

Abstract
MicroRNA miR-181a is down-regulated in the kidneys of hypertensive patients and hypertensive mice. In vitro, miR-181a is a posttranslational inhibitor of renin expression, but pleiotropic mechanisms by which miR-181a may influence blood pressure (BP) are unknown. Here we determined whether deletion of miR-181a/b-1 in vivo changes BP and the molecular mechanisms involved at the single-cell level. We developed a knockout mouse model lacking miR-181a/b-1 genes using CRISPR/Cas9 technology. Radio-telemetry probes were implanted in twelve-week-old C57BL/6J wild-type and miR-181a/b-1 knockout mice. Systolic and diastolic BP were 4-5mmHg higher in knockout compared with wild-type mice over 24-hours (P<0.01). Compared with wild-type mice, renal renin was higher in the juxtaglomerular cells of knockout mice. BP was similar in wild-type mice on a high (3.1%) versus low (0.3%) sodium diet (+0.4±0.8mmHg) but knockout mice showed salt sensitivity (+3.3±0.8mmHg, P<0.001). Since microRNAs can target several mRNAs simultaneously, we performed single-nuclei RNA-sequencing in 6,699 renal cells. We identified 12 distinct types of renal cells, all of which had genes that were dysregulated. This included genes involved in renal fibrosis and inflammation such as Stat4, Col4a1, Cd81, Flt3l, Cxcl16, Smad4. We observed up-regulation of pathways related to the immune system, inflammatory response, reactive oxygen species and nerve development, consistent with higher tyrosine hydroxylase in the kidney. In conclusion, downregulation of the miR-181a gene led to increased BP and salt sensitivity in mice. This is likely due to an increase in renin expression in juxtaglomerular cells, as well as microRNA-driven pleiotropic effects impacting renal pathways associated with hypertension.
Keywords: blood pressure, renin, microRNA, single-cell RNA-sequencing, salt, sodium
Introduction
MicroRNAs (miRs) are small non-coding RNAs known for their ability to regulate entire physiological pathways at the post-transcriptional level, usually by binding to the 3’ untranslated region (UTR) of specific messenger RNA (mRNA) molecules.1 MiRs have emerged in clinical medicine as therapeutic drugs and also as biomarkers of pathology.2 In the cardiovascular field, an important role for miRs has been suggested for hypertension,1 heart failure, arrhythmias, and acute coronary syndrome.3 Of particular interest is miR-181a, which can bind to the 3’UTR of renin mRNA and downregulate renin levels in vitro.4 MiR-181a is also downregulated in the kidneys of patients with hypertension compared with those with normal blood pressure (BP),4 and both circulating and renal miR-181a are correlated with levels of BP.5
Similar to humans, the kidneys of genetically hypertensive mice have lower levels of miR-181a and higher levels of renin compared with their normotensive counterparts.6 Moreover, in the human kidney, renin and miR-181a are co-localised in the epithelial cells of the collecting ducts.5 Contrary to juxtaglomerular renin, intratubular renin is not released into the circulation. Rather, it has been shown to act in an autocrine or paracrine manner,7 regulating the intra-renal renin-angiotensin system (RAS). Furthermore, hyperactivity of the intratubular RAS can affect both sodium sensitivity and BP.8 However, due to the pleiotropic capacity of miRs, it is likely that miR-181a also regulates BP via genes other than renin in a tissue as heterogeneous as the kidney. Thus, understanding the precise pathophysiological and molecular roles of miR-181a may provide a novel modality for preventing cardiovascular disease by treating hypertension.
In the current study, we deleted miR-181a/b-1 to determine its effects on BP and sensitivity to salt. To determine miR-181a mechanism of action we investigated the renal transcriptome at the single-cell level in mice. We describe the development of a new CRIPSR/Cas9 miR-181a/b-1 global knockout (KO) model, which has lower but not total absence of miR-181a, to replicate the levels of miR-181a observed in human hypertension.4 We combined physiological and molecular studies in the first report to describe the pleiotropic role of a miR at the single-cell level in control of BP.
Methods
Expanded methodology is available in the online-only supplement.
Animals
All experiments and surgical procedures were approved by the Alfred Medical Research Education Precinct Animal Ethics Committee (E/1617/2016/B) and conducted in accordance with the Australian Code for the Care and Use of Animals for Scientific Purposes, in line with international standards. Experiments were conducted on age-matched C57BL/6J (wild-type, WT, n=31) and homozygous KO (n=33) mice with global deletion of the gene miR-181a/b-1 (but miR-181a/b-2 gene was left intact). Food and water were accessible ad libitum (Chow pellets, “Irradiated Rat and Mouse Cubes” from Specialty Feeds, Glen Forrest, 0.2% sodium).
Salt sensitivity
To determine salt sensitivity, a separate group of WT and miR-181a/b-1 KO mice were fed ad libitum a low (0.03% sodium) or high (3.0% sodium) sodium diet (Specialty feeds, Glen Forrest). Animals were randomly allocated to receive each of the diets in turn for one week. Researchers were not blind to the food due to the need to change and monitor food regularly, as an animal ethics requirement. However, the data were analysed blindly.
Cardiovascular Measurements
Radiotelemetry transmitters (model TA11PA-C10; Data Sciences International, St Paul, MN) were implanted in 12-week old mice, as detailed in the online-only Data Supplement and per our previous studies (n=12 WT, n=15 KO).9, 10 Systolic (SAP), diastolic (DAP) and mean arterial blood pressure (MAP), heart rate (HR) and locomotor activity were measured in conscious, unrestrained mice from 10-days after surgery for a period of 48 hours. To further determine the contribution of the RAS versus the sympathetic nervous system (SNS), we also measured cardiovascular parameters for 30-minutes following administration of pentolinium (5mg/kg; Sigma-Aldrich) to mice pre-treated with enalaprilat (an angiotensin converting enzyme, ACE, inhibitor, at 1mg/kg; Merck & Co.) during the active (dark) and inactive (light) period as previously described.6 Sample size and power calculations were conducted prior to the study. These indicated that for an effect size d=1.67, we would need at least 11 mice per group to detect a difference of 5 mmHg in BP (power=0.95 and α=0.05).
Tyrosine hydroxylase staining
Kidneys were fixed with 10% neutral buffered formalin, dehydrated and embedded in paraffin. Tyrosine hydroxylase (TH) staining was performed on kidney sections from WT (n=6) and miR-181a/b-1 KO (n=5) mice. The percentage of TH staining in the cortical tubules was semi-quantitatively assessed as previously described6, 11 (see online-only Data Supplement).
Measurement and localisation of renin mRNA and protein in the kidney
Localisation of renin mRNA was determined in coronal tissue sections using the RNAScope 2.5 Brown Assay (Advanced Cell Diagnostics, Hayward, CA) according to manufacturer’s instructions. Localisation of renin protein was determined using immunostaining by the peroxidase technique. Briefly, formalin fixed mouse kidney sections (2-3 μm) were processed for renin specific immunolocalization as described previously.12 Renin kidney immunolocalization was assessed using a polyclonal rabbit renin antibody (provided as a gift by Dr. R. Ariel Gomez, University of Virginia, VA-USA) at a 1:200 dilution. Peroxidase activity was visualized with 0.1% 3,3’-diaminobenzidine tetrahydrochloride (DAB, Sigma, St. Louis, MO), followed by counterstaining and mounting with aqueous mounting media (Biomeda, Fisher Scientific). Negative control included omission of primary antibody from the staining reaction. Sections were imaged using bright field microscopy (Olympus BX53 Light Microscope, Japan). A macro (developed by C.J.) was used to identify the amount of renin and renin positive juxtaglomerular cells in the FIJI processing package. All juxtaglomerular apparatus (JGA) visible in the slide were counted (average 185 in WT and 192 in KO mice for renin protein and 163 in WT and 177 in KO for renin mRNA). However, due to technical difficulties and laboratory/border closures the sample size was small (n=2-4 per group), but number of medulla images assessed per sample were high (average 8.8 regions/sample).
Single-nuclei RNA-sequencing
Single-cell suspensions were prepared from the kidneys of male WT (n=4) and miR-181a/b-1 KO (n=4) mice. Mice were euthanised, perfused with PBS and the kidneys isolated. Kidneys were weighed and 0.25g was minced. The same region was selected for consistency, and represented a combination of medulla and cortex. The minced kidney tissue was digested with dispase II and collagenase IV, filtered and resuspended. Antibody staining was performed according to the online-only Data Supplement and live nucleated cells were sorted using fluorescence-activated cell sorting (FACS). Single-nuclei mRNA libraries were prepared per strain using the 10X Genomics pipeline. The Cell Ranger software version 3.1.0 (10X Genomics) was used to process the raw sequencing data before subsequent analyses. Downstream analysis on kidney single-nuclei RNA-sequencing (scRNA-seq) dataset was performed using Seurat R package 3.2.0.13 In order to explore the transcriptional heterogeneity and to undertake cell clustering, dimensionality was reduced using principal component analysis (PCA), selecting 40 PC. PC loadings were used as inputs for a graph-based approach to clustering of cells at a resolution of 1.2, and for t-distributed stochastic neighbour embedding (t-SNE) for two-dimensional visualization purposes. Identified clusters were manually annotated based on features corresponding to canonical cell-type genes before merging clusters corresponding to the same cell type14-16 (Table S1). Differentially expressed (DE) genes were identified, between WT and KO samples, by identifying the genes expressed in at least 10% of cells in at least one of the groups being compared. To test for differential expression, MAST was used including cellular detection rate as a covariate (MASTcpmDetRate). The complete scRNA-seq dataset is available upon request.
Statistical Analysis
All cardiovascular data were analysed using a mixed model, repeated measure (split-plot) analysis of variance (ANOVA) using Microsoft Excel 2016. The main effect of strain was further analysed using a series of non-orthogonal contrasts. A Bonferroni correction was used to correct for multiple comparisons with a Greenhouse-Geisser correction to adjust for violation of sphericity. GraphPad Prism (version 7) package was used for the statistical analyses and graphing of tissue weight, TH, miR-181a and renin expression. Two-tailed unpaired t-tests were used to compare single values from WT and KO mice. All data are presented as mean ± SEM. Two-tailed P<0.05 was considered statistically significant.
Results
Deletion of miR-181a increased blood pressure
The successful deletion of the miR-181a/b-1 gene was achieved using CRISPR-Cas9 genome editing (Figure 1). Mice were genotyped using genomic DNA which was confirmed by PCR. miRNA-181a abundance was 10-fold lower in miR-181a/b-1 KO mice than WT mice, while heterozygous mice had intermediate levels of miR-181a (P<0.001, Figure 1). Mice then underwent a 5-week experimental protocol (Figure 1). On average miR-181a/b-1 KO mice had consistently elevated BP over a 24-hour period (P<0.001, Figure 2). Average 24-hour mean arterial pressure (MAP) was 4.6mmHg higher in miR-181a/b-1 KO mice than WT mice (P<0.01, Figure 2). Similarly, miR-181a/b-1 KO mice had higher 24-hour SAP (mean difference 4.9mmHg, P<0.01) and DAP (mean difference 4.4mmHg, P<0.01) than WT mice (Figure 2). The difference in BP between the two strains was most prominent during the dark (active) period, when miRNA-181a/b-1 KO mice (n=15) had greater MAP (+ 5.6 mmHg, P=0.015), SAP (+6.0 mmHg, P=0.01), and DAP (+5.4 mmHg, P=0.02) than WT mice (n=12) (Figure 2). The MAP, SAP and DAP of KO mice were not significantly different to WT during the light inactive period (P>0.05, Figure 2). MiRNA-181a/b-1 KO mice had a greater heart rate (HR) than WT mice in the dark (active) period (P=0.05). Activity was 33% greater in miR-181a/b-1 KO mice than WT mice during the dark (active) period (+0.5, P=0.015, Figure S1. However, no detectable difference in activity was observed during the light (inactive) period or on average over 24-hours (Figure S1). The depressor response to ACE inhibition (enalaprilat) was similar in the two strains (Figure S2). Further, the depressor response to ganglion blockade by pentolinium, after pre-treatment with enalaprilat, was also similar in WT and KO mice during the dark (active) and light (inactive) periods (Figure S2). Deletion of miR-181a did not significantly affect responses to dirty cage, feeding or restraint stress challenges (Figure S2).
Figure 1. Development and validation of miR-181a knockout.
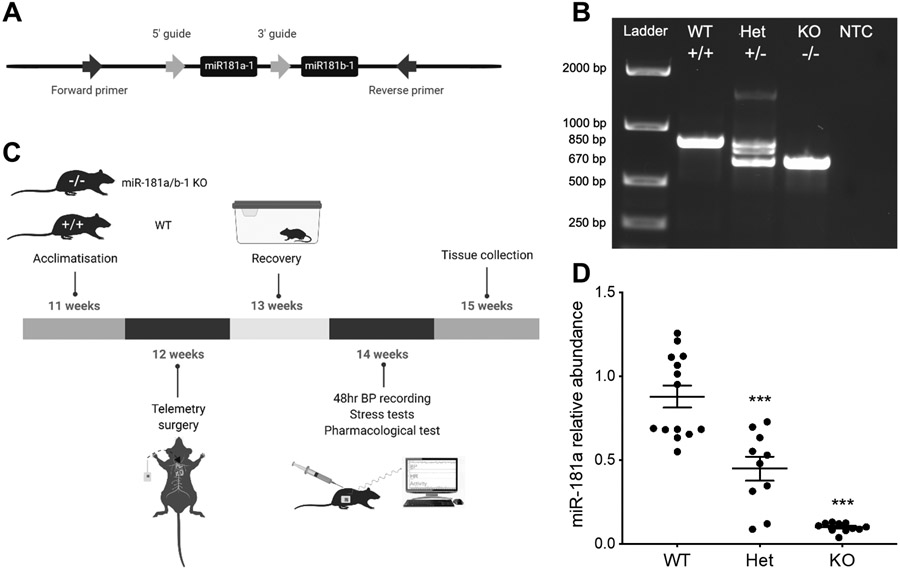
A, Example of genotyping strategy used to isolate the miR-181a-1 and miR-181b-1 loci on the wild type allele. B, Example of an agarose electrophoresis gel used to identify wild-type (WT), heterozygous (Het) and homozygous (KO) miR-181a/b-1 KO mice, including a non-template control (NTC). Genotypes were distinguished based on the presence of 850 base pair (bp) bands (WT) and 670 bp bands (KO). C, Schematic representation of the 5-week experimental protocol timeline. D, MicroRNA-181a relative abundance in WT (n=14), miR-181a/b-1 heterozygous (Het, n=10) and homozygous (KO, n=11) KO mice during the active period. Bars represent average values±SEM. Statistical analysis was conducted using one-way analysis of variance. Comparisons are between strains ***P<0.001 compared to WT.
Figure 2. Blood pressure of miR-181a/b-1 knockout.
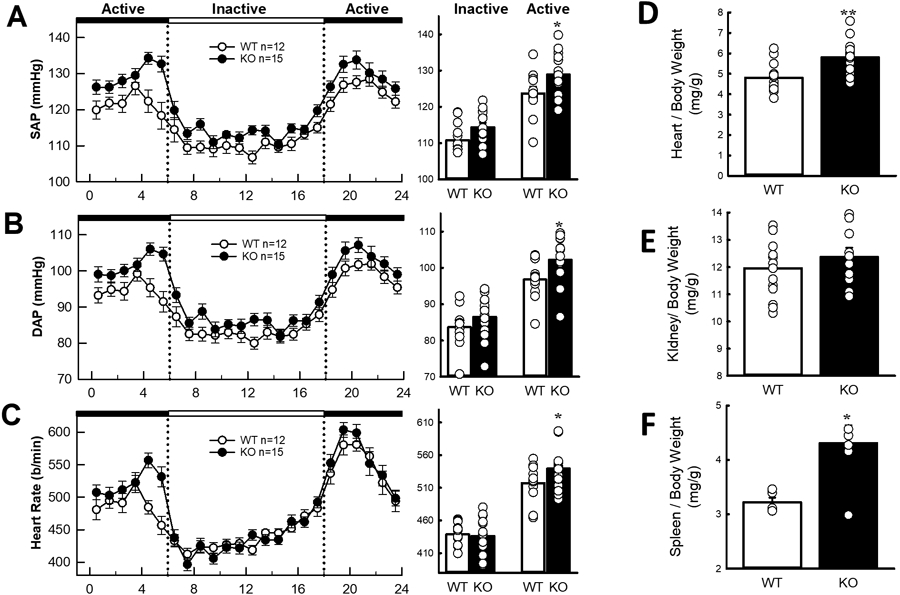
Average 24-hour A, systolic blood pressure (SAP), B, diastolic blood pressure (DAP) and C, heart rate (HR). The dotted lines show the transition from the active (dark, black panels) periods to the inactive (light, white panel) periods. Histograms (right) represent the average SAP, DAP and HR during the inactive and active periods in each mouse strain. D, Heart to body weight ratio (mg/g), E, Kidney to body weight ratio (mg/g), and F, Spleen to body weight ratio (mg/g). All values are mean ± SEM. A-C, statistical analysis was conducted using between groups, split plot analysis of variance with a Bonferroni and Greenhouse Geisser adjustment. D-F, 2 factor ANOVA with post hoc Bonferroni adjusted contrasts two-tailed unpaired t-test. *P < 0.05, **P < 0.01, ***P < 0.001 compared to WT.
Food and water intake did not differ significantly between WT and miR-181a/b-1 KO mice (Figure S3). Cardiac weight index, determined as heart to body weight ratio, was 21% greater in miR-181a/b-1 KO mice than WT mice (P<0.001, Figure 2). There were no detectable differences in (body-weight adjusted) kidney weight index between miR-181a/b-1 KO and WT mice, while KO mice had larger spleens (+34%, P<0.05, Figure 2). TH staining in the kidney, used as a marker of sympathetic innervation, was 2.5-fold greater in miR-18a/b-1 KO mice (5.2±1.1%) than WT mice (2.0±0.3%, P=0.03, Figure 3). Together, our data suggests miR-181a is involved in BP regulation, leads to cardiac hypertrophy, and increases renal sympathetic activity, but is not involved in the pressor responses to aversive stress.
Figure 3. Renal renin and tyrosine hydroxylase in the miR-181a/b-1 knockout.

A, Percentage area of the image positively stained for tyrosine hydroxylase (TH) in WT and miR-181a/b-1 KO mice. B, Representative micrograph showing TH staining (dark brown) in cortical tubules (scale bar=50 μm) of WT and miR-181a/b-1 KO mice. C, Representative images of in situ hybridisation (top images) and immunohistochemistry (bottom images), with renin shown as brown stain (scale bars=20μm). D, Percentage of renin mRNA positive juxtaglomerular apparatus (JGA) and renin protein in the JGA. Bars represent average values ± SEM. Statistical analysis was conducted using one-way analysis of variance. Comparisons are between strains *P<0.05 compared to WT. n=2-5/group.
Deletion of miR-181a increased renal renin
Since miR-181a binds to and down-regulates renin in vitro, it is likely that renin levels are affected. Accordingly, JGA in miR-181a/b-1 KO mice more frequently expressed renin mRNA (mean±SEM: WT 48.9±1.5% vs KO 58.3±2.1%, P=0.02, Figure 3) and protein (WT 32.1±4.4% vs KO 39.0±1.2%, P=0.14, Figure 3) than WT mice. Renin mRNA (WT 4.2±1.0% vs KO 8.7±3.1%, Figure S4) and protein (WT 1.6±0.03% vs KO 3.8±0.4%, P=0.23) tended to be greater in the medulla, but these apparent effects were not statistically significant.
Combined, these data suggest that renal renin, particularly in the JGA, is overexpressed in the miR-181a/b-1 KO mice.
Deletion of miR-181a increased salt sensitivity
WT mice had similar 24-hour MAP when fed a high or low salt diet (mean difference 0.4mmHg, Figure 4). In contrast, miR-181a/b-1 KO mice showed a degree of salt sensitivity (Figure 4). Most notably, DAP was 4.8mmHg greater during the dark (active) period and 3.2mmHg greater during the light (inactive) period in miR-181a/b-1 KO mice fed a high salt diet compared with a low salt diet (P<0.05, Figure 4). Similarly, a high salt diet led to increased SAP during the dark (active, +3.8mmHg, P<0.05) but not during the light (inactive, +2.0mmHg, P=0.08) period, compared with miR-181a/b-1 KO mice fed a low salt diet. There were no detectable differences in urine output, water and food consumption between miR-181a/b-1 KO and WT mice on any of the diets (Figure S3). However, as expected, when fed a high salt diet, both miR-181a/b-1 KO and WT mice had greater water consumption and urine output than when fed a low salt diet (P<0.001, Figure S3). Together, these results indicate that miR-181a/b-1 KO mice are more salt-sensitive than WT mice.
Figure 4. Blood pressure in the miR-181a/b-1 knockout response to salt.
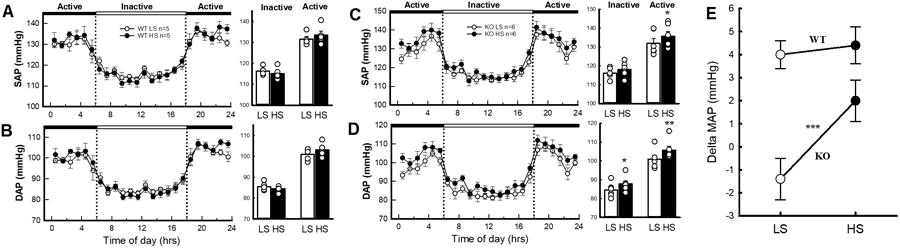
Average 24-hour systolic blood pressure (SAP) and diastolic blood pressure (DAP) in WT A, and B, and miR-181a/b-1 KO mice C, and D, fed low (white) and high (black) salt diets. The dotted lines show the transition from the active (dark, black panels) periods to the inactive (light, white panel) periods. Histograms (right) represent the average SAP and DAP during the inactive and active periods in each strain. E, Average change in mean arterial pressure (MAP) in WT and miR-181a/b-1 KO mice in response to low and high salt diets. Statistical analysis was conducted using between groups, split plot analysis of variance with a Bonferroni and Greenhouse Geisser adjustment *P<0.05.
Single-Nuclei RNA-sequencing
To determine the impact of miR-181a KO on renal cell types, we performed scRNA-seq, on nucleated kidney cells (see Online-only Methods) isolated and pooled from four WT and four miR-181a/b-1 KO mice. scRNA-seq yielded 6,699 cells (3,361 cells sequenced at 139,800 reads/cell for WT and 3,338 cells sequenced at 140,517 reads/cell for miR-181a/b-1 KO mice) that passed quality control in our analysis pipeline (Figure 5A and Figure S5A). Following cell clustering, we identified 12 distinct major cell populations in both WT and miR-181a/b-1 KO strains (Figure 5B) including 22 cell populations that corresponded to macrophages, endothelial cells, juxtaglomerular cells, cells of the proximal tubule (PT), dendritic cells (DC), natural killer cells (NK), T cells and B cells (Figure 5C and S5B, Table S1). Importantly, many of the most highly expressed genes in JGA cells were previously identified as part of human reninomas.17
Figure 5. Isolation and analysis of kidney cells by scRNA-seq.

A, schematic outline of procedure to isolate and analysing cell types from control and miR181a/b-1 KO mouse kidneys. B, t-SNE projection of kidney cell types identified by scRNA-seq. C, Top-five distinct genes for each cell type using an unsupervised approach (please see Table S1 for top 10 gene markers per cell population). Legend: Gran, granulocytes; PT, proximal tubules; NK cells, natural killer cells; DCs, dendritic cells tubule; JGC, juxtaglomerular cells; CD-PT, collecting duct principal cells.
Analysis of gene expression profiles revealed miR-181a/b-1 KO impacts multiple cell populations to varying degrees with macrophages and endothelial cells most impacted (Figure 6A and Table S2). However, it should be noted that sensitivity for discovering differentially expressed genes is reduced for less abundant cell types in our dataset (Figure S6), which is a common phenomenon in scRNA-seq.18 Examination of the top ten up- and down-regulated genes in miR-181a/b-1 KO mice showed that most genes are cell-specifically regulated (Figure 6 and Table S2). Amongst the top up-regulated genes were Stat4 in B cells (fold change=11, expression validated by real-time PCR in Figure S7) which is associated with inflammation,19 collagen genes associated with fibrosis such as Col4a1 (fold change=1.5) in endothelial cells, the marker of renal-resident macrophages Cd8120 (fold change=1.2), Flt3l found in natural killer cells (only expressed in miR-181a/b-1 KO mice) which exacerbates accumulation of T cells in the kidney,21 Malat1 found in macrophages (fold change=1.2) shown to be higher in hypertensive patients, while inhibition of Malat1 lowered BP in an animal model,22 Notch2 in proximal tubules (only expressed in miR-181a/b-1 KO mice) which is involved in nephron development,23 and Polr2f found in proximal tubules (fold change=2.2) previously associated with established hypertension in a meta-analysis.24 Several of the up-regulated genes have been previously demonstrated to have a role in renal injury, inflammation and fibrosis including Cxcl1625 found in distal convoluted tubule cells (fold change=2.1), and Smad4 found in collecting duct principal cells (fold change=1.3).26 The function of most of the down-regulated genes we identified is unknown at least in the context of hypertension and renal function. This is not surprising considering that when miR-181a is downregulated, genes that are directly controlled by this miR would be expected to be upregulated. We still identified a few relevant genes including Ormdl1 in fibroblasts (absent in KO) associated with BP in a genome-wide association study,27 and Tmem27 in proximal tubule cells (fold change=−1.5) which is an ACE2 isoform also involved in nitric oxide synthesis.28
Figure 6. Gene expression changes in miR181a/b-1 KO (knockout) kidney cell populations.

A, Lollipop plot summarizing the number of up- and downregulated genes (uncorrected P<0.01, see Table S2). B, Dot plot summarizing top 10 genes up- or downregulated within kidney cell types of miR181a/b-1 KO mice relative to control mice. Circle size indicates fold change; circle saturation indicates relative expression level (dark, high; clear, low); black dots at the center of circles indicate uncorrected P<0.01. C, Gene ontology pathways dysregulated in miR-181a/b-1 KO (please see Tables S3 and S4 for complete gene ontology). CD-PC indicates collecting duct principal cells; DC, dendritic cell; GO, gene ontology; Gran, granulocyte; JGC, juxtaglomerular cell; NK, natural killer; and PT, proximal tubule.
Gene ontology analyses were then performed to determine pathways altered by miR-181a/b-1 deficiency. Most identified pathways were down-regulated in dendritic cells, endothelial cells, macrophages and juxtaglomerular cells (Figure 6D, Tables S3 and S4). In juxtaglomerular cells, we observed the down-regulation of pathways involved in interleukin-10 (IL10) production, which is anti-inflammatory and limits BP increase.29 Many down-regulated pathways identified in the other cell types were involved in ribosomal units and translation. In endothelial cells, we observed the up-regulation of pathways related to axonogenesis and nerve development, which is consistent with our findings regarding higher levels of TH, which point to higher sympathetic activity. Genes in pathways associated with the activation of the immune system in macrophages, B and T cell populations, and inflammatory response (e.g., interleukin-6 secretion, T cell-mediated cytotoxicity) were also up-regulated. Moreover, there was an up-regulation of pathways involved in the regulation of reactive oxygen species (ROS) metabolic processes in macrophages and B cells. Overall, these data support an effect of miR-181a/b-1 on many mechanisms associated with hypertension including nerve activation, immune system activation, inflammation and ROS.
Discussion
Here we show that a 10-fold reduction in miR-181a/b, through the use of a new CRISPR/Cas9 KO model, resulted in higher BP, cardiac hypertrophy and caused a degree of salt sensitivity. Similar to our findings from kidneys obtained from humans with confirmed hypertension,4 we were able to reduce (but not completely remove) miR-181a expression in this model, while renal renin mRNA and protein appeared increased, particularly in juxtaglomerular cells. These data indicate that miR-181a exerts post-transcriptional control over renal renin expression. Our single-nuclei transcriptomic analyses, however, defined a pleiotropic effect of miR-181a in BP regulation by the kidneys that goes beyond renin regulation. Indeed, miR-181a also impacted pro-inflammatory IL6 and anti-inflammatory IL10 pathways, known to impact sodium handling. 30 Finally, the elevated TH staining observed in the kidneys of miR-181a/b-1 KO mice is consistent with increased nerve development identified by scRNA-seq and may point to a role for the SNS in BP regulation in this model. Together, our results support the hypothesis that deletion of miR-181a leads to elevated BP through multiple classical (RAS, SNS) and emerging (non-coding RNA, pro-inflammatory) mechanistic pathways.
We have previously shown that low levels of renal miR-181a are associated with increased BP in human and pre-clinical models.5, 6 Furthermore, circulating plasma levels of miR-181a were associated with BP in two independent cohorts.5 Likewise, expression of miR-181a in monocytes was found to be negatively correlated with systolic BP in obese patients.31 Others have demonstrated that mice lacking miR-181a exhibit cardiac hypertrophy and cardiac dysfunction.32 These observations and our current data showing cardiac hypertrophy in miR-181a/b-1 KO mice likely reflect the effects of higher BP, since expression of miR-181a in the heart is extremely low (data not shown). Our findings indicate a role of miR-181a in BP regulation in vivo and expand our knowledge about the mechanisms involved at the single-cell level in the kidney. Here, we show for the first time that global deletion of the miR-181a/b-1 genes in vivo results in chronically elevated BP over a 24-hour period and pinpoint several novel mechanisms in the kidney. Thus, we demonstrate that under-expression of miR-181a is not merely a consequence of high BP, but rather a driving factor leading to the development of elevated BP.
In the present study, we found that miR-181a/b-1 KO mice had significantly higher levels of renin in the JGA, but not in the renal medulla. The present study did not rule out the possibility that other post-transcriptional modifications to renin mRNA might be involved. We previously demonstrated in angiotensin (Ang) II-induced hypertension models that renin mRNA and protein in the medulla were differentially regulated from JG renin.31 A possible explanation is that, combined with miR-post-transcriptional regulation, medullary renin control differ from JGA renin.33 In addition to JGA, in the human kidney we previously reported that miR-181a co-localizes with renin in the distal nephron.5 Collecting duct renin is thought to act within the intrarenal RAS in an autocrine or paracrine manner rather than systemically.7, 34 In addition to renin, both ACE and angiotensinogen, components required for the synthesis of Ang II, are present in the lumen of renal tubules. Given that tubular Ang II increases epithelial sodium transport within the collecting duct, it has been suggested that overexpression of renin in this region can promote sodium reabsorption and consequent increases in BP. Indeed, mice which overexpress collecting duct renin showed increased BP and salt sensitivity.8 Conversely, mice with specific deletion of the Ren-1c gene in the collecting duct, or reduced local renin activity due to cell-type specific deficiency of the prorenin receptor (Atp6ap2) gene in collecting duct cells, both elicit attenuated Ang II-induced hypertension and decreased distal sodium reabsorption.35, 36 This hypothesis is supported by the observation in the present study that a high salt diet leads to increased BP in miR-181a/b-1 KO mice but not in WT mice. Further evidence that renin is acting in an autocrine manner is provided by the observed similar depressor response to the ACE inhibitor enalaprilat in WT and miR-181a/b-1 KO mice fed a control diet (Figure S1). Given that enalaprilat inhibits production of circulating Ang II, the equal decrease in MAP in both wild-type and miR-181a/b-1 KO mice suggests that the increased renin, which appears to be in the kidneys of miR-181a/b-1 KO mice, is not released into the circulation. Therefore, it is likely that medullary renin mRNA is promoting greater sodium retention, thus, increasing BP in miR-181a/b-1 KO mice. This also suggests that the increase in BP is not only due to miR-181a actions on renin, but that other targets are important. However, to confirm this hypothesis we would need to measure levels of circulating renin as well as quantify renin in the collecting ducts instead of the whole-medullary tissue, both of which were not possible in the current investigation.
Our previous findings indicate there may be a link between the SNS and miR-181a.6 Indeed, renal denervation lowered BP and normalised expression of miR-181a and renin in Schlager hypertensive (BPH/2J) mice with neurogenic hypertension, but not in normotensive control mice.37 Since BPH/2J mice and miR-181a/b-1 KO mice have lower miR-181a levels, one may expect to see altered sympathetic activity in the KO mice.6 However, on a normal-salt diet, the depressor response to ganglionic blockade in miR-181a/b-1 KO mice was similar to that of WT mice, both in the active and inactive period. Consistent with this, there were no detectable differences in the sympathetically mediated pressor responses to stress tests between WT and miR-181a/b-1 KO mice. Thus, it would appear that the deletion of the miR-181a gene does not affect regulation of the SNS. On the contrary, TH staining, a marker of sympathetic innervation, was greater in miR-181a/b-1 KO mice than WT mice, consistent with our scRNA-seq data. Combined with recent findings showing that renal denervation reduced BP and restored levels of miR-181a in the hypertensive mice,37 this may imply that the SNS has a role, either directly or indirectly, in the regulation of miR-181a.
We also identified juxtaglomerular cells characterised by the expression of Ren1, Myl9, Mgp, Rgs5 and Acta2, previously characterised in human reninomas,17 that had not been previously characterised by scRNA-seq. However, given that miRs can bind to and regulate many mRNA targets, a single miR can have pleiotropic effects on many different biological pathways above and beyond the renin gene Ren1.38 In the Results, we include information about the top genes up- and downregulated, and their previously identified roles in hypertension and cardiovascular disease. Out of these genes, 11 were predicted to be targeted by miR-181a (Table S5). Malat114 and Notch239 have been experimentally validated as having a binding site for miR-181a in their 3’UTR, while Col4a116 was co-regulated with miR-181a. Genes co-expressed with miR-181a were previously determined using next-generation RNA-sequencing.5 Renal miR-181a expression was co-expressed with increased expression of mRNAs common to signalling cascades of immunity and inflammation.5 In the past decade it has been well established that the pathogenesis of hypertension is due, in part, to immune system dysregulation.40 This is relevant as previously miR-181a has been shown to regulate development of T-cells,41, 42 consistent with the increased spleen size we observed. Furthermore, miR-181a has various immune targets including toll-like receptor 4,43 interleukin-1α, interleukin-1β, IL6 and TNFα.44, 45 For the first time, we have studied the hypertensive mouse kidney at a single-cell level, to further elucidate the pleiotropic effects of miR-181a, and validated that it affects some of these pathways including IL6 signalling pathways.
Activation of IL6 signalling in the kidneys could be a result of reactive oxygen species (ROS). The members of the miR-181 family (miR-181a/b/c/d) are considered ‘MitomiRs’, miRs of nuclear origin that can be found in the mitochondria.46, 47 Thus, these MitomiRs could lead to mitochondrial dysfunction, driving an increase in ROS and proinflammatory molecules. miR-181a and miR-181c share a common 3’-terminal motif (UGUCGGUGAGU),32 which may guide their translocation to the mitochondria.5 In human kidneys, miR-181a is inversely expressed with pathways associated with mitochondrial respiratory function.5 In the present study, we identified that miR-181a/b-1 KO mice had downregulated pathways related to mitochondrial respiratory chain complex assembly and organisation, and NADH dehydrogenase complex assembly in endothelial cells, particularly involving the genes Ndufb9, Ndufs5, Ndufa5, Uqcrb, Ndufs8, Ndufs7, Ndufa2, Ndufa6, Ndufa3, Ndufa1, and Cox17. These results support a pleotropic role of miR-181a in the regulation of BP via targeting several mechanisms and cell types in the kidney. Future studies may address how these mechanisms interact, such as miR-181a, renin and mitochondrial dysfunction, and result in higher BP. Future studies should test whether the genes and mechanisms described here, including changes in renin expression, are indeed driving changes to BP and salt-sensitivity in the miR-181a/b-1 KO model. Gene pathways were significant after adjustment for multiple comparisons, while some of the individual genes were not. This may suggest that at the individual gene level miR-181a has a small effect, but additive gene effects still significantly impact entire pathways that could have physiological roles, which is consistent with pleiotropic roles of miRs.
Besides a role in renal function, miR-181a is downregulated in the heart of diabetic patients and rats,48 and in later stages of heart failure in the transverse aortic constriction (TAC) mouse mode.49 miR-181a and miR-181b were upregulated in samples from the coronary sinus of patients with heart failure with those with healthy heart function,50 which may suggest that these miRs are being excreted from the failing heart. Consistent with our findings of the miR-181a/b-1 KO mouse having an enlarged heart, this model also has reduced fractional shortening and ejection fraction,32 particularly after myocardial infarction.51 Moreover, miR-181a was identified as a regulator of the aldosterone-mineralocorticoid receptor (Aldo-MR) pathway, via regulation of lipocalin-2 (LCN2/NGAL).51 Thus, the link between miR-181a and cardiac dysfunction may be via its role in mitochondrial function32 or the Aldo-MR pathway.51
We acknowledge our study has some limitations. Due to gene proximity, both miR-181a-1 and miR-181b-1 genes were deleted in mice. There are, however, two genes that code for mature miR-181a: miR-181a-1 and miR-181a-2.52 The global deletion of miR-181a/b-1 in mice has been found to have a greater impact on primary and mature miR-181a levels, as well as immune driven function, than KO of miR-181a/b-2.53 Given that complete deficiency of miR-181a/b-1 and miR-181a/b-2 in mice does not appear to be compatible with life,32 miR-181a/b-1 is probably the main gene responsible for miR-181a expression. Because low (but not total absence of) expression of miR-181a is observed in the kidneys of human hypertensive patients,4 the miR-181a/b-1 genes were selected for deletion in the current project. MiR-181a/b-1 was previously shown to regulate endothelial dysfunction via TGF-β signalling in vascular smooth muscle cells, leading to higher pulse wave velocity and systolic BP.54 However, human microarray data revealed that, unlike miR-181a, miR-181b is not differentially expressed in human hypertensive kidneys and appears to not regulate renin expression.4 Moreover, using a transient renal-specific miR-181a mimic, we were recently able to lower BP in BPH mice for a short-period.37 Therefore, it is anticipated that the changes in renin expression observed in the miR-181a/b-1 KO mice can be attributed predominantly to the deletion of the miR-181a-1 gene.
Moreover, we acknowledge that in this study we only analysed the BP and transcriptome of male mice. However, given that data previously obtained from two large cohorts of human hypertensive patients showed no sex differences in miR-181a expression,5 we would not expect to see a difference in our model. We also acknowledge that the single-cell RNA-seq data had high prevalence of certain cell populations (e.g. endothelial cells and macrophages) and low representation of others, including kidney-specific cell types (e.g. tubular cells). While the prevalence of different cells types was different from previously described,55 we also detected new cell types (e.g. JGC). This is likely due to the different methodologies for tissue preparation used, or methods to map cells to specific populations. To ensure a high percentage of live cells, we used FACS to sort live and nucleated cells, which is a strength of our protocol.
Supplementary Material
Novelty and Significance.
What Is New?
This is the first report to describe the pleiotropic role of a microRNA at the single-cell level to control blood pressure in vivo.
MicroRNA-181a/b deficiency in mice led to increased blood pressure and blood pressure response to sodium intake.
MicroRNA-181a/b deficiency modulated single-nuclei expression across all renal cell types.
What Is Relevant?
MicroRNA-181a is down-regulated in the kidneys of hypertensive patients and hypertensive mice.
Pleiotropic mechanisms by which microRNA-181a may influence blood pressure remain unknown.
Summary
Downregulation of the microRNA-181a/b gene led to increased blood pressure and salt sensitivity in mice.
This is likely due to an increase in renin expression in juxtaglomerular cells, as well as microRNA-driven pleiotropic effects impacting renal pathways associated with hypertension.
Perspectives.
Our findings demonstrate that deficiency of miR-181a/b via global deletion of miR-181a/b-1 gene leads to elevated BP and some salt sensitivity in mice, and the mechanisms involved at the single-nuclei level in the kidney. Our findings show that a single miRNA can influence a wide array of pathways in several cell types in a tissue as heterogeneous as the kidney, leading to a complex phenotype. While there are currently antihypertensive agents available which target the RAS, understanding how down-regulation of miR-181a can lead to elevated BP and salt sensitivity provides opportunities for the development of novel therapies to prevent, diagnose and treat hypertension.
Acknowledgments
We acknowledge the technical assistance of John-Luis Moretti. We thank Dr. R. Ariel Gomez from University of Virginia for the anti-renin antibody, and the Australian Genome Research Facility (AGRF) for high throughput sequencing, and the support it receives from the Commonwealth.
Sources of Funding
This work was supported by grants (GNT1104528, GNT1065714, GNT1188503) awarded to F.Z.M., R.E., F.J.C., A.R.P, and G.A.H from the National Health & Medical Research Council of Australia (NHMRC), and the National Institutes of Health (NIH-NIDDK, DK104375) and Tulane University Faculty Pilot Program awarded to M.C.P. K.J. and G.A.H. are supported by fellowships from the NHMRC, F.Z.M is supported by a National Heart Foundation Future Leader Fellowship. The Baker Heart & Diabetes Institute is supported in part by the Victorian Government's Operational Infrastructure Support Program.
Footnotes
Disclosures
None.
References
- 1.Marques FZ, Booth SA, Charchar FJ. The emerging role of non-coding rna in essential hypertension and blood pressure regulation. J Hum Hypertens. 2015;29:459–467 [DOI] [PubMed] [Google Scholar]
- 2.Hanna J, Hossain GS, Kocerha J. The potential for microrna therapeutics and clinical research. Front Genet. 2019;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jusic A, Devaux Y, Action EU-CC. Noncoding rnas in hypertension. Hypertension. 2019;74:477–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marques FZ, Campain AE, Tomaszewski M, Zukowska-Szczechowska E, Yang YH, Charchar FJ, Morris BJ. Gene expression profiling reveals renin mrna overexpression in human hypertensive kidneys and a role for micrornas. Hypertension. 2011;58:1093–1098 [DOI] [PubMed] [Google Scholar]
- 5.Marques FZ, Romaine SP, Denniff M, Eales J, Dormer J, Garrelds IM, Wojnar L, Musialik K, Duda-Raszewska B, Kiszka B, Duda M, Morris BJ, Samani NJ, Danser AJ, Bogdanski P, Zukowska-Szczechowska E, Charchar FJ, Tomaszewski M. Signatures of mir-181a on the renal transcriptome and blood pressure. Mol Med. 2015;21:739–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson KL, Marques FZ, Watson AM, Palma-Rigo K, Nguyen-Huu TP, Morris BJ, Charchar FJ, Davern PJ, Head GA. A novel interaction between sympathetic overactivity and aberrant regulation of renin by mir-181a in bph/2j genetically hypertensive mice. Hypertension. 2013;62:775–781 [DOI] [PubMed] [Google Scholar]
- 7.Prieto-Carrasquero MC, Botros FT, Kobori H, Navar LG. Collecting duct renin: A major player in angiotensin ii-dependent hypertension. J Am Soc Hypertens. 2009;3:96–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramkumar N, Ying J, Stuart D, Kohan DE. Overexpression of renin in the collecting duct causes elevated blood pressure. Am J Hypertens. 2013;26:965–972 [DOI] [PubMed] [Google Scholar]
- 9.Davern PJ, Nguyen-Huu TP, La Greca L, Abdelkader A, Head GA. Role of the sympathetic nervous system in schlager genetically hypertensive mice. Hypertension. 2009;54:852–859 [DOI] [PubMed] [Google Scholar]
- 10.Stevenson ER, Johns EM, Marques FZ, Jackson KL, Davern PJ, Evans RG, Head GA. Positive allosteric modulation of gabaa receptors attenuates high blood pressure in schlager hypertensive mice. J Hypertens. 2017;35:546–557 [DOI] [PubMed] [Google Scholar]
- 11.Watson AMD, Gould EAM, Penfold SA, Lambert GW, Pratama PR, Dai A, Gray SP, Head GA, Jandeleit-Dahm KA. Diabetes and hypertension differentially affect renal catecholamines and renal reactive oxygen species. Front Physiol. 2019;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prieto-Carrasquero MC, Harrison-Bernard LM, Kobori H, Ozawa Y, Hering-Smith KS, Hamm LL, Navar LG. Enhancement of collecting duct renin in angiotensin ii-dependent hypertensive rats. Hypertension. 2004;44:223–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen X, He L, Zhao Y, Li Y, Zhang S, Sun K, So K, Chen F, Zhou L, Lu L, Wang L, Zhu X, Bao X, Esteban MA, Nakagawa S, Prasanth KV, Wu Z, Sun H, Wang H. Malat1 regulates myogenic differentiation and muscle regeneration through modulating myod transcriptional activity. Cell Discov. 2017;3:17002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dweep H, Gretz N. Mirwalk2.0: A comprehensive atlas of microrna-target interactions. Nat Methods. 2015;12:697. [DOI] [PubMed] [Google Scholar]
- 16.Tian R, Wang L, Zou H, Song M, Liu L, Zhang H. Role of the xist-mir-181a-col4a1 axis in the development and progression of keratoconus. Mol Vis. 2020;26:1–13 [PMC free article] [PubMed] [Google Scholar]
- 17.Martini AG, Xa LK, Lacombe MJ, Blanchet-Cohen A, Mercure C, Haibe-Kains B, Friesema ECH, van den Meiracker AH, Gross KW, Azizi M, Corvol P, Nguyen G, Reudelhuber TL, Danser AHJ. Transcriptome analysis of human reninomas as an approach to understanding juxtaglomerular cell biology. Hypertension. 2017;69:1145–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLellan MA, Skelly DA, Dona MSI, Squiers GT, Farrugia GE, Gaynor TL, Cohen CD, Pandey R, Diep H, Vinh A, Rosenthal NA, Pinto AR. High-resolution transcriptomic profiling of the heart during chronic stress reveals cellular drivers of cardiac fibrosis and hypertrophy. Circulation. 2020;142:1448–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaplan MH. Stat4: A critical regulator of inflammation in vivo. Immunol Res. 2005;31:231–242 [DOI] [PubMed] [Google Scholar]
- 20.Zimmerman KA, Bentley MR, Lever JM, Li Z, Crossman DK, Song CJ, Liu S, Crowley MR, George JF, Mrug M, Yoder BK. Single-cell rna sequencing identifies candidate renal resident macrophage gene expression signatures across species. J Am Soc Nephrol. 2019;30:767–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu X, Rudemiller NP, Privratsky JR, Ren J, Wen Y, Griffiths R, Crowley SD. Classical dendritic cells mediate hypertension by promoting renal oxidative stress and fluid retention. Hypertension. 2020;75:131–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xue Y-Z, Li Z-J, Liu W-T, Shan J-J, Wang L, Su Q. Down-regulation of lncrna malat1 alleviates vascular lesion and vascular remodeling of rats with hypertension. Aging (Albany NY). 2019;11:5192–5205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng HT, Kim M, Valerius MT, Surendran K, Schuster-Gossler K, Gossler A, McMahon AP, Kopan R. Notch2, but not notch1, is required for proximal fate acquisition in the mammalian nephron. Development. 2007;134:801–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marques FZ, Campain AE, Yang YH, Morris BJ. Meta-analysis of genome-wide gene expression differences in onset and maintenance phases of genetic hypertension. Hypertension. 2010;56:319–324 [DOI] [PubMed] [Google Scholar]
- 25.Liang H, Ma Z, Peng H, He L, Hu Z, Wang Y. Cxcl16 deficiency attenuates renal injury and fibrosis in salt-sensitive hypertension. Sci Rep. 2016;6:28715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meng XM, Huang XR, Xiao J, Chung AC, Qin W, Chen HY, Lan HY. Disruption of smad4 impairs tgf-β/smad3 and smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int. 2012;81:266–279 [DOI] [PubMed] [Google Scholar]
- 27.Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, Glazer NL, Morrison AC, Johnson AD, Aspelund T, Aulchenko Y, Lumley T, Köttgen A, Vasan RS, Rivadeneira F, Eiriksdottir G, Guo X, Arking DE, Mitchell GF, Mattace-Raso FU, Smith AV, Taylor K, Scharpf RB, Hwang SJ, Sijbrands EJ, Bis J, Harris TB, Ganesh SK, O'Donnell CJ, Hofman A, Rotter JI, Coresh J, Benjamin EJ, Uitterlinden AG, Heiss G, Fox CS, Witteman JC, Boerwinkle E, Wang TJ, Gudnason V, Larson MG, Chakravarti A, Psaty BM, van Duijn CM. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41:677–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chu PL, Le TH. Role of collectrin, an ace2 homologue, in blood pressure homeostasis. Curr Hypertens Rep. 2014;16:490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lima VV, Zemse SM, Chiao CW, Bomfim GF, Tostes RC, Clinton Webb R, Giachini FR. Interleukin-10 limits increased blood pressure and vascular rhoa/rho-kinase signaling in angiotensin ii-infused mice. Life Sci. 2016;145:137–143 [DOI] [PubMed] [Google Scholar]
- 30.Norlander AE, Madhur MS. Inflammatory cytokines regulate renal sodium transporters: How, where, and why? Am J Physiol Renal Physiol. 2017;313:F141–F144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hulsmans M, Sinnaeve P, Van der Schueren B, Mathieu C, Janssens S, Holvoet P. Decreased mir-181a expression in monocytes of obese patients is associated with the occurrence of metabolic syndrome and coronary artery disease. J Clin Endocrinol Metab. 2012;97:E1213–1218 [DOI] [PubMed] [Google Scholar]
- 32.Das S, Kohr M, Dunkerly-Eyring B, Lee DI, Bedja D, Kent OA, Leung AK, Henao-Mejia J, Flavell RA, Steenbergen C. Divergent effects of mir-181 family members on myocardial function through protective cytosolic and detrimental mitochondrial microrna targets. J Am Heart Assoc. 2017;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalez AA, Liu L, Lara LS, Seth DM, Navar LG, Prieto MC. Angiotensin ii stimulates renin in inner medullary collecting duct cells via protein kinase c and independent of epithelial sodium channel and mineralocorticoid receptor activity. Hypertension. 2011;57:594–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension. 2011;57:355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prieto-Carrasquero MC, Reverte V, Mamenko M, Kuczeriszka M, Veiras LC, Rosales CB, McLellan M, Gentile O, Jensen VB, Ichihara A, McDonough AA, Pochynyuk OM, Gonzalez AA. Collecting duct prorenin receptor knockout reduces renal function, increases sodium excretion, and mitigates renal responses in ang ii-induced hypertensive mice. Am J Physiol Renal Physiol. 2017;313:F1243–f1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramkumar N, Stuart D, Rees S, Hoek AV, Sigmund CD, Kohan DE. Collecting duct-specific knockout of renin attenuates angiotensin ii-induced hypertension. Am J Physiol Renal Physiol. 2014;307:F931–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson KL, Gueguen C, Lim K, Eikelis N, Stevenson ER, Charchar FJ, Lambert GW, Burke SL, Paterson MR, Marques FZ, Head GA. Neural suppression of mirna-181a in the kidney elevates renin expression and exacerbates hypertension in schlager mice. Hypertens Res. 2020;43:1152–1164 [DOI] [PubMed] [Google Scholar]
- 38.Christopher AF, Kaur RP, Kaur G, Kaur A, Gupta V, Bansal P. Microrna therapeutics: Discovering novel targets and developing specific therapy. Perspect Clin Res. 2016;7:68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang S-X, Zhao Z-Y, Weng G-H, He X-Y, Wu C-J, Fu C-Y, Sui Z-Y, Ma Y-S, Liu T. Upregulation of mir-181a suppresses the formation of glioblastoma stem cells by targeting the notch2 oncogene and correlates with good prognosis in patients with glioblastoma multiforme. Biochem Biophys Res Commun. 2017;486:1129–1136 [DOI] [PubMed] [Google Scholar]
- 40.Drummond GR, Vinh A, Guzik TJ, Sobey CG. Immune mechanisms of hypertension. Nat Rev Immunol. 2019;19:517–532 [DOI] [PubMed] [Google Scholar]
- 41.Kroesen BJ, Teteloshvili N, Smigielska-Czepiel K, Brouwer E, Boots AM, van den Berg A, Kluiver J. Immuno-mirs: Critical regulators of t-cell development, function and ageing. Immunology. 2015;144:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ye Z, Li G, Kim C, Hu B, Jadhav RR, Weyand CM, Goronzy JJ. Regulation of mir-181a expression in t cell aging. Nat Commun. 2018;9:3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Du XJ, Lu JM, Sha Y. Mir-181a inhibits vascular inflammation induced by ox-ldl via targeting tlr4 in human macrophages. J Cell Physiol. 2018;233:6996–7003 [DOI] [PubMed] [Google Scholar]
- 44.Xie W, Li M, Xu N, Lv Q, Huang N, He J, Zhang Y. Mir-181a regulates inflammation responses in monocytes and macrophages. PLOS ONE. 2013;8:e58639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie W, Li Z, Li M, Xu N, Zhang Y. Mir-181a and inflammation: Mirna homeostasis response to inflammatory stimuli in vivo. Biochem Biophys Res Commun. 2013;430:647–652 [DOI] [PubMed] [Google Scholar]
- 46.Macgregor-Das AM, Das S. A microrna's journey to the center of the mitochondria. Am J Physiol Heart Circ Physiol. 2018;315:H206–H215 [DOI] [PubMed] [Google Scholar]
- 47.Jusic A, Devaux Y, Action EU-CC. Mitochondrial noncoding rna-regulatory network in cardiovascular disease. Basic Res Cardiol. 2020;115:23. [DOI] [PubMed] [Google Scholar]
- 48.Raut SK, Singh GB, Rastogi B, Saikia UN, Mittal A, Dogra N, Singh S, Prasad R, Khullar M. Mir-30c and mir-181a synergistically modulate p53-p21 pathway in diabetes induced cardiac hypertrophy. Mol Cell Biochem. 2016;417:191–203 [DOI] [PubMed] [Google Scholar]
- 49.Wang X, Song C, Zhou X, Han X, Li J, Wang Z, Shang H, Liu Y, Cao H. Mitochondria associated microrna expression profiling of heart failure. Biomed Res Int. 2017;2017:4042509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marques FZ, Vizi D, Khammy O, Mariani JA, Kaye DM. The transcardiac gradient of cardio-micrornas in the failing heart. Eur J Heart Fail. 2016;18:1000–1008 [DOI] [PubMed] [Google Scholar]
- 51.Garg A, Foinquinos A, Jung M, Janssen-Peters H, Biss S, Bauersachs J, Gupta SK, Thum T. Mirna-181a is a novel regulator of aldosterone-mineralocorticoid receptor-mediated cardiac remodelling. Eur J Heart Fail. 2020;22:1366–1377 [DOI] [PubMed] [Google Scholar]
- 52.Liu G, Min H, Yue S, Chen C-Z. Pre-mirna loop nucleotides control the distinct activities of mir-181a-1 and mir-181c in early t cell development. PLOS ONE. 2008;3:e3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fragoso R, Mao T, Wang S, Schaffert S, Gong X, Yue S, Luong R, Min H, Yashiro-Ohtani Y, Davis M, Pear W, Chen C-Z. Modulating the strength and threshold of notch oncogenic signals by mir-181a-1/b-1. PLOS Genetics. 2012;8:e1002855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hori D, Dunkerly-Eyring B, Nomura Y, Biswas D, Steppan J, Henao-Mejia J, Adachi H, Santhanam L, Berkowitz DE, Steenbergen C, Flavell RA, Das S. Mir-181b regulates vascular stiffness age dependently in part by regulating tgf-beta signaling. PLoS One. 2017;12:e0174108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, Li M, Barasch J, Susztak K. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science. 2018;360:758–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.