

Abstract
Liposomes, a nanoscale drug delivery system, are well known for their ability to improve pharmacokinetics and reduce drug toxicity. In this work, maltoheptaose (G7)-presenting glycoliposomes were synthesized and evaluated in the delivery of the antibiotic rifampicin. Two types of liposomes were prepared: nonfluid liposomes from l-α-phosphatidylcholine (PC) and cholesterol, and fluid liposomes from 1,2-dipalmitoyl-sn-glycero-3-phosphocholine and 1,2-dimyristoyl-sn-glycero-3-phospho-(1′-rac-glycerol). G7-derivatized glycolipid, G7-DPPE (DPPE: 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine), was incorporated into the liposomes at 21 and 14 μmol/mg to form nanoparticles of 75 ± 12 and 146 ± 14 nm for the nonfluid and fluid G7-glycoliposomes, respectively. The multivalent G7-glycoliposomes were characterized by lectin binding with concanavalin A (Con A). The dissociation constant Kd between Con A and the nonfluid or fluid G7-glycoliposomes was 0.93 or 0.51 μM, which represented ~900- or 1600-fold stronger affinity than the binding between Con A and G7. The G7-glycoliposomes were loaded with rifampicin at 6.6 and 16 wt % encapsulation for the nonfluid and fluid G7-glycoliposomes, respectively. Introducing a carbohydrate in the liposomes slowed down the release of rifampicin, with the G7-glycoliposomes having the slowest release rate and the lowest permeability coefficient among the liposome formulations. The fluid G7-glycoliposomes lowered the minimal inhibitory concentration (MIC) of rifampicin against E. coli ORN208 by about 3 times, whereas liposomes without G7 or Man (d-mannose)-glycoliposomes showed no improvement in MIC. The rifampicin-loaded fluid G7-glycoliposomes demonstrated the best sustained antibacterial activity against E. coli, with up to 2 log reduction in the colony forming units at 4 × MIC after 24 h. Fluorescence resonance energy transfer and confocal fluorescence microscopy revealed stronger interactions of the bacterium with the fluid G7-glycoliposomes than other liposome formulations.
Keywords: maltoheptaose, glycoliposomes, E. coli, rifampicin, drug delivery
Graphical Abstract
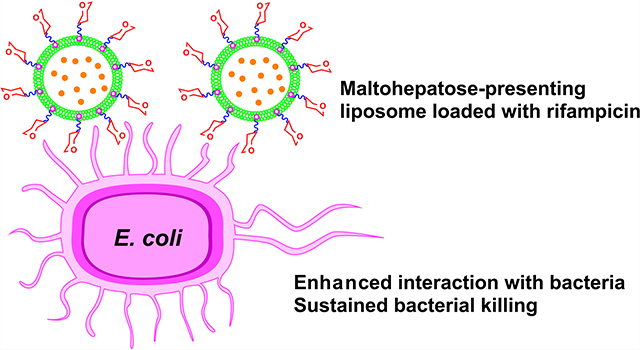
INTRODUCTION
Nanoparticle-based drug delivery has witnessed impressive progress in the past several decades owing to the unique physicochemical properties of nanoparticles.1 Nanoparticles can increase the uptake of drugs into target cells, leading to higher intracellular drug concentrations, higher therapeutic efficacy, and lower off-target toxicity than their small-molecule counterparts by altering the biodistribution of the drugs. Nanoparticle-based delivery platforms such as liposomes, polymeric nanoparticles, dendrimers, and inorganic nanoparticles have been used in formulating antimicrobial therapies, showing improved pharmacokinetic and therapeutic performance compared to the free drugs.2,3 Liposome drug delivery vehicles have been widely studied and are well known for their ability to improve pharmacokinetics and biodistribution and to reduce drug toxicity. Of the various nanoparticle-based drug delivery systems, liposomes and polymer nanoparticles constitute the vast majority of Food and Drug Administration (FDA)- and European Medicine Agency-approved formulations.4,5 For example, a liposomal formulation of amphotericin B, trademarked as AmBisome by Gilead Sciences, was approved by FDA in 1997 to treat a variety of serious fungal infections with reduced nephrotoxicity.6
Some liposomal formulations are also shown to fuse with the outer membranes (OMs) of Gram-negative bacteria and improve the penetration of the antimicrobial payloads into the bacterial cells. For example, tobramycin-encapsulated fluid liposomes prepared from 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1,2-dimyristoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DMPG) were shown to fuse with the OM of P. aeruginosa.7,8 The fusion process increased the antibiotic penetration to the bacterium, leading to increased intracellular drug concentration by overcoming the OM barrier. The fusion process was also observed in other liposome formulations.9,10 For example, Chen and co-workers showed that the efficiency of the fusion process played a key role in the antibacterial activity of tobramycin encapsulated in fluid liposomes, possibly involving cationic bridging between the bacterium and the DMPG phospholipid on the fluid liposome.11,12
The ability to target bacteria is another feature of the liposome-based antibiotic delivery systems. Targeting can be achieved by conjugating a specific targeting moiety to the liposome carrier that can bind to the receptor on the bacterium. Thus, antibiotics-loaded liposomes decorated with the targeting ligand could be directed to a particular bacterium or infected tissues, resulting in the local release of antibiotics on or into the bacterium.13–15 Carbohydrates are found on the surface of all cells. Through interactions with carbohydrate-binding proteins, cell surface carbohydrates mediate many cellular processes including bacterial and viral infections.16–18 As such, carbohydrates have also been used as targeting moieties in liposome-based antibiotic delivery.19,20 In this regard, bacterium-specific carbohydrates that can selectively target the bacterium in the presence of mammalian cells are especially useful in drug delivery.21 We have shown that trehalose-functionalized nanoparticles selectively targeted mycobacteria and had minimal interactions with macrophage or mammalian cells.22,23 Additionally, the antibiotic isoniazid, when encapsulated in mesoporous silica nanoparticles functionalized with trehalose, inhibited mycobacterial growth at over 8-folds better than the free antibiotic.24,25 Maltodextrin is another bacterium-specific carbohydrate. For example, Murthy and co-workers developed a maltohexaose-based fluorescence imaging probe to detect E. coli with high in vivo specificity.26 We have also shown that silica nanoparticles functionalized with G7 resulted in significantly increased surface binding with E. coli.27
In this work, we employed liposomes as the delivery system to test the hypothesis that G7 as the targeting ligand would increase the surface binding of G7-liposomes with E. coli and antibiotic-loaded G7-liposomes would have improved bacterial killing properties. Toward this end, a G7-conjugated glycolipid was synthesized and subsequently formulated into fluid and nonfluid liposomes by lipid film hydration or ethanol injection. Lectin interaction assays demonstrated the successful incorporation of G7-glycolipid into the liposomes, showing 2–3 orders of magnitude stronger binding affinity between lectin concanavalin A (Con A) and G7-glycoliposomes than the free G7. Rifampicin, an antibiotic that acts by inhibiting the DNA-dependent bacterial RNA polymerase and preventing RNA transcription, was chosen as a model antibiotic in this study.28,29 Rifampicin is a broad-spectrum semisynthetic antibiotic and is active against both Gram-negative and Gram-positive bacteria including several forms of mycobacteria. It is, however, less active against Gram-negative than the Gram-positive bacteria, attributed to the lower OM permeability coupled with active efflux pumps from Gram-negative bacteria like E. coli.30,31 Rifampicin is used at a relatively high oral dose (10 mg/kg daily), which causes side effects such as hepatotoxicity.32,33 Additionally, rifampicin is an amphiphilic molecule and is unstable in aqueous solution.34 As such, various formulations have been explored, including liposomes, to improve its storage stability, reduce systemic exposure, and minimize adverse effects.35,36 In this work, we found that rifampicin-loaded fluid liposomes exhibited higher antimicrobial activities than nonfluid liposomes against E. coli. The bacterial killing was also more efficient for fluid liposomes, for which the fluid G7-glycoliposomes showed the best sustained killing of E. coli over time. Fluorescence resonance energy transfer (FRET) and confocal fluorescence microscopy revealed enhanced interactions of fluid G7-glycoliposomes with the bacterium.
RESULTS AND DISCUSSION
Synthesis of Glycolipids and Glycoliposomes.
For comparison, another glycoliposome, Man-decorated glycoliposome, was also prepared. Man-conjugated glycolipid, Man-DPPE, was synthesized following the reaction sequence in Scheme 1. N-Hydroxysuccinimide (NHS)-activated mannoside 5 is the key intermediate in the synthesis of Man-DPPE and was prepared from the starting material methyl α-s-mannopyranoside in five steps.37,38 Briefly, methyl α-d-mannopyranoside was treated with benzyl bromide to give perbenzyl-protected mannoside 1. Subsequent selective deprotection of the anomeric OH under acidic conditions afforded compound 2, which then reacted with t-butyl bromoacetate in the presence of KOH and tetra-n-butylammonium bromide (TBAB) in toluene to give the α-anomer 3 in an 85% yield. Deprotection of t-butyloxycarbonyl (Boc) group in 3 by trifluoroacetic acid (TFA) gave compound 4, which reacted with NHS to afford mannoside 5. The product Man-DPPE was obtained by coupling 5 with DPPE-NH2 under mild basic conditions followed by deprotection of the benzyl ether (Bn) groups via palladium-catalyzed hydrogenation.
Scheme 1. Synthesis of Man-DPPEa.
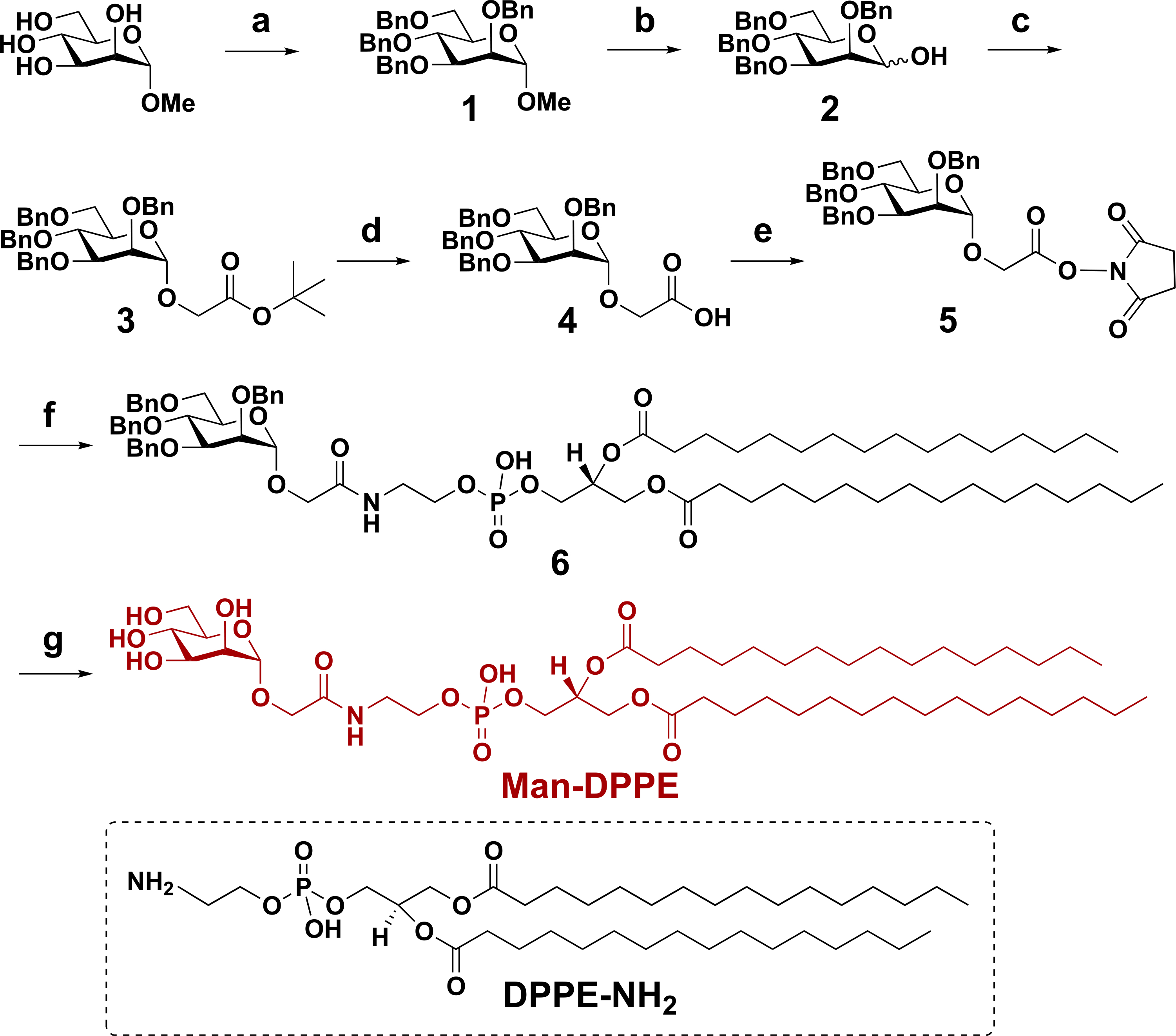
a(a) Benzyl bromide, NaH, tetra-n-butylammonium iodide, N,N-dimethylformamide (DMF), 0 °C, RT, 12 h (90%); (b) glacial acetic acid, HCl, 85 °C, 7 h (45%); (c) t-butyl 2-bromoacetate, KOH, TBAB, toluene, 0 °C, RT, 1 h (85%); (d) TFA, dichloromethane (DCM), RT, 45 min (83%); (e) NHS, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDAC), DCM, 24 h (90%); (f) DPPE-NH2, triethylamine, DMF, 16 h (88%); and (g) Pd/C, H2, THF/MeOH, 24 h (quantitative yield).
An S-linked G7-DPPE was designed as the synthetic protocol was more efficient than that of the O-linked analogue (Scheme 2).39,40 G7-DPPE was synthesized by coupling carboxyl-derivatized DPPE (17) with an amine-derivatized G7 (12), which was prepared from β-cyclodextrin (β-CD) in six steps following established protocols.23,41 Briefly, β-CD was acetylated with acetic anhydride catalyzed by FeCl3. Subsequent ring opening gave peracetylated G7 (7) in a combined 33% yield.42 Bromination of compound 7 at the anomeric position afforded glycosyl bromide 8, which yielded thioglycoside 9 after reaction with potassium thioacetate (KSAc) in acetone. Deacetylation of 9 gave maltoheptaose thiolate 10, which was further alkylated with an iodo azide 16 (Scheme S1; see SI for detailed synthesis)43 to afford compound 11. Catalytic hydrogenation of azide 11 gave the corresponding amine 12, which was coupled with compound 17 (Scheme S2; see SI for detailed synthesis) using EDAC to yield the final glycolipid G7-DPPE.
Scheme 2. Synthesis of G7-DPPEb.
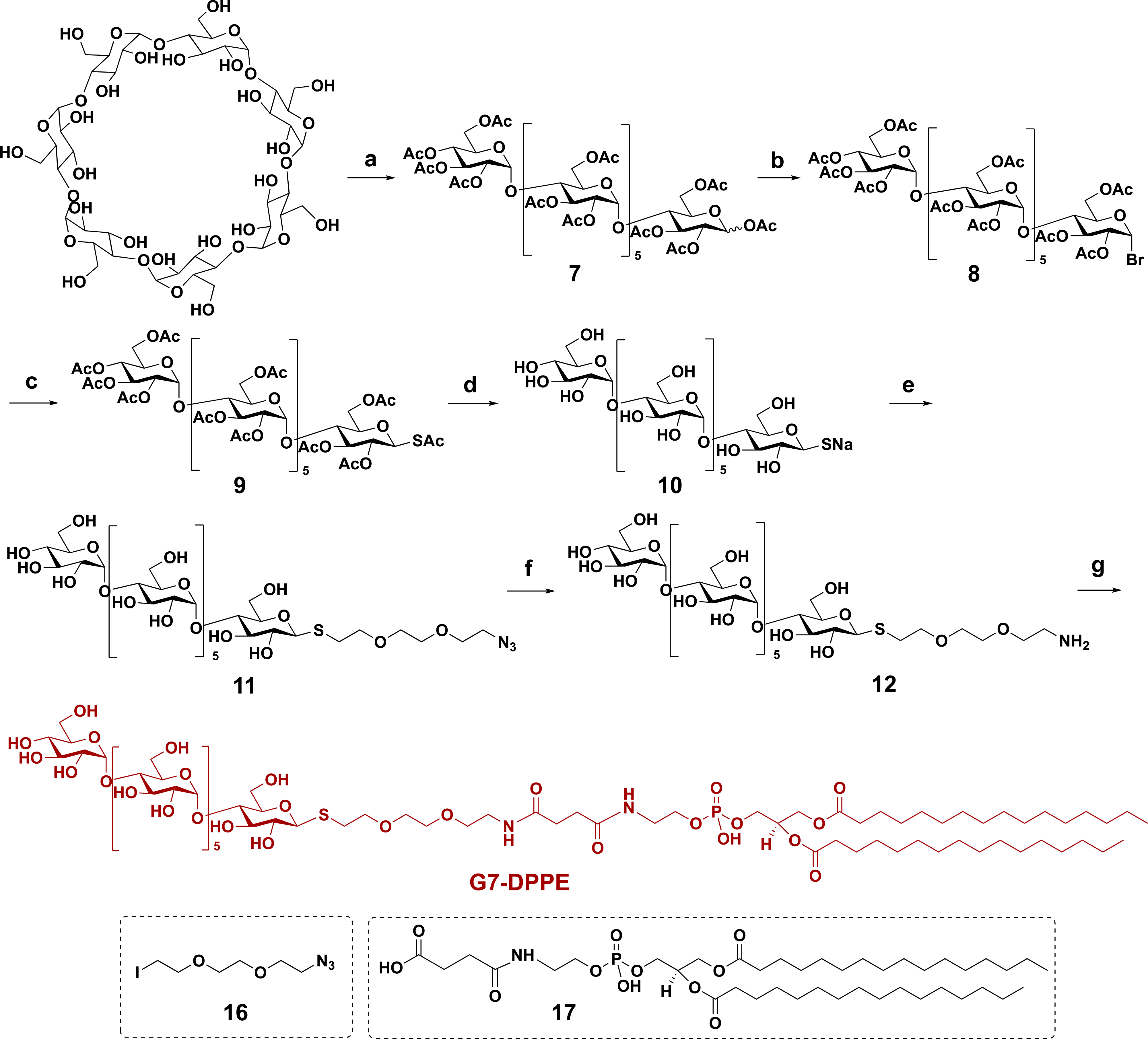
b(a) Acetic anhydride, FeCl3, RT, 70 °C, 4 h (33%); (b) acetic acid/HBr, 0 °C, RT, 2 h; (c) KSAc, acetone, RT, 5 h (66% from 7); (d) NaOMe, MeOH, RT, 3 h (94%); (e) 16, DMF, 2 h, 58%; (f) Pd/C, H2, MeOH/H2O, RT, 2 h (quantitative yield); and (g) 17, EDAC, DMF, 8 h (46%).
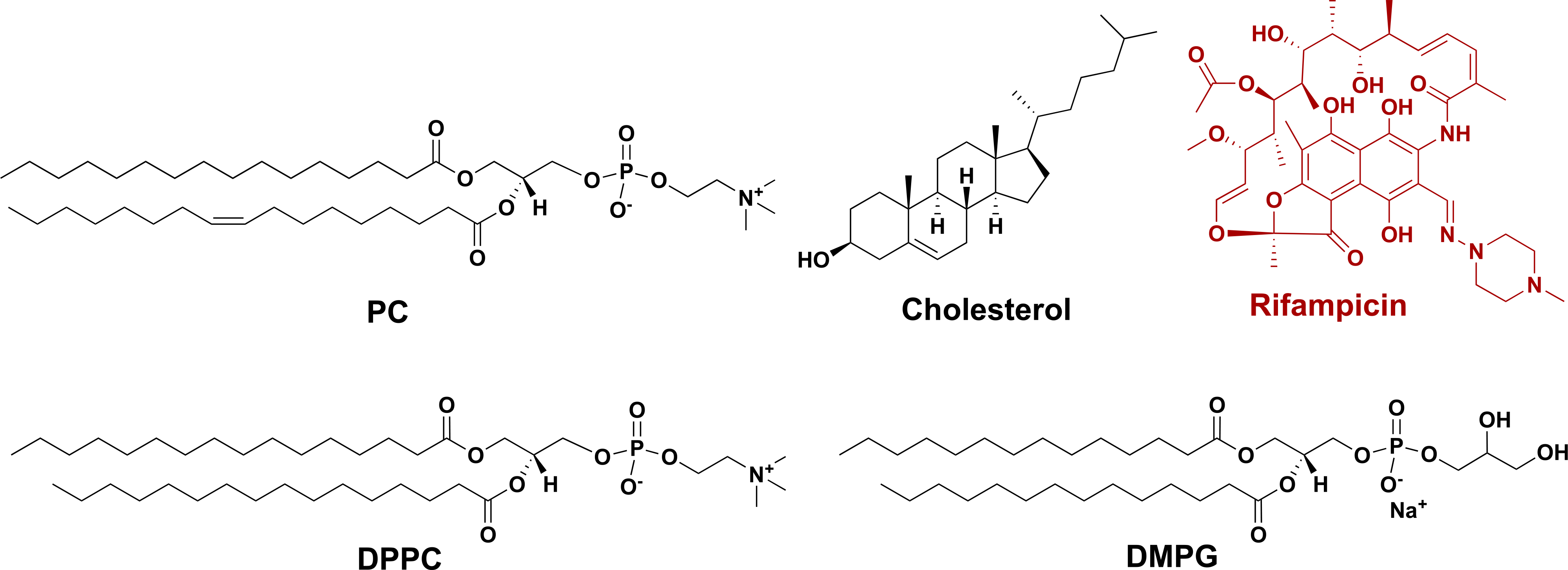
Carbohydrate-presenting liposomes were prepared by mixing the glycolipid G7-DPPE or Man-DPPE with the following lipids: PC and cholesterol (Chol) for nonfluid liposomes or DPPC and DMPG for fluid liposomes. Nonfluid liposomes made from PC and Chol are known to have high stability as well as good release profiles for drug molecules of different physicochemical characteristics.44 Fluid liposomes prepared from DPPC and DMPG are less rigid, as these lipids have low gel to liquid crystalline phase transition temperatures.44 Liposomes are generally prepared using two main methods: (1) thin-film hydration followed by extrusion through a porous membrane, which gives larger unilamellar vesicles (LUVs) or (2) ethanol injection, which gives smaller unilamellar vesicles (SUVs).45,46 We employed both methods to make liposomes with and without the antibiotic rifampicin (Rif; Scheme 3). To make liposomes by thin-film hydration, PC, Chol (or DMPG and DPPC; Scheme 3), together with G7-DPPE or Man-DPPE were mixed with 5:1 v/v chloroform/methanol, which was then rotary-evaporated to give a thin lipid film. The film was hydrated by water or the aqueous solution of rifampicin to give the crude liposomes, which were then extruded through a polycarbonate membrane of 100 nm pore size to afford relatively uniform liposomes. To make liposomes by ethanol injection, glycolipid G7-DPPE or Man-DPPE, rifampicin, PC, and Chol (or DMPG and DPPC) were dissolved in 10:1 v/v ethanol/DMSO, which was then diluted by injecting into water. The mixture was dialyzed at 4 °C to yield the purified liposomes.
Scheme 3. Structures of Lipids (PC, Cholesterol, DPPC, and DMPG) and Rifampicin.
The prepared liposomes were characterized by dynamic light scattering (DLS) to determine the hydrodynamic diameters and the size uniformity (i.e., polydispersity index (Đ); Table 1). The amount of rifampicin encapsulated in the liposomes was obtained by lysing the liposomes with dimethyl sulfoxide (DMSO), and the concentration was determined using UV–vis spectroscopy by comparing the absorbance at 475 nm with a standard calibration curve (Figure S1). The thin-film hydration technique produced liposomes of similar particle sizes after membrane extrusion, regardless of the nature of the lipids or the mole ratio (e.g., entry 1 vs 6). In all cases, liposomes prepared by thin-film hydration were more uniform in size (Đ: 0.15–0.20, entries 1 and 6–9) than those prepared by ethanol injection (Đ: 0.25–0.28, entries 2–5).
Table 1.
Characterization of Liposomes: Particle Diameter, Size Distribution, and Drug Loading
| entry | liposome composition (mole ratio of lipids) | type of liposomes | method of preparation | hydrodynamic diameter (nm)a | Ðb | Rif encapsulation yield (%)c |
Rif wt % in liposomed |
|---|---|---|---|---|---|---|---|
| 1 | PC/Chol (2:1) | nonfluid | thin-film hydration | 122 ± 10 | 0.16 | 0 | 0 |
| 2 | PC/Chol (8:1) | nonfluid | ethanol injection | 62 ± 11 | 0.28 | 0 | 0 |
| 3 | PC/Chol (8:1)/Rif | nonfluid | ethanol injection | 67 ± 9 | 0.25 | 3.2 | 8.7 |
| 4 | PC/Chol/Man-DPPE (8:1:0.8)/Rif | nonfluid | ethanol injection | 64 ± 8 | 0.26 | 2.2 | 6.6 |
| 5 | PC/Chol/G7-DPPE (8:1:0.8)/Rif | nonfluid | ethanol injection | 75 ± 12 | 0.25 | 2.7 | 7.1 |
| 6 | DPPE/DMPG (6:1) | fluid | thin-film hydration | 125 ± 8 | 0.20 | 0 | 0 |
| 7 | DPPE/DMPG (6:1)/Rif | fluid | thin-film hydration | 133 ± 11 | 0.15 | 1.1 | 7.9 |
| 8 | DPPE/DMPG/Man-DPPE (6:1:0.6)/Rif | fluid | thin-film hydration | 140 ± 14 | 0.19 | 2.1 | 14 |
| 9 | DPPE/DMPG/G7-DPPE (6:1:0.6)/Rif | fluid | thin-film hydration | 146 ± 14 | 0.16 | 2.4 | 16 |
Z-average size measured by DLS. Data are presented as average ± SD from three different measurements.
Ð: polydispersity index, measured by DLS.
Rif encapsulation yield (%) is the percent of rifampicin encapsulated in the liposomes versus the amount of Rif initially added.
Rif wt % is the percent of rifampicin encapsulated versus the total weight of the liposomes.
Entries 1–5 were nonfluid liposomes prepared from PC/Chol. To load rifampicin, a lower amount of cholesterol was used (8:1 PC/Chol) to formulate the liposomes, which has been shown to increase rifampicin encapsulation.47 As expected, liposomes prepared by thin-film hydration were larger (122 ± 10 nm, entry 1) than those prepared by ethanol injection (62 ± 11 nm, entry 2). Encapsulation of rifampicin increased the size of the liposomes slightly (67 ± 9 nm, entry 3). Incorporation of Man-DPPE did not cause much change in particle size (64 ± 8 nm, entry 4). For the nonfluid G7-glycoliposomes, the particle size increased to 75 ± 12 nm (entry 5). This is probably due to the hydration of the large G7 ligands in the liposomes, which led to an increase in the hydrodynamic volume. For nonfluid liposomes without the glycolipid, 3.2% of the initially added rifampicin was encapsulated (entry 3). The drug encapsulation decreased to 2.2% (entry 4) and 2.7% (entry 5) for the nonfluid Man- and G7-glycoliposomes. When considering the percent of rifampicin encapsulated versus the total weight of the liposomes, the drug loading decreased from 8.7 to 6.6 and 7.1 wt % for the nonfluid Man- and G7-glycoliposomes, respectively. For the fluid liposome formulations, on the other hand, the drug loading increased with the addition of Man-DPPE or G7-DPPE. The percent weights of rifampicin encapsulated were 14 wt % (entry 8) and 16 wt % (entry 9) for the fluid Man- and G7-glycoliposomes, respectively, which were more than twice higher than the corresponding nonfluid glycoliposomes (entries 4 and 5).
G7 Slows Down the Release of Rifampicin from Glycoliposomes.
The kinetics of rifampicin release from the liposomes was assessed by dialyzing the rifampicin-loaded liposomes in Milli-Q water at 37 °C and measuring the amount of rifampicin released at different time periods by UV–vis spectroscopy (Figure 1).48,49 For rifampicin loaded in the nonfluid liposome (PC/Chol), ~70% of rifampicin was released from the liposomes after 2 h, and the release was plateaued at ~80% after 4 h (Figure 1a). The addition of the glycolipid slowed down the release of rifampicin to 59 and 51% after 2 h for the nonfluid Man- and G7-glycoliposomes, respectively. In the case of the fluid liposome formulation (DPPC/DMPG), the rate of rifampicin release accelerated to 79% in the first 30 min and plateaued at ~90% after about 2 h (Figure 1b). The faster drug release from the fluid liposomes is consistent with their less ordered and lower-density lipid bilayer structures compared to that of the more rigid nonfluid liposomes. The rifampicin release decreased significantly with the addition of the glycolipid to the fluid liposomes. Only 52 and 43% of rifampicin were released in the first 30 min for the fluid Man- and G7-glycoliposomes, respectively. The duration of release was also longer. For example, the release of rifampicin from G7-glycoliposomes continued to 24 h.
Figure 1.
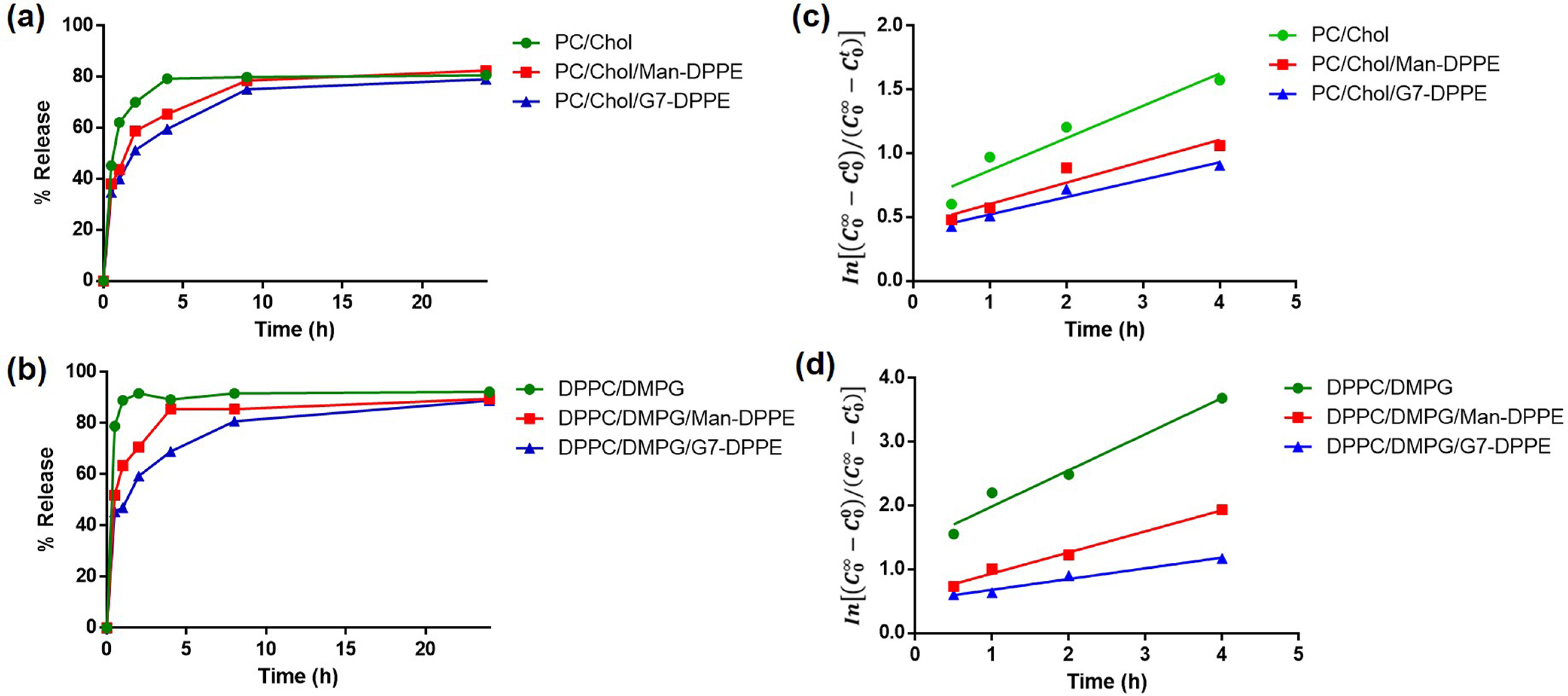
(a, b) Time course of rifampicin release from nonfluid (a) and fluid liposomes (b). (c, d) Kinetics of drug release for rifampicin encapsulated in nonfluid (c) and fluid liposomes (d). Liposomes were dialyzed in Milli-Q water at 37 °C while shaking. Concentrations of rifampicin were measured by UV–vis spectroscopy against a standard calibration curve (Figure S1).
To evaluate the kinetics of rifampicin release from the liposomes, a model by Anderson and co-workers was used.50 The model assumed that (1) the drug release from liposomes through the dialysis membrane follows a first-order kinetics governed by a simple diffusion process, (2) the liposome remains intact during the dialysis, and (3) there is no interaction between the drug and the dialysis membrane. and are the drug concentrations inside and outside the liposomes at time t, respectively. By plotting vs time (Figure 1c,d; see SI calculations), the apparent first-order rate constant kobs was obtained (Table 2). For both nonfluid and fluid liposomes, introducing Man-DPPE resulted in a decrease in kobs (entry 2 vs 1, entry 5 vs 4), and G7-DPPE further lowered kobs (entries 3 and 6). The apparent permeability coefficient Papp calculated from the hydrodynamic diameter (eq S5) showed the same trend that the glycolipid slowed down the release of rifampicin from the liposomes (Table 2). The decrease was the largest for the fluid G7-glycoliposomes, where Papp was reduced to less than half of that of the fluid liposomes without G7. One possible explanation for these results is that the hydrophilic carbohydrates presented on the liposomes served as an extra barrier to the diffusion of the nonpolar rifampicin in and out of the liposomes, with G7 having higher impact due to its large size. The Papp was 3.5 nm/h for PC/Chol liposomes, which is on the same order of magnitude as the permeability coefficient of 1.0 nm/h reported for glucose encapsulated in egg-lecithin LUV liposomes, and is 2–3 orders of magnitude larger than the more rigid LUV liposomes prepared from 50:50 DPPC/Chol.50
Table 2.
Apparent First-Order Rate Constant kobs and Permeability Coefficient Papp of Rifampicin Release from Liposomes
Ligand Density and Binding Affinity of G7-Glycoliposomes with Lectin Con A.
To confirm that the carbohydrate ligands were presented on the glycoliposomes, Man- and G7-glycoliposomes were treated with Con A, a lectin that binds both mannosides and glucosides. To quantify the binding affinity, the amount of Man-DPPE or G7-DPPE incorporated into the liposomes was determined by the anthrone–sulfuric acid colorimetry assay that has been widely used to quantify carbohydrate concentrations including carbohydrate-presenting nanoparticles.51,52 In this assay, the carbohydrate is first dehydrated by concentrated sulfuric acid to furfural, which then reacts with anthrone to give a blue-green hydroxyfurfural anthrone complex.53,54 The glycoliposomes were treated with a solution of 0.5 wt % anthrone in 98% sulfuric acid, and the absorbance at 620 nm was compared with the corresponding calibration curves obtained from Man-DPPE or G7-DPPE (Figure S2). The amount of Man-DPPE or G7-DPPE incorporated into the glycoliposomes was subsequently calculated (Table 3). Since the glycolipid can be incorporated at both the exterior and interior of the liposomes, the results from the anthrone–sulfuric acid assay would reflect the total amount of glycolipids in the liposomes. Results showed that at the same amount of glycolipid in the feed, 3–4 times more Man-DPPE than G7-DPPE was incorporated in the nonfluid (66 vs 21 μmol/mg, entries 1 and 2) and fluid liposomes (59 vs 14 μmol/mg, entries 3 and 4). This is likely due to the much larger and more sterically hindered G7 structure, giving lower incorporation of G7-DPPE into the glycoliposomes compared to that of the smaller Man-DPPE.
Table 3.
Ligand Density and Binding Affinity of Man- and G7-Glycoliposomes with Con A
| entry | glycoliposome composition | types of liposomes | glycolipid in glycoliposomes (μmol/mg)a | size of glycoliposomes (nm)b | Bmax (103 nm)c | h c | Kd (μM)c | affinity enhancementd |
|---|---|---|---|---|---|---|---|---|
| 1 | PC/Chol/Man-DPPE (2:1:0.2) | nonfluid | 66 | 128 ± 12 | 4.9 | 4.6 | 1.1 | 309 |
| 2 | PC/Chol/G7-DPPE (2:1:0.2) | nonfluid | 21 | 147 ± 11 | 6.4 | 7.0 | 0.93 | 892 |
| 3 | DPPC/DMPG/Man-DPPE (6:1:0.6) | fluid | 59 | 118 ± 15 | 8.7 | 5.1 | 2.9 | 117 |
| 4 | DPPC/DMPG/G7-DPPE (6:1:0.6) | fluid | 14 | 136 ± 8 | 7.0 | 2.8 | 0.51 | 1627 |
Con A has four binding sites for mannosides and glucosides.55,56 When Con A is treated with nanoparticles having mannosides or glucosides on the surface, interactions between the multivalent Con A and glyconanoparticles can form large agglomerates, leading to a concentration-dependent increase in the particle size.57–59 By monitoring the particle size versus the lectin concentration and fitting the data to a suitable binding isotherm, the binding affinity can be obtained.60,61 In this study, glycoliposomes were treated with varying concentrations of Con A at 20 °C for 2 h, and the hydrodynamic diameters of the resulting samples were measured by DLS. The increase in the particle size (ΔD), which is the difference between the hydrodynamic diameters of the glycoliposomes before and after treating with Con A, was plotted against the concentration of Con A (Figure 2). The data were then fitted with the Hill equation, eq 1
| 1 |
where Bmax is the maximum specific binding, h is the Hill coefficient, and Kd is the apparent dissociation constant of glycoliposomes with Con A. For liposomes that do not contain Man or G7, no change in the hydrodynamic diameters was observed after treating with Con A.
Figure 2.
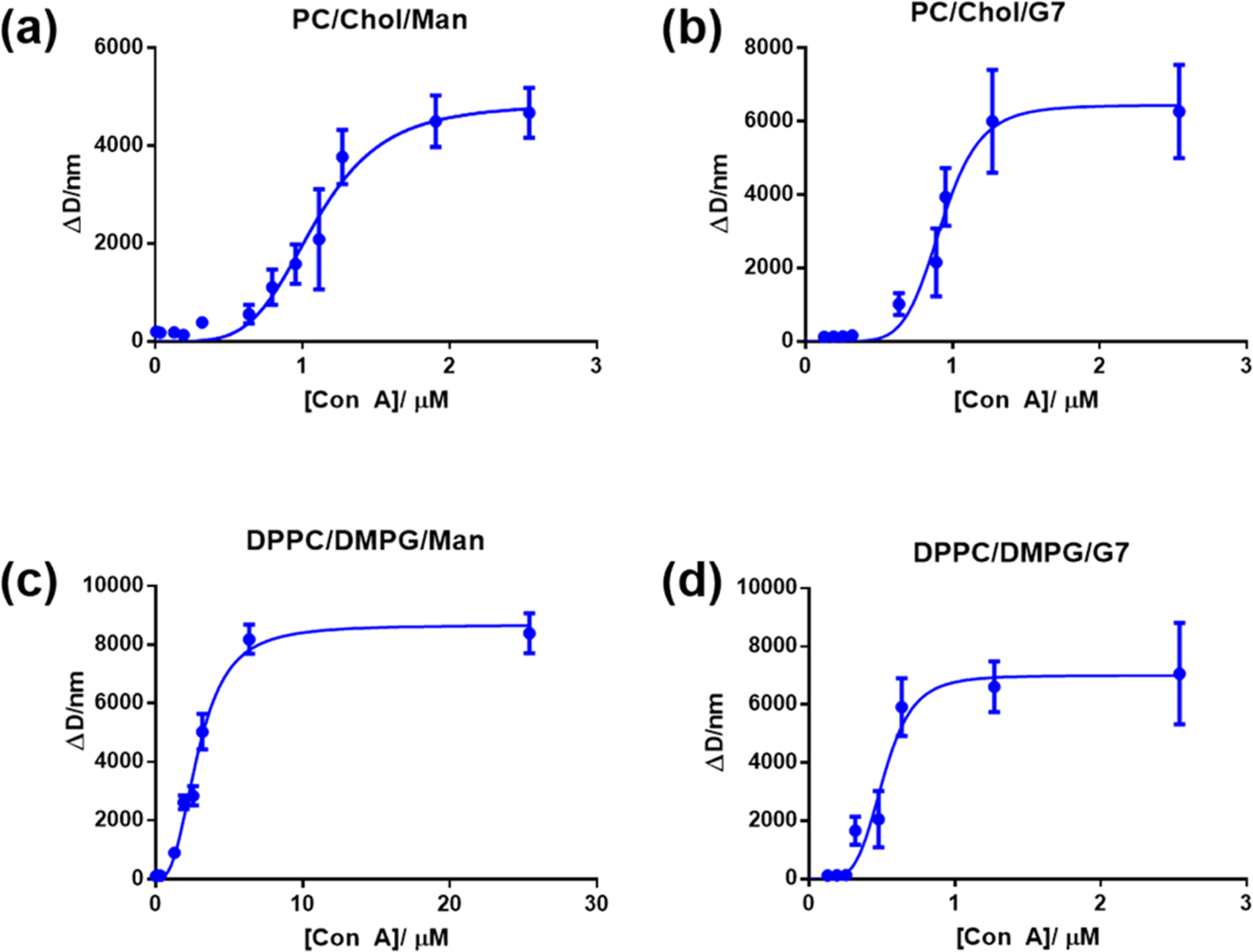
Changes in the hydrodynamic diameter (ΔD) of glycoliposomes vs the concentrations of Con A: experimental data (circles) and the corresponding Hill fitting curves (lines) for (a) nonfluid Man-glycoliposomes, (b) nonfluid G7-glycoliposomes, (c) fluid Man-glycoliposomes, and (d) fluid G7-glycoliposomes. Each data point was the average of two independent trials.
The nonfluid G7-glycoliposomes exhibited a slightly stronger binding with Con A (Kd = 0.93 μM, entry 2) than Man-glycoliposomes (Kd = 1.1 μM, entry 1; Table 3). Compared to the monovalent binding (KD = 0.34 and 0.83 mM between Con A and Man or G7, respectively, determined by thermostatic disc electrophoresis),62 the results showed 309- (entry 1) and 892-fold (entry 2) affinity enhancement for the nonfluid Man- and G7-glycoliposomes, respectively. Considering that the density of G7-DPPE, 21 μmol/mg, was ~3 times lower than that of Man-DPPE (66 μmol/mg) on the glycoliposomes, this represents ~9 times higher relative binding affinity of nonfluid G7-glycoliposomes than that of Man-glycoliposomes with Con A.
For the fluid liposomes, the binding affinity of Con A with the fluid G7-glycoliposomes (Kd = 0.51 μM, entry 4) was ~6 times higher than that of Man-glycoliposomes (Kd = 2.9 μM, entry 3). This represents 117- (entry 3) and 1627-fold (entry 4) affinity enhancement for Man- and G7-glycoliposomes compared to the monovalent binding with Man or G7. Considering the density of G7-DPPE and Man-DPPE on the glycoliposomes, 59 and 14 μmol/mg, respectively (entries 3 and 4), this represents 59 times higher relative binding affinity of the fluid G7-glycoliposomes than that of Man-glycoliposomes. The Hill coefficient h was greater than 1 in all cases, indicating positive cooperativity of the glycoliposomes in the interactions with Con A.
Antibacterial Activity of Rifampicin-Loaded Liposomes.
The minimal inhibitory concentration (MIC) of rifampicin-loaded liposomes against E. coli ORN208, a mutant strain of E. coli that lacks Man-binding fimbria I expression,63 was determined by the standard broth microdilution method. Neither the fluid nor the nonfluid liposomes alone showed any activity (entries 1 and 5; Table 4). The MIC of rifampicin loaded in the nonfluid liposomes was 6–12 μg/mL (entry 2), slightly worse than the free rifampicin (6 μg/mL, entry 9). Incorporation of the glycolipid lowered the MIC slightly to 4–8 and 7 μg/mL for the nonfluid Man- and G7-glycoliposomes, respectively (entries 3 and 4). For rifampicin loaded in fluid liposomes (6:1 DPPE/DMPG), the MIC was 3–6 μg/mL (entry 6), whereas rifampicin loaded in fluid Man-glycoliposomes showed a slightly higher MIC (9 μg/mL, entry 7) than the free rifampicin. The MIC of rifampicin loaded in fluid G7-glycoliposomes was 2 μg/mL (entry 8), lower than the liposomes without G7-DPPE (entry 6) or the free rifampicin. The drug loading alone is not the reason behind the increase in activity, as the amount of rifampicin encapsulated in the fluid G7-glycoliposomes (16 wt %) was only slightly higher than the fluid Man-glycoliposomes (14 wt %; Table 1).
Table 4.
MIC (μg/mL Based on Rifampicin) against E. coli ORN208a
| entry | samples | wt % Rif | MIC (μg/mL) |
|---|---|---|---|
| 1 | nonfluid liposomes (8:1 PC/Chol) | >250/>250 | |
| 2 | nonfluid liposomes (8:1 PC/Chol)/Rif | 8.7/8.7 | 12/6 |
| 3 | nonfluid Man-glycoliposomes (8:1:0.8 PC/Chol/Man-DPPE)/Rif | 6.6/6.6 | 8/4 |
| 4 | nonfluid G7-liposomes (8:1:0.8 PC/Chol/G7-DPPE)/Rif | 7.1/7.1 | 7/7 |
| 5 | nonfluid G7-liposomes (8:1:0.8 PC/Chol/G7-DPPE) | >250/>250 | |
| 6 | fluid liposomes (6:1 DPPC/DMPG)/Rif | 7.9/7.9 | 6/3 |
| 7 | fluid Man-glycoliposomes (6:1:0.6 DPPC/DMPG/Man-DPPE)/Rif | 14/14 | 9/9 |
| 8 | fluid G7-glycoliposomes (6:1:0.6 DPPC/DMPG/G7-DPPE)/Rif | 16/16 | 2/2 |
| 9 | rifampicin | 6/6 |
Assays were repeated twice, and both results are presented.
Time-Dependent Antimicrobial Activities of Rifampicin-Loaded Glycoliposomes.
The time-dependent antimicrobial activity of rifampicin-loaded liposomes was investigated using the time–kill assay by treating E. coli ORN208 with 1, 2, or 4 × MIC of rifampicin-loaded liposomes or the free rifampicin over a period of 24 h. The MICs of all samples were measured prior to the experiments. Samples were taken out at 2, 4, 8, and 24 h, and the colonies were counted after a 12 h incubation. Rifampicin is considered bacteriostatic to E. coli;64 thus, it is not surprising that it was not bactericidal even at 4 × MIC (Figure 3a). Rifampicin-loaded nonfluid liposomes (Figure 3b) and Man-glycoliposomes (Figure 3c) showed similar behavior as the free rifampicin, except that the bacterial population stayed static over the 24 h period for all concentrations tested. For rifampicin-loaded nonfluid G7-glycoliposomes, the bacterial counts were reduced by ~0.5 log unit at 1 × MIC after 8 h and by ~1 log unit at 2 × and 4 × MICs after 2 h (Figure 3d). The effect was more pronounced in the case of fluid liposomes. The rifampicin-loaded fluid liposome was not bactericidal (Figure 3e), similar to the rifampicin-loaded nonfluid liposome (Figure 3b). Unlike the rifampicin-loaded nonfluid Man-glycoliposomes that showed no inhibition of bacterial growth (Figure 3c), rifampicin-loaded fluid Man-glycoliposomes reduced bacterial colonies at all concentrations tested (Figure 3f). The bacterial counts were reduced by more than 1 log unit at 4 × MIC after 2 h. The fluid G7-glycoliposomes were the most effective, reducing the colonies by ~1 log unit at 2 × and 4 × MICs after 1 h and by ~2 log units at 24 h (Figure 3g). Thus, the rifampicin-loaded fluid G7-glycoliposomes were not only more active than free rifampicin and other liposome formulations, the antimicrobial activity was also sustained over time.
Figure 3.
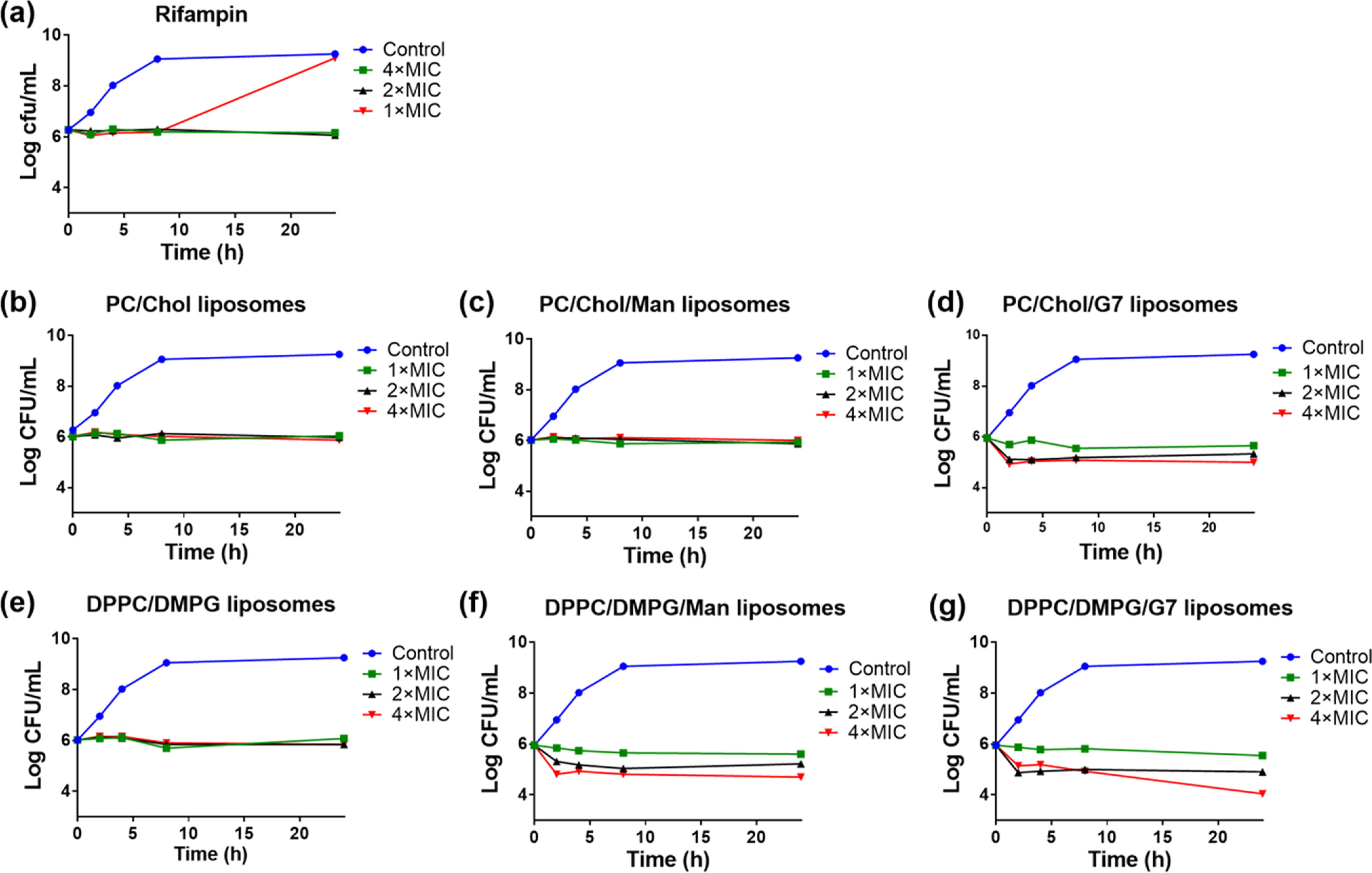
Time–kill kinetics of (a) rifampicin, (b) rifampicin-loaded nonfluid liposomes, (c) rifampicin-loaded nonfluid Man-glycoliposomes, (d) rifampicin-loaded nonfluid G7-glycoliposomes, (e) rifampicin-loaded fluid liposomes, (f) rifampicin-loaded fluid Man-glycoliposomes, and (g) rifampicin-loaded fluid G7-glycoliposomes. The control was E. coli ORN208 alone in the Mueller Hinton broth (MHB). Lines were drawn by connecting the data points to aid visualization.
Interactions of Liposomes with Bacterium by FRET and Confocal Microscopy.
FRET was employed to investigate the interaction between liposomes and the bacterium and to test whether fusion was involved in the interaction.65 Two fluorescent lipid dyes, the FRET donor NBD-PE and the FRET acceptor Rh-PE, were incorporated into the liposomes (Figure 4). When the two lipid dyes are at a distance within the Förster radius, FRET can occur where the emission of the donor NBD-PE (at 460 nm excitation) excites the acceptor Rh-PE, leading to the fluorescence emission of the acceptor at 590 nm. The FRET between these two dyes is highly efficient even at very low density in the liposome (<1%), owing to the significant overlap of the emission band of NBD-PE and the excitation band of Rh-PE.65 Since the efficiency of the fluorescence energy transfer is mainly dependent on the distance separating the donor and the acceptor, FRET can be conveniently used to study the fusion of liposomes with the cell membrane.66 If the NBD/Rh-labeled liposome fuses with the bacterial membrane, the concentration of the dyes in the liposome would decrease. This would increase the distance between the two lipid dyes, which would decrease the FRET efficiency and lower the Rh emission intensity at 590 nm. If NBD/Rh-labeled liposomes only bind but do not fuse with the bacterium, the emission intensity at 590 nm will either remain unchanged or increase if the two dyes become closer in distance.
Figure 4.
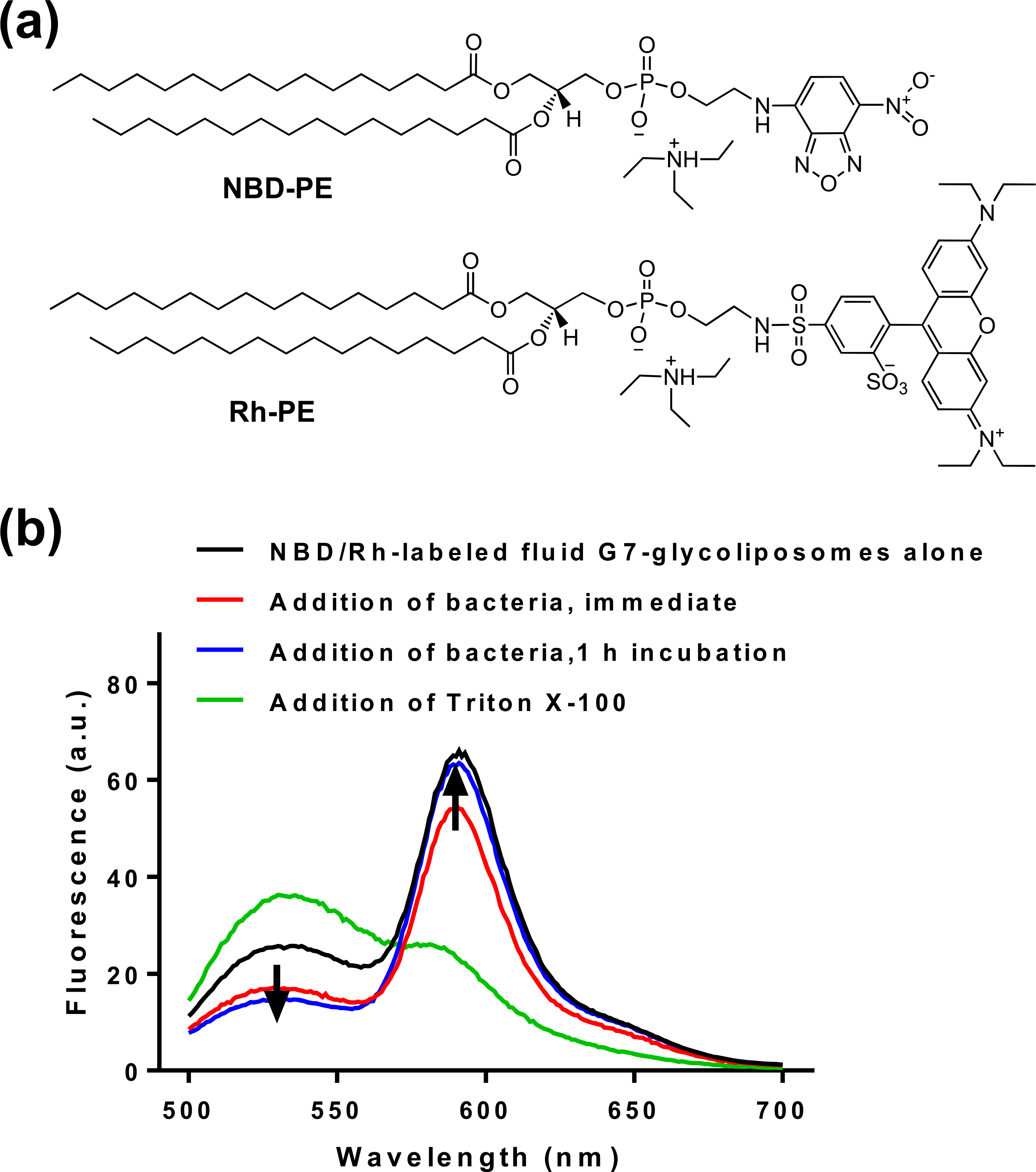
(a) Structures of FRET donor NBD-PE and acceptor Rh-PE. (b) Fluorescence spectra of NBD/Rh-labeled fluid G7-glycoliposomes alone (black), after the addition of Triton X-100 (green), or E. coli ORN208 immediately (red) and after 1 h incubation (blue). Excitation: 460 nm.
The dye-incorporated fluid G7-glycoliposomes were prepared by adding NBD-PE and Rh-PE (0.5 mol % of G7-DPPE each) to the fluid liposome formulation (DPPC/DMPG/G7-DPPE) using the thin-film hydration method. The fluorescence spectrum of NBD/Rh-labeled fluid G7-glycoliposomes contained two peaks at 536 nm and 590 nm, corresponding to the emissions of NBD-PE and Rh-PE, respectively (black curve, Figure 4). To confirm the presence of FRET, the liposomes were disintegrated by a detergent Triton X-100. This resulted in a large decrease in the emission intensity at 590 nm (Rh-PE) and an increase in the intensity at 536 nm (NBD-PE) (green curve, Figure 4). The intensity ratio of the two peaks at 590 and 536 nm, I590/I536, decreased from 2.5 for the liposome (entry 1) to 0.6 after the addition of Triton X-100 (entry 2, Table 5). This demonstrates that FRET indeed occurred in the NBD/Rh-labeled fluid G7-liposomes.
Table 5.
Fluorescence Intensity at 590 nm (Rh) and 536 nm (NBD) of NBD/Rh-Labeled Fluid G7-Glycoliposomes before and after Incubations with E. coli ORN208
| entry | sample | fluorescence intensity at 590 nm (au) | fluorescence intensity at 536 nm (au) | I590nm/I536nm |
|---|---|---|---|---|
| 1 | NBD/Rh-labeled fluid G7- glycoliposomes alone | 64.9 | 25.7 | 2.5 |
| 2 | addition of Triton X-100 | 23.4 | 36.2 | 0.6 |
| 3 | addition of bacteria, immediate | 54.8 | 16.8 | 3.3 |
| 4 | addition of bacteria, after 1 h incubation | 62.9 | 14.6 | 4.3 |
When NBD/Rh-labeled fluid G7-glycoliposomes were mixed with E. coli ORN208, the intensities at both 590 and 536 nm decreased (red curve, Figure 4) and the intensity ratio I590/I536 increased 32% from 2.5 to 3.3 (entry 3). After incubation for 1 h, the intensity at 590 nm increased, and at the same time, the intensity at 536 nm decreased (blue curve, Figure 4), resulting in an overall increase of I590/I536 to 4.3, a 72% increase from the G7-glycoliposomes prior to incubation with the bacteria (entry 4, Table 5). The result also excluded the possibility of fusion. If fusion occurred between the liposomes and the bacterium, the distance between Rh and NBD would increase due to fusion-induced dilution. This would lead to a decrease in the emission intensity at 590 nm and thus lower FRET efficiency.
The results in Table 5 showed that the FRET efficiency increased after mixing G7-glycoliposomes with the bacterium and increased further after incubation for 1 h. The higher-energy transfer implies that Rh-PE and NBD-PE became closer in distance when G7-glycoliposomes were mixed with the bacterium. Two possibilities can contribute to this: the liposomes interacted among themselves to form aggregates or with the bacterium. To investigate the possibility that liposomes form aggregates among themselves, NBD/Rh-labeled fluid G7-glycoliposomes incubated with the bacteria were imaged by confocal fluorescence microscopy. A series of snapshots were taken from a movie recorded (Figure S4). The images showed liposome nanoparticles on some bacterial cells, and the liposomes adhered to the cell surface despite the rapid movement of the bacteria. No large aggregates of the liposome particles were observed. These observations support the hypothesis that the increase in the FRET efficiency is due to the interaction of liposomes with the bacteria.
For NBD/Rh-labeled Man-glycoliposomes and NBD/Rh-labeled liposomes without glycolipid, I590/I536 also increased after incubation with the bacterium but the extent of increase in FRET efficiency was lower than that of G7-glycoliposomes (Figure S3 and Table S1). These results suggest stronger binding of G7-glycoliposomes to E. coli than that of Man-glycoliposomes and those without glycolipids.
CONCLUSIONS
In summary, we have successfully prepared nonfluid and fluid G7- and Man-glycoliposomes with uniform nanoparticle sizes. The multivalent display of G7 and Man on the nanoparticles was demonstrated by significantly enhanced binding of these glyconanoparticles toward Con A than that of the free ligands G7 and Man. The fluid G7-glycoliposomes exhibited the strongest binding with Con A, with more than 3 orders of magnitude higher affinity than free G7, despite having a lower amount of G7-DPPE incorporated into the glycoliposomes. The fluid liposome formulation also gave higher rifampicin encapsulation than the nonfluid liposomes, with the fluid glycoliposomes yielding the highest rifampicin loading, 16 wt % for the fluid G7-glycoliposomes. The presence of G7 on the liposomes also reduced the kinetics of rifampicin release, with G7-glycoliposomes having the slowest rate of release and lowest permeability coefficient. The rifampicin-loaded fluid G7-glycoliposomes showed 3 times lower MIC against E. coli ORN208 compared to all other liposome formulations and free rifampicin. The rifampicin-loaded fluid G7-glycoliposomes exhibited sustained bacterial killing, resulted in ~2 log unit reduction in bacterial colonies over 24 h at 4 × MIC. FRET experiments and confocal imaging suggested that a stronger binding of fluid G7-glycoliposomes to the bacterium may contribute to the sustained bacterial killing. The use of a bacterium-specific carbohydrate as the targeting agent in liposome formulations can be a promising strategy in antibiotic delivery.
EXPERIMENTAL SECTION
Materials and Instrumentation.
Rifampicin was purchased from MilliporeSigma (St. Louis, MO). Except for the glycolipids Man-DPPE and G7-DPPE, which were synthesized as shown below, all other lipids used in this study were purchased from Avanti Polar Lipids (Alabaster, Alabama). Con A was purchased from Vector Laboratories. Other reagents were purchased from either MilliporeSigma or Fisher Scientific (Hampton, NH) and were used without further purification unless otherwise noted. The experimental details for the syntheses of compounds 14–17 can be found in the Supporting Information. Polycarbonate membranes (0.1 μm, 19 mm) were purchased from Avanti Polar Lipid. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance Spectrospin-500 spectrometer or a Bruker Avance DPX 200 NMR spectrometer. 31P NMR spectra were recorded on a Bruker Avance DPX 200 NMR spectrometer using 85% H3PO4 (0.00 ppm) as the external reference. HRMS spectra were collected in the electrospray ionization (ESI) mode on a Bruker MicrOTOF II mass spectrometer at the University of Massachusetts Amherst. UV–vis spectra were collected on a Perkin Elmer LAMBDA 35 UV–vis spectrophotometer. Dynamic light scattering (DLS) measurements were carried out on a Nano Series Zen 40 03 Zetasizer (Malvern Instruments Ltd, Malvern, UK).
Synthesis of Glycolipid Man-DPPE (Scheme 1).
Methyl 2,3,4,6-Tetra-O-benzyl-α-D-mannopyranoside (1).
To a solution of methyl α-d-mannopyranoside (1.94 g, 10.0 mmol) in dry N,N-dimethylformamide (DMF, 60 mL), sodium hydride (NaH, 3.52 g, 88.1 mmol) and tetra-n-butylammonium iodide (TBAI, 0.37 g, 1.0 mmol) were added portion-wise at 0 °C. After 30 min, benzyl bromide (8.3 mL, 70.8 mmol) was added dropwise under Ar. After stirring at room temperature (RT) overnight, water (200 mL) was poured into the reaction and the mixture was extracted with 200 mL of ethyl acetate 3 times. The combined organic phase was washed with brine and dried over Na2SO4. After evaporation of the solvent, the crude product was vacuum-dried overnight and subsequently purified by column chromatography (hexanes/ethyl acetate, 9:1–5:1) to give compound 1 as a viscous solid (4.99 g, 90%). 1H NMR (500 MHz, CDCl3; Figure S5) δ 7.49–7.21 (m, 8H, aromatic), 7.17 (s, 2H, aromatic), 4.88 (d, J = 0.4 Hz, 1H, H-1), 4.85–4.48 (m, 8H, PhCH2O), 3.97 (t, J = 8.7 Hz, 1H, H-4), 3.88 (dd, J = 9.2, 2.8 Hz, 1H, H-3), 3.85–3.69 (m, 4H, H-2, H-5, H-6a, H-6b), 3.33 (s, 3H, OMe).
2,3,4,6-Tetra-O-benzyl-α/β-D-mannopyranose (2).
To a mixed solvent of glacial acetic acid (11 mL) and aqueous HCl (2 M, 9 mL), compound 1 (2.0 g, 3.6 mmol) was added, and the reaction mixture was stirred at 85 °C for 7 h. After diluting with cold water and dichloromethane (DCM), the organic fraction was washed with cold water once and then with aqueous NaHCO3 twice and dried over MgSO4. The filtrate was concentrated to give the crude product, which was purified by column chromatography (4:1 hexanes/ethyl acetate) to give compound 2 as a viscous solid (1.8 g, α/β = 10/1, 45%). α-Anomer: 1H NMR (500 MHz, CDCl3; Figure S6) δ 7.25–7.38 (m, 18 H, aromatic), 7.16–7.18 (m, 2H, aromatic), 5.26 (s, 1H, H-1), 4.43–4.96 (m, 8H, PhCH2O), 4.02–4.05 (m, 1H, H-5), 3.96 (dd, 1H, J = 3.0, 9.3 Hz, H-3), 3.87 (t, 1H, J = 9.6 Hz, H-4), 3.79–3.81 (m, 1H, H-2), 3.65–3.75 (m, 2H, H-6a, H-6b).
t-Butyloxycarbonylmethyl 2,3,4,6-Tetra-O-benzyl-α-D-mannopyranoside (3).
To a solution of 2 (560 mg, 1.04 mmol) and tetrabutylammonium bromide (TBAB, 17 mg, 0.053 mmol) in toluene (10 mL), fine powers of dry KOH (350 mg, 6.24 mmol) were added. The reaction was cooled in an ice bath, and t-butyl bromoacetate (470 μL, 1.57 mmol) was added dropwise. The reaction was stirred at RT for 30 min and was then diluted by ether and water. The organic fraction was separated, washed with water followed by brine, and dried over Na2SO4. After removing the solvent, the crude product was purified by column chromatography (7:1 hexanes/ethyl acetate) to give product 3 as a viscous solid (610 mg, 85%). 1H NMR (500 MHz, CDCl3; Figure S7) δ 7.42–7.20 (m, 18H, aromatic), 7.15 (d, J = 5.1 Hz, 2H, aromatic), 5.08 (s, 1H, H-1), 4.88 (d, J = 10.7 Hz, 1H, PhCH2O), 4.74 (s, 2H, PhCH2O), 4.70–4.44 (m, 5H, PhCH2O), 4.12 (d, J = 16.6 Hz, 1H, OCH2COO), 4.06–3.89 (m, 4H, OCH2COO, H-4, H-3, H-2), 3.87–3.67 (m, 3H, H-5, H-6a, H-6b), 1.45 (s, 9H, OC(CH3)3). 13C NMR (126 MHz, CDCl3; Figure S8) δ 169.26, 138.71, 138.67, 138.52, 138.48, 128.50, 128.13, 128.05, 127.95, 127.76, 127.68, 97.52, 82.05, 80.12, 75.29, 74.89, 74.63, 73.55, 72.81, 72.43, 72.16, 69.37, 63.64, 28.29, 28.29, 28.29.
Carboxymethyl 2,3,4,6-Tetra-O-benzyl-α-D-mannopyranoside (4).
To a solution of compound 3 (610 mg, 0.93 mmol) in dichloromethane (DCM, 7 mL) was added trifluoroacetic acid (TFA, 7 mL), and the reaction was stirred at RT for 30 min. The solvent was removed on a rotovap, and the remaining TFA was removed by coevaporation with toluene under reduced pressure. The crude product was purified by column chromatography (1:1 hexanes/ethyl acetate) to give compound 4 as a viscous solid (462 mg, 83%). 1H NMR (500 MHz, CDCl3; Figure S9) δ 7.41–7.22 (m, 18H, aromatic), 7.15 (dd, J = 7.0, 2.2 Hz, 2H, aromatic), 5.02 (d, J = 1.2 Hz, 1H, H-1), 4.86 (d, J = 10.8 Hz, 1H, PhCH2O), 4.75 (d, J = 12.4 Hz, 1H, PhCH2O), 4.71 (d, J = 12.4 Hz, 1H, PhCH2O), 4.66–4.58 (m, 3H, PhCH2O), 4.54 (d, J = 12.2 Hz, 1H, PhCH2O), 4.49 (d, J = 10.8 Hz, 1H, PhCH2O), 4.24 (d, J = 17.1 Hz, 1H, OCH2COO), 4.17 (d, J = 17.1 Hz, 1H, OCH2COO), 3.97 (t, J = 9.2 Hz, 1H, H-4), 3.94–3.89 (m, J = 3.0 Hz, 2H, H-2, H-3), 3.81–3.76 (m, 1H, H-5), 3.74 (dd, J = 10.6, 5.0 Hz, 1H, H-6a), 3.70 (dd, J = 10.5, 1.8 Hz, 1H, H-6b). 13C NMR (126 MHz, CDCl3; Figure S10) δ 174.55, 138.55, 138.46, 138.32, 138.24, 128.55, 128.16, 128.01, 127.82, 98.00, 79.84, 75.28, 74.86, 74.46, 73.56, 72.92, 72.63, 72.34, 69.30, 63.22.
N-Succinimidyloxycarbonylmethyl 2,3,4,6-Tetra-O-benzyl-α-D-mannopyranoside (5).
Compound 4 (112 mg, 0.187 mmol), N-hydroxysuccinimide (NHS, 26 mg, 0.225 mmol), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDAC, 180 mg, 0.935 mmol) were dissolved in dry DCM, and the reaction mixture was stirred at RT for 24 h. Water was then added, and the mixture was extracted twice with DCM. The combined organic fractions were washed with water, dried over MgSO4, filtered, and the filtrate was evaporated under vacuum. The crude product was purified by flash column chromatography (2:3 hexanes/ethyl acetate) to give compound 5 as a viscous solid (117 mg, 90%). 1H NMR (500 MHz, CDCl3; Figure S11) δ 7.39–7.22 (m, 18H, aromatic), 7.16 (dd, J = 7.4, 1.9 Hz, 2H, aromatic), 5.08 (d, J = 1.5 Hz, 1H, H-1), 4.86 (d, J = 10.8 Hz, 1H, PhCH2O), 4.70 (d, J = 13.0 Hz, 2H, PhCH2O), 4.66 (d, J = 12.2 Hz, 1H, PhCH2O), 4.61–4.42 (m, 6H, PhCH2O, OCH2COO), 4.02 (t, J = 8.9 Hz, 1H, H-4), 3.95–3.87 (m, 2H, H-3, H-2), 3.77 (m, 2H, H-5, H-6a), 3.71 (dd, J = 4.3 Hz, 12.7 Hz, 1H, H-6b). 13C NMR (126 MHz, CDCl3; Figure S12) δ 168.75, 165.63, 138.55, 138.53, 138.47, 138.24, 128.51, 128.10, 127.97, 127.82, 127.78, 98.29, 79.68, 75.23, 74.66, 74.33, 73.60, 72.95, 72.91, 72.80, 72.28, 69.17, 61.67, 25.70, 25.70.
Compound 6.
To a solution of compound 5 (97 mg, 0.14 mmol) and DPPE-NH2 (97 mg, 0.14 mmol) in chloroform (CHCl3, 20 mL) and DMF (1 mL), Et3N (100 μL) was added. After stirring overnight, the solvent was removed by rotovap. The crude product was purified by column chromatography (15:1 CHCl3/MeOH) to afford compound 6 (Rf = 0.32, 15:1 CHCl3/MeOH) as a viscous solid (156 mg, 88%). 1H NMR (500 MHz, CDCl3; Figure S13) δ 7.53–6.97 (m, 20H, aromatic), 5.18 (s, 1H, POCH2CHCH2O), 4.99 (s, 1H, H-1), 4.89–4.55 (m, 6H, PhCH2O), 4.46 (d, J = 9.9 Hz, 2H, PhCH2O), 4.32 (d, J = 10.36 Hz, 1H, OCH2COO), 4.27–3.80 (m, 10H, H-2, H-3, H-4, OCH2COO, CH2OP, POCH2, CHCH2OCO), 3.80–3.64 (m, 3H, H-5, H-6a, H-6b), 3.55 (s, 1H, NHCH2), 3.43 (s, 1H, NHCH2), 2.21 (d, J = 6.6 Hz, 4H, OOCCH2 × 2), 1.51 (s, 4H, OOCCH2CH2 × 2), 1.40–1.02 (m, 48H, aliphatic), 0.88 (t, J = 7.0 Hz, 6H, CH2CH3 × 2). 13C NMR (126 MHz, CDCl3; Figure S14) δ 173.66, 173.40, 173.35, 138.68, 138.59, 128.51, 128.12, 128.00, 127.91, 127.81, 98.64, 79.76, 75.17, 74.84, 74.73, 73.53, 72.84, 72.57, 72.24, 70.48, 64.12, 64.09, 62.89, 39.90, 34.40, 34.26, 32.14, 29.97, 29.90, 29.59, 29.43, 25.42, 25.13, 25.08, 22.90, 14.41, 14.36, 14.31. 31P NMR (81 MHz, CDCl3; Figure S15) δ −1.77.
Man-DPPE.
Compound 6 (156 mg, 0.123 mmol) was dissolved in 2:3 v/v THF/MeOH (10 mL), and Pd(OH)2/C (10%, 33 mg, 0.032 mmol) was added. The reaction flask was purged with Ar and then with H2. The reaction was stirred for 24 h, after which the mixture was filtered through celite. The filtrate was evaporated under reduced pressure to yield compound Man-DPPE as a white solid (114 mg, quantitative yield). 1H NMR (500 MHz, 7:4:0.1 v/v/v CDCl3/CD3OD/D2O; Figure S16) δ 5.24 (s, 1H, POCH2CHCH2O), 4.86 (s, 1H, H-1), 4.42 (d, J = 10.3 Hz, 1H, OCH2COO), 4.18 (d, J = 12.06 Hz, 2H, CHCH2OCO), 4.12–3.64 (m, 10H, H-2, H-3, H-4, H-5, H-6a, OCH2COO, CH2OP, POCH2), 3.54 (m, 3H, H-6b, NHCH2), 2.34 (s, 4H, OOCCH2 × 2), 1.61 (s, 4H, OOCCH2CH2 × 2), 1.27 (s, 48H, aliphatic), 0.89 (s, 6H, CH2CH3 × 2). 13C NMR (50 MHz, 7:4:0.1 v/v/v CDCl3/CD3OD/D2O; Figure S17) δ 173.73, 173.35, 170.01, 99.53, 77.87, 77.23, 76.59, 73.23, 70.33, 70.16, 69.95, 66.58, 65.51, 63.87, 63.49, 62.38, 60.73, 49.63, 49.20, 48.78, 48.35, 47.92, 47.50, 47.07, 39.40, 33.99, 33.84, 31.67, 29.44, 29.28, 29.10, 28.90, 24.68, 24.63, 22.39, 13.61. 31P NMR (81 MHz, 7:4:0.1 v/v/v CDCl3/CD3OD/D2O; Figure S18) δ 0.40.
Synthesis of Glycolipid G7-DPPE (Scheme 2).
D-Maltoheptaose Tricosaacetate (7).
To a stirred suspension of FeCl3 (200 mg, 1.23 mmol) in acetic anhydride (Ac2O, 12.5 mL, 132 mmol) in an ice bath, β-cyclodextrin (4.00 g, 3.52 mmol) was added slowly. The mixture was stirred at RT for 2.5 h followed by 70 °C for 3.5 h. After cooling to RT, the solution was poured into 500 mL of water. The precipitation was collected and washed with water followed by cold ethanol. The crude product was purified by column chromatography (4:1 ethyl acetate/hexanes) to give 7 as a white solid (2.24 g, 33%).
1-Bromo-docosa-O-acetyl-α-D-maltoheptaopyranose (8).
To a solution of compound 7 (4.7 g, 2.2 mmol) in dry DCM (40 mL) cooled to 0 °C, 9 mL of 33% HBr/AcOH solution was added dropwise. The reaction was slowly warmed to RT and stirred at RT for 2 h. It was then poured into 100 mL of ice water, and the aqueous layer was extracted with 100 mL of DCM 3 times. The combined organic fraction was washed sequentially with 1 M NaHCO3 (100 mL × 3), water, and brine. After drying over MgSO4 and removing the solvent from the filtrate, the crude compound 8 was obtained (4.5 g), which was used immediately in the next step without purification.
Docosa-O-acetyl-1-S-acetyl-1-thio-α-D-maltoheptaopyranose (9).
To a solution of crude compound 8 (4.5 g) in 50 mL of acetone, KSAc (900 mg, 7.89 mmol) was added. After stirring at RT for 5 h, the solvent was slowly removed under vacuum. Water (100 mL) was added, and the mixture was extracted with ethyl acetate (100 mL × 3). The combined organic fraction was washed with brine and dried over Na2SO4. After removing the solvent from the filtrate, the residue was purified by column chromatography (3:1 ethyl acetate/hexanes) to afford compound 9 as a colorless viscous solid (3.1 g, 66% combined yield for 2 steps from 7). 1H NMR (500 MHz, CDCl3; Figure S19) δ 5.47–5.24 (m, 14H), 5.06 (t, J = 9.8 Hz, 1H), 4.97 (t, J = 9.6 Hz, 1H), 4.85 (dd, J = 10.5, 3.9 Hz, 1H), 4.78–4.69 (m, 5H), 4.56–4.44 (m, 6H), 4.37–4.14 (m, 7H), 4.09–3.84 (m, 14H), 2.38 (s, 3H, SAc), 2.26–1.88 (m, 66H, OAc). 13C NMR (126 MHz, CDCl3; Figure S20) δ 191.79, 170.74, 170.68, 170.54, 170.46, 170.44, 170.41, 170.37, 170.36, 169.91, 169.75, 169.64, 169.54, 169.51, 169.47, 169.46, 169.45, 95.79, 95.76, 95.73, 95.64, 82.77, 79.78, 78.99, 76.57, 76.25, 73.53, 73.32, 73.27, 72.41, 71.73, 71.71, 71.61, 71.58, 70.54, 70.49, 70.44, 70.39, 70.06, 69.95, 69.38, 69.12, 69.00, 68.97, 68.95, 68.47, 67.98, 62.86, 62.50, 62.42, 62.23, 61.41, 30.83, 20.88, 20.87, 20.81, 20.68, 20.61, 20.59, 20.57.
1-Thio-β-D-maltoheptaopyranose Sodium Salt (10).
To a solution of compound 9 (2.0 g, 0.94 mmol) in MeOH (200 mL), a solution of NaOMe (81 mg, 1.5 mmol) in methanol (5 mL) was added dropwise. The reaction was stirred at RT for 3 h, after which a white precipitate was obtained. The solvent was slowly removed, and fresh MeOH (200 mL) was added. The mixture was further stirred at RT for 2 h. The precipitate was collected by filtration and washed with cold methanol to give product 10 as a white solid (1.1 g, 94%). 1H NMR (500 MHz, D2O; Figure S21) δ 5.42–5.35 (m, 6H), 4.55 (d, J = 9.1 Hz, 1H, H-1), 4.00–3.92 (m, 5H), 3.93–3.55 (m, 35H), 3.51 (ddd, J = 9.5, 5.5, 1.9 Hz, 1H), 3.42 (t, J = 9.5 Hz, 1H), 3.05 (t, J = 9.1 Hz, 1H). 13C NMR (126 MHz, D2O; Figure S22) δ 99.76, 99.69, 99.59, 84.11, 78.50, 78.23, 78.10, 77.32, 76.99, 76.95, 76.75, 73.33, 73.31, 72.88, 72.70, 71.73, 71.59, 71.20, 71.18, 71.11, 69.31, 61.25, 60.48, 60.41.
Compound 11.
Compounds 10 (470 mg, 0.395 mmol) and 16 (170 mg, 0.596 mmol) were dissolved in DMF (10 mL). After stirring at 50 °C for 4 h, the solvent was removed by low-pressure distillation. The residue was purified by column chromatography (1-butanol/ethanol/H2O, 3:5:0.1 to 3:5:1) to afford compound 11 as a pale yellow solid (239 mg, 58%). 1H NMR (500 MHz, D2O; Figure S23) δ 5.51–5.43 (m, 6H), 4.69 (d, J = 9.9 Hz, 1H, H-1), 4.08–3.64 (m, 48H), 3.61–3.56 (m, 2H, CH2N3), 3.49 (t, J = 9.5 Hz, 1H), 3.45–3.40 (m, 1H), 3.08 (dt, J = 12.8, 6.4 Hz, 1H, SH2), 2.99 (dt, J = 13.9, 6.4 Hz, 1H, SH2). 13C NMR (50 MHz, D2O; Figure S24) δ 100.23, 100.10, 85.68, 78.89, 78.01, 77.43, 73.77, 73.35, 73.17, 72.62, 72.21, 72.00, 71.66, 70.72, 69.99, 69.86, 69.80, 69.69, 61.29, 60.91, 29.51.
Compound 12.
Compound 11 (85 mg, 0.066 mmol) was dissolved in 2:1 v/v MeOH/H2O (4 mL), and Pd(OH)2/C (10%, 11 mg, 0.011 mmol) was added. The reaction flask was purged with Ar and then with H2. The reaction mixture was stirred at RT for 2 h and then filtered through celite. The filtrate was evaporated under reduced pressure to yield compound 12 as a white solid (83 mg, quantitative yield). 1H NMR (500 MHz, D2O) δ 5.39 (d, J = 4.1 Hz, 6H), 4.60 (d, J = 9.9 Hz, 1H), 4.02–3.55 (m, 48H), 3.42 (t, J = 9.4 Hz, 1H), 3.36 (t, J = 9.5 Hz, 1H), 3.12 (t, J = 5.1 Hz, 2H, CH2NH2), 3.01 (dt, J = 12.4, 6.2 Hz, 1H, SH2), 2.93 (dt, J = 13.5, 6.3 Hz, 1H, SH2).
G7-DPPE.
Compound 12 (147 mg, 0.113), compound 17 (121 mg, 0.136 mmol), and EDAC (65 mg, 0.339 mmol) were added to 5 mL of dry DMF, and the reaction was stirred at RT for 8 h. The solvent was removed by low-pressure distillation at 40 °C to give the crude product, which was then purified by column chromatography (CHCl3/MeOH/H2O, 7:4:0 to 7:4:0.8 to 7:4:1.2) to afford G7-DPPE as a white solid (108 mg, 46%). TLC of the purified product showed a single spot (Rf = 0.22, 7:4:0.5 CHCl3/MeOH/H2O) after staining with 5% sulfuric acid in ethanol. 1H NMR (500 MHz, 7:4 CDCl3/CD3OD; Figure S26) δ 5.32–4.97 (m, 7H), 4.43 (dd, J = 11.9, 2.0 Hz, 1H), 4.19 (dd, J = 12.0, 6.9 Hz, 1H), 4.09–3.26 (m, 60H), 3.02–2.84 (m, 2H), 2.55 (s, 4H), 2.33 (dd, J = 15.6, 8.1 Hz, 4H), 1.61 (dd, J = 13.8, 6.9 Hz, 4H), 1.46–1.12 (m, 48H), 0.89 (t, J = 6.9 Hz, 6H). 13C NMR (126 MHz, CDCl3/CD3OD = 7:4; Figure S27) δ 173.69, 173.32, 101.47, 77.42, 77.16, 76.90, 73.29, 71.96, 69.73, 63.29, 57.19, 48.80, 48.63, 48.46, 48.29, 48.12, 47.95, 47.78, 33.98, 33.83, 31.61, 29.40, 29.34, 29.28, 29.26, 29.10, 29.04, 28.89, 28.86, 24.68, 24.61, 22.33, 13.56. 13P NMR (81 MHz, CDCl3/CD3OD = 7:4; Figure S28) δ 2.29. High-resolution mass spectrometry (HRMS) calcd. for C89H161N2O47PS [M – H]−, m/z: 2071.9655, 2072.9688, 2073.9722; [M – 2H]2−, m/z: 1035.4791, 1035.9808, 1036.4825. Found: 2071.9679, 2072.9680, 2073.9733, 1035.4800, 1035.9821, 1036.4822 (Figure S29).
Preparation of Nonfluid Liposomes by Thin-Film Hydration and Extrusion (Entry 1, Table 1).
To a mixture of egg PC (15.8 mg, 21 μmol), cholesterol (3.8 mg, 10 μmol) in a 100 mL round-bottom flask was added 5:1 v/v chloroform/methanol (5 mL). After the solids were fully dissolved, the solvent was rotary-evaporated to yield a thin film around the flask, and the lipid film was further dried under vacuum overnight. Water (15 mL) was added to the dry film. The resulting mixture was sonicated for 2 min and then extruded 10 times through a 100 nm polycarbonate filter. The liposomes were then dialyzed at 4 °C for 24 h. The resulting liposomes were kept at 4 °C for further use.
Preparation of Nonfluid Liposomes by Ethanol Injection (Entries 2–5, Table 1).
To prepare the nonfluid liposomes alone (entry 2), 1 mL of 10:1 v/v methanol/DMSO was added to a 10 mL round-bottom flask containing egg PC (7.7 mg, 10 μmol) and cholesterol (0.5 mg, 1.3 μmol). After the solids were completely dissolved, the solution was added dropwise to a scintillation vial containing 10 mL of water under vigorously shaking. The liposomes were dialyzed at 4 °C for 24 h, and the purified liposomes were kept at 4 °C for further use.
Rifampicin-loaded nonfluid liposomes (entry 3) were prepared following the same procedure as entry 2 above, except rifampicin (3.4 mg, 4.1 μmol) was added.
For rifampicin-loaded nonfluid Man-glycoliposomes (entry 4) and G7-glycoliposomes (entry 5), Man-DPPE (1.0 mg, 1.1 μmol) or G7-DPPE (2 mg, 1.1 μmol) at a mole ratio of 8:1:0.8 PC/Chol/glycolipid was included in the liposome formulation in addition to rifampicin. The liposomes were purified by dialysis at 4 °C for 24 h and stored at 4 °C.
Preparation of Fluid Liposomes by Thin-Film Hydration and Extrusion (Entries 6–9).
To prepare the fluid liposomes alone (entry 6), 10 mL of 5:1 v/v chloroform/methanol was added to a mixture of DPPC (12 mg, 16 μmol) and DMPG (2.0 mg, 2.9 μmol) in a 50 mL round-bottom flask. After the solids were fully dissolved, the solvent was rotary-evaporated to yield a thin film around the flask, which was further dried under vacuum overnight. Water (10 mL) was added to the dried film, and the resulting mixture was sonicated for 2 min. It was then frozen at −80 °C and thawed at 55 °C, and this process was repeated 3 times. The resulting mixture was extruded through a 100 nm polycarbonate filter at 55 °C for 10 times. The liposomes were dialyzed at 4 °C for 24 h, and the purified liposomes were kept at 4 °C until further use.
To prepare the rifampicin-loaded fluid liposomes (entry 7), after DPPC (12 mg, 16 μmol) and DMPG (2.0 mg, 2.9 μmol) were fully dissolved, a solution of rifampicin (10 mg, 12 μmol) in chloroform (10 mL) was added. The solvent was then evaporated on a rotavap, and the thin lipid film around the inside of the flask was further dried under vacuum overnight. Hydration and purification followed the same procedure as entry 6 above.
To prepare the rifampicin-loaded fluid Man-glycoliposomes (entry 8) and G7-glycoliposomes (entry 9), Man-DPPE (1.8 mg, 2.0 μmol) or G7-DPPE (4 mg, 2.0 μmol) was added to DPPC (12 mg, 16 μmol) and DMPG (2.0 mg, 2.9 μmol) at a mole ratio of 6:1:0.6 DPPC/DMPG/glycolipid. The rest of the procedure followed the same as entry 7 above.
Determining the Amount of Rifampicin Encapsulated in Liposomes.
To determine the amount of rifampicin encapsulated in the rifampicin-loaded liposomes, the liposomes were lysed by adding 10 times the volume of DMSO to the purified liposomes. The absorbance of the resulting solution at 475 nm was measured on a UV–vis spectrometer. The concentration of rifampicin was determined by comparing the absorbance to the standard calibration curve of rifampicin, constructed by measuring the absorbances of varying concentrations of rifampicin solutions in DMSO at 475 nm (Figure S1).
Drug Release.
To determine the kinetics of the rifampicin release from the liposomes, rifampicin-loaded liposomes were dialyzed, and the amount of rifampicin released was monitored by measuring the concentration of rifampicin in the solution. A dialysis bag (MW cutoff 10 000) containing 2 mL of the purified rifampicin-loaded liposomes was placed in a beaker containing 100 mL of Milli-Q water. It was then placed in an incubator at 37 °C while shaking. An aliquot (1 mL) of the aqueous solution was taken out at 0.5, 1, 2, 4, 9, and 24 h. The concentration of rifampicin was determined by UV–vis spectroscopy by comparing it to the standard calibration curve of rifampicin (Figure S1).
Determining the Amount of Man-DPPE or G7-DPPE Incorporated into Liposomes.
A colorimetry assay based on anthrone/sulfuric acid was used to determine the amount of Man-DPPE or G7-DPPE incorporated into the liposomes.52,57,67 Dehydration of a hexose-like glucose or Man by the concentrated sulfuric acid gives a furfuraldehyde, which is then condensed with anthrone to form a yellowish-green furanylmethylene–anthrone product with a λmax of 620 nm. Standard calibration curves were first constructed using Man-DPPE and G7-DPPE. Stock solutions of Man-DPPE and G7-DPPE were prepared at 10 mg/mL in 7:4:0.1 v/v/v chloroform/MeOH/H2O. Aliquots of the stock solutions were added to scintillation vials, and the organic solvents were removed by an Ar flow. Ten milliliters of Milli-Q water was added to each vial, and the solutions were sonicated for 10 min to make the concentration series for Man-DPPE (50, 75, 100, 150, 200, 300, and 600 μg/mL) and G7-DPPE (20, 30, 50, 75, 100, 150, and 200 μg/mL). A freshly prepared solution of anthrone in concentrated H2SO4 (0.8 mL, 0.5 wt %) was slowly added to the solutions of Man-DPPE or G7-DPPE (0.4 mL) in an ice bath, and the solutions were heated to 100 °C for 10 min. After cooling to room temperature, the absorbances at 620 nm were recorded on a microplate reader (Infinite 200 PRO, TECAN). The data were plotted against the concentrations of Man-DPPE or G7-DPPE as the calibration curves (Figure S2). To determine the amount of Man-DPPE or G7-DPPE incorporated into the liposomes, the liposomes were treated with freshly prepared anthrone/sulfuric acid reagent following the same procedure described above. The absorbances at 620 nm were measured, and the data were compared to the calibration curves in Figure S2, from which the amounts of Man-DPPE or G7-DPPE were calculated.
Determination of Dissociation Constant Kd between Glycoliposomes and Con A by DLS.
G7- or Man-glycoliposomes (without rifampicin) were treated with a solution of various concentrations of Con A in 10 mM pH 7.2 HEPES buffer containing 1.0 mM of MnCl2 and CaCl2 for 2 h while shaking on a vortex mixer. For DLS measurements, the suspension was diluted to 2 mL using the HEPES buffer. Each DLS measurement was performed at 20 scans and repeated 3 times. The change in the hydrodynamic diameter (ΔD) of the liposomes vs Con A concentration was plotted, and the dissociation constant was calculated by fitting the curve to the Hill equation using GraphPad Prism.
Determination of MIC by Broth Microdilution.
E. coli ORN208 was grown in Mueller Hinton broth (MHB) at 37 °C overnight. The culture was diluted 5 times by MHB and incubated at 37 °C for an additional 3 h until the OD600 reached 0.3. The bacterial suspension was then diluted 100-folds by MHB, and 100 μL was added to a 96-well plate containing 2× serially diluted liposomes or rifampicin solutions (100 μL in each well, each in triplicates). Plates were incubated at 37 °C for 16 h with shaking at 180 rpm, AlamarBlue (20 μL) was added to each well, and the mixture was incubated for 2 more hours. The fluorescence intensity at 590 nm (excitation: 560 nm) was recorded using a microplate reader (Infinite 200 PRO, TECAN). The MIC was the lowest drug concentration that led to a ≥90% reduction in fluorescence relative to the untreated cells.
Purified liposome solutions in water were used directly for serial dilution. For rifampicin, a stock solution was prepared in DMSO (10 mg/mL) and then diluted with Milli-Q water to the final concentration series. The maximal DMSO concentration in the highest concentration rifampicin (256 μg/mL) was 2.5%, at which the bacterial cells were still viable. DMSO did not affect bacterial growth until reaching the concentration of 20%.
Time–Kill Study.
E. coli ORN208 was cultured according to the procedure above. The 100-fold diluted bacterial suspension (1.5 mL) was added to a 24-well plate containing 1.5 mL each of the rifampicin solutions or rifampicin-loaded liposomes at the concentrations of 1 × MIC or 2 × MIC or 4 × MIC, and the plate was then placed in an incubator at 37 °C while shaking at 180 rpm. Bacteria in MHB alone were the control. Bacterial colony forming unit (CFU) was estimated by the single plate-serial dilution spotting (SP-SDS) technique.68 An aliquot of 50 μL of bacterial suspension was taken out from each well at the time points of 0, 2, 4, 8, and 24 h, from which six dilutions were prepared (101–106) from each suspension. An aliquot of 20 μL of these six dilutions was applied as 10–15 microdrops over an MH agar plate that was divided into six sections. After overnight incubation at 37 °C, the sections that yielded 6–60 colonies were used to calculate the bacterial CFU.
FRET Experiment.
The liposomes containing NBD-PE and Rh-PE were prepared by thin-film hydration and extrusion, following the same procedure as described earlier. To a mixture of DPPC (6 mg, 8 μmol), DMPG (1 mg, 1.4 μmol), and Man-DPPE (0.9 mg, 1 μmol) or G7-DPPE (2 mg, 1 μmol) in a 50 mL round-bottom flask was added 10 mL of 2:1 v/v chloroform-methanol. After the solids were fully dissolved, NBD-PE (51 μg, 0.053 μmol) and Rh-PE (72 μg, 0.054 μmol) were added. The solvent was then evaporated, and the thin lipid film was further dried under vacuum overnight. Water (11 mL) was added to the dried film. The mixture was sonicated briefly, subjected to three freeze (−80 °C) and thaw (in 65 °C water bath) cycles, and then extruded through two stacked 100 nm polycarbonate membranes at 60 °C for 10 times. The purified liposomes were stored at 4 °C in the dark. For the FRET experiment, 1.8 mL of PBS buffer (pH 7.4, 10 mM), E. coli ORN208 (OD600 = 0.6 in PBS buffer), or the detergent (0.2 vol % Triton X-100) was mixed with 0.2 mL of the liposome solution in a cuvette. Initial mixing was done by repeatedly pipetting up and down or under shaking at 180 rpm at 37 °C for 1 h in the case of bacteria. Fluorescence spectra were recorded on an Agilent Cary Eclipse fluorescence spectrophotometer by scanning in the range of 500–700 nm at an excitation of 460 nm, the λmax of NBD-PE.
Confocal Fluorescence Microscopy Imaging.
E. coli ORN208 was grown in MHB to an OD600 of 0.3. The bacterial pellet was collected by centrifuging the broth culture at 5000 rpm for 10 min and resuspended in pH 7.4 PBS buffer to reach the same concentration. NBD/Rh-labeled liposomes (1 mL) were added to 1 mL of the bacterial suspension. The mixture was further incubated for 1 h. A drop (20 μL) of the suspension was mixed with 20 μL of glycerol, pipetted onto a glass coverslip, sealed with nail polish, and examined under a confocal fluorescence microscope (Olympus FV300).
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful for the financial support from NIH (R15GM128164).
Footnotes
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acsanm.1c01320
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsanm.1c01320.
Synthesis of compounds 16 and 17; standard calibration curves of rifampicin, Man-DPPE, and G7-DPPE; calculation of kobs and Papp; and bacterium–liposome interactions by FRET and confocal fluorescence microscopy (PDF)
Contributor Information
Bin Wu, Department of Chemistry, The University of Massachusetts Lowell, Lowell, Massachusetts 01854, United States.
William Ndugire, Department of Chemistry, The University of Massachusetts Lowell, Lowell, Massachusetts 01854, United States.
Xuan Chen, Department of Chemistry, The University of Massachusetts Lowell, Lowell, Massachusetts 01854, United States.
Mingdi Yan, Department of Chemistry, The University of Massachusetts Lowell, Lowell, Massachusetts 01854, United States.
REFERENCES
- (1).Mitchell MJ; Billingsley MM; Haley RM; Wechsler ME; Peppas NA; Langer R Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discovery 2020, 20, 101–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Gupta A; Landis RF; Rotello VM Nanoparticle-Based Antimicrobials: Surface Functionality is Critical. F1000Research 2016, 5, 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Rudramurthy G; Swamy M; Sinniah U; Ghasemzadeh A Nanoparticles: Alternatives Against Drug-Resistant Pathogenic Microbes. Molecules 2016, 21, 836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Anselmo AC; Mitragotri S A Review of Clinical Translation of Inorganic Nanoparticles. AAPS J 2015, 17, 1041–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Anselmo AC; Mitragotri S Nanoparticles in the clinic. Bioeng. Transl. Med 2016, 1, 10–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bobo D; Robinson KJ; Islam J; Thurecht KJ; Corrie SR Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm. Res 2016, 33, 2373–2387. [DOI] [PubMed] [Google Scholar]
- (7).Beaulac C; Sachetelli S; Lagace J In-vitro bactericidal efficacy of sub-MIC concentrations of liposome-encapsulated antibiotic against gram-negative and gram-positive bacteria. J. Antimicrob. Chemother 1998, 41, 35–41. [DOI] [PubMed] [Google Scholar]
- (8).Sachetelli S; Khalil H; Chen T; Beaulac C; Sénéchal S; Lagacé J Demonstration of a fusion mechanism between a fluid bactericidal liposomal formulation and bacterial cells. Biochim. Biophys. Acta, Biomembr 2000, 1463, 254–266. [DOI] [PubMed] [Google Scholar]
- (9).Drulis-Kawa Z; Dorotkiewicz-Jach A; Gubernator J; Gula G; Bocer T; Doroszkiewicz W The interaction between Pseudomonas aeruginosa cells and cationic PC:Chol:DOTAP liposomal vesicles versus outer-membrane structure and envelope properties of bacterial cell. Int. J. Pharm 2009, 367, 211–219. [DOI] [PubMed] [Google Scholar]
- (10).Ma Y; Wang Z; Zhao W; Lu T; Wang R; Mei Q; Chen T Enhanced bactericidal potency of nanoliposomes by modification of the fusion activity between liposomes and bacterium. Int. J. Nanomed 2013, 8, 2351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wang Z; Ma Y; Khalil H; Wang R; Lu T; Zhao W; Zhang Y; Chen J; Chen T Fusion between fluid liposomes and intact bacteria: study of driving parameters and in vitro bactericidal efficacy. Int. J. Nanomed 2016, 11, 4025–4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Vyas SP; Sihorkar V; Jain S Mannosylated liposomes for bio-film targeting. Int. J. Pharm 2007, 330, 6–13. [DOI] [PubMed] [Google Scholar]
- (13).Pinto-Alphandary H; Andremont A; Couvreur P Targeted delivery of antibiotics using liposomes and nanoparticles: research and applications. Int. J. Antimicrob. Agents 2000, 13, 155–168. [DOI] [PubMed] [Google Scholar]
- (14).Noble GT; Stefanick JF; Ashley JD; Kiziltepe T; Bilgicer B Ligand-targeted liposome design: challenges and fundamental considerations. Trends Biotechnol 2014, 32, 32–45. [DOI] [PubMed] [Google Scholar]
- (15).Allen TM; Cullis PR Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Delivery Rev. 2013, 65, 36–48. [DOI] [PubMed] [Google Scholar]
- (16).Lee YC; Lee RT Carbohydrate-Protein Interactions: Basis of Glycobiology. Acc. Chem. Res 1995, 28, 321–327. [Google Scholar]
- (17).Lasky LA Selectins: interpreters of cell-specific carbohydrate information during inflammation. Science 1992, 258, 964–969. [DOI] [PubMed] [Google Scholar]
- (18).Smith AE; Helenius A How Viruses Enter Animal Cells. Science 2004, 304, 237–242. [DOI] [PubMed] [Google Scholar]
- (19).Frenz T; Grabski E; Duran V; Hozsa C; Stepczynska A; Furch M; Gieseler RK; Kalinke U Antigen presenting cellselective drug delivery by glycan-decorated nanocarriers. Eur. J. Pharm. Biopharm 2015, 95, 13–17. [DOI] [PubMed] [Google Scholar]
- (20).Weingart JJ; Vabbilisetty P; Sun X-L Glyco-Functionalized Liposomes; John Wiley & Sons, Inc., 2016; pp 211–232. [Google Scholar]
- (21).Ramström O; Yan M Glyconanomaterials for combating bacterial infections. Chemistry 2015, 21, 16310–16317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Jayawardana KW; Jayawardena HS; Wijesundera SA; De Zoysa T; Sundhoro M; Yan M Selective targeting of Mycobacterium smegmatis with trehalose-functionalized nanoparticles. Chem. Commun 2015, 51, 12028–12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chen X; Wu B; Jayawardana KW; Hao N; Jayawardena HS; Langer R; Jaklenec A; Yan M Magnetic multivalent trehalose glycopolymer nanoparticles for the detection of mycobacteria. Adv. Healthcare Mater 2016, 5, 2007–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hao N; Chen X; Jeon S; Yan M Carbohydrate-conjugated hollow oblate mesoporous silica nanoparticles as nanoantibiotics to target mycobacteria. Adv. Healthcare Mater 2015, 4, 2797–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhou J; Jayawardana KW; Kong N; Ren Y; Hao N; Yan M; Ramstroem O Trehalose-Conjugated, Photofunctionalized Mesoporous Silica Nanoparticles for Efficient Delivery of Isoniazid into Mycobacteria. ACS Biomater. Sci. Eng 2015, 1, 1250–1255. [DOI] [PubMed] [Google Scholar]
- (26).Ning X-H; Lee S-J; Wang Z-R; Kim D-I; Stubblefield B; Gilbert E; Murthy N Maltodextrin-based imaging probes detect bacteria in vivo with high sensitivity and specificity. Nat. Mater 2011, 10, 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Jayawardena HSN; Jayawardana KW; Chen X; Yan M Maltoheptaose promotes nanoparticle internalization by Escherichia coli. Chem. Commun 2013, 49, 3034–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wehrli W Rifampin: mechanisms of action and resistance. Rev. Infect. Dis 1983, 5, S407–11. [DOI] [PubMed] [Google Scholar]
- (29).Wehrli W; Knusel F; Schmid K; Staehelin M Interaction of rifamycin with bacterial RNA polymerase. Proc. Natl. Acad. Sci. USA 1968, 61, 667–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Williams KJ; Piddock LJ Accumulation of rifampicin by Escherichia coli and Staphylococcus aureus. J. Antimicrob. Chemother 1998, 42, 597–603. [DOI] [PubMed] [Google Scholar]
- (31).Jin DJ; Gross CA Mapping and sequencing of mutations in the Escherichia colirpoB gene that lead to rifampicin resistance. J. Mol. Biol 1988, 202, 45–58. [DOI] [PubMed] [Google Scholar]
- (32).Attri S; Rana SV; Vaiphei K; Sodhi CP; Katyal R; Goel RC; Nain CK; Singh K Isoniazid- and rifampicin-induced oxidative hepatic injury - protection by N-acetylcysteine. Hum. Exp. Toxicol 2000, 19, 517–522. [DOI] [PubMed] [Google Scholar]
- (33).Santhosh S; Sini TK; Anandan R; Mathew PT Hepatoprotective activity of chitosan against isoniazid and rifampicin-induced toxicity in experimental rats. Eur. J. Pharmacol 2007, 572, 69–73. [DOI] [PubMed] [Google Scholar]
- (34).Changsan N; Chan HK; Separovic F; Srichana T Physicochemical characterization and stability of rifampicin liposome dry powder formulations for inhalation. J. Pharm. Sci 2009, 98, 628–639. [DOI] [PubMed] [Google Scholar]
- (35).Salem II; Flasher DL; Duzgunes N Liposome-encapsulated antibiotics. Methods Enzymol 2005, 391, 261–291. [DOI] [PubMed] [Google Scholar]
- (36).Khadka P; Dummer J; Hill PC; Das SC Considerations in preparing for clinical studies of inhaled rifampicin to enhance tuberculosis treatment. Int. J. Pharm 2018, 548, 244–254. [DOI] [PubMed] [Google Scholar]
- (37).Norberg O; Deng L; Aastrup T; Yan M; Ramstrom O Photo-Click Immobilization on Quartz Crystal Microbalance Sensors for Selective Carbohydrate-Protein Interaction Analyses. Anal. Chem 2011, 83, 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Norberg O; Lee IH; Aastrup T; Yan M; Ramstroem O Photogenerated lectin sensors produced by thiol-ene/yne photo-click chemistry in aqueous solution. Biosens. Bioelectron 2012, 34, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Deng L; Wang X; Uppalapati S; Norberg O; Dong H; Joliton A; Yan M; Ramstroem O Stereocontrolled 1-S-glycosylation and comparative binding studies of photoprobe-thiosaccharide conjugates with their O-linked analogs. Pure Appl. Chem 2013, 85, 1789–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Norberg O; Wu B; Thota N; Ge J-T; Fauquet G; Saur A-K; Aastrup T; Dong H; Yan M; Ramstroem O Synthesis and binding affinity analysis of α1–2- and α1–6-O/S-linked dimannosides for the elucidation of sulfur in glycosidic bonds using quartz crystal microbalance sensors. Carbohydr. Res 2017, 452, 35–42. [DOI] [PubMed] [Google Scholar]
- (41).Ndugire W; Wu B; Yan M Synthesis of carbohydrate-grafted glycopolymers using a catalyst-Free, perfluoroarylazide-mediated fast staudinger reaction. Molecules 2019, 24, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Farkas E; Jánossy L; Harangi J; Kandra L; Lipták A Synthesis of chromogenic substrates of α-amylases on a cyclodextrin basis. Carbohydr. Res 1997, 303, 407–415. [DOI] [PubMed] [Google Scholar]
- (43).Deng L; Norberg O; Uppalapati S; Yan M; Ramstrom O Stereoselective synthesis of light-activatable perfluorophenylazide-conjugated carbohydrates for glycoarray fabrication and evaluation of structural effects on protein binding by SPR imaging. Org. Biomol. Chem 2011, 9, 3188–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Briuglia ML; Rotella C; McFarlane A; Lamprou DA Influence of cholesterol on liposome stability and on in vitro drug release. Drug Delivery Transl. Res 2015, 5, 231–242. [DOI] [PubMed] [Google Scholar]
- (45).Jaafar-Maalej C; Diab R; Andrieu V; Elaissari A; Fessi H Ethanol injection method for hydrophilic and lipophilic drug-loaded liposome preparation. J. Liposome Res 2010, 20, 228–243. [DOI] [PubMed] [Google Scholar]
- (46).Batzri S; Korn ED Single bilayer liposomes prepared without sonication. Biochim. Biophys. Acta, Biomembr 1973, 298, 1015–1019. [DOI] [PubMed] [Google Scholar]
- (47).Manca ML; Sinico C; Maccioni AM; Diez O; Fadda AM; Manconi M Composition influence on pulmonary delivery of rifampicin liposomes. Pharmaceutics 2012, 4, 590–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Wang WT; Liu JG; Sun WH; Wang WY; Wang SL; Zhu NH Widely tunable single bandpass microwave photonic filter based on Brillouin-assisted optical carrier recovery. Opt. Express 2014, 22, 29304–29313. [DOI] [PubMed] [Google Scholar]
- (49).Calvo P; V-J J; Alonso MJ Comparative in vitro evaluation of several colloidal systems, nanoparticles, nanocapsules, and nanoemulsions, as ocular drug carriers. J. Pharm. Sci 1996, 85, 530–536. [DOI] [PubMed] [Google Scholar]
- (50).Xiang T; Xu Y; Anderson BD The barrier domain for solute permeation varies with lipid bilayer phase structure. J. Membr. Biol 1998, 165, 77–90. [DOI] [PubMed] [Google Scholar]
- (51).Wang X; Ramström O; Yan M A photochemically initiated chemistry for coupling underivatized carbohydrates to gold nanoparticles. J. Mater. Chem 2009, 19, 8944–8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Morris DL Quantitative determination of carbohydrates with dreywood’s anthrone reagent. Science 1948, 107, 254–255. [DOI] [PubMed] [Google Scholar]
- (53).Hurd CD; Isenhour LL Pentose Reactions. I. Furfural Formation. J. Am. Chem. Soc 1932, 54, 317–330. [Google Scholar]
- (54).Wolfrom ML; Schuetz RD; Cavalieri LF Chemical Interactions of Amino Compounds and Sugars. III.1 The Conversion of D-Glucose to 5-(Hydroxymethyl)-2-furaldehyde2. J. Am. Chem. Soc 1948, 70, 514–517. [DOI] [PubMed] [Google Scholar]
- (55).Hardman KD; Ainsworth CF Structure of the concanavalin A-methyl alpha-D-mannopyranoside complex at 6-A resolution. Biochemistry 1976, 15, 1120–1128. [DOI] [PubMed] [Google Scholar]
- (56).Harrop SJ; Helliwell JR; Wan TC; Kalb AJ; Tong L; Yariv J Structure solution of a cubic crystal of concanavalin A complexed with methyl alpha-D-glucopyranoside. Acta Crystallogr., Sect. D: Biol. Crystallogr 1996, 52, 143–55. [DOI] [PubMed] [Google Scholar]
- (57).Wang X; Ramstrom O; Yan M Quantitative analysis of multivalent ligand presentation on gold glyconanoparticles and the impact on lectin binding. Anal. Chem 2010, 82, 9082–9089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Wang X; Ramstrom O; Yan M Dye-doped silica nanoparticles as efficient labels for glycans. Chem. Commun 2011, 47, 4261–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Kong N; Shimpi MR; Park J; Ramstroem O; Yan M Carbohydrate conjugation through microwave-assisted functionalization of single-walled carbon nanotubes using perfluorophenyl azides. Carbohydr. Res 2015, 405, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Wang X; Ramstrom O; Yan M Dynamic light scattering as an efficient tool to study glyconanoparticle-lectin interactions. Analyst 2011, 136, 4174–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Liyanage SH; Yan M Quantification of binding affinity of glyconanomaterials with lectins. Chem. Commun 2020, 56, 13491–13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Takeo K; Suzuno R; Fujimoto M; Kuwahara A; Nakamura K Seibutsu Butsuri Kagaku 1982, 26, 173–177. [Google Scholar]
- (63).Harris SL; Spears PA; Havell EA; Hamrick TS; Horton JR; Orndorff PE Characterization of Escherichia coli type 1 pilus mutants with altered binding specificities. J. Bacteriol 2001, 183, 4099–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Lobritz MA; Belenky P; Porter CBM; Gutierrez A; Yang JH; Schwarz EG; Dwyer DJ; Khalil AS; Collins JJ Antibiotic efficacy is linked to bacterial cellular respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 8173–8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Struck DK; Hoekstra D; Pagano RE Use of resonance energy transfer to monitor membrane fusion. Biochemistry 1981, 20, 4093–4099. [DOI] [PubMed] [Google Scholar]
- (66).Marsden HR; Tomatsu I; Kros A Model systems for membrane fusion. Chem. Soc. Rev 2011, 40, 1572–85. [DOI] [PubMed] [Google Scholar]
- (67).Wang X; Ramstrom O; Yan M A photochemically initiated chemistry for coupling underivatized carbohydrates to gold nanoparticles. J. Mater. Chem 2009, 19, 8944–8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Thomas P; Sekhar AC; Upreti R; Mujawar MM; Pasha SS Optimization of single plate-serial dilution spotting (SP-SDS) with sample anchoring as an assured method for bacterial and yeast cfu enumeration and single colony isolation from diverse samples. Biotechnol. Rep 2015, 8, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.