

Abstract

The 1,5-HAT–1,2-(ester)alkyl radical migration (Surzur–Tanner rearrangement) radical/polar sequence triggered by alkoxyl radicals has been studied on a series of C-glycosyl substrates with 3-C-(α,β-d,l-glycopyranosyl)1-propanol and C-(α-d,l-glycopyranosyl)methanol structures prepared from chiral pool d- and l-sugar. The use of acetoxy and diphenoxyphosphatoxy as leaving groups provides an efficient construction of 10-deoxy-1,6-dioxaspiro[4.5]decane and 4-deoxy-6,8-dioxabicyclo[3.2.1]octane frameworks. The alkoxyl radicals were generated by the reaction of the corresponding N-alkoxyphthalimides with group 14 hydrides [n-Bu3SnH(D) and (TMS)3SiH], and in comparative terms, the reaction was also initiated by visible light photocatalysis using the Hantzsch ester/fac-Ir(ppy)3 procedure. Special attention was devoted to the influence of the relative stereochemistry of the centers involved in the radical sequence on the reaction outcome. The addition of BF3•Et2O as a catalyst to the radical sequence resulted in a significant increase in the yields of the desired bicyclic ketals.
Introduction
The development of synthetic methodologies for bicyclic 1,6-dioxaspiro[4.5]decane1 and 6,8-dioxabicyclo[3.2.1]octane (6,8-DOBCO)2 scaffolds is largely stimulated by their occurrence as the structural core of highly active insect pheromones.3 They can also be widely found as subunits4 in the structure of other more complex and biologically important natural products such as steroids,5 polyether ionophores,6 and marine toxins.7 In some cases, both structural motifs are present in the same natural skeleton, as occurs in pinnatoxins and the related pteriatoxins, potent neurotoxins of a dinoflagellate origin.8 Moreover, both bicyclic ketals have attracted much interest from synthetic chemists as versatile building blocks in fine organic synthesis.9
In the carbohydrate field, the preparation of spiro-heterocycles has been recently reviewed.10 Several naturally occurring 2,7-anhydro-β-d-glyco-hept-2-ulopyranose sugars with 6,8-dioxabicyclo[3.2.1]octane structures have been described. The most representative example is sedoheptulosan (2,7-anhydro-β-d-altro-hept-2-ulopyranose), although analogous compounds with d-gluco and d-manno stereochemistry are also known.11
In previous papers, we reported on a new procedure for the stereoselective construction of 1,6-dioxaspiro[4.5]decane12 and 6,8-dioxabicyclo[3.2.1]octane13 frameworks on carbohydrate models as described in Scheme 1. Under mild oxidative conditions (PhI(OAc)2/I2), the initially generated alkoxyl radicals (i.e., I and II, PGO) trigger a 1,5-hydrogen atom transfer (1,5-HAT)14–radical oxidation–nucleophilic cyclization through a radical/polar crossover sequence that ultimately leads to the desired bicycles (i.e., III and IV, respectively) in a single step. In some cases, [4.5] spiroketal systems with a kinetic nonanomeric unstable configuration at the spiro center can be preferentially obtained using this methodology. Also using this simple procedure, natural C-glycosyl compounds of a C-(1,6-anhydro-β-d-glyco-1-ulopyranosyl)methanol structure (i.e., IV) with rare stereochemistries d-ido, d-gulo, and d-altro can be obtained from readily available d-gluco, d-galacto, and d-manno chiral pool sugars, respectively.13
Scheme 1. 1,5-HAT Reactions of 3-C-(Glycopyranosyl)propan-1-O-yl and C-(Glycopyranosyl)methan-1-O-yl Radicals.

S–T = Surzur–Tanner; RPC = radical polar crossover; HAT = hydrogen atom transfer.
Otherwise, the generation of the above-mentioned alkoxyl radicals (i.e., I and II, PGO) under reductive conditions proceeds by a different mechanism that allows the preparation of interesting and highly versatile chiral synthons. Homolytic intermolecular allylmethalation of the intermediate C1 radical may lead to C-ketosides (i.e., V, R = All).15 The regioselective HAT by alkoxyl radicals of the H5 enables also the C5-allylation and the possibility of preparing C-ketosides on both sides of the pyranosyl ring oxygen.16 Although, at first glance, the homolytic reduction of the C1- and C5-radical intermediates might seem of little synthetic utility, it allows a diastereoselective interconversion between d- and l-C-glycosides (i.e., VI)17 and α- and β-C-glycosides (i.e., V, R = H), which is difficult to achieve using conventional methods.18
Since the discovery by Surzur and Teissier19 and by Tanner and Law20 in the 1960s that β-(acyloxy)alkyl radicals undergo a 1,2-suprafacial migration of their ester group, this rearrangement has attracted considerable mechanistic and synthetic attention.21 The use of β-(phosphatoxy)alkyl radicals22 with a better leaving group (LG) and complexation with Lewis acids23 notably increase the reaction rate and consequently its importance from a synthetic point of view.
In carbohydrate chemistry, this rearrangement has been exploited for a convenient synthesis of 2-deoxypyranoses from 1-pyranosyl radicals24 and in the stereoselective preparation of purine and pyrimidine α-nucleosides.25 This rearrangement is also involved in the DNA and RNA strand scission from 2’- and 4’-radicals via the cleavage of the β-phosphate.26
It is evident that if we end the above-mentioned 1,5-HAT sequences with a β-(acyloxy)alkyl radical (i.e., I and II, LG), a simple and versatile preparation of 2-deoxy-C-glycosides on a 1-ulopyranose ring system (i.e., VII and VIII) could be achieved, where the HAT and the vicinal deoxygenation through an alkene radical-cation intermediate would occur in the same synthetic step. In fact, we would gain access to a series of ketoses with 5-deoxy-non-4-ulopyranose (i.e., VII) and 5-deoxy-hept-6-ulopyranose (also named 3-deoxy-hept-2-ulopyranose) (i.e., VIII) structures by long-range selective oxidation at C1 and C5 ring carbon atoms, respectively. The synthetic interest is apparent; the 3-deoxy-hept-2-ulopyranose framework present in VIII is intimately related to the ring system of octulosonic (Kdo, Kdn) and sialic acids.27 Procedures for the preparation of analogous [4.5] spiroketals in 2-deoxy-pyranose systems using different methodologies have been described in previous publications.28 In general, deoxy-pyranoses are important targets and are frequently found in bioactive secondary metabolites of microbial origin.29
In this paper, the 1,5-HAT–Surzur–Tanner (S–T) radical/polar sequence has been studied principally on a series of C-glycosyl substrates with 3-C-(glyco)1-propanol (i.e., I, LG) and C-(glyco)methanol (i.e., II, LG) structures prepared from chiral pool d- and l-sugar and with α- and β-configurations at the anomeric center. The initial alkoxyl radicals were generated by homolytic cleavage of the corresponding N-alkoxyphthalimide derivatives using the n-Bu3SnH/AIBN protocol under several different conditions.30 In most cases, the reaction finishes with an intramolecular nucleophilic 5-cyclization at the cine position of the radical-cation–LG anion pair intermediate to give the expected bicyclic acetal (i.e., VII or VIII) with a deoxygenated carbon atom at the vicinal position.21e
To unambiguously determine the fate of the radical throughout the cascade sequence, the experiments will also be performed with n-Bu3SnD/AIBN. This will allow us, among other things, to detect whether in the last step of the sequence the β-elimination of the ester takes place by the expected radical-polar β-(ester)alkyl shift mechanism or by a competitive pure radical β-(ester)alkyl fragmentation.21 Additionally, the influence of boron trifluoride as a catalyst on the sequence outcome will be addressed. In comparative terms, the reaction was also initiated by visible light photocatalysis using the Hantzsch ester/fac-Ir(ppy)3 procedure.31 In all cases, the reactions were allowed to proceed until the complete consumption of the radical precursors as indicated by TLC.
Due to the stereochemical requirements for the HAT reaction transition state,32 much attention has been paid to the not always apparent conformation of the sugar rings in these C-glycosyl compounds. For this purpose, the 3JHCCH vicinal ring coupling constants were extracted from the experimental 1D 1H NMR spectra by iterative simulation33 and compared with the values calculated on minimized structures in 4C1 and 1C4 conformations [see Tables S1 and S2 in the Supporting Information (SI)].34
Previous examples of the HAT–S–T rearrangement sequence have been reported in the formation of tetrahydrofurans from β-(phosphatoxy)alkyl radicals35 and as a key step in the synthesis of cephalosporolide E.36 We have also described another example of this sequence during the reaction of methyl 2,3,4-tri-O-acetyl-6-deoxy-α-d-Talp-(1 → 4)-2,3-di-O-methyl-α-d-Glcp-6-O-yl disaccharide radical.37 The initial 1,8-HAT(6IO• → 5IIC•) between the two sugars generated a 4IIβ-(acetoxy)5II-alkyl radical that led finally to the formation of a rare eight-membered 4II-deoxy-1,3,5-trioxocane ring system. The use of n-Bu3SnD confirms that, at least in part, the last step of the sequence involves an S–T rearrangement through a cine 8-exo-substitution mechanism. On the other hand, unsuccessful attempts to trap the intermediate alkene radical cation intramolecularly by carboxylate anions have been reported.38
To obtain a complete picture of the stereochemical influence of the substituents in the course of the radical sequence, we have prepared 3-C-(glycopyranosyl)1-propoxyphthalimides with α,β-d-gluco (1–4), α,β-d-manno (5–8), α-l-fuco (9 and 10), and α,β-d-arabino (11 and 12) configurations (Scheme 4).39 A few examples of 3-C-(α-d-ribofuranosyl)1-propoxyphthalimides (13–15) have been included in this work to study the influence of the greater conformational flexibility of the five-membered ring (Scheme 5). Furthermore, C-(glycopyranosyl)N-methoxyphthalimides with α-d-gluco (16–19), α-d-galacto (20 and 21), α-l-rhamno (22), and α-l-fuco (23 and 24) configurations (Scheme 6) have also been synthesized (Schemes 4–6 are presented later in this work). In most of these models, it has been possible to investigate the differences between the migratory capabilities of acetoxy and diphenoxyphosphatoxy groups and how they affect the final result of the sequence.40
Scheme 4. Synthesis of 3-C-(Glycopyranosyl)1-propoxyphthalimide (1–12) Precursors of 1,6-Dioxaspiro[4.5]decane Structures.

Reagents and conditions: (a) (i) BH3•THF 1 M complex, THF, 0 °C to rt, 1 h. (ii) NaOH 3 M, H2O2 30%, 0 °C, 1 h. (b) HONPhth, Ph3P, DEAD, THF, 0 °C to rt, 1–4 h. (c) Ac2O, Py, DMAP, 0 °C to rt, 1 h. (d) ClPO(OPh)2, DMAP, CH2Cl2, 0 °C to rt, 2 h. (e) (i) I2, CH2Cl2, rt, 3 h. (ii) Zn dust, AcOH, Et2O:MeOH, rt, overnight. (f) (i) allyltrimethylsilane, BF3•Et2O, CH3CN, 0 °C to rt, 1.5 h. (ii) Na2CO3, MeOH, rt, 2.5 h. (iii) PhCH(OMe)2, CSA, DMF, rt, overnight. (g) (i) HONPhth, Ph3P, DEAD, THF, 0 °C to rt, 0.5 h. (ii) ClPO(OPh)2, DMAP, CH2Cl2, 0 °C to rt, 1.5 h. (h) TFA/H2O, CH2Cl2, 0 °C to rt, 1 h.
Values in parentheses are isolate yields.
Scheme 5. Synthesis of 3-C-(α-d-Ribofuranosyl)1-propoxyphthalimide (13–15) Precursors of 1,6-Dioxaspiro[4.4]nonane Structures.

Reagents and conditions: (a) DPSCl, imidazole, DMF, 0 °C, 0.5 h. (b) (i) BH3•THF 1 M complex, THF, 0 °C to rt, 2.5 h. (ii) NaHCO3, H2O2 30%, 0 °C, 1 h. (c) (i) HONPhth, Ph3P, DEAD, 50 °C, 2 h. (ii) Ac2O, DMAP, Py, rt, 1 h. (d) (i) HONPhth, Ph3P, DEAD, 50 °C, 2 h. (ii) Tf2O, Py, rt, 1 h. (e) 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane, Py, 0 °C, 20 h. (f) HONPhth, Ph3P, DEAD, 50 °C, overnight. (g) ClPO(OPh)2, DMAP, CH2Cl2, rt, 3 h.
Values in parentheses are isolate yields.
Scheme 6. Synthesis of C-(Glycopyranosyl)N-methoxyphthalimide Precursors of 6,8-Dioxabicyclo[3.2.1]heptane Structures.

Reagents and conditions: (a) Ac2O, Py, DMAP, rt, 0.5–1.5 h. (b) (i) propargyl trimethylsilane/Et2O 39% v/v, TMSOTf, CH3CN, sonication, rt, 1.5–3 h. (ii) DPSCl, imidazole, DMF, 0 °C, 2 h. (c) (i) O3, CH2Cl2–MeOH, −78 °C. (ii) NaBH4, 0 °C to rt, 1–3 h. (d) HONPhth, Ph3P, DEAD, 0 °C, 1.5 h–overnight. (e) (i) K2CO3, MeOH, rt, overnight. (ii) ClPO(OPh)2, DMAP, CH2Cl2, rt, 2.5–7 h. (f) (i) K2CO3, MeOH, rt, overnight. (ii) TsCl, Py, rt, overnight. (g) (i) DHP, p-TsOH•H2O, CH2Cl2, rt, 2 h. (ii) TBAF/THF 1 M, THF, rt, 3 h. (iii) K2CO3, MeOH, rt, 4 h. (iv) ClPO(OPh)2, Py, rt, overnight. (h) NaH 60%, p-TsO-CHD-Ph, DMF/CH2Cl2, rt, 1 h. (i) BF3•OEt2, TMSOTf, propargyl trimethylsilane, CH3CN, 0 °C to rt, 15 h. (j) (i) K2CO3, MeOH, rt, 3 h. (ii) 2,2-dimethoxypropane, p-TsOH•H2O, acetone, rt, 3 h. (iii) NaH 60%, BnBr, DMF, 0 °C, 3 h. (k) (i) TFA/H2O, rt, 2 h. (ii) NaH 60%, MeI, DMF, 0 °C, 1.5 h. (iii) O3, CH2Cl2–MeOH, −78 °C. (iv) NaBH4, 0 °C to rt, 0.75 h. (l) (i) H2, Pd/C 10%, EtOAc, rt, overnight. (ii) HONPhth, Ph3P, DEAD, 0 °C to rt, 3.5 h. (m) ClPO(OPh)2, DMAP, CH2Cl2, rt, 2 h. (n) (i) K2CO3, MeOH, rt, 3 h. (ii) 2,3-butanedione, (MeO)3CH, BF3•Et2O, MeOH, 60 °C, 4.5 h. (o) (i) NaH 60%, BnBr, DMF, 0 °C, 2 h. (ii) TFA/H2O, 40 °C, overnight. (iii) NaH 60%, MeI, DMF, 0 °C to rt, 2 h. (iv) O3, CH2Cl2–MeOH, −78 °C. (v) NaBH4, 0 °C to rt, 1 h. (p) (i) DPSCl, imidazole, DMF, rt, 3 h. (ii) H2, Pd/C 10%, EtOAc, rt, overnight. (iii) Ac2O, Py, DMAP, rt, 0.5 h. (iv) TBAF/THF, 1 M, THF, rt, 4 h. (q) (i) DPSCl, imidazole, DMF, rt, 3 h. (ii) H2, Pd/C 10%, EtOAc, rt, overnight. (iii) ClPO(OPh)2, DMAP, CH2Cl2, rt, 3.5 h. (iv) TBAF/THF, 1 M, THF, rt, 3.5 h.
Values in parentheses are isolate yields.
Results and Discussion
Synthesis of 10-Deoxy-1,6-dioxaspiro[4.5]decane and 9-Deoxy-1,6-dioxaspiro[4.4]nonane Scaffolds
The results of the study with 3-C-(α,β-d-Glcp)propan-1-O-yl radicals using 2-acetyl and 2-diphenoxyphosphoryl as LGs are summarized in Table 1. Initial experiments with 3-C-(2-O-acetyl-α-d-Glcp)1-propoxyphthalimide precursor 1 employing conditions optimized for the generation of alkoxyl radicals from N-alkoxyphthalimides using n-Bu3SnH (1 equiv) in a dilute solution (0.013 M) of toluene at reflux temperature and AIBN as the initiator gave a mixture of three compounds: 25, 26β, and 26α (Table 1, entry 1). The major product 25 was identified as the expected 1,5-HAT–S–T spiroketal. The minor components of the reaction are 26β, which is formed by hydrogen abstraction at C-1 and subsequent radical axial quenching with inversion of configuration, and isomeric alcohol 26α, which could arise either by abstraction and retention of the configuration or simply by premature reduction of the alkoxyl radical. In the latter case, a combination of both mechanisms could be operative and cannot be ruled out at the present stage of the work. The yield of the cyclized product 25 was increased to 50% by lowering the tin hydride concentration with a syringe pump; under these conditions, the C-glucosyl compound 26β could not be detected (Table 1, entry 2). Attempts to improve the yield using (TMS)3SiH (TTMSS), a group 14 hydride with a smaller hydrogen donor capacity,41 to avoid the reduction of radical intermediates met with no success.
Table 1. 1,5-HAT–S–T Sequence in 3-C-(α,β-d-Glcp)1-propoxyphthalimides 1–4a.

Reagents and conditions: method A: n-Bu3SnH (1 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method B: n-Bu3SnH (1 equiv/h), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method C: TTMSS (1 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; and method D: n-Bu3SnD (1 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux.
Values in parentheses are isolate yields; deuterium incorporation (2H/1H) is included in partially labeled compounds.
Considerable analytical and spectroscopic data were diagnostic of the spiroketal structure of 25, unambiguously expressing the presence of a quaternary ketal carbon and the additional methylene group as well as the disappearance of the acetyl group. In a minimized structure, the pyranose ring adopts preferentially a 4C1 chair conformation, from which the calculated coupling constants were in agreement with the experimental values (see Table S3 in the SI). The configuration of the spiro center was tentatively assigned as 1S, with the anomeric oxygen in an axial position,42 according to the downfield displacement observed for the H3 and H5 protons that in this conformation present 1,3-diaxial interactions with the C1–O bond. In addition, the absence of NOE interactions between H1’ and H5 and/or H3 that were present in previously reported analogous [4.5] spiroketals in 2-deoxy-pyranose systems with 1R stereochemistry may also support this assignment.28a
The use of n-Bu3SnD showed the quantitative monodeuteration for (2-2H)25 and for the inverted product (1-2H)26β (Table 1, entry 4). Moreover, the early reduction of the alkoxyl radical was solely responsible for the unlabeled 26α and no retention at C1 could be detected in this experiment. The diastereoselective ratio (2Hax/2Heq, 7:1) of deuterium at (2-2H)25 is mostly attributable to a β-facial preference for the radical quenching due to steric hindrance.
For comparative purposes, we have prepared C-(2-O-diphenoxyphosphoryl-α-d-Glcp)1-propoxyphthalimide 2. The rate constant of the β-(phosphatoxy)alkyl radical migration should be several orders of magnitude greater than that recorded for comparable acyloxy shifts.21d However, the reaction of 2 with n-Bu3SnH/AIBN afforded the 1,5-HAT–S–T substitution product 25 in a similar yield (44%) together with a mixture of alcohols that, after acetylation, were identified as 27β and 27α (Table 1, entry 5). The slow addition of n-Bu3SnH generates the bicycle 25 as a sole product in 53% yield (Table 1, entry 6). The reaction with n-Bu3SnD showed the complete monodeuteration for the spirocompound (2β-2H)25 achieved in a significantly better yield (62%) (Table 1, entry 7). The inseparable mixture of the complete labeling inverted product (1-2H)28β and the reduced unlabeled alcohol 28α was also obtained (20%, 1:2.1).
This protocol was also applied to C-(2-O-acetyl-β-d-Glcp)1-propoxyphthalimide 3, where the pyranose ring adopts preferentially a 4C1 conformation with the three-carbon tether in an equatorial position and, consequently, the abstractable hydrogen atom at C1 is axially oriented (see Table S1 in the SI). Unfortunately, treatment of 3 under the same conditions mentioned above did not increase the yield of 25 (Table 1, compare entries 8–10 with 1–3). Now, the principal compound is C-glucosyl compound 26β, and according to the reaction with n-Bu3SnD, approximately 50% is formed by prereduction of the alkoxyl radical {[1-2H]26β (2H/1H, 1:1)} (Table 1, entry 11). These results were rather unexpected since electrophilic radicals abstract axial hydrogen atoms much faster than the equatorial ones and the initial 1,5-HAT should be favored relative to our previous models 1 and 2.43 The different reactivity between 1 and 3 can be explained by a possible memory of chirality effect of the C1 radical after the 1,5-HAT reaction (Table 1, compare entries 1 and 8).44
Moreover, the migration of a phosphatoxy group contributed to a marked improvement in the yield of 25 as shown in model 4 (Table 1, entries 12–14). Under these conditions, no appreciable amounts of C-glucosyl compounds resulting from the reduction of intermediate radicals were detected. As observed in previous models, the yield of the spiroketal increased significantly when changing from a hydride donor 25 to a less reactive deuteride donor (2-2H)25 (Table 1, compare entries 12 and 14). These results probably reflect a kinetic isotope effect (KIE) in which a slower process permits the radical to reach the end of the sequence, avoiding prereduction and the formation of uncyclized products.
The reaction of an analogous series of 3-C-(α,β-d-Manp)propan-1-O-yl radicals using also 5-acetyl and 5-diphenoxyphosphatoxy as LGs is summarized in Table 2. The 3-C-(2-O-acetyl-α-d-Manp)1-propoxyphthalimide 5 under the classical tin hydride conditions afforded exclusively uncyclized compounds 29β and 29α, as confirmed by deuterium labeling experiments (Table 2, entries 1 and 2). The expected spiroketal 25 could not be detected.
Table 2. 1,5-HAT–S–T Sequence in 3-C-(α,β-d-Manp)1-propoxyphthalimides 5–8a.

Reagents and conditions: method A: n-Bu3SnH (1 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method B: n-Bu3SnH (1 equiv/h), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method C: TTMSS (1 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method D: n-Bu3SnD (1 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method E: n-Bu3SnD (1 equiv), BF3•Et2O (0.2 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method F: Hantzsch ester (1.1 equiv), fac-Ir(ppy)3 (0.01 equiv), THF (0.007 M), rt, blue LED; and method G: Hantzsch ester (0.37 equiv/h), fac-Ir(ppy)3 (0.01 equiv), THF (0.007 M), rt, blue LED.
Values in parentheses are isolate yields; deuterium incorporation (2H/1H) is included in partially labeled compounds.
The isomeric β-phthalimide 7 behaved similarly, with only 29β (97%) being obtained (Table 2, entry 7). Under tin deuteride conditions, the reaction gave [1-2H]29β (2H/1H, 5.8:1); the labeled compound originated by deuterium incorporation with retention after the 1,5-HAT and the unlabeled compound by direct reduction of the alkoxyl radical (Table 2, entry 10). Knowing that aluminum and scandium Lewis acid can efficiently enhance the rate of the S–T rearrangement,23 we envisioned an experiment that under the same conditions [n-Bu3SnD (1 equiv), AIBN (0.1 equiv), and PhCH3 (0.013 M) at 110 °C] contains a catalytic amount of BF3•Et2O (0.2 equiv). To our delight, the radical sequence now proceeded nicely to the end, furnishing the desired product [2-2H]25 in 50% yield along with a small amount of [1-2H]29β (19%) (Table 2, entry 11). The 1H NMR analysis of the deuterium incorporation at [2-2H]25 (2H/1H, 1.6:1) revealed that, in this case, the β-elimination of the ester could take place not only by the radical-polar β-(acyloxy)alkyl shift mechanism but also by a competitive pure radical β-(acyloxy)alkyl fragmentation.45 Alternatively, acid-catalyzed opening and recombination of the spiroketal ring through an unobserved glucal intermediate [3-C-(1,5-anhydro-2-deoxy-d-arabino-hex-1-enopyranosyl)propan-1-ol] may also account for the loss of deuterium detected.
The 3-C-(2-O-diphenoxyphosphoryl-α,β-d-Manp)1-propoxyphthalimide models 6 and 8 with a faster migratory group gave, under standard tin hydride conditions, spiroketal 25 in moderate yields (Table 2, entries 3 and 12, respectively). The yields of (2-2H)25 improved using tin deuteride (52% in both cases) and increased notably upon Lewis acid catalysis giving [2-2H]25 (65%, 2H/1H, 2.4:1) by partial labeling (Table 2, entries 4, 14, and 15). The difference in reactivity between 2-acetyl-d-gluco (1 and 3) and -d-manno derivatives (5 and 7) has been attributed to the observed lower migration efficiency of axial β-(acetoxy)alkyl radicals.24b,46
The formation of alkoxyl radicals from N-alkoxyphthalimide under photoredox catalysis conditions and their use in selective C(sp3)–H functionalization through 1,5-HAT have been recently reported.,31b47 As far as we know, this type of methodology has never been employed to initiate the 1,5-HAT–S–T sequence described in this paper. The blue LED irradiation of phthalimides 6 and 8 in the presence of a catalytic amount of fac-[Ir(ppy)3] and Hantzsch ester as the reductant afforded spirocycle 25 in a disappointingly low yield, with the prereduced alcohols 30α and 30β, respectively, being the major products (Table 2, entries 5 and 16). Although the yield of 25 increased slightly by slowly adding the Hantzsch ester to the reaction mixture using a syringe pump, it is still clearly inferior to the results obtained with the tin hydrides (Table 2, entries 6 and 17).
Next, this study was extended to the acetyl and diphenoxyphosphoryl 3-C-(α-l-Fucp)1-propoxyphthalimides 9 and 10, respectively, as described in Table 3. When the 2-acetyl precursor 9 was treated with n-Bu3SnH/AIBN, the main product was the expected spirocycle 31 (46%) together with an inseparable mixture (2:3, 15%) of two minor alcohols: 6-deoxy-d-altro32 and l-fuco33 (Table 3, entry 1). Although both diastereomers can be tentatively identified by NMR analysis of the mixture, additional support for these structures came from the complete separation and characterization of diphenoxyphosphoryl analogues 34 and 35 achieved during the reaction of phthalimide 10 (Table 3, entry 5).
Table 3. 1,5-HAT–S–T Sequence in 3-C-(α-l-Fucp)- and 3-C-(d-Arap)1-propoxyphthalimides 9–12a.

Reagents and conditions: method A: n-Bu3SnH (1 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method D: n-Bu3SnD (1 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method E: n-Bu3SnD (1 equiv), BF3•Et2O (0.2 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method F: Hantzsch ester (1.1 equiv), fac-Ir(ppy)3 (0.01 equiv), THF (0.007 M), rt, blue LED; and method G: Hantzsch ester (0.37 equiv/h), fac-Ir(ppy)3 (0.01 equiv), THF (0.007 M), rt, blue LED.
Values in parentheses are isolate yields; deuterium incorporation (2H/1H) is included in partially labeled compounds. dr = diastereomeric ratio; only the major isomer is shown.
The spiroketal 31 with a nonanomeric configuration at the spirocenter was isolated and contaminated with a small amount of the thermodynamic isomer (1S/1R, 85:15). In both isomers, the pyranosyl ring preferentially adopts a 1C4 chair conformation (see Table S3 in the SI). The 1H NMR spectrum of 31 shows a 4Jw coupling (1.3 Hz, calcd 1.3 Hz)48 between H2α and H4 equatorial hydrogens, which also supports the mentioned conformation (see Table S4 in the SI). The 1S configuration of the major isomer (shown in Table 3) was established based on the NOE interactions observed between H5 and H1’ and H2’. The downfield displacement observed for H3 (0.1 ppm) and H5 (0.3 ppm) in the 1H NMR spectrum of the minor 1R-isomer lends further evidence to the proposed spiroketal stereochemistry.
Several interesting conclusions can be drawn from the results obtained during the deuteration experiment (Table 3, entry 2). Compound [PhCH-2H]31 (D/H = 1.5:1; dr = 4:1) showed no significant incorporation of deuterium at the C2 site, but instead a partial deuteration of a benzylic proton at C4 could be detected. The quantitative incorporation of deuterium confirmed that the 6-deoxy-d-altrose derivative (5-2H)32 was formed by the reductive inversion of configuration of a 5-radical intermediate. Finally, the undeuterated alcohol 33 was formed exclusively by prereduction of the initial alkoxyl radical. All our attempts to obtain spirocyclic 31 by applying the photoredox conditions mentioned above were unsuccessful, with only prereduced alcohol 33 being isolated instead (Table 3, entries 3 and 4).
The reaction of 3-C-(2-diphenoxyphosphoryl-α-l-Fucp)1-propoxyphthalimide 10 provided the desired bicycle 31 in better yield (52%) together with small amounts of the 6-deoxy-d-altro34 and l-fuco35 derivatives that could now be conveniently characterized. The 6-deoxy-β-d-altropyranosyl ring in 34 exists preferentially in a 4C1 conformation with the two alkyl residues in an equatorial position, with the value of the 3J4,5 = 9.8 Hz (calcd = 8.4 Hz) confirming the inversion of configuration at C5. Furthermore, a new compound 36 with a 2,7-dioxabicyclo[4.4.0]decane skeleton, hitherto undetected in the reaction of previous models, was also isolated in 10% yield (Table 3, entry 5). The structure and stereochemistry of 36, a constitutional isomer of 31, were readily established by analytic and spectroscopic means. Most significantly, the 3J fucopyranosyl ring coupling constants extracted by DAISY from the experimental spectrum and NOE interactions of H1 with H3 and H5, and H2 with H1’ confirmed the trans-fused bis(pyran) proposed framework.49
A possible propagation cycle for the acetyl and diphenoxyphosphoryl 3-C-(α-l-Fucp)propan-1-O-yl radical chain reactions, employing tin deuteride as reductant, is outlined in Scheme 2. The electrophilic alkoxyl radical (I) triggers two competitive hydrogen atom transfer reactions by abstraction of stereochemically accessible 1H (1,5-HAT) and 5H (1,7-HAT). Many examples of 1,5-hydrogen translocations are known; however, their 1,7-HAT counterparts are comparatively very scarce.14,50 The 5-alkyl radical (II) leads finally to 3-C-(6-deoxy-β-d-altropyranosyl)1-propanol derivatives (5-2H)32 and (5-2H)34 with inversion of configuration. The 1-alkyl radical (III) continues the cascade sequence by the two mechanisms mentioned before: pure radical β-fragmentation to give unlabeled 31, through a non-isolated olefin, and S–T rearrangement through the radical-polar intermediate (IV).45 When phosphate is used as LG, the reaction is directed toward two competitive pathways: cine and ipso intramolecular cyclization by the primary alcohol that now acts as a nucleophile. The minor ipso cyclization affords the bis(pyran) (1-2H)36 through radical (V). Furthermore, the cine substitution provides 2-radical (VI) that regioselectively abstracts a benzylic hydrogen from the 4-OBn protecting group by means of another 1,5-HAT process. Consequently, no deuterium incorporation (within the limits of NMR detection) was observed at C2. Reductive quenching of radical VII leads to the quantitatively deuterated (PhCH-2H)31, isolated together with the unlabeled 31 formed by the β-fragmentation mechanism.
Scheme 2. Propagation Cycle of 3-C-(α-l-Fucp)propan-1-O-yl Radicals.

The prereduction of the alkoxyl radical and the initiation and termination steps are omitted for clarity.
The 1H and 13C{1H} NMR spectra of isolated [PhCH-2H]31 (D/H, 1.5:1; dr = 4:1) deserve some comments. The deuteration at 4-OBn is highly stereoselective, providing evidence for a steric hindered deuteride addition. Both diastereoisomeric deuterated benzyl ethers seem to adopt two different conformations that affect the chemical shift displacement of surrounding protons and carbons. Thus, for example, the 6-methyl group signal appears as three doublets of approximately the expected intensities (1.5:0.3:1.2): 1.130 ppm (J = 6.3 Hz, D major), 1.133 ppm (J = 6.6 Hz, D minor), and 1.135 ppm (J = 6.3 Hz, unlabeled) (see Table S5 and Figure S1 in the SI for details).
This anomalous behavior that may be attributable to the aromatic ring current effect can also be observed in its 13C{1H} NMR spectrum; the C4 atom appears as three signals: 74.97 ppm (D major), 75.02 ppm (D minor), and 75.09 ppm (unlabeled), also with intensities in accordance with the relative proportions (see Table S6 and Figure S3 in the SI for details).
This effect has not been detected in analogous monodeuterated 4-OBn compounds with a d-glucose configuration described in the literature.51 In an attempt to rationalize this unexpected NMR result, we prepared methyl 4-O-benzyl-6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-α-d-[4-O-PhCH-2H]galactopyranoside ([PhCH-2H]97) by the reaction of alcohol 96 with benzyl α-[2H]-4-methylbenzenesulfonate (Scheme 6).52 As expected for a d-sugar, [PhCH-2H]97 adopts a 4C1 conformation, while [PhCH-2H]31 exists preferentially in a 1C4 chair. Since the structures of d-galactose and l-fucose are in a pseudoenantiomeric relationship, 4-OBn would have a very similar stereochemical environment in [PhCH-2H]97 to that which it has in the structure of [PhCH-2H]31. Indeed, in the NMR spectra of labeled [PhCH-2H]97 (D/H, 7:1; dr = 1:1), it is also observed how both diastereoisomeric deuterated benzyl ethers affect the chemical displacement of the surrounding protons and carbons differently. For example, in the 13C{1H} NMR spectrum, the C4 atom analogously appears as three signals at 73.56 ppm (D1), 73.59 ppm (D2), and 73.64 ppm (unlabeled) with the expected intensities (see Table S6 and Figure S4 in the SI for details).
The effectiveness of this methodology was also tested on a d-pentose structure. Thus, the reaction with n-Bu3SnH/AIBN of 3-C-(2-O-diphenoxyphosphoryl-α,β-d-arabinopyranosyl)1-propoxyphthalimide derivatives 11 as a mixture of anomers and its deprotected diastereoisomeric pure β-diol 12 afforded exclusively the desired spirocycles 37 and 38, respectively (Table 3, entries 7 and 10). The 2-deoxy-arabinopyranosyl ring adopted preferentially a 1C4 conformation (see Table S3 in the SI). Compound 38 was previously described by an alternative glycosylation method using thermodynamic conditions, and a 1R anomeric stabilized configuration was assigned.28c Consequently, we have not found NOE interaction between H1’ and H3 and/or H5 as in previous thermodynamic spiroketals prepared in this work.
The analysis of the isotopic distribution in [2-2H]37 (2H/1H, 1:1) and [2-2H]38 (2H/1H, 2.3:1), obtained by reductive n-Bu3SnD/AIBN with or without the BF3•Et2O catalyst, showed a partial monodeuteration at C2, with the major isotopomer occupying the β-equatorial position (Table 3, entries 8, 9, and 11).
For the sake of completeness, this methodology was also extended to a series of furanosyl models derived from 3-C-(α-d-ribofuranosyl)1-propanol as described in Table 4. When the reaction of acetyl phthalimide 13 was carried out under the n-Bu3SnH(D)/AIBN conditions, no traces of any compound with a 1,6-dioxaspiro[4.4]nonane skeleton were detected. Only the alcohol 40 was obtained (Table 4, entries 1 and 2). The deuterium composition of [1-2H]40 (2H/1H, 2:1) indicates that a significant 1,5-HAT reaction has taken place, but the C1-radical intermediate is reduced before the S–T rearrangement occurs. An equimolecular mixture of spirocycles [2-2H]39 (2H/1H, 1.3:1) was achieved in moderate yield by adding a catalytic amount of BF3•Et2O to the reaction medium (Table 4, entry 3). Also in this case, a substantial loss of deuterium at C2 indicated the possibility of competitive mechanisms with the 1,2-β-(acyloxy)alkyl radical migration. A change to a better LG such as triflate 14 increased the rate of S–T rearrangement, and the sequence could now be completed under standard tin hydride conditions (Table 4, entries 5 and 6). However, adding BF3•Et2O to the reaction resulted in a very complex mixture containing alcohol 41 as the sole identifiable product. The initiation of the reaction under photoredox catalysis conditions on both phthalimides 13 and 14 afforded poorer results (Table 4, entries 4 and 8).
Table 4. 1,5-HAT–S–T Sequence in 3-C-(α-d-Ribf)1-propoxyphthalimides 13–15a.

Reagents and conditions: method A: n-Bu3SnH (1 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method D: n-Bu3SnD (1 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method E: n-Bu3SnD (1 equiv), BF3•Et2O (0.2 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; and method F: Hantzsch ester (1.1 equiv), fac-Ir(ppy)3 (0.01 equiv), THF (0.007 M), rt, blue LED.
Values in parentheses are isolate yields; deuterium incorporation (2H/1H) is included in partially labeled compounds. dr = diastereomeric ratio.
The use of diphenylphosphate as in 15 gave access exclusively to spirocycles 42 and 43, isolated as a separable mixture of anomers in 62% overall yield (Table 4, entries 9 and 10). Again, under photoredox conditions, lower yields of the spirocyclic compounds and significant amounts of prematurely reduced alcohol 44 were obtained (Table 4, entry 11).
Synthesis of 4-Deoxy-6,8-dioxabicyclo[3.2.1]octane Scaffolds
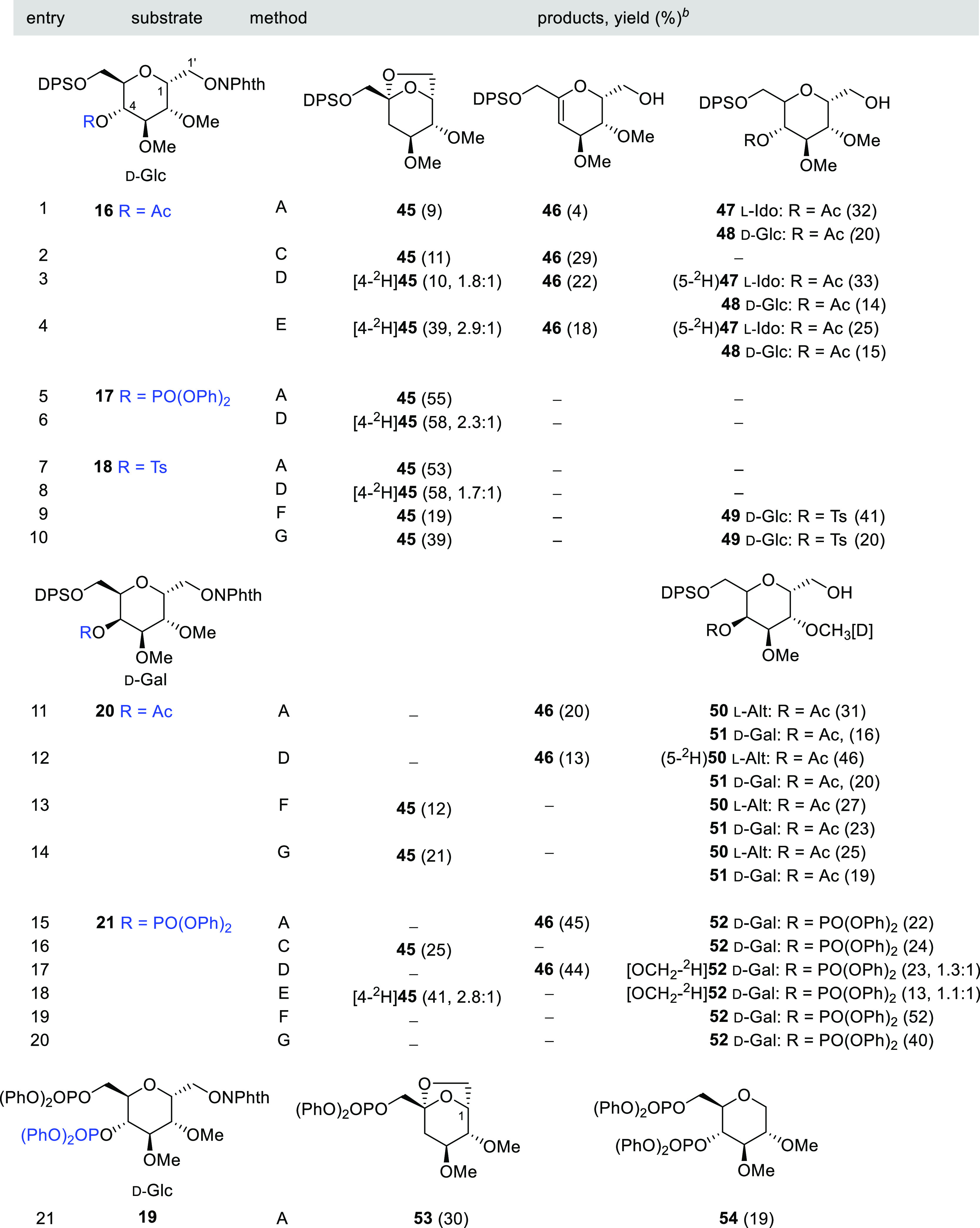
The objective of this section is the preparation of 4-deoxy carbohydrates with a 6,8-dioxabicyclo[3.2.1]octane skeleton by applying this 1,5-HAT–S–T rearrangement sequence to C-glycosyl compounds of a C-(α-d,l-glycopyranosyl)methanol general structure, and the results are included in Tables 5 and 6. The sequence was first attempted on the C-(4-O-acetyl-6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-α-d-glucopyranosyl)N-methoxyphthalimide (16) model. In this compound, the glucopyranosyl ring adopts preferentially a 4C1 chair conformation, and thus, the initial 1,5-HAT reaction should be favored (see Table S2 in the SI). However, the tin hydride conditions led to a mixture of four compounds, in which the desired bicycle 45 was isolated as a minor product. The other compounds were the unstable olefin 46 formed presumably by β-(acyloxy)alkyl fragmentation, and 47 and 48 generated by the premature reduction of intermediate radicals (Table 5, entry 1).
Table 5. 1,5-HAT–S–T Sequence in C-(α-d-Glcp)- and C-(α-d-Galp)N-methoxyphthalimides 16–21a.
Reagents and conditions: method A: n-Bu3SnH (1 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method C: TTMSS (1 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method D: n-Bu3SnD (1 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method E: n-Bu3SnD (1 equiv), BF3•Et2O (0.2 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method F: Hantzsch ester (1.1 equiv), fac-Ir(ppy)3 (0.01 equiv), THF (0.007 M), rt, blue LED; and method G: Hantzsch ester (0.37 equiv/h), fac-Ir(ppy)3 (0.01 equiv), THF (0.007 M), rt, blue LED.
Values in parentheses are isolate yields; deuterium incorporation (2H/1H) is included in partially labeled compounds.
Table 6. 1,5-HAT–S–T Sequence in C-(α-l-Fucp)- and C-(α-l-Rhap)N-methoxyphthalimides 22–24a.

Reagents and conditions: method A: n-Bu3SnH (1 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method C: TTMSS (1 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method D: n-Bu3SnD (1 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method E: n-Bu3SnD (1 equiv), BF3•Et2O (0.2 equiv), AIBN (0.1 equiv), PhCH3 (0.013 M), reflux; method F: Hantzsch ester (1.1 equiv), fac-Ir(ppy)3 (0.01 equiv), THF (0.007 M), rt, blue LED; and method G: Hantzsch ester (0.37 equiv/h), fac-Ir(ppy)3 (0.01 equiv), THF (0.007 M), rt, blue LED.
Values in parentheses are isolate yields; deuterium incorporation (2H/1H) is included in partially labeled compounds.
The structural and stereochemical assignment of these compounds rests on spectroscopic and analytical data. Conformational evidence was obtained by extracting the ring J-coupling from a simulated spectrum (see Table S7 in the SI). In addition, the long-range couplings 4J2,1’ (1.1 Hz, calcd 1.1 Hz)48 observed in the spectrum of 45 and 4J2,4 (1.2 Hz, calcd 1.2 Hz)48 in 47 confirms the 4C1 and 1C4 conformations, respectively, for the sugar rings (see Table S4 in the SI). Therefore, the main product 47 was assigned an l-Ido structure by the inversion of configuration at C5. Using TTMSS or n-Bu3SnD as reductants did not significantly improve the yield of the bicycle 45 but markedly increased the formation of the olefin 46 (Table 5, entries 2 and 3). As expected, the best yield was achieved by adding Lewis acid; [4-2H]45 (39%; 2H/1H, 2.9:1; 4R/4S, 1:1.2) was formed with a high deuterium content but low stereoselectivity (Table 5, entry 4).
As shown in Table 5 (entries 5–8), we need better LGs for the sequence of reactions to reach the end. In these experiments, the starting 4-O-diphenoxyphosphoryl 17 and 4-O-tosyl-phthalimide 18 were exclusively transformed into 45 or [4-2H]45 and no prereduction or β-fragmentation byproducts were detected. Under photoredox catalysis conditions, tosyl derivative 18 gave the bicycle 45 in poor yield, which could be substantially enhanced by adding the Hantzsch ester slowly via a syringe pump (Table 5, entries 9 and 10).
The reaction of the models C-(6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-α-d-galactopyranosyl)N-methoxyphthalimide (20 and 21) was then examined, and the results are presented in Table 5. Under the n-Bu3SnH(D) conditions and irrespective of whether the starting phthalimide was 20 or 21, no products with the 6,8-dioxabicyclo[3.2.1]octane skeleton were detected (Table 5, entries 11, 12, 15, and 17). In these experiments, the only relevant products isolated were olefin 46 and the l-altrose derivative 50, both generated by the radical quenching at C5 prior to the S–T rearrangement. Best results were ultimately attained using n-Bu3SnD under the Lewis acid catalyst, with compounds [4-2H]45 (41%; 2H/1H, 2.8:1; 4R/4S, 1:1.2) and [OCH2-2H]52 being obtained (Table 5, entry 18). Analogously to the reaction of 16, [4-2H]45 was formed with a high deuterium incorporation at C4 but with a low stereoselectivity (Table 5, compare entries 4 and 18). The incorporation of deuterium in [OCH2-2H]52 (2H/1H, 1.1:1) indicates a competitive abstraction of the H5 and the methoxyl group at C2 initiated by the alkoxyl radical through 1,5-HAT and 1,6-HAT processes, respectively. As would be expected, in this d-galactose model 21, which is less prone to undergo the 1,2-(ester)alkyl radical migration, the photoredox catalytic reaction gave only the prereduced alcohol 52, with compound 45 being undetectable by 1H NMR (Table 5, entries 19 and 20).
A propagation cycle for the acetyl and diphenoxyphosphoryl (α-d-Galp)methan-1-O-yl radical chain reactions, employing tin deuteride as the reductant, is shown in Scheme 3. The alkoxyl radical (I) initiated two competitive abstraction processes: 1,5-HAT of the 5H and 1,6-HAT of one hydrogen of the methoxyl group at C2. The radical II leads to (OCH2-2H)52, whereas the radical at C5 (III) may be stabilized by three different mechanisms: reduction with inversion of configuration giving rise to l-altrose derivative (5-2H)50 (path a), radical β-fragmentation of the LG that can explain the formation of olefin 46 and the unlabeled 45 (path b),45 or continuing the sequence by the radical–ionic mechanism that finally provided (4-2H)45 through the cine cyclization step (path c).
Scheme 3. Propagation Cycle of C-(α-d-Galp)methan-1-O-yl Radicals.

The prereduction of the alkoxyl radical and the initiation and termination steps are omitted for clarity.
A phthalimide precursor 19 having two plausible LGs at C4 and C6 was also included in this study (Table 5, entry 21). Since the endocyclic alkene radical cation intermediate should be more stable than the exocyclic alternative, it is not surprising that we only obtained the 4-deoxy-bicycle 53 by C4-OPO(OPh)2 migration. This product was accompanied by d-glucitol derivative 54 as a result of the competitive β-fragmentation of the primary alkoxyl radical, which had not previously been observed in other members of this series.,50c53
Examples with l-sugar frameworks such as α-l-Fucp (23 and 24) and α-l-Rhap (22) have also been accomplished, and the results are summarized in Table 6. Since α-l-Fucp and α-d-Galp have a pseudoenantiomeric relationship, an analogous reaction pattern could be expected. Indeed, the results obtained for the l-Fucp derivatives (23 and 24) are quite similar to those observed for the previously studied d-Galp phthalimides 20 and 21 (compare Table 5, entries 11–20 with Table 6, entries 1–11). Thus, neither the acetyl 23 nor diphenoxyphosphoryl 24 precursor gave the desired 6,8-dioxabicyclic compound when submitted to the n-Bu3SnH(D)/AIBN conditions in the absence of activating additives (Table 6, entries 1, 2, 6, and 8). In both cases, olefin 56 was the main product with a yield that reached a maximum of 70% using phosphatoxy as LG (Table 6, entry 6).
In the reaction of acetylphthalimide 23, the olefin 56 was always accompanied by small amounts of 57 with an inverted 6-deoxy-d-altrose structure (Table 6, entries 1–5). The conversion of phthalimides 23 and 24 into 6,8-dioxabicyclic compound [4-2H]55 was only possible in the presence of a catalytic amount of BF3•Et2O (Table 6, entries 3 and 9). Under these conditions, a new anhydro-alditol 3-O-acetyl-2,6-anhydro-1-deoxy-4,5-di-O-methyl-d-(6-2H)galactitol [(1-2H)60] was also isolated in a very low yield (3%), probably generated by the β-fragmentation of the alkoxyl radical at the beginning of the sequence (not shown in Table 6, entry 3). Parallel to what occurred for the d-Galp derivatives 20 and 21, surprisingly, under photoredox conditions, the acetyl precursor 23 afforded the 6,8-dioxabicyclo 55 although in low yields, while the diphenoxyphosphoryl precursor 24 yielded exclusively the reduced alcohol 59 (compare Table 6, entries 4, 5, 10, and 11 with Table 5, entries 13, 14, 19, and 20).
The phthalimides derived from α-l-Rhap22 and α-d-Glcp 17 have a very similar stereochemical arrangement to the atoms involved in the radical sequence, and consequently, an analogous behavior should be expected (compare Table 5, entries 5 and 6 with Table 6, entries 12 and 14). Indeed, the phthalimide 22 afforded exclusively the 6,8-dioxabicyclo 61 in good yield not only with tin hydride but also employing TTMSS or under the photoredox conditions.
Conclusions
In summary, the fate of the 3-C-(α,β-d,l-glycopyranosyl)1-propan-O-yl radical moves through the C-glycosyl skeleton by a 1,5-HAT–S–T rearrangement radical/polar sequence giving 1,4-anhydro-5-deoxy-non-4-ulopyranoses with a 10-deoxy-1,6-dioxaspiro[4.5]decane structure.
The reaction under the tin hydride conditions appears to be reasonably independent of the axial or equatorial configuration of the abstractable 1H but is highly influenced by the nature and stereochemistry of the LGs (2-acetoxy or 2-phosphatoxy) used in the S–T rearrangement at the end of the sequence.39 Thus, the 2-phosphatoxy LG in an equatorial position as in 3-C-(2-O-diphenoxyphosphoryl-α,β-d-Glcp)1-propoxyphthalimides 2 and 4 was found to provide the best results (Table 1, entries 5–7 and 12–14). With a poorer 2-acetoxy LG axially disposed as in C-(2-O-acetyl-α,β-d-Manp)1-propoxyphthalimides 5 and 7, the sequence did not reach the end and only the prereduced compound 29β was obtained (Table 2, entries 1, 2, and 7–10). In the two intermediate situations—equatorial 2-acetoxy phthalimides 1 and 3 (Table 1, entries 1–4 and 8–11) and axial 2-diphenoxyphosphoryl phthalimides 6 and 8 (Table 2, entries 3–6 and 12–17)—the spirocycle 25 is formed in significant amounts, indicating that the low migratory capacity of the LG can be compensated by favorable stereochemical effects and vice versa. A comparison of the best results obtained with the different LGs has been summarized in Table S9 at the SI.
Some other interesting facts can be culled from the data described in Tables 1 and 2. First, an expected KIE was observed during the formation of 25, with the yield increasing significantly in most cases when deuteride was used instead of hydride donors (e.g., Table 1, compare entries 8 and 11; see also Table S9 in the SI). The sequence yield was also dramatically improved by complexation with BF3•Et2O (Table 2, compare entries 10 and 11).
All these observations are in excellent agreement with experimental results obtained in the application of this methodology to C-glycopyranosyl models for the synthesis of either 10-deoxy-1,6-dioxaspiro[4.5]decane (Tables 1–3) or 4-deoxy-6,8-dioxabicyclo[3.2.1]octane frameworks (Tables 5–6). For example, 3-C-(α-l-Fucp)1-propoxyphthalimides 9 and 10 with the pyranosyl ring in a 1C4 conformation and the 2-acetoxy and 2-phosphatoxy LGs equatorially disposed afforded the spiroketal 31 in good yield (Table 3, entries 1, 2 and 5, 6). Nevertheless, C-(α-d-Galp)N-methoxyphthalimides 20 and 21 with the pyranosyl ring in a 4C1 conformation and the 4-acetoxy and 4-phosphatoxy LGs axially oriented did not give the expected bicyclic ketal 45, which was achieved only after the addition of BF3•Et2O to the reaction media, as evidenced in the case of 21 (Table 5, entry 18). Also in this line, C-(α-l-Rhap)N-methoxyphthalimide 22 (1C4, 4-phosphatoxy equatorially positioned) smoothly led to the desired compound 61 (Table 6, entries 12–15), whereas C-(α-l-Fucp)N-methoxyphthalimides 23 and 24 (1C4, axial LGs) reacted only under acid catalysis (Table 6, entries 3 and 9).
In the reaction of C-(d,l-Glyp)N-methoxyphthalimides, a new olefin with a C-(hex-4-enopyranosyl)methanol structure was formed (Tables 5 and 6), appearing exclusively when the S–T rearrangement is unfavored: with the 4-acetoxy group in the equatorial or axial disposition (compounds 16, 20, and 23; 4–31%) or with the 4-phosphatoxy group axially oriented (compounds 21 and 24; 44–70%). It is not detected in favored S–T rearrangements (4-phosphatoxy or 4-p-toluenesulfonyloxy equatorial) (compounds 17, 18, and 22). Presumably due to the highly strained dioxabicyclo[3.2.1]octane system, a pure radical β-(ester)alkyl fragmentation competes, in some cases very favorably, with a radical-polar β-(ester)alkyl shift mechanism.
The results observed when the reaction is applied to d-pentoses deserve special comments (Tables 3 and 4). With 3-C-(2-O-diphenoxyphosphoryl-α,β-d-Arap)1-propoxyphthalimides 11 and 12, the reaction behaved analogously and the expected spiroketals 37 and 38 were, respectively, formed in similar yields (Table 3, entries 7–11). Notwithstanding, some differences with these trends are observed during the reaction of d-pentofuranosyl substrates. The reaction of 3-C-(2-acetyl-α-d-Ribf)1-propoxyphthalimide 13 proceeds exclusively in the presence of BF3•Et2O and the use of a better LG is necessary, as observed in compounds 14 and 15 (Table 4, entries 5–11).
In these more flexible five-membered rings, the configuration of the LGs does not appear to be as important. A pseudo-rotational analysis of compounds 13, 14, and 15 shows that the most populated conformers appear at phase angles of P = 354–9° (3T2) in the northern region of the pseudo-rotational itinerary, leaving the LG in a pseudo-axial configuration (see Table S8 in the SI for details).
When the sequences were initiated by visible light photocatalysis, low yields were observed in all 3-C-(α,β-d,l-Glyp)1-propoxyphthalimides, which were in general lower than those obtained with tin hydride (Tables 2 and 3, methods F and G). The spirocycles were always accompanied by high percentages of prereduced products. A similar behavior was observed in most cases of C-(d,l-Glyp)N-methoxyphthalimides. Thus, in the reaction of 21, no traces of products resulting from the 1,5-HAT could be detected, with the prereduced alcohol 52 being formed exclusively (Table 5, entries 19 and 20). This means that, under these conditions, the six-membered TS of the 1,5-HAT cannot be reached probably due to conformational restrictions promoted by the bulky axially oriented diphenoxyphosphatoxy group. Paradoxically, with a poorer 2-acetoxy LG axially disposed as in C-(4-O-acetyl-α-d-Galp)N-methoxyphthalimide 20, the [3.2.1]bicyclic 45 and the inverted l-altro derivative 50 were obtained in a 46% combined yield (Table 5, entries 13 and 14). This is presumably due to the smaller steric demands of the acetoxy group. The same occurred with C-(α-l-Fucp)N-methoxyphthalimides 23 and 24 with which 20 and 21 present a pseudoenantiomeric relationship (Table 6, entries 4, 5 and 10, 11). We have also noted that, under these photoredox conditions, no 4-enopyranosyl olefins (i.e., 46 or 56) were detected (Tables 5–6, methods F and G).
In the best of situations, C-(4-O-diphenoxyphosphoryl-2,3-di-O-methyl-α-l-Rhap)N-methoxyphthalimide (22) (1C4, phosphatoxy equatorial) with the Hantzsch ester introduced slowly by a syringe pump, the bicycle 61 is produced in a yield (61%) comparable to that obtained with n-Bu3SnD/BF3•Et2O (Table 6, compare entries 15 and 17). These photocatalyzed reactions, carried out at room temperature, appear to be strongly influenced by the conformational equilibrium of the pyranosyl ring. However, under the tin hydride conditions (refluxing toluene, 110 °C), the TS required for the HAT reaction can be more readily attained.
Preparation of 3-C-(Glycopyranosyl)1-propanol and 3-C-(Glycofuranosyl)1-propanol Models
C-Glycosyl compounds of the 3-C-(α,β-d,l-glycopyranosyl)1-propene type 62, 65, 68, 71, and 75 were synthesized starting from perbenzylated d-glucose, d-mannose, or l-fucose, as required in each case, according to the procedure reported by Nicotra et al. (Scheme 4).54 Otherwise, for the d-arabinopyranose series, the allylation of 78 with allyltrimethylsilane and BF3•Et2O gave an inseparable anomeric mixture of allyl derivatives in 69% yield. The saponification of the acetyl groups and the selective acetal protection by treatment overnight with PhCH(OMe)2 and CSA gave access to β- and α-phenyl benzylidene substituted products as a mixture of anomers 79 (β/α, 3:1) and 80 (β/α, 3.5:1) with a free hydroxyl group at C2. Next, oxidative hydroboration of all the allyl compounds mentioned above gave efficiently the corresponding diols 63, 66, 69, 72, 76, and 81, whose primary hydroxyl groups were converted selectively to 3-C-(α,β-d,l-glycopyranosyl)N-propoxyphthalimides by the reaction with N-hydroxyphthalimide via Mitsunobu condensation yielding 64, 67, 70, 73, and 77.55 There only remains the subsequent protection of the free secondary hydroxyl group as a good LG. We thus prepared the acetyl derivatives 1, 3, 5, 7, and 9 and the phenyl phosphates 2, 4, 6, 8, 10, and 11. Finally, acid hydrolysis of the benzylidene acetal in the diastereoisomeric mixture 11 provides, after chromatographic purification, the pure major β-diastereomer 12.
In the furanose series, we prepared the corresponding allyl ribose derivative 82 following a similar strategy to that described before for the d-arabinopyranose model (Scheme 5).56 Saponification of the acetyl groups and treatment of the corresponding triol with DPSCl and imidazole in dichloromethane at 0 °C produced the diprotected product 83 in 36% yield together with the diol 84 obtained in 40% yield.13,57 On the other hand, the reaction of 82 with 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane in dry pyridine afforded the monoalcohol 86 in 69% yield. Once again, oxidative hydroboration of 83 and 86 gave the corresponding diols 85 and 87. The conversion of the primary alcohol to an N-alkoxyphthalimide and the introduction of an LG at C2 afforded the required models: the acetate 13, triflate 14, and phenyl phosphate 15.
Preparation of C-(Glycopyranosyl)methanol Models
The synthesis of 6,8-dioxabicyclo[3.2.1]heptane scaffolds commenced with the preparation of C-(4-O-acetyl-α-d,l-glycopyranosyl)allenes 91, 98, 101, 102, and 107 (Scheme 6). To achieve this with high α-diastereoselectivity, we employed the ultrasound-assisted C-glycosylation described by Murphy et al. using propargyl trimethylsilane and a Lewis acid catalyst.58 Next, LG was interchanged from OAc to PO(OPh)2, yielding 92 and 99, and to the tosyl group, giving access to 93, by saponification of the acetyl group and treatment with the corresponding acid chloride in basic media. Subsequent reductive ozonolysis afforded the C-(α-glycopyranosyl)methanol derivatives 48, 94, 49, 51, and 52 in good yields. Product 48 was also used as a precursor to prepare a diphosphate substrate 95 to analyze whether competitive migrations of the LGs at C4 and C6 could occur. First, it was necessary to protect the primary C1’-alcohol as a tetrahydropyranyl (THP) ether; then removal of both the silyl and the acyl protectors gave a diol, which was transformed to a diphosphate by treatment with ClPO(OPh)2 in pyridine overnight. Acid hydrolysis of the THP protector afforded 95 in a 37% overall yield (four-step). Finally, Mitsunobu condensation of all the primary alcohols mentioned above with N-hydroxyphthalimide yielded C-(α-glycopyranosyl)N-methoxyphthalimide derivatives 16, 17, 18, 19, 20, and 21 in good to excellent yields.
In the case of the l-rhamnose 101 and l-fucose 107 derivatives, it was found necessary to hydrolyze the acetyl groups at C2, C3, and C4 to protect selectively the C2 and C3 hydroxyl groups as cyclic acetals to enable the ulterior introduction of the LG at C4. Once the isopropylidene group was introduced for the l-rhamnose derivative, benzylation of the free alcohol at C4 afforded 103 in 54% yield. Acid hydrolysis of the transitory acetal assembly, methylation of both C2 and C3 hydroxyl groups, and reductive ozonolysis gave monoalcohol 104 in 45% overall yield. Afterward, palladium-catalyzed hydrogenolysis of the benzyl protective group gave the corresponding diol that was subsequently transformed into the N-alkoxyphthalimide 105 in 57% yield. The corresponding phenyl phosphate 22 was obtained efficiently after 2 h by treatment with ClPO(OPh)2 and DMAP at room temperature in dichloromethane.
For the l-fucose derivative, butane 2,3-bisacetal protection59 was selected to obtain 108 in 65% yield. Next, a similar strategy as described for the previous model was employed to afford monoalcohol 109 in 43% overall yield. Transient protection of the primary alcohol as a DPS ether allows the hydrogenolysis of the benzyl ether and the introduction of the LG at C4. Therefore, the formation of the acetate or phenyl phosphate followed by DPS removal (TBAF) allowed the generation of 58 and 59 in good overall yields. The conversion of the corresponding primary alcohols to N-alkoxyphthalimides occurred in 51% yield for both substrates to generate 23 and 24, respectively.
Experimental Section
General Information
Commercially available reagents and solvents were analytical grade or were purified by standard procedures prior to use. Solvents for starting material preparation and radical reactions were dried before use. The spray reagents for TLC analysis were conducted with 0.5% vanillin in H2SO4–EtOH (4:1) or, in some specific cases, with the Pancaldi reagent {(NH4)6MoO4, Ce(SO4)2, H2SO4, H2O}60 and further heating until the development of color. Melting points were determined with a hot-stage apparatus. Optical rotations were measured at the sodium line at the ambient temperature in CHCl3 solutions. IR spectra were measured as thin films on CHCl3 solutions. NMR spectra were determined at 500 or 400 MHz for 1H and at 125.7 or 100 MHz for 13C{1H} in CDCl3 or C6D6 as stated. The chemical shifts are given in parts per million (ppm) relative to TMS at δ 0.00 ppm or to residual CDCl3 at δ 7.26 ppm for proton spectra and relative to CDCl3 at δ 77.00 ppm for carbon spectra. NMR spectra were assigned with the aid of 1D and 2D techniques, including 13C DEPT-135, COSY, HSQC, HMBC, and NOESY. The DAISY program as implemented in the TopSpin 4.0.6 software package was used for the simulation of 1H NMR spectra. Low- and high-resolution mass spectra were recorded by using an electrospray (ESI+) and TOF analyzer. Flash column chromatography was performed on a Merck silica gel 60 PF (0.063–0.2 mm). For the chromatography of the radical reactions with n-Bu3SnH or n-Bu3SnD, 10% KF was added and mixed with the silica gel. Circular layers of 1 and 2 mm of the Merck silica gel 60 PF254 were used on a Chromatotron for centrifugally assisted chromatography. HPLC separations were undertaken using a semipreparative (10 × 250 mm) Ascentis Si normal-phase column. An ultrasonic bath was used (2510E-DTH, Branson) for the synthesis of the allene precursors and for the deoxygenation of the THF for the photocatalytic reactions. Photochemical reactions were carried out with 15 W blue LEDs (468 nm peak wavelength, 25 nm spectral half-wave width, composed of 15 LED units each with 1 W, 3 V, 300 mA, 5 cm distance from the light source to the irradiation vessel). For convenience, the atom-numbering system used along this section and in the assignments of the Experimental Section corresponds to the one depicted in structures of the schemes and tables, although an IUPAC systematic nomenclature has been used throughout this paper. The IUPAC nomenclature for deuterated carbohydrates (2-Carb-16.6, with the parentheses indicating substitution and square brackets for partial labeling) has been used throughout the manuscript.
General Methods for Radical and Photoredox Reactions (Tables 1–6)
Method A: Fast Addition of n-Bu3SnH
A solution of the phthalimide (1 mmol) in dry toluene (75 mL) was treated with n-Bu3SnH (269 μL, 1 mmol) and AIBN (16.4 mg, 0.1 mmol) and heated under reflux. Every hour, the same quantity of AIBN was added. In some cases, a supplementary addition of n-Bu3SnH (269 μL, 1 mmol) was required as indicated. When all the starting material was consumed, the reaction mixture was directly poured into a column chromatography on a silica gel with 10% KF (hexanes to hexanes–EtOAc) to give the corresponding products.
Method B: Slow Addition of n-Bu3SnH (1 equiv/h)
A solution of the phthalimide (1 mmol) in dry toluene (75 mL) was treated with AIBN (16.4 mg, 0.1 mmol), and n-Bu3SnH (269 μL, 1 mmol) was dropwise added during 1 h by means of a syringe pump under reflux. Every hour, the same quantity of AIBN was added. In some cases, a supplementary addition of n-Bu3SnH (269 μL, 1 mmol) was required as indicated. When all the starting material was consumed, the reaction mixture was directly poured into a column chromatography on a silica gel with 10% KF (hexanes to hexanes–EtOAc) to give the corresponding products.
Method C: Fast Addition of TTMSS
A solution of the phthalimide (1 mmol) in dry toluene (75 mL) was treated with AIBN (16.4 mg, 0.1 mmol) and TTMSS (308.5 μL, 1 mmol) and heated under reflux. Every hour, the same quantity of AIBN was added. In some cases, a supplementary addition of TTMSS (308.5 μL, 1 mmol) was required as indicated. When all the starting material was consumed, the reaction mixture was evaporated and purified by column chromatography (hexanes–EtOAc) to give the corresponding products.
Method D: Fast Addition of n-Bu3SnD
A solution of the phthalimide (1 mmol) in dry toluene (75 mL) was treated with n-Bu3SnD (270.4 μL, 1 mmol) and AIBN (16.4 mg, 0.1 mmol) and heated under reflux. Every hour, the same quantity of AIBN was added. In some cases, a supplementary addition of n-Bu3SnD (270.4 μL, 1 mmol) was required as indicated. When all the starting material was consumed, the reaction mixture was directly poured into a column chromatography on a silica gel with 10% KF (hexanes to hexanes–EtOAc) to give the corresponding products.
Method E: Fast Addition of n-Bu3SnD and BF3•Et2O
A solution of the phthalimide (1 mmol) in dry toluene (75 mL) was treated with n-Bu3SnD (270.4 μL, 1 mmol), BF3•Et2O (24.7 μL, 0.2 mmol), and AIBN (16.4 mg, 0.11 mmol) and heated at 100 °C. Every hour, the same quantity of AIBN was added. In some cases, a supplementary addition of n-Bu3SnD (270.4 μL, 1 mmol) and BF3•Et2O (24.7 μL, 0.2 mmol) was required as indicated. When all the starting material was consumed, the reaction mixture was directly poured into a column chromatography on a silica gel with 10% KF (hexanes to hexanes–EtOAc) to give the corresponding products.
Method F: Photoredox Conditions
A deoxygenated solution of the phthalimide (1 mmol), Hantzsch ester (278.6 mg, 1.1 mmol), and fac-Ir(ppy)3 (6.5 mg, 0.01 mmol) in dry THF (148.7 mL) was placed in a Schlenk tube under nitrogen and irradiated with blue LEDs at room temperature. The reaction mixture was concentrated and purified directly by chromatotron (hexanes–EtOAc) to give the corresponding products.
Method G: Photoredox Conditions with Slow Addition of Hantzsch Ester
A deoxygenated solution of the phthalimide (1 mmol) and fac-Ir(ppy)3 (6.5 mg, 0.01 mmol) in dry THF (116 mL) was placed in a Schlenk tube under nitrogen and irradiated with blue LEDs at room temperature. A solution of Hantzsch ester (279.1 mg, 1.1 mmol) in dry THF (34.9 mL) was then slowly added with a syringe pump over a period of 3 h. The reaction mixture was concentrated and purified directly by chromatotron (hexanes–EtOAc, 8:2 to 1:1) to give the corresponding products.
Synthesis of 10-Deoxy-1,6-dioxaspiro[4.5]decane Structures (Tables 1–3)
Radical Reactions of 1
Method A
Following the general procedure, starting from substrate 1 (62.8 mg, 0.092 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (25 μL, 0.092 mmol) was required. All the starting material was consumed after 3 h. Column chromatography (hexanes to hexanes–EtOAc, 1:1) gave (4S)-1,4-anhydro-6,7,9-tri-O-benzyl-2,3,5-trideoxy-d-arabino-non-4-ulopyranose (25) (18.6 mg, 0.039 mmol, 43%) as an amorphous solid, 3-C-(2-O-acetyl-3,4,6-tri-O-benzyl-β-d-glucopyranosyl)1-propanol (26β) (6.9 mg, 0.013 mmol, 14%) as a colorless oil, and 3-C-(2-O-acetyl-3,4,6-tri-O-benzyl-α-d-glucopyranosyl)1-propanol (26α) (4.1 mg, 0.008 mmol, 8%) as an amorphous solid. Compound 25: [α]D = +40.0 (c = 0.18, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.34–7.18 (m, 15H, Ar), 4.89 (d, J = 11.1 Hz, 1H, OBn), 4.66 (d, J = 11.7 Hz, 1H, OBn), 4.62 (d, J = 11.7 Hz, 1H, OBn), 4.61 (d, J = 12.3 Hz, 1H, OBn), 4.54 (d, J = 11.0 Hz, 1H, OBn), 4.51 (d, J = 12.3 Hz, 1H, OBn), 3.990 (ddd, J = 11.5, 8.9, 5.1 Hz, 1H, 3-H), 3.89 (ddd, J = 8.2, 8.2, 5.4 Hz, 1H, 3’-Hb), 3.83 (ddd, J = 8.2, 8.2, 6.3 Hz, 1H, 3’-Ha), 3.79 (ddd, J = 9.9, 4.3, 1.9 Hz, 1H, 5-H), 3.74 (dd, J = 10.8, 4.3 Hz, 1H, 6-Hb), 3.64 (dd, J = 10.8, 1.9 Hz, 1H, 6-Ha), 3.57 (dd, J = 9.9, 8.9 Hz, 1H, 4-H), 2.222 (dd, J = 12.7, 5.1 Hz, 1H, 2-Hb), 2.12–2.00 (m, 2H, 1’-Hb, 2’-Hb), 1.872 (dd, J = 12.7, 11.5 Hz, 1H, 2-Ha), 1.85 (m, 1H, 2’-Ha), 1.76 ppm (m, 1H, 1’-Ha). 13C{1H} NMR (100.6 MHz, CDCl3) δC 138.9 (2 × C, Ar), 138.5 (C, Ar), 128.34 (2 × CH, Ar), 128.28 (2 × CH, Ar), 128.2 (2 × CH, Ar), 127.76 (2 × CH, Ar), 127.74 (2 × CH, Ar), 127.6 (2 × CH, Ar), 127.5 (2 × CH, Ar), 127.4, (CH, Ar), 106.33 (C, C-1), 79.2 (CH, C-4), 78.51 (CH, C-3), 74.7 (CH2, OBn), 73.3 (CH2, OBn), 71.8 (CH2, OBn), 71.8 (CH, C-5), 69.4 (CH2, C-6), 67.3 (CH2, C-3’), 38.62 (CH2, C-2), 37.2 (CH2, C-1’), 23.5 ppm (CH2, C-2’). IR (CHCl3): ν = 3009, 2939, 1456, 1089 cm–1. MS (ESI) m/z (%) = 497 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C30H34NaO5 497.2304; found 497.2303. Anal. calcd for C30H34O5: C, 75.92; H, 7.12. Found: C, 76.07; H, 7.21. Compound 26β: [α]D = +12.3 (c = 0.43, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.34–7.15 (m, 15H, Ar), 4.887 (dd, J = 9.3, 9.3 Hz, 1H, 2-H), 4.81 (d, J = 11.4 Hz, 1H, OBn), 4.77 (d, J = 10.9 Hz, 1H, OBn), 4.65 (d, J = 11.4 Hz, 1H, OBn), 4.59 (d, J = 12.2 Hz, 1H, OBn), 4.53 (d, J = 12.2 Hz, 1H, OBn), 4.52 (d, J = 10.6 Hz, 1H, OBn), 3.68 (dd, J = 10.9, 1.9 Hz, 1H, 6-Hb), 3.66–3.59 (m, 5H, 3’-H2, 3-H, 4-H, 6-Ha), 3.46 (m, 1H, 5-H), 3.34 (ddd, J = 9.8, 9.8, 2.4 Hz, 1H, 1-H), 1.94 (s, 3H, OAc), 1.75–1.61 (m, 3H, 1’-Hb, 2’-H2), 1.509 ppm (m, 1H, 1’-Ha), 1H from OH is missing. 13C{1H} NMR (100.6 MHz, CDCl3) δC 169.9 (C, OAc), 138.4 (C, Ar), 138.1 (C, Ar), 138.0 (C, Ar), 128.42 (2 × CH, Ar), 128.41 (2 × CH, Ar), 128.37 (2 × CH, Ar), 128.0 (2 × CH, Ar), 127.8 (3 × CH, Ar), 127.70 (2 × CH, Ar), 127.67 (CH, Ar), 127.6 (CH, Ar), 84.7 (CH, C-3), 79.0 (CH, C-4), 78.5 (CH, C-5), 78.0 (CH, C-1), 75.2 (CH2, OBn), 75.0 (CH2, OBn), 73.81 (CH, C-2), 73.5 (CH2, OBn), 69.1 (CH2, C-6), 62.7 (CH2, C-3’), 28.84 (CH2, C-2’), 28.24 (CH2, C-1’), 20.9 ppm (CH3, OAc). IR (CHCl3): ν = 3430, 3014, 1738, 1229, 1039 cm–1. MS (ESI) m/z (%) = 557 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C32H38NaO7 557.2515; found 557.2513. Anal. calcd for C32H38O7: C, 71.89; H, 7.16. Found: C, 71.86; H, 7.37. Compound 26α: [α]D = +41.4 (c = 0.36, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.34–7.14 (m, 15H, Ar), 5.03 (dd, J = 8.5, 5.4 Hz, 1H, 2-H), 4.75 (d, J = 11.7 Hz, 1H, OBn), 4.70 (d, J = 11.0 Hz, 1H, OBn), 4.70 (d, J = 11.0 Hz, 1H, OBn), 4.59 (d, J = 12.0 Hz, 1H, OBn), 4.51 (d, J = 12.0 Hz, 1H, OBn), 4.48 (d, J = 11.1 Hz, 1H, OBn), 4.13 (m, 1H, 1-H), 3.81 (dd, J = 8.2, 8.2 Hz, 1H, 3-H), 3.75 (ddd, J = 8.2, 3.8, 3.8 Hz, 1H, 5-H), 3.71–3.63 (m, 4H, 3’-H2, 6-H2), 3.60 (dd, J = 8.2, 8.2 Hz, 1H, 4-H), 1.99 (s, 3H, OAc), 1.87 (br s, 1H, OH), 1.80 (m, 1H, 1’-Hb), 1.70–1.58 (m, 2H, 2’-H2), 1.51 ppm (m, 1H, 1’-Ha). 13C{1H} NMR (100.6 MHz, CDCl3) δC 170.0 (C, OAc), 138.3 (C, Ar), 138.0 (2 × C, Ar), 127.6–128.4 (15 × CH, Ar), 79.9 (CH, C-3), 77.5 (CH, C-4), 74.6 (CH2, OBn), 74.5 (CH2, OBn), 73.4 (CH2, OBn), 73.0 (CH, C-2), 72.4 (CH, C-1), 72.0 (CH, C-5), 69.1 (CH2, C-6), 62.2 (CH2, C-3’), 28.9 (CH2, C-2’), 22.5 (CH2, C-1’), 20.9 ppm (CH3, OAc). IR (CHCl3): ν = 3477, 3014, 2942, 1740, 1236, 1100 cm–1. MS (ESI) m/z (%) = 557 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C32H38NaO7 557.2515; found 557.2515. Anal. calcd for C32H38O7: C, 71.89; H, 7.16. Found: C, 71.57; H, 7.51.
Method B
Following the general procedure, starting from substrate 1 (123.9 mg, 0.18 mmol), after 2 h of reaction, two more equivalents of n-Bu3SnH (98 μL, 0.36 mmol) added by a syringe pump were required. All the starting material was consumed after 14 h. Column chromatography (hexanes to hexanes–EtOAc, 1:1) gave 25 (43.4 mg, 0.09 mmol, 50%) and 26α (14.1 mg, 0.026 mmol, 15%).
Method C
Following the general procedure, starting from substrate 1 (62.8 mg, 0.092 mmol), after 2 h of reaction, a supplementary addition of TTMSS (28.5 μL, 0.092 mmol) was required. All the starting material was consumed after 5 h. Column chromatography (hexanes–EtOAc, 9:1 to 6:4) gave 25 (8.4 mg, 0.018 mmol, 19%) and the reduced product 26α (1.5 mg, 0.003 mmol, 3%).
Method D
Following the general procedure, starting from substrate 1 (94 mg, 0.14 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnD (37 μL, 0.14 mmol) was required. All the starting material was consumed after 5 h. Column chromatography (hexanes to hexanes–EtOAc, 1:1) gave (4S)-1,4-anhydro-6,7,9-tri-O-benzyl-2,3,5-trideoxy-d-(5-2H)arabino-non-4-ulopyranose [(2-2H)25] (28.5 mg, 0.060 mmol, 43%, 2β-2H/2α-2H, 7:3) as an amorphous solid, 3-C-(2-O-acetyl-3,4,6-tri-O-benzyl-β-d-(1-2H)glucopyranosyl)1-propanol [(1-2H)26β] (4.8 mg, 0.009 mmol, 6%) as a colorless oil, and the prematurely reduced product 26α (3 mg, 0.006 mmol, 4%). Compound (2-2H)25: 1H NMR (400 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.35–7.18 (m, 15H, Ar). 4.89 (d, J = 11.1 Hz, 1H, OBn), 4.66 (d, J = 11.7 Hz, 1H, OBn), 4.61 (d, J = 12.3 Hz, 1H, OBn), 4.61 (d, J = 12.3 Hz, 1H, OBn), 4.54 (d, J = 11.0 Hz, 1H, OBn), 4.51 (d, J = 12.3 Hz, 1H, OBn), 3.987 [dd, J = 8.9, 5.1 Hz, 0.7H, 3-H (from 2β-2H)], 3.987 [dd, J = 11.5, 8.9 Hz, 0.3H, 3-H (from 2α-2H)], 3.89 (ddd, J = 8.2, 8.2, 5.3 Hz, 1H, 3’-Hb), 3.83 (ddd, J = 8.2, 8.2, 6.6 Hz, 1H, 3’-Ha), 3.79 (ddd, J = 9.9, 4.2, 1.7 Hz, 1H, 5-H), 3.74 (dd, J = 10.7, 4.2 Hz, 1H, 6-Hb), 3.65 (dd, J = 10.7, 1.7 Hz, 1H, 6-Ha), 3.57 (dd, J = 9.9, 8.9 Hz, 1H, 4-H), 2.200 (d, J = 5.1 Hz, 0.7H, 2α-H), 2.13–1.99 (m, 2H, 1’-Hb, 2’-Hb), 1.86 (m, 1H, 2’-Ha), 1.85 (d, J = 11.5 Hz, 0.3H, 2β-H), 1.74 ppm (m, 1H, 1’-Ha). 13C{1H} NMR (100.6 MHz, CDCl3) δC 138.9 (2 × C, Ar), 138.5 (C, Ar), 128.33 (2 × CH, Ar), 128.28 (2 × CH, Ar), 128.2 (2 × CH, Ar), 127.76 (2 × CH, Ar), 127.74 (2 × CH, Ar), 127.6 (2 × CH, Ar), 127.5 (2 × CH, Ar), 127.4, (CH, Ar), 106.30 (C, C-1), 79.1 (CH, C-4), 78.50 (CH, C-3), 74.7 (CH2, OBn), 73.3 (CH2, OBn), 71.8 (CH2, OBn), 71.8 (CH, C-5), 69.4 (CH2, C-6), 67.2 (CH2, C-3’), 38.26 (CHD, t, JCD = 19.7 Hz, C-2), 37.1 (CH2, C-1’), 23.5 ppm (CH2, C-2’). MS (ESI) m/z (%) = 498 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C30H332HNaO5 498.2367; found 498.2366. Compound (1-2H)26β: 1H NMR (400 MHz, CDCl3) δH 7.34–7.14 (m, 15H, Ar), 4.881 (d, J = 8.8 Hz, 1H, 2-H), 4.81 (d, J = 11.4 Hz, 1H, OBn), 4.77 (d, J = 10.8 Hz, 1H, OBn), 4.65 (d, J = 11.4 Hz, 1H, OBn), 4.59 (d, J = 12.2 Hz, 1H, OBn), 4.55 (d, J = 10.2 Hz, 1H, OBn), 4.52 (d, J = 10.6 Hz, 1H, OBn), 3.69 (dd, J = 10.7, 1.9 Hz, 1H, 6-Hb), 3.66–3.57 (m, 5H, 3’-H2, 3-H, 4-H, 6-Ha), 3.46 (m, 1H, 5-H), 1.94 (s, 3H, OAc), 1.75–1.61 (m, 3H, 1’-Hb, 2’-H2), 1.497 ppm (ddd, J = 13.4, 13.4, 6.4 Hz, 1H, 1’-Ha), 1H from OH is missing. 13C{1H} NMR (100.6 MHz, CDCl3) δC 169.9 (C, OAc), 138.4 (C, Ar), 138.1 (C, Ar), 138.0 (C, Ar), 128.42 (4 × CH, Ar), 128.38 (2 × CH, Ar), 128.0 (2 × CH, Ar), 127.8 (3 × CH, Ar), 127.70 (2 × CH, Ar), 127.67 (CH, Ar), 127.65 (CH, Ar), 84.6 (CH, C-3), 78.9 (CH, C-4), 78.5 (CH, C-5), 75.2 (CH2, OBn), 75.0 (CH2, OBn), 73.74 (CH, C-2), 73.5 (CH2, OBn), 69.1 (CH2, C-6), 62.7 (CH2, C-3’), 28.8 (CH2, C-2’), 28.13 (CH2, C-1’), 20.9 ppm (CH3, OAc). MS (ESI) m/z (%) = 558 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C32H372HNaO7 558.2578; found 558.2574.
Radical Reactions of 2
Method A
Following the general procedure, starting from substrate 2 (89.7 mg, 0.10 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (29 μL, 0.11 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes to hexanes–EtOAc, 1:1) gave 25 (20.8 mg, 0.044 mmol, 44%) and an inseparable mixture of alcohols (12.8 mg, 0.018 mmol, 16%) that was elucidated by the usual acetylation to obtain 3-C-(3,4,6-tri-O-benzyl-2-O-diphenoxyphosphoryl-β-d-glucopyranosyl)1-propyl acetate (27β) (3.9 mg, 0.005 mmol, 5% from 2) and 3-C-(3,4,6-tri-O-benzyl-2-O-diphenoxyphosphoryl-α-d-glucopyranosyl)1-propyl acetate (27α) (6.1 mg, 0.008 mmol, 7% from 2), both as colorless oils. Compound 27β: [α]D = +16.2 (c = 0.33, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.36–7.11 (m, 25H, Ar), 4.59 (d, J = 12.3 Hz, 1H, OBn), 4.52 (m, 1H, 2-H), 4.52 (d, J = 12.0 Hz, 1H, OBn), 4.51 (d, J = 12.0 Hz, 1H, OBn), 4.46 (d, J = 12.3 Hz, 1H, OBn), 4.43 (d, J = 12.0 Hz, 1H, OBn), 4.13 (d, J = 12.3 Hz, 1H, OBn), 4.09 (dd, J = 2.5, 2.5 Hz, 1H, 3-H), 4.01–3.92 (m, 3H, 5-H, 3’-H2), 3.78 (m, 1H, 1-H), 3.68 (dd, J = 10.1, 6.6 Hz, 1H, 6-Hb), 3.54 (dd, J = 10.1, 5.7 Hz, 1H, 6-Ha), 3.35 (br s, 1H, 4-H), 1.98 (s, 3H, OAc), 1.83–1.75 (m, 2H, 1’-Hb, 2’-Hb), 1.65–1.39 ppm (m, 2H, 1’-Ha, 2’-Ha). 13C{1H} NMR (125.7 MHz, CDCl3) δC 171.1 (C, OAc), 150.5 (2 × C, Ar), 138.3 (C, Ar), 137.7 (C, Ar), 137.4 (C, Ar), 120.0–129.7 (25 × CH, Ar), 77.2 (CH, C-2), 75.4 (CH, C-5), 74.6 (d, 3JPC = 6.4 Hz, CH, C-1), 73.4 (CH2, OBn), 72.3 (CH2, OBn), 71.7 (CH, C-3 or C-4), 71.6 (CH2, OBn), 71.5 (CH, C-3 or C-4), 69.8 (CH2, C-6), 64.3 (CH2, C-3’), 27.5 (CH2, C-1’ or C-2’), 24.9 (CH2, C-1’ or C-2’), 20.9 ppm (CH3, OAc). IR (CHCl3): ν = 2927, 1733, 1491, 1027 cm–1. MS (ESI) m/z (%) = 789 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C44H47NaO10P 789.2805; found 789.2831. Compound 27α: [α]D = +22.0 (c = 0.30, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.32–7.08 (m, 25H, Ar), 4.82 (d, J = 11.1 Hz, 1H, OBn), 4.79 (ddd, J = 8.3, 5.7 Hz, 3JPH = 8.3 Hz, 1H, 2-H), 4.74 (d, J = 10.7 Hz, 1H, OBn), 4.73 (d, J = 11.1 Hz, 1H, OBn), 4.60 (d, J = 12.0 Hz, 1H, OBn), 4.46 (d, J = 12.3 Hz, 1H, OBn), 4.45 (d, J = 10.5 Hz, 1H, OBn), 4.15 (m, 1H, 1-H), 4.02 (ddd, J = 10.7, 10.7, 6.3 Hz, 1H, 3’-Hb), 3.98 (ddd, J = 10.8, 10.8, 6.3 Hz, 1H, 3’-Ha), 3.86 (dd, J = 8.6, 8.6 Hz, 1H, 3-H), 3.70–3.60 (m, 4H, 4-H, 5-H, 6-H2), 2.00 (s, 3H, OAc), 1.77–1.69 (m, 2H, 1’-Hb, 2’-Hb), 1.62–1.49 ppm (m, 2H, 1’-Ha, 2’-Ha). 13C{1H} NMR (125.7 MHz, CDCl3) δC 171.1 (C, OAc), 150.5 (2 × C, Ar), 138.0 (C, Ar), 137.9 (C, Ar), 137.8 (C, Ar), 129.8 (2 × CH, Ar), 129.7 (2 × CH, Ar), 128.41 (2 × CH, Ar), 128.37 (2 × CH, Ar), 128.3 (2 × CH, Ar), 127.9 (2 × CH, Ar), 127.84 (3 × CH, Ar), 127.78 (2 × CH, Ar), 127.7 (CH, Ar), 127.6 (CH, Ar), 125.5 (CH, Ar), 125.3 (CH, Ar), 120.14 (CH, Ar), 120.10 (CH, Ar), 120.0 (CH, Ar), 119.9 (CH, Ar), 119.9–129.8 (25 × CH, Ar), 80.5 (d, 3JPC = 6.4 Hz, CH, C-3), 78.4 (d, 3JPC = 7.4 Hz, CH, C-1), 77.7 (CH, C-2), 75.1 (CH2, OBn), 74.9 (CH2, OBn), 73.5 (CH2, OBn), 73.4 (CH, C-5), 71.5 (CH, C-4), 68.6 (CH2, C-6), 64.1 (CH2, C-3’), 24.5 (CH2, C-1’ or C-2’), 24.5 (CH2, C-1’ or C-2’), 21.0 ppm (CH3, OAc). IR (CHCl3): ν = 3013, 2928, 1730, 1026 cm–1. MS (ESI) m/z (%) = 789 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C44H47NaO10P 789.2805; found 789.2823.
Method B
Following the general procedure, starting from substrate 2 (56 mg, 0.064 mmol), after 2 h of reaction, two more equivalents of n-Bu3SnH (35 μL, 0.128 mmol) added by a syringe pump were required. All the starting material was consumed after 11 h. Column chromatography (hexanes to hexanes–EtOAc, 1:1) gave 25 (16.2 mg, 0.034 mmol, 53%).
Method D
Following the general procedure, starting from substrate 2 (66.3 mg, 0.076 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnD (21 μL, 0.076 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes to hexanes–EtOAc, 1:1) gave (2-2H)25 (22.5 mg, 0.047 mmol, 62%) and the inseparable mixture of 3-C-(3,4,6-tri-O-benzyl-2-O-diphenoxyphosphoryl-β-d-(1-2H)glucopyranosyl)1-propanol [(1-2H)28β] and 3-C-(3,4,6-tri-O-benzyl-2-O-diphenoxyphosphoryl-α-d-glucopyranosyl)1-propanol (28α) (11.1 mg, 0.015 mmol, 20%, (1-2H)28β/28α, 1:2.1) as a colorless oil. Compounds (1-2H)28β and 28α: 1H NMR (500 MHz, CDCl3, selected resolved signals of (1-2H)28β from the mix spectrum) δH 4.74 (d, J = 11.0 Hz, 1H, OBn), 4.74 (d, J = 11.0 Hz, 1H, OBn), 4.59 (d, J = 12.0 Hz, 1H, OBn), 4.532 (d, J = 2.6 Hz, 1H, 2-H), 4.53 (d, J = 12.0 Hz, 1H, OBn), 4.46 (d, J = 11.0 Hz, 1H, OBn), 4.42 (d, J = 11.7 Hz, 1H, OBn), 4.10 (d, J = 12.2 Hz, 1H, OBn), 4.07 (dd, J = 2.6, 2.6 Hz, 1H, 3-H), 3.44 (dd, J = 10.1, 0.0 Hz, 1H, 6-Ha), 3.31 ppm (dd, J = 2.6, 1.0 Hz, 1H, 4-H). 13C{1H} NMR (125.7 MHz, CDCl3, selected resolved signals of (1-2H)28β from the mix spectrum) δC 150.4 (d, 2JPC = 7.0 Hz, 2 × C, Ar), 138.1 (C, Ar), 137.6 (C, Ar), 137.3 (C, Ar), 80.5 (d, 3JPC = 6.4 Hz, CH, C-3), 78.55 (d, 2JPC = 7.4 Hz, CH, C-2), 75.0 (CH, C-5), 73.5 (CH2, OBn), 72.2 (CH2, OBn), 71.5 (CH2, OBn), 71.5 (CH, C-4), 69.8 (CH2, C-6), 62.5 (CH2, C-3’), 29.68 (CH2, C-1’ or C-2’), 28.08 ppm (CH2, C-1’ or C-2’). 1H NMR (500 MHz, CDCl3, selected resolved signals of 28α from the mix spectrum) δH 4.80 (d, J = 11.0 Hz, 1H, OBn), 4.770 (ddd, J = 7.6, 4.5 Hz, 3JPH = 6.3 Hz, 1H, 2-H), 4.71 (d, J = 10.8 Hz, 1H, OBn), 4.58 (d, J = 12.0 Hz, 1H, OBn), 4.48 (d, J = 12.3 Hz, 1H, OBn), 4.44 (d, J = 11.1 Hz, 1H, OBn), 4.23 (ddd, J = 11.4, 5.7, 2.2 Hz, 1-H), 3.85 ppm (dd, J = 8.5, 8.5 Hz, 1H, 3-H). 13C{1H} NMR (125.7 MHz, CDCl3, selected resolved signals of 28α from the mix spectrum) δC 150.5 (d, 2JPC = 7.0 Hz, 2 × C, Ar), 138.0 (C, Ar), 137.8 (2 × C, Ar), 80.5 (d, 3JPC = 6.4 Hz, CH, C-3), 78.55 (d, 2JPC = 7.4 Hz, CH, C-2), 77.8 (CH, C-5), 75.1 (CH2, OBn), 74.8 (CH2, OBn), 73.6 (CH, C-1), 73.5 (CH2, OBn), 71.4 (CH, C-4), 68.8 (CH2, C-6), 61.8 (CH2, C-3’), 28.56 (CH2, C-1’ or C-2’), 20.69 ppm (CH2, C-1’ or C-2’). IR (CHCl3): ν = 3490, 3422, 2928, 1550, 1491, 1192 cm–1. MS (ESI) m/z (%) = 747 (99.6) [M + Na]+, 748 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C42H45NaO9P 747.2699; found 747.2697, [M + Na]+ calcd for C42H442HNaO9P 748.2762; found 748.2764.
Radical Reactions of 3
Method A
Following the general procedure, starting from substrate 3 (89.2 mg, 0.13 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (36 μL, 0.13 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes to hexanes–EtOAc, 6:4) gave 25 (13.9 mg, 0.029 mmol, 23%) and 26β (20.4 mg, 0.038 mmol, 29%).
Method B
Following the general procedure, starting from substrate 3 (110.2 mg, 0.16 mmol), after 2 h of reaction, two more equivalents of n-Bu3SnH (88 μL, 0.32 mmol) added by a syringe pump over 2 h were required. All the starting material was consumed after 5 h. Column chromatography (hexanes to hexanes–EtOAc, 6:4) gave 25 (22.4 mg, 0.047 mmol, 30%) and the reduced product 26β (16.4 mg, 0.031 mmol, 19%).
Method C
Following the general procedure, starting from substrate 3 (96.2 mg, 0.14 mmol), after 2 h of reaction, a supplementary addition of TTMSS (66 μL, 0.21 mmol) was required. All the starting material was consumed after 5 h. Column chromatography (hexanes–EtOAc, 9:1 to 6:4) gave 25 (10.2 mg, 0.022 mmol, 23%) and the reduced product 26β (3 mg, 0.006 mmol, 4%).
Method D
Following the general procedure, starting from substrate 3 (106.6 mg, 0.16 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnD (42 μL, 0.16 mmol) was required. All the starting material was consumed after 5 h. Column chromatography (hexanes to hexanes–EtOAc, 6:4) gave (2-2H)25 (25.2 mg, 0.053 mmol, 33%) and [1-2H]26β (8.3 mg, 0.016 mmol, 10%, 2H/1H 1:1).
Radical Reactions of 4
Method A
Following the general procedure, starting from substrate 4 (57.3 mg, 0.066 mmol), after 2 h of reaction and again after 4 h, a supplementary addition of n-Bu3SnH (18 μL, 0.066 mmol) was required. All the starting material was consumed after 6 h. Column chromatography (hexanes to hexanes–EtOAc, 8:2) gave 25 (14.2 mg, 0.030 mmol, 45%).
Method B
Following the general procedure, starting from substrate 4 (53 mg, 0.061 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (16 μL, 0.061 mmol) added by a syringe pump over 1 h was required. All the starting material was consumed after 6 h. Column chromatography (hexanes to hexanes–EtOAc, 8:2) gave 25 (12 mg, 0.025 mmol, 42%).
Method D
Following the general procedure, starting from substrate 4 (75 mg, 0.086 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnD (23 μL, 0.086 mmol) was required. All the starting material was consumed after 5 h. Column chromatography (hexanes to hexanes–EtOAc, 8:2) gave (2-2H)25 (22.5 mg, 0.047 mmol, 55%).
Radical Reactions of 5
Method A
Following the general procedure, starting from substrate 5 (40.2 mg, 0.059 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (16 μL, 0.059 mmol) was required. All the starting material was consumed after 7 h. Column chromatography (hexanes to hexanes–EtOAc, 6:4) gave an inseparable mixture of isomers 3-C-(2-O-acetyl-3,4,6-tri-O-benzyl-β-d-mannopyranosyl)1-propanol (29β) and 3-C-(2-O-acetyl-3,4,6-tri-O-benzyl-α-d-mannopyranosyl)1-propanol (29α) (16.1 mg, 0.030 mmol, 51%, 2.3:1) as a colorless oil. Compounds 29β and 29α: 1H NMR (500 MHz, selected resolved signals of 29β from the mix spectrum) δH 5.473 (dd, J = 2.2, 0.0 Hz, 1H, 2-H), 4.85 (d, J = 10.8 Hz, 1H, OBn), 4.74 (d, J = 11.4 Hz, 1H, OBn), 4.61 (d, J = 12.3 Hz, 1H, OBn), 4.54 (d, J = 12.0 Hz, 1H, OBn), 4.50 (d, J = 11.1 Hz, 1H, OBn), 4.49 (d, J = 11.4 Hz, 1H, OBn), 4.48 (d, J = 10.4 Hz, 1H, OBn), 2.17 ppm (s, 3H, OAc). 13C{1H} NMR (100.6 MHz, CDCl3, selected resolved signals of 29β from the mix spectrum) δC 170.9 (C, OAc), 138.3 (C, Ar), 138.2 (C, Ar), 137.8 (C, Ar), 81.9 (CH), 79.3 (CH), 77.2 (CH, C-1), 75.1 (CH2, OBn), 74.7 (CH), 73.5 (CH2, OBn), 71.6 (CH2, OBn), 69.6 (CH2, C-6), 69.56 (CH, C-2), 62.6 (CH2, C-3’), 29.5 (CH2, C-2’), 29.3 (CH2, C-2’), 28.17 (CH2, C-1’), 21.0 ppm (CH3, OAc). 1H NMR (500 MHz, CDCl3, selected resolved signals of 29α from the mix spectrum) δH 5.245 (dd, J = 2.8, 2.8 Hz, 1H, 2-H), 4.81 (d, J = 11.1 Hz, 1H, OBn), 4.65 (d, J = 11.4 Hz, 1H, OBn), 4.61 (d, J = 12.3 Hz, 1H, OBn), 4.51 (d, J = 11.7 Hz, 1H, OBn), 4.47 (d, J = 11.1 Hz, 1H, OBn), 3.99 (ddd, J = 10.7, 6.6, 3.8 Hz, 1H, 1-H), 3.85 (dd, J = 8.2, 3.2 Hz, 1H, 3-H), 2.13 ppm (s, 3H, OAc). 13C{1H} NMR (100.6 MHz, CDCl3, selected resolved signals of 29α from the mix spectrum) δC 170.6 (C, OAc), 138.3 (C, Ar), 138.2 (C, Ar) 137.8 (C, Ar), 77.7 (CH), 75.3 (CH2, OBn), 74.8 (CH), 74.7 (CH), 73.5 (CH2, OBn), 72.8 (CH), 71.9 (CH2, OBn), 70.9 (CH), 69.4 (CH2, C-6), 62.0 (CH2, C-3’), 24.95 (CH2, C-1’), 21.2 ppm (CH3, OAc). IR (CHCl3): ν = 3496, 3014, 2928, 1735, 1238, 1095 cm–1. MS (ESI) m/z (%) = 557 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C32H38NaO7 557.2515; found 557.2515. Anal. calcd for C32H38O7: C, 71.89; H, 7.16. Found: C, 71.55; H, 7.11.
Method D
Following the general procedure, starting from substrate 5 (30.2 mg, 0.044 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnD (12 μL, 0.044 mmol) was required. All the starting material was consumed after 5 h. Column chromatography (hexanes to hexanes–EtOAc, 7:3) gave the mixture of 3-C-(2-O-acetyl-3,4,6-tri-O-benzyl-β-d-(1-2H)mannopyranosyl)1-propanol [(1-2H)29β] and 29α (14.4 mg, 0.027 mmol, 61%, 2H/1H 1.8:1) as a colorless oil. Compounds (1-2H)29β and 29α: 1H NMR (500 MHz, CDCl3, selected resolved signals of (1-2H)29β from the mix spectrum) δH 4.74 (d, J = 11.0 Hz, 1H, OBn), 4.74 (d, J = 11.0 Hz, 1H, OBn), 4.59 (d, J = 12.0 Hz, 1H, OBn), 4.532 (d, J = 2.6 Hz, 1H, 2-H), 4.53 (d, J = 12.0 Hz, 1H, OBn), 4.46 (d, J = 11.0 Hz, 1H, OBn), 4.42 (d, J = 11.7 Hz, 1H, OBn), 4.10 (d, J = 12.2 Hz, 1H, OBn), 4.07 (dd, J = 2.6, 2.6 Hz, 1H, 3-H), 3.44 (dd, J = 10.1, 0.0 Hz, 1H, 6-Ha), 3.31 ppm (dd, J = 2.6, 1.0 Hz, 1H, 4-H). 13C{1H} NMR (125.7 MHz, CDCl3, selected resolved signals of (1-2H)29β from the mix spectrum) δC 150.4 (d, 2JPC = 7.0 Hz, 2 × C, Ar), 138.1 (C, Ar), 137.6 (C, Ar), 137.3 (C, Ar), 80.5 (d, 3JPC = 6.4 Hz, CH, C-3), 78.55 (d, 2JPC = 7.4 Hz, CH, C-2), 75.0 (CH, C-5), 73.5 (CH2, OBn), 72.2 (CH2, OBn), 71.5 (CH2, OBn), 71.5 (CH, C-4), 69.8 (CH2, C-6), 62.5 (CH2, C-3’), 29.68 (CH2, C-1’ or C-2’), 28.08 ppm (CH2, C-1’ or C-2’). IR (CHCl3): ν = 3490, 3422, 2928, 1550, 1491, 1192 cm–1. MS (ESI) m/z (%) = 747 (99.6) [M + Na]+, 748 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C42H45NaO9P 747.2699; found 747.2697, [M + Na]+ calcd for C42H442HNaO9P 748.2762; found 748.2764.
Radical Reactions of 6
Method A
Following the general procedure, starting from substrate 6 (39 mg, 0.045 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (12 μL, 0.045 mmol) was required. All the starting material was consumed after 6 h. Column chromatography (hexanes to hexanes–EtOAc, 1:1) gave 25 (9.7 mg, 0.020 mmol, 46%) and 3-C-(3,4,6-tri-O-benzyl-2-O-diphenoxyphosphoryl-β-d-mannopyranosyl)1-propanol (30α) (4 mg, 0.006 mmol, 12%) as a colorless oil. Compound 30α: [α]D = +1.6 (c = 0.77, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.36–7.11 (m, 25H, Ar), 4.90 (ddd, J = 3.2, 3.2 Hz, 3JPH = 6.3 Hz, 1H, 2-H), 4.74 (d, J = 11.4 Hz, 1H, OBn), 4.65 (d, J = 11.4 Hz, 1H, OBn), 4.56 (d, J = 12.0 Hz, 1H, OBn), 4.52 (d, J = 12.0 Hz, 1H, OBn), 4.47 (d, J = 11.4 Hz, 1H, OBn), 4.38 (d, J = 11.1 Hz, 1H, OBn), 4.04 (ddd, J = 10.1, 3.5, 3.5 Hz, 1H, 1-H), 3.85 (ddd, J = 8.2, 2.5 Hz, 4JPH = 2.5 Hz, 1H, 3-H), 3.76 (ddd, J = 8.5, 5.4, 3.5 Hz, 1H, 5-H), 3.69–3.56 (m, 5H, 3’-H2, 4-H, 6-H2), 1.78–1.47 ppm (m, 4H, 1’-H2, 2’-H2), 1H from OH is missing. Stereochemistry was assigned as 1α since the starting phthalimide 6 was obtained by the reaction with N-hydroxyphthalimide under Mitsunobu conditions. 13C{1H} NMR (125.7 MHz, CDCl3) δC 150.7 (C, Ar), 150.5 (C, Ar), 138.1 (C, Ar), 138.0 (C, Ar), 137.6 (C, Ar), 129.8 (2 × CH, Ar), 129.6 (2 × CH, Ar), 128.38 (2 × CH, Ar), 128.36 (2 × CH, Ar), 128.3 (2 × CH, Ar), 128.2 (2 × CH, Ar), 128.0 (2 × CH, Ar), 127.8 (4 × CH, Ar), 127.6 (CH, Ar), 125.4 (CH, Ar), 125.1 (CH, Ar), 120.4 (CH, Ar), 120.3 (CH, Ar), 120.23 (CH, Ar), 120.19 (CH, Ar), 77.7 (d, 2JPC = 6.4 Hz, CH, C-2), 77.6 (2 × CH, C-3, C-4), 74.5 (CH2, OBn), 74.4 (CH, C-1), 73.4 (CH2, OBn), 73.1 (CH, C-5), 72.1 (CH2, OBn), 69.1 (CH2, C-6), 61.8 (CH2, C-3’), 29.4 (CH2, C-1’ or C-2’), 29.1 ppm (CH2, C-1’ or C-2’). IR (CHCl3): ν = 3503, 2928, 1712, 1491, 1192 cm–1. MS (ESI) m/z (%) = 747 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C42H45NaO9P 747.2699; found 747.2692.
Method D
Following the general procedure, starting from substrate 6 (30.2 mg, 0.035 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnD (9 μL, 0.035 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes to hexanes–EtOAc, 1:1) gave (2-2H)25 (8.6 mg, 0.018 mmol, 52%) and 30α (4.7 mg, 0.005 mmol, 19%).
Method F
Following the general procedure, starting from substrate 6 (57.3 mg, 0.066 mmol), all the starting material was consumed after 1 h. Column chromatography (hexanes–EtOAc, 85:15 to 4:6) gave 25 (9 mg, 0.019 mmol, 29%) and 30α (24.2 mg, 0.033 mmol, 51%).
Method G
Following the general procedure, starting from substrate 6 (59 mg, 0.068 mmol), all the starting material was consumed after 3 h. Column chromatography (hexanes–EtOAc, 8:2 to 4:6) gave 25 (11.3 mg, 0.024 mmol, 35%) and 30α (22.1 mg, 0.031 mmol, 45%).
Radical Reactions of 7
Method A
Following the general procedure, starting from substrate 7 (74.6 mg, 0.11 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (30 μL, 0.11 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes to hexanes–EtOAc, 6:4) gave 3-C-(2-O-acetyl-3,4,6-tri-O-benzyl-β-d-mannopyranosyl)1-propanol (29β) (56.8 mg, 0.11 mmol, 97%) as an amorphous solid: [α]D = −27.6 (c = 0.87, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.35–7.15 (m, 15H, Ar), 5.474 (dd, J = 2.2, 0.0 Hz, 1H, 2-H), 4.85 (d, J = 10.7 Hz, 1H, OBn), 4.74 (d, J = 11.1 Hz, 1H, OBn), 4.61 (d, J = 12.0 Hz, 1H, OBn), 4.54 (d, J = 12.3 Hz, 1H, OBn), 4.48 (d, J = 11.0 Hz, 1H, OBn), 4.48 (d, J = 11.0 Hz, 1H, OBn), 3.74 (dd, J = 10.7, 1.9 Hz, 1H, 6-Hb), 3.70–3.63 (m, 5H, 3-H, 4-H, 5-H, 6-Ha, 3’-Hb), 3.51–3.46 (m, 2H, 1-H, 3’-Ha), 2.17 (s, 3H, OAc), 1.73–1.66 (m, 3H, 1’-Hb, 2’-H2), 1.572 ppm (m, 1H, 1’-Ha), 1H from OH is missing. 13C{1H} NMR (125.7 MHz, CDCl3) δC 170.9 (C, OAc), 138.3 (C, Ar), 138.2 (C, Ar), 137.8 (C, Ar), 127.5–128.3 (15 × CH, Ar), 81.8 (CH), 79.2 (CH), 77.1 (CH, C-1), 75.1 (CH2, OBn), 74.7 (CH), 73.4 (CH2, OBn), 71.5 (CH2, OBn), 69.49 (CH, C-2), 69.5 (CH2, C-6), 62.4 (CH2, C-3’), 29.3 (CH2, C-2’), 28.08 (CH2, C-1’), 20.9 ppm (CH3, OAc). IR (CHCl3): ν = 3430, 3015, 2936, 1735, 1091 cm–1. MS (ESI) m/z (%) = 557 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C32H38NaO7 557.2515; found 557.2519. Anal. calcd for C32H38O7: C, 71.89; H, 7.16. Found: C, 71.93; H, 7.08.
Method B
Following the general procedure, starting from substrate 7 (63.8 mg, 0.094 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (13 μL, 0.046 mmol) added by a syringe pump over 1 h was required. All the starting material was consumed after 5 h. Column chromatography (hexanes to hexanes–EtOAc, 6:4) gave 29β (38.3 mg, 0.072 mmol, 76%).
Method C
Following the general procedure, starting from substrate 7 (63.9 mg, 0.094 mmol), after 3 h of reaction, a supplementary addition of TTMSS (29 μL, 0.094 mmol) was required. All the starting material was consumed after 5 h. Column chromatography (hexanes–EtOAc, 7:3 to 1:1) gave 29β (28.8 mg, 0.054 mmol, 57%).
Method D
Following the general procedure, starting from substrate 7 (52.4 mg, 0.077 mmol), after 3 h of reaction, a supplementary addition of n-Bu3SnD (21 μL, 0.077 mmol) was required. All the starting material was consumed after 6 h. Column chromatography (hexanes to hexanes–EtOAc, 1:1) gave 3-C-(2-O-acetyl-3,4,6-tri-O-benzyl-β-d-[1-2H]mannopyranosyl)1-propanol ([1-2H]29β) (30.8 mg, 0.057 mmol, 75%, 2H/1H 5.8:1) as an amorphous solid: 1H NMR (500 MHz, CDCl3) δH 7.36–7.15 (m, 15H, Ar), 5.466 (d, J = 2.9 Hz, 1H, 2-H), 4.85 (d, J = 10.7 Hz, 1H, OBn), 4.74 (d, J = 11.1 Hz, 1H, OBn), 4.61 (d, J = 12.0 Hz, 1H, OBn), 4.54 (d, J = 12.3 Hz, 1H, OBn), 4.48 (d, J = 10.4 Hz, 1H, OBn), 4.48 (d, J = 10.4 Hz, 1H, OBn), 3.73 (dd, J = 10.8, 1.9 Hz, 1H, 6-Hb), 3.70–3.63 (m, 5H, 3-H, 4-H, 5-H, 6-Ha, 3’-Hb), 3.47 (ddd, J = 8.6, 6.0, 1.9 Hz, 1H, 3’-Ha), 2.18 (s, 3H, OAc), 1.71–1.66 (m, 3H, 1’-Hb, 2’-H2), 1.566 ppm (m, 1H, 1’-Ha), 1H from OH is missing. 13C{1H} NMR (125.7 MHz, CDCl3) δC 170.9 (C, OAc), 138.3 (C, Ar), 138.2 (C, Ar), 137.8 (C, Ar), 127.6–128.4 (15 × CH, Ar), 81.8 (CH), 79.2 (CH), 75.1 (CH2, OBn), 74.7 (CH), 73.4 (CH2, OBn), 71.5 (CH2, OBn), 69.50 (CH, C-2), 69.4 (CH2, C-6), 62.5 (CH2, C-3’), 29.4 (CH2, C-2’), 28.06 (CH2, C-1’), 21.0 ppm (CH3, OAc), C-1 was undetectable. MS (ESI) m/z (%) = 558 (100) [M + Na]+, 557 (16) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C32H372HNaO7 558.2578; found 558.2582, [M + Na]+ calcd for C32H38NaO7 557.2515; found 557.2511.
Method E
Following the general procedure, starting from substrate 7 (57.8 mg, 0.085 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnD (23 μL, 0.085 mmol) and BF3•Et2O (2 μL, 0.017 mmol) was required. All the starting material was consumed after 5 h. Column chromatography (hexanes to hexanes–EtOAc, 1:1) gave [2-2H]25 (20.2 mg, 0.043 mmol, 50%, 2H/1H 1.6:1) and [1-2H]29β (8.6 mg, 0.016 mmol, 19%, 2H/1H 3.5:1).
Radical Reactions of 8
Method A
Following the general procedure, starting from substrate 8 (60.4 mg, 0.07 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (19 μL, 0.07 mmol) was required. All the starting material was consumed after 6 h. Column chromatography (hexanes to hexanes–EtOAc, 8:2) gave 25 (12 mg, 0.025 mmol, 37%).
Method B
Following the general procedure, starting from substrate 8 (64.6 mg, 0.075 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (20 μL, 0.075 mmol) added by a syringe pump over 1 h was required. All the starting material was consumed after 11 h. Column chromatography (hexanes to hexanes–EtOAc, 8:2) gave 25 (13.7 mg, 0.029 mmol, 39%).
Method D
Following the general procedure, starting from substrate 8 (92.7 mg, 0.11 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnD (29 μL, 0.11 mmol) was required. All the starting material was consumed after 3 h. Column chromatography (hexanes to hexanes–EtOAc, 8:2) gave (2-2H)25 (26.3 mg, 0.055 mmol, 52%).
Method E
Following the general procedure, starting from substrate 8 (60 mg, 0.069 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnD (17 μL, 0.069 mmol) and BF3•Et2O (2 μL, 0.016 mmol) was required. All the starting material was consumed after 3 h. Column chromatography (hexanes to hexanes–EtOAc, 1:1) gave [2-2H]25 (20.2 mg, 0.043 mmol, 65%, 2H/1H 2.4:1).
Method F
Following the general procedure, starting from substrate 8 (33.6 mg, 0.039 mmol), all the starting material was consumed after 2 h. Column chromatography (hexanes–EtOAc, 8:2 to 1:1) gave 25 (4.1 mg, 8.6·10–3 mmol, 22%) and 3-C-(3,4,6-tri-O-benzyl-2-O-diphenoxyphosphoryl-β-d-mannopyranosyl)1-propanol (30β) (10.3 mg, 0.014 mmol, 37%) as a colorless oil. Compound 30β: [α]D = −27.9 (c = 0.10, CHCl3). 1H NMR (400 MHz, CDCl3) δH 7.39–7.05 (m, 25H, Ar), 5.01 (dd, J = 9.0, 2.1, 1H, 2-H), 4.90 (d, J = 11.3 Hz, 1H, OBn), 4.63 (d, J = 11.1 Hz, 1H, OBn), 4.60 (d, J = 12.5 Hz, 1H, OBn), 4.53 (d, J = 12.2 Hz, 1H, OBn), 4.51 (d, J = 11.4 Hz, 1H, OBn), 4.29 (d, J = 10.8 Hz, 1H, OBn), 3.67 (dd, J = 10.8, 1.8 Hz, 1H), 3.63–3.56 (m, 3H), 3.54–3.49 (m, 2H), 3.44–3.40 (m, 2H), 1.68–1.54 ppm (m, 4H, 1’-H2, 2’-H2), 1H from OH is missing. 13C{1H} NMR (100.6 MHz, CDCl3) δC 150.9 (d, 2JPC = 8.5 Hz, C, Ar), 150.7 (d, 2JPC = 6.4 Hz, C, Ar), 138.2 (C, Ar), 138.1 (C, Ar), 137.6 (C, Ar), 129.7 (2 × CH, Ar), 129.4 (2 × CH, Ar), 128.4 (2 × CH, Ar), 128.33 (4 × CH, Ar), 128.26 (2 × CH, Ar), 128.0 (2 × CH, Ar), 127.8 (2 × CH, Ar), 127.70 (CH, Ar), 127.65 (CH, Ar), 127.6 (CH, Ar), 125.3 (CH, Ar), 124.8 (CH, Ar), 120.4 (CH, Ar), 120.3 (CH, Ar), 120.22 (CH, Ar), 120.16 (CH, Ar), 81.8 (CH, C-3), 79.2 (CH, C-5), 77.3 (CH, C-2), 77.2 (d, 3JPC = 7.8 Hz, CH, C-1), 75.2 (CH2, OBn), 74.1 (CH, C-4), 73.4 (CH2, OBn), 71.7 (CH2, OBn), 69.3 (CH2, C-6), 62.4 (CH2, C-3’), 29.3 (CH2, C-1’ or C-2’), 28.1 ppm (CH2, C-1’ or C-2’). IR (CHCl3): ν = 3567, 2928, 2858, 1490, 1212 cm–1. MS (ESI) m/z (%) = 747 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C42H45NaO9P 747.2699; found 747.2709.
Method G
Following the general procedure, starting from substrate 8 (37.1 mg, 0.043 mmol), all the starting material was consumed after 3 h. Column chromatography (hexanes–EtOAc, 8:2 to 1:1) gave 25 (6.5 mg, 0.014 mmol, 32%) and 30β (7.8 mg, 0.011 mmol, 25%).
Radical Reactions of 9
Method A
Following the general procedure, starting from substrate 9 (64.4 mg, 0.11 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (30 μL, 0.11 mmol) was required. All the starting material was consumed after 5 h. Column chromatography (hexanes to hexanes–EtOAc, 6:4) gave (4S)-1,4-anhydro-6,7-di-O-benzyl-2,3,5,9-tetradeoxy-β-l-lyxo-non-4-ulopyranose (31S) contaminated with the thermodynamic isomer (4R)-1,4-anhydro-6,7-di-O-benzyl-2,3,5,9-tetradeoxy-α-l-lyxo-non-4-ulopyranose (31R) (19.2 mg, 0.05 mmol, 46%, S/R, 85:15) as an amorphous solid and an inseparable mixture of 3-C-(2-O-acetyl-3,4-di-O-benzyl-6-deoxy-β-d-altropyranosyl)1-propanol (32) and 3-C-(2-O-acetyl-3,4-di-O-benzyl-α-l-fucopyranosyl)1-propanol (33) (7.4 mg, 0.017 mmol, 15%, 1:1.7) as a colorless oil. Compound 31S: 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.41–7.25 (m, 10H, Ar), 4.955 (d, J = 12.0 Hz, 1H, OBn), 4.73 (d, J = 11.7 Hz, 1H, OBn), 4.63 (d, J = 12.0 Hz, 1H, OBn), 4.60 (d, J = 12.0 Hz, 1H, OBn), 3.93 (ddd, J = 12.1, 4.5, 2.7 Hz, 1H, 3-H), 3.87 (ddd, J = 8.2, 8.2, 5.4 Hz, 1H, 3’-Hb), 3.82 (dddd, J = 6.5, 6.5, 6.5, 1.7 Hz, 1H, 5-H), 3.81 (ddd, J = 8.2, 8.2, 6.6 Hz, 1H, 3’-Ha), 3.58 (ddd, J = 2.7, 1.7 Hz, 4J2a,4 = 1.3 Hz, 1H, 4-H), 2.32 (dd, J = 12.3, 12.1 Hz, 1H, 2-Hb), 2.13–2.00 (m, 2H, 1’-Hb, 2’-Hb), 1.92 (dd, J = 12.3, 4.5 Hz, 4J2a,4 = 1.3 Hz, 1H, 2-Ha), 1.90 (m, 1H, 2’-Ha), 1.75 (m, 1H, 1’-Ha), 1.14 ppm (d, J = 6.5 Hz, 3H, 6-H3), 1H NMR (500 MHz, C6D6, simulated ring coupling constants using DAISY) δΗ 7.44–7.42 (m, 2H, Ar), 7.38–7.35 (m, 2H, Ar), 7.22–7.13 (m, 6H, Ar), 5.05 (d, J = 11.5 Hz, 1H, C4-OBn), 4.59 (d, J = 11.5 Hz, 1H, C4-OBn), 4.44 (d, J = 12.1 Hz, 1H, C3-OBn), 4.40 (d, J = 12.1 Hz, 1H, C3-OBn), 4.02 (ddd, J = 12.0, 4.5, 2.7 Hz, 1H, 3-H), 3.87 (dddd, J = 6.5, 6.5, 6.5, 1.4 Hz, 1H, 5-H), 3.79–3.73 (m, 1H, 3’-Hb), 3.71–3.64 (m, 1H, 3’-Ha), 3.36 (ddd, J = 2.7, 1.4 Hz, 4J2a,4 = 1.2 Hz, 1H, 4-H), 2.49 (dd, J = 12.1, 12.0 Hz, 1H, 2-Hb), 2.03–2.00 (m, 1H), 1.94 (ddd, J = 12.1, 4.5 Hz, 4J2a,4 = 1.2 Hz, 1H, 2-Ha), 1.86–1.78 (m, 1H), 1.46–1.40 (m, 2H), 1.28 ppm (d, J = 6.5 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 139.0 (C, Ar), 138.8 (C, Ar), 128.5 (2 × CH, Ar), 128.4 (2 × CH, Ar), 128.1 (2 × CH, Ar), 127.2 (2 × CH, Ar), 127.2 (2 × CH, Ar), 106.8 (C, C-1), 77.3 (CH, C-3), 75.0 (CH, C-4), 74.16 (CH2, OBn), 70.5 (CH2, OBn), 67.6 (CH, C-5), 67.1 (CH2, C-3’), 37.5 (CH2, C-1’), 34.0 (CH2, C-2), 23.6 (CH2, C-2’), 17.4 ppm (CH3, C-6). 13C{1H} NMR (125.7 MHz, C6D6) δC 140.3 (C, Ar) some aromatic carbons were not observed, 140.0 (C, Ar), 107.4 (C, C-1), 78.0 (CH, C-3), 76.9 (CH, C-4), 75.3 (CH2, OBn), 70.8 (CH2, OBn), 68.5 (CH, C-5), 67.6 (CH2, C-3’), 38.2 (CH2, C-1’), 34.8 (CH2, C-2), 24.4 (CH2, C-2’), 18.1 ppm (CH3, C-6). IR (CHCl3): ν = 2930, 1226, 1206 cm–1. MS (ESI) m/z (%) = 391 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C23H28NaO4 391.1885; found 391.1891. Compounds 32 and 33: 1H NMR (500 MHz, CDCl3, selected signals of 32 from the mix spectrum) δH 4.95 (dd, J = 3.8, 1.6 Hz, 1H, 2-H), 4.72 (br s, 2H, OBn), 4.45 (d, J = 11.7 Hz, 1H, OBn), 4.34 (d, J = 11.7 Hz, 1H, OBn), 3.93 (dddd, J = 9.5, 6.3, 6.3, 6.3 Hz, 1H, 5-H), 3.88 (ddd, J = 9.2, 4.1, 1.3 Hz, 1H, 1-H), 3.248 (dd, J = 9.8, 3.2 Hz, 1H, 4-H), 1.286 ppm (d, J = 6.3 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3, selected signals of 32 from the mix spectrum) δC 170.4 (C, OAc), 138.0 (C, Ar), 137.7 (C, Ar), 77.67 (CH, C-4), 73.4 (CH, C-1), 72.6 (CH2, OBn), 71.7 (CH, C-3), 71.4 (CH, C-2), 71.3 (CH2, OBn), 71.2 (CH, C-5), 62.6 (CH2, C-3’), 27.6 (CH2, C-1’ or C-2’), 26.1 (CH2, C-1’ or C-2’), 8.19 ppm (CH3, C-6). 1H NMR (500 MHz, CDCl3, selected signals of 33 from the mix spectrum) δH 5.08 (dd, J = 5.1, 2.6 Hz, 1H, 2-H), 4.76 (d, J = 12.0 Hz, 1H, OBn), 4.67 (d, J = 12.0 Hz, 1H, OBn), 4.63 (d, J = 12.0 Hz, 1H, OBn), 4.54 (d, J = 12.0 Hz, 1H, OBn), 4.14–4.06 (m, 2H, 1-H, 5-H), 3.75 (dd, J = 4.8, 3.2 Hz, 1H, 4-H), 1.41 ppm (d, J = 6.9 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3, selected signals of 33 from the mix spectrum) δC 170.3 (C, OAc), 138.4 (C, Ar), 138.2 (C, Ar), 75.4 (CH, C-3), 74.2 (CH, C-4), 73.0 (CH2, OBn), 72.0 (CH2, OBn), 71.8 (CH, C-2), 70.0 (CH, C-5), 67.5 (CH, C-1), 62.6 (CH2, C-3’), 29.7 (CH2, C-1’ or C-2’), 29.4 (CH2, C-1’ or C-2’), 14.2 ppm (CH3, C-6). IR (CHCl3): ν = 3690, 3018, 1735, 1222 cm–1. MS (ESI) m/z (%) = 451 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C25H32NaO6 451.2097; found 451.2095.
Method D
Following the general procedure, starting from substrate 9 (59.2 mg, 0.10 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnD (28 μL, 0.10 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes to hexanes–EtOAc, 6:4) gave (4S)-1,4-anhydro-6,7-di-O-benzyl-2,3,5,9-tetradeoxy-β-l-[7-O-PhCH-2H]lyxo-non-4-ulopyranose ([PhCH-2H]31) (19.5 mg, 0.053 mmol, 53%, 2H/1H 1.5:1) and a mixture of 3-C-(2-O-acetyl-3,4-di-O-benzyl-6-deoxy-β-d-(5-2H)altropyranosyl)1-propanol (5-2H)32 and 33 (15.8 mg, 0.037 mmol, 36%, 2H/1H 1:2.8) as colorless oils. Compound [PhCH-2H]31: 1H NMR (500 MHz, CDCl3) δH 7.40–7.25 (m, 10H, Ar), 4.95 (d, J = 12.0 Hz, 0.4H, OBn), 4.93 (br s, 0.5H, O-CHD-Ph), 4.72 (d, J = 12.0 Hz, 0.4H, OBn), 4.71 (br s, 0.1H, O-CHD-Ph), 4.62 (d, J = 12.0 Hz, 1H, OBn), 4.59 (d, J = 12.0 Hz, 1H, OBn), 3.931 (ddd, J = 12.3, 4.4, 2.5 Hz, 0.5H, 3-H), 3.929 (ddd, J = 12.3, 5.0, 2.8 Hz, 0.5H, 3-H), 3.87 (ddd, J = 8.2, 8.2, 5.7 Hz, 1H, 3’-Hb), 3.82 (ddd, J = 8.2, 8.2, 6.6 Hz, 1H, 3’-Ha), 3.81 (m, 1H, 5-H), 3.57 (br s, 1H, 4-H), 2.32 (dd, J = 12.3, 12.3 Hz, 1H, 2-Hb), 2.13–1.99 (m, 2H, 1’-Hb, 2’-Hb), 1.92 (ddd, J = 12.3, 4.7 Hz, 4J2a,4 = 1.0 Hz, 1H, 2-Ha), 1.85 (m, 1H, 2’-Ha), 1.74 (ddd, J = 12.3, 10.4, 7.9 Hz, 1H, 1’-Ha), 1.135 (d, J = 6.3 Hz, 1.5H, 6-H3), 1.133 (d, J = 6.6 Hz, 0.3H, 6-H3), 1.130 ppm (d, J = 6.3 Hz, 1.2H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 139.0 (0.4C, Ar), 138.9 (0.6C, Ar), 138.8 (C, Ar), 128.49 (CH, Ar), 128.45 (CH, Ar), 128.4 (2 × CH, Ar), 128.1 (2 × CH, Ar), 127.4 (2 × CH, Ar), 127.2 (2 × CH, Ar), 106.8 (C, C-1), 77.7 (CH, C-3), 75.09 (0.4CH, C-4), 75.02 (0.1CH, C-4), 74.97 (0.5CH, C-4), 74.18 (0.4CH2, OBn), 73.59 (t, JCD = 22.1 Hz, 0.6CHD-Ph), 70.5 (CH2, OBn), 67.7 (CH, C-5), 67.1 (CH2, C-3’), 37.5 (CH2, C-1’), 34.0 (CH2, C-2), 23.6 (CH2, C-2’), 17.4 ppm (CH3, C-6). 1H NMR (500 MHz, C6D6) δH 7.44–7.42 (m, 2H, Ar), 7.36–7.35 (m, 2H, Ar), 7.22–7.10 (m, 6H, Ar), 5.046 (d, J = 11.6 Hz, 0.4H, OBn), 5.019 (br s, 0.5H, O-CHD-Ph), 4.59 (d, J = 11.5 Hz, 0.4H, OBn), 4.567 (br s, 0.1H, O-CHD-Ph), 4.44 (d, J = 12.1 Hz, 1H, OBn), 4.40 (d, J = 11.9 Hz, 1H, OBn), 4.02 (ddd, J = 12.1, 4.8, 2.8 Hz, 1H, 3-H), 3.89–3.85 (m, 1H, 5-H), 3.78–3.74 (m, 1H, 3’-Hb), 3.79–3.66 (m, 1H, 3’-Ha), 3.36 (m, 1H, 4-H), 2.496 (dd, J = 12.1, 12.1 Hz, 0.5H, 2-Hb), 2.494 (dd, J = 12.1, 12.1 Hz, 0.5H, 2-Hb), 2.02 (m, 1H), 1.94 (ddd, J = 12.6, 4.3 Hz, 4J2a,4 = 0.6 Hz, 1H, 2-Ha), 1.86–1.78 (m, 1H), 1.46–1.40 (m, 2H), 1.280 (d, J = 6.6 Hz, 1.5H, 6-H3), 1.276 ppm (d, J = 6.6 Hz, 1.5H, 6-H3). 13C{1H} NMR (125.7 MHz, C6D6) δC the aromatic carbons were not observed, 107.1 ppm (C, C-1), 77.7 (CH, C-3), 76.93 (0.4CH, C-4), 76.88 (0.1CH, C-4), 76.85 (0.5CH, C-4), 75.26 (0.4CH2, OBn), 74.85 (t, JCD = 22.1 Hz, 0.6CHD-Ph), 70.8 (CH2, OBn), 68.5 (CH, C-5), 67.6 (CH2, C-3’), 38.2 (CH2, C-1’), 34.8 (CH2, C-2), 24.4 (CH2, C-2’), 18.2 ppm (CH3, C-6). MS (ESI) m/z (%) = 392 (100) [M + Na]+, 391 (48) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C23H272HNaO4 392.1948; found 392.1936, [M + Na]+ calcd for C23H28NaO4 391.1885; found 391.1891. Mixture of (5-2H)32/33: 1H NMR (400 MHz, CDCl3, only the deuterated product (5-2H)32 is described) δH 7.40–7.22 (m, 10H, Ar), 4.95 (dd, J = 3.5, 1.2 Hz, 1H, 2-H), 4.72 (br s, 2H, OBn), 4.45 (d, J = 11.7 Hz, 1H, OBn), 4.34 (d, J = 11.7 Hz, 1H, OBn), 3.88 (ddd, J = 9.1, 5.3, 1.3 Hz, 1H, 1-H), 3.80 (m, 1H, 3-H), 3.69–3.58 (m, 2H, 3’-H2), 3.248 (d, J = 3.1 Hz, 1H, 4-H), 2.06 (s, 3H, OAc), 1.67–1.45 (m, 4H, 1’-H2, 2’-H2), 1.280 ppm (s, 3H, 6-H3), 1H from OH is missing. 13C{1H} NMR (100.6 MHz, CDCl3, only the deuterated product (5-2H)32 is described) δC 170.3 (C, OAc), 138.0 (C, Ar), 137.7 (C, Ar), 127.5–128.3 (10 × CH, Ar), 77.48 (CH, C-4), 73.3 (CH, C-1), 72.7 (CH2, OBn), 71.8 (CH, C-3), 71.4 (CH, C-2), 71.4 (CH2, OBn), 62.6 (CH2, C-3’), 27.6 (CH2, C-1’ or C-2’), 26.1 (CH2, C-1’ or C-2’), 21.0 (CH3, OAc), 18.07 ppm (CH3, C-6). MS (ESI) m/z (%) = 452 (100) [M + Na]+, 451 (75) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C25H312HNaO6 452.2159; found 452.2159, [M + Na]+ calcd for C25H32NaO6 451.2097; found 451.2098.
Method F
Following the general procedure, starting from substrate 9 (17.9 mg, 0.031 mmol), all the starting material was consumed after 3 h. Column chromatography (hexanes–EtOAc, 6:4 to 4:6) gave 33 (8.7 mg, 0.020 mmol, 65%).
Method G
Following the general procedure, starting from substrate 9 (18.2 mg, 0.032 mmol), all the starting material was consumed after 3 h. Column chromatography (hexanes–EtOAc, 6:4 to 4:6) gave 33 (13.1 mg, 0.031 mmol, 67%).
Radical Reactions of 10
Method A
Following the general procedure, starting from substrate 10 (95.8 mg, 0.13 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (15 μL, 0.07 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes to hexanes–EtOAc, 4:6) gave 31 (24.7 mg, 0.07 mmol, 52%, S/R, 83:17), 2,6:5,9-di-anhydro-3,4-di-O-benzyl-1,7,8-trideoxy-d-glycero-d-galacto-nonitol (36) (4.6 mg, 0.013 mmol, 10%), 3-C-(3,4-di-O-benzyl-6-deoxy-2-O-diphenoxyphosphoryl-β-d-altropyranosyl)1-propanol (34) (5.1 mg, 0.008 mmol, 6%), and 3-C-(3,4-di-O-benzyl-2-O-diphenoxyphosphoryl-α-l-fucopyranosyl)1-propanol (35) (9.7 mg, 0.016 mmol, 12%), all as colorless oils. Compound 36: [α]D = +2.2 (c = 0.27, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.42–7.27 (m, 10H, Ar), 4.98 (d, J = 11.7 Hz, 1H, OBn), 4.88 (d, J = 12.3 Hz, 1H, OBn), 4.71 (d, J = 11.7 Hz, 1H, OBn), 4.70 (d, J = 12.3 Hz, 1H, OBn), 3.98 (m, 1H, 3’-Hb), 3.629 (dd, J = 9.8, 9.1 Hz, 1H, 2-H), 3.62 (dd, J = 3.0, 1.3 Hz, 1H, 4-H), 3.52 (dd, J = 9.8, 3.0 Hz, 1H, 3-H), 3.51 (dddd, J = 6.4, 6.4, 6.4, 1.3 Hz, 1H, 5-H), 3.46 (m, 1H, 3’-Ha), 3.06 (ddd, J = 11.1, 9.1, 4.3 Hz, 1H, 1-H), 2.038 (m, 1H, 1’-Hb), 1.75–1.70 (m, 2H, 2’-H2), 1.611 (m, 1H, 1’-Ha), 1.16 ppm (d, J = 6.4 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 139.0 (C, Ar), 138.7 (C, Ar), 128.6 (2 × CH, Ar), 128.4 (2 × CH, Ar), 128.2 (2 × CH, Ar), 127.6 (CH, Ar), 127.52 (2 × CH, Ar), 127.47 (CH, Ar), 81.9 (CH, C-3), 79.60 (CH, C-2 or C-4), 77.88 (CH, C-2 or C-4), 76.4 (CH, C-1), 74.9 (CH, C-5), 74.9 (CH2, OBn), 73.0 (CH2, OBn), 67.9 (CH2, C-3’), 29.12 (CH2, C-1’), 25.6 (CH2, C-2’), 17.3 ppm (CH3, C-6). IR (CHCl3): ν = 3017, 1454, 1220 cm–1. MS (ESI) m/z (%) = 391 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C23H28NaO4 391.1885; found 391.1878. Compound 34: [α]D = +10.0 (c = 0.50, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.37–7.20 (m, 20H, Ar), 4.68 (d, J = 12.0 Hz, 1H, OBn), 4.62 (d, J = 12.0 Hz, 1H, OBn), 4.59 (m, 1H, 2-H), 4.21 (d, J = 11.7 Hz, 1H, OBn), 4.11 (d, J = 11.7 Hz, 1H, OBn), 4.03 (dd, J = 3.2, 3.2 Hz, 1H, 3-H), 3.90–3.81 (m, 2H, 1-H, 5-H), 3.57–3.54 (m, 2H, 3’-H2), 3.240 (dd, J = 9.8, 2.9 Hz, 1H, 4-H), 1.63–1.50 (m, 4H, 1’-H2, 2’-H2), 1.262 ppm (d, J = 6.3 Hz, 3H, 6-H3), 1H from OH is missing. 13C{1H} NMR (125.7 MHz, CDCl3) δC 150.5 (2 × C, Ar), 137.8 (2 × C, Ar), 129.9 (2 × CH, Ar), 129.8 (2 × CH, Ar), 128.4 (2 × CH, Ar), 128.3 (2 × CH, Ar), 128.1 (2 × CH, Ar), 127.9 (CH, Ar), 127.7 (3 × CH, Ar), 125.6 (2 × CH, Ar), 120.2 (CH, Ar), 120.1 (CH, Ar), 120.09 (CH, Ar), 120.06 (CH, Ar), 77.99 (CH, C-4), 77.3 (CH, C-2), 73.5 (d, 3JPC = 6.4 Hz, CH, C-1), 73.0 (CH2, OBn), 72.2 (CH, C-3), 71.5 (CH2, OBn), 71.1 (CH, C-5), 62.6 (CH2, C-3’), 29.3 (CH2, C-1’), 27.5 (CH2, C-2’), 18.23 ppm (CH3, C-6). IR (CHCl3): ν = 3426, 3022, 1490, 1220 cm–1. MS (ESI) m/z (%) = 641 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C35H39NaO8P 641.2280; found 641.2288. Compound 35: [α]D = −24.8 (c = 0.72, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.33–7.16 (m, 20H, Ar), 4.83 (ddd, J = 6.0, 2.8 Hz, 3JPH = 8.5 Hz, 1H, 2-H), 4.70 (d, J = 11.7 Hz, 1H, OBn), 4.63 (d, J = 11.7 Hz, 1H, OBn), 4.54 (d, J = 12.0 Hz, 1H, OBn), 4.41 (d, J = 11.7 Hz, 1H, OBn), 4.09–4.04 (m, 2H, 1-H, 5-H), 3.92 (dd, J = 5.7, 3.2 Hz, 1H, 3-H), 3.721 (dd, J = 3.8, 3.8 Hz, 1H, 4-H), 3.55 (br s, 2H, 3’-H2), 1.84 (br s, 1H, OH), 1.63–1.52 (m, 4H, 1’-H2, 2’-H2), 1.361 ppm (d, J = 6.9 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 150.5 (d, 2JPC = 7.4 Hz, C, Ar), 150.5 (d, 2JPC = 7.4 Hz, C, Ar), 138.2 (C, Ar), 138.0 (C, Ar), 129.81 (2 × CH, Ar), 129.76 (2 × CH, Ar), 128.4 (2 × CH, Ar), 128.3 (2 × CH, Ar), 127.7 (3 × CH, Ar), 127.62 (CH, Ar), 127.56 (2 × CH, Ar), 125.4 (2 × CH, Ar), 120.2 (CH, Ar), 120.12 (CH, Ar), 120.06 (CH, Ar), 120.0 (CH, Ar), 77.5 (CH, C-2), 75.9 (CH, C-3), 74.65 (CH, C-4), 73.2 (CH2, OBn), 72.4 (CH2, OBn), 69.6 (CH, C-5), 68.5 (d, 3JPC = 4.2 Hz, CH, C-1), 62.4 (CH2, C-3’), 29.7 (CH2, C-1’), 29.2 (CH2, C-2’), 14.44 ppm (CH3, C-6). IR (CHCl3): ν = 3454, 3015, 1490, 1205 cm–1. MS (ESI) m/z (%) = 641 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C35H39NaO8P 641.2280; found 641.2278.
Method D
Following the general procedure, starting from substrate 10 (85.2 mg, 0.11 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnD (15 μL, 0.07 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes to hexanes–EtOAc, 4:6) gave [PhCH-2H]31 (20.1 mg, 0.05 mmol, 49%, 2H/1H 1.7:1), 2,6:5,9-di-anhydro-3,4-di-O-benzyl-1,7,8-trideoxy-d-[6-2H]glycero-d-galacto-nonitol [(1-2H)36] (4.5 mg, 0.012 mmol, 11%, 2H/1H 8:1), and 3-C-(3,4-di-O-benzyl-6-deoxy-2-O-diphenoxyphosphoryl-β-d-(5-2H)altropyranosyl)1-propanol [(5-2H)34] (4.3 mg, 0.007 mmol, 6%) as colorless oils and 35 (6 mg, 0.01 mmol, 9%). Compound (1-2H)36: 1H NMR (500 MHz, only the deuterated product is described) δH 7.42–7.26 (m, 10H, Ar), 4.98 (d, J = 11.7 Hz, 1H, OBn), 4.88 (d, J = 12.3 Hz, 1H, OBn), 4.71 (d, J = 11.7 Hz, 1H, OBn), 4.70 (d, J = 12.3 Hz, 1H, OBn), 3.98 (m, 1H, 3’-Hb), 3.637 (d, J = 9.8 Hz, 1H, 2-H), 3.62 (dd, J = 2.8, 1.0 Hz, 1H, 4-H), 3.52 (dd, J = 9.8, 2.8 Hz, 1H, 3-H), 3.51 (dddd, J = 6.3, 6.3, 6.3, 1.3 Hz, 1H, 5-H), 3.46 (m, 1H, 3’-Ha), 2.029 (m, 1H, 1’-Hb), 1.74–1.70 (m, 2H, 2’-H2), 1.606 (m, 1H, 1’-Ha), 1.16 ppm (d, J = 6.7 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3, only the deuterated product is described) δC 138.9 (C, Ar), 138.6 (C, Ar), 128.5 (2 × CH, Ar), 128.3 (2 × CH, Ar), 128.1 (2 × CH, Ar), 127.54 (CH, Ar), 127.49 (2 × CH, Ar), 127.4 (CH, Ar), 81.9 (CH, C-3), 79.49 (CH, C-2), 77.84 (CH, C-4), 74.8 (CH, C-5), 74.8 (CH2, OBn), 73.0 (CH2, OBn), 67.9 (CH2, C-3’), 28.99 (CH2, C-1’), 25.6 (CH2, C-2’), 17.3 ppm (CH3, C-6). MS (ESI) m/z (%) = 392 (100) [M + Na]+, 391 (12) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C23H272HNaO4 392.1948; found 392.1954. Compound (5-2H)34: 1H NMR (500 MHz, CDCl3) δH 7.37–7.20 (m, 20H, Ar), 4.68 (d, J = 12.0 Hz, 1H, OBn), 4.62 (d, J = 12.0 Hz, 1H, OBn), 4.57 (m, 1H, 2-H), 4.20 (d, J = 11.7 Hz, 1H, OBn), 4.11 (d, J = 11.7 Hz, 1H, OBn), 4.02 (dd, J = 3.5, 3.5 Hz, 1H, 3-H), 3.83 (m, 1H, 1-H), 3.57–3.54 (m, 2H, 3’-H2), 3.235 (d, J = 2.9 Hz, 1H, 4-H), 2.00 (br s, 1H, OH), 1.64–1.50 (m, 4H, 1’-H2, 2’-H2), 1.248 ppm (s, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 150.5 (2 × C, Ar), 137.9 (2 × C, Ar), 129.9 (2 × CH, Ar), 129.8 (2 × CH, Ar), 128.4 (2 × CH, Ar), 128.3 (2 × CH, Ar), 128.1 (2 × CH, Ar), 127.9 (CH, Ar), 127.7 (2 × CH, Ar), 127.4 (CH, Ar), 125.5 (2 × CH, Ar), 120.15 (CH, Ar), 120.10 (CH, Ar), 120.08 (CH, Ar), 120.04 (CH, Ar), 77.91 (CH, C-4), 77.3 (CH, C-2), 73.5 (d, 3JPC = 6.4 Hz, CH, C-1), 73.0 (CH2, OBn), 72.2 (CH, C-3), 71.5 (CH2, OBn), 62.6 (CH2, C-3’), 29.3 (CH2, C-1’), 27.5 (CH2, C-2’), 18.10 ppm (CH3, C-6). MS (ESI) m/z (%) = 642 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C35H382HNaO8P 642.2280; found 642.2291.
Radical Reactions of 11
Method A
Following the general procedure, starting from substrate 11 (68.2 mg, 0.10 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (29 μL, 0.10 mmol) was required. All the starting material was consumed after 3 h. Column chromatography (hexanes to hexanes–EtOAc, 9:1) gave (4S)-1,4-anhydro-6,7-O-benzylidene-2,3,5-trideoxy-d-erythro-oct-4-ulopyranose (37) (15.8 mg, 0.06 mmol, 60%) as a colorless oil: [α]D = −109.3 (c = 0.28, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.48–7.46 (m, 2H, Ar), 7.40–7.35 (m, 3H, Ar), 6.20 (s, 1H, PhCH), 4.700 (ddd, J = 8.7, 6.3, 5.5 Hz, 1H, 3-H), 4.10 (ddd, J = 5.5, 2.4, 1.0 Hz, 1H, 4-H), 4.03 (dd, J = 13.3, 1.0 Hz, 1H, 5-Hb), 4.02 (dd, J = 13.3, 2.4 Hz, 1H, 5-Ha), 3.95 (ddd, J = 8.2, 8.2, 5.7 Hz, 1H, 3’-Hb), 3.88 (ddd, J = 8.2, 8.2, 6.3 Hz, 1H, 3’-Ha), 2.11 (dd, J = 13.5, 8.7 Hz, 1H, 2-Hb), 2.05 (dd, J = 13.5, 6.3 Hz, 1H, 2-Ha), 2.18–2.04 (m, 2H, 1’-Hb, 2’-Hb), 1.93 (m, 1H, 2’-Ha), 1.80 ppm (ddd, J = 12.3, 10.1, 7.9 Hz, 1H, 1’-Ha). 13C{1H} NMR (125.7 MHz, CDCl3) δC 139.3 (C, Ar), 128.9 (CH, Ar), 128.4 (2 × CH, Ar), 126.1 (2 × CH, Ar), 105.75 (C, C-1), 102.6 (CH, PhCH), 71.83 (CH, C-4), 71.72 (CH, C-3), 67.4 (CH2, C-3’), 60.6 (CH2, C-5), 37.9 (CH2, C-1’), 34.0 (CH2, C-2), 23.6 ppm (CH2, C-2’). IR (CHCl3): ν = 3024, 1458, 1210 cm–1. MS (ESI) m/z (%) = 285 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C15H18NaO4 285.1103; found 285.1103. Anal. calcd for C15H18O4: C, 68.68; H, 6.92. Found: C, 68.68; H, 7.00.
Method D
Following the general procedure, starting from substrate 11 (72.6 mg, 0.11 mmol), after 2 h, all the starting material was consumed. Column chromatography (hexanes to hexanes–EtOAc, 9:1) gave (4S)-1,4-anhydro-6,7-O-benzylidene-2,3,5-trideoxy-d-[5-2H]erythro-oct-4-ulopyranose ([2-2H]37) (15.7 mg, 0.06 mmol, 54%, 2H/1H 1:1) as a colorless oil: 1H NMR (500 MHz, CDCl3, only the deuterated compound is described) δH 7.48–7.46 (m, 2H, Ar), 7.40–7.34 (m, 3H, Ar), 6.20 (s, 1H, PhCH), 4.701 (dd, J = 8.6, 5.4 Hz, 1H, 3-H), 4.11 (ddd, J = 5.4, 2.3, 1.3 Hz, 1H, 4-H), 4.03 (dd, J = 13.6, 1.0 Hz, 1H, 5-Hb), 4.00 (dd, J = 13.3, 2.2 Hz, 1H, 5-Ha), 3.94 (ddd, J = 8.2, 8.2, 5.7 Hz, 1H, 3’-Hb), 3.88 (ddd, J = 8.2, 8.2, 6.3 Hz, 1H, 3’-Ha), 2.18–2.04 (m, 3H, 2-H, 1’-Hb, 2’-Hb), 1.92 (m, 1H, 2’-Ha), 1.79 ppm (ddd, J = 12.3, 10.1, 7.9 Hz, 1H, 1’-Ha). 13C{1H} NMR (125.7 MHz, CDCl3, only the deuterated compound is described) δC 139.3 (C, Ar), 128.9 (CH, Ar), 128.4 (2 × CH, Ar), 126.1 (2 × CH, Ar), 105.72 (C, C-1), 102.5 (CH, PhCH), 71.80 (CH, C-3 or C-4), 71.67 (CH, C-3 or C-4), 67.4 (CH2, C-3’), 60.6 (CH2, C-5), 37.9 (CH2, C-1’), 33.69 (t, JCD = 20.1 Hz, CHD, C-2), 23.6 ppm (CH2, C-2’). MS (ESI) m/z (%) = 286 (100) [M + Na]+, 285 (83) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C15H172HNaO4 286.1166; found 286.1154, [M + Na]+ calcd for C15H18NaO4 285.1103; found 285.1107.
Method E
Following the general procedure, starting from substrate 11 (60 mg, 0.091 mmol), after 2 h, all the starting material was consumed. Column chromatography (hexanes to hexanes–EtOAc, 9:1) gave [2-2H]37 (14.4 mg, 0.055 mmol, 60%, 2H/1H 1:1).
Radical Reactions of 12
Method A
Following the general procedure, starting from substrate 12 (65.2 mg, 0.11 mmol), all the starting material was consumed after 2 h. Column chromatography (DCM to DCM–MeOH, 97:3) gave (4S)-1,4-anhydro-2,3,5-trideoxy-d-erythro-oct-4-ulopyranose (38) (10.4 mg, 0.06 mmol, 54%) as a colorless oil: [α]D = −57.3 (c = 0.62, CHCl3). 1H NMR (400 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 4.055 (ddd, J = 11.7, 5.3, 3.5 Hz, 1H, 3-H), 3.90 (dd, J = 12.5, 1.6 Hz, 1H, 5-Hb), 3.96–3.83 (m, 2H, 3’-H2), 3.80 (br s, 1H, 4-H), 3.72 (dd, J = 12.5, 2.3 Hz, 1H, 5-Ha), 2.31 (br s, 1H, OH), 2.17 (br s, 1H, OH), 2.09–2.01 (m, 2H, 1’-Hb, 2’-Hb), 1.99 (dd, J = 12.8, 11.7 Hz, 1H, 2-Hb), 1.88 (m, 1H, 2’-Ha), 1.86 (dd, J = 12.8, 5.3 Hz, 1H, 2-Ha), 1.72 ppm (m, 1H, 1’-Ha). 13C{1H} NMR (125.7 MHz, CDCl3) δC 106.63 (C, C-1), 67.9 (CH, C-4), 67.5 (CH2, C-3’), 66.59 (CH, C-3), 63.7 (CH2, C-5), 37.5 (CH2, C-1’), 36.83 (CH2, C-2), 23.3 ppm (CH2, C-2’). IR (CHCl3): ν = 3685, 3571, 3020, 1056 cm–1. MS (ESI) m/z (%) = 197 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C8H14NaO4 197.0790; found 197.0789. Anal. calcd for C8H14O4: C, 55.16; H, 8.10. Found: C, 54.97; H, 7.92.
Method D
Following the general procedure, starting from substrate 12 (64.2 mg, 0.11 mmol), after 2 h, all the starting material was consumed. Column chromatography (DCM to DCM–MeOH, 97:3) gave (4S)-1,4-anhydro-2,3,5-trideoxy-d-[5-2H]erythro-oct-4-ulopyranose ([2-2H]38) (12.2 mg, 0.07 mmol, 63%, 2H/1H 2.3:1) as a colorless oil: 1H NMR (500 MHz, CDCl3, only the deuterated compound is described) δH 4.049 (m, 1H, 3-H), 3.95–3.85 (m, 2H, 3’-H2), 3.90 (dd, J = 12.6, 1.3 Hz, 1H, 5-Hb), 3.80 (br s, 1H, 4-H), 3.72 (dd, J = 12.6, 2.2 Hz, 1H, 5-Ha), 2.22 (br s, 1H, OH), 2.09–2.00 (m, 2H, 1’-Hb, 2’-Hb), 1.92–1.85 (m, 2H, 2-H, 2’-Ha), 1.72 ppm (m, 1H, 1’-Ha), 1H from OH is missing. 13C{1H} NMR (125.7 MHz, CDCl3, only the deuterated compound is described) δC 106.57 (C, C-1), 67.9 (CH2, C-4), 67.5 (CH2, C-3’), 66.52 (CH, C-3), 63.7 (CH, C-5), 37.4 (CH2, C-1’), 23.3 ppm (CH2, C-2’), C-2 was undetectable. MS (ESI) m/z (%) = 198 (100) [M + Na]+, 197 (44) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C8H132HNaO4 198.0853; found 198.0856, [M + Na]+ calcd for C8H14NaO4 197.0790; found 197.0796.
Synthesis of 9-Deoxy-1,6-dioxaspiro[4.4]nonane structures (Table 4)
Radical Reactions of 13
Method A
Following the general procedure, starting from substrate 13 (102 mg, 0.12 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (32 μL, 0.12 mmol) was required. All the starting material was consumed after 3 h. Column chromatography on a silica gel without KF (hexanes to hexanes–EtOAc, 8:2) gave 3-C-(2-O-acetyl-3,5-di-O-tert-butyldiphenylsilyl-α-d-ribofuranosyl)1-propanol (40) (51.2 mg, 0.072 mmol, 56%) as a colorless oil: [α]D = +46.0 (c = 0.61, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.65–7.26 (m, 20H, Ar), 5.12 (dd, J = 4.4, 4.4 Hz, 1H, 2-H), 4.61 (dd, J = 6.7, 4.5 Hz, 1H, 3-H), 4.03 (m, 1H, 4-H), 3.97 (m, 1H, 1-H), 3.66–3.60 (m, 3H, 5-Hb, 3’-H2), 3.32 (dd, J = 11.4, 3.5 Hz, 1H, 5-Ha), 2.13 (s, 3H, OAc), 1.69–1.53 (m, 4H, 1’-H2, 2’-H2), 1.02 (s, 9H, tBu), 0.91 ppm (s, 9H, tBu). 13C{1H} NMR (125.7 MHz, CDCl3) δC 170.4 (C, OAc). 135.78 (2 × CH, Ar), 135.72 (2 × CH, Ar), 135.60 (2 × CH, Ar), 135.55 (2 × CH, Ar), 133.3 (C, Ar), 133.24 (C, Ar), 133.22 (C, Ar), 132.7 (C, Ar), 129.98 (CH, Ar), 129.96 (CH, Ar), 129.5 (2 × CH, Ar), 127.76 (2 × CH, Ar), 127.72 (2 × CH, Ar), 127.56 (2 × CH, Ar), 127.54 (2 × CH, Ar), 82.8 (CH, C-4), 79.5 (CH, C-1), 75.0 (CH, C-2), 72.8 (CH, C-3), 63.6 (CH2, C-5), 62.6 (CH2, C-3’), 29.6 (CH2, C-1’ or C-2’), 26.8 (3 × CH3, tBu), 26.7 (3 × CH3, tBu), 26.5 (CH2, C-1’ or C-2’), 21.0 (CH3, OAc), 19.2 (C, tBu), 19.1 ppm (C, tBu). IR (CHCl3): ν = 3451, 2932, 1735, 1216, 1113 cm–1. MS (ESI) m/z (%) = 733 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C42H54NaO6Si2 733.3357; found 733.3360. Anal. calcd for C42H54O6Si2: C, 70.95; H, 7.66. Found: C, 71.05; H, 7.82.
Method D
Following the general procedure, starting from substrate 13 (98.4 mg, 0.12 mmol), after 2 h of reaction and again after 4 h, a supplementary addition of n-Bu3SnD (31 μL, 0.12 mmol) was required. All the starting material was consumed after 5 h. Column chromatography on a silica gel without KF (hexanes to hexanes–EtOAc, 8:2) gave 3-C-(2-O-acetyl-3,5-di-O-tert-butyldiphenylsilyl-α-d-[1-2H]ribofuranosyl)1-propanol ([1-2H]40) (46.1 mg, 0.065 mmol, 54%, 2H/1H 2:1) as a colorless oil: 1H NMR (500 MHz, CDCl3, only the deuterated compound is described) δH 7.65–7.26 (m, 20H, Ar), 5.117 (d, J = 4.7 Hz, 1H, 2-H), 4.61 (dd, J = 6.9, 4.7 Hz, 1H, 3-H), 4.03 (m, 1H, 4-H), 3.66–3.60 (m, 3H, 5-Hb, 3’-H2), 3.31 (dd, J = 11.4, 3.5 Hz, 1H, 5-Ha), 2.13 (s, 3H, OAc), 1.69–1.53 (m, 4H, 1’-H2, 2’-H2), 1.02 (s, 9H, tBu), 0.91 ppm (s, 9H, tBu). 13C{1H} NMR (100.6 MHz, CDCl3) δC 170.4 (C, OAc), 135.79 (2 × CH, Ar), 135.73 (2 × CH, Ar), 135.61 (2 × CH, Ar), 135.56 (2 × CH, Ar), 133.4 (C, Ar), 133.28 (C, Ar), 133.25 (C, Ar), 132.7 (C, Ar), 129.98 (CH, Ar), 129.95 (CH, Ar), 129.5 (2 × CH, Ar), 127.76 (2 × CH, Ar), 127.72 (2 × CH, Ar), 127.56 (2 × CH, Ar), 127.54 (2 × CH, Ar), 82.8 (CH, C-4), 74.96 (CH, C-2), 72.8 (CH, C-3), 63.7 (CH2, C-5), 62.6 (CH2, C-3’), 29.6 (CH2, C-1’ or C-2’), 26.8 (3 × CH3, tBu), 26.7 (3 × CH3, tBu), 26.5 (CH2, C-1’ or C-2’), 21.0 (CH3, OAc), 19.2 (C, tBu), 19.1 ppm (C, tBu). MS (ESI) m/z (%) = 734 (100) [M + Na]+, 733 (34) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C42H532HNaO6Si2 734.3419; found 734.3417, [M + Na]+ calcd for C42H54NaO6Si2 733.3357; found 733.3351.
Method E
Following the general procedure, starting from substrate 13 (128.6 mg, 0.15 mmol), after 2 h, a supplementary addition of n-Bu3SnD (41 μL, 0.15 mmol) and BF3•Et2O (4 μL, 0.03 mmol) was required. All the starting material was consumed after 5 h. Column chromatography on a silica gel without KF (hexanes to hexanes–EtOAc, 75:25) gave (4RS)-1,4-anhydro-5-O-acetyl-6,8-bis-O-tert-butyldiphenylsilyl-2,3-dideoxy-d-[5-2H]ribo-oct-4-ulofuranose ([2-2H]39) (39.1 mg, 0.06 mmol, 40%, 2H/1H 1.3:1, 1R/1S 1:1) as a colorless oil and (1-2H)40 (13.9 mg, 0.02 mmol, 13%). Compound [2-2H]39: 1H NMR (500 MHz, CDCl3, only nondeuterated products of both isomers are described) δH 7.72–7.25 (m, 40H, Ar), 4.44 (m, 2H, 3-H), 4.12–4.08 (m, 2H, 4-H), 4.00 (ddd, J = 8.5, 8.5, 5.4 Hz, 1H, 3’-Hb), 3.90 (ddd, J = 7.6, 7.6, 7.6 Hz, 1H, 3’-Ha), 3.83 (ddd, J = 8.2, 8.2, 4.5 Hz, 1H, 3’-Hb), 3.70 (ddd, J = 7.6, 7.6, 7.6 Hz, 1H, 3’-Ha), 3.62 (dd, J = 11.4, 2.6 Hz, 1H, 5-Hb), 3.50 (dd, J = 10.8, 5.4 Hz, 1H, 5-Hb), 3.45 (dd, J = 11.0, 5.7 Hz, 1H, 5-Ha), 3.40 (dd, J = 11.0, 3.5 Hz, 1H, 5-Ha), 2.22 (dd, J = 9.5, 9.5 Hz, 1H, 1’-Hb), 2.12 (m, 1H, 2’-Hb), 2.10–1.96 (m, 6H, 1’-Hb, 2 × 2’-H2, 2’-Ha, 2 × 2-Hb), 1.88–1.81 (m, 3H, 1’-Ha, 2 × 2-Ha), 1.73 (ddd, J = 12.0, 8.9, 8.9 Hz, 1H, 1’-Ha), 1.06 (s, 18H, tBu), 0.96 (s, 9H, tBu), 0.93 ppm (s, 9H, tBu). 13C{1H} NMR (125.7 MHz, CDCl3, only nondeuterated products of both isomers are described) δC 134.07 (C, Ar), 133.87 (C, Ar), 133.81 (C, Ar), 133.63 (2 × C, Ar), 133.61 (C, Ar), 133.59 (C, Ar), 133.5 (C, Ar), 127.5–135.9 (40 × CH, Ar), 114.8 (C, C-1), 114.2 (C, C-1), 86.9 (CH, C-4), 85.9 (CH, C-4), 74.1 (CH, C-3), 72.9 (CH, C-3), 67.4 (CH2, C-3’), 67.1 (CH2, C-3’), 65.0 (CH2, C-5), 63.4 (CH2, C-5), 44.3 (CH2, C-2’), 43.5 (CH2, C-2’), 36.8 (CH2, C-1’), 36.1 (CH2, C-1’), 27.0 (3 × CH3, tBu), 26.9 (3 × CH3, tBu), 26.8 (3 × CH3, tBu), 26.7 (3 × CH3, tBu), 24.22 (CH2, C-2), 24.20 (CH2, C-2), 19.19 (C, tBu), 19.16 (2 × C, tBu), 19.1 ppm (C, tBu). IR (CHCl3): ν = 2932, 1428, 1222, 1113 cm–1. MS (ESI) m/z (%) = 674 (100) [M + Na]+, 673 (33) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C40H492HNaO4Si2 674.3208; found 674.3206, [M + Na]+ calcd for C40H50NaO4Si2 673.3145; found 673.3163.
Method F
Following the general procedure, starting from substrate 13 (40.8 mg, 0.048 mmol), all the starting material was consumed after 75 h. Chromatotron chromatography (hexanes–EtOAc, 7:3) gave (4RS)-1,4-anhydro-5-O-acetyl-6,8-bis-O-tert-butyldiphenylsilyl-2,3-dideoxy-d-ribo-oct-4-ulofuranose (39) (0.6 mg, 9.6·10–4 mmol, 2%, 1R/1S 1:1) and 40 (22.8 mg, 0.032 mmol, 67%) as colorless oils. Compound 39: 1H NMR (500 MHz, CDCl3, descrived above for the [2-2H]39). 13C{1H} NMR (125.7 MHz, CDCl3, descrived above for the [2-2H]39). MS (ESI) m/z (%) = 673 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C40H50NaO4Si2: 673.3145 [M + Na]+; found 673.3146. C40H50NaO4Si2 (650.99): calcd. C 73.80, H 7.74; found: C 73.70, H 7.74.
Radical Reactions of 14
Method A
Following the general procedure, starting from substrate 14 (56.9 mg, 0.06 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (16 μL, 0.06 mmol) was required. All the starting material was consumed after 4 h. Column chromatography on a silica gel without KF (hexanes to hexanes–EtOAc, 7:3) gave 39 (18 mg, 0.028 mmol, 46%, 1R/1S 1.2:1).
Method D
Following the general procedure, starting from substrate 14 (54.5 mg, 0.06 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnD (16 μL, 0.06 mmol) was required. All the starting material was consumed after 5 h. Column chromatography on a silica gel without KF (hexanes to hexanes–EtOAc, 7:3) gave [2-2H]39 (13 mg, 0.02 mmol, 35%, 2H/1H 1:1.4).
Method E
Following the general procedure, starting from substrate 14 (49.8 mg, 0.05 mmol), after 4 h, the reaction was discarded since although the remaining starting material was present, several more polar products were detected in the TLC.
Method F
Following the general procedure, starting from substrate 14 (57.4 mg, 0.06 mmol), all the starting material was consumed after 0.75 h. Chromatotron chromatography (hexanes–EtOAc, 97:3 to 7:3) gave 39 (4.8 mg, 0.007 mmol, 12%, 1R/1S 1.2:1) and 3-C-(3,5-di-O-tert-butyldiphenylsilyl-2-O-trifluoromethanesulfonyl-α-d-ribofuranosyl)1-propanol (41) (20.3 mg, 0.025 mmol, 42%) as a colorless oil. Compound 41: [α]D = +18.8 (c = 1.3, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.45–7.22 (m, 20H, Ar), 5.26 (dd, J = 4.1, 4.1 Hz, 1H, 2-H), 4.67 (dd, J = 4.8, 4.8 Hz, 1H, 3-H), 4.17 (ddd, J = 10.1, 3.2, 3.2 Hz, 1H, 1-H), 3.95 (ddd, J = 5.4, 2.9, 2.9 Hz, 1H, 4-H), 3.65 (m, 2H, 3’-H2), 3.34 (dd, J = 11.7, 2.2 Hz, 1H, 5-Hb), 2.75 (dd, J = 11.7, 3.2 Hz, 1H, 5-Ha), 1.86 (m, 1H, 1’-Hb), 1.77–1.65 (m, 3H, 1’-Ha, 2’-H2), 1.06 (s, 9H, tBu), 0.87 ppm (s, 9H, tBu). 13C{1H} NMR (125.7 MHz, CDCl3) δC 135.9 (2 × CH, Ar), 135.8 (2 × CH, Ar), 135.5 (4 × CH, Ar), 133.1 (C, Ar), 132.96 (C, Ar), 132.95 (C, Ar), 131.7 (C, Ar), 130.15 (CH, Ar), 130.10 (CH, Ar), 129.6 (2 × CH, Ar), 127.9 (2 × CH, Ar), 127.8 (2 × CH, Ar), 127.6 (4 × CH, Ar), 89.1 (CH, C-2), 82.8 (CH, C-4), 78.4 (CH, C-1), 73.3 (CH, C-3), 63.7 (CH2, C-5), 62.4 (CH2, C-3’), 29.4 (CH2, C-1’), 26.7 (3 × CH3, tBu), 26.6 (3 × CH3, tBu), 26.5 (CH2, C-2’), 19.2 (C, tBu), 19.0 ppm (C, tBu), 1C from CF3 group is missing. IR (CHCl3): ν = 3694, 3429, 3020, 2933, 2254, 1778, 1740, 1224, 1113 cm–1. MS (ESI) m/z (%) = 823 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C41H51F3NaO7SSi2 823.2744; found 823.2750. Anal. calcd for C41H51F3O7SSi2: C, 61.47; H, 6.42; S, 4.00. Found: C, 61.20; H, 6.44; S, 3.62.
Radical Reactions of 15
Method A
Following the general procedure, starting from substrate 15 (93.8 mg, 0.12 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (31 μL, 0.12 mmol) was required. All the starting material was consumed after 3 h. Column chromatography on a silica gel without KF (hexanes to hexanes–EtOAc, 97:3) gave (4R)-1,4-anhydro-2,3,5-trideoxy-6,8-bis-O-(1,1,3,3-tetraisopropyldisiloxanyl)-d-erythro-oct-4-ulofuranose (42) (13 mg, 0.031 mmol, 27%) and (4S)-1,4-anhydro-2,3,5-trideoxy-6,8-bis-O-(1,1,3,3-tetraisopropyldisiloxanyl)-d-erythro-oct-4-ulofuranose (43) (16.8 mg, 0.040 mmol, 35%), both as colorless oils. Compound 42: [α]D = −56.6 (c = 0.53, CHCl3). 1H NMR (500 MHz, CDCl3) δH 4.64 (ddd, J = 8.9, 7.3, 5.4 Hz, 1H, 3-H), 3.95 (dd, J = 10.1, 2.5 Hz, 1H, 5-Hb), 3.89 (ddd, J = 8.2, 8.2, 5.4 Hz, 1H, 3’-Hb), 3.84–3.77 (m, 3H, 4-H, 5-Ha, 3’-Ha), 2.35 (dd, J = 12.3, 7.3 Hz, 1H, 2-Hb), 2.18 (dd, J = 12.7, 8.9 Hz, 1H, 2-Ha), 2.06 (ddd, J = 11.7, 11.7, 3.2 Hz, 1H, 1’-Hb), 2.02 (m, 1H, 2’-Hb), 1.93–1.82 (m, 2H, 1’-Ha, 2’-Ha), 1.10–0.99 ppm (m, 28H, iPr). 13C{1H} NMR (125.7 MHz, CDCl3) δC 113.1 (C, C-1), 84.4 (CH, C-4), 74.7 (CH, C-3), 67.3 (CH2, C-3’), 66.1 (CH2, C-5), 44.0 (CH2, C-2), 34.9 (CH2, C-1’), 23.9 (CH2, C-2’), 17.6 (CH3, iPr), 17.4 (3 × CH3, iPr), 17.3 (CH3, iPr), 17.1 (CH3, iPr), 17.02 (CH3, iPr), 16.99 (CH3, iPr), 13.4 (2 × CH, iPr), 12.8 (CH, iPr), 12.6 ppm (CH, iPr). IR (CHCl3): ν = 2947, 2868, 1464, 1136, 1035 cm–1. MS (ESI) m/z (%) = 439 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C20H40NaO5Si2 439.2312; found 439.2308. Anal. calcd for C20H40O5Si2: C, 57.65; H, 9.68. Found: C, 57.39; H, 9.46. Compound 43: [α]D = +20.8 (c = 0.89, CHCl3). 1H NMR (500 MHz, CDCl3) δH 4.33 (ddd, J = 8.2, 8.2, 6.9 Hz, 1H, 3-H), 3.99 (dd, J = 11.4, 2.2 Hz, 1H, 5-Hb), 3.94–3.89 (m, 2H, 3’-H2), 3.86–3.80 (m, 2H, 4-H, 5-Ha), 2.38 (dd, J = 13.3, 8.2 Hz, 1H, 2-Hb), 2.23 (dd, J = 13.2, 7.3 Hz, 1H, 2-Ha), 2.08–2.01 (m, 2H, 1’-Hb, 2’-Hb), 1.91–1.82 (m, 2H, 1’-Ha, 2’-Ha), 1.10–0.99 ppm (m, 28H, iPr). 13C{1H} NMR (125.7 MHz, CDCl3) δC 112.7 (C, C-1), 82.9 (CH, C-4), 71.5 (CH, C-3), 67.3 (CH2, C-3’), 62.3 (CH2, C-5), 43.3 (CH2, C-2), 36.5 (CH2, C-1’), 24.2 (CH2, C-2’), 17.5 (CH3, iPr), 17.36 (2 × CH3, iPr), 17.35 (CH3, iPr), 17.27 (CH3, iPr), 17.2 (CH3, iPr), 17.0 (CH3, iPr), 16.9 (CH3, iPr), 13.5 (CH, iPr), 13.2 (CH, iPr), 12.8 (CH, iPr), 12.6 ppm (CH, iPr). IR (CHCl3): ν = 2947, 2868, 1465, 1210, 1133, 1043 cm–1. MS (ESI) m/z (%) = 439 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C20H40NaO5Si2 439.2312; found 439.2312. Anal. calcd for C20H40O5Si2: C, 57.65; H, 9.68. Found: C, 57.39; H, 9.46.
Method D
Following the general procedure, starting from substrate 15 (93.7 mg, 0.12 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnD (31 μL, 0.12 mmol) was required. All the starting material was consumed after 6 h. Column chromatography on a silica gel without KF (hexanes to hexanes–EtOAc, 97:3) gave (4R)-1,4-anhydro-2,3,5-trideoxy-6,8-bis-O-(1,1,3,3-tetraisopropyldisiloxanyl)-β-d-[5-2H]erythro-oct-4-ulofuranose ([2-2H]42) (11.7 mg, 0.02 mmol, 24%, 2H/1H 1.2:1) and (4S)-1,4-anhydro-2,3,5-trideoxy-6,8-bis-O-(1,1,3,3-tetraisopropyldisiloxanyl)-β-d-[5-2H]erythro-oct-4-ulofuranose ([2-2H]43) (10 mg, 0.024 mmol, 21%, 2H/1H 1.5:1), which was obtained as a 1:1.2 mixture with [2-2H]42. Compound [2-2H]42: 1H NMR (500 MHz, CDCl3, only deuterated 2RS isomers are described) δH 4.64 (m, 1H, 3-H), 3.96–3.87 (m, 2H, 5-Hb, 3’-Hb), 3.84–3.77 (m, 3H, 4-H, 5-Ha, 3’-Ha), 2.336 (d, J = 7.3 Hz, 1H, 2-H, 2R isomer), 2.166 (d, J = 9.2 Hz, 1H, 2-H, 2S isomer), 2.06 (ddd, J = 11.4, 11.4, 2.9 Hz, 1H, 1’-Hb), 2.01 (m, 1H, 2’-Hb), 1.93–1.83 (m, 2H, 1’-Ha, 2’-Ha), 1.10–0.99 ppm (m, 28H, iPr). 13C{1H} NMR (125.7 MHz, CDCl3, only deuterated 2RS isomers are described) δC 113.1 (C, C-1), 84.4 (CH, C-4), 74.60 (CH, C-3), 67.3 (CH2, C-3’), 66.1 (CH2, C-5), 43.68 (t, JCD = 22.2 Hz, CHD, C-2), 34.9 (CH2, C-1’), 23.9 (CH2, C-2’), 17.6 (CH3, iPr), 17.4 (3 × CH3, iPr), 17.3 (CH3, iPr), 17.1 (CH3, iPr), 17.02 (CH3, iPr), 16.99 (CH3, iPr), 13.4 (2 × CH, iPr), 12.8 (CH, iPr), 12.6 ppm (CH, iPr). MS (ESI) m/z (%) = 440 (100) [M + Na]+, 439 (68) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C20H392HNaO5Si2 440.2375; found 440.2372, [M + Na]+ calcd for C20H40NaO5Si2 439.2312; found 439.2300. Compound [2-2H]43: 1H NMR (500 MHz, CDCl3, only deuterated 2RS isomers are described) δH 4.33 (ddd, J = 8.2, 8.2, 6.9 Hz, 1H, 3-H), 3.99 (dd, J = 11.4, 2.2 Hz, 1H, 5-Hb), 3.94–3.89 (m, 2H, 3’-H2), 3.86–3.80 (m, 2H, 4-H, 5-Ha), 2.365 (d, J = 8.5 Hz, 1H, 2-H), 2.228 (d, J = 6.9 Hz, 1H, 2-H), 2.08–2.01 (m, 2H, 1’-Hb, 2’-Hb), 1.91–1.82 (m, 2H, 1’-Ha, 2’-Ha), 1.10–0.99 ppm (m, 28H, iPr). 13C{1H} NMR (125.7 MHz, CDCl3) δC 112.7 (C, C-1), 82.9 (CH, C-4), 71.5 (CH, C-3), 67.3 (CH2, C-3’), 62.3 (CH2, C-5), 42.95 (t, JCD = 21.2 Hz, CHD, C-2), 36.5 (CH2, C-1’), 24.2 (CH2, C-2’), 17.5 (CH3, iPr), 17.4 (2 × CH3, iPr), 17.3 (2 × CH3, iPr), 17.2 (CH3, iPr), 17.0 (CH3, iPr), 16.9 (CH3, iPr), 13.5 (CH, iPr), 13.2 (CH, iPr), 12.8 (CH, iPr), 12.6 ppm (CH, iPr). IR (CHCl3): ν = 2947, 2868, 1465, 1210, 1133, 1043 cm–1. MS (ESI) m/z (%) = 440 (100) [M + Na]+, 439 (55) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C20H392HNaO5Si2 440.2375; found 440.2374, [M + Na]+ calcd for C20H40NaO5Si2 439.2312; found 439.2306.
Method F
Following the general procedure, starting from substrate 15 (53.8 mg, 0.066 mmol), all the starting material was consumed after 0.5 h. Chromatotron chromatography (hexanes–EtOAc, 95:5 to 1:1) gave 42 and 43 (7.3 mg, 0.018 mmol, 26%, 1R/1S 1:2.5), and 3-C-(2-O-diphenoxyphosphoryl-3,5-bis-O-(1,1,3,3-tetraisopropyldisiloxanyl)-α-d-ribofuranosyl)1-propanol (44) (9.5 mg, 0.014 mmol, 22%) as a colorless oil. Compound 44: [α]D = +12.9 (c = 0.71, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.33–7.15 (m, 10H, Ar), 5.13 (ddd, J = 3.8, 3.8 Hz, 3JPH = 7.9 Hz, 1H, 2-H), 4.47 (m, 1H, 3-H), 4.12 (m, 1H, 1-H), 4.00 (dd, J = 12.6, 2.8 Hz, 1H, 5-Hb), 3.95–3.91 (m, 2H, 4-H, 5-Ha), 3.51–3.49 (m, 2H, 3’-H2), 1.66–1.51 (m, 4H, 1’-H2, 2’-H2), 1.09–0.81 ppm (m, 28H, iPr), 1H from OH is missing. 13C{1H} NMR (125.7 MHz, CDCl3) δC 150.9 (d, 2JPC = 7.4 Hz, C, Ar), 150.6 (d, 2JPC = 7.4 Hz, C, Ar), 129.7 (2 × CH, Ar), 129.6 (2 × CH, Ar), 125.3 (CH, Ar), 125.1 (CH, Ar), 120.19 (CH, Ar), 120.14 (CH, Ar), 120.0 (CH, Ar), 119.9 (CH, Ar), 81.6 (d, 2JPC = 6.4 Hz, CH, C-2), 79.8 (CH, C-4), 79.5 (d, 3JPC = 6.3 Hz, CH, C-1), 71.6 (CH, C-3), 62.5 (CH2, C-3’), 60.9 (CH2, C-5), 29.2 (CH2, C-1’ or C-2’), 26.7 (CH2, C-1’ or C-2’), 17.4 (CH3, iPr), 17.28 (CH3, iPr), 17.27 (CH3, iPr), 17.25 (CH3, iPr), 17.0 (2 × CH3, iPr), 16.8 (CH3, iPr), 16.7 (CH3, iPr), 13.5 (CH, iPr), 13.1 (CH, iPr), 12.6 (CH, iPr), 12.4 ppm (CH, iPr). IR (CHCl3): ν = 3692, 3610, 3022, 2948, 1490.1210, 1039 cm–1. MS (ESI) m/z (%) = 689 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C32H51NNaO9PSi2 689.2707; found 689.2706. Anal. calcd for C32H51NO9PSi2: C, 57.63; H, 7.71. Found: C, 57.61; H, 8.07.
Synthesis of 4-Deoxy-6,8-dioxabicyclo[3.2.1]heptane Structures (Tables 5 and 6)
Radical Reactions of 16
Method A
Following the general procedure, starting from substrate 16 (49 mg, 0.076 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (20 μL, 0.076 mmol) was required. All the starting material was consumed after 5 h. Column chromatography (hexanes to hexanes–EtOAc, 6:4) gave (2S)-2,7-anhydro-1-O-tert-butyldiphenylsilyl-3-deoxy-4,5-di-O-methyl-β-d-xylo-hept-2-ulopyranose (45) (3.1 mg, 0.007 mmol, 9%), an inseparable mixture of 48 (7.6 mg, 0.015 mmol, 20%) and unstable C-(6-O-tert-butyldiphenylsilyl-4-deoxy-2,3-di-O-methyl-β-l-threo-hex-4-enopyranosyl)methanol (46) (1.5 mg, 0.03 mmol, 4%), and C-(4-O-acetyl-6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-β-l-idopyranosyl)methanol (47) (12.1 mg, 0.024 mmol, 32%), all as colorless oils. Compound 45: [α]D = +14.5 (c = 0.38, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.70–7.68 (m, 4H, Ar), 7.45–7.37 (m, 6H, Ar), 4.56 (ddd, J = 5.0, 4.3, 0.0 Hz, 1H, 1-H), 4.03 (dd, J = 7.6, 0.0 Hz, 1H, 1’-Hb), 3.75 (d, J = 11.0 Hz, 1H, 6-Hb), 3.73 (d, J = 10.7 Hz, 1H, 6-Ha), 3.68 (dd, J = 7.5, 5.0 Hz, 4J2,1’a = 1.1 Hz, 1H, 1’-Ha), 3.571 (ddd, J = 10.1, 8.2, 6.6 Hz, 1H, 3-H), 3.50 (s, 3H, OMe), 3.42 (s, 3H, OMe), 3.40 (ddd, J = 8.2, 4.4 Hz,4J2,1’a = 1.1 Hz, 1H, 2-H), 2.36 (dd, J = 13.0, 6.6 Hz, 1H, 4-Hb), 1.70 (dd, J = 13.0, 10.1 Hz, 1H, 4-Ha), 1.08 ppm (s, 9H, tBu). 13C{1H} NMR (100.6 MHz, CDCl3) δC 135.68 (2 × CH, Ar), 135.67 (2 × CH, Ar), 133.2 (2 × C, Ar), 129.7 (2 × CH, Ar), 127.7 (4 × CH, Ar), 107.94 (C, C-5), 81.0 (CH, C-2), 77.78 (CH, C-3), 73.7 (CH, C-1), 66.8 (CH2, C-6), 65.8 (CH2, C-1’), 58.4 (CH3, OMe), 57.2 (CH3, OMe), 37.08 (CH2, C-4), 26.8 (3 × CH3, DPS), 19.3 ppm (C, DPS). IR (CHCl3): ν = 2931, 1464, 1113 cm–1. MS (ESI) m/z (%) = 465 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C25H34NaO5Si 465.2073; found 465.2071. Anal. calcd for C25H34O5Si: C, 67.84; H, 7.74. Found: C, 67.63; H, 7.68. Compound 46: could not be purified perfectly due to its instability. 1H NMR (500 MHz, CDCl3, simulated coupling constants of the allylic system using DAISY) δH 7.74–7.34 (m, 10H, Ar), 5.23 (dddd, J = 4.9 Hz, 4J = 1.5, 1.5, 1.0 Hz, 1H, 4-H), 4.16 (ddd, J = 13.9 Hz, 4J = 1.0 Hz, 5J = 1.6 Hz, 1H, 6-Hb), 4.12 (ddd, J = 13.9 Hz, 4J = 1.5 Hz, 5J = 0.7 Hz, 1H, 6-Ha), 3.98–3.92 (m, 2H, 1-H, 1’-Hb), 3.81 (m, 1H, 1’-Ha), 3.72 (dddd, J = 4.9, 2.3 Hz, 5J = 1.6, 0.7 Hz, 1H, 3-H), 3.454 (s, 3H, OMe), 3.450 (m, 1H, 2-H), 3.42 (s, 3H, OMe), 1.08 ppm (s, 9H, tBu), 1H from OH is missing. 13C{1H} NMR (100.6 MHz, CDCl3) δC 156.0 (C, C-5), 135.58 (2 × CH, Ar), 135.56 (2 × CH, Ar), 133.3 (2 × C, Ar), 129.7 (2 × CH, Ar), 127.7 (4 × CH, Ar), 92.8 (CH, C-4), 76.5 (CH, C-3), 74.1 (CH, C-1), 69.4 (CH, C-2), 62.7 (2 × CH2, C-1, C-6), 58.0 (CH3, OMe), 55.4 (CH3, OMe), 26.8 (3 × CH3, DPS), 19.3 ppm (C, DPS). IR (CHCl3): ν = 3674, 3504, 2931, 1113 cm–1. MS (ESI) m/z (%) = 465 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C25H34NaO5Si 465.2073; found 465.2061. Compound 47: [α]D = +0.4 (c = 1.20, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.64–7.61 (m, 4H, Ar), 7.45–7.35 (m, 6H, Ar), 5.074 (ddd, J = 2.6, 1.9, 4J2,4 = 1.2 Hz, 1H, 4-H), 4.01 (ddd, J = 9.1, 5.2, 1.6 Hz, 1H, 5-H), 3.94 (dd, J = 11.7, 8.0 Hz, 1H, 1’-Hb), 3.81 (dd, J = 9.8, 5.2 Hz, 1H, 6-Hb), 3.79 (ddd, J = 8.0, 4.0, 1.6 Hz, 1H, 1-H), 3.78 (dd, J = 11.7, 9.1 Hz, 1H, 6-Ha), 3.74 (dd, J = 2.7, 2.6 Hz, 1H, 3-H), 3.63 (dd, J = 11.7, 4.0 Hz, 1H, 1’-Ha), 3.55 (s, 3H, OMe), 3.36 (s, 3H, OMe), 3.20 (ddd, J = 2.7, 1.6, 4J2,4 = 1.2 Hz, 1H, 2-H), 2.03 (s, 3H, OAc), 1.85 (br s, 1H, OH), 1.04 ppm (s, 9H, tBu). 13C{1H} NMR (100.6 MHz, CDCl3) δC 170.8 (C, OAc), 135.6 (2 × CH, Ar), 135.5 (2 × CH, Ar), 133.3 (C, Ar), 133.2 (C, Ar), 129.74 (CH, Ar), 129.72 (CH, Ar), 127.7 (4 × CH, Ar), 76.3 (CH, C-1 or C-2), 76.1 (CH, C-1 or C-2), 74.6 (CH, C-5), 71.7 (CH, C-3), 66.20 (CH, C-4), 62.6 (CH2, C-1’), 61.62 (CH2, C-6), 58.1 (CH3, OMe), 58.0 (CH3, OMe), 26.8 (3 × CH3, DPS), 21.0 (CH3, OAc), 19.1 ppm (C, DPS). IR (CHCl3): ν = 3675, 3594, 2933, 1731, 1103 cm–1. MS (ESI) m/z (%) = 525 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C27H38NaO7Si 525.2285; found 525.2276. Anal. calcd for C27H38O7Si: C, 64.51; H, 7.62. Found: C, 64.81; H, 7.86.
Method C
Following the general procedure, starting from substrate 16 (54.5 mg, 0.084 mmol), after 2 h of reaction, a supplementary addition of TTMSS (26 μL, 0.084 mmol) was required. All the starting material was consumed after 7 h. Column chromatography (hexanes–EtOAc, 9:1 to 7:3) gave 45 (4.1 mg, 0.009 mmol, 11%) and 46 (10.8 mg, 0.024 mmol, 29%).
Method D
Following the general procedure, starting from substrate 16 (69.9 mg, 0.11 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnD (29 μL, 0.11 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes to hexanes–EtOAc, 1:1) gave 2,7-anhydro-1-O-tert-butyldiphenylsilyl-3-deoxy-4,5-di-O-methyl-β-d-[3-2H]xylo-hept-2-ulopyranose ([4-2H]45) (5.3 mg, 0.011 mmol, 10%, 2H/1H 1.8:1, 4R/4S 1:1.2), an inseparable mixture of reduced alcohol 48 (7.6 mg, 0.015 mmol, 14%) and olefin 46 (10.7 mg, 0.024 mmol, 22%), and C-(4-O-acetyl-6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-β-l-(5-2H)idopyranosyl)methanol [(5-2H)47] (18.3 mg, 0.036 mmol, 33%), all as colorless oils. Compound [4-2H]45: 1H NMR (500 MHz, CDCl3, only deuterated 4RS isomers are described) δH 7.70–7.65 (m, 4H, DPS), 7.45–7.37 (m, 6H, DPS), 4.56 (ddd, J = 4.4, 4.4, 0.0 Hz, 1H, 1-H), 4.03 (dd, J = 7.3, 0.0 Hz, 1H, 1’-Hb), 3.75 (d, J = 11.0 Hz, 1H, 6-Hb), 3.73 (d, J = 11.0 Hz, 1H, 6-Ha), 3.70–3.66 (m, 1H, 1’-Ha), 3.60–3.54 (m, 1H, 3-H), 3.50 (s, 3H, OMe), 3.43 (s, 3H, OMe), 3.42 (dd, J = 8.8, 3.5 Hz, 1H, 2-H), 2.35 (d, J = 6.6 Hz, 1H, 4-H, 4R isomer), 1.69 (d, J = 10.1 Hz, 1H, 4-H, 4S isomer), 1.08 ppm (s, 18H, tBu). 13C{1H} NMR (100.6 MHz, CDCl3, only deuterated 4RS isomers are described) δC 135.68 (2 × CH, DPS), 135.66 (2 × CH, DPS), 133.27 (C, DPS), 133.20 (C, DPS), 129.7 (2 × CH, DPS), 127.7 (4 × CH, DPS), 107.90 (C, C-5), 81.0 (CH, C-2), 77.73 (CH, C-3, 4R or 4S isomer), 77.70 (CH, C-3, 4R or 4S isomer), 73.7 (CH, C-1), 66.8 (CH2, C-6), 65.8 (CH2, C-1’), 58.4 (CH3, OMe), 57.2 (CH3, OMe), 36.75 (t, JCD = 19.1 Hz, CHD, C-4), 26.8 (3 × CH3, DPS), 19.3 ppm (C, DPS). MS (ESI) m/z (%) = 466 (100) [M + Na]+, 465 (46) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C25H332HNaO5Si 466.2136; found 466.2141, [M + Na]+ calcd for C25H34NaO5Si 465.2073; found 465.2060. Compound (5-2H)47: 1H NMR (400 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.64–7.61 (m, 4H, Ar), 7.45–7.35 (m, 6H, Ar), 5.070 (dd, J = 2.7 Hz, 4J2,4 = 1.2 Hz, 1H, 4-H), 3.93 (dd, J = 11.7, 8.1 Hz, 1H, 1’-Hb), 3.81 (d, J = 9.6 Hz, 1H, 6-Ha), 3.79 (d, J = 9.6 Hz, 1H, 6-Hb), 3.79 (ddd, J = 8.1, 4.0, 1.6 Hz, 1H, 1-H), 3.73 (dd, J = 2.7, 2.7 Hz, 1H, 3-H), 3.62 (dd, J = 11.7, 4.0 Hz, 1H, 1’-Ha), 3.55 (s, 3H, OMe), 3.36 (s, 3H, OMe), 3.19 (ddd, J = 2.7, 1.6 Hz, 4J2,4 = 1.2 Hz, 1H, 2-H), 2.03 (s, 3H, OAc), 1.04 ppm (s, 9H, tBu), 1H from OH is missing. 13C{1H} NMR (100.6 MHz, CDCl3) δC 170.8 (C, OAc), 135.6 (2 × CH, Ar), 135.5 (2 × CH, Ar), 133.3 (C, Ar), 133.2 (C, Ar), 129.74 (CH, Ar), 129.71 (CH, Ar), 127.7 (4 × CH, Ar), 76.2 (CH, C-1 or C-2), 76.1 (CH, C-1 or C-2), 71.7 (CH, C-3), 66.14 (CH, C-4), 62.6 (CH2, C-1’), 61.54 (CH2, C-6), 58.1 (CH3, OMe), 58.0 (CH3, OMe), 26.8 (3 × CH3, DPS), 21.0 (CH3, OAc), 19.1 ppm (C, DPS). MS (ESI) m/z (%) = 526 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C27H372HNaO7Si 526.2347; found 526.2346.
Method E
Following the general procedure, starting from substrate 16 (38.3 mg, 0.059 mmol), after 2 h, a supplementary addition of n-Bu3SnD (16 μL, 0.059 mmol) and BF3•Et2O (2 μL, 0.012 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes to hexanes–EtOAc, 1:1) gave [4-2H]45 (10.3 mg, 0.023 mmol, 39%, 2H/1H 2.9:1, 4R/4S 1:1.2), an inseparable mixture of 48 (4.6 mg, 0.009 mmol, 15%) and unstable 46 (4.6 mg, 0.010 mmol, 18%), and (5-2H)47 (7.3 mg, 0.015 mmol, 25%).
Radical Reactions of 17
Method A
Following the general procedure, starting from substrate 17 (49 mg, 0.058 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (16 μL, 0.058 mmol) was required. All the starting material was consumed after 5 h. Column chromatography on a silica gel without KF (hexanes to hexanes–EtOAc, 9:1) gave 45 (14 mg, 0.032 mmol, 55%).
Method D
Following the general procedure, starting from substrate 17 (59.6 mg, 0.071 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnD (19 μL, 0.071 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes to hexanes–EtOAc, 9:1) gave [4-2H]45 (18.1 mg, 0.041 mmol, 58%, 2H/1H 2.3:1, 4R/4S 1:1.2).
Radical Reactions of 18
Method A
Following the general procedure, starting from substrate 18 (38 mg, 0.05 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (13 μL, 0.05 mmol) was required. All the starting material was consumed after 6 h. Column chromatography on a silica gel without KF (hexanes to hexanes–EtOAc, 9:1) gave 45 (11.7 mg, 0.027 mmol, 53%).
Method D
Following the general procedure, starting from substrate 18 (38.8 mg, 0.05 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnD (14 μL, 0.05 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes to hexanes–EtOAc, 9:1) gave [4-2H]45 (13 mg, 0.029 mmol, 58%, 2H/1H 1.7:1, 4R/4S 1:1.2).
Method F
Following the general procedure, starting from substrate 18 (12.4 mg, 0.016 mmol), all the starting material was consumed after 1.5 h. Chromatotron chromatography (hexanes–EtOAc, 8:2 to 4:6) gave 45 (1.4 mg, 0.003 mmol, 19%) and 49 (4.1 mg, 0.007 mmol, 41%).
Method G
Following the general procedure, starting from substrate 18 (14.1 mg, 0.019 mmol), all the starting material was consumed after 3 h. Chromatotron chromatography (hexanes–EtOAc, 8:2 to 4:6) gave 45 (3.2 mg, 0.007 mmol, 39%) and 49 (2.3 mg, 0.004 mmol, 20%).
Radical Reactions of 19
Method A
Following the general procedure, starting from substrate 19 (72.6 mg, 0.087 mmol), after 2 h, a supplementary addition of n-Bu3SnH (24 μL, 0.087 mmol) was required. All the starting material was consumed after 3 h. Column chromatography (hexanes to hexanes–EtOAc, 2:8) gave 2,7-anhydro-3-deoxy-1-O-diphenoxyfosforyl-4,5-di-O-methyl-β-d-xylo-hept-2-ulopyranose (53) (11.5 mg, 0.026 mmol, 30%) and 1,5-anhydro-4,6-bis-O-diphenoxyphosphoryl-2,3-di-O-methyl-d-glucitol (54) (11 mg, 0.017 mmol, 19%) as colorless oils. Compound 53: [α]D = +2.6 (c = 0.46, CHCl3). 1H NMR (400 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.36–7.17 (m, 10H, Ar), 4.54 (ddd, J = 5.1, 3.9, 0.0 Hz, 1H, 1-H), 4.29 (d, 3JPH = 8.2 Hz, 2H, 6-H2), 4.02 (dd, J = 7.7, 0.0 Hz, 1H, 1’-Hb), 3.65 (ddd, J = 7.5, 5.1 Hz, 4J2,1’a = 1.1 Hz, 1H, 1’-Ha), 3.528 (ddd, J = 10.0, 7.8, 6.5 Hz, 1H, 3-H), 3.47 (s, 3H, OMe), 3.36 (ddd, J = 7.8, 3.9 Hz, 4J2,1’a = 1.1 Hz, 1H, 2-H), 3.36 (s, 3H, OMe), 2.29 (dd, J = 12.9, 6.5 Hz, 1H, 4-Hb), 1.52 ppm (dd, J = 12.9, 10.0 Hz, 1H, 4-Ha). 13C{1H} NMR (125.7 MHz, CDCl3) δC 150.5 (d, 2JPC = 6.3 Hz, 2 × C, Ar), 120.1–129.8 (10 × CH, Ar), 105.65 (d, 3JPC = 7.4 Hz, C, C-5), 80.4 (CH, C-2), 77.31 (CH, C-3), 73.9 (CH, C-1), 69.3 (d, 2JPC = 5.3 Hz, CH2, C-6), 66.2 (CH2, C-1’), 58.5 (CH3, OMe), 57.1 (CH3, OMe), 36.76 ppm (CH2, C-4). IR (CHCl3): ν = 2929, 1490, 1232 cm–1. MS (ESI) m/z (%) = 459 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C21H25NaO8P 459.1185; found 459.1175. Compound 54: [α]D = +27.0 (c = 0.70, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.33–7.10 (m, 20H, Ar), 4.42 (ddd, J = 11.6, 2.0 Hz, 3JPH = 8.2 Hz, 1H, 6-Hb), 4.36 (ddd, J = 9.7, 9.1 Hz, 3JPH = 9.4 Hz, 1H, 4-H), 4.13 (ddd, J = 11.6, 5.9 Hz, 3JPH = 9.8 Hz, 1H, 6-Ha), 3.950 (dd, J = 11.3, 5.2 Hz, 1H, 1-Hb), 3.51 (ddd, J = 9.7, 5.9, 2.0 Hz, 1H, 5-H), 3.45 (s, 3H, OMe), 3.43 (s, 3H, OMe), 3.29 (dd, J = 9.1, 8.8 Hz, 1H, 3-H), 3.207 (ddd, J = 10.6, 8.8, 5.2 Hz, 1H, 2-H), 3.040 ppm (dd, J = 11.3, 10.6 Hz, 1H, 1-Ha). 13C{1H} NMR (125.7 MHz, CDCl3) δC 150.7 (d, 2JPC = 7.4 Hz, C, Ar), 150.6 (d, 2JPC = 7.4 Hz, C, Ar), 150.5 (d, 2JPC = 7.4 Hz, C, Ar), 150.4 (d, 2JPC = 7.4 Hz, C, Ar), 120.0–129.8 (20 × CH, Ar), 84.9 (CH, C-3), 79.82 (CH, C-2), 76.7 (CH, C-5), 75.8 (d, 2JPC = 6.3 Hz, CH, C-4), 67.5 (d, 2JPC = 6.4 Hz, CH2, C-6), 67.19 (CH2, C-1), 60.5 (CH3, OMe), 58.7 ppm (CH3, OMe). IR (CHCl3): ν = 3020, 2929, 1490, 1218 cm–1. MS (ESI) m/z (%) = 679 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C32H34NaO11P2 679.1474; found 679.1474.
Radical Reactions of 20
Method A
Following the general procedure, starting from substrate 20 (89 mg, 0.14 mmol), after 2 h of reaction, a supplementary addition of n-Bu3SnH (37 μL, 0.14 mmol) was required. All the starting material was consumed after 5 h. Column chromatography on a silica gel without KF (hexanes to hexanes–EtOAc, 75:25) gave an inseparable mixture of C-(4-O-acetyl-6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-β-l-altropyranosyl)methanol (50) and 51 (33 mg, 0.066 mmol, 47%, 2:1) as a colorless oil, and 46 (12.1 mg, 0.027 mmol, 20%). Compounds 50 and 51: 1H NMR (500 MHz, CDCl3, only 50 is described) δH 7.73–7.63 (m, 4H, Ar), 7.46–7.35 (m, 6H, Ar), 5.144 (dd, J = 10.1, 2.9 Hz, 1H, 4-H), 3.93–3.77 (m, 7H, 1-H, 3-H, 5-H, 6-H2, 1’-H2), 3.46 (s, 3H, OMe), 3.45 (s, 3H, OMe), 3.37 (dd, J = 3.8, 1.0 Hz, 1H, 2-H), 2.03 (s, 3H, OAc), 1.05 ppm (s, 9H, tBu), 1H from OH is missing. 13C{1H} NMR (100.6 MHz, CDCl3, only 50 is described) δC 169.9 (C, OAc), 135.8 (2 × CH, Ar), 135.6 (2 × CH, Ar), 133.9 (C, Ar), 133.6 (C, Ar), 129.5 (2 × CH, Ar), 127.6 (2 × CH, Ar), 127.5 (2 × CH, Ar), 77.5 (CH, C-2), 74.7 (CH, C-1 or C-5), 74.4 (CH, C-1 or C-5), 74.4 (CH, C-3), 68.55 (CH, C-4), 63.88 (CH2, C-6), 62.8 (CH2, C-1’), 59.2 (CH3, OMe), 58.2 (CH3, OMe), 26.7 (3 × CH3, DPS), 20.9 (CH3, OAc), 19.3 ppm (C, DPS). IR (CHCl3): ν = 3690, 3567, 2933, 1737, 1217 cm–1. MS (ESI) m/z (%) = 525 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C27H38NaO7Si 525.2285; found 525.2267. Anal. calcd for C27H38O7Si: C, 64.51; H, 7.62. Found: C, 64.58; H. 7.84.
Method D
Following the general procedure, starting from substrate 20 (38 mg, 0.06 mmol), after 2 h of reaction and again after 4 h, a supplementary addition of n-Bu3SnD (16 μL, 0.06 mmol) was required. All the starting material was consumed after 9 h. Column chromatography on a silica gel without KF (hexanes to hexanes–EtOAc, 8:2) gave an inseparable mixture of three compounds, (5-2H)50 and 51 (19.3 mg, 0.04 mmol, 66%, 2.3:1) and olefin 46 (3.3 mg, 0.007 mmol, 13%), as a colorless oil. Mixture of (5-2H)50/51/46: 1H NMR (400 MHz, CDCl3, only (5-2H)50 is described) δH 7.73–7.60 (m, 4H, Ar), 7.44–7.32 (m, 6H, Ar), 5.137 (d, J = 3.1 Hz, 1H, 4-H), 3.94–3.76 (m, 6H, 1-H, 3-H, 6-H2, 1’-H2), 3.45 (s, 6H, 2 × OMe), 3.37 (dd, J = 3.8, 1.0 Hz, 1H, 2-H), 2.03 (s, 3H, OAc), 1.05 ppm (s, 9H, tBu), 1H from OH is missing. 13C{1H} NMR (100.6 MHz, CDCl3, only (5-2H)50 is described) δC 169.9 (C, OAc), 135.8 (4 × CH, Ar), 133.4 (C, Ar), 133.3 (C, Ar), 129.5 (2 × CH, Ar), 127.5 (4 × CH, Ar), 77.6 (CH, C-2), 74.7 (CH, C-1), 74.5 (CH, C-3), 68.53 (CH, C-4), 63.84 (CH2, C-6), 62.7 (CH2, C-1’), 59.3 (CH3, OMe), 58.2 (CH3, OMe), 26.7 (3 × CH3, DPS), 20.9 (CH3, OAc), 19.3 ppm (C, DPS). MS (ESI) m/z (%) = 526 (100) [M + Na]+, 525 (48) [M + Na]+, 465 (54) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C27H372HNaO7Si 526.2358; found 526.2358, [M + Na]+ calcd for C27H38NaO7Si 525.2285; found 525.2282, [M + Na]+ calcd for C25H34NaO5Si 465.2073; found 465.2089.
Method F
Following the general procedure, starting from substrate 20 (57 mg, 0.088 mmol), all the starting material was consumed after 2 h. Chromatotron chromatography (hexanes–EtOAc, 8:2 to 7:3) gave 45 (4.67 mg, 0.011 mmol, 12%) and an inseparable mixture of 50 and 51 (22.1 mg, 0.044 mmol, 50%, 1.2:1).
Method G
Following the general procedure, starting from substrate 20 (43 mg, 0.066 mmol), all the starting material was consumed after 3 h. Chromatotron chromatography (hexanes–EtOAc, 8:2 to 7:3) gave 45 (6.1 mg, 0.014 mmol, 21%) and an inseparable mixture of 50 and 51 (14.6 mg, 0.029 mmol, 44%, 1.3:1).
Radical Reactions of 21
Method A
Following the general procedure, starting from substrate 21 (75.7 mg, 0.09 mmol), all the starting material was consumed after 2 h. Column chromatography on a silica gel without KF (hexanes to hexanes–EtOAc, 7:3) gave olefin 46 (18.1 mg, 0.041 mmol, 45%) and alcohol 52 (13.8 mg, 0.020 mmol, 22%).
Method C
Following the general procedure, starting from substrate 21 (73 mg, 0.087 mmol), after 2 h of reaction, a supplementary addition of TTMSS (27 μL, 0.087 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes–EtOAc, 8:2) gave 45 (9.6 mg, 0.022 mmol, 25%) and 52 (14.5 mg, 0.021 mmol, 24%).
Method D
Following the general procedure, starting from substrate 21 (74.3 mg, 0.089 mmol), all the starting material was consumed after 2 h. Column chromatography on a silica gel without KF (hexanes to hexanes–EtOAc, 1:1) gave C-(6-O-tert-butyldiphenylsilyl-4-O-diphenoxyphosphoryl-2,3-di-O-methyl-α-d-[2-OMe-2H]galactopyranosyl)methanol ([OCH2-2H]52) (14.2 mg, 0.020 mmol, 23%, 2H/1H 1.3:1) and olefin 46 (17.3 mg, 0.039 mmol, 44%), both as colorless oils. Compound [OCH2-2H]52: 1H NMR (500 MHz, CDCl3, only the deuterated product is described) δH 7.65–7.05 (m, 20H, Ar), 5.05 (ddd, J = 2.8, 2.2 Hz, 3JPH = 8.8 Hz, 1H, 4-H), 4.09 (ddd, J = 7.3, 5.7, 5.7 Hz, 1H, 1-H), 3.80–3.70 (m, 5H, 5-H, 6-H2, 1’-H2), 3.57 (dd, J = 8.5, 5.7 Hz, 1H, 2-H), 3.38 (m, 1H, 3-H), 3.372 (t, J = 1.6 Hz, 2H, OCH2D), 3.35 (s, 3H, OMe), 1.03 ppm (s, 9H, tBu), 1H from OH is missing. 13C{1H} NMR (125.7 MHz, CDCl3, only the deuterated product is described) δC 150.8 (d, 2JPC = 7.4 Hz, C, Ar), 150.4 (d, 2JPC = 7.4 Hz, C, Ar), 135.6 (2 × CH, Ar), 135.5 (2 × CH, Ar), 133.3 (C, Ar), 133.2 (C, Ar), 129.8 (CH, Ar), 129.72 (CH, Ar), 129.65 (2 × CH, Ar), 129.4 (2 × CH, Ar), 127.7 (4 × CH, Ar), 125.2 (CH, Ar), 125.1 (CH, Ar), 120.34 (CH, Ar), 120.30 (CH, Ar), 120.02 (CH, Ar), 119.98 (CH, Ar), 78.7 (CH, C-3), 77.0 (CH, C-2), 74.2 (d, 2JPC = 6.3 Hz, CH, C-4), 72.9 (d, 3JPC = 5.3 Hz, CH, C-5), 72.6 (CH, C-1), 62.2 (CH2, C-6), 59.3 (CH2, C-1’), 59.26 (CH2D), 57.7 (CH3, OMe), 26.7 (3 × CH3, DPS), 19.1 ppm (C, DPS). MS (ESI) m/z (%) = 716 (100) [M + Na]+, 715 (67) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C37H442HNaO9PSi 716.2531; found 716.2554, M + Na]+ calcd for C37H45NaO9PSi 715.2468; found 715.2471.
Method E
Following the general procedure, starting from substrate 21 (81.8 mg, 0.098 mmol), after 2 h, a supplementary addition of n-Bu3SnD (26 μL, 0.098 mmol) and BF3•Et2O (3 μL, 0.024 mmol) was required. All the starting material was consumed after 3 h. Column chromatography on a silica gel without KF (hexanes to hexanes–EtOAc, 1:1) gave [4-2H]45 (17.8 mg, 0.040 mmol, 41%, 2H/1H 2.8:1, 4R/4S 1:1.2) and [OCH2-2H]52 (8.6 mg, 0.012 mmol, 13%, 2H/1H 1.1:1).
Method F
Following the general procedure, starting from substrate 21 (36.9 mg, 0.044 mmol), all the starting material was consumed after 3 h. Chromatotron chromatography (hexanes–EtOAc, 8:2 to 4:6) gave 52 (15.8 mg, 0.023 mmol, 52%).
Method G
Following the general procedure, starting from substrate 21 (40.5 mg, 0.048 mmol), all the starting material was consumed after 3 h. Chromatotron chromatography (hexanes–EtOAc, 8:2 to 4:6) gave 52 (13.5 mg, 0.019 mmol, 40%).
Radical Reactions of 22
Method A
Following the general procedure, starting from substrate 22 (106.8 mg, 0.18 mmol), after 2 h, a supplementary addition of n-Bu3SnH (49 μL, 0.18 mmol) was required. All the starting material was consumed after 5 h. Column chromatography (hexanes to hexanes–EtOAc, 1:1) gave 2,7-anhydro-1,3-dideoxy-4,5-di-O-methyl-β-l-ribo-hept-2-ulopyranose (61) (18.9 mg, 0.10 mmol, 56%) as a colorless oil. [α]D = +0.02 (c = 0.34, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 4.75 (ddd, J = 5.8, 2.8, 0.9 Hz, 1H, 1-H), 3.82 (dd, J = 7.7, 5.8 Hz, 1H, 1’-Hb), 3.67 (dd, J = 7.7, 0.9 Hz, 1H, 1’-Ha), 3.626 (ddd, J = 11.1, 6.0, 4.1 Hz, 1H, 3-H), 3.46 (dd, J = 4.1, 2.8 Hz, 1H, 2-H), 3.56 (s, 3H, OMe), 3.38 (s, 3H, OMe), 2.121 (dd, J = 12.5, 6.0 Hz, 1H, 4-Hb), 1.816 (dd, J = 12.5, 11.1 Hz, 1H, 4-Ha), 1.51 ppm (s, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 106.96 (C, C-5), 75.4 (CH, C-2), 74.1 (CH, C-1), 73.78 (CH, C-3), 65.8 (CH2, C-1’), 57.9 (CH3, OMe), 56.2 (CH3, OMe), 38.69 (CH2, C-4), 23.8 ppm (CH3, C-6). IR (CHCl3): ν = 3015, 2934, 1389, 1198 cm–1. MS (ESI) m/z (%) = 211 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C9H16NaO4 211.0946; found 211.0948. Anal. calcd for C9H16O4: C, 57.43; H, 8.57. Found: C, 57.63; H, 8.63.
Method C
Following the general procedure, starting from substrate 22 (58.8 mg, 0.10 mmol), after 2 h of reaction, a supplementary addition of TTMSS (31 μL, 0.10 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes–EtOAc, 9:1 to 6:4) gave 61 (9 mg, 0.048 mmol, 48%).
Method D
Following the general procedure, starting from substrate 22 (61.5 mg, 0.105 mmol), after 2 h, a supplementary addition of n-Bu3SnD (29 μL, 0.105 mmol) was required. All the starting material was consumed after 5 h. Column chromatography (hexanes to hexanes–EtOAc, 1:1) gave 2,7-anhydro-1,3-dideoxy-4,5-di-O-methyl-β-l-[4-2H]ribo-hept-2-ulopyranose ([4-2H]61) (8.7 mg, 0.046 mmol, 44%, 2H/1H 1.8:1, 4R/4S 1:1.3) as a colorless oil: 1H NMR (500 MHz, CDCl3, only deuterated isomers are described) δH 4.75 (ddd, J = 5.7, 2.8, 0.0 Hz, 1H, 1-H), 3.82 (dd, J = 7.6, 5.7 Hz, 1H, 1’-Hb), 3.67 (dd, J = 7.9, 0.0 Hz, 1H, 1’-Ha), 3.63–3.60 (m, 1H, 3-H), 3.55 (s, 3H, OMe), 3.47–3.44 (m, 1H, 2-H), 3.38 (s, 3H, OMe), 2.101 (d, J = 5.7 Hz, 1H, 4-H, 4S isomer), 1.793 (d, J = 11.1 Hz, 1H, 4-H, 4R isomer), 1.51 ppm (s, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3, only deuterated isomers are described) δC 106.92 (C, C-5), 75.4 (CH, C-2), 74.1 (CH, C-1), 73.69 (CH, C-3), 65.8 (CH2, C-1’), 57.8 (CH3, OMe), 56.2 (CH3, OMe), 38.52 (t, JCD = 20.1 Hz, CHD, C-4), 23.8 ppm (CH3, C-6). MS (ESI) m/z (%) = 212 (100) [M + Na]+, 211 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C9H152HNaO4 212.1009; found 212.1005, [M + Na]+ calcd for C9H16NaO4 211.0946; found 211.0948.
Method E
Following the general procedure, starting from substrate 22 (77 mg, 0.13 mmol), after 2 h, a supplementary addition of n-Bu3SnD (35 μL, 0.13 mmol) and BF3·Et2O (3.3 μL, 0.026 mmol) was required. All the starting material was consumed after 7 h. Column chromatography (hexanes to hexanes–EtOAc, 6:4) gave [4-2H]61 (16.2 mg, 0.086 mmol, 66%, 2H/1H 1.3:1, 4R/4S 1:1.3).
Method F
Following the general procedure, starting from substrate 22 (28.9 mg, 0.05 mmol), all the starting material was consumed after 1.5 h. Chromatotron chromatography (hexanes–EtOAc, 4:6 to 3:7) gave 61 (5.2 mg, 0.028 mmol, 55%).
Method G
Following the general procedure, starting from substrate 22 (29.1 mg, 0.050 mmol), all the starting material was consumed after 3 h. Chromatotron chromatography (hexanes–EtOAc, 4:6 to 3:7) gave 61 (5.7 mg, 0.030 mmol, 61%).
Radical Reactions of 23
Method A
Following the general procedure, starting from substrate 23 (88 mg, 0.22 mmol), after 2 h, a supplementary addition of n-Bu3SnH (60 μL, 0.22 mmol) was required. All the starting material was consumed after 6 h. Column chromatography (hexanes to hexanes–EtOAc, 4:6) gave C-(4,6-dideoxy-2,3-di-O-methyl-β-d-threo-hex-4-enopyranosyl)methanol (56) (8.3 mg, 0.044 mmol, 20%) and C-(4-O-acetyl-2,3-6-deoxy-di-O-methyl-β-d-altropyranosyl)methanol (57) (4.4 mg, 0.018 mmol, 8%) as colorless oils, and 58 (7.8 mg, 0.031 mmol, 14%). Compound 56: [α]D = −119.1 (c = 0.45, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 4.78 (br ddd, J = 5.3 Hz, 4J = 2.0, 0.9 Hz, 1H, 4-H), 4.01 (dd, J = 11.4, 6.4 Hz, 1H, 1’-Hb), 3.94 (ddd, J = 6.4, 4.1, 1.5 Hz, 1H, 1-H), 3.86 (br dd, J = 11.4, 4.1 Hz, 1H, 1’-Ha), 3.60 (ddd, J = 5.3, 2.0 Hz, 5J = 1.0 Hz, 1H, 3-H), 3.45 (s, 3H, OMe), 3.40 (ddd, J = 2.0, 1.5 Hz, 4J = 2.0 Hz, 1H, 2-H), 3.39 (s, 3H, OMe), 2.25 (br s, 1H, OH), 1.86 ppm (dd, 5J = 1.0 Hz, 4J = 0.9 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 154.7 (C, C-5), 93.7 (CH, C-4), 75.7 (CH, C-2), 73.8 (CH, C-1), 69.7 (CH, C-3), 63.0 (CH2, C-1’), 58.1 (CH3, OMe), 55.4 (CH3, OMe), 20.0 ppm (CH3, C-6). IR (CHCl3): ν = 3691, 3602, 3013, 2933, 1672, 1226 cm–1. MS (ESI) m/z (%) = 211 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C9H16NaO4 211.0946; found 211.0942. Anal. calcd for C9H16O4: C, 57.43; H, 8.57. Found: C, 57.10; H, 8.27. Compound 57: [α]D = +50.3 (c = 0.35, CHCl3). 1H NMR (500 MHz, CDCl3) δH 4.799 (dd, J = 10.1, 3.2 Hz, 1H, 4-H), 3.92–3.84 (m, 4H, 1-H, 2-H, 5-H, 1’-Hb), 3.67 (m, 1H, 1’-Ha), 3.464 (s, 3H, OMe), 3.462 (s, 3H, OMe), 3.40 (dd, J = 3.8, 1.0 Hz, 1H, 3-H), 2.12 (s, 3H, OAc), 1.201 ppm (d, J = 6.4 Hz, 3H, 6-H3), 1H from OH is missing. 13C{1H} NMR (125.7 MHz, CDCl3) δC 170.2 (C, OAc), 77.7 (CH, C-3), 74.7 (CH, C-2), 74.2 (CH, C-1), 73.40 (CH, C-4), 69.68 (CH, C-5), 62.7 (CH2, C-1’), 59.3 (CH3, OMe), 58.4 (CH3, OMe), 21.1 (CH3, OAc), 17.79 ppm (CH3, C-6). IR (CHCl3): ν = 3690, 3603, 3018, 2935, 1734, 1220 cm–1. MS (ESI) m/z (%) = 271 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C11H20NaO6 271.1158; found 271.1167. Anal. calcd for C11H20O6: C, 53.21; H, 8.12. Found: C, 52.92; H, 7.97.
Method D
Following the general procedure, starting from substrate 23 (62 mg, 0.16 mmol), after 2 h, a supplementary addition of n-Bu3SnD (43 μL, 0.16 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes to hexanes–EtOAc, 3:7) gave 56 (9.4 mg, 0.05 mmol, 31%), C-(4-O-acetyl-2,3-6-deoxy-di-O-methyl-β-d-(5-2H)altropyranosyl)methanol [(5-2H)57] (7.6 mg, 0.031 mmol, 19%), and C-(4-O-acetyl-2,3-di-O-methyl-α-d-[5-2H]fucopyranosyl)methanol (5-2H]58) (8.4 mg, 0.034 mmol, 21%, 2H/1H 2.4:1) as colorless oils. Compound (5-2H)57: 1H NMR (500 MHz, CDCl3) δH 4.796 (d, J = 2.9 Hz, 1H, 4-H), 3.92–3.84 (m, 3H, 1-H, 2-H, 1’-Hb), 3.67 (m, 1H, 1’-Ha), 3.463 (s, 3H, OMe), 3.461 (s, 3H, OMe), 3.40 (dd, J = 3.5, 1.0 Hz, 1H, 3-H), 2.12 (s, 3H, OAc), 1.192 ppm (s, 3H, 6-H3), 1H from OH is missing. 13C{1H} NMR (125.7 MHz, CDCl3) δC 170.2 (C, OAc), 77.7 (CH, C-3), 74.7 (CH, C-2), 74.2 (CH, C-1), 73.33 (CH, C-4), 69.26 (t, JCD = 21.2 Hz, C, C-5), 62.7 (CH2, C-1’), 59.3 (CH3, OMe), 58.4 (CH3, OMe), 21.1 (CH3, OAc), 17.66 ppm (CH3, C-6). MS (ESI) m/z (%) = 272 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C11H192HNaO6 272.1220; found 272.1219. Compound [5-2H]58: 1H NMR (500 MHz, CDCl3, only the deuterated product is described) δH 5.323 (d, J = 3.2 Hz, 1H, 4-H), 4.23 (m, 1H, 1-H), 3.91–3.83 (m, 2H, 1’-H2), 3.70 (dd, J = 9.1, 6.0 Hz, 1H, 2-H), 3.50 (dd, J = 9.1, 3.5 Hz, 1H, 3-H), 3.50 (s, 3H, OMe), 3.42 (s, 3H, OMe), 2.17 (s, 3H, OAc), 1.169 ppm (s, 3H, 6-H3), 1H from OH is missing. 13C{1H} NMR (125.7 MHz, CDCl3, only the deuterated product is described) δC 170.7 (C, OAc). 78.8 (CH, C-3), 77.1 (CH, C-2), 73.4 (CH, C-1), 69.46 (CH, C-4), 59.7 (CH2, C-1’), 59.4 (CH3, OMe), 57.5 (CH3, OMe), 20.8 (CH3, OAc), 16.37 ppm (CH3, C-6). MS (ESI) m/z (%) = 272 (71) [M + Na]+, 271 (28) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C11H192HNaO6 272.1220; found 272.1222, [M + Na]+ calcd for C11H20NaO6 271.1158; found 271.1164.
Method E
Following the general procedure, starting from substrate 23 (126 mg, 0.32 mmol), after 2 h, a supplementary addition of n-Bu3SnD (87 μL, 0.32 mmol) and BF3•Et2O (8 μL, 0.064 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes to hexanes–EtOAc, 3:7) gave (5-2H)57 (9.5 mg, 0.038 mmol, 12%), [5-2H]58 (11.4 mg, 0.046 mmol, 14%, 2H/1H 1:2), the unstable and volatile 2,7-anhydro-1,3-dideoxy-4,5-di-O-methyl-β-l-[3-2H]xylo-hept-2-ulopyranose ([4-2H]55) (13 mg, 0.069 mmol, 21%, 2H/1H 5.4:1), and 3-O-acetyl-2,6-anhydro-1-deoxy-4,5-di-O-methyl-d-(6-2H)galactitol [(1-2H)60] (2 mg, 0.009 mmol, 3%) as colorless oils. Compound [4-2H]55: 1H NMR (500 MHz, CDCl3) δH 4.51 (ddd, J = 4.7, 4.7, 0.0 Hz, 1H, 1-H), 3.99 (dd, J = 7.6, 0.0 Hz, 1H, 1’-Hb), 3.74 (dd, J = 7.6, 5.4 Hz, 1H, 1’-Ha), 3.53–3.48 (m, 1H, 3-H), 3.48 (s, 3H, OMe), 3.40 (s, 3H, OMe), 3.35 (dd, J = 8.2, 4.1 Hz, 1H, 2-H), 2.33 (dd, J = 13.2, 6.7 Hz, 1H, 4-Hb), 2.32 (d, J = 6.6 Hz, 1H, 4-HD), 1.44 (m, 1H, 4-Ha), 1.49 ppm (s, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 107.0 (C, C-5), 80.7 (CH, C-2), 77.3 (CH, C-3), 73.5 (CH, C-1), 65.8 (CH2, C-1’), 58.3 (CH3, OMe), 57.1 (CH3, OMe), 36.5 (CH2, C-4 reduced product), 23.5 ppm (CH3, C-6), expected triplet for C-4 was imperceptible for the deuterated product. IR (CHCl3): ν = 3022, 2929, 1226 cm–1. MS (ESI) m/z (%) = 212 (100) [M + Na]+, 211 (18) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C9H152HNaO4 212.1009; found 212.1015, [M + Na]+ calcd for C9H16NaO4 211.0946; found 211.0950. Compound (1-2H)60: [α]D = −11.1 (c = 0.45, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 5.34 (dd, J = 3.4, 1.31 Hz, 1H, 4-H), 4.10 (br d, J = 4.4 Hz, 1H, 1-H), 3.58 (dddd, J = 6.6, 6.6, 6.6, 1.1 Hz, 1H, 5-H), 3.52 (m, 1H, 2-H), 3.50 (s, 3H, OMe), 3.43 (s, 3H, OMe), 3.24 (dd, J = 9.3, 3.4 Hz, 1H, 3-H), 2.18 (s, 3H, OAc), 1.17 ppm (d, J = 6.6 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 170.8 (C, OAc), 82.8 (CH, C-3), 75.4 (CH, C-2), 73.5 (CH, C-5), 69.4 (CH, C-4), 59.0 (CH3, OMe), 57.4 (CH3, OMe), 20.8 (CH3, OAc), 16.8 ppm (CH3, C-6), C-1 was imperceptible. IR (CHCl3): ν = 3016, 2932, 1226 cm–1. MS (ESI) m/z (%) = 242 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C10H172HNaO5 242.1115; found 242.1111.
Method F
Following the general procedure, starting from substrate 23 (13.9 mg, 0.035 mmol), all the starting material was consumed after 3 h. Chromatotron chromatography (hexanes–EtOAc, 4:6 to 0:1) gave 57 (1 mg, 0.004 mmol, 11%), 58 (2.2 mg, 0.09 mmol, 25%), and product 55 (0.7 mg, 0.004 mmol, 11%).
Method G
Following the general procedure, starting from substrate 23 (13.7 mg, 0.035 mmol), all the starting material was consumed after 3 h. Chromatotron chromatography (hexanes–EtOAc, 4:6 to 0:1) gave 57 (1.3 mg, 0.030 mmol, 15%), 58 (1.3 mg, 0.005 mmol, 15%), and product 55 (1.5 mg, 0.008 mmol, 23%).
Radical Reactions of 24
Method A
Following the general procedure, starting from substrate 24 (80.5 mg, 0.18 mmol), after 2 h, a supplementary addition of n-Bu3SnH (50 μL, 0.18 mmol) was required. All the starting material was consumed after 3 h. Column chromatography (hexanes to hexanes–EtOAc, 4:6) gave 56 (23.6 mg, 0.126 mmol, 70%) and 59 (10.2 mg, 0.023 mmol, 13%).
Method C
Following the general procedure, starting from substrate 24 (62 mg, 0.11 mmol), after 2 h of reaction, a supplementary addition of TTMSS (33 μL, 0.11 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes–EtOAc, 6:4 to 3:7) gave 56 (12 mg, 0.064 mmol, 58%) and 59 (3.7 mg, 0.008 mmol, 8%).
Method D
Following the general procedure, starting from substrate 24 (50 mg, 0.086 mmol), after 2 h, a supplementary addition of n-Bu3SnD (23 μL, 0.086 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes to hexanes–EtOAc, 3:7) gave 56 (12 mg, 0.064 mmol, 58%) and 59 (7.2 mg, 0.016 mmol, 19%).
Method E
Following the general procedure, starting from substrate 24 (37.7 mg, 0.066 mmol), after 2 h, a supplementary addition of n-Bu3SnD (17 μL, 0.066 mmol) and BF3•Et2O (2 μL, 0.016 mmol) was required. All the starting material was consumed after 4 h. Column chromatography (hexanes to hexanes–EtOAc, 3:7) gave 59 (4.2 mg, 0.009 mmol, 15%) and [4-2H]55 (5.1 mg, 0.027 mmol, 41%, 2H/1H 3.1:1).
Method F
Following the general procedure, starting from substrate 24 (53 mg, 0.091 mmol), all the starting material was consumed after 2 h. Chromatotron chromatography (hexanes–EtOAc, 3:7) gave 59 (19.7 mg, 0.045 mmol, 49%).
Method G
Following the general procedure, starting from substrate 24 (61 mg, 0.105 mmol), all the starting material was consumed after 3 h. Chromatotron chromatography (hexanes–EtOAc, 3:7) gave 59 (22.9 mg, 0.052 mmol, 50%).
3-C-(3,4-Di-O-benzyl-α-l-fucopyranosyl)1-propene (75)
3-C-(2,3,4-Tri-O-benzyl-α-l-fucopyranosyl)1-propene (74)61 (1.56 g, 3.41 mmol) was dissolved in dry CH2Cl2 (68 mL) under a N2 atmosphere, and I2 (8.6 g, 34.1 mmol) was added. The mixture was stirred at room temperature for 3 h, and then it was poured over an aqueous solution of Na2S2O3 and extracted with CH2Cl2. The combined extracts were dried over Na2SO4 and concentrated under reduced pressure. The resulting crude was dissolved in Et2O/MeOH (1:1) (35 mL), and Zn dust (2.04 g, 31.2 mmol) and AcOH (357 μL) were subsequently added, with the mixture stirred overnight at room temperature. Then, it was filtered over Celite, evaporated, poured over a saturated aqueous solution of NaHCO3, and extracted with CH2Cl2. The organic layers were dried over Na2SO4 and concentrated under reduced pressure. Column chromatography of the residue (hexanes–EtOAc, 8:2) gave 75 (791.8 mg, 2.15 mmol, 63%) as an amorphous solid: [α]D = −57.1 (c = 0.42, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.35–7.24 (m, 10H, Ar), 5.81 (dddd, J = 17.1, 10.1, 6.7, 6.7 Hz, 1H, 2’-H), 5.10 (dd, J = 17.1, 1.0 Hz, 1H, 3’-Hb), 5.05 (dd, J = 10.1, 0.0 Hz, 1H, 3’-Ha), 4.78 (d, J = 12.0 Hz, 1H, OBn), 4.73 (d, J = 12.0 Hz, 1H, OBn), 4.59 (d, J = 12.0 Hz, 1H, OBn), 4.58 (d, J = 11.9 Hz, 1H, OBn), 4.11 (m, 1H, 1-H), 4.04 (br s, 1H, 2-H), 3.94 (m, 1H, 5-H), 3.77 (dd, J = 3.2, 3.2 Hz, 1H, 4-H), 3.73 (dd, J = 7.0, 2.9 Hz, 1H, 3-H), 2.33 (dd, J = 7.6, 7.6 Hz, 2H, 1’-H2), 2.18 (br s, 1H, OH), 1.31 ppm (d, J = 6.6 Hz, 3H, 6-H3). 13C{1H} NMR (100.6 MHz, CDCl3) δC 138.4 (C, Ar), 138.3 (C, Ar), 135.0 (CH, C-2’), 128.4 (2 × CH, Ar), 128.3 (2 × CH, Ar), 127.7 (3 × CH, Ar), 127.6 (3 × CH, Ar), 116.7 (CH2, C-3’), 78.8 (CH, C-3), 75.0 (CH, C-4), 73.0 (CH2, OBn), 72.6 (CH2, OBn), 70.8 (CH, C-1), 69.1 (CH, C-5), 68.9 (CH, C-2), 31.9 (CH2, C-1’), 15.4 ppm (CH3, C-6). IR (CHCl3): ν = 3580, 3021, 1210 cm–1. MS (ESI) m/z (%) = 391 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C23H28NaO4 391.1885; found 391.1889.
3-C-(3,4-Di-O-benzylidene-α,β-d-arabinopyranosyl)1-propene (79 and 80)
Tetra-O-acetyl-d-arabinopyranose (78)62 (3.35 g, 10.54 mmol) was dissolved in dry CH3CN (129 mL) under a N2 atmosphere, and allyltrimethylsilane (9.8 mL, 61.51 mmol) and BF3•Et2O (6.2 mL, 49.2 mmol) were dropwise added at 0 °C. Then, the mixture was stirred at room temperature for 1.5 h. Subsequently, the solution was poured over a saturated aqueous solution of NaCl, extracted with EtOAc, dried over Na2SO4, and evaporated. Column chromatography of the residue (hexanes–EtOAc, 7:3) gave the allyl derivative (2.18 g, 7.27 mmol, 69%, 1S/1R isomers 6.8:1) as a colorless oil, which was subsequently dissolved in dry MeOH (34 mL), and Na2CO3 (1.23 g, 11.60 mmol) was added. The mixture was stirred at room temperature for 2.5 h, and then it was filtered, neutralized with the Amberlyst 15 H+ ion exchange resin, and evaporated. The crude was submitted to the benzylidene protection by treatment overnight with PhCH(OMe)2 (1.5 mL, 10.91 mmol) and CSA (17 mg, 0.07 mmol) in dry DMF (7.3 mL) at room temperature under a N2 atmosphere. The reaction was evaporated in a high vacuum rotovap and purified by column chromatography (hexanes–EtOAc, 8:2) to give 79 (228 mg, 0.87 mmol, 12%, d.r., 3:1) and 80 (780.8 g, 2.98 mmol, 41%, d.r., 3.5:1) as colorless oils. Compound 79: 1H NMR (500 MHz, CDCl3, only the major isomer is described) δH 7.48–7.34 (m, 5H, Ar), 6.23 (s, 1H, PhCH), 5.86 (dddd, J = 17.0, 10.1, 7.0, 7.0 Hz, 1H, 2’-H), 5.18 (dd, J = 17.0, 1.6 Hz, 1H, 3’-Hb), 5.13 (dd, J = 10.1, 1.6 Hz, 1H, 3’-Ha), 4.57 (ddd, J = 9.2, 6.7, 5.1 Hz, 1H, 4-H), 4.30 (dd, J = 5.1, 2.9 Hz, 1H, 3-H), 4.10 (dd, J = 12.0, 6.7 Hz, 1H, 5-Hb), 3.97 (br d, J = 5.4 Hz, 1H, 2-H), 3.73 (ddd, J = 7.9, 6.7, 1.6 Hz, 1H, 1-H), 3.55 (dd, J = 12.0, 9.1 Hz, 1H, 5-Ha), 2.48 (m, 1H, 1’-Hb), 2.36 ppm (m, 1H, 1’-Ha), 1H from OH is missing. 13C{1H} NMR (125.7 MHz, CDCl3, only the major isomer is described) δC 139.0 (C, Ar), 134.0 (CH, C-2’), 129.1 (CH, Ar), 128.4 (2 × CH, Ar), 125.9 (2 × CH, Ar), 117.6 (CH2, C-3’), 103.3 (CH, PhCH), 75.7 (CH, C-3), 75.0 (CH, C-1), 70.1 (CH, C-4), 67.5 (CH, C-2), 60.0 (CH2, C-5), 34.6 ppm (CH2, C-1’). IR (CHCl3): ν = 3567, 3452, 1643, 1457, 1100 cm–1. MS (ESI) m/z (%) = 285 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C15H18NaO4 285.1103; found 285.1099. Compound 80: 1H NMR (500 MHz, CDCl3, only the major isomer is described) δH 7.52–7.39 (m, 5H, Ar), 5.95 (s, 1H, PhCH), 5.84 (dddd, J = 17.4, 10.1, 7.3, 7.3 Hz, 1H, 2’-H), 5.18 (br d, J = 17.2 Hz, 1H, 3’-Hb), 5.11 (br d, J = 10.1 Hz, 1H, 3’-Ha), 4.43 (m, 1H, 4-H), 4.36 (dd, J = 5.7, 2.9 Hz, 1H, 3-H), 4.08 (dd, J = 12.0, 6.3 Hz, 1H, 5-Hb), 4.00 (dd, J = 2.5, 1.9 Hz, 1H, 2-H), 3.73 (ddd, J = 8.2, 6.6, 1.9 Hz, 1H, 1-H), 3.50 (dd, J = 12.0, 8.5 Hz, 1H, 5-Ha), 2.45 (m, 1H, 1’-Hb), 2.34 ppm (m, 1H, 1’-Ha), 1H from OH is missing. 13C{1H} NMR (125.7 MHz, CDCl3, only the major isomer is described) δC 137.1 (C, Ar), 134.0 (CH, C-2’), 129.1 (CH, Ar), 128.4 (2 × CH, Ar), 126.4 (2 × CH, Ar), 117.6 (CH2, C-3’), 104.3 (CH, PhCH), 77.7 (CH, C-3), 75.0 (CH, C-1), 69.2 (CH, C-4), 68.0 (CH2, C-5), 67.5 (CH, C-2), 34.8 ppm (CH2, C-1’). IR (CHCl3): ν = 3567, 3422, 3023, 1643, 1459, 1068 cm–1. MS (ESI) m/z (%) = 285 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C15H18NaO4 285.1103; found 285.1097.
3-C-(3,5-Di-O-tert-butyldiphenylsilyl-α-d-ribofuranosyl)1-propene (83)
To a solution of 3-C-(α-d-ribofuranosyl)1-propene (82)56,57 (5.24 g, 30.11 mmol, 87%) in dry CH2Cl2 (145 mL) at 0 °C were sequentially added imidazole (3.07 g, 45.17 mmol) and DPSCl (7.72 mL, 30.11 mmol). The resulting mixture was stirred at 0 °C for 3 h, treated with saturated aqueous NH4Cl, and extracted with CH2Cl2. Purification by column chromatography (hexanes–EtOAc, 97:3 to 6:4) afforded monoalcohol 83 (7.11 g, 10.94 mmol, 36%) and known diol 3-C-(5-O-tert-butyldiphenylsilyl-α-d-ribofuranosyl)1-propene (84)13,57 (4.92 g, 11.94 mmol, 40%) as colorless oils. Compound 83: [α]D = +33.1 (c = 0.58, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.57–7.15 (m, 20H, Ar), 5.77 (dddd, J = 17.0, 10.1, 7.0, 7.0 Hz, 1H, 2’-H), 5.04 (br d, J = 17.4 Hz, 1H, 3’-Hb), 4.97 (br d, J = 10.1 Hz, 1H, 3’-Ha), 4.49 (dd, J = 5.4, 5.4 Hz, 1H, 3-H), 3.87 (m, 1H, 4-H), 3.84 (m, 1H, 1-H), 3.77 (dd, J = 4.7, 4.7 Hz, 1H, 2-H), 3.50 (dd, J = 11.5, 2.2 Hz, 1H, 5-Hb), 3.14 (dd, J = 11.4, 3.2 Hz, 1H, 5-Ha), 2.76 (br s, 1H, OH), 2.38 (dd, J = 6.9, 6.9 Hz, 2H, 1’-H2), 1.00 (s, 9H, tBu), 0.80 ppm (s, 9H, tBu). 13C{1H} NMR (125.7 MHz, CDCl3) δC 135.7 (2 × CH, Ar), 135.63 (2 × CH, Ar), 135.61 (CH, C-2’), 135.5 (2 × CH, Ar), 135.1 (2 × CH, Ar), 133.4 (C, Ar), 133.2 (C, Ar), 132.6 (C, Ar), 132.4 (C, Ar), 130.20 (CH, Ar), 130.16 (CH, Ar), 129.5 (2 × CH, Ar), 128.0 (2 × CH, Ar), 127.8 (2 × CH, Ar), 127.5 (4 × CH, Ar), 116.6 (CH2, C-3’), 83.1 (CH, C-4), 81.0 (CH, C-1), 74.5 (CH, C-3), 72.7 (CH, C-2), 64.0 (CH2, C-5), 34.1 (CH2, C-1’), 26.9 (3 × CH3, tBu), 26.7 (3 × CH3, tBu), 19.2 (C, tBu), 19.0 ppm (C, tBu). IR (CHCl3): ν = 3673, 3541, 2932, 2860, 1428, 1113 cm–1. MS (ESI) m/z (%) = 673 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C40H50NaO4Si2 673.3145; found 673.3141.
3-C-(3,5-Di-O-1,1,3,3-tetraisopropyldisiloxanyl-α-d-ribofuranosyl)1-propene (86)
1,3-Dichloro-1,1,3,3-tetraisopropyldisiloxane (1.1 mL, 3.44 mmol) was added to a stirred solution of triol 82(56,57) (300 mg, 1.72 mmol) in dry pyridine (53 mL) at 0 °C. The reaction mixture was stirred at room temperature for 20 h, and then the pyridine was evaporated under reduced pressure. The residue was poured over 10% HCl and extracted with EtOAc. The combined organic extracts were washed with a saturated solution of NaHCO3, dried over Na2SO4, filtered, and evaporated. The crude was subjected to chromatography (hexanes–EtOAc, 95:5) to afford monoalcohol 86 (496.4 mg, 1.19 mmol, 69%) as a colorless oil: [α]D = −17.6 (c = 0.51, CHCl3). 1H NMR (500 MHz, CDCl3) δH 5.86 (1H, dddd, J = 17.1, 10.1, 7.0, 7.0 Hz, 1H, 2’-H), 5.15 (br d, J = 17.1 Hz, 1H, 3’-Hb), 5.06 (br d, J = 10.1 Hz, 1H, 3’-Ha), 4.37 (dd, J = 7.3, 4.8 Hz, 1H, 3-H), 4.10 (dd, J = 4.4, 4.4 Hz, 1H, 2-H), 4.01–3.97 (m, 2H, 1-H, 5-Hb), 3.93 (ddd, J = 7.0, 7.0, 3.5 Hz, 1H, 4-H), 3.83 (dd, J = 11.7, 6.3 Hz, 1H, 5-Ha), 2.52 (ddd, J = 14.2, 6.9, 6.9 Hz, 1H, 1’-Hb), 2.42 (ddd, J = 14.5, 7.3, 7.3 Hz, 1H, 1’-Ha), 1.10–0.89 ppm (m, 28H, iPr), 1H from OH is missing. 13C{1H} NMR (125.7 MHz, CDCl3) δC 134.6 (CH, C-2’), 117.1 (CH2, C-3’), 80.5 (CH, C-1 or C-4), 80.3 (CH, C-1 or C-4), 74.9 (CH, C-3), 72.4 (CH, C-2), 63.4 (CH2, C-5), 33.8 (CH2, C-1’), 17.5 (CH3, iPr), 17.38 (CH3, iPr), 17.35 (2 × CH3, iPr), 17.2 (CH3, iPr), 17.1 (2 × CH3, iPr), 17.0 (CH3, iPr), 13.4 (CH, iPr), 13.2 (CH, iPr), 12.9 (CH, iPr), 12.6 ppm (CH, iPr). IR (CHCl3): ν = 3671, 3540, 2949, 1732, 1643, 1465, 1120 cm–1. MS (ESI) m/z (%) = 439 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C20H40NaO5Si2 439.2312; found 439.2310.
General Procedure of Hydroboration to Give 63, 66, 69, 72, 76, 81, 85, and 87
The corresponding allyl derivative (1 mmol) was dissolved in dry THF (10.5 mL). BH3·THF 1 M complex (4 mL, 4 mmol) was added under a N2 atmosphere at 0 °C, and then the reaction was stirred at room temperature for 1 h. At 0 °C, an aqueous solution of NaOH 3 M (20 mL) was dropwise added followed by H2O2 30% (20 mL) and stirring was continued during 1 h at that temperature. The reaction was poured into brine and extracted with CH2Cl2. The organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography (hexanes–EtOAc) to give the corresponding alcohol.
3-C-(3,4,6-Tri-O-benzyl-α-d-glucopyranosyl)1-propanol (63)
Following the general procedure for the hydroboration, starting from 3-C-(3,4,6-tri-O-benzyl-α-d-glucopyranosyl)1-propene (62)54 (700 mg, 1.48 mmol) and purification by column chromatography (hexanes–EtOAc, 8:2), the alcohol 63 (478 mg, 0.97 mmol, 66%) was obtained as a crystalline solid: mp 101.5–102.3 °C (n-hexane–EtOAc); [α]D = +31.6 (c = 0.31, CHCl3). 1H NMR (400 MHz, CDCl3) δH 7.35–7.20 (m, 15H, Ar), 4.67 (d, J = 11.7 Hz, 1H, OBn), 4.63 (d, J = 11.4 Hz, 1H, OBn), 4.59 (d, J = 11.8 Hz, 1H, OBn), 4.54 (d, J = 12.2 Hz, 1H, OBn), 4.53 (d, J = 11.2 Hz, 1H, OBn), 4.50 (d, J = 12.1 Hz, 1H, OBn), 4.00 (ddd, J = 5.1, 5.1, 5.1 Hz, 1H, 5-H), 3.90 (ddd, J = 9.5, 3.2, 3.2 Hz, 1H, 1-H), 3.79 (dd, J = 10.1, 6.0 Hz, 1H, 6-Hb), 3.73 (dd, J = 5.8, 5.8 Hz, 1H, 3-H), 3.68–3.61 (m, 4H, 3’-H2, 2-H, 6-Ha), 3.58 (dd, J = 5.4, 5.4 Hz, 1H, 4-H), 2.96 (br s, 1H, OH), 2.15 (br s, 1H, OH), 1.80–1.61 ppm (m, 4H, 1’-H2, 2’-H2). 13C{1H} NMR (100.6 MHz, CDCl3) δC 138.0 (2 × C, Ar), 137.4 (C, Ar), 127.6–128.5 (15 × CH, Ar), 78.1 (CH, C-3), 75.2 (CH, C-4), 73.5 (CH2, OBn), 73.3 (CH2, OBn), 73.3 (CH, C-5), 73.0 (CH2, OBn), 71.9 (CH, C-1), 70.0 (CH, C-2), 68.2 (CH2, C-6), 62.6 (CH2, C-3’), 29.2 (CH2, C-2’), 24.8 ppm (CH2, C-1’), IR (CHCl3): ν = 3496, 2938, 1455, 1086 cm–1. MS (ESI) m/z (%) = 515 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C30H36NaO6 515.2410; found 515.2407. Anal. calcd for C30H36O6: C, 73.15; H, 7.37. Found: C, 72.90; H, 7.26.
3-C-(3,4,6-Tri-O-benzyl-β-d-glucopyranosyl)1-propanol (66)
Following the general procedure for the hydroboration, starting from 3-C-(3,4,6-tri-O-benzyl-β-d-glucopyranosyl)1-propene (65)50c (103 mg, 0.22 mmol) and purification by column chromatography (hexanes–EtOAc, 7:3), the alcohol 66 (77.3 mg, 0.16 mmol, 73%) was obtained as a crystalline solid: mp 112.0–112.7 °C (n-hexane–EtOAc); [α]D = +35.7 (c = 0.63, CHCl3). 1H NMR (400 MHz, CDCl3) δH 7.28–7.18 (m, 13H, Ar), 7.12–7.10 (m, 2H, Ar), 4.88 (d, J = 11.6 Hz, 1H, OBn), 4.71 (d, J = 10.8 Hz, 1H, OBn), 4.67 (d, J = 11.7 Hz, 1H, OBn), 4.53 (d, J = 12.2 Hz, 1H, OBn), 4.49 (d, J = 10.8 Hz, 1H, OBn), 4.45 (d, J = 12.1 Hz, 1H, OBn), 3.61 (ddd, J = 10.8, 10.8, 2.2 Hz, 1H, 5-H), 3.58–3.55 (m, 3H, 3’-H2, 6-Hb), 3.51 (dd, J = 9.3, 9.3 Hz, 1H, 4-H), 3.39 (dd, J = 8.9, 8.9 Hz, 1H, 3-H), 3.37 (m, 1H, 6-Ha), 3.26 (dd, J = 9.2, 9.2 Hz, 1H, 2-H), 3.15 (ddd, J = 8.5, 8.5, 2.4 Hz, 1H, 1-H), 2.22 (br s, 2H, 2 × OH), 1.92 (m, 1H, 1’-Hb), 1.68–1.62 (m, 2H, 2’-H2), 1.49 ppm (dddd, J = 7.7, 7.7, 7.7, 7.7 Hz, 1H, 1’-Ha), 13C{1H} NMR (100.6 MHz, CDCl3) δC 138.6 (C, Ar), 138.0 (2 × C, Ar), 128.6 (2 × CH, Ar), 128.4 (2 × CH, Ar), 128.3 (2 × CH, Ar), 127.89 (3 × CH, Ar), 127.84 (2 × CH, Ar), 127.79 (2 × CH, Ar), 127.75 (CH, Ar), 127.6 (CH, Ar), 86.8 (CH, C-3), 79.4 (CH, C-4), 78.8 (CH, C-5), 78.4 (CH, C-1), 75.2 (CH2, OBn), 74.7 (CH2, OBn), 73.7 (CH, C-2), 73.5 (CH2, OBn), 69.0 (CH2, C-6), 62.7 (CH2, C-3’), 28.8 (CH2, C-2’), 28.5 ppm (CH2, C-1’). IR (CHCl3): ν = 3588, 3500, 2928, 1455, 1052 cm–1. MS (ESI) m/z (%) = 515 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C30H36NaO6 515.2410; found 515.2412.
3-C-(3,4,6-Tri-O-benzyl-α-d-mannopyranosyl)1-propanol (69)
Following the general procedure for the hydroboration, starting from 3-C-(3,4,6-tri-O-benzyl-α-d-mannopyranosyl)1-propene (68)54,63 (675.6 mg, 1.42 mmol) and purification by column chromatography (hexanes–EtOAc, 1:1 to 3:7), the alcohol 69 (368.4 mg, 0.75 mmol, 53%) was obtained as an amorphous solid: [α]D = +34.0 (c = 0.45, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.35–7.25 (m, 13H, Ar), 7.22–7.20 (m, 2H, Ar), 4.71 (d, J = 11.1 Hz, 1H, OBn), 4.61 (d, J = 11.7 Hz, 1H, OBn), 4.57 (d, J = 10.1 Hz, 1H, OBn), 4.55 (d, J = 11.1 Hz, 1H, OBn), 4.52 (d, J = 11.4 Hz, 1H, OBn), 4.51 (d, J = 12.0 Hz, 1H, OBn), 3.89 (m, 1H, 1-H), 3.82–3.80 (m, 2H, 2-H, 3-H), 3.77–3.75 (m, 2H, 4-H, 5-H), 3.70 (dd, J = 10.1, 5.4 Hz, 1H, 6-Hb), 3.68–3.60 (m, 3H, 3’-H2, 6-Ha), 2.25 (br s, 2H, 2 × OH), 1.76–1.59 ppm (m, 4H, 1’-H2, 2’-H2). 13C{1H} NMR (100.6 MHz, CDCl3) δC 138.1 (2 × C, Ar), 137.6 (C, Ar), 127.6–128.6 (15 × CH, Ar), 79.2 (CH, C-3), 75.1 (CH, C-1), 74.2 (CH, C-5), 74.0 (CH2, OBn), 73.4 (CH2, OBn), 72.8 (CH, C-4), 72.3 (CH2, OBn), 69.4 (CH, C-2), 69.0 (CH2, C-6), 62.2 (CH2, C-3’), 29.2 (CH2, C-2’), 25.8 ppm (CH2, C-1’). IR (CHCl3): ν = 3562, 3500, 2933, 1094 cm–1. MS (ESI) m/z (%) = 515 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C30H36NaO6 515.2410; found 515.2403. Anal. calcd for C30H36O6: C, 78.15; H, 7.87. Found C, 78.07; H, 7.60.
3-C-(3,4,6-Tri-O-benzyl-β-d-mannopyranosyl)1-propanol (72)
Following the general procedure for the hydroboration, starting from 3-C-(3,4,6-tri-O-benzyl-β-d-mannopyranosyl)1-propene (71)54,63 (106 mg, 0.22 mmol) and purification by column chromatography (hexanes–EtOAc, 7:3), the alcohol 72 (72.6 mg, 0.15 mmol, 67%) was obtained as a colorless oil: [α]D = +2.1 (c = 0.48, CHCl3). 1H NMR (400 MHz, CDCl3) δH 7.37–7.17 (m, 15H, Ar), 4.83 (d, J = 10.8 Hz, 1H, OBn), 4.71 (d, J = 11.6 Hz, 1H, OBn), 4.64 (d, J = 11.7 Hz, 1H, OBn), 4.57 (d, J = 12.1 Hz, 1H, OBn), 4.52 (d, J = 12.1 Hz, 1H, OBn), 4.49 (d, J = 10.8 Hz, 1H, OBn), 3.90 (dd, J = 2.9, 0.0 Hz, 1H, 2-H), 3.74 (dd, J = 9.6, 9.6 Hz, 1H, 4-H), 3.70 (dd, J = 8.0, 1.6 Hz, 1H, 6-Hb), 3.63 (m, 3H, 6-Ha, 3’-H2), 3.57 (dd, J = 9.0, 3.2 Hz, 1H, 3-H), 3.40 (ddd, J = 9.8, 5.4, 1.9 Hz, 1H, 5-H), 3.35 (ddd, J = 9.2, 3.8, 0.0 Hz, 1H, 1-H), 2.94 (br s, 2H, 2 × OH), 1.88 (m, 1H, 1’-Hb), 1.75–1.64 ppm (m, 3H, 1’-Ha, 2’-H2). 13C{1H} NMR (100.6 MHz, CDCl3) δC 138.2 (C, Ar), 138.1 (C, Ar), 137.8 (C, Ar), 127.6–128.5 (15 × CH, Ar), 83.4 (CH, C-3), 79.0 (CH, C-1), 78.1 (CH, C-5), 75.1 (CH2, OBn), 74.7 (CH, C-4), 73.4 (CH2, OBn), 71.6 (CH2, OBn), 69.4 (CH2, C-6), 68.7 (CH, C-2), 62.6 (CH2, C-3’), 29.4 (CH2, C-2’), 28.0 ppm (CH2, C-1’). IR (CHCl3): ν = 3461, 2869, 1455, 1093 cm–1. MS (ESI) m/z (%) = 515 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C30H36NaO6 515.2410; found 515.2403. Anal. calcd for C30H36O6: C, 73.15; H, 7.37. Found: C, 73.36; H, 7.67.
3-C-(3,4-Di-O-benzyl-α-l-fucopyranosyl)1-propanol (76)
Following the general procedure for the hydroboration, starting from 75 (744.3 mg, 2.02 mmol) and purification by column chromatography (hexanes–EtOAc, 2:8), the alcohol 76 (442.9 mg, 1.15 mmol, 57%) was obtained as a colorless oil: [α]D = −51.4 (c = 0.52, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.35–7.25 (m, 10H, Ar), 4.78 (d, J = 11.7 Hz, 1H, OBn), 4.75 (d, J = 11.7 Hz, 1H, OBn), 4.60 (d, J = 11.7 Hz, 1H, OBn), 4.58 (d, J = 11.7 Hz, 1H, OBn), 4.07–4.06 (m, 2H, 1-H, 2-H), 3.91 (m, 1H, 5-H), 3.76 (dd, J = 3.2, 3.2 Hz, 1H, 4-H), 3.71 (dd, J = 6.6, 2.5 Hz, 1H, 3-H), 3.67–3.61 (m, 2H, 3’-H2), 2.40 (br s, 1H, OH), 2.21 (br s, 1H, OH), 1.69–1.62 (m, 4H, 1’-H2, 2’-H2), 1.31 ppm (d, J = 6.6 Hz, 3H, 6-H3). 13C{1H} NMR (100.6 MHz, CDCl3) δC 138.4 (C, Ar), 138.2 (C, Ar), 128.5 (2 × CH, Ar), 128.3 (2 × CH, Ar), 127.7 (3 × CH, Ar), 127.6 (3 × CH, Ar), 79.0 (CH, C-3), 75.1 (CH, C-4), 73.2 (CH2, OBn), 72.6 (CH2, OBn), 71.8 (CH, C-1 or C-2), 69.1 (CH, C-5), 68.9 (CH, C-1 or C-2), 62.6 (CH2, C-3’), 23.6 (2 × CH2, C-1’, C-2’), 15.6 ppm (CH3, C-6). IR (CHCl3): ν = 3585, 3422, 3016, 1228 cm–1. MS (ESI) m/z (%) = 409 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C23H30NaO5 409.1991; found 409.1990.
3-C-(3,4-Di-O-benzylidene-α,β-d-arabinopyranosyl)1-propanol (81)
Following the general procedure for the hydroboration, starting from 79 (180 mg, 0.69 mmol) and purification by column chromatography (hexanes–EtOAc, 2:8), the alcohol 81 (106.1 mg, 0.38 mmol, 55%) was obtained as a colorless oil: 1H NMR (500 MHz, CDCl3, only the major 1S isomer is described) δH 7.47–7.37 (m, 5H, Ar), 6.22 (br s, 1H, PhCH), 4.57 (ddd, J = 8.9, 6.4, 5.1 Hz, 1H, 4-H), 4.29 (dd, J = 5.1, 2.3 Hz, 1H, 3-H), 4.08 (dd, J = 11.7, 6.6 Hz, 1H, 5-Hb), 3.95 (br s, 1H, 2-H), 3.70–3.66 (m, 3H, 3’-H2, 1-H), 3.54 (dd, J = 11.7, 8.8 Hz, 1H, 5-Ha), 2.5 (br s, 1H, OH), 2.66 (br s, 1H, OH), 1.82–1.66 ppm (m, 4H, 1’-H2, 2’-H2). 13C{1H} NMR (125.7 MHz, CDCl3, only the major 1S isomer is described) δC 139.0 (C, Ar), 129.1 (CH, Ar), 128.4 (2 × CH, Ar), 125.9 (2 × CH, Ar), 103.3 (CH, PhCH), 75.7 (CH, C-1 or C-3), 75.6 (CH, C-1 or C-3), 70.1 (CH, C-4), 68.0 (CH, C-2), 65.9 (CH2, C-5), 62.6 (CH2, C-3’), 28.9 (CH2, C-1’ or C-2’), 26.7 ppm (CH2, C-1’ or C-2’). IR (CHCl3): ν = 3613, 3417, 2927, 1208, 1070 cm–1. MS (ESI) m/z (%) = 303 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C15H20NaO5 303.1208; found 303.1203.
3-C-(3,5-Di-O-tert-butyldiphenylsilyl-α-d-ribofuranosyl)1-propanol (85)
The general procedure for the hydroboration starting from 83 (3.55 g, 5.46 mmol) but adding dropwise at 0 °C an aqueous saturated solution of NaHCO3 (35.5 mL) instead of the NaOH solution followed by H2O2 30% (18 mL) gave, after purification by column chromatography (hexanes–EtOAc, 97:3 to 7:3), the alcohol 85 (1.28 mg, 1.92 mmol, 35%) as a colorless oil: [α]D = +23.0 (c = 0.64, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.65–7.26 (m, 20H, Ar), 4.53 (dd, J = 5.4, 5.4 Hz, 1H, 3-H), 3.97 (m, 1H, 4-H), 3.88 (m, 1H, 1-H), 3.82 (dd, J = 5.1, 5.1 Hz, 1H, 2-H), 3.71–3.62 (m, 2H, 3’-H2), 3.57 (dd, J = 11.4, 2.2 Hz, 1H, 5-Hb), 3.25 (dd, J = 11.4, 3.8 Hz, 1H, 5-Ha), 1.82–1.77 (m, 2H, 1’-H2), 1.71–1.66 (m, 2H, 2’-H2), 1.61 (br s, 2H, OH), 1.08 (s, 9H, tBu), 0.91 ppm (s, 9H, tBu). 13C{1H} NMR (125.7 MHz, CDCl3) δC 135.67 (2 × CH, Ar), 135.62 (2 × CH, Ar), 135.58 (2 × CH, Ar), 135.53 (2 × CH, Ar), 133.3 (C, Ar), 133.2 (C, Ar), 132.5 (C, Ar), 132.3 (C, Ar), 130.25 (CH, Ar), 130.20 (CH, Ar), 129.5 (2 × CH, Ar), 128.0 (2 × CH, Ar), 127.9 (2 × CH, Ar), 127.6 (4 × CH, Ar), 83.0 (CH, C-4), 81.5 (CH, C-1), 74.6 (CH, C-3), 73.0 (CH, C-2), 64.0 (CH2, C-5), 62.8 (CH2, C-3’), 29.6 (CH2, C-2’), 26.9 (3 × CH3, tBu), 26.7 (3 × CH3, tBu), 26.1 (CH2, C-1’), 19.2 (C, tBu), 19.1 ppm (C, tBu). IR (CHCl3): ν = 3532, 2932, 1428, 1206, 1113 cm–1. MS (ESI) m/z (%) = 691 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C40H52NaO5Si2 691.3251; found 691.3250.
3-C-(3,5-Di-O-1,1,3,3-tetraisopropyldisiloxanyl-α-d-ribofuranosyl)1-propanol (87)
The general procedure for the hydroboration starting from 86 (228 mg, 0.55 mmol) but adding dropwise at 0 °C an aqueous saturated solution of NaHCO3 (3.1 mL) instead of the NaOH solution followed by H2O2 30% (1.6 mL) gave, after purification by column chromatography (hexanes–EtOAc, 1:1), the alcohol 87 (150.47 mg, 0.35 mmol, 63%) as a colorless oil: [α]D = −10.8 (c = 0.62, CHCl3). 1H NMR (500 MHz, CDCl3) δH 4.38 (dd, J = 7.3, 4.8 Hz, 1H, 3-H), 5.00 (dd, J = 4.7, 4.7 Hz, 1H, 2-H), 4.00–3.96 (m, 2H, 1-H, 5-Hb), 3.91 (ddd, J = 7.3, 7.3, 3.5 Hz, 1H, 4-H), 3.64 (dd, J = 12.0, 6.3 Hz, 1H, 5-Ha), 3.69–3.63 (m, 2H, 3’-H2), 2.23 (br s, 2H, OH), 1.84 (ddd, J = 14.2, 7.6, 7.6 Hz, 1H, 1’-Hb), 1.79–1.64 (m, 3H, 1’-Ha, 2’-H2), 1.11–0.95 ppm (m, 28H, iPr). 13C{1H} NMR (125.7 MHz, CDCl3) δC 80.8 (CH, C-1), 80.3 (CH, C-4), 74.8 (CH, C-3), 72.8 (CH, C-2), 63.2 (CH2, C-5), 62.7 (CH2, C-3’), 29.3 (CH2, C-2’), 25.8 (CH2, C-1’), 17.4 (CH3, iPr), 17.33 (CH3, iPr), 17.30 (2 × CH3, iPr), 17.19 (CH3, iPr), 17.02 (2 × CH3, iPr), 16.93 (CH3, iPr), 13.4 (CH, iPr), 13.2 (CH, iPr), 12.8 (CH, iPr), 12.6 ppm (CH, iPr). IR (CHCl3): ν = 3622, 3528, 2948, 2870, 1465, 1041 cm–1. MS (ESI) m/z (%) = 457 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C20H42NaO6Si2 457.2418; found 457.2413.
General Procedure for the Mitsunobu Reaction to Give Phthalimide Derivatives 11, 13, 14, 64, 67, 70, 73, 77, and 88
DEAD (449 μL, 2.58 mmol) was added dropwise to a stirred solution of the alcohol (1 mmol), N-hydroxyphthalimide (420 mg, 2.58 mmol), and PPh3 (670 mg, 2.58 mmol) in dry THF (10.3 mL), and the resulting solution was stirred at 0 °C for 1–4 h. Then, the solvent was removed and the crude was quenched with water and extracted with CHCl3. The combined extracts were dried over Na2SO4 and concentrated under reduced pressure. Column chromatography of the residue (hexanes–EtOAc) gave the corresponding phthalimide.
3-C-(3,4-Di-O-benzylidene-2-O-diphenoxyphosphoryl-α,β-d-arabinopyranosyl)1-propoxyphthalimide (11)
Following the general procedure starting from alcohol 81 (95.2 mg, 0.34 mmol) stirring at 0 °C for 0.5 h, after purification by column chromatography (hexanes–EtOAc, 6:4), a phthalimide intermediate (202.4 mg) was obtained as a yellow oil. The crude (202.4 mg) was dissolved in dry CH2Cl2 (10.2 mL) under a N2 atmosphere. ClPO(OPh)2 (324 μL, 1.6 mmol) and DMAP (195.5 mg, 1.6 mmol) were added at 0 °C, and after 5 min, the mixture was stirred at room temperature for 1.5 h. The reaction was quenched with a saturated aqueous NH4Cl solution and extracted with CH2Cl2. The organic extracts were dried over Na2SO4 and concentrated under reduced pressure. Column chromatography of the residue (PhCH3–EtOAc, 9:1) gave 11 (149.4 mg, 0.23 mmol, 67%, 1S/1R 4.7:1) as a colorless oil. 1H NMR (500 MHz, CDCl3, only the major 1S isomer is described, simulated ring coupling constants using DAISY) δH 7.84–7.81 (m, 2H, Ar), 7.75–7.72 (m, 2H, Ar), 7.40–7.14 (m, 15H, Ar), 6.20 (s, 1H, PhCH), 4.88 (ddd, J = 3.3, 2.0 Hz, 3JPH = 9.2 Hz, 1H, 2-H), 4.38 (ddd, J = 7.9, 6.0, 5.2 Hz, 1H, 4-H), 4.32 (dd, J = 5.2, 3.3 Hz, 1H, 3-H), 4.13 (ddd, J = 6.7, 6.7, 1.3 Hz, 2H, 3’-H2), 4.04 (dd, J = 12.2, 6.0 Hz, 1H, 5-Hb), 3.83 (m, 1H, 1-H), 3.59 (dd, J = 12.2, 7.9 Hz, 1H, 5-Ha), 1.92 (m, 1H, 2’-Hb), 1.85–1.76 (m, 2H, 1’-Hb, 2’-Ha), 1.67 ppm (m, 1H, 1’-Ha). 13C{1H} NMR (125.7 MHz, CDCl3, only the major 1S isomer is described) δC 163.6 (2 × C, CO), 150.4 (d, 2JPC = 8.4 Hz, C, Ar), 150.3 (d, 2JPC = 7.4 Hz, C, Ar), 138.6 (C, Ar), 134.4 (2 × CH, Ar), 129.9 (2 × CH, Ar), 129.8 (2 × CH, Ar), 129.1 (2 × C, Ar), 129.0 (CH, Ar), 128.4 (2 × CH, Ar), 126.0 (2 × CH, Ar), 125.58 (CH, Ar), 125.55 (CH, Ar), 123.5 (2 × CH, Ar), 120.2 (2 × CH, Ar), 120.1 (2 × CH, Ar), 103.4 (CH, PhCH), 77.9 (CH2, C-3’), 75.3 (d, 2JPC = 6.3 Hz, CH, C-2), 73.6 (d, 3JPC = 5.3 Hz, CH, C-1 or C-3), 73.5 (d, 3JPC = 2.1 Hz, CH, C-1 or C-3), 70.5 (CH, C-4), 64.6 (CH2, C-5), 25.8 (CH2, C-1’), 24.5 ppm (CH2, C-2’). IR (CHCl3): ν = 3018, 1791, 1734, 1226 cm–1. MS (ESI) m/z (%) = 680 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C35H32NNaO10P 680.1662; found 680.1661. Anal. calcd for C35H32NO10P: C, 63.92; H, 4.90: N, 2.13. Found: C, 64.10; H, 5.16; N, 2.33.
3-C-(2-O-Acetyl-3,5-di-O-tert-butyldiphenylsilyl-α-d-ribofuranosyl)1-propoxyphthalimide (13)
Following the general procedure starting from alcohol 85 (604 mg, 0.90 mmol) and stirring at 50 °C for 2 h, after purification by column chromatography (hexanes–EtOAc, 8:2), a phthalimide intermediate was obtained (715.4 mg, 0.88 mmol, 98%) as a colorless oil. Phthalimide (715.4 mg, 0.88 mmol) was dissolved in dry pyridine (3.4 mL), and Ac2O (1.15 mL) and DMAP (1.1 mg, 9.0·10–3 mmol) were added. The reaction was stirred at room temperature for 1 h, and then it was evaporated in a high vacuum rotovap, quenched with an aqueous solution of HCl 10%, and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. Column chromatography of the residue (hexanes–EtOAc, 8:2) gave 13 (589.9 mg, 0.69 mmol, 78%) as a white solid. mp 42.3–43.7 °C (n-hexane–EtOAc); [α]D = +33.2 (c = 0.76, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.82–7.71 (m, 4H, Ar), 7.67–7.27 (m, 20H, Ar), 5.17 (dd, J = 4.7, 3.4 Hz, 1H, 2-H), 4.64 (dd, J = 6.7, 4.7 Hz, 1H, 3-H), 4.22–4.17 (m, 2H, 3’-H2), 4.03–3.98 (m, 2H, 1-H, 4-H), 3.61 (dd, J = 11.4, 2.2 Hz, 1H, 5-Hb), 3.31 (dd, J = 11.4, 3.2 Hz, 1H, 5-Ha), 2.15 (s, 3H, OAc), 1.95 (m, 1H, 2’-Hb), 1.81–1.68 (m, 3H, 1’-H2, 2’-Ha), 1.04 (s, 9H, tBu), 0.90 ppm (s, 9H, tBu). 13C{1H} NMR (125.7 MHz, CDCl3) δC 170.4 (C, OAc), 163.6 (2 × C, CO), 135.84 (2 × CH, Ar), 135.79 (2 × CH, Ar), 135.64 (2 × CH, Ar), 135.59 (2 × CH, Ar), 134.4 (2 × CH, Ar), 133.5 (C, Ar), 133.4 (C, Ar), 133.3 (C, Ar), 132.8 (C, Ar), 129.98 (CH, Ar), 129.95 (CH, Ar), 129.5 (2 × CH, Ar), 129.0 (2 × C, Ar), 127.78 (2 × CH, Ar), 127.74 (2 × CH, Ar), 127.57 (2 × CH, Ar), 127.55 (2 × CH, Ar), 123.5 (2 × CH, Ar), 82.8 (CH, C-4), 79.0 (CH, C-1), 78.3 (CH2, C-3’), 74.9 (CH, C-2), 72.8 (CH, C-3), 63.6 (CH2, C-5), 26.8 (3 × CH3, tBu), 26.7 (3 × CH3, tBu), 26.1 (CH2, C-1’), 25.0 (CH2, C-2’), 21.0 (CH3, OAc), 19.2 (C, tBu), 19.1 ppm (C, tBu). IR (CHCl3): ν = 2932, 2860, 1791, 1731, 1428, 1242, 1113 cm–1. MS (ESI) m/z (%) = 878 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C50H57NNaO8Si2 878.3520; found 878.3530.
3-C-(3,5-Di-O-tert-butyldiphenylsilyl-2-O-trifluoromethylsulfonyl-α-d-ribofuranosyl)1-propoxyphthalimide (14)
Following the general procedure starting from alcohol 85 (604 mg, 0.90 mmol) and stirring at 50 °C for 2 h, after purification by column chromatography (hexanes–EtOAc, 8:2), a phthalimide intermediate was obtained (715.4 mg, 0.88 mmol, 98%) as a colorless oil. Phthalimide (715.4 mg, 0.88 mmol) was dissolved in dry pyridine (0.26 mL), and Tf2O (22 μL, 0.13 mmol) was added. The reaction was stirred at room temperature for 1 h, and then it was evaporated in a high vacuum rotovap, quenched with an aqueous solution of HCl 10%, and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. Column chromatography of the residue (hexanes–EtOAc, 8:2) gave 14 (48.4 mg, 0.051 mmol, 78%) as a white solid. mp 42.6–43.9 °C (n-hexane–EtOAc); [α]D = +20.8 (c = 0.63, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.44–7.42 (m, 4H, Ar), 7.41–7.24 (m, 20H, Ar), 5.30 (dd, J = 4.6, 4.1 Hz, 1H, 2-H), 4.71 (dd, J = 5.7, 4.6 Hz, 1H, 3-H), 4.25–4.16 (m, 3H, 1-H, 3’-H2), 3.95 (m, 1H, 4-H), 3.35 (dd, J = 11.6, 2.0 Hz, 1H, 5-Hb), 2.76 (dd, J = 11.6, 3.1 Hz, 1H, 5-Ha), 2.05–1.92 (m, 3H, 1’-H2 or 2’-H2, 1’-Hb or 2’-Hb), 1.82 (dddd, J = 12.9, 12.9, 6.3, 6.3 Hz, 1H, 1’-Ha or 2’-Ha), 1.06 (s, 9H, tBu), 0.85 ppm (s, 9H, tBu). 13C{1H} NMR (125.7 MHz, CDCl3) δC 163.5 (2 × C, CO), 135.9 (2 × CH, Ar), 135.8 (2 × CH, Ar), 135.5 (4 × CH, Ar), 134.4 (2 × CH, Ar), 133.2 (C, Ar), 133.03 (C, Ar), 132.97 (C, Ar), 131.6 (C, Ar), 130.13 (CH, Ar), 130.06 (CH, Ar), 129.58 (CH, Ar), 129.56 (CH, Ar), 129.0 (2 × C, Ar), 127.9 (2 × CH, Ar), 127.7 (2 × CH, Ar), 127.55 (4 × CH, Ar), 123.5 (2 × CH, Ar), 89.3 (CH, C-2), 82.6 (CH, C-4), 77.84 (CH, C-1), 77.81 (CH2, C-3’), 73.1 (CH, C-3), 63.5 (CH2, C-5), 26.65 (3 × CH3, tBu), 26.63 (3 × CH3, tBu), 25.9 (CH2, C-1’ or C-2’), 24.8 (CH2, C-1’ or C-2’), 19.2 (C, tBu), 19.0 ppm (C, tBu), 1C from CF3 group is missing. IR (CHCl3): ν = 2932, 1791, 1734, 1113 cm–1. MS (ESI) m/z (%) = 968 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C49H54F3NNaO9SSi2 968.2908; found 968.2907. Anal. calcd for C49H54F3NO9SSi2: C, 62.20; H, 5.75; N, 1.48; S, 3.39. Found: C, 62.11; H, 5.97; N, 1.52; S, 3.19.
3-C-(3,4,6-Tri-O-benzyl-α-d-glucopyranosyl)1-propoxyphthalimide (64)
Following the general procedure starting from alcohol 63 (85.6 mg, 0.18 mmol) and purification by column chromatography (hexanes–Et2O, 1:1), product 64 (107 mg, 0.17 mmol, 93%) was obtained as a colorless oil: [α]D = +12.3 (c = 0.26, CHCl3). 1H NMR (400 MHz, CDCl3) δH 7.82–7.71 (m, 4H, Ar), 7.34–7.21 (m, 15H, Ar), 4.69 (d, J = 11.5 Hz, 1H, OBn), 4.63 (d, J = 11.4 Hz, 1H, OBn), 4.62 (d, J = 11.7 Hz, 1H, OBn), 4.56 (d, J = 12.4 Hz, 1H, OBn), 4.56 (d, J = 12.4 Hz, 1H, OBn), 4.48 (d, J = 12.0 Hz, 1H, OBn), 4.26–4.21 (m, 2H, 3’-H2), 4.02–3.96 (m, 2H, 1-H, 2-H), 3.82 (dd, J = 10.2, 5.7 Hz, 1H, 6-Hb), 3.77 (dd, J = 5.8, 5.8 Hz, 1H, 4-H), 3.73–3.68 (m, 2H, 5-H, 6-Ha), 3.63 (dd, J = 5.3, 5.3 Hz, 1H, 3-H), 1.99–1.76 ppm (m, 4H, 1’-H2, 2’-H2), 1H from OH is missing. 13C{1H} NMR (100.6 MHz, CDCl3) δC 163.6 (2 × C, CO), 138.1 (2 × C, Ar), 137.5 (C, Ar), 134.3 (2 × CH, Ar), 129.0 (2 × C, Ar), 128.5 (2 × CH, Ar), 128.4 (2 × CH, Ar), 128.3 (2 × CH, Ar), 127.84 (3 × CH, Ar), 127.79 (CH, Ar), 127.7 (2 × CH, Ar), 127.58 (2 × CH, Ar), 127.56 (CH, Ar), 123.4 (2 × CH, Ar), 78.3 (CH2, C-3’), 78.1 (CH, C-3), 75.3 (CH, C-4), 73.5 (CH2, OBn), 73.3 (CH2, OBn), 73.3 (CH, C-5), 72.9 (CH2, OBn), 71.4 (CH, C-1), 69.8 (CH, C-2), 68.3 (CH2, C-6), 24.5 (CH2, C-1’), 24.3 ppm (CH2, C-2’). IR (CHCl3): ν = 3514, 2935, 2871, 1792, 1737, 1372, 1083 cm–1. MS (E/I 70 eV): m/z (%) = 546 (6) [M – C7H7]+, 529 (51) [M – C7H8O]+, 91 (100) [C7H7]+. HRMS (E/I): m/z: [M – C7H8O]+ calcd for C31H31NO7 529.2101; found 529.2122.
3-C-(3,4,6-Tri-O-benzyl-β-d-glucopyranosyl)1-propoxyphthalimide (67)
Following the general procedure starting from alcohol 66 (810 mg, 1.64 mmol) and purification by column chromatography (hexanes–EtOAc, 1:1), product 67 (790 mg, 1.24 mmol, 76%) was obtained as a colorless oil: [α]D = +14.5 (c = 0.53, CHCl3). 1H NMR (400 MHz, CDCl3) δH 7.82–7.79 (m, 2H, Ar), 7.75–7.72 (m, 2H, Ar), 7.37–7.18 (m, 15H, Ar), 4.95 (d, J = 11.6 Hz, 1H, OBn), 4.80 (d, J = 11.0 Hz, 1H, OBn), 4.80 (d, J = 11.0 Hz, 1H, OBn), 4.61 (d, J = 12.2 Hz, 1H, OBn), 4.58 (d, J = 10.7 Hz, 1H, OBn), 4.53 (d, J = 12.3 Hz, 1H, OBn), 4.24 (ddd, J = 9.5, 6.6, 1.2 Hz, 2H, 3’-H2), 3.71 (br s, 2H, 6-H2), 3.61 (dd, J = 9.2, 9.2 Hz, 1H, 4-H), 3.52 (dd, J = 8.6, 8.6 Hz, 1H, 3-H), 3.42 (ddd, J = 9.6, 2.9, 2.9 Hz, 1H, 5-H), 3.39 (dd, J = 9.1, 9.1 Hz, 1H, 2-H), 3.28 (ddd, J = 8.2, 8.2, 2.2 Hz, 1H, 1-H), 2.14–1.98 (m, 2H, 1’-Hb, 2’-Hb), 1.87 (m, 1H, 2’-Ha), 1.71 ppm (m, 1H, 1’-Ha), 1H from OH is missing. 13C{1H} NMR (100.6 MHz, CDCl3) δC 163.7 (2 × C, CO), 138.7 (C, Ar), 138.2 (2 × C, Ar), 134.8 (2 × CH, Ar), 129.4 (2 × C, Ar), 129.0 (2 × CH, Ar), 128.8 (2 × CH, Ar), 128.7 (2 × CH, Ar), 128.31 (2 × CH, Ar), 128.25 (2 × CH, Ar), 128.2 (CH, Ar), 128.14 (2 × CH, Ar), 128.11 (CH, Ar), 127.9 (CH, Ar), 123.9 (2 × CH, Ar), 87.0 (CH, C-3), 79.1 (CH, C-1 or C-5), 78.9 (CH, C-1 or C-5), 78.6 (CH2, C-3’), 78.4 (CH, C-4), 75.2 (CH2, OBn), 74.8 (CH2, OBn), 73.9 (CH, C-2), 73.5 (CH2, OBn), 69.1 (CH2, C-6), 27.7 (CH2, C-1’), 24.0 ppm (CH2, C-2’). IR (CHCl3): ν = 3565, 2926, 2860, 1791, 1737, 1370, 1097 cm–1. MS (ESI) m/z (%) = 660 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C38H39NNaO8 660.2573; found 660.2558.
3-C-(3,4,6-Tri-O-benzyl-α-d-mannopyranosyl)1-propoxyphthalimide (70)
Following the general procedure starting from alcohol 69 (178.8 mg, 0.36 mmol) and purification by column chromatography (hexanes–EtOAc, 1:1), product 70 (206 mg, 0.32 mmol, 90%) was obtained as a colorless oil: [α]D = +13.5 (c = 1.50, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.83–7.81 (m, 2H, Ar), 7.74–7.73 (m, 2H, Ar), 7.34–7.20 (m, 15H, Ar), 4.73 (d, J = 11.4 Hz, 1H, OBn), 4.65 (d, J = 12.3 Hz, 1H, OBn), 4.63 (d, J = 11.7 Hz, 1H, OBn), 4.56 (d, J = 12.0 Hz, 1H, OBn), 4.53 (d, J = 11.4 Hz, 1H, OBn), 4.50 (d, J = 12.0 Hz, 1H, OBn), 4.27–4.19 (m, 2H, 3’-H2), 3.97 (m, 1H, 1-H), 3.89 (dd, J = 3.6, 2.9 Hz, 1H, 2-H), 3.83 (dd, J = 7.2, 3.1 Hz, 1H, 3-H), 3.81 (dd, J = 6.6, 6.6 Hz, 1H, 4-H), 3.77 (m, 1H, 5-H), 3.73 (dd, J = 10.4, 5.1 Hz, 1H, 6-Hb), 3.68 (dd, J = 10.4, 3.5 Hz, 1H, 6-Ha), 1.94 (m, 1H, 2’-Hb), 1.88–1.78 ppm (m, 3H, 1’-H2, 2’-Ha), 1H from OH is missing. 13C{1H} NMR (100.6 MHz, CDCl3) δC 163.6 (2 × C, CO), 138.3 (C, Ar), 138.2 (C, Ar), 137.7 (C, Ar), 134.4 (2 × CH, Ar), 129.0 (2 × C, Ar), 128.6 (2 × CH, Ar), 128.4 (2 × CH, Ar), 128.3 (2 × CH, Ar), 128.0 (CH, Ar), 127.9 (4 × CH, Ar), 127.73 (2 × CH, Ar), 127.68 (CH, Ar), 127.5 (CH, Ar), 123.5 (2 × CH, Ar), 79.4 (CH, C-3 or C-4), 78.0 (CH2, C-3’), 74.8 (CH, C-1), 74.3 (CH, C-5), 74.1 (CH2, OBn), 73.4 (CH2, OBn), 72.8 (CH, C-3 or C-4), 72.3 (CH2, OBn), 69.3 (CH, C-2), 69.1 (CH2, C-6), 25.5 (CH2, C-1’ or C-2’), 24.6 ppm (CH2, C-1’ or C-2’). IR (CHCl3): ν = 3559, 2930, 1789, 1731, 1082 cm–1. MS (ESI) m/z (%) = 660 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C38H39NNaO8 660.2573; found 660.2578.
3-C-(3,4,6-Tri-O-benzyl-β-d-mannopyranosyl)1-propoxyphthalimide (73)
Following the general procedure starting from alcohol 72 (72.6 mg, 0.15 mmol) and purification by column chromatography (hexanes–Et2O, 1:1), product 73 (80 mg, 0.13 mmol, 84%) was obtained as a colorless oil: [α]D = +5.3 (c = 0.34, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.83–7.80 (m, 2H, Ar), 7.75–7.72 (m, 2H, Ar), 7.38–7.18 (m, 15H, Ar), 4.85 (d, J = 10.7 Hz, 1H, OBn), 4.75 (d, J = 11.7 Hz, 1H, OBn), 4.67 (d, J = 11.7 Hz, 1H, OBn), 4.60 (d, J = 12.3 Hz, 1H, OBn), 4.53 (d, J = 10.4 Hz, 1H, OBn), 4.53 (d, J = 12.3 Hz, 1H, OBn), 4.29–4.20 (m, 2H, 3’-H2), 3.98 (dd, J = 2.9, 0.6 Hz, 1H, 2-H), 3.78 (dd, J = 9.4, 9.2 Hz, 1H, 4-H), 3.72 (dd, J = 11.3, 1.9 Hz, 1H, 6-Hb), 3.68 (dd, J = 11.3, 5.0 Hz, 1H, 6-Ha), 3.63 (dd, J = 9.2, 2.9 Hz, 1H, 3-H), 3.47 (ddd, J = 7.9, 4.1, 0.6 Hz, 1H, 1-H), 3.41 (ddd, J = 9.4, 5.0, 1.9 Hz, 1H, 5-H), 2.31 (br s, 1H, OH), 2.04–1.94 (m, 2H, 1’-Hb, 2’-Hb), 1.92–1.84 ppm (m, 2H, 1’-Ha, 2’-Ha). 13C{1H} NMR (100.6 MHz, CDCl3) δC 163.6 (2 × C, CO), 138.4 (2 × C, Ar), 137.9 (C, Ar), 134.4 (2 × CH, Ar), 129.0 (2 × C, Ar), 128.5 (2 × CH, Ar), 128.30 (2 × CH, Ar), 128.26 (2 × CH, Ar), 127.93 (2 × CH, Ar), 127.88 (3 × CH, Ar), 127.8 (2 × CH, Ar), 127.6 (CH, Ar), 127.5 (CH, Ar), 123.4 (2 × CH, Ar), 83.6 (CH, C-3), 79.2 (CH, C-1), 78.3 (CH2, C-3’), 77.3 (CH, C-5), 75.1 (CH2, OBn), 74.8 (CH, C-4), 73.4 (CH2, OBn), 71.6 (CH2, OBn), 69.5 (CH2, C-6), 68.4 (CH, C-2), 27.1 (CH2, C-2’), 24.7 ppm (CH2, C-1’). IR (CHCl3): ν = 3569, 2927, 2862, 1792, 1737, 1118 cm–1. MS (ESI) m/z (%) = 660 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C38H39NNaO8 660.2573; found 660.2576. Anal. calcd for C38H39NO8: C, 71.57; H, 6.16; N, 2.20. Found: C, 71.62; H, 6.30; N, 1.95.
3-C-(3,4-Di-O-benzyl-α-l-fucopyranosyl)1-propoxyphthalimide (77)
Following the general procedure starting from alcohol 76 (413 mg, 1.07 mmol) and purification by column chromatography (hexanes–EtOAc, 7:3), product 77 (536.9 mg, 1.01 mmol, 94%) was obtained as a colorless oil: [α]D = −14.4 (c = 0.80, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.83–7.81 (m, 2H, Ar), 7.74–7.73 (m, 2H, Ar), 7.37–7.24 (m, 10H, Ar), 4.79 (d, J = 11.7 Hz, 1H, OBn), 4.76 (d, J = 11.9 Hz, 1H, OBn), 4.62 (d, J = 12.0 Hz, 1H, OBn), 4.61 (d, J = 12.0 Hz, 1H, OBn), 4.28–4.21 (m, 2H, 3’-H2), 4.14–4.07 (m, 2H, 1-H, 2-H), 3.94 (m, 1H, 5-H), 3.79 (dd, J = 2.8, 2.8 Hz, 1H, 4-H), 3.76 (dd, J = 7.3, 2.9 Hz, 1H, 3-H), 2.52 (br s, 1H, OH), 2.04–1.77 (m, 4H, 1’-H2, 2’-H2), 1.30 ppm (d, J = 6.7 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 163.6 (2 × C, CO), 138.5 (C, Ar), 138.3 (C, Ar), 128.9 (2 × C, Ar), 128.5 (2 × CH, Ar), 128.4 (3 × CH, Ar), 128.2 (2 × CH, Ar), 127.7 (2 × CH, Ar), 127.6 (CH, Ar), 127.5 (2 × CH, Ar), 123.4 (2 × CH, Ar), 79.2 (CH, C-3), 78.3 (CH2, C-3’), 75.3 (CH, C-4), 73.2 (CH2, OBn), 72.5 (CH2, OBn), 71.9 (CH, C-2), 68.7 (CH, C-1), 68.6 (CH, C-5), 24.9 (CH2, C-1’ or C-2’), 22.8 (CH2, C-1’ or C-2’), 15.7 ppm (CH3, C-6). IR (CHCl3): ν = 3675, 3574, 3015, 1790, 1733, 1120 cm–1. MS (ESI) m/z (%) = 554 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C31H33NNaO7 554.2155; found 554.2147.
3-C-(3,5-Di-O-1,1,3,3-tetraisopropyldisiloxanyl-α-d-ribofuranosyl)1-propoxyphthalimide (88)
Following the general procedure starting from alcohol 87 (350 mg, 0.81 mmol) and purification by column chromatography (hexanes–EtOAc, 8:2), product 88 (457.9 mg, 0.79 mmol, 98%) was obtained as a colorless oil: [α]D = −13.0 (c = 0.73, CHCl3). 1H NMR (400 MHz, CDCl3) δH 7.83–7.72 (m, 4H, Ar), 4.36 (dd, J = 7.2, 4.8 Hz, 1H, 3-H), 4.25–4.20 (m, 2H, 3’-H2), 4.11 (dd, J = 4.8, 3.4 Hz, 1H, 2-H), 4.04 (m, 1H, 1-H), 3.95 (dd, J = 11.4, 3.2 Hz, 1H, 5-Hb), 3.89 (ddd, J = 6.1, 6.1, 3.4 Hz, 1H, 4-H), 3.83 (dd, J = 11.6, 6.1 Hz, 1H, 5-Ha), 1.96–1.80 (m, 4H, 1’-H2, 2’-H2), 1.09–0.97 ppm (m, 28H, iPr), 1H from OH is missing. 13C{1H} NMR (125.7 MHz, CDCl3) δC 163.6 (2 × C, CO), 134.4 (2 × CH, Ar), 129.0 (2 × C, Ar), 123.4 (2 × CH, Ar), 80.2 (CH, C-1 or C-4), 80.2 (CH, C-1 or C-4), 78.3 (CH2, C-3’), 74.6 (CH, C-3), 72.5 (CH, C-2), 63.1 (CH2, C-5), 25.3 (CH2, C-1’ or C-2’), 24.7 (CH2, C-1’ or C-2’), 17.43 (CH3, iPr), 17.31 (3 × CH3, iPr), 17.19 (CH3, iPr), 17.02 (2 × CH3, iPr), 16.94 (CH3, iPr), 13.4 (CH, iPr), 13.2 (CH, iPr), 12.8 (CH, iPr), 12.6 ppm (CH, iPr). IR (CHCl3): ν = 3546, 2948, 1791, 1733, 1467, 1039 cm–1. MS (ESI) m/z (%) = 602 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C28H45NNaO8Si2 602.2581; found 602.2585.
General Procedure to Give Acetyl Derivatives 1, 3, 5, 7, and 9
The phthalimide (1 mmol) was dissolved in dry pyridine (3.83 mL), and acetyl anhydride (1.1 mL) and DMAP (12.6 mg, 0.1 mmol) were added at 0 °C under a N2 atmosphere. The mixture was stirred at room temperature for 1 h. Then, the reaction was evaporated on the high vacuum rotovap, and the crude was quenched with HCl 10% and extracted with CH2Cl2. The organic extracts were washed with a saturated aqueous NaHCO3 solution, dried over Na2SO4, and concentrated under reduced pressure. Column chromatography of the residue (hexanes–EtOAc) gave the corresponding acetyl compound.
3-C-(2-O-Acetyl-3,4,6-tri-O-benzyl-α-d-glucopyranosyl)1-propoxyphthalimide (1)
Following the general procedure starting from phthalimide 64 (39.2 mg, 0.06 mmol) and purification by column chromatography (hexanes–EtOAc, 85:15), product 1 (30 mg, 0.04 mmol, 72%) was obtained as a crystalline solid: mp 99.7–100.5 °C (n-hexane–EtOAc); [α]D = +46.8 (c = 0.31, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.83–7.80 (m, 2H, Ar), 7.74–7.71 (m, 2H, Ar), 7.34–7.15 (m, 15H, Ar), 5.08 (dd, J = 9.0, 5.5 Hz, 1H, 2-H), 4.78 (d, J = 11.7 Hz, 1H, OBn), 4.75 (d, J = 11.1 Hz, 1H, OBn), 4.74 (d, J = 11.4 Hz, 1H, OBn), 4.60 (d, J = 12.3 Hz, 1H, OBn), 4.50 (d, J = 11.1 Hz, 1H, OBn), 4.48 (d, J = 12.0 Hz, 1H, OBn), 4.24 (dd, J = 6.0, 6.0 Hz, 2H, 3’-H2), 4.17 (ddd, J = 11.0, 5.5, 3.1 Hz, 1H, 1-H), 3.87 (dd, J = 9.0, 7.8 Hz, 1H, 3-H), 3.73–3.65 (m, 4H, 4-H, 5-H, 6-H2), 2.04 (s, 3H, OAc), 1.99–1.90 (m, 2H, 1’-Hb, 2’-Hb), 1.84–1.73 ppm (m, 2H, 1’-Ha, 2’-Ha). 13C{1H} NMR (100.6 MHz, CDCl3) δC 170.1 (C, OAc), 163.5 (2 × C, CO), 138.5 (C, Ar), 138.1 (2 × C, Ar), 134.4 (2 × CH, Ar), 129.0 (2 × C, Ar), 128.4 (2 × CH, Ar), 128.34 (2 × CH, Ar), 128.31 (2 × CH, Ar), 127.9 (2 × CH, Ar), 127.73 (2 × CH, Ar), 127.69 (CH, Ar), 127.61 (CH, Ar), 127.59 (2 × CH, Ar), 127.5 (CH, Ar), 123.4 (2 × CH, Ar), 80.1 (CH, C-3), 77.9 (CH2, C-3’), 77.6 (CH, C-4 or C-5), 74.8 (CH2, OBn), 74.6 (CH2, OBn), 73.5 (CH2, OBn), 73.0 (CH, C-2), 72.1 (CH, C-1), 71.9 (CH, C-4 or C-5), 69.0 (CH2, C-6), 24.3 (CH2, C-1’ or C-2’), 22.1 (CH2, C-1’ or C-2’), 20.9 ppm (CH3, OAc). IR (CHCl3): ν = 3013, 2870, 1790, 1734, 1236 cm–1. MS (ESI) m/z (%) = 702 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C40H41NNaO9 702.2679; found 702.2680. Anal. calcd for C40H41NO9: C, 70.68; H, 6.08; N, 2.06. Found C, 70.55; H, 6.07; N, 2.24.
3-C-(2-O-Acetyl-3,4,6-tri-O-benzyl-β-d-glucopyranosyl)1-propoxyphthalimide (3)
Following the general procedure starting from phthalimide 67 (790 mg, 1.26 mmol) and purification by column chromatography (hexanes–EtOAc, 7:3 to 1:1), product 3 (600 mg, 0.88 mmol, 71%) was obtained as a colorless oil: [α]D = +17.7 (c = 0.62, CHCl3). 1H NMR (400 MHz, CDCl3) δH 7.82–7.78 (m, 2H, Ar), 7.74–7.70 (m, 2H, Ar), 7.33–7.16 (m, 15H, Ar), 4.90 (dd, J = 9.5, 9.5 Hz, 1H, 2-H), 4.82 (d, J = 11.6 Hz, 1H, OBn), 4.78 (d, J = 10.8 Hz, 1H, OBn), 4.67 (d, J = 11.4 Hz, 1H, OBn), 4.60 (d, J = 12.3 Hz, 1H, OBn), 4.55 (d, J = 12.6 Hz, 1H, OBn), 4.51 (d, J = 12.3 Hz, 1H, OBn), 4.25 (m, 1H, 3’-Hb), 4.17 (m, 1H, 3’-Ha), 3.74–3.64 (m, 4H, 3-H, 4-H, 6-H2), 3.44 (ddd, J = 8.0, 4.0, 2.2 Hz, 1H, 5-H), 3.38 (ddd, J = 9.2, 9.2, 1.9 Hz, 1H, 1-H), 2.01 (m, 1H, 2’-Hb), 1.99 (s, 3H, OAc), 1.89–1.81 (m, 2H, 1’-Hb, 2’-Ha), 1.60 ppm (m, 1H, 1’-Ha). 13C{1H} NMR (100.6 MHz, CDCl3) δC 170.0 (C, OAc), 163.5 (2 × C, CO), 138.4 (C, Ar), 138.2 (C, Ar), 138.1 (C, Ar), 134.3 (2 × CH, Ar), 129.0 (2 × C, Ar), 128.4 (2 × CH, Ar), 128.33 (2 × CH, Ar), 128.28 (2 × CH, Ar), 127.9 (2 × CH, Ar), 127.7 (CH, Ar), 127.64 (4 × CH, Ar), 127.56 (CH, Ar), 127.5 (CH, Ar), 123.4 (2 × CH, Ar), 84.7 (CH, C-2), 79.1 (CH, C-3), 78.4 (CH, C-4), 78.1 (CH2, C-3’), 77.4 (CH, C-5), 75.1 (CH2, OBn), 74.9 (CH2, OBn), 73.8 (CH, C-1), 73.4 (CH2, OBn), 69.0 (CH2, C-6), 27.5 (CH2, C-1’), 24.0 (CH2, C-2’), 20.9 ppm (CH3, OAc). IR (CHCl3): ν = 3032, 2926, 2863, 1792, 1737, 1455, 1371, 1231, 1103 cm–1. MS (ESI) m/z (%) = 702 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C40H41NNaO9 702.2679; found 702.2689. Anal. calcd for C40H41NO9: C, 70.68; H, 6.08; N, 2.06. Found: C, 70.69; H, 6.20; N, 2.33.
3-C-(2-O-Acetyl-3,4,6-tri-O-benzyl-α-d-mannopyranosyl)1-propoxyphthalimide (5)
Following the general procedure starting from phthalimide 70 (99.2 mg, 0.16 mmol) and purification by column chromatography (hexanes–EtOAc, 6:4), product 5 (90.7 mg, 0.13 mmol, 86%) was obtained as a colorless oil: [α]D = +5.4 (c = 0.43, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.83–7.81 (m, 2H, Ar), 7.74–7.72 (m, 2H, Ar), 7.33–7.17 (m, 15H, Ar), 5.30 (dd, J = 3.4, 2.6 Hz, 1H, 2-H), 4.82 (d, J = 11.0 Hz, 1H, OBn), 4.69 (d, J = 11.4 Hz, 1H, OBn), 4.63 (d, J = 12.0 Hz, 1H, OBn), 4.54 (d, J = 11.4 Hz, 1H, OBn), 4.50 (d, J = 12.3 Hz, 1H, OBn), 4.49 (d, J = 11.1 Hz, 1H, OBn), 4.27–4.19 (m, 2H, 3’-H2), 4.03 (ddd, J = 10.9, 4.1, 2.6 Hz, 1H, 1-H), 3.93 (dd, J = 8.7, 3.4 Hz, 1H, 3-H), 3.85 (dd, J = 8.7, 8.4 Hz, 1H, 4-H), 3.78–3.69 (m, 3H, 5-H, 6-H2), 2.14 (s, 3H, OAc), 2.01–1.91 (m, 2H, 1’-H2), 1.87–1.77 ppm (m, 2H, 2’-H2). 13C{1H} NMR (100.6 MHz, CDCl3) δC 170.6 (C, OAc), 163.6 (2 × C, CO), 138.4 (2 × C, Ar), 137.9 (C, Ar), 134.4 (2 × CH, Ar), 129.0 (2 × C, Ar), 128.4 (2 × CH, Ar), 128.31 (2 × CH, Ar), 128.29 (2 × CH, Ar), 128.1 (2 × CH, Ar), 127.9 (2 × CH, Ar), 127.8 (CH, Ar), 127.7 (2 × CH, Ar), 127.6 (CH, Ar), 127.5 (CH, Ar), 123.5 (2 × CH, Ar), 77.9 (CH, C-3), 77.8 (CH2, C-3’), 75.0 (CH, C-4), 74.8 (CH, C-5), 74.7 (CH2, OBn), 73.5 (CH2, OBn), 72.8 (CH, C-1), 72.0 (CH2, OBn), 70.7 (CH, C-2), 69.4 (CH2, C-6), 25.0 (CH2, C-1’ or C-2’), 24.8 (CH2, C-1’ or C-2’), 21.2 ppm (CH3, OAc). IR (CHCl3): ν = 3034, 2929, 1790, 1734, 1189 cm–1. MS (ESI) m/z (%) = 702 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C40H41NNaO9 702.2679; found 702.2687. Anal. calcd for C40H41NO9: C, 70.68; H, 6.08; N, 2.06. Found: C, 70.74; H, 6.12; N, 2.04.
3-C-(2-O-Acetyl-3,4,6-tri-O-benzyl-β-d-mannopyranosyl)1-propoxyphthalimide (7)
Following the general procedure starting from phthalimide 73 (77 mg, 0.11 mmol) and purification by column chromatography (hexanes–EtOAc, 8:2), product 7 (60 mg, 0.09 mmol, 80%) was obtained as a colorless oil: [α]D = −16.7 (c = 0.63, CHCl3). 1H NMR (500 MHz, CDCl3, simulated coupling constants using DAISY) δH 7.82–7.79 (m, 2H, Ar), 7.74–7.72 (m, 2H, Ar), 7.34–7.16 (m, 15H, Ar), 5.52 (dd, J = 3.3, 1.0 Hz, 1H, 2-H), 4.86 (d, J = 10.8 Hz, 1H, OBn), 4.77 (d, J = 11.1 Hz, 1H, OBn), 4.64 (d, J = 12.3 Hz, 1H, OBn), 4.52 (d, J = 11.7 Hz, 1H, OBn), 4.50 (d, J = 10.4 Hz, 1H, OBn), 4.50 (d, J = 10.4 Hz, 1H, OBn), 4.25–4.18 (m, 2H, 3’-H2), 3.77 (dd, J = 9.8, 9.3 Hz, 1H, 4-H), 3.74 (m, 2H, 6-H2), 3.70 (dd, J = 9.3, 3.3 Hz, 1H, 3-H), 3.63 (ddd, J = 8.3, 4.7, 1.0 Hz, 1H, 1-H), 3.47 (ddd, J = 9.8, 5.3, 2.1 Hz, 1H, 5-H), 2.19 (s, 3H, OAc), 1.97 (m, 1H, 2’-Hb), 1.90–1.83 (m, 2H, 1’-Hb, 2’-Ha), 1.72 ppm (m, 1H, 1’-Ha). 13C{1H} NMR (125.7 MHz, CDCl3) δC 170.8 (C, OAc), 163.6 (2 × C, CO), 138.4 (C, Ar), 138.3 (C, Ar), 137.9 (C, Ar), 134.4 (2 × CH, Ar), 128.9 (2 × C, Ar), 128.34 (2 × CH, Ar), 128.26 (4 × CH, Ar), 128.1 (2 × CH, Ar), 127.9 (2 × CH, Ar), 127.8 (2 × CH, Ar), 127.7 (CH, Ar), 127.6 (CH, Ar), 127.5 (CH, Ar), 123.4 (2 × CH, Ar), 81.9 (CH, C-3), 79.4 (CH, C-5), 78.0 (CH2, C-3’), 76.2 (CH, C-1), 75.1 (CH2, OBn), 74.6 (CH, C-4), 73.4 (CH2, OBn), 71.5 (CH2, OBn), 69.4 (CH2, C-6), 69.2 (CH, C-2), 27.2 (CH2, C-1’ or C-2’), 24.4 (CH2, C-1’ or C-2’), 21.0 ppm (CH3, OAc). IR (CHCl3): ν = 3033, 2951, 2866, 1792, 1737, 1237, 1120 cm–1. MS (ESI) m/z (%) = 702 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C40H41NNaO9 702.2679; found 702.2675. Anal. calcd for C40H41NO9: C, 70.68; H, 6.08; N, 2.06. Found: C, 70.77; H, 6.05; N, 2.10.
3-C-(2-O-Acetyl-3,4-di-O-benzyl-α-l-fucopyranosyl)1-propoxyphthalimide (9)
Following the general procedure starting from phthalimide 77 (247.4 mg, 0.46 mmol) and purification by column chromatography (hexanes–EtOAc, 9:1 to7:3), product 9 (153.6 mg, 0.27 mmol, 58%) was obtained as a colorless oil: [α]D = −21.6 (c = 0.74, CHCl3). 1H NMR (500 MHz, CDCl3, simulated coupling constants using DAISY) δH 7.84–7.81 (m, 2H, Ar), 7.74–7.73 (m, 2H, Ar), 7.39–7.25 (m, 10H, Ar), 5.16 (dd, J = 5.9, 3.0 Hz, 1H, 2-H), 4.75 (d, J = 12.0 Hz, 1H, OBn), 4.70 (d, J = 12.0 Hz, 1H, OBn), 4.66 (d, J = 12.0 Hz, 1H, OBn), 4.55 (d, J = 12.0 Hz, 1H, OBn), 4.22 (dd, J = 5.0, 5.0 Hz, 2H, 3’-H2), 4.15 (ddd, J = 9.4, 4.1, 3.0 Hz, 1H, 1-H), 4.06 (dddd, J = 6.7, 6.7, 6.7, 4.5 Hz, 1H, 5-H), 3.82 (dd, J = 5.9, 3.2 Hz, 1H, 3-H), 3.74 (dd, J = 4.5, 3.2 Hz, 1H, 4-H), 2.08 (s, 3H, OAc), 1.91 (m, 1H, 2’-Hb), 1.81–1.74 (m, 2H, 1’-Hb, 2’-Ha), 1.66 (m, 1H, 1’-Ha), 1.36 ppm (d, J = 6.7 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 170.3 (C, OAc). 163.6 (2 × C, CO), 138.4 (C, Ar), 138.3 (C, Ar), 134.4 (2 × CH, Ar), 128.9 (2 × C, Ar), 128.28 (2 × CH, Ar), 128.26 (2 × CH, Ar), 127.7 (2 × CH, Ar), 127.6 (CH, Ar), 127.51 (CH, Ar), 127.46 (2 × CH, Ar), 123.4 (2 × CH, Ar), 78.1 (CH2, C-3’), 75.6 (CH, C-3), 74.6 (CH, C-4), 72.9 (CH2, OBn), 72.2 (CH2, OBn), 71.4 (CH, C-2), 69.5 (CH, C-5), 67.6 (CH, C-1), 24.9 (CH2, C-1’), 24.7 (CH2, C-2’), 21.0 (CH3, OAc), 14.6 ppm (CH3, C-6). IR (CHCl3): ν = 3029, 1791, 1734, 1214 cm–1. MS (ESI) m/z (%) = 596 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C33H35NNaO8 596.2260; found 596.2264. Anal. calcd for C33H35NO8: C, 69.10; H, 6.15; N, 2.44. Found: C, 69.25; H, 6.41; N, 2.58.
General Procedure to Give Diphenoxyphosphoryl Derivatives 2, 4, 6, 8, 10, and 15
The phthalimide (1 mmol) was dissolved in dry CH2Cl2 (7.5 mL) under a N2 atmosphere. ClPO(OPh)2 (1 mL, 4.7 mmol) and DMAP (580 mg, 4.75 mmol) were added at 0 °C, and after 5 min, the mixture was stirred at room temperature for 2 h. The reaction was quenched with a saturated aqueous NH4Cl solution and extracted with CH2Cl2. The organic extracts were dried over Na2SO4 and concentrated under reduced pressure. Column chromatography of the residue (hexanes–EtOAc) gave the phosphatyl precursor.
3-C-(3,4,6-Tri-O-benzyl-2-O-diphenoxyphosphoryl-α-d-glucopyranosyl)1-propoxyphthalimide (2)
Following the general procedure starting from phthalimide 64 (129.8 mg, 0.20 mmol) and purification by column chromatography (hexanes–EtOAc, 75:25), product 2 (132.6 mg, 0.15 mmol, 76%) was obtained as a colorless oil: [α]D = +40.5 (c = 0.44, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.83–7.81 (m, 2H, Ar), 7.74–7.72 (m, 2H, Ar), 7.30–7.10 (m, 25H, Ar), 4.83 (d, J = 11.0 Hz, 1H, OBn), 4.80 (ddd, J = 10.5, 5.5 Hz, 3JPH = 8.6 Hz, 1H, 2-H), 4.75 (d, J = 11.0 Hz, 1H, OBn), 4.74 (d, J = 11.1 Hz, 1H, OBn), 4.60 (d, J = 12.0 Hz, 1H, OBn), 4.46 (d, J = 11.0 Hz, 1H, OBn), 4.45 (d, J = 12.0 Hz, 1H, OBn), 4.20–4.15 (m, 2H, 3’-H2), 4.12 (ddd, J = 9.4, 7.2, 5.5 Hz, 1H, 1-H), 3.91 (ddd, J = 10.5, 5.7 Hz, 4JPH = 2.9 Hz, 1H, 3-H), 3.71–3.64 (m, 4H, 4-H, 5-H, 6-H2), 1.97 (m, 1H, 1’-Hb or 2’-Hb), 1.90 (m, 1H, 1’-Hb or 2’-Hb), 1.72 (m, 1H, 1’-Ha or 2’-Ha), 1.63 ppm (m, 1H, 1’-Ha or 2’-Ha). 13C{1H} NMR (125.7 MHz, CDCl3) δC 163.5 (2 × C, CO). 150.5 (d, 2JPC = 7.0 Hz, C, Ar), 150.4 (d, 2JPC = 6.4 Hz, C, Ar), 138.1 (C, Ar), 138.0 (C, Ar), 137.9 (C, Ar), 134.4 (2 × CH, Ar), 129.8 (2 × CH, Ar), 129.7 (2 × CH, Ar), 128.9 (2 × C, Ar), 128.3 (4 × CH, Ar), 128.2 (2 × CH, Ar), 127.8 (6 × CH, Ar), 127.7 (CH, Ar), 127.6 (CH, Ar), 127.5 (CH, Ar), 125.4 (CH, Ar), 125.2 (CH, Ar), 123.4 (2 × CH, Ar), 120.22 (CH, Ar), 120.18 (CH, Ar), 119.99 (CH, Ar), 119.95 (CH, Ar), 80.5 (d, 3JPC = 6.3 Hz, CH, C-3), 78.4 (d, 2JPC = 7.4 Hz, CH, C-2), 77.9 (CH2, C-3’), 77.8 (CH, C-1), 75.1 (CH2, OBn), 74.8 (CH2, OBn), 73.7 (CH, C-4 or C-5), 73.5 (CH2, OBn), 71.4 (CH, C-4 or C-5), 68.9 (CH2, C-6), 24.3 (CH2, C-1’ or C-2’), 21.1 ppm (CH2, C-1’ or C-2’). IR (CHCl3): ν = 3021, 2946, 1790, 1734, 1213 cm–1. MS (ESI) m/z (%) = 892 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C50H48NNaO11P 892.2863; found 892.2856. Anal. calcd for C50H48NO11P: C, 69.04; H; 5.56; N, 1.61. Found: C, 69.39; H, 5.74; N, 1.71.
3-C-(3,4,6-Tri-O-benzyl-2-O-diphenoxyphosphoryl-β-d-glucopyranosyl)1-propoxyphthalimide (4)
Following the general procedure starting from phthalimide 67 (327 mg, 0.51 mmol) and purification by column chromatography (hexanes–EtOAc, 7:3), product 4 (224 mg, 0.26 mmol, 51%) was obtained as a colorless oil: [α]D = +9.9 (c = 0.83, CHCl3). 1H NMR (500 MHz, CDCl3, simulated coupling constants using DAISY) δH 7.83–7.80 (m, 2H, Ar), 7.74–7.70 (m, 2H, Ar), 7.30–7.06 (m, 25H, Ar), 4.88 (d, J = 10.8 Hz, 1H, OBn), 4.83 (d, J = 10.8 Hz, 1H, OBn), 4.74 (d, J = 11.1 Hz, 1H, OBn), 4.58 (d, J = 12.3 Hz, 1H, OBn), 4.54 (d, J = 10.7 Hz, 1H, OBn), 4.49 (d, J = 12.3 Hz, 1H, OBn), 4.43 (ddd, J = 9.5, 8.9 Hz, 3JPH = 8.2 Hz, 1H, 2-H), 4.13 (ddd, J = 8.8, 7.6, 6.3 Hz, 1H, 3’-Hb), 4.04 (ddd, J = 8.8, 7.6, 6.6 Hz, 1H, 3’-Ha), 3.78 (dd, J = 9.2, 9.2 Hz, 1H, 3-H), 3.72–3.66 (m, 2H, 6-H2), 3.70 (dd, J = 9.5, 9.3 Hz, 1H, 4-H), 3.48 (dddd, J = 9.5, 8.2, 8.1, 2.5 Hz, 1H, 1-H), 3.45 (ddd, J = 9.5, 3.8, 1.8 Hz, 1H, 5-H), 2.00 (m, 1H, 2’-Hb), 1.89 (m, 1H, 2’-Ha), 1.83 (m, 1H, 1’-Hb), 1.57 ppm (m, 1H, 1’-Ha). 13C{1H} NMR (100.6 MHz, CDCl3) δC 163.4 (2 × C, CO), 150.6 (d, 2JPC = 7.1 Hz, C, Ar), 150.5 (d, 2JPC = 7.1 Hz, C, Ar), 138.3 (C, Ar), 138.1 (C, Ar), 138.0 (C, Ar), 134.3 (2 × CH, Ar), 129.6 (2 × CH, Ar), 129.5 (2 × CH, Ar), 129.0 (2 × C, Ar), 128.28 (2 × CH, Ar), 128.26 (2 × CH, Ar), 128.0 (2 × CH, Ar), 127.7 (2 × CH, Ar), 127.6 (3 × CH, Ar), 127.5 (3 × CH, Ar), 127.2 (CH, Ar), 125.3 (CH, Ar), 125.0 (CH, Ar), 123.3 (2 × CH, Ar), 120.4 (CH, Ar), 120.3 (CH, Ar), 120.1 (CH, Ar), 120.0 (CH, Ar), 84.7 (d, 3JPC = 2.1 Hz, CH, C-3), 80.6 (d, 2JPC = 7.7 Hz, CH, C-2), 79.0 (CH, C-4), 78.5 (CH, C-5), 78.0 (CH2, C-3’), 77.8 (d, 3JPC = 4.9 Hz, CH, C-1), 75.0 (CH2, OBn), 74.8 (CH2, OBn), 73.4 (CH2, OBn), 68.8 (CH2, C-6), 27.2 (CH2, C-1’), 23.9 ppm (CH2, C-2’). IR (CHCl3): ν = 3013, 2870, 1776, 1736, 1240, 1090 cm–1. MS (ESI) m/z (%) = 892 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C50H48NNaO11P 892.2863; found 892.2879. Anal. calcd for C50H48NO11P: C, 69.04; H, 5.56; N, 1.61. Found: C, 69.15; H, 5.64; N, 1.99.
3-C-(3,4,6-Tri-O-benzyl-2-O-diphenoxyphosphoryl-α-d-mannopyranosyl)1-propoxyphthalimide (6)
Following the general procedure starting from phthalimide 70 (109.2 mg, 0.17 mmol) and purification by column chromatography (hexanes–EtOAc, 65:35), product 6 (110.6 mg, 0.13 mmol, 74%) was obtained as a colorless oil: [α]D = −10.7 (c = 0.54, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.80–7.79 (m, 2H, Ar), 7.74–7.70 (m, 2H, Ar), 7.34–7.06 (m, 25H, Ar), 4.96 (ddd, J = 2.9, 2.9 Hz, 3JPH = 8.2 Hz, 1H, 2-H), 4.81 (d, J = 11.1 Hz, 1H, OBn), 4.67 (d, J = 11.0 Hz, 1H, OBn), 4.57 (d, J = 12.0 Hz, 1H, OBn), 4.53 (d, J = 11.4 Hz, 1H, OBn), 4.49 (d, J = 12.3 Hz, 1H, OBn), 4.40 (d, J = 11.0 Hz, 1H, OBn), 4.21–4.16 (m, 2H, 3’-H2), 4.07 (ddd, J = 9.8, 3.8, 3.8 Hz, 1H, 1-H), 3.92 (m, 1H, 3-H), 3.74–3.66 (m, 4H, 4-H, 5-H, 6-H2), 1.94–1.85 (m, 2H, 1’-Hb, 2’-Hb), 1.81–1.70 ppm (m, 2H, 1’-Ha, 2’-Ha). 13C{1H} NMR (125.7 MHz, CDCl3) δC 163.5 (2 × C, CO), 150.7 (d, 2JPC = 7.4 Hz, C, Ar), 150.5 (d, 2JPC = 7.4 Hz, C, Ar), 138.3 (C, Ar), 138.1 (C, Ar), 137.7 (C, Ar), 134.3 (2 × CH, Ar), 129.7 (2 × CH, Ar), 129.5 (2 × CH, Ar), 128.9 (2 × C, Ar), 128.3 (2 × CH, Ar), 128.24 (4 × CH, Ar), 128.19 (2 × CH, Ar), 127.9 (2 × CH, Ar), 127.64 (2 × CH, Ar), 127.59 (2 × CH, Ar), 127.4 (CH, Ar), 125.2 (CH, Ar), 125.0 (CH, Ar), 123.4 (2 × CH, Ar), 120.3 (CH, Ar), 120.24 (CH, Ar), 120.18 (CH, Ar), 120.1 (CH, Ar), 77.8 (d, 3JPC = 3.2 Hz, CH, C-3), 77.7 (CH2, C-3’), 77.5 (d, 2JPC = 6.4 Hz, CH, C-2), 74.9 (br s, CH, C-1), 74.5 (CH2, OBn), 74.3 (CH, C-4 or C-5), 73.3 (CH2, OBn), 73.0 (CH, C-4 or C-5), 72.1 (CH2, OBn), 69.1 (CH2, C-6), 24.8 (CH2, C-1’ or C-2’), 24.6 ppm (CH2, C-1’ or C-2’). IR (CHCl3): ν = 2929, 2858, 1793, 1737, 1491, 1191 cm–1. MS (ESI) m/z (%) = 892 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C50H48NNaO11P 892.2863; found 892.2864. Anal. calcd for C50H48NO11P: C, 69.04; H, 5.56; N, 1.61. Found: C, 68.91; H, 5.95; N, 1.91.
3-C-(3,4,6-Tri-O-benzyl-2-O-diphenoxyphosphoryl-β-d-mannopyranosyl)1-propoxyphthalimide (8)
Following the general procedure starting from phthalimide 73 (220 mg, 0.34 mmol) and purification by column chromatography (hexanes–EtOAc, 7:3), product 8 (242.7 mg, 0.28 mmol, 81%) was obtained as a colorless oil: [α]D = −16.6 (c = 0.53, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.83–7.80 (m, 2H, Ar), 7.76–7.72 (m, 2H, Ar), 7.39–7.12 (m, 25H, Ar), 5.09 (br dd, J = 1.9 Hz, 3JPH = 8.8 Hz, 1H, 2-H), 4.94 (d, J = 11.4 Hz, 1H, OBn), 4.63 (d, J = 10.7 Hz, 1H, OBn), 4.62 (d, J = 12.9 Hz, 1H, OBn), 4.52 (d, J = 11.7 Hz, 1H, OBn), 4.52 (d, J = 11.7 Hz, 1H, OBn), 4.31 (d, J = 10.8 Hz, 1H, OBn), 4.08 (dd, J = 6.4, 6.4 Hz, 2H, 3’-H2), 3.68 (br d, J = 3.2 Hz, 2H, 6-H2), 3.63 (m, 2H, 3-H, 4-H), 3.56 (m, 1H, 1-H), 3.42 (m, 1H, 5-H), 1.86 (m, 1H, 2’-Hb), 1.81–1.73 (m, 2H, 1’-Hb, 2’-Ha), 1.68 ppm (m, 1H, 1’-Ha). 13C{1H} NMR (100.6 MHz, CDCl3) δC 163.5 (2 × C, CO), 150.9 (d, 2JPC = 7.8 Hz, C, Ar), 150.7 (d, 2JPC = 7.0 Hz, C, Ar), 138.5 (C, Ar), 138.4 (C, Ar), 137.8 (C, Ar), 134.4 (2 × CH, Ar), 129.7 (2 × CH, Ar), 129.3 (2 × CH, Ar), 129.0 (2 × C, Ar), 128.4 (2 × CH, Ar), 128.3 (4 × CH, Ar), 128.2 (2 × CH, Ar), 127.9 (2 × CH, Ar), 127.7 (2 × CH, Ar), 127.6 (CH, Ar), 127.5 (CH, Ar), 127.4 (CH, Ar), 125.2 (CH, Ar), 124.7 (CH, Ar), 123.4 (2 × CH, Ar), 120.4 (CH, Ar), 120.3 (CH, Ar), 120.23 (CH, Ar), 120.18 (CH, Ar), 82.0 (CH, C-3), 79.5 (CH, C-5), 77.9 (CH2, C-3’), 77.0 (CH, C-2), 76.4 (d, 3JPC = 5.6 Hz, CH, C-1), 75.1 (CH2, OBn), 74.2 (CH, C-4), 73.4 (CH2, OBn), 71.8 (CH2, OBn), 69.3 (CH2, C-6), 27.2 (CH2, C-1’), 24.3 ppm (CH2, C-2’). IR (CHCl3): ν = 3013, 2869, 1789, 1733, 1193 cm–1. MS (ESI) m/z (%) = 892 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C50H48NNaO11P 892.2863; found 892.2861. Anal. calcd for C50H48NO11P: C, 69.04; H, 5.56; N, 1.61. Found: C, 68.94; H, 5.83; N, 1.67.
3-C-(3,4-Di-O-benzyl-2-O-diphenoxyphosphoryl-α-l-fucopyranosyl)1-propoxyphthalimide (10)
Following the general procedure starting from phthalimide 77 (253 mg, 0.48 mmol) and purification by column chromatography (hexanes–EtOAc, 7:3), product 10 (205 mg, 0.27 mmol, 56%) was obtained as a colorless oil: [α]D = −19.6 (c = 0.43, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.83–7.80 (m, 2H, Ar), 7.75–7.73 (m, 2H, Ar), 7.37–7.13 (m, 20H, Ar), 4.91 (ddd, J = 6.3, 3.3 Hz, 3JPH = 7.5 Hz, 1H, 2-H), 4.69 (d, J = 12.0 Hz, 1H, OBn), 4.66 (d, J = 12.0 Hz, 1H, OBn), 4.57 (d, J = 11.7 Hz, 1H, OBn), 4.42 (d, J = 11.7 Hz, 1H, OBn), 4.17–4.09 (m, 3H, 1-H, 3’-H2), 4.01 (dddd, J = 6.7, 6.7, 6.7, 4.0 Hz, 1H, 5-H), 3.95 (dd, J = 6.3, 3.1 Hz, 1H, 3-H), 3.72 (dd, J = 4.1, 3.1 Hz, 1H, 4-H), 1.90–1.76 (m, 2H, 1’-H2 or 2’-H2), 1.74–1.60 (m, 2H, 1’-H2 or 2’-H2), 1.31 ppm (d, J = 6.7 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 163.5 (2 × C, CO), 150.52 (d, 2JPC = 7.4 Hz, C, Ar), 150.48 (d, 2JPC = 7.4 Hz, C, Ar), 138.3 (C, Ar), 138.1 (C, Ar), 134.4 (2 × CH, Ar), 129.7 (4 × CH, Ar), 128.9 (2 × C, Ar), 128.3 (2 × CH, Ar), 128.2 (2 × CH, Ar), 127.6 (4 × CH, Ar), 127.5 (2 × CH, Ar), 125.4 (CH, Ar), 125.3 (CH, Ar), 123.4 (2 × CH, Ar), 120.2 (CH, Ar), 120.12 (CH, Ar), 120.08 (CH, Ar), 120.0 (CH, Ar), 78.1 (CH2, C-3’), 77.2 (CH, C-2), 76.1 (CH, C-3), 75.0 (CH, C-4), 73.1 (CH2, OBn), 72.6 (CH2, OBn), 69.2 (CH, C-5), 68.9 (CH, C-1), 24.7 (CH2, C-1’), 24.1 (CH2, C-2’), 14.8 ppm (CH3, C-6). IR (CHCl3): ν = 3027, 1791, 1733, 1214 cm–1. MS (ESI) m/z (%) = 786 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C43H42NNaO10P 786.2444; found 786.2448. Anal. calcd for C43H42NO10P: C, 67.62; H, 5.54; N, 1.83. Found: C, 67.81; H, 5.88; N, 1.48.
3-C-(2-O-Diphenoxyphosphoryl-3,5-di-O-1,1,3,3-tetraisopropyldisiloxanyl-α-d-ribofuranosyl)1-propoxyphthalimide (15)
Following the general procedure starting from phthalimide 88 (351 mg, 0.61 mmol) and purification by column chromatography (hexanes–EtOAc, 8:2 to 7:3), product 15 (443 mg, 0.55 mmol, 90%) was obtained as a colorless oil: [α]D = +15.8 (c = 0.83, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.83–7.73 (m, 4H, Ar), 7.32–7.12 (m, 10H, Ar), 5.15 (ddd, J = 3.9, 2.9 Hz, 3JPH = 7.9 Hz, 1H, 2-H), 4.47 (ddd, J = 9.1, 3.9 Hz, 4JPH = 1.5 Hz, 1H, 3-H), 4.19 (m, 1H, 1-H), 4.08 (br dd, J = 6.3, 6.3 Hz, 2H, 3’-H2), 3.99 (dd, J = 12.8, 3.1 Hz, 1H, 5-Hb), 3.94–3.90 (m, 2H, 4-H, 5-Ha), 1.88 (m, 1H, 2’-Hb), 1.71 (m, 1H, 2’-Ha), 1.65–1.57 (m, 2H, 1’-H2), 1.10–0.97 ppm (m, 28H, iPr). 13C{1H} NMR (125.7 MHz, CDCl3) δC 163.6 (2 × C, CO), 150.9 (d, 2JPC = 7.4 Hz, C, Ar), 150.6 (d, 2JPC = 6.4 Hz, C, Ar), 134.4 (2 × CH, Ar), 129.7 (2 × CH, Ar), 129.6 (2 × CH, Ar), 129.0 (2 × C, Ar), 125.2 (CH, Ar), 125.0 (CH, Ar), 123.4 (2 × CH, Ar), 120.23 (CH, Ar), 120.18 (CH, Ar), 120.05 (CH, Ar), 120.00 (CH, Ar), 81.5 (d, 2JPC = 6.4 Hz, CH, C-2), 79.6 (CH, C-4), 78.9 (d, 3JPC = 5.3 Hz, CH, C-1), 77.9 (CH2, C-3’), 71.7 (CH, C-3), 61.0 (CH2, C-5), 26.1 (CH2, C-1’), 24.6 (CH2, C-2’), 17.4 (CH3, iPr), 17.3 (CH3, iPr), 17.28 (CH3, iPr), 17.26 (CH3, iPr), 17.04 (CH3, iPr), 17.01 (CH3, iPr), 16.8 (CH3, iPr), 16.7 (CH3, iPr), 13.5 (CH, iPr), 13.1 (CH, iPr), 12.7 (CH, iPr), 12.4 ppm (CH, iPr). IR (CHCl3): ν = 2948, 1791, 1733, 1490, 1212 cm–1. MS (ESI) m/z (%) = 834 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C40H54NNaO11PSi2 834.2871; found 834.2874. Anal. calcd for C40H54NO11PSi2: C, 59.17; H, 6.70; N, 1.72. Found: C, 59.00; H, 6.94; N, 1.67.
3-C-(2-O-Diphenoxyphosphoryl-β-d-arabinopyranosyl)1-propoxyphthalimide (12)
Compound 11 (227 mg, 0.35 mmol) was dissolved in CH2Cl2 (1.6 mL), and TFA/H2O (230 μL, 80%) was dropwise added at 0 °C. After 1 h, the mixture was neutralized with a saturated aqueous solution of NaHCO3 and extracted with CH2Cl2. The organic extracts were dried over Na2SO4 and concentrated under reduced pressure. Column chromatography of the residue (hexanes–EtOAc, 3:7) gave 12 (161.7 mg, 0.28 mmol, 81%) as a colorless oil: [α]D = +4.6 (c = 0.56, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.83–7.80 (m, 2H, Ar), 7.75–7.72 (m, 2H, Ar), 7.35–7.17 (m, 10H, Ar), 4.60 (ddd, J = 4.1, 1.3 Hz, 3JPH = 8.2 Hz, 1H, 2-H), 4.16–4.05 (m, 3H, 3-H, 3’-H2), 3.86–3.81 (m, 2H, 1-H, 4-H), 3.74 (dd, J = 11.1, 5.1 Hz, 1H, 5-Hb), 3.48 (dd, J = 10.7, 10.7 Hz, 1H, 5-Ha), 2.75 (br d, J = 8.2 Hz, 1H, OH), 2.01 (br s, 1H, OH), 1.86 (m, 1H, 2’-Hb), 1.75 (m, 1H, 2’-Ha), 1.68 (m, 1H, 1’-Hb), 1.58 ppm (m, 1H, 1’-Ha). 13C{1H} NMR (125.7 MHz, CDCl3) δC 163.6 (2 × C, CO), 150.3 (d, 2JPC = 7.4 Hz, C, Ar), 150.2 (d, 2JPC = 8.4 Hz, C, Ar), 134.4 (2 × CH, Ar), 129.9 (4 × CH, Ar), 128.9 (2 × C, Ar), 125.7 (CH, Ar), 125.6 (CH, Ar), 123.5 (2 × CH, Ar), 120.09 (CH, Ar), 120.06 (CH, Ar), 120.04 (CH, Ar), 120.00 (CH, Ar), 79.1 (d, 2JPC = 6.4 Hz, CH, C-2), 78.0 (CH2, C-3’), 72.4 (d, 3JPC = 6.4 Hz, CH, C-1), 68.0 (CH, C-3), 65.9 (CH2, C-5), 64.2 (CH, C-4), 26.0 (CH2, C-1’), 24.5 ppm (CH2, C-2’). IR (CHCl3): ν = 3688, 3557, 3393, 3026, 1790, 1733, 1211 cm–1. MS (ESI) m/z (%) = 592 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C28H28NNaO10P 592.1349; found 592.1341. Anal. calcd for C28H28NO10P: C, 59.05; H, 4.96; N, 2.46. Found: C, 59.26; H, 4.99; N, 2.48.
General Procedure to Give Acetyl Derivatives 90 and 97
The corresponding alcohol (1 mmol) was dissolved in dry pyridine (3.7 mL), and Ac2O (1.2 mL) and DMAP (13.7 mg, 0.11 mmol) were added. The reaction was stirred at room temperature for 0.5 h, and then it was evaporated in a high vacuum rotovap, quenched with an aqueous solution of HCl 10%, and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. Column chromatography of the residue (hexanes–EtOAc) gave the acetyl derivative.
Methyl 4-O-Acetyl-6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-α-d-glucopyranoside (90)
Following the general procedure starting from methyl 6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-α-d-glucopyranoside (89)64 (119.7 mg, 0.27 mmol) and purification by column chromatography (hexanes–EtOAc, 7:3), product 90 (122.7 mg, 0.25 mmol, 94%) was obtained as a colorless oil: [α]D = +65.3 (c = 0.80, CHCl3). 1H NMR (400 MHz, CDCl3) δH 7.69–7.67 ppm (m, 4H, Ar), 7.45–7.36 (m, 6H, Ar), 4.87 (d, J = 3.5 Hz, 1H, 1-H), 4.87 (dd, J = 9.5, 9.5 Hz, 1H, 4-H), 3.75 (ddd, J = 10.1, 5.7, 2.5 Hz, 1H, 5-H), 3.71–3.62 (m, 2H, 6-H2), 3.58 (dd, J = 9.5, 9.5 Hz, 1H, 3-H), 3.54 (s, 3H, OMe), 3.51 (s, 3H, OMe), 3.47 (s, 3H, OMe), 3.30 (dd, J = 9.6, 3.7 Hz, 1H, 2-H), 1.95 (s, 3H, OAc), 1.05 ppm (s, 9H, tBu). 13C{1H} NMR (100.6 MHz, CDCl3) δC 169.5 (C, OAc), 135.7 (2 × CH, Ar), 135.6 (2 × CH, Ar), 133.4 (C, Ar), 133.3 (C, Ar), 129.62 (CH, Ar), 129.60 (CH, Ar), 127.62 (2 × CH, Ar), 127.59 (2 × CH, Ar), 97.3 (CH, C-1), 81.5 (CH, C-2 or C-5), 81.1 (CH, C-2 or C-5), 70.6 (CH, C-3 or C-4), 70.5 (CH, C-3 or C-4), 63.2 (CH2, C-6), 60.6 (CH3, OMe), 59.1 (CH3, OMe), 55.1 (CH3, OMe), 26.7 (3 × CH3, DPS), 20.8 (CH3, OAc), 19.2 ppm (C, DPS). IR (CHCl3): ν = 2932, 1748, 1233, 1046 cm–1. MS (ESI) m/z (%) = 525 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C27H38NaO7Si 525.2285; found 525.2272. Anal. calcd for C27H38O7Si: C, 64.51; H, 7.62. Found: C, 64.45; H, 7.85.
Methyl 4-O-Acetyl-6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-α-d-galactopyranoside (97)
Following the general procedure starting from methyl 6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-α-d-galactopyranoside (96)64 (2.15 g, 4.80 mmol) and purification by column chromatography (hexanes–EtOAc, 7:3), product 97 (2.19 g, 4.36 mmol, 91%) was obtained as a colorless oil: [α]D = +65.5 (c = 1.45, CHCl3). 1H NMR (400 MHz, CDCl3) δH 7.69–7.64 (m, 4H, Ar), 7.45–7.36 (m, 6H, Ar), 5.58 (dd, J = 3.2, 1.0 Hz, 1H, 4-H), 4.88 (d, J = 3.6 Hz, 1H, 1-H), 3.93 (ddd, J = 6.9, 6.9, 0.9 Hz, 1H, 5-H), 3.69 (dd, J = 10.2, 6.3 Hz, 1H, 6-Hb), 3.64 (dd, J = 10.4, 7.0 Hz, 1H, 6-Ha), 3.60 (dd, J = 10.1, 3.4 Hz, 1H, 3-H), 3.51 (s, 3H, OMe), 3.50 (dd, J = 10.1, 6.4 Hz, 1H, 2-H), 3.43 (s, 3H, OMe), 3.40 (s, 3H, OMe), 2.03 (s, 3H, OAc), 1.06 ppm (s, 9H, tBu). 13C{1H} NMR (100.6 MHz, CDCl3) δC 169.9 ppm (C, OAc), 135.5 (4 × CH, Ar), 133.2 (C, Ar), 133.1 (C, Ar), 129.7 (CH, Ar), 129.6 (CH, Ar), 127.7 (2 × CH, Ar), 127.6 (2 × CH, Ar), 97.7 (CH, C-1), 78.1 (CH, C-2 or C-5), 77.3 (CH, C-2 or C-5), 69.2 (CH, C-3 or C-4), 66.9 (CH, C-3 or C-4), 62.2 (CH2, C-6), 59.0 (CH3, OMe), 57.6 (CH3, OMe), 55.2 (CH3, OMe), 26.7 (3 × CH3, DPS), 20.6 (CH3, OAc), 19.0 ppm (C, DPS). IR (CHCl3): ν = 2934, 1741, 1239, 1106 cm–1. MS (ESI) m/z (%) = 525 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C27H38NaO7Si 525.2285; found 525.2279. Anal. calcd for C27H38O7Si: C, 64.51; H, 7.62. Found: C, 64.72; H, 7.50.
General Procedure to Give Allenyl Derivatives 91, 98, and 107
The precursor (1 mmol) was dissolved in CH3CN (11 mL) under a N2 atmosphere, and freshly prepared propargyl trimethylsilane/Et2O65 39% v/v (0.77 mL, 2 mmol) and TMSOTf (0.4 mL, 2.27 mmol) were dropwise added. The reaction was sonicated in an ultrasonic bath for 1.5–3 h, and then it was poured over a saturated aqueous solution of NaHCO3 and extracted with EtOAc. The organic phase was dried over Na2SO4, filtered, and evaporated. The crude was dissolved in DMF (2 mL), and DPSCl (176 μL, 38.68 mmol) and imidazole (102 mg, 22.42 mmol) were added. The reaction was stirred at room temperature for 2 h, evaporated under reduced high pressure, and purified by column chromatography (hexanes–EtOAc) to give the expected allenyl derivative.
C-(4-O-Acetyl-6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-α-d-glucopyranosyl)allene (91)
Following the general procedure starting from 90 (2.23 g, 4.55 mmol) and purification by column chromatography (hexanes–EtOAc, 9:1), product 91 (991.3 mg, 1.79 mmol, 39%) was obtained as a colorless oil: [α]D = +91.4 (c = 0.80, CHCl3). 1H NMR (400 MHz, CDCl3) δH 7.70–7.66 (m, 4H, Ar), 7.44–7.35 (m, 6H, Ar), 5.38 (ddd, J = 6.7, 6.7, 6.7 Hz, 1H, 1’-H), 4.93 (dd, J = 9.2, 9.2 Hz, 1H, 3-H), 4.87 (dd, J = 6.7, 0.0 Hz, 1H, 3’-Hb), 4.86 (dd, J = 6.7, 0.0 Hz, 1H, 3’-Ha), 4.78 (m, 1H, 1-H), 3.85–3.80 (m, 2H, 4-H, 6-Hb), 3.69–3.67 (m, 2H, 5-H, 6-Ha), 3.51 (s, 3H, OMe), 3.48 (s, 3H, OMe), 3.43 (dd, J = 9.5, 6.3 Hz, 1H, 2-H), 1.96 (s, 3H, OAc), 1.06 ppm (s, 9H, tBu). 13C{1H} NMR (100.6 MHz, CDCl3) δC 209.5 (C, C-2’), 169.5 (C, OAc), 135.7 (4 × CH, Ar), 133.6 (C, Ar), 133.4 (C, Ar), 129.6 (CH, Ar), 129.5 (CH, Ar), 127.57 (2 × CH, Ar), 127.55 (2 × CH, Ar), 85.5 (CH, C-1’), 81.0 (2 × CH, C-2, C-5), 76.7 (CH2, C-3’), 72.8 (CH, C-4), 71.1 (CH, C-1), 70.5 (CH, C-3), 63.6 (CH2, C-6), 60.0 (CH3, OMe), 58.4 (CH3, OMe), 26.8 (3 × CH3, DPS), 20.9 (CH3, OAc), 19.2 ppm (C, DPS). IR (CHCl3): ν = 3024, 2934, 1956, 1736, 1208 cm–1. MS (ESI) m/z (%) = 533 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C29H38NaO6Si 533.2335; found 533.2333.
C-(4-O-Acetyl-6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-α-d-galactopyranosyl)allene (98)
Following the general procedure starting from 97 (1.91 g, 3.80 mmol) and purification by column chromatography (hexanes–EtOAc, 8:2), product 98 (1.27 g, 2.49 mmol, 59%) was obtained as a yellow oil: [α]D = +101.0 (c = 0.81, CHCl3). 1H NMR (400 MHz, CDCl3) δH 7.68–7.63 (m, 4H, Ar), 7.45–7.35 (m, 6H, Ar), 5.67 (dd, J = 2.9, 1.2 Hz, 1H, 4-H), 5.33 (m, 1H, 1’-H), 4.85–4.79 (m, 3H, 1-H, 3’-H2), 3.98 (ddd, J = 7.6, 5.7, 1.5 Hz, 1H, 5-H), 3.67 (dd, J = 10.1, 5.8 Hz, 1H, 6-Hb), 3.66 (dd, J = 10.5, 5.7 Hz, 1H, 2-H), 3.59 (dd, J = 9.8, 7.9 Hz, 1H, 6-Ha), 3.47 (s, 3H, OMe), 3.46 (s, 3H, OMe), 3.40 (dd, J = 9.9, 3.6 Hz, 1H, 3-H), 2.06 (s, 3H, OAc), 1.06 ppm (s, 9H, tBu). 13C{1H} NMR (100.6 MHz, CDCl3) δC 209.0 (C, C-2’). 169.8 (C, OAc), 135.60 (2 × CH, Ar), 135.56 (2 × CH, Ar), 133.4 (C, Ar), 133.2 (C, Ar), 129.72 (CH, Ar), 129.67 (CH, Ar), 127.7 (2 × CH, Ar), 127.6 (2 × CH, Ar), 85.2 (CH, C-1’), 78.8 (CH, C-5), 77.1 (CH, C-2), 76.9 (CH2, C-3’), 71.5 (CH, C-4), 71.0 (CH, C-1), 66.6 (CH, C-3), 61.8 (CH2, C-6), 58.5 (CH3, OMe), 57.7 (CH3, OMe), 26.7 (3 × CH3, DPS), 20.7 (CH3, OAc), 19.1 ppm (C, DPS). IR (CHCl3): ν = 3016, 2934, 1955, 1741, 1208 cm–1. MS (ESI) m/z (%) = 533 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C29H38NaO6Si 533.2335; found 533.2328. Anal. calcd for C29H38O6Si: C, 68.20; H, 7.50. Found: C, 68.24; H, 7.70.
C-(2,3,4-Tri-O-acetyl-α-l-fucopyranosyl)allene (107)
Following the general procedure starting from peracetyl l-fucose (106) (877 mg, 2.64 mmol) and purification by column chromatography (hexanes–EtOAc, 8:2), product 107 (601.8 mg, 1.93 mmol, 73%) was obtained as a yellow oil: [α]D = −170.0 (c = 0.92, CHCl3). 1H NMR (500 MHz, C6D6) δH 5.68 (dd, J = 11.0, 6.0 Hz, 1H, 2-H), 5.46 (dd, J = 11.0, 3.5 Hz, 1H, 3-H), 5.37 (dd, J = 3.5, 1.3 Hz, 1H, 4-H), 5.17 (ddd, J = 6.9, 6.9, 6.9 Hz, 1H, 1′-H), 5.04 (dddd, J = 6.1, 6.1, 3.2, 3.2 Hz, 1H, 1-H), 4.59 (ddd, J = 11.3, 6.6, 3.2 Hz, 1H, 3′-Hb), 4.55 (ddd, J = 11.7, 6.9, 3.5 Hz, 1H, 3′-Ha), 3.67 (dddd, J = 6.3, 6.3, 6.3, 1.0 Hz, 1H, 5-H), 1.75 (s, 3H, OAc), 1.65 (s, 3H, OAc), 1.59 (s, 3H, OAc), 0.97 ppm (d, J = 6.3 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, C6D6) δC 209.8 (CH, C-2’), 170.2 (C, OAc), 169.8 (C, OAc), 169.4 (C, OAc), 85.4 (CH, C-1’), 76.6 (CH2, C-3’), 71.7 (CH, C-1), 71.5 (CH, C-4), 69.1 (CH, C-3), 68.4 (CH, C-2), 66.6 (CH, C-5), 20.4 (CH3, OAc), 20.2 (CH3, OAc), 20.0 (CH3, OAc), 16.3 ppm (CH3, C-6). IR (CHCl3): ν = 2989, 2944, 1957, 1750, 1373, 1230, 1062 cm–1. MS (ESI) m/z (%) = 335 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C15H20NaO7 335.1107; found 335.1101. Anal. calcd for C15H20O7: C, 57.69; H, 6.45. Found: C, 58.07; H, 6.79.
C-(2,3,4-Tri-O-acetyl-α-l-rhamnopyranosyl)allene (101) and C-(2,3,4-Tri-O-acetyl-β-l-rhamnopyranosyl)allene (102)
BF3•OEt2 (6.40 mL, 51.86 mmol) and TMSOTf (6.20 mL, 34.66 mmol) were dropwise added to a solution of peracetyl l-rhamnose (100) (6 g, 18.0 mmol) and fresly prepared propargyl trimethylsilane (5.11 mL, 34.28 mmol) in dry CH3CN (39 mL) at 0 °C, and the mixture was stirred at room temperature for 15 h. Then, it was poured over HCl 10% and extracted with EtOAc. The organic extracts were washed with saturated aqueous solutions of NaHCO3 and NaCl, dried over Na2SO4, and concentrated to dryness under reduced pressure. Column chromatography of the residue (hexanes–EtOAc, 95:5) gave 101 (3.80 g, 12.18 mmol, 67%) and 102 (1.40 g, 4.49 mmol, 25%) that could not be completely purified and was isolated as a mixture (1:1) with 101. Compound 101: colorless oil, [α]D = −16.7 (c = 0.42, CHCl3). 1H NMR (500 MHz, CDCl3) δH 5.40 (dd, J = 3.3, 2.0 Hz, 1H, 2-H), 5.26 (dd, J = 9.8, 3.5 Hz, 1H, 3-H), 5.21 (ddd, J = 6.8, 6.8, 4.4 Hz, 1H, 1′-H), 5.07 (dd, J = 9.5, 9.5 Hz, 1H, 4-H), 5.05 (ddd, J = 12.3, 6.9, 4.1 Hz, 1H, 3′-Hb), 4.99 (ddd, J = 11.4, 6.9, 4.4 Hz, 1H, 3′-Ha), 4.57 (dddd, J = 4.1, 4.1, 4.1, 1.9 Hz, 1H, 1-H), 3.84 (dddd, J = 9.5, 6.2, 6.2, 6.2 Hz, 1H, 5-H), 2.14 (s, 3H, OAc), 2.04 (s, 3H, OAc), 1.97 (s, 3H, OAc), 1.21 ppm (d, J = 6.0 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 207.7 (CH, C-2’), 170.3 (C, OAc), 170.0 (C, OAc), 169.9 (C, OAc), 87.8 (CH, C-1’), 78.7 (CH2, C-3’), 73.3 (CH, C-1), 71.6 (CH, C-4), 70.3 (CH, C-2), 69.3 (CH, C-3), 68.8 (CH, C-5), 21.0 (CH3, OAc), 20.8 (CH3, OAc), 20.6 (CH3, OAc), 17.6 ppm (CH3, C-6). IR (CHCl3): ν = 2981, 2933, 1955, 1746, 1369, 1221 cm–1. MS (ESI) m/z (%) = 335 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C15H20NaO7 335.1107; found 335.1107. Anal. calcd for C15H20O7: C, 57.69; H, 6.45. Found: C, 58.07; H, 6.79. Compound 102: 1H NMR (500 MHz, CDCl3, signals taken from a spectrum of the mix of 101 and 102) δH 5.39 (dd, J = 2.9, 1.3 Hz, 1H, 2-H), 5.14 (ddd, J = 6.6, 6.6, 6.6 Hz, 1H, 1′-H), 5.05 (m, 1H, 3-H), 4.98 (dd, J = 6.7, 4.4 Hz, 1H, 4-H), 4.86 (ddd, J = 11.4, 6.6, 2.2 Hz, 1H, 3′-Hb), 4.81 (ddd, J = 11.7, 6.9, 2.2 Hz, 1H, 3′-Ha), 4.19 (dddd, J = 6.9, 2.2, 2.2, 1.3 Hz, 1H, 1-H), 3.54 (dddd, J = 9.1, 6.3, 6.3, 6.3 Hz, 1H, 5-H), 2.15 (s, 3H, OAc), 2.04 (s, 3H, OAc), 1.96 (s, 3H, OAc), 1.25 ppm (d, J = 6.3 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3, signals taken from a spectrum of the mix of 101 and 102) δC 208.4 (CH, C-2’), 170.4 (C, OAc), 170.3 (C, OAc), 169.9 (C, OAc), 87.3 (CH, C-1’), 77.4 (CH2, C-3’), 75.1 (CH, C-1), 74.6 (CH, C-5), 72.3 (CH, C-4), 70.6 (CH, C-3), 70.4 (CH, C-2), 21.0 (CH3, OAc), 20.7 (CH3, OAc), 20.6 (CH3, OAc), 17.7 ppm (CH3, C-6). IR (CHCl3): ν = 2981, 2937, 1959, 1750, 1373, 1225, 1054 cm–1. MS (ESI) m/z (%) = 335 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C15H20NaO7 335.1107; found 335.1107.
C-(4-O-Benzyl-2,3-O-isopropylidene-α-l-rhamnopyranosyl)allene (103)
To a solution of 101 (1.89 g, 6.06 mmol) in dry MeOH (45 mL) was added K2CO3 (1.35 g, 9.78 mmol), and the mixture was stirred at room temperature for 3 h. Then, it was filtered and neutralized with the Amberlyst 15 H+ ion exchange resin. It was filtered again under vacuum and evaporated. The resulting organic crude was dissolved in acetone (60 mL), and 2,2-dimethoxypropane (1.9 mL, 15.15 mmol) and p-TsOH·H2O (692 mg, 3.64 mmol) were subsequently added while stirring at room temperature for 3 h. The acetone was evaporated, and the residue was poured over a saturated aqueous solution of NaHCO3 and extracted with CH2Cl2. The organic phase was dried over Na2SO4 and concentrated to dryness under reduced pressure. The organic crude was dissolved in dry DMF (73 mL) under a N2 atmosphere, and NaH 60% in mineral oil (364 mg, 9.09 mmol) was slowly added at 0 °C. After 20 min, BnBr (1.4 mL, 12.12 mmol) was dropwise added and stirring was continued at 0 °C for 3 h. Ice-water was used to destroy the excess of NaH, and the mixture was evaporated in a high vacuum rotovap, poured over a saturated solution of NH4Cl, and extracted with CH2Cl2. The combined extracts were dried over Na2SO4 and concentrated to dryness under reduced pressure. Column chromatography of the residue (hexanes–EtOAc, 95:5) gave 103 (1.03 g, 3.27 mmol, 54%) as a yellow oil: [α]D = +37.0 (c = 0.74, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.36–7.24 (m, 5H, Ar), 5.23 (ddd, J = 7.0, 7.0, 5.1 Hz, 1H, 1’-H), 4.90–4.86 (m, 3H, OBn, 3’-H2), 4.73 (m, 1H, 1-H), 4.62 (d, J = 11.7 Hz, 1H, OBn), 4.29 (dd, J = 5.4, 1.6 Hz, 1H, 2-H), 4.19 (dd, J = 7.0, 5.6 Hz, 1H, 3-H), 3.61 (dddd, J = 9.5, 6.0, 6.0, 6.0 Hz, 1H, 5-H), 3.26 (dd, J = 9.8, 7.3 Hz, 1H, 4-H), 1.52 (s, 3H, Me), 1.37 (s, 3H, Me), 1.26 ppm (d, J = 6.3 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 208.0 (C, C-2’), 138.2 (C, Ar), 128.1 (2 × CH, Ar), 127.9 (2 × CH, Ar), 127.5 (CH, Ar), 108.6 (C, isopropylidene), 89.5 (CH, C-1’), 81.5 (CH, C-4), 78.0 (CH, C-3), 77.4 (CH2, C-3’), 75.5 (CH, C-2), 72.9 (CH2, OBn), 70.7 (CH, C-1), 67.0 (CH, C-5), 28.0 (CH3, Me), 26.3 (CH3, Me), 18.0 ppm (CH3, C-6). IR (CHCl3): ν = 3016, 2993, 1955, 1228, 1074 cm–1. MS (ESI) m/z (%) = 339 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C19H24NaO4 339.1572; found 339.1567. Anal. calcd for C19H24O4: C, 72.13; H, 7.65. Found: C, 72.06; H, 7.74.
C-(2,3-O-[(2S,3S)-2,3-Dimethoxybutane-2,3-diyl]-α-l-fucopyranosyl)allene (108)
To a solution of 107 (5.07 g, 16.25 mmol) in dry MeOH (244 mL) was added K2CO3 (3.6 g, 26 mmol), and the mixture was stirred at room temperature for 3 h. Then, it was filtered and neutralized with the Amberlyst 15 H+ ion exchange resin. It was filtered again under vacuum and evaporated. The resulting organic crude was dissolved in dry MeOH (81.3 mL), and 2,3-butanedione (2.85 mL, 32.5 mmol), (MeO)3CH (7.1 mL, 65 mmol), and BF3•Et2O (3.4 mL, 30.88 mmol) were added dropwise. The mixture was stirred at 60 °C for 4.5 h. A few pipettes of Et3N were added while stirring at room temperature for 15 min. Then, it was evaporated to dryness. Column chromatography of the residue (hexanes–EtOAc, 7:3) gave 108 (3.18 g, 10.6 mmol, 65%) as a colorless oil: [α]D = −16.9 (c = 1.15, CHCl3). 1H NMR (500 MHz, CDCl3) δH 5.38 (ddd, J = 6.8, 6.8, 4.4 Hz, 1H, 1’-H), 4.85 (dd, J = 6.9, 4.1 Hz, 2H, 3’-H2), 4.61 (dddd, J = 6.0, 4.1, 4.1, 4.1 Hz, 1H, 1-H), 4.29 (dd, J = 10.6, 6.0 Hz, 1H, 2-H), 4.00 (dddd, J = 6.3, 6.3, 6.3, 0.0 Hz, 1H, 5-H), 3.85 (dd, J = 10.6, 3.1 Hz, 1H, 3-H), 3.72 (br s, 1H, 4-H), 3.24 (s, 3H, OMe), 3.23 (s, 3H, OMe), 2.39 (br s, 1H, OH), 1.31 (s, 3H, Me), 1.26 (d, J = 6.3 Hz, 3H, 6-H3), 1.25 ppm (s, 3H, Me). 13C{1H} NMR (125.7 MHz, CDCl3) δC 208.7 (C, C-2’), 100.1 (C), 99.8 (C), 85.8 (CH, C-1’), 77.0 (CH2, C-3’), 71.5 (CH, C-1), 70.8 (CH, C-4), 68.4 (CH, C-5), 67.9 (CH, C-3), 64.0 (CH, C-2), 47.9 (2 × CH3, 2 × OMe), 17.6 (2 × CH3, Me, C-6), 16.5 ppm (CH3, Me). IR (CHCl3): ν = 3672, 3583, 3010, 2942, 1957, 1226 cm–1. MS (ESI) m/z (%) = 323 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C15H24NaO6 323.1471; found 323.1476. Anal. calcd for C15H24O6: C, 59.98; H, 8.05. Found: C, 59.90; H, 8.35.
General Procedure to Give Diphenoxyphosphoryl Derivatives 92, 95, and 99
The corresponding alcohol (1 mmol) in dry CH2Cl2 (58 mL) was treated with DMAP (562 mg, 4.61 mmol) and ClPO(OPh)2 (0.95 mL, 4.61 mmol) at room temperature for 2 h. The reaction was quenched with a saturated aqueous solution of NH4Cl and extracted with CH2Cl2. The organic phase was dried over Na2SO4, filtered, and evaporated. Column chromatography of the residue (hexanes–EtOAc) gave the diphenoxyphosphoryl derivatives.
C-(6-O-tert-Butyldiphenylsilyl-4-O-diphenoxyphosphoryl-2,3-di-O-methyl-α-d-glucopyranosyl)allene (92)
To a solution of the allene 91 (97.4 mg, 0.19 mmol) in dry MeOH (0.95 mL) was added K2CO3 (2.1 mg, 0.015 mmol), and the mixture was stirred at room temperature overnight. The residue was evaporated to afford the intermediate alcohol that was submitted to the general procedure to give the diphenoxylphosphoryl derivative for 2.5 h. Column chromatography (hexanes–EtOAc, 85:15) gave 92 (96.5 mg, 0.14 mmol, 73%) as a colorless oil: [α]D = +45.6 (c = 0.45, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.68–7.07 (m, 20H, Ar), 5.36 (ddd, J = 8.3, 8.3, 8.3 Hz, 1H, 1’-H), 4.84 (dd, J = 8.6, 0.0 Hz, 1H, 3’-Hb), 4.83 (dd, J = 8.4, 0.0 Hz, 1H, 3’-Ha), 4.74 (m, 1H, 1-H), 4.62 (ddd, J = 8.5, 8.5 Hz, 3JPH = 8.5 Hz, 1H, 4-H), 3.96–3.87 (m, 2H, 5-H, 6-Hb), 3.79 (dd, J = 14.9, 6.7 Hz, 1H, 6-Ha), 3.57 (dd, J = 10.9, 8.3 Hz, 1H, 3-H), 3.46 (m, 1H, 2-H), 3.45 (s, 6H, OMe), 1.04 ppm (s, 9H, tBu). 13C{1H} NMR (100.6 MHz, CDCl3) δC 209.6 (C, C-2’), 150.8 (d, 2JPC = 7.0 Hz, C, Ar), 150.6 (d, 2JPC = 7.8 Hz, C, Ar), 133.6 (2 × C, Ar), 119.9–135.7 (20 × CH, Ar), 85.2 (CH, C-1’), 81.6 (CH, C-2), 81.1 (CH, C-3), 76.7 (CH2, C-3’), 76.6 (CH, C-4), 72.8 (d, 3JPC = 7.0 Hz, CH, C-5), 70.9 (CH, C-1), 63.0 (CH2, C-6), 60.2 (CH3, OMe), 58.2 (CH3, OMe), 26.7 (3 × CH3, DPS), 19.3 ppm (C, DPS). IR (CHCl3): ν = 3015, 2933, 1955, 1592, 1490, 1191 cm–1. MS (ESI) m/z (%) = 723 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C39H45NaO8PSi 723.2519; found 723.2510. Anal. calcd for C39H45O8Psi: C, 66.84; H, 6.47. Found: C, 66.90; H, 6.42.
C-(4,6-Bis-O-diphenoxyphosphoryl-2,3-di-O-methyl-α-d-glucopyranosyl)methanol (95)
Compound 48 (620 mg, 1.24 mmol) was dissolved in CH2Cl2 (6.2 mL), and DHP (282 μL, 3.09 mmol) and p-TsOH·H2O (15 mg, 0.08 mmol) were added while stirring at room temperature for 2 h. The reaction was poured over a saturated aqueous solution of NaHCO3 and extracted with CH2Cl2. The organic extract was dried over Na2SO4, filtered, and evaporated. The crude in dry THF (25 mL) was treated with TBAF/THF 1 M solution (1.9 mL, 1.9 mmol) for 3 h at room temperature. Then, the mixture was evaporated to dryness and quickly chromatographed (hexanes–EtOAc 3:7) to obtain the corresponding alcohol (315 mg, 0.91 mmol, 73%) as an orange oil that was saponified with K2CO3 (25 mg, 0.18 mmol) in MeOH (4.6 mL) at room temperature for 4 h, filtered over a pad of Celite, and concentrated. The resulting diol was treated with ClPO(OPh)2 (1.7 mL, 8.19 mmol) and dry pyridine (9.1 mL, 112.5 mmol) at room temperature overnight. The reaction was evaporated in a high vacuum rotovap, quenched with an aqueous solution of HCl 10%, and extracted with CH2Cl2. The organic phase was washed with a saturated aqueous solution of NaHCO3, dried over Na2SO4, and evaporated. Finally, the THP protecting group was hydrolyzed by treatment with p-TsOH·H2O (17.3 mg, 0.091 mmol) in MeOH (1.8 mL) at room temperature for 2 h. The mixture was poured over a saturated aqueous solution of NaHCO3 and extracted with CH2Cl2. The organic phase was dried over Na2SO4, filtered, and evaporated. Column chromatography of the residue (hexanes–EtOAc, 4:6) gave 95 (315.3 mg, 0.46 mmol, 37% overall yield) as a colorless oil: [α]D = +20.6 (c = 1.25, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.34–7.11 (m, 20H, Ar), 4.33–4.20 (m, 3H, 4-H, 6-H2), 4.14–4.09 (m, 2H, 1-H, 5-H), 3.90 (dd, J = 12.6, 8.8 Hz, 1H, 1’-Hb), 3.70 (dd, J = 12.6, 3.5 Hz, 1H, 1’-Ha), 3.51 (dd, J = 8.2, 8.2 Hz, 1H, 3-H), 3.42 (s, 6H, 2 × OMe), 3.40 ppm (dd, J = 8.2, 5.7 Hz, 1H, 2-H), 1H from OH is missing. 13C{1H} NMR (125.7 MHz, CDCl3) δC 150.7 (d, 2JPC = 7.4 Hz, C, Ar), 150.6 (d, 2JPC = 7.4 Hz, C, Ar), 150.5 (d, 2JPC = 7.4 Hz, C, Ar), 150.3 (d, 2JPC = 7.4 Hz, C, Ar), 120.0–129.8 (20 × CH, Ar), 80.4 (2 × CH, C-2, C-3), 75.9 (d, 2JPC = 6.4 Hz, CH, C-4), 74.1 (CH, C-1), 70.9 (CH, C-5), 68.3 (d, 2JPC = 7.4 Hz, CH2, C-6), 60.3 (CH3, OMe), 58.8 (CH3, OMe), 58.3 ppm (CH2, C-1’). IR (CHCl3): ν = 3690, 3620, 3024, 2401, 1491, 1226 cm–1. MS (ESI) m/z (%) = 709 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C33H36NaO12P2 709.1580; found 709.1582. Anal. calcd for C33H36O12P2: C, 57.73; H, 5.28. Found: C, 57.82; H, 5.62.
C-(6-O-tert-Butyldiphenylsilyl-4-O-diphenoxyphosphoryl-2,3-di-O-methyl-α-d-galactopyranosyl)allene (99)
To a solution of the acetyl derivative 98 (749.5 mg, 1.47 mmol) in MeOH (7.3 mL) was added K2CO3 (16.2 mg, 0.12 mmol), and it was stirred at room temperature overnight. The mixture was filtered, evaporated, and submitted to the general procedure to give the diphenoxyphosphoryl derivative. Column chromatography (hexanes–EtOAc, 8:2) of the residue afforded 99 (928.6 mg, 1.32 mmol, 90%) as a colorless oil: [α]D = +74.6 (c = 0.46, CHCl3). 1H NMR (400 MHz, CDCl3) δH 7.08–7.65 (m, 20H, Ar), 5.28–5.24 (m, 2H, 1-H, 1’-H), 4.79–4.67 (m, 3H, 4-H, 3’-H2), 3.85 (m, 1H, 5-H), 3.79 (dd, J = 10.1, 7.4 Hz, 1H, 6-Hb), 3.69 (dd, J = 10.1, 6.1 Hz, 1H, 6-Ha), 3.61 (dd, J = 9.6, 5.6 Hz, 1H, 3-H), 3.41 (s, 3H, OMe), 3.38 (s, 3H, OMe), 3.32 (dd, J = 9.6, 1.3 Hz, 1H, 2-H), 1.03 ppm (s, 9H, tBu). 13C{1H} NMR (100.6 MHz, CDCl3) δC 208.8 (C, C-2’), 150.8 (d, 2JPC = 7.8 Hz, C, Ar), 150.7 (d, 2JPC = 7.1 Hz, C, Ar), 135.61 (2 × CH, Ar), 135.56 (2 × CH, Ar), 133.5 (C, Ar), 133.3 (C, Ar), 129.8 (CH, Ar), 129.7 (CH, Ar), 129.6 (2 × CH, Ar), 129.3 (2 × CH, Ar), 127.64 (2 × CH, Ar), 127.59 (2 × CH, Ar), 125.0 (CH, Ar), 124.9 (CH, Ar), 120.52 (CH, Ar), 120.45 (CH, Ar), 120.12 (CH, Ar), 120.07 (CH, Ar), 85.3 (CH, C-1’), 79.1 (CH, C-2), 76.7 (CH2, C-3’), 76.7 (CH, C-3), 74.5 (d, 2JPC = 6.3 Hz, CH, C-4), 71.7 (d, 3JPC = 5.6 Hz, CH, C-5), 71.3 (CH, C-1), 62.1 (CH2, C-6), 59.0 (CH3, OMe), 57.5 (CH3, OMe), 26.7 (3 × CH3, DPS), 19.1 ppm (C, DPS). IR (CHCl3): ν = 3016, 2933, 1956, 1592, 1490, 1112 cm–1. MS (ESI) m/z (%) = 723 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C39H45NaO8PSi 723.2519; found 723.2513. Anal. calcd for C39H45O8PSi: C, 66.84; H, 6.47. Found: C, 66.80; H, 6.48.
C-(6-O-tert-Butyldiphenylsilyl-2,3-di-O-methyl-4-O-tosyl-α-d-glucopyranosyl)allene (93)
To a solution of the allene 91 (301 mg, 0.59 mmol) in dry MeOH (3 mL), K2CO3 (6 mg, 0.04 mmol) was added and the mixture was stirred at room temperature overnight. The residue was evaporated to afford the intermediate alcohol that was dissolved in dry pyridine (6 mL) and treated with TsCl (343 mg, 1.8 mmol) overnight. The reaction was evaporated at high vacuum rotovap, quenched with HCl 10%, and extracted with CH2Cl2. The organic phase was washed with a saturated aqueous solution of NaHCO3, dried over Na2SO4, filtered, and evaporated. Column chromatography (hexanes–EtOAc, 9:1) gave 93 (165.1 mg, 0.27 mmol, 45%) as a colorless oil: [α]D = +69.6 (c = 0.46, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.78–7.18 (m, 14H, Ar), 5.31 (ddd, J = 6.7, 6.7, 6.7 Hz, 1H, 1’-H), 4.82 (dd, J = 6.7, 0.0 Hz, 1H, 3’-Hb), 4.81 (dd, J = 6.9, 0.0 Hz, 1H, 3’-Ha), 4.75–4.70 (m, 2H, 1-H, 3-H), 3.88 (dd, J = 11.4, 2.2 Hz, 1H, 6-Hb), 3.82 (m, 1H, 4-H), 3.76 (dd, J = 11.0, 4.7 Hz, 1H, 6-Ha), 3.46 (s, 3H, OMe), 3.42–3.40 (m, 2H, 2-H, 5-H), 3.23 (s, 3H, OMe), 2.39 (s, 3H, OTs), 1.09 ppm (s, 9H, tBu). 13C{1H} NMR (125.7 MHz, CDCl3) δC 209.5 (C, C-2’), 144.2 (C, Ar), 135.0 (C, Ar), 133.7 (C, Ar), 133.6 (C, Ar), 127.5–135.9 (14 × CH, Ar), 85.3 (CH, C-1’), 81.7 (CH, C-2 or C-5), 80.7 (CH, C-2 or C-5), 77.9 (CH, C-1), 76.8 (CH2, C-3’), 72.4 (CH, C-4), 70.8 (CH, C-3), 62.8 (CH2, C-6), 60.3 (CH3, OMe), 58.4 (CH3, OMe), 26.8 (3 × CH3, DPS), 21.6 (CH3, OTs), 19.4 ppm (C, DPS). IR (CHCl3): ν = 3015, 2933, 1955, 1599, 1373, 1112 cm–1. MS (ESI) m/z (%) = 645 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C34H42NaO7SSi 645.2318; found 645.2321. Anal. calcd for C34H42O7SSi: C, 65.56; H, 6.80. Found: C, 65.40; H, 7.05.
General Procedure for the Synthesis of the Hydroxymethyl Derivatives 48, 49, 51, 52, 58, 59, 94, 104, and 109
A solution of the allene (1 mmol) in CH2Cl2–MeOH (30 mL, 4:1) was cooled to −78 °C, and ozone was bubbled into the solution until it became blue. Then, nitrogen was introduced through the mixture to expel the excess of ozone, and it was heated to 0 °C. Afterward, NaBH4 (75.3 mg, 1.99 mmol) was added slowly and the solution was stirred for 2 h at room temperature. The reaction mixture was then poured into brine, extracted with CH2Cl2, dried over Na2SO4, and concentrated. Column chromatography (hexanes–EtOAc) of the residue afforded the title alcohol.
C-(4-O-Acetyl-6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-α-d-glucopyranosyl)methanol (48)
Following the general procedure starting from allene 91 (524 mg, 1.03 mmol) and purification by column chromatography (hexanes–EtOAc, 25:75), alcohol 48 (332.7 mg, 0.66 mmol, 64%) was obtained as a colorless oil: [α]D = +19.0 (c = 0.39, CHCl3). 1H NMR (500 MHz CDCl3, 2-H and 3-H are strongly coupled; therefore, 4-H and 1-H show virtual coupling effects; the chemical shifts and coupling constants shown below were obtained using DAISY) δH 7.70–7.65 ppm (m, 4H, Ar), 7.45–7.36 (m, 6H, Ar), 4.89 (dd, J = 8.0, 5.8 Hz, 1H, 4-H), 4.13 (ddd, J = 8.6, 4.8, 4.6 Hz, 1H, 1-H), 3.91 (dd, J = 12.0, 8.6 Hz, 1H, 1’-Hb), 3.77 (dd, J = 12.0, 4.6 Hz, 1H, 1’-Ha), 3.76 (ddd, J = 8.0, 5.8, 4.1 Hz, 1H, 5-H), 3.72 (dd, J = 10.8, 4.1 Hz, 1H, 6-Hb), 3.71 (dd, J = 10.8, 5.8 Hz, 1H, 6-Ha), 3.480 (s, 3H, OMe), 3.478 (s, 3H, OMe), 3.467 (dd, J = 7.8, 7.1 Hz, 1H, 3-H), 3.467 (dd, J = 7.1, 4.8 Hz, 1H, 2-H), 1.98 (s, 3H, OAc), 1.06 ppm (s, 9H, tBu), 1H from OH is missing. 1H NMR (500 MHz, C6D6) δH 7.85–7.82 (m, 4H, Ar), 7.26–7.21 (m, 6H, Ar), 5.21 (dd, J = 8.5, 8.5 Hz, 1H, 4-H), 4.07 (ddd, J = 5.0, 5.0, 8.3 Hz, 1H, 1-H), 3.84–3.79 (m, 5H, 1’-H2, 5-H, 6-H2), 3.44 (dd, J = 8.4, 8.4 Hz, 1H, 3-H), 3.31 (s, 3H, OMe), 3.26 (dd, J = 8.5, 5.9 Hz, 1H, 2-H), 2.99 (s, 3H, OMe), 1.63 (s, 3H, OAc), 1.23 ppm (s, 9H, tBu). 13C{1H} NMR (100.6 MHz, CDCl3) δC 169.7 (C, OAc), 135.7 (2 × CH, Ar), 135.6 (2 × CH, Ar), 133.3 (C, Ar), 133.2 (C, Ar), 129.72 (2 × CH, Ar), 129.70 (2 × CH, Ar), 127.69 (2 × CH, Ar), 127.67 (2 × CH, Ar), 79.9 (CH, C-2 or C-5), 79.8 (CH, C-2 or C-5), 73.4 (CH, C-4), 72.5 (CH, C-1), 69.6 (CH, C-3), 63.2 (CH2, C-6), 59.8 (CH3, OMe), 59.6 (CH2, C-1’), 58.9 (CH3, OMe), 26.8 (3 × CH3, DPS), 20.9 (CH3, OAc), 19.2 ppm (C, DPS). IR (CHCl3): ν = 3680, 3553, 2934, 1742, 1217 cm–1. MS (ESI) m/z (%) = 525 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C27H38NaO7Si 525.2285; found 525.2286.
C-(6-O-tert-Butyldiphenylsilyl-2,3-di-O-methyl-4-O-tosyl-α-d-glucopyranosyl)methanol (49)
Following the general procedure starting from allene 93 (226.2 mg, 0.36 mmol) and purification by column chromatography (hexanes–EtOAc, 6:4), alcohol 49 (147.9 mg, 0.24 mmol, 67%) was obtained as a colorless oil: [α]D = +15.2 (c = 0.54, CHCl3). 1H NMR (500 MHz, CDCl3, simulated coupling constants using DAISY) δH 7.74–7.17 (m, 14H, Ar), 4.64 (dd, J = 7.6, 6.8 Hz, 1H, 4-H), 4.04 (ddd, J = 8.5, 5.1, 4.4 Hz, 1H, 1-H), 3.834 (dd, J = 10.9, 3.7 Hz, 1H, 6-Hb), 3.831 (dd, J = 12.1, 8.5 Hz, 1H, 1’-Hb), 3.73 (ddd, J = 7.6, 6.0, 3.7 Hz, 1H, 5-H), 3.69 (dd, J = 12.1, 4.4 Hz, 1H, 1’-Ha), 3.66 (dd, J = 10.9, 6.0 Hz, 1H, 6-Ha), 3.51 (dd, J = 7.2, 6.8 Hz, 1H, 3-H), 3.47 (s, 3H, OMe), 3.43 (dd, J = 7.2, 5.1 Hz, 1H, 2-H), 3.29 (s, 3H, OMe), 2.36 (s, 3H, OTs), 1.05 ppm (s, 9H, tBu), 1H from OH is missing. 13C{1H} NMR (100.6 MHz, CDCl3) δC 144.5 (C, OTs), 134.3 (C, OTs), 133.3 (C, Ar), 133.2 (C, Ar), 127.6–135.6 (14 × CH, Ar), 79.8 (CH, C-2), 79.1 (CH, C-3), 76.3 (CH, C-4), 73.0 (CH, C-5), 72.2 (CH, C-1), 62.5 (CH2, C-6), 59.8 (CH3, OMe), 59.7 (CH2, C-1’), 58.7 (CH3, OMe), 26.7 (3 × CH3, DPS), 21.6 (CH3, OTs), 19.2 ppm (C, DPS). IR (CHCl3): ν = 3694, 3574, 3024, 2934, 1600, 1104 cm–1. MS (ESI) m/z (%) = 637 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C32H42NaO8SSi 637.2267; found 637.2257. Anal. calcd for C32H42O8SSi: C, 62.51; H, 6.89; S, 5.22. Found: C, 62.29; H, 7.09; S, 4.86.
C-(4-O-Acetyl-6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-α-d-galactopyranosyl)methanol (51)
Following the general procedure starting from allene 98 (461.5 mg, 0.90 mmol) and purification by column chromatography (hexanes–EtOAc, 25:75), alcohol 51 (316.3 mg, 0.63 mmol, 70%) was obtained as a colorless oil: [α]D = +30.6 (c = 0.66, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.67–7.64 (m, 4H, Ar), 7.46–7.37 (m, 6H, Ar), 5.52 (dd, J = 3.3, 1.9 Hz, 1H, 4-H), 4.20 (ddd, J = 8.4, 6.1, 4.8 Hz, 1H, 1-H), 3.83 (ddd, J = 6.6, 6.2, 1.9 Hz, 1H, 5-H), 3.82 (dd, J = 12.0, 8.4 Hz, 1H, 1’-Hb), 3.77 (dd, J = 12.0, 5.1 Hz, 1H, 1’-Ha), 3.73 (dd, J = 10.4, 6.6 Hz, 1H, 6-Hb), 3.66 (dd, J = 9.1, 6.1 Hz, 1H, 2-H), 3.61 (dd, J = 10.4, 6.2 Hz, 1H, 6-Ha), 3.48 (s, 3H, OMe), 3.42 (dd, J = 9.1, 3.3 Hz, 1H, 3-H), 3.41 (s, 3H, OMe), 2.02 (s, 3H, OAc), 1.06 ppm (s, 9H, tBu), 1H from OH is missing. 13C{1H} NMR (100.6 MHz, CDCl3) δC 169.9 (C, OAc), 135.6 (4 × CH, Ar), 133.2 (C, Ar), 133.0 (C, Ar), 129.8 (2 × CH, Ar), 127.7 (4 × CH, Ar), 79.0 (CH, C-2 or C-5), 77.5 (CH, C-2 or C-5), 73.4 (CH, C-4), 71.9 (CH, C-1), 66.6 (CH, C-3), 62.2 (CH2, C-6), 59.4 (CH3, OMe), 59.1 (CH2, C-1’), 57.7 (CH3, OMe), 26.8 (3 × CH3, DPS), 20.7 (CH3, OAc), 19.1 ppm (C, DPS). IR (CHCl3): ν = 3686, 3620, 3015, 2975, 1742, 1229 cm–1. MS (ESI) m/z (%) = 525 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C27H38NaO7Si 525.2285; found 525.2284. Anal. calcd for C27H38O7Si: C, 64.51; H, 7.62. Found: C, 64.20; H, 7.65.
C-(6-O-tert-Butyldiphenylsilyl-4-O-diphenoxyphosphoryl-2,3-di-O-methyl-α-d-galactopyranosyl)methanol (52)
Following the general procedure starting from allene 99 (896.2 g, 1.28 mmol) and purification by column chromatography (hexanes–EtOAc, 6:4), alcohol 52 (567.3 mg, 0.82 mmol, 64%) was obtained as a colorless oil: [α]D = +25.0 (c = 0.32, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.65–7.05 (m, 20H, Ar), 5.06 (ddd, J = 2.6, 1.6 Hz, 3JPH = 8.5 Hz, 1H, 4-H), 4.09 (ddd, J = 7.3, 5.7, 5.7 Hz, 1H, 1-H), 3.80–3.68 (m, 5H, 5-H, 6-H2, 1’-H2), 3.57 (dd, J = 8.8, 5.6 Hz, 1H, 2-H), 3.389 (s, 3H, OMe), 3.38 (dd, J = 8.8, 2.6 Hz, 1H, 3-H), 3.35 (s, 3H, OMe), 1.93 (br s, 1H, OH), 1.03 ppm (s, 9H, tBu). 13C{1H} NMR (125.7 MHz, CDCl3) δC 150.8 (d, 2JPC = 7.4 Hz, C, Ar), 150.5 (d, 2JPC = 7.4 Hz, C, Ar), 135.6 (2 × CH, Ar), 135.5 (2 × CH, Ar), 133.3 (C, Ar), 133.2 (C, Ar), 129.74 (CH, Ar), 129.72 (CH, Ar), 129.6 (2 × CH, Ar), 129.4 (2 × CH, Ar), 127.7 (4 × CH, Ar), 125.2 (CH, Ar), 125.1 (CH, Ar), 120.33 (CH, Ar), 120.28 (CH, Ar), 120.00 (CH, Ar), 119.96 (CH, Ar), 78.9 (CH, C-3), 77.0 (CH, C-2), 74.2 (d, 2JPC = 6.3 Hz, CH, C-4), 72.8 (d, 3JPC = 6.3 Hz, CH, C-5), 72.6 (CH, C-1), 62.2 (CH2, C-6), 59.58 (CH3, OMe), 59.2 (CH2, C-1’), 57.7 (CH3, OMe), 26.7 (3 × CH3, DPS), 19.0 ppm (C, DPS). IR (CHCl3): ν = 3690, 3546, 3023, 2934, 1592, 1206 cm–1. MS (ESI) m/z (%) = 715 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C37H45NaO9PSi 715.2468; found 715.2475. Anal. calcd for C37H45O9PSi: C, 64.14; H, 6.55. Found: C, 64.02; H, 6.67.
C-(4-O-Acetyl-2,3-di-O-methyl-α-d-fucopyranosyl)methanol (58)
Alcohol 109 (200 mg, 0.68 mmol) was dissolved in dry DMF (2.7 mL), and DPSCl (238 μL, 1.01 mmol) and imidazole (138 mg, 2.03 mmol) were added at room temperature. After 3 h, the reaction was evaporated in a high vacuum rotovap, quenched with H2O, and extracted with CH2Cl2. The organic phase was dried over Na2SO4, filtered, and evaporated. To a solution of the resulting crude in dry EtOAc (16 mL) was added Pd/C 10% (152 mg), and the mixture was submitted to H2 atmosphere overnight. The reaction was filtered over a pad of Celite and evaporated. The alcohol was protected by treatment with dry pyridine (2.6 mL), Ac2O (0.9 mL), and DMAP (8.3 mg, 0.068 mmol) for 0.5 h. The mixture was evaporated in a high vacuum rotovap, quenched with aqueous HCl 10%, and extracted with CH2Cl2. The organic phase was washed with a saturated aqueous solution of NaHCO3, dried with Na2SO4, filtered, and concentrated to dryness. Finally, the silyl group was deprotected by treatment with a 1 M solution of TBAF/THF (1.4 mL, 1.4 mmol) in dry THF (13.6 mL) for 4 h at room temperature. The mixture was evaporated, and the residue was chromatographed in a silica gel column (hexanes–EtOAc, 4:6 to 2:8) to give 58 (111 mg, 0.45 mmol, 66%) as a colorless oil: [α]D = −76.0 (c = 0.65, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 5.33 (dd, J = 3.5, 2.0 Hz, 1H, 4-H), 4.23 (ddd, J = 8.0, 5.9, 4.7 Hz, 1H, 1-H), 3.96 (dddd, J = 6.5, 6.5, 6.5, 2.0 Hz, 1H, 5-H), 3.89 (dd, J = 12.1, 8.0 Hz, 1H, 1’-Hb), 3.85 (dd, J = 12.1, 4.7 Hz, 1H, 1’-Ha), 3.71 (dd, J = 8.2, 5.9 Hz, 1H, 2-H), 3.50 (dd, J = 8.2, 3.5 Hz, 1H, 3-H), 3.50 (s, 3H, OMe), 3.42 (s, 3H, OMe), 2.17 (s, 3H, OAc), 2.10 (br s, 1H, OH), 1.17 ppm (d, J = 6.5 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 170.6 (C, OAc), 78.7 (CH, C-3), 77.1 (CH, C-2), 73.4 (CH, C-1), 69.5 (CH, C-4), 67.0 (CH, C-5), 59.5 (CH2, C-1’), 59.3 (CH3, OMe), 57.5 (CH3, OMe), 20.8 (CH3, OAc), 16.5 ppm (CH3, C-6). IR (CHCl3): ν = 3677, 3502, 3018, 2939, 1739, 1239 cm–1. MS (ESI) m/z (%) = 271 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C11H20NaO6 271.1158; found 271.1159.
C-(4-O-Diphenoxyphosphoryl-2,3-di-O-methyl-α-d-fucopyranosyl)methanol (59)
Alcohol 109 (1.4 g, 4.73 mmol) was dissolved in dry DMF (18.9 mL), and imidazole (960 mg, 14.1 mmol) and DPSCl (1.7 mL, 7.25 mmol) were added at room temperature. After 3 h, the reaction was evaporated in a high vacuum rotovap, quenched with H2O, and extracted with CH2Cl2. The organic phase was dried over Na2SO4, filtered, and evaporated. To a solution of the resulting crude in dry EtOAc (111 mL) was added Pd/C 10% (1.06 g), and the mixture was submitted to a H2 atmosphere overnight. The reaction was filtered over a pad of Celite and evaporated. The alcohol was protected without purification by treatment with DMAP (2.66 g, 21.7 mmol) and ClPO(OPh)2 (2.82 mL, 21.7 mmol) in dry CH2Cl2 (110 mL) for 3.5 h. The mixture was quenched with a saturated aqueous NH4Cl solution and extracted with CH2Cl2. The organic phase was dried with Na2SO4, filtered, and concentrated to dryness. Finally, the silyl group was deprotected by treatment with a 1 M solution of TBAF/THF (9.46 mL, 9.46 mmol) in dry THF (95 mL) for 3.5 h at room temperature. The mixture was evaporated, and the residue was chromatographed on a silica gel column (hexanes–EtOAc, 1:1) to give 59 (1.24 g, 2.84 mmol, 60%) as a colorless oil: [α]D = −41.8 (c = 0.49, CHCl3). 1H NMR (400 MHz, CDCl3, simulated coupling constants using DAISY) δH 7.37–7.17 (m, 10H, Ar), 4.92 (ddd, J = 3.0, 2.5 Hz, 3JPH = 9.1 Hz, 1H, 4-H), 4.14 (ddd, J = 7.5, 5.7, 5.2 Hz, 1H, 1-H), 3.96 (ddddd, J = 6.5, 6.5, 6.5, 2.5 Hz, 4JPH = 1.9 Hz, 1H, 5-H), 3.87–3.77 (m, 2H, 1’-H2), 3.60 (dd, J = 8.4, 5.2 Hz, 1H, 2-H), 3.53 (ddd, J = 8.4, 3.0, Hz, 4JPH = 1.0 Hz, 1H, 3-H), 3.43 (s, 3H, OMe), 3.41 (s, 3H, OMe), 1.22 ppm (d, J = 6.5 Hz, 3H, 6-H3), 1H from OH is missing. 13C{1H} NMR (125.7 MHz, CDCl3) δC 150.8 (d, 2JPC = 8.5 Hz, C, Ar), 150.6 (d, 2JPC = 7.4 Hz, C, Ar), 129.7 (2 × CH, Ar), 129.3 (2 × CH, Ar), 125.3 (CH, Ar), 125.1 (CH, Ar), 120.32 (CH, Ar), 120.28 (CH, Ar), 120.11 (CH, Ar), 120.07 (CH, Ar), 78.7 (CH, C-3), 76.9 (CH, C-2), 76.8 (d, 3JPC = 6.3 Hz, CH, C-4), 72.3 (CH, C-1), 67.9 (d, 3JPC = 5.3 Hz, CH, C-5), 60.1 (CH2, C-1’), 59.5 (CH3, OMe), 57.8 (CH3, OMe), 15.9 ppm (CH3, C-6). IR (CHCl3): ν = 3694, 3018, 2938, 1490, 1218 cm–1. MS (ESI) m/z (%) = 461 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C21H27NaO8P 461.1341; found 461.1342. Anal. calcd for C21H27O8P: C, 57.53; H, 6.21. Found: C, 57.62; H, 6.49.
C-(6-O-tert-Butyldiphenylsilyl-4-O-diphenoxyphosphoryl-2,3-di-O-methyl-α-d-glucopyranosyl)methanol (94)
Following the general procedure starting from allene 92 (1.08 g, 1.54 mmol) and purification by column chromatography (hexanes–EtOAc, 6:4), alcohol 94 (556.2 mg, 0.80 mmol, 52%) was obtained as a colorless oil: [α]D = +30.3 (c = 0.33, CHCl3). 1H NMR (500 MHz, CDCl3, simulated coupling constants using DAISY) δH 7.65–7.07 (m, 20H, Ar), 4.56 (ddd, J = 8.5, 7.6 Hz, 3JPH = 9.4 Hz, 1H, 4-H), 4.13 (ddd, J = 8.8, 5.7, 4.6 Hz, 1H, 1-H), 3.91 (dd, J = 11.0, 32.8 Hz, 1H, 6-Hb), 3.90 (dd, J = 12.1, 8.8 Hz, 1H, 1’-Hb), 3.83 (ddd, J = 8.5, 5.9, 2.8 Hz, 1H, 5-H), 3.76 (dd, J = 11.0, 5.9 Hz, 1H, 6-Ha), 3.75 (dd, J = 12.1, 4.6 Hz, 1H, 1’-Ha), 3.57 (dd, J = 7.9, 7.6 Hz, 1H, 3-H), 3.50 (dd, J = 7.9, 5.7 Hz, 1H, 2-H), 3.45 (s, 3H, OMe), 3.42 (s, 3H, OMe), 1.03 ppm (s, 9H, tBu), 1H from OH is missing. 13C{1H} NMR (100.6 MHz, CDCl3) δC 150.7 (d, 2JPC = 7.0 Hz, C, Ar), 150.5 (d, 2JPC = 7.1 Hz, C, Ar), 133.3 (2 × C, Ar), 120.0–135.7 (20 × CH, Ar), 80.6 (CH, C-2 or C-3), 80.4 (CH, C-2 or C-3), 75.7 (d, 2JPC = 6.3 Hz, CH, C-4), 73.3 (d, 3JPC = 6.3 Hz, CH, C-5), 72.6 (CH, C-1), 62.9 (CH2, C-6), 59.9 (CH3, OMe), 59.3 (CH2, C-1’), 58.8 (CH3, OMe), 26.8 (3 × CH3, DPS), 19.2 ppm (C, DPS). IR (CHCl3): ν = 3676, 3532, 3016, 2934, 1591, 1490 cm–1. MS (ESI) m/z (%) = 715 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C37H45NaO9PSi 715.2468; found 715.2469. Anal. calcd for C37H45O9PSi: C, 64.14; H, 6.55. Found: C, 64.06; H, 6.36.
C-(4-O-Benzyl-2,3-di-O-methyl-α-l-rhamnopyranosyl)methanol (104)
Allene 103 (154 mg, 0.49 mmol) in TFA/H2O (4.5 mL, 4:6) was stirred at room temperature for 2 h. The solution was evaporated in a high vacuum rotovap, poured over a saturated aqueous solution of NaHCO3, extracted with CH2Cl2, dried over Na2SO4, and concentrated. The crude was dissolved in dry DMF (5.9 mL) under a N2 atmosphere, and NaH 60% in mineral oil (58.8 mg, 1.47 mmol) was slowly added at 0 °C. After 20 min, MeI (122 μL, 1.96 mmol) was dropwise added and stirring was continued at 0 °C for 1 h. Ice-water was used to destroy the excess of NaH, and the mixture was evaporated in a high vacuum rotovap, poured over a saturated solution of NH4Cl, and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated to dryness under reduced pressure. The organic residue was then submitted to the general procedure for the synthesis of hydroxymethyl derivatives. Column chromatography of the residue (hexanes–EtOAc, 4:6) gave 104 (65.4 mg, 0.22 mmol, 45%, three steps) as a colorless oil: [α]D = −22.4 (c = 0.293, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.37–7.29 (m, 5H, Ar), 4.70 (d, J = 11.7 Hz, 1H, OBn), 4.65 (d, J = 11.7 Hz, 1H, OBn), 3.99 (m, 1H, 1-H), 3.89 (dddd, J = 6.6, 6.6, 6.6, 4.7 Hz, 1H, 5-H), 3.80 (dd, J = 11.7, 7.0 Hz, 1H, 1’-Hb), 3.70 (dd, J = 11.4, 4.5 Hz, 1H, 1’-Ha), 3.60–3.56 (m, 2H, 2-H, 3-H), 3.52 (dd, J = 5.4, 4.7 Hz, 1H, 4-H), 3.45 (s, 3H, OMe), 3.44 (s, 3H, OMe), 2.07 (br s, 1H, OH), 1.34 ppm (d, J = 7.0 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 138.2 (C, Ar), 128.4 (2 × CH, Ar), 127.8 (3 × CH, Ar), 78.4 (CH, C-2 or C-3), 78.0 (CH, C-4), 75.7 (CH, C-2 or C-3), 73.4 (CH2, OBn), 70.7 (CH, C-1), 70.6 (CH, C-5), 61.7 (CH2, C-1’), 58.0 (CH3, OMe), 57.4 (CH3, OMe), 17.2 ppm (CH3, C-6). IR (CHCl3): ν = 3587, 3478, 2587, 2478, 2015, 2935, 1455, 1088 cm–1. MS (ESI) m/z (%) = 319 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C16H24NaO5 319.1521; found 319.1525. Anal. calcd for C16H24O5: C, 64.84; H, 8.16. Found: C, 64.61; H, 8.37.
C-(4-O-Benzyl-2,3-di-O-methyl-α-d-fucopyranosyl)methanol (109)
Alcohol 108 (251.4 mg, 0.84 mmol) was dissolved in dry DMF (10 mL) under a N2 atmosphere, and NaH 60% in mineral oil (50.4 mg, 1.26 mmol) was slowly added at 0 °C. After 20 min, BnBr (200 μL, 1.68 mmol) was dropwise added and stirring was continued at 0 °C for 2 h. Ice-water was used to destroy the NaH in excess, and the mixture was evaporated in a high vacuum rotovap, poured over a saturated solution of NH4Cl, and extracted with CH2Cl2. The combined extracts were dried over Na2SO4 and concentrated to dryness under reduced pressure. The crude was dissolved in TFA/H2O (7.7 mL, 4:6) and stirred at 40 °C overnight. The solution was evaporated in a high vacuum rotovap, quenched with a saturated aqueous solution of NaHCO3, and extracted with CH2Cl2. The organic residue was submitted to methyl protection by treatment with NaH 60% (100.8 mg, 2.52 mmol) and MeI (209 μL, 3.36 mmol) in DMF (10 mL) for 2 h from 0 °C until room temperature. Ice-water was used to destroy the excess of NaH, and the mixture was evaporated in a high vacuum rotovap, poured over a saturated solution of NH4Cl, and extracted with CH2Cl2. The organic phase was dried over Na2SO4 and concentrated to dryness under reduced pressure. Finally, the crude was submitted to the general procedure to give hydroxymethyl derivatives. Column chromatography of the residue (hexanes–EtOAc, 4:6) gave 109 (106.6 mg, 0.36 mmol, 43%) as a colorless oil: [α]D = −30.2 (c = 1.35, CHCl3). 1H NMR (400 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.39–7.27 (m, 5H, Ar), 4.77 (d, J = 11.8 Hz, 1H, OBn), 4.63 (d, J = 11.8 Hz, 1H, OBn), 4.13 (ddd, J = 7.9, 4.7, 4.2 Hz, 1H, 1-H), 3.97 (dddd, J = 6.7, 6.7, 6.7, 3.6 Hz, 1H, 5-H), 3.83 (dd, J = 11.6, 7.9 Hz, 1H, 1’-Hb), 3.76 (dd, J = 3.6, 2.9 Hz, 1H, 4-H), 3.74 (dd, J = 11.6, 4.7 Hz, 1H, 1’-Ha), 3.69 (dd, J = 7.0, 4.2 Hz, 1H, 2-H), 3.53 (dd, J = 7.0, 2.9 Hz, 1H, 3-H), 3.50 (s, 3H, OMe), 3.45 (s, 3H, OMe), 2.11 (br s, 1H, OH), 1.28 ppm (d, J = 6.7 Hz, 3H, 6-H3). 13C{1H} NMR (100.6 MHz, CDCl3) δC 138.4 (C, Ar), 128.3 (2 × CH, Ar), 128.0 (2 × CH, Ar), 127.7 (CH, Ar), 79.4 (CH, C-3), 78.1 (CH, C-2), 75.0 (CH, C-4), 73.4 (CH2, OBn), 70.7 (CH, C-1), 69.4 (CH, C-5), 60.8 (CH2, C-1’), 58.9 (CH3, OMe), 58.6 (CH3, OMe), 15.5 ppm (CH3, C-6). IR (CHCl3): ν = 3676, 3588, 3012, 2937, 1101 cm–1. MS (ESI) m/z (%) = 319 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C16H24NaO5 319.1521; found 319.1519. Anal. calcd for C16H24O5: C, 64.84; H, 8.16; found: C, 64.89; H, 8.20.
General Procedure for the Synthesis of Phthalimide Derivatives 16, 17, 18, 19, 20, 21, 22, 23, 24, and 105
DEAD (394 μL, 2.50 mmol) was added dropwise to a stirred solution of the alcohol (1 mmol), N-hydroxyphthalimide (408 mg, 2.5 mmol), and PPh3 (656 mg, 2.5 mmol) in dry THF (10 mL), and the resulting solution was stirred at 0 °C for 0.5–2.5 h. Then, the solvent was removed and the crude was quenched with water and extracted with CHCl3. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. Column chromatography of the residue (hexanes–EtOAc) gave the corresponding phthalimides.
C-(4-O-Acetyl-6-O-tert-Butyldiphenylsilyl-2,3-di-O-methyl-α-d-glucopyranosyl)N-methoxyphthalimide (16)
Following the general procedure starting from alcohol 48 (332 mg, 0.66 mmol) and purification by column chromatography (hexanes–EtOAc, 8:2), product 16 (281 mg, 0.43 mmol, 66%) was obtained as an amorphous solid: [α]D = +10.4 (c = 0.53, CHCl3). 1H NMR (500 MHz, CDCl3, simulated coupling constants using DAISY) δH 7.74–7.62 (m, 8H, Ar), 7.43–7.34 (m, 6H, Ar), 5.17 (dd, J = 6.8, 6.5 Hz, 1H, 4-H), 4.58 (dd, J = 10.8, 7.5 Hz, 1H, 1’-Hb), 4.48 (ddd, J = 7.5, 4.6, 3.9 Hz, 1H, 1-H), 4.37 (dd, J = 10.8, 3.9 Hz, 1H, 1’-Ha), 3.88 (ddd, J = 6.8, 5.0, 4.6 Hz, 1H, 5-H), 3.77 (dd, J = 11.0, 4.6 Hz, 1H, 6-Hb), 3.71 (dd, J = 11.0, 5.0 Hz, 1H, 6-Ha), 3.52 (dd, J = 7.1, 4.6 Hz, 1H, 2-H), 3.51 (s, 3H, OMe), 3.49 (s, 3H, OMe), 3.46 (dd, J = 7.1, 6.5 Hz, 1H, 3-H), 2.03 (s, 3H, OAc), 1.03 ppm (s, 9H, tBu). 13C{1H} NMR (125.7 MHz, CDCl3) δC 169.7 (C, OAc), 163.1 (2 × C, CO), 135.64 (2 × CH, Ar), 135.60 (2 × CH, Ar), 134.3 (2 × CH, Ar), 133.4 (C, Ar), 133.3 (C, Ar), 129.5 (CH, Ar), 129.4 (CH, Ar), 128.8 (2 × C, Ar), 127.5 (4 × CH, Ar), 123.4 (2 × CH, Ar), 78.3 (CH, C-2 or C-3), 78.0 (CH, C-2 or C-3), 74.5 (CH2, C-1’), 73.6 (CH, C-5), 70.3 (CH, C-1), 68.2 (CH, C-4), 62.3 (CH2, C-6), 59.3 (CH3, OMe), 58.7 (CH3, OMe), 26.7 (3 × CH3, DPS), 21.0 (CH3, OAc), 19.1 ppm (C, DPS). IR (CHCl3): ν = 2934, 1792, 1735, 1236 cm–1. MS (ESI) m/z (%) = 670 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C35H41NNaO9Si 670.2448; found 670.2441. Anal. calcd for C35H41NO9Si: C, 64.89; H, 6.38; N, 2.16. Found: C, 65.05; H, 6.40; N, 2.18.
C-(6-O-tert-Butyldiphenylsilyl-4-O-diphenoxyphosphoryl-2,3-di-O-methyl-α-d-glucopyranosyl)N-methoxyphthalimide (17)
Following the general procedure starting from alcohol 94 (507.2 mg, 0.73 mmol) and purification by column chromatography (hexanes–EtOAc, 75:25), product 17 (331.3 mg, 0.40 mmol, 54%) was obtained as a an amorphous solid: [α]D = +14.9 (c = 0.47, CHCl3). 1H NMR (500 MHz, CDCl3, simulated coupling constants using DAISY) δH 7.73–7.11 (m, 24H, Ar), 4.87 (ddd, J = 7.0, 7.0 Hz, 3JPH = 9.4 Hz, 1H, 4-H), 4.58 (dd, J = 10.9, 7.6 Hz, 1H, 1’-Hb), 4.48 (ddd, J = 7.6, 5.3, 3.6 Hz, 1H, 1-H), 4.36 (dd, J = 10.9, 3.6 Hz, 1H, 1’-Ha), 3.95 (ddd, J = 7.0, 4.0, 4.0 Hz, 1H, 5-H), 3.77 (dd, J = 4.0, 4.0 Hz, 2H, 6-H2), 3.60 (dd, J = 7.4, 7.0 Hz, 1H, 3-H), 3.51 (dd, J = 7.4, 5.3 Hz, 1H, 2-H), 3.48 (s, 3H, OMe), 3.44 (s, 3H, OMe), 1.00 ppm (s, 9H, tBu). 13C{1H} NMR (100.6 MHz, CDCl3) δC 163.2 (2 × C, CO), 150.7 (d, 2JPC = 7.0 Hz, C, Ar), 150.6 (d, 2JPC = 7.8 Hz, C, Ar), 133.5 (C, Ar), 133.4 (C, Ar), 128.8 (2 × C, Ar), 119.9–135.7 (24 × CH, Ar), 79.3 (CH, C-3), 78.8 (CH, C-2), 74.5 (d, 2JPC = 7.0 Hz, CH, C-4), 74.0 (CH2, C-1’), 73.7 (d, 3JPC = 7.1 Hz, CH, C-5), 70.6 (CH, C-1), 62.1 (CH2, C-6), 59.6 (CH3, OMe), 58.7 (CH3, OMe), 26.7 (3 × CH3, DPS), 19.2 ppm (C, DPS). IR (CHCl3): ν = 2934, 1792, 1735, 1490, 1190 cm–1. MS (ESI) m/z (%) = 860 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C45H48NNaO11PSi 860.2632; found 860.2628. Anal. calcd for C45H48NO11PSi: C, 64.50; H, 5.77; N, 1.67. Found: C, 64.42; H, 5.61; N, 1.82.
C-(6-O-tert-Butyldiphenylsilyl-2,3-di-O-methyl-4-O-tosyl-α-d-glucopyranosyl)N-methoxyphthalimide (18)
Following the general procedure starting from alcohol 49 (124.5 mg, 0.20 mmol) and purification by column chromatography (hexanes–EtOAc, 75:25), phthalimide 18 (121.5 mg, 0.16 mmol, 80%) was obtained as a colorless oil: [α]D = +5.8 (c = 0.36, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.78–7.13 (m, 18H, Ar), 4.96 (dd, J = 6.2, 5.8 Hz, 1H, 4-H), 4.50 (dd, J = 11.0, 7.4 Hz, 1H, 1’-Hb), 4.39 (ddd, J = 7.4, 4.3, 3.8 Hz, 1H, 1-H), 4.32 (dd, J = 11.0, 3.8 Hz, 1H, 1’-Ha), 3.86–3.80 (m, 2H, 5-H, 6-Hb), 3.71 (m, 1H, 6-Ha), 3.60 (dd, J = 6.4, 5.8 Hz, 1H, 3-H), 3.51 (s, 3H, OMe), 3.48 (dd, J = 6.4, 4.3 Hz, 1H, 2-H), 3.36 (s, 3H, OMe), 2.35 (s, 3H, OTs), 1.05 ppm (s, 9H, tBu). 13C{1H} NMR (100.6 MHz, CDCl3) δC 163.1 (2 × C, CO), 144.3 (C, OTs), 134.3 (C, OTs), 133.5 (C, Ar), 133.2 (C, Ar), 128.7 (2 × C, Ar), 123.4–135.6 (18 × CH, Ar), 77.8 (CH, C-2), 77.6 (CH, C-3), 74.8 (CH2, C-1’), 74.7 (CH, C-4), 73.5 (CH, C-5), 69.9 (CH, C-1), 61.7 (CH2, C-6), 59.3 (CH3, OMe), 58.5 (CH3, OMe), 26.7 (3 × CH3, DPS), 21.5 (CH3, OTs), 19.2 ppm (C, DPS). IR (CHCl3): ν = 3024, 2934, 1792, 1735, 1215, 1106 cm–1. MS (ESI) m/z (%) = 782 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C40H45NNaO10SSi 782.2431; found 782.2434. Anal. calcd for C40H45NO10SSi: C, 63.22; H, 5.97; N, 1.84; S, 4.22. Found: C, 63.35; H, 6.17; N, 1.75; S, 4.43.
C-(4,6-Bis-O-diphenoxyphosphoryl-2,3-di-O-methyl-α-d-glucopyranosyl)N-methoxyphthalimide (19)
Following the general procedure starting from alcohol 95 (315 mg, 0.46 mmol) and purification by column chromatography (hexanes–EtOAc, 1:1), product 19 (235.8 mg, 0.28 mmol, 62%) was obtained as a colorless oil: [α]D = +13.5 (c = 1.90, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.83–7.81 (m, 2H, Ar), 7.74–7.73 (m, 2H, Ar), 7.33–7.11 (m, 20H, Ar), 4.53 (ddd, J = 7.9, 7.9 Hz, 3JPH = 7.9 Hz, 1H, 4-H), 4.49 (dd, J = 10.1, 6.6 Hz, 1H, 1’-Hb), 4.42–4.33 (m, 3H, 1-H, 6-Hb, 1’-Ha), 4.26 (ddd, J = 12.3, 4.7 Hz, 3JPH = 7.9 Hz, 1H, 6-Ha), 4.14 (m, 1H, 5-H), 3.58 (dd, J = 7.3, 7.3 Hz, 1H, 3-H), 3.44 (s, 3H, OMe), 3.43 (s, 3H, OMe), 3.41 ppm (dd, J = 7.6, 4.7 Hz, 1H, 2-H). 13C{1H} NMR (125.7 MHz, CDCl3) δC 163.3 (2 × C, CO), 150.6 (d, 2JPC = 6.3 Hz, 2 × C, Ar), 150.5 (d, 2JPC = 7.4 Hz, C, Ar), 150.4 (d, 2JPC = 8.5 Hz, C, Ar), 128.8 (2 × C, Ar), 120.0–134.5 (24 × CH, Ar), 79.3 (CH, C-3), 78.4 (CH, C-2), 74.4 (d, 2JPC = 6.4 Hz, CH, C-4), 74.1 (CH2, C-1’), 72.8 (dd, 3JPC = 6.1, 6.1 Hz, CH, C-5), 70.4 (CH, C-1), 66.5 (d, 2JPC = 6.3 Hz, CH2, C-6), 59.8 (CH3, OMe), 58.8 ppm (CH3, OMe). IR (CHCl3): ν = 3018, 2937, 1736, 1214 cm–1. MS (ESI) m/z (%) = 854 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C41H39NNaO14P2: 854.1744; found 854.1743. Anal. calcd for C41H39NO14P2: C, 59.21; H, 4.73; N, 1.68. Found: C, 58.99; H, 4.89; N, 2.08.
C-(4-O-Acetyl-6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-α-d-galactopyranosyl)N-methoxyphthalimide (20)
Following the general procedure starting from alcohol 51 (285.9 mg, 0.57 mmol) and purification by column chromatography (hexanes–EtOAc, 75:25), product 20 (318.2 mg, 0.49 mmol, 86%) was obtained as a crystalline solid: mp 48.9–50.0 °C (n-hexane–EtOAc); [α]D = +6.7 (c = 0.43, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.74–7.61 (m, 8H, Ar), 7.43–7.35 (m, 6H, Ar), 5.68 (dd, J = 3.2, 2.1 Hz, 1H, 4-H), 4.62–4.58 (m, 2H, 1-H, 1’-Hb), 4.38 (m, 1H, 1’-Ha), 4.09 (ddd, J = 8.1, 5.7, 2.1 Hz, 1H, 5-H), 3.69 (dd, J = 9.1, 5.4 Hz, 1H, 2-H), 3.64 (dd, J = 10.0, 5.7 Hz, 1H, 6-Hb), 3.54 (dd, J = 10.0, 8.1 Hz, 1H, 6-Ha), 3.50 (s, 3H, OMe), 3.46 (dd, J = 9.1, 3.2 Hz, 1H, 3-H), 3.44 (s, 3H, OMe), 2.02 (s, 3H, OAc), 1.03 ppm (s, 9H, tBu). 13C{1H} NMR (125.7 MHz, CDCl3) δC 169.7 (C, OAc), 163.2 (2 × C, CO), 135.60 (2 × CH, Ar), 135.56 (2 × CH, Ar), 134.4 (2 × CH, Ar), 133.3 (C, Ar), 133.1 (C, Ar), 129.7 (CH, Ar), 129.6 (CH, Ar), 128.8 (2 × C, Ar), 127.67 (2 × CH, Ar), 127.66 (2 × CH, Ar), 123.4 (2 × CH, Ar), 78.9 (CH, C-3), 76.3 (CH, C-2), 72.8 (CH2, C-1’), 72.2 (CH, C-1), 71.5 (CH, C-5), 66.1 (CH, C-4), 61.1 (CH2, C-6), 59.3 (CH3, OMe), 57.7 (CH3, OMe), 26.7 (3 × CH3, DPS), 20.8 (CH3, OAc), 19.0 ppm (C, DPS). IR (CHCl3): ν = 2933, 1792, 1734, 1226 cm–1. MS (ESI) m/z (%) = 670 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C35H41NNaO9Si: 670.2448; found 670.2443. Anal. calcd for C35H41NO9Si: C, 64.89; H, 6.38; N, 2.16. Found: C, 64.67; H, 6.41; N, 2.23.
C-(6-O-tert-Butyldiphenylsilyl-4-O-diphenoxyphosphoryl-2,3-di-O-methyl-α-d-galactopyranosyl)N-methoxyphthalimide (21)
Following the general procedure starting from alcohol 52 (510 mg, 0.74 mmol) (327 mg, 0.51 mmol) and purification by column chromatography (hexanes–EtOAc, 8:2), product 21 (574 mg, 0.68 mmol, 93%) was obtained as a colorless oil: [α]D = −9.1 (c = 0.67, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.74–7.11 (m, 24H, Ar), 5.24 (ddd, J = 2.9, 2.7 Hz, 3JPH = 8.7 Hz, 1H, 4-H), 4.47–4.41 (m, 2H, 1-H, 1’-Hb), 4.33 (m, 1H, 1’-Ha), 4.03 (dddd, J = 6.7, 5.8, 2.7 Hz, 4JPH = 2.3 Hz, 1H, 5-H), 3.77 (dd, J = 10.6, 5.8 Hz, 1H, 6-Hb), 3.74 (dd, J = 10.6, 6.7 Hz, 1H, 6-Ha), 3.66 (dd, J = 8.2, 5.6 Hz, 1H, 2-H), 3.42 (s, 3H, OMe), 3.39 (dd, J = 8.2, 2.9 Hz, 1H, 3-H), 3.35 (s, 3H, OMe), 1.00 ppm (s, 9H, tBu). 13C{1H} NMR (100.6 MHz, CDCl3) δC 163.2 (2 × C, CO), 150.7 (d, 2JPC = 7.4 Hz, C, Ar), 150.6 (d, 2JPC = 6.4 Hz, C, Ar), 135.58 (2 × CH, Ar), 135.55 (2 × CH, Ar), 134.4 (2 × CH, Ar), 133.5 (C, Ar), 133.3 (C, Ar), 129.7 (2 × CH, Ar), 129.6 (CH, Ar), 129.5 (CH, Ar), 129.4 (2 × CH, Ar), 128.8 (2 × C, Ar), 127.7 (2 × CH, Ar), 127.6 (2 × CH, Ar), 125.1 (CH, Ar), 125.0 (CH, Ar), 123.4 (2 × CH, Ar), 120.4 (CH, Ar), 120.3 (CH, Ar), 120.1 (CH, Ar), 120.0 (CH, Ar), 78.3 (CH, C-3), 75.7 (CH, C-2), 73.8 (d, 2JPC = 6.3 Hz, CH, C-4), 73.5 (CH2, C-1’), 73.1 (CH, C-1), 70.6 (br s, CH, C-5), 60.9 (CH2, C-6), 59.5 (CH3, OMe), 57.8 (CH3, OMe), 26.7 (3 × CH3, DPS), 19.1 ppm (C, DPS). IR (CHCl3): ν = 3015, 2933, 1792, 1730, 1490, 1190 cm–1. MS (ESI) m/z (%) = 860 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C45H48NNaO11PSi: 860.2632; found 860.2648. Anal. calcd for C45H48NO11PSi: C, 64.50; H, 5.77; N, 1.67. Found: C, 64.82; H, 6.01; N, 1.92.
C-(4-O-Diphenoxyphosphoryl-2,3-di-O-methyl-α-l-rhamnopyranosyl)N-methoxyphthalimide (22)
Alcohol 105 (231.7 mg, 0.66 mmol) in dry CH2Cl2 (38 mL) was treated with DMAP (371 mg, 3.04 mmol) and ClPO(OPh)2 (0.63 mL, 3.04 mmol) for 2 h. The reaction was quenched with a saturated aqueous solution of NH4Cl and extracted with CH2Cl2. The organic phase was dried over Na2SO4, filtered, and evaporated. Column chromatography of the residue (hexanes–EtOAc, 6:4) gave the compound 22 (333 mg, 0.55 mmol, 84%) as an amorphous solid: [α]D = −0.1 (c = 0.41, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.84–7.82 (m, 2H, Ar), 7.75–7.73 (m, 2H, Ar), 7.37–7.17 (m, 10H, Ar), 4.63 (ddd, J = 5.2, 3.2 Hz, 3JPH = 9.2 Hz, 1H, 4-H), 4.46 (dd, J = 10.9, 3.2 Hz, 1H, 1’-Hb), 4.38 (dd, J = 10.9, 6.2 Hz, 1H, 1’-Ha), 4.23 (ddd, J = 7.6, 6.2, 3.2 Hz, 1H, 1-H), 4.01 (dddd, J = 6.9, 6.9, 6.9, 3.7 Hz, 1H, 5-H), 3.71 (dd, J = 5.2, 3.2 Hz, 1H, 3-H), 3.61 (dd, J = 7.6, 3.2 Hz, 1H, 2-H), 3.42 (s, 3H, OMe), 3.35 (s, 3H, OMe), 1.34 ppm (d, J = 6.9 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 163.2 (2 × C, CO), 150.5 (d, 2JPC = 7.4 Hz, C, Ar), 150.3 (d, 2JPC = 7.4 Hz, C, Ar), 134.4 (2 × CH, Ar), 129.8 (2 × CH, Ar), 129.7 (2 × CH, Ar), 128.9 (2 × C, Ar), 125.42 (CH, Ar), 125.37 (CH, Ar), 123.4 (2 × CH, Ar), 120.2 (CH, Ar), 120.12 (CH, Ar), 120.11 (CH, Ar), 120.07 (CH, Ar), 77.4 (d, 2JPC = 6.4 Hz, CH, C-4), 77.2 (CH2, C-1’), 76.2 (d, 3JPC = 3.2 Hz, CH, C-3), 73.9 (CH, C-2), 71.6 (d, 3JPC = 5.3 Hz, CH, C-5), 68.2 (CH, C-1), 58.3 (CH3, OMe), 57.2 (CH3, OMe), 16.2 ppm (CH3, C-6). IR (CHCl3): ν = 3015, 2939, 1792, 1736, 1490, 1209 cm–1. MS (ESI) m/z (%) = 606 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C29H30NNaO10P: 606.1505; found 606.1516. Anal. calcd for C29H30NO10P: C, 59.69; H, 5.18; N, 2.40. Found: C, 59.37; H, 5.40; N, 2.68.
C-(4-O-Acetyl-2,3-di-O-methyl-α-l-fucopyranosyl)N-methoxyphthalimide (23)
Following the general procedure starting from 58 (65.6 mg, 0.26 mmol) and purification by column chromatography (hexanes–Et2O, 4:6), product 23 (52.4 mg, 0.13 mmol, 51%) was obtained as a white crystalline solid: mp 37.8–38.5 °C (n-hexane–EtOAc). [α]D = −42.5 (c = 0.72, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.86–7.84 (m, 2H, Ar), 7.78–7.76 (m, 2H, Ar), 5.31 (dd, J = 3.4, 2.0 Hz, 1H, 4-H), 4.67 (ddd, J = 9.0, 5.9, 1.9 Hz, 1H, 1-H), 4.63 (dd, J = 10.7, 9.0 Hz, 1H, 1’-Hb), 4.39 (dd, J = 10.7, 1.9 Hz, 1H, 1’-Ha), 4.09 (dddd, J = 6.4, 6.4, 6.4, 2.0 Hz, 1H, 5-H), 3.71 (dd, J = 9.3, 5.9 Hz, 1H, 2-H), 3.51 (s, 3H, OMe), 3.40 (s, 3H, OMe), 3.36 (dd, J = 9.3, 3.4 Hz, 1H, 3-H), 2.16 (s, 3H, OAc), 1.11 ppm (d, J = 6.4 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 170.6 (C, OAc), 163.3 (2 × C, CO), 134.5 (2 × CH, Ar), 128.8 (2 × C, Ar), 123.5 (2 × CH, Ar), 78.6 (CH, C-3), 76.1 (CH, C-2), 72.8 (CH2, C-1’), 71.8 (CH, C-1), 69.4 (CH, C-4), 67.0 (CH, C-5), 59.2 (CH3, OMe), 57.6 (CH3, OMe), 20.8 (CH3, OAc), 16.1 ppm (CH3, C-6). IR (CHCl3): ν = 3018, 2936, 1791, 1744, 1239 cm–1. MS (ESI) m/z (%) = 416 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C19H23NNaO8: 416.1321; found 416.1328. Anal. calcd for C19H23NO8: C, 58.01; H, 5.89; N, 3.56. Found: C, 58.33; H, 5.95; N, 3.83.
C-(4-O-Diphenoxyphosphoryl-2,3-di-O-methyl-α-l-fucopyranosyl)N-methoxyphthalimide (24)
Following the general procedure starting from 59 (232 mg, 0.53 mmol) and purification by column chromatography (hexanes–EtOAc, 6:4), product 24 (52.4 mg, 0.13 mmol, 51%) was obtained as a colorless oil: [α]D = −19.0 (c = 0.57, CHCl3). 1H NMR (500 MHz, CDCl3, simulated ring coupling constants using DAISY) δH 7.84–7.81 (m, 2H, Ar), 7.76–7.74 (m, 2H, Ar), 7.34–7.16 (m, 10H, Ar), 4.91 (ddd, J = 2.5, 3.3 Hz, 3JPH = 9.0 Hz, 1H, 4-H), 4.55–4.50 (m, 2H, 1-H, 1’-Hb), 4.35 (m, 1H, 1’-Ha), 4.08 (ddddd, J = 6.5, 6.5, 6.5, 2.5 Hz, 4JPH = 2.0 Hz, 1H, 5-H), 3.61 (dd, J = 7.9, 5.0 Hz, 1H, 2-H), 3.43 (dd, J = 7.9, 3.3 Hz, 1H, 3-H), 3.42 (s, 3H, OMe), 3.39 (s, 3H, OMe), 1.19 ppm (d, J = 6.5 Hz, 3H, 6-H3). 13C{1H} NMR (125.7 MHz, CDCl3) δC 163.3 (2 × C, CO), 150.7 (d, 2JPC = 7.4 Hz, C, Ar), 150.5 (d, 2JPC = 6.4 Hz, C, Ar), 134.5 (2 × CH, Ar), 129.7 (2 × CH, Ar), 129.5 (2 × CH, Ar), 128.8 (2 × C, Ar), 125.2 (CH, Ar), 125.1 (CH, Ar), 123.5 (2 × CH, Ar), 120.3 (CH, Ar), 120.2 (CH, Ar), 120.1 (CH, Ar), 120.0 (CH, Ar), 78.4 (CH, C-3), 76.5 (d, 2JPC = 6.4 Hz, CH, C-4), 75.9 (CH, C-2), 73.8 (CH2, C-1’), 70.3 (CH, C-1), 68.1 (d, 3JPC = 6.3 Hz, CH, C-5), 59.4 (CH3, OMe), 57.9 (CH3, OMe), 15.4 ppm (CH3, C-6). IR (CHCl3): ν = 3026, 2938, 1792, 1734, 1210 cm–1. MS (ESI) m/z (%) = 606 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C29H30NNaO10P: 606.1505; found 606.1500. Anal. calcd for C29H30NO10P: C, 59.69; H, 5.18; N, 2.40. Found: C, 59.61; H, 5.23; N, 2.70.
C-(2,3-Di-O-methyl-α-l-rhamnopyranosyl)N-methoxyphthalimide (105)
To a solution of 104 (570 mg, 1.92 mmol) in dry EtOAc (45 mL) was added Pd/C 10% (430 mg), and the mixture was submitted to a H2 atmosphere overnight. The reaction was filtered over a pad of Celite and evaporated. The residue was then submitted to the general procedure to give phthalimide 105 (383 mg, 1.09 mmol, 57%) as a colorless oil: [α]D = −22.6 (c = 1.34, CHCl3). 1H NMR (500 MHz, CDCl3) δH 7.84–7.82 (m, 2H, Ar), 7.76–7.74 (m, 2H, Ar), 4.53 (dd, J = 11.0, 4.9 Hz, 1H, 1’-Hb), 4.32 (dd, J = 11.0, 4.7 Hz, 1H, 1’-Ha), 4.24 (ddd, J = 4.7, 4.7, 4.7 Hz, 1H, 1-H), 4.07 (dd, J = 5.1, 3.3 Hz, 1H, 2-H), 3.70–3.67 (m, 2H, 4-H, 5-H), 3.60 (dd, J = 7.2, 3.3 Hz, 1H, 3-H), 3.52 (s, 3H, OMe), 3.50 (s, 3H, OMe), 1.28 ppm (d, J = 6.3 Hz, 3H, 6-H3), 1H from OH is missing. 13C{1H} NMR (125.7 MHz, CDCl3) δC 163.4 (2 × C, CO), 134.6 (2 × CH, Ar), 128.7 (2 × C, Ar), 123.6 (2 × CH, Ar), 79.5 (CH, C-3), 76.4 (CH2, C-1’), 73.6 (CH, C-2), 72.8 (CH, C-4 or C-5), 70.5 (CH, C-4 or C-5), 70.3 (CH, C-1), 57.7 (CH3, OMe), 57.5 (CH3, OMe), 16.9 ppm (CH3, C-6). IR (CHCl3): ν = 3674, 3501, 3022, 2937, 1792, 1735, 1212 cm–1. MS (ESI) m/z (%) = 374 (100) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C17H21NNaO7: 374.1216; found 374.1213. Anal. calcd for C17H21NO7: C, 58.11; H, 6.02; N, 3.99. Found: C, 58.22; H, 6.18; N, 4.11.
Methyl 4-O-Benzyl-6-O-tert-butyldiphenylsilyl-2,3-di-O-methyl-α-d-[4-O-PhCH-2H]galactopyranoside ([PhCH-2H]97)
A mixture of 96 (47.4 mg, 0.10 mmol) and benzyl α-[2H]-4-methylbenzenesulfonate52 (31.6 mg, 0.12 mmol, 2H/1H 6.9:1) in DMF (0.2 mL) and CH2Cl2 (0.2 mL) was cooled to 0 °C. Then, sodium hydride (60% dispersion in mineral oil, 8 mg, 0.20 mmol) was added to the mixture, and the reaction was gradually warmed up to room temperature. After stirring for 1 h, the Dowex 50WX4-200 was added to quench the reaction. The mixture was filtered through a pad of Celite, and the filtrate was evaporated under reduced pressure. The crude residue was purified by column chromatography (hexanes–EtOAc, 8:2 to 7:3) to obtain the product [PhCH-2H]97 (28.6 mg, 0.05 mmol, 50%, 2H/1H 7:1) as a colorless oil: 1H NMR (500 MHz, CDCl3) δH 7.67–7.60 (m, 4H, Ar), 7.46–7.32 (m, 6H, Ar), 7.31–7.21 (m, 5H, Ar), 4.91 (d, J = 11.4 Hz, 0.1H, O-CH2-Ph), 4.900 (br s, 0.4H, O-CHD-Ph), 4.84 (d, J = 3.6 Hz, 1H, 1-H), 4.60 (d, J = 11.5 Hz, 0.1H, O-CH2-Ph), 4.589 (br s, 0.4H, O-CHD-Ph), 4.01 (br s, 1H, 4-H), 3.79–3.69 (m, 4H, 6-H2, 2-H, 5-H), 3.56 (dd, J = 10.1, 2.8 Hz, 1H, 3-H), 3.51 (s, 3H, 3-OMe), 3.51 (s, 3H, 2-OMe), 3.32 (s, 3H, 1-OMe), 1.06 ppm (s, 9H). 13C{1H} NMR (125.7 MHz, CDCl3) δC 138.6 (C, Ar), 135.56 (2 × CH, Ar), 135.53 (2 × CH, Ar), 133.4 (2 × C, Ar), 129.74 (CH, Ar), 129.71 (CH, Ar), 128.2 (2 × CH, Ar), 128.1 (2 × CH, Ar), 127.71 (2 × CH, Ar), 127.70 (2 × CH, Ar), 127.5 (CH, Ar), 97.7 (CH, C-1), 80.8 (CH, C-3), 78.1 (CH, C-2), 74.73 (0.12CH2, O-CH2-Ph), 74.35 (0.88CH, t, JCD = 22.1 Hz, O-CHD-Ph), 73.64 (0.12CH, C-4), 73.59 (0.44CH, C-4), 73.56 (0.44CH, C-4), 70.7 (CH, C-5), 62.8 (CH2, C-6), 58. 8 (CH3, OMe), 58.3 (CH3, OMe), 55.0 (CH3, OMe), 26.9 (3 × CH3, DPS), 19.2 ppm (C, DPS). 1H NMR (500 MHz, C6D6) δH 7.84–7.79 (m, 2H, Ar), 7.79–7.74 (m, 2H, Ar), 7.35 (m, 2H, Ar), 7.23–7.07 (m, 9H, Ar), 5.07 (d, J = 11.4 Hz, 0.1H, O-CH2-Ph), 5.05 (br s, 0.4H, O-CHD-Ph), 4.82 (d, J = 3.6 Hz, 1H, 1-H), 4.61 (d, J = 11.4 Hz, 0.1H, O-CH2-Ph), 4.58 (br s, 0.4H, O-CHD-Ph), 4.105 (dd, J = 10.0, 6.6 Hz, 0.5H, 6-H), 4.107 (dd, J = 10.1, 6.5 Hz, 0.5H, 6-H), 4.06 (dd, J = 10.1, 3.7 Hz, 0.5H, 6-H), 4.05 (dd, J = 10.2, 3.8 Hz, 0.5H, 6-H), 3.97–3.90 (m, 3H, 5-H, 4-H, 2-H), 3.73 (dd, J = 10.0, 2.9 Hz, 1H, 3-H), 3.35 (s, 3H, 3-OMe), 3.232 (s, 1.5H, 2-OMe), 3.230 (s, 1.5H, 2-OMe), 3.21 (s, 3H, 1-OMe), 1.19 ppm (s, 9H). 13C{1H} NMR (125.7 MHz, C6D6) δC 139.98 (C, Ar), 136.41 (2 × CH, Ar), 136.31 (2 × CH, Ar), 134.33 (C, Ar), 134.16 (C, Ar), 130.42 (CH, Ar), 130.40 (CH, Ar), 98.93 (CH, C-1), 81.71 (CH, C-3), 79.40 (CH, C-2), 75.55 (0.12CH, C-4), 75.51 (0.44CH, C-4), 75.48 (0.44CH, C-4), 75.45 (0.12CH2, O-CH2-Ph), 75.08 (0.44CH, t, JCD = 22.1 Hz, O-CHD-Ph), 75.05 (0.44CH, t, JCD = 21.1 Hz, O-CHD-Ph), 71.89 (CH, C-5), 64.14 (CH2, C-6), 59.00 (CH3, OMe), 58.59 (CH3, OMe), 55.26 (CH3, OMe), 27.46 (3 × CH3, DPS), 19.81 ppm (C, DPS), some aromatic carbons were not observed. IR (CHCl3): ν = 3020, 2932, 1471, 1428, 1220, 1103 cm–1. MS (ESI) m/z (%) = 574 (100) [M + Na]+, 573 (13) [M + Na]+. HRMS (ESI) m/z: [M + Na]+ calcd for C32H412HNaO6Si 574.2711; found 574.2716; [M + Na]+ calcd for C32H42NaO6Si 573.2648; found 573.2653.
Acknowledgments
The authors gratefully acknowledge the financial support from the Investigation Programs of the Ministerio de Economia y Competitividad (CTQ2010-18244), Fundación CajaCanarias (2015-BIO08), and the Gobierno de Canarias (ProID2017010017). A.S.M. is grateful to the CSIC JAE Predoc Program.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c01376.
Tables of calculated ring coupling constants (3JH,H) of starting phthalimides and final bicyclic ketals; calculated long-range 4Jw coupling constants; selected signals of 1H and 13C{1H} NMR spectra of labeled [PhCH-2H]31 and [PhCH-2H]97; reactivity differences between LGs in the 1,5-hydrogen atom transfer/Surzur–Tanner rearrangement sequence; and copies of the 1H and 13C{1H} NMR spectra of all new compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Gillard R. M.; Brimble M. A. Benzannulated Spiroketal Natural Products: Isolation, Biological Activity, Biosynthesis, and Total Synthesis. Org. Biomol. Chem. 2019, 17, 8272–8307. 10.1039/C9OB01598A. [DOI] [PubMed] [Google Scholar]; b Zhang F.-M.; Zhang S.-Y.; Tu Y.-Q. Recent Progress in the Isolation, Bioactivity, Biosynthesis, and Total Synthesis of Natural Spiroketals. Nat. Prod. Rep. 2018, 35, 75–104. 10.1039/C7NP00043J. [DOI] [PubMed] [Google Scholar]; c Quach R.; Furkert D. P.; Brimble M. A. Gold Catalysis: Synthesis of Spiro, Bridged, and Fused Ketal Natural Products. Org. Biomol. Chem. 2017, 15, 3098–3104. 10.1039/C7OB00496F. [DOI] [PubMed] [Google Scholar]; d Brimble M. A.; Stubbing L. A.. Synthesis of 5,6- and 6,6-Spirocyclic Compounds. In Synthesis of saturated oxygenated heterocycles I; Springer: Berlin, Heidelberg, 2014, 35, 189–267, 10.1007/978-3-642-41473-2_5. [DOI] [Google Scholar]; e Palmes J.; Aponick A. Strategies for Spiroketal Synthesis Based on Transition-Metal Catalysis. Synthesis 2012, 44, 3699–3721. 10.1055/s-0032-1317489. [DOI] [Google Scholar]; f Aho J. E.; Pihko P. M.; Rissa T. K. Nonanomeric Spiroketals in Natural Products: Structures, Sources, and Synthetic Strategies. Chem. Rev. 2005, 105, 4406–4440. 10.1021/cr050559n. [DOI] [PubMed] [Google Scholar]; g Perron F.; Albizati K. F. Chemistry of Spiroketals. Chem. Rev. 1989, 89, 1617–1661. 10.1021/cr00097a015. [DOI] [Google Scholar]
- a Zhang W.; Tong R. Synthetic Approaches To Construct the 6,8-DOBCO Framework in Natural Products. J. Org. Chem. 2016, 81, 2203–2212. 10.1021/acs.joc.6b00246. [DOI] [PubMed] [Google Scholar]; b Milroy L.-G.; Zinzalla G.; Prencipe G.; Michel P.; Ley S. V.; Gunaratnam M.; Beltran M.; Neidle S. Chemical Variation of Natural-Product-Like Scaffolds: Design, Synthesis, and Biological Activity of Fused Bicyclic Acetal Derivatives. Angew. Chem., Int. Ed. 2007, 46, 2493–2496. 10.1002/anie.200604688. [DOI] [PubMed] [Google Scholar]; c Jun J.-G. Introduction of Bicyclic Ketal Chemistry: Synthesis and Transformation Reaction of 6,8-Dioxabicyclo[3.2.1]Octane Skeletal System. Synlett 2003, 1759–1777. 10.1055/s-2003-41415. [DOI] [Google Scholar]; d Mundy B. P.; Lipkowitz K. B.; Dirks G. W. Chemistry of the 6,8-Dioxabicyclo[3.2.1]Octane Series. Sources, Synthesis, Structures and Reactions. Heterocycles 1977, 51–76. 10.3987/R-1977-01-0051. [DOI] [Google Scholar]; For recent articles, see; e Zeng R.; Li J. L.; Zhang X.; Liu Y. Q.; Jia Z. Q.; Leng H. J.; Huang Q. W.; Liu Y.; Li Q. Z. Diastereoselective Construction of 6,8-Dioxabicyclo[3.2.1]Octane Frameworks from Vinylethylene Carbonates via Palladium-Organo Relay Catalysis. ACS Catal. 2019, 9, 8256–8262. 10.1021/acscatal.9b02598. [DOI] [Google Scholar]; f Hohol R. E.; Arcure H.; Witczak Z. J.; Bielski R.; Kirschbaum K.; Andreana P.; Mencer D. E. One-Pot Synthesis of Carbohydrate Exo-Cyclic Enones and Hemiketals with 6,8-Dioxabicyclo-[3.2.1]Octane Moieties. Serendipitous Formation of a Spironolactone When 2-Pyridinecarboxaldehyde Is Used as the Reactant. Part II. Tetrahedron 2018, 74, 7303–7309. 10.1016/j.tet.2018.10.049. [DOI] [Google Scholar]; g Schmidt E. Y.; Tatarinova I. V.; Semenova N. V.; Protsuk N. I.; Ushakov I. A.; Trofimov B. A. Exploring Acetylene Chemistry: A Transition Metal-Free Route to Dienyl 6,8-Dioxabicyclo[3.2.1]Octanes from Ketones and Acetylenes. J. Org. Chem. 2018, 83, 10272–10280. 10.1021/acs.joc.8b01449. [DOI] [PubMed] [Google Scholar]; h Schmidt E. Y.; Bidusenko I. A.; Ushakov I. A.; Trofimov B. A. Unfolding the Frontalin Chemistry: A Facile Selective Hydrogenation of 7-Methylidene-6,8-Dioxabicyclo[3.2.1]Octanes, 2:2 Ensembles of Ketones and Acetylene. Mendeleev Commun. 2018, 28, 513–514. 10.1016/j.mencom.2018.09.021. [DOI] [Google Scholar]; i Schmidt E. Y.; Bidusenko I. A.; Ushakov I. A.; Protsuk N. I.; Trofimov B. A. An Easy Access to Sulfur Derivatives of 6,8-Dioxabicyclo[3.2.1]Octanes, Naturally Abundant Scaffolds. Synthesis 2018, 50, 2624–2630. 10.1055/s-0036-1591990. [DOI] [Google Scholar]
- a Mori K. Protective Group-Free Syntheses of (±)-Frontalin, (±)-endo-Brevicomin, (±)-exo-Brevicomin, and (±)-3,4-Dehydro-exo-Brevicomin: Racemic Pheromones with a 6,8-Dioxabicyclo[3.2.1]Octane Ring. Biosci., Biotechnol., Biochem. 2011, 75, 976–981. 10.1271/bbb.110071. [DOI] [PubMed] [Google Scholar]; b Booth Y. K.; Kitching W.; De Voss J. J. Biosynthesis of Insect Spiroacetals. Nat. Prod. Rep. 2009, 26, 490–525. 10.1039/b717392j. [DOI] [PubMed] [Google Scholar]; c Francke W.; Kitching W. Spiroacetals in Insects. Curr. Org. Chem. 2001, 5, 233–251. 10.2174/1385272013375652. [DOI] [Google Scholar]; d Mori K.The Synthesis of Insect Pheromones. In The Total Synthesis of Natural Products; 1981; pp. 1–183, 10.1002/9780470129678.ch1. [DOI] [Google Scholar]
- For a study of the abundance of bicyclic acetals in natural products, seeLenci E.; Menchi G.; Saldívar-Gonzalez F. I.; Medina-Franco J. L.; Trabocchi A. Bicyclic Acetals: Biological Relevance, Scaffold Analysis, and Applications in Diversity-Oriented Synthesis. Org. Biomol. Chem. 2019, 17, 1037–1052. 10.1039/C8OB02808G. [DOI] [PubMed] [Google Scholar]
- a For recent reviews, seeIglesias-Arteaga M. A.; Morzycki J. W.. Cephalostatins and Ritterazines. In The Alkaloids Chemistry and Biology; 2013; pp. 153–279, 10.1016/B978-0-12-407774-4.00002-9. [DOI] [PubMed] [Google Scholar]; b Lee S.; LaCour T. G.; Fuchs P. L. Chemistry of Trisdecacyclic Pyrazine Antineoplastics: The Cephalostatins and Ritterazines. Chem. Rev. 2009, 109, 2275–2314. 10.1021/cr800365m. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Simmons-Boyce J. L.; Tinto W. F. Steroidal Saponins and Sapogenins from the Agavaceae Family. Nat. Prod. Commun. 2007, 2, 99–114. 10.1177/1934578X0700200120. [DOI] [Google Scholar]
- Liu H.; Lin S.; Jacobsen K. M.; Poulsen T. B. Chemical Syntheses and Chemical Biology of Carboxyl Polyether Ionophores: Recent Highlights. Angew. Chem., Int. Ed. 2019, 58, 13630–13642. 10.1002/anie.201812982. [DOI] [PubMed] [Google Scholar]
- Carroll A. R.; Copp B. R.; Davis R. A.; Keyzers R. A.; Prinsep M. R. Marine Natural Products. Nat. Prod. Rep. 2020, 37, 175–223. 10.1039/C9NP00069K. [DOI] [PubMed] [Google Scholar]; See also previous reviews of this series.
- a Beaumont S.; Ilardi E. A.; Tappin N. D. C.; Zakarian A. Marine Toxins with Spiroimine Rings: Total Synthesis of Pinnatoxin A. Eur. J. Org. Chem. 2010, 2010, 5743–5765. 10.1002/ejoc.201000842. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Matsuura F.; Peters R.; Anada M.; Harried S. S.; Hao J.; Kishi Y. Unified Total Synthesis of Pteriatoxins and Their Diastereomers. J. Am. Chem. Soc. 2006, 128, 7463–7465. 10.1021/ja0618954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ren J.; Wang J.; Tong R. Asymmetric Total Synthesis of (+)-Attenol B. Org. Lett. 2015, 17, 744–747. 10.1021/acs.orglett.5b00038. [DOI] [PubMed] [Google Scholar]; b Ren J.; Tong R. Asymmetric Total Synthesis of (+)-Didemniserinolipid B via Achmatowicz Rearrangement/Bicycloketalization. J. Org. Chem. 2014, 79, 6987–6995. 10.1021/jo501142q. [DOI] [PubMed] [Google Scholar]; c Schmidt E. Y.; Trofimov B. A.; Zorina N. V.; Mikhaleva A. I.; Ushakov I. A.; Skital’tseva E. V.; Kazheva O. N.; Alexandrov G. G.; Dyachenko O. A. Synthesis of Functionalized 3,4-Dihydropyrans via Rearrangement of the Products of a One-Pot Diastereoselective Assembly of Ketones and Acetylene. Eur. J. Org. Chem. 2010, 2010, 6727–6730. 10.1002/ejoc.201001229. [DOI] [Google Scholar]; d Jong-Gab Jun J.-G.; Shin D. G.; Shin H. S.; Kim S. H. Transformation of Bicyclic Ketal Compound to 1,2-Cyclopentanediol via 1,5-Diketone. Bull. Korean Chem. Soc. 1992, 13, 176–179. [Google Scholar]
- Somsák L., Ed. Carbohydrate-Spiro-Heterocycles; Springer International Publishing, 2019. [Google Scholar]
- a Chen Y.; Wang X.; Wang J.; Yang Y. Synthesis of D-Manno-Heptulose via a Cascade Aldol/Hemiketalization Reaction. Beilstein J. Org. Chem. 2017, 13, 795–799. 10.3762/bjoc.13.79. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Subrizi F.; Cárdenas-Fernández M.; Lye G. J.; Ward J. M.; Dalby P. A.; Sheppard T. D.; Hailes H. C. Transketolase Catalysed Upgrading of L-Arabinose: The One-Step Stereoselective Synthesis of L-Gluco-Heptulose. Green Chem. 2016, 18, 3158–3165. 10.1039/C5GC02660A. [DOI] [Google Scholar]; c Jacobsen A.; Thiem J. Contemporary Syntheses of 2-Ketoheptoses and Derivatives. Curr. Org. Chem. 2014, 18, 2833–2841. 10.2174/1385272819666141016215205. [DOI] [Google Scholar]; d Matsuda S.; Yamanoi T.; Watanabe M. Syntheses of a Partially Benzylated Derivative of the Anhydro-D-Altro-Heptulose Found in Coriaria Japonica A and of Its Analogs. Tetrahedron 2008, 64, 8082–8088. 10.1016/j.tet.2008.06.068. [DOI] [Google Scholar]
- a Martín A.; Salazar J. A.; Suárez E. Synthesis of Chiral Spiroacetals from Carbohydrates. J. Org. Chem. 1996, 61, 3999–4006. 10.1021/jo960060g. [DOI] [PubMed] [Google Scholar]; b Dorta R. L.; Martín A.; Salazar J. A.; Suárez E.; Prangé T. Syntheses of Chiral Dispiroacetals from Carbohydrates. J. Org. Chem. 1998, 63, 2251–2261. 10.1021/jo972023a. [DOI] [Google Scholar]
- Francisco C. G.; Herrera A. J.; Suárez E. Intramolecular Hydrogen Abstraction Reaction Promoted by Alkoxy Radicals in Carbohydrates. Synthesis of Chiral 2,7-Dioxabicyclo[2.2.1]Heptane and 6,8-Dioxabicyclo[3.2.1]Octane Ring Systems. J. Org. Chem. 2002, 67, 7439–7445. 10.1021/jo026004z. [DOI] [PubMed] [Google Scholar]
- For a recent review on HAT reactions, seeNechab M.; Mondal S.; Bertrand M. P. 1,n-Hydrogen-Atom Transfer (HAT) Reactions in Which n ≠5: An Updated Inventory. Chem. - Eur. J. 2014, 20, 16034–16059. 10.1002/chem.201403951. [DOI] [PubMed] [Google Scholar]; and references cited therein.
- a For the interest of C-ketosides as versatile chiral synthons, seePérez-Martín I.; Suárez E.. Radicals and Carbohydrates. In Encyclopedia of Radicals in Chemistry, Biology and Materials; John Wiley & Sons, Ltd: Chichester, UK, 2012; pp. 1131–1174, 10.1002/9781119953678.rad031. [DOI] [Google Scholar]; For selected examples, see; b Pal A. P. J.; Mallick A.; Reddy Y. S.; Vankar Y. D. Molecular Iodine-Promoted N- and C-Glycosylation of 1-C-Alkyl (or Phenyl)-Glycopyranoses. Tetrahedron Lett. 2010, 51, 6334–6337. 10.1016/j.tetlet.2010.09.124. [DOI] [Google Scholar]; c Malapelle A.; Coslovi A.; Doisneau G.; Beau J. M. An Expeditious Synthesis of N-Acetylneuraminic Acid α-C-Glycosyl Derivatives (“α-C-Glycosides)” from the Anomeric Acetates. Eur. J. Org. Chem. 2007, 2007, 3145–3157. 10.1002/ejoc.200700181. [DOI] [Google Scholar]; d Majumder U.; Cox J. M.; Johnson H. W.; Rainier J. D. Total Synthesis of Gambierol: The Generation of the A–C and F–H Subunits by Using a C-Glycoside Centered Strategy. Chem. - Eur. J. 2006, 12, 1736–1746. 10.1002/chem.200500993. [DOI] [PubMed] [Google Scholar]; e Roberts S. W.; Rainier J. D. Substitution and Remote Protecting Group Influence on the Oxidation/Addition of α-Substituted 1,2-Anhydroglycosides: A Novel Entry into C-Ketosides. Org. Lett. 2005, 7, 1141–1144. 10.1021/ol0501469. [DOI] [PubMed] [Google Scholar]
- León E. I.; Martín Á.; Pérez-Martín I.; Quintanal L. M.; Suárez E. C-C Bond Formation by Sequential Intramolecular Hydrogen Atom Transfer/Intermolecular Radical Allylation Reaction in Carbohydrate Systems. Eur. J. Org. Chem. 2012, 2012, 3818–3829. 10.1002/ejoc.201200300. [DOI] [Google Scholar]
- a Takahashi H.; Shida T.; Hitomi Y.; Iwai Y.; Miyama N.; Nishiyama K.; Sawada D.; Ikegami S. Divergent Synthesis of L-Sugars and L-Iminosugars from D-Sugars. Chem. - Eur. J. 2006, 12, 5868–5877. 10.1002/chem.200600268. [DOI] [PubMed] [Google Scholar]; b Hollingsworth R.; Song X. A Facile and General Synthesis of Rare L-Sugar Lactones. Synlett 2007, 2007, 1247–1250. 10.1055/s-2007-977451. [DOI] [Google Scholar]; c Ermolenko L.; Sasaki N. A. Diastereoselective Synthesis of All Eight L-Hexoses from L-Ascorbic Acid. J. Org. Chem. 2006, 71, 693–703. 10.1021/jo0521192. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Shao H.; Lacroix E.; Wu S.-H.; Jennings H. J.; Zou W. Epimerization of 2‘-Carbonylalkyl-C-Glycosides via Enolation, β-Elimination and Intramolecular Cycloaddition. J. Org. Chem. 2003, 68, 8097–8105. 10.1021/jo034446k. [DOI] [PubMed] [Google Scholar]; And references cited therein.
- a Surzur J. M.; Teissier P. Addition Radicalaire de Esters Sur Les Alcools Ethyleniques. C. R. Acad. Sci. Fr. Ser. C 1967, 264, 1981–1984. [Google Scholar]; b Surzur J. M.; Teissier P. Radical Addition to Unsaturated Alcohols. III. Addition on Ethylene Acetate: 1,2-Radical Migration of Acetoxy Group. Bull. Soc. Chim. Fr. 1970, 3060–3070. [Google Scholar]
- Tanner D. D.; Law F. C. P. Free-Radical Acetoxy Group Migration. J. Am. Chem. Soc. 1969, 91, 7535–7537. 10.1021/ja01054a068. [DOI] [Google Scholar]
- a Beckwith A. L. J.; Duggan P. J. The Mechanism of the β-(Acyloxy)alkyl Radical Rearrangement: Substituent and Solvent Effects. J. Am. Chem. Soc. 1996, 118, 12838–12839. 10.1021/ja963153o. [DOI] [Google Scholar]; b Zipse H. [1,2]-Acyloxy Shifts in Radicals. A Computational Investigation of Substituent and Solvent Effects. J. Am. Chem. Soc. 1997, 119, 1087–1093. 10.1021/ja963249i. [DOI] [Google Scholar]; c Zipse H.; Bootz M. 1,2-Migration in β-(Acyloxy)ethyl Radicals Revisited–Concerted or Stepwise?. J. Chem. Soc., Perkin Trans. 2 2001, 2, 1566–1572. 10.1039/b103324g. [DOI] [Google Scholar]; For reviews, see; d Beckwith A. L. J.; Crich D.; Duggan P. J.; Yao Q. Chemistry of β-(Acyloxy)Alkyl and β-(Phosphatoxy)Alkyl Radicals and Related Species: Radical and Radical Ionic Migrations and Fragmentations of Carbon–Oxygen Bonds. Chem. Rev. 1997, 97, 3273–3312. 10.1021/cr950207o. [DOI] [PubMed] [Google Scholar]; e Crich D.Radical Rearrangements of Esters. In Radicals in Organic Synthesis; Renaud P.; Sibi M. P. Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2001; pp. 188–206, 10.1002/9783527618293.ch35. [DOI] [Google Scholar]; f Sasaki K.; Cumpstey I.; Crich D.. Radical Rearrangements: Ester-Substituted Radicals and Hydrogen Atom Migrations. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu C., Studer A. Eds.; John Wiley & Sons, Ltd: Chichester, UK, 2012; pp. 125–146, 10.1002/9781119953678.rad036. [DOI] [Google Scholar]
- a Crich D.; Yao Q. The β-(Phosphonooxy)Alkyl Radical Rearrangement. J. Am. Chem. Soc. 1993, 115, 1165–1166. 10.1021/ja00056a060. [DOI] [Google Scholar]; b Koch A.; Lamberth C.; Wetterich F.; Giese B. Radical Rearrangement of 2-O-(Diphenylphosphoryl)Glycosyl Bromides. A New Synthesis for 2-Deoxy Disaccharides and 2-Deoxy Ribonucleosides. J. Org. Chem. 1993, 58, 1083–1089. 10.1021/jo00057a020. [DOI] [Google Scholar]
- Lacôte E.; Renaud P. Rate Enhancement of the Radical 1,2-Acyloxy Shift (Surzur-Tanner Rearrangement) by Complexation with Lewis Acids. Angew. Chem., Int. Ed. 1998, 37, 2259–2262. . [DOI] [PubMed] [Google Scholar]
- a Giese B.; Gröninger K. S. 1,3,4,6-Tetra-O-Acetyl-2-Deoxy-α-D-Glucopyranose. Org. Synth. 1990, 69, 66–69. 10.15227/orgsyn.069.0066. [DOI] [Google Scholar]; b Giese B.; Gilges S.; Groninger K. S.; Lamberth C.; Witzel T. Synthesis of 2-Deoxy Sugars. Liebigs Ann. der Chem. 1988, 615–617. 10.1002/jlac.198819880625. [DOI] [Google Scholar]
- Gimisis T.; Ialongo G.; Chatgilialoglu C. Generation of C-1′ Radicals through a β-(Acyloxy)Alkyl Rearrangement in Modified Purine and Pyrimidine Nucleosides. Tetrahedron 1998, 54, 573–592. 10.1016/S0040-4020(97)10317-9. [DOI] [Google Scholar]
- a Paul R.; Greenberg M. M. Mechanistic Studies on RNA Strand Scission from a C2′-Radical. J. Org. Chem. 2016, 81, 9199–9205. 10.1021/acs.joc.6b01760. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Taverna Porro M. L.; Greenberg M. M. DNA Double Strand Cleavage via Interstrand Hydrogen Atom Abstraction. J. Am. Chem. Soc. 2013, 135, 16368–16371. 10.1021/ja409513q. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Resendiz M. J. E.; Pottiboyina V.; Sevilla M. D.; Greenberg M. M. Direct Strand Scission in Double Stranded RNA via a C5-Pyrimidine Radical. J. Am. Chem. Soc. 2012, 134, 3917–3924. 10.1021/ja300044e. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Kerwin S. M.DNA Damage Due to Diradical-Generating Cyclizations. In Radical and Radical Ion Reactivity in Nucleic Acid Chemistry; Greenberg M. M., Ed.; John Wiley & Sons, 2009; pp. 389–443. [Google Scholar]; e Giese B. Long-Distance Charge Transport in DNA: The Hopping Mechanism. Acc. Chem. Res. 2000, 33, 631–636. 10.1021/ar990040b. [DOI] [PubMed] [Google Scholar]; f Pogozelski W. K.; Tullius T. D. Oxidative Strand Scission of Nucleic Acids: Routes Initiated by Hydrogen Abstraction from the Sugar Moiety. Chem. Rev. 1998, 98, 1089–1108. 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]; and references cited therein.
- a For a recent review, seeSchauer R.; Kamerling J. P. Exploration of the Sialic Acid World. Adv. Carbohydr. Chem. Biochem. 2018, 75, 1–213. 10.1016/bs.accb.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; For chemical synthesis of Kdo and its derivatives, see; b Feng Y.; Dong J.; Xu F.; Liu A.; Wang L.; Zhang Q.; Chai Y. Efficient Large Scale Syntheses of 3-Deoxy-D-Manno-2-Octulosonic Acid (Kdo) and Its Derivatives. Org. Lett. 2015, 17, 2388–2391. 10.1021/acs.orglett.5b00901. [DOI] [PubMed] [Google Scholar]; and references cited therein. For chemical synthesis of sialic acids, see; c Ishizawa K.; Majima S.; Wei X. F.; Mitsunuma H.; Shimizu Y.; Kanai M. Copper(I)-Catalyzed Stereodivergent Propargylation of N-Acetyl Mannosamine for Protecting Group Minimal Synthesis of C3-Substituted Sialic Acids. J. Org. Chem. 2019, 84, 10615–10628. 10.1021/acs.joc.9b00887. [DOI] [PubMed] [Google Scholar]; and references cited therein.
- a Sun Z.; Winschel G. A.; Borovika A.; Nagorny P. Chiral Phosphoric Acid-Catalyzed Enantioselective and Diastereoselective Spiroketalizations. J. Am. Chem. Soc. 2012, 134, 8074–8077. 10.1021/ja302704m. [DOI] [PubMed] [Google Scholar]; b Audrain H.; Thorhauge J.; Hazell R. G.; Jørgensen K. A. A Novel Catalytic and Highly Enantioselective Approach for the Synthesis of Optically Active Carbohydrate Derivatives. J. Org. Chem. 2000, 65, 4487–4497. 10.1021/jo9918596. [DOI] [PubMed] [Google Scholar]; c Izquierdo Cubero I.; Plaza López-Espinosa M. T.; Richardson A. C.; Aamlid K. H. Enantiospecific Synthesis of (R)-1,6-Dioxaspiro[4.5]Decane from a Derivative of D-Fructose. Carbohydr. Res. 1993, 242, 281–286. 10.1016/0008-6215(93)80042-D. [DOI] [Google Scholar]
- a Elshahawi S. I.; Shaaban K. A.; Kharel M. K.; Thorson J. S. A Comprehensive Review of Glycosylated Bacterial Natural Products. Chem. Soc. Rev. 2015, 44, 7591–7697. 10.1039/C4CS00426D. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kirschning A.; Jesberger M.; Schöning K. U. Concepts for the Total Synthesis of Deoxy Sugars. Synthesis 2001, 2001, 507–540. 10.1055/s-2001-12342. [DOI] [Google Scholar]; c Hanessian S. Deoxy Sugars. Adv. Carbohydr. Chem. 1967, 21, 143–207. 10.1016/S0096-5332(08)60317-3. [DOI] [PubMed] [Google Scholar]
- For early reports on the use of n-Bu3SnH/AIBN for the generation of alkoxyl radicals from N-alkoxyphthalimides, see:; a Kim S.; Lee T. A.; Song Y. Facile Generation of Alkoxy Radicals from N-Alkoxyphthalimides. Synlett 1998, 1998, 471–472. 10.1055/s-1998-1711. [DOI] [Google Scholar]; b Okada K.; Okamoto K.; Oda M. A New and Practical Method of Decarboxylation: Photosensitized Decarboxylation of N-Acyloxyphthalimides via Electron-Transfer Mechanism. J. Am. Chem. Soc. 1988, 110, 8736–8738. 10.1021/ja00234a047. [DOI] [Google Scholar]; c Martín A.; Rodríguez M. S.; Suárez E. Synthesis of Alditols by Reductive Radical Fragmentation of N-Phthalimido Glycosides. Preparation of Chiral Synthetic Intermediates. Tetrahedron Lett. 1999, 40, 7525–7528. 10.1016/S0040-4039(99)01482-3. [DOI] [Google Scholar]
- a Jia K.; Chen Y. Visible-Light-Induced Alkoxyl Radical Generation for Inert Chemical Bond Cleavage/Functionalization. Chem. Commun. 2018, 54, 6105–6112. 10.1039/C8CC02642D. [DOI] [PubMed] [Google Scholar]; b Hu X.-Q.; Chen J.-R.; Xiao W.-J. Controllable Remote C–H Bond Functionalization by Visible-Light Photocatalysis. Angew. Chem., Int. Ed. 2017, 56, 1960–1962. 10.1002/anie.201611463. [DOI] [PubMed] [Google Scholar]; c Studer A.; Curran D. P. Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem., Int. Ed. 2016, 55, 58–102. 10.1002/anie.201505090. [DOI] [PubMed] [Google Scholar]; d Huang W.; Cheng X. Hantzsch Esters as Multifunctional Reagents in Visible-Light Photoredox Catalysis. Synlett 2017, 28, 148–158. 10.1055/s-0036-1588129. [DOI] [Google Scholar]; e Shaw M. H.; Twilton J.; MacMillan D. W. C. Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016, 81, 6898–6926. 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dorigo A. E.; McCarrick M. A.; Loncharich R. J.; Houk K. N. Transition Structures for Hydrogen Atom Transfers to Oxygen. Comparisons of Intermolecular and Intramolecular Processes, and Open- and Closed-Shell Systems. J. Am. Chem. Soc. 1990, 112, 7508–7514. 10.1021/ja00177a009. [DOI] [Google Scholar]; b Dorigo A. E.; Houk K. N. The Relationship between Proximity and Reactivity. An Ab Initio Study of the Flexibility of the OH• + CH4 Hydrogen Abstraction Transition State and a Force-Field Model for the Transition States of Intramolecular Hydrogen Abstractions. J. Org. Chem. 1988, 53, 1650–1664. 10.1021/jo00243a011. [DOI] [Google Scholar]; c Dorigo A. E.; Houk K. N. Transition Structures for Intramolecular Hydrogen-Atom Transfers: The Energetic Advantage of Seven-Membered over Six-Membered Transition Structures. J. Am. Chem. Soc. 1987, 109, 2195–2197. 10.1021/ja00241a056. [DOI] [Google Scholar]
- DAISY program simulator as implemented in TOPSPIN, version 4.0.6, for Bruker.
- Coupling constants were calculated from a generalization of the Karplus equation established by Haasnoot as implemented in Maestro version 9.0, Schrödinger, LLC, New York, NY, 2009.Haasnoot C. A. G.; de Leeuw F. A. A. M.; Altona C. The Relationship between Proton-Proton NMR Coupling Constants and Substituent Electronegativities-I: An Empirical Generalization of the Karplus Equation. Tetrahedron 1980, 36, 2783–2792. 10.1016/0040-4020(80)80155-4. [DOI] [Google Scholar]
- Crich D.; Huang X.; Newcomb M. Inter- and Intramolecular Pathways for the Formation of Tetrahydrofurans from β-(Phosphatoxy)Alkyl Radicals. Evidence for a Dissociative Mechanism. J. Org. Chem. 2000, 65, 523–529. 10.1021/jo991570o. [DOI] [PubMed] [Google Scholar]
- Cortezano-Arellano O.; Quintero L.; Sartillo-Piscil F. Total Synthesis of Cephalosporolide E via a Tandem Radical/Polar Crossover Reaction. The Use of the Radical Cations under Nonoxidative Conditions in Total Synthesis. J. Org. Chem. 2015, 80, 2601–2608. 10.1021/jo502757c. [DOI] [PubMed] [Google Scholar]
- Martín A.; Pérez-Martín I.; Quintanal L. M.; Suárez E. Intramolecular 1,8-Hydrogen Atom Transfer. Stereoselectivity of the Hexopyranos-5′-yl Radical Reactions in Hexp-(1→4)-Hexp Disaccharide Systems. J. Org. Chem 2008, 73, 7710–7720. 10.1021/jo801499d. [DOI] [PubMed] [Google Scholar]
- Crich D.; Suk D.-H. The β-(Acyloxy)alkyl Radical Rearrangement Revisited. Can. J. Chem. 2004, 82, 75–79. 10.1139/v03-148. [DOI] [Google Scholar]
- For previous studies on migratory differences between axial and equatorial diphenoxyphosphatoxy groups in isolated S-T rearrangement, see; a Koch A.; Giese B. Radical Rearrangements of 2-O-(Diphenoxyphosphoryl)Glycosyl Bromides. Helv. Chim. Acta 1993, 76, 1687–1701. 10.1002/hlca.19930760426. [DOI] [Google Scholar]; b Crich D.; Suk D.-H.; Sun S. Highly Diastereoselective Radical Cyclization of a Glucose-Derived Enol Ether Radical Cation/Phosphate Anion Pair. Tetrahedron: Asymmetry 2003, 14, 2861–2864. 10.1016/S0957-4166(03)00538-X. [DOI] [Google Scholar]
- For comparison between the displacement of acetoxy and diphenoxyphosphatoxy groups in the S-T rearrangement, see Ref (21d).
- a Chatgilialoglu C.; Lalevée J. Recent Applications of the (TMS)3SiH Radical-Based Reagent. Molecules 2012, 17, 527–555. 10.3390/molecules17010527. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chatgilialoglu C.; Ferreri C.; Landais Y.; Timokhin V. I. Thirty Years of (TMS)3SiH: A Milestone in Radical-Based Synthetic Chemistry. Chem. Rev. 2018, 118, 6516–6572. 10.1021/acs.chemrev.8b00109. [DOI] [PubMed] [Google Scholar]; c Ballestri M.; Chatgilialoglu C.; Clark K. B.; Griller D.; Giese B.; Kopping B. Tris(Trimethylsilyl)Silane as a Radical-Based Reducing Agent in Synthesis. J. Org. Chem. 1991, 56, 678–683. 10.1021/jo00002a035. [DOI] [Google Scholar]
- a Deslongchamps P.; Rowan D. D.; Pothier N.; Sauvé G.; Saunders J. K. 1,7-Dioxaspiro[5.5]Undecanes. An Excellent System for the Study of Stereoelectronic Effects (Anomeric and Exo-Anomeric Effects) in Acetals. Can. J. Chem. 1981, 59, 1105–1121. 10.1139/v81-164. [DOI] [Google Scholar]; b Deslongchamps P.Stereoelectronic Effects in Organic Chemistry; Pergamon Press: New York, 1983. [Google Scholar]
- a Beckwith A. L. J.; Easton C. J. Stereoelectronic Effects in Hydrogen Atom Abstraction from Substituted 1,3-Dioxanes. J. Am. Chem. Soc. 1981, 103, 615–619. 10.1021/ja00393a019. [DOI] [Google Scholar]; b Hayday K.; McKelvey R. D. An Anomeric Effect in Photochemical Hydrogen Abstraction Reactions of Tetrahydropyranyl Ethers. J. Org. Chem. 1976, 41, 2222–2223. 10.1021/jo00874a037. [DOI] [Google Scholar]
- Chatgilialoglu C.; Studer A.. Encyclopedia of Radicals in Chemistry, Biology and Materials; John Wiley & Sons, Ltd: Chichester, UK, 2012; pp. 655–692, 10.1002/9781119953678.rad019. [DOI] [Google Scholar]
- a Although, examples of pure radical β-(ester)alkyl fragmentation with concomitant decarboxylation are scarce, we have used it as a simple mechanism to explain the partial loss of deuterium found in some of the 1,5-HAT–Surzur-Tanner rearrangement products. Alternative mechanisms like adventitious acid-catalyzed opening and recombination of the spiroketal ring or the use of n-Bu3SnH(D) as a potential external nucleophile for the trapping of the alkene radical-cation intermediate can also be considered. For references of pure radical β-(ester)alkyl fragmentation, see:Motherwell W. B.; Imboden C.. Decarboxylation via O-Acyl Thiohydroxamates. In Radicals in Organic Synthesis; Renaud P.; Sibi M. P., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2001; pp. 109–134, 10.1002/9783527618293.ch7. [DOI] [Google Scholar]; b Barton D. H. R.; Dowlatshahi H. A.; Motherwell W. B.; Villemin D. A New Radical Decarboxylation Reaction for the Conversion of Carboxylic-Acids into Hydrocarbons. J. Chem. Soc., Chem. Commun. 1980, 732–733. 10.1039/c39800000732. [DOI] [Google Scholar]; c Crich D.; Mo X.-S. Free-Radical Chemistry of Lactones: Fragmentation of β-Lactones. The Beneficial Effect of Catalytic Benzeneselenol on Chain Propagation. J. Org. Chem. 1997, 62, 8624–8625. 10.1021/jo971712j. [DOI] [Google Scholar]; d Crich D.; Mo X.-S. Free Radical Chemistry of β-Lactones. Arrhenius Parameters for the Decarboxylative Cleavage and Ring Expansion of 2-Oxetanon-4-ylcarbinyl Radicals. Facilitation of Chain Propagation by Catalytic Benzeneselenol. J. Am. Chem. Soc. 1998, 120, 8298–8304. 10.1021/ja980447w. [DOI] [Google Scholar]; See also Ref (21e).
- Bruyère I.; Tóth Z.; Benyahia H.; Xue J. L.; Praly J.-P. NaBH3CN and other Systems as Substitutes of Tin and Silicon Hydrides in the Light or Heat-initiated Reduction of Halosugars: a Tunable Access to either 2-Deoxy Sugars or 1,5-Anhydro-itols. Tetrahedron 2013, 69, 9656–9662. 10.1016/j.tet.2013.09.032. [DOI] [Google Scholar]
- For a pioneer report, see:Zhang J.; Li Y.; Zhang F.; Hu C.; Chen Y. Generation of Alkoxyl Radicals by Photoredox Catalysis Enables Selective C(Sp 3)–H Functionalization under Mild Reaction Conditions. Angew. Chem., Int. Ed. 2016, 55, 1872–1875. 10.1002/anie.201510014. [DOI] [PubMed] [Google Scholar]
- Abraham R. J.; Gottschalck H.; Paulsen H.; Thomas W. A. The Proton Magnetic Resonance Spectra and Conformations of Cyclic Compounds. Part II. The p.m.r. Spectra of the Conduritols. J. Chem. Soc. 1965, 6268–6277. 10.1039/jr9650006268. [DOI] [Google Scholar]
- For the synthesis of an analogous trans-fused bis(pyran) compound from a sugar, see:Oguri H. Designed Hapten Aimed at Anti-Ciguatoxin Monoclonal Antibody: Synthesis, Immunization and Discrimination of the C2 Configuration. Synthesis 1999, 1999, 1431–1436. 10.1055/s-1999-3646. [DOI] [Google Scholar]
- a Guyenne S.; León E. I.; Martín A.; Pérez-Martín I.; Suárez E. Intramolecular 1,8-Hydrogen Atom Transfer Reactions in Disaccharide Systems Containing Furanose Units. J. Org. Chem. 2012, 77, 7371–7391. 10.1021/jo301153u. [DOI] [PubMed] [Google Scholar]; b León E. I.; Martín A.; Peréz-Martín I.; Quintanal L. M.; Suárez E. Hydrogen Atom Transfer Experiments Provide Chemical Evidence for the Conformational Differences between C- and O-Disaccharides. Eur. J. Org. Chem. 2010, 2010, 5248–5262. 10.1002/ejoc.201000470. [DOI] [Google Scholar]; c Boto A.; Hernández D.; Hernández R.; Suárez E. β-Fragmentation of Primary Alkoxyl Radicals versus Hydrogen Abstraction: Synthesis of Polyols and α,ω-Differently Substituted Cyclic Ethers from Carbohydrates. J. Org. Chem. 2003, 68, 5310–5319. 10.1021/jo034442f. [DOI] [PubMed] [Google Scholar]
- a Lee I.-C.; Zulueta M. M. L.; Shie C.-R.; Arco S. D.; Hung S.-C. Deuterium-Isotope Study on the Reductive Ring Opening of Benzylidene Acetals. Org. Biomol. Chem. 2011, 9, 7655–7658. 10.1039/c1ob06056b. [DOI] [PubMed] [Google Scholar]; b Johnsson R.; Ohlin M.; Ellervik U. Reductive Openings of Benzylidene Acetals Revisited: A Mechanistic Scheme for Regio- and Stereoselectivity. J. Org. Chem. 2010, 75, 8003–8011. 10.1021/jo101184d. [DOI] [PubMed] [Google Scholar]; c Shie C.-R.; Tzeng Z.-H.; Kulkarni S. S.; Uang B.-J.; Hsu C.-Y.; Hung S.-C. Cu(OTf)2 as an Efficient and Dual-Purpose Catalyst in the Regioselective Reductive Ring Opening of Benzylidene Acetals. Angew. Chem., Int. Ed. 2005, 44, 1665–1668. 10.1002/anie.200462172. [DOI] [PubMed] [Google Scholar]
- Barnett D. W.; Panigot M. J.; Curley R. W. Stereoselective Route to 15N-Labeled-β-Deuterated Amino Acids: Synthesis of (2S,3R)-[3-2H,15N]-Phenylalanine. Tetrahedron: Asymmetry 2002, 13, 1893–1900. 10.1016/S0957-4166(02)00487-1. [DOI] [Google Scholar]
- For references on the β-fragmentation of primary alkoxyl radicals in carbohydrates, see; a Sánchez-Eleuterio A.; Quintero L.; Sartillo-Piscil F. High 1,3-trans Stereoselectivity in Nucleophilic Substitution at the Anomeric Position and β-Fragmentation of the Primary Alkoxyl Radical in 3-Amino-3-deoxy-ribofuranose Derivatives: Application to the Synthesis of 2-epi-(−)-Jaspine B. J. Org. Chem. 2011, 76, 5466–5471. 10.1021/jo200639t. [DOI] [PubMed] [Google Scholar]; b Hernández-García L.; Quintero L.; Sánchez M.; Sartillo-Piscil F. Beneficial Effect of Internal Hydrogen Bonding Interactions on the β-Fragmentation of Primary Alkoxyl Radicals. Two-Step Conversion of d-Xylo- and d-Ribofuranoses into l-Threose and d-Erythrose, Respectively. J. Org. Chem. 2007, 72, 8196–8201. 10.1021/jo0709551. [DOI] [PubMed] [Google Scholar]
- Cipolla L.; Lay L.; Nicotra F. New and Easy Access to C-Glycosides of Glucosamine and Mannosamine. J. Org. Chem. 1997, 62, 6678–6681. 10.1021/jo970127f. [DOI] [Google Scholar]
- a Mitsunobu O. The Use of Diethyl Azodicarboxylate and Triphenylphosphine in Synthesis and Transformation of Natural Products. Synthesis 1981, 1981, 1–28. 10.1055/s-1981-29317. [DOI] [Google Scholar]; b Grochowski E.; Jurczak J. A New Synthesis of O-Alkylhydroxylamines. Synthesis 1976, 1976, 682–684. 10.1055/s-1976-24157. [DOI] [Google Scholar]
- a Bennek J. A.; Gray G. R. An Efficient Synthesis of Anhydroalditols and Allylic-Glycosides. J. Org. Chem. 1987, 52, 892–897. 10.1021/jo00381a030. [DOI] [Google Scholar]; b Kozikowski A. P.; Sorgi K. L.; Wang B. C.; Xu Z. An Improved Method for the Synthesis of Anomerically Allylated C-Glycopyranosides and C-Glycofuranosides. Tetrahedron Lett. 1983, 24, 1563–1566. 10.1016/S0040-4039(00)81710-4. [DOI] [Google Scholar]
- Li X.; Li J.; Mootoo D. R. Synthesis of the ABCD Trioxadispiroketal Subunit of Azaspiracid-1: An Iodoetherification–Dehydroiodination Strategy for Complex Spiroketals. Org. Lett. 2007, 9, 4303–4306. 10.1021/ol701866v. [DOI] [PubMed] [Google Scholar]
- a Jarikote D. V.; O’Reilly C.; Murphy P. V. Ultrasound-Assisted Synthesis of C-Glycosides. Tetrahedron Lett. 2010, 51, 6776–6778. 10.1016/j.tetlet.2010.10.113. [DOI] [Google Scholar]; b Zhu Y.-H.; Vogel P. Synthesis of a C-Disaccharide Analog of the Thomsen-Friedenreich (T) Epitope. Synlett 2001, 2001, 79–81. 10.1055/s-2001-9698. [DOI] [Google Scholar]; c Schäfer A.; Thiem J. Synthesis of Novel Donor Mimetics of UDP-Gal, UDP-GlcNAc, and UDP-GalNAc as Potential Transferase Inhibitors. J. Org. Chem. 2000, 65, 24–29. 10.1021/jo990766l. [DOI] [PubMed] [Google Scholar]
- a Grice P.; Ley S. V.; Pietruszka J.; Priepke H. W. M.; Warriner S. L. Preparation, Structure, Derivatisation and NMR Data of Cyclohexane-1,2-Diacetal Protected Carbohydrates. J. Chem. Soc., Perkin Trans. 1 1997, 351–364. 10.1039/a605851e. [DOI] [Google Scholar]; b Hense A.; Ley S. V.; Osborn H. M.; Owen D. R.; Poisson J. F.; Warriner S. L.; Wesson K. E. Direct Preparation of Diacetals from 1,2-Diketones and Their Use as 1,2-Diol Protecting Groups. J. Chem. Soc. Perkin Trans. 1 1997, 1, 2023–2032. 10.1039/a702497e. [DOI] [Google Scholar]; c Ley S. V.; Boons G.-J.; Leslie R.; Woods M.; Hollinshead D. M. Dispiroketals in Synthesis (Part 3): 1 Selective Protection of Diequatorial Vicinal Diols in Carbohydrates. Synthesis 1993, 1993, 689–692. 10.1055/s-1993-25923. [DOI] [Google Scholar]
- a Pirrung M. C.The Synthetic Organic Chemist’s Companion; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007, pp. 171, 10.1002/9780470141045. [DOI] [Google Scholar]; b Long D. D.; Smith M. D.; Martín A.; Wheatley J. R.; Watkin D. G.; Müller M.; Fleet G. W. J. Complex Tetrahydrofurans from Carbohydrate Lactones: THF Amino Acids as Building Blocks for Unnatural Biopolymers. J. Chem. Soc., Perkin Trans. 1 2002, 1982–1998. 10.1039/B111258A. [DOI] [Google Scholar]
- La Ferla B.; Russo L.; Airoldi C.; Nicotra F. Solid-Phase Supported Mimic of GDP-L-Galactose. Tetrahedron: Asymmetry 2009, 20, 744–745. 10.1016/j.tetasy.2009.03.005. [DOI] [Google Scholar]
- Allavudeen S. S.; Kuberan B.; Loganathan D. A Method for Obtaining Equilibrium Tautomeric Mixtures of Reducing Sugars via Glycosylamines Using Nonaqueous Media. Carbohydr. Res. 2002, 337, 965–968. 10.1016/S0008-6215(02)00075-7. [DOI] [PubMed] [Google Scholar]
- Beignet J.; Tiernan J.; Woo C. H.; Kariuki B. M.; Cox L. R. Stereoselective Synthesis of Allyl-C-Mannosyl Compounds: Use of a Temporary Silicon Connection in Intramolecular Allylation Strategies with Allylsilanes. J. Org. Chem. 2004, 69, 6341–6356. 10.1021/jo049061w. [DOI] [PubMed] [Google Scholar]
- Martín A.; Pérez-Martín I.; Quintanal L. M.; Suárez E. Intramolecular 1,8- versus 1,6-Hydrogen Atom Transfer between Pyranose Units in a (1→4)-Disaccharide Model Promoted by Alkoxyl Radicals. Conformational and Stereochemical Requirements. Org. Lett. 2007, 9, 1785–1788. 10.1021/ol070496q. [DOI] [PubMed] [Google Scholar]
- a Kobertz W. R.; Bertozzi C. R.; Bednarski M. D. An Efficient Method for the Synthesis of α- and β-C-Glycosyl Aldehydes. Tetrahedron Lett. 1992, 33, 737–740. 10.1016/S0040-4039(00)77703-3. [DOI] [Google Scholar]; b Brueckner C.; Holzinger H.; Reissig H. U. Diastereoselective Syntheses of Highly Substituted Methyl Tetrahydrofuran-3-Carboxylates by Reaction of γ-Lactols with Silylated Nucleophiles. J. Org. Chem. 1988, 53, 2450–2456. 10.1021/jo00246a011. [DOI] [Google Scholar]; c Babirad S. A.; Wang Y.; Kishi Y. Synthesis of C-Disaccharides. J. Org. Chem. 1987, 52, 1370–1372. 10.1021/jo00383a045. [DOI] [Google Scholar]; d Pornet J.; Miginiac L.; Jaworski K.; Randrianoelina B. Regiospecific Allenylation of Acetals with Propargyltrimethylsilanes Catalyzed by a Lewis Acid: Synthesis of α-Allenyl Ethers and 3-Silylated 3,4-Dihydrofurans. Organometallics 1985, 4, 333–338. 10.1021/om00121a024. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.