

Abstract
Non-alcoholic fatty liver disease (NAFLD) has emerged as the most prevalent liver disease in the world, yet there are still no approved pharmacological therapies to prevent or treat this condition. NAFLD encompasses a spectrum of severity, ranging from simple steatosis to non-alcoholic steatohepatitis (NASH). Although NASH is linked to an increased risk of hepatocellular carcinoma and cirrhosis and has now become the leading cause of liver failure-related transplantation, the majority of patients with NASH will ultimately die as a result of complications of type 2 diabetes mellitus (T2DM) and cardiometabolic diseases. Importantly, NAFLD is closely linked to obesity and tightly interrelated with insulin resistance and T2DM. Thus, targeting these interconnected conditions and taking a holistic attitude to the treatment of metabolic disease could prove to be a very beneficial approach. This Review will explore the latest relevant literature and discuss the ongoing therapeutic options for NAFLD focused on targeting intermediary metabolism, insulin resistance and T2DM to remedy the global health burden of these diseases.
Non-alcoholic fatty liver disease (NAFLD) is defined as ectopic lipid accumulation in the liver in the absence of excessive alcohol intake or other attributable cause; 25% of the world’s population might have NAFLD and direct costs are estimated at US$100 billion annually in the USA alone1,2. NAFLD is tightly linked to obesity and, while diet and exercise have proven to be effective in the treatment of NAFLD3, the long-term sustainability of these interventions is poor. Furthermore, there is no licensed drug therapy for non-alcoholic steatohepatitis (NASH) and there is a critical need for the development of effective treatments for this global health problem.
NAFLD encompasses a spectrum of disease severity (FIG. 1) ranging from simple steatosis to more advanced disease characterized by hepatocyte death, inflammation and the development of fibrotic lesions (NASH). Simple steatosis is characterized by lipid accumulation in hepatocytes with minimal hepatocellular injury but is associated with an increased risk of other metabolic disorders, including insulin resistance, type 2 diabetes mellitus (T2DM), dyslipidaemia and hypertension4,5. Epidemiological studies show that NAFLD is more prevalent in men than in premenopausal women6 and premenopausal women are less likely to develop fibrosis than men7,8. By contrast, the protective effect of the female sex is not observed after menopause. It is estimated that NASH is present in ~60% of patients with NAFLD who undergo a liver biopsy and 41% of patients with NASH exhibit considerable fibrosis1. In patients with advanced fibrosis, ~22% go on to develop cirrhosis and ~2% of patients with cirrhosis develop hepatocellular carcinoma (HCC) within 3 years1,9 (FIG. 1). HCC and cirrhotic liver failure can only be cured with an extreme treatment — liver transplantation. Liver disease secondary to NASH has become a leading cause for liver transplantation in the USA due to increasing rates of obesity-related NASH and the increasing availability of alternative curative therapies for other diseases, such as hepatitis C virus infection10,11. Unfortunately, most patients with liver failure will ultimately not receive a transplant and will die while waiting for a donor organ12. Further, the majority of patients with NASH will die from cardiometabolic disease rather than from a liver-related cause due to the tight relationship between NAFLD and T2DM. Thus, therapeutics that target NAFLD, T2DM and cardiometabolic risk factors might be the most effective treatment.
Fig. 1 |. The pathological spectrum of NAFLD.
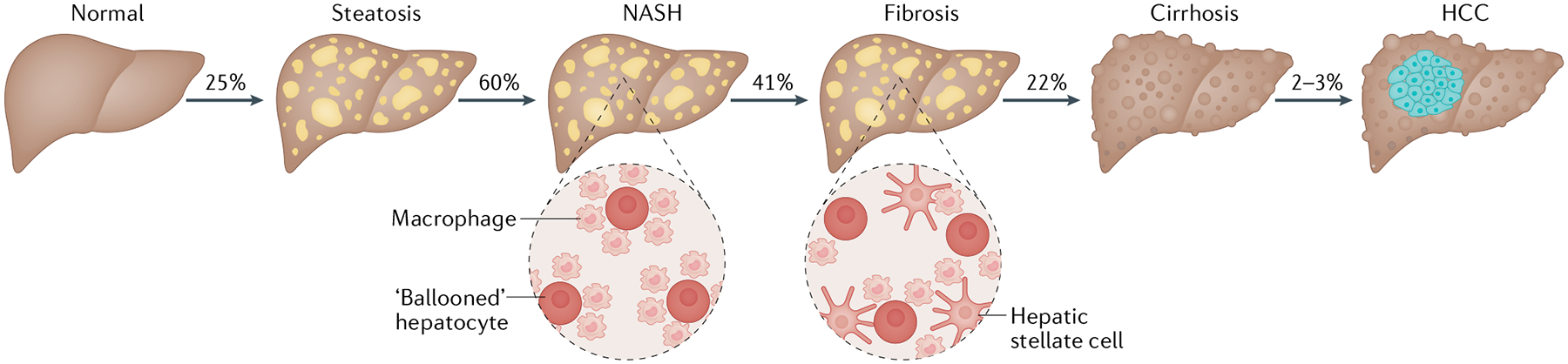
The excess accumulation of liver lipids, mostly in the form of triglycerides, is referred to as steatosis, which is defined as the presence of >5% lipid accumulation within hepatocytes4. It is estimated that approximately 25% of the global population has some form of non-alcoholic fatty liver disease (NAFLD)1. Eventually, excess lipid accumulation might overwhelm the liver’s capacity for the proper storage or disposal of fatty acids, leading to the generation of toxic lipid species. Lipotoxicity might produce endoplasmic reticulum stress, oxidative stress and mitochondrial dysfunction, resulting in inflammation and hepatocyte degeneration (ballooning), which are the defining characteristics of non-alcoholic steatohepatitis (NASH). NASH is estimated to be present in up to 60% of patients with NAFLD who have undergone a liver biopsy1. Over time, hepatocyte death, inflammation and immune cell activation might promote hepatic stellate cell activation. Stellate cells differentiate into fibrogenic myofibroblasts that migrate to sites of hepatic injury and are the major drivers of fibrosis28. Detectable fibrosis is present in approximately 41% of patients with NASH1 and 22% of individuals with advanced fibrosis go on to develop hepatic scarring, referred to as cirrhosis9. Lastly, ~2% of patients with cirrhosis will probably develop hepatocellular carcinoma (HCC) within 3 years1. Percentages in the figure are representative of the proportion of the subpopulation that will probably present or progress to more advanced liver disease.
Pathophysiology of NAFLD
The liver serves many functions, particularly through the regulation, mobilization and storage of nutrients. Hepatocytes have important roles in gluconeogenesis, ketogenesis, amino acid metabolism, and triglyceride and lipoprotein metabolism. While the liver is not a primary organ for lipid storage, ectopic lipid accumulation in the liver can occur under a variety of conditions (FIG. 2). In patients with NAFLD, it has been determined that several sources contribute to intrahepatic triglyceride accumulation, including hepatic uptake from the plasma of non-esterified fatty acids (NEFAs; 59%) and dietary fat (15%) as well as de novo lipogenesis (DNL; 26%)13. Insulin resistance promotes adipose tissue lipolysis leading to increased NEFA levels in the blood, which are taken up by the liver in a concentration-dependent manner13–15. Other metabolic perturbations common in insulin resistance and T2DM, such as hyperinsulinaemia and hyperglycaemia, drive the increased hepatic conversion of carbohydrates into fatty acids through DNL16 and rates of hepatic DNL are markedly elevated in patients with NAFLD17. The expression and/or activity of several enzymes involved in DNL or the storage of fatty acids in triglycerides are known to be increased in patients with NAFLD18 and targeting these enzymes, including ketohexokinase, acetyl-CoA carboxylase, fatty acid synthase, stearoyl-CoA desaturase and diacylglycerol acyltransferase, has been therapeutically explored (FIG. 3). Given the contributing effects of insulin resistance on adipose tissue lipolysis and hepatic DNL, agents that improve systemic insulin sensitivity are being tested for their efficacy in patients with NAFLD.
Fig. 2 |. Role of insulin resistance in NAFLD.
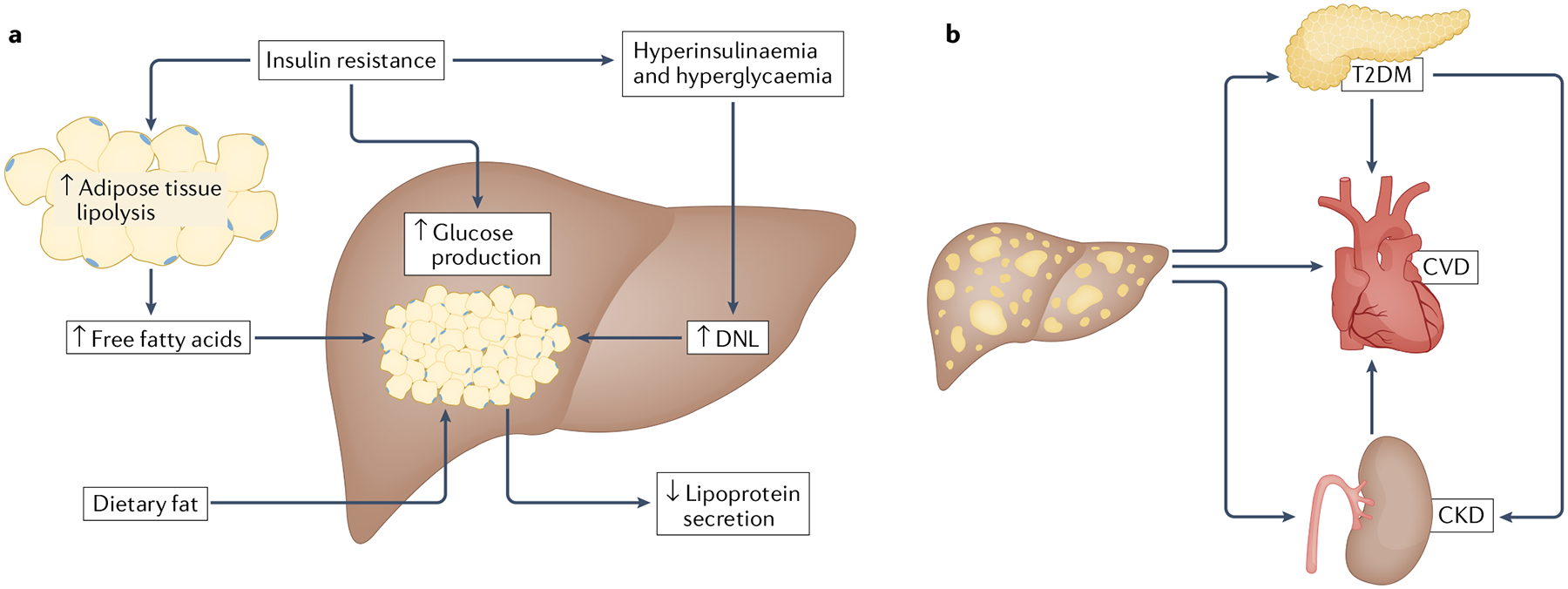
a | Insulin resistance occurs when tissues inadequately respond to the hormone insulin23. In adipose tissue, the normal ability of insulin to suppress lipolysis is impaired, resulting in the excess release of free fatty acids, which are subsequently taken up by the liver in addition to fat obtained from the diet13–15. The inability of tissues to properly respond to insulin results in abnormally elevated glucose levels (hyperglycaemia), which leads to the production and secretion of surplus insulin levels to those needed to maintain blood levels of glucose (hyperinsulinaemia)23. Both hyperglycaemia and hyperlipidaemia stimulate de novo lipogenesis (DNL), which further elevates hepatic lipid accumulation16,17. Normally, appropriate hepatic levels of lipids would be maintained by their increased export through lipoprotein secretion; however, lipoprotein secretion is not adequately elevated in non-alcoholic fatty liver disease (NAFLD)167. b | NAFLD is also linked to several extrahepatic comorbidities. Patients with NAFLD have an increased risk of developing type 2 diabetes mellitus (T2DM)42 and cardiovascular disease (CVD)168. In addition, T2DM is an independent risk factor for NAFLD5,35 and is also correlated with an increased risk of heart disease47. Lastly, NAFLD is associated with an elevated risk of developing chronic kidney disease (CKD), which is also highly correlated to T2DM and CVD49.
Fig. 3 |. Targeting of intermediary metabolism for NAFLD therapy.
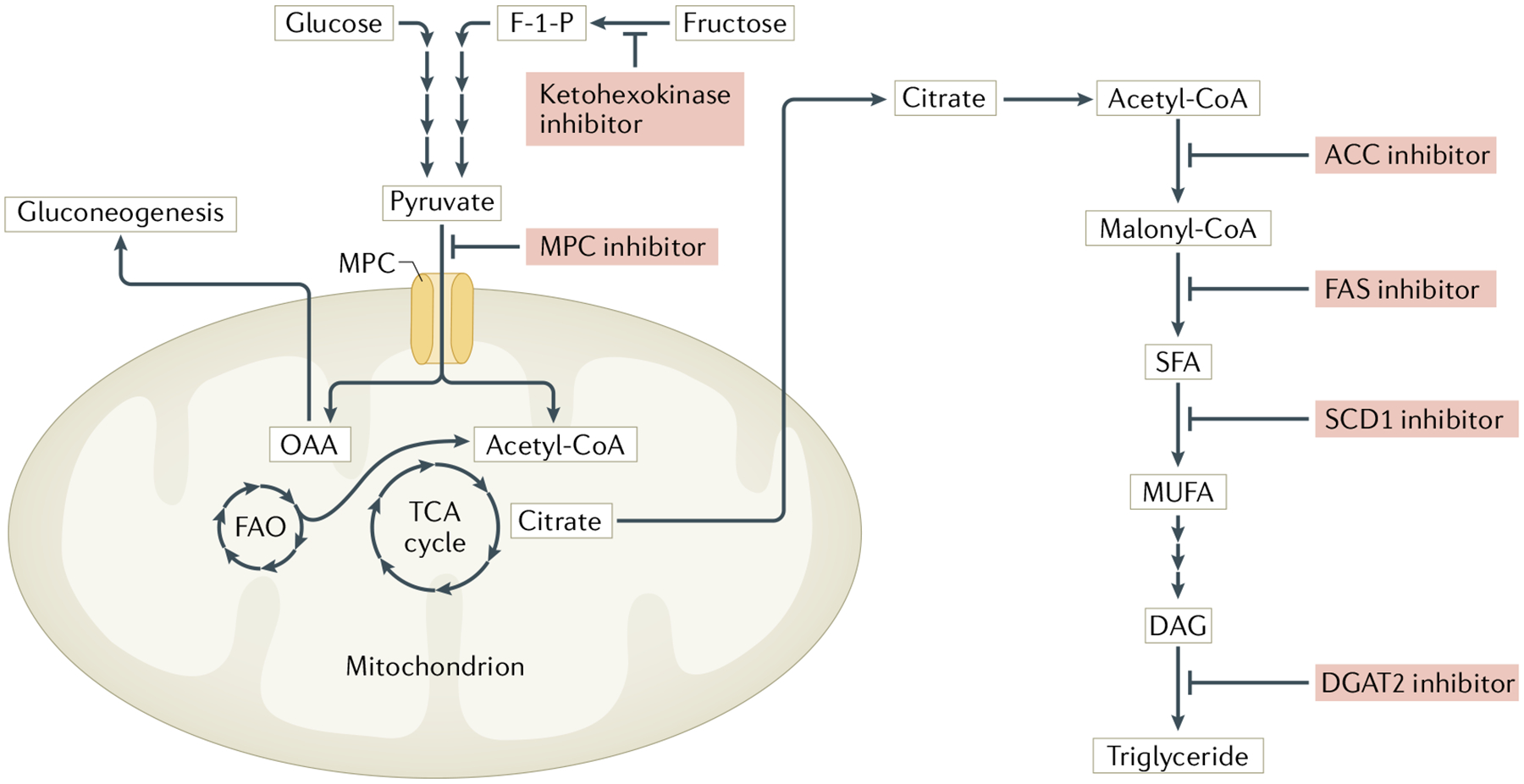
Mechanistic illustration of investigational drugs currently in clinical trials. Inhibitors of ketohexokinase block the rate-limiting step of fructose metabolism, an inducer of de novo lipogenesis149. Mitochondrial pyruvate carrier (MPC) inhibitors act downstream to prevent the import of pyruvate into the mitochondria, where it can be metabolized to intermediates that are used for gluconeogenesis (OAA) or fatty acid synthesis (citrate)85,152. Lastly, several inhibitors are being investigated that target various enzymes (such as ACC, FAS, SCD1 and DGAT2) involved in the synthesis of triglycerides18. ACC, acetyl-CoA carboxylase; DAG, diacylglycerol; DGAT2, diacylglycerol acyltransferase 2; F-1-P, fructose-1-phosphate; FAO, fatty acid oxidation; FAS, fatty acid synthase; MUFA, monounsaturated fatty acid; OAA, oxaloacetate; SCD1, stearoyl-CoA desaturase 1; SFA, saturated fatty acid; TCA, tricarboxylic acid cycle.
Although impairments in the mitochondrial metabolism of fatty acids could also contribute to lipid accumulation, mitochondrial dysfunction seems to happen only in the later stages of NASH progression19. Nevertheless, compounds that stimulate mitochondrial oxidative metabolism, including thyroid hormone receptor agonists20, ligands for nuclear receptors that control the expression of mitochondrial genes21 and inhibitors of mitochondrial pyruvate import22, have shown efficacy in the treatment of NASH.
Triglycerides comprise the primary form of storage lipid and are generally considered inert. However, the accumulation of toxic lipids, such as NEFAs, ceramides and diacylglycerol, is positively correlated with insulin resistance23,24. These lipids might also provoke endoplasmic reticulum stress, oxidative stress and mitochondrial dysfunction, which promotes inflammation and hepatocyte degeneration (ballooning)25,26. The ‘fatty’ liver pheno-type is really an amalgam of these various lipids and is generally quantified by histology-based or MRI-based approaches that do not distinguish between triglycerides or other forms of neutral lipids. Therefore, we will refer to the therapeutic effects as reducing liver ‘steatosis’ or ‘lipid’ content as an endpoint that might represent the reduced accumulation of some or all of these species.
Various immune cell types are present in the liver, including macrophages, neutrophils and T cells27. These cells perform a variety of functions that can be protective (for example, remodelling damaged tissue) or deleterious (exacerbating hepatic inflammation). Inflammation and hepatocyte damage lead to the activation of hepatic stellate cells, which migrate to sites of liver injury and differentiate into myofibroblasts28,29. Stellate cells produce collagen and other components of fibrotic lesions and the development of fibrosis is the best predictor of future liver failure, need for transplantation and mortality30,31.
The current standard for a diagnosis of NASH requires a liver biopsy and histological confirmation of hepatocyte ballooning, inflammatory infiltrates and fibrosis4. To be judged as beneficial in phase III clinical trials, NAFLD therapeutics must lead to a resolution of steatohepatitis without worsening fibrosis or reduce fibrosis with no worsening of steatohepatitis32. However, liver biopsies are invasive, can cause internal bleeding, and considerable variability exists in histological evaluations between observers or upon repeat assessments33. The development of less invasive and more consistent standards could speed up the approval of effective therapeutics.
NAFLD and T2DM: pathophysiological links
NAFLD progression is closely associated with insulin resistance and T2DM34 (FIG. 2). Available evidence also suggests that a bidirectional relationship exists between NAFLD (or NASH) and T2DM (TABLE 1). Both T2DM and obesity have been independently associated with an increased risk of developing NAFLD5,35 and improvements in insulin sensitivity have been positively associated with histological improvements in NASH and fibrosis regression36. A baseline diagnosis of T2DM was the strongest independent risk factor for patients with NASH going on to develop cirrhosis and HCC37,38 and was positively associated with overall mortality and liver-related outcomes in patients with NAFLD30.
Table 1 |.
Conditions associated with NAFLD
| Condition | Prevalence in NAFLD (%) | Prevalence in NASH (%) | Ref. |
|---|---|---|---|
| Obesity | 51 | 82 | 1 |
| Type 2 diabetes mellitus | 23 | 44 | 1 |
| Dyslipidaemia | 69 | 72 | 1 |
| Hypertriglyceridaemia | 41 | 83 | 1 |
| Hypertension | 39 | 68 | 1 |
| Metabolic syndrome | 43 | 71 | 1 |
| CKD | 20–55 | NA | 49 |
CKD, chronic kidney disease; NA, not available; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis.
Hepatic lipid accumulation is associated with impaired insulin sensitivity, which is exacerbated in patients with NASH and might be a predictor of fibrosis (TABLE 1)15,39,40. The accumulation of specific lipids, such as diacylglycerol or ceramides, can impair hepatic insulin signalling, which leads to a pathological increase in hepatic glucose production23. Elevations in circulating levels of transaminases (a surrogate marker of liver injury and possible NAFLD) that are not explained by other diagnoses are positively linked to an increased future risk of developing T2DM41 and patients with NAFLD have a greater than two-fold increased risk of developing T2DM42. Similarly, the global prevalence of NAFLD in patients with T2DM is estimated to be more than 55%, with NASH expected to occur in 37% of patients with T2DM43. Due to this complex interrelationship, it has been suggested that NAFLD and NASH should be considered in the treatment of T2DM44 and a study published in 2020 has highlighted that the non-invasive screening for NAFLD is cost-effective in patients with T2DM in the USA45.
Cardiovascular disease (CVD) is the primary cause of mortality in patients with T2DM, accounting for up to two-thirds of all deaths46, and increased HbA1c levels are strongly correlated to an increased risk of heart disease and overall mortality47. Accordingly, a meta-analysis published in 2020 demonstrated that several glucose-lowering strategies, particularly those that reduced body weight, decrease the risk of major adverse cardiovascular events, which demonstrates the usefulness of diabetic therapies in providing beneficial cardiovascular effects48. Not only would a reduction in CVD — the leading cause of mortality in patients with NAFLD — be valuable but, as stated previously, a loss of body weight is also effective in resolving the features of NASH3.
In addition to CVD-related macrovascular complications, patients with NAFLD are at increased risk of developing microvascular disease, particularly chronic kidney disease, which is also highly associated with CVD and T2DM. An increased histological severity of NAFLD correlates with progressively lower renal function, which is independent of other comorbidities, including T2DM, hypertension, dyslipidaemia and adiposity49. Patients with NASH who had improvements in fibrosis and/or a resolution of NASH due to lifestyle interventions also had a statistically significant increase in renal function50.
In the past few years, the term ‘metabolic-associated fatty liver disease’ has been proposed to more accurately describe patients who have liver disease coincident with metabolic dysfunction. For example, a patient with hepatic steatosis in combination with obesity and/or T2DM or an individual with a normal BMI who has at least two metabolic risk factors51. Although not yet universally accepted, this new terminology seeks to include a larger population of patients than the term NAFLD, who probably share many underlying and possibly undiagnosed comorbidities, to help expand the collaboration between hepatologists and diabetologists to advance growth in our understanding and treatment of this population of patients51–53. Due to the interplay between NAFLD, NASH and T2DM, it is likely that targeting insulin sensitivity and hyperglycaemia and adopting a holistic attitude towards the treatment of metabolic disease in patients with NAFLD might prove advantageous.
Effects of anti-diabetic agents on NAFLD
Given the tight link between T2DM and NAFLD, numerous anti-diabetic drugs have been tested as NASH therapeutics in experimental models and we will highlight the available evidence for therapeutic agents that have advanced to clinical trials. The first-line T2DM therapy, metformin, has not been shown to have beneficial effects on NASH in people and will not be discussed. However, there are a number of clinical trials for modulators of glucagon-like peptide 1 (GLP1) activity, insulin-sensitizing thiazolidinediones and inhibitors of the sodium–glucose cotransporter 2 (SGLT2), which will be discussed in this section.
GLP1 modulators
GLP1 is an endogenous intestinal hormone, acting through the G protein-coupled GLP1 receptor (GLP1R), that directly stimulates the production and release of insulin and indirectly inhibits glucagon secretion and reduces food intake54. The circulating half-life of GLP1 is only 1–2 minutes due to the action of dipeptidyl peptidase 4 (DPP4), which cleaves and inactivates GLP1. Given the positive effects of GLP1 on glucose homeostasis, engaging its receptor or optimizing its activity is a logical therapeutic target for the treatment of hyperglycaemia. Multiple GLP1R agonists (exenatide, liraglutide, dulaglutide and semaglutide) are approved for the treatment of T2DM54.
GLP1R agonists reduce hepatic steatosis and markers of liver damage in genetically induced and diet-induced diabetic mice, rats and rhesus macaques55,56. The treatment of patients with T2DM with either exenatide, liraglutide or semaglutide has demonstrated efficacy in reducing hepatic lipid content as well as levels of liver enzymes and inflammatory markers, an effect associated with improved HbA1c levels and body-weight loss (TABLE 2)57–60. Furthermore, compared with control individuals given placebo, a greater percentage of patients with NASH who were treated with semaglutide achieved resolution of NASH with no worsening of fibrosis61. Additionally, semaglutide is being tested in patients with NASH and compensated cirrhosis (NCT03987451) to determine its efficacy in the reduction of fibrosis score without a worsening of NASH. Many of the beneficial aspects of GLP1R agonism on NAFLD endpoints are closely associated with weight loss and improved metabolic indexes. GLP1R is not expressed in the liver at appreciable levels62,63 and, thus, the effects of activating this receptor on NASH endpoints are probably not due to direct hepatic actions but rather due to systemic improvements in metabolism.
Table 2 |.
Major clinical trial outcomes of anti-diabetic agents in NAFLD
| Drug target | Drug name (duration) | Study population | Hepatic outcomes reported | Diabetic outcomes reported | Refs |
|---|---|---|---|---|---|
| GLP1R agonist | Exenatide (26 weeks) | Obesity, T2DMa, NAFLD (95%) | ↓ Hepatic triglyceride | ↓ Weight | 57 |
| Liraglutide (meta-analysis) | Obesity, T2DM | ↓ ALT | Not reported | 166 | |
| Liraglutide (12 weeks) | Obesity, NASH | ↓ ALT, AST, DNL | ↓ Weight and adipose mass, ↓ HbA1c and serum levels of glucose, ↓ LDL cholesterol, ↑ insulin sensitivity (clamp studies) | 58 | |
| Liraglutide (48 weeks) | Obesity, NASH | ↑ NASH resolution, ↔ fibrosis worsening | ↓ Weight, ↓ HbA1c and serum levels of glucose, ↑ HDL cholesterol | 59 | |
| Semaglutide (meta-analysis) | Obesity, with or without T2DM | ↓ ALT | Not reported | 60 | |
| Semaglutide (72 weeks) | Obesity, NASH (F1–F3 fibrosis) | ↑ NASH resolution, ↓ ALT, ASTc | ↓ Weightc, ↓ HbA1cc | 61 | |
| DPP4 inhibitor | Sitagliptin (24 weeks) | Obesity, pre-diabetes, early T2DM | ↔ No changes | ↔ No changes | 67 |
| Sitagliptin (24 weeks) | Obesity, T2DM, NASH | ↔ No changes | ↔ No changes | 68 | |
| Thiazolidinedione | Rosig1itazoneb (48 weeks; single arm) | Obesity, NASH | ↓ ALT and AST, ↓ steatosis and inflammatory score | ↓ HbA1c and fasting insulin, ↑ glucose tolerance, ↑ weight | 77 |
| Rosiglitazone (52 weeks) | Obesity, NASH | ↓ ALT, ↓ steatosis | ↓ Fasting glucose and insulin, ↑ weight | 76 | |
| Pioglitazone (24 weeks) and a hypocaloric diet | Obesity, T2DM, NASH | ↓ ALT and AST, ↓ steatosis and inflammatory score | ↓ Fasting glucose and insulin, ↑ insulin sensitivity, ↑ HDL cholesterol, ↑ weight | 79 | |
| Pioglitazone (96 weeks) | Obesity, NASH | ↓ ALT and AST, ↓ NAS, ↑ NASH resolution | ↓ Fasting glucose, insulin and triglyceride, ↑ insulin sensitivity, ↑ HDL cholesterol, ↑ weight | 80,81 | |
| SGLT2 inhibitor | Canagliflozin (24 weeks) | Obesity, T2DM | ↔ No changes | ↓ Weight, ↓ HbA1c, fasting glucose and insulin, ↑ hepatic insulin sensitivity | 88 |
| Dapagliflozin (8 weeks) | Obesity, T2DM | ↓ Liver lipids, ↓ liver stiffness | ↓ Weight, ↓ HbA1c, fasting glucose | 89 | |
| Dapagliflozin (24 weeks) | T2DM, NAFLD | ↓ ALT | ↓ Weight, ↓ HOMA-IR | 90 | |
| Empagliflozin (24 weeks) | Obesity, T2DM | ↓ Liver lipids | ↓ Weight, ↓ glucose | 91 | |
| Empagliflozin (20 weeks) | T2DM, NAFLD | ↓ ALT, ↓ liver lipids | ↔ No differences relative to control | 92 | |
| FXR | Obeticholic acid (FLINT) (72 weeks) | Obesity, T2DM, NASH | ↓ ALT and AST, ↓ steatosis and NAS, ↓ fibrosis | ↓ Weight, ↑ insulin and HOMA-IR, ↑ LDL cholesterol, ↓ HDL cholesterol | 105 |
| Obeticholic acid (REGENERATE) (72 weeks) | NASH (F1–F3 fibrosis) | ↓ ALT and AST, ↓ fibrosis | Not analysed | 106 | |
| Cilofexor (GS-9674) (24 weeks) | Obesity, NASH | ↓ Liver lipids | No significant changes | 111 |
Unless otherwise stated, reported outcomes are differences from the placebo or control group. For non-alcoholic steatohepatitis (NASH), patients were diagnosed with NASH via histological analysis. Non-alcoholic fatty liver disease (NAFLD) is considered to be the presence of steatosis (histological or imaging (that is, MRI)) and/or elevated alanine transaminase (ALT) or AST (aspartate transaminase) levels. Steatosis is based on liver histology. ↓, decrease; ↑, increase; ↔, no change; DNL, de novo lipogenesis; DPP4, dipeptidyl peptidase 4; FXR, farnesoid X receptor; GLP1R, G protein-coupled GLP1 receptor; HOMA-IR, Homeostatic Model Assessment of Insulin Resistance; NAS, NAFLD activity score; SGLT2, sodium–glucose cotransporter 2; T2DM, type 2 diabetes mellitus.
Uncontrolled T2DM: patients with T2DM not effectively managed with oral anti-diabetic therapy.
Results are comparisons within treatment group, relative to baseline.
Results not statistically analysed. Liver lipids are based on MRI-PDFF (or ultrasonography).
The inhibition of DPP4 has also been targeted therapeutically to enhance GLP1 activity. Patients with NAFLD, T2DM and obesity have increased DPP4 levels, which is positively correlated with hepatocyte apoptosis and fibrosis64,65. Currently, four DPP4 inhibitors (alogliptin, linagliptin, saxagliptin and sitagliptin) are approved for treatment in T2DM. DPP4 inhibitors reduced hepatic inflammation, fibrosis and liver tumour development in mouse models of NASH64,66. By contrast, sitagliptin treatment failed to provide any improvement in hepatic lipid content, despite a statistically significant reduction in HbA1c (REFS67,68). Therefore, these inhibitors might not be an effective strategy for the treatment of patients with NAFLD.
Thiazolidinedione insulin sensitizers
Pioglitazone and rosiglitazone treat T2DM by sensitizing the body to the effects of insulin and are identifiable by their thiazolidinedione ring. Pioglitazone and rosiglitazone are potent activators of the nuclear receptor PPARγ, which is most highly expressed in adipose tissue and has a critical role in adipocyte differentiation and lipid and glucose metabolism as well as in suppressing inflammation69. Thiazolidinediones probably improve peripheral insulin sensitivity by stimulating the release of adipokines, promoting triglyceride storage in adipose tissue and enhancing the suppressive action of insulin on lipolysis, resulting in reduced plasma levels of free fatty acids and decreased hepatic lipid accretion70. Additionally, thiazolidinedione treatment directly reduces hepatic stellate cell activation and fibrosis in rats71. Thus, thiazolidinediones might improve hepatic and peripheral insulin sensitivity to attenuate NASH and might also have direct effects on hepatic pathophysiology. However, the use of thiazolidinediones in people is now limited by PPARγ-driven adverse effects, including weight gain, oedema and the risk of bone fracture72. In addition, a meta-analysis published in 2007 found that rosiglitazone use was associated with an increased risk of myocardial infarction and cardiovascular death compared with placebo73, resulting in a black-box warning and restriction of rosiglitazone use by the FDA. However, the warning and restriction were later removed after additional studies failed to replicate the findings regarding cardiovascular risks74. Nonetheless, at present, rosiglitazone is used very rarely to treat patients with T2DM.
Of the two approved thiazolidinediones, rosiglitazone is the most potent activator of PPARγ75. Studies have demonstrated that patients with NASH receiving rosiglitazone had enhanced insulin sensitivity and reduced hepatic steatosis; however, considerable weight gain and oedema in the lower extremities were reported (TABLE 2)76,77. As the clinical use of rosiglitazone as an insulin sensitizer has largely been discontinued, there has been very little further evaluation of the drug for the treatment of NASH subsequent to the studies conducted almost 15 years ago. Pioglitazone is also an effective insulin sensitizer but is a less potent PPARγ agonist and is associated with fewer adverse effects than rosiglitazone75,78. Treatment with pioglitazone in patients with T2DM and NASH resulted in improved insulin sensitivity and reduced inflammation and hepatocyte degeneration as well as in reduced fibrosis relative to pretreatment measurements, yet there was no difference in the resolution of fibrosis compared with the placebo group79. Additionally, patients with NASH but no T2DM receiving pioglitazone had improved fasting glucose levels and a greater resolution of NASH, though with no improvement in fibrosis, and increased weight gain versus patients receiving placebo80,81. A meta-analysis of randomized controlled trials using the thiazolidinediones, either pioglitazone or rosiglitazone, demonstrated that thiazolidinedione treatment led to improved liver histology, including reduced steatosis, inflammation and degeneration82. However, a statistically significant decrease in fibrosis was only seen in studies using pioglitazone versus placebo, suggesting that pioglitazone might be more efficacious than rosiglitazone for the resolution of fibrosis. Further examination demonstrated that responders to pioglitazone had increased plasma concentrations of pioglitazone83 and genetic analysis identified a variant of CYP2C8 (the gene that encodes the predominant pioglitazone-metabolizing enzyme) that was associated with fibrosis score84. Collectively, these studies indicate the usefulness of pioglitazone in the resolution of NASH and highlight the importance of pharmacogenetics, which can drastically alter drug metabolism and response to treatment.
Given the greater efficacy of pioglitazone than rosiglitazone, despite a markedly lower affinity for PPARγ, it seems reasonable to explore whether the therapeutic pharmacology of thiazolidinediones might not be entirely dependent on PPARγ activation. Indeed, other molecular targets of thiazolidinediones have been identified85 and one will be discussed in greater detail in subsequent sections.
SGLT2 inhibitors
SGLT2 is a sodium-dependent glucose transporter primarily expressed in the proximal tubule epithelium of the kidney and is responsible for the majority (>90%) of filtered glucose reabsorption86. Inhibitors of SGLT2 (canagliflozin, dapagliflozin, empagliflozin and ertugliflozin) lead to the increased urinary excretion of glucose and are approved anti-hyperglycaemic agents for T2DM. In addition, SGLT2 inhibitors induce weight loss in many people and reduce the risk of major cardiovascular events86. In a mouse model of T2DM, SGLT2 inhibition significantly reduced levels of liver enzymes, steatosis, hepatocyte damage and fibrosis87.
Several trials have tested the efficacy of SGLT2 inhibitors in human NAFLD and T2DM. Patients treated with either canagliflozin, dapagliflozin or empagliflozin showed reduced hyperglycaemia and improvements in hepatic lipid content, liver enzymes and liver stiffness (TABLE 2)88–92. Notably, as with GLP1R agonists, SGLT2 inhibitors caused weight loss, which strongly correlated with the decrease in hepatic lipid content88. However, a large retrospective study comparing canagliflozin, dapagliflozin, liraglutide or sitagliptin added to previous T2DM treatment (insulin, metformin or sulfonylurea), demonstrated that only the SGLT2 inhibitors canagliflozin and dapagliflozin resulted in significantly improved levels of liver enzymes independently of body-weight loss and HbA1c (REF.93). As with GLP1R, SLGT2 is not expressed in the liver86 and treatment-induced weight loss or metabolic improvements might indirectly result in reduced liver lipid content. However, SGLT2 inhibitor use is associated with an increased risk of lower limb amputation and diabetic ketoacidosis compared with GLP1R agonist treatment94. Overall, SGLT2 inhibitors are effective in managing glycaemic control and lowering hepatic lipid content but histological analyses are needed to evaluate their usefulness in NASH.
Targeting intermediary metabolism in NAFLD
While the previous discussion detailed the experimental use of approved diabetic drugs for the treatment of NASH, the following section will highlight the metabolic therapeutics in development that might improve NAFLD as well as address diabetic and cardiometabolic disease endpoints. Due to space limitations and the lack of positive data, we will not discuss clinical trials evaluating the use of statins4 or ezetimibe95 (an agent that lowers levels of LDL cholesterol by inhibiting intestinal cholesterol absorption) in patients with NASH. Furthermore, other drug classes targeting apoptosis, inflammation and fibrosis, which might reduce insulin resistance, hepatic inflammation and fibrosis in mice fed a high-fat diet96,97, have few reported outcomes on metabolic parameters in clinical trials and will not be discussed. Due to the number of agents tested only in preclinical models, the discussion will be centred on experimental agents that have progressed to clinical trials.
Nuclear receptor modulators
FXR agonists, bile acids and synthetic bile acids.
Bile acids are cholesterol metabolites, synthesized in the liver, that facilitate dietary lipid absorption. The synthesis of bile acids is elevated in T2DM98 and altered bile acid concentrations correlate with histological features in patients with NASH99. The use of nor-ursodeoxycholic acid, a synthetic bile acid homologue, reduces inflammation and fibrosis in mice100. Additionally, nor-ursodeoxycholic acid demonstrated a dose-dependent decrease in liver enzymes in patients with NAFLD101 and is currently being tested in patients with NASH.
In addition to playing a role in lipid absorption, bile acids act as lipid signalling mediators. The farnesoid X receptor (FXR) is a nuclear receptor that is activated by selected bile acids. FXR is highly expressed in the liver and intestine and regulates the expression of genes involved in cholesterol and bile acid synthesis98. In mice, FXR activation reduced blood levels of glucose and plasma levels of lipids, but the long-term administration of FXR agonists led to glucose intolerance102,103. The use of obeticholic acid, a bile acid derivative that activates FXR, increased insulin sensitivity and reduced markers of liver fibrosis and inflammation in patients with NAFLD and T2DM104. More recent studies demonstrated improvements in histological markers of NASH and fibrosis with obeticholic acid treatment (TABLE 2)105,106. However, patients exhibited increased levels of LDL cholesterol, elevated insulin concentrations and pruritus as adverse effects105,106. The elevations in insulin and LDL cholesterol concentrations could lead to an increased risk of developing T2DM and CVD. While obeticholic acid might eventually be approved for the treatment of NASH, additional data on its safety and efficacy are necessary.
Interestingly, a secondary analysis of the FLINT trial demonstrated that those who responded to obeticholic acid treatment, defined as patients with a ≥30% reduction in liver lipid content, had substantially increased odds of improved steatosis and reduced cell death upon histological analysis107. Further examination in patients with NASH demonstrated that various clinical parameters could be integrated into a model to predict a patient’s histological response to treatment108.
Other FXR agonists, including EDP-305 and tropifexor, have proven effective in resolving fibrosis in mouse models of NASH109,110. However, glucose homeostasis outcomes were not reported. The non-steroidal FXR agonist cilofexor caused a reduction in liver lipid content in patients with NASH without altering blood levels of lipids or indicators of insulin resistance111. The differences between obeticholic acid and cilofexor outcomes reported in trials are possibly due to the site-specific activation of FXR. While cilofexor primarily targets intestinal FXR, obeticholic acid is available systemically. The restriction of FXR activation to the intestine led to weight loss in mice by promoting the secretion of fibroblast growth factors (discussed below) to normalize glucose metabolism without increasing plasma levels of lipids112. Ultimately, intestine-specific FXR agonists might reduce adverse effects and ongoing trials (NCT03449446 and NCT03987074) in patients with NASH should be insightful with regards to their usefulness.
Experimental PPAR agonists.
The PPAR family of nuclear receptor transcription factors (PPARα, PPARδ and PPARγ) are characterized by large, promiscuous ligand-binding pockets and are known to regulate numerous aspects of metabolism through gene transcription. Whereas PPARγ is adipocyte enriched and tends to regulate lipid storage, PPARα and PPARδ are expressed most highly in oxidative tissues and regulate genes encoding enzymes involved in mitochondrial biogenesis and metabolism, fatty acid oxidation, ketogenesis, fatty acid uptake, and triglyceride turnover21. Numerous natural and synthetic PPAR agonists have been studied for therapeutic efficacy, including agonists that can activate two or three PPAR isoforms21.
Elafibranor (GFT505), a dual PPARα–PPARδ agonist, reduced hepatic steatosis, inflammation and fibrosis in both mouse and rat models of NAFLD113. Patients with obesity who were treated with elafibranor had reduced levels of liver enzymes and improved insulin sensitivity114. In a phase IIb clinical trial (GOLDEN-505), elafibranor promoted moderate resolution of NASH; however, some patients developed elevated serum levels of creatinine and renal impairment115. A larger phase III trial is under way but interim results showed no statistically significant improvement in histological NASH endpoints compared with placebo116. The use of lanifibranor (IVA337), a pan-PPAR agonist, improved glucose metabolism, steatosis and hepatocyte injury in multiple mouse models of NASH117. A phase II trial might demonstrate that lanifibranor was effective in promoting the resolution of NASH with improved fibrosis in patients with T2DM and NASH118; however, the peer-reviewed publication for this work is yet to be published. A dual PPARα/γ agonist, saroglitazar, reduced hepatic steatosis and inflammation and prevented fibrosis in mice119 and improved insulin sensitivity in patients with T2DM120. Trials in patients with NASH are ongoing (NCT03061721 and NCT03863574). Collectively, experimental PPAR agonists demonstrate some efficacy in NAFLD but their use might be hobbled by adverse effects attributable to PPARγ and PPARδ activation.
THRβ agonists.
The thyroid hormone receptor (THR) is a nuclear receptor that mediates various functions important for growth and metabolism. Two THR isoforms, THRα and THRβ, are encoded by the genes THRA and THRB, respectively121. While excess THRα activation might lead to cardiac abnormalities122, specific targeting of THRβ, the primary liver isoform, increases hepatic fatty acid oxidation and reduces steatosis and hyperlipidaemia in rats and mice with genetically induced or diet-induced T2DM20. The liver-targeted THRβ agonist MGL-3196 produced statistically significant reductions in hepatic lipid content and levels of liver enzymes and plasma lipids in patients with NASH but no change was observed in glucose or insulin levels123. Another THRβ agonist, VK2809, reduced hepatic steatosis in mice124 and led to a decreased liver lipid content in patients with NAFLD125. While no changes in glucose homeostasis were observed, the beneficial effects of THRβ on liver lipid content and enzymes, coupled with the potential to improve cardiometabolic outcomes, has warranted an ongoing clinical trial (NCT04173065) to determine its efficacy in the resolution of NASH.
Inhibitors of DNL
ACC inhibitors.
Acetyl-CoA carboxylases (ACCs) catalyse the committed step in DNL through the conversion of acetyl-CoA to malonyl-CoA (FIG. 3). Furthermore, the product of this reaction, malonyl-CoA, is a signalling molecule that suppresses fatty acid oxidation through the inhibition of carnitine palmitoyltransferase 1. Thus, ACC inhibition could reduce hepatic lipid accumulation by suppressing DNL and stimulating fatty acid oxidation. ACC inhibitors improve hepatic steatosis and insulin sensitivity but increase plasma concentrations of triglyceride in mice126, rats127 and humans126. The hypertriglyceridaemia is probably secondary to reduced long-chain polyunsaturated fatty acid concentrations, which promotes VLDL secretion126. In patients with NAFLD, the ACC inhibitors firsocostat (GS-0976) and PF-05221304 resulted in reduced liver lipid content, stiffness and enzyme levels, yet both resulted in hypertriglyceridaemia128–131. Although somewhat efficacious, ACC inhibitor use might be derailed by the secondary consequence of causing hypertriglyceridaemia, but ongoing trials (NCT04399538) using combination therapies are exploring whether these adverse effects can be circumvented.
FAS inhibitors.
Fatty acid synthase (FAS) is a critical enzyme in the DNL pathway that uses malonyl-CoA, synthesized by ACC, to form saturated long-chain fatty acids (FIG. 3). The hepatic expression and activity of FAS are upregulated in NAFLD and preclinical studies in obese mice demonstrate that FAS inhibition lowers liver levels of triglycerides and improves insulin sensitivity132. A phase I trial showed that the FAS inhibitor TVB-2640 reduced DNL, hepatic accumulation of lipids and blood alanine transaminase levels, when compared with baseline values after 10 days of treatment133. Unlike ACC inhibition, targeting the downstream enzyme FAS did not induce increased plasma levels of triglycerides in this early-stage trial. Findings released in 2020 from a phase II trial indicate that TVB-2640 reduces liver lipid content in patients with NASH without treatment-related adverse effects134, but the full results have not yet been reported.
SCD1 inhibitors.
Stearoyl-CoA desaturase 1 (SCD1) is a fatty acid desaturase, highly expressed in lipogenic tissues, such as adipose and liver, that converts saturated fatty acids, such as palmitate and stearate, to monounsaturated fatty acids135 (FIG. 3). SCD1 activity was elevated in proportion to liver lipid content in patients with NAFLD136 and the genetic knockout of hepatic SCD1 expression effectively reduced fatty liver and insulin resistance in high-fat diet-fed mice and rats137. The liver-targeted SCD1 inhibitor, aramchol, reduced hepatic oxidative stress, inflammation and fibrosis in a mouse model of NASH138 and decreased levels of lipids in the liver, liver enzymes and HbA1c in patients with NAFLD139,140. A phase III trial is currently evaluating the efficacy of aramchol in patients with NASH and fibrosis.
DGAT inhibitors.
The final step in triglyceride synthesis, esterifying a fatty acyl-CoA moiety to diacylglycerol, is catalysed by diacylglycerol acyltransferase (DGAT) (FIG. 3). Two isoforms of this enzyme have been identified, DGAT1 and DGAT2, which have distinct patterns of expression and substrate specificity141. Early studies, using antisense oligonucleotides to reduce DGAT2 expression in diabetic db/db mice have shown reduced levels of steatosis and glucose but hepatocyte damage was exacerbated by lipotoxicity due to increased hepatic levels of free fatty acids142. By contrast, mice with liver-specific deletion of DGAT2 showed decreased liver levels of triglycerides without worsening hepatic inflammation143. Interestingly, the beneficial effects on hepatic steatosis were not associated with decreased plasma levels of glucose or insulin in this insulin-resistant mouse model. Clinically, an antisense oligonucleotide inhibitor of hepatic DGAT2 expression, IONIS-DGAT2Rx, reduces liver levels of lipids in patients with T2DM and NAFLD but had no effects on the plasma levels of glucose or lipids144,145. A small-molecule DGAT2 inhibitor, PF-06865571, has also been shown to reduce levels of lipids in the liver in phase I trials146. Unfortunately, many patients receiving PF-06865571 experienced minor gastrointestinal events, such as diarrhoea, probably due to fat malabsorption. If this effect can be overcome, there seems to be great potential for DGAT2 inhibition as a treatment for NAFLD.
Ketohexokinase inhibitors.
Ketohexokinase is an enzyme that catalyses the phosphorylation of fructose to fructose-1-phosphate, an important step in fructose metabolism (FIG. 3). Fructose overconsumption leads to elevated ketohexokinase expression, impaired fatty acid oxidation, increased DNL, hepatic steatosis and impaired insulin signalling, but this effect can be mitigated by the liver-specific deletion of ketohexokinase in high fructose-fed mice147,148. In early stage clinical trials, patients receiving the ketohexokinase inhibitor PF-06835919 had reduced lipid content in the liver but no effects on markers of insulin resistance were seen149,150. Longer phase II trials are under way to evaluate its efficacy in patients with NAFLD and T2DM (NCT03969719).
MPC inhibitors.
In the past decade, the mitochondrial pyruvate carrier (MPC) was identified as a heterodimeric complex of two proteins, MPC1 and MPC2, which functions to transport pyruvate from the cytosol across the inner mitochondrial membrane151 (FIG. 3). This is a required step in the conversion of carbohydrate to fatty acids via DNL. Unexpectedly, the MPC has been identified as a target of the potent insulin-sensitizing thiazolidinediones85 and it has been postulated that thiazolidinedione-mediated inhibition of the MPC might mediate some of the beneficial pharmacology of these agents. The hepatocyte-specific deletion of MPC2 in mice, which also leads to the destabilization and degradation of MPC1, protects against hyperglycaemia by reducing gluconeogenesis and ameliorates fibrosis in a mouse model of NASH22,152. MSDC-0602K, a PPARγ-sparing thiazolidinedione with inhibitory effects on the MPC85, reduces the severity of liver injury and fibrosis in mice22. In a clinical trial, MSDC-0602K did not achieve primary histological endpoints but resulted in reduced hepatic steatosis, circulating levels of liver enzymes and other NASH biomarkers, with a markedly improved insulin sensitivity compared with placebo control in patients with biopsy-confirmed NASH153. Although the efficacy of this approach for reversing NASH is still unclear, this PPARγ-sparing thiazolidinedione might have beneficial effects on a variety of cardiometabolic risk factors with fewer adverse effects than traditional PPARγ-activating thiazolidinediones.
Fibroblast growth factors
FGF19.
Fibroblast growth factor 19 (FGF19; mouse orthologue is FGF15) is a gastrointestinal hormone that regulates bile acid synthesis, glucose metabolism and hepatic fatty acid oxidation, and is known to be a target of FXR154. The overexpression or administration of FGF19 in mice reduces adiposity and hepatic steatosis and improves glucose homeostasis by reducing lipogenic gene expression and increasing energy expenditure155. Levels of FGF19 are reduced in patients with NASH and the administration of an FGF19 analogue, NGM282, effectively decreased levels of hepatic lipid content and liver enzymes with no effect on blood levels of glucose156. However, NGM282 elevated levels of LDL cholesterol156 and could potentially enhance the risk of CVD in these patients. Elevated LDL cholesterol levels are possibly a result of FGF19-mediated suppression of cholesterol 7α-hydroxylase (CYP7A1)157, the rate-limiting enzyme that promotes cholesterol disposal through its conversion to bile acids. This adverse effect is similar to that seen with FXR agonists but the dual use of NGM282 with a statin produced adequate management of cholesterol levels in patients with NASH in a clinical trial158. Though histological improvements have been observed in patients in a small study159, the long-term safety and effectiveness of NGM282 remains to be determined.
FGF21.
FGF21 acts as a hormone to regulate food preference, intermediary metabolism and energy expenditure154. The expression of FGF21 is activated by the PPARs but is also induced in response to a variety of stressors, including high levels of fructose, liver injury or long-term fasting154. Interestingly, patients with NAFLD have elevated FGF21 levels that are inversely correlated with impaired insulin sensitivity154,160, suggesting an FGF21-resistant state161. However, high doses of recombinant FGF21 effectively reduced lipid content in the liver, plasma concentrations of lipid and body weight, and improved glucose and insulin intolerance in obese mice and rhesus macaques162,163. These effects were largely driven by the enhanced energy expenditure and diminished hepatic expression of lipogenic enzymes. In patients with NASH, treatment with recombinant PEGylated FGF21 (BMS-986036) improved plasma levels of glucose and lipids, decreased steatosis scoring and lowered markers of liver injury compared with placebo control164,165. The added benefit of reduced cardiometabolic parameters with improvements in liver steatosis and injury make this FGF21 analogue an attractive option and it is currently being investigated in patients with NASH and advanced fibrosis (NCT03486912 and NCT03486899).
Conclusions
NAFLD is the most common form of liver disease yet there are no drugs licensed for its treatment. Due to the close association between NAFLD and T2DM, many agents that are currently prescribed for hyperglycaemia have yielded positive results on NASH biomarkers. Experimental agents that target aspects of intermediary metabolism have also proven beneficial to varying degrees but adverse effects might limit their use. The overall benefit of metabolic-targeted therapies for NAFLD and NASH is attractive as these drugs might also reduce other cardiometabolic risk factors that contribute to CVD and complications of T2DM, which are the leading cause of mortality in patients with NAFLD and NASH.
Key points.
Non-alcoholic fatty liver disease (NAFLD) has become the most common liver disease globally, yet there are currently no approved therapies.
While NAFLD progression to non-alcoholic steatohepatitis is becoming the leading cause of end-stage liver failure, the leading causes of death in patients with NAFLD are complications of cardiometabolic disease.
A tight relationship exists between NAFLD, insulin resistance and type 2 diabetes mellitus.
It is likely that developing therapeutics that target both NAFLD and cardiometabolic risk factors might be extremely beneficial.
Acknowledgements
Work in the authors’ lab is supported by grants from the National Institutes of Health (R01 DK104735 and R01 DK117657). D.F. is supported by an NIH training grant, T32 DK007120.
Footnotes
Competing interests
B.N.F. is a stockholder and member of the scientific advisory board of Cirius Therapeutics Inc., which is developing MSDC-0602 for the treatment of NASH. D.F. declares no competing interests.
References
- 1.Younossi ZM et al. Global epidemiology of nonalcoholic fatty liver disease — Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Younossi ZM et al. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 64, 1577–1586 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Vilar-Gomez E et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 149, 367–378 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Chalasani N et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67, 328–357 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Younossi ZM et al. Epidemiology of chronic liver diseases in the USA in the past three decades. Gut 69, 564–568 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Lonardo A et al. Sex differences in nonalcoholic fatty liver disease: state of the art and identification of research gaps. Hepatology 70, 1457–1469 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang JD et al. Gender and menopause impact severity of fibrosis among patients with nonalcoholic steatohepatitis. Hepatology 59, 1406–1414 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DiStefano JK NAFLD and NASH in postmenopausal women: implications for diagnosis and treatment. Endocrinology 161, bqaa134 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanyal AJ et al. The natural history of advanced fibrosis due to nonalcoholic steatohepatitis: data from the simtuzumab trials. Hepatology 70, 1913–1927 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Sheka AC et al. Nonalcoholic steatohepatitis: a review. JAMA 323, 1175–1183 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Younossi Z Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol 15, 11–20 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Ivanics T, Abreu P, De Martin E & Sapisochin G Changing trends in liver transplantation: challenges and solutions. Transplantation 105, 743–756 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Donnelly KL et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest 115, 1343–1351 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fabbrini E et al. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 134, 424–431 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korenblat KM, Fabbrini E, Mohammed BS & Klein S Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology 134, 1369–1375 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith GI et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Invest 130, 1453–1460 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lambert JE, Ramos-Roman MA, Browning JD & Parks EJ Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 146, 726–735 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samuel VT & Shulman GI Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 27, 22–41 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koliaki C Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 21, 739–746 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Cable EE et al. Reduction of hepatic steatosis in rats and mice after treatment with a liver-targeted thyroid hormone receptor agonist. Hepatology 49, 407–417 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Cheng HS et al. Exploration and development of PPAR modulators in health and disease: an update of clinical evidence. Int. J. Mol. Sci 20, 5055 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCommis KS et al. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology 65, 1543–1556 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petersen MC & Shulman GI Mechanisms of insulin action and insulin resistance. Physiol. Rev 98, 2133–2223 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samuel VT & Shulman GI Nonalcoholic fatty liver disease, insulin resistance, and ceramides. N. Eng. J. Med 381, 1866–1869 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Rinella ME Nonalcoholic fatty liver disease: a systematic review. JAMA 313, 2263–2273 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Marra F & Svegliati-Baroni G Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol 68, 280–295 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Kubes P & Jenne C Immune responses in the liver. Ann. Rev. Immunol 36, 247–277 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Tsuchida T & Friedman SL Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol 14, 397–411 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Schwabe RF, Tabas I & Pajvani UB Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology 158, 1913–1928 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Angulo P Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 149, 389–397 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taylor RS et al. Association between fibrosis stage and outcomes of patients with nonalcoholic fatty liver disease: a systematic review and meta-analysis. Gastroenterology 58, 1611–1625 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Rinella ME, Tacke F, Sanyal AJ & Anstee QM Report on the AASLD/EASL joint workshop on clinical trial endpoints in NAFLD. J. Hepatol 71, 823–833 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Davison BA et al. Suboptimal reliability of liver biopsy evaluation has implications for randomized clinical trials. J. Hepatol 73, 1322–1332 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Cotter TG & Rinella M Nonalcoholic fatty liver disease 2020: the state of the disease. Gastroenterology 158, 1851–1864 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Zheng Y, Ley SH & Hu FB Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol 14, 88–98 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Kleiner DE et al. Association of histologic disease activity with progression of nonalcoholic fatty liver disease. JAMA Netw. Open 2, e1912565 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alexander M et al. Risks and clinical predictors of cirrhosis and hepatocellular carcinoma diagnoses in adults with diagnosed NAFLD: real-world study of 18 million patients in four European cohorts. BMC Med. 17, 95 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang JD et al. Diabetes is associated with increased risk of hepatocellular carcinoma in patients with cirrhosis from nonalcoholic fatty liver disease. Hepatology 71, 907–916 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lomonaco R et al. Metabolic impact of nonalcoholic steatohepatitis in obese patients with type 2 diabetes. Diabetes Care 39, 632–638 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Musso G et al. Nonalcoholic steatohepatitis versus steatosis: adipose tissue insulin resistance and dysfunctional response to fat ingestion predict liver injury and altered glucose and lipoprotein metabolism. Hepatology 56, 933–942 (2012). [DOI] [PubMed] [Google Scholar]
- 41.De Silva NMG et al. Liver function and risk of type 2 diabetes: bidirectional mendelian randomization study. Diabetes 68, 1681–1691 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mantovani A, Byrne CD, Bonora E & Targher G Nonalcoholic fatty liver disease and risk of incident type 2 diabetes: a meta-analysis. Diabetes Care 41, 372–382 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Younossi ZM et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta-analysis. J. Hepatol 71, 793–801 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Cusi K Time to include nonalcoholic steatohepatitis in the management of patients with type 2 diabetes. Diabetes Care 43, 275–279 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Noureddin M et al. Screening for non-alcoholic fatty liver disease in persons with type 2 diabetes in the U.S. is cost effective: a comprehensive cost-utility analysis. Gastroenterology 159, 1985–1987 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Low Wang CC, Hess CN, Hiatt WR & Goldfine AB Clinical update: cardiovascular disease in diabetes mellitus: atherosclerotic cardiovascular disease and heart failure in type 2 diabetes mellitus — mechanisms, management, and clinical considerations. Circulation 133, 2459–2502 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Selvin E et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N. Eng. J. Med 362, 800–811 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ghosh-Swaby OR et al. Glucose-lowering drugs or strategies, atherosclerotic cardiovascular events, and heart failure in people with or at risk of type 2 diabetes: an updated systematic review and meta-analysis of randomised cardiovascular outcome trials. Lancet Diabetes Endocrinol. 8, 418–435 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Byrne CD & Targher G NAFLD as a driver of chronic kidney disease. J. Hepatol 72, 785–801 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Vilar-Gomez E et al. Improvement in liver histology due to lifestyle modification is independently associated with improved kidney function in patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther 45, 332–344 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Eslam M, Sanyal AJ & George J MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 158, 1999–2014 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Tilg H & Effenberger M From NAFLD to MAFLD: when pathophysiology succeeds. Nat. Rev. Gastroenterol. Hepatol 17, 387–388 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Younossi ZM et al. From NAFLD to MAFLD: implications of a premature change in terminology. Hepatology 73, 1194–1198 (2021). [DOI] [PubMed] [Google Scholar]
- 54.Gimeno RE, Briere DA & Seeley RJ Leveraging the gut to treat metabolic disease. Cell Metab. 31, 679–698 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Young AA et al. Glucose-lowering and insulin-sensitizing actions of exendin-4: studies in obese diabetic (ob/ob, db/db) mice, diabetic fatty Zucker rats, and diabetic rhesus monkeys (Macaca mulatta). Diabetes 48, 1026–1034 (1999). [DOI] [PubMed] [Google Scholar]
- 56.Tølbøl KS et al. Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet-induced obese mouse models of biopsy-confirmed nonalcoholic steatohepatitis. World J. Gastroenterol 24, 179–194 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dutour A et al. Exenatide decreases liver fat content and epicardial adipose tissue in patients with obesity and type 2 diabetes: a prospective randomized clinical trial using magnetic resonance imaging and spectroscopy. Diabetes Obes. Metab 18, 882–891 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Armstrong MJ et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J. Hepatol 64, 399–408 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Armstrong MJ et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 387, 679–690 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Newsome P et al. Effect of semaglutide on liver enzymes and markers of inflammation in subjects with type 2 diabetes and/or obesity. Aliment. Pharmacol. Ther 50, 193–203 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Newsome PN et al. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Eng. J. Med 384, 1113–1124 (2021). [DOI] [PubMed] [Google Scholar]
- 62.Muller TD et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab 30, 72–130 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ferrannini E Sodium-glucose co-transporters and their inhibition: clinical physiology. Cell Metab. 26, 27–38 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Watt MJ, Miotto PM, De Nardo W & Montgomery MK The liver as an endocrine organ-linking NAFLD and insulin resistance. Endocr. Rev 40, 1367–1393 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Williams KH et al. Circulating dipeptidyl peptidase-4 activity correlates with measures of hepatocyte apoptosis and fibrosis in non-alcoholic fatty liver disease in type 2 diabetes mellitus and obesity: a dual cohort cross-sectional study. J. Diabetes 7, 809–819 (2015). [DOI] [PubMed] [Google Scholar]
- 66.Kawakubo M et al. Dipeptidyl peptidase-4 inhibition prevents nonalcoholic steatohepatitis-associated liver fibrosis and tumor development in mice independently of its anti-diabetic effects. Sci. Rep 10, 983 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cui J et al. Sitagliptin vs. placebo for non-alcoholic fatty liver disease: a randomized controlled trial. J. Hepatol 65, 369–376 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Joy TR et al. Sitagliptin in patients with non-alcoholic steatohepatitis: a randomized, placebo-controlled trial. World J. Gastroenterol 23, 141–150 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gross B, Pawlak M, Lefebvre P & Staels B PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol 13, 36–49 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Mayerson AB et al. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes 51, 797 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Galli A et al. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology 122, 1924–1940 (2002). [DOI] [PubMed] [Google Scholar]
- 72.Musso G, Cassader M, Paschetta E & Gambino R Thiazolidinediones and advanced liver fibrosis in nonalcoholic steatohepatitis: a meta-analysis. JAMA Inter. Med 177, 633–640 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nissen SE & Wolski K Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Eng. J. Med 356, 2457–2471 (2007). [DOI] [PubMed] [Google Scholar]
- 74.Hickson RP, Cole AL & Dusetzina SB Implications of removing rosiglitazone’s black box warning and restricted access program on the uptake of thiazolidinediones and dipeptidyl peptidase-4 inhibitors among patients with type 2 diabetes. J. Manag. Care Spec. Pharm 25, 72–79 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lehmann JM et al. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J. Biol. Chem 270, 12953–12956 (1995). [DOI] [PubMed] [Google Scholar]
- 76.Ratziu V et al. Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 135, 100–110 (2008). [DOI] [PubMed] [Google Scholar]
- 77.Neuschwander-Tetri BA, Brunt EM, Wehmeier KR, Oliver D & Bacon BR Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand rosiglitazone. Hepatology 38, 1008–1017 (2003). [DOI] [PubMed] [Google Scholar]
- 78.Juurlink DN et al. Adverse cardiovascular events during treatment with pioglitazone and rosiglitazone: population based cohort study. BMJ 339, b2942 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Belfort R et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Eng. J. Med 355, 2297–2307 (2006). [DOI] [PubMed] [Google Scholar]
- 80.Sanyal AJ et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med 362, 1675–1685 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cusi K et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus a randomized trial. Ann. Inter. Med 165, 305–315 (2016). [DOI] [PubMed] [Google Scholar]
- 82.Boettcher E, Csako G, Pucino F, Wesley R & Loomba R Meta-analysis: pioglitazone improves liver histology and fibrosis in patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther 35, 66–75 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kawaguchi-Suzuki M, Bril F, Kalavalapalli S, Cusi K & Frye RF Concentration-dependent response to pioglitazone in nonalcoholic steatohepatitis. Aliment. Pharmacol. Ther 46, 56–61 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kawaguchi-Suzuki M et al. A genetic score associates with pioglitazone response in patients with non-alcoholic steatohepatitis. Front. Pharmacol 9, 752 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Colca JR et al. Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT)–relationship to newly identified mitochondrial pyruvate carrier proteins. PLoS One 8, e61551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Heerspink HJ, Perkins BA, Fitchett DH, Husain M & Cherney DZ Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation 134, 752–772 (2016). [DOI] [PubMed] [Google Scholar]
- 87.Tahara A, Takasu T, Yokono M, Imamura M & Kurosaki E Characterization and comparison of SGLT2 inhibitors: part 3. Effects on diabetic complications in type 2 diabetic mice. Eur. J. Pharmacol 809, 163–171 (2017). [DOI] [PubMed] [Google Scholar]
- 88.Cusi K et al. Effect of canagliflozin treatment on hepatic triglyceride content and glucose metabolism in patients with type 2 diabetes. Diabetes Obes. Metab 21, 812–821 (2019). [DOI] [PubMed] [Google Scholar]
- 89.Latva-Rasku A et al. The SGLT2 inhibitor dapagliflozin reduces liver fat but does not affect tissue insulin sensitivity: a randomized, double-blind, placebo-controlled study with 8-week treatment in type 2 diabetes patients. Diabetes Care 42, 931–937 (2019). [DOI] [PubMed] [Google Scholar]
- 90.Shimizu M et al. Evaluation of the effects of dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on hepatic steatosis and fibrosis using transient elastography in patients with type 2 diabetes and non-alcoholic fatty liver disease. Diabetes Obes. Metab 21, 285–292 (2019). [DOI] [PubMed] [Google Scholar]
- 91.Kahl S et al. Empagliflozin effectively lowers liver fat content in well-controlled type 2 diabetes: a randomized, double-blind, phase 4, placebo-controlled trial. Diabetes Care 43, 298–305 (2020). [DOI] [PubMed] [Google Scholar]
- 92.Kuchay MS et al. Effect of empagliflozin on liver fat in patients with type 2 diabetes and nonalcoholic fatty liver disease: a randomized controlled trial (E-LIFT Trial). Diabetes Care 41, 1801–1808 (2018). [DOI] [PubMed] [Google Scholar]
- 93.Bajaj HS et al. SGLT2 inhibitors and incretin agents: associations with alanine aminotransferase activity in type 2 diabetes. Diabetes Metab. 44, 493–499 (2018). [DOI] [PubMed] [Google Scholar]
- 94.Ueda P et al. Sodium glucose cotransporter 2 inhibitors and risk of serious adverse events: nationwide register based cohort study. BMJ 363, k4365 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Loomba R et al. Ezetimibe for the treatment of nonalcoholic steatohepatitis: assessment by novel magnetic resonance imaging and magnetic resonance elastography in a randomized trial (MOZART trial). Hepatology 61, 1239–1250 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li P et al. Hematopoietic-derived galectin-3 causes cellular and systemic insulin resistance. Cell 167, 973–984 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang Z, Park H & Bae EJ Efficacy of evogliptin and cenicriviroc against nonalcoholic steatohepatitis in mice: a comparative study. Korean J. Physiol. Pharmacol 23, 459–466 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ahmad TR & Haeusler RA Bile acids in glucose metabolism and insulin signalling — mechanisms and research needs. Nat. Rev. Endocrinol 15, 701–712 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Puri P et al. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology 67, 534–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Beraza N et al. Nor-ursodeoxycholic acid reverses hepatocyte-specific nemo-dependent steatohepatitis. Gut 60, 387–396 (2011). [DOI] [PubMed] [Google Scholar]
- 101.Traussnigg S et al. Norursodeoxycholic acid versus placebo in the treatment of non-alcoholic fatty liver disease: a double-blind, randomised, placebo-controlled, phase 2 dose-finding trial. Lancet Gastroenterol. Hepatol 4, 781–793 (2019). [DOI] [PubMed] [Google Scholar]
- 102.Zhang Y et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl Acad. Sci. USA 103, 1006–1011 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Watanabe M Lowering bile acid pool size with a synthetic farnesoid X receptor (FXR) agonist induces obesity and diabetes through reduced energy expenditure. J. Biol. Chem 286, 26913–26920 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mudaliar S Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 145, 574–582 (2013). [DOI] [PubMed] [Google Scholar]
- 105.Neuschwander-Tetri BA et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 385, 956–965 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Younossi ZM et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 394, 2184–2196 (2019). [DOI] [PubMed] [Google Scholar]
- 107.Loomba R et al. Multicenter validation of association between decline in MRI-PDFF and histologic response in nonalcoholic steatohepatitis. Hepatology 72, 1219–1229 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Loomba R et al. Factors associated with histologic response in adult patients with nonalcoholic steatohepatitis. Gastroenterology 156, 88–95 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hernandez ED et al. Tropifexor-mediated abrogation of steatohepatitis and fibrosis is associated with the antioxidative gene expression profile in rodents. Hepatol. Commun 3, 1085–1097 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.An P et al. A novel non-bile acid FXR agonist EDP-305 potently suppresses liver injury and fibrosis without worsening of ductular reaction. Liver Int. 40, 1655–1669 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Patel K et al. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology 72, 58–71 (2020). [DOI] [PubMed] [Google Scholar]
- 112.Fang S et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med 21, 159–165 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Staels B et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 58, 1941–1952 (2013). [DOI] [PubMed] [Google Scholar]
- 114.Cariou B et al. Dual peroxisome proliferator-activated receptor α/δ agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 36, 2923–2930 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ratziu V et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 150, 1147–1159 (2016). [DOI] [PubMed] [Google Scholar]
- 116.Genfit. GENFIT: Announces Results from Interim Analysis of RESOLVE-IT Phase 3 Trial of Elafibranor in Adults with NASH and Fibrosis https://ir.genfit.com/news-releases/news-release-details/genfit-announces-results-interim-analysis-resolve-it-phase-3 (2020).
- 117.Wettstein G et al. The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol. Commun 1, 524–537 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Inventiva. Inventiva’s lanifibranor meets the primary and key secondary endpoints in the Phase IIb NATIVE clinical trial in non-alcoholic steatohepatitis (NASH) https://inventivapharma.com/inventivas-lanifibranor-meets-the-primary-and-key-secondary-endpoints-in-the-phase-iib-native-clinical-trial-in-non-alcoholic-steatohepatitis-nash/ (2020).
- 119.Jain MR et al. Dual PPARα/γ agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver Int. 38, 1084–1094 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jain N et al. Effect of a dual PPAR α/γ agonist on insulin sensitivity in patients of type 2 diabetes with hypertriglyceridemia- randomized double-blind placebo-controlled trial. Sci. Rep 9, 19017 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sinha RA, Singh BK & Yen PM Direct effects of thyroid hormones on hepatic lipid metabolism. Nat. Rev. Endocrinol 14, 259–269 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wikström L et al. Abnormal heart rate and body temperature in mice lacking thyroid hormone receptor alpha 1. EMBO J. 17, 455–461 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Harrison SA et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 394, 2012–2024 (2019). [DOI] [PubMed] [Google Scholar]
- 124.Zhou J et al. A liver-specific thyromimetic, vk2809, decreases hepatosteatosis in glycogen storage disease type Ia. Thyroid 29, 1158–1167 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Loomba R et al. VK2809, a novel liver-directed thyroid receptor beta agonist, significantly reduces liver fat with both low and high doses in patients with non-alcoholic fatty liver disease: a phase 2 randomized, placebo-controlled trial. J. Hepatol 70, e150–e151 (2019). [Google Scholar]
- 126.Kim CW et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 26, 394–406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Harriman G et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc. Natl Acad. Sci. USA 113, E1796–E1805 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Loomba R et al. GS-0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease. Gastroenterology 155, 1463–1473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lawitz EJ et al. Acetyl-CoA carboxylase inhibitor GS-0976 for 12 weeks reduces hepatic de novo lipogenesis and steatosis in patients with nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol 16, 1983–1991 (2018). [DOI] [PubMed] [Google Scholar]
- 130.Esler W et al. Partial inhibition of de novo lipogenesis with the acetyl-CoA carboxylase inhibitor PF-05221304 does not increase circulating triglycerides in humans and is sufficient to lower steatosis in rats. J. Hepatol 70, e69 (2019). [Google Scholar]
- 131.Amin N et al. PF-05221304 (PF’1304), a liver-targeted acetylcoa carboxylase inhibitor (ACCI), in adults with nonalcoholic fatty liver disease (NAFLD) demonstrates robust reductions in liver fat and alt-phase 2a, dose-ranging study. Hepatology 70 (Suppl. 1), 21A–22A (2019). [Google Scholar]
- 132.Wu M et al. Antidiabetic and antisteatotic effects of the selective fatty acid synthase (FAS) inhibitor platensimycin in mouse models of diabetes. Proc. Natl Acad. Sci. USA 108, 5378–5383 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Syed-Abdul MM et al. First-in-class fatty acid synthase inhibitor TVB-2640 reduces hepatic de novo lipogenesis in males with metabolic abnormalities. Hepatology 72, 103–118 (2020). [DOI] [PubMed] [Google Scholar]
- 134.Sagimet. Sagimet announces positive topline results in 12-week nash phase 2 clinical trial of FASN inhibitor TVB-2640 https://sagimet.com/sagimet-announces-positive-topline-results-in-12-week-nash-phase-2-clinical-trial-of-fasn-inhibitor-tvb-2640/ (2020).
- 135.Aljohani AM, Syed DN & Ntambi JM Insights into stearoyl-CoA desaturase-1 regulation of systemic metabolism. Trends Endocrinol. Metab 28, 831–842 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kotronen A et al. Hepatic stearoyl-CoA desaturase (SCD)-1 activity and diacylglycerol but not ceramide concentrations are increased in the nonalcoholic human fatty liver. Diabetes 58, 203–208 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gutiérrez-Juárez R et al. Critical role of stearoyl-CoA desaturase-1 (SCD1) in the onset of diet-induced hepatic insulin resistance. J. Clin. Invest 116, 1686–1695 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Iruarrizaga-Lejarreta M et al. Role of Aramchol in steatohepatitis and fibrosis in mice. Hepatol. Commun 1, 911–927 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Safadi R et al. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol 12, 2085–2091 (2014). [DOI] [PubMed] [Google Scholar]
- 140.Ratziu V, Ladron-De-Guevara L, Safadi R & Poordad F One-year results of the global phase 2b randomized placebo-controlled arrest trial of aramchol, a stearoyl CoA desaturase inhibitor, in patients with nash. Hepatology 68, 1448A–1449A (2018).29604231 [Google Scholar]
- 141.Yen CLE, Stone SJ, Koliwad S, Harris C & Farese RV Jr DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res 49, 2283–2301 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yamaguchi K et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 45, 1366–1374 (2007). [DOI] [PubMed] [Google Scholar]
- 143.Gluchowski NL et al. Hepatocyte deletion of triglyceride-synthesis enzyme acyl CoA: diacylglycerol acyltransferase 2 reduces steatosis without increasing inflammation or fibrosis in mice. Hepatology 70, 1972–1985 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Loomba R et al. An international, randomized, placebo-controlled phase 2 trial demonstrates novel effects of DGAT2 antisense inhibition in reducing steatosis without causing hypertriglyceridemia in T2DM patients. J. Hepatol 70, e67–e68 (2019). [Google Scholar]
- 145.Loomba R et al. Novel antisense inhibition of diacylglycerol O-acyltransferase 2 for treatment of non-alcoholic fatty liver disease: a multicentre, double-blind, randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol. Hepatol 5, 829–838 (2020). [DOI] [PubMed] [Google Scholar]
- 146.Saxena A, Chidsey K, Somayaji V, Ogden A & Duvvuri S Diacylglycerol acyltransferase 2 (DGAT2) inhibitor PF-06865571 reduces liver fat by MRI-PDFF after 2 weeks in adults with NAFLD. Hepatology 70 (Suppl. 1), 1260A (2019). [Google Scholar]
- 147.Jang C et al. The small intestine shields the liver from fructose-induced steatosis. Nat. Metab 2, 586–593 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Andres-Hernando A et al. Deletion of fructokinase in the liver or in the intestine reveals differential effects on sugar-induced metabolic dysfunction. Cell Metab. 32, 117–127 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Softic S et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Invest 127, 4059–4074 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Calle R, Bergman A, Somayaji V, Chidsey K & Kazierad D PS-110 Ketohexokinase inhibitor PF-06835919 administered for 6 weeks reduces whole liver fat as measured by magnetic resonance imaging-proton density fat fraction in subjects with non-alcoholic fatty liver disease. J. Hepatol 70, e69–e70 (2019). [Google Scholar]
- 151.Bricker DK et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337, 96–100 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.McCommis KS et al. Loss of mitochondrial pyruvate carrier 2 in the liver leads to defects in gluconeogenesis and compensation via pyruvate-alanine cycling. Cell Metab. 22, 682–694 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Harrison SA et al. Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: a randomized, double-blind, placebo-controlled phase IIb study. J. Hepatol 72, 613–626 (2020). [DOI] [PubMed] [Google Scholar]
- 154.Degirolamo C, Sabbà C & Moschetta A Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat. Rev. Drug. Discov 15, 51–69 (2016). [DOI] [PubMed] [Google Scholar]
- 155.Fu L et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 145, 2594–2603 (2004). [DOI] [PubMed] [Google Scholar]
- 156.Harrison SA et al. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 391, 1174–1185 (2018). [DOI] [PubMed] [Google Scholar]
- 157.Luo J et al. A nontumorigenic variant of FGF19 treats cholestatic liver diseases. Sci. Transl. Med 6, 247ra100 (2014). [DOI] [PubMed] [Google Scholar]
- 158.Rinella ME et al. Rosuvastatin improves the FGF19 analogue NGM282-associated lipid changes in patients with non-alcoholic steatohepatitis. J. Hepatol 70, 735–744 (2019). [DOI] [PubMed] [Google Scholar]
- 159.Harrison SA et al. NGM282 improves liver fibrosis and histology in 12 weeks in patients with nonalcoholic steatohepatitis. Hepatology 71, 1198–1212 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chavez AO et al. Circulating fibroblast growth factor-21 is elevated in impaired glucose tolerance and type 2 diabetes and correlates with muscle and hepatic insulin resistance. Diabetes Care 32, 1542–1546 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Fisher FM et al. Obesity is a fibroblast growth factor 21 (FGF21)-resistant state. Diabetes 59, 2781–2789 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Véniant MM et al. Long-acting FGF21 has enhanced efficacy in diet-induced obese mice and in obese rhesus monkeys. Endocrinology 153, 4192–4203 (2012). [DOI] [PubMed] [Google Scholar]
- 163.Xu J et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 58, 250–259 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Charles ED et al. Pegbelfermin (BMS-986036), PEGylated FGF21, in patients with obesity and type 2 diabetes: results from a randomized phase 2 study. Obesity 27, 41–49 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sanyal A et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 392, 2705–2717 (2019). [DOI] [PubMed] [Google Scholar]
- 166.Armstrong MJ et al. Safety and efficacy of liraglutide in patients with type 2 diabetes and elevated liver enzymes: individual patient data meta-analysis of the LEAD program. Aliment. Pharmacol. Ther 37, 234–242 (2013). [DOI] [PubMed] [Google Scholar]
- 167.Fujita K et al. Dysfunctional very-low-density lipoprotein synthesis and release is a key factor in nonalcoholic steatohepatitis pathogenesis. Hepatology 50, 772–780 (2009). [DOI] [PubMed] [Google Scholar]
- 168.Targher G, Byrne CD & Tilg H NAFLD and increased risk of cardiovascular disease: clinical associations, pathophysiological mechanisms and pharmacological implications. Gut 69, 1691–1705 (2020). [DOI] [PubMed] [Google Scholar]