

Abstract
Hi-C is a genome-wide assay based on Chromosome Conformation Capture and high-throughput sequencing to decipher 3D chromatin organization in the nucleus. However, computational methods to detect functional interactions utilizing Hi-C data face challenges including the correction for various sources of biases and the identification of functional interactions with low counts of interacting fragments. We present Chrom-Lasso, a lasso linear regression model that removes complex biases assumption-free and identifies functional interacting loci with increased power by combining information of local reads distribution surrounding the area of interest. We showed that interacting regions identified by Chrom-Lasso are more enriched for 5C validated interactions and functional GWAS hits than that of GOTHiC and Fit-Hi-C. To further demonstrate the ability of Chrom-Lasso to detect interactions of functional importance, we performed time-series Hi-C and RNA-seq during T cell activation and exhaustion. We showed that the dynamic changes in gene expression and chromatin interactions identified by Chrom-Lasso were largely concordant with each other. Finally, we experimentally confirmed Chrom-Lasso’s finding that Erbb3 was co-regulated with distinct neighboring genes at different states during T cell activation. Our results highlight Chrom-Lasso’s utility in detecting weak functional interaction between cis-regulatory elements, such as promoters and enhancers.
Keywords: 3D genomics, Hi-C data analysis, lasso regression, functional chromatin interactions
Introduction
Chromatin interactions are spatial structures that can bring distal regulatory elements to spatial proximity to each other or gene promoters and thereby affect gene transcription [1]. The formation of interactions is precisely regulated and is essential for the normal cellular process [2]. Previous studies demonstrated that transcription factors (TFs), such as estrogen receptors, can induce promoter–enhancer interactions upon binding to enhancers distal to the downstream genes and subsequently activate the expression of these genes [3]. Interactions add a new layer of complexity to the already complex process of transcriptional regulation.
Chromosome Conformation Capture (3C), which involves cross-linking and fragmentation of chromatin followed by PCR amplification, has revolutionized the investigation of chromatin interactions [4]. Hi-C integrates 3C and high-throughput sequencing to enable unbiased profiling of the genome-wide spatial proximity [5]. Sequencing data generated by the Hi-C experiment are composed of hybrid fragments, the two ends of which are mapped to two distant genomic locations. A hybrid fragment is indicative of spatial proximity of the two locations the two ends mapped to and most existing statistical models to identify interactions using Hi-C data are to find a higher-than-expected number of hybrid fragments linking a pair of genomic loci [6, 7].
Many challenges remain in the statistic modeling of Hi-C data. For example, (i) biases from different sources, including the efficiency of endonuclease digestion, the efficiency of ligation in the Hi-C experiment and GC content biases in the subsequent high-throughput sequencing [8]. (ii) The signal-to-noise ratio of the sequencing data is not ideal. Hi-C requires hundreds of millions, in some cases billions of reads to reach saturation in identifying genome-wide interactions, and suffers from low sensitivity when sequencing depth is insufficient [8]. (iii) Currently, the analysis of Hi-C data mostly focuses on the detection of structural interactions such as the interactions between the boundaries of topologically associated domains (TADs) because these interactions are more stable across the entire cell population, therefore, easier to be detected using the Hi-C assay [9]. However, the fine-tuning of gene expression is usually mediated by regulatory elements, such as enhancers through their interactions with gene promoters. Functional interactions between regulatory elements are generally less stable and only exist in a small fraction of cells at a given snapshot [2]. Hybrid fragments linking regulatory elements are therefore less likely to be identified.
Analysis of Hi-C genomic data requires computational methods that can distinguish fragments generated by spatial proximity ligations from random ligations and linear proximity ligations, mitigate complex sources of biases and identify functional interactions with high statistical power. Though a handful of methods exist to detect interactions based on a variety of mathematical models, the comparison among these methods demonstrated that each method has its advantages and disadvantages that may be suitable for distinct tasks [10].
We present Chrom-Lasso, a lasso regression-based model to identify interactions from Hi-C data. Chrom-Lasso corrects the complex confounding factors at each locus using an assumption-free approach based on the number of inter-chromosomal hybrid fragments (hybrid fragments occur between different chromosomes) detected at that locus. Because the vast majority of the inter-chromosomal hybrid fragments are formed by random ligations and any bias at a specific genomic locus can be reflected in the number of inter-chromosome hybrid fragments associated with that locus [11]. Another important feature of Chrom-Lasso is that, instead of testing the number of hybrid fragments in each genomic locus independently, it models the distribution of hybrid fragments surrounding a pair of interacting loci as power-law distribution, in which the expected hybrid fragment decreases as the distance between the location of the fragments and the focal loci increases [12, 13]. Thus, it can utilize not only the fragments within a specific region but also fragments nearby to increase the statistical power of detecting interactions and avoid calling an artificial spike at a single locus as a signal. Chrom-Lasso also takes advantage of the linearity of log-transformed power-law distribution and converts the deconvolution of the complex signals in Hi-C data to a conventional feature selection problem in multiple linear regression, which is subsequently carried out using lasso regression. Last but not least, Chrom-Lasso generates a background P-value distribution using millions of randomly picked genomic loci to calculate false discovery rate (FDR).
We assessed the performance of Chrom-Lasso by comparing its results with that of GOTHiC [7] and Fit-Hi-C [6]. A previous comprehensive evaluation across different Hi-C data analysis methods (HiCCUPS [14], GOTHiC [7], HOMER [15], diffHic [16], HIPPIE [17] and Fit-Hi-C [6]) showed that the interactions identified by GOTHiC were the most reproducible and GOTHiC recovered the largest number of true-positive interactions [10]. Although another popular Hi-C data analysis tool, FitHiC2 [18], added an inter-chromosomal interaction calling function and a merging filter module compared to Fit-Hi-C, the core interaction calling algorithm remains similar. We showed that Chrom-Lasso detected more 5C validated interactions compared to GOTHiC and identified more interactions with potential biological significance than that of GOTHiC and Fit-Hi-C. Moreover, we applied the method to study the dynamics of chromatin interactions during the process of CD8+ T cell activation. The results showed that the changes in chromatin interactions detected by Chrom-Lasso were consistent with the changes in gene expression and with the state of the T cells. We experimentally validated the interactions involved Erbb3, which shared co-regulation with distinct neighboring genes via chromatin loops at the different stages of T cell activation, demonstrating the power of Chrom-Lasso in detecting functional interactions associated with transcriptional regulation.
Materials and methods
The statistical framework of Chrom-Lasso
See Supplementary File: The mathematical specification of the Chrom-Lasso algorithm.
The comparison between Chrom-Lasso and other software
To compare the results between GOTHiC/Fit-Hi-C and Chrom-Lasso, we downloaded the GOTHiC/Fit-Hi-C analysis results of 15 samples of the GM12878 Hi-C dataset ([19], Supplementary Table S1 available online at https://academic.oup.com/bib). We chose GM12878 dataset because it has the highest number of 5C identified intra-chromosomal interactions (interactions occur within the same chromosome), which we took as true-positives events. The significant interactions identified by GOTHiC were selected by FDR < 0.05 and read counts >10, the significant interactions identified by Fit-Hi-C were selected by FDR < 0.05 [10] and the significant interactions identified by Chrom-Lasso were selected by FDR < 0.05 for further analysis. To focus on long-range interactions, we set 20 000 bp as the minimum genomic distance between two interacting loci identified by both GOTHiC/Fit-Hi-C and Chrom-Lasso.
Unlike GOTHiC or Fit-Hi-C, which divides the genome into bins with fixed size and chromatin interactions are defined as two bins with a significantly elevated number of hybrid fragments linking them, Chrom-Lasso outputs a pair of genomic positions (1 bp in length) as the most likely centers of the two interacting regions based on the distribution of the surrounding hybrid fragments. Because the downstream comparison between Chrom-Lasso and GOTHiC/Fit-Hi-C relies heavily on overlapping interaction loci with known functional elements in the genome, for a fair comparison, we expanded the interacting loci identified by Chrom-Lasso to match the bin size of GOTHiC/Fit-Hi-C.
Overview of interactions detected by different methods
We first compared the total number of significant intra-chromosomal interactions detected by the three methods in different samples. The significant intra-chromosomal interactions identified by GOTHiC were selected using the following criteria, FDR < 0.05 and read counts >10. The significant intra-chromosomal interactions identified by Fit-Hi-C and Chrom-Lasso were selected with FDR < 0.05 [10].
Overlapping with 5C cis interactions
We downloaded cis interactions (interactions occur within the same chromosome) identified experimentally via 5C technology in GM12878 cells from a public database [20], and then we set a 2500 bp range for both directions of each interacting loci for Chrom-Lasso results to match the 5000 bp bin size used in GOTHiC/Fit-Hi-C. We considered a Hi-C interaction overlapping with a 5C interaction if both of the interacting regions called from Hi-C had an overlapping segment with interacting regions identified using 5C. When comparing Chrom-Lasso with the other method, we first selected the same number of significant interactions detected by Chrom-Lasso as that detected by the method we are comparing it to under an FDR cutoff of 0.05. We also calculated the proportions of interactions overlap with 5C in all detected interactions by the three methods.
Overlapping with GWAS hits and eQTLs
We came up with three strategies to examine whether the interacting regions found by Hi-C data analysis methods were enriched with functional genomic loci discovered by large-scale population studies. (i) We tested which of the two methods identified more interacting regions overlapped with disease-related single-nucleotide polymorphisms (SNPs) [21]. We chose to test SNPs associated with autoimmune disease, cancer and all kinds of diseases. For finding interacting regions related to disease-associated SNPs, we used GoShifter [22], a tool developed to find enriched SNPs for a given list of genomic loci. (ii) We sought to investigate if the chromatin interactions overlapped with eQTL SNPs, which we downloaded from the GTEx consortium [23–25]. We chose eQTLs for the spleen because GM12878 is a lymphocyte-derived cell line. For the spleen eQTL records, we counted the total number of unique eQTL SNP loci overlapping interacting regions detected by Chrom-Lasso or GOTHiC/Fit-Hi-C in each sample. To focus our analysis on long-range functional interactions, we removed interactions with a distance of less than 20 000 bp. (iii) We counted the number of interactions with both interacting loci located in gene promoter regions. We used the first base of the first exon as the transcription start site (TSS) and the promoter regions are defined as upstream and downstream 1000 bp from the TSS of genes based on the genome ‘gtf’ file from GENCODE [26], we then searched for interactions with both loci associated with promoter regions. When comparing Chrom-Lasso with the other method, we first selected the same number of significant interactions detected by Chrom-Lasso as that detected by the method we are comparing it to under an FDR cutoff of 0.05. We also calculated the proportions of promoter–promoter interactions in all detected interactions by the three methods.
Assessing reproducibility
We calculated the total number of interactions detected in domains based on general human domain files for each replicate in the GM12878 dataset [27] and then calculated the correlation between different samples to evaluate the reproducibility of interaction calling in different methods. We analyzed the GM12878 dataset consisted of 15 samples ([19], Supplementary Table S1 available online at https://academic.oup.com/bib) treated with different restriction endonucleases (MboI or DpnII) to assess the reproducibility of Chrom-Lasso between Hi-C samples treated with the same restriction endonuclease or different restriction endonucleases.
The data analysis of in vitro mouse CD8+ T cell activation model
For analyzing Hi-C data, the preprocessing of raw sequencing data followed the preprocessing protocol of Juicer [14], a tool developed to identify chromatin interactions using Hi-C data. And for further identifying interactions from Hi-C data, we used Chrom-Lasso, and all source code and test data were uploaded to GitHub with a detailed tutorial (see Availability).
To process RNA-seq data, all sequencing data from each sample were aligned to the ‘mm10’ reference genome using the HISAT2 aligner tool [28], and the transformation from raw reads to gene counts was done by HTSeq [29]. The differentially expressed gene analysis was done via R package ‘DESeq2’ [30], and the gene set enrichment analysis (GSEA) [31] was done with R package ‘fgsea’ [32].
Overlapping with ChIP-seq peaks
To evaluate the overlap between interacting regions with TF and histone modification, we first downloaded the ChIP-seq peaks of different TF and histone modifications from the ENCODE project [33, 34]. We then defined a score to evaluate the enrichment of interacting loci overlapping with these ChIP-seq signals. For a specific TF or histone modification, the numerator of the enrichment score was the length of the ChIP-seq regions overlapping interacting regions divided by the total length of interacting regions, and the denominator of enrichment score was the total length of all ChIP-seq regions divided by the length of the whole genome. If the enrichment score for a TF or histone modification in a sample was above 1, we inferred that this functional element was enriched in the interacting regions of this sample; otherwise, the functional element was deemed as depleted in the interacting regions. We used significant interactions (FDR < 0.05) for this analysis.
Analyzing changes in gene expression level
The demonstration of RNA-seq analysis results was the combination of differentially expressed gene analysis and GSEA. For each of the three transitions of cell states during the in vitro mouse CD8+ T cell activation, from naïve T cell (Tn) to short-term activated effector T cell (Teff1), from short-term activated effector T cell (Teff1) to long-term activated effector T cell (Teff2) and from long-term activated effector T cell (Teff2) to exhausted T cell (Tex), we did differentially expressed gene analysis and then ranked all genes according to their log2 fold change, followed by pre-ranked GSEA using the ranked gene list to calculate their enrichment on KEGG pathways [35] associated with the immune system and cell cycle. We finally derived the normalized enrichment score (NES) from the GSEA results for each transition then drew the heat map to show the change of gene expression level of related pathways during each transition.
Analyzing changes in chromatin interactions
To study the changes in the strength of interactions during this process, we proposed a strategy that we counted the total number of interactions related to genes involved in pathways. We analyzed the same KEGG pathways as in RNA-seq data analysis. We defined that interactions with one end anchored within a 5000 bp range centered by the TSS of a gene as interactions associated with promoter regions, here we relaxed the criteria for defining promoters to find more promoter-linked interactions to better illustrate the strength of interactions of a pathway. For each Hi-C sample, we first counted the total number of significant interactions (FDR < 0.05) involved in each pathway gene set, which was the sum of significant interactions associated with all genes in this pathway. Then, we divided the total number of significant interactions associated with this pathway by the total number of significant interactions detected in this sample to assess the strength of interaction for this pathway in this sample.
Screening functional interactions for experimental validation
The selection of interactions for validation was by the following strategy: we first counted the total number of interactions involved in the promoter region of the genes in the four samples, and we preserved genes that were the intersection of genes in Tn, Teff1 and Teff2 as our highly interactive genes. After intensive study of literature, we picked interactions with one end anchored near gene Erbb3, which plays an important role in the regulation of cell proliferation and differentiation [36].
Mice
C57BL/6 mice were purchased from Vital River Laboratories and maintained under specific pathogen-free conditions in the Animal Facility of Tsinghua University. All mice used in the experiments were 8-week-old female mice. All studies were approved by the Animal Care and Use Committee of Tsinghua University.
Isolation and in vitro activation of naïve CD8+ T cell
Naïve CD8+ T cells were isolated from single-cell suspensions of splenocytes using EasySep™ Mouse Naïve CD8+ T Cell Isolation Kit (STEMCELL technologies, 19858) according to the manufacturer’s instructions. Freshly purified naïve CD8+ T cells were stimulated with anti-mouse CD3e (10 μg/ml) (BioLegend, 100314) and anti-mouse CD28 (10 μg/ml) (BioLegend, 102112). Overnight, recombinant mouse IL-2 (PeproTech, AF212-12-20) was added to the culture at 300 U/ml. T cells were used for further experiments after 2 or 5 days of in vitro activation.
Cell sorting
Cells were sorted on a BD influx (BD Biosciences). Single-cell suspensions of freshly purified naïve CD8+ T cells and activated CD8+ T cells were incubated with PBS containing 1% FBS and then stained with the indicated antibodies for 30 min on ice. Staining reagents included FITC anti-CD8 (53-6.7) (BD, 553031), eFluor 450 anti-CD44 (IM7) (eBioscience, 48-0441-82), APC anti- CD62L (MEL-14) (eBioscience, 47-0621-82), APC anti-PD-1 (J43) (BD, 562671) and PE anti-TIM-3 (5D12) (BD, 566346). Dead cells and cell aggregates were excluded from analyses by Fixable Viability Dyes eFluor™ 506 (eBioscience, 65-0866-18) and FSC-H/FSC-A characteristics. Gating criteria were as follows: for Tn: CD8+, CD44−, CD62L+; for Teff1 and Teff2: CD8+, PD-1+, TIM-3−; for Tex: CD8+, PD-1+, TIM-3+.
Hi-C library generation and sequencing
All the procedures are performed as in situ Hi-C protocol with minor modifications [19, 37]. Briefly, 0.2 million T cells from fluorescence-activated cell sorting were fixed with 1% formaldehyde at room temperature (RT) for 10 min. Formaldehyde was quenched with glycine (a final concentration of 0.2 M) for 10 min at RT. Then, T cells were washed once with cold 1× PBS and lysed in 150 μl lysis buffer (10 mM Tris-HCl pH 8.0, 10 mM NaCl, 0.2% Igepal CA630 and proteinase inhibitor) on ice for 50 min. Pelleted nuclei were washed once with 250 μl of ice-cold Hi-C lysis buffer, and the supernatant was discarded carefully with a pipette. Chromatin was solubilized in 25 μl 0.5% SDS and incubated at 62°C for 10 min. 72.5 μl of water and 12.5 μl of 10% Triton X-100 (Sigma, 93443) were added to quench the SDS at 37°C for 20 min. Then, the chromatin was digested with 50 U MboI at 37°C overnight with rotation with a total volume of 125 μl. MboI was then inactivated at 62°C for 20 min. To fill in the restriction fragment overhangs and mark the DNA ends with biotin, 0.75 μl 10 mM dCTP, 0.75 μl 10 mM dGTP, 0.75 μl 10 mM dTTP, 18.75 μl 0.4 mM biotin-14-dATP and 20 U Klenow were added to the solution and the reaction was incubated at 37°C for 1.5 h with rotation. After adding 450 μl ligation mix (331.5 μl water, 60 μl 10× NEB T4 DNA ligase buffer, 50 μl 10% Triton X-100, 6 μl 10 mg/ml BSA and 2.5 μl 400 U/μl T4 DNA ligase), the fragments were ligated at RT for 4 h with rotation. This was followed by a reversal of crosslinking and DNA purification. DNA was sheared to 300–500 bp with Covaris M220. The biotin-labeled DNA was then pulled down with 75 μl Dynabeads M-280 Streptavidin (Thermo Fisher Scientific, 11205D). Sequencing library preparation was performed on beads, including end repair, dATP tailing, adaptor ligation and PCR amplification. Twelve cycles of PCR amplification were performed with Q5® High-Fidelity DNA Polymerase (NEB, M0491S). Finally, size selection was done with AMPure XP beads and fragments ranging from 200 to 1000 bp were selected. All the libraries were sequenced on Illumina HiseqXten-PE150 (Novogene) according to the manufacturer’s instruction.
RNA sequencing library preparation and sequencing
RNA of T cells (0.2 million cells per sample) was extracted using the Monarch Total RNA Miniprep Kit (NEB, T2010S). After quality analysis, mRNA enrichment was carried out with NEBNext Poly(A) mRNA Magnetic Isolation Module kit (NEB, E7490L) and bulk RNA-seq libraries were constructed using NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, E7530L). All experimental procedures followed the manufacturer’s specification. All libraries were sequenced on an Illumina Hiseq Xten-PE150 (Novogene) according to the manufacturer’s instruction.
PCR analysis of the ligation products
Three or four primer pairs were designed to test if the ligation events occurred across five interacting regions in different states of the T cell activation time-course. To improve the sensitivity and accuracy of detection, a nested PCR method was performed. The first-round PCR was performed using 0.5 ng Hi-C library products; 1 of 100 of the first-round PCR product was then used in the second-round PCR amplification. For PCR reactions, each well was mixed with 25 μl NEBNext High-Fidelity 2X PCR Master Mix (NEB, M0541L) and 5 μl of 10 μM corresponding paired primers (Supplementary Tables S2 and S3 available online at https://academic.oup.com/bib). All amplification reactions were carried out using the following thermocycling program: 98°C for 30 s, 25 cycles of (98°C for 10 s, 60°C for 30 s, 72°C for 1 min) and a final 72°C for 5 min. All second-round PCR products were resolved by electrophoresis on 1.5% agarose gels.
Results
A lasso linear regression model to identify interactions from Hi-C data
Existing algorithms for identifying chromatin interactions usually divide the genome into bins with fixed length and build a statistical model to find the pairs of bins having higher than expected numbers of hybrid fragments linking the two bins [6]. Random distribution of background hybrid fragments is usually modeled as negative binomial distribution, Poisson distribution or a mixture of these distributions [7, 11, 38]. The vast majority of these models test individual bins independently using a local or a global background without considering the distribution of the reads of their neighbors. The probability of two genomic loci interacting with each other and joint by the ligase during a Hi-C experiment decreases as the genomic distance between the two loci increases, and it has been shown that the relationship between the probability of interaction and the genomic distance is best described as a power-law distribution [12, 13]. A great property of the power-law distribution is that it can be transformed into a linear relationship by taking the logarithm of both the independent and dependent variables. We took advantage of the special relationship and model the distribution of hybrid fragments surrounding the two interacting loci to explain the ligation probability of the restriction endonuclease cutting sites (RECSs) at various distances to the two interacting loci (Supplementary File, 3 available online at https://academic.oup.com/bib).
In theory, all hybrid fragments are formed by ligating two distant RECSs during the Hi-C experiment. Thus, we mapped each end of the hybrid fragments to its nearby cutting site and discarded hybrid fragments with one end mapped to a genomic locus that is 500 bp away from any consensus RECS. A previous study showed that genomic loci located in different TADs seldom interact with each other [27]. Therefore, to limit the number of statistical tests, instead of testing the number of hybrid fragments linking every possible pairwise genomic locus across the genome against the expected number under the null hypothesis, we only considered pairwise locus within the same TADs and excluded testing the interactions between genomic loci located in different TADs.
The strategy Chrom-Lasso takes to identify interaction can be briefly described as follows (Figure 1). After filtering hybrid fragments produced via self-ligation and random-ligation as introduced in a previous study [39], Chrom-Lasso attempts to remove biases in the data in a single step utilizing the variation in the number of inter-chromosomal hybrid fragments mapping to different RECSs. Due to the low signal-to-noise ratio of the Hi-C experiment and the scarcity of inter-chromosomal interactions, the vast majority of the inter-chromosomal hybrid fragments are formed by random ligations. Thus, the variation in the number of inter-chromosomal hybrid fragments linked to different RECSs can reflect the biases in the Hi-C data generated at various steps of the experiment [11]. A normalization factor for each RECS is calculated based on the number of inter-chromosomal hybrid fragments linked to that RECS. The normalization factors of the two RECSs under test are then multiplied to calculate the random ligation probability between them (Supplementary File, 4 available online at https://academic.oup.com/bib).
Figure 1 .
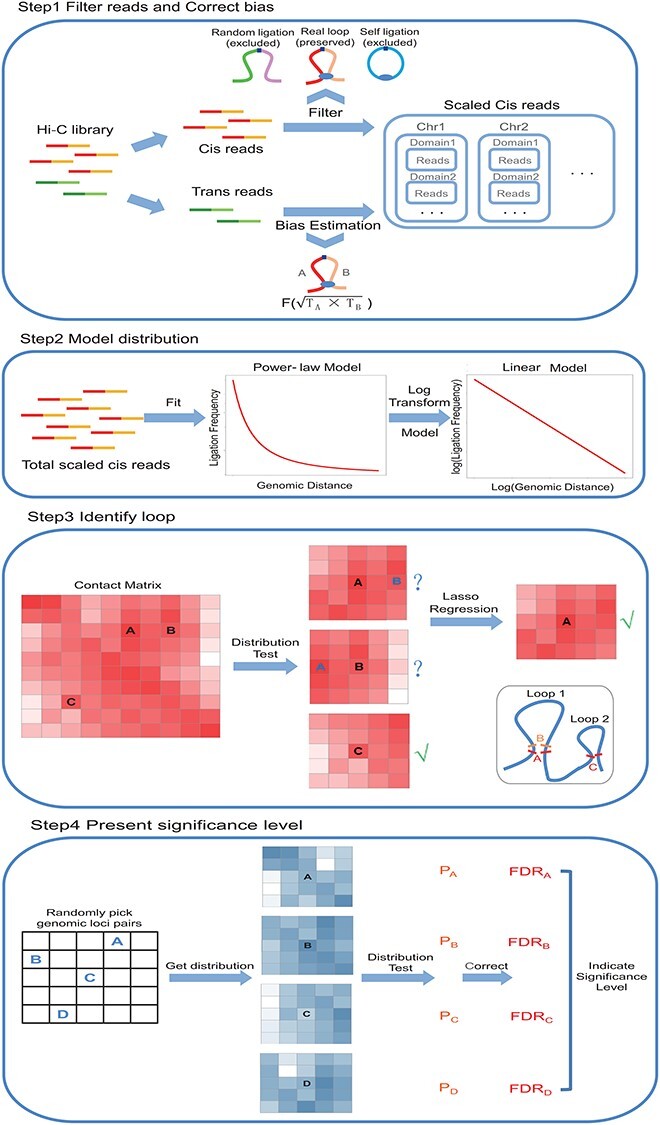
Schematic overview of Chrom-Lasso. The overall analysis strategy of Chrom-Lasso involves four steps. Step1 is to filter the random-ligation reads and self-ligation reads from the Hi-C library and to take the trans (inter-chromosomal) reads as the reflection of biases resulting from the Hi-C experiment and high throughput sequencing to estimate biases for further detecting functional interactions. Step2 focuses on modeling the distribution between ligation frequency and genomic distance which fits linear distribution after log transformation. Step3 detects true chromatin interactions based on testing whether the potential interacting center and its surrounding reads fit the distribution modeled in Step2, and when two or more potential interaction centers are detected within a user-defined neighborhood region (e.g. interaction center A and B), it performs lasso regression to determine the iinteracting center(s) fit the model the best. Step4 is to calculate the FDR for interactions according to a background P-value distribution inferred by randomly picking genomic loci pairs and testing their reads distribution.
The log transformation of the probability density function of the power-law distribution is a linear function. Therefore, if an interaction exists between two genomic loci (B, E) (Supplementary File, 2, figure b available online at https://academic.oup.com/bib), the log transformation of the counts of hybrid fragments linking two nearby genomic loci (C, D) linearly decreases as the log transformation of the summed distance from C, D to the interacting loci B, E increases (Supplementary File, 6 available online at https://academic.oup.com/bib). Instead of testing if the number of hybrid fragments linking to two genomic loci is higher than expected or not given a genome-wide or a local background distribution, Chrom-Lasso defines its null hypothesis as no linear relationship exists between the log-transformed number of hybrid fragments linking two surrounding regions of the interacting loci and the log-transformed distance from the two loci to the interacting loci (Supplementary File, 7 available online at https://academic.oup.com/bib). Accordingly, the alternative hypothesis of Chrom-Lasso is that there exists a linear relationship. The parameters of the linear model are estimated with lasso regression with the ‘L1’ penalty using the R package ‘nnlasso’ [40].
We model the observed log counts of hybrid fragments as the dependent variable and consider it as a mixture of hybrid fragments generated from ligations of all pairs of potentially interacting loci within a certain region. The testing regions are centered on one pair of potentially interacting loci and extended by five restriction cutting sites in both directions. The expected distribution of hybrid fragments of two pairs of closely positioned and potentially interacting loci can be highly correlated; thus, the parameters can be difficult to estimate if regular linear regression is used. Lasso regression can help select the true interacting centers among the closely positioned potentially interacting loci for the reason that it penalizes the sum of the absolute value of the coefficients of all pairs of potentially interacting loci (Supplementary File, 8 available online at https://academic.oup.com/bib). As a result of its modeling strategy, Chrom-Lasso can achieve higher accuracy by utilizing information of the surrounding regions of potentially interacting loci in contrast with testing each pair of loci independently. Another advantage of Chrom-Lasso is that the estimated coefficient for each pair of interacting loci can be interpreted as the proportion of cells having the interaction at the snapshot of the experiment. Finally, Chrom-Lasso calculates the FDR for the potential interacting loci based on the distribution of P-values generated by fitting random pairs of genomic loci to the linear model (Supplementary File, 9 available online at https://academic.oup.com/bib).
Chrom-Lasso identified more functional interactions than existing software
GOTHiC is a Hi-C data analysis method based on a binomial probabilistic model that resolves different sources of biases and identifies true interactions [7]. And it also applies the ratio of observed-over-expected counts to measure the strength of the interaction. This model takes no consideration of genomic distance and yields a well-controlled FDR. Fit-Hi-C computes confidence estimates for Hi-C data by capturing the relationship between genomic distance and ligation probability without any parametric assumption [6]. Another newly developed method HiC-DC [41] calls a significant overlap of long-range contacts with Fit-Hi-C [18].
A previous study quantitatively compared the performance of six algorithms to detect interactions using Hi-C data and showed that GOTHiC found the most cis interactions in the majority of the tested datasets. The same study also reported that GOTHiC called more reproducible interactions and recovered the highest number of true-positive interactions [10]. Based on the results of significant intra-chromosomal interactions (FDR < 0.05) detected by different methods from this study, we noticed that Chrom-Lasso called a comparable number of interactions with GOTHiC and significantly more than Fit-Hi-C (Figure 2A). We visualized the significant interactions detected by different methods in the same chromosomal region, showing that Chrom-Lasso detects interactions anchored at RECS map without the bin size limitation (Supplementary Figure S1 available online at https://academic.oup.com/bib). Considering GOTHiC and Fit-Hi-C’s overall performance compared to other existing methods, we decided to compare our method, Chrom-Lasso, to GOTHiC and Fit-Hi-C to assess its performance. The comparison was made using the publicly available data of in situ Hi-C assay in the GM12878 cells ([19], Supplementary Table S1 available online at https://academic.oup.com/bib). To make a fair comparison, we focused on long-range interactions with a distance between the two interacting loci above 20 kb and preserved significant interactions with the same total number (see Materials and methods).
Figure 2 .
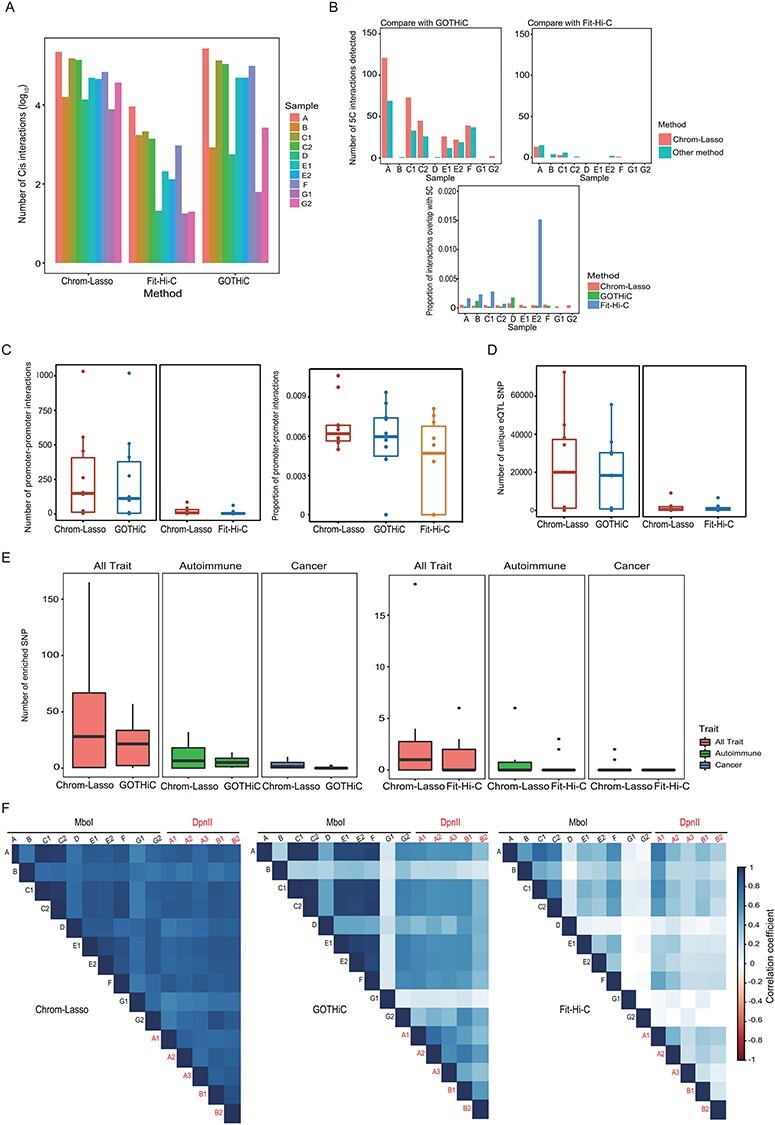
Comparison between Chrom-Lasso and other software. (A) The total number of significant cis interactions (interactions occur within the same chromosome, FDR < 0.05) detected by different methods. (B) Bar plots demonstrate the number of 5C interactions detected by the tested methods in indicated replicates when comparing same number of top significant interactions (upper panels), and the proportion of interactions overlap with 5C in all interactions identified (the lower panel). (C) Box plots show the number of promoter–promoter interactions found in the top significant interactions (left), and the proportion of promoter–promoter interactions in all interactions identified (right). (D) Box plots show the number of unique eQTL SNPs involving interactions found by tested methods. (E) Box plots compare the number of disease-associated SNPs involving interacting loci in different replicates. The comparison was done in three GWAS categories: SNPs associated with all diseases (red), SNPs associated with autoimmune diseases (green) and SNPs associated with cancer (blue). (F) Correlation matrices show the correlation between different replicates (treated with MboI or DpnII) based on the number of interactions detected in each TAD.
To compare the power to recall true-positive interactions between Chrom-Lasso and other methods, we used interactions detected using 5C technology in the GM12878 cell line as true-positive interactions [10, 19]. We demonstrated that Chrom-Lasso identified more true-positive interactions than GOTHiC, which detected the largest number of true-positive interactions among the six widely used Hi-C interaction calling methods [10], in most samples. Chrom-Lasso also had more stable performance of detecting 5C interactions among samples with different total sequenced reads than Fit-Hi-C (Figure 2B, detailed methods see Materials and methods). We then tested the methods’ performance in detecting promoter–promoter interactions. Promoters are more likely to involve in long-range interactions than a set of random loci in the genome and promoter–promoter interactions play important role in recruiting genes to shared transcription factories [42], so the performance of detecting promoter–promoter interactions reflects the power of detecting potential functional interactions through different methods. Chrom-Lasso is more likely to detect promoter–promoter interactions than other methods (Figure 2C, detailed methods see Materials and methods).
To further evaluate the functional relevance of interactions, we overlapped the interacting loci with expression quantitative trait loci (eQTL) SNPs [24] and disease-associated SNPs [21]. GWAS/eQTL SNPs are, on average, more likely to form functional interactions than a set of random genomic loci. Functional interactions that connect regulatory elements with genes play important role in transcriptional regulation, and the GWAS/eQTL SNPs located in the regulatory elements region can influence gene expression through the interaction with target genes [43–45]. Our results showed that interactions detected by Chrom-Lasso were more likely to overlap with eQTL SNPs (Figure 2D, detailed methods see Materials and methods). For disease-associated SNPs, we examined three sets of SNPs, all disease-associated SNPs, cancer-associated SNPs and autoimmune disease-associated SNPs. Similar to the eQTL SNPs, we found that interacting regions detected by Chrom-Lasso were more enriched for disease-associated SNPs (Figure 2E).
To assess the reproducibility between replicates, we demonstrated that the numbers of interactions in TADs detected by Chrom-Lasso were more correlated among the replicates than that detected by GOTHiC and Fit-Hi-C (Figure 2F). In addition, we showed that Chrom-Lasso produced more consistent contact maps and interactions between biological replicates treated with two different restriction endonucleases than that of GOTHiC and Fit-Hi-C (Figure 2F).
Our results highlight the ability of Chrom-Lasso in detecting functional interactions. The increased statistical power is likely due to borrowed information from nearby regions, leading to the detection of a larger number of functionally relevant interactions with high reproducibility.
Hi-C and Chrom-Lasso capture the dynamics of functional interactions during the process of CD8+ T cell activation
To further validate the ability of Chrom-Lasso to investigate biological function related to chromosome organization, we applied Chrom-Lasso to study the dynamic changes of interactions during the process of CD8+ T cell activation.
Cytotoxic CD8+ T cells are the main effector cells of the adaptive immune system responding to infections and diseases [46]. Activation of CD8+ T cells involves profound changes in the gene regulatory networks [47]. Accumulate evidence has demonstrated that spatial chromatin organization formed by interactions added a new perspective to the understanding functionality of transcriptional regulation [48]. Here, we sought to investigate the dynamic changes of interactions and their impact on transcriptional regulation throughout CD8+ T cell activation by generating Hi-C and RNA-seq data in cells at four different states during the process.
The in vitro CD8+ T cell activation model started from naïve CD8+ T cells separated from mouse spleen (day 0) to exhausted CD8+ T cells marked by the expression of Tim-3 on day 5 after constant anti-CD3/CD28 and IL2 stimulation. We collected cells for Hi-C experiments at four different states, including naïve T cells (Tn, day 0), the short-term activated effector T cells (Teff1, day 2), the long-term activated effector T cells (Teff2, day 5) and exhausted T cells (Tex, day 5). We also performed RNA-seq in cells at these four states with three replicates for each state (see Materials and methods). We preprocessed the raw Hi-C sequencing data following the Juicer preprocessing protocol [14], and then we identified interactions using Chrom-Lasso.
To assess the performance of detecting interactions associated with cis-regulatory elements that involve transcriptional regulation, we overlapped interacting loci of significant interactions (FDR < 0.05) identified by Chrom-Lasso with the mouse spleen ChIP-seq peaks of different TFs and histone modifications from ENCODE (see Materials and methods). We found that the interacting loci identified at four different cell states were enriched with binding sites of insulator protein such as CTCF similar to previous reported [49], showing Chrom-Lasso’s ability to find known structural interacting loci. Our results also suggested that the interacting loci were enriched with histone markers related to active chromatin state, enhancers (H3K4me1 and H3K27ac) and promoters (H3K4me3), consistent with the expectation that functional interactions preferentially involve promoters and distal regulatory elements such as enhancers (Figure 3A). However, the enrichment level of histone markers for transcribed regions (H3K36me3) and the Polycomb repression (H3K27me3) was weaker than that of the functional regulatory elements (Figure 3A). Together, these results highlight the strong capability of Chrom-Lasso in identifying interactions associated with functional elements that may impact transcriptional regulation.
Figure 3 .
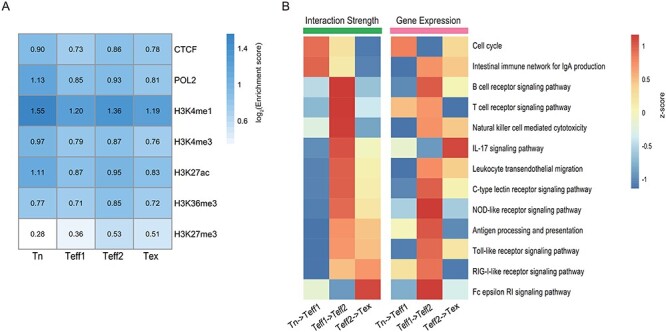
Widespread changes in gene expression and chromatin interactions during the process of CD8+ T cell activation. (A) Heat map represents the log2 enrichment score of the indicated ChIP-seq targets in four samples. (B) Heat maps show the changes in gene expression and interaction strength in the cell cycle and immune-related KEGG pathways during CD8+ T cell activation. The left panel shows the scaled log2 fold change of interaction strength measured the proportion of significant interactions (FDR < 0.05) involved in the promoter region of genes in the indicated pathway, and the right panel shows the scaled log2 enrichment score of pre-ranked GSEA of the same pathway.
Next, we sought to study the dynamic changes of interactions during the process of CD8+ T cell activation and their impact on the function of biological pathways. We defined interaction strength for a certain pathway as the proportion of significant interactions (FDR < 0.05) involved with promoter regions of all genes belong to that pathway in the total number of detected significant interactions (see Materials and methods). We found that the interaction strength of the cell cycle pathway increased from the naïve state to the short-term activated effector state (Figure 3B, left panel). However, the interaction strength for the immune response-related pathways, such as the T cell receptor signaling pathway, initially decreased from the naïve state to the short-term activated effector state and then increased after long-term activation (Figure 3B, left panel). These findings remained consistent under different FDR cutoffs (Supplementary Figure S2 available online at https://academic.oup.com/bib). These results suggest that cell proliferation dominated the initial stage of T cell activation, leading to a rapid expansion of the T cell population, which performs the effector function in combating infections.
To investigate the regulatory function of chromatin interactions on gene expression, we examined the correlation of changes in the interaction strength and changes in gene expression during T cell activation. The principal component analysis of all RNA-seq samples demonstrated that long-term activated effector cells shared similar characteristics with exhausted cells in gene expression profile (Supplementary Figure S3 available online at https://academic.oup.com/bib). Therefore, we mainly focused our analysis on the process from the naïve state to the long-term activated effector state.
For each transition from one state to the subsequent state, we ranked all genes according to their log2 fold change in expression. Then, we used these pre-ranked genes to perform GSEA on cell cycle and immune system-related KEGG pathways and finally derived the NES to represent the change of activity of pathways during the activation process (see Materials and methods). The GSEA results demonstrated that the expression level of genes in the cell cycle pathway was upregulated from the naïve state to the short-term activated effector state, yet most of the immune response-related pathways were not significantly activated at this initial stage (Figure 3B, right panel). However, we discovered a significant upregulation of the expression level of genes in most immune response-related pathways from the short-term activated effector state to the long-term activated effector state (Figure 3B, right panel).
These results were in line with the dynamic changes in the interaction strength. The change of gene expression level in cell cycle and immune system-related pathways combined with the change of interaction strength implied that the in vitro activation of mouse CD8+ T cell was mainly composed of two stages, the first stage was the cell proliferation stage to enlarge the T cell population, and the second stage was the cytotoxic stage to perform immune response. The consistency between the change of interaction strength and change of gene expression level in cell cycle and immune system-related pathways suggests that Chrom-Lasso performed well in identifying functional interactions that regulate gene expression via three-dimensional genome organization.
Chromatin interactions facilitated co-regulation between Erbb3 and neighboring genes
After confirming the capability of Chrom-Lasso to detect functional interactions genome-wide, we selected specific interactions that might play important role in regulating gene transcription during T cell activation to perform experimental validation. Interactions with both ends overlapped genes are of particular interest because such interactions may mediate the co-regulation of the genes at the two ends through 3D chromatin organization [1]. Therefore, we searched for genes involved in a high number of interactions and sought to validate interactions between such genes and neighboring genes (see Materials and methods).
We noticed that gene Erbb3, which is known to play an essential role in the regulation of cell proliferation and differentiation [36], interacted with different neighboring genes in Tn, Teff1 and Teff2 cells. The interactions in the region centered on the Erbb3 locus undergone significant reorganization during T cell activation (Figure 4A). We found that the interactions nearby Erbb3 were sparsely distributed in Tn cells, and the interaction intensity of the region upstream of Erbb3 was significantly increased in Teff1 and Teff2 cells (Figure 4A, Figure 4B). Moreover, a new domain boundary at the middle of the region is increasingly apparent in Teff2 and Tex cells, as shown in the decrease of the proportion of interactions with two ends across subdomain A and subdomain B through four states (Figure 4C), and this new boundary may be a major cause of the dissociation between Erbb3 and genes in subdomain A. The Chrom-Lasso identified interactions showed that Erbb3 interacted with Rab5b, an intracellular membrane trafficking-related gene [50], at the naïve state. Erbb3 was then connected with Cdk2, a cell cycle-related gene [51], and Rnf41, a gene involved in cytokine receptor signaling [52], at the short-term activated effector state. Erbb3 was subsequently connected with Rab5b at the long-term activated effector state, and Ikzf4, a gene associated with an inhibitory function of T cells [53], at the exhausted state. The dynamic changes in interactions nearby the Erbb3 locus were consistent with the notion that T cells first went through the cell cycle and expanded in number then turned on their cytotoxic function during the activation process.
Figure 4 .
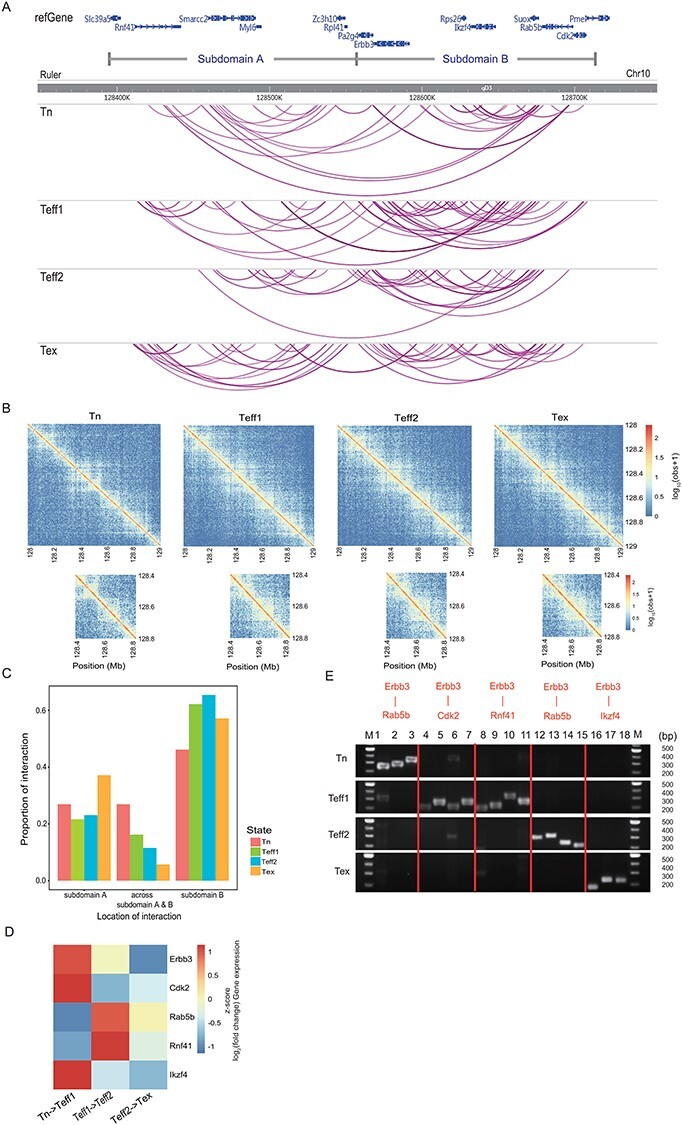
The PCR validation of Erbb3 involved interactions. (A) An overview of all interactions in the genomic region on chr10. The top panel shows the loci of genes involved in this region. We also separate this region into two potential subdomains A and B based on the changes in the proportion of interactions across these two subdomains, and the four panels below represent the distribution of all interactions detected in four samples in this region. Each purple line stands for an interaction. (B) Contact matrices demonstrate the log10 hybrid-fragment counts from the four Hi-C libraries at chr10: 128 000 000–129 000 000 at 5 kb resolution. (C) The proportion of interactions located in subdomain A, subdomain B and across these two subdomains in four samples. (D) Heat map depicts the scaled log2 fold change of averaged gene expression level. (E) PCR validation results of selected interactions. For each interaction detected by Chrom-Lasso, we captured the hybrid fragments surrounding the interacting loci from the Hi-C library and then designed primers based on the sequence of hybrid fragments for PCR. The result showed that the hybrid fragments are only detected in the corresponding Hi-C library, not in other Hi-C libraries.
More importantly, we found that when two genes are interacting with each other, their expression levels tend to change in the same direction when cells transit from one state to the next, suggesting that the expression level of two interacting genes may be co-regulated. For example, the interaction between Erbb3 and Cdk2 emerged in Teff1 cells; meanwhile, we found an upregulation of the expression level of both Erbb3 and Cdk2 in Teff1 cells compared to Tn cells (Figure 4D). Similarly, the interaction between Erbb3 and Rab5b appeared in Teff2 cells and the expression level of both genes increased compared to that in Teff1 cells (Figure 4D). The interaction between Erbb3 and Ikzf4 was detected in Tex cells and the expression level of both interacting genes was downregulated compared to that in Teff2 cells (Figure 4D). The consistency between the emergence of interaction and the co-regulation of the connected genes further demonstrated the ability of Chrom-Lasso to detect functional interactions.
Finally, we validated the gene–gene interactions identified by Chrom-Lasso by confirming the enrichment of the hybrid fragments linking the genes in the respective Hi-C libraries (see Materials and methods). PCR results demonstrated that the captured hybrid fragments for each Erbb3 involved gene–gene interaction only existed in the corresponding Hi-C library that identified the interaction (Figure 4E, Supplementary Table S2 available online at https://academic.oup.com/bib), and we also selected another two interactions with both interacting loci located in the gene promoter regions in each sample for PCR validation (Supplementary Figure S4, Supplementary Table S3 available online at https://academic.oup.com/bib), highlighting the high accuracy and specificity of Chrom-Lasso.
Discussion
The analysis of Hi-C data remains challenging for the following reasons. (i) Various biases exist in the Hi-C data, including experimental biases such as the cutting efficiency at restriction sites, ligation efficiency between pairs of cutting sites and sequencing biases such as GC content and mappability of reads. (ii) The real interactions captured by Hi-C include both functional interactions that bring regulatory elements into spatial proximity such as interactions linking gene promoter with distant enhancer and structural interactions such as CTCF-related interactions which are involved in the boundaries of TADs. Higher statistical power is required to detect functional interactions, because unlike structural interactions, functional interactions are transient and unstable, perhaps only exist in a small portion of cells at a given time.
Chrom-Lasso estimates the overall biases from inter-chromosomal hybrid fragments because the vast majority of the inter-chromosomal hybrid fragments are formed by random ligations and the differences in the probability of inter-chromosomal ligation at different cutting sites reflect the combined effect of various sources of biases. Therefore, the probability of inter-chromosomal ligation at different cutting sites can be used to correct the biases. Moreover, Chrom-Lasso borrows power from hybrid fragments surrounding the interacting loci via a lasso regression model. The application of lasso regression improved accuracy and resolution in detecting true interacting centers by effectively removes correlated interactions. Furthermore, Chrom-Lasso randomly picks pairs of genomic loci and tests if the log frequency of hybrid fragments decreases linearly as the log genomic distance between the two ends of the hybrid fragments increases. The distribution of P-values generated by such tests is used to estimate the FDR of the interactions. Last but not least, Chrom-Lasso provides a beta coefficient value for each pair of interacting loci to represent the relative strength of interaction, which implies the relative proportion of cells that has the interaction (Supplementary Figure S5 available online at https://academic.oup.com/bib).
Chrom-Lasso detected significantly more interactions identified by 5C experiments than GOTHiC, which was shown to outperform five other existing methods in this measure [10], highlighting the efficiency and accuracy of Chrom-Lasso in identifying true-positive functional interactions. We demonstrated that interacting loci identified by Chrom-Lasso has a higher rate of overlapping with eQTL SNPs, disease-associated SNPs and promoter–promoter co-regulations when compared to GOTHiC and Fit-Hi-C, which was reported to perform better in identifying functional interactions. We also highlighted that Chrom-Lasso reproduced very consistent significant interactions despite the biases caused by different experimental conditions. In conclusion, we presented Chrom-Lasso as an approach for Hi-C data analysis and demonstrated its efficacy in detecting long-range functional interactions with high reproducibility.
To further assess the performance of Chrom-Lasso in capturing the dynamic changes in functional interactions, we used Hi-C to investigate the changes in interaction strength during CD8+ T cell activation. We discovered intense interaction involved with genes in the cell cycle pathway in the early stage of CD8+ T cell activation and interaction strength of immune system-related pathways such as T cell receptor signaling pathway enhanced in the subsequent stage. The changes in the interaction strength of these pathways were consistent with the changes in gene expression we observed. Finally, we experimentally validated the dynamic changes in the interactions facilitating the co-regulation of Erbb3 and its neighboring genes at different stages of T cell activation. Interestingly, we observed a dynamic domain boundary formation, which potentially dissociated the interaction between Erbb3 and its downstream genes in long-term stimulated effector or exhausted T cells.
The comparison between Chrom-Lasso and GOTHiC or Fit-Hi-C and the application of Chrom-Lasso to analyze the in vitro time-course T cell activation data demonstrated the strong statistical power of Chrom-Lasso in detecting weak functional interactions from a noisy background within TADs. Combined with tools that can accurately define boundary domains [54, 55], we expect our strategy to provide researchers with a powerful tool for a broader range of variants of the Hi-C assay, such as single-cell Hi-C, Capture Hi-C and BL-Hi-C [56–58], which also generate data following a power-law distribution.
Availability
Chrom-Lasso is an open-source Hi-C interaction calling tool available in the GitHub repository (https://github.com/Lan-lab/Chrom-Lasso). The computational time of Chrom-Lasso is mainly determined by the density of cutting sites of restriction endonuclease in the genome. Chrom-Lasso can analyze a whole-genome Hi-C dataset treated with MboI in ~40 h via a single core of a 2.4GHz Intel(R) Xeon(R) CPU E5-2620 v3 on a server equipped with LINUX.
Accession Numbers
The Hi-C experimental data of GM12878 cell line used in the comparison between Chrom-Lasso and other methods were downloaded from the NCBI Gene Expression Omnibus (GEO) under accession number GSE63525. The Hi-C experimental data and RNA-seq data of the mouse model have been submitted to the NCBI GEO under accession number GSE158375.
Key Points
The vast majority of inter-chromosomal hybrid fragments (ICHFs) are formed by random ligations; thus, all biases at a specific genomic locus can be reflected in the number of ICHFs associated with that locus. Therefore, Chrom-Lasso corrects the complex biases assumption-free based on the number of ICHFs detected at each locus.
Chrom-Lasso takes advantage of the linearity of log-transformed power-law distribution, which converted the deconvolution of the complex signals in Hi-C data to a conventional feature selection problem in multiple linear regression.
Because Chrom-Lasso models the distribution of hybrid fragments in a region of multiple bins, not the counts in single genomic bins, it increased the statistical power for detecting weak signals and decreased the chance of calling an artificial spike in a signal bin as a signal.
Supplementary Material
Acknowledgements
We would like to thank Dr Xiang Zhou for his advice on the statistical modeling of the data and Drs Silvio Bicciato, Francesco Ferrari and Mattia Forcato for the great help in providing the detailed results in their study, which are used in the comparison between Chrom-Lasso and other methods. We are grateful to all our ‘dry lab’ group members for testing Chrom-Lasso. Finally, we thank all our ‘wet lab’ group members for the helpful discussion.
Jingzhe Lu is a PhD candidate student at the School of Medicine, Tsinghua University, Beijing, China. Her expertise is in bioinformatics.
Xu Wang is a PhD candidate student at the School of Medicine and the Tsinghua-Peking Center for Life science, Tsinghua University, Beijing, China. Her expertise is in tumor immunology.
Keyong Sun is a PhD candidate student at the School of Medicine and the Tsinghua-Peking Center for Life science, Tsinghua University, Beijing, China. His expertise is in techniques of single-cell sequencing and tumor immunology.
Xun Lan is an assistant professor at the School of Medicine and the Tsinghua-Peking Center for Life science, Tsinghua University, Beijing, China. His expertise is in bioinformatics and tumor immunology.
Contributor Information
Jingzhe Lu, School of Medicine, Tsinghua University, Beijing, China.
Xu Wang, School of Medicine and the Tsinghua-Peking Center for Life science, Tsinghua University, Beijing, China.
Keyong Sun, School of Medicine and the Tsinghua-Peking Center for Life science, Tsinghua University, Beijing, China.
Xun Lan, School of Medicine and the Tsinghua-Peking Center for Life science, Tsinghua University, Beijing, China.
Funding
This work was supported by the Tsinghua University-Peking University Jointed Center for Life Science (Grant No. 61020100119 to X.L.); National Thousand Young Talents Program of China (Grant No. 042021011 to X.L.).
References
- 1. Li G, Ruan X, Auerbach RK, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 2012;148(1-2):84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang Y, Wong CH, Birnbaum RY, et al. Chromatin connectivity maps reveal dynamic promoter-enhancer long-range associations. Nature 2013;504(7479):306–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carroll JS, Liu XS, Brodsky AS, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005;122(1):33–43. [DOI] [PubMed] [Google Scholar]
- 4. Dekker J, Rippe K, Dekker M, et al. Capturing chromosome conformation. Science (New York, NY) 2002;295(5558):1306–11. [DOI] [PubMed] [Google Scholar]
- 5. Lieberman-Aiden E, Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (New York, NY) 2009;326(5950):289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ay F, Bailey TL, Noble WS. Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts. Genome Res 2014;24(6):999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mifsud B, Martincorena I, Darbo E, et al. GOTHiC, a probabilistic model to resolve complex biases and to identify real interactions in Hi-C data. PLoS One 2017;12(4):e0174744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yaffe E, Tanay A. Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nat Genet 2011;43(11):1059–65. [DOI] [PubMed] [Google Scholar]
- 9. Chen F, Li G, Zhang MQ, et al. HiCDB: a sensitive and robust method for detecting contact domain boundaries. Nucleic Acids Res 2018;46(21):11239–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Forcato M, Nicoletti C, Pal K, et al. Comparison of computational methods for Hi-C data analysis. Nat Methods 2017;14(7):679–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cairns J, Freire-Pritchett P, Wingett SW, et al. CHiCAGO: robust detection of DNA looping interactions in Capture Hi-C data. Genome Biol 2016;17(1):127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rosa A, Becker NB, Everaers R. Looping probabilities in model interphase chromosomes. Biophys J 2010;98(11):2410–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bohn M, Heermann DW. Diffusion-driven looping provides a consistent framework for chromatin organization. PLoS One 2010;5(8):e12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Durand NC, Shamim MS, Machol I, et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Systems 2016;3(1):95–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010;38(4):576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lun AT, Smyth GK. diffHic: a Bioconductor package to detect differential genomic interactions in Hi-C data. BMC Bioinformatics 2015;16(1):258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hwang YC, Lin CF, Valladares O, et al. HIPPIE: a high-throughput identification pipeline for promoter interacting enhancer elements. Bioinformatics (Oxford, England) 2015;31(8):1290–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaul A, Bhattacharyya S, Ay F. Identifying statistically significant chromatin contacts from Hi-C data with FitHiC2. Nat Protoc 2020;15(3):991–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rao SS, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014;159(7):1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Teng L, He B, Wang J, et al. 4DGenome: a comprehensive database of chromatin interactions. Bioinformatics (Oxford, England) 2016;32(17):2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science (New York, NY) 2012;337(6099):1190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Trynka G, Westra HJ, Slowikowski K, et al. Disentangling the effects of colocalizing genomic annotations to functionally prioritize non-coding variants within complex-trait loci. Am J Hum Genet 2015;97(1):139–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lonsdale J, Thomas J, Salvatore M, et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013;45(6):580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sun W, Hu Y. eQTL mapping using RNA-seq data. Stat Biosci 2013;5(1):198–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. The Genotype-Tissue Expression (GTEx) pilot analysis . Multitissue gene regulation in humans. Science (New York, NY) 2015;348(6235):648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Frankish A, Diekhans M, Ferreira AM, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 2019;47(D1):D766–d773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012;485(7398):376–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim D, Paggi JM, Park C, et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 2019;37(8):907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics (Oxford, England) 2015;31(2):166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Korotkevich G, Sukhov V, Sergushichev A. Fast gene set enrichment analysis. bioRxiv 2019;060012. doi: 10.1101/060012. [DOI] [Google Scholar]
- 33. ENCODE Project Consortium. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 2011;9(4):e1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dunham I, Kundaje A, Aldred SF, et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489(7414):57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kanehisa M, Goto S, Kawashima S, et al. The KEGG resource for deciphering the genome. Nucleic Acids Res 2004;32(Database issue):D277–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Riese DJ, 2nd, Stern DF. Specificity within the EGF family/ErbB receptor family signaling network. BioEssays 1998;20(1):41–8. [DOI] [PubMed] [Google Scholar]
- 37. du Z, Zheng H, Huang B, et al. Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature 2017;547(7662):232–5. [DOI] [PubMed] [Google Scholar]
- 38. Hu M, Deng K, Selvaraj S, et al. HiCNorm: removing biases in Hi-C data via Poisson regression. Bioinformatics (Oxford, England) 2012;28(23):3131–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lan X, Witt H, Katsumura K, et al. Integration of Hi-C and ChIP-seq data reveals distinct types of chromatin linkages. Nucleic Acids Res 2012;40(16):7690–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mandal BN, Ma J. l1 regularized multiplicative iterative path algorithm for non-negative generalized linear models. Comput Stat Data Anal 2016;101:289–99. [Google Scholar]
- 41. Carty M, Zamparo L, Sahin M, et al. An integrated model for detecting significant chromatin interactions from high-resolution Hi-C data. Nat Commun 2017;8:15454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sexton T, Umlauf D, Kurukuti S, et al. The role of transcription factories in large-scale structure and dynamics of interphase chromatin. Semin Cell Dev Biol 2007;18(5):691–7. [DOI] [PubMed] [Google Scholar]
- 43. Dryden NH, Broome LR, Dudbridge F, et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Res 2014;24(11):1854–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ahmadiyeh N, Pomerantz MM, Grisanzio C, et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc Natl Acad Sci U S A 2010;107(21):9742–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Martin P, McGovern A, Orozco G, et al. Capture Hi-C reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci. Nat Commun 2015;6(1):10069–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Verdeil G, Fuertes Marraco SA, Murray T, et al. From T cell "exhaustion" to anti-cancer immunity. Biochim Biophys Acta 2016;1865(1):49–57. [DOI] [PubMed] [Google Scholar]
- 47. Saenz L, Lozano JJ, Valdor R, et al. Transcriptional regulation by poly(ADP-ribose) polymerase-1 during T cell activation. BMC Genomics 2008;9:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pancaldi V, Carrillo-de-Santa-Pau E, Javierre BM, et al. Integrating epigenomic data and 3D genomic structure with a new measure of chromatin assortativity. Genome Biol 2016;17(1):152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet 2014;15(4):234–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Banworth MJ, Li G. Consequences of Rab GTPase dysfunction in genetic or acquired human diseases. Small GTPases 2018;9(1-2):158–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Matsumoto Y, Hayashi K, Nishida E. Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr Biol 1999;9(8):429–32. [DOI] [PubMed] [Google Scholar]
- 52. Wauman J, De Ceuninck L, Vanderroost N, et al. RNF41 (Nrdp1) controls type 1 cytokine receptor degradation and ectodomain shedding. J Cell Sci 2011;124(Pt 6):921–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Petukhova L, Duvic M, Hordinsky M, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature 2010;466(7302):113–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Weinreb C, Raphael BJ. Identification of hierarchical chromatin domains. Bioinformatics (Oxford, England) 2016;32(11):1601–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Filippova D, Patro R, Duggal G, et al. Identification of alternative topological domains in chromatin. Algorithms Mol Biol 2014;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nagano T, Lubling Y, Stevens TJ, et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 2013;502(7469):59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mifsud B, Tavares-Cadete F, Young AN, et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet 2015;47(6):598–606. [DOI] [PubMed] [Google Scholar]
- 58. Liang Z, Li G, Wang Z, et al. BL-Hi-C is an efficient and sensitive approach for capturing structural and regulatory chromatin interactions. Nat Commun 2017;8(1):1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.