

Abstract
Background
Ripretinib 150 mg once daily (QD) is indicated for advanced gastrointestinal stromal tumors (GISTs) as at least fourth‐line therapy. In INVICTUS, ripretinib intrapatient dose escalation (IPDE) to 150 mg b.i.d. was allowed after progressive disease (PD) on 150 mg QD by blinded independent central review using modified RECIST 1.1. We report the efficacy and safety of ripretinib IPDE to 150 mg b.i.d. after PD among patients randomized to ripretinib 150 mg QD in the INVICTUS study.
Materials and Methods
Tumor imaging was performed every 28‐day cycle for the first four cycles in the ripretinib 150 mg QD period and then every other cycle, including the 150 mg b.i.d. period. Among the ripretinib IPDE patients, progression‐free survival (PFS)1 was the time from randomization until PD; PFS2 was the time from the first dose of ripretinib 150 mg b.i.d. to PD or death.
Results
Among 43 ripretinib IPDE patients, median PFS1 was 4.6 months (95% confidence interval [CI], 2.7–6.4) and median PFS2 was 3.7 months (95% CI, 3.1–5.3). Median overall survival was 18.4 months (95% CI, 14.5–not estimable). Ripretinib 150 mg b.i.d. (median duration of treatment 3.7 months) was well tolerated with new or worsening grade 3–4 treatment‐emergent adverse events (TEAEs) of anemia in six (14%) and abdominal pain in three (7%) patients. Ripretinib 150 mg b.i.d. was discontinued because of TEAEs in seven (16%) patients.
Conclusion
Ripretinib 150 mg b.i.d. after PD on 150 mg QD may provide additional clinically meaningful benefit with an acceptable safety profile in patients with at least fourth‐line GISTs.
Implications for Practice
Of the 85 patients with advanced gastrointestinal stromal tumor having received at least three prior anticancer therapies randomized to ripretinib 150 mg once daily (QD) in the phase III INVICTUS study, 43 underwent ripretinib intrapatient dose escalation (IPDE) to 150 mg b.i.d. after progressive disease (PD). Median progression‐free survival was 4.6 months before and 3.7 months after ripretinib IPDE. The safety profile of ripretinib 150 mg b.i.d. was acceptable. These findings indicate ripretinib IPDE to 150 mg b.i.d. may provide additional clinical benefit in patients with PD on ripretinib 150 mg QD, for whom limited treatment options exist.
Keywords: Gastrointestinal stromal tumors, Ripretinib, KIT, Platelet‐derived growth factor receptors, Protein‐tyrosine kinases
Short abstract
This article presents further results from the INVICTUS study, focusing on patients who received ripretinib 150 mg QD who received intrapatient dose escalation to 150 mg b.i.d. after progressive disease.
Introduction
Activating mutations in receptor tyrosine kinase proto‐oncogene, receptor tyrosine kinase (KIT), or platelet‐derived growth factor receptor α (PDGFRA) drive altered cell proliferation, differentiation, apoptosis, and survival in most gastrointestinal stromal tumors (GISTs) [1, 2, 3]. Targeting of KIT or PDGFRA with tyrosine‐kinase inhibitors (TKIs) has revolutionized treatment for patients with advanced/metastatic GISTs. Standard therapy for advanced/metastatic GISTs is imatinib (first‐line), sunitinib (second‐line), regorafenib (third‐line), and ripretinib (fourth‐line) [3, 4]. In addition, avapritinib is approved for advanced GISTs with PDGFRA exon 18 mutations [4]. Despite significant improvement in outcomes with TKI therapy in advanced GIST, progressive disease (PD) is inevitable in most patients following approved treatments. Progression largely occurs because of the emergence of secondary mutations in KIT or PDGFRA [5, 6, 7, 8].
Ripretinib, a switch‐control TKI, inhibits a broad range of known primary and secondary KIT and PDGFRA mutations that drive drug resistance by regulating the kinase switch pocket and activation loop [9]. Clinical data suggest the efficacy of ripretinib in advanced GIST as second‐line, third‐line, and at least fourth‐line therapy [10, 11]. In the dose‐escalation phase of the ripretinib phase I study (NCT02571036), the maximum tolerated dose (MTD) was not reached among the doses tested, including ripretinib 200 mg b.i.d. Based on the safety, pharmacokinetic (PK), and pharmacodynamic results of the phase I study, ripretinib 150 mg once daily (QD) was established as the recommended phase II dose (RP2D) [10]. Of note, ripretinib 150 mg b.i.d. in the dose‐escalation phase of the phase I study was well tolerated, without significant dose‐limiting toxicity (DLT) in patients with advanced GISTs.
Given the acceptable safety profile of ripretinib 150 mg b.i.d., patients in the phase III INVICTUS study (NCT03353753) were offered the option of ripretinib intrapatient dose escalation (IPDE) to 150 mg b.i.d. after PD on ripretinib 150 mg QD [11]. The primary analysis of the INVICTUS study has previously been reported [11]. Here, we present results focused on patients randomized to ripretinib 150 mg QD in the INVICTUS study who received ripretinib IPDE to 150 mg b.i.d. after PD assessed by blinded independent central review (BICR).
Materials and Methods
Study Design
INVICTUS was a double‐blind, randomized, placebo‐controlled, phase III study conducted at 29 hospitals in 12 countries across North America, Europe, and the Asia‐Pacific region. Detailed methods have previously been described [11]. Briefly, patients with advanced GISTs who had received at least three prior anticancer therapies were randomly assigned (2:1) to receive either ripretinib 150 mg QD or placebo in 28‐day cycles. Patients, investigators, research staff, and the sponsor study team were masked to the treatment allocation until confirmation of PD assessed by BICR using modified RECIST v1.1 (mRECIST 1.1).
At the time of PD as determined by BICR, patients in the ripretinib arm were offered the options of ripretinib IPDE to 150 mg b.i.d., continue ripretinib 150 mg QD if showing clinical benefit, or discontinue ripretinib. Dose interruptions or modifications of ripretinib due to adverse events (AEs) were permitted at the discretion of the investigator. In patients receiving ripretinib 150 mg QD, the first dose reduction was to 100 mg QD and the second reduction was to 50 mg QD. Patients requiring a dose of <50 mg QD were discontinued from the study. In patients receiving ripretinib 150 mg b.i.d., the first dose reduction was to 100 mg b.i.d. and the second reduction was to 150 mg QD. Patients randomized to placebo who crossed over to ripretinib 150 mg QD and had PD by investigator assessment using mRECIST were given the options of ripretinib IPDE to 150 mg b.i.d., continue ripretinib 150 mg QD if showing clinical benefit, or discontinue ripretinib. The results of ripretinib IPDE to 150 mg b.i.d. among patients in the placebo arm who crossed over to ripretinib are not reported in this article.
The safety and efficacy of patients randomized to ripretinib 150 mg QD in the INVICTUS study who received ripretinib IPDE to 150 mg b.i.d. after PD are presented in this article. This study was conducted in accordance with the Declaration of Helsinki and the International Council for Harmonisation Guidelines for Good Clinical Practice. All patients provided written informed consent to participate in the study. The protocol, protocol amendments, and informed consent documents were approved at each site by the institutional review board or ethics committee.
Participants
Patients 18 years or older with a diagnosis of GIST (with at least one measurable lesion according to mRECIST 1.1) and progression on at least imatinib, sunitinib, and regorafenib, or documented intolerance to any of these treatments despite dose modifications were eligible.
Patients randomized to ripretinib 150 mg QD in the INVICTUS study who underwent dose escalation to ripretinib 150 mg b.i.d. following PD by BICR according to mRECIST 1.1 are referred to as receiving ripretinib IPDE. The decision to initiate ripretinib IPDE after PD on ripretinib 150 mg QD was at the discretion of the investigator.
Procedures
Tumor evaluations were performed using computed tomography scans every 28‐day cycle for the first four cycles in the ripretinib 150 mg QD period and then every other cycle (56 days), including the ripretinib 150 mg b.i.d. period. AEs were monitored continuously from the signing of informed consent to 30 days after the last ripretinib dose. Safety evaluations included all treatment‐emergent adverse events (TEAEs) and dose modifications. To assess the correlation between ripretinib IPDE and AEs, TEAEs in the ripretinib 150 mg b.i.d. period represent new and worsening AEs after IPDE; TEAEs continuing over from the ripretinib 150 mg QD period were not counted.
Statistical Analysis
Progression‐free survival (PFS) after ripretinib IPDE to 150 mg b.i.d. was an exploratory endpoint of the INVICTUS study. Among the patients receiving ripretinib IPDE, PFS1 was the interval between the date of randomization to PD by BICR; PFS2 was the interval between the date of the first dose of 150 mg b.i.d. to PD by BICR or death and analyzed using the Kaplan‐Meier (KM) method. Overall survival (OS) was calculated from the date of randomization to the date of death from any cause. Survival between groups was compared using the KM method and stratified log‐rank test. Hazard ratio (HR) was calculated using a Cox regression model, and the 95% confidence interval (CI) was based on the Wald method. Descriptive statistics were used to describe continuous variables, and discrete variables were summarized using frequencies and percentages. Statistical analyses were done with SAS version 9.4 (SAS Institute, Cary, NC).
Results
Patients
Between Feb 27, 2018, and Nov 16, 2018, 129 of 154 assessed patients were randomized to either ripretinib 150 mg QD (n = 85) or placebo (n = 44). As of Aug 10, 2020, 65 patients randomized to ripretinib 150 mg QD had PD by BICR. Of these, 43 patients received ripretinib IPDE to 150 mg b.i.d., and 22 patients either continued ripretinib 150 mg QD or discontinued study treatment (supplemental online Fig. 1). Baseline characteristics at study entry of patients receiving ripretinib IPDE (n = 43) versus not receiving ripretinib IPDE (n = 22) were similar and are provided in Table 1. At the time of PD while on ripretinib 150 mg QD, patients undergoing ripretinib IPDE had better Eastern Cooperative Oncology Group (ECOG) performance status than those not undergoing ripretinib IPDE (ECOG 0, 51% vs. 18%; ECOG 1, 37% vs. 64%, respectively; supplemental online Table 1). The median duration of treatment with ripretinib 150 mg b.i.d. was 3.7 months (range, 1 day–18.6 months), and 26% (11 of 43 patients) received ripretinib 150 mg b.i.d. for 6 months or longer. PK analysis (n = 33) showed that ripretinib IPDE to 150 mg b.i.d. from 150 mg QD resulted in an approximately twofold increase in the steady‐state trough concentration of ripretinib (supplemental online Fig. 2).
Figure 1.
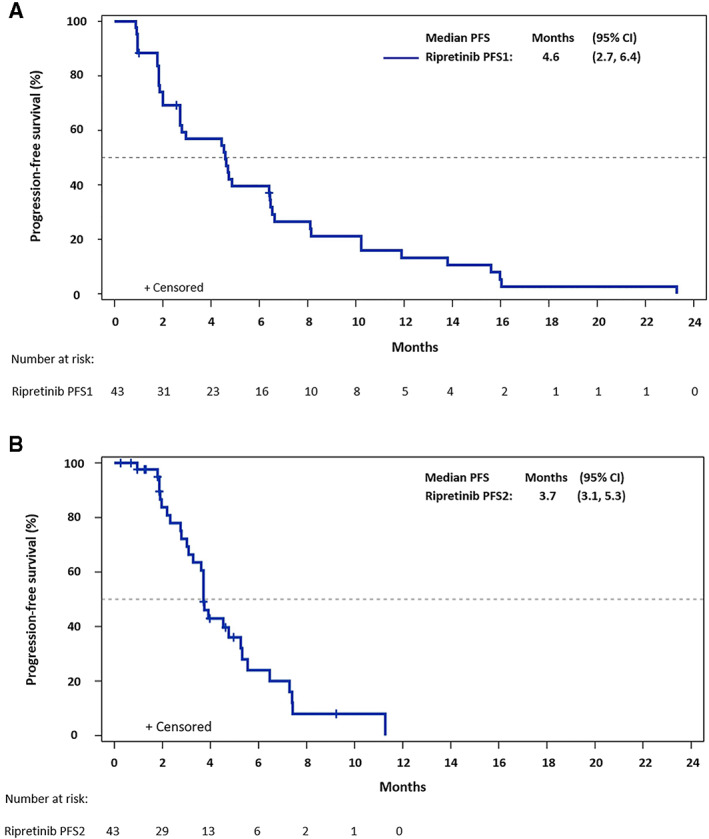
Kaplan‐Meier plot of progression‐free survival among patients receiving ripretinib intrapatient dose escalation (IPDE) to 150 mg b.i.d. (A): PFS in the ripretinib 150 mg once daily (QD) period (PFS1). (B): PFS in the ripretinib 150 mg b.i.d. period (PFS2). Of the 43 ripretinib IPDE patients, 3 with progressive disease during ripretinib 150 mg QD were censored due to new anticancer therapy or surgery/radiation; 10 were censored during ripretinib 150 mg b.i.d. period for multiple end‐of‐treatment reasons. Abbreviations: CI, confidence interval; PFS, progression‐free survival.
Table 1.
Baseline characteristics at study entry of patients with advanced GISTs randomized to ripretinib in INVICTUS
| Characteristics | Patients with PD receiving ripretinib IPDE to 150 mg b.i.d. (n = 43) | Patients with PD not receiving ripretinib IPDE (n = 22) |
|---|---|---|
| Age at study entry, median (range), yr | 59 (36–79) | 57 (40–82) |
| 18–64 | 27 (63) | 18 (82) |
| 65–74 | 12 (28) | 1 (5) |
| ≥75 | 4 (9) | 3 (14) |
| Sex | ||
| Male | 25 (58) | 13 (59) |
| Female | 18 (42) | 9 (41) |
| Race | ||
| White | 35 (81) | 14 (64) |
| Non‐White | 2 (5) | 7 (32) |
| Not reported | 6 (14) | 1 (5) |
| Region | ||
| U.S. | 18 (42) | 10 (45) |
| Non‐U.S. | 25 (58) | 12 (55) |
| ECOG performance status | ||
| 0 | 21 (49) | 8 (36) |
| 1 | 17 (40) | 11 (50) |
| 2 | 5 (12) | 3 (14) |
| Number of previous systemic therapies a | ||
| 3 | 28 (65) | 12 (55) |
| 4–7 | 15 (35) | 10 (45) |
| Median sum of longest diameters of target lesions (range), mm | 111 (46–495) | 123 (21–365) |
| Primary mutation (central testing of tumor tissue) | ||
| KIT exon 11 | 25 (58) | 12 (55) |
| KIT exon 9 | 7 (16) | 3 (14) |
| Other KIT | 1 (2) | 0 |
| PDGFRA | 1 (2) | 2 (9) |
| KIT wild type/PDGFRA wild type | 3 (7) | 2 (9) |
| Not available b or not done c | 6 (14) | 3 (14) |
Data are presented as n (%) unless otherwise noted.
An anticancer therapy in neoadjuvant/adjuvant/first‐line metastatic/rechallenge was all counted as one prior line of therapy.
Tumor tissue analyzed for baseline mutations, but analysis failed.
Biopsy completed per protocol, but sample not received for analysis.
Abbreviations: ECOG, Eastern Cooperative Oncology Group; GIST, gastrointestinal stromal tumor; IPDE, intrapatient dose escalation; PD, progressive disease; PDGFRA, platelet‐derived growth factor receptor α; QD, once daily.
Figure 2.
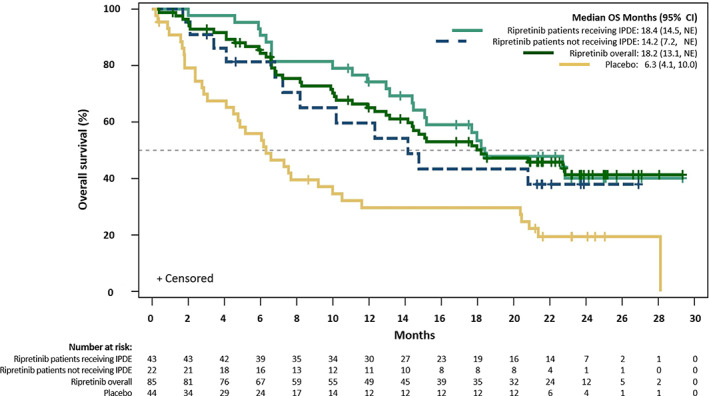
Kaplan‐Meier estimates of overall survival in patients with advanced gastrointestinal stromal tumors in INVICTUS. Abbreviations: CI, confidence interval; IPDE, intrapatient dose escalation; NE, not estimable; OS, overall survival.
Efficacy
As of Aug 10, 2020, among the 43 patients in the ripretinib arm receiving IPDE, the median PFS1 (mPFS1) was 4.6 months (95% CI, 2.7–6.4 months) and median PFS2 (mPFS2) was 3.7 months (95% CI, 3.1–5.3 months) (Fig. 1). The ratio of mPFS2/mPFS1 was 80%. At the time of data cutoff, seven patients who received ripretinib IPDE were still on treatment, including four who showed PD per BICR (supplemental online Fig. 3). Although three patients receiving ripretinib IPDE had a confirmed partial response when receiving 150 mg QD before PD, there were no further responses by mRECIST in the 150 mg b.i.d. period. The utility of continuing ripretinib 150 mg QD after PD could not be assessed given the limited number of patients and multiple reasons for continuing ripretinib 150 mg QD (which were not collected during the study) after PD.
Median OS (mOS) was 18.4 months (95% CI, 14.5–not estimable) in patients randomized to ripretinib 150 mg QD with PD and receiving IPDE to 150 mg b.i.d. (n = 43) and 14.2 months (95% CI, 7.2–not estimable) in those randomized to ripretinib 150 mg QD with PD and not receiving IPDE (n = 22) (HR, 0.74; 95% CI, 0.37–1.49; Fig. 2). Among the intention‐to‐treat population of INVICTUS, mOS was 18.2 months (95% CI, 13.1–not estimable) in the ripretinib group (n = 85) versus 6.3 months (95% CI, 4.1–10 months) in the placebo group (n = 44) (HR 0.42; 95% CI, 0.27–0.67; Fig. 2).
Safety
Ripretinib was well tolerated during both the 150 mg QD and b.i.d. period among the ripretinib IPDE patients. A comparison of the most frequently reported TEAEs (>10% of patients) and the corresponding grade 3–4 TEAEs in the ripretinib IPDE population in the 150 mg QD and b.i.d. period are shown in Table 2. The most common TEAEs of any grade reported as new or worsening in >15% of patients during the ripretinib 150 mg b.i.d. period were abdominal pain, decreased appetite, anemia, nausea, increased blood bilirubin levels, constipation, diarrhea, fatigue, myalgia, and palmar‐plantar erythrodysesthesia syndrome. The most frequent grade 3–4 TEAEs (>5% of patients) during the ripretinib 150 mg b.i.d. period were anemia (6 [14%] of 43 patients) and abdominal pain (3 [7%] patients). Serious TEAEs of grade 3–4 occurring in >4% of patients receiving ripretinib 150 mg b.i.d. were anemia (4 [9%] patients), abdominal pain (2 [5%] patients), and gastrointestinal hemorrhage (2 [5%] patients; supplemental online Table 2).
Table 2.
Treatment‐emergent adverse events reported in >10% of patients with advanced GISTs receiving ripretinib IPDE to 150 mg b.i.d
| Preferred term, n (%) | Ripretinib 150 mg QD period (n = 43) | Ripretinib 150 mg b.i.d. period (n = 43) a | ||
|---|---|---|---|---|
| All grades | Grade 3–4 | All grades | Grade 3–4 | |
| Abdominal pain | 18 (42) | 2 (5) | 13 (30) | 3 (7) |
| Decreased appetite | 13 (30) | 1 (2) | 11 (26) | 2 (5) |
| Anemia | 5 (12) | 1 (2) | 10 (23) | 6 (14) |
| Nausea | 13 (30) | 1 (2) | 10 (23) | 1 (2) |
| Blood bilirubin increased | 8 (19) | 0 | 8 (19) | 0 |
| Constipation | 16 (37) | 0 | 8 (19) | 0 |
| Diarrhea | 11 (26) | 0 | 7 (16) | 0 |
| Fatigue | 19 (44) | 1 (2) | 7 (16) | 2 (5) |
| Myalgia | 15 (35) | 1 (2) | 7 (16) | 0 |
| Palmar‐plantar erythrodysesthesia syndrome | 8 (19) | 0 | 7 (16) | 1 (2) |
| Alopecia | 26 (60) | — | 6 (14) | — |
| Asthenia | 7 (16) | 0 | 6 (14) | 1 (2) |
| Dyspnea | 4 (9) | 0 | 6 (14) | 1 (2) |
| Vomiting | 7 (16) | 1 (2) | 6 (14) | 1 (2) |
| Muscle spasms | 6 (14) | 0 | 5 (12) | 0 |
| Edema peripheral | 7 (16) | 0 | 5 (12) | 0 |
| Weight loss | 9 (21) | 0 | 5 (12) | 0 |
—indicates that no data were captured per adverse event grade ratings.
Data represent new or worsening TEAEs in the ripretinib 150 mg b.i.d. period. The ongoing TEAEs from the ripretinib 150 mg QD period were not included if they remained at the same or lower grade.
Abbreviations: GIST, gastrointestinal stromal tumor; QD, once daily; TEAEs, treatment‐emergent adverse events.
A summary of dose modifications among patients receiving ripretinib IPDE during both ripretinib 150 mg QD and b.i.d. dosing periods are presented in Table 3. In the ripretinib 150 mg QD period, any dose interruption or reduction occurred in 6 (14%) and 2 (5%) patients, respectively. While in the ripretinib 150 mg b.i.d. period, any dose interruption or reduction occurred in 11 (26%) and 8 (19%) patients, respectively. There were 10 TEAEs in seven (16%) patients in the ripretinib 150 mg b.i.d. period leading to treatment discontinuation; three patients each reported one of the following events: grade 3 fatigue (possibly treatment‐related), grade 3 hematemesis (possibly treatment‐related), and grade 1 anemia (treatment‐related). The remaining seven TEAEs were considered unlikely (one) or not related (six) to ripretinib.
Table 3.
Dose modifications occurring in patients with advanced GIST receiving ripretinib IPDE to 150 mg b.i.d
| Parameters, n (%) | Ripretinib 150 mg QD period (n = 43) | Ripretinib 150 mg b.i.d. period (n = 43) a |
|---|---|---|
| Any dose interruption | 6 (14) | 11 (26) |
| Any dose reduction | 2 (5) | 8 (19) |
| Any TEAE leading to treatment discontinuation | N/A | 7 b (16) |
Data only include dose interruption/dose reduction/treatment discontinuation in the ripretinib 150 mg b.i.d. period.
Seven patients had 10 TEAEs leading to treatment discontinuation.
Abbreviations: GIST, gastrointestinal stromal tumor; IPDE, intrapatient dose escalation; N/A, not applicable; QD, once daily; TEAE, treatment‐emergent adverse event.
Discussion
Results of the present analysis from the INVICTUS study showed that in a proportion of patients with advanced GISTs and PD on the ripretinib dose of 150 mg QD, dose escalation to 150 mg b.i.d. was associated with an additional PFS (mPFS2) of 3.7 months. Despite an average doubling of drug exposure, the safety profile of ripretinib 150 mg b.i.d. remained acceptable, and approximately 25% of the patients continued therapy for ≥6 months.
The primary results of the INVICTUS study showed that ripretinib 150 mg QD as at least fourth‐line therapy for patients with advanced GISTs significantly improved mPFS (6.3 months) versus placebo (1 month) [11]. The MTD of ripretinib was not reached in the phase I study among the doses tested. The RP2D of ripretinib 150 mg QD was based on in vitro and in vivo pharmacology studies predicting a threshold of 10,000 ng × h/mL for efficacy for ripretinib and PK analysis confirming an exposure above this threshold in >90% of patients with the ripretinib 150 mg QD dose in the phase I study [10]. Because the starting dose of ripretinib 150 mg b.i.d. was well tolerated without DLT in the phase I study, the efficacy of ripretinib IPDE to 150 mg b.i.d. among patients who progressed on BICR while on ripretinib 150 mg QD was assessed to test whether ripretinib IPDE might provide additional clinical benefit.
Reported treatment options for patients with advanced GIST, independent of the tumor genotype, after the failure of approved TKI therapies include avapritinib, cabozantinib, pazopanib, and imatinib rechallenge [4]. In a recently published analysis of the phase I NAVIGATOR trial, avapritinib as at least fourth‐line therapy for advanced GISTs with KIT or non‐D842V PDGFRA mutations showed a mPFS of 3.7 months, objective response rate (ORR) of 17%, and mOS of 11.6 months [12]. Also, cabozantinib as third‐line therapy showed a mPFS of 5.5 months, ORR of 14%, and mOS of 18.2 months in a single‐arm phase II study [13]. Historically, the mPFS, ORR, and mOS with imatinib rechallenge after the failure of at least two lines of TKI therapy were 1.8 months, 0%, and 8.2 months, respectively [14]. Pazopanib has been evaluated as at least third‐line therapy with an mPFS of 3.4 months, ORR of 0%, and mOS of 17.8 months [15]. In this study, ripretinib IPDE to 150 mg b.i.d. showed an mPFS2 of 3.7 months, ORR of 0%, and mOS of 18.4 months (including the OS in the 150 mg QD period). Of note, the mPFS2 (measured from the first date of ripretinib 150 mg b.i.d.) could potentially be underestimated, given the slight delay in the interval between PD on ripretinib 150 mg QD and initiation of ripretinib IPDE. Notwithstanding the limitations of a direct cross‐study comparison, the additional clinical benefit of ripretinib IPDE observed in this study confirms the results of the phase I study evaluating ripretinib IPDE across multiple lines of therapy and is similar to those previously reported for other TKIs in the heavily pretreated setting [16]. Not achieving objective response with ripretinib IPDE in greater than fourth line of treatment does not preclude a clinical benefit in asymptomatic patients, particularly since objective responses are rare with any agent in this clinical setting. Importantly, both mPFS1 and mPFS2 in patients receiving ripretinib IPDE were longer than the observed mPFS with placebo (4.6 months vs. 3.7 months vs. 1 month), underscoring the clinical benefit of ripretinib at both doses. Because the discontinuation of any TKI therapy is associated with immediate progression and ultimate fatal outcome without additional therapy, ripretinib IPDE for treatment after PD in this subset of patients beyond doubt slows down the progression and may provide a survival benefit. However, it is yet to be determined if reintroduction of a previously tolerated TKI or continuation of the standard dose of ripretinib would have a similar effect.
Despite the dose doubling, ripretinib 150 mg b.i.d. dosing showed an acceptable safety profile in this study. Grade 3–4 anemia and abdominal pain occurred more frequently after ripretinib IPDE. Also, ripretinib dose interruption, reduction, and treatment discontinuation due to TEAEs during ripretinib IPDE occurred in 11, 8, and 7 patients, respectively. The data on AEs and treatment modifications during ripretinib IPDE should be interpreted carefully given the confounding factors of disease progression, subjective clinical decision, and the longer duration of patient follow‐up.
A similar strategy of TKI dose escalation after PD on a lower dose has been evaluated previously with imatinib after first‐line therapy in advanced GISTs [17, 18]. In the EORTC 62005 study, 133 patients crossed over to imatinib 400 mg b.i.d. after PD on 400 mg QD with a median PFS of 2.7 months after crossover. The severity of anemia and fatigue increased significantly in the 400 mg b.i.d. period, and 51% discontinued therapy <6 months after dose escalation, largely because of PD. In the S0033 trial, 118 patients crossed over to imatinib 400 mg b.i.d. with mPFS of 5.0 months after crossover. Based on the findings of the two studies, dose escalation of imatinib to 400 mg b.i.d. is suggested as a reasonable option for patients progressing on imatinib 400 mg QD in the clinical practice guidelines for GISTs [4, 19].
A significant clinical challenge in the management of patients with advanced GISTs is the increasing rate of PD over time despite continued TKI therapy. The most common mechanism of resistance to TKIs is the emergence of secondary mutations in association with the original KIT mutation, which interferes with drug binding of ATP‐binding KIT inhibitors. This resistance is mostly clonal as evidenced by intratumoral nodules that occur in patients with advanced GISTs after initial stabilization and regression of the disease [20]. While preclinical studies indicate potent activity of ripretinib (IC50 in the low nanomolar range) against most secondary resistance mutations, there might be differential activity against some mutations and ripretinib IPDE may be a viable strategy in that setting [9]. Also, additional mutations that independently activate KIT‐downstream signaling such as v‐raf murine sarcoma viral oncogene homolog B1(BRAF) have been reported in pretreated patients with GISTs and may emerge more often following treatment with a broad‐spectrum KIT inhibitor such as ripretinib [21]. Therefore, a higher dose of ripretinib may be effective in inhibiting other kinases such as BRAF [9].
Limitations
This analysis has several limitations. This was an exploratory analysis of a subset of patients receiving ripretinib IPDE in the INVICTUS study. The decision to initiate ripretinib IPDE after PD by BICR on ripretinib 150 mg QD was at the discretion of the investigator. Although tumor measurements by BICR possibly reduced variation in the mRECIST assessment, it led to slight variability in the interval between PD on ripretinib 150 mg QD and initiation of ripretinib IPDE. An additional possibility is the failure of mRECIST, an anatomic tumor response criterion, to identify patients with clinically relevant progression. The mechanism of ripretinib resistance in advanced GISTs remains unknown and additional analyses are needed to address if ripretinib IPDE can be guided based on tumor genotype or other factors. Despite the shortcomings, the results are hypothesis‐generating regarding the role of ripretinib IPDE in the management of PD in patients with advanced GISTs. The additional clinical benefit observed with ripretinib IPDE is important given the high unmet need for treatment options in patients with advanced GISTs refractory to all currently approved TKIs.
Conclusion
These findings from the INVICTUS study suggest that in patients with at least fourth‐line advanced GISTs, ripretinib IPDE to 150 mg b.i.d. after progressive disease on a ripretinib dose of 150 mg QD may provide additional clinical benefit with an acceptable safety profile.
Author Contributions
Conception/design: Michael C. Heinrich, Suzanne George, Sebastian Bauer, Rodrigo Ruiz‐Soto, Margaret von Mehren, Jean‐Yves Blay
Provision of study material or patients: John R. Zalcberg, Michael C. Heinrich, Suzanne George, Sebastian Bauer, Patrick Schöffski, César Serrano, Hans Gelderblom, Robin L. Jones, Steven Attia, Gina D'Amato, Ping Chi, Peter Reichardt, Neeta Somaiah, Margaret von Mehren, Jean‐Yves Blay
Collection and/or assembly of data: John R. Zalcberg, Michael C. Heinrich, Suzanne George, Sebastian Bauer, Patrick Schöffski, César Serrano, Hans Gelderblom, Robin L. Jones, Steven Attia, Gina D'Amato, Ping Chi, Peter Reichardt, Neeta Somaiah, Julie Meade, Vienna Reichert, Kelvin Shi, Matthew L. Sherman, Rodrigo Ruiz‐Soto, Margaret von Mehren, Jean‐Yves Blay
Data analysis and interpretation: John R. Zalcberg, Michael C. Heinrich, Suzanne George, Sebastian Bauer, Patrick Schöffski, César Serrano, Hans Gelderblom, Robin L. Jones, Steven Attia, Gina D'Amato, Ping Chi, Peter Reichardt, Neeta Somaiah, Julie Meade, Vienna Reichert, Kelvin Shi, Matthew L. Sherman, Rodrigo Ruiz‐Soto, Margaret von Mehren, Jean‐Yves Blay
Manuscript writing: John R. Zalcberg, Michael C. Heinrich, Suzanne George, Sebastian Bauer, Patrick Schöffski, César Serrano, Hans Gelderblom, Robin L. Jones, Steven Attia, Gina D'Amato, Ping Chi, Peter Reichardt, Neeta Somaiah, Julie Meade, Vienna Reichert, Kelvin Shi, Matthew L. Sherman, Rodrigo Ruiz‐Soto, Margaret von Mehren, Jean‐Yves Blay
Final approval of manuscript: John R. Zalcberg, Michael C. Heinrich, Suzanne George, Sebastian Bauer, Patrick Schöffski, César Serrano, Hans Gelderblom, Robin L. Jones, Steven Attia, Gina D'Amato, Ping Chi, Peter Reichardt, Neeta Somaiah, Julie Meade, Vienna Reichert, Kelvin Shi, Matthew L. Sherman, Rodrigo Ruiz‐Soto, Margaret von Mehren, Jean‐Yves Blay
Disclosures
John R. Zalcberg: Pfizer, Merck Sharp & Dohme, Merck, Specialized Therapeutics, Targovax, Halozyme, Gilead Sciences, Bayer, Novella (H), Bayer, Merck, Roche, Bristol‐Myers Squibb, Pfizer, AstraZeneca, Specialized Therapeutics, Baxalta/Shire, Eli Lilly & Co., Boehringer‐Ingelheim, Merck Sharp & Dohme (RF), Merck, AstraZeneca, Merck Sharp & Dohme, Deciphera, Sirtex (travel fees), GW Pharmaceuticals, Aimmune, Vertex, Alnylam, BioMarin, Amarin, Freq Therapeutics, Global Blood Therapeutics, Gilead, Uniqure, Sangamo, Acceleron, Orphazyme, Moderna, Myokardia, Myovant, Concert Pharmaceuticals, Madrigal Pharmaceuticals, Zogenix (OI); Michael C. Heinrich: Deciphera Pharmaceuticals, Novartis, Blueprint Medicines, Molecular MD (C/A), Molecular MD (OI), a patent “Treatment of Gastrointestinal Stromal Tumors” licensed to Novartis, a patent “Activating Mutations of PDGFRA” issued (IP); Suzanne George: Blueprint Medicines, Deciphera Pharmaceuticals, Eli Lilly & Co., Bayer, AstraZeneca, Daiichi Sankyo, Exelixis, Alliance for Clinical Trial in Oncology Foundation (C/A), Blueprint Medicines, Deciphera Pharmaceuticals, Bayer, Pfizer, Novartis, Daiichi Sankyo, Springworks, Merck, Eisai (RF), Abbott Labs, Allergan, UpToDate [Wolters Kluwer Health, licensing royalties] (OI); Sebastian Bauer: Deciphera Pharmaceuticals, Blueprint Medicines, ADC Therapeutics, Nanobiotix, Bayer, Exelixis, Daiichi Sankyo, Roche, Eli Lilly & Co., Novartis (SAB), Incyte, Blueprint Medicines, Novartis (RF), Pharmamar (H, travel fees), Eli Lilly & Co., Novartis, Pharmamar (CME); Patrick Schöffski: Deciphera Pharmaceuticals (personal fees), Exelixis, Plexxikon, Eisai, Loxo, Eli Lilly & Co., Blueprint Medicines, Ellipses Pharma, Deciphera Pharmaceuticals, Merck, Servier, Genmab, Adaptimmune, Intellisphere, Transgene (institutional support); César Serrano: Karyopharm, Pfizer, Deciphera Pharmaceuticals, Bayer AG (RF), Immunicum AB, Deciphera Pharmaceuticals, Blueprint Medicines (C/A), Bayer AG, Blueprint Medicines (payment for lectures), PharmaMar, Pfizer, Bayer AG, Novartis, Eli Lilly & Co. (travel fees); Hans Gelderblom: Deciphera Pharmaceuticals (C/A); Robin L. Jones: Adaptimmune Therapeutics, Athenex, Inc.; Steven Attia: Desmoid Tumor Research Foundation (RF), ABScience, TRACON Pharma, CytRx Corporation, Bayer, Novartis, Daiichi Sankyo, Desmoid Tumor Research Foundation, Lilly, Immune Design, Karyopharm Therapeutics, Epizyme, Blueprint Medicines, Genmab, CBA Pharma, Merck, Philogen, Gradalis, Deciphera, Takeda, Incyte, Springworks, Adaptimmune, Advenchen Laboratories, Bavarian Nordic, BTG, PTC Therpaeutics,GlaxoSmithKline, FORMA Therapeutics (RF ‐ institutional); Gina D'Amato: Lilly Pharmaceuticals, Blueprint, Janssen, Deciphera, Epizyme, Diiachi Sankyo (C/A), Lilly, Janssen, Eisai (speaker bureau) Blueprint Medicines, Clinigen Group, Daichii Sankyo, Deciphera Pharmaceuticals, Inc., Eisai, Eli Lilly & Co., Epizyme, Helsinn Healthcare, Immune Design Corporation, Merck, PharmaMar, Tracon Pharmaceuticals, UpToDate (C/A); Ping Chi: Deciphera, Zailab, Novartis, Newbay (C/A), Array Biopharm/Pfizer, Deciphera, Newbay (RF), ORIC Pharmaceuticals, Deciphera (OI, IP), Deciphera (E); Peter Reichardt: Bayer, Clinigen, Roche, Deciphera, Mundibiopharma, Merck Sharp & Dohme, PharmaMar, Blueprint (C/A), PharmaMar, Eli Lilly & Co. (H); Neeta Somaiah: Deciphera, Bayer, Blueprint (C/A), Deciphera, Ascentage, Daiichi Sankyo, AstraZeneca, GlaxoSmithKline, Karyopharm (RF); Julie Meade: Deciphera (E, OI); Vienna Reichert: Deciphera (E, OI); Kelvin Shi: Deciphera (E), Alnylam, Immunogen, Karyopharm, Albireo, Avidity, AstraZeneca, Spectrum (OI); Matthew L. Sherman: Deciphera (E, OI); Rodrigo Ruiz‐Soto: Deciphera Pharmaceuticals (E, OI); Margaret von Mehren: Deciphera, Exelexis, Epizyme (SAB, H), Deciphera, Blueprint Medicines, Exelexis (C/A), ASCO, Blueprint, Deciphera, Gradalis, Genmab, Novartis (RF ‐ institutional); Jean‐Yves Blay: Deciphera, Bayer, Blueprint Medicines (RF), Deciphera, Bayer (H).
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Figure 1 Study flow
Supplemental Figure 2. Plasma trough concentrations of ripretinib, DP‐5439 (active metabolite of ripretinib) and combined (ripretinib + DP‐5439) at ripretinib 150 mg QD (pre‐IPDE) and 150 mg BID (post‐IPDE) among 33 pharmacokinetics evaluable patients receiving ripretinib IPDE to 150 mg BID after PD on 150 mg QD
Supplemental Figure 3. Total duration of ripretinib treatment among patients in the INVICTUS study who received IPDE to 150 mg BID after PD on 150 mg QD
Supplemental Table 1 Patient characteristics at the time of progressive disease while on ripretinib 150 mg QD in INVICTUS
Supplemental Table 2. Serious treatment‐emergent adverse events in >4% of patients receiving ripretinib IPDE to 150 mg BID
Acknowledgments
We thank the patients, their families and caregivers, the investigators, and the investigational site staff of the INVICTUS study. Medical writing was provided by Uma Chandrasekaran, Ph.D. (Deciphera Pharmaceuticals, LLC); editorial support was provided by AlphaBioCom, LLC (King of Prussia, PA, USA) and was funded by Deciphera Pharmaceuticals, LLC in compliance with international good publication practice guidelines.
The INVICTUS study was funded by Deciphera Pharmaceuticals, LLC. Dr. Heinrich received partial salary support from a VA Merit Award Grant (2I01BX000338‐05).
This study was designed by Deciphera Pharmaceuticals, LLC with input from the investigators. Deciphera Pharmaceuticals analyzed the data collected by the investigators and interpreted jointly with all the authors. The authors had access to the data to verify the completeness and accuracy of the data reported and for the adherence of the study to the protocol. The corresponding author had full access to all data in this study and had final responsibility for the decision to submit for publication.
Qualified scientific and medical researchers can make requests for individual participant data that underlie the results reported in this article, after de‐identification, at info@deciphera.com. Proposals for data will be evaluated and approved by Deciphera in its sole discretion. All approved researchers must sign a data access agreement before accessing the data. Data will be available as soon as possible but no later than within 1 year of the acceptance of the article for publication, and for 3 years after article publication. Deciphera will not share data from identified participants or a data dictionary.
Disclosures of potential conflicts of interest may be found at the end of this article.
References
- 1. Hirota S, Isozaki K, Moriyama Y et al. Gain‐of‐function mutations of c‐kit in human gastrointestinal stromal tumors. Science 1998;279:577–580. [DOI] [PubMed] [Google Scholar]
- 2. Heinrich MC, Corless CL, Duensing A et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299:708–710. [DOI] [PubMed] [Google Scholar]
- 3. Lostes‐Bardaji MJ, Garcia‐Illescas D, Valverde C et al. Ripretinib in gastrointestinal stromal tumor: the long‐awaited step forward. Ther Adv Med Oncol 2021;13:1758835920986498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Gastrointestinal Stromal Tumors. Version 1.2021. Plymouth Meeting, PA: National Comprehensive Cancer Center, 2021. https://www.nccn.org/professionals/physician_gls/pdf/gist.pdf. Accessed January 25, 2021. [Google Scholar]
- 5. Heinrich MC, Corless CL, Blanke CD et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol 2006;24:4764–4774. [DOI] [PubMed] [Google Scholar]
- 6. Liegl B, Kepten I, Le C et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol 2008;216:64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Serrano C, Marino‐Enriquez A, Tao DL et al. Complementary activity of tyrosine kinase inhibitors against secondary kit mutations in imatinib‐resistant gastrointestinal stromal tumours. Br J Cancer 2019;120:612–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grunewald S, Klug LR, Muhlenberg T et al. Resistance to avapritinib in PDGFRA‐driven GIST is caused by secondary mutations in the PDGFRA kinase domain. Cancer Discov 2021;11:108–125. [DOI] [PubMed] [Google Scholar]
- 9. Smith BD, Kaufman MD, Lu WP et al. Ripretinib (DCC‐2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug‐resistant KIT and PDGFRA variants. Cancer Cell 2019;35:738–751.e9. [DOI] [PubMed] [Google Scholar]
- 10. Janku F, Abdul Razak AR, Chi P et al. Switch control inhibition of KIT and PDGFRA in patients with advanced gastrointestinal stromal tumor: A phase I study of ripretinib. J Clin Oncol 2020;38:3294–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blay JY, Serrano C, Heinrich MC et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): A double‐blind, randomised, placebo‐controlled, phase 3 trial. Lancet Oncol 2020;21:923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. George S, Jones RL, Bauer S et al. Avapritinib in patients with advanced gastrointestinal stromal tumors following at least three prior lines of therapy. The Oncologist 2021;26:e639–e649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schoffski P, Mir O, Kasper B et al. Activity and safety of the multi‐target tyrosine kinase inhibitor cabozantinib in patients with metastatic gastrointestinal stromal tumour after treatment with imatinib and sunitinib: European Organisation for Research and Treatment of Cancer phase II trial 1317 'CaboGIST'. Eur J Cancer 2020;134:62–74. [DOI] [PubMed] [Google Scholar]
- 14. Kang YK, Ryu MH, Yoo C et al. Resumption of imatinib to control metastatic or unresectable gastrointestinal stromal tumours after failure of imatinib and sunitinib (RIGHT): A randomised, placebo‐controlled, phase 3 trial. Lancet Oncol 2013;14:1175–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mir O, Cropet C, Toulmonde M et al. Pazopanib plus best supportive care versus best supportive care alone in advanced gastrointestinal stromal tumours resistant to imatinib and sunitinib (PAZOGIST): A randomised, multicentre, open‐label phase 2 trial. Lancet Oncol 2016;17:632–641. [DOI] [PubMed] [Google Scholar]
- 16. Janku F, Chi P, Heinrich M et al. Ripretinib intra‐patient dose escalation (IPDE) following disease progression provides clinically meaningful progression‐free survival (PFS) in gastrointestinal stromal tumor (GIST) in phase I study. Ann Oncol 2020;31(suppl 4):S974–S975. [Google Scholar]
- 17. Zalcberg JR, Verweij J, Casali PG et al. Outcome of patients with advanced gastro‐intestinal stromal tumours crossing over to a daily imatinib dose of 800 mg after progression on 400 mg. Eur J Cancer 2005;41:1751–1757. [DOI] [PubMed] [Google Scholar]
- 18. Blanke CD, Rankin C, Demetri GD et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol 2008;26:626–632. [DOI] [PubMed] [Google Scholar]
- 19. Casali PG, Abecassis N, Aro HT et al. Gastrointestinal stromal tumours: ESMO‐EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol 2018;29(suppl 4):iv68–iv78. [DOI] [PubMed] [Google Scholar]
- 20. Wardelmann E, Merkelbach‐Bruse S, Pauls K et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res 2006;12:1743–1749. [DOI] [PubMed] [Google Scholar]
- 21. Muhlenberg T, Ketzer J, Heinrich MC et al. KIT‐dependent and KIT‐independent genomic heterogeneity of resistance in gastrointestinal stromal tumors ‐ TORC1/2 inhibition as salvage strategy. Mol Cancer Ther 2019;18:1985–1996. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Figure 1 Study flow
Supplemental Figure 2. Plasma trough concentrations of ripretinib, DP‐5439 (active metabolite of ripretinib) and combined (ripretinib + DP‐5439) at ripretinib 150 mg QD (pre‐IPDE) and 150 mg BID (post‐IPDE) among 33 pharmacokinetics evaluable patients receiving ripretinib IPDE to 150 mg BID after PD on 150 mg QD
Supplemental Figure 3. Total duration of ripretinib treatment among patients in the INVICTUS study who received IPDE to 150 mg BID after PD on 150 mg QD
Supplemental Table 1 Patient characteristics at the time of progressive disease while on ripretinib 150 mg QD in INVICTUS
Supplemental Table 2. Serious treatment‐emergent adverse events in >4% of patients receiving ripretinib IPDE to 150 mg BID