

Summary
Cancer-associated fibroblasts (CAFs) are highly heterogeneous. With the lack of a comprehensive understanding on CAFs’ functional distinctions, it remains unclear how cancer treatments could be personalized based on CAFs in a patient’s tumor. We have established a living biobank of CAFs derived from biopsies of patients’ non-small lung cancer (NSCLC) that encompasses a broad molecular spectrum of CAFs in clinical NSCLC. By functionally interrogating CAFs heterogeneity using the same therapeutics received by patients, we identify three functional subtypes: 1) robustly protective of cancers and highly expressing HGF and FGF7, 2) moderately protective of cancers and highly expressing FGF7, and 3) those providing minimal protection. These functional differences among CAFs are governed by their intrinsic TGF-β signaling which suppresses HGF and FGF7 expression. This CAF functional classification correlates with patients’ clinical response to targeted therapies and also associates with the tumor immune microenvironment, therefore provides an avenue to guide personalized treatment.
Graphical Abstract
eTOC Blurb
Hu et al. identify that cancer-associated fibroblasts (CAFs) derived from non-small cell lung cancer (NSCLC) patients are functionally heterogeneous. These functional distinctions directly impact response to clinical anticancer treatment and associate with the tumor immune microenvironment. Thus, CAFs functional heterogeneity defines a unique parameter for designing more personalized treatments.
Introduction
The current personalized cancer treatment paradigm is largely based on molecular features, e.g., oncogenic mutations, of cancer cells (Herbst et al., 2018). However, patients rarely achieve complete remissions and both the depth and the duration of patients’ responses vary widely (Jänne et al., 2015; Sequist et al., 2015). Reasons underlying these differences are not fully understood. It is yet to be determined if functional differences among cancer-associated fibroblasts (CAFs) play a major role in the diverse clinical responses observed and if analyzing CAFs heterogeneity can improve cancer therapy (Junttila and de Sauvage, 2013).
CAFs constitute a substantial part of the tumor microenvironment and are recognized as an important component of the cancer’s ecosystem (Kalluri and Zeisberg, 2006; Sahai et al., 2020). Recent single-cell RNA-sequencing (scRNA-seq) studies of solid tumors, including non-small cell lung cancer (NSCLC), suggest that CAFs are a collection of cells with diverse molecular features (Bartoschek et al., 2018; Elyada et al., 2019; Lambrechts et al., 2018; Li et al., 2017; Qian et al., 2020). Furthermore, a few rare but biologically unique fibroblasts subtypes have been reported in breast and pancreatic cancers (Costa et al., 2018; Dominguez et al., 2020; Öhlund et al., 2017; Su et al., 2018). This diversity in CAFs raises increasing interest in exploiting CAFs to improve personalized cancer treatment. However, to what extent CAFs are functionally distinct and what clinical impacts different CAFs may have remain largely unknown. The lack of a comprehensive understanding of CAFs’ functional heterogeneity has hindered the development of more personalized cancer treatment, with previous studies based on various CAF definitions yielding inconsistent results (Koliaraki et al., 2015; Pallangyo et al., 2015; Wagner, 2016). Similarly, previous attempts to universally target and broadly deplete CAFs rarely improved patient’s outcome (Catenacci et al., 2015; Hofheinz et al., 2003; E. J. Kim et al., 2014; Narra et al., 2014). Thus, it remains unclear whether and how improved treatment strategies could be developed based on CAFs’ heterogeneity. In order to answer these questions, it is crucial to fully understand functional underpinnings of the broad spectrum of CAFs in a specific cancer type and their impacts on the current treatments of that cancer type.
Currently, the broad survey and characterization of the landscape of CAFs’ functions are challenging. Flow cytometry-based studies require predetermined CAF markers for CAFs differentiation, but a growing number of studies show that canonical CAF markers are often insufficient to distinguish CAFs’ functions (Dominguez et al., 2020; Eckert et al., 2019; Su et al., 2018). Although scRNA-seq studies have clearly demonstrated various molecular phenotypes among CAFs, they are less effective in demonstrating CAFs’ different functions. Notably, however, results from scRNA-seq studies challenge how we should investigate CAFs biology, i.e. whether a small collection of CAF models would be sufficient to represent different groups of CAFs in a specific cancer type (Bartoschek et al., 2018; Elyada et al., 2019; Lambrechts et al., 2018; Li et al., 2017; Qian et al., 2020). Furthermore, most current fibroblast models available for cancer research are not obtained from patients in the same clinical context, e.g., fibroblasts derived from fetus tissue or CAFs obtained from tumors with unrelated oncogenic backgrounds or from patients receiving unrelated treatments. Thus, it is often difficult to validate the clinical importance of the findings.
In order to address these critical needs, we have established a biobank of CAFs derived from biopsies of patients’ NSCLC harboring specific oncogenic alterations, such as EGFR mutations and ALK fusions. This large CAFs collection allows us to adequately recapitulate a broad spectrum of NSCLC CAFs with diverse molecular features. Here we have functionally characterized the landscape of NSCLC CAFs, revealed how they function differently, and demonstrated their potential clinical utilities.
Results
Establishment of a PDFs library adequately capturing NSCLC CAFs heterogeneity
To capture the diversity of CAFs in NSCLC for functional studies, we prospectively established a large collection of patient-derived fibroblast (PDF) cultures from biopsies of NSCLC harboring targetable oncogenic drivers such as EGFR mutations or ALK fusions (Figure 1A, S1A-B). We successfully derived PDF cultures from about 80% of biopsies (Figure S1C, Methods). Notably, it has been shown that fibroblasts’ phenotype can be affected by factors such as aging and site-of-origin (Chang et al., 2002; LeBleu and Kalluri, 2018; Mahmoudi et al., 2019; Pereira et al., 2019). The high success rate in culture establishment enabled us to encompass a large variety of unique demographic features (Figure 1B) in our PDF collection (n=60, Table S1). After lineage confirmation of the cultures (Figure S2A-B, Methods), we immortalized the early PDF cultures with hTERT as previously described (Morales et al., 1999) (Figure S2C) to allow faithful long-term culture and prevent senescence-associated secretory phenotype emergence (Coppe et al., 2010). Immortalized PDFs maintained good fidelity with their parental lines as demonstrated by the expression of established CAF markers (Figure S2D) and were used for subsequent studies.
Figure 1: Establishment of a living biobank adequately capturing NSCLC CAFs heterogeneity.
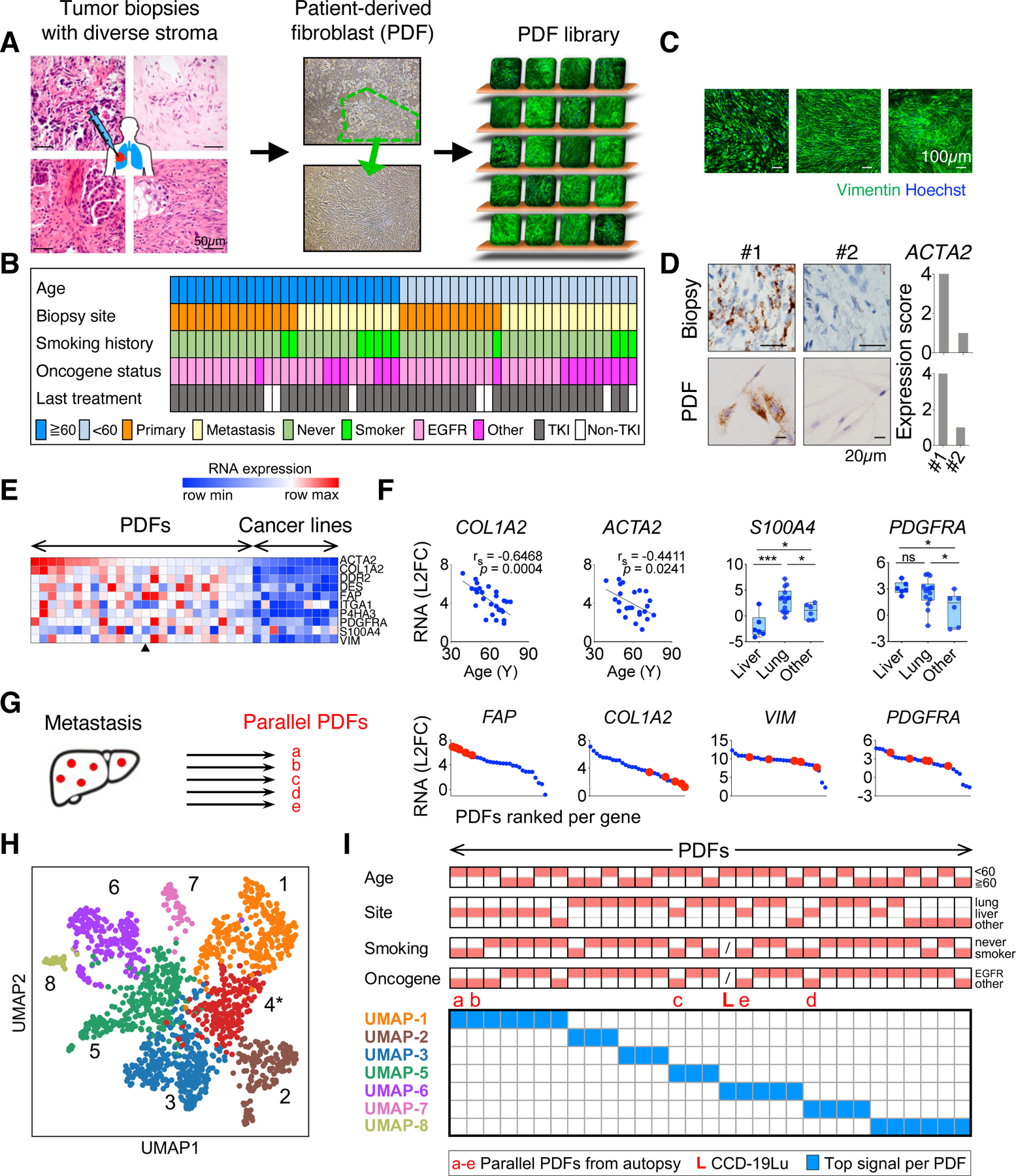
A. Workflow of patient-derived fibroblast (PDF) development. PDF library is symbolized with different PDFs (staining of Vimentin) on the shelf. B. Clinical features of patients whose tumors were used for developing PDFs. C. Images from immunofluorescence staining of Vimentin and Hoechst of representative PDFs. D. Images and quantification of αSMA (encoded by ACTA2) mRNA in two EGFR+ lung cancer samples and their corresponding PDFs detected by using RNAscope. E. mRNA levels of canonical CAF markers in PDFs and in lung cancer cell lines measured by qRT-PCR. The arrowhead indicates the average expression level of the five PDFs in (G). F. Correlations between the mRNA level of COL1A2 or ACTA2 and patients’ age at the time of biopsy across PDFs (left two graphs) and the mRNA level of S100A4 or PDGFRA according to the site of tumor biopsy. * p < 0.05, *** p < 0.001, Spearman’s r and two-tailed t-test are used. G. Expression of indicated CAF markers (red) in PDFs established from liver metastases in an autopsy case. H. Uniform Manifold Approximation and Projection (UMAP) analysis of 1,465 single fibroblasts in NSCLC (from Lambrechts et al., 2018) showing seven molecular classes, excluding UMAP-4 (*) due to poor quality cells. I. PDFs are mapped based on their top UMAP signal. Red blocks on the top indicate clinical features of the corresponding PDFs. See also Figures S1-S3 and Table S1.
We first examined the degree of phenotypic heterogeneity in NSCLC CAFs. Similar to variable pathohistological presentations of stroma across different tumors (Figure 1A), PDFs exhibited considerable variability with respect to their morphology and growth patterns (Figure 1C). Molecularly, CAFs’ expression of the myofibroblast marker αSMA (encoded by ACTA2) (Kalluri and Zeisberg, 2006) was found to be divergent across different biopsies, and this diversity was conserved in the corresponding PDF cultures as tested by RNAscope (Figure 1D, S2E). Similar results were observed for another CAF marker S100A4 (Kalluri and Zeisberg, 2006) (Figure S2E). Thus, we do not observe a systematic change in previously defined fibroblast activation markers between a PDF culture and the biopsy from which it was derived from. To gain more insight into the variety and fidelity of these models, we characterized the expression of 10 CAF markers among these PDF models (Figure 1E). While transcriptional regulatory networks dictating fibroblasts identity are not well understood (De Jaeghere et al., 2019), previous studies suggested that fibroblast state can be affected by clinical factors such as patients’ age or site-of-origin of tumors (Chang et al., 2002; LeBleu and Kalluri, 2018; Mahmoudi et al., 2019; Pereira et al., 2019). Intriguingly, the expression of collagen type 1 alpha 2 (COL1A2, a subunit of type I collagen heterotrimers) and of αSMA strongly correlated with patients’ age, reminiscent of the normal aging process (Figure 1F), whereas the expression of PDGFRA and of S100A4 correlated with the site of tumor biopsy (Figure 1F, S2F), consistent with previous findings (Lawson et al., 2005; Österreicher et al., 2011). We did not observe a systematic variation in these markers between EGFR and non-EGFR (mostly ALK) tumor CAFs (Figure S2F), similar to the lung CAFs in previous datasets (Figure S2G) (N. Kim et al., 2020; Laughney et al., 2020; Maynard et al., 2020).
A growing number of studies have indicated that canonical fibroblast markers are insufficient to dissect fibroblasts’ functional differences (Dominguez et al., 2020; Eckert et al., 2019; Su et al., 2018). Our findings support this observation. For example, although PDFs derived from five independent liver metastases of an autopsy case show consistent expression of some CAF markers (e.g., FAP), they also show profound variation in the expression of others (e.g., PDGFRA) (Figure 1G, S2H), suggesting that factors beyond site-of-origin and a patient’s genetics are responsible for these differences. The phenotypic presentation, mechanistic cause, as well as biological impact of such variations of CAFs may not be predictable with the current knowledge. Therefore, the PDF library provides a valuable resource for investigating these key questions to understand functional CAFs heterogeneity.
Although the PDF library adequately captures a wide range of clinical and pathological varieties with a focus on EGFR and ALK NSCLCs (mostly adenocarcinoma in non-smokers), we attempted to compare the phenotypic heterogeneity of PDFs with that of NSCLC CAFs in real-world clinical datasets. We analyzed 1,465 fibroblast cells from a representative scRNA-seq dataset of resected NSCLCs, including CAFs in lung adenocarcinoma, squamous cell carcinoma, and large cell carcinoma (Lambrechts et al., 2018). Uniform Manifold Approximation and Projection (UMAP) revealed seven molecular classes, UMAP 1–8 with UMAP-4 being excluded due to poor quality single cells designation in the dataset (Figure 1H). Intriguingly, UMAP 8 was marked by the expression of type II human leukocyte antigen (HLA) and resembles a recently reported antigen presenting CAFs (apCAFs) (Figure S3A-B) (Chen et al., 2021; Elyada et al., 2019; Xing et al., 2021). We then mapped PDFs to these UMAP classes based on their expression of top expressed genes specific to each UMAP class (Figure S3A-B, Methods). These analyses demonstrated that PDFs differentially expressed unique UMAP marker genes (Figure S3C) and together recapitulated all seven molecular classes of clinical NSCLC CAFs (Figure 1I). Further supporting that PDFs faithfully recapitulate the overall molecular repertoire of CAFs, the signature proximity among PDF UMAP classes mirrored that among CAF single cells (Figure S3D). Together, this PDF library largely captures the molecular heterogeneity of CAFs existed in NSCLC in patients.
NSCLC CAFs heterogeneity determines TKI treatment efficacy
Oncogenic mutations in EGFR and fusions in ALK are present in about 20% and 5%, respectively, of advanced NSCLC and tyrosine kinase inhibitors (TKIs) constitute the backbone of the standard-of-care of patients having these tumors (Herbst et al., 2018). However, patients whose tumors carrying the same genetic alteration do not benefit equally from the same treatment, only a few achieve maximum remission whereas the majority of patients show different degrees of response or even no response (Jänne et al., 2015; Sequist et al., 2015). While mechanisms intrinsic to malignant cells in determining treatment response are important, the role of CAFs is less clear. In order to decipher whether and to what degree CAFs affect response to EGFR TKI treatment, we co-cultured the EGFR mutant (herein EGFR+) cancer cell line MGH707 with various PDFs. In the absence of PDFs, the EGFR TKI osimertinib (EGFRi) potently blocked the cancer cells’ growth (Figure 2A, top). Co-culturing with certain PDFs substantially maintained the proliferation of cancer cells upon EGFRi treatment, while other PDFs provided only modest to no protection (Figure 2A, top). The diversity of PDFs’ impact can be sufficiently phenocopied by the corresponding PDFs’ conditioned media (Figure 2A, bottom), which was confirmed in a cohort of seven PDFs coupled with six cancer models (overall Spearman correlation, rs=0.80, Figure 2B). These results suggest that PDFs play a significant role in determining the overall efficacy of EGFRi treatment. However, this effect is heterogeneous and is PDF-dependent, supporting the diversity in PDFs’ function. These results also indicate that the rescue effect of PDFs is mainly mediated by soluble factors, whereby we leveraged conditioned media for high throughput analysis on PDFs.
Figure 2: CAFs determine the TKI treatment efficacy on NSCLC.
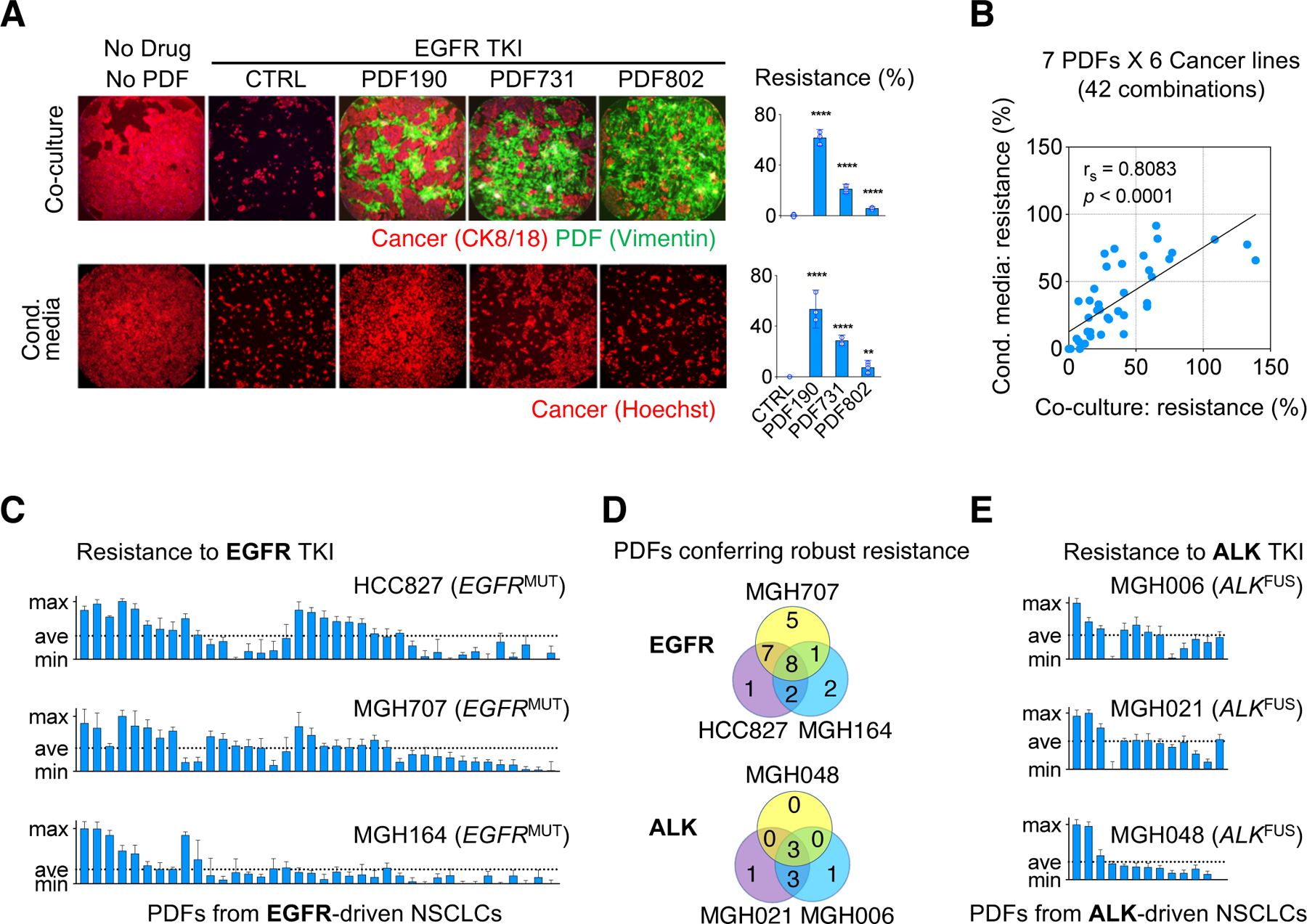
A. Representative images and quantification of rescue % of MGH707 NSCLC cells co-cultured with indicated PDFs (top) or PDF conditioned media (bottom)..B. The viability outcome of cancer cells (n=6) evaluated in the presence of EGFRi and either a PDF co-culture (n=7) or a PDF conditioned media. Rescue obtained in the two settings are plotted against each other. C. Viability rescuing effect against EGFRi across three EGFR+ NSCLC cell lines by conditioned media from PDFs derived from 38 EGFR+ NSCLC. Each bar corresponds to a PDF’s effect tested in four replicates, mean values and 95% CI are plotted. D. Venn diagram showing PDFs conferring robust resistance (above average level per cancer model) in EGFR+ (C) and ALK+ (E) tumor-derived PDFs. E. Viability rescuing effect against ALKi across different ALK+ NSCLC cell lines by conditioned media from PDFs derived from 13 ALK+ NSCLC. Results shown as in (C).
We further surveyed 38 PDFs derived from EGFR+ NSCLC biopsies and assessed their ability to rescue three EGFR+ cancer models treated with EGFRi (Figure 2C). We found that there was a wide range of rescue afforded by these PDFs. Moreover, PDFs that provided strong rescue (rescue above the average level across all tested PDFs) also varied among cancer models. Specifically, 8 PDFs conferred strong rescue to all three cancer cell lines, 18 PDFs rescued only 1 or 2 of cancer cell lines, and 12 PDFs had a negligible effect on all three cancer cell lines (Figure 2C and Figure 2D, top). PDFs from NSCLCs with ALK-fusions (herein ALK+) similarly demonstrated a range of rescue effect to ALK+ NSCLCs cell lines treated with the ALK TKI lorlatinib (ALKi), wherein a few PDFs rescued all ALK+ cancer models and other PDFs rescued only some of ALK+ cancer cell lines (Figure 2D, bottom and Figure 2E). Several cancer-CAF crosstalk machineries have been reported to affect different aspects of cancer biology (Ghesquière et al., 2014; Gieniec et al., 2019; Quail and Joyce, 2013; Sethi and Kang, 2011). Our data here support that cancer cells have differential capacities and preferences to utilize CAF-derived secreted factors. Importantly, the heterogeneity in CAFs function is a key factor determining the specific context and the strength of the cancer-CAF crosstalk that contributes to the overall TKI response.
NSCLC CAFs recurrently rescue EGFR+ cancers via activating MET and/or FGFR
To better characterize the functional landscape of NSCLC CAFs and to unveil the potential cancer-CAF crosstalk related to TKI response, we used two approaches. First, we performed a large-scale secretome profiling of PDF cultures by using a 448-analyte multiplexed ELISA (Figure S4A, Table S2). Second, we compiled functional analysis on PDFs (n=38 EGFR+, n=22 non-EGFR+) with a large panel of patient-derived cancer models (n=12 EGFR+, n=4 non-EGFR+) aiming to recapitulate a myriad of unique cancer-CAF combinations that may exist in patients.
Multiplexed ELISA revealed a number of factors with diverse expression across PDFs (Figure S4A, Table S2). By comparing secreted factors with PDFs’ rescue capacities, we found that top correlates were highly enriched for growth factors, among which the MET ligand HGF ranked the highest (Figure 3A, S4B). Notably, activation of the MET receptor tyrosine kinase by gene amplification is known to mediate EGFRi resistance in the clinic (Engelman et al., 2007; Piotrowska et al., 2018; Sequist et al., 2011b). We confirmed that HGF is heterogeneously expressed by NSCLC PDFs in cell cultures (Figure S4C) as well as in clinical biopsies (Figure S4D). Similar to previous reports (Straussman et al., 2012; Turke et al., 2010; Wang et al., 2009; Wilson et al., 2012), we confirmed that exogeneous HGF could activate cancer cells’ MET and downstream signaling, such as PI3K-AKT and MAPK, despite inhibition of EGFR (Figure S4E). These results indicate that CAFs may protect cancer cells by activating tyrosine kinase receptor-mediated signaling to bypass the requirement for EGFR signaling (Figure S4F). Indeed, HGFhigh PDFs (i.e., PDFs secreting above the medium level of HGF among PDFs assessed by ELISA) conferred higher rescue than HGFlow PDFs (Figure S4G). Interestingly, however, combining the MET inhibitor INC280 (METi, Figure S4E) with EGFRi fully reversed resistance driven by recombinant HGF but only partially reversed the resistance driven by PDFs (Figure 3B, S4H). Notably, the addition of the METi abrogated rescue provided by HGFhigh PDFs more prominently, but a widespread HGF/MET-independent rescue effect remained across PDFs (Figure 3B, Figure S5A-B). Together, these results showed that only some CAFs mediated EGFRi resistance through activating MET, whereas many CAFs could also mediate EGFRi resistance through other soluble factor(s).
Figure 3: NSCLC CAFs recurrently rescue EGFR cancers via bypass signaling.
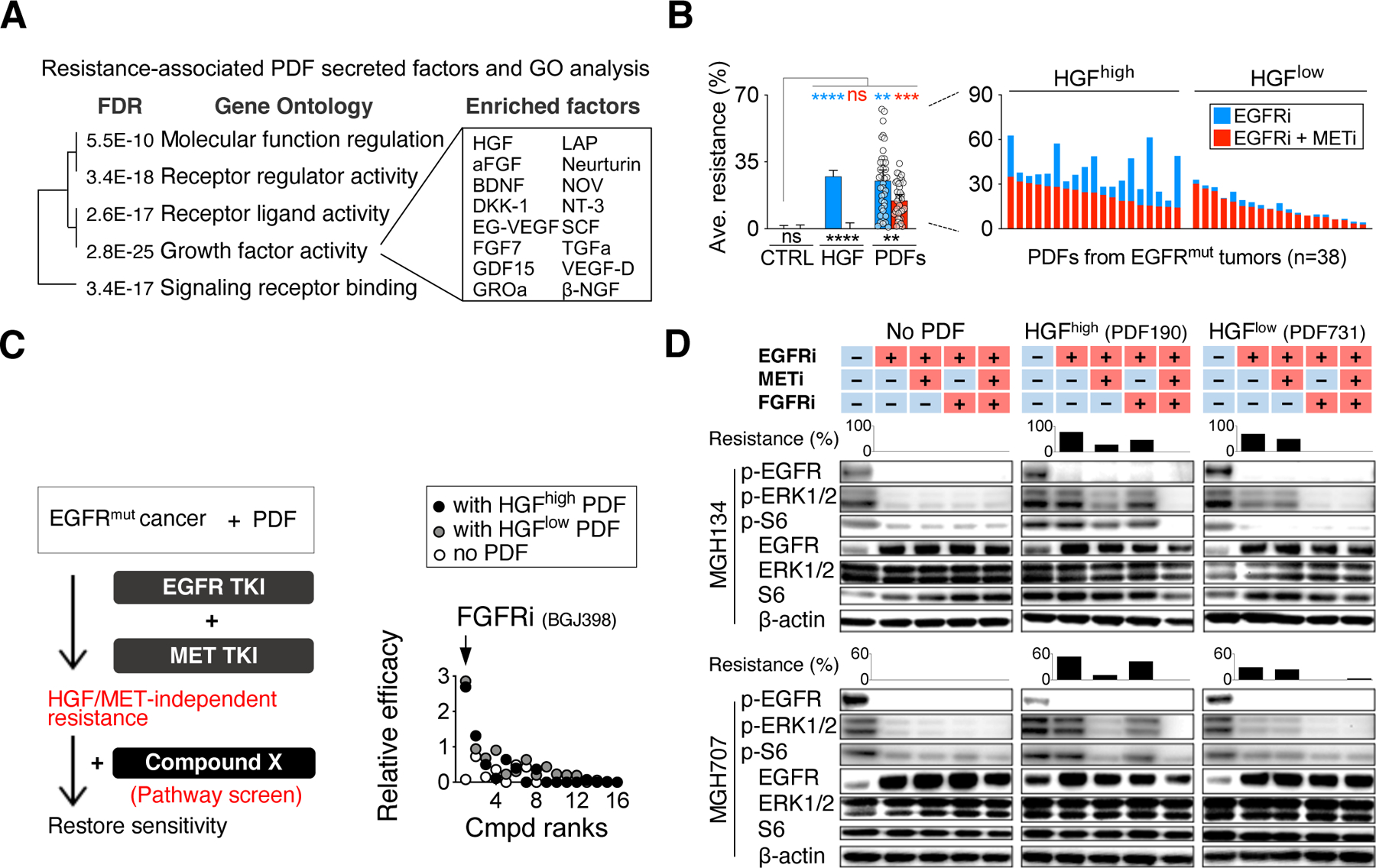
A. PDF-secreted factors profiled by a 448-analyte multiplexed ELISA, and the gene ontology of the top 30 PDF rescue correlates (ranked by Spearman’s r). B. Effects of METi on diminishing rhHGF-driven (10ng/ml) and PDF conditioned media-driven EGFRi resistance. PDFs’ rescue against EGFRi + METi treatment (red bars) is superimposed over PDFs’ rescue against EGFRi (blue bars). Effect of each PDF conditioned medium (dots on the left and bars on the right) is tested across 12 EGFR+ cancer cells. HGFhigh and HGFlow indicate PDF conditioned media with HGF level above and below the median value, respectively. Mean with 95% CI. ns, not significant; **, p < 0.01, ***, p < 0.001, ****, p < 0.0001, two-tailed t test. C. A screening across 16 compounds to identify pathway-specific inhibitors that can negate HGF/MET-independent resistance. Relative efficacy is measured by comparing cancer cells’ response to the indicated compound alone and their response to the compound in the presence of dual EGFR and MET inhibition (IC50 shift). Two cancer models (MGH134 and MGH707, average is shown) are used and are tested both in the absence and presence of conditioned media from two different PDFs (HGFhigh and HGFlow). D. Western blotting in two cancer cell lines showing rescue of ERK and S6 phosphorylation by PDFs and the effect of the addition of FGFRi and METi on cancer cell signaling. Bars correspond to the matched resistance effect in the presence of the indicated inhibitors. See also Figures S4-S5 and Table S2-S3.
To uncover PDF-derived factors other than HGF that protect cells from EGFRi, we interrogated cancer cells in both the absence of presence of PDF-conditioned media in a focused drug combination screen (Figure 3C). Among the 16 compounds targeting key cancer signaling pathways tested (Table S3), the pan-fibroblast growth factor receptors (FGFR) 1–3 inhibitor BGJ398 (FGFRi) was most effective at negating HGF-independent rescue by PDFs (Figure 3C, S5A-D). FGFRi and METi together blocked the reactivated downstream signaling mediated by PDF190 (HGFhigh) upon EGFRi, and FGFRi alone was sufficient to suppress the downstream signaling in EGFRi resistance induced by PDF731 (HGFlow) (Figure 3D). Indeed, we found the same CAF have the potential to promote MET and/or FGFR activation, dependent on the cancer cell; conversely, the same cancer cell may be rescued by either MET or FGFR, dependent on the CAF (Figure S5E). Together, these data revealed that FGFR in addition to MET is an essential bypass signaling pathway activated in cancer cells by CAFs in NSCLC.
To identify if there’s other recurrent cancer-CAF crosstalk contributing to EGFR TKI resistance, we surveyed 38 EGFR+ tumor-derived PDFs and tested them across 12 EGFR+ cancer cell lines. This matrix, comprising 456 unique PDF-cancer combinations, uncovered widespread PDF-driven EGFRi resistance that was only partially overcome by METi for HGFhigh PDFs (Figure 4A). The FGFRi-based combination had an even broader impact across most of the PDFs, and almost fully reversed resistance by HGFlow PDFs (Figure 4A). In a few cancer cell lines, the rescue by HGFhigh PDFs was more impacted by the FGFRi combination than the METi combination, suggesting that some EGFR+ cancers may be more poised to take advantage of FGFR-mediated resistance. The PDFs’ overall rescue is highly consistent with the additive effects of FGFR-mediated plus MET-mediated rescue (Figure S5F); and combining EGFRi with both FGFRi and METi abrogated PDF-mediated resistance in almost all PDF-cancer combinations (Figure 4A). Collectively, these data elucidate that MET and FGFR recurrently and potently mediate CAF-driven EGFRi resistance. CAFs heterogeneously activate one or both of these pathways, whose combined effect determines the CAFs overall rescue levels.
Figure 4: HGF-MET and FGF-FGFR are two mainstream CAF-cancer crosstalk contributing to resistance.
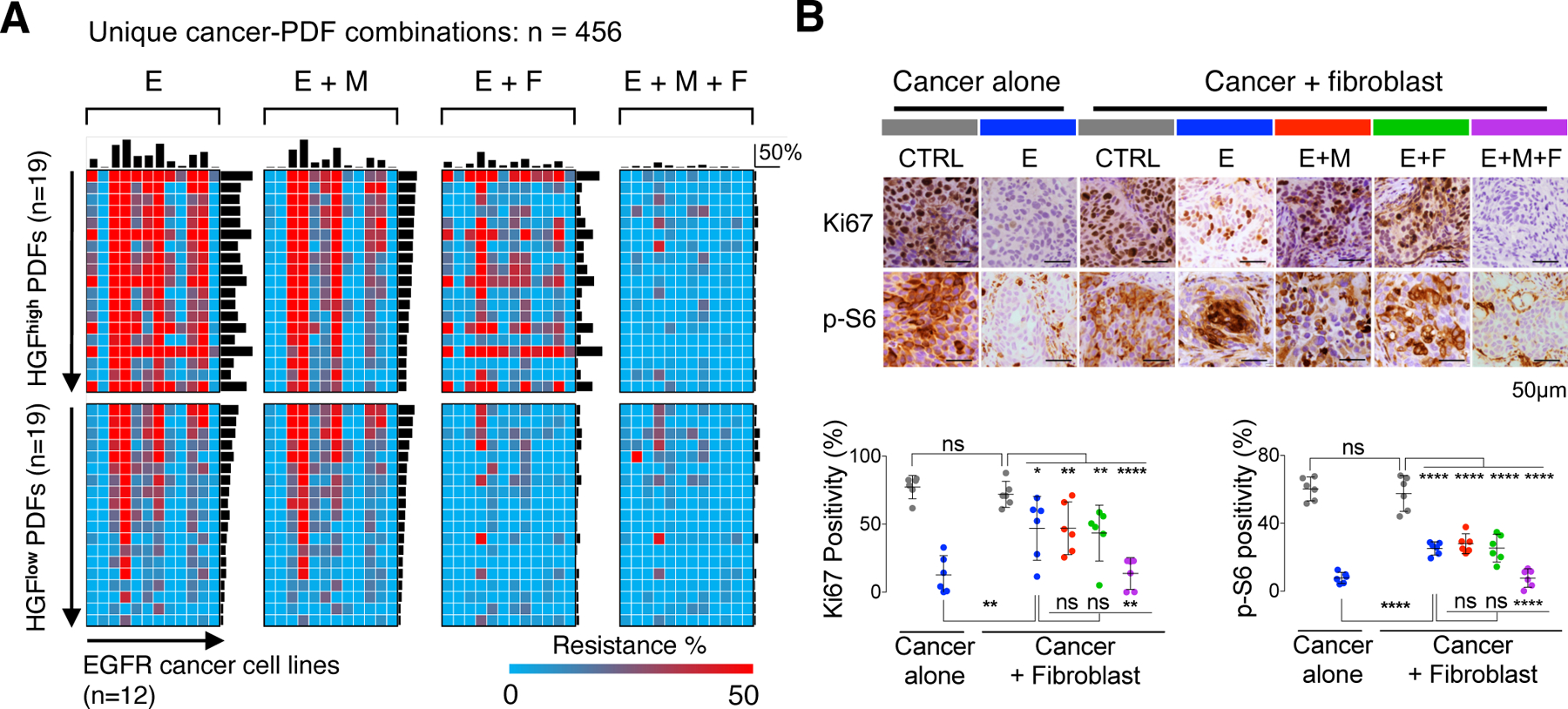
A. A set of 38 PDFs (all derived from EGFR+ NSCLC) conditioned media was tested across 12 EGFR+ cancer cell lines in the presence of EGFRi (E) alone or EGFRi in combination with METi (M), FGFRi (F) or both. Top bars are average resistance level of a given cancer cell line tested across all PDFs, and side bars are average rescue effect of a given PDF tested across all cancer lines. B. Nude mice implanted with MGH707 cancer cells alone or together with CCD19-Lu fibroblasts were treated as indicated 10 days after injection for 3 days. Given the extra tumor volume due to fibroblasts, Ki67 and phospho-S6 IHC staining in cancer cells, instead of the tumor size, were measured. Six xenograft tumors were quantified in each treatment group. * p < 0.05, ** p < 0.01, **** p < 0.0001, two-tailed t test. See also Figures S5.
Next, we validated if CAF-derived activation of MET and FGFR pathways were sufficient to mediate EGFRi resistance in vivo. Given that human fibroblasts do not persist in mice (references (Blomme et al., 2018; Cassidy et al., 2015; Olsen et al., 2010) and Figure S5G), we examined the effect of fibroblasts on signaling and cell cycle of co-injected cancer cells in response to TKIs. Cancer cells (MGH707) were implanted either alone or together with CCD19-Lu, a fast-growing human lung fibroblast line that rescued cancer cells from EGFR inhibition robustly in vitro (Figure S5H). Co-injection of fibroblasts mitigated EGFRi-mediated suppression of cancer cell proliferation (Ki67) and downstream signaling (phosho-S6) (Figure 4B). METi or FGFRi added separately to EGFRi had no significant impact on cell proliferation or downstream signaling. In contrast, the triple combination significantly reduced both proliferation and signaling (Figure 4B). These data show proof of concept that fibroblasts can activate both MET and FGFR to confer EGFRi resistance in vivo and that in some cases inhibition of both is necessary for re-sensitization, which provides a starting point to identify more effective treatment by further tailoring it based on the impact of CAFs.
Expression of HGF and FGF7 define three subtypes of CAFs marked with distinct therapeutic strategies
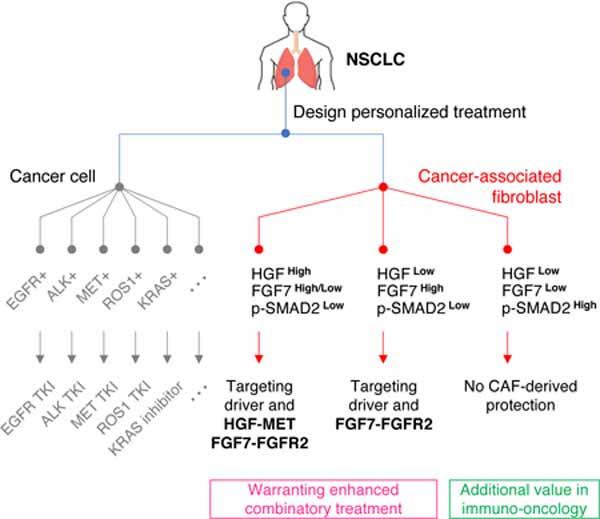
Because MET and FGFR recurrently and potently mediated CAF-driven EGFRi resistance, we next classified PDFs based on their capacities to activate these pathways. We first tested the PDF rescue capacity in another genetic context (ALK+) and found that PDFs conferring substantial resistance to EGFRi, regardless of their original tumor’s oncogenic background, are also robust rescuers of ALKi (Figure S6A). We therefore analyzed the EGFR TKI rescue profile of all 60 PDFs, including 22 PDFs from non-EGFR+ NSCLCs (Table S1) and identified three distinctive functional subtypes of CAFs: subtype I robustly and broadly rescued EGFRi via MET, with or without involving FGFR (including CCD19-Lu), subtype II conferred more modest rescue primarily via FGFR, and subtype III had a minimal rescue effect (Figure 5A-B, Methods). By analyzing the PDF-matched patients, we found this functional classification of CAFs was independent with patients’ age, biopsy site, or tumor oncogene status (Figure S6B), suggesting this classification is not only limited to EGFR+ lung cancer. Therefore, we also assessed whether the CAF functional subtypes affected other NSCLC targeted therapies such as ALKi in ALK+ NSCLCs. Using a cohort of 19 PDFs (including 13 from ALK+ tumors), we consistently found that subtype I and II PDFs conferred rescue to ALK+ cancers upon ALKi whereas subtype III PDFs permitted a better response (Figure 5C).
Figure 5: Expression of HGF and FGF7 define three subtypes of CAFs marked with distinct therapeutic strategies.
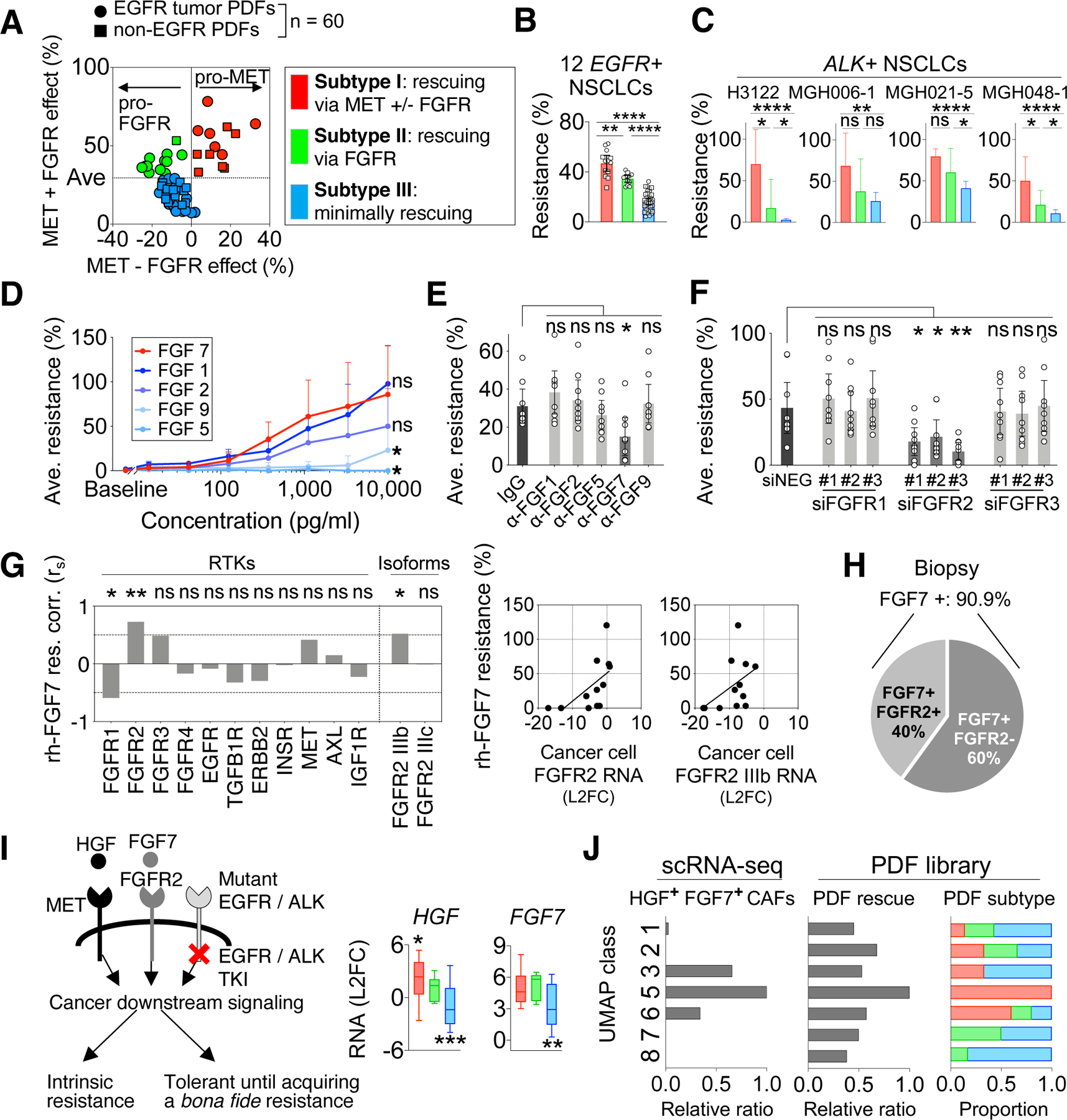
A. Sixty PDFs (dots) are classified according to their rescue effect mediated by MET and FGFR: MET-predominant rescue (red), FGFR-predominant rescue (green), and minimum rescue (blue). “MET – FGFR effect” (x-axis) is calculated by MET effect (resistance to EGFRi+FGFRi) minus FGFR effect (resistance to EGFRi+METi). “MET+FGFR effect” (y-axis) is calculated by MET effect plus FGFR effect. B. The overall EGFRi resistance (plain effect against EGFRi) conferred by PDFs is then plotted based on the functional subtypes defined in (A). (A-B), Effect of each PDF (dot) is tested across 12 EGFR+ NSCLC cancers. C. The rescue level of 19 PDFs on ALK+ NSCLC cell lines against ALKi. Results are shown by PDFs’ functional subtypes defined in (A). D. The average effect of indicated recombinant FGF on resistance to EGFRi across 5 cancer cell lines. E. The effect of neutralizing indicated FGF in PDF conditioned media on diminishing cancer cells’ resistance to EGFRi plus METi (HGF-independent resistance). F. The effect of knockdown FGFR1, FGFR2 and FGFR3 in cancer cells on diminishing cancer cells’ resistance to EGFRi plus METi in the presence of PDF conditioned media. (E-F), Effect of each PDFs (dots, n = 9) is tested across 5 cancer models. (B-F), Mean with 95% CI are shown. G. Correlations between cancer cells’ expression of indicated receptors and their resistance level conferred by recombinant FGF7 (10ng/mL). Two-tailed Spearman’s r is used. H. Prevalence of FGF7 and FGFR2 expression in EGFR+ NSCLC biopsies (n=11). I. Schematics showing that HGF and FGF7 mediate the bypass activation of cancer downstream signaling and resistance (left). HGF and FGF7 RNA levels are assessed in PDFs based on their functional subtypes by qRT-PCR (right). Whiskers are maximum and minimum values, two-tailed t test based on single group compared to all other PDFs. J. Comparison between CAF molecular classes defined by scRNA-seq analysis and CAF functional subtypes revealed by PDF analysis. The PDFs’ functional profiles are plotted by the UMAP classes (right). * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, two-tailed t test. See also Figures S6.
The three functionally defined CAF subtypes illustrate distinct treatment strategies to overcome CAF-mediated therapy resistance: MET plus FGFR pathway blockade is needed to counter the impact of subtype I CAFs, FGFR pathway blockade is needed for subtype II CAFs, and no combination is needed for subtype III CAFs (Figure S6C). While EGFRi plus METi combination is currently being investigated in clinical trials and is well tolerated (Scagliotti et al., 2015; Sequist et al., 2020; 2011a), the feasibility of combining the pan-FGFR inhibitor, especially in the setting of the triple TKI combination, is less clear. To inform more precise, and potentially more clinically tolerable, targeting of CAF-mediated protection, we determined if there were specific FGF and FGFR isoforms responsible for mediating EGFRi resistance. Among 18 FGF ligands (Ornitz and Itoh, 2015), FGF7 was the most prevalently expressed one in lung tumor stroma (Figure S6D) and was highly expressed by PDFs (FigureS6E). Compared with other FGFs, FGF7’s level in PDFs best correlated with FGFR-mediated rescue (Figure S6F-G). Recombinant FGF7 significantly maintained cancer cell growth (Figure 5D) and restored downstream signaling (Figure S6H) upon EGFRi treatment. While nonselective FGFR ligands FGF1 and FGF2 (Ornitz and Itoh, 2015) also had the potential to rescue cancer cell growth (Figure 5D), only specific targeting of FGF7, but not other FGFs, in conditioned media with blocking antibodies substantially alleviated PDF-mediated rescue (Figure 5E). Consistent with EGFR+ cancers, recombinant HGF and FGF7 were also protective to ALKi in ALK+ cancers (Figure S6I). Furthermore, the expression of HGF and FGF7 were also detected in the xenograft model used above (Figure S6J), confirming that the expression of HGF and FGF7 is valid in both cell cultures and in vivo. Together, these data point to FGF7 as a key CAF-derived rescue factor, in addition to HGF.
FGFR2, and more specifically the IIIb isoform of FGFR2, has been identified as the major receptor for FGF7 (Ornitz and Itoh, 2015). Indeed, only knocking down FGFR2, but no other FGFRs, broadly abrogated rescue by PDFs (Figure 5F). Consistently, we found the expression of FGFR2 IIIb, but not other FGFRs, in cancer cells correlated with FGF7’s rescue effect (Figure 5G). In EGFR+ tumors specifically, FGF7 was present in 90% of the biopsy cohort, and the expression of FGFR2 was detected in 40% of these samples (Figure 5H, S6K), further supporting that this FGF7-FGFR2 axis could be prominent in the clinic. Moreover, some cancer cells upregulate FGFR2 in response to EGFRi treatment when co-cultured with matched PDF derived from the same patient biopsy (Figure S6L). Additionally, some cancer cells alone also increase FGFR2/3 expression upon EGFRi addition (Ware et al., 2010). Together, there is a considerable potential impact of this FGF7-FGFR2 bypass signaling on designing more precise and tolerable treatments (eg, FGF7 or FGFR2 specific blockade) to overcome CAF-mediated TKI resistance.
In accordance with the functional classification, subtype I PDFs expressed the highest HGF and subtype I and II PDFs expressed high FGF7, in contrast to subtype III PDFs (Figure 5I). As an independent validation, the proportion of the three functionally defined PDFs was similar to that of lung CAFs with high HGF, low HGF/high FGF7, and low HGF/low FGF7 levels in previous NSCLC scRNA-seq datasets (Figure S6M) (N. Kim et al., 2020; Lambrechts et al., 2018; Laughney et al., 2020; Maynard et al., 2020; Travaglini et al., 2020). We next examined the overlap between molecular classifications in scRNA-seq and CAFs’ functional subtypes. Based on the scRNA-seq analysis, UMAP-5 fibroblasts expressed higher HGF and FGF7 (Figure 5J). Indeed, PDFs having the UMAP-5 signature best matched the subtype I PDFs (high HGF and FGF7) and had the highest EGFRi rescue capacity (Figure 5J). By analyzing the canonical CAF markers, the subtype I PDFs had higher expression of PDGFRA but lower expression of ITGA1, which was also observed in UMAP-5 single cells (Figure S6N-O). Conversely, UMAP categories other than UMAP-5 did not sufficiently delineate differential CAFs rescuing phenotypes. This observation reveals the limitations of using molecular phenotypes alone to decipher CAFs heterogeneity (Dominguez et al., 2020; Eckert et al., 2019; Su et al., 2018) and highlights the value of our functional approach to directly delineate different CAFs’ therapeutic effects.
Intrinsic TGF-beta signaling contributes to CAF functional heterogeneity by suppressing HGF and FGF7 expression
In order to investigate the mechanism underlining HGF and FGF7 overexpression in subtypes I and II PDFs in contrast to subtype III PDFs, we performed RNA sequencing of 21 PDFs and carried out unguided clustering based on the top 1000 variably expressed genes. The result showed that subtypes I and II PDFs were more closely clustered together whereas the majority of subtype III PDFs were evidently distinct from others (Figure 6A, Data S1), suggesting that the overexpression of HGF and FGF7 in subtype I and subtype II PDFs (Figure 6B, Table S4) was likely a part of a global transcriptional variation. To identify potential signaling pathways associated with HGF and FGF7 overexpression, we annotated the genes overexpressed in subtypes I and II PDFs by Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. Three pathways were identified commonly related with genes overexpressed in subtype I and II PDFs compared to subtype III, including transforming growth factor beta (TGF-β) signaling pathway that showed a high statistical significance (Figure 6C, Table S4). Intriguingly, we found that it was mainly the genes upstream, but not downstream, of TGF-β1 that were overexpressed in subtypes I and II PDFs, including decorin (DCN), fibromodulin (FMOD), and latent-transforming growth factor beta-binding protein 1 (LTBP1) (Figure 6D), all of which were known to suppress TGF-β1 activation in the extracellular space (Costanza et al., 2017). Of note, TGF-β1 itself was expressed similarly among all three subtypes of CAFs (Figure 6D, S7A). Thus, we surmised that subtypes I and II PDFs may had lower TGF-β signaling activity given their overexpression of these suppressors.
Figure 6: Intrinsic TGF-beta signaling contributes to CAF functional heterogeneity by suppressing HGF and FGF7 expression.
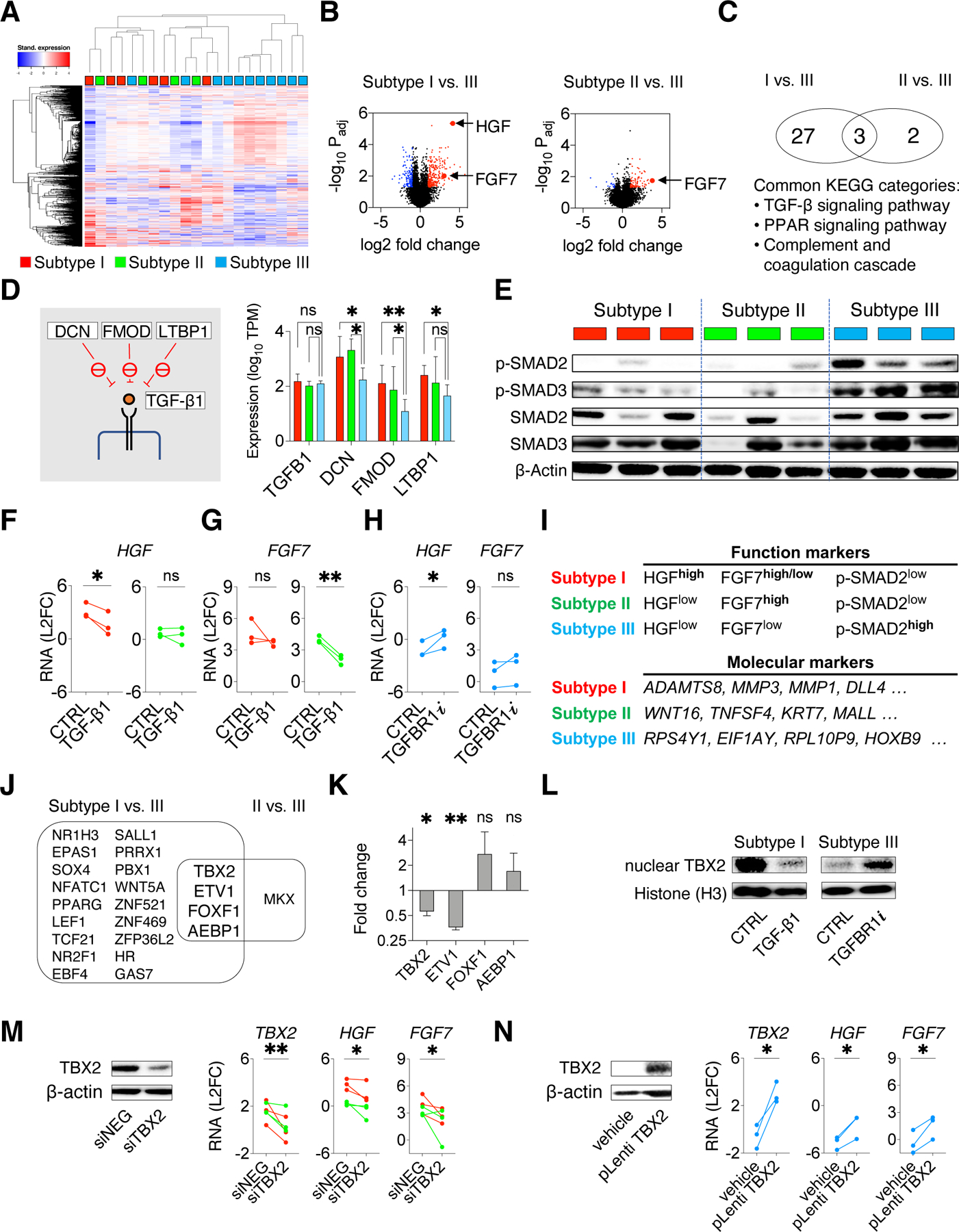
A. Heatmap with unsupervised clustering showing the top 1000 differentially expressed genes across a total of 21 PDFs. B. Volcano plots show the over-expressed genes (red) and under-expressed genes (blue) in subtype I or II PDFs compared with subtype III PDFs. C. Venn diagram showing pathways (KEGG annotation) related with genes over-expressed in subtypes I and II PDFs. D. RNAseq expressions of TGF-β1 and TGF-β1 upstream suppressors DCN, FMOD, and LTBP1(schematics on the left) in subtypes I (red), II (green), and III PDFs (blue). Mean with 95% CI. Two-tailed t test is used. E. Western blotting shows TGF-β signaling (phospho-SMAD2/SMAD3) in PDFs. Lysates were also probed in Figure S7B. F-G. HGF (F) and FGF7 (G) RNA expression measured by qRT-PCR in subtypes I (red) and II (green) PDFs upon activating TGF-β signaling using TGF-β1 (10ng/mL) for 24 hours. H. HGF and FGF7 RNA expression in subtype III (blue) PDFs after TGFBR1 inhibitor vactosertib (1μM) treatment for 24 hours. I. Function markers (HGF, FGF7, and phospho-SMAD2) and molecular markers (most variably expressed genes identified by PDF RNA sequencing, top four genes are shown) to distinguish CAF functional subtypes. J. Venn diagram shows transcription factor genes commonly over-expressed in subtype I and subtype II PDFs. K. RNA expression change of the indicated transcription factors genes in subtype I PDFs after treating with TGF-β1 for 24 hours. Mean with standard error are shown. L. Western blotting shows the nuclear TBX2 in a subtype I PDF upon TGF-β1 treatment and in a subtype III PDF upon TGFBR1i treatment. Histone H3 is used as a loading control. M. HGF and FGF7 expression in subtypes I (red) and II (green) PDFs upon TBX2 knockdown (siRNA pool). N. HGF and FGF7 expression in subtype III (blue) PDFs upon ectopic expression of TBX2. (M-N), knockdown and overexpression are confirmed by western blotting (left) and qRT-PCR (right),. (F-H, K, M-N), Paired one-tailed t-test is used. * p < 0.05, ** p < 0.01. See also Figures S7 and Table S4.
Signaling profiling confirmed that phospho-SMAD2 and phospho-SMAD3, two key mediators of TGF-β signaling, were prominently present in subtype III PDFs but only minimal in subtypes I and II PDFs (Figure 6E). Interestingly, subtypes I and II PDFs also seem to have higher variability in total SMAD2 and SMAD3 levels compared to subtype III PDFs (Figure 6E). TGF-β signaling was previously identified to mediate the fibroblast to myofibroblast transformation during the wound healing process (Kalluri, 2016; Midgley et al., 2013). Additionally, pancreatic stellate cells may be transformed into inflammatory fibroblast (iCAF) phenotype upon activation of JAK/STAT pathway or myofibroblast (mCAF) phenotype if TGF-β signaling is alternatively activated (Biffi et al., 2019). Nonetheless, despite exogeneous TGF-β1 could potentiate its downstream signaling activity and enhance αSMA expression in all NSCLC CAFs, we found the baseline αSMA (ACTA2) level was not significantly different across the three subtypes of NSCLC CAFs (Figure S6N, S7B). Furthermore, we found phospho-STAT3 level was not significantly associated with any particular CAF functional subtype either (Figure S7B). Collectively, these data support that the NSCLC CAFs functional classification is associated with an intrinsically regulated TGF-β signaling and that this functional distinction is independent of the canonical myofibroblast status identified in the wound healing process or the iCAF-mCAF classification in pancreatic stellate cell studies. Next, we examined if TGF-β signaling governed HGF and FGF7 expression. Loss of TGF-β signaling in fibroblasts is associated with increased HGF secretion and paracrine MET activation in mice (Bhowmick et al., 2004). In human lung CAFs, TGF-β1 treatment suppressed HGF and FGF7 expression in subtypes I and II PDFs (Figure 6F-G) and diminished their capacity to confer EGFRi resistance (Figure S7C). Conversely, treating subtype III PDFs with the TGF-β receptor type I inhibitor vactosertib (TGFBR1i) increased the expression of HGF and, to a lesser extent, FGF7 (Figure 6H). Therefore, CAFs’ intrinsic TGF-β signaling has an important role in determining HGF and FGF7 levels, and HGF, FGF7, and phospho-SMAD2 can be used as functional markers to differentiate CAF subtypes (Figure 6I, S7D). As a complementary approach, analysis of the most variably expressed genes from PDF RNA sequencing data, also identified molecular markers to distinguish CAF functional subtypes (Figure 6I, Table S4).
We speculated that transcription factors may be involved in regulating the expression of growth factors. Further interrogation of CAFs differentially expressed genes, we identified four transcription factors to be potentially overexpressed in subtypes I and II PDFs compared to subtype III PDFs (Figure 6J). We confirmed the down regulation of TBX2 and ETV1 in CAFs exposed to TGF-β1 (Figure 6K). ETV1 has been reported to be suppressed by TGF-β signaling and to contribute to HGF expression in dermal fibroblasts (Bordignon et al., 2019). Consistently, we showed that knocking down ETV1 reduced HGF expression in subtypes I and II PDFs whereases overexpression of ETV1 enhanced HGF expression in subtype III PDFs (Figure S7E-G). The role of TBX2 in fibroblasts is still unclear. TGF-β1 treatment reduced not only TBX2 mRNA level (Figure 6K) but also TBX2 nuclear concentration in subtype I PDF (Figure 6L). Conversely, the TBX2 nuclear concentration was increased upon TGFBR1i treatment in subtype III PDFs (Figure 6L). Additionally, knocking down TBX2 suppressed HGF and FGF7 expression in subtypes I and II PDFs (Figure 6M, S7H), whereas overexpressing TBX2 increased HGF and FGF7 levels in subtype III PDFs (Figure 6N). Meanwhile, knockdown of ETV1/TBX2 reduced, although not fully abolished PDF-driven resistance (Figure S7I), suggesting that additional TFs altering CAF function may exist and warrant further studies in the future. Collectively, these data show that CAFs’ intrinsic TGF- β signaling contributes to fibroblast functional heterogeneity, at least in part via transcriptional networks including ETV1 and TBX2.
CAFs functional classification correlates with patients’ clinical outcome
We next examined whether functional subtypes of CAFs correlated with patients’ clinical response by analyzing three patient cohorts. In the first cohort, we obtained the secretome in the conditioned media from the first week of culturing pre-treatment biopsies (thus, containing the secretory profile more similar to the original tumor’s) from 12 EGFR+ NSCLC patients who subsequently received covalent EGFR TKI treatment (Figure 7A, Table S5). In line with our findings, patients with poor response to EGFR TKI were associated with higher HGF and FGF7 levels in their tumor secretome (Figure 7A). For the second cohort of 13 patients (Figure 7B, Table S5), we established PDFs from their biopsies before their initial treatment with covalent EGFR TKI. By comparing the PDFs’ ex vivo rescue phenotype with the corresponding patient’s clinical response, we found that patients whose tumors harbored subtype III CAFs were more likely to respond to EGFR TKI than patients whose PDFs were classified as subtype I or II (Figure 7B). For the third independent EGFR+ NSCLC cohort, we analyzed a public RNAseq dataset obtained from biopsies both before and after osimertinib treatment from 11 patients (Roper et al., 2020). We further confirmed that NSCLC patients with limited response (progression-free survival, PFS < 12 months) had higher expression of HGF and FGF7 in their pre-osimertinib biopsies (Figure 7C). Intriguingly, for patients with a better response (PFS > 12 months), HGF and FGF7 expressions often increased in the post-osimertinib (resistance) biopsies (Figure S7J). Consistently, we found that stromal abundances of HGF and FGF7 were dynamic over the course of disease and treatment (Figure 7D). These findings suggest an evolution of tumor microenvironment in response to treatment pressure and/or disease progression. Thus, we further analyzed PDFs derived from longitudinal biopsies from 6 patients who received at least one line of TKI treatment between two biopsies. Interestingly, PDFs derived from later biopsies were generally stronger rescuers than PDFs from the initial biopsies (Figure 7E), supporting a plausible functional evolution of CAFs either selected for or modified by the treatment and/or disease progression. Further work will be needed to validate these findings in additional patients and to distinguish between these possibilities.
Figure 7: CAFs functional classification correlates with patients’ clinical outcome.
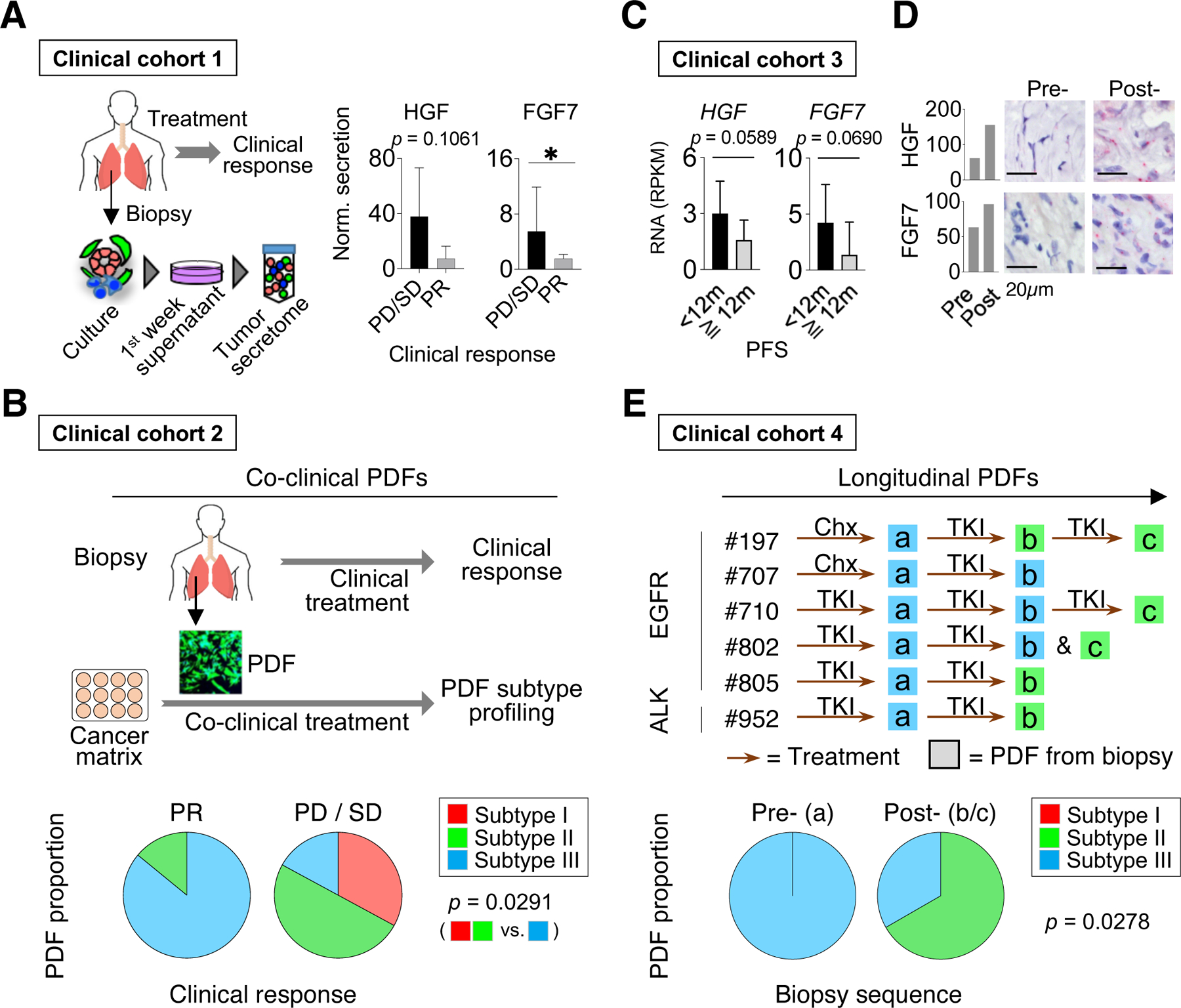
A. Normalized FGF7 and HGF secretion in 12 tumor secretome samples derived from EGFR+ NSCLC biopsies before the covalent EGFR TKI (osimertinib or equivalent) treatment. Results are compared based on patients’ clinical response, progressive disease (PD)/stable disease (SD) vs. partial response (PR). Average with 95% CI are shown, one-tailed Mann-Whitney U test. B. The functional subtypes of PDFs established from 13 NSCLC patients before receiving a covalent EGFR TKI treatment (osimertinib or equivalent) are plotted against patients’ response to their treatment. C. RNAseq data of pre-osimertinib biopsies from 11 EGFR+ NSCLC patients (from Roper et al., 2020). The HGF and FGF7 RNA levels are shown based on patients’ progression-free survival (PFS) on the treatment. Average and 95% CI are shown, one-tailed t-test. D. RNA expression of HGF and FGF7 are stained by RNAscope in pre- and post-treatment biopsy samples from two patients. E. The functional heterogeneity in a collection of PDFs established from longitudinal biopsies from same patients. PDFs are colored by functional subtypes. Bottom: the proportion of PDF subtypes according to early (a) and later biopsies (b/c). (B and E), two-tailed Fisher’s exact test is used. See also Figures S7 and Table S5.
Subtype III CAFs are also chemoattractant to immune cells
Immune checkpoint blockade has become a prominent option for NSCLCs. Asides from cancer cells’ intrinsic factors (e.g. mutation burden), fibroblast is also implied to affect tumor’s immune background (De Jaeghere et al., 2019; Sahai et al., 2020). We investigated whether CAFs functional classification was associated with the tumor immune status in patients. EGFR+ and ALK+ NSCLC have significantly lower tumor mutation burden (Willis et al., 2019) and the immune cell infiltration in these tumors is generally low (Gainor et al., 2016). Thus, investigating EGFR+ and ALK+ NSCLCs may provide a cleaner background to understand CAFs’ impact on immune cell infiltration.
To this end, we analyzed the CD8+ tumor infiltrating lymphocyte (TIL) status in a cohort of EGFR+ NSCLC (Table S5) and compared them with PDF models established from corresponding tumors. We found that all TIL+ biopsies had subtype III PDFs whereas subtypes I and II PDFs were present only in TIL- biopsies, although the difference is not statistically significant probably due to small sample size (Figure 8A). To gain insight into CAFs’ impact on immune cell migration, we performed an immune focused ELISA array assay consisted of 100 immune related factors across the three subtypes of PDFs. Interestingly, compared to subtypes I and II PDFs, subtype III PDFs expressed multiple chemokines with chemoattractant properties for T-lymphocytes and monocytes, including CXCL11, CXCL12, CCL14, CCL17, and CCL20, at higher levels (Figure 8B, Table S6). In order to functionally analyze and validate CAFs’ impact, we devised an ex vivo microfluidic assay to recapitulate the immune cell migration process. In short, PDFs were embedded in type I collagen, the major type of collagen expressed in lung cancer tissue (Naba et al., 2016); and then the immune cells were suspension cultured in a peripheral channel in mimicry of tumors capillaries (Figure 8C-D). This assay allowed us to monitor immune cells penetrating through collagen to enter the matrix following the chemogradients generated by fibroblasts. Using peripheral blood mononuclear cells (PBMC) and peripheral blood CD8+ T cells from two healthy donors, we found that the subtype III PDFs exhibited the most chemoattraction (Figure 8E-F). Similarly, subtype III PDFs also demonstrated superior capacities to recruit Jurkat cells (T-cell lineage leukemia) and THP-1 cells (monocytic leukemia model) (Figure S7K-L). While the mechanism underlining the difference in PDFs’ chemotaxis capacities is still to be determined in additional models and immune contexts, one plausible explanation is that subtypes I and II PDFs express higher peroxisome proliferator activated receptor (PPAR) γ and multiple PPAR pathway effectors (Figure 6C), which are known to inhibit the expression of inflammatory cytokines and to direct the differentiation of immune cells towards anti-inflammatory phenotypes (Straus and Glass, 2007). Collectively, these data show that CAFs functional classification is also associated with different levels of immune cell infiltration, in addition to determining targeted therapy response (Figure 8G). With further understanding on its biological and clinical impact, the phenomenon observed here could also aid in designing cancer immune therapies.
Figure 8: Subtype III CAFs are chemoattractant to immune cells.
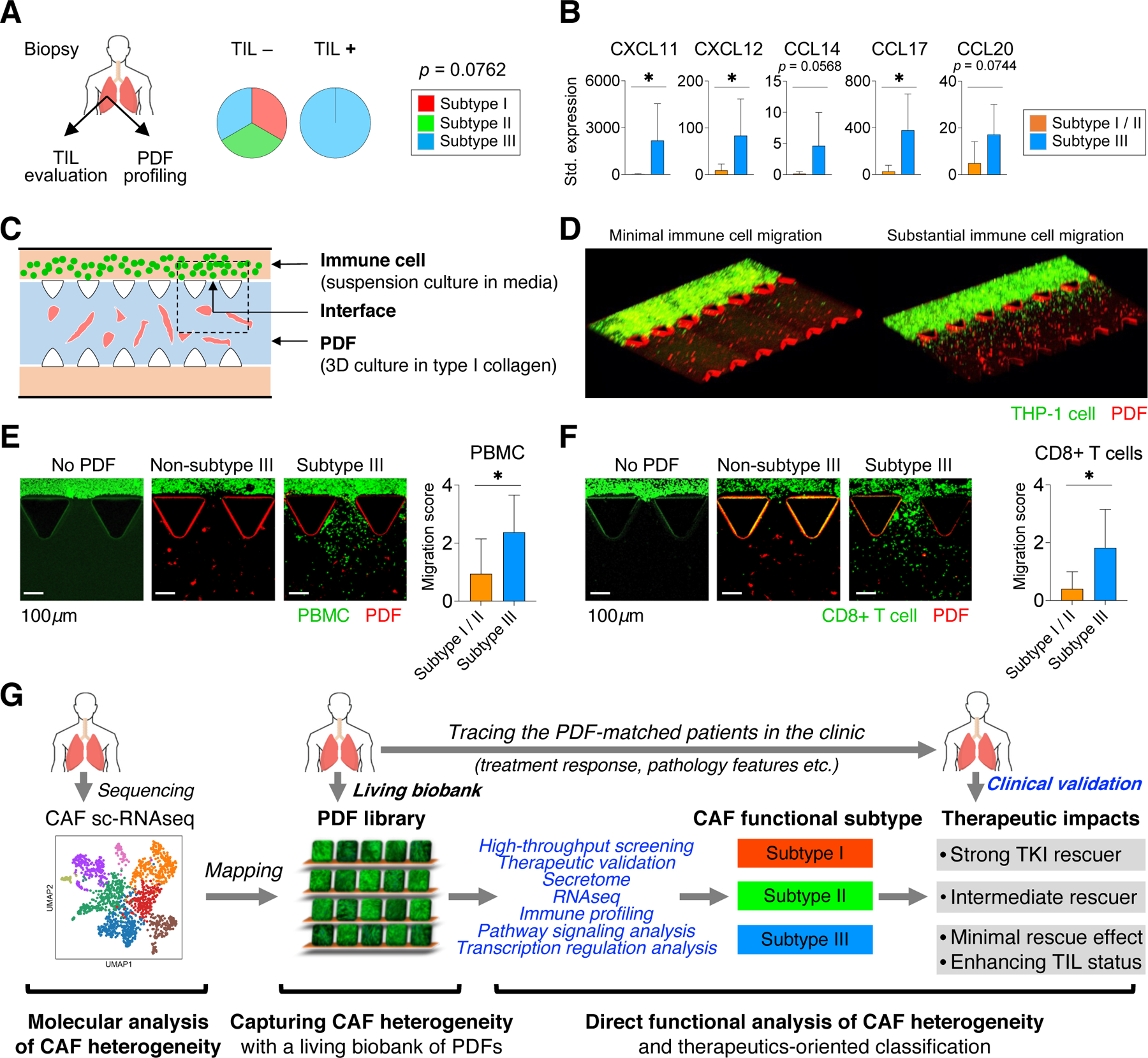
A. The status of tumor-infiltrating lymphocytes, based on CD8 staining, in EGFR+ NSCLC (n=10) according to functional subtypes of PDFs. Two-tailed Fisher’s exact test is used. B. The expression of indicated chemokines with chemoattractant properties for T-lymphocytes and monocytes in subtype III PDFs compared with subtypes I and II PDFs. Mean with 95% CI. * p < 0.05, two-tailed t test is used. C. Schematics of an ex vivo microfluidic assay to recapitulate the immune cell migration process. D. Representative images showing minimal (left) and substantial (right) immune cell migration in the microfluidic chip. E-F. Example images and summary of non-subtype III PDFs (subtypes I and II, n=4, example of a subtype I PDF is shown) and subtype III PDFs (n=4) in chemoattracting peripheral blood mononuclear cells (PBMC) (E) and peripheral blood CD8+ T cells (F) from two healthy donors. One of the representative interface areas is shown. Average level with 95%CI is shown. *, p < 0.05, one-tailed t-test is used. G. A graphic summary of the current study. See also Figure S7 and Table S5-6.
Discussion
Herein, we report establishing a living biobank of CAFs that enables us to recapitulate a broad spectrum of NSCLC CAFs with diverse molecular phenotypes and allows us to systematically survey CAFs’ functions in an unbiased manner (Figure 8G). By leveraging this PDF library, we identify three major functional subtypes of CAFs that exhibit distinct impacts on treatments using EGFR and ALK TKIs. Further, we are able to demonstrate a link between a NSCLC patient’s clinical response and the functional classification of CAFs from that patient’s tumors, thus providing evidence supporting that this CAFs functional classification may have considerable value in future clinical management of cancer patients.
Unlike cancer cells that can be readily distinguished based on genomic aberrations, the characterization of CAFs heterogeneity has been challenging given their variations in both molecular features and functions. Importantly, mechanisms leading to CAFs’ heterogeneity are still largely unclear. Leveraging this collection of patient-derived CAF models, we show that CAFs’ phenotypic and functional presentations have intriguing connections with several patients’ clinical characteristics, including age, anatomic site, smoking status, and even treatment history. However, we did not observe a significant difference in CAFs’ molecular and functional features between tumors with different oncogene backgrounds. This is perhaps not surprising given that our current PDF library is mostly representative of EGFR+ and ALK+ NSCLC, which share similar demographic features, such as adenocarcinoma in non-smokers, and signaling pathway activation. Therefore, this PDF library provides valuable insights in understanding the causes of CAFs heterogeneity and can be further expanded to include CAFs from tumors with additional oncogene and histological types in the future.
Reactivation of signaling downstream of EGFR and ALK, such as PI3K-AKT and MAPK, independent of these receptors is a major mechanism of resistance to EGFR and ALK TKI treatments, respectively (Gainor and Shaw, 2013). A growing number of growth factors with this bypass potential have been identified, including HGF Wang 2009 and Wilson 2012 and FGF7 discovered in this study. Beyond the effect of these factors, the current study provides substantial evidence supporting that the CAFs functional category determines the overall TKI response. Thus, in cases with subtype I CAFs, combined suppression of both MET and FGFR2 is necessary to successfully control the disease, as we demonstrated both in vitro and in vivo. EGFR TKI in combination with MET TKI has proven clinically well tolerable but the feasibility of additionally adding a pan-FGFR TKI is less clear. The FGF7-FGFR2 signaling axis identified in this study, alone or together with the HGF-MET signaling axis, may help to mediate adaptive resistance up front or nurture cancer cells until full acquired resistance develops. Thus, it is important to design more precise and tolerable treatments, such as FGFR isoform specific inhibition.
We demonstrate that high HGF and FGF7 expression in subtypes I and II PDFs is a part of a global transcription variation, at least in part, governed by an intrinsically regulated TGF-β signaling and downstream transcription factors. Moreover, HGF and FGF7 are also key factors in fetal lung development (Chang et al., 2002; Ohmichi et al., 1998) and are readily prevalent in a subset of normal lung fibroblasts as evident in previous scRNA-seq studies (N. Kim et al., 2020; Laughney et al., 2020; Travaglini et al., 2020), suggesting that the lung fibroblasts heterogeneity could be present even before oncogenesis. Therefore, these three functional CAF subtypes extends the understanding of fibroblasts heterogeneity beyond the canonical classifications, such as the myofibroblast status discovered in the wound healing process (Kalluri, 2016; Midgley et al., 2013) and the iCAF-mCAF classification identified in pancreatic stellate cells (Biffi et al., 2019).
Apart from targeted therapy, we show that this CAF classification also has a potential for evaluating patients in the context of immune therapy and may also aid in the research in other aspects of cancer biology. It is possible that the current definition of functional subtypes of CAFs may be less applicable in the context of other treatment regiments. However, the platform described here can be used as a paradigm and that additional CAF classification depending on the treatment of choice can be adapted from direct functional studies. Additionally, we mainly focus on characterizing the secretion function of CAFs in this study. It’s possible that other factors such as extracellular matrix (Lo et al., 2015) and the metabolism process (Eckert et al., 2019) may have additional impacts on TKI response. However, the CAFs’ impact identified in vitro using conditioned media can be validated in vivo, suggesting that CAFs secretion may play a predominant role. Notably, fibroblast heterogeneity has also become increasingly apparent as revealed by reports on other cancers (Bartoschek et al., 2018; Elyada et al., 2019; Lambrechts et al., 2018; Li et al., 2017; Qian et al., 2020) and other diseases, such as fibrotic diseases (Peyser et al., 2019; Shook et al., 2018; Xie et al., 2018), rheumatologic diseases (Croft et al., 2019; Wohlfahrt et al., 2019), and aging (Mahmoudi et al., 2019). Our approach in exploring and exploiting fibroblast heterogeneity may also provide a valuable paradigm for these disciplines to further improve clinical patient management.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources, reagents and samples should be directed to and will be fulfilled by the Lead Contact, Haichuan Hu (hhu5@mgh.harvard.edu).
Materials availability
Materials and reagents used in this study are listed in the Key resources table. Reagents generated in our laboratory in this study are available upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa Fluor 488 Goat anti-Rabbit secondary antibody | ThermoScientific | Cat# A11008 |
| Alexa Fluor 647 Donkey anti-Mouse secondary antibody | ThermoScientific | Cat# A-31571 |
| anti-FGF1 neutralizing antibody | R&D System | Cat# AF232 |
| anti-FGF2 neutralizing antibody | R&D System | Cat# AF-233-NA |
| anti-FGF5 neutralizing antibody | R&D System | Cat# AF-237-NA |
| anti-FGF7 neutralizing antibody | R&D System | Cat# AF-251-NA |
| anti-FGF9 neutralizing antibody | R&D System | Cat# MAB273–100 |
| anti-Mouse IgG HRP-linked secondary antibody | Cell Signaling Technology | Cat# 7076 |
| anti-Rabbit IgG HRP-linked secondary antibody | Cell Signaling Technology | Cat# 7074 |
| CD8 | Leica Biosystems | RTU Clone 4B11 |
| Cytokeratin 8/18 | Dako | Cat# M3652 |
| EGFR | Santa Cruz | Cat# sc-373746 |
| Erk1/2 | Cell Signaling Technology | Cat# 9102 |
| Histone H3 | Cell Signaling Technology | Cat# 4499 |
| Ki67 | Leica Biosystems | IVT Clone K2 |
| Normal IgG Control | R&D System | Cat# AB-108-C |
| Normal IgG Control | R&D System | Cat# MAB002 |
| phospho-EGFR (Y1068) | Abcam | Cat# ab5644 |
| phospho-Erk 1/2 (Thr202/Tyr204) | Cell Signaling Technology | Cat# 9101 |
| phospho-MET (Tyr1234/1235) | Cell Signaling Technology | Cat# 3129 |
| phospho-S6 (Ser240/244) | Cell Signaling Technology | Cat# 5364 |
| phospho-SMAD2 (Ser465/467) | Cell Signaling Technology | Cat# 3108 |
| phospho-STAT3 (Tyr705) | Cell Signaling Technology | Cat# 9145 |
| phosphor-SMAD3 (Ser423/425) | Invitrogen | Cat# 710756 |
| S6 | Cell Signaling Technology | Cat# 2217 |
| SMAD2 | Cell Signaling Technology | Cat# 3103 |
| SMAD3 | Abcam | Cat# ab40854 |
| TBX2 | Abnova | Cat# H00006909-M01 |
| Vimentin | Dako | Cat# M0725 |
| α-SMA | Abcam | Cat# ab5694 |
| β-Actin | Cell Signaling Technology | Cat# 4970 |
| Biological samples | ||
| NSCLC biopsy tissue | Massachusetts General Hospital, Boston, MA | IRB #13–416 |
| Human peripheral blood mononuclear cell | STEMCELL | Cat# 70025 |
| Human peripheral blood CD8+ T-cells | STEMCELL | Cat# 200–0164 |
| Chemicals, peptides, and recombinant proteins | ||
| Osimertinib | Selleckchem | Cat# S7297 |
| INC280 | Selleckchem | Cat# S2788 |
| BGJ398 | Selleckchem | Cat# S2183 |
| Lorlatinib | Selleckchem | Cat# S7536 |
| SH-4–54 | Selleckchem | Cat# S7337 |
| GDC0941 | Selleckchem | Cat# S1065 |
| R406 | Selleckchem | Cat# S2194 |
| SB431542 | Selleckchem | Cat# S1067 |
| ABT263 | Selleckchem | Cat# S1001 |
| AEW541 | Selleckchem | Cat# S1034 |
| AZD0530 | Selleckchem | Cat# S1006 |
| LEE011 | Selleckchem | Cat# S7440 |
| MLN8237 | Selleckchem | Cat# S1133 |
| Ruxolitinib | Selleckchem | Cat# S1378 |
| TAE226 | Selleckchem | Cat# S2820 |
| TP0903 | Selleckchem | Cat# S7846 |
| Rapamycin | Selleckchem | Cat# S1039 |
| Trametinib | Selleckchem | Cat# S2673 |
| Vactosertib | Selleckchem | Cat# S7530 |
| Cilengitide | MedChemExpress | Cat# HY-16141 |
| HGF | Peprotech | Cat# 100–39 |
| FGF1 | Peprotech | Cat# 100–17A |
| FGF2 | Peprotech | Cat# 100–18C |
| FGF5 | Peprotech | Cat# 100–34 |
| FGF7 | Peprotech | Cat# 100–19 |
| FGF9 | Peprotech | Cat# 100–23 |
| TGF-β1 | Peprotech | Cat# 100–21 |
| IL-2 | STEMCELL | Cat# 78036.1 |
| DNase I | STEMCELL | Cat# 07900 |
| Type-I collagen | Nitta Gelatin | Cat# 631–00651 |
| Liberase | Roche | Cat# LIBDH-RO |
| Blasticidin | Gibico | Cat# A1113903 |
| Critical commercial assays | ||
| Tumor Dissociation Kit, human | Miltenyi Biotec | Cat# 130–095-929 |
| Anti-fibroblast microbeads, human | Miltenyi Biotec | Cat# 130–050-601 |
| pLenti6/V5 Directional TOPO Cloning Kit | Invitrogen | Cat# K495510 |
| ViraPower Lentiviral Directional TOPO Expression Kit | Invitrogen | Cat# K495000 |
| Lenti-X Concentrator | TaKaRa | Cat# 631232 |
| 3D Cell Culture Chips | AIMbiotech | Cat# DAX-1, HOL-1 |
| CellTracker Green CMFDA Dye | Invitrogen | Cat# C7025 |
| CellTracker Red CMTPX Dye | Invitrogen | Cat# C34552 |
| Hoechst 33342 | ThermoScientific | Cat# H3570 |
| BioCoat Transwell (0.4 µm) | Corning | Cat# 354570 |
| RNeasy Micro Kit | Qiagen | Cat# 74004 |
| TRAPeze Telomerase Detection Kit | Millipore | Cat# S7700 |
| Nunc Lab-Tek Chamber Slide System | ThermoScientific | Cat# 177429PK |
| RNAscope 2.5 HD Detection Kit-Brown | Advanced Cell Diagnostics | Cat# 322370 |
| RNAscope 2.5 HD Duplex Reagent Kit | Advanced Cell Diagnostics | Cat# 322430 |
| Nuclear Extraction Kit | Abcam | Cat# ab113474 |
| ELISA tests for HGF, FGF7 | RayBiotech | Cat# ELH-HGF-1, EHFGF7 |
| ELISA tests for IGFBP6 | Invitrogen | Cat# EHIGFBP6 |
| Customized quantitative proteomics array | RayBiotech | NA |
| Deposited data | ||
| NSCLC sc-RNAseq | (Lambrechts et al., 2018) | https://gbiomed.kuleuven.be/english/research/50000622/laboratories/54213024/scrnaseq_tutorial/fibroblasts |
| NSCLC sc-RNAseq | (N. Kim et al., 2020) | GSE131907 |
| NSCLC sc-RNAseq | (Laughney et al., 2020) | GSE123904 |
| NSCLC sc-RNAseq | (Maynard et al., 2020) | PRJNA591860 |
| Normal lung sc-RNAseq | (Travaglini et al., 2020) | EGAS00001004344 |
| RNAseq of NSCLC biopsies | (Roper et al., 2020) | NA |
| Stromal score in TCGA LUAD tumors | (Yoshihara et al., 2013) | NA |
| TCGA lung adenocarcinoma RNAseq (LUAD) | cBioPortal | http://www.cbioportal.org |
| Experimental models: Cell lines | ||
| 293FT | Invitrogen | Cat# R70007 |
| CCD-19Lu | ATCC | Cat# CCL-210 |
| Jurkat | ATCC | Cat# TIB-152 |
| THP-1 | ATCC | Cat# TIB-202 |
| NCI-H1975 | MGH Center for Molecular Therapeutics Cell Bank | NA |
| NCI-H3122 | MGH Center for Molecular Therapeutics Cell Bank | NA |
| HCC4006 | MGH Center for Molecular Therapeutics Cell Bank | NA |
| HCC827 | MGH Center for Molecular Therapeutics Cell Bank | NA |
| PC9 | MGH Center for Molecular Therapeutics Cell Bank | NA |
| MGH006–1 | MGH patient-derived cell line (Crystal et al., 2014) | NA |
| MGH021–5 | MGH patient-derived cell line (Crystal et al., 2014) | NA |
| MGH048–1 | MGH patient-derived cell line | NA |
| MGH119–1 | MGH patient-derived cell line (Jia et al., 2016) | NA |
| MGH121–1 | MGH patient-derived cell line (Jia et al., 2016; Niederst et al., 2015) | NA |
| MGH134–1 | MGH patient-derived cell line (Jia et al., 2016) | NA |
| MGH154–1 | MGH patient-derived cell line | NA |
| MGH164–1 | MGH patient-derived cell line | NA |
| MGH707–1 | MGH patient-derived cell line (Kodack et al., 2017) | NA |
| MGH708–1 | MGH patient-derived cell line | NA |
| MGH805–1 | MGH patient-derived cell line | NA |
| PDF cell lines | MGH patient-derived cell line | NA |
| Experimental models: Organisms/strains | ||
| Female athymic nude (Nu/Nu) mice | MGH Gnotobiotic Mouse Core | NA |
| Oligonucleotides | ||
| Individual siRNA for FGFR1 | Ambion | Cat# s5164, s5165, s5166 |
| Individual siRNA for FGFR2 | Ambion | Cat# s5173, s5174, s5175 |
| Individual siRNA for FGFR3 | Ambion | Cat# s5167, s5168, s5169 |
| Negative control siRNA | Ambion | Cat# 4390844 |
| ON-TARGETplus Human ETV1 siRNA Pool | Horizon | Cat# L-003801–00-0005 |
| ON-TARGETplus Human TBX2 siRNA Pool | Horizon | Cat# L-012196–00-0005 |
| ON-TARGETplus Non-targeting Control Pool | Horizon | Cat# D-001810–10-05 |
| Individual siRNA for ETV1 | Qiagen | Cat# 1027416-GS2115 |
| Individual siRNA for TBX2 | Qiagen | Cat# 1027416-GS6909 |
| Negative control siRNA | Qiagen | Cat# 1022076 |
| RNAscope probe for ACTA2 | Advanced Cell Diagnostics | Cat# 311811 |
| RNAscope probe for S100A4 | Advanced Cell Diagnostics | Cat# 422071 |
| RNAscope probe for HGF | Advanced Cell Diagnostics | Cat# 310761 |
| RNAscope probe for FGF7 | Advanced Cell Diagnostics | Cat# 443441 |
| RNAscope probe for FGFR2 | Advanced Cell Diagnostics | Cat# 311171 |
| RNAscope probe for KRT18 | Advanced Cell Diagnostics | Cat# 310211 |
| q-PCR primers | This paper (Table S7) | NA |
| Recombinant DNA | ||
| hTERT (NM_198253) | Applied Biological Materials | Cat# LV808298 |
| pLenti6/V5-hTERT | This paper | NA |
| Lenti ORF clone: TBX2 | Origene | Cat# RC208558 |
| Lenti ORF clone: ETV1 | Origene | Cat# RC210533 |
| Lenti ORF clone: vehicle | Origene | Cat# PS100001 |
| Software and algorithms | ||
| HALO ISH v2.2 | Indica Labs | NA |
| HALO Cytonuclear IHC v1.6 | Indica Labs | NA |
| iDEP.91 | (Ge et al., 2018) | http://bioinformatics.sdstate.edu/idep |
| ShinyGo v0.61 | (Ge et al., 2020) | http://bioinformatics.sdstate.edu/go/ |
| GraphPad Prism v7.0 | GraphPad Software Inc. | NA |
| Other | ||
| Bel-Art™ Cloning Cylinders | FisherScientific | Cat# 07–907-10 |
| Polybrene | Millipore | Cat# TR-1003-G |
| Lipofectamine RNAiMAX | Invitrogen | Cat# 13778150 |
| T-cell Media | STEMCELL | Cat# 10981 |
| 40μm Strainer | FisherScientific | Cat# 08–771-1 |
Data and code availability
PDF RNAseq data (Data S1) and PDF secretome data are provided in the supplementary of this paper. A Jupyter notebook (Data S2) is provided to reproduce the UMAP analysis of NSCLC CAF sc-RNAseq shown in this paper.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
PDF cell line establishment
A systematic approach was used to culture the cancer-associated fibroblasts from biopsies (Figure S1C). This allowed a high success rate in fibroblasts establishment (80% in biopsies except for fluid samples, Figure S1C). Specifically, biopsy samples were received from patients at Massachusetts General Hospital (MGH) and processed as previously described (Kodack et al., 2017). All patients signed informed consent to participate in a protocol approved by the Institutional Review Board giving permission for research to be performed on their samples. Processing in the laboratory began approximately 30 minutes after sample collection. Biopsies were mechanically minced with sterile disposable scalpels and enzymatically digested with 25 mg/mL liberase for 1 hour in a 37°C MultiTherm shaker (Benchmark Scientific) set at 1,000 rpm. Larger samples (such as those from resection procedures) were dissociated according to Miltenyi Biotec’s Tumor Dissociation Kit with the gentleMACs instrument. After digestion or lysis, in most cases (76%), cells were seeded on dishes coated with mitotically inactivated feeder cells (irradiated with 5,000 rad and are unable to undergo division (Shamblott et al., 1998; Siegfried et al., 1991)) with nutrient enriched media(Kodack et al., 2017), or also attempted on non-feeder cell-based culture media only for larger tissue samples. To enable fibroblast separation in different cultures, cell growth for each sample was monitored to determine if separation techniques were necessary to isolate a pure fibroblast population. In most cases (78%), the fibroblast cells were naturally selected as they grew over and competed out other cell types. Additional methods included differential trypsinization, which took advantage of the observation that fibroblasts trypsinized from and adhered to plates faster than other cell types; in some experiments, to acquire purer fibroblast populations, we employed magnetic-activated cell sorting (MASC) with anti-fibroblast microbeads or single clone selections using cloning cylinders. Once a pure fibroblast population grew to confluence and were transitioned into R10 on a normal plastic plate (85% of cultures takes 5 passages or less from initial plating to reach this stage), the cells were then used for immortalization. To verify that the PDFs were not epithelial-to-mesenchymal transformed cancer cells, the finished PDF cell lines were then Sanger sequenced to confirm the absence of oncogenic mutations (e.g. EGFR activating mutation for PDFs derived from EGFR mutant biopsy samples and ALK translocation for PDFs derived from ALK rearranged biopsies), and PDF RNA were further tested by qRT-PCR to confirm they were negative for epithelial marker expression, but positive for CAF markers (Figure S2A-B).
PDF cell line immortalization and lentiviral infection
PDF immortalization was performed by ectopic expression of telomerase (hTERT) because it is efficient and does not result in changes typically associated with malignant transformation (Morales et al., 1999). The hTERT (NM_198253, plasmid from Applied Biological Materials) coding sequence was PCR amplified for ligation into the pLenti6/V5-D-TOPO vector including a Blasticidin resistance gene for mammalian cell selection. Virus was made by transfecting the pLenti6/V5 constructs along with helper plasmids (ViraPower Lentiviral Directional TOPO Expression Kit) in 293FT cells. Filtered (0.45µm) and concentrated (Lenti-X Concentrator) virus was used for PDF infection in the presence of 8µg/mL Polybrene. The infected PDF cells were selected beginning at least 48 hours after infection using Blasticidin (10 µg/mL). The hTERTinfected, Blasticidin-selected PDFs were then expanded on a 1:2–1:4 ratio for each passage. In general, immortalized cell cultures of the first ten passages, and no more than the first twenty passages (for the best growers), were used for further experiments. The same lentivirus generation approach was used for overexpressing ETV1, TBX2, and vehicle plasmid in the immortalized PDFs. These infected cells were then allowed to rest for at least 72 hours before further use.
Cancer cell culture
A total of 12 EGFR mutant, osimertinib sensitive lung cancer cell lines were used. These included four commercial cancer cell lines H1975, HCC4006, HCC827, PC9, and eight cancer cell lines established from patients at MGH: MGH119–1, MGH121–1, MGH134–1, MGH154–1, MGH164–1, MGH707–1, MGH708–1, and MGH805–1. Also a total of 4 ALK rearranged, lorlatinib sensitive lung cancer cell lines were used. These included a commercial cancer model H3122 and three patient-derived models: MGH006–1, MGH021–5, and MGH048–1. The patient-derived cancer models were developed under the same IRB-approved protocol for PDF establishment. All experiments were performed in R10. All cells were routinely tested and verified to be free of mycoplasma contamination.
TRAPeze Telomerase Activity Detection
Briefly, telomerase extension was performed by adding 200 ng of fibroblast cell extract per sample and incubating at 30°C for 30 minutes; then the reaction mix was subjected to 3-step PCR at 94 °C/15 seconds, 59 °C/30 seconds, and 72 °C/1 minute for 30 cycles. The resultant PCR products were separated on a 12% polyacrylamide gel and stained with 0.5 mg/ml ethidium bromide (Sigma) for 30 minutes before imaging.
PDF immunofluorescence staining
PDFs were seeded at a density of 500 cells/well in 384-well plates. After growing in R10 for 7 days, the PDFs were fixed in 3.7% formaldehyde. Then the PDFs were stained with anti-Vimentin (1:100), and on the next day Alexa Fluor 647 Donkey anti-Mouse secondary antibody (1:100), both of which were incubated overnight at 4°. All antibodies were prepared in 1% BSA, 0.1% Triton-X. Finally, cell nuclei were stained with 4 mg/mL Hoechst 33342 and images were acquired with the ImageXpress Micro XL High-Content Imager from Molecular Devices.
In situ fibroblast marker gene expression analysis
Formalin-fixed paraffin-embedded (FFPE) clinical biopsy samples were sectioned at 4µm and kept at −80 °C before use. Corresponding PDF cultures were prepared according to the Advanced Cell Diagnostics’ protocol for cultured adherent cells using RNAscope analysis. Cells were seeded in plastic chamber slides for 24 hours. The chamber walls were then torn down, and the slides were fixed with 10% neutral buffered formalin, serially dehydrated with ethanol, and kept at −20 °C before use. The FFPE sections and PDF culture slides were then subjected to detection for ACTA2 and S100A4 mRNA expression by RNAscope (RNAscope 2.5 HD Detection Kit-Brown), and counter stained with Hematoxylin. The stromal area of the FFPE clinical sections and PDF culture slides were then further scored based on Advanced Cell Diagnostics’ semi-quantitative criteria: 0: No staining or <1 dot/10 cells; 1: 1–3 dots/cell; 2: 4–9 dots/cell and none or very few dot clusters; 3: 10–15 dots/cell and <10% dots are in clusters; 4: >15 dots/cell and >10% dots are in clusters. At least six 40X fields were randomly captured and evaluated to generate the final score for each sample.
PDF-mediated EGFR TKI resistance screening via co-culturing with cancer cells
Patient-derived fibroblasts (PDFs) were seeded at a density of 500 cells/well in 384-well plates, cancer cells were seeded at equal density 24 hours later, then the co-cultures were incubated overnight before drugging. Cells were drugged with EGFRi (osimertinib, 200nM) using a Tecan D300e drug dispenser. Six days after treatment, cells were fixed in 3.7% formaldehyde and stained with anti-Cytokeratin 8/18 (1:100) and anti-Vimentin (1:100) overnight at 4°. Secondary antibodies were added the next day: Alexa Fluor 488 Goat anti-Rabbit IgG (1:100) and Alexa Fluor 647 Donkey anti-Mouse IgG (1:100) and incubated overnight at 4°. All antibodies were prepared in 1% BSA, 0.1% Triton-X. Finally, cell nuclei were stained with 4 mg/mL Hoechst 33342. Fluorescent images were automatically acquired with the ImageXpress Micro XL High-Content Imager (Molecular Devices) for channels corresponding to Cytokeratin 8/18, Vimentin, and nuclei staining. Image analysis was performed with Molecular Devices’ MetaXpress software as previously described (Kodack et al., 2017), and the number of cancer cells (Hoechst+ and Cytokeratin 8/18+ cell count, denoted as N) were quantified. PDF-mediated growth rescue was calculated as:
All screens were carried out in quadruplicate.
PDF-mediated TKI resistance screening via conditioned media (CM)
To prepare CM, PDF cells were cultured with R10 in 10cm or 15 cm plates, dependent on their proliferation rates. CM was harvested six days after media refreshment, when plates were 70–90% confluent. One day before treatment, cancer cells were seeded at a density of 1,000 cells/well in 384-well plates. On the day of treatment, for conditions with PDF CM, CM was added so that cancer cells were in 50% PDF CM / 50% R10. Human recombinant HGF was prepared in R10 and added to a final concentration of 10ng/mL. For controls, only R10 was added so that cells were in 100% R10. Based on the treatment conditions, EGFRi (osimertinib, 200nM), METi (INC280, 200nM), and FGFRi (BGJ398, 500nM) were added alone or in combination via the drug dispenser as previously described. For the testing of ALK rearranged cancer models, ALKi (Lorlatinib, 300nM) was used under the same culture condition with PDF CM, or with recombinant HGF and FGF7 added to a final concentration of 10ng/mL. After 72 hours of treatment, cells were fixed in 3.7% formaldehyde and cell nuclei were stained with 4 mg/mL Hoechst 33342. Plate imaging was performed as described in the co-culture screen, and the Hoechst+ nuclei counts were treated as the total number of cells (N). Similar to the co-culture, PDF-mediated growth rescue was calculated by:
All screens were carried out in quadruplicate. By surveying a total of 60 PDFs and their impacts on EGFR TKI resistance (assessed across 12 EGFR+ NSCLC cancer cell models), Subtype I PDFs are defined as those strongly conferring EGFR TKI resistance predominantly via activating HGF-MET signaling (i.e., Resistance % to EGFRi+FGFRi > Resistance % to EGFRi+METi, and the additive effect of the two is above average level); Subtype II PDFs are defined as those strongly conferring EGFR TKI resistance predominantly via activating FGF7-FGFR2 signaling (i.e., Resistance % to EGFRi+METi > Resistance % to EGFRi+FGFRi, and the additive effect of the two is above average level); and Subtype III PDFs are defined as those conferring no resistance or below the average level.
Recombinant FGF-mediated EGFR TKI resistance
Experiments were set up according to the protocol previously described in the conditioned media experiment, with the following changes: 1) On the day of treatment, recombinant human FGF proteins were serially diluted with R10 and added with a final concentration from 0 to 10ng/mL. 2) Cancer cells were drugged with EGFRi (osimertinib, 200nM). After 72 hours of treatment, cells were fixed, stained, and imaged. Rescue was calculated using the same method as above.
Neutralizing antibodies blocking PDF CM-mediated resistance
Experiments were set up according to the protocol previously described in the conditioned media experiment, with the following changes: 1) On the day of treatment, after adding in PDF CM, all neutralizing antibodies were added at a concentration of 3 mg/mL (based on previous neutralization titration tests, antibodies at this concentration block at least 50% of the EGFR TKI resistance mediated by recombinant FGFs (10ng/mL)). 2) To interrogate the HGF-independent resistance, cancer cells were treated with EGFRi + METi combo (osimertinib + INC280, both 200nM). After 72 hours of treatment, cells were fixed, stained, and imaged. Rescue was calculated using the same method as above.
Knocking down experiments using siRNA
FGFRs knocking down experiments were set up according to the protocol previously described in the conditioned media experiment, with the following changes: 1) After seeded for 24 hours, cancer cells were transfected with 10nM siRNAs by using Lipofectamine RNAiMAX and incubated overnight before treatment. 2) On the day of treatment, media was refreshed with 50% PDF CM / 50% R10, or 100% R10. 3) To interrogate the HGF-independent resistance, cancer cells were treated with EGFRi + METi combo (osimertinib + INC280, both 200nM). After 72 hours of treatment, cells were fixed, stained, and imaged. Rescue was calculated using the same method as above. ETV1, and TBX2 knocking down experiments using the ON-TARGETplus siRNA Pool system and individual siRNAs from Qiagen was performed on PDFs per the manufactuer’s instruction with negative siRNAs used as the control. PDFs were treated with 50nM of the siRNA Pool or 25nM of each individual siRNAs, then media was refreshed 24 hours after siRNA treatement, and RNA was prepped 72 hours after treatment.
Drug combination screening to identify by-pass signaling in cancer cells
Experiments were set up according to the protocol previously described in the conditioned media experiment, with the following changes: 1) PDF CM was added on the drugging day so that cells were in 50% CM / 50% R10 or in 100% R10. 2) Cancer cells were treated with serial doses of test drug alone or in combination with EGFRi + METi combo (osimertinib + INC280, both 200nM). The top concentration for each compound was set based on known activity and the concentrations decreased in half-log increments. The top concentration for each compound was 32 uM (SH-4–54), 10uM (Cilengitide, GDC0941, R406, SB431542), 3.2uM (ABT263, AEW541, AZD0530, BGJ398, LEE011, MLN8237, Ruxolitinib, TAE226, TP0903), or 320nM (Rapamycin, Trametinib). After 72 hours of treatment, cells were fixed, stained, and imaged using the same protocol as above. The cancers’ dose-response to the test drug with or without EGFRi + METi was fit to a nonlinear regression model using a three-parameter analytic method in GraphPad Prism 7.0. The half maximal inhibitory concentration (IC50) for each drug was extracted, and the relative efficacy was calculated by subtracting the IC50 with EGFRi + METi from the IC50 without EGFRi and METi (∆IC50). All screens were carried out in quadruplicate.
Western blotting and antibodies
Cells were seeded in six-well plates overnight before treatment (see treatment conditions below). After 24 hours of drug treatment, lysates were collected, and protein levels were quantified via BCA assay (Thermo Scientific). Protein electrophoresis was performed using 4–12% NuPAGE Bis-Tris gels (Thermo Scientific) in MOPS SDS running buffer (Thermo Scientific) before transferring to nitrocellulose membranes. In conditions using PDF CM, media was changed to 50% PDF CM / 50% R10 on the day of treatment. Growth factors (HGF, FGF7, TGF-β1) are used as 10ng/mL unless specified. Small molecule inhibitors are used as EGFRi (200nM), METi (200nM), FGFRi (500nM), and TGFBR1i (1μM) unless specified. Primary antibodies were prepared at a 1:1,000 dilution and were then probed with HRP-linked secondary antibody (1:50,000). Nuclear extraction is performed per the manufacturer’s instruction.
Tumor secretome (biopsy early primary culture supernatant) collection
Supernatant was collected from early primary cultures of biopsy tissues. The primary culture condition is the same as described in the cell line establishment session, and the supernatant was collected after the first week of culture. As a control, media from the same seeding conditions were also banked.
Enzyme-linked immunosorbent assay (ELISA)
The HGF and FGF7 levels in PDF conditioned media and tumor secretome (early primary culture supernatant) samples were measured via ELISA. Given that early primary cultures from biopsies have divergent number of cells, the HGF and FGF7 were further normalized to IGFBP-6, a stably expressed cytokine with levels directly proportional to the raw count of CAF cells.
Quantitative proteomics array
Quantitative proteomics array was performed by RayBiotech (Norcross, GA) using a Quantibody array, with the analytes listed in Table S2. Background levels in a media control were subtracted from those in test samples to calculate protein concentrations. The result is then further normalized by the total number of cells (ng/mL per 100,000 cells) evaluated by fibroblast cells seeded in parallel in a 96-well plated and stained with Hoechst for nuclei counting. An immune-focused array is performed in the same manner and listed in Table S6.
In situ analysis of FGF and HGF RNA expression
FFPE clinical biopsy samples and xenograft samples were both sectioned at 4µm and were subjected to detection for HGF, FGF7, FGFR2, and/or KRT18 RNA expression by RNAscope (RNAscope 2.5 HD Duplex Assay), and counter stained with Hematoxylin. At least six 40X regions per slide were randomly captured and analyzed using HALO software (ISH v2.2 algorithms). Cells with one or more detectable FGF7 dots and no detectable FGFR2 dots were considered as FGF7+ cells; cells with one or more detectable FGFR2 dots regardless of the FGF7 status were considered as FGFR2+ cells. Samples with more than 10% FGF7+ cells were considered as FGF7 positive, and samples with more than 10% FGFR2+ cells were considered as FGFR2 positive.
In vivo study
All mouse studies were approved by the Institutional Animal Care and Use Committee at Massachusetts General Hospital in accordance with institutional guidelines. For generating tumor bearing mouse models, ten million MGH707 cancer cells were injected subcutaneously with or without CCD19Lu fibroblast cells in a 1:2 ratio into flanks of 6–8 weeks old athymic nude mice. Treatment was started 10 days after injections when palpable tumors were formed. Mice bearing cancer alone xenografts were randomized into two groups to receive vehicle or EGFRi (10mg/kg osimertinib, once daily(Cross et al., 2014)) treatment; and mice bearing cancer plus fibroblast xenografts were randomized into five groups to receive vehicle, EGFRi, EGFRi + METi (10mg/kg INC280, twice daily(Jia et al., 2016)), EGFRi + FGFRi (15mg/kg BGJ398, once daily(Guagnano et al., 2012)), and EGFRi + METi + FGFRi treatment. On the third day of treatment, mice were sacrificed 3 hours after the last drug administration and tumors were harvested and fixed in 10% neutral buffered formalin for immunohistochemistry (IHC) analysis. IHC analysis was performed at the Histopathology Research Core at Massachusetts General Hospital by sectioning FFPE tumor samples at 4µm and staining for phospho-S6 and Ki67. At least six 20X regions per slide were randomly captured and analyzed by using HALO software (Cytonuclear IHC v1.6 algorithms). The algorithm for calling a positively-stained cancer cell was trained in a pre-test and was applied to all the samples assessed in the experiment. The number of positively-stained cancer cells was then divided by the total number of cancer cells analyzed to generate the percentage of pS6- or Ki67-expressing cells per sample.
Single cell analysis of lung cancer fibroblast database to uncover subpopulations and marker genes
Lambrechts et al. analyzed resected NSCLC tumor tissues in 5 patients via single cell RNA sequencing (scRNA-seq) (Lambrechts et al., 2018), whereby their t-Distributed Stochastic Neighbor Embedding (tSNE) analysis identified 7 fibroblast molecular subpopulations and proposed limited marker genes for each subpopulations. We reanalyze the data by using Scanpy with Uniform Manifold Approximation and Projection (UMAP) visualization, and we found the new analysis had improved capacity to distinguish molecular subpopulations (i.e. we identified UMAP-7 and UMAP-8 which are two new fibroblast molecular classes that were not discovered by the reported tSNE analysis). Therefore, we decided to leverage this new analysis to systematically identify key marker genes per each molecular subpopulation for analyzing the PDF models as described in the following.
The processed fibroblasts scRNA-seq dataset of 1465 cells and 33693 genes was downloaded, and cell annotations and raw count matrix were extracted from the loom file. We used Scanpy (v1.4.3) (Wolf et al., 2018) to perform single cell clustering analysis and to visualize the results with UMAP. More specifically, genes expressed in less than three cells were first filtered out. Then for each cell, its expression matrix was normalized by the total read count. The normalized gene expression matrix was further multiplied by the scale factor 10,000 and log-transformed. We selected highly variable genes and regressed out effects of total counts per cell and the percentage of mitochondrial genes expressed. Then principal component analysis (PCA) was performed and top 30 principal components (PCs) were chosen to compute the neighborhood graph (n_neighbors = 15). A community-detection-based method Louvain (Blondel et al., 2008) was used to cluster the neighborhood graph of cells (resolution = 0.5). Finally, the graph of cells was embedded in two dimensions using UMAP (McInnes et al., 2018). Based on Louvain clustering solution, the cells within each group (cluster) were contrasted with the rest of cells. Then t-test was used to rank genes within each group to identify marker genes. A Jupyter notebook (Data S2) is provided to reproduce the analysis results shown in this paper.
The processed scRNA-seq dataset of fibroblasts in lung lesions (normal and tumor) from another four studies (N. Kim et al., 2020; Laughney et al., 2020; Maynard et al., 2020; Travaglini et al., 2020) are downloaded from the reported portal, respectively.
Mapping PDFs based on UMAP classification and validation
Based on the top marker genes per UMAP class, we assessed the RNA express of the top 3 marker genes in every UMAP class (21 marker genes in total, UMAP-4 was excluded from analysis as poor quality cells) in PDFs via qRT-PCR. The expression of each marker gene (normalized by reference gene) was then standardized across the PDFs. The average level of the top 3 marker genes per each UMAP class was then calculated and used to represent the signal of the corresponding UMAP class in each PDF. By comparing the signal between UMAP classes in each PDF, the PDF is mapped based on the highest UMAP signal it expressed (standardized value).
Validation of the PDF mapping was first performed based on comparing key marker genes’ expression in PDF-UMAP classes (assessed by mean value of PDFs mapped to the same UMAP class, qRT-PCR) and in single-cell-UMAP classes (assessed by mean value of single cells in the same UMAP class, log10 transformed scRNA-seq counts). In further validation, the proximity between PDF-UMAP classes (data matrix of PDF expression, UMAP signal per UMAP class per PDF is the mean value of the top 3 marker genes, qRT-PCR) was also compared with the proximity between single-cell-UMAP classes (data matrix of fibroblast single cells, UMAP signal per UMAP class per single cell is the mean value of the top 3 marker genes, log transformed scRNA-seq counts). Unguided hierarchical clustering on UMAP classes in PDFs and single cells was performed based on Spearman’s r.
Cancer-PDF pair co-culture
MGH805–1 (cancer) and PDF805–1 (PDF) are paired cancer and PDF lines derived from the same biopsy tissue using the same platform (see above PDF cell line establishment section). Cancer cells (at a density of 150,000/ chamber) and PDF cells (at a density of 200,000/ chamber) are seeded separately one day before the treatment in the top and bottom chambers of a 6-well transwell system, respectively. The 0.4µm transwell pore size allows the exchange of secreted soluble factors but prevents cells migration from contaminating the counterpart. On the day of treatment, the cancer cells (top chamber) are either left alone or co-cultured with PDFs (bottom chamber in the same unit), and treated with no drug (R10 media) or osimertinib (EGFR TKI added to a final concentration of 200nM) for 5 days with two biological replicates per each condition. Then the cancer cells are immediately separated from the co-culture system by unloading the top chambers for total RNA extraction (RNeasy Mini Kit). RNA library preparation and next-generation sequencing are performed and analyzed by BGI America (Cambridge, Massachusetts) by using DNBseq platform, resulting in approximately 46.8 million reads per sample on average. HISAT2 was used to map sequencing reads to transcripts in the human hg19 reference genome. Based on the gene expression level, differentially expression genes (DEG) between osimertinib-treated cancer cell samples with and without PDF co-culture were reported by using the DEseq2 algorithms (Love et al., 2014).
PDF RNAseq analysis
We performed bulk RNAseq on a total of 21 PDFs with each one in duplicates. Average expression of the PDFs are used in performing unguided clustering, and the dupilicates are used for identifying differentially expressed genes (DEG) analysis. Analysis is done by using iDEP.91 tool (Ge et al., 2018) with all parameters set to default. In specific, the DEG genes are considered by false discovery rate less than 0.1, and minimal fold change more than 2. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis on the DEG genes are performed by using ShinyGo v0.61 too1(Ge et al., 2020).
qRT-PCR
Total RNA from PDF and cancer cell line cultures was extracted as per standard protocols (RNeasy Mini Kit, Qiagen) and reverse-transcribed into cDNA with the Biorad iScript Reverse Transcription Supermix. Reactions were run on a LightCycler 480 instrument (Roche) for quantitation. Primers used for each gene of interest are listed below. The housekeeper gene TATA-box binding protein gene (TBP) was selected as a reference gene because it was reported to have the most reliably identical and stable expression between tumor and normal lung tissues (Hu et al., 2016; Søes et al., 2013), as well as stable expression in human pulmonary fibroblasts (Stock et al., 2011). The FGFR2 isoform specific primers were selected according to Drugan et al. (Drugan et al., 1998). The mRNA expression for each gene was calculated by –∆Ct = – (Ct gene of interest – Ct housekeeper gene). When comparing gene expression of PDF lines to cancer cell lines, –∆∆Ct [(–∆Ct PDF lines) – (–∆Ct Cancer cell lines)] was used to quantify the expression level in PDF lines relative to cancer cell lines. The primers used for qRT-PCR are listed in Table S7.
In silico analysis of the stromal enrichment of FGFs
To analyze the correlation between the abundance of tumor stroma and gene expression, stromal score was extracted from Yoshihara et al.’s previous work (Yoshihara et al., 2013), in which the level of infiltrating stromal cells was calculated by the “ESTIMATE” method. The corresponding TCGA tumors’ RNA expression data (RNAseq V2, RSEM) was extracted from the cBioPortal (Cerami et al., 2012). A total of 228 TCGA lung adenocarcinoma samples available for both RNA expression and stromal score were included in this analysis.
Immune profiling and immune cell migration assay
Tumor infiltrating lymphocyte (TIL) profiling is performed as previously described (Gainor et al., 2016). CD8 staining by immunohistochemistry is used for immune cell enumeration. CD8+ TILs were semiquantitatively evaluated on a scale of 0 to 3 based on the extent of positive lymphocytes infiltrating within tumor cells. Each score was defined on the basis of the fraction of tumor cells on top of which CD8+ T cells were present: score 0, none or rare; score 1, <5%; score 2, >5% and <25%; and score 3, >25%. Then the scores were dichotomized into positive (scores 2–3) and negative (scores 0–1) TIL status.
Frozen human peripheral blood mononuclear cell (PBMC) and peripheral blood CD8+ T cells are thawed out two days before the experiment and culture in T-cell media supplemented with 100IU/mL IL-2. On the day of experiment, both cells are treated with DNase I (0.1mg/mL), and filterd through a 40μm strainer.
In the ex vivo assay to assess PDFs chemoattractant capacities to immune cells, a microfluidic chip (AIM biotech) was used to recapitulate the immune cell migration process. PDFs and immune cells were first stained with 3μM CellTracker in green (CMFDA) or in red (CMTPX) at 37 for 30 minutes. After washing with PBS, the PDFs were three-dimensionally embedded in type I collagen (2mg/mL) at a final concentration of 1 million/mL. Allowing collagen polymerization for 30 minutes, the peripheral channels were primed with R10 media. Then a total of 120,000 PBMC, CD8+ T cells, Jurkat cells, or THP-1 cells were reconstituted in R10 and injected to a peripheral channel. After 7 days of culture, the immune cell (green fluorescence) was imaged with fluorescent microscope. The immune migration is scored based on the depth of migration (0–3, from passing the interface to passing the middle line), as well as the scale of migration (all interfaces were evaluated and a final score was the average of all evaluations). Migration score is assessed across all interfaces in the chip and averaged by three replicates.
Clinical cohorts
Clinical cohort 1 (n=12, Table S5) was consisted of patients meeting the following criteria: the patient subsequently received a third-generation EGFR TKI treatment (osimertinib or equivalent); no other bona fide resistance mechanism is identified prior to the next third-generation EGFR TKI treatment (e.g. MET amplification). Tumor secretomes are derived from biopsies that precedes the patient’s treatment. Clinical response of the tumor to treatment is based on the Response Evaluation Criteria in Solid Tumors (Eisenhauer et al., 2009). A distinctive clinical cohort 2 (n=13, Table S5) was consisted of patients meeting the following criteria: the patient receives their first third-generation EGFR TKI treatment (osimertinib or equivalent); no other bona fide resistance mechanism is identified prior to the third-generation EGFR TKI treatment (e.g. MET amplification). PDFs models are derived from biopsies that precedes the patient’s treatment. An independent clinical cohort 3 consisted with EGFR+ NSCLC cases were also analyzed. This public RNAseq dataset contains RNAseq result from biopsies both before and after osimertinib treatment from 11 patients (Roper et al. (Roper et al., 2020)). For cases with multiple samples at the same timepoint (eg, post-psimertinib biopsy), the average value is used.
Statistics
Statistical analysis was performed by using GraphPad Prism 7.0. The following analysis, and format of data presentation were applied unless specified: Mean value comparison in-between groups were analyzed using an unpaired two-tailed Student’s t-test; correlation between two datasets was achieved by Spearman’s r method and p < 0.05 was considered to be statistically significant.
Supplementary Material
Highlights.
A living biobank of CAFs from NSCLC patients recapitulates clinical CAF heterogeneity
Therapeutic profiling of the NSCLC CAFs reveals three distinctive functional subtypes
Subtype I and II CAFs have high HGF and FGF7 expression and protect cancer cells
Subtype III CAFs associate with better clinical response and immune cell migration
Acknowledgements
We thank Khaleda Haider, Benjamin Drapkin, Chendi Li, Eliane Cortez, and Emily Niederst for support and discussions. We also thank Kristin Dionne, Kuan Chee Mun, Lim Sei Hien, Xunqin Yin, Maria T. Gianatasio, and Lorin Ferris for technical support. This manuscript was edited at Life Science Editors.
Funding: This study was funded by support from The Lung Cancer Research Foundation (HH); NIH 2R01CA137008 (LVS), R01CA164273 (ATS), K08CA197389 (ANH), K23HL132120 (LPH); Wellcome Trust 102696 (CHB); MGH Cancer Center Excellence Awards (HH); LUNGevity (ZP); LUNGevity/Upstage Lung Cancer (LPH); National Human Genome Research Institute (NHGRI) Genomic Innovator Award R35HG010717 (LP), Guangzhou Health Care Collaborative Innovation Major Projects (RPH). Additional support was provided by Lungstrong, the Kevin Hoffman family, Targeting for a Cure for Lung Cancer, The Susanne E. Coyne fund, and Be a Piece of the Solution.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
ZP receives commercial research support from Novartis, Tesaro, Spectrum, AstraZeneca and Takeda; and serves as a consultant/advisory board member for AstraZeneca, Takeda, Novartis, ImmunoGen, Guardant Health and Spectrum. LVS serves as a compensated consultant or received honoraria from AstraZeneca, Janssen, Merrimack, and Genentech; and receives institutional research funding from AstraZeneca, Boehringer Ingelheim, Novartis, Genentech, Merrimack, Blueprint Medicines and LOXO. CHB laboratory received support for research from Novartis, Amgen, and Araxes. ATS is an employee of Novartis and a paid consultant for Pfizer, Genentech/Roche, Ariad/Takeda, Syros, Blueprint Medicine, KSQ Therapeutics, TP Therapeutics, Chugai, Daiichi-Sankyo, LOXO/ Bayer, Achilles, Archer, Foundation Medicine, and Guardant. MMK serves as a consultant for Merrimack Pharmaceuticals and H3 Biomedicine. ANH receives commercial research grants from Amgen, Novartis, Relay Therapeutics, Pfizer and Roche/Genentech. RSH receives consulting honoraria from Boehringer Ingelheim, Tarveda, Apollomics; and receives institutional research funding from Daichii Sankyo, Agios, Novartis, Corvus, Mirati, Genentech Roche, Incyte, Abbvie, Celgene, and Exelixis. LP has financial interests in Edilytics. L.P.’s interests were reviewed and are managed by Massachusetts General Hospital and Partners Health Care in accordance with their conflict-of-interest policies. JJW, YQM, and RPH are employees of RayBiotech Inc. MJN is a Novartis employee and equity holder. DPK, CHB, and JAE are Novartis employees (contribution at MGH). The other authors declare no competing interests.
References
- Bartoschek M, Oskolkov N, Bocci M, Lövrot J, Larsson C, Sommarin M, Madsen CD, Lindgren D, Pekar G, Karlsson G, Ringnér M, Bergh J, Björklund Å, Pietras K, 2018. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat Commun 9, 5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL, 2004. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 303, 848–851. [DOI] [PubMed] [Google Scholar]
- Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, Tuveson DA, 2019. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov 9, 282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomme A, Van Simaeys G, Doumont G, Costanza B, Bellier J, Otaka Y, Sherer F, Lovinfosse P, Boutry S, Palacios AP, De Pauw E, Hirano T, Yokobori T, Hustinx R, Bellahcène A, Delvenne P, Detry O, Goldman S, Nishiyama M, Castronovo V, Turtoi A, 2018. Murine stroma adopts a human-like metabolic phenotype in the PDX model of colorectal cancer and liver metastases. Oncogene 37, 1237–1250. [DOI] [PubMed] [Google Scholar]
- Blondel VD, Guillaume J-L, Lambiotte R, Lefebvre E, 2008. Fast unfolding of communities in large networks. J. Stat. Mech 2008, P10008. [Google Scholar]
- Bordignon P, Bottoni G, Xu X, Popescu AS, Truan Z, Guenova E, Kofler L, Jafari P, Ostano P, Röcken M, Neel V, Dotto GP, 2019. Dualism of FGF and TGF-β Signaling in Heterogeneous Cancer-Associated Fibroblast Activation with ETV1 as a Critical Determinant. Cell Rep 28, 2358–2372.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy JW, Caldas C, Bruna A, 2015. Maintaining Tumor Heterogeneity in Patient-Derived Tumor Xenografts. Cancer Res 75, 2963–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catenacci DVT, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, Marsh R, Wallace J, Kozloff M, Rajdev L, Cohen D, Wade J, Sleckman B, Lenz H-J, Stiff P, Kumar P, Xu P, Henderson L, Takebe N, Salgia R, Wang X, Stadler WM, de Sauvage FJ, Kindler HL, 2015. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. JCO 33, 4284–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N, 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Chi J-T, Dudoit S, Bondre C, van de Rijn M, Botstein D, Brown PO, 2002. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc. Natl. Acad. Sci. U.S.A 99, 12877–12882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Kim J, Yang S, Wang H, Wu C-J, Sugimoto H, LeBleu VS, Kalluri R, 2021. Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe J-P, Desprez P-Y, Krtolica A, Campisi J, 2010. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5, 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, Sirven P, Magagna I, Fuhrmann L, Bernard C, Bonneau C, Kondratova M, Kuperstein I, Zinovyev A, Givel A-M, Parrini M-C, Soumelis V, Vincent-Salomon A, Mechta-Grigoriou F, 2018. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 1–28. [DOI] [PubMed] [Google Scholar]
- Costanza B, Umelo I, Bellier J, Castronovo V, Turtoi A, 2017. Stromal Modulators of TGF-β in Cancer. J. Clin. Med 6, 7–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, Savary L, Wehmeyer C, Naylor AJ, Kemble S, Begum J, Dürholz K, Perlman H, Barone F, McGettrick HM, Fearon DT, Wei K, Raychaudhuri S, Korsunsky I, Brenner MB, Coles M, Sansom SN, Filer A, Buckley CD, 2019. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 570, 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DAE, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MRV, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, Red-Brewer M, Ichihara E, Sun J, Jin H, Ballard P, Al-Kadhimi K, Rowlinson R, Klinowska T, Richmond GHP, Cantarini M, Kim D-W, Ranson MR, Pao W, 2014. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 4, 1046–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A, Greninger P, Lee D, Kalsy A, Gomez-Caraballo M, Elamine L, Howe E, Hur W, Lifshits E, Robinson HE, Katayama R, Faber AC, Awad MM, Ramaswamy S, Mino-Kenudson M, Iafrate AJ, Benes CH, Engelman JA, 2014. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 346, 1480–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jaeghere EA, Denys HG, De Wever O, 2019. Fibroblasts Fuel Immune Escape in the Tumor Microenvironment. TRENDS in CANCER: TRECAN 5, 704–723. [DOI] [PubMed] [Google Scholar]
- Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, Breart B, Foreman O, Bainbridge TW, Castiglioni A, Şenbabaoğlu Y, Modrusan Z, Liang Y, Junttila MR, Klijn C, Bourgon R, Turley SJ, 2020. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15 +Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov 10, 232–253. [DOI] [PubMed] [Google Scholar]
- Drugan CS, Paterson IC, Prime SS, 1998. Fibroblast growth factor receptor expression reflects cellular differentiation in human oral squamous carcinoma cell lines. Carcinogenesis 19, 1153–1156. [DOI] [PubMed] [Google Scholar]
- Eckert MA, Coscia F, Chryplewicz A, Chang JW, Hernandez KM, Pan S, Tienda SM, Nahotko DA, Li G, Blaženović I, Lastra RR, Curtis M, Yamada SD, Perets R, McGregor SM, Andrade J, Fiehn O, Moellering RE, Mann M, Lengyel E, 2019. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 569, 723–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J, 2009. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer [DOI] [PubMed] [Google Scholar]
- Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, Sivajothi S, Armstrong TD, Engle DD, Yu KH, Hao Y, Wolfgang CL, Park Y, Preall J, Jaffee EM, Califano A, Robson P, Tuveson DA, 2019. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov 9, 1102–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale C-M, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Jänne PA, 2007. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Gainor JF, Shaw AT, 2013. Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. J. Clin. Oncol 31, 3987–3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, Huynh TG, Zhao L, Fulton L, Schultz KR, Howe E, Farago AF, Sullivan RJ, Stone JR, Digumarthy S, Moran T, Hata AN, Yagi Y, Yeap BY, Engelman JA, Mino-Kenudson M, 2016. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: A Retrospective Analysis. Clin. Cancer Res 22, 4585–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge SX, Jung D, Yao R, 2020. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge SX, Son EW, Yao R, 2018. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinformatics 19, 534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghesquière B, Wong BW, Kuchnio A, Carmeliet P, 2014. Metabolism of stromal and immune cells in health and disease. Nature 511, 167–176. [DOI] [PubMed] [Google Scholar]
- Gieniec KA, Butler LM, Worthley DL, Woods SL, 2019. Cancer-associated fibroblasts-heroes or villains? Br. J. Cancer 121, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guagnano V, Kauffmann A, Wöhrle S, Stamm C, Ito M, Barys L, Pornon A, Yao Y, Li F, Zhang Y, Chen Z, Wilson CJ, Bordas V, Le Douget M, Gaither LA, Borawski J, Monahan JE, Venkatesan K, Brümmendorf T, Thomas DM, García-Echeverría C, Hofmann F, Sellers WR, Graus-Porta D, 2012. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov 2, 1118–1133. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Morgensztern D, Boshoff C, 2018. The biology and management of non-small cell lung cancer. Nature 553, 446–454. [DOI] [PubMed] [Google Scholar]
- Hofheinz RD, Al-Batran SE, Research FH, 2003. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 26, 44–48. [DOI] [PubMed] [Google Scholar]
- Hu H, Sun Z, Li Y, Zhang Y, Li H, Zhang Y, Pan Y, Shen L, Wang R, Sun Y, Chen H, 2016. The Histologic Classifications of Lung Adenocarcinomas Are Discriminable by Unique Lineage Backgrounds. J Thorac Oncol 11, 2161–2172. [DOI] [PubMed] [Google Scholar]
- Jänne PA, Yang JC-H, Kim D-W, Planchard D, Ohe Y, Ramalingam SS, Ahn M-J, Kim S-W, Su W-C, Horn L, Haggstrom D, Felip E, Kim J-H, Frewer P, Cantarini M, Brown KH, Dickinson PA, Ghiorghiu S, Ranson M, 2015. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med 372, 1689–1699. [DOI] [PubMed] [Google Scholar]
- Jia Y, Juarez J, Li J, Manuia M, Niederst MJ, Tompkins C, Timple N, Vaillancourt M-T, Pferdekamper AC, Lockerman EL, Li C, Anderson J, Costa C, Liao D, Murphy E, DiDonato M, Bursulaya B, Lelais G, Barretina J, McNeill M, Epple R, Marsilje TH, Pathan N, Engelman JA, Michellys P-Y, McNamara P, Harris J, Bender S, Kasibhatla S, 2016. EGF816 Exerts Anticancer Effects in Non-Small Cell Lung Cancer by Irreversibly and Selectively Targeting Primary and Acquired Activating Mutations in the EGF Receptor. Cancer Res 76, 1591–1602. [DOI] [PubMed] [Google Scholar]
- Junttila MR, de Sauvage FJ, 2013. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501, 346–354. [DOI] [PubMed] [Google Scholar]
- Kalluri R, 2016. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Zeisberg M, 2006. Fibroblasts in cancer. Nat. Rev. Cancer 6, 392–401. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Sahai V, Abel EV, Griffith KA, Greenson JK, Takebe N, Khan GN, Blau JL, Craig R, Balis UG, Zalupski MM, Simeone DM, 2014. Pilot Clinical Trial of Hedgehog Pathway Inhibitor GDC-0449 (Vismodegib) in Combination with Gemcitabine in Patients with Metastatic Pancreatic Adenocarcinoma. Clin. Cancer Res 20, 5937–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Kim HK, Lee K, Hong Y, Cho JH, Choi JW, Lee J-I, Suh Y-L, Ku BM, Eum HH, Choi S, Choi Y-L, Joung J-G, Park W-Y, Jung HA, Sun J-M, Lee S-H, Ahn JS, Park K, Ahn M-J, Lee H-O, 2020. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun 11, 2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodack DP, Farago AF, Dastur A, Held MA, Dardaei L, Friboulet L, Flotow von, F., Damon LJ, Lee D, Parks M, Dicecca R, Greenberg M, Kattermann KE, Riley AK, Fintelmann FJ, Rizzo C, Piotrowska Z, Shaw AT, Gainor JF, Sequist LV, Niederst MJ, Engelman JA, Benes CH, 2017. Primary Patient-Derived Cancer Cells and Their Potential for Personalized Cancer Patient Care. Cell Rep 21, 3298–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koliaraki V, Pasparakis M, Kollias G, 2015. IKKβ in intestinal mesenchymal cells promotes initiation of colitis-associated cancer. J. Exp. Med 212, 2235–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, Bassez A, Decaluwé H, Pircher A, Van den Eynde K, Weynand B, Verbeken E, De Leyn P, Liston A, Vansteenkiste J, Carmeliet P, Aerts S, Thienpont B, 2018. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med 24, 1277–1289. [DOI] [PubMed] [Google Scholar]
- Laughney AM, Hu J, Campbell NR, Bakhoum SF, Setty M, Lavallée V-P, Xie Y, Masilionis I, Carr AJ, Kottapalli S, Allaj V, Mattar M, Rekhtman N, Xavier JB, Mazutis L, Poirier JT, Rudin CM, Pe’er D, Massagué J, 2020. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat. Med 26, 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson WE, Polosukhin VV, Zoia O, Stathopoulos GT, Han W, Plieth D, Loyd JE, Neilson EG, Blackwell TS, 2005. Characterization of Fibroblast-specific Protein 1 in Pulmonary Fibrosis. Am J Respir Crit Care Med 171, 899–907. [DOI] [PubMed] [Google Scholar]
- LeBleu VS, Kalluri R, 2018. A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis. Model. Mech 11, dmm029447–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, Kong SL, Chua C, Hon LK, Tan WS, Wong M, Choi PJ, Wee LJK, Hillmer AM, Tan IB, Robson P, Prabhakar S, 2017. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet [DOI] [PubMed] [Google Scholar]
- Lo A, Wang L-CS, Scholler J, Monslow J, Avery D, Newick K, O’Brien S, Evans RA, Bajor DJ, Clendenin C, Durham AC, Buza EL, Vonderheide RH, June CH, Albelda SM, Puré E, 2015. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res 75, 2800–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S, 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi S, Mancini E, Xu L, Moore A, Jahanbani F, Hebestreit K, Srinivasan R, Li X, Devarajan K, Prélot L, Ang CE, Shibuya Y, Benayoun BA, Chang ALS, Wernig M, Wysocka J, Longaker MT, Snyder MP, Brunet A, 2019. Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing. Nature 574, 553–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard A, McCoach CE, Rotow JK, Harris L, Haderk F, Kerr DL, Yu EA, Schenk EL, Tan W, Zee A, Tan M, Gui P, Lea T, Wu W, Urisman A, Jones K, Sit R, Kolli PK, Seeley E, Gesthalter Y, Le DD, Yamauchi KA, Naeger DM, Bandyopadhyay S, Shah K, Cech L, Thomas NJ, Gupta A, Gonzalez M, Do H, Tan L, Bacaltos B, Gomez-Sjoberg R, Gubens M, Jahan T, Kratz JR, Jablons D, Neff N, Doebele RC, Weissman J, Blakely CM, Darmanis S, Bivona TG, 2020. Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell 182, 1232–1251.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L, Healy J, Melville J, 2018. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction [Google Scholar]
- Midgley AC, Rogers M, Hallett MB, Clayton A, Bowen T, Phillips AO, Steadman R, 2013. Transforming growth factor-β1 (TGF-β1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J. Biol. Chem 288, 14824–14838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales CP, Holt SE, Ouellette M, Kaur KJ, Yan Y, Wilson KS, White MA, Wright WE, Shay JW, 1999. Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nat. Genet 21, 115–118. [DOI] [PubMed] [Google Scholar]
- Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA, Hynes RO, 2016. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol 49, 10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narra K, Mullins SR, Lee H-O, Strzemkowski-Brun B, Magalong K, Christiansen VJ, McKee PA, Egleston B, Cohen SJ, Weiner LM, Meropol NJ, Cheng JD, 2014. Phase II trial of single agent Val-boroPro (talabostat) inhibiting fibroblast activation protein in patients with metastatic colorectal cancer. Cancer Biology & Therapy 6, 1691–1699. [DOI] [PubMed] [Google Scholar]
- Niederst MJ, Hu H, Mulvey HE, Lockerman EL, Garcia AR, Piotrowska Z, Sequist LV, Engelman JA, 2015. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin. Cancer Res 21, 3924–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmichi H, Koshimizu U, Matsumoto K, Nakamura T, 1998. Hepatocyte growth factor (HGF) acts as a mesenchyme-derived morphogenic factor during fetal lung development. Development 125, 1315–1324. [DOI] [PubMed] [Google Scholar]
- Olsen CJ, Moreira J, Lukanidin EM, Ambartsumian NS, 2010. Human mammary fibroblasts stimulate invasion of breast cancer cells in a three-dimensional culture and increase stroma development in mouse xenografts. BMC Cancer 10, 444–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Itoh N, 2015. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol 4, 215–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA, Lee EJ, Chio IIC, Hwang C-I, Tiriac H, Baker LA, Engle DD, Feig C, Kultti A, Egeblad M, Fearon DT, Crawford JM, Clevers H, Park Y, Tuveson DA, 2017. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med 214, 579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Österreicher CH, Penz-Österreicher M, Grivennikov SI, Guma M, Koltsova EK, Datz C, Sasik R, Hardiman G, Karin M, Brenner DA, 2011. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc. Natl. Acad. Sci. U.S.A 108, 308–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallangyo CK, Ziegler PK, Greten FR, 2015. IKKβ acts as a tumor suppressor in cancer-associated fibroblasts during intestinal tumorigenesis. J. Exp. Med 212, 2253–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira BA, Vennin C, Papanicolaou M, Chambers CR, Herrmann D, Morton JP, Cox TR, Timpson P, 2019. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. TRENDS in CANCER: TRECAN 5, 724–741. [DOI] [PubMed] [Google Scholar]
- Peyser R, MacDonnell S, Gao Y, Cheng L, Kim Y, Kaplan T, Ruan Q, Wei Y, Ni M, Adler C, Zhang W, Devalaraja-Narashimha K, Grindley J, Halasz G, Morton L, 2019. Defining the Activated Fibroblast Population in Lung Fibrosis Using Single-Cell Sequencing. Am J Respir Cell Mol Biol 61, 74–85. [DOI] [PubMed] [Google Scholar]
- Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, Marcoux N, Banwait MK, Digumarthy SR, Su W, Yoda S, Riley AK, Nangia V, Lin JJ, Nagy RJ, Lanman RB, Dias-Santagata D, Mino-Kenudson M, Iafrate AJ, Heist RS, Shaw AT, Evans EK, Clifford C, Ou S-HI, Wolf B, Hata AN, Sequist LV, 2018. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov 8, 1529–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Olbrecht S, Boeckx B, Vos H, Laoui D, Etlioglu E, Wauters E, Pomella V, Verbandt S, Busschaert P, Bassez A, Franken A, Bempt MV, Xiong J, Weynand B, van Herck Y, Antoranz A, Bosisio FM, Thienpont B, Floris G, Vergote I, Smeets A, Tejpar S, Lambrechts D, 2020. A Pan-cancer Blueprint of the Heterogeneous Tumour Microenvironment Revealed by Single-Cell Profiling. bioRxiv 572, 2020.04.01.019646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Joyce JA, 2013. Microenvironmental regulation of tumor progression and metastasis. Nat. Med 19, 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper N, Brown A-L, Wei JS, Pack S, Trindade C, Kim C, Restifo O, Gao S, Sindiri S, Mehrabadi F, Meskini El, R., Ohler ZW, Maity TK, Venugopalan A, Cultraro CM, Akoth E, Padiernos E, Chen H, Kesarwala A, Smart DK, Nilubol N, Rajan A, Piotrowska Z, Xi L, Raffeld M, Panchenko AR, Sahinalp C, Hewitt S, Hoang CD, Khan J, Guha U, 2020. Clonal Evolution and Heterogeneity of Osimertinib Acquired Resistance Mechanisms in EGFR Mutant Lung Cancer. Cell Rep Med 1, 100007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, Hynes RO, Jain RK, Janowitz T, Jørgensen C, Kimmelman AC, Kolonin MG, Maki RG, Powers RS, Puré E, Ramirez DC, Scherz-Shouval R, Sherman MH, Stewart S, Tlsty TD, Tuveson DA, Watt FM, Weaver V, Weeraratna AT, Werb Z, 2020. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 20, 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scagliotti G, Pawel von, J., Novello S, Ramlau R, Favaretto A, Barlesi F, Akerley W, Orlov S, Santoro A, Spigel D, Hirsh V, Shepherd FA, Sequist LV, Sandler A, Ross JS, Wang Q, Roemeling von, R., Shuster D, Schwartz B, 2015. Phase III Multinational, Randomized, Double-Blind, Placebo-Controlled Study of Tivantinib (ARQ 197) Plus Erlotinib Versus Erlotinib Alone in Previously Treated Patients With Locally Advanced or Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J. Clin. Oncol 33, 2667–2674. [DOI] [PubMed] [Google Scholar]
- Sequist LV, Han J-Y, Ahn M-J, Cho BC, Yu H, Kim S-W, Yang JC-H, MD JSL, MD W-CS, MD DK, PhD SO, MD MC, PhD RBV, MD AM, MD LO, MSc PF, PhD XO, Oxnard G, 2020. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: interim results from a multicentre, open-label, phase 1b study. Lancet Oncology 21, 373–386. [DOI] [PubMed] [Google Scholar]
- Sequist LV, Pawel von, J., Garmey EG, Akerley WL, Brugger W, Ferrari D, Chen Y, Costa DB, Gerber DE, Orlov S, Ramlau R, Arthur S, Gorbachevsky I, Schwartz B, Schiller JH, 2011a. Randomized Phase II Study of Erlotinib Plus Tivantinib Versus Erlotinib Plus Placebo in Previously Treated Non–Small-Cell Lung Cancer. JCO 29, 3307–3315. [DOI] [PubMed] [Google Scholar]
- Sequist LV, Soria J-C, Goldman JW, Wakelee HA, Gadgeel SM, Varga A, Papadimitrakopoulou V, Solomon BJ, Oxnard GR, Dziadziuszko R, Aisner DL, Doebele RC, Galasso C, Garon EB, Heist RS, Logan J, Neal JW, Mendenhall MA, Nichols S, Piotrowska Z, Wozniak AJ, Raponi M, Karlovich CA, Jaw-Tsai S, Isaacson J, Despain D, Matheny SL, Rolfe L, Allen AR, Camidge DR, 2015. Rociletinib in EGFR-mutated non-small-cell lung cancer. N. Engl. J. Med 372, 1700–1709. [DOI] [PubMed] [Google Scholar]
- Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, Akhavanfard S, Heist RS, Temel J, Christensen JG, Wain JC, Lynch TJ, Vernovsky K, Mark EJ, Lanuti M, Iafrate AJ, Mino-Kenudson M, Engelman JA, 2011b. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3, 75ra26–75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi N, Kang Y, 2011. Unravelling the complexity of metastasis - molecular understanding and targeted therapies. Nat. Rev. Cancer 11, 735–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamblott MJ, Axelman J, Wang S, Bugg EM, Littlefield JW, Donovan PJ, Blumenthal PD, Huggins GR, Gearhart JD, 1998. Derivation of pluripotent stem cells from cultured human primordial germ cells. Proc. Natl. Acad. Sci. U.S.A 95, 13726–13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shook BA, Wasko RR, Rivera-Gonzalez GC, Salazar-Gatzimas E, López-Giráldez F, Dash BC, Muñoz-Rojas AR, Aultman KD, Zwick RK, Lei V, Arbiser JL, Miller-Jensen K, Clark DA, Hsia HC, Horsley V, 2018. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegfried JM, Resau JH, Miura I, Testa JR, 1991. Primary culture of solid lung tumors for chromosomal analysis, in: Adolph KW (Ed.), Advanced Techniques in Chromosome Research New York. [Google Scholar]
- Stock C, Leoni P, Shi-Wen X, Abraham D, Nicholson A, Wells A, Renzoni E, Lindahl G, 2011. Identification of stable housekeeping genes for real-time PCR in human pulmonary fibroblasts. European Respiratory Journal 38, p3805. [Google Scholar]
- Straus DS, Glass CK, 2007. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol 28, 551–558. [DOI] [PubMed] [Google Scholar]
- Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, Cooper ZA, Chapman PB, Solit DB, Ribas A, Lo RS, Flaherty KT, Ogino S, Wargo JA, Golub TR, 2012. Tumour microenvironment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 487, 500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S, Chen J, Yao H, Liu J, Yu S, Lao L, Wang M, Luo M, Xing Y, Chen F, Huang D, Zhao J, Yang L, Liao D, Su F, Li M, Liu Q, Song E, 2018. CD10+GPR77+Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 172, 841–856.e16. [DOI] [PubMed] [Google Scholar]
- Søes S, Sørensen BS, Alsner J, Overgaard J, Hager H, Hansen LL, Kristensen LS, 2013. Identification of accurate reference genes for RT-qPCR analysis of formalin-fixed paraffin-embedded tissue from primary non-small cell lung cancers and brain and lymph node metastases. Lung Cancer 81, 180–186. [DOI] [PubMed] [Google Scholar]
- Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, Chang S, Conley SD, Mori Y, Seita J, Berry GJ, Shrager JB, Metzger RJ, Kuo CS, Neff N, Weissman IL, Quake SR, Krasnow MA, 2020. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turke AB, Zejnullahu K, Wu Y-L, Song Y, Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L, Lindeman NI, Murphy C, Akhavanfard S, Yeap BY, Xiao Y, Capelletti M, Iafrate AJ, Lee C, Christensen JG, Engelman JA, Jänne PA, 2010. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 17, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EF, 2016. Cancer: Fibroblasts for all seasons. Nature 530, 42–43. [DOI] [PubMed] [Google Scholar]
- Wang W, Li Q, Yamada T, Matsumoto K, Matsumoto I, Oda M, Watanabe G, Kayano Y, Nishioka Y, Sone S, Yano S, 2009. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin. Cancer Res 15, 6630–6638. [DOI] [PubMed] [Google Scholar]
- Ware KE, Marshall ME, Heasley LR, Marek L, Hinz TK, Hercule P, Helfrich BA, Doebele RC, Heasley LE, 2010. Rapidly acquired resistance to EGFR tyrosine kinase inhibitors in NSCLC cell lines through de-repression of FGFR2 and FGFR3 expression. PLoS ONE 5, e14117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis C, Fiander M, Tran D, Korytowsky B, Thomas J-M, Calderon F, Zyczynski TM, Brixner D, Stenehjem DD, 2019. Tumor mutational burden in lung cancer: a systematic literature review. Oncotarget 10, 6604–6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, Ribas A, Li J, Moffat J, Sutherlin DP, Koeppen H, Merchant M, Neve R, Settleman J, 2012. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 487, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlfahrt T, Rauber S, Uebe S, Luber M, Soare A, Ekici A, Weber S, Matei A-E, Chen C-W, Maier C, Karouzakis E, Kiener HP, Pachera E, Dees C, Beyer C, Daniel C, Gelse K, Kremer AE, Naschberger E, Stürzl M, Butter F, Sticherling M, Finotto S, Kreuter A, Kaplan MH, Jüngel A, Gay S, Nutt SL, Boykin DW, Poon GMK, Distler O, Schett G, Distler JHW, Ramming A, 2019. PU.1 controls fibroblast polarization and tissue fibrosis. Nature 566, 344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, Theis FJ, 2018. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, Wang Y, Deng N, Huang G, Taghavifar F, Geng Y, Liu N, Kulur V, Yao C, Chen P, Liu Z, Stripp B, Tang J, Liang J, Noble PW, Jiang D, 2018. Single-Cell Deconvolution of Fibroblast Heterogeneity in Mouse Pulmonary Fibrosis. Cell Rep 22, 3625–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing X, Yang F, Huang Q, Guo H, Li J, Qiu M, Bai F, Wang J, 2021. Decoding the multicellular ecosystem of lung adenocarcinoma manifested as pulmonary subsolid nodules by single-cell RNA sequencing. Sci Adv 7, eabd9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, Carter SL, Getz G, Stemke-Hale K, Mills GB, Verhaak RGW, 2013. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
PDF RNAseq data (Data S1) and PDF secretome data are provided in the supplementary of this paper. A Jupyter notebook (Data S2) is provided to reproduce the UMAP analysis of NSCLC CAF sc-RNAseq shown in this paper.