

Abstract
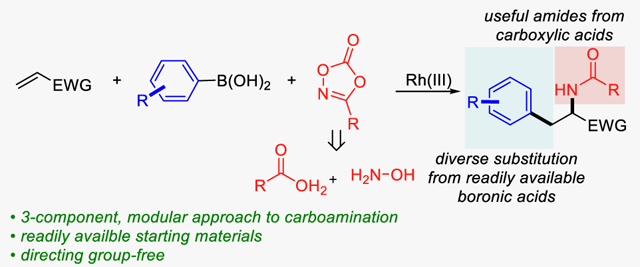
Herein we report a Rh(III)-catalyzed three-component carboamination of alkenes from readily available aryl boronic acids as a carbon source and dioxazolones as nitrogen electrophiles. This protocol provides facile access to valuable amine products including α-amino acid derivatives in good yield and regioselectivity without the need for a directing functionality. A series of experiments suggest a mechanism in which the Rh(III) catalyst undergoes transmetalation with the aryl boronic acid followed by turnover limiting, alkene migratory insertion into the Rh(III)-aryl bond. Subsequently, fast Rh-nitrene formation provides the syn-carboamination product selectively after reductive elimination and proto-demetalation. Importantly, the protocol provides 3-component coupling products in preference to a variety of 2-component undesired by-products.
Keywords: Carboamination, Rh(III) catalysis, α-amino acid synthesis, alkene difunctionalization, directing group-free
Graphical Abstract
The simultaneous installation of two functional groups across the ubiquitous alkene double bond in a stereoselective manner is a powerful transformation in organic synthesis1 and provides an efficient way of rapidly increasing molecular complexity from readily available starting materials. Among various potential difunctionalizations, the carboamination of alkenes offers direct access to valuable and pharmaceutically important amine products by forming both C-C and C-N bonds in a single step.2 A handful of powerful annulations,3 or intramolecular carboaminations,4 have been developed, which are currently limited to the synthesis of cyclic products.
In 2015, we described a Rh(III)-catalyzed stereospecific, syn-carboamination of activated alkenes using N-enoxyphthalimide as both carbon and nitrogen source of the reaction to furnish acyclic amine products (Scheme 1a).5 Subsequently, Glorius reported the carboamination of acrylates initiated by Cp*Co(III)-catalyzed Csp2−H activation of N-phenoxyamides (Scheme 1b),6 followed more recently by Cramer’s demonstration of an asymmetric version of this reaction.7 Recently, Ellman reported the synthesis of α-branched amine through the Rh(III)-catalyzed three-component 1,1-carboamination of terminal alkenes that also initiated by directing group assisted C-H activation (Scheme 1c).8
Scheme 1.

Intermolecular carboamination of alkenes.
Other significant contributions towards the linear carboamination of alkenes have been made utilizing Pd or Ni catalysis with olefins bearing covalently linked aminoquinoline directing groups,9 as well as through single electron pathways involving nitrogen or carbon centered radicals.2b,10
Despite recent progress in the field, the current state-of-the-art in carboamination involves additional steps to install and remove the often-undesired directing functionality, an inherent loss in stereocontrol imparted through open shell intermediates, or synthetically taxing substrates that limit the chemical space available to their practical application.
In searching for a more general solution to this problem, we became interested in developing an intermolecular 3-component carboamination that uses readily accessible carbon and nitrogen sources. Such a modular approach would enable the rapid synthesis of a diverse library of functionalized amine products by simply switching coupling partners. As a reaction design to achieve this goal (Scheme 1d), we envisioned that a carbon nucleophile coordinates to Cp*Rh(III) complex after transmetalation or ligand displacement then undergoes highly regioselective migratory insertion with activated alkenes in the absence of directing group. Subsequent reaction with nitrene precursors forms Rh-nitrene intermediates which will undergo reductive elimination to form a C-N bond and deliver desired carboamination products.
The challenge is that there are several undesired side product pathways as the reaction becomes a multi-component system. For example, after transmetalation, it can undergo dimerization of carbon source or direct C-N coupling if subsequent alkene migratory insertion is slow. Also, after migratory insertion of Rh-carbon bond into the alkenes, undesired β-hydride elimination or protodemetalation will result in Heck-type and hydroarylation products. Last, hydroamination side product also can be formed if the nitrogen source of the reaction directly reacts with the alkene coupling partner.
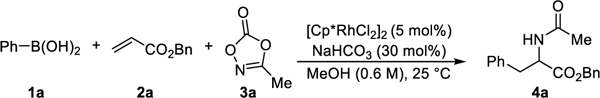
With this hypothesis in mind, we initiated a systematic investigation of carbon nucleophiles such as organoboron and organostannanes, with nitrene precursors such as azides, hydroxamates, and dioxazolones, under various reaction conditions. An initial hit was identified when we combined phenylboronic acid (1a), benzyl acrylate (2a), and 3-methyl-1,4,2-dioxazol-5-one (3a) with [Cp*RhCl2]2 as a catalyst in methanol at room temperature delivering the desired carboamination product (4a) in 8% yield, with the mass balance comprised of Heck-type, hydroamination, and direct C-N coupling products. Inspired by a large library of commercially available boronic acids and readily accessible dioxazolones which can be easily prepared from carboxylic acids,11 we decided to optimize the reaction and gratifyingly, achieved a 77% yield of the desired carboamination product (4a) (Table 1, entry 1, see supporting information for details).
Table 1.
Reaction Optimization.
| ||
|---|---|---|
|
| ||
| entry | deviation from standard conditions | yield of 4aa (%) |
|
| ||
| 1 | None | 77 (68)b |
| 2 | Other nitrene precursors instead of 3a | - |
| 3 | [Cp*Rh(MeCN)3](SbF6)2 instead of [Cp*RhCI2]2 | - |
| 4 | [Rh(COD)2CI]2 instead of [Cp*RhCI2]2 | - |
| 5 | NaOAc instead of NaHCO3 | 5 |
| 6 | 60 °C instead of 25 °C | 42 |
| 7 | 1a (2 equiv)/ 2a (1 equiv)/ 3a (1 equiv) | 64 |
| 8 | 1 mmol scale with 2.5 mol% [Cp*RhCI2]2 | 78 (69)b |
Reactions were conducted on a 0.1 mmol scale using 1a (2.5 equiv), 2a (3.0 equiv), and 3a (1 equiv).
Determined by analysis of 1H NMR of the unpurified reaction mixture.
Isolated yield.
Other nitrene precursors12 that are frequently used for amination chemistry with Cp* group 9 catalysis are completely ineffective for this chemistry. In the cases of Ts-N3, Ts-NH-OPiv, 5,5-dimethyl-1,4,2-dioxazole, and 1,4,2-dioxazol-5-thione, no conversion of starting materials is observed. When N-(pivaloyloxy)amides are used as nitrogen source of the reaction, direct C-N coupling product was observed as major side product and N-(tetrafluorophenoxy)amides give Heck-type and hydroarylation side products in high yield. (entry 2, see supporting information for details, Table S1). Also, the choice of the catalyst is important since cationic Rh(III) catalysts such as [Cp*Rh(MeCN)3](SbF6)2 (entry 3) or an in situ generated cationic Rh(III) complex does not lead to the desired product. Several Rh(I) catalysts such as [Rh(COD)2Cl]2 (entry 4) that are commonly used for conjugate addition chemistry with arylboronic acids13 or other Cp* group 9 catalysts such as [Cp*CoCl2]2 or [Cp*IrCl2]2 failed to deliver the carboamination products.
Screening of base additives shows that bicarbonate, carbonate, or fluoride generally gives a higher reaction yield compared to acetates, and NaHCO3 was selected as the optimal additive. When acetates are used as additive (entry 5), the reaction yield is significantly decreased, and Heck-type product becomes major product of the reaction in low yield. One possible explanation is that acetates act as bidentate ligand and prevent coordination of dioxazolone. The reaction proceeds smoothly at room temperature and tends to give a lower yield at a higher temperature, as shown with the 42% yield obtained when the reaction was conducted at 60 °C (entry 6). When both alkene (2a) and dioxazolone (3a) are used as the limiting reagent of the reaction, the reaction gives synthetically useful a 64% yield (entry 7), and a 77% yield was achieved when 1a (2.5 equiv), 2a (3 equiv), and 3a (1 equiv) was used for the reaction (See supporting information for details, Table S4). Lastly, the reaction can be performed on a 1.0 mmol scale with 2.5 mol% catalyst loading without loss of the product yield (entry 8).
Having optimized the reaction conditions, we next sought to explore the scope of the transformation (Scheme 2). The reaction with aryl boronic acids containing a variety of functional groups at the para position of phenyl ring proceeds smoothly, delivering products with -CF3 (4b), -F (4c), -Br (4d), -OMe (4e), hydroxy (4f), silyl ether (4g), alkene (4h), alkyl (4i), nitrile (4j), and aldehyde (4k) functional groups in good yield. Aryl boronic acids with a substituent in the meta position (4l), having multiple substituents (4o, 4p), naphthyl boronic acid (4q), and 3,4-methylenedioxyphenylboronic acid (4r) also work well in this reaction. In the case of vinyl boronic acids such as 1-penten-1-ylboronic acid or alkyl boronic acids, the reactions are unsuccessful. (See supporting information for limitations).
Scheme 2. Substrate scope.

Reactions were conducted on a 0.1 mmol scale using 1 (2.5 equiv), 2 (3.0 equiv), and 3 (1.0 equiv). Yields of isolated products after purification by chromatography are reported. Diastereoselectivity was determined by analysis of 1H NMR of the unpurified reaction mixture.
For alkene substrate scope, the reaction with monosubstituted acrylates gives corresponding phenylalanine derivatives (5b-5d) in good yield. Acrylonitrile (2e) also works well in this reaction, providing a potential for further functional group derivatization. Secondary acrylamides are also good alkene coupling partners, delivering carboamination products (5f-5h) containing two amide bonds. The reaction with bridged bicyclic alkenes such as norbornene (2i), oxabenzonorbornadiene (2j), and aza-benzonorbornadiene (2k) gives syn-carboamination products (5i-5k) in good yield and diastereoselectivity. In the case of the reactions with cyclopropenes (2l-2m), the reaction occurs cis to the methyl group, presumably because of a smaller steric demand during the alkene migratory insertion step, giving cyclopropyl amine products (5l-5m) in high diastereoselectivity.
Internal electron-deficient alkenes such as ethyl crotonate, ethyl cinnamate, or cyclopentenone and less-strained cyclic alkenes such as cyclohexene or 3,4-dihydropyran do not lead to the product formation and direct N-phenylation (9) products are observed as a major side product of the reaction in high yield presumably due to the slow alkene migratory insertion (see supporting information for details).
Lastly, a variety of functional groups can be installed at the amide side chain using dioxazolones with different substituents at the 3 position (3b-3h) prepared from corresponding carboxylic acids. The reactions with dioxazolone 3d or 3f containing allylic or benzylic C-H bonds give the carboamination products selectively (6d, 6f). These are noteworthy as they have been used for intramolecular C-H amination chemistry.14 The carboamination occurs intermolecularly in preference to an intramolecular C-H insertion suggesting a mechanistic dichotomy with Chang’s Ir-based system.
We next designed a series of experiments to interrogate the mechanism of the reaction. First, we conducted the reaction under standard conditions, but without phenylboronic acid (Scheme 3a). As a result, neither N-acyl aziridine nor aziridine ring-opening products15 are observed after the reaction. This strongly indicates that the Rh(III)-catalyzed alkene aziridination and subsequent ring-opening with phenylboronic acid is unlikely as the mechanism of the reaction. During the optimization study, we observed Heck-type (7) and hydroarylation products (8) as side products of the reaction, presumably formed through β-hydride elimination and proto-demetallation after the alkene migratory insertion.
Scheme 3. Mechanistic investigation.

aStandard conditions: [Cp*RhCl2]2 (5 mol%), NaHCO3 (30 mol%), MeOH (0.6 M), dioxazolone (1 equiv), arylboronic acid (2.5 equiv), alkene (3 equiv), 25 °C. b1) 6 M HCl; 2) SOCl2, MeOH; 3) Picolinic acid, EDCI, HOBt, DCM; 4) Pd(OAc)2, PhI(OAc)2, DCM.
To determine whether these side products give the desired carboamination product (4), both benzyl cinnamate (7) and benzyl 3-phenylpropanoate (8) were subjected to the reaction using 1-naphthyl boronic acid (2q) as the carbon source of the reaction (Scheme 3b). While both reactions give the desired product (4q) from 1-naphthyl boronic acid (2q) in good yield, carboamination products (4a) from these side products (7 or 8) are not observed. These experiments suggest 7 and 8 are off-cycle side products that do not re-enter the catalytic cycle. In addition, monitoring the reaction progress with 1H NMR indicates that the reaction with dioxazolone that leads to product formation is faster than β-hydride elimination at the beginning of the reaction. The amount of β-hydride elimination product increases after consumption of limiting dioxazolone (3a) (see supporting information for the details).
In order to determine the turnover limiting step of the reaction, we investigated the alkene migratory insertion step with benzyl acrylate (2a) and deuterated benzyl acrylate (2a-d2). The inverse secondary kinetic isotope effect (KIE) value of 0.72 measured from the intermolecular competition experiment (Scheme 3c) or 0.55 calculated by comparing the initial rate of two separate reactions, one with benzyl acrylate (2a) and one with deuterated benzyl acrylate (2a-d2) (see supporting information for details) suggest sp2 to sp3 hybridization change occurs during the turnover limiting step of the reaction (Scheme 3c).16
In order to determine the relative stereochemistry of the product, we conducted the reaction with mono-deuterated benzyl acrylate (2a-d1, 85:14 E/Z) (Scheme 3d). Interestingly, the reaction gives the product (4a-d1) containing almost identical deuterium incorporation with starting alkene (2a-d1) which suggests the carboamination is highly diastereoselective. The product was cyclized by Pd-catalyzed intramolecular C-H amidation,17 which confirmed syn-carboamination of the alkene.
Based on these experiments, we propose the following mechanism for the reaction (Scheme 4). First, the active monomeric Rh complex I undergoes transmetalation with phenylboronic acid (1a) to give Rh-phenyl complex II. Next, turnover limiting migratory insertion into the Rh-aryl bond forms Rh(III) intermediate IV. Subsequently, dioxazolone (3a) coordinates to the electron-rich alkyl Rh(III) complex IV followed by Rh(V)-nitrene formation with the exclusion of CO2. The desired syn-carboamination product (4a) is formed after reductive elimination and proto-demetallation with the regeneration of active Rh catalyst I.
Scheme 4.

Proposed mechanism.
In summary, we have developed Rh(III)-catalyzed 3-component regioselective syn-carboamination of alkenes. The reaction shows a broad scope with a variety of commercially available arylboronic acids and dioxazolones easily prepared from carboxylic acids. Mechanistic investigations suggest alkene migratory insertion to be the turnover limiting step supported by secondary KIE. In addition, deuterium labeling experiments provide experimental evidence for stereoselective syn-carboamination process. This method provides a rapid access to valuable amines including non-natural α-amino acids from abundant alkenes with good regioselectivity and high diastereoselectivity with internal cyclic alkenes.
Supplementary Material
Acknowledgments
Funding Sources
We gratefully acknowledge NIGMS (GM80442) for support.
ABBREVIATIONS
- Cp*
pentamethylcyclopentadienyl
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental procedures, detailed optimization table, characterization, copies of 1H, 13C, and 19F NMR spectra for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Kolb HC; VanNieuwenhze MS; Sharpless KB Catalytic Asymmetric Dihydroxylation. Chem. Rev 1994, 94, 2483–2547. [Google Scholar]; (b) McDonald RI; Liu G; Stahl SS Palladium(II)-Catalyzed Alkene Functionalization via Nucleopalladation: Stereochemical Pathways and Enantioselective Catalytic Applications. Chem. Rev 2011, 111, 2981–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jensen KH; Sigman MS Mechanistic approaches to palladium-catalyzed alkene difunctionalization reactions. Org. Biomol. Chem 2008, 6, 4083–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chemler SR The enantioselective intramolecular aminative functionalization of unactivated alkenes, dienes, allenes and alkynes for the synthesis of chiral nitrogen heterocycles. Org. Biomol. Chem 2009, 7, 3009–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Romero RM; Wöste TH; Muñiz K. Vicinal Difunctionalization of Alkenes with Iodine(III) Reagents and Catalysts. Chem. – An Asian J 2014, 9, 972–983. [DOI] [PubMed] [Google Scholar]; (f) Cardona F; Goti A. Metal-catalysed 1,2-diamination reactions. Nat. Chem 2009, 1, 269–275. [DOI] [PubMed] [Google Scholar]
- (2).(a) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]; (b) Jiang H; Studer A. Intermolecular radical carboamination of alkenes. Chem. Soc. Rev 2020, 49, 1790–1811. [DOI] [PubMed] [Google Scholar]
- (3).(a) Coldham I; Hufton R. Intramolecular Dipolar Cycloaddition Reactions of Azomethine Ylides. Chem. Rev 2005, 105, 2765–2810. [DOI] [PubMed] [Google Scholar]; (b) Nakamura I; Yamamoto Y. Transition-Metal-Catalyzed Reactions in Heterocyclic Synthesis. Chem. Rev 2004, 104, 2127–2198. [DOI] [PubMed] [Google Scholar]
- (4).(a) Wolfe JP, Synthesis of Saturated Heterocycles via Metal-Catalyzed Alkene Carboamination or Carboalkoxylation Reactions. In Synthesis of Heterocycles via Metal-Catalyzed Reactions that Generate One or More Carbon-Heteroatom Bonds, Wolfe JP, Ed. Springer Berlin Heidelberg: Berlin, Heidelberg, 2013; pp 1–37. [Google Scholar]; (b) Mai DN; Wolfe JP Asymmetric Palladium-Catalyzed Carboamination Reactions for the Synthesis of Enantiomerically Enriched 2-(Arylmethyl)- and 2-(Alkenylmethyl)pyrrolidines. J. Am. Chem. Soc 2010, 132, 12157–12159. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zeng W; Chemler SR Copper(II)-Catalyzed Enantioselective Intramolecular Carboamination of Alkenes. J. Am. Chem. Soc 2007, 129, 12948–12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Piou T; Rovis T. Rhodium-catalysed syn-carboamination of alkenes via a transient directing group. Nature 2015, 527, 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Lerchen A; Knecht T; Daniliuc CG; Glorius F. Unnatural Amino Acid Synthesis Enabled by the Regioselective Cobalt(III)-Catalyzed Intermolecular Carboamination of Alkenes. Angew. Chem. Int. Ed 2016, 55, 15166–15170. [DOI] [PubMed] [Google Scholar]; (b) Wang X; Gensch T; Lerchen A; Daniliuc CG; Glorius F. Cp*Rh(III)/Bicyclic Olefin Cocatalyzed C–H Bond Amidation by Intramolecular Amide Transfer. J. Am. Chem. Soc 2017, 139, 6506–6512. [DOI] [PubMed] [Google Scholar]
- (7).Ozols K; Onodera S; Woźniak Ł; Cramer N. Cobalt(III)-Catalyzed Enantioselective Intermolecular Carboamination by C−H Functionalization. Angew. Chem. Int. Ed 2021, 60, 655–659. [DOI] [PubMed] [Google Scholar]
- (8).(a) Maity S; Potter TJ; Ellman JA α-Branched amines by catalytic 1,1-addition of C–H bonds and aminating agents to terminal alkenes. Nat. Catal 2019, 2, 756–762. During the preparation of this manuscript, two conceptually related transformations involving a directing group assisted C-H activation strategy were reported: see: [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brandes DS; Sirvent A; Mercado BQ; Ellman JA Three-Component 1,2-Carboamidation of Bridged Bicyclic Alkenes via RhIII-Catalyzed Addition of C–H Bonds and Amidating Reagents. Org. Lett 2021, 23, 2836–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mi R; Zhang X; Wang J; Chen H; Lan Y; Wang F; Li X. Rhodium-Catalyzed Regio-, Diastereo-, and Enantioselective Three-Component Carboamination of Dienes via C–H Activation. ACS Catal. 2021, 11, 6692–6697. [Google Scholar]
- (9).(a) Liu Z; Wang Y; Wang Z; Zeng T; Liu P; Engle KM Catalytic Intermolecular Carboamination of Unactivated Alkenes via Directed Aminopalladation. J. Am. Chem. Soc 2017, 139, 11261–11270. [DOI] [PubMed] [Google Scholar]; (b) van der Puyl VA; Derosa J; Engle KM Directed, Nickel-Catalyzed Umpolung 1,2-Carboamination of Alkenyl Carbonyl Compounds. ACS Catal. 2019, 9, 224–229. [Google Scholar]
- (10).(a) Wang D; Wu L; Wang F; Wan X; Chen P; Lin Z; Liu G. Asymmetric Copper-Catalyzed Intermolecular Aminoarylation of Styrenes: Efficient Access to Optical 2,2-Diarylethylamines. J. Am. Chem. Soc 2017, 139, 6811–6814. [DOI] [PubMed] [Google Scholar]; (b) Gockel SN; Buchanan TL; Hull KL Cu-Catalyzed Three-Component Carboamination of Alkenes. J. Am. Chem. Soc 2018, 140, 58–61. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Monos TM; McAtee RC; Stephenson CRJ Arylsulfonylacetamides as bifunctional reagents for alkene aminoarylation. Science 2018, 361, 1369–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kennedy-Ellis JJ; Boldt ED; Chemler SR Synthesis of Benzylureas and Related Amine Derivatives via Copper-Catalyzed Three-Component Carboamination of Styrenes. Org. Lett 2020, 22, 8365–8369. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Gockel SN; Lee S; Gay BL; Hull KL Oxidative Three-Component Carboamination of Vinylarenes with Alkylboronic Acids. ACS Catal. 2021, 11, 5166–5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Park Y; Park KT; Kim JG; Chang S. Mechanistic Studies on the Rh(III)-Mediated Amido Transfer Process Leading to Robust C–H Amination with a New Type of Amidating Reagent. J. Am. Chem. Soc 2015, 137, 4534–4542. [DOI] [PubMed] [Google Scholar]; (b) van Vliet KM; de Bruin B. Dioxazolones: Stable Substrates for the Catalytic Transfer of Acyl Nitrenes. ACS Catal. 2020, 10, 4751–4769. [Google Scholar]
- (12).(a) Shin K; Kim H; Chang S. Transition-Metal-Catalyzed C–N Bond Forming Reactions Using Organic Azides as the Nitrogen Source: A Journey for the Mild and Versatile C–H Amination. Acc. Chem. Res 2015, 48, 1040–1052. [DOI] [PubMed] [Google Scholar]; (b) Park Y; Kim Y; Chang S. Transition Metal-Catalyzed C–H Amination: Scope, Mechanism, and Applications. Chem. Rev 2017, 117, 9247–9301. [DOI] [PubMed] [Google Scholar]; (c) Lee S; Lei H; Rovis T. A Rh(III)-Catalyzed Formal [4+1] Approach to Pyrrolidines from Unactivated Terminal Alkenes and Nitrene Sources. J. Am. Chem. Soc 2019, 141, 12536–12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Fagnou K; Lautens M. Rhodium-Catalyzed Carbon−Carbon Bond Forming Reactions of Organometallic Compounds. Chem. Rev 2003, 103, 169–196. [DOI] [PubMed] [Google Scholar]; (b) Hayashi T; Yamasaki K. Rhodium-Catalyzed Asymmetric 1,4-Addition and Its Related Asymmetric Reactions. Chem. Rev 2003, 103, 2829–2844. [DOI] [PubMed] [Google Scholar]; (c) Edwards HJ; Hargrave JD; Penrose SD; Frost CG Synthetic applications of rhodium catalysed conjugate addition. Chem. Soc. Rev 2010, 39, 2093–2105. [DOI] [PubMed] [Google Scholar]
- (14).Hong SY; Park Y; Hwang Y; Kim YB; Baik M-H; Chang S. Selective formation of γ-lactams via C–H amidation enabled by tailored iridium catalysts. Science 2018, 359, 1016. [DOI] [PubMed] [Google Scholar]
- (15).Lee S; Jang YJ; Phipps EJT; Lei H; Rovis T. Rhodium(III)-Catalyzed Three-Component 1,2-Diamination of Unactivated Terminal Alkenes. Synthesis 2020, 52, 1247–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).(a) Gómez-Gallego M; Sierra MA Kinetic Isotope Effects in the Study of Organometallic Reaction Mechanisms. Chem. Rev 2011, 111, 4857–4963. [DOI] [PubMed] [Google Scholar]; (b) Simmons EM; Hartwig JF On the Interpretation of Deuterium Kinetic Isotope Effects in C-H Bond Functionalizations by Transition-Metal Complexes. Angew. Chem. Int. Ed 2012, 51, 3066–3072. [DOI] [PubMed] [Google Scholar]
- (17).He G; Lu C; Zhao Y; Nack WA; Chen G. Improved Protocol for Indoline Synthesis via Palladium-Catalyzed Intramolecular C(sp2)–H Amination. Org. Lett 2012, 14, 2944–2947. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.