

SUMMARY
The electron transport chain promotes aspartate synthesis, which is required for cancer cell proliferation. However, it is unclear whether aspartate is limiting in normal stem cells. We found that mouse hematopoietic stem cells (HSCs) depend entirely on cell-autonomous aspartate synthesis, which increases upon HSC activation. Over-expression of the glutamate/aspartate transporter, Glast, or deletion of glutamic-oxaloacetic transaminase 1 (Got1) each increased aspartate levels in hematopoietic stem/progenitor cells and increased the function of HSCs but not colony-forming progenitors. Conversely, deletion of glutamic-oxaloacetic transaminase 2 (Got2) reduced aspartate levels and the function of HSCs but not colony-forming progenitors. Deletion of Got1 and Got2 eliminated HSCs. Isotope tracing showed aspartate was used to synthesize asparagine and purines. Both contributed to increased HSC function as deletion of asparagine synthetase or treatment with 6-mercaptopurine attenuated the increased function of GLAST over-expressing HSCs. HSC function is thus limited by aspartate, purine, and asparagine availability during hematopoietic regeneration.
Keywords: Hematopoietic stem cell, mitochondria, metabolism, aspartate, asparagine, purine
Graphical Abstract
eTOC blurbs:
Mitochondrial function promotes aspartate synthesis, a critical building block for proteins and nucleotides. Morrison and colleagues show hematopoietic stem cells depend exclusively on cell-autonomously synthesized aspartate and HSC function is limited by aspartate availability. During regeneration, HSCs increase aspartate synthesis, which promotes HSC function by increasing asparagine and purine synthesis.
INTRODUCTION
Most cells depend upon aspartate that is cell-autonomously synthesized. Aspartate is synthesized from glutamate and oxaloacetate in mitochondria by glutamic-oxaloacetic transaminase 2 (GOT2) and is consumed in the cytosol by glutamic-oxaloacetic transaminase 1 (GOT1), though both reactions are reversible (Figure S1A). Electron transport chain function promotes the synthesis of aspartate by promoting the generation of oxaloacetate in the tricarboxylic acid (TCA) cycle (Birsoy et al., 2015, Sullivan et al., 2015). Exogenous aspartate can rescue the proliferation of cancer cells in the absence of electron transport chain function (Alkan et al., 2018, Bailis et al., 2019, Birsoy et al., 2015, Garcia-Bermudez et al., 2018, Gui et al., 2016, Krall et al., 2021, Sullivan et al., 2015, Sullivan et al., 2018). This limitation can be partly rescued by supplementing cells with nucleosides (Alkan et al., 2018, Garcia-Bermudez et al., 2018, Sullivan et al., 2015, Sullivan et al., 2018) or asparagine (Krall et al., 2021). This suggests that one of the most important functions of the electron transport chain is to promote aspartate synthesis, which is used to synthesize nucleotides and asparagine.
Stem cells are thought to depend mainly on glycolysis (Ito and Suda, 2014, Nakamura-Ishizu et al., 2020, Simsek et al., 2010, Suda et al., 2011, Takubo et al., 2013), though this has not yet been tested by metabolic flux analysis and mitochondrial function has only been studied to a limited extent in stem cells (Guitart et al., 2016, Luchsinger et al., 2016, Maryanovich et al., 2015, Shapira and Christofk, 2020, Snoeck, 2017). Electron transport chain activity is often, but not always, required for stem cell function (van den Ameele and Brand, 2019, Zhang et al., 2020) including HSCs (Ansó et al., 2017, Bejarano-García et al., 2016, Hinge et al., 2020, Liang et al., 2020, Mansell et al., 2021, Morganti et al., 2019). Conditional deletion of the mitochondrial transcription factor A (Tfam), which is required for mitochondrial DNA replication and expression of electron transport chain components, impairs hair follicle morphogenesis but has little effect on the intrafollicular epidermis (Baris et al., 2011, Kloepper et al., 2015). Conversely, loss of mitochondrial pyruvate carrier function is sufficient to increase intestinal stem cell frequency and promote hair follicle stem cell activation, despite the fact that it would be expected to reduce TCA cycle and electron transport chain activity (Flores et al., 2017, Schell et al., 2017). Mitochondrial function thus appears to have complex effects that differ among stem cells.
Aspartate is used by cells for the synthesis of proteins, other amino acids (arginine, asparagine, glutamate and N-acetylaspartate), and nucleotides (purines and pyrimidines) (Figure S1A). Aspartate can also be used to fuel the TCA cycle (Figure S1A). Acute myeloid leukemia cells require aspartate for pyrimidine synthesis and may derive aspartate from Leptin Receptor+ stromal cells in the bone marrow (van Gastel et al., 2020), which are a key component of the HSC niche (Ding and Morrison, 2013, Ding et al., 2012). Some proliferating normal cells are also limited by aspartate synthesis, including T cells (Bailis et al., 2019) and pulmonary endothelial cells (Bertero et al., 2016). The aspartate-glutamate carrier 1, which transports aspartate from mitochondria to the cytosol, is required for the proliferation of oligodendrocyte progenitors (Petralla et al., 2019).
Some stem cells express the glutamate/aspartate transporter, GLAST (Slc1a3), including neural stem cells (Liu et al., 2006, Mich et al., 2014, Mori et al., 2006), rendering them able to take up extracellular aspartate. Knockdown of malate dehydrogenase 1, a component of the malate-aspartate shuttle, reduces the reconstituting capacity of fetal liver HSCs in irradiated mice (Gu et al., 2020). These results raise the question of whether aspartate levels limit stem cell function.
One limitation that has made it difficult to study stem cell metabolism is the challenge of quantifying metabolites in small numbers of cells isolated directly from tissues. To address this, we developed a metabolomic method that detects approximately 160 metabolites in 10,000 flow cytometrically-isolated HSCs (Agathocleous et al., 2017, DeVilbiss et al., 2021). Using this method, the levels of most metabolites do not change during cell isolation.
RESULTS
GLAST over-expression increases aspartate levels and function in HSCs
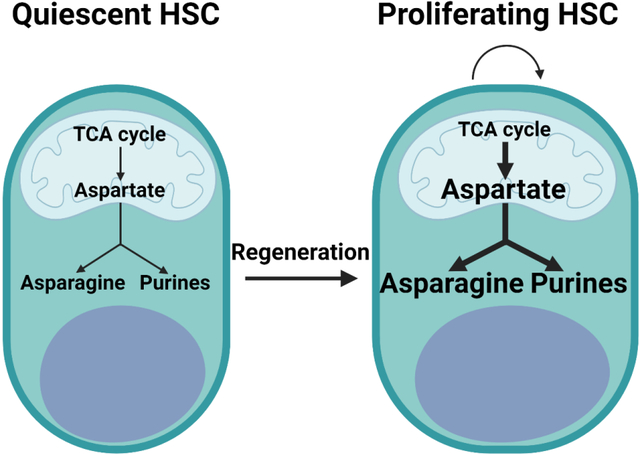
In contrast to endothelial protein C receptor (Epcr, also known as Procr), which is expressed by HSCs and multipotent progenitors (MPPs) (Balazs et al., 2006) (Figure 1A), the aspartate transporters Slc1a1, Slc1a2, Slc1a3 (Glast), Slc1a6, and Slc1a7 were not detected in HSCs or other hematopoietic progenitors from the bone marrow (Figures 1B–1F). We generated GLAST over-expressing mice to confer upon hematopoietic cells the capacity to take up extracellular aspartate (Figure S1B). This allele included a loxp-Stop-loxp cassette, such that Cre-mediated recombination was required for GLAST expression. Bone marrow cells from Vav1-Cre; Rosa26LSL-Glast, but not wild-type, mice exhibited strong GLAST expression (Figure 1G). These mice were born in expected mendelian frequencies (Figure S1C) and were grossly normal in size and appearance (Figures 1H and 1I). They had normal blood cell counts (Figure 1J) and cellularity in the bone marrow, spleen, and thymus (Figure 1K).
Figure 1. GLAST over-expression increases aspartate levels and HSC function.
(A-F) FPKM values of Procr (A) and known aspartate transporters (B-F) in hematopoietic stem and progenitor cells. (G) Western blots with antibodies against GLAST and Histone H3 using Vav1-cre; Rosa26LSL-Glast and wild-type bone marrow cells. Glast-OE refers to Vav1-cre; Rosa26LSL-Glast mice throughout the figure. (H and I) Vav1-cre; Rosa26LSL-Glast and control mice did not differ in size or appearance. (J) White blood cell, red blood cell, and platelet counts in the blood of Vav1-cre; Rosa26LSL-Glast and control mice. (K) Cellularity of the bone marrow from one femur and one tibia, the spleen, and the thymus. (L) Uptake of labelled aspartate by HSCs cultured from Vav1-cre; Rosa26LSL-Glast or control bone marrow. (M and N) Aspartate (M) and glutamate (N) levels in HSCs/MPPs, HPCs, LK cells, and CD45+ cells from Vav1-cre; Rosa26LSL-Glast or control bone marrow. (O-Q) Numbers of HSCs (O), restricted progenitors (P), and differentiated hematopoietic cells (Q) in the bone marrow from one femur and one tibia. (R and S) Numbers and sizes of colonies formed by 10,000 bone marrow cells (G: granulocyte, M: macrophage, E: erythrocyte). (T and U) Donor-derived CD45+, myeloid, B, and T cells in the blood (T) as well as HSCs and restricted hematopoietic progenitors in the bone marrow (U) of mice competitively transplanted with Vav1-cre; Rosa26LSL-Glast or control donor bone marrow cells (n=30 recipient mice total from 6 independent experiments with 6 donors per genotype). All data represent mean ± standard deviation (* P< 0.05; ** P<0.01; *** P<0.001). The number of mice analyzed per genotype is shown in each panel. See also Figure S1 and Table S1.
To test if hematopoietic cells could take up aspartate, we cultured wild-type or GLAST over-expressing HSCs for 7 days under conditions that promote HSC self-renewal (Wilkinson et al., 2019) and supplemented the cultures with physiological levels (20 μM) of universally13C-labelled aspartate (Bunpo et al., 2008, Newgard et al., 2009). Based on long-term reconstitution assays, HSCs were sustained in these cultures (Figure S1E) and 78±7.2% of cells from these cultures were able to form colonies (Figure S1D). These cultures thus contained a mixture of HSCs and primitive hematopoietic progenitors but few differentiated cells. Only 0.008±0.006% of aspartate was universally labelled in wild-type cells but 47±1.3% of aspartate was universally labelled in Vav1-Cre; Rosa26LSL-Glast cells (Figure 1L). Hematopoietic stem/progenitor cells thus have little or no capacity to take up aspartate but GLAST over-expression conferred this ability.
To better understand the effects of GLAST over-expression, we isolated HSCs and MPPs, hematopoietic progenitor cells (HPCs), myeloid progenitors (Lin−c-kit+ (LK) cells) and unfractionated hematopoietic cells (CD45+ cells) from Vav1-Cre; Rosa26LSL-Glast and control bone marrow (Table S1 shows the markers used to isolate each cell population characterized in this study). Metabolomic analysis detected 102 to 105 metabolites above background in each cell population. Only 3 metabolites significantly differed between Vav1-Cre; Rosa26LSL-Glast and control HSCs/MPPs, including aspartate and glutamate, which were significantly higher in Vav1-Cre; Rosa26LSL-Glast HSCs/MPPs, consistent with GLAST over-expression (Figures 1M and 1N). The only other significant difference was that glutamine was increased in GLAST over-expressing as compared to control HSCs/MPPs (Figure S1F). GLAST over-expression in hematopoietic cells, thus, preferentially increased aspartate levels in HSCs/MPPs.
Vav1-Cre; Rosa26LSL-Glast mice had a significantly increased number of HSCs in the bone marrow as compared to control mice (Figure 1O). We did not observe any significant differences in the numbers of MPPs, restricted progenitors, or differentiated hematopoietic cells in Vav1-Cre; Rosa26LSL-Glast versus control bone marrow (Figures 1P and 1Q). We did not observe any significant differences in the numbers or sizes of colonies formed in culture by Vav1-Cre; Rosa26LSL-Glast versus control bone marrow cells (Figures 1R and 1S). Finally, we did not detect any difference in the numbers of HSCs, restricted progenitors, or differentiated hematopoietic cells in the spleen or thymus (Figures S1G–S1J). GLAST over-expression in hematopoietic cells, thus, appeared to preferentially increase the number of HSCs in the bone marrow.
Vav1-Cre; Rosa26LSL-Glast bone marrow cells gave significantly higher levels of donor cell reconstitution in all lineages as compared to control cells in the blood (Figure 1T) and bone marrow (Figure 1U). Secondary transplantation of the bone marrow cells from these primary recipients again gave higher levels of reconstitution in all lineages by Vav1-Cre; Rosa26LSL-Glast as compared to control donor cells (Figure S1K). GLAST over-expression thus increased HSC function during hematopoietic regeneration.
Got1 deficiency increases aspartate levels and HSC function
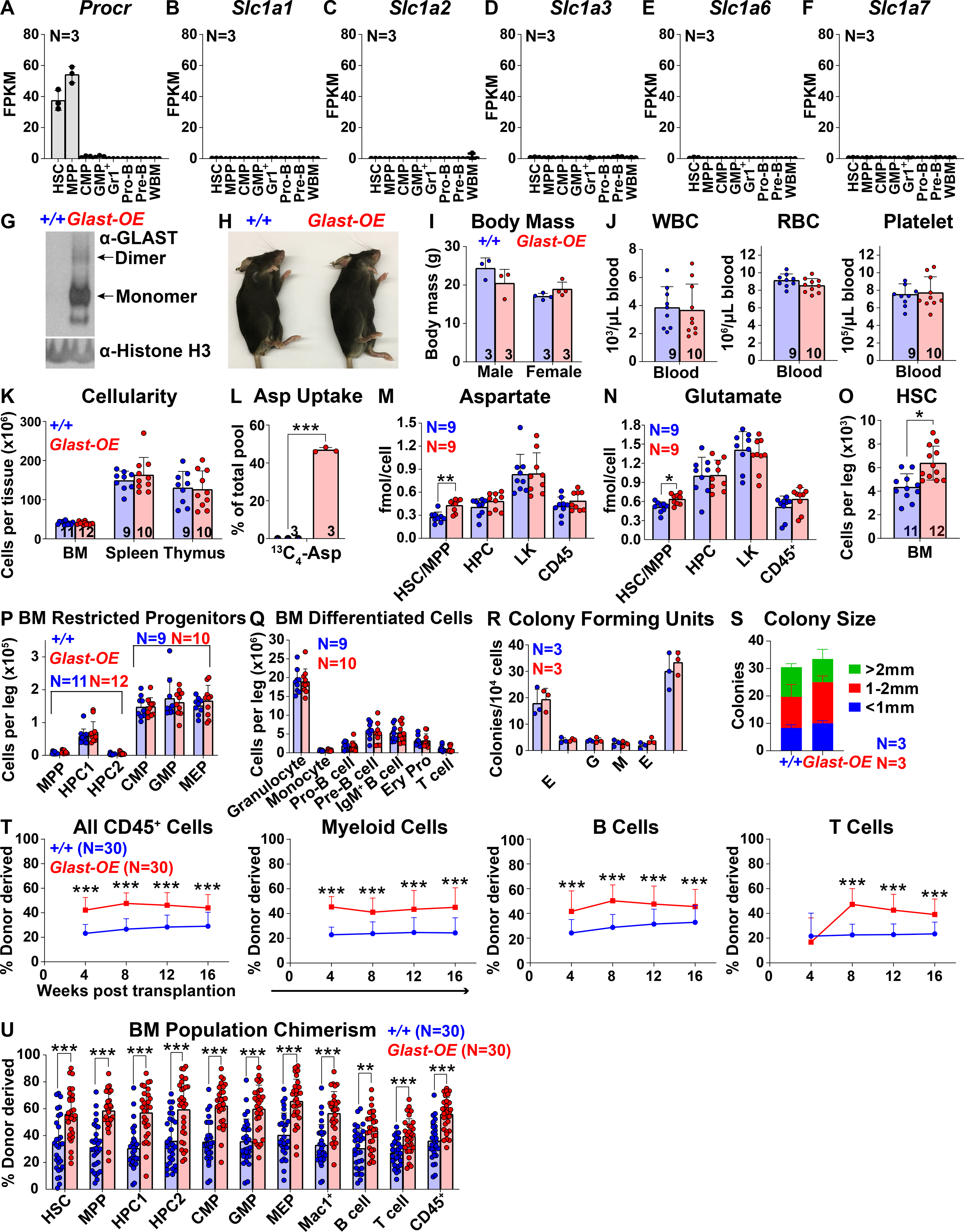
To test if the increased HSC function in Vav1-Cre; Rosa26LSL-Glast bone marrow was caused by the increase in aspartate levels we took an orthogonal approach to modulate aspartate levels. We generated a floxed allele of Got1 (Figure S2A). Conditional deletion of Got1 from hematopoietic cells would be expected to increase aspartate levels (Birsoy et al., 2015, Safer, 1975, Son et al., 2013) (Figure S1A). We treated Mx1-Cre; Got1fl/fl mice and littermate controls with polyinosine-polycytidylic acid (pIpC) (Kühn et al., 1995, Yilmaz et al., 2006) at 5 to 6 weeks of age to induce recombination and then assessed hematopoiesis 2 to 4 weeks later. We confirmed the loss of GOT1 protein from Mx1-Cre; Got1fl/fl bone marrow cells (Figure 2A). The pIpC-treated Mx1-Cre; Got1fl/fl mice were grossly normal in size and appearance (Figures 2B and 2C), with normal blood cell counts (Figure 2D) and cellularity in the bone marrow, spleen, and thymus (Figure 2E).
Figure 2. Got1 deficiency increases aspartate levels and HSC function.
(A) Western blots with antibodies against GOT1 and Histone H3 were performed using bone marrow cells from Mx1-cre; Got1fl/fl and control mice (* denotes nonspecific bands). (B and C) Mx1-cre; Got1fl/fl and control mice did not differ in size or appearance. (D) White blood cell, red blood cell, and platelet counts in the blood of Mx1-cre; Got1fl/fl and control mice. (E) Cellularity of the bone marrow from one femur and one tibia, the spleen, and the thymus. (F and G) Aspartate (F) and glutamate (G) levels in HSCs/MPPs, HPCs, LK cells, and CD45+ cells from Mx1-cre; Got1fl/fl and control bone marrow. (H-J) Numbers of HSCs (H), restricted progenitors (I), and differentiated cells (J) in the bone marrow from one femur and one tibia. (K and L) Numbers and sizes of colonies formed by 10,000 bone marrow cells. (M) Donor-derived CD45+, myeloid, B, and T cells in the blood of mice competitively transplanted with Mx1-cre; Got1fl/fl or control bone marrow cells (n=28–30 recipient mice total from 6 independent experiments with 6 donors per genotype). (N and O) Aspartate levels in HSCs/MPPs, HPCs, LK cells, and CD45+ cells from cyclophosphamide/G-CSF (N) or pIpC (O) treated versus untreated control bone marrow. (P and Q) In vivo isotope tracing in hematopoietic stem and progenitor cell populations isolated from cyclophosphamide/G-CSF (P) or pIpC (Q) treated as compared to untreated control mice infused with 15N-glutamine: fractional enrichment of 15N-aspartate normalized to 15N-glutamine. (R) Aspartate levels in HSCs/MPPs, HPCs, LK cells, and CD45+ cells: data was pooled from Figures 1M, 2F, 2N and 2O. All data represent mean ± standard deviation (* P< 0.05; ** P<0.01; *** P<0.001). The number of mice analyzed per genotype is shown in each panel. See also Figure S2, Tables S1 and S2.
Metabolomic analysis of HSCs/MPPs, HPCs, LK cells, and CD45+ cells from the bone marrow of Mx1-Cre; Got1fl/fl mice and littermate controls detected 129 to 133 metabolites above background. Twelve metabolites significantly differed between Mx1-Cre; Got1fl/fl and control HSCs/MPPs (Table S2). The only metabolite that significantly changed in the same direction as in Vav1-Cre; Rosa26LSL-Glast versus control HSCs/MPPs, was aspartate, which was significantly higher in Mx1-Cre; Got1fl/fl as compared to control HSCs/MPPs (Figure 2F). In contrast to Vav1-Cre; Rosa26LSL-Glast bone marrow, glutamate levels did not significantly differ between any cell populations in Mx1-Cre; Got1fl/fl as compared to control bone marrow (Figure 2G).
The numbers of HSCs, MPPs, HPCs, restricted progenitors, differentiated cells, and colony-forming progenitors did not significantly differ between Mx1-Cre; Got1fl/fl and littermate control bone marrow (Figures 2H–2L). There were no significant differences in the numbers of HSCs, restricted progenitors, or differentiated hematopoietic cells in the spleen or thymus (Figures S2B–S2E). However, competitive transplantation into irradiated mice revealed significantly higher levels of donor cell reconstitution in all lineages from Mx1-Cre; Got1fl/fl as compared to control bone marrow cells (Figure 2M). Got1 deficiency thus increased aspartate levels and HSC function during hematopoietic regeneration.
HSC activation increases aspartate synthesis
Bone marrow cells from GLAST over-expressing and Got1-deficient mice gave higher levels of reconstitution in irradiated mice (Figures 1T and 2M) but little or no increase in HSC frequency under steady-state conditions (Figures 1O and 2H). This suggested that aspartate levels limited HSC function during hematopoietic regeneration. To test if aspartate synthesis increased in response to HSC activation, we treated mice with cyclophosphamide plus G-CSF (Morrison et al., 1997) or pIpC (Essers et al., 2009). Aspartate levels were significantly higher in HSCs/MPPs from the bone marrow of mice treated with either cyclophosphamide/G-CSF or pIpC as compared to untreated control mice (Figures 2N and 2O). Got1 and Got2, but not Slc1a3, expression also increased in HSCs/MPPs from mice treated with cyclophosphamide/G-CSF or pIpC as compared to untreated control mice (Figures S2F–S2K). Isotope tracing from α−15N-glutamine in vivo showed that aspartate synthesis significantly increased in HSCs/MPPs and HPCs from cyclophosphamide/G-CSF or pIpC treated as compared to control mice (Figures 2P and 2Q). Aspartate synthesis increases upon HSC activation.
Aspartate levels significantly increased in HPCs and LK myeloid progenitors as compared to HSCs but declined as these cells differentiated (Figure 2R). Aspartate synthesis from 15N-glutamine was also significantly higher in myeloid progenitors as compared to HSCs/MPPs in vivo (Figures 2P and 2Q). Aspartate synthesis from glutamine thus increases in rapidly dividing myeloid progenitors as compared to quiescent HSCs, suggesting that aspartate synthesis rates vary among hematopoietic stem/progenitor cells in concert with anabolic demands.
Got2 deficiency decreases aspartate levels and HSC function
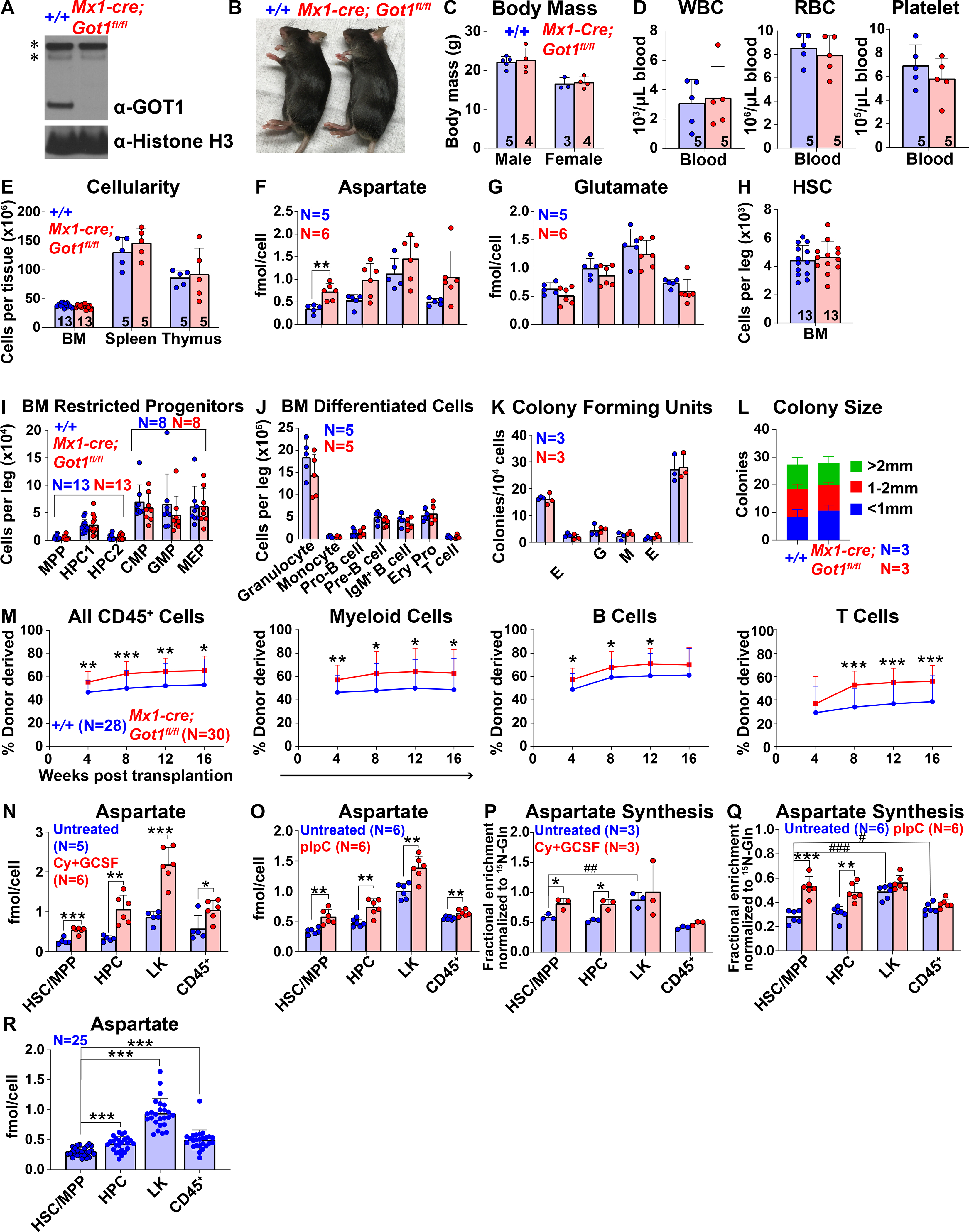
To test if decreased aspartate levels reduced HSC function we generated a floxed allele of Got2 (Figure S3A). Conditional deletion of this enzyme would be expected to decrease aspartate levels (Safer, 1975, Son et al., 2013). Mx1-Cre; Got2fl/fl mice were born in expected mendelian frequencies (Figure S3B). We treated Mx1-Cre; Got2fl/fl mice and littermate controls with pIpC at 5 to 6 weeks of age and then assessed hematopoiesis 2 to 4 weeks later. We confirmed the loss of GOT2 protein from Mx1-Cre; Got2fl/fl bone marrow cells (Figure 3A). The Mx1-Cre; Got2fl/fl mice were grossly normal in size and appearance (Figures 3B and 3C) and had normal white blood cell and platelet counts but reduced red blood cell counts (Figure 3D). Bone marrow cellularity was significantly reduced in Mx1-Cre; Got2fl/fl as compared to control mice while spleen and thymus cellularity were normal (Figure 3E).
Figure 3. Got2 deficiency decreases aspartate levels and HSC function.
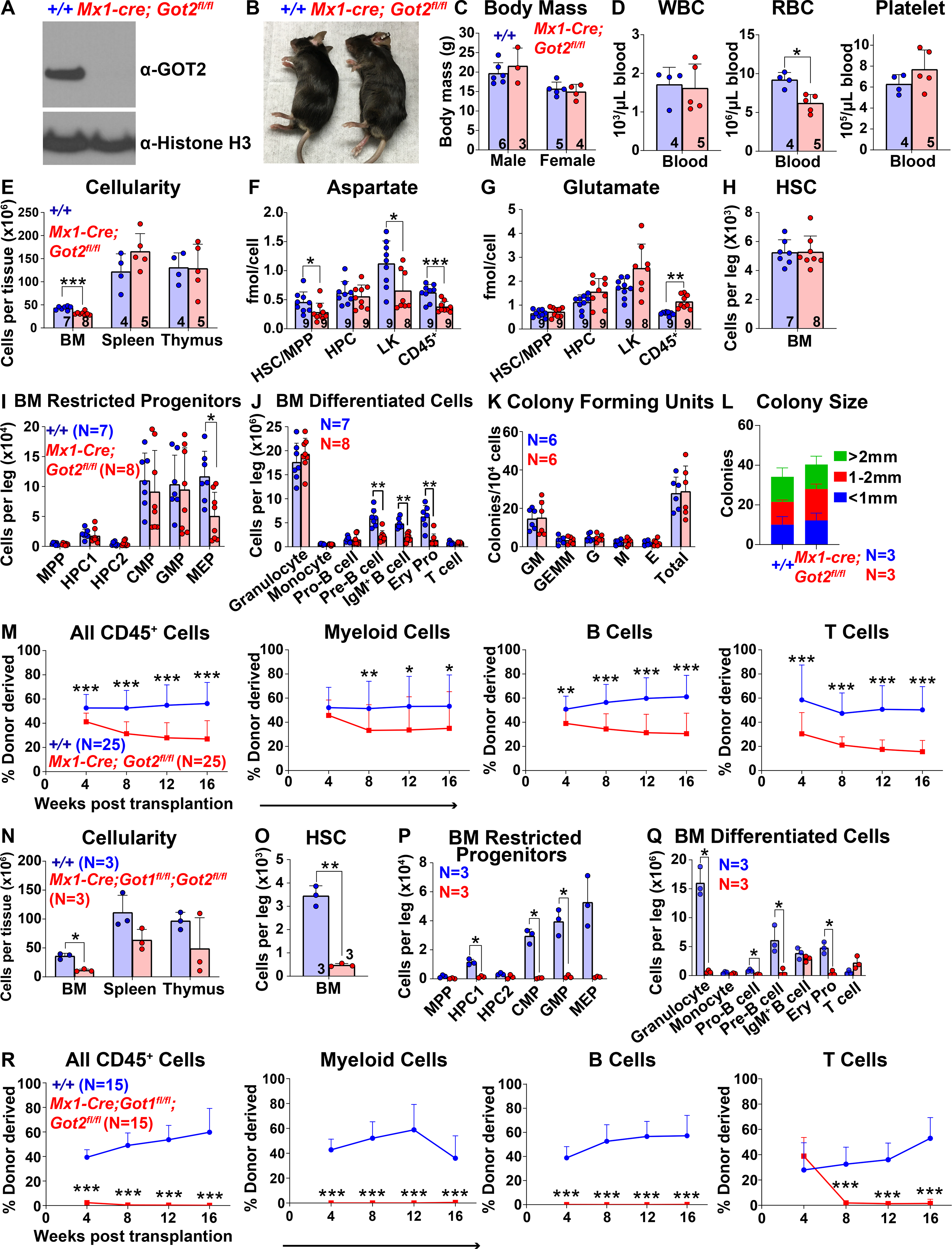
(A) Western blots with antibodies against GOT2 and Histone H3 were performed using bone marrow cells from Mx1-cre; Got2fl/fl and control wild-type mice. (B and C) Mx1-cre; Got2fl/fl and control mice did not differ in size or appearance. (D) White blood cell, red blood cell, and platelet counts in the blood of Mx1-cre; Got2fl/fl and control mice. (E) Cellularity of the bone marrow from one femur and one tibia, the spleen, and the thymus. (F and G) Aspartate (F) and glutamate (G) levels in HSCs/MPPs, HPCs, LK cells, and CD45+ cells from Mx1-cre; Got2fl/fl and control bone marrow. (H-J) Numbers of HSCs (H), restricted hematopoietic progenitors (I), and differentiated hematopoietic cells (J) in the bone marrow from one femur and one tibia. (K and L) Numbers and sizes of colonies formed by 10,000 bone marrow cells. (M) Donor-derived CD45+, myeloid, B, and T cells in the blood of mice competitively transplanted with Mx1-cre; Got2fl/fl or control donor bone marrow cells (n=25 recipient mice total from 5 independent experiments with 5 donors per genotype). (N) Cellularity of the bone marrow from one femur and one tibia, the spleen, and the thymus of Mx1-cre; Got1fl/fl; Got2fl/fl or control wild-type mice. (O-Q) Numbers of HSCs (O), restricted hematopoietic progenitors (P), and differentiated hematopoietic cells (Q) in the bone marrow from one femur and one tibia. (R) Donor-derived CD45+, myeloid, B, and T cells in the blood of mice competitively transplanted with Mx1-cre; Got1fl/fl; Got2fI/fI or control donor bone marrow cells (n=15 recipient mice total from 3 independent experiments with 3 donors per genotype). All data represent mean ± standard deviation (* P< 0.05; ** P<0.01; *** P<0.001). The number of mice analyzed per genotype is shown in each panel. See also Figure S3, Tables S1 and S3.
Metabolomic analysis of HSCs/MPPs, HPCs, LK myeloid progenitors, and CD45+ cells from the bone marrow of Mx1-Cre; Got2fl/fl mice and littermate controls detected 156 metabolites above background in each cell population. Six metabolites significantly differed between Mx1-Cre; Got2fl/fl and control HSCs/MPPs (Table S3). The only metabolite that significantly changed in the opposite direction as compared to GLAST over-expressing and Got1-deficient HSCs was aspartate, which was significantly lower in Mx1-Cre; Got2fl/fl as compared to control HSCs/MPPs, as expected (Figure 3F). Aspartate levels were also significantly lower in Mx1-Cre; Got2fl/fl as compared to control LK and CD45+ cells. Glutamate levels were significantly increased in CD45+ cells, but not in HSCs/MPPs, HPCs or LK cells from Mx1-Cre; Got2fl/fl as compared to control bone marrow (Figure 3G).
The numbers of HSCs, most restricted hematopoietic progenitors, and most differentiated hematopoietic cells did not significantly differ in the bone marrow of Mx1-Cre; Got2fl/fl as compared to littermate controls (Figures 3H–3J). The numbers and sizes of colonies formed by bone marrow cells in culture did not significantly differ between Mx1-Cre; Got2fl/fl and control mice (Figures 3K and 3L). The numbers of HSCs, granulocytes and erythroid progenitors were significantly increased in the spleens of Mx1-Cre; Got2fl/fl as compared to control mice (Figures S3C and S3E), apparently reflecting the induction of extramedullary hematopoiesis in response to reduced red blood cell counts (Figure 3D). Other restricted hematopoietic progenitors and differentiated hematopoietic cells did not significantly differ among the spleens and thymuses of Mx1-Cre; Got2fl/fl and control mice (Figures S3D–S3F). Competitive transplantation into irradiated mice revealed significantly lower levels of donor cell reconstitution in all lineages from Mx1-Cre; Got2fl/fl as compared to control bone marrow cells (Figure 3M). Got2 deficiency thus decreased aspartate levels and HSC function during hematopoietic regeneration.
Since GOT1 and GOT2 catalyze the same reversible reaction between aspartate and glutamate (Figure S4C), we hypothesized that these enzymes might compensate for each other in Got1 deficient or Got2 deficient mice (Garcia-Bermudez et al., 2021). To test this, we generated Mx1-Cre; Got1fl/fl; Got2fl/fl double mutant mice, treated them with pIpC at 5 to 6 weeks of age, and assessed hematopoiesis 1 week later. Blood cell counts (Figure S3G) and bone marrow cellularity (Figure 3N) were significantly reduced in Mx1-Cre; Got1fl/fl; Got2fl/fl as compared to control mice. There was a trend toward reduced spleen and thymus cellularity as well, though the differences were not statistically significant (Figure 3N). HSCs were profoundly depleted in the bone marrow of Mx1-Cre; Got1fl/fl; Got2fl/fl as compared to control mice, as were many populations of restricted hematopoietic progenitors and differentiated hematopoietic cells (Figures 3O–3Q) in spleens and thymuses (Figures S3H–S3K). Bone marrow cells from Mx1-Cre; Got1fl/fl; Got2fl/fl mice failed to give long-term multilineage reconstitution upon competitive transplantation into irradiated mice (Figure 3R). Loss of endogenous aspartate synthesis thus leads to severe defects in HSC maintenance and function.
Overall, GLAST over-expression and Got1 deficiency increased aspartate levels and HSC function while Got2 deficiency reduced aspartate levels and HSC function. While Got1 deficiency and Got2 deficiency had opposite effects on aspartate levels, both would be expected to disrupt the malate-aspartate shuttle, which transports malate, aspartate, and glutamate across the mitochondrial membrane (Figure S1A). Since Got1 deficiency and Got2 deficiency had opposite effects on HSC function, the changes in HSC function appear to be caused by changes in aspartate levels, not by the disruption of the malate-aspartate shuttle. Moreover, aspartate was the only metabolite that consistently changed in opposite directions in GLAST over-expressing and Got1-deficient as compared to Got2-deficient HSCs.
Aspartate can be used in the TCA cycle and to make nucleotides and asparagine
Aspartate can be used for the synthesis of proteins, purines, pyrimidines, asparagine, N-acetylaspartate (NAA), and arginine, and as an anaplerotic substrate for the TCA cycle (Figure S1A). We assessed the rate of protein synthesis in HSCs by assessing the rate of O-propargyl-puromycin incorporation in vivo (Signer et al., 2014). We observed no difference in the rate of protein synthesis by GLAST over-expressing as compared to control HSCs in adult bone marrow during either steady-state conditions or 15 days after transplantation into irradiated mice (Figures S4A and S4B). This suggests aspartate levels are not limiting for protein synthesis by quiescent or proliferating HSCs, consistent with the observation that HSCs have unusually low rates of protein synthesis relative to other hematopoietic cells (Signer et al., 2014).
To assess other potential fates of aspartate, we performed isotope tracing of universally 13C or 15N-labelled aspartate in GLAST over-expressing HSCs cultured for 7 days in medium that promotes HSC self-renewal (Wilkinson et al., 2019) (Figures S1D and S1E). We added [U-13C] (m+4) or 15N (m+1)-labelled aspartate to the cultures and lysed the cells for analysis 2 and 8 hours later. Negative control samples without labelled aspartate had fractional enrichments of 13C or 15N-labelled metabolites consistent with natural abundance. We corrected for natural abundance when analyzing experimental samples. The fractional enrichments of 13C (m+4) versus 15N (m+1) labelled aspartate were 64±1.2% and 60±1.2%, respectively, at 2 hours in GLAST over-expressing hematopoietic stem/progenitor cells (Figure 4A).
Figure 4. Tracing of isotopically labelled aspartate in hematopoietic stem/progenitor cells.
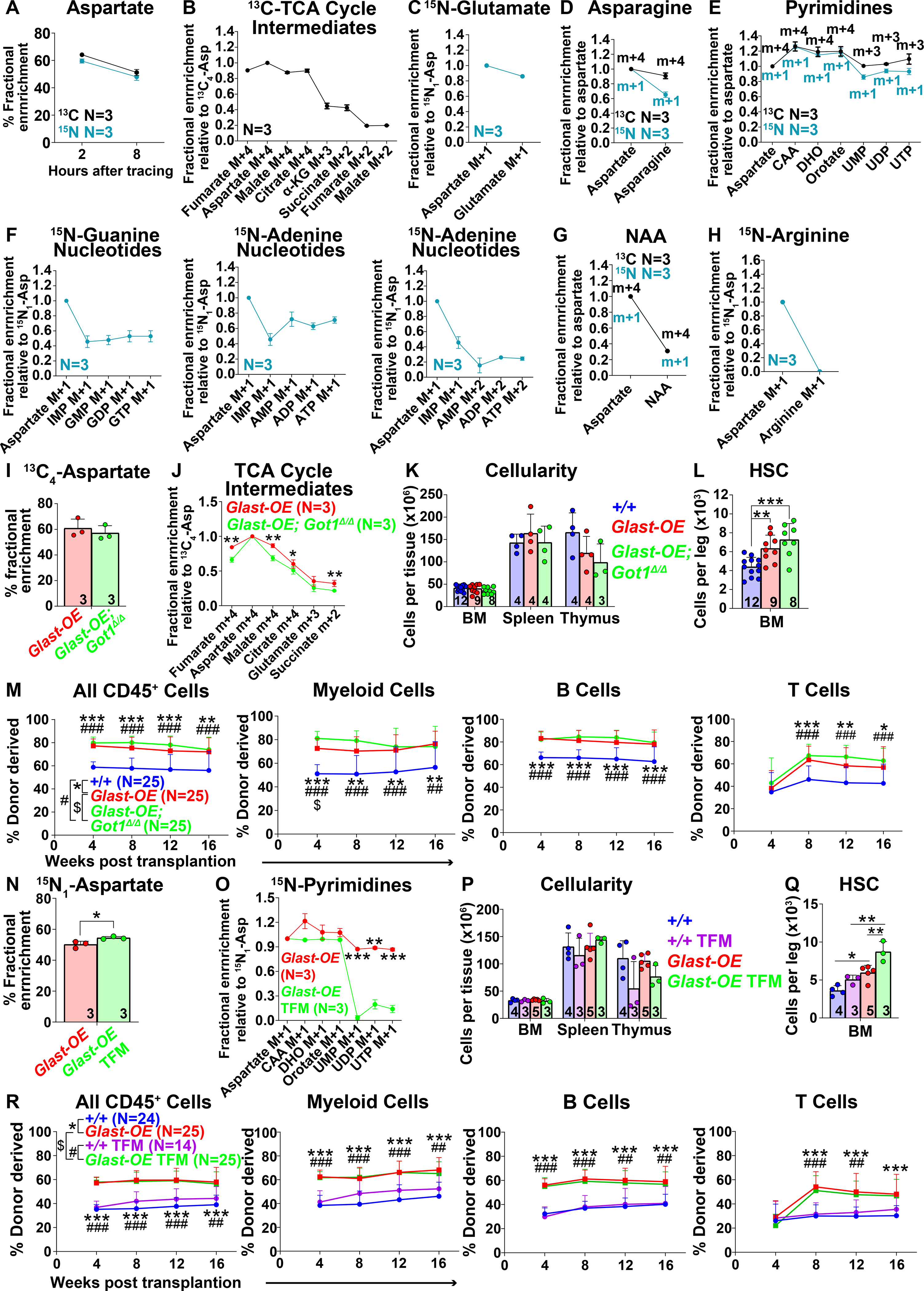
(A-G) Isotope tracing in GLAST over-expressing hematopoietic stem/progenitor cells cultured with 200μM universally 13C (black) or 15N (blue) labelled aspartate. Fractional enrichment of aspartate (A), TCA cycle intermediates (B), glutamate (C), asparagine (D), pyrimidines (E), purines (F), NAA (G) and arginine (H). The fractional enrichment of arginine (H) was measured in cells cultured in arginine-free medium. The natural abundance of isotopes was corrected using the AccuCor method (Su et al., 2017). The fractional enrichment was shown at 2 hours after adding labelled aspartate to culture for TCA cycle intermediates (B) and glutamate (C), and after 8 hours for the other pathways (D-H). (α-KG: α-ketoglutarate, CAA: carbamoyl aspartic acid, DHO: dihydroorotate). (I and J) Fractional enrichment of aspartate (I) and TCA cycle intermediates (J) in hematopoietic stem/progenitor cells cultured from Vav1-cre; Rosa26LSL-GIast (Glast-OE) or Vav1-cre; Rosa26LSL-Glast; Got1fl/fl (Glast-OE; Got1Δ/Δ) mice and supplemented with universally 13C-labelled aspartate for 2 hours. (K) Cellularity of the bone marrow from one femur and one tibia, the spleen, and the thymus. (L) Number of HSCs in the bone marrow from one femur and one tibia. (M) Donor-derived CD45+, myeloid, B, and T cells in the blood of mice competitively transplanted with Glast-OE; Got1Δ/Δ, Glast-OE, or control donor bone marrow cells (n=25 recipient mice total from 5 independent experiments with 5 donors per genotype). (N and O) Fractional enrichment of aspartate (N) and pyrimidine synthesis intermediates (O) in Glast-OE stem/progenitor cells cultured with 15N-labelled aspartate for 8 hours, with or without teriflunomide (TFM). (P) Cellularity of the bone marrow from one femur and one tibia, the spleen, and the thymus from Glast-OE or wild-type control mice treated with vehicle or TFM. (Q) Number of HSCs in the bone marrow from one femur and one tibia. (R) Donor-derived CD45+, myeloid, B, and T cells in the blood of mice competitively transplanted with Glast-OE or control donor bone marrow cells, and with or without TFM treatment of the recipient mice (n=14–25 recipient mice total from 5 independent experiments with 3–5 donors per condition). All data represent mean ± standard deviation (* P< 0.05; ** P<0.01; *** P<0.001). The number of mice analyzed per genotype is in each panel. See also Figure S4 and Table S1.
13C-labelled aspartate (m+4) was incorporated into TCA cycle intermediates with the highest fractional enrichments in species proximal to those that can be exchanged across the mitochondrial membrane by the malate-aspartate shuttle (Figure 4B). Consistent with this, 15N-labelled aspartate gave rise to 15N-labelled glutamate (Figure 4C), which can also enter mitochondria through the malate-aspartate shuttle (Figure S1A). 15N-labelled and 13C-labelled aspartate traced into asparagine (Figure 4D) as well as pyrimidines (Figures 4E and S4D) and purines (Figures 4F and S4E) and to a much lesser extent NAA (Figure 4G). Thus, exogenous aspartate can contribute to the TCA cycle or be used to synthesize nucleotides and asparagine. We did not detect labelled arginine in our experiments, even when we cultured the cells in arginine-free medium (Figure 4H), suggesting that aspartate is not used for arginine synthesis by hematopoietic stem/progenitor cells.
The TCA cycle
Since aspartate contributed to the TCA cycle (Figure 4B), we tested if this contributed to the increase in HSC function in GLAST over-expressing HSCs. We reasoned that Got1 deficiency would reduce the contribution of aspartate to the TCA cycle by reducing the generation of malate. To test this, we cultured HSCs from Vav1-cre; Rosa26LSL-Glast; Got1fl/fl mice for 5 or 6 days, added [U-13C] (m+4) aspartate to the culture and analyzed by mass spectrometry 2 hours later. Got1 deficiency did not significantly affect the fractional enrichment of aspartate in GLAST over-expressing as compared to control stem/progenitor cells (Figure 4I). However, it did significantly reduce the fractional enrichments in TCA cycle intermediates (Figure 4J). An approximately 2/3 reduction in the flux of label from aspartate to malate would be required to achieve this effect if the rates of other reactions were unaffected by Got1 deficiency (see Figure S4F for an explanation).
Given that Got1 deficiency reduced the entry of carbon from aspartate into the TCA cycle, we tested if it eliminated the increase in HSC function in GLAST over-expressing mice. Vav1-cre; Rosa26LSL-Glast; Got1fl/fl mice did not significantly differ from Vav1-cre; Rosa26LSL-Glast or wild-type controls with respect to bone marrow, spleen, or thymus cellularity (Figure 4K), blood cell counts (Figure S4G), or the numbers of restricted hematopoietic progenitors or most differentiated hematopoietic cells in the bone marrow, spleen, or thymus (Figures S4H–S4M). GLAST over-expression was associated with significantly higher numbers of HSCs in the bone marrow (Figure 4L) and multilineage reconstituting activity upon transplantation of bone marrow cells into irradiated mice (Figure 4M) but Got1 deficiency did not attenuate either of these increases. Indeed, there was a trend toward higher HSC numbers and reconstituting activity in the bone marrow of Vav1-cre; Rosa26LSL-Glast; Got1fl/fl as compared to Vav1-cre; Rosa26LSL-Glast mice (Figures 4L and 4M) despite the reduced contribution of aspartate to the TCA cycle (Figure 4J). Therefore, there was no evidence that the contribution of aspartate to the TCA cycle increased HSC number or function.
Pyrimidine synthesis
We cultured GLAST over-expressing HSCs for 7 days and then inhibited de novo pyrimidine synthesis using teriflunomide, a selective inhibitor of dihydroorotate dehydrogenase (Figure S4D), before adding 15N-labelled aspartate to the cultures for eight hours. Teriflunomide slightly increased the fractional enrichment of aspartate in hematopoietic stem/progenitor cells (Figure 4N) and substantially reduced uridine synthesis (Figure 4O). Teriflunomide treatment also reduced de novo pyrimidine synthesis in the bone marrow in vivo (Figure S4N).
We treated GLAST over-expressing or wild-type control mice with teriflunomide or vehicle for 16–17 days. GLAST over-expressing mice did not significantly differ from wild-type controls, regardless of teriflunomide treatment, in terms of bone marrow, spleen, or thymus cellularity (Figure 4P), blood cell counts (Figure S4O), or the numbers of most restricted progenitors and differentiated hematopoietic cells in the bone marrow, spleen, or thymus (Figures S4P–S4U). GLAST over-expression was associated with significantly higher numbers of HSCs in the bone marrow (Figure 4Q) and teriflunomide treatment further increased HSC numbers in GLAST over-expressing mice (Figure 4Q). GLAST over-expressing bone marrow cells also gave significantly higher levels of donor cell reconstitution in all lineages as compared to control cells and this was not affected by teriflunomide treatment (Figure 4R). Therefore, GLAST did not appear to promote HSC function by increasing pyrimidine synthesis.
NAA synthesis
Aspartate can be acetylated by N-acetyltransferase 8 like (NAT8L) to form NAA (Wiame et al., 2009). To test if NAA regulates HSC function we conditionally deleted Nat8l using Vav1-cre (Maier et al., 2015). NAA was profoundly depleted in bone marrow cells after Nat8l deletion (Figure 5A). Vav1-Cre; Nat8lfl/fl mice were grossly normal in size and appearance (Figures 5B and 5C) and were born in expected mendelian frequencies (Figure S5A). They had normal blood cell counts (Figure 5D) and cellularity in the bone marrow, spleen, and thymus (Figure 5E). The numbers of hematopoietic stem and progenitor cells as well as differentiated cells in the bone marrow (Figures 5F–5H), spleen and thymus (Figures S5B–S5E) were also normal. Vav1-Cre; Nat8lfl/fl and control bone marrow cells gave similar levels of long-term donor cell reconstitution upon competitive transplantation into irradiated mice (Figure 5I). NAA, thus, is not required for HSC function under normal conditions.
Figure 5. N-acetylaspartate (NAA) synthesis does not mediate the effects of GLAST over-expression on HSC function.
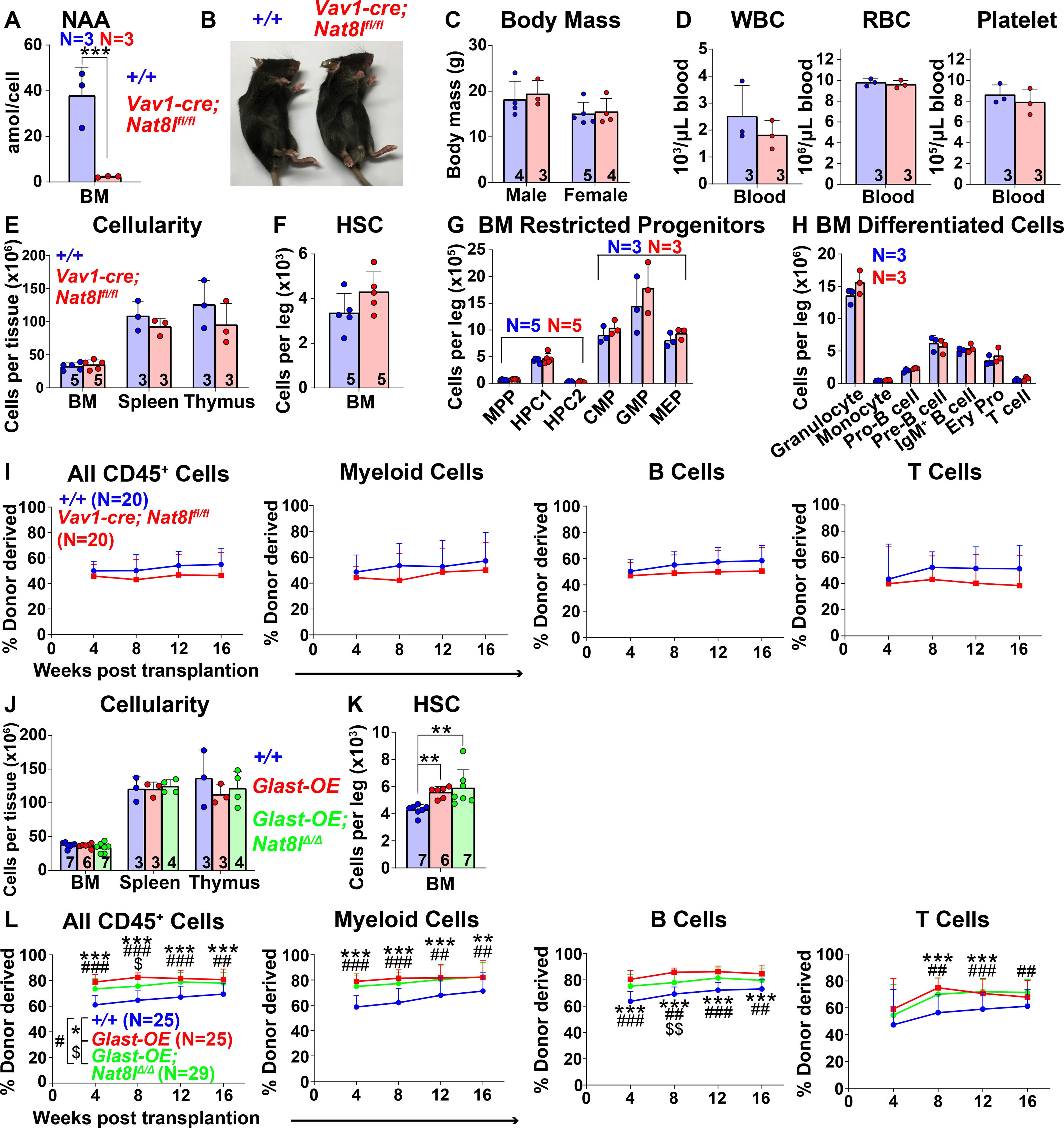
(A) NAA was severely depleted in bone marrow cells from Vav1-cre; Nat8lfl/fl mice. (B and C) Vav1-cre; Nat8lfl/fl and control mice did not differ in size or appearance. (D) White blood cell, red blood cell, and platelet counts in Vav1-cre; Nat8lfl/fl and control blood. (E) Cellularity of the bone marrow from one femur and one tibia, the spleen, and the thymus. (F-H) Numbers of HSCs (F), restricted progenitors (G), and differentiated hematopoietic cells (H) in the bone marrow from one femur and one tibia. (I) Donor-derived CD45+, myeloid, B, and T cells in the blood of mice competitively transplanted with Vav1-cre; Nat8lfl/fl or control bone marrow cells (n=20 recipient mice total from 4 independent experiments using 4 donors per genotype). (J) Cellularity of the bone marrow from one femur and one tibia, the spleen, and the thymus of Vav1-cre; Rosa26LSL-Glast (Glast-OE), Vav1-cre; Rosa26LSL-Glast; Nat8lfl/fl (Glast-OE; Nat8lΔ/Δ), and control mice. (K) Number of HSCs in the bone marrow from one femur and one tibia. (L) Donor-derived CD45+, myeloid, B, and T cells in the blood of mice competitively transplanted with Glast-OE; Nat8lΔ/Δ; Glast-OE or control bone marrow cells (n=25–29 recipient mice total from 5 independent experiments with 5–6 donors per genotype). All data represent mean ± standard deviation (* P< 0.05; ** P<0.01; *** P<0.001). The number of mice analyzed per genotype is shown in each panel. See also Figure S5 and Table S1.
Vav1-cre; Rosa26LSL-Glast; Nat8lfl/fl mice did not significantly differ from Vav1-cre; Rosa26LSL-Glast or wild-type controls with respect to bone marrow, spleen, or thymus cellularity (Figure 5J), blood cell counts (Figure S5F), or the numbers of hematopoietic progenitors and differentiated cells in the bone marrow, spleen, or thymus (Figures S5G–S5L). Bone marrow from GLAST over-expressing mice had significantly higher numbers of HSCs (Figure 5K) and multilineage reconstituting activity upon transplantation into irradiated mice (Figure 5L) but Nat8l deficiency did not attenuate these increases. NAA synthesis, therefore, did not appear to contribute to the effect of GLAST over-expression on HSC function.
Asparagine synthesis contributes to HSC function
Since hematopoietic stem/progenitor cells used aspartate partly for asparagine synthesis (Figure 4D), we tested if this contributed to the increase in HSC function in GLAST over-expressing mice. We generated Vav1-cre; Asnsfl/fl mice to delete the gene that encodes asparagine synthetase from hematopoietic cells (Hope et al., 2020, Ruzzo et al., 2013). We cultured HSCs from Vav1-cre; Rosa26LSL-Glast and Vav1-cre; Rosa26LSL-Glast; Asnsfl/fl mice for 5 or 6 days, added [U-13C] (m+4) aspartate to culture, and analyzed by mass spectrometry 2 hours later. Asns deficiency modestly increased the fractional enrichment of aspartate in GLAST over-expressing cells (Figure 6A) and completely abolished 13C-labeling in asparagine (Figure 6B).
Figure 6. Asparagine synthesis contributes to the effects of GLAST on HSC function.
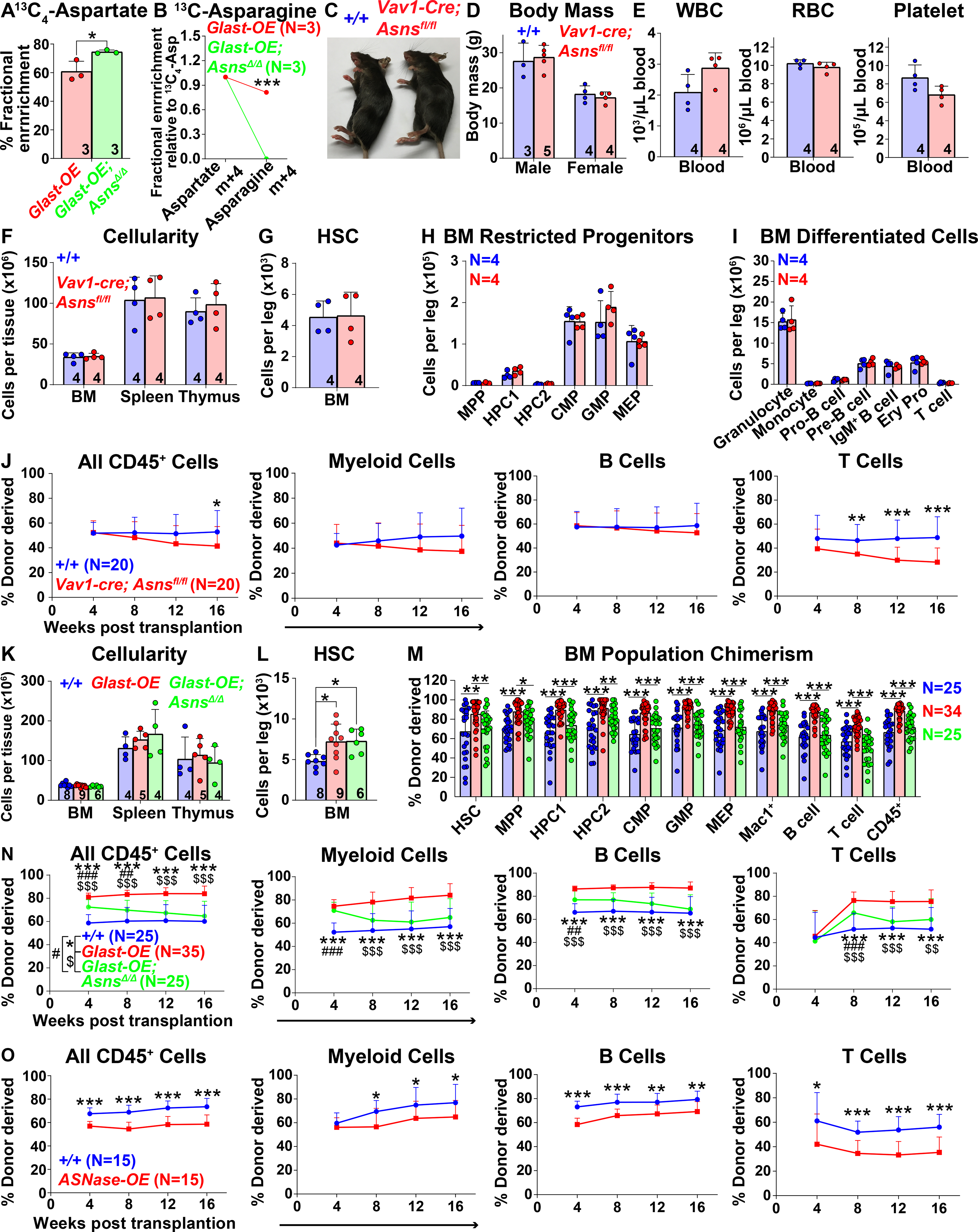
(A and B) Isotope tracing in hematopoietic stem/progenitor cells from Vav1-cre; Rosa26LSL-Glast (Glast-OE) or Vav1-cre; Rosa26LSL-Glast; Asnsfl/fl (Glast-OE; AsnsΔ/Δ) mice cultured with [U-13C] (m+4)-labelled aspartate. Fractional enrichment of aspartate (A), and asparagine (B). These data are from the same experiment as in Figures 4I and 4J. (C and D) Vav1-cre; Asnsfl/fl and control mice did not differ in size or appearance. (E) White blood cell, red blood cell, and platelet counts in the blood of Vav1-cre; Asnsfl/fl and control mice. (F) Cellularity of the bone marrow from one femur and one tibia, the spleen, and the thymus. (G-I) Number of HSCs (G), restricted progenitors (H) and differentiated hematopoietic cells (I) in the bone marrow from one femur and one tibia. (J) Donor-derived CD45+, myeloid, B, and T cells in the blood of mice competitively transplanted with Vav1-cre; Asnsfl/fl or control bone marrow cells (n=20 recipient mice total from 4 donors per genotype in 4 independent experiments). (K) Cellularity of the bone marrow from one femur and one tibia, the spleen, and the thymus of Glast-OE, Glast-OE; AsnsΔ/Δ and control mice. (L) Number of HSCs in the bone marrow from one femur and one tibia. (M and N) Donor-derived HSCs and restricted hematopoietic progenitors in the bone marrow (M) as well as CD45+, myeloid, B, and T cells in the blood (N) of mice competitively transplanted with Glast-OE; Asns Δ/Δ, Glast-OE, and control bone marrow cells (n=25–35 recipient mice total from 5–7 donors per genotype in 5 independent experiments). (O) Donor-derived CD45+, myeloid, B, and T cells in the blood of mice competitively transplanted with Vav1-cre; Rosa26LSL-gpASNase1 (ASNase-OE) or control donor bone marrow cells (n=15 recipient mice total from 3 donors per genotype in 3 independent experiments). All data represent mean ± standard deviation (* P< 0.05; ** P<0.01; *** P<0.001). The number of mice analyzed per genotype is in each panel. See also Figure S6 and Table S1.
Vav1-cre; Asnsfl/fl mice were grossly normal in body mass and appearance (Figures 6C and 6D) and were born in expected mendelian ratios (Figures S6A). Vav1-cre; Asnsfl/fl mice did not significantly differ from wild-type controls with respect to blood cell counts (Figure 6E), bone marrow, spleen, or thymus cellularity (Figure 6F), or the numbers of HSCs (Figure 6G), or other hematopoietic progenitors and differentiated cells in the bone marrow (Figures 6H and 6I), spleen, or thymus (Figures S6B–S6E). Bone marrow cells from Vav1-cre; Asnsfl/fl and control mice gave similar levels of donor cell reconstitution upon transplantation into irradiated mice with the exception of T cells (Figure 6J). Asparagine synthetase was, therefore, not required by HSCs or restricted hematopoietic progenitors, perhaps because they can readily take up asparagine from circulating pools (Pavlova et al., 2018). The requirement for asparagine synthetase in T cells is consistent with prior studies (Hope et al., 2020, Wu et al., 2021).
Aspartate levels can limit asparagine synthesis (Sun et al., 2019, Zhang et al., 2014). We, therefore, wondered if GLAST over-expression promoted HSC function by increasing asparagine synthesis. To test this, we compared Vav1-cre; Rosa26LSL-GLAST; Asnsfl/fl mice with Vav1-cre; Rosa26LSL-GLAST and wild-type controls. These mice did not differ in terms of bone marrow, spleen, or thymus cellularity (Figure 6K), blood cell counts (Figure S6F), or the numbers of hematopoietic progenitors or differentiated cells in the bone marrow, spleen, or thymus (Figures S6G–S6L). Asns deficiency did not restore normal numbers of HSCs in the bone marrow of GLAST over-expressing mice (Figure 6L) but it did significantly reduce the levels of donor myeloid, B, and T cells in the blood (Figure 6N) and stem/progenitor cells in the bone marrow of recipient mice (Figure 6M). Asns deficiency also reduced reconstitution by GLAST over-expressing cells upon secondary transplantation (Figure S6M). Therefore, asparagine synthesis contributed to the increased function of GLAST over-expressing HSCs.
We hypothesized that the normal HSC function in the absence of asparagine synthetase reflected the ability of HSCs to take up exogenous asparagine to compensate for the lack of endogenous synthesis. Therefore, to test if asparagine levels limit the function of normal HSCs,we conditionally over-expressed guinea pig asparaginase 1 (Rosa26LSL-gpASNase1), which degrades intracellular asparagine (Figure S6N) (Sullivan et al., 2015, Sullivan et al., 2018). Bone marrow cells from Vav1-cre; Rosa26LSL-gpASNase1 mice had substantially reduced asparagine levels as compared to control cells (Figure S6O). We did not observe any differences in the HSC number or cellularity of bone marrow, spleen or thymus from asparaginase over-expressing versus control mice (Figures S6P and S6Q). However, Vav1-cre; Rosa26LSL-gpASNase1 bone marrow cells gave significantly lower levels of donor cell reconstitution in all lineages as compared to control bone marrow cells when competitively transplanted into irradiated mice (Figure 6O). Asparagine levels, thus, limit HSC function during hematopoietic regeneration but can be increased either through synthesis or uptake of exogenous asparagine.
Purine synthesis contributes to HSC function
Aspartate is required at two stages of purine synthesis, for de novo purine synthesis and for the conversion of inosine monophosphate (IMP) to adenosine monophosphate (AMP) (Figures 4F and S4E). To test if this contributed to the increase in HSC number or function in GLAST over-expressing mice we cultured GLAST over-expressing HSCs for 7 days and then inhibited purine synthesis using 6-mercaptopurine (6MP; Figure S4E) (Atkinson et al., 1964, Scholar et al., 1972, Tay et al., 1969), before adding 15N-labelled aspartate to the cultures for eight hours. 6MP did not significantly affect the fractional enrichment of aspartate in GLAST over-expressing hematopoietic stem/progenitor cells (Figure 7A) but it significantly reduced the fractional enrichment in guanine and adenine nucleotides relative to IMP (Figure 7B), consistent with inhibition of purine synthesis from IMP.
Figure 7. Purine synthesis contributes to the effects of GLAST on HSC function.
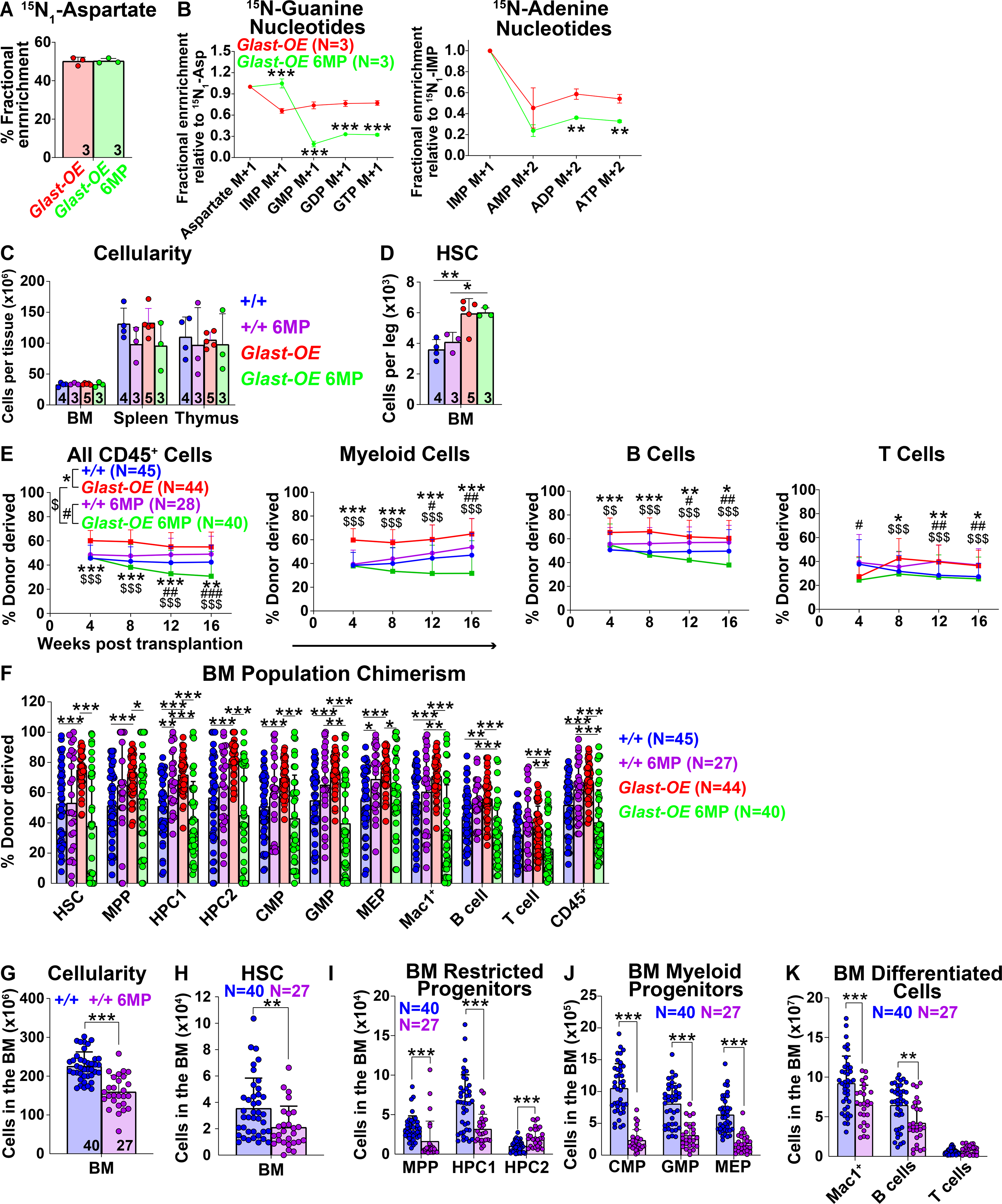
(A and B) Fractional enrichment of aspartate (A) and purine synthesis intermediates (B) in Vav1-cre; Rosa26LSL-Glast (Glast-OE) hematopoietic stem/progenitor cells cultured with 15N-labelled aspartate for 8 hours, with or without 6MP. These data are from the same experiment shown in Figures 4N and 4O. (C) Cellularity of the bone marrow from one femur and one tibia, the spleen, and the thymus of Glast-OE or wild-type control mice treated with vehicle or 6MP. (D) Number of HSCs in the bone marrow from one femur and one tibia. Data in panels C and D are from the same experiment shown in Figures 4P and 4Q. (E and F) Donor-derived CD45+, myeloid, B, and T cells in the blood (E) as well as HSCs and restricted hematopoietic progenitors in the bone marrow (F) of mice competitively transplanted with Glast-OE or control donor bone marrow cells, and with or without 6MP treatment of the recipient mice (n=27–45 recipient mice total from 6–9 donors per condition in 8 independent experiments). (G) Bone marrow cellularity (two femurs, two tibias and the spine) in transplant recipients treated for 16 weeks with 6MP versus untreated controls. (H-K) Number of HSCs (H), restricted progenitors (I), myeloid progenitors (J) and differentiated hematopoietic cells (K) in the same bone marrow specimens. Data in panels G-K are from the same experiments shown in Figure 7F. All data represent mean ± standard deviation (* P< 0.05; ** P<0.01; *** P<0.001). The number of mice analyzed per genotype is shown in each panel. See also Figure S7 and Table S1.
We treated GLAST over-expressing or wild-type control mice with 6MP or vehicle for 16 or 17 days. GLAST over-expressing mice did not significantly differ from wild-type controls, regardless of 6MP treatment, with respect to bone marrow, spleen, or thymus cellularity (Figure 7C), blood cell counts (Figure S7A), or the numbers of most restricted progenitors and differentiated cells in the bone marrow, spleen, or thymus (Figures S7B–S7G). 6MP treatment reduced the numbers of some myeloid and megakaryocyte/erythroid progenitors in the bone marrow and spleen regardless of genotype (Figures S7B and S7E). 6MP treatment did not reduce the number of HSCs in the bone marrow of GLAST over-expressing mice (Figure 7D).
We competitively transplanted GLAST over-expressing or control bone marrow cells into irradiated mice and then treated half of the recipient mice with 6MP. 6MP treatment did not reduce the frequency of donor cells produced by control bone marrow cells in either the blood (Figure 7E) or the bone marrow (Figure 7F); however, 6MP did significantly reduce the frequency of donor cells produced by GLAST over-expressing bone marrow cells in the blood (Figure 7E) and bone marrow in primary (Figure 7F) and secondary recipients (Figure S7H). 6MP treatment, thus, eliminated the competitive advantage of GLAST over-expressing as compared to control cells. This suggests purine synthesis contributed to the effect of GLAST over-expression on HSC function.
The lack of effect of 6MP treatment on donor cell reconstitution by control bone marrow cells in the competitive reconstitution assay was hard to interpret because 6MP would be expected to equally inhibit purine synthesis in both the donor and competitor control cells. To test if 6MP treatment reduced the reconstituting capacity of wild-type HSCs we performed a non-competitive reconstitution assay. The 6MP-treated recipients had significantly lower numbers of total cells, HSCs, restricted progenitors, and differentiated cells in their bone marrow as compared to untreated mice (Figures 7G–7K). Purine synthesis thus promotes the function of wild-type HSCs in addition to GLAST over-expressing HSCs during hematopoietic regeneration.
DISCUSSION
We found that aspartate synthesis increases upon HSC activation (Figures 2P and 2Q) and that aspartate levels limit HSC function during hematopoietic regeneration (e.g. Figure 1T and 2M). Our results suggest that HSCs increase their synthesis of aspartate, asparagine, and purines during hematopoietic regeneration to satisfy increased anabolic demands. Aspartate did not appear to be limiting for the function of most restricted hematopoietic progenitors (Figures 1R and 1S). This raises the possibility that aspartate is preferentially limiting for the function of stem cells and other cells with extensive self-renewal potential such as cancer cells and T cells (Alkan et al., 2018, Bailis et al., 2019, Birsoy et al., 2015, Garcia-Bermudez et al., 2018, Gui et al., 2016, Krall et al., 2021, Sullivan et al., 2015, Sullivan et al., 2018).
The dependence upon endogenous aspartate synthesis may be a tumor suppressor mechanism. Therapies that inhibit asparagine (L-asparaginase) or purine (6MP) synthesis are used to treat leukemia (Batool et al., 2016, Schmiegelow et al., 2014). Some myeloid leukemias activate GLAST expression (NCBI Gene Expression Omnibus GSE3725; (Krivtsov et al., 2006, van Gastel et al., 2020)). However, GLAST over-expression was not sufficient to transform hematopoietic cells and had a limited effect on HSC frequency and function, consistent with the fact that HSCs are regulated by redundant tumor suppressor mechanisms (He et al., 2009). GLAST over-expression might promote self-renewal to a greater extent if combined with the loss of other tumor suppressors.
Aspartate availability is well-suited to limit cell proliferation given its requirement for both protein and nucleotide synthesis. The importance of mitochondrial function for aspartate synthesis (Bailis et al., 2019, Birsoy et al., 2015, Garcia-Bermudez et al., 2018, Gui et al., 2016, Sullivan et al., 2015, Sullivan et al., 2018) suggests that the reliance upon endogenously produced aspartate is a checkpoint that prevents cells from dividing in the absence of adequate mitochondrial function. This may be a general mechanism that ensures proteins and nucleotides are not synthesized at levels beyond those that can be sustained by mitochondrial function.
Limitations of study
We were not able to perform isotope tracing from labelled aspartate in HSCs in vivo due to their rarity and the low fractional enrichment of aspartate in HSCs that could be achieved after infusion of aspartate into mice. In addition, several metabolites downstream of aspartate are difficult to detect by metabolomic analysis in small numbers of cells. It is possible that the isotope tracing performed in cultured HSCs might yield results that differ from what occurs under physiological conditions in vivo. We performed isotope tracing of labelled aspartate in cultured HSCs under conditions that allow self-renewal (Wilkinson et al., 2019) but the progeny of HSCs in these cultures are a mixture of HSCs and restricted hematopoietic progenitors. Consequently, the results reflect the fate of aspartate in stem and progenitor cells not in pure HSCs. Nonetheless, we functionally interrogated all of the pathways that consumed aspartate by experiments that assessed HSC function in vivo. Therefore, our conclusions are supported by functional studies of highly purified HSCs in vivo.
STAR METHODS
Lead Contact
Further information and requests for resources and reagents should be directed and will be fulfilled by the lead contact, Sean J. Morrison: Sean.Morrison@UTSouthwestern.edu.
Materials Availability
Material transfer agreements are required for Rosa26LSL-Glast , Rosa26LSL-gpASNase1, Got1fl and Got2fl allele due to requirement by University of Texas Southwestern Medical Center.
Data and Code Availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
To generate Rosa26LSL-Glast or Rosa26LSL-gpAsnase1 mice, Alt-R S.p. Cas9 Nuclease V3 (Integrated DNA Technologies), sgRNA (Integrated DNA Technologies, Table S4), TracrRNA (Integrated DNA Technologies) and recombineering plasmids (modified from Chu et al., 2016) containing the mouse slc1a3 or guinea pig asparaginase 1 coding sequences (Table S4) were microinjected into C57BL/6 zygotes. To generate Got1fl and Got2fl mice, Alt-R S.p. Cas9 Nuclease V3 (Integrated DNA Technologies), sgRNA (Integrated DNA Technologies, Table S4), TracrRNA (Integrated DNA Technologies) and donor oligos (Integrated DNA Technologies, Table S4) were microinjected into C57BL/6 zygotes. Chimeric mice were genotyped by sequencing the targeted locus and by polymerase chain reaction (PCR) analysis. Founders were backcrossed with C57BL/Ka mice for at least three generations prior to analysis. Mx1-cre (Kühn et al., 1995) mice, Vav1-cre (De Boer et al., 2003) mice and Flp (Rodríguez, et al., 2000) mice were obtained from Jackson Laboratory. Asnsfl mice were ordered from the European Mouse Mutant Archive. Nat8lfl mice were previously described (Maier et a., 2015). These mice were also backcrossed at least three times onto a C57Bl/Ka background and were maintained on this background. We used 6–16 week old male or female mice in all experiments, including littermate or age-matched controls. To induce Cre expression in Mx1-Cre mice, both Mx1-Cre and control mice received an intraperitoneal injection of 20 μg of Polyinosinic:polycytidylic acid dissolved in PBS every other day for 9 days (5 injections total) starting at 5–6 weeks of age. C57BL/Ka-Thy-1.2 (CD45.1) and C57BL/Ka-Thy-1.1/Thy-1.2 (CD45.1/CD45.2) mice were used as recipient mice and as a source of competitive bone marrow cells in transplantation experiments. Mice were treated with 6MP in the drinking water: 6MP (Toronto Research Chemicals) was dissolved in tap water at 10 mg/L (pH=7), filtered, provided to untransplanted or transplanted mice (starting on the day of the transplant), and changed twice a week. Mice were also treated with teriflunomide in the drinking water: teriflunomide (Medchem Express) was dissolved in tap water containing 0.6% Tween 80 at 60 mg/L (pH=7), filtered, provided to untransplanted or transplanted mice (starting on the day of the transplant), and changed once a week. Control animals received drinking water containing 0.6% Tween 80. To induce HSC proliferation, mice were injected intraperitoneally with a single dose of 4 mg cyclophosphamide dissolved in saline followed by 2 daily subcutaneous injections of 5μg G-CSF dissolved in 5% glucose and sacrificed the following day. Alternatively, mice received a single intraperitoneal injection of 2.5 ug/g body mass of polyinosinic:polycytidylic acid (GE Healthcare) dissolved in PBS and sacrificed 72 hours later. Mice were randomly assigned into control or treatment groups. All mice were housed in AAALAC-accredited, specific-pathogen-free animal care facilities at the University of Texas Southwestern Medical Center (UTSW). All procedures were approved by the UTSW Institutional Animal Care and Use Committee.
Primary cell culture
For colony forming assays, 1×104 bone marrow cells (6 well plates) or single cells sorted from HSC cultures (96 well plates) were seeded per well in Methocult GM M3434 medium (Stemcell Technologies) supplemented with 10ng/ml of recombinant thrombopoietin (Peprotech) and penicillin/streptomycin (Fisher Scientific). Colonies were counted after 10 days of culture at 37°C.
METHOD DETAILS
Flow cytometric analysis and sorting of hematopoietic cells
Bone marrow was flushed from two tibias and two femurs using staining medium (HBSS supplemented with 2% heat inactivated bovine serum) and dissociated into a single cell suspension by gently triturating with a 23-gauge needle. Spleens and thymuses were mechanically dissociated by crushing and triturating. Cells were resuspended in staining medium and filtered through a 40 μm cell strainer. Cells were counted, and then stained with antibodies at 4°C for 30 minutes (except for anti-CD34 antibody staining, which was for 90 minutes). For staining of HSCs and restricted hematopoietic progenitors, cells were first stained with biotin-conjugated antibodies against CD34, washed with staining medium and then resuspended in staining medium containing fluorophore-conjugated antibodies against lineage markers (CD2, CD3, CD5, CD8a, Gr1, Ter119 and B220), c-kit, Sca1, CD150, CD48, CD16/32 and PE-Cy7-conjugated streptavidin. Details about the antibodies used are in the key resources table. For analysis of differentiated cells, cells were stained with fluorophore-conjugated antibodies against Mac-1, Gr1, B220, CD43, IgM, CD3, Ter119 and CD71. For analysis of thymocytes, cells were stained with fluorophore-conjugated antibodies against CD4, CD8, CD25 and CD44. Cells were then analyzed using a FACS Canto RUO (BD Biosciences), LSRFortessa (BD Biosciences), FACSAria II SORP (BD Biosciences) or a FACSAria Fusion SORP (BD Biosciences) cytometer. Dead cells were identified and gated out of all analyses by including 1 μg/ml 4’,6-diamidino-2-phenylindole (DAPI) or propidium iodide in the staining medium used to resuspend cells for flow cytometry. Flow cytometry data were analyzed using BD FACSDiva (BD Biosciences) or Flowjo (Flowjo).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| α-mouse Gr1 Biotin | Tonbo Biosciences | Clone RB6–8C5; RRID:AB_2621652 |
| α-mouse Gr1 FITC | Tonbo Biosciences | Clone RB6–8C5; RRID:AB_2621721 |
| α-mouse Gr1 PE-Cy7 | Tonbo Biosciences | Clone RB6–8C5; RRID:AB_2621870 |
| α-mouse Gr1 APC | Tonbo Biosciences | Clone RB6–8C5; RRID:AB_2621610 |
| α-mouse Gr1 PE | Tonbo Biosciences | Clone RB6–8C5; RRID:AB_2621803 |
| α-mouse CD2 Biotin | Biolegend | Clone RM2–5; RRID:AB_312650 |
| α-mouse CD2 FITC | Tonbo Biosciences | Clone RM2–5; RRID:AB_2621657 |
| α-mouse CD2 PE | Tonbo Biosciences | Clone RM2–5; RRID:AB_2621728 |
| α-mouse CD2 APC | Biolegend | Clone RM2–5; RRID:AB_2563090 |
| α-mouse CD5 Biotin | Biolegend | Clone 53–7.3; RRID:AB_312732 |
| α-mouse CD5 FITC | Biolegend | Clone 53–7.3; RRID:AB_312734 |
| α-mouse CD5 PE | Biolegend | Clone 53–7.3; RRID:AB_312736 |
| α-mouse CD5 APC | Biolegend | Clone 53–7.3; RRID:AB_2563929 |
| α-mouse CD3 Biotin | Biolegend | Clone 17A2; RRID:AB_2563946 |
| α-mouse CD3 FITC | Biolegend | Clone 17A2; RRID:AB_312661 |
| α-mouse CD3 PE | Tonbo Biosciences | Clone 17A2; RRID:AB_2621731 |
| α-mouse CD3 APC | Tonbo Biosciences | Clone 17A2; RRID:AB_2621538 |
| α-mouse CD8a Biotin | Tonbo Biosciences | Clone 53–6.7; RRID:AB_2621638 |
| α-mouse CD8a FITC | Tonbo Biosciences | Clone 53–6.7; RRID:AB_2621671 |
| α-mouse CD8a PE | Tonbo Biosciences | Clone 53–6.7; RRID:AB_2621741 |
| α-mouse CD8a APC | Biolegend | Clone 53–6.7; RRID:AB_312751 |
| α-human/mouse B220 Biotin | Tonbo Biosciences | Clone RA3–6B2; RRID:AB_2621644 |
| α-human/mouse B220 FITC | Tonbo Biosciences | Clone RA3–6B2; RRID:AB_2621690 |
| α-human/mouse B220 APC | Tonbo Biosciences | Clone RA3–6B2; RRID:AB_2621574 |
| α-human/mouse B220 Percp-Cy5.5 | Tonbo Biosciences | Clone RA3–6B2; RRID:AB_2621892 |
| α-human/mouse B220 PE | Tonbo Biosciences | Clone RA3–6B2; RRID:AB_2621764 |
| α-mouse CD45.1 FITC | Biolegend | Clone A20; RRID:AB_313494 |
| α-mouse CD45.1 PE-Cy7 | Tonbo Biosciences | Clone A20; RRID:AB_2621850 |
| α-mouse CD45.2 VioletFluor 450 | Tonbo Biosciences | Clone 104; RRID:AB_2621950 |
| α-mouse Ter119 Biotin | Tonbo Biosciences | Clone TER-119; RRID:AB_2621651 |
| α-mouse Ter119 FITC | Tonbo Biosciences | Clone TER-119; RRID:AB_2621720 |
| α-mouse Ter119 PE | Tonbo Biosciences | Clone TER-119; RRID:AB_2621802 |
| α-mouse Ter119 Brilliant Violet 510 | BD Biosciences | Clone TER-119; RRID:AB_2738532 |
| α-mouse Ter119 APC | Tonbo Biosciences | Clone TER-119; RRID:AB_2621609 |
| α-mouse CD71 FITC | eBioscience | Clone R17217; RRID:AB_465124 |
| α-mouse c-kit APC-eFluor780 | eBioscience | Clone 2B8; RRID:AB_1272177 |
| α-mouse CD117 MicroBeads | Miltenyi Biotec | Clone 3C1; RRID:AB_2753213 |
| α-mouse CD45 MicroBeads | Miltenyi Biotec | Clone 30F11; RRID:AB_2877061 |
| Anti-APC magnetic microbeads | Miltenyi Biotec | RRID:AB_244367 |
| α-mouse Sca1 Percp-Cy5.5 | Thermo Fisher Scientific | Clone D7; RRID:AB_914372 |
| α-mouse CD150 PE | Biolegend | Clone TC15–12F12.2; RRID:AB_313682 |
| α-mouse CD150 PE-Cy7 | Biolegend | Clone TC15–12F12.2; RRID:AB_439796 |
| α-mouse CD150 APC | Biolegend | Clone TC15–12F12.2; RRID:AB_493461 |
| α-mouse CD48 APC | Biolegend | Clone HM48–1; RRID:AB_571996 |
| α-mouse CD48 PE-Cy7 | Biolegend | Clone HM48–1; RRID:AB_2075049 |
| α-mouse CD48 Alexa Fluor 700 | Biolegend | Clone HM48–1; RRID:AB_10612754 |
| α-mouse CD34 Biotin | Thermo Fisher Scientific | Clone RAM34; RRID:AB_466426 |
| α-mouse CD43 PE | BD Biosciences | Clone S7; RRID:AB_394748 |
| α-mouse IgM APC | Thermo Fisher Scientific | Clone Il/41; RRID:AB_469458 |
| α-mouse Mac-1 (CD11b) APC-efluor780 | Thermo Fisher Scientific | Clone M1/70; RRID:AB_1603193 |
| α-mouse CD16/32 (FcγRIII/II) BV510 | Biolegend | Clone 93; RRID:AB_2563692 |
| α-mouse CD45 FITC | Tonbo Biosciences | Clone 30-F11; RRID:AB_2621689 |
| Streptavidin PE-Cy7 | Fisher Scientific | BDB557598 |
| α-mouse CD4 APC | Biolegend | Clone GK1.5; RRID:AB_312697 |
| α-mouse/human CD44 PE-Cy7 | Biolegend | Clone IM7; RRID:AB_830787 |
| α-mouse CD25 PerCP-Cy5.5 | Tonbo Biosciences | Clone PC61.5; RRID:AB_2621889 |
| α-GLAST APC | Miltenyi Biotec | Clone ACSA-1; RRID:AB_2660783 |
| α-GOT1 | Abcam | Clone C1; Cat. ab239487 |
| α-GOT2 | Proteintech | Polyclonal; RRID:AB_2247898 |
| α-Histone H3 | Abcam | Polyclonal; RRID:AB_302613 |
| α-mouse IgG, HRP-linked | Cell Signaling Technology | Polyclonal; RRID:AB_330924 |
| α-rabbit IgG, HRP-linked | Cell Signaling Technology | Polyclonal; RRID:AB_2099233 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MethoCult™ GF M3434 | STEMCELL Technologies | Cat. M3434 |
| Cyclophosphamide | Amneal Pharmaceuticals | N/A |
| G-CSF (Neupogen) | Amgen | N/A |
| HI bovine serum | Thermo Fisher Scientific | Cat. 26170043 |
| Penicillin-Streptomycin solution | Fisher Scientific | Cat. SV30010 |
| Penicillin-Streptomycin-Glutamine (100X) | Thermo Fisher Scientific | Cat. 10378016 |
| DMEM, high glucose | Thermo Fisher Scientific | Cat. 11965118 |
| DMEM, low glucose | Thermo Fisher Scientific | Cat. 11885–092 |
| Silac DMEM Flex Media, no glucose, no phenol red | Thermo Fisher Scientific | Cat. A2493901 |
| Ham's F-12 Nutrient Mix | Thermo Fisher Scientific | Cat. 11765062 |
| Leibovitz L-15 medium, no phenol red | Life Technologies | Cat. 21083–027 |
| Neurobasal medium | Thermo Fisher Scientific | Cat. 21103049 |
| Insulin-Transferrin-Selenium-Ethanolamine (ITS -X) (100X) | Thermo Fisher Scientific | Cat. 51500056 |
| HEPES (1 M) | Life Technologies | Cat. 15630–080 |
| Poly(vinyl alcohol),87–90% hydrolyzed, average mol wt 30,000–70,000 | Sigma-Aldrich | Cat. P8136–250G |
| Hank's Balanced Salt Solution (HBSS) | Thermo Fisher Scientific | Cat. MT21022CV |
| Recombinant murine thrombopoietin (TPO) | Peprotech | Cat. 315–14 |
| Recombinant murine SCF | Peprotech | Cat. 250–03 |
| Recombinant human bFGF | R&D Systems | Cat. 233-FB-025 |
| Recombinant Human EGF Protein, CF | R&D Systems | Cat. 236-EG-200 |
| Recombinant Human IGF-I Protein, CF | R&D Systems | Cat. 291-G1–200 |
| BioCoat™ Fibronectin 6-well Clear Flat Bottom TC-Treated Multiwell Plate | Corning | Cat. 354402 |
| BioCoat™ Fibronectin 96-well Clear Flat Bottom TC-treated Microplates | Corning | Cat. 354409 |
| Corning ultra low attachment surface culture dish, 6-well plate | Corning | Cat. 07200601 |
| Acetonitrile, Optima LC/MS Grade | Fisher Scientific | Cat. A955–500 |
| Methanol, Optima LC/MS Grade | Fisher Scientific | Cat. A456–4 |
| Water, Optima LC/MS Grade | Fisher Scientific | Cat. W6–4 |
| L-Aspartic acid (13C4, 99%) | Cambridge Isotope Laboratories | Cat. CLM-1801-H-PK |
| L-Aspartic acid-15N | Sigma-Aldrich | Cat. 332135–100MG |
| L-Aspartic acid (1,4-13C2, 99%) | Cambridge Isotope Laboratories | Cat. CLM-4455-PK |
| L-Asparagine:H2O (15N2, 98%) | Cambridge Isotope Laboratories | Cat. NLM-3286–0.5 |
| N-Acetyl-L-aspartic acid-1,2,3,4-13C4 | Sigma-Aldrich | Cat. 683647 |
| DL-Glutamic acid (2,3,3,4,4-D5, 97%) | Cambridge Isotope Laboratories | Cat. DLM-357-PK |
| L-GLUTAMINE (ALPHA-15N, 98%) | Cambridge Isotope Laboratories | Cat. NLM-1016–1 |
| Teriflunomide | MedChem Express | Cat. HY-15405 |
| 6-Mercaptopurine Monohydrate | Toronto Research Chemicals | Cat. M225450 |
| Tween 80 | Sigma-Aldrich | Cat. P1754–1L |
| polyinosinic:polycytidylic acid (pIpC) | GE Healthcare | Cat. 27–4732-01 |
| Alt-R® S.p. Cas9 Nuclease V3 | Integrated DNA Technologies | Cat. 1081059 |
| 4’,6-diamidino-2-phenylindole | Sigma-Aldrich | Cat. D8417–10MG |
| Propidium iodide | Sigma-Aldrich | Cat. P4170–25MG |
| Alexa Fluor™ 555 Azide, Triethylammonium Salt | Thermo Fisher Scientific | Cat. a20012 |
| O-propargyl-puromycin | Medchem Source | Custom synthesis |
| O-propargyl-puromycin | Thermo Fisher Scientific | Cat. C10459 |
| Paraformaldehyde | Fisher Scientific | Cat. AAJ19943K2 |
| Fetal Bovine Serum | Gemini Bio-Products | Cat. 900–208 |
| Saponin | Sigma-Aldrich | Cat. 47036–50G-F |
| Urea (powder, BioReagent, for molecular biology) | Sigma-Aldrich | Cat. U5378–100G |
| Triton X-100 | Fisher Scientific | Cat. BP151–100 |
| DTT | Sigma-Aldrich | Cat. 10197777001 |
| NuPAGE LDS Sample buffer (4X) | Life Technologies | Cat. NP0007 |
| Membrane, Immun-Blot PVDF | Biorad | Cat. 1620177 |
| NuPAGE® MES SDS Running Buffer (20X) | Life Technologies | Cat. NP0002 |
| Nupage Transfer Buffer (20x) | Life Technologies | Cat. NP00061 |
| SuperSignal West Pico Chemiluminescent Substrate | Thermo Fisher Scientific | Cat. 34579 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat. 34095 |
| Restore™ Western Blot Stripping Buffer | Thermo Fisher Scientific | Cat. 21059 |
| Trypsin, Bovine Pancreas | EMD Millipore | Cat. 6502–2.5MU |
| Trypsin inhibitor from Glycine max (soybean) | Sigma-Aldrich | Cat. T6522–100MG |
| DNase I (grade II, from bovine pancreas) | Sigma-Aldrich | Cat. 10104159001 |
| N-2 Supplement (100X) | Thermo Fisher Scientific | Cat. 17502048 |
| B-27™ Supplement (50X), serum free | Thermo Fisher Scientific | Cat. 17504044 |
| Bovine serum albumin | Sigma-Aldrich | Cat. A7030 |
| 2-mercaptoethanol | Sigma-Aldrich | Cat. M6250–100ML |
| TRIzol LS Reagent | Thermo Fisher Scientific | Cat. 10296–010 |
| Iscript Reverse Transcription Supermix | Bio-Rad | Cat. 1708841 |
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat. 172–5124 |
| Critical Commercial Assays | ||
| Click-iT® Cell Reaction Buffer Kit | Life Technologies | Cat. C10269 |
| LS Columns | Miltenyi Biotec | Cat. 130–042-401 |
| Deposited Data | ||
| RNAseq analysis | Signer et al., 2016 | SRA: PRJNA699097 |
| Microarray analysis | Krivtsov et al., 2006 | GEO: GSE3725 |
| Experimental Models: Organisms/Strains | ||
| Rosa26 LSL-Glast | this paper | N/A |
| Rosa26 LSL-gpASNase1 | this paper | N/A |
| Got1 fl | this paper | N/A |
| Got2 fl | this paper | N/A |
| Nat8l fl | Maier et al., 2015 | N/A |
| Asnsfl (C57BL/6N-Asnstm1a(EUCOMM)Wtsi/H) | European Mouse Mutant Archive | RRID:IMSR_EM: 05307 |
| Mx1-cre (B6.Cg-Tg(Mx1-cre)1Cgn/J) | Kühn et al., 1995 | RRID:IMSR_JAX: 003556 |
| Vav1-cre (B6.Cg-Commd10Tg(Vav1-icre)A2Kio/J) | De Boer et al., 2003 | RRID:IMSR_JAX:008610 |
| Flp (B6;SJL-Tg(ACTFLPe)9205Dym/J) | Rodriguez et al., 2000 | RRID:IMSR_JAX:003800 |
| Oligonucleotides | ||
| genotyping primers, qPCR primers, sgRNA and donor oligos | Integrated DNA technologies | Table S4 |
| Alt-R® CRISPR-Cas9 tracrRNA | Integrated DNA technologies | Cat. 1072532 |
| Recombinant DNA | ||
| pR26 CAG AsiSI/Mlui cloning vector | Chu et al., 2016 | RRID:Addgene_74286 |
| TCM1004 -MGC premier cDNA Clone for Slc1a3 | Transomics | Cat. BC066154 |
| Guinea pig Asparaginase 1 coding sequence cloning template | Integrated DNA technologies | Table S4 |
| Software and Algorithms | ||
| Graphpad Prism v.9.0.1 | Graphpad | RRID:SCR_002798 |
| Biorender | Biorender | RRID:SCR_018361 |
| ImageJ v1.52a | Wayne Rasband | RRID:SCR_003070 |
| Chemdraw v.19.1 | PerkinElmer | RRID:SCR_016768 |
| Flowjo | Flowjo | RRID:SCR_008520 |
| BD FACSDiva | BD Biosciences | RRID:SCR_001456 |
| MultiQuant v.4.1.1 | SCIEX | N/A |
| TraceFinder v.4.1 or v.5.0 | Thermo Scientific | N/A |
To sort HSCs, tibias, femurs, pelvises, and spines were crushed using a mortar and pestle. The bone marrow cells were then gently triturated and filtered, as above, to obtain a single cell suspension. Cells were stained with APC-efluo780 conjugated anti-c-kit antibody and c-kit+ cells were enriched using either anti-APC paramagnetic microbeads (Miltenyi Biotec) or enriched with anti-CD117 microbeads directly (Miltenyi Biotec). Cells were then stained with fluorophore-conjugated antibodies against lineage markers (CD2, CD3, CD5, CD8a, B220, Ter119, and Gr1), Sca1, c-kit, CD150 and CD48. Cells were isolated by two successive rounds of sorting to ensure purity using a FACSAria II or a FACSAria Fusion SORP cytometer. For peripheral blood analysis, 100–300 μl of blood was collected from the tail vein or from the heart after euthanasia and mixed with 5μl of 500mM EDTA to prevent clotting. White blood cell, red blood cell, and platelet counts were determined using a Hemavet HV950 (Drew Scientific).
Flow cytometric sorting for metabolomic analysis
We used published methods to perform metabolomic analysis on flow cytometrically isolated cells (Agathocleous et al., 2017, DeVilbiss et al., 2021). Cells were kept cold at all times to minimize metabolic changes, were isolated quickly and sorted directly into lysis buffer. Using these methods, the levels of most metabolites do not significantly change during cell preparation and sorting (Agathocleous et al., 2017, DeVilbiss et al., 2021). Mice were euthanized by cervical dislocation and bone marrow cells were obtained by crushing femurs, tibias, vertebrae and pelvic bones with an ice-cold mortar and pestle on ice in 3 ml of HBSS. Cells were filtered through a 40 μm cell strainer into 50 ml tubes on ice. To sort HSCs/MPPs, HPCs, or LK cells, anti-CD117 microbeads (Miltenyi Biotech) were added to each sample, followed 10 minutes later by the fluorophore-conjugated antibodies described above. Then c-kit+ cells were enriched using a QuadroMACS manual separator (Miltenyi Biotech) in the cold room and centrifuged once to remove unbound microbeads. To sort CD45+ cells, bone marrow cells was incubated with anti-CD45 microbeads (Miltenyi Biotech), followed 10 minutes later by adding FITC-conjugated anti-CD45 antibody, and then enriched with a QuadroMACS manual separator (Miltenyi Biotech) in the cold room. The cells were centrifuged and washed once to remove unbound microbeads. Cells were sorted using a FACSAria or FACSFusion cytometer running with a sheath fluid of 0.5x PBS, prepared fresh using Milli-Q water (Millipore), and a 70-μm nozzle in a four-way purity sort mode to minimize the volume of sorted drops, so as to eliminate ion suppression of mass spectrometry signals from salts in the sheath fluid. The sheath tank was washed with Milli-Q deionized water before the experiment to eliminate contaminants and dedicated glassware was used to make all buffers. Each cell sample was suspended in HBSS and kept at 4 °C during sorting, which took about 20 minutes per sample. Similar numbers of cells from each population were directly sorted in each experiment (usually 5,000–10,000 cells) into 40 μl of cold acetonitrile (final concentration 80%) pre-chilled on ice and maintained at 4°C during sorting. If possible, two technical replicates were sorted for each cell population from each mouse. After sorting, each sample was processed for metabolite extraction immediately. Metabolites were extracted by vigorous vortexing and centrifuged at 17,000g for 15 min at 4°C. The supernatant was transferred to autosampler vials and frozen at −80°C until analysis.
Metabolite extraction from abundant samples
To extract metabolites from abundant samples (>1×105 cells), such as unfractionated bone marrow cells, the cells were pelleted by centrifugation and washed with ice-cold saline twice. The cell pellet was resuspended in 50–100 μl of 80% acetonitrile, except for NAA quantification in Figure 5A where 80% methanol was used. Metabolites were extracted by vigorous vortexing and centrifuged at 17,000×g for 15 min at 4°C. Supernatant in acetonitrile was transferred to autosampler vials and frozen at −80°C until analysis. Supernatant in methanol was lyophilized using a Speedvac (Thermo Scientific). Dried metabolites were reconstituted in 50–100 μl of 0.03% formic acid, vortexed, centrifuged at 17,000×g for 5 minutes at 4°C and the supernatant was transferred to autosampler vials immediately before analysis.
Isotope tracing of labelled aspartate
We sorted 5000–25000 HSCs from mice of the indicated genotypes into each well of fibronectin-coated 6-well plates (Corning) containing HSC self-renewal medium (Wilkinson et al., 2019) and cultured them for 5–7 days. The medium contained Ham’s F-12 nutrient mixture (Thermo Fisher Scientific), 1% Penicillin-Streptomycin-Glutamine (Thermo Fisher Scientific), 1% Insulin-Transferrin-Selenium-Ethanolamine (Thermo Fisher Scientific), 0.1% Polyvinyl alcohol (Sigma-Aldrich), 10mM HEPES (Life Technologies), 10 ng/ml recombinant murine stem cell factor (SCF) (Peprotech), and 100 ng/ml recombinant murine thrombopoietin (Peprotech). Cells were split when the density reached 80% confluency. On the day of tracing, 200,000–500,000 cells were transferred into medium in which the Ham’s F-12 was replaced with DMEM (Thermo Fisher Scientific) to eliminate unlabelled aspartate. For analysis of fractional enrichment in arginine (Figure 4H), DMEM was replaced with arginine-free DMEM. 20μM or 200μM of either universally 13C-labelled L-aspartate (Cambridge Isotope Laboratories) or 200μM 15N-labelled L-aspartate (Sigma-Aldrich) was added into the culture medium for the indicated period of time before the cells were washed and lysed for mass spectrometry. In some experiments 6MP (2 μM), teriflunomide (50 μM) or DMSO alone were added to the cells 1 hour before isotope-labelled aspartate was added. Metabolites were extracted as described above.
Metabolomic analysis
To quantitate aspartate, glutamate, NAA, and asparagine levels, labelled internal standards were added to the cell extracts: 5 pmoles of L-aspartic acid (1,4- 13C2) (Cambridge Isotope Laboratories), 5 pmols of DL-glutamic acid (2,3,3,4,4-D5) (Cambridge Isotope Laboratories), 0.05 pmoles of NAA (1,2,3,4- 13C4) (Sigma-Aldrich) and 0.2–1 pmole of L-asparagine (15N2) (Cambridge Isotope Laboratories) were added to the 40 μl acetonitrile before sorting.
All metabolomic analyses were performed using a Millipore Sigma ZIC-pHILIC column (2.1 × 150, 5 μm) with a binary solvent gradient, except for the NAA quantification in Figure 5A (see details below). Mobile phase A was water containing 10 mM ammonium acetate, pH 9.8 with ammonium hydroxide; mobile phase B was 100% acetonitrile. Gradient separation proceeded as follows: from 0 to 15 minutes mobile phase B was ramped linearly from 90% to 30%; from 15 minutes to 18 minutes, mobile phase B was held at 30%; from 18 minutes to 19 minutes, mobile phase B was ramped linearly from 30% to 90%; mobile phase B was held at 90% from 19 minutes to 27 minutes to regenerate the initial chromatographic environment. Throughout, the solvent flow rate was kept at 250 μl/minute and the column temperature was maintained at 25°C. For low abundance samples, 20 μl of sample was injected onto the column. For high abundance samples, 10 μl was injected. Most mass spectrometry data were acquired using a Thermo Scientific (Bremen, Germany) QExactive HF-X or Orbitrap Fusion Lumos mass spectrometer (LC-MS/MS). A polarity-switching MS1 only acquisition method was used. Each polarity was acquired at a resolving power of 120,000 full width at half maximum (FWHM); the automatic gain control (AGC) target was set to 1,000,000 with a maximum inject time of 50 milliseconds. The scan range was set to 80–1200 Daltons. A small number of metabolomic analyses were performed using a SCIEX 6500+ Q-Trap mass spectrometer (SCIEX) coupled to the same HILIC method. The mass spectrometry analysis was operated in MRM mode monitoring the following transitions for aspartate, glutamate, their respective internal standards and asparagine, in positive mode (aspartate 134/74, aspartate internal standard (ISTD) 136/75 and asparagine 133/74) or negative mode (glutamate 146/102 and glutamate ISTD 151/107). Transitions and source parameters were optimized by infusion before analysis.
For the NAA quantification in Figure 5A, an AB 5500 Q-TRAP triple quadrupole mass spectrometer (SCIEX) was coupled to a Scherzo SM-C18 column (100 × 2mm, 3 μm, Imtakt USA) with a binary solvent gradient. Mobile phase A was water containing 0.03% formic acid; mobile phase B was 100% acetonitrile containing 0.03% formic acid. Gradient separation proceeded as follows: from 0 to 3 minutes only mobile phase A was running; From 3 to 15 minutes, mobile phase B was ramped linearly from 0% to 100%; from 15 minutes to 16.7 minutes, mobile phase B was held at 100%; from 16.7 minutes to 17.2 minutes, mobile phase B was ramped linearly from 100% to 0%; mobile phase B was held at 0% from 17.2 minutes to 20 minutes to regenerate the initial chromatographic environment. Throughout, the solvent flow rate was kept at 500 μl/minute and the column temperature was 35°C. The mass spectrometry analysis was operated in MRM mode monitoring NAA 176/134 or NAA ISTD 180/138 in positive mode. Transitions and source parameters were optimized by infusion before analysis.
Data were manually reviewed and peaks were integrated using TraceFinder 4.1 or 5.0 (Thermo Scientific) software for samples analyzed using the QExactive HF-X or Orbitrap Fusion Lumos mass spectrometer and MultiQuant 4.1.1 (SCIEX) for samples analyzed using the SCIEX 6500+ Q-Trap or SCIEX AB 5500 Q-TRAP mass spectrometer. For absolute quantification (moles per cell), the signal intensity of unlabelled metabolites was normalized to known quantities of isotope-labelled internal controls and cell number in each sample.
We developed an R tool (https://git.biohpc.swmed.edu/CRI/ODA) for the analysis of metabolite peak intensity data as previously described (DeVilbiss et al., 2021). To assess the statistical significance of differences in metabolite levels between two samples, we normalized the cell samples using the Relative Log Expression (RLE) method (Anders and Huber, 2010). Metabolite abundance was then normalized to average signal intensity of the corresponding metabolite in wild-type unfractionated bone marrow samples in each experiment.
For isotope tracing experiments, the theoretical masses of 13C and 15N labelled metabolites were calculated and added to a library of predicted isotopes in TraceFinder (Thermo Scientific). These masses were then searched with a 5-ppm tolerance and integrated only if the peak apex showed less than a 1% difference in retention time from the unlabelled monoisotopic mass in the same chromatogram. The natural abundance of 13C and 15N isotopes was corrected using the AccuCor method (Su et al., 2017).
Bone marrow reconstitution assays
Recipient mice (CD45.1/CD45.2 or CD45.1) were irradiated using an XRAD 320 X-ray irradiator (Precision X-Ray Inc.) with two doses of 540 rad at least 3h apart. For bone marrow transplantation, unfractionated bone marrow cells from donor (CD45.2) and competitor mice (CD45.1 or CD45.1/CD45.2) were mixed and injected intravenously through the retro-orbital venous sinus. For secondary transplantation, 5 million unfractionated bone marrow cells were injected intravenously through the retro-orbital venous sinus into 1–2 secondary recipient mice per primary recipient mouse. For transplantation of cultured cells, all of the cells in a well were mixed with competitor cells and injected intravenously into the retro-orbital venous sinus of irradiated recipient mice, one well of donor cells per recipient. Every 4 weeks until 16 weeks after transplantation, 50–100 μl of blood was collected from the tail vein and mixed with 200 μl of 10 mM EDTA in PBS to prevent clotting. Cells were subjected to ammonium-chloride potassium chloride red cell lysis. Cells were then stained with antibodies against CD45.1, CD45.2, Mac-1, Gr1, B220, and CD3 to evaluate the levels of donor cell engraftment in the myeloid, B, and T cell lineages. Levels of donor cell engraftment were evaluated in the bone marrow of recipient mice at least 16 weeks after transplantation (tibias, femurs and spines were collected and crushed using a mortar and pestle). Cells were resuspended in staining medium, filtered through a 40 μm cell strainer, stained with antibodies as described above, and analyzed using a FACS Canto RUO or a LSRFortessa cytometer.
Measuring the rate of protein synthesis
O-propargyl-puromycin (50 mg/kg body mass; pH 6.4−6.6 in PBS, Medchem Source or Thermo Fisher Scientific) was injected intraperitoneally. One hour later, the mice were euthanized. Bone marrow was harvested and 10 million cells were stained with antibodies against HSC cell surface markers as described above. After washing, cells were fixed in 0.5 ml of 1% paraformaldehyde (Fisher Scientific) in PBS for 15 minutes on ice. The cells were washed in PBS, then permeabilized in 200 μl PBS supplemented with 3% fetal bovine serum (Gemini Bio-Products) and 0.1% saponin (Sigma) for 5 minutes at room temperature and washed once. The azide-alkyne cycloaddition was performed using the Click-iT Cell Reaction Buffer Kit (Life Technologies) and azide conjugated to Alexa Fluor 555 (Thermo Fisher Scientific) at 5 μM final concentration. After 30 minutes, the cells were washed twice in PBS supplemented with 3% fetal bovine serum and 0.1% saponin, then resuspended in PBS supplemented with DAPI (4 μg/ml final concentration) and analyzed by flow cytometry.
Protein extraction and western blot analysis
Equal numbers of bone marrow cells from each mouse were added into trichloroacetic acid (TCA, Sigma-Aldrich) and the TCA concentration was adjusted to 10%. Samples were incubated on ice for 10min, centrifuged at 16,000×g for 15 min, then precipitates were washed twice with cold acetone and dried. Samples were solubilized in 9 M urea, 2% Triton X-100 (Fisher Scientific) and 1% DTT (Sigma-Aldrich). NuPAGE LDS Sample buffer (Life Technologies) was added. Samples were heated at 70°C for 10 min, separated on 4–12% NuPAGE Bis-Tris polyacrylamide gels (Life Technologies), and transferred to 0.2 μm PVDF membranes (BioRad) by wet transfer using NuPAGE transfer buffer (Life Technologies). Western blots were performed using antibodies against GLAST (ACSA-1, Miltenyi Biotech, RRID:AB 2660783), Histone H3 (polyclonal, Abcam, RRID:AB_302613), GOT1 (clone C1, Abcam, Cat. Ab239487), GOT2 (polyclonal, Proteintech, RRID:AB_2247898), HRP-linked anti-rabbit IgG antibody (polyclonal, Cell Signaling Technology, RRID:AB_2099233) and HRP-linked anti-mouse IgG antibody (polyclonal, Cell Signaling Technology, RRID:AB_330924). Signals were detected using SuperSignal West Pico or West Femto chemiluminescence kits (Thermo Scientific). Blots were stripped with Western Blot Stripping Buffer (Thermo Fisher Scientific) before re-probing.
RNA extraction and real time qPCR
5,000–10,000 cells were sorted directly into Trizol LS reagent (Thermo Fisher Scientific). Neurospheres were grown from 6 week-old wild-type mice as described (Mich et al., 2014) and then resuspended in Trizol LS reagent as a positive control for Slc1a3 expression. Total RNA was extracted and reverse transcribed using iScript Reverse Transcription Supermix (Bio-Rad) per the manufacturers’ instructions. Real time PCR was performed using iTaq Universal SYBR Green Supermix (Bio-Rad) using a CFX384 Real-Time PCR machine (Bio-Rad). Primers used for qPCR are listed in Table S4. Transcript levels were normalized to Actin (Actb) using the ΔCt method.
Statistical methods
Data in all figures were obtained in at least three independent experiments as indicated in the figure legends. In isotope tracing experiments cells from three different mice were sorted into culture on one or two different days. Data are shown as mean ± standard deviation. Mice were allocated to experiments randomly and samples processed in an arbitrary order, but formal randomization techniques were not used. No formal blinding was applied when performing the experiments or analyzing the data. Sample sizes were not predetermined based on statistical power calculations but were based on our experience with these assays. No data were excluded. When mice died after transplantation, blood chimerism data that had already been collected were included. The statistical methods are specified in the figure legends.
Before analyzing the statistical significance of differences among treatments, we tested for normality in the data distributions using the D’Agostino-Pearson omnibus test for samples with n >= 20 or the Shapiro-Wilk normality test for smaller samples. We also tested for similar variability among treatments using an F-test (when comparing two samples) or Levene’s median test (when comparing more than two samples). When the data were normally distributed and variability was similar among treatments, we used parametric tests to assess statistical significance. When the data significantly deviated from normality or variability significantly different among treatments, we log2-transformed the data and tested again. If the transformed data no longer significantly deviated from normality and equal variability, we then performed parametric tests on the transformed data. If the transformed data still significantly deviated from normality or equal variability, we performed non-parametric tests on the non-transformed data.
When data or log2-transformed data were normal and equally variable, statistical analyses were performed using Student’s t-tests (when there were two genotypes or treatment conditions), one-way ANOVAs (when there were more than two genotypes or treatment conditions), two-way matched samples ANOVAs (when there were multiple genotypes and multiple cell populations), or mixed effects models (when there were missing values but the data were otherwise consistent with the assumptions for a two-way matched samples ANOVA). When the data or log2-transformed data were normal but unequally variable, statistical analyses were performed using Welch’s t-tests (when there were two genotypes) or Welch’s one-way ANOVAs (when there were more than two genotypes). When the data and log2-transformed data were abnormal or unequally variable, statistical analysis was performed using Mann-Whitney (when there were two genotypes) or Kruskal-Wallis tests (when there were more than two genotypes). P-values from multiple comparisons were adjusted using Tukey’s (when there were three genotypes and all of the comparisons were of interest) or Sidak’s method (when there were more than three genotypes and planned comparisons) after ANOVAs and mixed effects models, Dunnett’s T3 method after Welch’s one-way ANOVAs, or Dunn’s method after Kruskal-Wallis tests. The Benjamini-Hochberg method for multiple comparisons adjustment was used when comparing multiple cell populations in the same tissue among mice with different genotypes, the same metabolite in multiple cell populations among mice with different genotypes, or multiple metabolites between two samples. Fisher’s exact tests were used to assess whether frequencies of progeny deviated from expected mendelian ratios. All statistical tests were two-sided. All data represent mean ± standard deviation. Statistical tests were performed using GraphPad Prism V9.0.1 or R 4.0.2.
The data in Figures 4I, 4J, 6A and 6B were obtained in the same experiments and data from the GLAST-OE control samples were reused in each of these figure panels. The data in Figures 4N, 4O, 7A and 7B were obtained in the same experiments and data from the untreated control samples were reused in each of these figure panels. The data in Figures 4P, 4Q, 7C, 7D, S4N–S4T and S7A–S7G were obtained in the same experiments and data from the untreated control mice were reused in each of these figure panels.
Supplementary Material
Highlights:
Hematopoietic stem cells (HSCs) depend exclusively on the aspartate they synthesize
Aspartate synthesis in HSCs increases during hematopoietic regeneration
HSC function is limited by aspartate availability during hematopoietic regeneration
Aspartate increases HSC function by increasing asparagine and purine synthesis
ACKNOWLEDGMENTS
S.J.M. is a Howard Hughes Medical Institute Investigator, the Mary McDermott Cook Chair in Pediatric Genetics, the Kathryn and Gene Bishop Distinguished Chair in Pediatric Research, the director of the Hamon Laboratory for Stem Cells and Cancer, and a Cancer Prevention and Research Institute of Texas Scholar. L.Q. was supported by a Howard Hughes Medical Institute International Student Research Fellowship (59108352). M.A. was supported by a Cancer Prevention and Research Institute of Texas Scholar grant (RR180007) and an American Society of Hematology Faculty Scholar grant. This work was funded by the National Institute on Aging (R37 AG024945), the National Institute of Diabetes and Digestive and Kidney diseases (R01 DK11875), and the Cancer Prevention and Research Institute of Texas (RP180778). The BioHPC high performance computing cloud at UTSW was used for data analysis and storage as well as metabolomics data analysis (ODA) software deployment. We thank N. Loof and the Moody Foundation Flow Cytometry Facility, L. Zacharias for help with metabolomic analysis and M. Nitcher for help with mouse colony management. We thank Y. Zhang, H. Zhu, M. Nguyen and R. Hammer for Crispr/Cas9 microinjections into mouse embryos.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Agathocleous M, Meacham CE, Burgess RJ, Piskounova E, Zhao Z, Crane GM, Cowin BL, Bruner E, Murphy MM, Chen W, et al. (2017). Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi K, Traver D, Miyamoto T & Weissman IL (2000). A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404, 193–7. [DOI] [PubMed] [Google Scholar]
- Alkan HF, Walter KE, Luengo A, Madreiter-Sokolowski CT, Stryeck S, Lau AN, Al-Zoughbi W, Lewis CA, Thomas CJ, Hoefler G, et al. (2018). Cytosolic Aspartate Availability Determines Cell Survival When Glutamine Is Limiting. Cell Metab 28, 706–720.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S & Huber W (2010). Differential expression analysis for sequence count data. Genome Biol 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansó E, Weinberg SE, Diebold LP, Thompson BJ, Malinge S, Schumacker Paul t., Liu X, Zhang Y, Shao Z, Steadman M, et al. (2017). The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol 19, 614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson MR, Morton RK & Murray AW (1964). Inhibition of adenylosuccinate synthetase and adenylosuccinate lyase from Ehrlich ascites-tumour cells by 6-thioinosine 5’-phosphate. Biochem. J 92, 398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailis W, Shyer JA, Zhao J, Canaveras JCG, Al Khazal FJ, Qu R, Steach HR, Bielecki P, Khan O, Jackson R, et al. (2019). Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 571, 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balazs AB, Fabian AJ, Esmon CT & Mulligan RC (2006). Endothelial protein C receptor (CD201) explicitly identifies hematopoietic stem cells in murine bone marrow. Blood 107, 2317–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baris OR, Klose A, Kloepper JE, Weiland D, Neuhaus JF, Schauen M, Wille A, Müller A, Merkwirth C, Langer T, et al. (2011). The mitochondrial electron transport chain is dispensable for proliferation and differentiation of epidermal progenitor cells. Stem Cells 29, 1459–68. [DOI] [PubMed] [Google Scholar]
- Batool T, Makky EA, Jalal M & Yusoff MM (2016). A Comprehensive Review on L-Asparaginase and Its Applications. Appl. Biochem. Biotechnol 178, 900–23. [DOI] [PubMed] [Google Scholar]
- Bejarano-García JA, Millán-Uclés Á, Rosado IV, Sánchez-Abarca LI, Caballero-Velázquez T, Durán-Galván MJ, Pérez-Simón JA, & Piruat JI (2016). Sensitivity of hematopoietic stem cells to mitochondrial dysfunction by SdhD gene deletion. Cell Death Dis 7, e2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, Zhao J, Tai Y, Tang Y, Zhang Y-Y, et al. (2016). Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Invest 126, 3313–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M & Sabatini DM (2015). An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162, 540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunpo P, Murray B, Cundiff J, Brizius E, Aldrich CJ & Anthony TG (2008). Alanyl-glutamine consumption modifies the suppressive effect of L-asparaginase on lymphocyte populations in mice. J. Nutr 138, 338–43. [DOI] [PubMed] [Google Scholar]
- Chu VT, Weber T, Graf R, Sommermann T, Petsch K, Sack U, Volchkov P, Rajewsky K & Kühn R (2016). Efficient generation of Rosa26 knock-in mice using CRISPR/Cas9 in C57BL/6 zygotes. BMC Biotechnol 16, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ, et al. (2003). Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur. J. Immunol 33, 314–325. [DOI] [PubMed] [Google Scholar]
- Devilbiss AW, Zhao Z, Martin-Sandoval MS, Ubellacker JM, Tasdogan A, Agathocleous M, Mathews TP & Morrison SJ (2021). Metabolomic profiling of rare cell populations isolated by flow cytometry from tissues. eLife 10, e61980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L & Morrison SJ (2013). Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 495, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Saunders TL, Enikolopov G & Morrison SJ (2012). Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores A, Schell J, Krall AS, Jelinek D, Miranda M, Grigorian M, Braas D, White AC, Zhou JL, Graham NA, et al. (2017). Lactate dehydrogenase activity drives hair follicle stem cell activation. Nat. Cell Biol 19, 1017–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bermudez J, Baudrier L, La K, Zhu XG, Fidelin J, Sviderskiy VO, Papagiannakopoulos T, Molina H, Snuderl M, Lewis CA, et al. (2018). Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol 20, 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bermudez J, Prasad S, Baudrier L, Badgley MA, Liu Y, La K, Soula M, Williams RT, Yamaguchi N, Hwang RF, et al. (2021). Adaptive stimulation of macropinocytosis overcomes aspartate limitation in cancer cells under hypoxia. bioRxiv. 2021.02.02.429407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey DI, Kennedy J, Suda T & Zlotnik A (1993). A developmental pathway involving four phenotypically and functionally distinct subsets of CD3-CD4-CD8-triple-negative adult mouse thymocytes defined by CD44 and CD25 expression. J. Immunol 150, 4244–52. [PubMed] [Google Scholar]
- Gu H, Chen C, Hao X, Su N, Huang D, Zou Y, Lin S-H, Chen X, Zheng D, Liu L, et al. (2020). MDH1-mediated malate-aspartate NADH shuttle maintains the activity levels of fetal liver hematopoietic stem cells. Blood 136, 553–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, Davidson SM, Freinkman E, Thomas CJ & Vander Heiden MG (2016). Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab 24, 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guitart AV, Panagopoulou TI, Villacreces A, Vukovic M, Sepulveda C, Allen L, Carter RN, van de Lagemaat LN, Morgan M, Giles P et al. (2017). Fumarate hydratase is a critical metabolic regulator of hematopoietic stem cell functions. J. Exp. Med 214(3), 719–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy RR, Carmack CE, Shinton SA, Kemp JD & Hayakawa K (1991). Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J. Exp. Med 173, 1213–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Nakada D and Morrison SJ (2009). Mechanisms of stem cell self-renewal. Annu. Rev. Cell Dev. Biol 25, 377–406. [DOI] [PubMed] [Google Scholar]
- Hinge A, He J, Bartram J, Javier J, Xu J, Fjellman E, Sesaki H, Li T, Yu J, Wunderlich M, et al. (2020). Asymmetrically Segregated Mitochondria Provide Cellular Memory of Hematopoietic Stem Cell Replicative History and Drive HSC Attrition. Cell Stem Cell 26, 420–430.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope HC, Brownlie RJ, Steele L & Salmond RJ (2020). Coordination of asparagine uptake and asparagine synthetase expression is required for T cell activation. bioRxiv 10.1101/2020.02.28.969774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K & Suda T (2014). Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol 15, 243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz H, Iwashita T, Yilmaz OH, Terhorst C & Morrison SJ (2005). SLAM Family Receptors Distinguish Hematopoietic Stem and Progenitor Cells and Reveal Endothelial Niches for Stem Cells. Cell 121, 1109–1121. [DOI] [PubMed] [Google Scholar]
- Kloepper JE, Baris OR, Reuter K, Kobayashi K, Weiland D, Vidali S, Tobin DJ, Niemann C, Wiesner RJ & Paus R (2015). Mitochondrial Function in Murine Skin Epithelium Is Crucial for Hair Follicle Morphogenesis and Epithelial–Mesenchymal Interactions. J. Invest. Dermatol 135, 679–689. [DOI] [PubMed] [Google Scholar]
- Krall AS, Xu S, Graeber TG, Braas D & Christofk HR (2016). Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun 7, 11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall AS, Mullen PJ, Surjono F, Momcilovic M, Schmid EW, Halbrook CJ, Thambundit A, Mittelman SD, Lyssiotis CA, Shackelford DB, et al. (2021). Asparagine couples mitochondrial respiration to ATF4 activity and tumor growth. Cell Metab (In press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. (2006). Transformation from committed progenitor to leukaemia stem cell initiated by MLL–AF9. Nature 442, 818–822. [DOI] [PubMed] [Google Scholar]
- Kühn R, Schwenk F, Aguet M & Rajewsky K (1995). Inducible gene targeting in mice. Science 269, 1427–9. [DOI] [PubMed] [Google Scholar]
- Liang R, Arif T, Kalmykova S, Kasianov A, Lin M, Menon V, Qiu J, Bernitz JM, Moore K, Lin F, et al. (2020). Restraining Lysosomal Activity Preserves Hematopoietic Stem Cell Quiescence and Potency. Cell Stem Cell 26, 359–376.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Bolteus AJ, Balkin DM, Henschel O & Bordey A (2006). GFAP-expressing cells in the postnatal subventricular zone display a unique glial phenotype intermediate between radial glia and astrocytes. Glia 54, 394–410. [DOI] [PubMed] [Google Scholar]
- Luchsinger LL, De Almeida MJ, Corrigan DJ, Mumau M & Snoeck H-W (2016). Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature 529, 528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier H, Wang-Eckhardt L, Hartmann D, Gieselmann V & Eckhardt M (2015). N-Acetylaspartate Synthase Deficiency Corrects the Myelin Phenotype in a Canavan Disease Mouse Model But Does Not Affect Survival Time. J. Neurosci 35, 14501–14516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansell E, Sigurdsson V, Deltcheva E, Brown J, James C, Miharada K, Soneji S, Larsson J & Enver T (2021). Mitochondrial Potentiation Ameliorates Age-Related Heterogeneity in Hematopoietic Stem Cell Function. Cell Stem Cell 28, 241–256.e6. [DOI] [PubMed] [Google Scholar]
- Maryanovich M, Zaltsman Y, Ruggiero A, Goldman A, Shachnai L, Zaidman SL, Porat Z, Golan K, Lapidot T & Gross A (2015). An MTCH2 pathway repressing mitochondria metabolism regulates haematopoietic stem cell fate. Nat. Commun 6, 7901. [DOI] [PubMed] [Google Scholar]
- Mich JK, Signer RAJ, Nakada D, Pineda A, Burgess RJ, Vue TY, Johnson JE & Morrison SJ (2014). Prospective identification of functionally distinct stem cells and neurosphere-initiating cells in adult mouse forebrain. eLife 3, e02669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti C, Bonora M, Ito K & Ito K (2019). Electron transport chain complex II sustains high mitochondrial membrane potential in hematopoietic stem and progenitor cells. Stem Cell Res 40, 101573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T, Tanaka K, Buffo A, Wurst W, Kühn R & Götz M (2006). Inducible gene deletion in astroglia and radial glia—A valuable tool for functional and lineage analysis. Glia 54, 21–34. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Wright DE & Weissman IL (1997). Cyclophosphamide/granulocyte colony-stimulating factor induces hematopoietic stem cells to proliferate prior to mobilization. Proc. Natl. Acad. Sci. U.S.A 94, 1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura-Ishizu A, Ito K & Suda T (2020). Hematopoietic Stem Cell Metabolism during Development and Aging. Dev. Cell 54, 239–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, et al. (2009). A Branched-Chain Amino Acid-Related Metabolic Signature that Differentiates Obese and Lean Humans and Contributes to Insulin Resistance. Cell Metab 9, 311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro H, Ding L & Morrison SJ (2013). SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell stem cell 13, 102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, Hui S, Ghergurovich JM, Fan J, Intlekofer AM, White RM, Rabinowitz JD, Thompson CB & Zhang J (2018). As Extracellular Glutamine Levels Decline, Asparagine Becomes an Essential Amino Acid. Cell Metab 27, 428–438.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralla S, Peña-Altamira LE, Poeta E, Massenzio F, Virgili M, Barile SN, Sbano L, Profilo E, Corricelli M, Danese A, et al. (2019). Deficiency of Mitochondrial Aspartate-Glutamate Carrier 1 Leads to Oligodendrocyte Precursor Cell Proliferation Defects Both In Vitro and In Vivo. Int. J. Mol. Sci 20, 4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF & Dymecki SM (2000). High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat. Genet 25, 139–40. [DOI] [PubMed] [Google Scholar]
- Ruzzo EK, Capo-Chichi J-M, Ben-Zeev B, Chitayat D, Mao H, Pappas AL, Hitomi Y, Lu Y-F, Yao X, Hamdan FF, et al. (2013). Deficiency of asparagine synthetase causes congenital microcephaly and a progressive form of encephalopathy. Neuron 80, 429–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safer B (1975). The Metabolic Significance of the Malate-Aspartate Cycle in Heart. Circ. Res 37, 527–533. [DOI] [PubMed] [Google Scholar]
- Schell JC, Wisidagama DR, Bensard C, Zhao H, Wei P, Tanner J, Flores A, Mohlman J, Sorensen LK, Earl CS, et al. (2017). Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat. Cell Biol 19, 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiegelow K, Nielsen SN, Frandsen TL & Nersting J (2014). Mercaptopurine/Methotrexate Maintenance Therapy of Childhood Acute Lymphoblastic Leukemia: Clinical Facts and Fiction. J. Pediatr. Hematol. Oncol 36:503–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholar EM, Brown PR & Parks RE (1972). Synergistic Effect of 6-Mercaptopurine and 6-Methylmercaptopurine Ribonucleoside on the Levels of Adenine Nucleotides of Sarcoma 180 Cells. Cancer Res 32, 259–269. [PubMed] [Google Scholar]
- Shapira SN & Christofk HR (2020). Metabolic Regulation of Tissue Stem Cells. Trends Cell Biol 30, 566–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signer R. a. J., Magee JA, Salic A & Morrison SJ (2014). Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signer R. a. J., Qi L, Zhao Z, Thompson D, Sigova AA, Fan ZP, Demartino GN, Young RA, Sonenberg N & Morrison SJ (2016). The rate of protein synthesis in hematopoietic stem cells is limited partly by 4E-BPs. Genes Dev 30:1698–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, Schneider JW, Zhang CC & Sadek HA (2010). The Distinct Metabolic Profile of Hematopoietic Stem Cells Reflects Their Location in a Hypoxic Niche. Cell Stem Cell 7, 380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoeck H-W (2017). Mitochondrial regulation of hematopoietic stem cells. Curr. Opin. Cell Biol 49, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Lu W & Rabinowitz JD (2017). Metabolite Spectral Accuracy on Orbitraps. Anal. Chem 89, 5940–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Takubo K & Semenza G. l. (2011). Metabolic Regulation of Hematopoietic Stem Cells in the Hypoxic Niche. Cell Stem Cell 9, 298–310. [DOI] [PubMed] [Google Scholar]
- Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E & Vander heiden MG (2015). Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Luengo A, Danai LV, Bush LN, Diehl FF, Hosios AM, Lau AN, Elmiligy S, Malstrom S, Lewis CA & Vander Heiden MG (2018). Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol 20, 782–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Nagel R, Zaal EA, Ugalde AP, Han R, Proost N, Song J-Y, Pataskar A, Burylo A, Fu H, et al. (2019). SLC1A3 contributes to L-asparaginase resistance in solid tumors. EMBO J 38, e102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, Goda N, Rahimi Y, Johnson RS, Soga T, et al. (2013). Regulation of Glycolysis by Pdk Functions as a Metabolic Checkpoint for Cell Cycle Quiescence in Hematopoietic Stem Cells. Cell Stem Cell 12, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay BS, Lilley RM, Murray AW & Atkinson MR (1969). Inhibition of phosphoribosyl pyrophosphate amidotransferase from Ehrlich ascites-tumour cells by thiopurine nucleotides. Biochem. Pharmacol 18, 936–938. [DOI] [PubMed] [Google Scholar]
- Van Den Ameele J & Brand AH (2019). Neural stem cell temporal patterning and brain tumour growth rely on oxidative phosphorylation. eLife 8, e47887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gastel N, Spinelli JB, Sharda A, Schajnovitz A, Baryawno N, Rhee C, Oki T, Grace E, Soled HJ, Milosevic J, et al. (2020). Induction of a Timed Metabolic Collapse to Overcome Cancer Chemoresistance. Cell Metab 32, 391–403.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiame E, Tyteca D, Pierrot N, Collard F, Amyere M, Noel G, Desmedt J, Nassogne MC, Vikkula M, Octave JN, et al. (2009). Molecular identification of aspartate N-acetyltransferase and its mutation in hypoacetylaspartia. Biochem. J 425, 127–36. [DOI] [PubMed] [Google Scholar]
- Wilkinson AC, Ishida R, Kikuchi M, Sudo K, Morita M, Crisostomo RV, Yamamoto R, Loh KM, Nakamura Y, Watanabe M, et al. (2019). Long-term ex vivo haematopoietic-stemcell expansion allows nonconditioned transplantation. Nature 571, 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Li G, Li L, Li D, Dong Z & Jiang P (2021). Asparagine enhances LCK signalling to potentiate CD8+ T-cell activation and anti-tumour responses. Nat. Cell Biol 23, 75–86. [DOI] [PubMed] [Google Scholar]
- Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H & Morrison SJ (2006). Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441, 475–82. [DOI] [PubMed] [Google Scholar]
- Zhang F, Pirooznia M & Xu H (2020). Mitochondria regulate intestinal stem cell proliferation and epithelial homeostasis through FOXO. Mol. Biol. Cell 31, 1538–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Fan J, Venneti S, Cross JR, Takagi T, Bhinder B, Djaballah H, Kanai M, Cheng EH, Judkins AR, et al. (2014). Asparagine Plays a Critical Role in Regulating Cellular Adaptation to Glutamine Depletion. Mol. Cell 56, 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.