

Abstract
Background
Cardiovascular disease is the number one cause of death globally. According to the World Health Organization (WHO), 7.4 million people died from ischaemic heart disease in 2012, constituting 15% of all deaths. Beta‐blockers are recommended and are often used in patients with heart failure after acute myocardial infarction. However, it is currently unclear whether beta‐blockers should be used in patients without heart failure after acute myocardial infarction. Previous meta‐analyses on the topic have shown conflicting results. No previous systematic review using Cochrane methods has assessed the effects of beta‐blockers in patients without heart failure after acute myocardial infarction.
Objectives
To assess the benefits and harms of beta‐blockers compared with placebo or no treatment in patients without heart failure and with left ventricular ejection fraction (LVEF) greater than 40% in the non‐acute phase after myocardial infarction.
Search methods
We searched CENTRAL, MEDLINE, Embase, LILACS, Science Citation Index ‐ Expanded, BIOSIS Citation Index, the WHO International Clinical Trials Registry Platform, ClinicalTrials.gov, European Medicines Agency, Food and Drug Administration, Turning Research Into Practice, Google Scholar, and SciSearch from their inception to February 2021.
Selection criteria
We included all randomised clinical trials assessing effects of beta‐blockers versus control (placebo or no treatment) in patients without heart failure after myocardial infarction, irrespective of publication type and status, date, and language. We excluded trials randomising participants with diagnosed heart failure at the time of randomisation.
Data collection and analysis
We followed our published protocol, with a few changes made, and methodological recommendations provided by Cochrane and Jakobsen and colleagues. Two review authors independently extracted data. Our primary outcomes were all‐cause mortality, serious adverse events, and major cardiovascular events (composite of cardiovascular mortality and non‐fatal myocardial reinfarction). Our secondary outcomes were quality of life, angina, cardiovascular mortality, and myocardial infarction during follow‐up. We assessed all outcomes at maximum follow‐up. We systematically assessed risks of bias using seven bias domains and we assessed the certainty of evidence using the GRADE approach.
Main results
We included 25 trials randomising a total of 22,423 participants (mean age 56.9 years). All trials and outcomes were at high risk of bias. In all, 24 of 25 trials included a mixed group of participants with ST‐elevation myocardial infarction and non‐ST myocardial infarction, and no trials provided separate results for each type of infarction. One trial included participants with only ST‐elevation myocardial infarction. All trials except one included participants younger than 75 years of age. Methods used to exclude heart failure were various and were likely insufficient. A total of 21 trials used placebo, and four trials used no intervention, as the comparator. All patients received usual care; 24 of 25 trials were from the pre‐reperfusion era (published from 1974 to 1999), and only one trial was from the reperfusion era (published in 2018). The certainty of evidence was moderate to low for all outcomes.
Our meta‐analyses show that beta‐blockers compared with placebo or no intervention probably reduce the risks of all‐cause mortality (risk ratio (RR) 0.81, 97.5% confidence interval (CI) 0.73 to 0.90; I² = 15%; 22,085 participants, 21 trials; moderate‐certainty evidence) and myocardial reinfarction (RR 0.76, 98% CI 0.69 to 0.88; I² = 0%; 19,606 participants, 19 trials; moderate‐certainty evidence). Our meta‐analyses show that beta‐blockers compared with placebo or no intervention may reduce the risks of major cardiovascular events (RR 0.72, 97.5% CI 0.69 to 0.84; 14,994 participants, 15 trials; low‐certainty evidence) and cardiovascular mortality (RR 0.73, 98% CI 0.68 to 0.85; I² = 47%; 21,763 participants, 19 trials; low‐certainty evidence). Hence, evidence seems to suggest that beta‐blockers versus placebo or no treatment may result in a minimum reduction of 10% in RR for risks of all‐cause mortality, major cardiovascular events, cardiovascular mortality, and myocardial infarction. However, beta‐blockers compared with placebo or no intervention may not affect the risk of angina (RR 1.04, 98% CI 0.93 to 1.13; I² = 0%; 7115 participants, 5 trials; low‐certainty evidence).
No trials provided data on serious adverse events according to good clinical practice from the International Committee for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH‐GCP), nor on quality of life.
Authors' conclusions
Beta‐blockers probably reduce the risks of all‐cause mortality and myocardial reinfarction in patients younger than 75 years of age without heart failure following acute myocardial infarction. Beta‐blockers may further reduce the risks of major cardiovascular events and cardiovascular mortality compared with placebo or no intervention in patients younger than 75 years of age without heart failure following acute myocardial infarction. These effects could, however, be driven by patients with unrecognised heart failure. The effects of beta‐blockers on serious adverse events, angina, and quality of life are unclear due to sparse data or no data at all. All trials and outcomes were at high risk of bias, and incomplete outcome data bias alone could account for the effect seen when major cardiovascular events, angina, and myocardial infarction are assessed. The evidence in this review is of moderate to low certainty, and the true result may depart substantially from the results presented here. Future trials should particularly focus on patients 75 years of age and older, and on assessment of serious adverse events according to ICH‐GCP and quality of life. Newer randomised clinical trials at low risk of bias and at low risk of random errors are needed if the benefits and harms of beta‐blockers in contemporary patients without heart failure following acute myocardial infarction are to be assessed properly. Such trials ought to be designed according to the SPIRIT statement and reported according to the CONSORT statement.
Plain language summary
Benefits and harms of beta‐blockers versus placebo or no intervention in the non‐acute phase of a myocardial infarction
Background
According to the World Health Organization, 7.4 million people died from ischaemic heart disease in 2012. This represents 15% of all global deaths. Patients with a heart attack but without heart failure may receive beta‐blockers as non‐acute treatment. Beta‐blockers inhibit beta‐receptors. This can result in a reduction in oxygen needed by the heart and may reduce complications associated with a heart attack.
Study characteristics
We searched scientific databases from their beginning to February 2021 and found 25 randomised clinical trials. People had the same chance to be allocated to groups receiving beta‐blockers or control. In 21 trials, the control was a placebo. In four trials, the control was no intervention. Trials included 22,423 adults with mean age of 56.9 years (range 50 to 63 years).
Key results
Patients without heart failure after a heart attack receiving beta‐blockers compared with placebo or no intervention probably have lower risk of death and of a new heart attack and may have reduced risk of major cardiovascular events and death of any heart‐related cause, but likely not of angina pectoris. The effects of beta‐blockers on serious adverse events and on quality of life were uncertain due to lack of data.
Certainty of evidence
The evidence should be interpreted with caution, as certainty was judged to be moderate to low for all outcomes. Reasons mainly include high risk of bias for all included trials, limitations in design and execution, and risk of random error. This means that results might overestimate the beneficial effects of beta‐blockers and underestimate the harmful effects. Therefore, the above mentioned results may change in the future, after additional well‐designed randomised clinical trials are conducted.
Summary of findings
Summary of findings 1. Summary of findings at maximum follow‐up.
| Beta‐blockers compared with placebo or no intervention for patients after an acute myocardial infarction | ||||||
|
Patient or population: patients without heart failure Settings: any setting Intervention: beta‐blockers Comparison: placebo or no intervention | ||||||
| Outcomes | Illustrative comparative risks* | Relative effect | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo or no intervention | Beta‐blockers | |||||
| All‐cause mortality at maximum follow‐up (mean follow‐up 24.9 months; range 9 to 60 months) | Study population |
RR 0.81 97.5% CI (0.73 to 0.90) |
22,085 (21 RCTs) |
⊕⊕⊕⊝ Moderatea | Absolute risk for mortality at maximum follow‐up was 8.7% in the beta‐blocker group compared with 10.9% in the control group, and the NNT was 46 participants | |
| 109 per 1000 | 87 per 1000 (81 to 97) | |||||
| Major cardiovascular events (cardiovascular mortality or non‐fatal myocardial infarction) at maximum follow‐up (mean follow‐up 26.3 months; range 9 to 48 months) | Study population |
RR 0.72 97.5% CI (0.62 to 0.83) |
14,994 (15 RCTs) |
⊕⊕⊝⊝ Lowa,b | Absolute risk for major cardiovascular events at maximum follow‐up was 10.3% in the beta‐blocker group compared with 14.0% in the control group, and the NNT was 23 participants | |
| 140 per 1000 | 103 per 1000 (97 to 118) | |||||
| Serious adverse events at maximum follow‐up | No trials reported serious adverse events according to ICH‐GCP | |||||
| Quality of life | No trials reported quality of life on a continuous or any other scale | |||||
|
Angina pectoris at maximum follow‐up (mean follow‐up 10 months; range 12 to 47 months) |
Study population |
RR 1.04 98% CI (0.95 to 1.13) |
7715 (5 RCTs) |
⊕⊕⊝⊝ Lowa,c | Absolute risk for angina at maximum follow‐up was 26.4% in the beta‐blocker group compared with 25.6% in the control group, and the NNT was 125 participants | |
| 256 per 1000 | 264 per 1000 (238 to 289) | |||||
| Cardiovascular mortality at maximum follow‐up (mean follow‐up 28.8 months; range 9 to 48 months) | Study population |
RR 0.73 98% CI (0.61 to 0.88) |
21,763 (19 RCTs) |
⊕⊕⊝⊝ Lowa,b | Absolute risk for cardiovascular mortality at maximum follow‐up was 6.0% in the beta‐blocker group compared with 8.0% in the control group, and the NNT was 50 participants | |
| 80 per 1000 | 60 per 1000 (54 to 68) | |||||
| Myocardial infarction at maximum follow‐up (mean follow‐up 33.3 months; range 9 to 48 months) | Study population |
RR 0.76 98% CI (0.67 to 0.86) |
19,606 (19 RCTs) |
⊕⊕⊕⊝ Moderatea | Absolute risk for myocardial infarction at maximum follow‐up was 5.9% in the beta‐blocker group compared with 7.8% in the control group, and the NNT was 53 participants | |
| 78 per 1000 | 59 per 1000 (54 to 69) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 97.5% or 98% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 97.5% or 98% CI). CI: confidence interval; ICH‐GCP: International Committee for Harmonization of Technical Requirements for Pharmaceuticals for Human Use; NNT: number needed to treat; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence. High certainty: further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: we are very uncertain about the estimate. | ||||||
aDowngraded by one level due to risk of bias. All included trials were at high risk of bias due to unclear or high risk in several bias domains. None of the included trials were at low risk of bias.
bDowngraded by one level due to risk of inconsistency based on a moderate I²(30% to 60%) with a small P value (P < 0.05) when the statistical test was assessed for heterogeneity.
cDowngraded by one level due to risk of imprecision based on optimal information size not being reached, absolute and relative 98% CIs showing both appreciable benefit and harm, and sample size being small.
Background
Description of the condition
Cardiovascular disease is the number one cause of death globally (Cooper 2000; Lloyd‐Jones 2010; Nichols 2014; Rosamond 2008; Schmidt 2012). Ischaemic heart disease accounts for almost 50% of the disease burden of the cardiovascular diseases (Nichols 2014). According to the World Health Organization (WHO), 7.4 million people died from ischaemic heart disease in 2012 (WHO 2015).
Ischaemic heart disease is caused by different underlying mechanisms: (1) atherosclerotic plaque‐related obstruction of the coronary arteries; (2) focal or diffuse spasms of normal or plaque‐diseased arteries; (3) microvascular dysfunction; or (4) left ventricular dysfunction caused by acute myocardial necrosis or ischaemic cardiomyopathy (Montalescot 2013). Ischaemic heart disease increases risks of stable angina pectoris (see later) and of acute coronary syndrome (see later).
Stable angina pectoris is defined as episodes of reversible myocardial demand or supply mismatch leading to ischaemia or hypoxia of the heart muscle. These processes lead to transient chest discomfort or pain that is precipitated by activities such as walking, or by emotion or stress, with no to minimal symptoms at rest and with beneficial effects of sublingual nitroglycerin on pain (Montalescot 2013).
Acute coronary syndrome is a collective term for (1) unstable angina pectoris (chest pain during rest related to ischaemia or hypoxia of the heart muscle (Roffi 2016)); (2) non‐ST‐elevation myocardial infarction (NSTEMI); or (3) ST‐elevation myocardial infarction (STEMI) (Roffi 2016; Steg 2012). Myocardial infarction is caused by death of cardiac myocytes (myocardial necrosis) due to ischaemia (O'Gara 2013; Steg 2012). Myocardial infarction is clinically defined as elevated serum levels of cardiac biomarkers (cardiac‐specific troponins and the myocardial band (MB) isoenzyme of creatine kinase (CK‐MB) among others) and changes in the ST‐segment on an electrocardiogram (ECG) (STEMI and NSTEMI) or symptoms of cardiac ischaemia (Roffi 2016; Steg 2012).
Diagnosis of myocardial infarction is dependent on elevation of serum levels of cardiac‐specific troponin I, troponin T, or CK‐MB, among others (Roffi 2016; Steg 2012). However, these enzymes often are not detectable before 8 to 24 hours after the first symptoms of myocardial infarction. Beta‐blockers accordingly may be commenced as an intervention for people with suspected myocardial infarction or may be commenced as an intervention for people with confirmed diagnosis of myocardial infarction at a later time.
Pathologically, the classification of myocardial infarction is based on size (microscopic (focal necrosis) ‐ small (10% of the left ventricular myocardium), moderate (10% to 30% of the left ventricular myocardium), or large (> 30% of the left ventricular myocardium)) ‐ and on location (anterior, lateral, inferior, posterior, septal, or a combination of locations) (Thygesen 2007).
Furthermore, the pathological appearance can be used to classify the temporal development of myocardial infarction as evolving (< 6 hours), acute (6 hours to 7 days), healing (7 days to 28 days), or healed (> 4 weeks) (Thygesen 2007; Thygesen 2020).
An acute myocardial infarction occurring within 28 days of the first episode of myocardial infarction is defined as a reinfarction; a myocardial infarction occurring after 28 days of the first episode of the event is defined as a recurrent myocardial infarction (Mendis 2011).
Clinically, the causes of myocardial infarction are generally divided into five main classes based on morphological changes in the coronary arteries and/or on clinical history (Thygesen 2012; Thygesen 2018).
Type 1: spontaneous myocardial infarction related to atherosclerotic plaque rupture, ulceration, fissuring, erosion, or dissection, with resulting intraluminal thrombus in one or more of the coronary arteries, often caused by coronary artery disease.
Type 2: myocardial infarction secondary to an ischaemic imbalance such as coronary artery spasm, coronary embolism, anaemia, arrhythmia, hypertension, or hypotension.
Type 3: myocardial infarction with symptoms suggestive of myocardial ischaemia and resulting in sudden unexpected cardiac death when biomarker values are unavailable or could not be obtained before death.
Type 4a: myocardial infarction associated with percutaneous coronary intervention (PCI).
Type 4b: myocardial infarction associated with stent thrombosis as documented by angiography or at autopsy.
Type 5: myocardial infarction associated with coronary artery bypass graft (CABG).
Major complications associated with myocardial infarction include the following.
Life‐threatening ventricular arrhythmia caused by changes in electrophysiological characteristics of the myocyte, electrolyte imbalance, continuous ischaemia, and variations in heart rate ‐ all due to obstruction and hence reduced flow to the myocardium and myocardial necrosis (Brieger 2009; Stevenson 1989).
Mechanical complications caused by necrosis of the myocardium such as ventricular wall rupture, septum rupture, and papillary muscle rupture (Brieger 2009; Pohjola‐Sintonen 1989; Stevenson 1989).
Cardiogenic shock caused by failure of the ventricle to pump an adequate amount of blood, leading to systemic hypotension (Brieger 2009; Stevenson 1989).
Acute decompensated heart failure caused by impairment in systolic and diastolic function due to myocardial ischaemia (Brieger 2009).
Depression (Thombs 2006).
Description of the intervention
The discovery of the difference between adrenergic receptors by Raymond Ahlquist in 1948 led Sir James Black to develop the first clinically useful beta‐receptor blocker (propranolol) in 1964 (Ahlquist 1948; Black 1964). This discovery was awarded the Nobel Prize in 1988 (Quirke 2006). Beta‐blockers are classified as non‐selective beta‐blockers or selective beta‐blockers according to their selectivity for one of the three subtypes of beta‐receptors.
-
The beta₁‐receptor is mainly located in:
the heart, where it induces positive chronotropic effects (increases heart rate) and positive inotropic effects (increases contractility of the myocardium); and
in the kidneys, where activation of the beta₁‐receptor results in increased release of renin, which in turn increases blood pressure, among other effects (Golan 2012; Marlin 1975; Singh 1975).
The beta₂‐receptor is mainly located in smooth muscle cells, where it promotes relaxation; in skeletal muscle cells, where it promotes tremor and increased glycogenolysis; and in the liver, where it increases glycogenolysis (Golan 2012).
The beta₃‐receptor is mainly located in adipose tissue, where it primarily induces lipolysis (Golan 2012).
Beta‐blockers may be administered both intravenously and orally.
Three different classes of beta‐blockers are available.
First‐generation non‐selective beta‐blockers (e.g. propranolol, oxprenolol, sotalol, timolol), affecting all beta‐receptors.
Second‐generation selective beta‐blockers (e.g. metoprolol, bisoprolol, acebutolol, atenolol, esmolol), mainly affecting the heart.
Third‐generation beta‐blockers, which have combined non‐selective beta‐blocking effects and alpha‐blocking effects (e.g. carvedilol), affecting all beta‐receptors plus alpha‐receptors in the vessels, lowering blood pressure.
Oral beta‐blockers may be used as secondary prevention, especially in the non‐acute phase, defined as more than seven days after an acute myocardial infarction (Smith 2011). Furthermore, over the past few decades, in the so‐called reperfusion era, reperfusion strategies have played a central role in secondary prevention after a myocardial infarction (Ibanaz 2015; Roffi 2016).
How the intervention might work
The beta‐receptor is an adrenergic Gs heterodimeric G‐protein‐coupled receptor located throughout the body. Beta‐receptors are stimulated by the sympathetic nervous system, with catecholamines epinephrine (adrenaline) and norepinephrine (noradrenaline) as their primary endogenous agonists. The role of non‐acute treatment with beta‐blockers in people with myocardial infarction rests on their inhibition of chronotropic and inotropic effects of the beta‐receptor, leading to lowering of intracellular levels of cyclic adenosine monophosphate and calcium (Lubbe 1992). This may result in a reduction in heart rate, in cardiac contractility, and in systemic arterial pressure, thereby decreasing the oxygen demand of the heart, and consequently reducing ischaemic chest pain and improving left ventricular compliance (Lopez‐Sendon 2004). Additionally, this inhibition of the beta‐receptor is thought to decrease recurrent ischaemia and might decrease the risk of life‐threatening ventricular arrhythmias and other complications associated with myocardial infarction by prolonging the ventricular refractory period (Grandi 2019; Roffi 2016; Steg 2012).
Why it is important to do this review
The prevalence of ischaemic heart disease is considerable, and former meta‐analyses have shown conflicting results (see later). According to the WHO, 7.4 million people died of ischaemic heart disease in 2012 (Lloyd‐Jones 2010; Nichols 2014; Rosamond 2008; WHO 2015). Therefore, the right treatment may result in a considerable reduction in disease burden and healthcare costs.
The role of beta‐blockers in the present as well as in other clinical settings is still debated.
Beta‐blockers used to be contraindicated in people with congestive heart failure (Bristow 2000). Non‐selective combined alpha‐ and beta‐blockers are now a part of standard treatment for congestive heart failure (Chatterjee 2013; Yancy 2013). Beta‐blockers are also considered an option for treatment of hypertension but are rarely used as first‐line treatment (Mancia 2013). A recent Cochrane Review found that beta‐blockers were inferior when compared with other antihypertensive drugs (Wiysonge 2012). Non‐selective beta‐blockers are used for treatment of anxiety due to their effects on decreasing tremor and tachycardia (Turner 1994).
Perioperative beta‐blockade for major non‐cardiac surgery in people with risk factors for ischaemic heart disease has been tested in several randomised clinical trials (Bangalore 2008; Devereaux 2008; Juul 2006), and it seems to increase 30‐day all‐cause mortality as well as the occurrence of stroke, although non‐fatal myocardial infarction is reduced (Bangalore 2008). Perioperative usage of beta‐blockers remains a controversial topic (Wang 2020).
Beta‐blockers may cause both cardiac adverse effects and non‐cardiac adverse effects. Among the most serious cardiac adverse effects is exacerbation of heart failure in people with acute decompensated heart failure due to the need for sympathetic activity to maintain cardiac output (Taylor 1982). In addition, beta‐blocker withdrawal has been shown to cause exacerbation of ischaemic symptoms and to precipitate acute myocardial infarction in people with ischaemic heart disease (Houston 1981).
Case studies have suggested that depression, fatigue, and sexual dysfunction are among beta‐blocker‐induced non‐cardiac adverse effects (Greenblatt 1974; Waal 1967; Warren 1977). However, a meta‐analysis comparing beta‐blockers versus placebo showed no difference in depressive symptoms and only a minor increase in sexual dysfunction and fatigue among participants randomised to beta‐blockers compared with placebo (Ko 2002).
Although beta‐blockers are considered standard treatment for people with diagnosed heart failure (Chatterjee 2013; Yancy 2013), it remains unclear whether beta‐blockers have a beneficial effect in the non‐acute phase following acute myocardial infarction for people without heart failure (Collet 2020; Roffi 2016). This Cochrane Review will be the first to specifically assess effects of treatment with beta‐blockers in the non‐acute phase after myocardial infarction.
Evidence on effects of beta‐blockers following acute myocardial infarction in patients without heart failure
Two meta‐analyses including randomised controlled trials conducted before 2000 compared the effects of any type of beta‐blocker versus no beta‐blocker on long‐term outcomes among participants with suspected or diagnosed myocardial infarction (Freemantle 1999; Yusuf 1985). Both showed a beneficial effect of beta‐blockers for mortality; however, all included trials were performed before the reperfusion era, when heart failure was a common finding in these patients. A more recent meta‐analysis including randomised controlled trials from both pre‐reperfusion and reperfusion eras found a beneficial effect on mortality only in trials from the pre‐reperfusion era, in which participants had not received revascularisation (percutaneous coronary intervention or coronary artery bypass graft) nor thrombolytics (e.g. streptokinase) (Bangalore 2014). Bangalore 2014 also found beneficial effects of beta‐blockers on symptoms of angina and risk of recurrent myocardial infarction regardless of whether or not participants received intervention for reperfusion. However, this study also shows that beta‐blockers seemed to increase the severity of heart failure among participants receiving intervention for reperfusion (Bangalore 2014). It must be noted that Bangalore 2014 included a larger number of trials than Freemantle 1999 and Yusuf 1985, and only Bangalore 2014 included trials after reperfusion strategies had been implemented. It is important to keep in mind that implementation of rapid reperfusion in the treatment of myocardial infarction has had beneficial effects on both survival and development of heart failure because of reduced infarct size (Ibanaz 2015). Thus, it is of utmost importance to identify the effects of beta‐blockers specifically in the modern reperfusion era. However, none of these meta‐analyses systematically assessed trials from the reperfusion era including only participants without heart failure and with preserved left ventricular ejection fraction (LVEF) in the non‐acute phase following acute myocardial infarction.
Despite the lack of recent randomised controlled trials from the current reperfusion era assessing long‐term effects of beta‐blockers in patients without heart failure following acute myocardial infarction, a large number of recent observational and registry studies assessing this issue have been conducted. A recent meta‐analysis including 16 observational studies from 2000 to 2017 found a beneficial effect on reduction of all‐cause mortality among almost 170,000 patients without heart failure following acute myocardial infarction (rate ratio 0.74, 95% confidence interval (CI) 0.64 to 0.85) (Dahl 2019). However, with control for bias due to the presence of publication and small‐study effect bias, the beneficial effects of beta‐blocker therapy disappeared. Another meta‐analysis consisting of 10 observational studies with a total of 40,000 patients assessed effects of oral beta‐blocker therapy after PCI treatment for patients following acute myocardial infarction with follow‐up of at least three months; researchers found a beneficial effect on all‐cause mortality only in those with reduced LVEF (unadjusted relative risk 0.58, 95% CI 0.48 to 0.71) (Huang 2015). However, no significant beneficial effects were found in the subgroup of patients with preserved LVEF (relative risk 0.79, 95% CI 0.59 to 1.07), and potential beneficial effects of beta‐blocker therapy appeared to gradually disappear after more than one year of follow‐up after myocardial infarction. These findings were further supported by a cohort study following 3145 patients for a total of one‐year follow‐up after STEMI, where patients with reduced LVEF had a better prognosis, and fewer patients reached the primary outcome of all‐cause mortality or hospital re‐admission for a cardiovascular event (hazard ratio (HR) 0.431, 95% CI 0.262 to 0.703; P = 0.001) (Ferreira 2021). However, in patients with preserved (HR 0.73, 95% CI 0.51 to 1.04; P = 0.081) and mid‐range (HR 1.01, 95% CI 0.64 to 1.61; P = 0.959) LVEF, no such beneficial effect was seen compared to the non‐beta‐blocker group. In contrast to these findings, a meta‐analysis of seven observational studies totaling 11,000 patients, assessed effects of long‐term beta‐blocker therapy (> 6 months) among STEMI patients with LVEF greater than 40% who underwent primary PCI and found long‐term beta‐blocker therapy to be associated with decreased all‐cause mortality (combined hazard ratio 0.79, 95% CI 0.65 to 0.97) (Misumida 2016).
Guideline recommendations
Clear inconsistency between international guidelines is evident in their recommendations for the use of beta‐blockers in patients without heart failure and a preserved left ventricular ejection fraction after myocardial infarction (Amsterdam 2014; Antman 2004; Collet 2020; Ibanez 2017; NICE 2013; O'Gara 2013; Steg 2012; Roffi 2016).
According to European Society of Cardiology (ESC) guidelines, oral beta‐blocker therapy is recommended during hospital stay and should be continued thereafter for all STEMI patients without contraindications with a class IIa recommendation (level of evidence (LoE) B), with no further specification of patient group. However, for patients with heart failure and/or left ventricular ejection fraction dysfunction (LVEF < 40%), oral treatment with beta‐blockers is indicated with a class I recommendation (LoE A) (Ibanez 2017; Steg 2012).
American College of Cardiology Foundation/American Heart Association (ACCF/AHA) guidelines from 2004 initially recommended beta‐blocker therapy as a class I recommendation (LoE A) for all STEMI patients except those at low risk (defined as normal or near‐normal ventricular function, successful reperfusion, absence of significant ventricular arrhythmias) and those with contraindications. Treatment was suggested to begin within a few days of the event, if not initiated acutely, and to continue indefinitely. However, it was argued that it would be reasonable to prescribe beta‐blockers for low‐risk patients after STEMI but only with a class IIa recommendation (LoE A) (Antman 2004). More recent ACCF/AHA guidelines from 2013 suggest that oral beta‐blockers should be initiated in the first 24 hours for patients with STEMI who do not have any of the following: signs of heart failure, evidence of a low‐output state, increased risk of cardiogenic shock, or other contraindications with a class I recommendation (LoE B). Furthermore, it was suggested that therapy should be continued during and after hospitalisation for all patients with STEMI with a class I recommendation (LoE B). This recommendation was further extended to suggest a three‐year treatment course for patients without heart failure while acknowledging lack of data on the long‐term effects of beta‐blocker therapy (O'Gara 2013).
For patients with non‐ST elevation after myocardial infarction, the use of beta‐blockers is provided with a class I recommendation (LoE A), according to ESC guidelines, but only in the context of heart failure and LVEF less than 40% (Roffi 2016). However, no recommendations have been made for NSTEMI patients without heart failure and normal left ventricular function, as it appears that this has not yet been investigated in contemporary randomised clinical trials according to ESC guidelines (Roffi 2016). The latest ESC guidelines from 2020 further reinforce the need for studies evaluating the value of long‐term therapy with beta‐blockers for patients with LVEF 40% or greater (Collet 2020). In contrast to this, ACCF/AHA guidelines appear to be based on enough data to find it reasonable to continue beta‐blocker therapy with a class IIa recommendation (LoE C) for NSTEMI patients with normal left ventricular function (Amsterdam 2014).
Last, the 2013 National Institute for Health and Care Excellence (NICE) guideline recommends continuing beta‐blocker therapy for at least one year after myocardial infarction for patients without heart failure or reduced LVEF (NICE 2013).
Hence, meta‐analyses and guidelines have shown conflicting results and recommendations, and no former reviews have used Cochrane methods to systematically assess effects of beta‐blockers among people without heart failure and with LVEF 40% or greater after myocardial infarction (Higgins 2011a). The present Cochrane Review will be the first to assess risk of systematic errors ('bias'), design errors, and random errors ('play of chance') (Higgins 2011a), including trials irrespective of outcome, duration of follow‐up, number of participants, language, and publication status.
Objectives
To assess the benefits and harms of beta‐blockers compared with placebo or no treatment in patients without heart failure and with left ventricular ejection fraction (LVEF) greater than 40% in the non‐acute phase after myocardial infarction.
Methods
Criteria for considering studies for this review
Types of studies
We included all randomised clinical trials irrespective of publication type, reported outcomes, publication status, publication date, and language. We did not include cluster randomised trials due to the inferiority of these trials compared to individually randomised clinical trials. We did not specifically search for non‐randomised studies.
Types of participants
We included any participants, irrespective of age, whom trialists described as receiving a diagnosis of myocardial infarction in the non‐acute and stable phase after acute myocardial infarction. We excluded trials assessing effects of beta‐blockers among patients with post‐myocardial infarction heart failure, defined as clinically overt heart failure with New York Heart Association (NYHA) Class III/IV and LVEF less than 40% at the time of discharge from the hospital. However, we included participants with Killip class I/II and NYHA Class I/II heart failure and with acute heart failure as defined by the trialist during hospitalisation with acute myocardial infarction.
Types of interventions
We included three types of trials.
Beta‐blocker compared with placebo.
Beta‐blocker added to a co‐intervention compared with a similar co‐intervention.
Beta‐blocker compared with no treatment.
We accepted any co‐intervention (any medical therapy or any revascularisation strategy) provided it was planned to be delivered similarly to experimental and control groups. We assumed that no interaction between effects of co‐interventions would ‘even out’ in both groups, so possible effects of beta‐blockers would be reflected in the results.
We accepted any type of beta‐blocker as the experimental intervention (non‐selective beta‐blockers (e.g. propanolol, oxprenolol, sotalol, timolol), selective beta₁‐blockers (e.g. metoprolol, bisoprolol, acebutolol, atenolol, esmolol), and beta‐blockers that are combined alpha‐ and non‐selective beta‐blockers (e.g. carvedilol)).
Our analysis included trials assessing effects of any type of secondary prevention beta‐blockers commenced in the non‐acute phase after myocardial infarction.
We accepted trialists' definitions of whether beta‐blockers were administered in an acute/subacute phase or in a 'non‐acute phase', and we included only trials in which beta‐blockers were administered in the non‐acute phase following acute myocardial infarction. However, as a rule of thumb, we considered beta‐blockers administered more than seven days after myocardial infarction as 'administered in a non‐acute phase'. We also included trials in which beta‐blockers were administered within seven days after a myocardial infarction if trialists described participants as fully recovered after acute myocardial infarction, assuming beta‐blockers were then administered in the non‐acute phase. We assessed these trials in a post hoc subgroup analysis comparing trials in which beta‐blockers were administered more than seven days after myocardial infarction to trials in which beta‐blockers were administered within seven days after myocardial infarction (see Subgroup analysis and investigation of heterogeneity).
The effects of beta‐blockers administered in the acute/subacute phase are assessed in another review (Safi 2019).
Types of outcome measures
Primary outcomes
All‐cause mortality
Major cardiovascular event (MACE) defined as a composite outcome consisting of cardiovascular mortality (as defined by trialists) and non‐fatal myocardial infarction (as defined by trialists). Additionally, we assessed cardiovascular mortality and myocardial infarction separately as secondary outcomes (see later). We reported MACE (cardiovascular mortality + non‐fatal myocardial infarction) only if we were confident that there was no risk of double‐counting, and if the trialist had clearly defined and reported cardiovascular mortality and non‐fatal infarction
Serious adverse event defined as any untoward medical occurrence that was life‐threatening, resulted in death, or was persistent or led to significant disability; or prolonged hospitalisation or any medical event that had jeopardised the participant or required intervention to prevent it (ICH‐GCP 2015). Because we expected reporting of serious adverse events in many trials to be very heterogeneous and not done strictly according to recommendations regarding good clinical practice from the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH‐GCP) (ICH‐GCP 2015), we planned to include the event as a serious adverse event if trial authors used the term ‘serious adverse event' without referring to ICH‐GCP. However, no trials specifically assessed serious adverse events according to the definition provided by ICH‐GCP. Instead, trials reported one specific serious adverse event, which was already included in one of the other outcomes in this review, or a composite of several different events without referring to the actual proportions of participants
Secondary outcomes
Quality of life measured on any valid scale, such as Short Form Health Survey (SF‐36) (Ware 1992). However, no trials reported this outcome
Angina measured on any valid scale, such as Canadian Cardiovascular Society (CCS) Angina Score (Campeau 1976)
Cardiovascular mortality
Myocardial infarction
We planned to narratively report adverse events, presenting them in a table. However, because most of the included trials did not extensively report adverse events, we did not do this.
We estimated all outcomes at maximum follow‐up, which was our outcome of primary interest. When data at different time periods were presented, we included only the longest follow‐up result. Furthermore, we used a subgroup analysis to assess different follow‐up durations included in the trials (please see Subgroup analysis and investigation of heterogeneity).
Search methods for identification of studies
Electronic searches
We searched the following electronic databases on 23 February 2021 to identify relevant trials (Royle 2003).
Cochrane Central Register of Controlled Trials (CENTRAL; 2021, Issue 2 of 12), in the Cochrane Library.
Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, MEDLINE Daily, and MEDLINE Ovid (1946 to 22 February 2021).
Embase Ovid (1974 to 22 February 2021).
Latin American and Caribbean Health Science Information database (LILACS) Bireme (1982 to 22 February 2021).
Science Citation Index ‐ Expanded on the Web of Science (SCI‐EXPANDED) (Clarivate Analytics, 1900 to 23 February 2021).
BIOSIS on the Web of Science (Clarivate Analytics, 1926 to 23 February 2021).
The preliminary search strategy for MEDLINE (Ovid) was adapted for use with the other databases (Appendix 1). We applied the Cochrane sensitivity‐precision maximising randomised controlled trial filter to MEDLINE (Ovid) and adaptations of it to the other databases, except CENTRAL (Lefebvre 2011).
Additionally, we searched the WHO International Clinical Trials Registry Platform (WHO ICTRP) (apps.who.int/trialsearch), US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (www.clinicaltrials.gov), European Medicines Agency (EMA) (www.ema.europa.eu/ema/), Food and Drug Administration (FDA) (www.fda.gov), Turning Research Into Practice (TRIP), Google Scholar, and SciSearch on 23 February 2021 for finished trials as well as ongoing trials.
We searched all databases from their inception to the present, and we did not impose any restriction on language of publication or publication status. If we identified any papers in a language not known by the review author group, we sought help in our network and acknowledged this in the Acknowledgements section.
Searching other resources
We identified additional trials by searching the bibliographies of review articles, references of papers of included studies, and any relevant retraction statements and errata for included studies and included them when relevant.
Data collection and analysis
We used Review Manager 5 (RevMan 5) (RevMan 2014), and, if needed, we planned to use STATA 16.1 (STATA 2016), to perform the analyses. However, we did not include any analyses for which STATA 16.1 was needed.
Selection of studies
Two review authors (SS and NJS) assessed each identified trial independently. If a trial was identified as relevant by one review author, but not by another, we discussed the reasoning behind each decision. If no agreement could be reached, we involved a third review author (JCJ) to resolve the discussion. Included trials are reported in Characteristics of included studies.
We reported excluded trials that a reader might feasibly have expected to see as included trials in Characteristics of excluded studies.
Data extraction and management
We used a data collection form for trial characteristics and outcome data for all 25 trials included in the review. Two review authors (SS and NJS) independently extracted trial characteristics and outcome data from included trials as follows.
Methods: duration of trial, details of any 'run‐in' period, date of publication.
Participants: number randomised, number analysed, mean age, sex, inclusion criteria, exclusion criteria.
Interventions: intervention, comparison, concomitant medications; excluded medications.
Outcomes: primary and secondary outcomes specified and collected, time points reported.
Notes: funding for trial, notable conflicts of interest for trial authors.
We resolved disagreements by reaching consensus or by involving another review author (JCJ). One review author (SS) transferred data into the RevMan 5 file (RevMan 2014), and another (NJS) double‐checked the data entry. We double‐checked that data were entered correctly by comparing data presented in the systematic review with those provided in study reports. A second review author (NJS) spot‐checked trial characteristics for accuracy against the trial report.
Assessment of risk of bias in included studies
We used instructions given in the Cochrane Handbook for Systematic Reviews of Interventions in our evaluation of methods and of risk of bias of included trials (Higgins 2011b). Two review authors (SS and NJS) assessed included trials independently. We evaluated risks of bias in random sequence generation, allocation concealment, blinding of participants and treatment providers, blinding of outcome assessment, incomplete outcome data, selective outcome reporting, and other sources of bias. We did this because these domains enable classification of randomised clinical trials at low risk of bias and of randomised clinical trials at unclear or high risk of bias. The latter trials often overestimate benefits and underestimate harms (Gluud 2006; Kjaergard 2001; Lundh 2012; Moher 1998; Savovic 2012; Savovic 2012a; Savovic 2018; Schulz 1995; Wood 2008). For additional details on how risk of bias was assessed, see Appendix 2.
Overall risk of bias
Low risk of bias: outcome result was classified at overall 'low risk of bias' only if all bias domains described in the above paragraphs were classified at low risk of bias
High risk of bias: outcome result was classified at 'high risk of bias' if any of the bias risk domains described above were classified at unclear or high risk of bias.
We assessed blinding of outcome assessment, incomplete outcome data, and selective outcome reporting domains for each outcome. Thus, we were able to assess bias risk for each result in addition to each trial.
Measures of treatment effect
Dichotomous outcomes
We calculated risk ratios (RRs) with 97.5% confidence intervals (CIs) for our primary dichotomous outcomes, and 98% CIs for our secondary dichotomous outcomes.
Continuous outcomes
We planned to calculate mean differences (MDs) with 95% CIs for continuous outcomes. We planned to use standardised mean differences (SMDs) when all trials assessed the same outcome but measured it in a variety of ways, for example, when they used different scales (Deeks 2011). However, we did not include any continuous outcomes and we calculated no MD or SMD.
Unit of analysis issues
In the case of multi‐armed trials, we planned to split the number of participants in the control group by the number of arms to avoid double‐counting. We further planned to report the number of comparisons in addition to the number of trials when reporting results. However, none of the included trials that contributed data to our review were multi‐arm trials.
If we found any cross‐over trials, we planned to include data only from the first treatment period (Elbourne 2002). However, we included only one cross‐over trial that did not contribute any data to our review (Barvik 1992).
Dealing with missing data
We contacted all trial authors to request missing data. Many of these authors did not respond to our emails. See Characteristics of included studies.
If included trials had used rigorous methods (i.e. reporting on outcomes for all participants or multiple imputation to deal with missing data), we used these data in our primary analysis (Sterne 2009). We did not impute missing values for any outcomes in our primary analysis. Additionally, for continuous outcomes, if standard deviations (SDs) were not reported, we planned to calculate SDs using data from the trial when possible. However, no trials reported continuous outcomes. We did not use intention‐to‐treat data if the original report did not contain such data.
In our sensitivity analysis for dichotomous outcomes, we imputed data (see below and Sensitivity analysis). We also planned this approach for continuous outcomes; however, no trials reported any continuous outcomes.
Best‐worst and worst‐best case scenarios
To assess the potential impact of missing data for dichotomous outcomes, we performed the following sensitivity analyses.
'Best‐worst case' scenario
We assumed that all participants lost to follow‐up in the experimental group survived and had no serious adverse event, major cardiovascular event, or reinfarction, and that all those in the control group with missing outcomes did not survive or had a serious adverse event, a major cardiovascular event, or a reinfarction.
'Worst‐best case' scenario
We assumed that all participants lost to follow‐up in the experimental group did not survive or had a serious adverse event, a major cardiovascular event, or a reinfarction, and that all those in the control group with missing outcomes survived and had no serious adverse event, major cardiovascular event, or reinfarction.
We presented results from both scenarios in our publication.
To assess the potential impact of missing SDs for continuous outcomes, we planned to perform the following sensitivity analysis.
When SDs were missing and could not be calculated, we planned to impute SDs from trials with similar populations and low risk of bias. If we found no such trials, we planned to impute SDs from trials with a similar population. As the final option, we planned to impute SDs from all trials. However, no trials reported any continuous outcomes.
Assessment of heterogeneity
We primarily investigated forest plots to visually assess any sign of heterogeneity. We then assessed the presence of statistical heterogeneity by performing the Chi² test (threshold P < 0.10) and measured the quantities of heterogeneity by using the I² statistic (Higgins 2002; Higgins 2003). We followed the recommendations for threshold in the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011).
0% to 40%: might not be important.
30% to 60%: may represent moderate heterogeneity.
50% to 90%: may represent substantial heterogeneity.
75% to 100%: considerable heterogeneity.
We investigated possible heterogeneity by performing subgroup and sensitivity analyses and determined whether a meta‐analysis should be avoided (Deeks 2011). However, we found no such meta‐analyses to be excluded.
Assessment of reporting biases
We used a funnel plot to assess reporting bias if we included 10 or more trials. Using asymmetry of the funnel plot, we assessed risk of bias. If the funnel plot showed significant asymmetry, we planned to assess this further using the Harbord test if Tau² was less than 0.1 (Harbord 2006), or the Rücker test if Tau² was greater than 0.1 (Higgins 2011d; Rücker 2008). We used the odds ratio when conducting the Harbord test. This was relevant only for all‐cause mortality.
For continuous outcomes, we planned to use the regression asymmetry test (Egger 1997). However, we included no continuous outcomes.
Data synthesis
Meta‐analysis
We planned to accept both end‐scores and change‐from‐baseline scores when analysing continuous outcomes. If both end‐scores and change‐from‐baseline scores were reported, we planned to use end‐scores. If only change‐from‐baseline scores were reported, we planned to analyse these results together with end‐scores in the same meta‐analyses (Higgins 2011c). However, we included no continuous outcomes.
We undertook this systematic review according to recommendations stated in the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011). We used the statistical software RevMan 5 as provided by Cochrane to meta‐analyse data (RevMan 2014). We planned to use STATA in case of zero event trials (STATA 2016), when RevMan 5 zero event handling (replacing zero with a constant of 0.5) was not sufficient, for example, in cases with skewed numbers of participants between groups (which we planned to handle according to Sweeting 2004 and in cases for which meta‐regression (post hoc) was needed. However, we included no zero event trials and meta‐regression was not needed.
Assessment of significance
We assessed our intervention effects using both random‐effects model meta‐analyses and fixed‐effect model meta‐analyses (DerSimonian 1986; DeMets 1987). We used the more conservative point estimate of the two (Jakobsen 2014). The more conservative point estimate was the estimate closest to no effect (highest P value). If the two estimates were equal, we used the estimate with the widest confidence interval. We assessed three primary outcomes, and due to the risk of multiplicity, we calculated a P value less than 0.025 and a 97.5% CI for the primary outcomes (Jakobsen 2014). We assessed four secondary outcomes and calculated a P value less than 0.02 and a 98% CI for secondary outcomes (Jakobsen 2014). We used the statistical software STATA version 16.1 (command:meta) for CI‐adjusted meta‐analyses (STATA 2016).
We included all trials in our analyses, and we planned to conduct a sensitivity analysis of trials at low risk of bias. If results were similar, we would base our primary conclusions at maximum follow‐up on the overall analysis. If they differed, we would base our primary conclusions on trials with low risk of bias. However, we did not include any trials at low risk of bias, and our primary conclusions were based on the overall analysis.
Subgroup analysis and investigation of heterogeneity
We planned to perform the following subgroup analyses.
Trials in which participants received intervention for reperfusion (coronary artery bypass graft, percutaneous coronary intervention, or thrombolytics) compared to trials in which participants did not receive intervention for reperfusion. Additionally, we planned to assess if there seemed to be a difference between different reperfusion strategies. However, no trials reported data on participants who received any intervention for reperfusion, and we could not carry out this subgroup analysis.
-
Trials in which the experimental group received different types of beta‐blockers.
Acebutolol.
Alprenolol.
Atenolol.
Carvedilol.
Metoprolol.
Oxprenolol.
Pindolol.
Practolol.
Propranolol.
Sotalol.
Timolol.
-
Trials with different follow‐up.
Six months or less.
Between six months and 12 months.
Between one year and three years.
Three years or longer.
-
Trials with different age of participants.
Aged 0 to 18 years.
Aged 18 to 75 years.
Aged 75 years or older. However, no trials reported data on participants aged 0 to 18 years and 75 years or older, and we could not carry out this subgroup analysis.
Trials that randomise men compared to trials that randomise women. However, this subgroup analysis could not be carried out due to no data.
-
Trials with different clinical trial registration status.
Pre‐registration.
Post‐registration.
No registration.
-
Comparison of effects of beta‐blockers versus placebo or no intervention between trials including different types of acute myocardial infarction.
Unstable angina pectoris.
NSTEMI.
STEMI. However, no trials reported exact data on participants with different types of acute myocardial infarction, and we could not carry out this subgroup analysis.
Post hoc subgroup analyses
-
Comparison of effects between trials with different funding
Industry‐funded trials or trial with unknown funding
Non‐industry‐funded trials
Comparison of trials in which beta‐blockers were administered more than seven days after acute myocardial infarction (non‐acute phase) to trials in which beta‐blockers were administered within seven days after acute myocardial infarction (subacute phase)
Comparison of trials specifically excluding heart failure participants to trials specifically excluding heart failure participants but likely not adhering to this. We had, in our protocol, planned to exclude trials specifically randomising participants with heart failure. However, several trials specifically excluded heart failure participants but reported some percentage of participants with heart failure in the baseline table. We chose to include these trials but decided to perform a post hoc subgroup analysis comparing these trials to trials without any heart failure participants
Sensitivity analysis
We planned to assess the potential impact of bias by performing a sensitivity analysis from which we would exclude trials with overall high risk of bias. However, because all included trials were at high risk of bias, we did not do this.
To assess the potential impact of missing data for dichotomous outcomes, we performed best‐worst and worst‐best case scenarios (see Dealing with missing data).
If any post hoc analysis was included, we regarded these primarily as hypothesis‐generating. A post hoc test for subgroup differences comparing trials specifically excluding heart failure participants to trials specifically excluding heart failure participants but likely not adhering to this was included in the review.
Summary of findings and assessment of the certainty of the evidence
We used the GRADE system to assess the certainty of the body of evidence associated with each of the primary outcomes (all‐cause mortality, major cardiovascular events, serious adverse events) and with four secondary outcomes (quality of life, angina, cardiovascular mortality, myocardial infarction) (Guyatt 2008). We constructed a 'Summary of findings' (SoF) table using the GRADEpro Guideline Development Tool (www.gradepro.org). The GRADE approach appraises the certainty of a body of evidence based on the extent to which one can be confident that an estimate of effect or association reflects the item being assessed. The certainty measure of a body of evidence considers within‐study risk of bias, directness of evidence, heterogeneity of data, precision of effect estimates, and risk of publication bias. Two review authors (SS and NJS) assessed the certainty of evidence independently and decided on downgrading. If no agreement could be reached, a third review author (JCJ) resolved the discussion. We justified all decisions to downgrade the certainty of trials using footnotes, and we made comments to aid the reader's understanding of the review when necessary. We included all trials in our analyses, and we planned to conduct a sensitivity analysis with trials at low risk of bias; however, we found no trials to be at low risk of bias. If results had been similar, we would base our primary SoF table and primary conclusions (from our primary time point of interest at maximum follow‐up) on the overall analysis. If results had differed, we would base our primary SoF table and primary conclusions on trials at low risk of bias.
Results
Description of studies
We assessed all trials according to the Cochrane Handbook for Systematic Reviews of Interventions and the protocol for this review (Higgins 2011a; Nielsen 2017). Characteristics of each trial can be found in Characteristics of included studies; Characteristics of excluded studies; and Characteristics of ongoing studies sections.
Results of the search
Through our initial search, we identified 19,140 references at the Cochrane Central Register of Controlled Trials (CENTRAL), in the Cochrane Library (n = 2799), MEDLINE (n = 2471), Embase (n = 6152), Science Citation Index ‐ Expanded (n = 5008), BIOSIS (n = 2615), and the Latin American Caribbean Health Sciences Literature (LILACS) (n = 95). The search strategy is presented in Appendix 1. We found five potentially relevant references when searching clinical trials registers, Google Scholar, and reference lists of included trials, as well as previous systematic reviews and other types of reviews. After removing duplicates, we screened 12,889 records. We deemed 133 records relevant and obtained full texts for further evaluation. We then excluded 72 studies based on review of titles, abstracts, or full texts (see Figure 1). Five records reported ongoing trials (see Characteristics of ongoing studies). Reasons for exclusion for six studies that most closely missed our inclusion criteria are listed in the Characteristics of excluded studies table. The remaining 56 full‐text articles reported on 25 completed randomised clinical trials, which we included in the review according to our pre‐defined inclusion and exclusion criteria.
1.
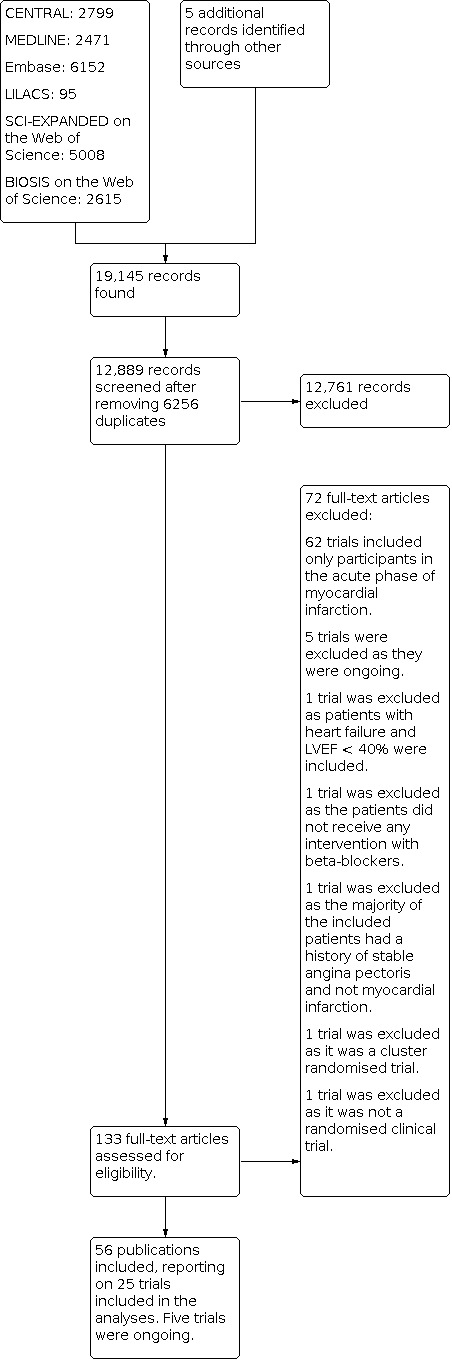
Study flow diagram.
Included studies
We included 56 references reporting on 25 trials comparing beta‐blockers versus control (Figure 1), as well as five ongoing trials (BETAMI 2018; DANBLOCK 2018; MINOCA‐BAT 2021; REBOOT‐CNIC2018; The AβYSS Study 2018). Most trials were conducted between 1974 and 1999, and only one trial occurred in 2018 (Capital‐RCT 2018). These trials (often multi‐national) were conducted at sites in 19 different countries: Australia, China, Denmark, France, Germany, Holland, Hungary, Italy, Japan, New Zealand, Norway, Russia, Scotland, South Africa, Sweden, Switzerland, United Kingdom, United States, and Yugoslavia. For further details on included trials and baseline characteristics of included participants, see Characteristics of included studies.
Only one trial exclusively included participants with STEMI (Capital‐RCT 2018), and no included trials reported outcomes with STEMI, NSTEMI, or unstable angina status on presentation separately. Hence, determination of benefits in these subgroups could not be assessed separately. It is important to note that most trials were conducted in an era that preceded coronary reperfusion (coronary artery bypass graft, percutaneous coronary intervention, or thrombolytics). Thus, determination of benefits in these subgroups could not be sufficiently assessed separately. It is similarly important to know that a large proportion of trials did not describe the use of lipid‐lowering medication, including statins. Fifteen trials excluded participants with heart failure, and 10 trials were equivocal and specifically excluded participants with heart failure but reported some heart failure participants in the baseline table (Ahnve 1980; APSI 1997; Australien & Swedish 1983; Baber 1980; BHAT 1982; E.I.S. 1984; Julian 1982; LIT 1987; NMS 1981; NPT 1982). Mazzuero 1987 was a multi‐armed trial (see Characteristics of included studies), and four trials did not report any useful data to be extracted (Ades 1987; Barvik 1992; Curtis 1991; Mazzuero 1987); hence, available data for analysis were reported in the remaining 21 trials.
At maximum follow‐up (median 43.3 months, range 9 to 60 months), 21 of 25 trials reported all‐cause mortality, 15 of 25 reported major cardiovascular events, 5 of 25 reported angina pectoris, 19 of 25 reported cardiovascular mortality, and 19 of 25 reported myocardial reinfarction. None of the trials reported serious adverse events according to ICH‐GCP nor quality of life. For further details, see Table 1. Three trials included stabilised participants for whom administration of beta‐blockers was started within seven days following acute myocardial infarction (Capital‐RCT 2018; NPT 1982; Poulsen 1999). Remaining included trials administered beta‐blockers after at least seven days following acute myocardial infarction and continued this treatment for a minimum of nine months or for years thereafter.
Participants
A total of 22,423 participants were randomised in the 25 included trials. However, given that only 21 of the included trials contributed useful data, our analyses are based on 22,085 participants. The smallest of the trials included only 28 participants (Barvik 1992), and the largest trial enrolled 3837 participants (MIS 1975). Mean age in the 25 trials reporting age was 56.9 years (range 50 to 63 years). The mean proportion of women was 17.3% (2/25 trials did not report the sex distribution). The mean proportion of participants with a former myocardial infarction was 12.6% (12/25 trials did not report previous myocardial infarctions).
Experimental intervention
Eleven different beta‐blockers were assessed in the included trials: six with metoprolol, six with propranolol, three with oxprenolol, two with alprenolol, two with timolol, and one each with acebutolol, atenolol, carvedilol, pindolol, practolol, and sotalol.
Control intervention
We included 21 trials in which the control group received placebo and four trials in which the control group received no intervention other than the co‐intervention or usual care (Ahlmark 1976; BCSG 1997; Capital‐RCT 2018; Mazur 1984).
Co‐interventions
A total of 15 of 25 trials did not describe any co‐intervention. Seven trials received standard medical therapy as co‐intervention consisting of digitalis, diuretics, vasodilators, antiarrhythmics, anticoagulants, nitrates, and aspirin (Ades 1987; Ahlmark 1976; APSI 1997; Australien & Swedish 1983; Capital‐RCT 2018; E.I.S. 1984; Olsson 1985). One trial described the co‐intervention as conventional therapy without further specification (BCSG 1997); one trial added tranquilliser, potassium, antihypertensive, dipyridamole, insulin, hormonal, oral hypoglycaemic, sulphinpyrazone, and lipid‐lowering drugs to standard medical therapy as the co‐intervention (BHAT 1982); and participants in one trial received long‐acting nitrates and nifedipine as co‐intervention when needed (Schwartz 1992). As stated, only Capital‐RCT 2018 was from the reperfusion era, and remaining included trials did not include statins or invasive cardiology interventions. For further details, see Characteristics of included studies.
Summary of funding sources
Five trials did not report the source of funding (Ahlmark 1976; Amsterdam Metoprolol Trial 1983; Barvik 1992; Mazur 1984; Mazzuero 1987). Five trials were funded by charity or by academic institutions (BCSG 1997; Capital‐RCT 2018; Curtis 1991; Olsson 1985; Poulsen 1999); 14 studies were funded by pharmaceutical companies (Ades 1987; Ahnve 1980; APSI 1997; Australien & Swedish 1983; Baber 1980; E.I.S. 1984; Julian 1982; LIT 1987; MIS 1975; NMS 1981; NPT 1982; Schwartz 1992; Taylor 1982; Wilhelmsson 1974); and one study received free study medication for a trial that had otherwise been planned and conducted independently and was funded largely by public means (BHAT 1982). For further details, see Characteristics of included studies.
Excluded studies
We excluded 67 trials after full‐text assessment based on our inclusion and exclusion criteria. Of these trials, 62 included only participants in the acute phase of myocardial infarction and were therefore included in Safi 2019 and were excluded from this review. Only five of these 67 trials are reported in the Characteristics of excluded studies table. One trial was excluded as it included patients with heart failure; another was excluded as patients did not receive any intervention with beta‐blockers; one was excluded as most included patients had a history of stable angina pectoris ‐ not myocardial infarction; another was excluded as it was a cluster randomised trial; and one was excluded as it was not a randomised clinical trial. For further details, see Characteristics of excluded studies.
Risk of bias in included studies
Based on information that we collected from published reports and from study authors, we considered all 25 trials to be at high risk of bias. We judged many trials to be at unclear risk of bias in several domains when we could not obtain additional information from study authors when contacted. Additional information can be found in the risk of bias summary (Figure 2), the risk of bias graph (Figure 3), the 'Summary of findings' table (Table 1), and the Characteristics of included studies section.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
3.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Generation of the random sequence was at low risk of bias in three trials (APSI 1997; BHAT 1982; Capital‐RCT 2018). In the remaining 23 trials, the method used for allocation concealment was insufficiently described and we therefore judged these trials to be at unclear risk of bias.
The method used to conceal allocation was at low risk of bias in four trials (APSI 1997; E.I.S. 1984; MIS 1975; Schwartz 1992). In the remaining 21 trials, the method of allocation concealment was insufficiently described and we therefore judged these trials to be at unclear risk of bias.
Blinding
Blinding of participants and personnel was at low risk of bias in seven trials (APSI 1997; BHAT 1982; E.I.S. 1984; MIS 1975; Olsson 1985; Schwartz 1992; Taylor 1982), and it was at high risk of bias in two trials (Ahlmark 1976; Capital‐RCT 2018). In the remaining 16 trials, the method for blinding of participants and personnel was insufficiently described and we therefore judged these trials to be at unclear risk of bias.
Blinding of outcome assessors was at low risk of bias in 12 trials (Ahnve 1980; APSI 1997; BHAT 1982; Capital‐RCT 2018; Curtis 1991; E.I.S. 1984; Mazzuero 1987; NMS 1981; Olsson 1985; Schwartz 1992; Taylor 1982; Wilhelmsson 1974). In the remaining 13 trials, methods used to blind outcome assessors were insufficiently described and we therefore judged these trials to be at unclear risk of bias.
Incomplete outcome data
Incomplete outcome data were at low risk of bias in eight trials (Australien & Swedish 1983; Baber 1980; BHAT 1982; Capital‐RCT 2018; E.I.S. 1984; Julian 1982; Olsson 1985; Schwartz 1992). Six trials did not properly deal with incomplete outcome data and were at high risk of bias (Ahlmark 1976; APSI 1997; Barvik 1992; Curtis 1991; Taylor 1982; Wilhelmsson 1974). In the remaining 11 trials, incomplete outcome data were insufficiently described and we therefore judged these trials to be at unclear risk of bias.
Selective reporting
Three trials reported results of outcomes stated in their respective protocols, or they reported our primary outcomes, resulting in low risk of bias (BHAT 1982; Capital‐RCT 2018; E.I.S. 1984). Six trials had no protocol and did not report our primary outcomes sufficiently, resulting in high risk of bias (Ahlmark 1976; Australien & Swedish 1983; Baber 1980; Curtis 1991; Julian 1982; LIT 1987). For the remaining 16 trials, no protocol could be found, but some of our primary outcomes were reported, resulting in unclear risk of bias.
Other potential sources of bias
One trial was prematurely terminated and was judged to be at unclear risk of bias (Capital‐RCT 2018); all other trials were judged to be at low risk of bias when other potential sources of bias were assessed.
Effects of interventions
See: Table 1
Primary outcomes
All‐cause mortality
In all, 21 of 25 trials with a total of 22,085 participants and mean maximum follow‐up of 24.9 months (range 9 to 60 months) reported all‐cause mortality. A total of 976 of 11,236 participants receiving beta‐blockers died versus 1182 of 10,849 control participants. Random‐effects meta‐analysis shows that beta‐blockers probably reduce the risk of all‐cause mortality compared with placebo or no intervention (risk ratio (RR) 0.81, 97.5% confidence interval (CI) 0.73 to 0.90; I² = 15%; 22,085 participants, 21 trials; moderate‐certainty evidence; Figure 4). The point estimate of the meta‐analysis result corresponds to 87 of 1000 beta‐blocker patients dying compared with 109 of 1000 control participants dying, or a number needed to treat (NNT) of 46 participants. Absolute risk for mortality at maximum follow‐up was 8.7% in the beta‐blocker group compared with 10.9% in the control group. Optimal information size according to the GRADE Handbook using a proportion of 10.9% in the control group, relative risk reduction (RRR) of 10%, alpha of 2.5%, and beta of 10% was estimated to be 54,272 participants, and we included only 22,085 participants (see Table 1).
4.
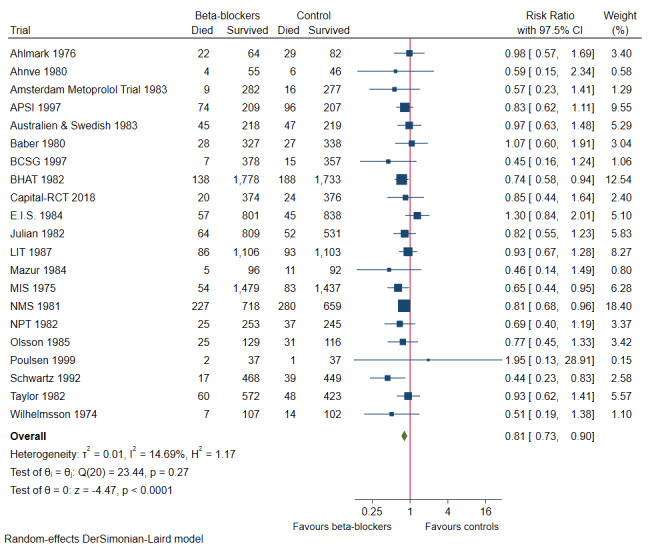
Forest plot of comparison: 1 All‐cause mortality at maximum follow‐up, outcome: 1.1 All‐cause mortality.
Heterogeneity
Visual inspection of the forest plot and tests for statistical heterogeneity (I²= 15%; P = 0.27) indicated no important heterogeneity (Figure 4).
Risk of bias and sensitivity analyses
We assessed risk of bias of this outcome result as high.
Best‐worst and worst‐best case meta‐analyses showed that incomplete outcome data bias alone did not have the potential to influence the meta‐analysis result (best‐worst meta‐analysis: RR 0.70, 95% CI 0.59 to 0.83; I² = 71%; 22,309 participants, 21 trials; moderate‐certainty evidence; Analysis 1.8); worst‐best meta‐analysis: RR 0.89, 95% CI 0.72 to 1.09; I² = 83%; 22,309 participants, 21 trials; moderate‐certainty evidence; Analysis 1.9). Data were imputed for four trials.
1.8. Analysis.

Comparison 1: All‐cause mortality at maximum follow‐up, Outcome 8: All‐cause mortality ‐ 'Best‐worst case scenario'
1.9. Analysis.

Comparison 1: All‐cause mortality at maximum follow‐up, Outcome 9: All‐cause mortality ‐ 'Worst‐best case scenario'
Visual inspection of the funnel plot showed some signs of asymmetry (see Figure 5). Based on visual inspection of the funnel plot, we assessed the risk of publication bias as high. However, we found no signs of small‐study effect when using the Harbord test (P = 0.46) or the Egger test (P = 0.31).
5.

Funnel plot of comparison: 1 All‐cause mortality at maximum follow‐up, outcome: 1.1 All‐cause mortality.
Subgroup analyses
Testing for subgroup differences showed no evidence of a difference in subgroup analyses according to different types of beta‐blockers administered (P = 0.89; Analysis 1.2), different follow‐up periods (P = 0.73; Analysis 1.3), and varying clinical registration status (P = 0.66; Analysis 1.4). However, due to an uneven covariate distribution between different subgroups, these analyses may not have been able to detect any subgroup differences. Remaining tests for subgroup differences were not possible due to lack of data for these subgroups: reperfusion or no reperfusion; different age of participants; men compared to women; and different types of acute coronary syndrome (NSTEMI, STEMI, or unstable angina pectoris (UAP)).
1.2. Analysis.

Comparison 1: All‐cause mortality at maximum follow‐up, Outcome 2: All‐cause mortality ‐ Type of beta‐blocker
1.3. Analysis.

Comparison 1: All‐cause mortality at maximum follow‐up, Outcome 3: All‐cause mortality ‐ Different follow‐up
1.4. Analysis.

Comparison 1: All‐cause mortality at maximum follow‐up, Outcome 4: All‐cause mortality ‐ Registration status
Post hoc subgroup analyses
Testing for subgroup differences showed no evidence of a difference in our post hoc subgroup analyses, suggesting that whether trials received industry versus non‐industry funding (P = 1.00; Analysis 1.5), beta‐blockers were administered within or after seven days following acute myocardial infarction (P = 0.84; Analysis 1.6), or heart failure patients were specifically excluded from trials (P = 0.12; Analysis 1.7), effects of beta‐blockers on all‐cause mortality were not modified when compared to placebo or no intervention. However, due to an uneven covariate distribution between different subgroups, these analyses may not have been able to detect any subgroup differences.
1.5. Analysis.

Comparison 1: All‐cause mortality at maximum follow‐up, Outcome 5: All‐cause mortality ‐ Industry vs non‐industry funding
1.6. Analysis.

Comparison 1: All‐cause mortality at maximum follow‐up, Outcome 6: All‐cause mortality ‐ Subacute vs non‐acute phase
1.7. Analysis.

Comparison 1: All‐cause mortality at maximum follow‐up, Outcome 7: All‐cause mortality ‐ Heart failure vs no heart failure
Major cardiovascular events
In all, 15 of 25 trials with a total of 14,994 participants and mean maximum follow‐up of 26.3 months (range 9 to 48 months) reported major cardiovascular events (composite outcome of cardiovascular mortality and non‐fatal myocardial infarction). A total of 796 of 7701 participants receiving beta‐blockers had a major cardiovascular event versus 1020 of 7293 control participants. Random‐effects meta‐analysis showed that beta‐blockers may reduce the risk of major cardiovascular events compared with placebo or no intervention (RR 0.72, 97.5% CI 0.62 to 0.83; I² = 42%; 14,994 participants, 15 trials; low‐certainty evidence; Figure 6). The point estimate of the meta‐analysis result corresponds to 103 of 1000 beta‐blocker patients dying compared with 140 of 1000 control participants dying, or NNT of 23 participants. Absolute risk for major cardiovascular events at maximum follow‐up was 10.3% in the beta‐blocker group compared with 14.0% in the control group. Optimal information size according to the GRADE Handbook using a proportion of 14.0% in the control group, RRR of 10%, alpha of 2.5%, and beta of 10% was estimated to be 62,729 participants, and we included only 14,994 participants (see Table 1).
6.

Forest plot of comparison: 2 Major adverse cardiovascular events (MACE) at maximum follow‐up, outcome: 2.1 MACE (major cardiovascular events).
Heterogeneity
Visual inspection of the forest plot and tests for statistical heterogeneity (I²= 42%; P = 0.04) indicated moderate heterogeneity (Figure 6).
Risk of bias and sensitivity analyses
We assessed risk of bias of the outcome result as high.
Best‐worst and worst‐best case meta‐analyses showed that incomplete outcome data bias alone had the potential to influence the meta‐analysis result (best‐worst meta‐analysis: RR 0.64, 95% CI 0.50 to 0.81; I² = 76%; 15,225 participants, 15 trials; low‐certainty evidence; Analysis 2.8; worst‐best meta‐analysis: RR 0.92, 95% CI 0.68 to 1.25; I² = 87%; 15,225 participants, 15 trials; low‐certainty evidence; Analysis 2.9). Data were imputed for four trials.
2.8. Analysis.

Comparison 2: Major adverse cardiovascular events (MACE) at maximum follow‐up, Outcome 8: MACE (major cardiovascular events) ‐ 'Best‐worst case scenario'
2.9. Analysis.

Comparison 2: Major adverse cardiovascular events (MACE) at maximum follow‐up, Outcome 9: MACE (major cardiovascular events) ‐ 'Worst‐best case scenario'
Visual inspection of the funnel plot revealed some signs of asymmetry (see Figure 7). Based on visual inspection of the funnel plot, we assessed the risk of publication bias as high. However, we found no signs of small‐study effect when using the Harbord test (P = 0.10) or the Egger test (P = 0.06).
7.

Funnel plot of comparison: 2 Major adverse cardiovascular events (MACE) at maximum follow‐up, outcome: 2.1 MACE (major cardiovascular events).
Subgroup analyses
Testing for subgroup differences showed no evidence of a difference when trials were compared according to the different types of beta‐blocker administered (P = 0.46; Analysis 2.2), different follow‐up periods reported (P = 0.52; Analysis 2.3), and varying clinical registration status (P = 0.91; Analysis 2.4). However, due to an uneven covariate distribution between different subgroups, these analyses may not have been able to detect any subgroup differences. Remaining tests for subgroup differences were not possible due to lack of data for these subgroups: reperfusion or no reperfusion; different age of participants; men compared to women; and different types of acute coronary syndrome (NSTEMI, STEMI, or UAP).
2.2. Analysis.

Comparison 2: Major adverse cardiovascular events (MACE) at maximum follow‐up, Outcome 2: MACE (major cardiovascular events) ‐ Type of beta‐blocker
2.3. Analysis.

Comparison 2: Major adverse cardiovascular events (MACE) at maximum follow‐up, Outcome 3: MACE (major cardiovascular events) ‐ Different follow‐up
2.4. Analysis.

Comparison 2: Major adverse cardiovascular events (MACE) at maximum follow‐up, Outcome 4: MACE (major cardiovascular events) ‐ Registration status
Post hoc subgroup analyses
Testing for subgroup differences showed no evidence of a difference in our post hoc subgroup analyses, suggesting that whether trials received industry versus non‐industry funding (P = 0.26; Analysis 2.5), beta‐blockers were administered within or after seven days following acute myocardial infarction (P = 1.00; Analysis 2.6), or heart failure patients were specifically excluded from trials (P = 0.80; Analysis 2.7), effects of beta‐blockers on major cardiovascular mortality were not modified when compared to placebo or no intervention. However, due to an uneven covariate distribution between different subgroups, these analyses may not have been able to detect any subgroup differences.
2.5. Analysis.

Comparison 2: Major adverse cardiovascular events (MACE) at maximum follow‐up, Outcome 5: MACE (major cardiovascular events) ‐ Industry vs non‐industry funding
2.6. Analysis.

Comparison 2: Major adverse cardiovascular events (MACE) at maximum follow‐up, Outcome 6: MACE (major cardiovascular events) ‐ Subacute vs non‐acute phase
2.7. Analysis.

Comparison 2: Major adverse cardiovascular events (MACE) at maximum follow‐up, Outcome 7: MACE (major cardiovascular events) ‐ Heart failure vs no heart failure
Serious adverse events
No trials specifically assessed or reported serious adverse events according to ICH‐GCP. Instead, trials reported one specific serious adverse event, which was already included in one of the other outcomes in this review, or a composite of several different events without referring to actual proportions of participants.
Secondary outcomes
Quality of life
No trials reported quality of life on a continuous or any other scale at any time point.
Angina pectoris
Five trials reported angina with mean maximum follow‐up of 10 months (range 12 to 47 months) (Ahlmark 1976; APSI 1997; BHAT 1982; Capital‐RCT 2018; E.I.S. 1984). Different definitions and ways of measuring angina were used: Ahlmark 1976 and APSI 1997 reported angina without further definitions, BHAT 1982 reported angina by using the Rose Questionnaire, Capital‐RCT 2018 reported 'vasospastic angina', and E.I.S. 1984 reported angina as 'angina pectoris requiring change in treatment'.
Five trials randomising a total of 7115 participants reported on the proportion of participants with angina (Ahlmark 1976; APSI 1997; BHAT 1982; Capital‐RCT 2018; E.I.S. 1984). A total of 932 of 3526 participants receiving beta‐blockers developed angina compared with 918 of 3589 control participants. Fixed‐effect meta‐analysis showed that beta‐blockers likely may not affect the risk of angina compared with placebo or no intervention (RR 1.04, 98% CI 0.95 to 1.13; I² = 0%; 7115 participants, 5 trials; low‐certainty evidence; Figure 8). The point estimate of the meta‐analysis result corresponds to 264 of 1000 beta‐blocker patients having angina pectoris compared with 255 of 1000 control participants, or NNT of 125 participants. Absolute risk for angina at maximum follow‐up was 26.4% in the beta‐blocker group compared with 25.6% in the control group. Optimal information size according to the GRADE Handbook using a proportion of 25.6% in the control group, RRR of 10%, alpha of 2.0%, and beta of 10% was estimated to be 14,623 participants, and we included only 7115 participants (see Table 1).
8.

Forest plot of 3.1 Angina pectoris on a dichotomous scale.
*E.I.S 1984: angina defined as angina events that required a change in treatment.
Best‐worst and worst‐best case meta‐analyses showed that incomplete outcome data bias alone had the potential to influence the meta‐analysis result (best‐worst meta‐analysis: RR 0.89, 95% CI 0.83 to 0.96; I² = 96%; 7372 participants, 5 trials; low‐certainty evidence; Analysis 3.2; worst‐best meta‐analysis: RR 1.19, 95% CI 1.11 to 1.27; I² = 96%; 7372 participants, 5 trials; low‐certainty evidence; Analysis 3.3). Data were imputed for two trials.
3.2. Analysis.

Comparison 3: Angina pectoris at maximum follow‐up, Outcome 2: Angina pectoris on a dichotomous scale ‐ 'Best‐worst case scenario'
3.3. Analysis.

Comparison 3: Angina pectoris at maximum follow‐up, Outcome 3: Angina pectoris on a dichotomous scale ‐ 'Worst‐best case scenario'
No further analyses were conducted due to sparse data.
Cardiovascular mortality
In all, 19 of 25 trials with a total of 21,763 participants and mean maximum follow‐up of 28.8 months (range 9 to 48 months) reported cardiovascular mortality. A total of 661 of 11,080 participants receiving beta‐blockers died because of a cardiovascular event versus 857 of 10,683 control participants. Random‐effects meta‐analysis showed that beta‐blockers may reduce the risk of cardiovascular mortality compared with placebo or no intervention (RR 0.73, 98% CI 0.61 to 0.88; I² = 47%; 21,763 participants, 19 trials; low‐certainty evidence; Figure 9). The point estimate of the meta‐analysis result corresponds to 60 of 1000 beta‐blocker patients having a cardiovascular death compared with 80 of 1000 control participants, or NNT of 50 participants. Absolute risk for cardiovascular mortality at maximum follow‐up was 6.0% in the beta‐blocker group compared with 8.0% in the control group. Optimal information size according to the GRADE Handbook using a proportion of 8.0% in the control group, RRR of 10%, alpha of 2.0%, and beta of 10% was estimated to be 137,885 participants, and we included only 21,763 participants (see Table 1).
9.
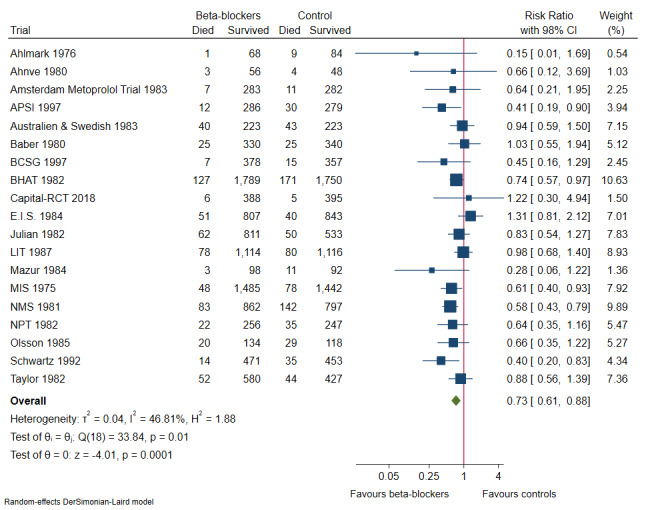
Forest plot of comparison: 4 Cardiovascular mortality at maximum follow‐up, outcome: 4.1 Cardiovascular mortality.
Heterogeneity
Visual inspection of the forest plot and tests for statistical heterogeneity (I²= 47%; P = 0.01) indicated moderate heterogeneity (Figure 9).
Risk of bias and sensitivity analyses
We assessed risk of bias of the outcome result as high.
Best‐worst and worst‐best case meta‐analyses showed that incomplete outcome data bias alone had no potential to influence the meta‐analysis result (best‐worst meta‐analysis: RR 0.71, 95% CI 0.60 to 0.83; I² = 50%; 21,770 participants, 19 trials; low‐certainty evidence; Analysis 4.8; worst‐best meta‐analysis: RR 0.75, 95% CI 0.63 to 0.90; I² = 61%; 21,770 participants, 19 trials; low‐certainty evidence; Analysis 4.9). Data were imputed for only two trials.
4.8. Analysis.

Comparison 4: Cardiovascular mortality at maximum follow‐up, Outcome 8: Cardiovascular mortality ‐ 'Best‐worst case scenario'
4.9. Analysis.

Comparison 4: Cardiovascular mortality at maximum follow‐up, Outcome 9: Cardiovascular mortality ‐ 'Worst‐best case scenario'
Visual inspection of the funnel plot revealed some signs of asymmetry (see Figure 10). Based on visual inspection of the funnel plot, we assessed risk of publication bias as high. However, we found no signs of small‐study effect when using the Harbord test (P = 0.13) or the Egger test (P = 0.07).
10.

Funnel plot of comparison: 4 Cardiovascular mortality at maximum follow‐up, outcome: 4.1 Cardiovascular mortality.
Subgroup analyses
Testing for subgroup differences showed no evidence of a difference in subgroup analyses according to different types of beta‐blockers administered (P = 0.14; Analysis 4.2), different follow‐up periods (P = 0.47; Analysis 4.3), and varying clinical registration status (P = 0.67; Analysis 4.4). However, due to an uneven covariate distribution between different subgroups, these analyses may not have been able to detect any subgroup differences. Remaining tests for subgroup differences were not possible due to lack of data for these subgroups: reperfusion or no reperfusion; different age of participants; men compared to women; and different types of acute coronary syndrome (NSTEMI, STEMI, or UAP).
4.2. Analysis.

Comparison 4: Cardiovascular mortality at maximum follow‐up, Outcome 2: Cardiovascular mortality ‐ Type of beta‐blocker
4.3. Analysis.

Comparison 4: Cardiovascular mortality at maximum follow‐up, Outcome 3: Cardiovascular mortality ‐ Different follow‐up
4.4. Analysis.

Comparison 4: Cardiovascular mortality at maximum follow‐up, Outcome 4: Cardiovascular mortality ‐ Registration status
Post hoc subgroup analyses
Testing for subgroup differences showed no evidence of a difference in our post hoc subgroup analyses, suggesting that whether trials received industry versus non‐industry funding (P = 0.22; Analysis 4.5), beta‐blockers were administered within or after seven days following acute myocardial infarction (P = 0.98; Analysis 4.6), or heart failure patients were specifically excluded from trials (P = 0.20; Analysis 4.7), effects of beta‐blockers on cardiovascular mortality were not modified when compared to placebo or no intervention. However, due to an uneven covariate distribution between different subgroups, these analyses may not have been able to detect any subgroup differences.
4.5. Analysis.

Comparison 4: Cardiovascular mortality at maximum follow‐up, Outcome 5: Cardiovascular mortality ‐ Industry vs non‐industry funding
4.6. Analysis.

Comparison 4: Cardiovascular mortality at maximum follow‐up, Outcome 6: Cardiovascular mortality ‐ Subacute vs non‐acute phase
4.7. Analysis.

Comparison 4: Cardiovascular mortality at maximum follow‐up, Outcome 7: Cardiovascular mortality ‐ Heart failure vs no heart failure
Myocardial infarction
In all, 19 of 25 trials with a total of 19,606 participants and mean maximum follow‐up of 33.3 months (range 9 to 48 months) reported myocardial infarction. A total of 594 of 10,003 participants receiving beta‐blockers had a reinfarction versus 746 of 9603 control participants. Fixed‐effect meta‐analysis showed that beta‐blockers probably reduce the risk of myocardial reinfarction compared with placebo or no intervention (RR 0.76, 98% CI 0.67 to 0.86; I² = 0%; 19,606 participants, 19 trials; moderate‐certainty evidence; Figure 11). The point estimate of the meta‐analysis result corresponds to 59 of 1000 beta‐blocker patients having a new myocardial infarction during follow‐up compared with 78 of 1000 control participants, or NNT of 53 participants. Absolute risk for myocardial infarction at maximum follow‐up was 5.9% in the beta‐blocker group compared with 7.8% in the control group. Optimal information size according to the GRADE Handbook using a proportion of 7.8% in the control group, RRR of 10%, alpha of 2.0%, and beta of 10% was estimated to be 58,717 participants, and we included only 19,606 participants (see Table 1).
11.
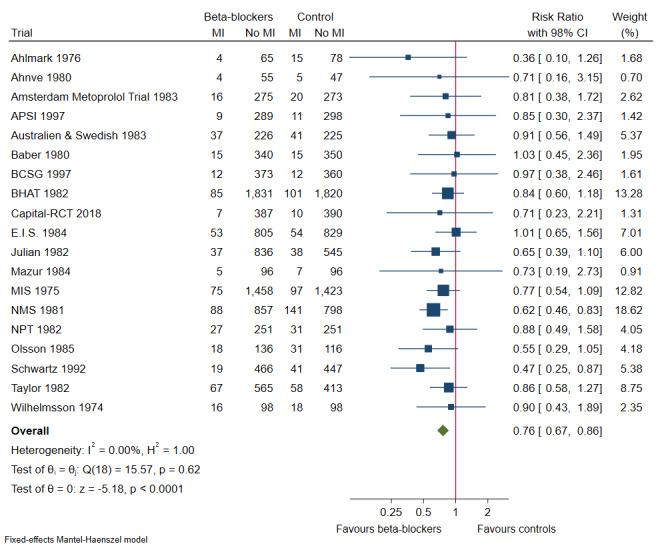
Forest plot of comparison: 5 Myocardial reinfarction at maximum follow‐up, outcome: 5.1 Myocardial infarction.
Heterogeneity
Visual inspection of the forest plot and tests for statistical heterogeneity (I²= 0%; P = 0.62) indicated no heterogeneity (Figure 11).
Risk of bias and sensitivity analyses
We assessed risk of bias of the outcome result as high.
Best‐worst and worst‐best case meta‐analyses showed that incomplete outcome data bias alone had the potential to influence the meta‐analysis result (best‐worst meta‐analysis: RR 0.67, 95% CI 0.54 to 0.84; I² = 70%; 19,837 participants, 19 trials; moderate‐certainty evidence; Analysis 5.8; worst‐best meta‐analysis: RR 0.93, 95% CI 0.71 to 1.23; I² = 84%; 19,837 participants, 19 trials; moderate‐certainty evidence; Analysis 5.9). Data were imputed for three trials.
5.8. Analysis.

Comparison 5: Myocardial reinfarction at maximum follow‐up, Outcome 8: Myocardial infarction ‐ 'Best‐worst case scenario'
5.9. Analysis.

Comparison 5: Myocardial reinfarction at maximum follow‐up, Outcome 9: Myocardial infarction ‐ 'Worst‐best case scenario'
Visual inspection of the funnel plot revealed no signs of asymmetry (see Figure 12). Based on visual inspection of the funnel plot, we assessed risk of publication bias as low.
12.

Funnel plot of comparison: 5 Myocardial reinfarction at maximum follow‐up, outcome: 5.1 Myocardial infarction.
Subgroup analyses
Testing for subgroup differences showed no evidence of a difference in subgroup analyses according to different types of beta‐blockers administered (P = 0.74; Analysis 5.2), different follow‐up periods (P = 0.63; Analysis 5.3), and varying clinical registration status (P = 0.68; Analysis 5.4). However, due to an uneven covariate distribution between different subgroups, these analyses may not have been able to detect any subgroup differences. Remaining tests for subgroup differences were not possible due to lack of data for these subgroups: reperfusion or no reperfusion; different age of participants; men compared to women; and different types of acute coronary syndrome (NSTEMI, STEMI, or UAP).
5.2. Analysis.

Comparison 5: Myocardial reinfarction at maximum follow‐up, Outcome 2: Myocardial infarction ‐ Type of beta‐blocker
5.3. Analysis.

Comparison 5: Myocardial reinfarction at maximum follow‐up, Outcome 3: Myocardial infarction ‐ Different follow‐up
5.4. Analysis.

Comparison 5: Myocardial reinfarction at maximum follow‐up, Outcome 4: Myocardial infarction ‐ Registration status
Post hoc subgroup analyses
Testing for subgroup differences showed no evidence of a difference in our post hoc subgroup analyses, suggesting that whether trials received industry versus non‐industry funding (P = 0.40; Analysis 5.5), beta‐blockers were administered within or after seven days following acute myocardial infarction (P = 0.64; Analysis 5.6), or heart failure patients were specifically excluded from trials (P = 0.32; Analysis 5.7), effects of beta‐blockers on cardiovascular mortality were not modified when compared to placebo or no intervention. However, due to an uneven covariate distribution between different subgroups, these analyses may not have been able to detect any subgroup differences.
5.5. Analysis.

Comparison 5: Myocardial reinfarction at maximum follow‐up, Outcome 5: Myocardial infarction ‐ Industry vs non‐industry funding
5.6. Analysis.

Comparison 5: Myocardial reinfarction at maximum follow‐up, Outcome 6: Myocardial infarction ‐ Subacute vs non‐acute phase
5.7. Analysis.

Comparison 5: Myocardial reinfarction at maximum follow‐up, Outcome 7: Myocardial reinfarction ‐ Heart failure vs no heart failure
Discussion
Summary of main results
We included a total of 25 trials consisting of 22,423 participants (mean age 56.9 years, range 50 to 63 years). However, four trials did not contribute any useful data. All trials and outcome results were at high risk of bias. The certainty of evidence according to GRADE was moderate to low for all outcome results. Most trials included a mix of participants with ST‐elevation myocardial infarction (STEMI) and non‐ST‐elevation myocardial infarction (NSTEMI), and no trials provided separate results for each type of myocardial infarction. Only one trial specifically assessed participants with STEMI. We included 21 trials in which the control group received placebo and four trials in which the control group received no intervention. We included six trials in which participants were observed for up to 12 months, 12 trials in which participants were observed for one to three years, and three trials in which participants were observed for three years or longer. We assume that beta‐blockers were administered during these periods. Methods used to exclude heart failure were varying and likely were insufficient because they did not diagnose all degrees of heart failure.
Our meta‐analyses show moderate‐certainty evidence that beta‐blockers compared with placebo or no intervention probably reduce the risk of all‐cause mortality and myocardial reinfarction. When major cardiovascular events and cardiovascular mortality were assessed, meta‐analyses yielded low‐certainty evidence suggesting that beta‐blockers compared with placebo or no intervention may reduce the risk. Hence, evidence seems to suggest that beta‐blockers versus placebo or no treatment may result in a minimum 10% reduction in risk ratio for all‐cause mortality, major cardiovascular events, cardiovascular mortality, and myocardial infarction. However, when angina was assessed, meta‐analysis yielded low‐certainty evidence suggesting that beta‐blockers compared with placebo or no intervention may not affect the risk.
No data were provided on serious adverse events according to International Conference on Harmonization ‐ Good Clinical Practice Guidelines (ICH‐GCP) nor on quality of life.
Testing for subgroup differences showed no evidence of a difference in our pre‐defined and post hoc subgroup analyses according to different types of beta‐blockers administered, different follow‐up periods provided, varying clinical registration status, differences in industry funding status, beta‐blockers administered within or after seven days following acute myocardial infarction, or heart failure patients specifically excluded from trials, for any of these outcomes. However, due to an uneven covariate distribution between different subgroups, these analyses may not have been able to detect any subgroup differences. Tests for subgroup differences were not possible for reperfusion or no reperfusion; different age of participants; men compared to women; and different types of acute coronary syndrome (NSTEMI, STEMI, or unstable angina pectoris (UAP)) due to lack of data.
Overall completeness and applicability of evidence
This review provides the most contemporary appraisal of evidence to date on beta‐blockers for patients without heart failure and with left ventricular ejection fraction (LVEF) greater than 40% following acute myocardial infarction. We searched for published and unpublished trials, irrespective of publication type, publication status, publication date, and language. We also searched for trials in bibliographies of both Cochrane and non‐Cochrane reviews. All trials and results were at high risk of bias. Hence, there is a risk that our results may overestimate benefits and underestimate harms of beta‐blockers.
We included all participants without heart failure after myocardial infarction, irrespective of age, sex, type of beta‐blocker used, and type of control group intervention received (placebo or no intervention). However, because methods used to exclude heart failure varied and were likely insufficient, we cannot exclude the possibility that any benefits could have been driven by such participants (Bhatt 2017; Biglane 2017; Kotecha 2017). We found no to moderate signs of statistical heterogeneity, which indicates that pooling of these diverse participants and interventions was appropriate.
All trials except one included participants younger than 75 years of age. All patients received usual care, but 24 of 25 trials were from the pre‐reperfusion era (published from 1974 to 1999) and only one trial was from the reperfusion era (published in 2018). Therefore, we are dealing with a select group of participants, which potentially limits the generalisability of our findings to present‐day patients (please see later).
We found no data on effects of beta‐blockers versus placebo or no intervention on serious adverse events according to ICH‐GCP or quality of life. Therefore, effects of beta‐blockers on serious adverse events and quality of life remain unclear.
Quality of the evidence
We assessed the certainty of evidence for results of each outcome using GRADE. Our GRADE assessment generally showed that evidence was of moderate to low certainty for all outcomes (see Table 1).
Risk of systematic error ('bias')
We found no trials and no outcome results at low risk of bias. All included trials were at high risk of bias due to unclear or high risk in several bias domains (see Figure 3). Therefore, the risk that our results may overestimate the beneficial effects and underestimate the harmful effects of beta‐blockers is high (Gluud 2006; Hrobjartsson 2012; Hrobjartsson 2013; Hrobjartsson 2014; Hrobjartsson 2014a; Kjaergard 2001; Moher 1998; Savovic 2012; Savovic 2018; Schulz 1995; Wood 2008). Hence, we downgraded all outcomes by one level due to risk of bias.
Imprecision
We further downgraded angina by one level due to risk of imprecision because the optimal information size (OIS) was not reached, absolute and relative 98% confidence intervals (CIs) showed both appreciable benefit and harm, and sample size was small. However, we downgraded none of the other outcomes for imprecision, although the OIS was not reached, large numbers of events were reported, sample sizes were large, and 97.5% or 98% CIs were fairly narrow.
Inconsistency
We assessed statistical heterogeneity in our primary analyses to be of no to moderate importance. We downgraded major cardiovascular events (MACE) and cardiovascular mortality by one level due to risk of inconsistency based on a moderate I² (30% to 60%) and a small P value (P < 0.05) when the statistical test for heterogeneity was used for assessment.
Indirectness
We downgraded no outcomes for indirectness.
Publication bias
Funnel plots assessing all‐cause mortality, MACE, and myocardial infarction at maximum follow‐up showed some signs of asymmetry. However, we detected no signs of small‐study effect when we used the Harbord test or the Egger test (Harbord 2006). Hence, there was no strong suspicion of small‐study or publication bias, and we downgraded none of the outcomes.
Potential biases in the review process
Strengths
Our review has several strengths. None of the review authors had any conflicts of interests. We conducted this review according to the methods outlined in the Cochrane Handbook for Systematic Reviews of Interventions and by Jakobsen and colleagues (Higgins 2011; Jakobsen 2014). We followed our peer‐reviewed protocol, which was published before the literature search began (Nielsen 2017), and we described a few exceptions under Differences between protocol and review. We included trials regardless of language of publication and whether they reported data on the outcomes we had planned to assess. Two independent review authors double‐extracted data, minimising the risk of inaccurate data extraction; we assessed risk of bias in all trials according to the Cochrane Handbook for Systematic Reviews of Interventions and risk of industry influence according to Lundh 2017. We contacted all relevant trial authors if we needed additional information. However, only one trial author replied and provided us with relevant information on our pre‐defined outcome measures. We included more participants than were included in any previous systematic review on this topic, which gives us increased power and precision to detect any significant differences between intervention and control groups. We tested the robustness of our results by using GRADE to assess the certainty of evidence and sensitivity analyses (best‐worst and worst‐best) to test the potential impact of incomplete outcome data bias. Hence, this review considered both risks of random errors and risks of systematic errors, which adds robustness to our results and conclusions.
Limitations
Our systematic review has several limitations. Our findings, interpretations, and conclusions are affected by the quality and quantity of the trials included in this review.
Furthermore, by including only randomised clinical trials and ignoring harms assessed in quasi‐randomised studies and observational studies, we run the risk of focusing overly on potential benefits at the cost of overlooking late and rare harms.
Risk of bias assessment
Our risk of bias assessment shows that all trials were at high risk of bias. Therefore, it is highly probable that our review results are also biased (i.e. there is great risk that our results overestimate benefit and underestimate harms) (Gluud 2006; Hrobjartsson 2012; Hrobjartsson 2013; Hrobjartsson 2014; Hrobjartsson 2014a; Kjaergard 2001; Moher 1998; Savovic 2012; Savovic 2012a; Savovic 2018; Schulz 1995; Wood 2008). This is the primary limitation of our review.
Incomplete outcome data
Incomplete outcome data were at low risk of bias in eight trials. Six trials did not properly deal with incomplete outcome data and were at high risk of bias. In the remaining 11 trials, incomplete outcome data were insufficiently described; we therefore judged these trials to be at unclear risk of bias. However, our best‐worst and worst‐best analyses show that risk of incomplete outcome data bias was high when major cardiovascular mortality, myocardial infarction, and angina were assessed.
Ahlmark 1976 included 393 participants in the original round of randomisation but followed up on only 162 of these participants and included them in one of two groups. Participants were excluded from the trial if, after randomisation, they were found not to have acute infarction, to have died in the hospital, to not belong to the hospital's catchment area, or to have persisting contraindications to beta‐blockade. This gives rise to high incomplete outcome data bias. Given that a large proportion of trials were performed more than 30 years ago, one might argue that trialists may not have properly approached or reported incomplete outcome data, and our best‐worst and worst‐best sensitivity analyses might highly underestimate the potential impact of missing data because we used available trial data even if the number of participants included in the assessment was unclear based on the publication. Incomplete outcome data bias might have greater bias impact than our best‐worst and worst‐best case scenarios show (i.e. the ’true’ difference between actually observed cases and the intention‐to‐treat population might be greater than our data suggest).
Assessed time points
In our protocol, we pre‐defined the time point closest to 12 months (range 6 to 18 months) as our primary assessment time point, and maximum follow‐up as a secondary time point of interest. However, after further consideration, we decided to change our primary assessment time point to maximum follow‐up to achieve more power and precision, and, because we already had conducted a pre‐defined subgroup analysis looking at different follow‐up time points, we did not find it necessary to also assess outcomes at a secondary time point.
Continuous outcomes
We included quality of life and angina on any valid scale. However, none of the included trials reported data on quality of life, and trials provided only limited dichotomous data on angina. This is a great limitation, as we found insufficient information on important subjective patient‐relevant outcomes.
Clinical heterogeneity
Beta‐blockers used in the experimental group and co‐interventions used in different trials differed. Our results show no to moderate signs of statistical heterogeneity; this is a limitation of our review because subsequent transferability into a specific clinical context may be impaired.
Indirectness of evidence
It is important to note that less than 20% of participants included in this review were women, indicating the risk for evidence being indirect. However, when we considered the overall certainty of evidence, we did not find further indications suggesting significant variation in the study population. Hence, we did not find it necessary to decrease certainty for indirectness.
Assessed age range
In all, 17 of 25 studies included only participants younger than 70 years of age, six studies included participants between 70 and 75 years of age, and only one study included participants older than 75 years of age (Capital‐RCT 2018). The limited age range of patients included in this review and absence of data on the elderly, for whom risk‐benefit ratios may differ substantially, are important limitations that ought to be taken under serious consideration when our results are viewed, because we lack any evidence on effects of beta‐blockers for patients older than 75 years of age without heart failure and with preserved ejection fraction. This is especially of concern when it is considered how patients have gotten older in the past few decades, and how the number of older patients and especially of cardiac patients will continue to increase (Rodgers 2019). Therefore, it is of utmost importance to uncover in future clinical and research studies the impact of beta‐blockers for patients older than 75 years of age without heart failure and with preserved ejection fraction after myocardial infarction, to improve outcomes for the older population.
Composite outcome
We included two composite outcomes ‐ 'major cardiovascular events' (MACE) (defined as a composite outcome of cardiovascular mortality and non‐fatal myocardial infarction) and 'serious adverse events' according to ICH‐GCP. A potential limitation when composite outcomes are used is that each component of the composite outcome will not necessarily have similar degrees of severity (Garattini 2016). This might bias results for these outcomes (Garattini 2016). For example, if certain more severe serious adverse events occur in one of the intervention groups and other less severe serious adverse events occur in another intervention group, there is a risk of overlooking actual severity differences between compared groups on these composite outcomes (Garattini 2016). Furthermore, a potential limitation of a composite outcome as 'serious adverse events' is that heterogeneity arises when different events are compared. However, none of the included trials specifically assessed serious adverse events according to the recommendations of ICH‐GCP. Instead, trials reported one specific serious adverse event, which was already included in one of the other outcomes in this review, or a composite of several different events without referring to actual proportions of participants. When MACE was assessed, most trials successfully managed to report both cardiovascular mortality and non‐fatal myocardial infarction. Three trials did not differentiate between non‐fatal and fatal myocardial infarction when reporting data for myocardial infarction (BCSG 1997; E.I.S. 1984; NMS 1981), and two trials reported only cardiovascular mortality or myocardial infarction (LIT 1987; Wilhelmsson 1974). One trial did not specify the definition of myocardial infarction used in the trial, but we contacted the trial author, who clarified that only non‐fatal myocardial infarctions were reported (Capital‐RCT 2018). Hence, when possible, and when there was no risk of double‐counting patients, we calculated MACE as a composite of cardiovascular mortality and non‐fatal myocardial infarction. We believe that the clinical relevance of composite outcomes such as 'major cardiovascular events' and ‘serious adverse events’ and the resulting increased statistical power justify use of these composite outcomes as a primary outcome. However, the interpretative limitations ought to be considered.
External validity
Management of myocardial infarction has been evolving greatly in the past two decades with the introduction of early coronary reperfusion strategies leading to reduced mortality and improved LVEF by immediate restoration of myocardial blood flow and lowering of sympathetic activation following acute myocardial infarction. Since the 1980s, survival from myocardial infarction has improved by approximately 25% in Western countries ‐ an effect attributable not only to the introduction of reperfusion strategies but also to major advancements in medical therapy consisting of antiplatelet therapies, angiotensin‐converting enzyme (ACE) inhibitors/angiotensin‐receptor blockers (ARBs), and statins substantially improving the prognosis for patients following acute myocardial infarction (Noble 2017). Antiplatelets and statins have been shown to reduce the risk of reinfarction by reducing atherothrombosis, and ACE inhibitors and ARBs have been found to prevent adverse ventricular remodelling and development of severe heart failure (Antithrombotic Trialists' Collaboration 2002; Køber 1995; Pfeffer 1997; Steinhubl 2009). Hence, these therapies, which currently constitute standard care for myocardial infarction, have been shown to have the same positive effects as beta‐blockers on pathophysiological consequences following a myocardial infarction (Qamar 2020). However, most of the trials included in this review were conducted between 1974 and 1999 ‐ an era in which the above mentioned therapies were not available or were routinely practiced, and only one trial was from 2018 (Capital‐RCT 2018). Therefore, the findings of this review may not be compatible with the present management of myocardial infarction in the non‐acute phase following acute myocardial infarction. Furthermore, conduct and reporting of trials have immensely improved over the last two decades, which may have influenced the results of this review. This emphasises the importance of the five ongoing trials assessing long‐term effects of beta‐blockers for patients without heart failure and with preserved LVEF following acute myocardial infarction (see Characteristics of ongoing studies).
Definition of included patients
One important limitation of our review is that most of the included trials were conducted in an era in which the definition of heart failure and assessment and differentiation of STEMI/NSTEMI patients and LVEF differed from the current clinical approach. Most trials included a mix of participants with STEMI and NSTEMI, and no trials provided separate results for each type of myocardial infarction, except one trial, which specifically assessed participants with STEMI (Capital‐RCT 2018). Only three trials assessed LVEF and specifically included patients with preserved LVEF greater than 40% (BCSG 1997; Capital‐RCT 2018; Poulsen 1999). Most of the included trials stated that heart failure participants were excluded but provided no further specification on the diagnosis nor on the definition of heart failure. Some trials might have included participants with reduced LVEF but without clinical overt heart failure; other trials might have included participants with clinical overt heart failure but with estimated preserved LVEF, etc. Based on these trials, European Society of Cardiology (ESC) guidelines recommend the use of beta‐blockers for both STEMI and NSTEMI patients with heart failure and LVEF less than 40% with a class I recommendation. However, the newest ESC guidelines (from 2020) highlight the need for studies evaluating the value of long‐term therapy with beta‐blockers for patients with LVEF greater than 40%, and American College of Cardiology Foundation/American Heart Association (ACCF/AHA) guidelines recommend beta‐blockers for patients with both STEMI and NSTEMI following acute myocardial infarction.
In our protocol, we planned to exclude trials specifically randomising participants with post‐myocardial infarction heart failure, defined as clinically overt New York Heart Association (NYHA) Class III/IV heart failure and LVEF less than 40% at the time of discharge from the hospital. Several trials specifically excluded heart failure participants but reported some percentage of patients with heart failure in the baseline table or some percentage of patients discharged on digitalis and diuretics (for a detailed description, see Table 2). We chose to include these trials but decided to perform a post hoc subgroup analysis comparing trials specifically excluding heart failure participants to trials specifically excluding heart failure participants but likely not adhering to this. Testing for subgroup differences showed no evidence of a difference when these two subgroups were compared. However, because most trials did not specifically assess LVEF, the results of this review might be overestimated by potential inclusion of patients with reduced LVEF. Hence, the external validity of our results may refer only to patients post myocardial infarction and without severe heart failure, covering 70% to 80% of the heart failure patient group. Results may not reflect true intervention effects for the last 20% to 30% of patients with some degree of mild heart failure that was not detectable when trials were conducted but that would have been excluded today based on newer diagnostic techniques. Future trials ought to assess the effects of beta‐blockers separately in STEMI and NSTEMI patients while using a clear definition of no heart failure with preserved LVEF greater than 40%.
1. Detailed description of heart failure in the included trials.
| Study name | Definition of heart failure in the exclusion criteria | Digitalis and diuretics at discharge | Number of patients with HF included |
| Ades 1987 | Clinically overt heart failure (raies, third heart sound) | 3 patients on digitalis | NR |
| Ahlmark 1976 | Cardiac decompensation despite adequate therapy | On discharge, 50% in the alprenolol group and 46% in the control group were receiving digitalis, and 26% in the alprenolol group and 31% in the control group were taking diuretics |
NR |
| Ahnve 1980 | Severe heart failure (no further description given) | 15 patients from the experimental group and 11 patients from the control group were on digitalis at discharge or during follow‐up | NR |
| Amsterdam Metoprolol Trial 1983 | Excluded patients with NYHA Class III/IV | NR | NR |
| APSI 1997 | Acute heart failure that required treatment with > 2 drugs of different classes (e.g. diuretics, vasodilators). If the condition disappeared before the twenty‐second day, the patient could be included | No description of patients taking diuretics and digitalis; only nifedipine was allowed | Around 50% of patients had acute heart failure at the time of inclusion in the study |
| Australien & Swedish 1983 | Uncontrolled heart failure (no further description given) | 86/266 from the control group and 81/263 from the experimental group were on digitalis at discharge. 111/266 from the control group and 120/263 from the experimental group were on diuretics at the time of discharge | 160/266 from the control group and 162/263 from the experimental group had left ventricular failure during hospitalisation and before randomisation |
| Baber 1980 | Persistent heart failure (no further description given) | NR | Around 20% of patients in both groups had cardiac failure in the acute phase. Patients with heart failure were withdrawn from the study |
| Barvik 1992 | Clinical signs of congestive heart failure. Included patients with NYHA Class I/II | NR | NR |
| BCSG 1997 | Patients with contraindications to beta‐blockers. However, all patients had LVEF > 40% | NR | NR |
| BHAT 1982 | History of severe congestive heart failure (no further description given) | Around 17% of patients were on diuretics and 12.5% were on digitalis on the time of randomisation | Around 14.5% of patients in each group had congestive heart failure during hospitalisation and before randomisation |
| Capital‐RCT 2018 | Reduced LVEF (LVEF < 40%), symptomatic HF. Included Killip class I/II with preserved ejection fraction | No patients received digitalis or diuretics | NR |
| Curtis 1991 | Patients were excluded if they could not safely undergo exercise testing due to persistent heart failure | NR | NR |
| E.I.S. 1984 | Heart failure (no further description given) | NR | 13% of patients in each group had left heart failure during the acute myocardial infarction, but none were noted to have heart failure at the time of randomisation |
| Julian 1982 | Clinical evidence of heart failure at the 12th post‐infarction day | NR | Around 21.5% in each group had heart failure during the acute phase |
| LIT 1987 | Congestive heart failure (no further description given) | NR | Around 6% in each group had moderate to severe CHF between admission and pre‐entry |
| Mazur 1984 | Contraindications to beta‐blockers. No further definition given. However, Bangalore 2014, which excluded trials with post‐myocardial infarction heart failure or left ventricular systolic dysfunction, included this trial | NR | NR |
| Mazzuero 1987 | Excluded patients treated with digitalis and NYHA III/IV | NR | NR |
| MIS 1975 | Evidence of congestive heart failure at proposed date of entry | NR | Patients with cardiac failure during follow‐up were withdrawn from trial medication |
| NMS 1981 | Uncontrolled cardiac failure. Included Killip class I/II | NR | Around 33% of patients in each group had heart failure at randomisation and before treatment initiation. Heart failure and pulmonary oedema were reasons for withdrawal only if treatment with digitalis and diuretics did not effect satisfactory improvement |
| NPT 1982 | Patients who presented with heart failure on admission or during the initial phase of infarction were included if signs of failure had disappeared at the time of randomisation. Patients with severe heart failure ‐ that is, cardiogenic shock or pulmonary oedema ‐ and patients who still presented with signs of heart failure at the time of randomisation, although treated with digitalis and furosemide 40 to 80 mg/d, were excluded | NR | Around 40% in each group presented with left ventricular failure in the acute phase |
| Olsson 1985 | Severe cardiac failure not responding to conventional treatment with digitalis and diuretic drugs | Around 23.5% in each group were on digitalis at discharge. Around 45% in each group were on diuretics at discharge | NR |
| Poulsen 1999 | Severe uncontrolled congestive heart failure. LVEF < 40%, because it appears that all patients had LVEF > 40% at baseline | NR | NR |
| Schwartz 1992 | Clinically overt heart failure (NYHA III and IV) | NR | NR |
| Taylor 1982 | Cardiac contraindications to beta‐blockade ‐ i.e. radiographic evidence of heart failure (cardiothoracic ratio > 0.50) or pulmonary venous congestion, resting heart rate < 50 beats per minute, or any grade of heart block | NR | NR |
| Wilhelmsson 1974 | Cardiac decompensation despite treatment with optimum doses of digitalis and diuretics | NR | NR |
LVEF: left ventricular ejection fraction.
NR: not reported.
NYHA: New York Heart Association.
Subgroup analyses
We planned a large number of subgroup analyses, and this may significantly increase the risk of type I errors (Jakobsen 2014).
Post hoc analyses
After completing the protocol, we added three important post hoc subgroup analyses to assess differences in effect in:
trials funded by industry compared to non‐industry‐funded trials;
trials that administered beta‐blockers within compared to after seven days following acute myocardial infarction (subacute compared to non‐acute phase); and
trials specifically excluding heart failure participants compared to trials specifically excluding heart failure participants but likely not adhering to this.
Different types of beta‐blockers
We accepted any type of beta‐blocker as an experimental intervention (non‐selective beta‐blockers, selective beta₁‐blockers, and combined non‐selective alpha‐ and beta‐blockers). Theoretically, these different types of beta‐blockers have different effects. We systematically assessed the degree of heterogeneity in all meta‐analyses, and we carefully planned subgroup analyses comparing effects of different types of beta‐blockers. None of the subgroup analyses comparing different types of beta‐blockers at maximum follow‐up showed any evidence of a difference when different types of beta‐blockers were compared. However, it is a potential limitation that the different types of beta‐blockers included might have different effects, and that these different effects might bias our results.
Cluster randomised trials
We did not include cluster randomised trials because such results will never have the same validity as results from individually randomised clinical trials. However, it is a limitation of our review that we did not include cluster randomised trials because if such trials exist at low risk of bias, it would have been important to include these results in our review. Therefore, in future updates of our review, we will include cluster randomised trials.
Agreements and disagreements with other studies or reviews
We have identified several reviews assessing the effects of beta‐blockers versus control in patients without heart failure after myocardial infarction. Only one of these reviews systematically assessed risks of random errors and employed adequate assessments of risks of bias using some of the Cochrane domains (Bangalore 2014); the rest performed no risk of bias assessment ‐ Lewis 1982; Yusuf 1985 ‐ or only a limited risk of bias assessment (Freemantle 1999).
Lewis 1982 assessed patients with myocardial infarction and without heart failure and distinguished between early (within 48 hours) and late intervention. This review showed evidence of a highly beneficial effect of beta‐blockade on all‐cause mortality when late‐entry trials were assessed (risk ratio (RR) 0.74, 95% confidence interval (CI) 0.65 to 0.83; P < 0.00001). Review authors did not control for risks of random error.
Yusuf 1985 assessed effects of beta‐blockers on all‐cause mortality at short‐term and long‐term treatment with beta‐blockers for patients without heart failure. However, two trials in which participants had heart failure were included in the short‐term treatment. This review showed evidence of a beneficial effect of long‐term treatment with beta‐blockers on total mortality and sudden death. Furthermore, a reduction in non‐fatal reinfarction was found at long‐term treatment with beta‐blockers (odds ratio (OR) 0.74, 95% CI 0.66 to 0.83; P < 0.0001). Review authors did not control for risks of random error.
Freemantle 1999 assessed patients who had had a myocardial infarction with or without heart failure for whom treatment with any type of beta‐blocker was started at any stage before or after the myocardial infarction. This review found evidence of a beneficial effect on mortality (OR 0.77, 95% CI 0.69 to 0.85) and reinfarction at long‐term treatment with beta‐blockers. Review authors did not control for risks of random error.
Bangalore 2014 assessed participants with myocardial infarction and without heart failure in the acute and subacute phases of a myocardial infarction. This review distinguished between pre‐reperfusion and reperfusion eras (when > 50% of patients received reperfusion with thrombolytics or with revascularisation or aspirin/statin), and all included trials conducted in the reperfusion era included participants in the acute phase of a myocardial infarction. In the pre‐reperfusion era, beta‐blockers were associated with a statistically significant reduction in mortality (incidence rate ratio (IRR) 0.79, 95% CI 0.70 to 0.89), cardiovascular mortality, and myocardial infarction in post‐myocardial infarction trials. In the reperfusion era, beta‐blockers were associated with no beneficial effect except for myocardial infarction. However, a significant increase in heart failure and drug discontinuation was found in both eras in post‐myocardial infarction trials.
Our present review results, when all‐cause mortality was assessed, are in agreement with those of Lewis 1982, Yusuf 1985, and Freemantle 1999 ‐ all showing that beta‐blockers seem to have a beneficial effect on the risk of all‐cause mortality. The same is seen in Bangalore 2014 for the pre‐reperfusion era, where a beneficial effect on all‐cause mortality was found when post‐myocardial infarction trials were assessed. As only one of the included trials was conducted in the reperfusion era and included participants undergoing percutaneous coronary intervention (PCI), we could not assess the effects of beta‐blockers in the reperfusion era.
No other review assessed our composite outcome 'major cardiovascular events'. However, when we assessed each component separately, our review result was in agreement with those of Yusuf 1985, Freemantle 1999, and Bangalore 2014 ‐ all showing that beta‐blockers seem to have beneficial effects on cardiovascular mortality and myocardial infarction. However, Bangalore 2014 did not find this beneficial effect in the reperfusion era.
Authors' conclusions
Implications for practice.
Beta‐blockers probably reduce risks of all‐cause mortality and myocardial reinfarction compared with placebo or no intervention in patients younger than 75 years of age without heart failure following acute myocardial infarction. Beta‐blockers may further reduce the risks of major cardiovascular events and cardiovascular mortality compared with placebo or no intervention in patients younger than 75 years of age without heart failure following acute myocardial infarction. However, these effects could be driven by patients with unrecognised heart failure. The effects of beta‐blockers on serious adverse events, angina, and quality of life are unclear due to sparse data or no data at all. All trials and outcomes were at high risk of bias, and incomplete outcome data bias alone could account for the effects seen when major cardiovascular events, angina, and myocardial infarction were assessed. The evidence in this review is of moderate to low certainty, and the true result may depart substantially from the results presented here.
Implications for research.
Due to a paradigm shift in the management of acute myocardial infarction, future trials should revisit the beta‐blocker hypothesis in the post‐reperfusion era for patients without heart failure and with preserved LVEF. These trials should particularly focus on assessing serious adverse events according to ICH‐GCP and quality of life. Furthermore, future trials ought to assess effects of beta‐blockers on older patients (e.g. 75 years of age and older) and should use standard high‐quality measures to exclude participants with heart failure before randomisation. With regard to blinding, the effects of beta‐blockers on pulse rate make these trials difficult to blind, and blinded outcome assessors should therefore be widely involved. Newer randomised clinical trials at low risk of bias and at low risk of random error are needed if the benefits and harms of beta‐blockers in patients without heart failure following acute myocardial infarction are to be assessed properly. Such trials ought to be designed according to the SPIRIT statement and reported according to the CONSORT statement (Chan 2013; Schulz 2010).
History
Protocol first published: Issue 2, 2017
Acknowledgements
We thank Managing Editor Dimitrinka Nikolova for her assistance in translating Russian trials.
We thank Dr. Kristian Thygesen for his expertise and contribution to clarification of the definition of the non‐acute phase following acute myocardial infarction.
We are grateful for the input of peer reviewers Jeffrey Goldberger and Mark Y Chan.
Appendices
Appendix 1. Search strategies
CENTRAL
#1 MeSH descriptor: [Adrenergic beta‐Antagonists] explode all trees
#2 betablock*
#3 beta‐block*
#4 b‐block*
#5 (beta near/3 (antagonist* or receptor* or adrenergic* or block*))
#6 (beta‐adrenoreceptor near/3 block*)
#7 beta‐adrenergic*
#8 beta‐antagonist*
#9 (beta‐receptor adj3 block*)
#10 acebutolol
#11 alprenolol
#12 atenolol
#13 betaxolol
#14 bisoprolol
#15 brevibloc
#16 bupranolol
#17 butoxamine
#18 carteolol
#19 cartrol
#20 carvedilol
#21 celiprolol
#22 coreg
#23 corgard
#24 dihydroalprenolol
#25 esmolol
#26 inderal
#27 inderide
#28 innopran
#29 iodocyanopindolol
#30 kerlone
#31 labetalol
#32 levatol
#33 levobunolol
#34 lopressor
#35 metipranolol
#36 metoprolol
#37 nadolol
#38 nebivolol
#39 normodyne
#40 oxprenolol
#41 penbutolol
#42 pindolol
#43 practolol
#44 propranolol
#45 sectral
#46 sotalol
#47 tenoretic
#48 tenormin
#49 tertatolol
#50 timolol
#51 toprol
#52 trandate
#53 visken
#54 zebeta
#55 ziac
#56 #1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 or #9 or #10 or #11 or #12 or #13 or #14 or #15 or #16 or #17 or #18 or #19 or #20 or #21 or #22 or #23 or #24 or #25 or #26 or #27 or #28 or #29 or #30 or #31 or #32 or #33 or #34 or #35 or #36 or #37 or #38 or #39 or #40 or #41 or #42 or #43 or #44 or #45 or #46 or #47 or #48 or #49 or #50 or #51 or #52 or #53 or #54 or #55
#57 MeSH descriptor: [Myocardial Infarction] explode all trees
#58 (myocardial near/2 infarct*)
#59 heart attack*
#60 heart infarct*
#61 #57 or #58 or #59 or #60
#62 #56 and #61
MEDLINE Ovid
1. exp Adrenergic beta‐Antagonists/
2. betablock*.tw.
3. beta‐block*.tw.
4. b‐block*.tw.
5. (beta adj3 (antagonist* or receptor* or adrenergic* or block*)).tw.
6. (beta‐adrenoreceptor adj3 block*).tw.
7. beta‐adrenergic*.tw.
8. beta‐antagonist*.tw.
9. (beta‐receptor adj3 block*).tw.
10. acebutolol.tw.
11. alprenolol.tw.
12. atenolol.tw.
13. betaxolol.tw.
14. bisoprolol.tw.
15. brevibloc.tw.
16. bupranolol.tw.
17. butoxamine.tw.
18. carteolol.tw.
19. cartrol.tw.
20. carvedilol.tw.
21. celiprolol.tw.
22. coreg.tw.
23. corgard.tw.
24. dihydroalprenolol.tw.
25. esmolol.tw.
26. inderal.tw.
27. inderide.tw.
28. innopran.tw.
29. iodocyanopindolol.tw.
30. kerlone.tw.
31. labetalol.tw.
32. levatol.tw.
33. levobunolol.tw.
34. lopressor.tw.
35. metipranolol.tw.
36. metoprolol.tw.
37. nadolol.tw.
38. nebivolol.tw.
39. normodyne.tw.
40. oxprenolol.tw.
41. penbutolol.tw.
42. pindolol.tw.
43. practolol.tw.
44. propranolol.tw.
45. sectral.tw.
46. sotalol.tw.
47. tenoretic.tw.
48. tenormin.tw.
49. tertatolol.tw.
50. timolol.tw.
51. toprol.tw.
52. trandate.tw.
53. visken.tw.
54. zebeta.tw.
55. ziac.tw.
56. 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 or 14 or 15 or 16 or 17 or 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 or 26 or 27 or 28 or 29 or 30 or 31 or 32 or 33 or 34 or 35 or 36 or 37 or 38 or 39 or 40 or 41 or 42 or 43 or 44 or 45 or 46 or 47 or 48 or 49 or 50 or 51 or 52 or 53 or 54 or 55
57. exp Myocardial Infarction/
58. (myocardial adj2 infarct$).tw.
59. heart attack$.tw.
60. heart infarct*.tw.
61. 57 or 58 or 59 or 60
62. 56 and 61
63. randomized controlled trial.pt.
64. controlled clinical trial.pt.
65. randomized.ab.
66. placebo.ab.
67. clinical trials as topic.sh.
68. randomly.ab.
69. trial.ti.
70. 63 or 64 or 65 or 66 or 67 or 68 or 69
71. exp animals/ not humans.sh.
72. 70 not 71
73. 62 and 72
Embase Ovid
1. exp beta adrenergic receptor blocking agent/
2. betablock*.tw.
3. beta‐block*.tw.
4. b‐block*.tw.
5. (beta adj3 (antagonist* or receptor* or adrenergic* or block*)).tw.
6. (beta‐adrenoreceptor adj3 block*).tw.
7. beta‐adrenergic*.tw.
8. beta‐antagonist*.tw.
9. (beta‐receptor adj3 block*).tw.
10. acebutolol.tw.
11. alprenolol.tw.
12. atenolol.tw.
13. betaxolol.tw.
14. bisoprolol.tw.
15. brevibloc.tw.
16. bupranolol.tw.
17. butoxamine.tw.
18. carteolol.tw.
19. cartrol.tw.
20. carvedilol.tw.
21. celiprolol.tw.
22. coreg.tw.
23. corgard.tw.
24. dihydroalprenolol.tw.
25. esmolol.tw.
26. inderal.tw.
27. inderide.tw.
28. innopran.tw.
29. iodocyanopindolol.tw.
30. kerlone.tw.
31. labetalol.tw.
32. levatol.tw.
33. levobunolol.tw.
34. lopressor.tw.
35. metipranolol.tw.
36. metoprolol.tw.
37. nadolol.tw.
38. nebivolol.tw.
39. normodyne.tw.
40. oxprenolol.tw.
41. penbutolol.tw.
42. pindolol.tw.
43. practolol.tw.
44. propranolol.tw.
45. sectral.tw.
46. sotalol.tw.
47. tenoretic.tw.
48. tenormin.tw.
49. tertatolol.tw.
50. timolol.tw.
51. toprol.tw.
52. trandate.tw.
53. visken.tw.
54. zebeta.tw.
55. ziac.tw.
56. 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 or 14 or 15 or 16 or 17 or 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 or 26 or 27 or 28 or 29 or 30 or 31 or 32 or 33 or 34 or 35 or 36 or 37 or 38 or 39 or 40 or 41 or 42 or 43 or 44 or 45 or 46 or 47 or 48 or 49 or 50 or 51 or 52 or 53 or 54 or 55
57. exp heart infarction/
58. (myocardial adj2 infarct*).tw.
59. heart attack*.tw.
60. heart infarct*.tw.
61. 57 or 58 or 59 or 60
62. 56 and 61
63. random$.tw.
64. factorial$.tw.
65. crossover$.tw.
66. cross over$.tw.
67. cross‐over$.tw.
68. placebo$.tw.
69. (doubl$ adj blind$).tw.
70. (singl$ adj blind$).tw.
71. assign$.tw.
72. allocat$.tw.
73. volunteer$.tw.
74. crossover procedure/
75. double blind procedure/
76. randomized controlled trial/
77. single blind procedure/
78. 63 or 64 or 65 or 66 or 67 or 68 or 69 or 70 or 71 or 72 or 73 or 74 or 75 or 76 or 77
79. (animal/ or nonhuman/) not human/
80. 78 not 79
81. 62 and 80
LILACS
betablock$ or beta‐block$ or b‐block$ or "beta block$" or beta‐adrenergic$ or beta‐antagonist$ or "beta‐adrenoreceptor block$" or "beta antagnoist$" or "beta receptor$" or "beta adrenergic$" [Words] or acebutolol or alprenolol or atenolol or betaxolol or bisoprolol or brevibloc or bupranolol or butoxamine or carteolol or cartrol or carvedilol or celiprolol or coreg or corgard or dihydroalprenolol or esmolol or inderal or inderide or innopran or iodocyanopindolol or kerlone or labetalol or levatol or levobunolol or lopressor or metipranolol or metoprolol or nadolol or nebivolol or normodyne or oxprenolol or penbutolol or pindolol or practolol or propranolol or sectral or sotalol or tenoretic or tenormin or tertatolol or timolol or toprol or trandate or visken or zebeta or ziac [Words] and "myocardial infarct$" or "heart attack$" or "heart infarct$" [Words]
SCI‐EXPANDED and BIOSIS
# 18 #17 AND #16
# 17 TS=(random* or blind* or allocat* or assign* or trial* or placebo* or crossover* or cross‐over*)
# 16 #15 AND #11
# 15 #14 OR #13 OR #12
# 14 TS=heart infarct*
# 13 TS=heart attack*
# 12 TS=(myocardial near/2 infarct*)
# 11 #10 OR #9 OR #8 OR #7 OR #6 OR #5 OR #4 OR #3 OR #2 OR #1
# 10 TS=(acebutolol or alprenolol or atenolol or betaxolol or bisoprolol or brevibloc or bupranolol or butoxamine or carteolol or cartrol or carvedilol or celiprolol or coreg or corgard or dihydroalprenolol or esmolol or inderal or inderide or innopran or iodocyanopindolol or kerlone or labetalol or levatol or levobunolol or lopressor or metipranolol or metoprolol or nadolol or nebivolol or normodyne or oxprenolol or penbutolol or pindolol or practolol or propranolol or sectral or sotalol or tenoretic or tenormin or tertatolol or timolol or toprol or trandate or visken or zebeta or ziac)
# 9 TS=(beta‐receptor near/3 block*)
# 8 TS=beta‐antagonist*
# 7 TS=beta‐adrenergic*
# 6 TS=(beta‐adrenoreceptor near/3 block*)
# 5 TS=(beta near/3 (antagonist* or receptor* or adrenergic* or block*))
# 4 TS=b‐block*
# 3 TS=beta‐block*
# 2 TS= betablock*
# 1 TS=Adrenergic beta‐Antagonists
US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (www.clinicaltrials.gov);World Health Organization International Clinical Trials Registry Platform (WHO ICTRP) (apps.who.int/trialsearch); European Medicines Agency (EMA) (www.ema.europa.eu/ema/); the Food and Drug Administration (FDA) (www.fda.gov); Turning Research Into Practice (TRIP); Google Scholar; and SciSearch:
# Beta‐blockers
# Betablockers
# Myocardial infarction
# Interventional
# Randomised controlled trial
# Non‐acute myocardial infarction
Appendix 2. Details on assessment of risk of bias
We will classify each trial according to the domains below for each outcome.
Random sequence generation
Low risk: if sequence generation is achieved using computer random number generator or a random numbers table. Drawing lots, tossing a coin, shuffling cards, and throwing dice are also considered adequate if performed by an independent adjudicator
Unclear risk: if the method of randomisation is not specified
High risk: if the allocation sequence is not randomised or is only quasi‐randomised
Allocation sequence concealment
Low risk: if allocation of participants is performed by a central independent unit, an on‐site locked computer, identical‐looking numbered sealed opaque envelopes, or drug bottles or containers prepared by an independent investigator. There must be no risk of the investigator knowing the sequence
Unclear risk: if the trial is classified as randomised but the allocation concealment process is not described
High risk: if the allocation sequence is known to the investigators who assigned participants
Blinding of participants and personnel
Low risk: if participants and the personnel are blinded to treatment allocation and this is described
Unclear risk: if the procedure of blinding is insufficiently described or is not described at all
High risk: if blinding of participants and personnel is not performed
Blinding of outcome assessment
Low risk: if trial investigators performing outcome assessments, analyses, and calculations are blinded to the intervention
Unclear risk: if the procedure of blinding is insufficiently described or is not described at all
High risk: if blinding of outcome assessment is not performed
Incomplete outcome data
Low risk: (1) there are no dropouts or withdrawals for all outcomes, or (2) numbers and reasons for withdrawals and dropouts for all outcomes are clearly stated and can be described as similar in both groups, and the trial handles missing data appropriately in intention‐to‐treat analysis using proper methods (e.g. multiple imputations). As a general rule, the trial is judged as having low risk of bias due to incomplete outcome data if the number of dropouts is less than 5%. However, the 5% cutoff is not definitive
Unclear risk: numbers and reasons for withdrawals and dropouts are not clearly stated
High risk: the pattern of dropouts can be described as different in the two intervention groups, or the trial uses improper methods in dealing with missing data (e.g. last observation carried forward).
Selective outcome reporting
Low risk: a protocol is published before or at the time the trial is begun and the outcomes called for in the protocol are reported on. If there is no protocol or the protocol is published after the trial has begun, reporting of the primary outcomes will grant the trial a grade of low risk of bias
Unclear risk: if there is no protocol and the primary outcomes are not reported on
High risk: if the outcomes that are called on in a protocol are not reported on
Other bias risk
Low risk of bias: the trial appears to be free of other components (e.g. academic bias, for‐profit bias) that could put it at risk of bias
Unclear risk of bias: the trial may or may not be free of other components that could put it at risk of bias
High risk of bias: there are other factors in the trial that could put it at risk of bias (e.g. authors have conducted trials on the same topic, for‐profit bias)
Data and analyses
Comparison 1. All‐cause mortality at maximum follow‐up.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 All‐cause mortality | 21 | 22085 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.74, 0.89] |
| 1.2 All‐cause mortality ‐ Type of beta‐blocker | 21 | 22085 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.74, 0.89] |
| 1.2.1 Alprenolol | 2 | 427 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.42, 1.45] |
| 1.2.2 Acebutolol | 1 | 586 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.64, 1.07] |
| 1.2.3 Atenolol | 1 | 757 | Risk Ratio (M‐H, Random, 95% CI) | 0.45 [0.19, 1.09] |
| 1.2.4 Carvedilol | 1 | 794 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.48, 1.51] |
| 1.2.5 Metoprolol | 5 | 3461 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.67, 1.06] |
| 1.2.6 Oxprenolol | 3 | 3817 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.49, 1.45] |
| 1.2.7 Pindolol | 1 | 529 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.67, 1.40] |
| 1.2.8 Practolol | 1 | 3053 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.46, 0.90] |
| 1.2.9 Propranolol | 4 | 5321 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.63, 0.90] |
| 1.2.10 Sotalol | 1 | 1456 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.58, 1.17] |
| 1.2.11 Timolol | 1 | 1884 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.69, 0.94] |
| 1.3 All‐cause mortality ‐ Different follow‐up | 21 | 22085 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.74, 0.89] |
| 1.3.1 Between 6 months and 12 months | 6 | 7607 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.67, 1.13] |
| 1.3.2 Between 1 year and 3 years | 12 | 11214 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.67, 0.90] |
| 1.3.3 3 years or longer | 3 | 3264 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.72, 0.92] |
| 1.4 All‐cause mortality ‐ Registration status | 21 | 22085 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.74, 0.89] |
| 1.4.1 Post‐registration | 1 | 3837 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.60, 0.91] |
| 1.4.2 No registration | 19 | 17454 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.74, 0.91] |
| 1.4.3 Pre‐registration | 1 | 794 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.48, 1.51] |
| 1.5 All‐cause mortality ‐ Industry vs non‐industry funding | 21 | 22085 | Odds Ratio (M‐H, Random, 95% CI) | 0.78 [0.70, 0.87] |
| 1.5.1 Industry‐funded trials or unknown funding | 17 | 20327 | Odds Ratio (M‐H, Random, 95% CI) | 0.78 [0.68, 0.89] |
| 1.5.2 Non‐industry‐funded trials | 4 | 1758 | Odds Ratio (M‐H, Random, 95% CI) | 0.78 [0.59, 1.02] |
| 1.6 All‐cause mortality ‐ Subacute vs non‐acute phase | 21 | 22085 | Odds Ratio (M‐H, Random, 95% CI) | 0.78 [0.70, 0.87] |
| 1.6.1 Beta‐blockers administered within 7 days following acute myocardial infarction (subacute phase) | 3 | 1431 | Odds Ratio (M‐H, Random, 95% CI) | 0.75 [0.50, 1.11] |
| 1.6.2 Beta‐blockers administered after 7 days following acute myocardial infarction (non‐acute phase) | 18 | 20654 | Odds Ratio (M‐H, Random, 95% CI) | 0.78 [0.69, 0.88] |
| 1.7 All‐cause mortality ‐ Heart failure vs no heart failure | 21 | 22085 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.74, 0.89] |
| 1.7.1 Trials specifically excluding heart failure participants | 11 | 8273 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.60, 0.85] |
| 1.7.2 Trials specifically excluding heart failure participants but likely not adhering to this | 10 | 13812 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.77, 0.93] |
| 1.8 All‐cause mortality ‐ 'Best‐worst case scenario' | 21 | 22309 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.59, 0.83] |
| 1.9 All‐cause mortality ‐ 'Worst‐best case scenario' | 21 | 22309 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.72, 1.09] |
1.1. Analysis.

Comparison 1: All‐cause mortality at maximum follow‐up, Outcome 1: All‐cause mortality
Comparison 2. Major adverse cardiovascular events (MACE) at maximum follow‐up.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 MACE (major cardiovascular events) | 15 | 14994 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.63, 0.81] |
| 2.2 MACE (major cardiovascular events) ‐ Type of beta‐blocker | 15 | 14994 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.69, 0.85] |
| 2.2.1 Alprenolol | 1 | 162 | Risk Ratio (M‐H, Random, 95% CI) | 0.36 [0.12, 1.04] |
| 2.2.2 Acebutolol | 1 | 607 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.22, 0.79] |
| 2.2.3 Carvedilol | 1 | 794 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.42, 1.82] |
| 2.2.4 Metoprolol | 3 | 996 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.48, 0.90] |
| 2.2.5 Oxprenolol | 2 | 2076 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.36, 1.22] |
| 2.2.6 Pindolol | 1 | 529 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.63, 1.40] |
| 2.2.7 Practolol | 1 | 3053 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.57, 1.03] |
| 2.2.8 Propranolol | 4 | 5321 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.65, 0.94] |
| 2.2.9 Sotalol | 1 | 1456 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.58, 1.18] |
| 2.3 MACE (major cardiovascular events) ‐ Different follow‐up | 15 | 14994 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.69, 0.85] |
| 2.3.1 Between 6 months and 12 months | 5 | 3431 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.67, 1.06] |
| 2.3.2 Between 1 year and 3 years | 8 | 10162 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.64, 0.86] |
| 2.3.3 3 years or longer | 2 | 1401 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.28, 1.24] |
| 2.4 MACE (major cardiovascular events) ‐ Registration status | 15 | 14994 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.69, 0.85] |
| 2.4.1 Post‐registration | 1 | 3837 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.60, 0.93] |
| 2.4.2 No registration | 13 | 10363 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.66, 0.86] |
| 2.4.3 Pre‐registration | 1 | 794 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.42, 1.82] |
| 2.5 MACE (major cardiovascular events) ‐ Industry vs non‐industry funding | 15 | 14994 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.63, 0.77] |
| 2.5.1 Industry‐funded trials or unknown funding | 12 | 13292 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.64, 0.78] |
| 2.5.2 Non‐industry‐funded trials | 3 | 1702 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.40, 0.81] |
| 2.6 MACE (major cardiovascular events) ‐ Subacute vs non‐acute phase | 15 | 14994 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.63, 0.77] |
| 2.6.1 Beta‐blockers administered within 7 days following acute myocardial infarction (subacute phase) | 2 | 1354 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.47, 1.02] |
| 2.6.2 Beta‐blockers administered after 7 days following acute myocardial infarction (non‐acute phase) | 13 | 13640 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.62, 0.77] |
| 2.7 MACE (major cardiovascular events) ‐ Heart failure vs no heart failure | 15 | 14994 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.69, 0.85] |
| 2.7.1 Trials specifically excluding heart failure participants | 8 | 7174 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.62, 0.86] |
| 2.7.2 Trials specifically excluding heart failure participants but likely not adhering to this | 7 | 7820 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.67, 0.91] |
| 2.8 MACE (major cardiovascular events) ‐ 'Best‐worst case scenario' | 15 | 15225 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.50, 0.81] |
| 2.9 MACE (major cardiovascular events) ‐ 'Worst‐best case scenario' | 15 | 15225 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.68, 1.25] |
2.1. Analysis.

Comparison 2: Major adverse cardiovascular events (MACE) at maximum follow‐up, Outcome 1: MACE (major cardiovascular events)
Comparison 3. Angina pectoris at maximum follow‐up.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Angina pectoris on a dichotomous scale | 5 | 7115 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.96, 1.11] |
| 3.2 Angina pectoris on a dichotomous scale ‐ 'Best‐worst case scenario' | 5 | 7372 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.83, 0.96] |
| 3.3 Angina pectoris on a dichotomous scale ‐ 'Worst‐best case scenario' | 5 | 7372 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.19 [1.11, 1.27] |
3.1. Analysis.

Comparison 3: Angina pectoris at maximum follow‐up, Outcome 1: Angina pectoris on a dichotomous scale
Comparison 4. Cardiovascular mortality at maximum follow‐up.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Cardiovascular mortality | 19 | 21763 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.63, 0.85] |
| 4.2 Cardiovascular mortality ‐ Type of beta‐blocker | 19 | 21763 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.63, 0.85] |
| 4.2.1 Atenolol | 1 | 757 | Risk Ratio (M‐H, Random, 95% CI) | 0.45 [0.19, 1.09] |
| 4.2.2 Acebutolol | 1 | 607 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.22, 0.79] |
| 4.2.3 Alprenolol | 1 | 162 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.02, 1.15] |
| 4.2.4 Carvedilol | 1 | 794 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.37, 3.96] |
| 4.2.5 Metoprolol | 4 | 3383 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.67, 1.10] |
| 4.2.6 Oxprenolol | 3 | 3817 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.45, 1.46] |
| 4.2.7 Pindolol | 1 | 529 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.63, 1.40] |
| 4.2.8 Practolol | 1 | 3053 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.43, 0.87] |
| 4.2.9 Propranolol | 4 | 5321 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.56, 0.97] |
| 4.2.10 Sotalol | 1 | 1456 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.58, 1.18] |
| 4.2.11 Timolol | 1 | 1884 | Risk Ratio (M‐H, Random, 95% CI) | 0.58 [0.45, 0.75] |
| 4.3 Cardiovascular mortality ‐ Different follow‐up | 19 | 21763 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.63, 0.85] |
| 4.3.1 Between 6 months and 12 months | 7 | 5778 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.60, 1.08] |
| 4.3.2 Between 1 year and 3 years | 11 | 15191 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.57, 0.83] |
| 4.3.3 3 years or longer | 1 | 794 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.37, 3.96] |
| 4.4 Cardiovascular mortality ‐ Registration status | 19 | 21763 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.63, 0.85] |
| 4.4.1 Post‐registration | 1 | 3837 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.60, 0.93] |
| 4.4.2 No registration | 17 | 17132 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.60, 0.86] |
| 4.4.3 Pre‐registration | 1 | 794 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.37, 3.96] |
| 4.5 Cardiovascular mortality ‐ Industry vs non‐industry funding | 19 | 21763 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.64, 0.80] |
| 4.5.1 Industry‐funded trials or unknown funding | 16 | 20061 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.65, 0.81] |
| 4.5.2 Non‐industry‐funded trials | 3 | 1702 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.36, 0.85] |
| 4.6 Cardiovascular mortality ‐ Subacute vs non‐acute phase | 19 | 21763 | Odds Ratio (M‐H, Random, 95% CI) | 0.71 [0.60, 0.84] |
| 4.6.1 Beta‐blockers administered within 7 days following acute myocardial infarction (subacute phase) | 2 | 1354 | Odds Ratio (M‐H, Random, 95% CI) | 0.70 [0.40, 1.22] |
| 4.6.2 Beta‐blockers administered after 7 days following acute myocardial infarction (non‐acute phase) | 17 | 20409 | Odds Ratio (M‐H, Random, 95% CI) | 0.71 [0.59, 0.84] |
| 4.7 Cardiovascular mortality ‐ Heart failure vs no heart failure | 19 | 21763 | Risk Ratio (M‐H, Random, 95% CI) | 0.73 [0.63, 0.85] |
| 4.7.1 Trials specifically excluding heart failure participants | 9 | 7930 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.48, 0.79] |
| 4.7.2 Trials specifically excluding heart failure participants but likely not adhering to this | 10 | 13833 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.66, 0.96] |
| 4.8 Cardiovascular mortality ‐ 'Best‐worst case scenario' | 19 | 21770 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.60, 0.83] |
| 4.9 Cardiovascular mortality ‐ 'Worst‐best case scenario' | 19 | 21770 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.63, 0.90] |
4.1. Analysis.

Comparison 4: Cardiovascular mortality at maximum follow‐up, Outcome 1: Cardiovascular mortality
Comparison 5. Myocardial reinfarction at maximum follow‐up.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Myocardial infarction | 19 | 19606 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.69, 0.84] |
| 5.2 Myocardial infarction ‐ Type of beta‐blocker | 19 | 19606 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.68, 0.83] |
| 5.2.1 Alprenolol | 2 | 392 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.40, 1.15] |
| 5.2.2 Acebutolol | 1 | 607 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.36, 2.02] |
| 5.2.3 Atenolol | 1 | 757 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.44, 2.12] |
| 5.2.4 Carvedilol | 1 | 794 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.27, 1.85] |
| 5.2.5 Metoprolol | 3 | 996 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.45, 0.97] |
| 5.2.6 Oxprenolol | 3 | 3817 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.62, 0.99] |
| 5.2.7 Pindolol | 1 | 529 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.61, 1.38] |
| 5.2.8 Practolol | 1 | 3053 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.57, 1.03] |
| 5.2.9 Propranolol | 4 | 5321 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.69, 1.08] |
| 5.2.10 Sotalol | 1 | 1456 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.36, 0.96] |
| 5.2.11 Timolol | 1 | 1884 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.48, 0.80] |
| 5.3 Myocardial infarction ‐ Different follow‐up | 19 | 19606 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.68, 0.83] |
| 5.3.1 Between 6 months and 12 months | 7 | 5779 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.66, 1.03] |
| 5.3.2 Between 1 year and 3 years | 11 | 13033 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.65, 0.82] |
| 5.3.3 3 years or longer | 1 | 794 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.27, 1.85] |
| 5.4 Myocardial infarction ‐ Registration status | 19 | 19606 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.68, 0.83] |
| 5.4.1 Post‐registration | 1 | 3837 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.64, 1.12] |
| 5.4.2 No registration | 17 | 14975 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.66, 0.83] |
| 5.4.3 Pre‐registration | 1 | 794 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.27, 1.85] |
| 5.5 Myocardial infarction ‐ Industry vs non‐industry funding | 19 | 19606 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.66, 0.83] |
| 5.5.1 Industry‐funded trials or unknown funding | 16 | 17904 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.67, 0.84] |
| 5.5.2 Non‐industry‐funded trials | 3 | 1702 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.39, 0.97] |
| 5.6 Myocardial infarction ‐ Subacute vs non‐acute phase | 19 | 19606 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.66, 0.83] |
| 5.6.1 Beta‐blockers administered within 7 days following acute myocardial infarction (subacute phase) | 2 | 1354 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.51, 1.33] |
| 5.6.2 Beta‐blockers administered after 7 days following acute myocardial infarction (non‐acute phase) | 17 | 18252 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.66, 0.83] |
| 5.7 Myocardial reinfarction ‐ Heart failure vs no heart failure | 19 | 19606 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.68, 0.83] |
| 5.7.1 Trials specifically excluding heart failure participants | 10 | 8161 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.62, 0.85] |
| 5.7.2 Trials specifically excluding heart failure participants but likely not adhering to this | 9 | 11445 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.67, 0.89] |
| 5.8 Myocardial infarction ‐ 'Best‐worst case scenario' | 19 | 19837 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.54, 0.84] |
| 5.9 Myocardial infarction ‐ 'Worst‐best case scenario' | 19 | 19837 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.71, 1.23] |
5.1. Analysis.

Comparison 5: Myocardial reinfarction at maximum follow‐up, Outcome 1: Myocardial infarction
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ades 1987.
| Study characteristics | ||
| Methods | Randomised clinical trial at a single site in the United States | |
| Participants | 36 participants meeting at least 2 of 3 criteria for diagnosis of AMI: myocardial ischaemic pain lasting 30 minutes or longer, an increase in serum creatine kinase levels to twice the normal range with MB isoenzyme present, appearance of new Q waves or evolutionary ST changes, younger than 75 years and diagnosed Male:female = 30:6 Mean age = 53 years Exclusion criteria: age 75 years or older, post‐AMI infarction chest pain in the 4 days before treadmill testing, clinically overt heart failure (raies, third heart sound), severe obstructive lung disease, long‐term beta‐adrenergic blockade for systemic hypertension or exercise‐limiting peripheral vascular disease |
|
| Interventions | Experimental group: metoprolol (100 mg 1 tablet twice daily for a minimum of 5 doses before initial submaximal treadmill test (performed around 8 days after AMI)) Control group: matching placebo Co‐intervention: other anti‐ischaemic medications, such as nitrates or calcium antagonist drugs, were allowed but were kept steady for the duration of the protocol. Nine patients were taking calcium antagonist drugs, 16 nitrates, and 3 digitalis Excluded medication: not described |
|
| Outcomes | Outcomes: exercise parameters, rest haemodynamic variables Time points reported: Day 6 |
|
| Notes | Study author was contacted at Philip.Ades@uvmhealth.org on 20‐06‐2017. No response was received Study was described as a double‐blinded, placebo‐controlled, cross‐over trial. However, It did not report how many participants were included in each group, and no useful data could be extracted from this trial Study was funded by Geigy, Inc., which provided study medication and matching placebo medication |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | This was a double‐blinded, placebo‐controlled, cross‐over trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | This was a double‐blinded, placebo‐controlled, cross‐over trial |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not described |
| Selective reporting (reporting bias) | High risk | No protocol could be found, and mortality and SAEs were not reported |
| Other bias | Low risk | No other biases were found |
Ahlmark 1976.
| Study characteristics | ||
| Methods | Randomised clinical trial at a single site in Sweden. Duration not mentioned | |
| Participants | 162 participants younger than 66 years admitted to the Medical Department of Falu Hospital with diagnosed myocardial infarction were included in the trial Male:female = 142:20 Mean age = 57 years Exclusion criteria: older than 66 years, contraindications to beta‐blockade: cardiac decompensation despite adequate therapy, bradycardia (heart rate < 50 per minute), atrioventricular block I to III (PQ > 0.24 seconds), bronchial asthma, labile insulin‐treated diabetes mellitus, serious mental disease |
|
| Interventions | Experimental group: alprenolol (400 mg/d) (n = 69) Control group: no intervention other than the co‐intervention (n = 93) Co‐intervention: digitalis, diuretics, antiarrhythmics Excluded medication: not described |
|
| Outcomes | Outcomes: death. sudden death, fatal + non‐fatal myocardial reinfarction Time point reported: 24 months |
|
| Notes | Email was not found 393 participants were originally randomised to 2 groups; however, 231 participants were excluded due to death, diagnosis not verified, contraindications, or foreign catchment area. Only 162 participants were followed up. When counting the total number of participants in each group, we considered death participants to be included in the total number of participants. The rest of the excluded participants were accounted for in 'best‐worst and worst‐best' analyses MACE was calculated and reported as a composite of 'non‐fatal reinfarction + sudden death (within 24 hours of symptom onset)' Funding was not described |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described sufficiently, other than "Patients were randomly allocated to the alprenolol group or the control group at the time of admission to hospital" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 124 from the experimental group and 107 from the control group were withdrawn from the study after randomisation |
| Selective reporting (reporting bias) | Unclear risk | No protocol was found; however mortality was reported |
| Other bias | Low risk | No other biases were found |
Ahnve 1980.
| Study characteristics | ||
| Methods | Randomised clinical trial at a single site in Sweden between May 1977 and December 1978 | |
| Participants | 111 participants younger than 70 years of age who had had a recent acute myocardial infarction and were in sinus rhythm when discharged alive from the hospital were included in the trial Male:female = 88:23 Mean age = 60 years Exclusion criteria: complete bundle branch block, severe heart failure, hypotension, bronchial asthma |
|
| Interventions | Experimental group: metoprolol (100 mg b.i.d.) (n = 59) Control group: placebo (n = 52) Co‐intervention: not described Excluded medication: not described |
|
| Outcomes | Outcomes: fatal + non‐fatal myocardial reinfarction and sudden death Time points reported: 6 and 12 months after the acute event |
|
| Notes | Email was not found Mean time from myocardial infarction to randomisation is not reported; however, participants had to be discharged alive from the hospital after acute myocardial infarction to be included. Hence, we assume participants were randomised in the non‐acute phase after myocardial infarction 10 patients died; however, only the number of sudden deaths is reported for each group (3 in metoprolol group, 4 in placebo group). Reported data on the 10 deaths was found in Yusuf 1985, where the trial was included and was reported with the help of personal communication MACE was calculated and was reported as a composite of 'non‐fatal reinfarction + sudden death (presumably arrhythmic or cardiac rupture)' Study was funded by grants from the Swedish National Association against Heart and Chest Diseases and the Hässle Company (pharmaceutical company) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Prior to discharge the patients were stratified and randomised" |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as double‐blinded; however no further information was reported |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Salient clinical data were registered by code on special charts and transferred to punch‐cards for computer analysis. The QTc intervals were measured retrospectively by one of us (S.A.) without knowledge of the patients' clinical data" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be found; however mortality was reported |
| Other bias | Low risk | No other biases were found |
Amsterdam Metoprolol Trial 1983.
| Study characteristics | ||
| Methods | Randomised clinical trial at a single site in Sweden. Duration not mentioned | |
| Participants | 584 participants younger than 70 years of age, NYHA Class I/II, and 3 to 5 weeks after MI were included in the trial Male:female = not mentioned Mean age = not mentioned Exclusion criteria: not mentioned |
|
| Interventions | Experimental group: metoprolol (100 mg b.i.d.) (n = 291) Control group: placebo (n = 293) Co‐intervention: not mentioned Excluded medication: not mentioned |
|
| Outcomes | Outcomes: mortality, non‐fatal myocardial reinfarction, arrhythmias Time point reported: 24 months |
|
| Notes | Email was not found The original study could not be found. Therefore, we used data from Yusuf 1985 (study referred to as 'Manger Cats'), which Bangalore 2014 has referred to as well MACE was calculated and was reported as a composite of 'non‐fatal reinfarction + sudden death (presumably arrhythmic or cardiac rupture)' Funding not described |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be found; however mortality was reported |
| Other bias | Low risk | No other biases were found |
APSI 1997.
| Study characteristics | ||
| Methods | Randomised multi‐centre trial at 44 clinical centres in France between April 1987 and September 1988 | |
| Participants | 607 patients met the following inclusion criteria: (1) had > 2 of the 3 classic signs of AMI, that is, typical chest pain of 11 hours in duration, typical Q waves, and significant release of cardiac enzyme(s); (2) were admitted for this acute event > 2 and < 22 days before; and presented with > 6 of the secondary risk factors of the selection algorithm, including “major” secondary risk factor (i.e. before documented AMI, history of dyspnoea when walking on flat ground, documented atrial fibrillation, ventricular fibrillation, ventricular tachycardia, overt heart failure or sinusal tachycardia during the reference event, recurrent AMI or angina pectoris before the eighth day) Male:female = 443:164 Mean age = 62.9 years Exclusion criteria: heart rate < 45 beats/min; complete auriculoventricular block and acute heart failure that required treatment with > 2 drugs of different classes (e.g. diuretics, vasodilators) (if the condition disappeared before the twenty‐second day, the patient could be included); contraindication to beta‐blocking treatment; non‐cardiac disease with poor prognosis; impossibility to participate; indication for beta‐blocking treatment; age > 75 years; death; malignancy; valvular disease; coma; asthma; chronic broncho‐pneumopathy; Raynaud syndrome; participation in another study; enrolled in APSI before |
|
| Interventions | Experimental group: acebutolol (200 mg twice daily for 12 months) (n = 298) Control group: placebo (n = 309) Co‐intervention: 30% of patients had taken aspirin regularly; one‐half were given oral anticoagulants; one‐third were given diuretics, whereas 10% received digitalis. Finally, 39.3% in the acebutolol group and 37.9% in the placebo group received nifedipine Excluded medication: not described |
|
| Outcomes | Primary: total death (reported at a median of 5 years of follow‐up) Secondary: cardiovascular death (sudden death, fatal + non‐fatal myocardial reinfarction, cardiac failure, stroke or cerebral haemorrhage) (reported at 12 months' follow‐up) Time points reported: median of 5 years and 12 months |
|
| Notes | Study author was contacted on 10‐02‐2017 at jpb@upcl.univ‐lyon1.fr and jean‐pierre.boissel@novadiscovery.com. No response was received Mean time from onset of symptoms to randomisation was 10.5 days In the first publication from 1990, total mortality in the placebo group is 34, and in the second publication from 1997, the same mortality at 1 year (37) was reported in Table 1. However, because 34 deaths were reported in 2 out of 3 publications, this is the number we used MACE was calculated and was reported as a composite of 'non‐fatal reinfarction + cardiovascular mortality' Study was funded by a grant from SPECIA Pharmaceuticals, Paris, France. However, it is said that "the Policy Board monitored the trial results on an ongoing basis. Its voting members were not otherwise involved in the trial and had no connection with the sponsor". Furthermore, the study was initiated by the French Society of Cardiology |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Central randomisation was achieved with a Minitel@, a terminal linked to the telephone system allowing an on‐line remote data entry" |
| Allocation concealment (selection bias) | Low risk | "After editing of the eligibility criteria by the central computer, the treatment number was shown on the screen. Random permutations with blocks of 6 were used for randomisation. Treatment numbers were not sequential" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Tablets of acebutolol and placebo were indistinguishable. A round‐the‐clock telephone service was available for investigators who needed to know the treatment. The code could only be broken in case of life‐threatening conditions, the care of which required to know the treatment given to the patient" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The coordinating and data handling center was in charge of managing the trial, editing collected data, controlling compliance by both investigators and patients, randomising the treatment through an on‐line computerized procedure and analysing the results" Nobody but the voting members of this committee and 4 individuals in the coordinating centre were aware of the interim results. The critical events committee met periodically to classify the causes of death. Its members were not otherwise involved in the trial and were kept blind vis‐a‐vis study treatment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | None of the 607 included and randomised patients were lost to follow‐up at 12 months' follow‐up; however, at maximum follow‐up, which is our primary time point of interest, 15/298 (5.0%) from the acebutolol group and 6/309 (1.9%) from the placebo group were lost to follow‐up |
| Selective reporting (reporting bias) | Unclear risk | No protocol was found; however mortality was reported |
| Other bias | Low risk | No other biases were found |
Australien & Swedish 1983.
| Study characteristics | ||
| Methods | Randomised clinical trial at 5 sites in Sweden and at 2 sites in Australia between February 1978 and January 1980 | |
| Participants | 529 participants, of both sexes, aged up to the end of their 69th year, with clinical diagnosis of acute myocardial infarction associated with electrical and/or mechanical complications 1 to 21 days after onset of symptoms were eligible to enter the study Male:female = 439:90 Mean age = 58 years Exclusion criteria: medical contraindications to use of pindolol; uncontrolled heart failure; unrelated heart disease; persistent heart block of second or third degree; persistent bradycardia < 50 beats/min; obstructive airways disease; uncontrollable insulin‐dependent diabetes; known hypersensitivity to beta‐blocking drugs; other diseases serious enough to worsen the short‐term prognosis irrespective of the myocardial infarction; pregnancy; necessity to use beta‐blocking drugs or calcium antagonists. Patients who were unable to return for regular control were also excluded |
|
| Interventions | Experimental group: pindolol (15 mg/d orally). Could be changed to half a tablet or extra up to 20 mg/d if necessary (n = 263) Control group: placebo (n = 266) Co‐intervention: digitalis, diuretics, vasodilators (nitrates), antiarrhythmics, and anticoagulants Excluded medication: not described |
|
| Outcomes | Primary: death Other: cardiac death, non‐cardiac death, sudden death, fatal + non‐fatal myocardial reinfarction Time point reported: 2 years |
|
| Notes | Email was not found More than 400 participants were included later than 1 week after myocardial infarction MACE was calculated and was reported as a composite of non‐fatal reinfarction + cardiac death Study was funded by Sandoz Ltd., Basle, which also coordinated the study and processed the data |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Numbers and reasons for withdrawals and dropouts for all outcomes are clearly stated ‐ more or less similar in both groups ‐ and the trial handles missing data appropriately in intention‐to‐treat analysis 76 patients from the pindolol group and 50 from the placebo group were withdrawn. However, they were followed up regarding mortality |
| Selective reporting (reporting bias) | High risk | No protocol was found, and the trial did not report serious adverse events |
| Other bias | Low risk | No other biases were found |
Baber 1980.
| Study characteristics | ||
| Methods | Randomised clinical trial at 49 sites in the United Kingdom, Italy, and Yugoslavia. Duration not mentioned | |
| Participants | 720 participants younger than 70 years of age who survived an anterior myocardial infarction were included within 2 to 14 days after myocardial infarction (mean 8.5 days) Male:female = 609:111 Mean age = 54.9 years Exclusion criteria: (1) bronchospasm; (2) atrioventricular block greater than first degree; (3) sinus bradycardia (< 55/min); (4) persistent heart failure; (5) beta‐blockade at the time of infarction |
|
| Interventions | Experimental group: propranolol (120 mg/d) (n = 355) Control group: placebo (n = 365) Co‐intervention: not described Excluded medication: not described |
|
| Outcomes | Primary: mortality, non‐fatal reinfarction Time points reported: 1 and 9 months |
|
| Notes | Email was not found MACE was calculated and was reported as a composite of non‐fatal reinfarction + cardiac death Study was funded by ICI Pharmaceuticals Division |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described sufficiently; however the trial is described as double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 88 (24%) participants from the placebo group and 82 (23%) from the propranolol group were withdrawn; however numbers and reasons for withdrawals and dropouts for all outcomes are clearly stated and can be described as similar in both groups; the trial handles missing data appropriately in intention‐to‐treat analysis (i.e. every randomised patient was followed up in relation to mortality and reinfarction) |
| Selective reporting (reporting bias) | Unclear risk | No protocol was found; however mortality was reported |
| Other bias | Low risk | No other biases were found |
Barvik 1992.
| Study characteristics | ||
| Methods | Randomised clinical cross‐over trial at a single site in Norway. Duration not mentioned | |
| Participants | 32 participants with a history of documented myocardial infarction at least 1 year before inclusion, beta‐blocker therapy after myocardial infarction for secondary prophylaxis, and NYHA I were included Male:female = 32:0 Mean age = 63 years Exclusion criteria: effort angina, electrocardiographic evidence of ischaemia, clinical signs of congestive heart failure |
|
| Interventions | This was a cross‐over trial: 4 weeks on therapy, followed by 1 week of gradual withdrawal of study medication. Patients were tested after 4 weeks without treatment (second baseline test). Patients were then crossed over to alternative treatment in a blinded fashion. After 4 weeks in this second treatment period, patients underwent a fourth test. Total study duration was 18 weeks for each patient. We included only data from the first 9 weeks before the cross‐over Experimental group: timolol (10 mg twice daily) for 4 weeks Control group: matching placebo for 4 weeks Co‐intervention: not described Excluded medication: not described |
|
| Outcomes | Outcome: cardiopulmonary exercise performance during a bicycle test Time point reported: 9 weeks |
|
| Notes | Email was not found Study was a cross‐over study, and no useful data were reported. Furthermore it is unknown how many participants were included in placebo and timolol groups for the first baseline test No data were extracted Funding was not described |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as double‐blinded; however, the method used is not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Described as double‐blinded; however, the method used is not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 4 patients were excluded after initial randomisation |
| Selective reporting (reporting bias) | High risk | No protocol could be found, and both mortality and SAEs were not reported |
| Other bias | Low risk | No other biases were found |
BCSG 1997.
| Study characteristics | ||
| Methods | Randomised clinical trial at 7 sites in China between October 1991 and March 1995 | |
| Participants | 1106 participants younger than 75 years with diagnosed acute myocardial infarction within 2 to 4 weeks and LVEF > 40% were included in the trial Male:female = 790:316 (2.8:1) Mean age = 59 years Exclusion criteria: contraindications to the respective trial medication |
|
| Interventions | In this 3‐arm study (atenolol, enalapril, and control), drugs were administered 2 to 4 weeks after onset of AMI when patients were stable. However, only 2 arms are relevant to our review Experimental group: atenolol (25 mg/tab for 28 months) (n = 385) Control group: no intervention other than co‐intervention (n = 372) Co‐intervention: conventional therapy + 50 mg of aspirin per day Excluded medication: not described |
|
| Outcomes | Outcomes: cardiac events (sudden cardiac death, heart failure death, total cardiac deaths, myocardial reinfarction), non‐cardiac death Time point reported: 19 months |
|
| Notes | Email was not found. Dr. Wu Ning, Department of Cardiology, PUMC Hospital, CAMS, Beijing, China Study reported data as ‰/pr month for each group. Therefore, we calculated the data as (((event rate / 1000) * 19) * number of patients). With regard to myocardial reinfarction, it is unclear whether this was based only on non‐fatal reinfarction or on both fatal and non‐fatal reinfarction. Furthermore, data were reported as total numbers for all 3 groups; however, it was stated that the incidence was the same in all 3 groups MACE was reported by the trialist as 'total cardiac events: consisting of sudden cardiac death, heart failure leading to mortality, myocardial reinfarction, and total cardiac death'. However, this was reported only as total numbers for all 3 groups ‐ not separately for each group. Furthermore, because it is unclear whether the outcome 'myocardial reinfarction' was based on only non‐fatal reinfarction or fatal + non‐fatal reinfarction, we were not able to calculate MACE Study was funded by the Ministry of Health |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described sufficiently, other than "patients were divided randomly..." |
| Allocation concealment (selection bias) | Unclear risk | Not described sufficiently, other than "patients were divided randomly..." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | No protocol was found; however mortality and serious adverse events were reported |
| Other bias | Low risk | No other biases were found |
BHAT 1982.
| Study characteristics | ||
| Methods | Randomised clinical trial at 31 sites in the United States between June 1978 and October 1980 | |
| Participants | 3837 participants, of both sexes, between 30 and 69 years of age, who were hospitalised with a definite acute myocardial infarction documented by appropriate symptoms, ECG, and enzymatic changes were included in the trial 5 to 21 days after hospitalisation Male:female = 3240:597 Mean age = 54.8 years Exclusion criteria: contraindications to propranolol, such as marked bradycardia; history of severe congestive heart failure or asthma as an adult; life‐threatening illness other than CHD; had undergone or were likely to undergo cardiac surgery; already taking or were likely to take beta‐blockers prescribed for them |
|
| Interventions | Experimental group: propranolol (initial dose of 20 mg orally increased to 40 mg every 8 hours). Afterwards, the maintenance dosis was 180 or 240 mg/d (n = 1916) Control group: placebo (n = 1921) Co‐interventions: vasodilator, diuretic, tranquilliser, digitalis, aspirin, antiarrhythmic, potassium, antihypertensive, anticoagulant, dipyridamole, insulin, hormonal agent, oral hypoglycaemic, sulphinpyrazone, lipid‐lowering drug Excluded medication: not described |
|
| Outcomes | Primary: all‐cause mortality Seconday: coronary heart disease mortality; sudden cardiac death; combined incidence of coronary heart disease mortality and non‐fatal MI; non‐fatal MI; adverse effects of propranolol Time points reported: 1 month, 1.5 months, 3 months, and every 3 months. Mean maximal follow‐up was 3 years |
|
| Notes | No email was found Mean time in the hospital before randomisation was 13.8 days Trialists reported MACE as 'non‐fatal reinfarction plus fatal arterioslerotic heart disease (i.e. total fatal and non‐fatal coronary heart disease)'. We reported this outcome as our MACE Study was funded by National Heart, Lung, and Blood Institute. Propranolol and matching placebo were prepared and donated by Ayerst Laboratories |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Not described sufficiently, other than "the coordination center randomly assigned either propranolol or placebo to eligible patients in a double‐blind manner" This was accomplished by telephone communication with the coordinating centre after verification of the patient's eligibility. Randomisation was blocked by clinical centre with separate schedules used for each centre |
| Allocation concealment (selection bias) | Unclear risk | Not described sufficiently, other than "the coordination center randomly assigned either propranolol or placebo to eligible patients in a double‐blind manner" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Trial is described as double‐blinded. "To maintain the "blind," patients taking placebo were also assigned to either 180‐ or 240‐mg daily dosage schedules. Dosage assignments were made through the coordination centre and remained in effect for the duration of the trial" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "All deaths were classified by the mortality classification subcommittee without knowledge of the treatment assignment. Study data were reviewed periodically by a policy and data monitoring board, the members of which were not investigators in the BHAT. Death certificates and in‐hospital information were analysed by the Mortality Classification Committee members and the deaths were coded without knowledge of whether patients had been taking propranolol or placebo. A Nonfatal Events Subcommitee, using pre‐determined definitions, performs a similar function for nonfatal myocardial infarction" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing data for 4 from the placebo group and 8 from the experimental group |
| Selective reporting (reporting bias) | Low risk | Protocol published + reported on pre‐defined outcomes. NCT number: NCT00000492 |
| Other bias | Low risk | No other biases were found |
Capital‐RCT 2018.
| Study characteristics | ||
| Methods | Multi‐centre randomised controlled trial at 67 centres in Japan between August 2010 and May 2014 | |
| Participants | Patients > 18 years old who underwent primary PCI within 24 hours after onset of STEMI successfully and had preserved left ventricular ejection fraction (LVEF > 40%) as assessed by echocardiography were included in the trial within 7 days Male:female = 639:155 Mean age = 64 years old Exclusion criteria: reduced LVEF (LVEF < 40%), prior cardioverter‐defibrillator implantation, contraindications to beta‐blocker therapy such as unstable haemodynamic status, bradyarrhythmias, symptomatic HF, severe bronchial asthma and/or chronic obstructive lung disease | |
| Interventions | Experimental group: carvedilol (oral, maximal dose of 20 mg) Control group: no intervention other than the co‐intervention Co‐intervention: administration of other standard medications for STEMI patients such as aspirin, thienopyridines, statins, and inhibitors of the renin‐angiotensin system were left to the physician's decision Excluded medication: not reported | |
| Outcomes | Primary: composite of all‐cause death, myocardial infarction, hospitalisation for acute coronary syndrome (ACS), and hospitalisation for HF Secondary outcomes: individual components of the primary endpoint as well as cardiac death, non‐cardiac death, stroke, vasospastic angina, major bleeding, definite stent thrombosis (ST), target‐lesion revascularisation (TLR), and any coronary revascularisation. 3 composite endpoints including cardiac death/MI/ACS/HF, cardiovascular death/MI/stroke, and death/MI/stroke/ACS/HF/any coronary revascularisation Time points reported: at 3 months, 1 year, and 3.9 years of follow‐up | |
| Notes | Study author was contacted at taketaka@kuhp.kyoto‐u.ac.jp on 28 February 2021 to clarify the definition of myocardial infarction used in the study. According to the study author, the definition of myocardial infarction included both fatal and non‐fatal cases; however, in reality, there were no fatal myocardial infarctions. Hence, reported myocardial infarction included only non‐fatal myocardial infarction cases Participants were randomised within 1 to 7 days after their successfully performed primary PCI; hence, beta‐blockers were administered to stabilised patients in the non‐acute phase following acute myocardial infarction MACE was not reported in accordance with our definition; however, after study authors clarified the outcome 'myocardial infarction' as reported in the study representing only non‐fatal myocardial infarction, we calculated and reported MACE as a composite of non‐fatal reinfarction + cardiac death Study was supported by an educational grant from the Research Institute for Production Development (Kyoto, Japan) ClinicalTrials.gov Identifier: NCT01155635 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed centrally through the electronic data capture system with a stochastic minimisation algorithm to balance treatment assignment |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | An independent clinical event committee adjudicated both primary and secondary endpoints in a fashion blinded to the assigned treatment group |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 29 participants in total were lost to follow‐up without further description. However, this was less than 5% of the total number of participants included in the analysis |
| Selective reporting (reporting bias) | Low risk | Registered at clinicaltrials.gov before randomisation (NCT01155635). All outcomes in the protocol are mentioned in the paper |
| Other bias | Unclear risk | Trial was prematurely terminated due to slow enrolment, which could lead to bias |
Curtis 1991.
| Study characteristics | ||
| Methods | Randomised clinical trial at a single site in the United States | |
| Participants | 31 participants hospitalised with recent acute myocardial infarction and with a positive initial exercise test (performed 1 to 2 weeks after myocardial infarction) were included in the trial Male:female = 16:12 Mean age = not described Exclusion criteria: left bundle branch block or left ventricular hypertrophy on ECG, if they refused, if they were unable to exercise due to weakness, deconditioning, or musculoskeletal problems. Patients were also excluded if they were unable to discontinue all antianginal therapy (nitrates, beta‐adrenergic blocking drugs, and calcium antagonists) before exercise testing, or if they could not safely undergo exercise testing due to postinfarction angina or persistent heart failure |
|
| Interventions | Experimental group: propranolol (240 mg/d) (n =19) Control group: placebo (n = 12) Co‐intervention: not described Excluded medication: antianginal drugs, nitrates, calcium antagonists, beta‐adrenergic blocking drugs for at least 24 hours before exercise |
|
| Outcomes | Outcomes: treadmill test using Bruce protocol Time point reported: not reported |
|
| Notes | Study authors were contacted at kirkwood_adams@med.unc.edu on 20‐06‐2017. No response was received Time from inclusion to randomisation was 1 to 2 weeks after myocardial infarction No useful data could be extracted from the study Study was funded by the American Heart Association, North Carolina Affiliate, Inc., and by a research grant from the General Clinical Research Centers branch of the Division of Research Resources, United States Public Health Service |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Electrocardiograms were interpreted independently by 2 separate cardiologists, blinded to treatment assignment from the randomised exercise tests, with disagreements, if any, resolved by discussion |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 2 (10.5%) participants from the experimental group and 1 (8.3%) from the placebo group were excluded from the trial |
| Selective reporting (reporting bias) | High risk | No protocol was found, and the trial did not report all‐cause mortality or serious adverse events |
| Other bias | Low risk | No other biases were found |
E.I.S. 1984.
| Study characteristics | ||
| Methods | Randomised trial at 57 sites in West Germany, United Kingdom, and Switzerland between July 1979 and July 1981 | |
| Participants | 1741 patients, aged 35 to 69 years, who had survived 14 to 36 days after acute myocardial infarction were included Male:female = 1458:283 Mean age = 54.8 years Exclusion criteria: heart failure; uncontrolled hypertension; angina pectoris; bronchospasm; advanced peripheral vascular disease; unstable diabetes mellitus; persistent bradycardia ≤ 50 beats/min; severe extracardiac condition; post or scheduled cardiac surgery; coincidental treatment with beta‐blockers |
|
| Interventions | Experimental group: oxprenolol (160 mg orally 1 tablet per day) for 12 months (n = 858) Control group: matching placebo (n = 883) Co‐intervention: digitalis; diuretics; aldosterone antagonists; aspirin or sulfinpyrazone; anticoagulants; nitrates; calcium antagonists |
|
| Outcomes | Primary: effect on survival; total morality; cardiac mortality; non‐fatal cardiac events; fatal + non‐fatal myocardial reinfarction; angina (events that required a change in treatment) Time point reported: 12 months' follow‐up |
|
| Notes | Study authors were contacted at rolf.schroeder@uk‐erlangen.de on 20‐06‐2017. No response was received MACE was not calculated nor reported because there was risk of double‐counting when non‐fatal reinfarction was added to cardiovascular death: "if a patient had a non‐fatal, and independently, a fatal event, each was computed". Even though a patient had a non‐fatal reinfarction, the same patient could have experienced a cardiovascular death and this could have been reported twice. Therefore, we reported only cardiovascular death and myocardial reinfarction separately Study was funded by the Ciba‐Geigy A.G. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | Sealed treatment code. It is not described whether the letter was opaque |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study was described as double‐blinded; it is said, "to ensure that the trial remained blind, each patient's sealed treatment code was sent to the Data Handling and Monitoring Centre at the end of his participation in the study" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Critical Events Committee was responsible for validation of all critical events. The members of this Committee were not otherwise involved in the trial and had no access to the trial code, i.e. validation was blind" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No patient was lost to follow‐up for mortality and non‐fatal cardiac events |
| Selective reporting (reporting bias) | Low risk | A pre‐published protocol was made. This protocol reported the outcomes later used in the final trial |
| Other bias | Low risk | No other biases were found |
Julian 1982.
| Study characteristics | ||
| Methods | Randomised trial at 21 sites in Scotland between January 1978 and August 1980 | |
| Participants | 1456 participants, surviving for 5 to 14 days after onset of acute myocardial infarction and between 30 and 69 years of age, were included Male:female = 1156:300 Mean age = 55.3 years Exclusion criteria: heart block greater than first degree; heart rate < 54 per minute; women of child‐bearing potential; history of asthma or obstructive airways disease; insulin‐dependent diabetes; clinical evidence of heart failure at the 12th post‐infarction day; systolic blood pressure persistently < 100 mmHg; positive antinuclear factor; other cardiac or non‐cardiac conditions thought to be serious enough to worsen the short‐term prognosis; lack of cooperation by the patient or inability to follow up with the patient for psychological or geographical reasons. Patients previously on a beta‐adrenoceptor blocking agent were entered only if they had been off this therapy for at least 5 days |
|
| Interventions | Experimental group: sotalol (320 mg orally daily) (n = 873) Control group: placebo (n = 583) Co‐intervention: in patients for whom anginal symptoms could not be controlled by regulation of physical activity and treatment with trinitrin and/or long‐acting nitrates, additional open administration of sotalol was permissible, irrespective of the randomised study medication Excluded medication: quinidine, procainamide, diphenylhydantoin, mexiletine, disopyramide, monoamine oxidase inhibitors, tricyclic antidepressant drugs, adrenoceptor blocking agents, calcium antagonists |
|
| Outcomes | Primary: death; confirmed or suspected fatal + non‐fatal myocardial reinfarction Time point reported: 12 months |
|
| Notes | No email was found Mean time from acute myocardial infarction to time of randomisation was 8.2 days MACE was calculated and was reported as "confirmed or suspected reinfarction plus cardiovascular death" Study was funded by Bristol‐Myers International Division, which also supplied the study medication (sotalol and placebo) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described sufficiently, other than "Randomisation was undertaken separately for each centre in blocks of ten, each block containing six allocations to sotalol and four to placebo" |
| Allocation concealment (selection bias) | Unclear risk | Not described sufficiently; however it is said that "the time of randomisation was determined as that when the next available numbered package was opened and the first dose given to the patient" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts were described. 340 (222 on sotalol and 121 on placebo) were withdrawn from the trial; however they were included in the analysis following the intention‐to‐treat method |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained; however mortality and serious adverse events were reported |
| Other bias | Low risk | No other biases were found |
LIT 1987.
| Study characteristics | ||
| Methods | Randomised clinical trial at 70 centres in the United States between August 1979 and April 1982 | |
| Participants | 2395 participants between 45 and 74 years of age and hospitalised 5 to 15 days after myocardial infarction were included Male:female = 1988:407 Mean age = 58 years Exclusion criteria: history of coronary artery bypass surgery; permanent pacemaker; contraindication to beta‐blocker therapy (heart rate < 50 beats/min, systolic blood pressure < 95 mmHg, congestive heart failure, second‐ or third‐degree heart block, severe intermittent claudication, bronchospastic disease likely to require treatment with bronchodilators, insulin‐dependent diabetes); condition likely to require beta‐blocker therapy (e.g. post‐infarction angina, moderate to severe hypertension); administration of any beta‐blocker within 3 days before the start of the pre‐entry evaluation; planned therapy with aspirin, sulfinpyrazone, clofibrate, or dipyridamole; life‐threatening condition other than coronary heart disease; condition likely to affect protocol compliance; history of adverse reaction to metoprolol or its analogues |
|
| Interventions | Experimental group: metoprolol (initial test dose of 25 mg or 50 mg gradually titrated over a 2 to 7‐day period to a maintenance dose of 100 mg twice daily for a year) (n = 1195) Control group: placebo (n = 1200) Co‐intervention: none mentioned |
|
| Outcomes | Primary: overall mortality (at 7, 12, and 18 months' follow‐up) Secondary: cardiac mortality, in particular, sudden cardiac death (at 7 and 12 months' follow‐up) Time points reported: 7, 12, and 18 months' follow‐up |
|
| Notes | No email was found Mean time from onset of symptoms to randomisation was 9.5 days MACE was not reported nor calculated because only data on cardiovascular mortality were available Funding of the trial was not described. However, the trial was conducted under the supervision of CIBA‐GEIGY personnel |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described sufficiently; however treatment is described as "double‐blinded" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 4 patients (1 on placebo and 3 on metoprolol) were lost to follow‐up at maximum follow‐up |
| Selective reporting (reporting bias) | High risk | No protocol could be obtained, and the trial did not report serious adverse events |
| Other bias | Low risk | No other biases were found |
Mazur 1984.
| Study characteristics | ||
| Methods | Randomised trial in Moscow, Russia. Duration not mentioned | |
| Participants | 204 participants, younger than 62 years of age, who no longer than 3 months before initiation of the study had a possible or definite myocardial infarction Male:female = 204:0 Mean age = 50 years Exclusion criteria: contraindications to beta‐blockers; however, not specified |
|
| Interventions | Experimental group: propranolol (40 to 320 mg/d ‐ mean dose = 150 mg/d) (n = 101) Control group: no intervention other than usual medical care provided at district policlinics (n = 103) Co‐intervention: none mentioned |
|
| Outcomes | Outcomes: death from all causes; coronary death; sudden coronary death; non‐fatal reinfarction Time point reported: median of 18 months' follow‐up |
|
| Notes | No email was found Study did not write whether patients with heart failure were excluded MACE was calculated and was reported as non‐fatal reinfarction + cardiovascular death (defined as death from ischaemic heart disease) Funding was not described |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised according to the matched pairs principle, taking into account patients' age (decades), category of myocardial infarction (possible or definite), presence of angina pectoris of effort, maximal grades of ventricular extrasystoles (occasional polytopic or frequent, including those with polytopic extrasystoles and grades 4 and 5), and arterial hypertension (arterial pressure > 160/95 mmHg) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 20 patients dropped out from the experimental group, but data were available for dropouts with regard to all‐cause mortality and cardiovascular death |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained; however mortality and serious adverse events were reported |
| Other bias | Low risk | No other biases were found |
Mazzuero 1987.
| Study characteristics | ||
| Methods | Randomised clinical trial at a single site in Italy. Duration not described | |
| Participants | 64 participants between 32 and 67 years of age and with a history of Q‐wave infarction, and clinical state that would allow changes in drug therapy and functional classes I or II according to the NYHA, were included. Patients were studied from 21 to 113 days (mean 44 days) after acute myocardial infarction Male:female = 64:0 Mean age = 51 years Exclusion criteria: absence of spontaneous angina and life‐threatening arrhythmias; no treatment with digitalis or amiodarone; no contraindications to beta‐blockers: heart rate > 50 beats/min; absence of atrioventricular or intraventricular conduction disturbances, bronchial asthma, clinically evident peripheral arterial disease, or uncompensated diabetes mellitus; resting blood pressure ≤ 150/90 mmHg; no contraindications to benzodiazepines; absence of psychopathological traits or disorders |
|
| Interventions | This was a 4‐armed study. We included only 3 of the 4 intervention arms (2 experimental groups and 1 placebo group) Experimental group 1: propranolol 120 mg daily orally (40 mg 3 times a day) (n = 16) Experimental group 2: atenolol 100 mg daily orally (n = 16) Control group: placebo (1 pill twice a day) (n = 8) Co‐intervention: none mentioned Excluded medication: nitrates, calcium channel blockers, antiarrhythmic agents, bezodiazepines |
|
| Outcomes | Outcomes: ECG, other cardiovascular parameters Time point reported: 48 hours |
|
| Notes | Study author was contacted at giorgio.mazzuero@fsm.it on 20‐06‐2017; however, this email could not be reached No useful data were reported Funding was not described |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Recordings were blindly analysed by a cardiologist (electrocardiogram) and a psychologist (all others) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It was not described whether any participants were lost to follow‐up |
| Selective reporting (reporting bias) | High risk | No protocol was found, and the trial did not report all‐cause mortality |
| Other bias | Low risk | No other biases were found |
MIS 1975.
| Study characteristics | ||
| Methods | Randomised clinical trial at 67 sites in the United Kingdom, South Africa, Hungary, New Zealand, Holland, and Australia. The trial started early in 1972, but the exact duration is not clarified | |
| Participants | 3053 patients, younger than 70 years of age, recovering from an acute myocardial infarction were included 1 to 4 weeks after the acute attack Male:female = 2628:410 Mean age = 55 years Exclusion criteria: age 70 and older; evidence of congestive heart failure at the proposed date of entry; heart block greater than first degree; heart rate < 60/min; history of bronchial asthma; any concurrent condition that the clinician considered to contraindicate participation in the trial |
|
| Interventions | Experimental group: practolol (200 mg twice daily) (n = 1533) Control group: placebo (lactose and excipients + 1 mg chloroquine matched for appearance) (n = 1520) Co‐intervention: not described Excluded medication: drugs considered to interact with beta‐adrenoreceptor antagonists were not permitted |
|
| Outcomes | Primary: death, non‐fatal reinfarction Secondary: effects of treatment on blood pressure, angina pectoris, and arrhythmia Time point reported: 14.2 months |
|
| Notes | No email was found MACE was reported as 'all cardiac events (death + non‐fatal myocardial reinfarction)' by the trialist in Table II in the publication from 1977. Because this number is also achieved by adding cardiac death to non‐fatal reinfarction, we believe it is the proportion of participants that has been reported on ‐ not the number of events. Furthermore, the trialist clarifies that if patients sustains more than 1 non‐fatal reinfarction during follow‐up, they are included only once Study was funded by I.C.I. Pharmaceuticals |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients in hospital 7 to 28 days after infarction were allocated to treatment with drug or placebo by a randomised code of numbers |
| Allocation concealment (selection bias) | Low risk | A sealed copy of the tablet identity code was sent to each hospital pharmacist and physician in charge for consultation in emergency |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Each clinician's containers were in sets numbered sequentially and were allocated randomly to drug or placebo Special 100‐mg tablets of practolol (white) were supplied in 250‐tablet containers, with similar containers of placebo tablets (lactose and excipients) matched for appearance |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It is not described whether any participants were lost to follow‐up |
| Selective reporting (reporting bias) | Unclear risk | No protocol was found; however mortality and serious adverse events were reported |
| Other bias | Low risk | No other biases were found |
NMS 1981.
| Study characteristics | ||
| Methods | Randomised clinical trial at 20 clinical centres in Norway between January 1978 and October 1979 | |
| Participants | 1884 stable patients with diagnosed acute myocardial infarction within 7 to 28 days were included in the trial Male:female = 1488:396 Mean age = 61 years Exclusion criteria: (1) any contraindication to beta‐adrenergic blockade on the day of evaluation — i.e. uncontrolled cardiac failure, resting heart rate < 50 beats per minute, second‐degree or third‐degree atrioventricular block, sinoatrial block, systolic blood pressure < 100 mmHg whether patient was standing or supine, unstable diabetes mellitus, chronic obstructive pulmonary disease, severe intermittent claudication, severe renal or hepatic impairment, adverse reactions during previous administration of beta‐adrenergic blocking agents; (2) any condition likely to hinder or confuse follow‐up or endpoint evaluation — i.e. concurrent serious disorders such as neoplasm, alcoholism, drug addiction, or psychiatric disease; (3) any need for treatment with a beta‐adrenergic blocking agent; (4) any indication for antiarrhythmic agents, lipid‐reducing agents, salicylates, or anticoagulants that was expected to last longer than 3 months (treatment with digitalis and diuretics was allowed); (5) various administrative problems — e.g. admission to hospital later than 48 hours after onset of symptoms, residence outside the study area, unwillingness to participate |
|
| Interventions | Experimental group: timolol (10 mg twice daily) (n = 945) Control group: placebo (n = 939) Co‐intervention: not described |
|
| Outcomes | Outcomes: all‐cause mortality (reported at 60 months' follow‐up); fatal and non‐fatal reinfarction (reported at 17 months' follow‐up) Time points reported: 12 to 33 months' follow‐up (mean 17), 60 months' follow‐up |
|
| Notes | No email was found MACE was not reported nor was it calculated because no data were available for non‐fatal reinfarction; therefore, it was not possible to calculate MACE without risk of double‐counting Study was funded by Merck Sharp & Dohme Laboratories |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described sufficiently; however the following is said about randomisation: "the ethical‐review committee, which had no connection with Merck Sharp & Dohme, possessed the randomization code from the beginning of the study and scrutinized the ethical and safety aspects throughout the study, thus ensuring proper handling of data. Only the members of this committee, none of whom was a study investigator, and six persons involved in data processing had access to decoded interim study information" "Patients were randomly assigned in preset multiples of 10 to treatment with either timolol or placebo" |
| Allocation concealment (selection bias) | Unclear risk | Not described sufficiently; however the following is said about allocation: "within each center and each of the risk groups, patients were randomly assigned in preset multiples of 10 to treatment with either timolol or placebo" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described sufficiently; however it is said that "the study was conducted in a double‐blind manner". Furthermore, it is said that "placebo and timolol tablets were similar in shape, size, and color but slightly different in taste", which could mean that participants were blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All deaths were classified by the steering committee according to a manual of classification and without knowledge of the study medication received |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not described sufficiently |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained; however mortality was reported |
| Other bias | Low risk | No other biases were found |
NPT 1982.
| Study characteristics | ||
| Methods | Randomised clinical trial at 12 sites in Norway between December 1977 and July 1980 | |
| Participants | 560 high‐risk survivors, 35 to 70 years of age, were included 4 to 6 days after their acute myocardial infarction Male:female = 476:84 Mean age = 58.4 years Exclusion criteria: severe heart failure (cardiogenic shock or pulmonary oedema); persistent signs of heart failure; good‐risk patients; need for beta‐blockade; diabetes mellitus; AV block II to III or SA block; hypotension; need for antiarrhythmics |
|
| Interventions | Experimental group: propranolol (160 mg/d, 40 mg 4 times daily) (n = 278) Control group: placebo (identical tablets) (n = 282) Co‐intervention: none mentioned |
|
| Outcomes | Primary: sudden cardiac death; total death; fatal and non‐fatal reinfarction; total number of cardiac events Time point reported: 12 months' follow‐up |
|
| Notes | No email was found Study included participants 4 to 6 days after recovery from acute myocardial infarction; hence, these participants are assumed to be in the non‐acute phase of myocardial infarction Study included patients who presented with heart failure on admission or during the initial phase of the infarction but for whom signs of failure had disappeared at the time of randomisation MACE was reported by the trialist and was defined as "total number of cardiac events: total number of sudden cardiac deaths, fatal and non‐fatal reinfarctions, and other cardiac deaths" Study was funded by Bio‐Science Laboratories. Grants from the Norwegian Council for Cardiovascular Diseases and the National Centre for Medical Products Control were also received. Imperial Chemical Industries Ltd. provided test tablets for the study and ICI‐Pharma |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The 2 risk groups were randomised separately at each participating centre in balanced blocks of 10, according to a double‐blind design |
| Allocation concealment (selection bias) | Unclear risk | Not described sufficiently |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described sufficiently; however, the study was described as double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described sufficiently; however, the study was described as double‐blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No dropouts were described |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained; however mortality was reported |
| Other bias | Low risk | No other biases were found |
Olsson 1985.
| Study characteristics | ||
| Methods | Randomised clinical trial at 2 sites in Sweden between May 1976 and December 1980 | |
| Participants | 301 participants who had suffered an acute myocardial infarction, were younger than 70 years old, lived in the catchment areas, were admitted to the CCU within 48 hours of onset of symptoms and development of myocardial infarction according to WHO criteria, were in sinus rhythm without complete bundle branch block, and were discharged 4 days before from the hospital were included 1 to 2 weeks after the acute event
Male:female = 242:59 Mean age = 59.5 years Exclusion criteria: systolic blood pressure < 100 mmHg; severe cardiac failure not responding to conventional treatment with digitalis and diuretic drugs; severe intermittent claudication; obstructive pulmonary disease; need for beta‐adrenoceptor blockade (i.e. severe angina pectoris or symptomatic arrhythmias responding to beta‐adrenergic blockade); other major disease; unwillingness to participate |
|
| Interventions | Experimental group: metoprolol (100 mg given as half a tablet 3 times daily for 3 days, then 1 tablet twice daily) for 36 months (n = 154) Control group: matching placebo (n = 147) Co‐intervention: digitalis was given to roughly a third and diuretics to about half of all patients throughout follow‐up. Class 1 antiarrhythmic drugs were given long term to 1 of the placebo‐treated and 3 of the metoprolol‐treated patients Excluded medication: not described |
|
| Outcomes | Primary: ventricular arrhythmias, all‐cause mortality, non‐fatal reinfarction, adverse events, sudden cardiac death Time points reported: 12 months, 36 months |
|
| Notes | Email was not found Mean time from onset of symptoms to randomisation is not clearly stated; however, participants were stratified according to the type of ventricular arrhythmias detected by a 6‐hour electrocardiographic recording performed 4 days before discharge and 1 to 2 weeks after the acute event. Hence, participants must have been randomised at least 1 week after acute myocardial infarction Trial reported total numbers of deaths and non‐fatal reinfarctions after 12 months' follow‐up in Olsson 1984. However, it is not reported how many events occurred in each randomised group separately. Therefore, we calculated non‐fatal reinfarction, cardiac mortality, and MACE with the help of cumulative Figures 1 and 6 in the Olsson 1985 publication. However, the total number of deaths was not found at 12 months' follow‐up MACE was reported by the trialist and was defined as 'major cardiac events' (i.e. either cardiac death or non‐fatal reinfarction) Study was funded by grants from The Swedish National Association Against Heart and Chest Diseases and AB Hassle, Molndal, Sweden |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described, other than "the patients were randomly assigned to receive..." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | All tablets were identical in shape, size, and colour. Patients thereafter were randomly allocated to double‐blind treatment with metoprolol or placebo. The double‐blind design is applicable also to beta‐adrenergic blocking agent studies, and the level of investigator blindness in the present study may be illustrated by the relatively large number of dose reductions occurring in the placebo group. Codes were not broken until patients had completed 3 years of follow‐up |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All endpoint evaluation as well as determination of sudden death was conducted without knowledge of the treatment group |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No patient had missing mortality or morbidity data during the 36‐month follow‐up period |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be found; however, mortality was reported |
| Other bias | Low risk | No other biases were found |
Poulsen 1999.
| Study characteristics | ||
| Methods | Randomised clinical trial at a single site in Denmark between September 1993 and January 1995 | |
| Participants | 77 participants with acute MI defined as creatinine kinase ≥ 210 IU and creatinine kinase B ≥ 20 IU or electrocardiographic evidence of MI (ST elevation > 1 mm in contiguous leads or subendocardial injury pattern) and typical chest pain were eligible for the study. Furthermore, all patients had LVEF > 40% at baseline and were between 40 and 75 years of age Male:female = 58:19 Mean age = 61.5 years Exclusion criteria: ongoing treatment with beta‐blockers, systolic blood pressure < 100 mmHg, heart rate < 50 beats/min, intermittent claudication, significant valvular heart disease, severe obstructive lung disease, appearance of atrioventricular block of second or third degree, uncontrolled diabetes mellitus, severe uncontrolled congestive heart failure, other life‐threatening disease |
|
| Interventions | Treatment begun 5 to 7 days after admission Experimental group: metoprolol 200 mg for 12 months (n = 39) Control group: placebo for 12 months (n = 38) Co‐intervention: none mentioned Excluded medication: none mentioned |
|
| Outcomes | Outcomes: cardiac mortality, ejection fraction, cardiovascular parameters Time points reported: 3, 6, and 12 months |
|
| Notes | Study authors were contacted at steepoul@rm.dk on 20‐06‐2017. No response was received Randomisation took place 5 to 7 days after admission with acute myocardial infarction Study reported only 'sudden cardiac mortality'; because no other deaths were reported, we used this as our all‐cause mortality MACE was not reported nor calculated because no data were available Study was funded by the Danish Heart Foundation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described sufficiently, other than "patients were randomly assigned to receive either metoprolol (200 mg) or placebo" |
| Allocation concealment (selection bias) | Unclear risk | Not described sufficiently, other than "patients were randomly assigned to receive either metoprolol (200 mg) or placebo" |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described; however, the study was described as double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Echocardiographic data were analyzed by one author (S.H.P.) blinded to the patients’ clinical data" However, assessment of mortality was not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It is not described whether any participants were lost to follow‐up. Only withdrawals were described |
| Selective reporting (reporting bias) | Unclear risk | No protocol was found; however mortality and serious adverse events were reported |
| Other bias | Low risk | No other biases were found |
Schwartz 1992.
| Study characteristics | ||
| Methods | Randomised clinical trial at 32 centres in Italy. Duration was not mentioned. However, the study was completed on December 1983 | |
| Participants | 1013 participants younger than 65 years of age who had been admitted to the coronary care unit with diagnosed myocardial infarction 20 to 40 days after onset of symptoms were included in the trial. The diagnosis of acute MI was made when the following criteria were present: (1) typical electrocardiographic findings with development of pathological Q waves and/or ST‐segment evolutive changes in the anterior leads; and (2) typical changes in creatinine phosphokinase, lactic dehydrogenase, and glutamic oxaloacetic transaminase serum levels Male:female = not reported for the entire group of randomised patients, but the proportion of male:female in the high‐risk group was 132:12 Mean age = not reported for the entire group of randomised patients, but mean age in the high‐risk group was 51.4 years Exclusion criteria: age > 65 years; heart rate < 50 beats/min; clinically overt heart failure (NYHA III and IV); first‐degree heart block with PQ > 0.24 seconds or second‐ or third‐degree heart block; sick sinus syndrome; insulin‐dependent diabetes; history of asthma or chronic pulmonary obstructive disease needing treatment of IV theophylline, or systemic cortisone or beta‐2 stimulants; intermittent claudication with necrotic lesions; haemodynamically significant valvular disease; need for 'open' treatment with beta‐blockers; other non‐cardiac condition serious enough to worsen the short‐term prognosis, or to confuse endpoint evaluations, such as concurrent neoplastic disease, alcoholism, drug addiction, or psychiatric disease; inability to follow up with patients for administrative reasons; unlikely patient cooperation with regards to treatment compliance and adherence to scheduled follow‐up visits |
|
| Interventions | Study randomised patients into high‐ and low‐risk groups according to whether participants experienced at least 1 episode of ventricular fibrillation, ventricular flutter, or ventricular tachycardia. The high‐risk group was further divided into 3 groups: oxprenolol, placebo, or left cardiac sympathetic denervation (LCSD), and the low‐risk group was divided into oxprenolol or placebo. When data were reported in our review, oxprenolol and placebo groups in high‐ and low‐risk groups were pooled Experimental group: oxprenolol (160 mg 1 tablet given once daily in the morning) (n = 485) Control group: placebo (1 tablet given once in the morning) (n = 488) Co‐intervention: long‐acting nitrates and nifedipine Excluded medication: beta‐blockers, amiodarone, verapamil, prenylamine, antiplatelet drugs |
|
| Outcomes | Outcomes: cardiac death, total death, fatal and non‐fatal reinfarction Time point reported: 22 months |
|
| Notes | Email was not found Mean time from being qualified for inclusion to randomisation was 30 days MACE was calculated and was reported (total cardiac deaths + non‐fatal reinfarctions) Ciba‐Geigy was involved in the trial |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients in the low‐risk group were randomly assigned, under double‐blind conditions and in equal proportions..." "Patients who gave their informed written consent entered the study and were randomised according to which of the two risk groups they belonged. For each group, randomization was balanced within each center in preset blocks of six patients..." |
| Allocation concealment (selection bias) | Low risk | Treatment allocation was obtained by opening the treatment allocation envelope sent from the Study Coordination Committee |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blindness was maintained at all stages for pharmacologically treated patients "...and for keeping the randomization code that had to be opened only if necessary; as a matter of fact, the Policy Committee never opened the randomization code throughout the study" "...the pharmacologic treatments were started immediately under double blind conditions..." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All data were processed by the Medical Department of Ciba‐Geigy Italy. Periodic random checks of the consistency of recorded and stored data were made by a statistician from the University of Milan, not otherwise involved in the study "...the End Points Evaluation Committee scrutinized the data of those patients who reached an end point and classified the events while maintaining blindness" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropout was reported |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be found; however mortality was reported |
| Other bias | Low risk | No other biases were found |
Taylor 1982.
| Study characteristics | ||
| Methods | Randomised clinical trial at 13 sites in England between January 1973 and December 1979 | |
| Participants | 1103 participants between 35 and 65 years of age with acute myocardial infarction between 1 and 90 months previously were included in the trial Male:female = 1103:0 Mean age = 51 years Exclusion criteria: (1) cardiac contraindications to beta‐blockade, i.e. radiographic evidence of heart failure (cardiothoracic ratio > 0.50) or pulmonary venous congestion, resting heart rate < 50 beats per minute, or any grade of heart block; (2) symptomatic obstructive airways disease or history of bronchial asthma; (3) diabetes mellitus requiring medication; (4) hypertension (diastolic blood pressure > 100 mmHg (Korotkoff phase IV); (5) intercurrent treatment with antidysrhythmics, beta‐blockers, salicylates, anticoagulants, antiplatelet drugs, or positive inotropic agents; (6) other serious systemic illness; (7) valvular or other non‐ischaemic heart disease; (8) administrative difficulties, e.g. residence geographically remote from study centres, language difficulties, antisocial activities, unreliability |
|
| Interventions | Experimental group: oxprenolol (40 mg given twice a day) (n = 632) Control group: placebo matched for size, shape, and colour (n = 471) Co‐intervention: not described Excluded medication: not described |
|
| Outcomes | Outcomes: total death, cardiac death, non‐fatal reinfarction Time point reported: 48 months (range 6 to 84 months) |
|
| Notes | Email was not found Mean time from myocardial infarction to randomisation was 13.5 months MACE was reported by the trialist as "cardiac events (death + non‐fatal reinfarction)" Study was funded by Ciba‐Geigy Pharmaceuticals Division (Horsham, England) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "After each patient had undergone outpatient assessment and given informed consent to participate, he received the trial number next in sequence, as well as the corresponding medication..." |
| Allocation concealment (selection bias) | Unclear risk | "After each patient had undergone outpatient assessment and given informed consent to participate, he received the trial number next in sequence, as well as the corresponding medication..." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The medication was centrally packaged, and sealed copies of the randomization code were held by the pharmacy in each hospital. No knowledge of the randomization of the trial population was available to the technical committee responsible for critical‐event verification at the end of the study. The electro‐cardiograms were evaluated in blinded fashion using the...." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "No knowledge of randomisation of the trial population was available to the technical committee responsible for critical event verification at the end of the study. Electrocardiograms were evaluated in blinded fashion using the...." "Information, including the total number of critical events and the distribution of these between the two randomized groups, was made available by the data‐processing unit to an independent monitoring committee that had no connection with Ciba‐Geigy or the investigators taking part in the study. The trial code and the distribution of events between the randomized groups were not disclosed to any investigator at any time" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 22 patients in the oxprenolol group and 9 in the placebo group were lost to follow‐up |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be found; however mortality was reported |
| Other bias | Low risk | No other biases were found |
Wilhelmsson 1974.
| Study characteristics | ||
| Methods | Randomised clinical trial at a single site in Sweden. Duration not described | |
| Participants | 230 participants discharged from hospital alive after myocardial infarction were included in the trial Male:female = 187:87 Mean age = not described Exclusion criteria: contraindications to beta‐blockade, namely, cardiac decompensation despite treatment with optimum doses of digitalis and diuretics; bradycardia (heart rate < 50 per minute); atrioventricular (AV) block I (PQ > 0.24 second); chronic obstructive lung disease requiring continuous treatment; systolic blood pressure < 110 mmHg in supine or standing position; labile diabetes treated with insulin; hepatic insufficiency or uraemia; chronic alcoholism or drug addiction |
|
| Interventions | Experimental group: alprenolol (400 mg/d) (n = 114) Control group: placebo (n = 116) Co‐intervention: not described Excluded medication: not described |
|
| Outcomes | Outcomes: death, sudden death, non‐fatal reinfarction Time point reported: 24 months |
|
| Notes | Email was not found MACE was not reported nor calculated because the definition of sudden death (death occurring within 24 hours of onset of any symptoms were defined as sudden) implies that mortality can be of any cause ‐ not only cardiac causes ‐ and because we have only data on non‐fatal reinfarction, we could not calculate MACE Study was funded by Astra Pharmaceuticals |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described sufficiently, other than "patients were randomly allocated to placebo or active treatment..." |
| Allocation concealment (selection bias) | Unclear risk | Not described sufficiently, other than "patients were randomly allocated to placebo or active treatment..." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described sufficiently; however, the study is reported as double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The 2 physicians who performed the study were never involved in the diagnosis of infarction nor in determination of the cause of death; these data were obtained from the myocardial infarction register |
| Incomplete outcome data (attrition bias) All outcomes | High risk | A total of 16 patients, 8 (7%) in each group, were excluded owing to defined contraindications. They were not included in the follow‐up. |
| Selective reporting (reporting bias) | Unclear risk | No protocol was found; however mortality and serious adverse events were reported |
| Other bias | Low risk | No other biases were found |
AMI: acute myocardial infarction.
CHD: coronary heart disease.
ECG: electrocardiogram.
HF: heart failure.
LVEF: left ventricular ejection fraction.
MACE: major cardiovascular event.
MI: myocardial infarction.
NYHA: New York Heart Association.
PCI: percutaneous coronary intervention.
SAE: serious adverse event.
STEMI: ST‐elevation myocardial infarction.
WHO: World Health Organization.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| CAPRICORN 2001 | Participants with heart failure and LVEF < 40% were included |
| COMMIT 2005 | Patients were followed only during the acute phase after myocardial infarction; hence, follow‐up was limited to 1 month |
| Mazur 1994 | This was not a randomised controlled trial |
| Mickley 1991 | Patients did not receive any intervention with beta‐blockers |
| Park 2013 | Most included patients had a history of stable angina pectoris ‐ not myocardial infarction |
| Smith 2008 | This was a cluster randomised trial |
LVEF: left ventricular ejection fraction.
Characteristics of ongoing studies [ordered by study ID]
BETAMI 2018.
| Study name | BEtablocker Treatment After Acute Myocardial Infarction in Patients Without Reduced Left Ventricular Systolic Function (BETAMI) |
| Methods | Prospective, randomised, open‐blinded endpoint (PROBE) study |
| Participants | Patients with AMI will be randomised 1 to 8 days following PCI or thrombolysis, and will be allocated to prescription of a BB or to no such prescription. They will be followed for at least 2 years with respect to primary and secondary endpoints |
| Interventions | Control group: no beta‐blocker will be administered. Patients randomised to no beta‐blockade will be discouraged from using beta‐blockade as long as there is no other indication than strictly secondary prevention after myocardial infarction. Any other treatment or management is to be given as per usual care Experimental group: a beta‐blocker will be administered. To reflect contemporary management, which this study is designed to test, there will not be a defined minimum dosage. Type and dose of BB will be left to the discretion of the PI. Generic drug and accepted dosages will be:
Any other treatment or management is to be given as per usual care |
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | 1 October 2018 |
| Contact information | John Munkhaugen, MD, PhD; johmun@vestreviken.no Vidar Ruddox, MD, PhD; vidar.ruddox@siv.no |
| Notes | NCT number: NCT03646357 |
DANBLOCK 2018.
| Study name | Danish Trial of Beta Blocker Treatment After Myocardial Infarction Without Reduced Ejection Fraction (DANBLOCK) |
| Methods | Randomised, controlled clinical trial |
| Participants | Patients with a history of acute myocardial infarction within the past 14 days and preserved ejection fraction (LVEF > 40) |
| Interventions | Control group
Experimental group
|
| Outcomes |
Primary outcome measures
Secondary outcome measures
|
| Starting date | 1 October 2018 |
| Contact information | Contact: Thomas SG Sehested, MD; thomas.steen.gyldenstierne.sehested@regionh.dk |
| Notes | Estimated completion date: 01.10.2022 NCT number: NCT03778554 |
MINOCA‐BAT 2021.
| Study name | Randomized Evaluation of Beta Blocker and Angiotensin Converting Enzyme Inhibitor (ACEI)/Angiotensin Receptor Blocker (ARB) Treatment in MINOCA Patients |
| Methods | Multi‐national, multi‐centre, pragmatic randomised clinical trial |
| Participants | Patients > 18 years of age with clinical diagnosis of MINOCA within the last 30 days, left ventricular ejection fraction ≥ 40% measured with echocardiography, MRI or left ventriculography after admission and before randomisation |
| Interventions | Experimental: beta‐blocker and ACEI/ARB Experimental: beta‐blocker alone Experimental: ACEI/ARB alone No intervention: no beta‐blocker and no ACEI/ARB |
| Outcomes |
Primary outcomes • Time to death of any cause • Time to re‐admission because of AMI, ischaemic stroke, or heart failure Secondary outcomes • All‐cause death • Cardiovascular death • Re‐admission because of AMI • Re‐admission because of ischaemic stroke • Re‐admission because of heart failure • Re‐admission because of unstable angina pectoris • Re‐admission because of atrial fibrillation |
| Starting date | 16 December 2018 |
| Contact information | Terese Karlin, MSc Pharm; +46 18 617 04 36; terese.karlin@ucr.uu.se |
| Notes | Estimated study completion date: 1 October 2025 NCT number: NCT03686696 |
REBOOT‐CNIC2018.
| Study name | TREatment With Beta‐blockers After myOcardial Infarction withOut Reduced Ejection fracTion |
| Methods | Randomised, open‐label, blinded endpoint clinical trial |
| Participants | Patients being discharged after an acute MI, with or without ST‐segment elevation, and with left ventricular ejection fraction > 40%, without history of heart failure (HF) before study inclusion |
| Interventions | Experimental: beta‐blocker therapy: atenolol, bisoprolol, carvedilol, metoprolol, nebivolol Control: no beta‐blocker therapy |
| Outcomes |
Primary outcome • Composite of "all‐cause death, nonfatal reinfarction, or heart failure hospitalisation" Secondary outcomes • Incidence rate of individual components of the primary outcome • Incidence rate of cardiac mortality • Incidence rate of cardiac arrhythmias • Incidence of subsequent revascularisations • Incidence of ICD (including CRT) insertion |
| Starting date | 23 July 2018 |
| Contact information | Borja Ibañez, MD, PhD, FESC; bibanez@cnic.es |
| Notes | Estimated primary completion date: 15 November 2024 NCT number: NCT03596385 |
The AβYSS Study 2018.
| Study name | Assessment of βeta‐Blocker Interruption After Uncomplicated Myocardial Infarction on Safety and Symptomatic Cardiac Events Requiring Hospitalization: The AβYSS Study |
| Methods | Multi‐centre, randomised, open‐label trial |
| Participants | Patients > 18 years of age with current treatment with beta‐blockers and prior documented acute myocardial infarction 6 months or more before randomisation |
| Interventions | Experimental: discontinuation of beta‐blockers (1850 post‐MI patients treated with chronic beta‐blocker treatment will undergo withdrawal of their beta‐blocker treatment) Active comparator: continuation of beta‐blockers (1850 post‐MI patients treated with chronic beta‐blocker treatment will be continued under their usual beta‐blocker treatment without modification) |
| Outcomes |
Primary outcome • Composite of major adverse cardiovascular events (MACE) including all‐cause death, stroke, myocardial infarction, hospitalisation for other cardiovascular (CV) reason Secondary outcomes • All individual parameters of MACE (all‐cause death, heart failure, arrhythmia, syncope, conduction disorder, or pacemaker implantation; high blood pressure) • Stroke • Myocardial infarction • Hospitalisation for other cardiovascular reasons |
| Starting date | 29 August 2018 |
| Contact information | Johanne SILVAIN, MD; 33 1 42 16 30 01; johanne.silvain@aphp.fr Gilles MONTALESCOT, MD, PhD; 33 1 42 16 30 07; gilles.montalescot@aphp.fr |
| Notes | Estimated study completion date: 29 August 2023 NCT number: NCT03498066 |
AMI: acute myocardial infarction.
BB: beta‐blocker.
CRT: cardiac re‐synchronisation therapy.
EQ5D: EuroQoL Group Quality of Life Questionnaire based on 5 dimensions.
ICD: implantable cardioverter‐defibrillator.
LVEF: left ventricular ejection fraction.
MI: myocardial infarction.
PCI: percutaneous coronary intervention.
PI: principal investigator.
Differences between protocol and review
The title was changed from "Beta‐blockers for non‐acute treatment after myocardial infarction" in the protocol to "Beta‐blockers for patients without heart failure after myocardial infarction", as this title describes our review more appropriately.
We searched for finished trials as well as ongoing trials on the European Medicines Agency (EMA) (www.ema.europa.eu/ema/) and Food and Drug Administration (FDA) (www.fda.gov) websites.
In our protocol, we pre‐defined the time points closest to 12 months' follow‐up as our primary assessment time point, and maximum follow‐up as a secondary time point of interest. However, we decided to remove the time point of closest to 12 months' follow‐up because we already had pre‐defined a subgroup analysis that would look at different follow‐up periods. Therefore, we used the time point of maximum follow‐up as our primary outcome, which gave us the most power and precision.
We updated the "Evidence on the effects of beta‐blockers for myocardial infarction" section in the Review Background and added information about differences between guideline recommendations and reports on newer cohort studies.
We reported only the excluded trials that a reader might feasibly have expected to see as included trials.
We did not use last observation carried forward to handle missing data or when the proportion of dropouts was less than 5%. Several publications show that this method should not be used (Jakobsen 2017).
When testing for small study effects, we used both the Harbord test and the Egger test to increase the robustness of our analysis.
We specified only in the published protocol that we would include randomised clinical trials. We did not specify that we would exclude cluster randomised clinical trials. We did not include cluster randomised trials in the present review because of the inferior methodological quality of cluster randomised trial compared to individually randomised clinical trials.
We did not report Trial Sequential Analysis (TSA) and the TSA‐adjusted CI in accordance with our pre‐published protocol (http://www.ctu.dk/tsa/) due to requests from the Cochrane Heart Group.
To assess imprecision, we estimated the optimal information size according to the GRADE Handbook using a relative risk ratio (RRR) of 10%; incidence based on the meta‐analysis; alpha of 2.5% when our primary outcomes were assessed and 2.0% when our secondary outcomes were assessed; and beta of 10%.
We modified our definition of our composite outcome 'major cardiovascular events' as suggested by the Cochrane Heart Group, so that the composite of both cardiovascular mortality and non‐fatal myocardial infarction during follow‐up was used ‐ not a composite of either cardiovascular mortality or non‐fatal myocardial reinfarction during follow‐up, as described in our protocol. If trialists did not report a pre‐defined 'MACE', we calculated our own MACE by adding cardiovascular mortality with non‐fatal myocardial infarction only if we were certain that there was no risk of double‐counting participants.
We added three post hoc subgroup analyses to assess:
potential differences in effect based on trials specifically excluding heart failure participants compared to trials specifically excluding heart failure participants but likely not adhering to this. In our protocol, we planned to exclude trials specifically randomising participants with heart failure. However, several trials specifically excluded heart failure participants but reported some percentage of participants with heart failure in the baseline table. We chose to include these trials but decided to perform a post hoc subgroup analysis comparing these trials to trials with no heart failure participants;
potential differences in effect based on industry funding; we added a subgroup comparing effects in trials that were sponsored by industry or had unclear sponsorship compared to trials that were not sponsored by industry; and
potential differences in effect based on trials in which beta‐blockers were administered more than seven days after acute myocardial infarction (non‐acute phase) and trials in which beta‐blockers were administered within seven days after myocardial infarction (subacute phase).
Contributions of authors
Sanam Safi (SS): involved in screening including trials and data extraction. Conceived, designed, and drafted the review.
Naqash J Sethi (NSJ): involved in screening including trials and data extraction. Provided general advice and revised the review.
Steven Kwasi Korang (SKK): provided general advice, made statistical analyses, and revised the review.
Emil Eik Nielsen (EEN): provided general advice and revised the review.
Joshua Feinberg (JF): provided general advice and revised the review.
Christian Gluud (CG): provided methodological and statistical advice and revised the review.
Janus C Jakobsen (JCJ): provided methodological, statistical, and clinical advice and revised the review.
All review authors agreed on the final review version.
Sources of support
Internal sources
No sources of support provided
External sources
-
National Institute for Health Research (NIHR), UK
This project was supported by the National Institute for Health Research via Cochrane Infrastructure, Cochrane Programme Grant, or Cochrane Incentive funding to the Cochrane Heart Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS, or the Department of Health
-
Northwestern University, USA
The Cochrane Heart Group US Satellite is supported by intramural support from the Northwestern University Feinberg School of Medicine and the Northwestern University Clinical and Translational Science (NUCATS) Institute (UL1TR000150)
Declarations of interest
The performance of this review is free of any real or perceived bias introduced by receipt of any benefit in cash or kind, on any subsidy derived from any source that may have been or was perceived to have an interest in the outcomes of the review.
Sanam Safi (SS): no conflicts of interest.
Naqash J Sethi (NSJ): no conflicts of interest.
Emil Eik Nielsen (EEN): no conflicts of interest.
Steven Kwasi Korang (SKK): no conflicts of interest.
Joshua Feinberg (JF): no conflicts of interest.
Christian Gluud (CG): member of The Copenhagen Trial Unit task force for developing Trial Sequential Analysis methods, manual, and software.
Januc C Jakobsen (JCJ): no conflicts of interest.
New
References
References to studies included in this review
Ades 1987 {published data only}
- Ades PA, Thomas JD, Hanson JS, Shapiro SM, LaMountain J. Effect of metoprolol on the submaximal stress test performed early after acute myocardial infarction. American Journal of Cardiology 1987;60(13):963-6. [DOI] [PubMed] [Google Scholar]
Ahlmark 1976 {published data only}
- Ahlmark G, Saetre H, Korsgren M. Reduction of sudden deaths after myocardial infarction. Lancet 1974;2(7896):1563. [DOI] [PubMed] [Google Scholar]
- Ahlmark G, Saetre H. Long-term treatment with beta-blockers after myocardial infarction. European Journal of Clinical Pharmacology 1976;10(2):77-83. [DOI] [PubMed] [Google Scholar]
- Ahlmark G, Saetre H. Positon of myocardial infarct and results of alprenolol treatment. BMJ 1976;1(6013):837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ahnve 1980 {published data only}
- Ahnve S, Erhardt L, Lundman T, Rehnqvist N, Sjögren A. Effect of metoprolol on QTc intervals after acute myocardial infarction. Acta Medica Scandinavica 1980;208(3):223-8. [DOI] [PubMed] [Google Scholar]
Amsterdam Metoprolol Trial 1983 {published data only}
- Yusuf S, Peto R, Lewis J, Collins R, Sleight P. Beta blockade during and after myocardial infarction: an overview of the randomized trials. Progress in Cardiovascular Diseases 1985;27(5):335-71. [DOI] [PubMed] [Google Scholar]
APSI 1997 {published data only}
- Boissel JP, Leizorovicz A, Picolet H, Ducruet T. Efficacy of acebutolol after acute myocardial infarction (the APSI trial). American Journal of Cardiology 1990;66(9):24C-31C. [DOI] [PubMed] [Google Scholar]
- Boissel JP, Leizorovicz A, Picolet H, Peyrieux JC. Secondary prevention after high-risk acute myocardial infarction with low-dose acebutolol. American Journal of Cardiology 1990;66(3):251-60. [DOI] [PubMed] [Google Scholar]
- Cucherat M, Boissel JP, Leizorovicz A. Persistent reduction of mortality for five years after one year of acebutolol treatment initiated during acute myocardial infarction. American Journal of Cardiology 1997;79(5):587-9. [DOI] [PubMed] [Google Scholar]
Australien & Swedish 1983 {published data only}
- Australian and Swedish Pindolol Study Group. The effect of pindolol on the two years mortality after complicated myocardial infarction. European Heart Journal 1983;4(6):367-75. [PubMed] [Google Scholar]
Baber 1980 {published data only}
- Baber NS, Evans DW, Howitt G, Thomas M, Wilson C, Lewis JA, et al. Multicentre post-infarction trial of propranolol in 49 hospitals in the United Kingdom, Italy, and Yugoslavia. British Heart Journal 1980;44(1):96-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Barvik 1992 {published data only}
- Barvik S, Dickstein K, Aarsland T, Vik-Mo H. Effect of timolol on cardiopulmonary exercise performance in men after myocardial infarction. American Journal of Cardiology 1982;69(3):163-8. [DOI] [PubMed] [Google Scholar]
BCSG 1997 {published data only}
- Wu N, Fan Z. Secondary prevention of cardiac events following myocardial infarction: effects of atenolol and enalapril. American Journal of Cardiology 1997;110(8):602-6. [PubMed] [Google Scholar]
BHAT 1982 {published data only}
- Beta-blocker Heart Attack Trial Research Group. A randomized trial of propranolol in patients with acute myocardial infarction. II. Morbidity results. JAMA 1983;250(20):2814-9. [DOI] [PubMed] [Google Scholar]
- Byington RP, Beta-Blocker Heart Attack Trial Research Group. Beta-blocker heart attack trial: design, methods, and baseline results. Control Clinical Trials 1984;5(4):382-437. [DOI] [PubMed] [Google Scholar]
- Gheorghiade M, Schultz L, Tilley B, Kao W, Goldstein S. Effects of propranolol in non-Q-wave acute myocardial infarction in the beta blocker heart attack trial. American Journal of Cardiology 1990;66(2):129-33. [DOI] [PubMed] [Google Scholar]
- Gheorghiade M, Schultz L, Tilley B, Kao W, Goldstein S. Natural history of the first non-Q wave myocardial infarction in the placebo arm of the Beta-Blocker Heart Attack Trial. American Heart Journal 1991;122(6):1548-53. [DOI] [PubMed] [Google Scholar]
- Gheorghiade M, Shivkumar K, Schultz L, Jafri S, Tilley B, Goldstein S. Prognostic significance of electrocardiographic persistent ST depression in patients with their first myocardial infarction in the placebo arm of the Beta-Blocker Heart Attack Trial. American Heart Journal 1993;126(2):271-8. [DOI] [PubMed] [Google Scholar]
- Goldstein S, Byington R. The Beta Blocker Heart Attack Trial: recruitment experience. Controlled Clinical Trials 1987;8(4 Suppl):79S-85S. [DOI] [PubMed] [Google Scholar]
- Kostis JB, Byington R, Friedman LM, Goldstein S, Furberg C. Prognostic significance of ventricular ectopic activity in survivors of acute myocardial infarction. Journal of the American College of Cardiology 1987;10(2):231-42. [DOI] [PubMed] [Google Scholar]
- National Heart, Lung, and Blood Institute. Beta-blocker heart attack trial study protocol [https://babel.hathitrust.org/cgi/pt?id=pur1.32754081210092;view=1up;seq=3]. U.S. Depeartment of Health and Human Services. Public Health Service, National Institues of Health 1978;81-2209. [Google Scholar]
- Peters RW, Byington R, Arensberg D, Friedman LM, Romhilt DW, Barker A, et al. Mortality in the beta blocker heart attack trial: circumstances surrounding death. Journal of Chronic Diseases 1987;40(1):75-82. [DOI] [PubMed] [Google Scholar]
- Shivkumar K, Schultz L, Goldstein S, Gheorghiade M. Effects of propanolol in patients entered in the Beta-Blocker Heart Attack Trial with their first myocardial infarction and persistent electrocardiographic ST-segment depression. American Heart Journal 1998;135(2 Pt 1):261-7. [DOI] [PubMed] [Google Scholar]
- The Beta-blocker Heart Attack Trial (BHAT). A randomized trial of propranolol in patients with acute myocardial infarction. JAMA 1982;247(12):1707-14. [DOI] [PubMed] [Google Scholar]
- The Beta Blocker Heart Attack Trial Research Group. Beta blocker heart attack trial: design features. Controlled Clinical Trials 1981;2(4):275-85. [DOI] [PubMed] [Google Scholar]
Capital‐RCT 2018 {published data only}
- Watanabe H, Ozasa N, Morimoto T, Shiomi H, Bingyuan B, Suwa S, et al. Long-term use of carvedilol in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. PloS One 2018;13(8):e0199347. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Curtis 1991 {published data only}
- Curtis JL, Houghton JL, Patterson JH, Koch G, Bradley DA, Adams KF Jr. Propranolol therapy alters estimation of potential cardiovascular risk derived from submaximal postinfarction exercise testing. American Heart Journal 1991;121(6 Pt 1):1655-64. [DOI] [PubMed] [Google Scholar]
E.I.S. 1984 {published data only}
- Bethge KP, Andresen D, Boissel JP, Leitner ER, Peyrieux JC, Schröder R, et al. Effect of oxprenolol on ventricular arrhythmias: the European Infarction Study experience. Journal of the American College of Cardiology 1985;6(5):963-72. [DOI] [PubMed] [Google Scholar]
- The European Infarction Study Group. A secondary prevention study with slow release oxprenolol after myocardial infarction: morbidity and mortality. European Heart Journal 1984;5:189-202. [PubMed] [Google Scholar]
- The European Infarction Study Group. A secondary prevention study with slow release oxprenolol after myocardial infarction. European Heart Journal 1982;3:583-6. [PubMed] [Google Scholar]
Julian 1982 {published data only}
- Julian DG, Prescott RJ, Jackson FS, Szekely P. Controlled trial of sotalol for one year after myocardial infarction. Lancet 1982;1(8282):1142-7. [DOI] [PubMed] [Google Scholar]
LIT 1987 {published data only}
- Lopressor Intervention Trial Research Group. The Lopressor Intervention Trial: multicentre study of metoprolol in survivors of acute myocardial infarction. European Heart Journal 1987;8:1056-64. [PubMed] [Google Scholar]
Mazur 1984 {published data only}
- Mazur NA, Kulginskaya IV, Ivanova LA, Ostrov Skaya TP, Smirnova TM, Svet EA, et al. Results of long-term propranolol treatment in myocardial infarction survivors with advanced grades of ventricular extrasystoles. Cor et Vasa 1984;26(4):241-7. [PubMed] [Google Scholar]
Mazzuero 1987 {published data only}
- Mazzuero G, Galdangelo F, Zotti AM, Bertolotti G, Tavazzi L. Effects of propranolol, atenolol, and chlordesmethyldiazepam on response to mental stress in patients with recent myocardial infarction. Clinical Cardiology 1987;10(6):293-302. [DOI] [PubMed] [Google Scholar]
MIS 1975 {published data only}
- A Multicentre International Study Group. Improvement in prognosis of myocardial infarction by long-term beta-adrenoreceptor blockade using practolol. BMJ 1975;3:735-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Multicentre International Study Group. Reduction in mortality after myocardial infarction with long-term beta-adrenoceptor blockade. BMJ 1977;2:419-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
NMS 1981 {published data only}
- Bellin EY, The Nowergian Multicenter Study Group. Six-year follow-up of the Norwegian Multicenter Study on timolol after myocardial infarction. 1986;314(16):1052-3. [DOI] [PubMed]
- Lund-Johansen, The Norwegian Multicenter Study Group. The Norwegian multicenter study on timolol after myocardial infarction. Part II. Effect in different risk groups, causes of death, heart arrest, reinfarctions, rehospitalizations and adverse experiences. Acta Medica Scandinavica 1981;651:243-52. [PubMed] [Google Scholar]
- Pedersen TR, The Norwegian Multicenter Study Group. Six-year follow-up of the Norwegian Multicenter Study on Timolol after Acute Myocardial Infarction. New England Journal of Medicine 1985;313(17):1055-8. [DOI] [PubMed] [Google Scholar]
- The Norwegian Multicenter Study Group. The Norwegian multicenter study on timolol after myocardial infarction. Acta Medica Scandinavica 1981;651:235-41. [DOI] [PubMed] [Google Scholar]
- The Norwegian Multicenter Study Group. Timolol-induced reduction in mortality and reinfarction in patients surviving acute myocardial infarction. New England Journal of Medicine 1981;304(14):801-7. [DOI] [PubMed] [Google Scholar]
NPT 1982 {published data only}
- Hansteen V, Møinichen E, Lorentsen E, Andersen A, Strøm O, Søiland K, et al. One year's treatment with propranolol after myocardial infarction: preliminary report of Norwegian multicentre trial. BMJ 1982;284:155-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansteen V. Beta blockade after myocardial infarction: the Norwegian propranolol study in high-risk patients. Circulation 1983;67(Suppl 1):I57-60. [PubMed] [Google Scholar]
- Hansteen V. The Norwegian propranolol trial in selected patients. British Journal of Clinical Pharmacology 1982;14:9S-12S. [DOI] [PMC free article] [PubMed] [Google Scholar]
Olsson 1985 {published data only}
- Olsson G, Levin LA, Rehnqvist N. Economic consequences of postinfarction prophylaxis with beta blockers: cost effectiveness of metoprolol. British Medical Journal (Clinical Research Edition) 1987;294(6568):339-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson G, Lubsen J, Es GA, Rehnqvist N. Quality of life after myocardial infarction: effect of long term metoprolol on mortality and morbidity. British Medical Journal (Clinical Research Edition) 1986;292(6534):1491-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson G, Odén A, Johansson L, Sjögren A, Rehnqvist N. Prognosis after withdrawal of chronic postinfarction metoprolol treatment: a 2-7 year follow-up. European Heart Journal 1988;9(4):365-72. [DOI] [PubMed] [Google Scholar]
- Olsson G, Rehnqvist N, Lundmand T, Melcher A. Metoprolol treatment after acute myocardial infarction - Effects on ventricular arrhythmias and exercise tests during 6 months. Acta Medica Scandinavica 1981;210(1-2):59-65. [PubMed] [Google Scholar]
- Olsson G, Rehnqvist N, Sjögren A, Erhardt L, Lundman T. Long-term treatment with metoprolol after myocardial infarction: effect on 3 year mortality and morbidity. Journal of the American College of Cardiology 1985;5(6):1428-37. [DOI] [PubMed] [Google Scholar]
Poulsen 1999 {published data only}
- Poulsen SH, Jensen SE, Egstrup K. Effects of long-term adrenergic beta-blockade on left ventricular diastolic filling in patients with acute myocardial infarction. American Heart Journal 1999;138(4 Pt 1):710-20. [DOI] [PubMed] [Google Scholar]
- Poulsen SH, Jensen SE, Egstrup K. Improvement of exercise capacity and left ventricular diastolic function with metoprolol XL after acute myocardial infarction. American Heart Journal 2000;140(1):E6-11. [DOI] [PubMed] [Google Scholar]
Schwartz 1992 {published data only}
- Schwartz PJ, Motolese M, Pollavini G, Lotto A, Ruberti U, Trazzi R, et al. Prevention of sudden cardiac death after a first myocardial infarction by pharmacologic or surgical antiadrenergic interventions. Journal of Cardiovascular Electrophysiology 1992;3(1):2-16. [Google Scholar]
Taylor 1982 {published data only}
- Taylor SH, Silke B, Ebbutt A, Sutton GC, Prout BJ, Burley DM. A long-term prevention study with oxprenolol in coronary heart disease. New England Journal of Medicine 1982;307(21):1293-301. [DOI] [PubMed] [Google Scholar]
Wilhelmsson 1974 {published data only}
- Vedin A, Wilhelmsson C, Werkö L. Chronic alprenolol treatment of patients with acute myocardial infarction after discharge from hospital. Acta Medica Scandinavica Supplementum 1975;575:3-56. [PubMed] [Google Scholar]
- Wihelmsson C, Vedin A, Wilhelmsen L, Tibblin G, Werkö L, Wedel H. Deaths and non-fatal reinfarctions during two years' follow-up. Acta Medica Scandinavica Supplementum 1975;575:19-24. [DOI] [PubMed] [Google Scholar]
- Wilhelmsson C, Vedin JA, Wilhelmsen L, Tibblin G, Werkö L. Reduction of sudden deaths after myocardial infarction by treatment with alprenolol. Preliminary results. Lancet 1974;2(7890):1157-60. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
CAPRICORN 2001 {published data only}
- Dargie HJ. Effect of carvedilol on outcome after myocardial infarction in patients with left-ventricular dysfunction: the CAPRICORN randomised trial. Lancet 2001;357(9266):1385-90. [DOI] [PubMed] [Google Scholar]
COMMIT 2005 {published data only}
- Chen ZM, Pan HC, Chen YP, Peto R, Collins R, Jiang LX, et al. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet (London, England) 2005;366(9497):1622-32. [PMID: ] [DOI] [PubMed] [Google Scholar]
Mazur 1994 {published data only}
- Mazur NA, Pavlova TS, Vasil'eva NN, Kul'ginskaia IV. [The comparative efficacy of verapamil, nifedipine and propranolol in patients with IHD and an enhanced risk of a fatal outcome] [Sravnitel'naia éffektivnost' verapamila, nifedipina i propranolola u bol'nykh IBS s povyshennym riskom letal'nogo iskhoda]. Terapevticheskii Arkhiv 1994;66(8):26-30. [PMID: ] [PubMed] [Google Scholar]
Mickley 1991 {published data only}
- Mickley H, Pless P, Nielsen JR, Møller M. Circadian variation of transient myocardial ischemia in the early out-of-hospital period after first acute myocardial infarction. American Heart Journal 1991;67(11):927-32. [DOI] [PubMed] [Google Scholar]
Park 2013 {published data only}
- Park H, Otani H, Noda T, Sato D, Okazaki T, Ueyama T, et al. Intracoronary followed by intravenous administration of the short-acting β-blocker landiolol prevents myocardial injury in the face of elective percutaneous coronary intervention. International Journal of Cardiology 2013;167(4):1547-51. [DOI] [PubMed] [Google Scholar]
Smith 2008 {published data only}
- Smith DH, Kramer JM, Perrin N, Platt R, Roblin DW, Lane K, et al. A randomized trial of direct-to-patient communication to enhance adherence to beta-blocker therapy following myocardial infarction. Archives of Internal Medicine 2008;168(5):477-83. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
BETAMI 2018 {published data only}
- Munkhaugen J, Ruddox V, Halvorsen S, Dammen T, Fagerland MW, Hernæs KH, et al. BEtablocker Treatment After acute Myocardial Infarction in revascularized patients without reduced left ventricular ejection fraction (BETAMI): rationale and design of a prospective, randomized, open, blinded end point study. American Heart Journal 2019;208:37-46. [PMID: ] [DOI] [PubMed] [Google Scholar]
DANBLOCK 2018 {published data only}
- Kristensen AMD, Bovin A, Zwisler AD, Cerquira C, Torp-Pedersen C, Bøtker HE, et al. Design and rationale of the Danish trial of beta-blocker treatment after myocardial infarction without reduced ejection fraction: study protocol for a randomized controlled trial. Trials 2020;21(1):415. [DOI] [PMC free article] [PubMed] [Google Scholar]
MINOCA‐BAT 2021 {published data only}
- Nordenskjöld AM, Agewall S, Atar D, Baron T, Beltrame J, Bergström O, et al. Randomized evaluation of beta blocker and ACE-inhibitor/angiotensin receptor blocker treatment in patients with myocardial infarction with non-obstructive coronary arteries (MINOCA-BAT): rationale and design. American Heart Journal 2021;231:96-104. [PMID: ] [DOI] [PubMed] [Google Scholar]
REBOOT‐CNIC2018 {published data only}
- Rossello X, Raposeiras-Roubin S, Latini R, Dominguez-Rodriguez A, Barrabés JA, Sánchez PL, et al. Rationale and design of the pragmatic clinical trial tREatment with Beta-blockers after myOcardial infarction withOut reduced ejection fracTion (REBOOT). European Heart Journal. Cardiovascular Pharmacotherapy 2021;August:pvab060. [PMID: ] [DOI] [PubMed] [Google Scholar]
The AβYSS Study 2018 {published data only}
- Assessment of βeta-Blocker Interruption After Uncomplicated Myocardial Infarction on Safety and Symptomatic Cardiac Events Requiring Hospitalization: The AβYSS Study. Ongoing study. 29 August 2018. Contact author for more information.
Additional references
Ahlquist 1948
- Ahlquist RP. A study of the adrenotropic receptors. American Journal of Physiology 1948;153(3):586-600. [PMID: ] [DOI] [PubMed] [Google Scholar]
Amsterdam 2014
- Amsterdam EA, Wenger NK, Brindis RG, Casey DE Jr, Ganiats TG, Holmes DR Jr, et al. 2014 AHA/ACC Guideline for the Management of Patients with Non-ST-Elevation Acute Coronary Syndromes: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Journal of the American College of Cardiology 2014;64(24):e139-228. [PMID: ] [DOI] [PubMed] [Google Scholar]
Antithrombotic Trialists' Collaboration 2002
- Antithrombotic Trialists' Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ (Clinical research ed.) 2002;324(7329):71-86. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Antman 2004
- Antman EM, Anbe DT, Armstrong PW, Bates ER, Green LA, Hand M, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction - executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to revise the 1999 guidelines for the management of patients with acute myocardial infarction). Journal of the American College of Cardiology 2004;44(3):671-719. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bangalore 2008
- Bangalore S, Wetterslev J, Pranesh S, Sawhney S, Gluud C, Messerli FH. Perioperative beta blockers in patients having non-cardiac surgery: a meta-analysis. Lancet 2008;372(9654):1962-76. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bangalore 2014
- Bangalore S, Makani H, Radford M, Thakur K, Toklu B, Katz SD, et al. Clinical outcomes with beta-blockers for myocardial infarction: a meta-analysis of randomized trials. American Journal of Medicine 2014;127(10):939-53. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bhatt 2017
- Bhatt AS, DeVore AD, DeWald TA, Swedberg K, Mentz RJ. Achieving a maximally tolerated beta-blocker dose in heart failure patients: is there room for improvement? Journal of the American College of Cardiology 2017;69(20):2542-50. [PMID: ] [DOI] [PubMed] [Google Scholar]
Biglane 2017
- Biglane JB, Becnel MF, Ventura HO, Krim SR. Pharmacologic therapy for heart failure with reduced ejection fraction: closing the gap between clinical guidelines and practice. Progress in Cardiovascular Diseases 2017;60(2):187-97. [PMID: ] [DOI] [PubMed] [Google Scholar]
Black 1964
- Black JW, Crowther AF, Shanks RG, Smith LH, Dornhorst AC. A new adrenergic beta receptor antagonist. Lancet 1964;1(7342):1080-1. [PMID: ] [DOI] [PubMed] [Google Scholar]
Brieger 2009
- Brieger D, Fox KA, Fitzgerald G, Eagle KA, Budaj A, Avezum A, et al. Predicting freedom from clinical events in non-ST-elevation acute coronary syndromes: the Global Registry of Acute Coronary Events. Heart (British Cardiac Society) 2009;95(11):888-94. [DOI] [PubMed] [Google Scholar]
Bristow 2000
- Bristow MR. Beta-adrenergic receptor blockade in chronic heart failure. Circulation 2000;101(5):558-69. [DOI] [PubMed] [Google Scholar]
Campeau 1976
- Campeau L. Letter: grading of angina pectoris. Circulation 1976;54(3):522-3. [PMID: ] [PubMed] [Google Scholar]
Chan 2013
- Chan A-W, Tetzlaff JM, Altman DG, Laupacis A, Gøtzsche PC, Krleža-Jerić K, et al. SPIRIT 2013 Statement: defining standard protocol items for clinical trials. Annals of Internal Medicine 2013;158(3):200-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chatterjee 2013
- Chatterjee S, Biondi-Zoccai G, Abbate A, D'Ascenzo F, Castagno D, Van Tassell B, et al. Benefits of beta blockers in patients with heart failure and reduced ejection fraction: network meta-analysis. BMJ (Clinical Research Ed) 2013;346:f55. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Collet 2020
Cooper 2000
- Cooper R, Cutler J, Desvigne-Nickens P, Fortmann SP, Friedman L, Havlik R, et al. Trends and disparities in coronary heart disease, stroke, and other cardiovascular diseases in the United States: findings of the national conference on cardiovascular disease prevention. Circulation 2000;102(25):3137-47. [PMID: ] [DOI] [PubMed]
Dahl 2019
- Dahl Aarvik M, Sandven I, Dondo TB, Gale CP, Ruddox V, Munkhaugen J, et al. Effect of oral β-blocker treatment on mortality in contemporary post-myocardial infarction patients: a systematic review and meta-analysis. European Heart Journal. Cardiovascular Pharmacotherapy 2019;5(1):12-20. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JP, Altman DG (editors). Chapter 9. Analysing data and undertaking meta-analyses. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
DeMets 1987
- DeMets DL. Methods for combining randomized clinical trials: strengths and limitations. Statistics in Medicine 1987;6(3):341-50. [PMID: ] [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta-analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177-88. [PMID: ] [DOI] [PubMed] [Google Scholar]
Devereaux 2008
- Devereaux PJ, Yang H, Yusuf S, Guyatt G, Leslie K, Villar JC, et al. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet 2008;371(9627):1839-47. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ (Clinical Research Ed) 1997;315(7109):629-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta-analyses involving cross-over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ferreira 2021
- Ferreira JA, Baptista RM, Monteiro SR, Gonçalves LM. Usefulness of universal beta-blocker therapy in patients after ST-elevation myocardial infarction. Medicine 2021;100(3):e23987. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Freemantle 1999
- Freemantle N, Cleland J, Young P, Mason J, Harrison J. Beta blockade after myocardial infarction: systematic review and meta regression analysis. BMJ (Clinical Research Ed) 1999;318(7200):1730-7. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Garattini 2016
- Garattini S, Jakobsen JC, Wetterslev J, Bertele V, Banzi R, Rath A, et al. Evidence-based clinical practice: overview of threats to the validity of evidence and how to minimise them. European Journal of Internal Medicine 2016;32:13-21. [DOI] [PubMed] [Google Scholar]
Gluud 2006
- Gluud LL. Bias in clinical intervention research. American Journal of Epidemiology 2006;163(6):493-501. [DOI] [PubMed] [Google Scholar]
Golan 2012
- Golan DE, Tashjian AH Jr, Armstrong EJ, Armstrong AW. Principles of Pharmacology - The Pathophysiologic Basis of Drug Therapy. 3rd edition. Philadelphia, PA: Lippincott Williams & Wilkins, 2012. [Google Scholar]
Grandi 2019
- Grandi E, Ripplinger CM. Antiarrhythmic mechanisms of beta blocker therapy. Pharmacological Research 2019;146:104274. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Greenblatt 1974
- Greenblatt DJ, Koch-Weser J. Adverse reactions to beta-adrenergic receptor blocking drugs: a report from the Boston collaborative drug surveillance program. Drugs 1974;7(1):118-29. [PMID: ] [DOI] [PubMed] [Google Scholar]
Guyatt 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck-Ytter Y, Alonso-Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ (Clinical Research Ed) 2008;336(7650):924-6. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Harbord 2006
- Harbord RM, Egger M, Sterne JA. A modified test for small-study effects in meta-analyses of controlled trials with binary endpoints. Statistics in Medicine 2006;25(20):3443-57. [PMID: ] [DOI] [PubMed] [Google Scholar]
Higgins 2002
- Higgins JT, Thompson SG. Quantifying heterogeneity in a meta-analysis. Statistics in Medicine 2002;21(11):1539-58. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003;327(7414):557-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JT, Altman DG, Sterne JAC (editors). Chapter 8. Assessing risk of bias in included studies. In: Higgins JT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011c
- Higgins JT, Whitehead A, Simmonds M. Sequential methods for random-effects meta-analysis. Statistics in Medicine 2011;30(9):903-21. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011d
- Higgins JT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0. The Cochrane Collaboration, 2011. Available from handbook.cochrane.org (updated March 2011):10.4.3.1 Recommendations on testing for funnel plot asymmetry. [Google Scholar]
Houston 1981
- Houston MC. Abrupt cessation of treatment in hypertension: consideration of clinical features, mechanisms, prevention and management of the discontinuation syndrome. American Heart Journal 1981;102(3 Pt 1):415-30. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hrobjartsson 2012
- Hrobjartsson A, Thomsen AS, Emanuelsson F, Tendal B, Hilden J, Boutron I, et al. Observer bias in randomised clinical trials with binary outcomes: systematic review of trials with both blinded and non-blinded outcome assessors. BMJ (Clinical Research Ed.) 2012;344:e1119. [DOI] [PubMed] [Google Scholar]
Hrobjartsson 2013
- Hrobjartsson A, Thomsen ASS, Emanuelsson F, Tendal B, Hilden J, Boutron I, et al. Observer bias in randomized clinical trials with measurement scale outcomes: a systematic review of trials with both blinded and nonblinded assessors. Canadian Medical Association Journal 2013;185(4):E201-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hrobjartsson 2014
- Hrobjartsson A, Emanuelsson F, Skou Thomsen AS, Hilden J, Brorson S. Bias due to lack of patient blinding in clinical trials. A systematic review of trials randomizing patients to blind and nonblind sub-studies. International Journal of Epidemiology 2014;43(4):1272-83. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hrobjartsson 2014a
- Hrobjartsson A, Thomsen AS, Emanuelsson F, Tendal B, Rasmussen JV, Hilden J, et al. Observer bias in randomized clinical trials with time-to-event outcomes: systematic review of trials with both blinded and non-blinded outcome assessors. International Journal of Epidemiology 2014;43(3):937-48. [DOI] [PubMed] [Google Scholar]
Huang 2015
- Huang BT, Huang FY, Zuo ZL, Liao YB, Heng Y, Wang PJ, et al. Meta-analysis of relation between oral β-blocker therapy and outcomes in patients with acute myocardial infarction who underwent percutaneous coronary intervention. American Journal of Cardiology 2015;115(11):1529-38. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ibanaz 2015
- Ibanaz B, Heusch G, Ovize M, Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. Journal of the American College of Cardiology 2015;65(14):1454-71. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ibanez 2017
- Ibanez B, James S, Agewall S, Antunes MJ, Bucciarelli-Ducci C, Bueno H, et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: the Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). European Heart Journal 2018;39(2):119-77. [DOI] [PubMed] [Google Scholar]
ICH‐GCP 2015
- ICH harmonised guideline: integrated addemdum to ICH E6(R1): Guideline for Good Clinical Practice E6(R2) ICH Consensus Guideline (ICH-GCP). Federal Register 2015.
Jakobsen 2014
- Jakobsen JC, Wetterslev J, Winkel P, Lange T, Gluud C. Thresholds for statistical and clinical significance in systematic reviews with meta-analytic methods. BMC Medical Research Methodology 2014;14:120. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jakobsen 2017
- Jakobsen JC, Gluud C, Wetterslev J, Winkel P. When and how should multiple imputation be used for handling missing data in randomised clinical trials - a practical guide with flowcharts. BMC Medical Research Methodology 2017;17(1):162. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Juul 2006
- Juul AB, Wetterslev J, Gluud C, Kofoed-Enevoldsen A, Jensen G, Callesen T, et al. Effect of perioperative beta blockade in patients with diabetes undergoing major non-cardiac surgery: randomised placebo controlled, blinded multicentre trial. BMJ (Clinical Research Ed) 2006;332(7556):1482. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kjaergard 2001
- Kjaergard LL, Villumsen J, Gluud C. Reported methodologic quality and discrepancies between large and small randomized trials in meta-analyses. Annals of Internal Medicine 2001;135(11):982-9. [DOI] [PubMed] [Google Scholar]
Ko 2002
- Ko DT, Hebert PR, Coffey CS, Sedrakyan A, Curtis JP, Krumholz HM. Beta-blocker therapy and symptoms of depression, fatigue, and sexual dysfunction. JAMA 2002;288(3):351-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kotecha 2017
- Kotecha D, Flather MD, Altman DG, Holmes J, Rosano G, Wikstrand J, et al. Heart rate and rhythm and the benefit of beta-blockers in patients with heart failure. Journal of the American College of Cardiology 2017;69(24):2885-96. [PMID: ] [DOI] [PubMed] [Google Scholar]
Køber 1995
- Køber L, Torp-Pedersen C, Carlsen JE, Bagger H, Eliasen P, Lyngborg K, et al. A clinical trial of the angiotensin-converting-enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction. Trandolapril Cardiac Evaluation (TRACE) Study Group. New England Journal of Medicine 1995;333(25):1670-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6. Searching for studies. In: Higgins JT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Lewis 1982
- Lewis JA. Beta-blockade after myocardial infarction - a statistical view. British Journal of Clinical Pharmacology 1982;14 Suppl 1:15S-21S. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lloyd‐Jones 2010
- Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, et al. Executive summary: heart disease and stroke statistics - 2010 update: a report from the American Heart Association. Circulation 2010;121(7):948-54. [PMID: ] [DOI] [PubMed] [Google Scholar]
Lopez‐Sendon 2004
- Lopez-Sendon J, Swedberg K, McMurray J, Tamargo J, Maggioni AP, Dargie H, et al. Expert consensus document on beta-adrenergic receptor blockers. European Heart Journal 2004;25(15):1341-62. [PMID: ] [DOI] [PubMed] [Google Scholar]
Lubbe 1992
- Lubbe WF, Podzuweit T, Opie LH. Potential arrhythmogenic role of cyclic adenosine monophosphate (AMP) and cytosolic calcium overload: implications for prophylactic effects of beta-blockers in myocardial infarction and proarrhythmic effects of phosphodiesterase inhibitors. Journal of the American College of Cardiology 1992;19(7):1622-33. [PMID: ] [DOI] [PubMed] [Google Scholar]
Lundh 2012
- Lundh A, Sismondo S, Lexchin J, Busuioc OA, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2012, Issue 12. Art. No: MR000033. [DOI: 10.1002/14651858.MR000033.pub2] [DOI] [PubMed] [Google Scholar]
Lundh 2017
- Lundh A, Lexchin J, Mintzes B, Schroll JB, Bero L, Sismondo S, et al. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2017, Issue 2. Art. No: MR000033. [DOI: 10.1002/14651858.MR000033.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mancia 2013
- Mancia G, Fagard R, Narkiewicz K, Redon J, Zanchetti A, Böhm M, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). European Heart Journal 2013;34(28):2159-219. [DOI] [PubMed] [Google Scholar]
Marlin 1975
- Marlin GE, Kumana CR, Kaye CM, Smith DM, Turner P. An investigation into the cardiac and pulmonary beta-adrenoceptor blocking activity of ICI 66,082 in man. British Journal of Clinical Pharmacology 1975;2(2):151-7. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mendis 2011
- Mendis S, Thygesen K, Kuulasmaa K, Giampaoli S, Mähönen M, Ngu Blackett K, et al. World Health Organization definition of myocardial infarction: 2008-09 revision. International Journal of Epidemiology 2011;40(1):139-46. [PMID: ] [DOI] [PubMed] [Google Scholar]
Misumida 2016
- Misumida N, Harjai K, Kernis S, Kanei Y. Does oral beta-blocker therapy improve long-term survival in ST-segment elevation myocardial infarction with preserved systolic function? A meta-analysis. Journal of Cardiovascular Pharmacology and Therapeutics 2016;21(3):280-5. [PMID: ] [DOI] [PubMed] [Google Scholar]
Moher 1998
- Moher D, Pham B, Jones A, Cook DJ, Jadad AR, Moher M, et al. Does quality of reports of randomised trials affect estimates of intervention efficacy reported in meta-analyses? Lancet 1998;352(9128):609-13. [DOI] [PubMed] [Google Scholar]
Montalescot 2013
- Montalescot G, Sechtem U, Achenbach S, Andreotti F, Arden C, Budaj A. 2013 ESC guidelines on the management of stable coronary artery disease: the Task Force on the management of stable coronary artery disease of the European Society of Cardiology. European Heart Journal 2013;34(38):2949-3003. [DOI] [PubMed] [Google Scholar]
NICE 2013
- National Clinical Guideline Centre (UK) MI - Secondary Prevention: Secondary Prevention in Primary and Secondary Care for Patients Following a Myocardial Infarction: Partial Update of NICE CG48. London: Royal College of Physicians (UK) 2013;PMID: 25340229.. [PubMed] [Google Scholar]
Nichols 2014
- Nichols M, Townsend N, Scarborough P, Rayner M. Cardiovascular disease in Europe 2014: epidemiological update. European Heart Journal 2014;35(42):2950-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Nielsen 2017
- Nielsen EE, Feinberg J, Safi S, Sethi NJ, Gluud C, Jakobsen JC. Beta-blockers for non-acute treatment after myocardial infarction. Cochrane Database of Systematic Reviews Feb 2017, Issue 1. Art. No: CD012565. [DOI: 10.1002/14651858.CD012565] [DOI] [Google Scholar]
Noble 2017
- Noble S, Roffi M. Routine beta-blocker administration following acute myocardial infarction: why still an unsolved issue? Journal of Thoracic Disease 2017;9(11):4191-4. [PMID: ] [DOI] [PMC free article] [PubMed]
O'Gara 2013
- O'Gara PT, Kushner FG, Ascheim DD, Casey DE, Chung MK, Lemos JA, et al. 2013 ACCF/AHA Guideline for the Management of ST-Elevation Myocardial Infarction. Journal of the American College of Cardiology 2013;61(4):104. [DOI] [PubMed] [Google Scholar]
Pfeffer 1997
- Pfeffer MA, Greaves SC, Arnold JM, Glynn RJ, LaMotte FS, Lee RT, et al. Early versus delayed angiotensin-converting enzyme inhibition therapy in acute myocardial infarction. The healing and early afterload reducing therapy trial. Circulation 1997;95(12):2643-51. [PMID: ] [DOI] [PubMed] [Google Scholar]
Pohjola‐Sintonen 1989
- Pohjola-Sintonen S, Muller JE, Stone PH, Willich SN, Antman EM, Davis VG, et al. Ventricular septal and free wall rupture complicating acute myocardial infarction: experience in the Multicenter Investigation of Limitation of Infarct Size. American Heart Journal 1989;117(4):809-18. [DOI] [PubMed] [Google Scholar]
Qamar 2020
Quirke 2006
- Quirke V. Theory into practice: James Black, receptor theory and the development of the beta-blockers at ICI, 1958-1978. Medical History 2006;50(1):69-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: The Nordic Cochrane Centre: The Cochrane Collaboration, 2014.
Rodgers 2019
- Rodgers JL, Jones J, Bolleddu SI, Vanthenapalli S, Rodgers LE, Shah K, et al. Cardiovascular risks associated with gender and aging. Journal of Cardiovascular Development and Disease 2019;6(2):19. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Roffi 2016
- Roffi M, Patrono C, Collet JP, Mueller C, Valgimigli M, Andreotti F, et al. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: Task Force for the Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation of the European Society of Cardiology (ESC). European Heart Journal 2016;37(3):267-315. [DOI] [PubMed] [Google Scholar]
Rosamond 2008
- Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, et al. Heart disease and stroke statistics - 2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2008;117(4):e25-146. [PMID: ] [DOI] [PubMed] [Google Scholar]
Royle 2003
- Royle P, Milne R. Literature searching for randomized controlled trials used in Cochrane Reviews: rapid versus exhaustive searches. International Journal of Technology Assessment in Health Care 2003;19(4):591-603. [DOI] [PubMed] [Google Scholar]
Rücker 2008
- Rücker G, Schwarzer G, Carpenter J. Arcsine test for publication bias in meta-analyses with binary outcomes. Statistics in Medicine 2008;27(5):746-63. [PMID: ] [DOI] [PubMed] [Google Scholar]
Safi 2019
- Safi S, Sethi NJ, Nielsen EE, Feinberg J, Gluud C, Jakobsen JC. Beta-blockers for suspected or diagnosed acute myocardial infarction. Cochrane Database of Systematic Reviews 2019;(12). [DOI: 10.1002/14651858.CD012484.pub2] [CD012484] [DOI] [PMC free article] [PubMed]
Savovic 2012
- Savovic J, Jones HE, Altman DG, Harris RJ, Juni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomized, controlled trials. Annals of Internal Medicine 2012;157(6):429-38. [DOI] [PubMed] [Google Scholar]
Savovic 2012a
- Savovic J, Jones H, Altman D, Harris R, Juni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomised controlled trials: combined analysis of meta-epidemiological studies. Health Technology Assesment 2012;16(35):1-82. [DOI] [PubMed] [Google Scholar]
Savovic 2018
- Savovic J, Turner RM, Mawdsley D, Jones HE, Beynon R, Higgins JT, et al. Association between risk-of-bias assessments and results of randomized trials in Cochrane reviews: the ROBES meta-epidemiologic study. American Journal of Epidemiology 2018;187(5):1113-22. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schmidt 2012
- Schmidt M, Jacobsen JB, Lash TL, Botker HE, Sorensen HT. 25 year trends in first time hospitalisation for acute myocardial infarction, subsequent short and long term mortality, and the prognostic impact of sex and comorbidity: a Danish nationwide cohort study. BMJ (Clinical Research Ed) 2012;344:e356. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273(5):408-12. [DOI] [PubMed] [Google Scholar]
Schulz 2010
- Schulz KF, Altman DG, Moher D, CONSORT Group. CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials. Annals of Internal Medicine 2010;152(11):726-32. [DOI] [PubMed] [Google Scholar]
Singh 1975
- Singh BN, Nisbet HE, Harris EA, Whitlock RM. A comparison of the actions of ICI66082 and propranolol on cardiac and peripheral beta-adrenoceptors. European Journal of Pharmacology 1975;34(1):75-86. [PMID: ] [DOI] [PubMed] [Google Scholar]
Smith 2011
- Smith SCJ, Benjamin EJ, Bonow RO, Braun LT, Creager MA, Franklin BA et al. AHA/ACCF secondary prevention and risk reduction therapy for patients with coronary and other atherosclerotic vascular disease: 2011 update: a guideline from the American Heart Association and American College of Cardiology Foundation. Circulation 2011;124:2458–73. [DOI] [PubMed] [Google Scholar]
STATA 2016 [Computer program]
- StataCorp; Stata Statistical Software. College Station: TX: StataCorp LLC, 2016. http://www.stata.com.
Steg 2012
- Steg PG, James SK, Atar D, Badano LP, Blomstrom-Lundqvist C, Borger MA, et al. ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. European Heart Journal 2012;33(20):2569-619. [DOI] [PubMed] [Google Scholar]
Steinhubl 2009
- Steinhubl SR, Bhatt DL, Brennan DM, Montalescot G, Hankey GJ, Eikelboom JW, et al. Aspirin to prevent cardiovascular disease: the association of aspirin dose and clopidogrel with thrombosis and bleeding. Annals of Internal Medicine 2009;150(6):379-86. [PMID: ] [DOI] [PubMed] [Google Scholar]
Sterne 2009
- Sterne JA, White IR, Carlin JB, Spratt M, Royston P, Kenward MG, et al. Multiple imputation for missing data in epidemiological and clinical research: potential and pitfalls. BMJ (Clinical Research Ed) 2009;338:b2393. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Stevenson 1989
- Stevenson WG, Linssen GC, Havenith MG, Brugada P, Wellens HJ. The spectrum of death after myocardial infarction: a necropsy study. American Heart Journal 1989;118(6):1182-8. [DOI] [PubMed] [Google Scholar]
Sweeting 2004
- Sweeting MJ, Sutton AJ, Lambert PC. What to add to nothing? Use and avoidance of continuity corrections in meta-analysis of sparse data. Statistics in Medicine 2004;23(9):1351-75. [PMID: ] [DOI] [PubMed] [Google Scholar]
Taylor 1982
- Taylor SH, Silke B, Lee PS. Intravenous beta-blockade in coronary heart disease: is cardioselectivity or intrinsic sympathomimetic activity hemodynamically useful? New England Journal of Medicine 1982;306(11):631-5. [PMID: ] [DOI] [PubMed] [Google Scholar]
Thombs 2006
- Thombs BD, Bass EB, Ford DE, Stewart KJ, Tsilidis KK, Patel U, et al. Prevalence of depression in survivors of acute myocardial infarction: review of the evidence. Journal of General Internal Medicine 2006;21(1):30-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Thygesen 2007
- Thygesen K, Alpert JS, White HD. Universal definition of myocardial infarction. European Heart Journal 2007;28(20):2525-38. [PMID: ] [DOI] [PubMed] [Google Scholar]
Thygesen 2012
- Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD. Third universal definition of myocardial infarction. Global Heart 2012;7(4):275-95. [PMID: ] [DOI] [PubMed] [Google Scholar]
Thygesen 2018
- Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, et al. Fourth universal definition of myocardial infarction. Journal of the American College of Cardiology 2018;72(18):2231-64. [PMID: ] [DOI] [PubMed] [Google Scholar]
Thygesen 2020
Turner 1994
- Turner SM, Beidel DC, Jacob RG. Social phobia: a comparison of behavior therapy and atenolol. Journal of Consulting and Clinical Psychology 1994;62(2):350-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Waal 1967
- Waal HJ. Propranolol-induced depression. British Medical Journal 1967;2(5543):50. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2020
- Wang X, Fernandez Robles C, Kertai MD. Perioperative β-blocker use: what is new in 2020? Current Opinion in Anaesthesiology 2020;33(3):417-22. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ware 1992
- Ware JEJ, Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Medical Care 1992;30(6):473-83. [PubMed] [Google Scholar]
Warren 1977
WHO 2015
- World Health Organization (WHO). Cardiovascular disease. www.who.int/cardiovascular_diseases/en/ (date accessed 7 February 2017). January 2015.
Wiysonge 2012
- Wiysonge CS, Bradley HA, Volmink J, Mayosi BM, Mbewu A, Opie LH, et al. Beta-blockers for hypertension. Cochrane Database of Systematic Reviews 2012;11:CD002003. [PMID: ] [DOI] [PubMed] [Google Scholar]
Wood 2008
- Wood L, Egger M, Gluud LL, Schulz KF, Juni P, Altman DG, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta-epidemiological study. BMJ (Clinical Research Ed) 2008;336(7644):601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yancy 2013
- Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DEJ, Drazner MH, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013;128:e240–e327. [DOI] [PubMed] [Google Scholar]
Yusuf 1985
- Yusuf S, Peto R, Lewis J, Collins R, Sleight P. Beta blockade during and after myocardial infarction: an overview of the randomized trials. Progress in Cardiovascular Diseases 1985;27(5):335-71. [PMID: ] [DOI] [PubMed] [Google Scholar]