholar]
- 51.Kranich R, Busemann AS, Bock D, et al. Rational design of novel, potent small molecule pan-selectin antagonists. J Med Chem. 2007;50(6):1101-1115. [DOI] [PubMed] [Google Scholar]
- 52.Watz H, Bock D, Meyer M, et al. Inhaled pan-selectin antagonist Bimosiamose attenuates airway inflammation in COPD. Pulm Pharmacol Ther. 2013;26(2):265-270. [DOI] [PubMed] [Google Scholar]
- 53.Leppänen A, Yago T, Otto VI, McEver RP, Cummings RD. Model glycosulfopeptides from P-selectin glycoprotein ligand-1 require tyrosine sulfation and a core 2-branched O-glycan to bind to L-selectin. J Biol Chem. 2003;278(29):26391-26400. [DOI] [PubMed] [Google Scholar]
- 54.Leppänen A, Mehta P, Ouyang YB, et al. A novel glycosulfopeptide binds to P-selectin and inhibits leukocyte adhesion to P-selectin. J Biol Chem. 1999;274(35):24838-24848. [DOI] [PubMed] [Google Scholar]
- 55.Leppänen A, White SP, Helin J, McEver RP, Cummings RD. Binding of glycosulfopeptides to P-selectin requires stereospecific contributions of individual tyrosine sulfate and sugar residues. J Biol Chem. 2000;275(50):39569-39578. [DOI] [PubMed] [Google Scholar]
- 56.Ushiyama S, Laue TM, Moore KL, Erickson HP, McEver RP. Structural and functional characterization of monomeric soluble P-selectin and comparison with membrane P-selectin. J Biol Chem. 1993;268(20):15229-15237. [PubMed] [Google Scholar]
- 57.Mehta P, Cummings RD, McEver RP. Affinity and kinetic analysis of P-selectin binding to P-selectin glycoprotein ligand-1. J Biol Chem. 1998;273(49):32506-32513. [DOI] [PubMed] [Google Scholar]
- 58.Japp AG, Chelliah R, Tattersall L, et al. Effect of PSI-697, a novel P-selectin inhibitor, on platelet-monocyte aggregate formation in humans. J Am Heart Assoc. 2013;2(1):e006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Telen MJ, Wun T, McCavit TL, et al. Randomized phase 2 study of GMI-1070 in SCD: reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood. 2015;125(17):2656-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.U.S. Food and Drug Administration Center for Drug Evaluation and Research . ADAKVEO (Crizanlizumab-tmca): BLA Multi-Disciplinary Review and Evaluation (BLA 761128). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761128Orig1s000MultidisciplineR.pdf Accessed 20 February 2021.
- 61.Krishnamurthy VR, Sardar MYR, Ying Y, et al. Glycopeptide analogues of PSGL-1 inhibit P-selectin in vitro and in vivo. Nat Commun. 2015;6(1):6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Diaz JA, Alvarado CM, Wrobleski SK, et al. The electrolytic inferior vena cava model (EIM) to study thrombogenesis and thrombus resolution with continuous blood flow in the mouse. Thromb Haemost. 2013;109(6):1158-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Culmer DL, Dunbar ML, Hawley AE, et al. E-selectin inhibition with GMI-1271 decreases venous thrombosis without profoundly affecting tail vein bleeding in a mouse model. Thromb Haemost. 2017;117(6):1171-1181. [DOI] [PubMed] [Google Scholar]
- 64.Kim KH, Barazia A, Cho J. Real-time imaging of heterotypic platelet-neutrophil interactions on the activated endothelium during vascular inflammation and thrombus formation in live mice. J Vis Exp. 2013;(74):50329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hicks AER, Leppänen A, Cummings RD, McEver RP, Hellewell PG, Norman KE. Glycosulfopeptides modeled on P-selectin glycoprotein ligand 1 inhibit P-selectin-dependent leukocyte rolling in vivo. FASEB J. 2002;16(11):1461-1462. [DOI] [PubMed] [Google Scholar]
- 66.Evangelista V, Manarini S, Sideri R, et al. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: role of PSGL-1 as a signaling molecule. Blood. 1999;93(3):876-885. [PubMed] [Google Scholar]
- 67.Ay C, Jungbauer LV, Sailer T, et al. High concentrations of soluble P-selectin are associated with risk of venous thromboembolism and the P-selectin Thr715 variant. Clin Chem. 2007;53(7):1235-1243. [DOI] [PubMed] [Google Scholar]
- 68.Ay C, Simanek R, Vormittag R, et al. High plasma levels of soluble P-selectin are predictive of venous thromboembolism in cancer patients: results from the Vienna Cancer and Thrombosis Study (CATS). Blood. 2008;112(7):2703-2708. [DOI] [PubMed] [Google Scholar]
- 69.Rectenwald JE, Myers DD Jr, Hawley AE, et al. D-dimer, P-selectin, and microparticles: novel markers to predict deep venous thrombosis. A pilot study. Thromb Haemost. 2005;94(6):1312-1317. [DOI] [PubMed] [Google Scholar]
- 70.Vandy FC, Stabler C, Eliassen AM, et al. Soluble P-selectin for the diagnosis of lower extremity deep venous thrombosis. J Vasc Surg Venous Lymphat Disord. 2013;1(2):117-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Diaz JA, Obi AT, Myers DD Jr, et al. Critical review of mouse models of venous thrombosis. Arterioscler Thromb Vasc Biol. 2012;32(3):556-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Budnik I, Brill A. Immune factors in deep vein thrombosis initiation. Trends Immunol. 2018;39(8):610-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou J, May L, Liao P, Gross PL, Weitz JI. Inferior vena cava ligation rapidly induces tissue factor expression and venous thrombosis in rats. Arterioscler Thromb Vasc Biol. 2009;29(6):863-869. [DOI] [PubMed] [Google Scholar]
- 74.Diaz JA, Saha P, Cooley B, et al. Choosing a mouse model of venous thrombosis: a consensus assessment of utility and application. J Thromb Haemost. 2019;17(4):699-707. [DOI] [PubMed] [Google Scholar]
- 75.Brooks EG, Trotman W, Wadsworth MP, et al. Valves of the deep venous system: an overlooked risk factor. Blood. 2009;114(6):1276-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dowdy SF. Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol. 2017;35(3):222-229. [DOI] [PubMed] [Google Scholar]
- 77.Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376(5):429-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bossard M, Vicent M. Polymer-Protein Conjugates: From Pegylation and Beyond. Amsterdam, The Netherlands: Elsevier Science; 2019. [Google Scholar]
- 79.Greenwald RB. PEG drugs: an overview. J Control Release. 2001;74(1-3):159-171. [DOI] [PubMed] [Google Scholar]
- 80.de Moraes NV, Moretti RAC, Furr EB III, McCurdy CR, Lanchote VL. Determination of mitragynine in rat plasma by LC-MS/MS: application to pharmacokinetics. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877(24):2593-2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fernández Ocaña M, James IT, Kabir M, et al. Clinical pharmacokinetic assessment of an anti-MAdCAM monoclonal antibody therapeutic by LC-MS/MS. Anal Chem. 2012;84(14):5959-5967. [DOI] [PubMed] [Google Scholar]
- 82.Chang L, Ren Y, Cao L, et al. Simultaneous determination and pharmacokinetic study of six flavonoids from Fructus Sophorae extract in rat plasma by LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;904:59-64. [DOI] [PubMed] [Google Scholar]
- 83.Wring SA, Randolph R, Park S, et al. Preclinical pharmacokinetics and pharmacodynamic target of SCY-078, a first-in-class orally active antifungal glucan synthesis inhibitor, in murine models of disseminated candidiasis. Antimicrob Agents Chemother. 2017;61(4):e02068-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stähli BE, Tardif J-C, Carrier M, et al. Effects of P-selectin antagonist inclacumab in patients undergoing coronary artery bypass graft surgery. J Am Coll Cardiol. 2016;67(3):344-346. [DOI] [PubMed] [Google Scholar]
- 85.Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sehnal D, Rose AS, Kovca J, Burley SK, Velankar S. Mol*: Towards a common library and tools for web molecular graphics. In Byska J, Krone M, Sommers B, eds. MolVa: Workshop on Molecular Graphics and Visual Analysis of Molecular Data 2018. https://diglib.eg.org/bitstream/handle/10.2312/molva20181103/029-033.pdf. Accessed 21 February 2021.
- 87.Somers WS, Tang J, Shaw GD, Camphausen RT. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLe(X) and PSGL-1. Cell. 2000;103(3):467-479. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.