

Abstract
Peters plus syndrome, characterized by defects in eye and skeletal development with isolated cases of ventriculomegaly/hydrocephalus, is caused by mutations in the β3-glucosyltransferase (B3GLCT) gene. In the endoplasmic reticulum, B3GLCT adds glucose to O-linked fucose on properly folded thrombospondin type 1 repeats (TSRs). The resulting glucose–fucose disaccharide is proposed to stabilize the TSR fold and promote secretion of B3GLCT substrates, with some substrates more sensitive than others to loss of glucose. Mouse B3glct mutants develop hydrocephalus at high frequency. In this study, we demonstrated that B3glct mutant ependymal cells had fewer cilia basal bodies and altered translational polarity compared to controls. Localization of mRNA encoding A Disintegrin and Metalloproteinase with ThromboSpondin type 1 repeat 20 (ADAMTS20) and ADAMTS9 suggested that reduced function of these B3GLCT substrates contributed to ependymal cell abnormalities. In addition, we showed that multiple B3GLCT substrates (Adamts3, Adamts9 and Adamts20) are expressed by the subcommissural organ, that subcommissural organ-spondin ((SSPO) also known as SCO-spondin) TSRs were modified with O-linked glucose–fucose and that loss of B3GLCT reduced secretion of SSPO in cultured cells. In the B3glct mutant, intracellular levels of SSPO were reduced and BiP levels increased, suggesting a folding defect. Secreted SSPO colocalized with BiP, raising the possibility that abnormal extracellular assembly of SSPO into Reissner’s fiber also contributed to impaired CSF flow in mutants. Combined, these studies underscore the complexity of the B3glct mutant hydrocephalus phenotype and demonstrate that impaired cerebrospinal fluid (CSF) flow likely stems from the collective effects of the mutation on multiple processes.
Keywords: C-mannosylation, hydrocephalus, O-fucosylation, SCO-spondin, thrombospondin
Introduction
Mutations in the β3-glucosyltransferase (B3GLCT) gene cause Peters plus syndrome in humans (PTRPLS, MiM#261540), a developmental disorder characterized by abnormalities in eye, craniofacial and skeletal development (Lesnik Oberstein et al. 2006, 2007; Khatri et al. 2019 Oct 8 [Updated 24 Aug 2017], Van Schooneveld et al. 1984; Reis et al. 2008; Weh et al. 2014). B3GLCT mutations can also be associated with high frequency of stillbirth with multiple congenital defects (Van Schooneveld et al. 1984; Canda et al. 2018). Notably, some PTRPLS patients develop ventriculomegaly or hydrocephalus and central nervous system abnormalities (Reis et al. 2008; Khatri et al. 2019). In the endoplasmic reticulum (ER), B3GLCT functions in concert with protein O-fucosyltransferase 2 (POFUT2) to add an O-linked glucose-β1-3fucose disaccharide to properly folded thrombospondin type 1 repeat (TSR) domains (Kozma et al. 2006; Luo, Koles, et al. 2006a; Luo, Nita-Lazar, et al. 2006b; Sato et al. 2006). TSRs fall into two groups with different disulfide bonding patterns. POFUT2 recognizes both groups and adds O-linked fucose to the serine or threonine (S/T) in a consensus sequence (Group 1 TSRs, C1-X-X-(S/T)-C2; Group 2 TSRs, C2-X-X-(S/T)-C3) (Kozma et al. 2006; Sato et al. 2006; Vasudevan et al. 2015; Holdener and Haltiwanger 2019). B3GLCT then transfers glucose to the O-fucosylated TSR to form the disaccharide (Kozma et al. 2006; Sato et al. 2006). The O-linked disaccharide is proposed to stabilize the TSR fold and promote trafficking of POFUT2/B3GLCT-modified proteins through the secretory pathway (Holdener and Haltiwanger 2019). In this regard, B3GLCT functions in a highly selective manner to ensure the efficient folding of up to 49 substrate proteins containing between one to 25 cysteine-rich TSRs (Schneider et al. 2017). Nearly half of these substrates are members of the A Disintegrin And Metalloproteinase with ThromboSpondin type 1 repeat (ADAMTS) family (Du et al. 2010; Vasudevan and Haltiwanger 2014; Schneider et al. 2017; Mead and Apte 2018). The O-linked fucose appears essential for trafficking POFUT2/B3GLCT substrates as evidenced by early embryonic lethality of mouse Pofut2 mutants (Du et al. 2010; Benz et al. 2016), and loss of secretion of substrate proteins in cell-based secretion assays using POFUT2 siRNA knockdown or CRISPR-Cas9 knockout cell lines (Vasudevan et al. 2015; Benz et al. 2016; Holdener et al. 2019; Zhang et al. 2020). In contrast, loss of B3GLCT has varying effects on trafficking of POFUT2-modified substrates (Hubmacher et al. 2017; Holdener et al. 2019). For example, although ADAMTS9 and ADAMTS20 share similar domain structures and each has 12 of 15 TSRs modified by POFUT2/B3GLCT, in cell-based secretion assays ADAMTS9 secretion is only partially reduced by loss of B3GLCT, whereas secretion of ADAMTS20 is blocked by loss of B3GLCT (Holdener et al. 2019).
In mice, B3glct mutations cause similar craniofacial and skeletal abnormalities to PTRPLS patients (Holdener et al. 2019). In addition, over 50% of B3glct mutants develop hydrocephalus with rounded or domed cranium and enlarged brain ventricles (Holdener et al. 2019). Hydrocephalus results from the accumulation of cerebrospinal fluid in the brain ventricular system and can be caused by abnormalities in cilia of the brain ventricles (Ohata et al. 2014; Takagishi et al. 2017; Abdelhamed et al. 2018), aqueduct stenosis (Vio et al. 2000; Fernandez-Llebrez et al. 2004; Dietrich et al. 2009; Nakajima et al. 2011), overproduction of CSF by the choroid plexus (Yang et al. 2019) and defects in the subcommissural organ (Vio et al. 2000; Perez-Figares et al. 2001; Somera and Jones 2004; Baas et al. 2006; Martínez-Peña y Valenzuela et al. 2006; Meiniel 2007; Carmona-Calero et al. 2009; Huh et al. 2009).
We predict that hydrocephalus in B3glct mutants is caused by impaired function of one or more B3GLCT substrates that are sensitive to loss of the glucose modification. Since both B3glct and Adamts20 mutants display pigmentation abnormalities and a high frequency of hydrocephalus (Holdener et al. 2019), we predict that reduced function of ADAMTS20 likely contributes significantly to hydrocephalus in B3glct mutants. Using histological as well as molecular analysis of B3glct mutant brain, we present evidence that loss of B3GLCT decreases CSF flow dynamics. Results from these analyses suggest that impaired CSF flow likely results from the effects of the B3glct mutation on ADAMTS20 function during ependymal cell polarization as well as from abnormalities in subcommissural organ function that include defects in ADAMTS20 and/or subcommissural organ spondin (SCO-spondin) secretion, processing and/or assembly.
Results
Cerebrospinal fluid flow is impaired in mouse B3glct mutants
To determine whether CSF bulk flow was impaired in 5-week-old B3glct mutant mice, we injected 4% Evans blue dye into the left lateral ventricle of control and mutant brains and analyzed the distribution of dye in the cerebral ventricles of bisected brains (Figure 1). In control mice, the dye traveled to the fourth ventricle and labeled the perivascular spaces on the brain surface within 10 min of injection (Figure 1A, B, a–d). In contrast, the B3glct mutants had reduced staining in the fourth ventricle (Figure 1C, D, e–h), and a mouse with more severe hydrocephalus also displayed reduced staining in the ventral aspect of the third ventricle (Figure 1E, F, i–l). Consistent with reduced dye flow to the posterior, the B3glct mutant brains also displayed reduced perivascular labeling around the pons, medulla oblongata and pyramid, suggesting possible altered CSF flow dynamics in the mutant.
Fig. 1.
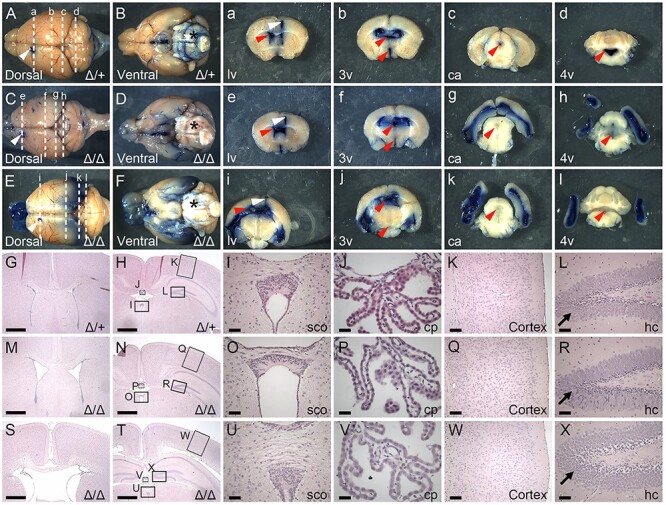
Loss of B3glct impairs CSF flow. (A–F and a–l) Representative images taken 10 min following injection of Evans Blue dye into the anterior horn of the left lateral ventricle (lv) (White arrowhead) at 5 weeks of age. (A, C, E) Dorsal and (B, D, F) ventral whole brain views with anterior to left and posterior to right. (a–l) Thick coronal sections of whole brains with approximate locations of the plane of sectioning indicated by dashed lines in A–F. White arrowhead indicates dye injection site. Red arrowheads indicate respective ventricles or central aqueduct. (A, B and a–d) In control brains (Δ/+) (n = 5), Evans blue dye flowed through the ventricular system and was detected in the lateral (lv), third (3v), central aqueduct (ca) and fourth (4v), as well as in the perivascular space (*) on the surface of the brain. (C–F and e–l) In contrast in B3glct knockout (Δ/Δ) (n = 4) brains, dye accumulated in the lateral ventricles with reduced flow to the third and fourth ventricles in moderate ventriculomegaly (C, D and e–h) and severe hydrocephalus (E, F and i–l) brains. (G–X) Hematoxylin and eosin staining of coronal sections from postnatal day 21 (P21) control (Δ/+) (G–L), B3glct knockout (Δ/Δ) with mild ventriculomegaly (M–R) and B3glct knockout (Δ/Δ) with severe hydrocephalus (S–X) animals. (G, M, S) Sections taken through lv and (H, N, T) sections through 3v. Arrows in panels L, R and X indicate granular layer in dentate gyrus in hippocampus. Boxes in panels H, N and T indicate regions expanded in panels I–L, O–R and U–X. Abbreviations: cp, choroid plexus; sco, subcommissural organ; hc, hippocampus. Scale bars: panels G, H, M, N, S and T 1 mm; panels I, O, U, L, R, X 100 μm; panels J, P, V 50 μm; and K, Q, W 200 μm.
To determine whether abnormalities detectable at the tissue level could be responsible for impairing CSF flow, we compared hematoxylin and eosin–stained histological sections from P21 B3glct control (Figure 1G–L) and B3glct mutant (Figure 1M–X) brains. We saw a range of lateral ventricle enlargement in B3glct mutants (Figure 1M and S) compared to control (Figure 1G) brains. B3glct mutants with significant ventricle enlargement exhibited organized but loosely arranged granular layer in the dentate gyrus of hippocampus (Figure 1R and X). These changes were likely secondary to hydrocephalus, but we cannot exclude the possibility of altered B3GLCT substrate in this tissue. Overall, loss of B3glct did not significantly alter the organization of other brain tissues (Figure 1N–Q and T–W) compared to controls (Figure 1H–L).
To identify which specific brain tissues could be affected by loss of B3glct, we employed RNAscope® to determine where B3glct was expressed at embryonic day 16 (E16) (Figure 2A–E″). Although, B3glct transcripts were ubiquitously detected at low levels throughout brain tissues such as the choroid plexus (Figure 2D, D′) and third ventricle (3 V) (Figure 2E, E″), transcripts were modestly enriched in the cortex ventricular zone (Figure 2B, B′, C, C′) and subcommissural organ (Figure 2E and E′). At P4, B3glct transcripts were detected at reduced levels in the lateral ventricle, choroid plexus and subcommissural organ (Figure 2F–G′). Pofut2 transcripts were similarly distributed at E16 and P4 (Supplemental Figure 1A–G′). Expression of B3glct in the ventricular zone and subcommissural organ raised the possibility that abnormalities in these tissues could contribute to defects in CSF flow.
Fig. 2.
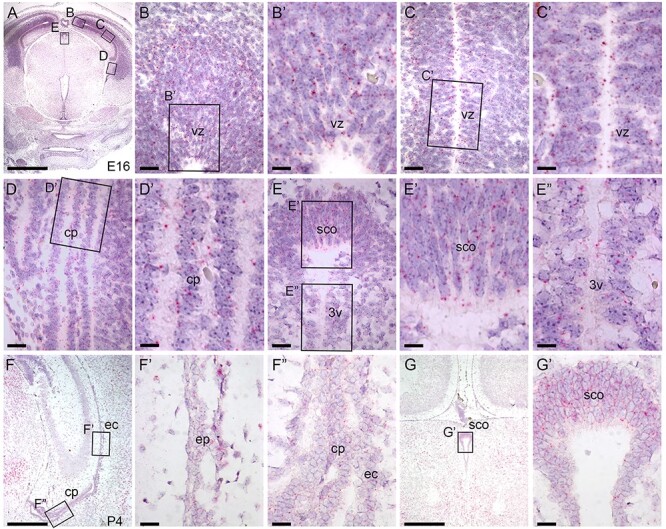
B3glct is expressed ubiquitously in brain. RNAscope® analysis of B3glct mRNA expression. Localization of B3glct mRNA appears as red dots overlying cells counterstained with hematoxylin (purple). (A–E″) At E 16.5, B3glct transcripts were localized in the ventricular zone (vz) of the cortex (B–C′), choroid plexus (cp) (D, D′), subcommissural organ (sco) (E, E′) and ependyma of third ventricle (3v) (E″). (F–G′) At P4, B3glct mRNA was localized to ependymal cells (ec) of the lateral ventricle (F′), choroid plexus (cp) (F″) and subcommissural organ (sco) (G, G′). Rectangles indicate the magnified regions of brain sections. Scale bars: panel A, 1 mm; panel F, G, 500 μm; panels B, C, D, E, F′, F″ and G′, 50 μm; and panels B′, C′, D′, E′ and E″, 20 μm.
Loss of B3glct reduces number of basal bodies and disrupts translational polarity of ependymal cells
Ependymal cells in the brain lateral ventricles are multiciliated and polarized in the epithelial plane (Del Bigio 1995; Mirzadeh et al. 2010). Translational polarity of ependymal cells is the coordinated displacement of cilia across the cells of the ependymal layer (Mirzadeh et al. 2010; Wallingford 2010; Takagishi et al. 2017). In mice, disrupted translational polarity of ependymal cilia is directly related to reduced CSF flow and is one major cause of hydrocephalus (Mirzadeh et al. 2010; Ohata et al. 2014; Takagishi et al. 2017; Abdelhamed et al. 2018). To determine if altered translational polarity of ependymal cells contributes to hydrocephalus in B3glct mutants, whole mounts of lateral ventricle walls from P21 brains were stained with γ-tubulin (labels basal bodies) and β-catenin (labels cell periphery) to measure the number and average displacement of the cilia basal bodies per cell and to assess the coordinated displacement of basal bodies across the tissue (Figure 3).
Fig. 3.
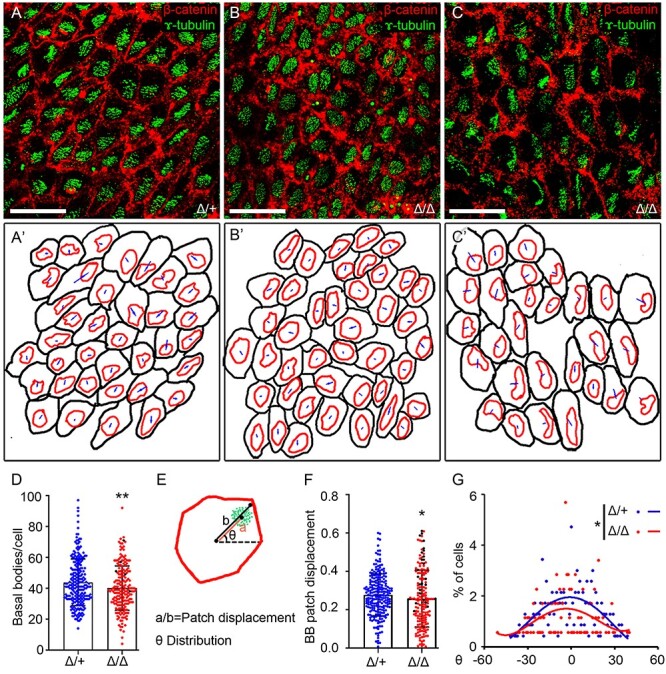
Loss of B3glct decreases number of basal bodies and alters translational polarity in ependymal cell. Analysis of cell polarity in P21 lateral ventricles. (A–C) Representative maximum projection images of P21 whole-mount brain lateral ventricular walls stained with γ-tubulin (green) to detect basal bodies and β-catenin (red) to identify cell borders in (A) B3glct control (Δ/+), (B) and B3glct knockouts (Δ/Δ) with mild ventriculomegaly, or (C) severe hydrocephalus. (A′–C′) Representative tracings of cells in maximum projection images used to calculate basal body patch displacement and theta distribution. (D) Total basal bodies per ependymal cells. (E) Cartoon depicting cell measurements used to calculate basal body patch displacement (a/b) (F) and theta angle (G). (D, F, G) Blue indicates B3glct control (Δ/+), red indicates B3glct knockout (Δ/Δ) with mild ventriculomegaly and black indicates B3glct knockout (Δ/Δ) with severe hydrocephalus. Data were evaluated for statistical significance using unpaired, two-tailed t-test (D), Mann–Whitney test (F) and Watson’s two-sample U2 test (G). *P ≤ 0.05, **P ≤ 0.01. Scale bars: panel A–C 20 μm.
The asymmetric localization of basal bodies in both control and B3glct mutants demonstrated that B3glct was not essential for postnatal transitioning of polarized cells to multiciliated ependymal cells (Figure 3A–C). However, in the B3glct mutants, the number of basal bodies per cell was significantly reduced compared to controls (Figure 3D); albeit, these numbers fell within the reported normal range (Mirzadeh et al. 2010). Despite being polarized, the patch displacement (ratio of the distance from the apical center of the cell to basal body patch [a] to the distance from the apical center of the cell to the edge of the cell [b]) was reduced in the B3glct mutants compared to controls (Figure 3E–F). Moreover, whereas in control lateral ventricles, most basal body patches were oriented to the same side of the cell (Figure 3A and A′), and the basal body patches in mutants with mild (Figure 3B and B′) or severe hydrocephalus (Figure 3C and C′) were not consistently oriented.
The orientation of basal bodies across the tissue was quantified by measuring the theta angle for each basal body patch within the plane of field (Figure 3E). In control animals, the coordinated orientation of basal bodies relative to the field of cells was reflected in the Poisson distribution of theta angles centered at zero (reflecting similar orientation) (Figure 3G). In contrast, the theta angles in B3glct mutants were more widely distributed with a lower percentage of cells exhibiting coordinated orientation (Figure 3G) and was consistent with the more disorganized appearance of the ependymal cell orientations (Figure 3B and C). Combined, these observations suggest that loss of B3glct contributed to defects in CSF flow by impacting either the number of cilia or translational polarity of ependymal cells.
SSPO is a novel B3GLCT substrate and loss of B3GLCT significantly reduced SSPO secretion in cultured cells
B3glct was also strongly expressed in the subcommissural organ, situated at the opening of the central aqueduct of the developing brain (Figure 2G and G′). Defects in development of the subcommissural organ or secretion of SSPO from this organ can cause hydrocephalus (Somera and Jones 2004; Baas et al. 2006; Meiniel 2007). We predicted that SSPO, with 17 POFUT2 consensus sites, could be impacted by loss of B3GLCT (Gonçalves-Mendes et al. 2003; Meiniel and Meiniel 2007) (Figure 4A). To evaluate whether SSPO was modified by POFUT2 and B3GLCT, we expressed and purified an SSPO fragment (TSRs 6–9) in HEK293T cells, digested with proteases and analyzed the resulting peptides by mass spectrometry to determine if the predicted POFUT2 consensus sequences were modified (Figure 4B, Supplemental Figure 2). A small fragment of SSPO was used because we were unsuccessful in expressing any larger portions of SSPO in HEK293T cells. This is likely because of the large number of cysteine-rich domains in SSPO including TSRs, EGF domains and CTCK domains (Figure 4A). Extracted ion chromatograms (EICs) of ions corresponding to the different glycoforms of peptides from TSRs 6, 7, 8 and 9 containing the POFUT2 consensus sequence revealed that all four TSRs were modified efficiently by POFUT2 and B3GLCT (Figure 4B, Supplemental Figure 2). In addition to O-fucose modifications, each of these TSRs contains W-X-X-W/C consensus sequences for C-mannosylation. These sites were also heavily modified on peptides detected from each of the TSRs (Figure 4C, Supplemental Figure 3) indicating that both the O-fucose and C-mannose modification sites on SSPO TSRs were modified.
Fig. 4.
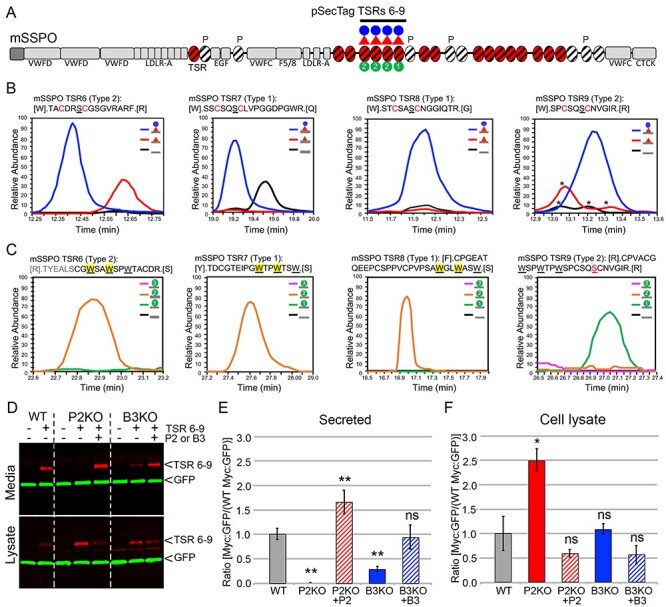
SSPO is a novel B3GLCT substrate and requires B3GLCT for efficient trafficking. (A) Domain structure of SCO-spondin (SSPO). The 25 SSPO TSRs are depicted by ovals. Red ovals identify 17 TSRs that have a consensus sequence for POFUT2 mediated O-fucosylation. White ovals are TSRs that lack a serine or threonine (S/T) in the consensus sequences for O-fucosylation, and P above a TSR indicates a proline in the position usually occupied by S/T. TSRs with consensus for modification with C-mannose are marked with diagonal black lines. In this study, we used the pSecTag2-SSPO TSRs 6–9 construct to confirm the presence of the glucose–fucose disaccharide (red triangle indicating fucose and blue circle glucose) and one or two C-mannose residues (green circles with #) on TSRs 6, 7, 8 and 9. Other SSPO domains include VWFD, von Willebrand factor–type D domain; LDLR-A, low density lipoprotein receptor class A domain; EGF, epidermal growth factor-like (EGF) domain; F5/8, factor V/VIII type C-like domain; VWFC, von Willebrand factor C repeats; CTCK, C-terminal cysteine knot domain. (B) Extracted ion chromatograms (EIC) of ions corresponding to the different glycoforms of peptides from TSRs 6, 7, 8 and 9 containing the POFUT2 consensus sequence. MS2 spectra for the major glycoforms and masses of ions used to generate the EICs can be found in Supplemental Figure 2. Although the score for the O-fucose glycopeptide from TSR6 was low, the mass of the parent ion matches the predicted mass of the glycopeptide and was used to generate the EIC. Black lines, unmodified peptide; red lines, fucose-modified peptide; blue lines, glucose–fucose–modified peptides. * indicate ions that match the mass of the relevant peptide but do not match the MS2 fragmentation pattern. (C) EICs of ions corresponding to the glycopeptides from TSRs 6, 7, 8 and 9 containing the C-mannosylation W-X-X-W motif. MS2 spectra for the major glycoforms and masses of ions used to generate the EICs can be found in Supplemental Figure 3. Black lines, unmodified peptide; green lines, Hex-modified peptide; orange lines, Hex–Hex modified peptides; pink lines, Hex–Hex–Hex modified peptides. The modified W is indicated by yellow shading for TSRs 6, 7 and 8. The MS2 data for the peptide from TSR9 are of insufficient quality to determine the location of the C-mannose. (D–F) Cell-based secretion assays were used to measure the effects of POFUT2 and B3GLCT mutations on trafficking of SSPO TSRs 6–9-Myc-His6 (TSRs 6–9, underlined in panel A) in wild-type HEK293T (WT), CRISPR-Cas9 mutagenized POFUT2 (P2 KO) or B3GLCT (B3 KO) HEK293T cells, or mutagenized cells rescued by cotransfection with plasmids encoding wild-type POFUT2 (+P2) or B3GLCT (+B3). (D) Representative western blot of the medium (above) and cell lysate (below). TSRs 6–9 were detected with anti-Myc (red), and GFP (green) was used as an internal transfection control. (E, F) Quantification of western blots of medium (E) and cell lysate (F). Data were evaluated for statistical significance using unpaired, one-tailed t-test. *P ≤ 0.05, **P ≤ 0.01, ns not significant. P ≤ 0.05).
To test if loss of the glucose–fucose disaccharide affected trafficking of SSPO in vitro, we evaluated the secretion of SSPO TSRs 6–9 in HEK293T POFUT2 or B3GLCT knockout cells (Figure 4D–F). SSPO TSRs 6–9 was secreted efficiently from the wild-type HEK293T cells. In contrast, loss of POFUT2 blocked the secretion of TSRs 6–9 (with intracellular accumulation of TSRs 6–9) and was rescued by cotransfection with POFUT2, suggesting that SSPO TSRs 6–9 required O-fucose for secretion. Loss of B3GLCT significantly reduced secretion of SSPO TSRs 6–9 (without significant accumulation of TSRs 6–9), and this reduction was rescued by cotransfection with B3GLCT (Figure 4D and E). These observations suggested that efficient secretion of SSPO TSRs 6–9 requires the O-linked disaccharide modification of its TSRs.
Secreted SSPO remains associated with BiP in B3glct mutants
To test whether O-linked glucose–fucose modification of SSPO TSRs was required in vivo, we examined the effects of the B3glct mutation on SSPO trafficking in the subcommissural organ of B3glct mutant mice. The subcommissural organ is pseudostratified and characterized by tall elongated kinociliated ependymal cells oriented such that their apical side faces the third ventricle/central aqueduct and their basal side the posterior commissure (Meiniel 2007) (Figure 5A, F, K). These cells have large ER cisternae where SSPO is synthesized and stored and subsequently transported through the Golgi apparatus to secretory vesicles formed in trans-Golgi. SSPO is ultimately released into the CSF where in many species processed forms of SSPO make up the major protein component of Reissner’s fiber (Munoz et al. 2019). Soluble forms of SSPO fragments are also found in the CSF (reviewed in Nualart et al. 1991; Munoz et al. 2019).
Fig. 5.
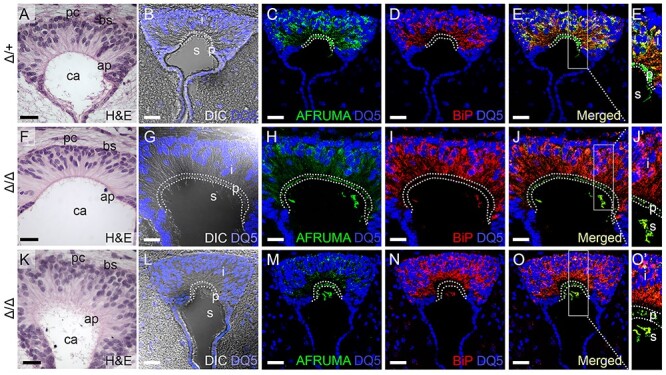
AFRUMA is secreted in B3glct mutant subcommissural organ. (A–O′) Analysis of SSPO trafficking in subcommissural organs from P21 wild-type (Δ/+) (A–E′) and B3glct knockout (Δ/Δ) with mild ventriculomegaly (F–J′) or severe hydrocephalus (K–O′). (A, F, K) Hematoxylin and eosin (H&E)-stained subcommissural organ sections. pc, posterior commissure; bs, basal side; ap, apical side; and ca, central aqueduct. (B, G, L) DRAQ5 (DQ5) staining of subcommissural organ sections merged with differential interference contrast (DIC) image to indicate boundaries of intracellular (i), presecretory (p) and secreted (s) regions of the subcommissural organ. (C–E′, H–J′, and M–O′) Maximum projection images of immunostained subcommissural organ sections stained with AFRUMA (green) and DRAQ5 (blue) (C, H, M) or anti-BiP (red) and DRAQ5 (blue) (D, I, N). Merged channels are shown in (E–E′, J–J′, and O–O′). Rectangles in (E, J, O) indicate regions digitally expanded to the right (E′, J′, O′). Dotted curves demarcate intracellular, presecretory and secreted regions in subcommissural organs. See Figure 6 for quantification of immunofluorescence signals and colocalization of signals. Scale bars: panels A, F, K, 50 μm, and panels B–E, G–J, L–O, 20 μM.
To determine how loss of B3glct affects trafficking of SSPO in vivo, we stained subcommissural organs with antibodies that recognize either Reissner’s fiber comprised primarily of SSPO (AFRUMA, green) or with antibodies that recognize the ER chaperone GRP78/binding immunoglobulin protein (BiP) (red). BiP facilitates protein folding in the ER and is often elevated under cellular stress resulting from protein misfolding (Wang et al. 2009; Adams et al. 2019). Sections were counterstained with DRAQ5 (blue) to identify nuclei and merged with differential interference contrast (DIC) images to delineate the cell margins (Figure 5B–E′, G–J′, L–O′). Quantification of AFRUMA and BiP corrected total cell fluorescence (CTCF) and AFRUMA:BiP colocalization are shown in Figure 6. In control animals, the AFRUMA signal was strongly detected in the intracellular perinuclear and cytoplasmic areas of the subcommissural organ (Figure 5C). The perinuclear AFRUMA staining overlapped significantly with BiP (Figures 5D–E′ and 6C) and is consistent with previous reports that intracellular SSPO resides for extended periods of time in the ER of normal subcommissural cells (Nualart et al. 1991, Munoz et al. 2019). In the apical presecretory region, AFRUMA was detected in granule-like structures devoid of BiP (Figure 5C–E′, dotted area and Figure 6A–C), consistent with previously described secretory granules containing the SSPO (Munoz et al. 2019). Once SSPO is secreted into the central aqueduct, AFRUMA staining (also devoid of BiP) detects fiber-like structures consistent with Reissner’s fiber (green next to “s” in Figures 5E′ and 6A–C).
Fig. 6.
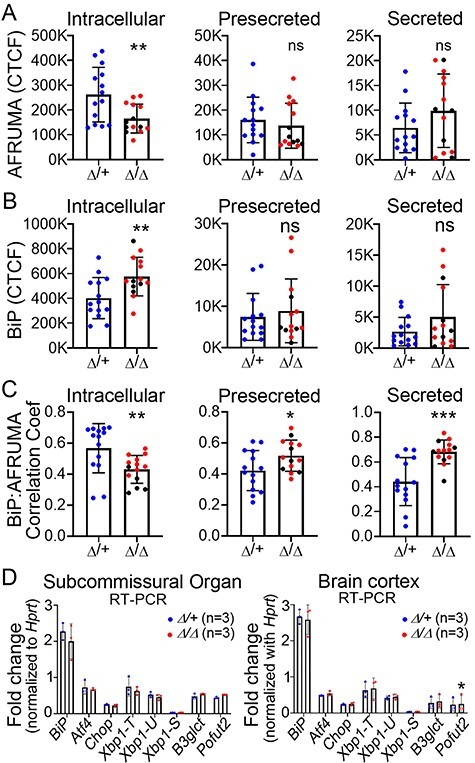
Secreted AFRUMA colocalizes with BiP in B3glct mutants, but UPR not activated. (A–C) Quantitation of corrected total cell fluorescence (CTCF) for AFRUMA (A) or BiP (B), and correlation coefficient for BiP and AFRUMA (C) in the intracellular and presecretory regions of the subcommissural organ and central canal (secreted). Representative images are shown in Figure 5. Fluorescence was measured from 14 sections located in the middle part of the subcommissural organ from five animals from each group (n = 5). Measurements for B3glct heterozygous controls (Δ/+) are plotted in blue dots; B3glct homozygotes (Δ/Δ) are plotted as red dots (mild ventriculomegaly) and black dots (severe hydrocephalus). (D) qRT-PCR analysis of UPR genes in subcommissural organ and brain cortex. Xbp1-T, -U and -S indicate total, unspliced and spliced form of Xbp1, respectively. Data were evaluated for statistical significance using unpaired, two-tailed t-test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ns not significant.
In contrast, AFRUMA staining in the intracellular region of B3glct mutants was significantly reduced and intracellular BiP staining was elevated (Figures 5H–J′, M–O′ and 6A–C). Unexpectedly, we observed that the AFRUMA signal in presecretory and secreted regions was comparable to controls, with BiP levels in these regions trending higher than controls (Figures 5C, H, M and D, I, N and 6A–C). Overall, in B3glct mutants, we observed a decrease in intracellular colocalization of AFRUMA:BiP and an increase in AFRUMA:BiP colocalization in the presecretory and secreted regions relative to controls (quantification in Figure 6C). The decreased intracellular levels of AFRUMA (quantification in Figure 6A) suggested that loss of B3GLCT led to degradation of some SSPO (presumably misfolded) in the ER. Moreover, the high colocalization of BiP with AFRUMA in the presecretory and secreted regions (quantification in Figure 6A–C) suggested that secreted SSPO in B3glct mutants was aberrantly folded or had defects in intra- or intermolecular disulfide bonding and/or processing.
To minimize the consequences of misfolded protein, cells activate the unfolded protein response (UPR) mediated by transcriptional regulators IRE1, ATF6 and PERK (Xu et al. 2005; Adams et al. 2019). We used reverse transcription quantitative PCR (RT-qPCR) to test whether the UPR was activated in the B3glct mutant subcommissural organ or other brain tissues (Figure 6D). Transcript levels of BiP, Atf4, Chop, Xbp1 (total, spliced and unspliced), B3glct (primers upstream of exon 4 deletion) and Pofut2 were unaffected in B3glct mutants relative to controls. These results suggested that loss of glucose on TSRs was not sufficient to trigger the UPR and did not trigger compensatory transcription of Pofut2.
Adamts9 and Adamts20 are expressed by lateral ventricle ependymal cells as well as the subcommissural organ
Previously, Adamts3 was shown to be expressed in the developing subcommissural organ (E 17.5) (Le Goff et al. 2006), raising the possibility that B3GLCT substrates other than SSPO could also be affected in B3glct mutants. We used RNAscope® to identify brain tissues expressing Adamts20 and the closely related Adamts9 (also a B3GLCT substrate; Dubail et al. 2016) at E16 and at P4 when radial glial cells lining the ventricles are transitioning to the polarized multiciliated ependymal cells (Figure 7). At E16, both Adamts9 and Adamts20 were expressed in radial glial cells lining the lateral ventricles and subcommissural organ and had complementary expression patterns in the developing choroid plexus with Adamts9 in the endothelial cells and Adamts20 in the epithelial layer (Figure 7A–D and I–L).
Fig. 7.
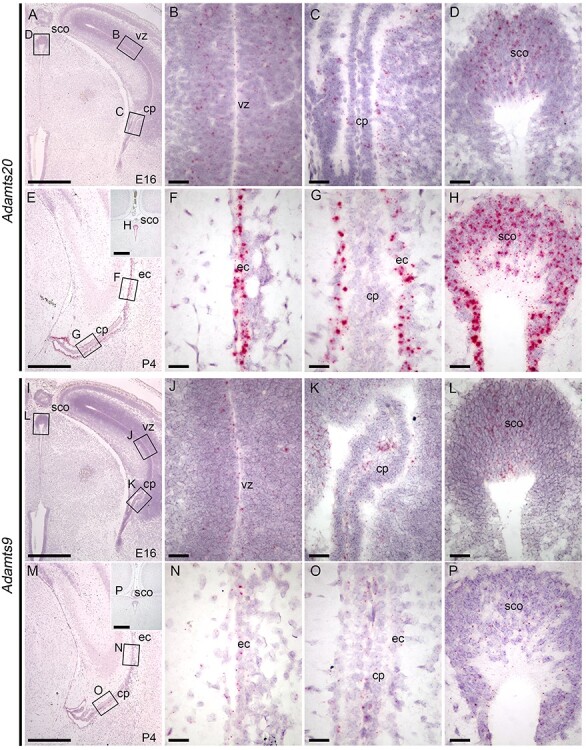
Adamts9 and Adamts20 mRNA localization in brain at E16 and P4. RNAscope™ analysis of (A–H) Adamts20 and (I–P) Adamts9 mRNA localization at E16 (A–D, I–L) and P4 (E–H, M–P). Areas indicated by black rectangles in panels A and E are expanded in panels B–D and F–H, respectively. Areas indicated by black rectangles in I and M are expanded in J–L and N–P, respectively. Scale bars: panels A, E, I, M, and insets, 500 μm; panels B–D, F–H, J–L and N–P, 50 μm. Abbreviations: vz, ventricular zone of cortex; cp, choroid plexus; ec, ependymal cells of lateral ventricle; sco, subcommissural organ.
By P4, Adamts20 was strongly expressed in the ependymal cells of the lateral ventricle, ependymal cells lining the central aqueduct and subcommissural organ, with no detectable expression in the choroid plexus of the lateral ventricle (Figure 7E–H). In contrast, Adamts9 was expressed at low levels in the ependymal cells of the lateral ventricle, endothelial cells of the choroid plexus and subcommissural organ (Figure 7M–P). We confirmed Adamts3 expression in the brain cortex, ependymal cells of the developing lateral ventricle and the choroid plexus at E16 and identified low levels of Adamts3 expression at P4 in lateral ventricle and subcommissural organ (Supplemental Figure 4A–H). In addition, we demonstrated that F-spondin (Spon1), also a B3GLCT substrate, was weakly expressed in the cells lining the lateral ventricle, subcommissural organ and choroid plexus at E16, with diminished expression at P4 (Supplemental Figure 4I–P). These data provided correlative evidence that hydrocephalus in B3glct mutants resulted from the combined effects of the mutation on multiple substrate proteins expressed by ependymal cells and the subcommissural organ.
Discussion
Maintaining CSF homeostasis is necessary to ensure normal brain physiology and requires a balance of CSF production, flow and absorption (Kaur et al. 2016; Benveniste et al. 2019). Optimal CSF flow requires unobstructed development of the brain ventricular system and coordinated movement of cilia on ependymal cells (Kahle et al. 2016). Here, we demonstrated that hydrocephalus in mouse B3glct mutants likely resulted from impaired CSF flow resulting from the combined effects of the mutation on numbers of cilia and translational polarity of ependymal cells, and abnormalities in subcommissural organ/Reissner’s fiber assembly. High incidence of hydrocephalus in Adamts20 mutants (Holdener et al. 2019), combined with the observation that Adamts9 and Adamts20 were coexpressed in both the ependymal cells and subcommissural organ (Figure 7) raised the possibility that reduced function of these B3GLCT substrates contributed to both defects in polarity of ependymal cells and subcommissural organ function. In addition, we demonstrated that SSPO was a novel B3GLCT substrate and that loss of B3glct not only impacted trafficking but also potentially impacted extracellular function of SSPO.
Impact of B3glct mutation on ependymal cell polarity
The planar polarity of ependymal cells coordinates ciliary beating and consequently regulates the direction of CSF flow (Mirzadeh et al. 2010). Polarization of ependymal cells starts during embryonic development and is completed by 2 weeks postnatally (reviewed in Redmond et al. (2019)). During embryogenesis, the radial glial precursors of the ependymal cells become polarized with a single asymmetrically localized cilium. By P5, a subpopulation of radial glial cells lining the brain ventricles differentiate into polarized multiciliated ependymal cells with basal bodies displaced to one side of the cell. In general, the basal body patches in these multiciliated ependymal cells are similarly displaced across the tissue (termed translational polarity), but the basal bodies of individual cilia are not yet oriented relative to one another within the cell and are thought to produce a weak ependymal flow. Between P4 and P20, the basal bodies within each cell orient parallel to the CSF flow, establishing rotational polarity of the ependymal cells and promoting maximum CSF flow (Mirzadeh et al. 2010; Ohata and Alvarez-Buylla 2016). Altered translational and rotational polarity of ependymal cilia causes hydrocephalus in Daple (Dvl-associated protein with high leucine content (Takagishi et al. 2017), Dishevelled (Ohata et al. 2014) and Kif3a (Mirzadeh et al. 2010). Some mouse mutants like nonmuscle myosin II (Hirota et al. 2010) and Mmp12 (Shan et al. 2018) reported change in cilia polarity and the former also develops hydrocephalus.
Given the elevated levels of Adamts20 mRNA at P4, when cells lining the ventricular zone transition to polarized multiciliated ependymal cells, and the high incidence of hydrocephalus in ADAMTS20 mutants (Holdener et al. 2019), we predict that ADAMTS20 is a major player in ependymal cell function (Figure 8A). ADAMTS9 and ADAMTS20 are secreted metalloproteinases with key roles reported in remodeling extracellular matrix during development (McCulloch et al. 2009; Enomoto et al. 2010; Nandadasa et al. 2015) and were recently shown to play an important noncanonical role in primary ciliogenesis by modulating basal body maturation and ciliary vesicle growth (Nandadasa et al. 2019). For this reason, loss of B3glct could alter ependymal cell polarity by impacting extracellular matrix properties or primary cilia development, or alternatively through an as yet unrecognized effect on multiciliogenesis. However, the lack of laterality defects or strong hedgehog phenotypes in B3glct mutants suggested that if primary cilia development were impacted by loss of B3GLCT, the effect would be subtle. Changes in extracellular matrix properties could result from altered proteolysis of ADAMTS9 and/or ADAMTS20 substrates including versican (Wu et al. 2002; Schwartz and Domowicz 2004; Enomoto et al. 2010). ADAMTS9 also likely influences fibronectin fibril turnover through direct cleavage at sites shared with other metalloproteinases including MMP2 (Doucet and Overall 2011; Shi and Sottile 2011; Wang et al. 2019). Notably, ependymal cell translational polarity is also affected by extracellular matrix changes in Mmp2 (Shan et al. 2018), dystroglycan (McClenahan et al. 2016) and MT-MMP mutants (Jiang et al. 2020) where the latter two exhibited hydrocephalus. Future studies will need to evaluate whether these molecules are substrates for ADAMTS9/20 and whether the effects of the B3glct mutation affect the rotational polarity of ependymal cells, cilia elongation and/or CSF flow dynamics.
Fig. 8.
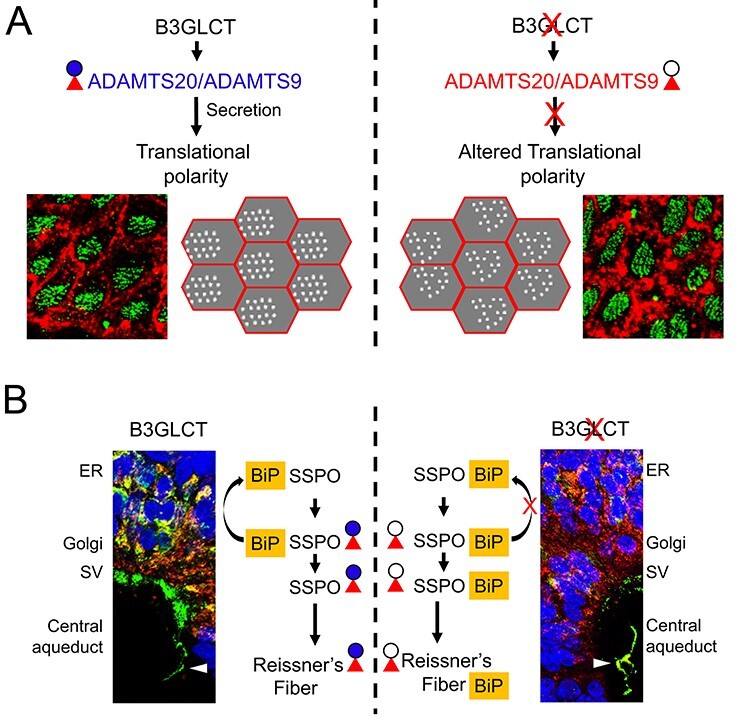
O-linked glucose-β1-3fucose disaccharide modification of TSRs is important for secretion and extracellular assembly of B3GLCT substrates. Impaired CSF flow in B3glct mutants likely results from the combined effects of the mutation on multiple substrates by impacting the efficiency of substrate secretion and/or extracellular assembly/function of substrates. (A) B3GLCT/POFUT2 modification facilitates stabilization of ADAMTS20 and ADAMTS9 TSR folds and promotes efficient secretion of these substrates, especially ADAMTS20 (Holdener et al. 2019). (A, top left) We propose that postnatal expression of Adamts20 and Adamts9 in the ependymal cells promotes translational polarity of the ependymal cell cilia by altering properties of the matrix and/or a noncanonical role in regulating cilia. (A, top right) In B3glct mutants, defects in ADAMTS20 and ADAMTS9 secretion (Holdener et al. 2019) result in impaired translational polarity of ependymal cells in B3glct mutants. (B) We propose that B3GLCT/POFUT2 modification of SSPO is not only important for efficient trafficking of SSPO but is also likely important for extracellular assembly of SSPO. (B, left) In wild-type subcommissural organ cells, SSPO and BiP colocalize within the ER. As SSPO transits to the Golgi, BiP is returned to the ER, and SSPO is packaged into secretory vesicles, processed and secreted. Secreted SSPO is found both as soluble fragments in the CSF and also assembles via intermolecular and intramolecular disulfide bonds (Munoz et al. 2019). (B, right) In B3glct mutants, intracellular levels of SSPO are reduced consistent with a role for the O-linked disaccharide in stabilizing the TSR fold. However, the continued association of SSPO with BiP in secretory vesicles and Reissner’s fiber suggests a defect in intra- or intermolecular assemble of SSPO, raising the possibility for an alternate role for the disaccharide in protecting the TSR disulfide bond from inappropriate extracellular intermolecular isomerization. Red triangle and blue circle indicate fucose and glucose, respectively, and black circle indicates glucose is absent. Abbreviations: ER, endoplasmic reticulum; SV, secretory vesicles.
In contrast to mouse B3glct mutants (Holdener et al. 2019), hydrocephalus/ventriculomegaly is less common in human PTRPLS patients (Reis et al. 2008) and can be associated with severe brain malformations (Schoner et al. 2013). Although the human and mouse ventricular systems are structurally similar, anatomical variations such as topology and CSF flow through the third ventricle likely make the mouse more susceptible to mutations that impact cilia function (Eichele et al. 2020). This prediction is consistent with the observation that hydrocephalus is observed at lower frequencies in human patients with cilia motility defects (Ibanez-Tallon et al. 2004) or significantly reduced number of cilia (Boon et al. 2014; Amirav et al. 2016). These results suggest that hydrocephalus or ventriculomegaly in PTRPLS patients (Reis et al. 2008) likely results from the impact of the mutation on multiple substrates in more than one tissue.
Impact of B3glct mutation on substrates in the subcommissural organ
Here, we demonstrated that multiple B3GLCT substrates including ADAMTS9, ADAMTS20 and ADAMTS3 as well as SSPO are expressed by the subcommissural organ and defects in folding one or a combination of these substrates could be responsible for the elevated levels of BiP protein in B3glct mutants. SSPO, with 17 of 25 TSRs containing POFUT2/B3GLCT consensus sites, is a major protein secreted by the subcommissural organ, situated on the dorsal side of the brain third ventricle at the entrance to the central aqueduct (Rodríguez et al. 1992; Rodriguez et al. 1998; Munoz et al. 2019). In many vertebrates, secreted SSPO assembles into Reissner’s fiber that extends from the subcommissural organ to the posterior of the spinal cord (Rodríguez et al. 1992, Rodriguez et al. 1998, Munoz et al. 2019), with soluble SSPO fragments also detected in the CSF (Vio et al. 2008).
Colocalization of SSPO with BiP in the presecretory vesicles and central canal in B3glct mutants suggested that defects in SSPO trafficking or assembly of Reissner’s fiber could contribute to development of hydrocephalus in mouse B3glct mutants (Figure 8B). Consistent with this prediction, hydrocephalus correlates with agenesis of the subcommissural organ in mouse mutants lacking Hdh, Msx1, Rfx3 or Psen1 or overexpressing Pac1 in the subcommissural organ and can also be associated with aqueductal stenosis (Ramos et al. 2004; Baas et al. 2006; Lang et al. 2006; Dietrich et al. 2009; Nakajima et al. 2011). Moreover, in rat, reduced secretion of SSPO in the spontaneously hypertensive and fetal-onset (H-Tx) hydrocephalus models precedes development of hydrocephalus (Somera and Jones 2004; Martínez-Peña y Valenzuela et al. 2006). Consistent with the role of SSPO/Reissner’s fiber in facilitating normal CSF flow, maternal delivery of anti-Reissner’s fiber antibodies to the rat fetus blocks the Reissner’s fiber formation (Vio et al. 2000) and led to the development of hydrocephalus. The lowered frequency of hydrocephalus in PTRPLS patients could stem from differences in subcommissural organ/SSPO function as this secretory organ undergoes regressive development during the early years of life (Rodriguez et al. 2001).
In contrast to mouse B3glct mutants or mutations affecting development of the mouse subcommissural organ, hydrocephalus was not reported in zebrafish b3glct or adamts9 loss-of-function mutants (Weh et al. 2017; Gray et al. 2021), scospondin (SSPO homologue) loss-of-function mutants (Cantaut-Belarif et al. 2018) or scospondin hypomorphic mutants that lead to disassembly of Reissner’s fiber (Weh et al. 2017; Orts-Del'Immagine et al. 2020; Troutwine et al. 2020). The lack of hydrocephalus in these fish mutants could result from species differences in CSF flow as well as differences in the effects of heartbeat or body movement on ventricle/aqueduct development (Olstad et al. 2019).
Role of O-linked glucose–fucose and C-mannosylation for SSPO secretion
SSPO, with 10 low-density lipoprotein receptor-A (LDLR-A) repeats and 25 TSRs (each with three disulfide bonds) (Figure 4A), presents a formidable challenge for the protein folding machinery. In the ER, POFUT2 and B3GLCT recognize and modify correctly folded TSRs, where the disaccharide is predicted to stabilize the TSR fold and accelerate the overall rate of TSR folding (Vasudevan et al. 2015; Holdener and Haltiwanger 2019). The range of phenotypes in human PTRPLS patients and mouse B3glct mutants suggests that loss of B3GLCT impacts some substrates more than others (Holdener et al. 2019). We previously demonstrated that ADAMTS20 was particularly sensitive to loss of glucose on the O-linked fucose. Reduced SSPO TSRs 6–9 trafficking in B3GLCT mutant cell lines (Figure 4) and reduced intracellular SSPO in B3glct mutants (Figure 5) suggested that SSPO trafficking was moderately impacted by loss of B3GLCT.
One difference between ADAMTS20 and SSPO TSRs that could account for this difference in trafficking efficiency is the presence of multiple C-mannosylation consensus sites (W-X-X-W or W-X-X-C) in all 25 SSPO TSRs (with confirmed modifications for TSRs 6–9 Figure 4A and C) vs. only one W-X-X-C site in 15 ADAMTS20 TSRs. Recently, it was shown that C-mannosylation on UNC-5 TSR2 stabilizes the native TSR fold and accelerates the rate of TSR folding (Shcherbakova et al. 2019). For this reason, we predict that in the absence of B3GLCT, the numerous C-mannosylation modifications on SSPO TSRs contributed to more efficient trafficking of SSPO (Figure 4) compared to ADAMTS20 (Holdener et al. 2019). Although this raises the possibility that secretion of other B3GLCT substrates with conserved C-mannosylation sites will be less sensitive to loss of B3GLCT, a counterexample exists. The TSRs of ADAMTS9, a close homolog of ADAMTS20, are not modified by C-mannose (Dubail et al. 2016), and secretion of ADAMTS9 TSRs 2–8 is only partially reduced in HEK293T cells lacking B3GLCT, similar to the effect on SSPO TSRs 6–9. Thus, there must be factors in addition to C-mannosylation that contribute to whether a POFUT2 substrate requires B3GLCT for secretion.
Conclusions
The results of this study identified defects in B3GLCT substrates and tissues that likely contribute to development of ventriculomegaly/hydrocephalus in B3glct mutant mice and provide additional evidence that failure to extend O-fucose on TSRs differentially affects B3GLCT substrates. It is unclear whether these differences in sensitivity are specific to the substrate or could be impacted by the tissues expressing the substrates. The altered dentate gyrus granular layer in mouse B3glct mutants underscores the complexity of the B3glct mutant phenotype and raises the possibility that defects in other B3GCLT substrates required for neurogenesis/neural pathfinding (i.e. SEMA5A, 5B, ADAMTS3, 4, 5) and/or blood vessel/lymphatic function (i.e. THSD1, ADAMTS1, 3, 8) could also be impacted by loss of B3GLCT and contribute to development of ventriculomegaly/hydrocephalus in mice and/or neurological changes in human PTRPLS patients (Lett et al. 2009; Hisanaga et al. 2012; Duan et al. 2014; Rodriguez-Manzaneque et al. 2015; Janssen et al. 2016; Ogino et al. 2017; Bradshaw et al. 2020; Nandadasa et al. 2020).
Materials and methods
Ethics statement
All animal work was conducted according to relevant national and international guidelines and under approved protocols. Stony Brook University animal facilities were approved by the NIH Office of Laboratory Animal Welfare (OLAW), assurance number D-16-00006. The animal studies were approved by the Stony Brook University Institutional Animal Care and Use Committee (IACUC), which follows all the guidance set forth in: Public Health Service Policy on Humane Care and Use of Laboratory Animals distributed by Office of Laboratory Animal Welfare, NIH; Animal Welfare Act and Animal Welfare Regulations distributed by United States Department of Agriculture and Guide for the Care and Use of Laboratory Animals distributed by the National Research Council. Stony Brook University animal facilities are accredited by Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC International).
Animals
All mice were housed and maintained under controlled temperature and light (22°C, 12 h light and 12 h dark) with access to sufficient food and water. At weaning, diet was supplemented with DietGel® 76A (ClearH20; Cat# 72-07-5022) to ensure that mutant animals received adequate hydration and nutrition. Generation of B3glctTm1bKOMP (B3glct-Δ4) mice was described previously (Holdener et al. 2019). These mice were maintained by backcrossing the B3glct-Δ4/+ males with C57BL/6J females at Stony Brook University. Mice backcrossed for 4–10 generations were used in this study.
Systemic CSF flow analysis
The patterns of CSF flow in the ventricular system was assessed and analyzed as described previously (Abdelhamed et al. 2018). Briefly, mice at approximately 5 weeks of age were deeply anesthetized with intraperitoneal administration of Ketamine (100 mg/Kg) and Xylazine (10 mg/Kg). Evans Blue dye (4% in PBS) was injected into the left lateral ventricle at 5 μL/min using the following coordinates: −1.8 mm deep, −0.8 mm left and −1.7 mm posterior from the bregma. Mice were euthanized after 10 min of injection and brains were dissected out and fixed in ice-cold 4% paraformaldehyde (PFA) overnight. Fixed brains were cut into 2 mm-thick coronal slices and photographed.
Whole-mount immunostaining of brain lateral ventricle wall
Whole-mount immunohistochemistry of the lateral ventricle wall was adapted from previous studies (Mirzadeh et al. 2010; Shan et al. 2018). Briefly, mouse brains were dissected to expose the lateral ventricle walls and fixed in 4% PFA in PBS at 4°C overnight. Tissues were then washed with 1X PBS and blocked in blocking solution (2.5% Goat serum; Vector Laboratories, Cat no. s1012) at room temperature for 1 h, followed by incubation with primary antibodies overnight at 4°C (1:400, mouse anti-β-catenin, BD Transduction Laboratories, Cat no. 6101154, and 1:400, rabbit polyclonal anti-γ-tubulin, Sigma-Aldrich, Cat no. T-5192) incubation. Secondary antibody incubation was performed at room temperature for 2 h (1:500; goat antirabbit IgG H&L Alexa Fluor® 488, Abcam, Cat. no. ab150077 and 1:500 goat antimouse IgG H&L Alexa Fluor® 647, Abcam, Cat. no. ab150115). Antibodies were diluted in blocking solution. Then, the tissues were washed with PBS and lateral ventricle walls were further dissected and mounted on glass slides with DAPI Fluoromount-G® (SouthernBiotech, Cat. no. 0100-20) and coverslipped. Confocal images were taken at 100× using Leica SP5 confocal (Leica, Germany).
ImageJ (http://imagej.net/) was used to analyze the basal bodies per cell, basal bodies patch displacement and basal bodies patch angle theta distribution from maximum projected images. Briefly, we first used the freehand selection tool to define regions of interest (ROIs) for the cell boundaries (β-catenin signal) and region of basal bodies within the cell (γ-tubulin signal). To calculate the number of basal bodies per cell, we used the analyze particle function to determine the number of green dots (basal bodies) per cell boundary ROI. To determine the basal bodies patch displacement and angle distribution per cell, we used the measure function to identify the centroids of the cell and the basal bodies patch and drew a line connecting the centroids and intersecting the cell boundary. These three coordinates, centroids of cell, centroids of basal bodies patch and the cell boundary intersection point, were exported to Microsoft Excel to determine the patch displacement and basal bodies patch angle (theta angle). For control (n = 6 mice), a total of 21 fields (3–4 per animal) were used with a total of 238 cells used for basal body number analysis and 233 cells for basal bodies patch displacement and theta distribution analyses. A total of 24 fields were used for analysis of 6 B3glct mutants (n = 6) with 204 cells used for basal body number analysis and 178 cells used for basal bodies patch displacement and theta distribution analyses. Data were evaluated for significance using unpaired, two-tailed t-test, Mann–Whitney (two-tailed) (GraphPad Prism 8) and Watson’s two sample U2 test (Oriana 4 version 4.02; Kovach Computing Services) for basal bodies count, patch displacement and theta distribution, respectively.
Brain histology and immunohistochemistry of subcommissural organ
Deeply anesthetized P21 mice were perfused with 4% PFA and brains were then postmortem fixed with 4% PFA for several days or until processed further. Fixed brains were rinsed with PBS and processed for paraffin embedding. Briefly, brain tissues were dehydrated by serially immersing into 30%, 50%, 70%, 80%, 90%, 95% and 100% ethanol and then xylene. The tissues were then infiltrated with and embedded in paraffin. The embedded tissue was sectioned at 5 μm thickness using a microtome.
Brain sections were routinely stained with Mayer’s hematoxylin and eosin (H&E) staining. Briefly, sections were deparaffinized using xylenes and rehydrated using ethanol (100%, 95%, 80% and 70%), washed with reverse osmosis water and stained in Mayer’s hematoxylin (Sigma, Cat. no. MHS32-1 L) for 10 min followed by running tap water washing for another 20 min. The sections were then passed through 70% ethanol for 2 min and counterstained with eosin (Fisher Scientific Cat. no. E511–25) for a minute. The sections were dehydrated for 2 min each by passing through 70% and 95% ethanol, which was followed by three washes with 100% ethanol and finally cleared in xylenes before mounting. Sections were mounted using Secure Mount™ (Fisher Scientific, Cat. no. 23–022208) solutions and covered with a coverslip.
For immunohistochemistry, the sections were deparaffinized using xylenes and rehydrated using ethanol (100%, 95%, 80% and 70%), rinsed with water and finally with 1X PBS. Endogenous peroxidase activity was blocked by incubation with 3% hydrogen peroxide in methanol for 10 min. Sections were then rinsed with 1X PBS and permeabilized with 0.1% Triton-X-100 in PBS for 30 min. The sections were blocked with 2.5% goat serum (Vector laboratories, Cat no. s1012) for 30 min and incubated overnight with anti-GRP78 BiP antibody (Abcam, Cat. no. ab21685) (1:1000) or rabbit polyclonal raised against bovine Reissner’s fiber (AFRUMA). AFRUMA (with recommended use at 1:1000) was a gift from Prof. Dr. Jesús Mateos Grondona, Universidad de Málaga, Spain, and was generated as described for AFRU in Rodríguez et al. 1984 (Rodríguez et al. 1984). The immunoreactivity was detected by using Goat Anti-Rabbit IgG H&L (Alexa Fluor® 488) (Abcam, Cat. no. ab150077) (1:500) or Cy3 conjugated Affinipure donkey antirabbit IgG (H + L) (Jackson ImmunoResearch Laboratories Inc., Cat. no. 711–165-152) (1:500). Then the slides were mounted either with DAPI Fluormount-G® (SouthernBiotech, Cat. no. 0100–20) or DRAQ5 (Abcam, Cat. no. ab108410) (1:1000) in SlowFade™ Gold Antifade Mountant (Invitrogen, Cat. no. S36937) and coverslipped.
Histological sections were photographed using a Nikon Optiphot microscope, AxioCam MRc camera and AxioVisionLE program (Zeiss). Fluorescent images were taken at 100× using Leica SP5 confocal (Leica, Germany). ImageJ (http://imagej.net/) was used to measure the fluorescence of the images as described previously (Gavet and Pines 2010). Fluorescence was measured in sections obtained from five animals (n = 5) for each group with a total of fourteen sections per animal located in the midsection of the subcommissural organ were evaluated. Data were evaluated for significance using unpaired, two-tailed t-test.
Analysis of SSPO O-fucosylation and C-mannosylation
Since SSPO is a very large protein, we sought to produce a portion containing several TSRs with O-fucose and C-mannose consensus sequences (Figure 4A) to confirm that the sites were being modified. We synthesized (GenScript) the DNA encoding TSRs 4–10 (Uniprot: Q8CG65 from 2336 to 2919; 584 amino acids, ENA mRNA: AJ491857.1 from 7006 to 8757; 1752 bps) with HindIII and XhoI restriction sites at 5′ and 3′, respectively. This sequence was subcloned into pSecTag2/Hygro C (Invitrogen), which encodes an N-terminal signal peptide and a C-terminal Myc-His6 tag. Upon transfection into HEK293T cells, the protein did not secrete well. Truncation mutants were generated eliminating one TSR at a time, and each was tested for secretion. The pSecTag2-SSPO TSRs 6–9 (generated using primers were 5′-TCCTGTGGCTGGTCTGCC-3′ and 5′-CAAAGCTTCGTACGTACGGCG-3′) were efficiently expressed and used for all experiments. pSecTag2-SSPO TSRs 6–9 were transfected into HEK293T cells, and the cells were cultured in Opti-MEM® Reduced Serum Medium (Gibco®). After 3 days, medium was collected and SSPO (TSRs 6–9) was purified with Ni-NTA chelating chromatography resin (G-Biosciences, St. Louis, MO) as described (Kakuda and Haltiwanger 2014). Purified protein was subjected to in-solution digestion as described (Kakuda and Haltiwanger 2014), and the resulting peptides were injected into an EASY-nLC 1000 HPLC system (Thermo Fisher Scientific) for peptide separation using a C18 reverse-phase column attached to a Q Exactive Plus Orbitrap mass spectrometer (Thermo Fisher Scientific). Separation of (glyco) peptides was performed using a 30 min binary gradient consisting of solvent A (5% acetonitrile in 0.1% formic acid) and solvent B (80% acetonitrile in 0.1% formic acid) with a constant flow rate of 300 nl/min. Spectra were recorded with a resolution of 70,000 in the positive polarity mode over the range of m/z 400–2000 and automatic gain control target value was 1 × 106. The 10 most prominent precursor ions in each full scan were isolated for higher energy collisional dissociation-tandem mass spectrometry (HCD-MS/MS) fragmentation with normalized collision energy of 27%, and automatic gain control target of 1 × 105, an isolation window of m/z 1.2, dynamic exclusion enabled and fragment resolution of 17,500. Raw data were analyzed using Xcalibur and Proteome Discoverer v.42.1.0.27.1981 (Thermo Fisher Scientific) and Byonic™ v.2.10.5 (Protein Metrics). Extracted ion chromatograms of all identified (glyco) peptides were generated using Xcalibur™ v.4.0.27.19 (Thermo Fisher Scientific).
Cell-based secretion assays and western blot analyses
Cell-based secretion assays for Mouse SSPO TSRs 6–9 were performed in wild-type HEK293T and CRISPR-Cas9–generated POFUT2 and B3GLCT knockout HEK293T cells as previously described (Holdener et al. 2019). Briefly, cells were transfected with a total of 1.94 μg of DNA, which included 1.6 μg of pSecTag2-SSPO TSRs 6–9 or 1.6 μg of empty vector, plus 0.24 μg of pcDNA4-POFUT2 (Benz et al. 2016) or 0.24 μg of pcDNA3.1-B3GLCT (Sato et al. 2006) or 0.24 μg of either pcDNA4 or pcDNA3.1 empty vectors, respectively, plus 0.1 μg of pEGFP-N1 (encoding Green Fluorescent Protein) (Schneider et al. 2018). Polyethylenimine (PEI) was used as a transfection reagent (1 mg/mL) at a ratio of 1:6 (DNA:PEI) for each well (Thomas et al. 2005). After a 48-h incubation, media was harvested and cells were lysed in buffer consisting of 1% Triton-X 100, 25 mM Tris, pH 8.0 containing a complete™ protease inhibitor cocktail tablet (Roche, Cat. No. 11836145001). Three biological replicates were performed for each condition.
For western blot analysis of media and cell lysates, proteins were resolved on a 4–20% gradient gel, transferred to a 0.45 μm nitrocellulose membrane and blocked in 5% milk. Membranes were detected for SSPO TSRs 6–9 with anti-Myc antibody (9E10, Stony Brook University Cell Culture/Hybridoma facility) and Alexa Fluor IRDye®680RD goat antimouse (LI-COR, Cat. No. 926–68070). GFP loading control was detected with anti-GFP antibody (Cell Signaling Technologies, Cat. No. 2555S) and Alexa Fluor IRDye® 800CW goat antirabbit (LI-COR, Cat. No. 926–32211). Membranes were incubated in primary antibodies at 4°C overnight at a 1:2000 dilution and then incubated in secondary antibodies at room temperature for 1 h in the dark at a 1:10,000 dilution. The membranes were imaged and quantified using the Odyssey® CLx system and analyzed with Image Studio Lite software (LI-COR Biosciences).
RNA extraction and real-time quantitative RT-PCR (qRT-PCR)
Mouse subcommissural organs and brain cortex were separately dissected out from the brain and total RNA was extracted using RNeasy® Mini Kit (Qiagen, ID 74104) according to manufacturer’s instructions. Total of 2 μg RNA from each RNA sample was reverse transcribed using SuperScript™ VILO™ cDNA Synthesis Kit (Invitrogen, Cat. no. 11754050). Real-time PCR was carried out using PowerTrack™ SYBR Green Master Mix (Thermo Fisher Scientific, Cat. no. A46109) on StepOnePlus™ Real-Time PCR System. The qRT-PCR results from control (n = 3; 3 replicates per sample) and mutant (n = 3; 3 replicates per sample) animals for each gene were normalized to those of Hprt and expressed as fold change, and the data are expressed as means ± standard deviations. The mean expression levels were compared between the control and the mutant groups using Student’s t-test. The primers of genes used were listed in Supplemental Table I.
RNAScope® analyses of mRNA localization
Mouse brain at embryonic day 16 (E16) and postnatal day 4 (P4) was fixed overnight in 4% PFA and processed for paraffin sectioning (5 μm). RNAScope® assay was carried out according to Advanced Cell Diagnostics’ instructions using B3glct (440301), Adamts3 (400431), Adamts9 (400441), Adamts20 (400541), Spon1 (578261) and Pofut2 (465281) probes and ACD HybEZ™ II Oven (Advanced Cell Diagnostics, Inc., Hayward, CA). Extended boiling with target retrieval reagent was followed. Probe binding was visualized by using RNAscope® 2.5 HD Detection Kit (Red) using Mayer’s hematoxylin as a counterstain. Bacterial gene encoding dihydrodipicolinate reductase (DapB; 310043) and Mus musculus peptidylprolyl isomerase B (Ppib; 313911) were used as negative and positive controls, respectively.
Supplementary Material
Acknowledgements
We would like to thank Professor Dr. Jesús Mateos Grondona, Universidad de Málaga, Spain, for generously providing AFRUMA antibody, Sulan Xu for assistance with histology, Andrea Jasmine Arreguin for assistance in mouse brain ventricle dissection and whole-mount immunostaining, Richard C. Grady and Daniel C. Cameron for their assistance with animal colony maintenance and constructive suggestions, and Drs. Kelvin Luther and Ao Zhang for useful comments on the manuscript.
Contributor Information
Sanjiv Neupane, Department of Biochemistry and Cell Biology, Stony Brook University, 450 Life Science Building, Stony Brook, NY 11794-5215, USA.
June Goto, Division of Neurosurgery, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Avenue, Cincinnati, OH 45229, USA.
Steven J Berardinelli, Department of Biochemistry and Molecular Biology, Complex Carbohydrate Research Center, 315 Riverbend Road, Athens, GA 30602, USA.
Atsuko Ito, Department of Biochemistry and Molecular Biology, Complex Carbohydrate Research Center, 315 Riverbend Road, Athens, GA 30602, USA.
Robert S Haltiwanger, Department of Biochemistry and Molecular Biology, Complex Carbohydrate Research Center, 315 Riverbend Road, Athens, GA 30602, USA.
Bernadette C Holdener, Department of Biochemistry and Cell Biology, Stony Brook University, 450 Life Science Building, Stony Brook, NY 11794-5215, USA.
Funding
Supported by grants from National Institutes of Child Health and Human Development (R01 HD090156 and R01 HD096030 to R.S.H. and B.C.H) and the 2019 Hydrocephalus Association Innovator Award to (B.C.H.).
Conflict of interest statement
Appropriate since authors have no conflicts of interest to declare.
Abbreviations
- ADAMTS
A Disintegrin and Metalloproteinase with ThromboSpondin type 1 repeat
- AFRUMA
Rabbit antibovine Reissner’s fiber antibody raised in Málaga
- B3GLCT
β3-glucosyltransferase
- BiP
Binding immunoglobulin protein (BiP) also known as (GRP-78), heat shock 70 kDa protein 5 (HSPA5), or (Byun1)
- CSF
Cerebrospinal fluid
- CTCF
Corrected total cell fluorescence
- ER
Endoplasmic reticulum
- O-linked glucose–fucose disaccharide
Oxygen-linked glucose-β1-3fucose disaccharide
- POFUT2
Protein O-fucosyltransferase 2
- PTRPLS
Peters plus syndrome (MIM #261540)
- SCO
Subcommissural organ
- SSPO
Subcommissural organ-spondin
- SPON1
F-spondin
- TSR
Thrombospondin type 1 repeat
References
- Abdelhamed Z, Vuong SM, Hill L, Shula C, Timms A, Beier D, Campbell K, Mangano FT, Stottmann RW, Goto J. 2018. A mutation in Ccdc39 causes neonatal hydrocephalus with abnormal motile cilia development in mice. Development. 145:dev154500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams CJ, Kopp MC, Larburu N, Nowak PR, Ali MMU. 2019. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front Mol Biosci. 6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amirav I, Wallmeier J, Loges NT, Menchen T, Pennekamp P, Mussaffi H, Abitbul R, Avital A, Bentur L, Dougherty GW, et al. 2016. Systematic analysis of CCNO variants in a defined population: Implications for clinical phenotype and differential diagnosis. Hum Mutat. 37:396–405. [DOI] [PubMed] [Google Scholar]
- Baas D, Meiniel A, Benadiba C, Bonnafe E, Meiniel O, Reith W, Durand B. 2006. A deficiency in RFX3 causes hydrocephalus associated with abnormal differentiation of ependymal cells. Eur J Neurosci. 24:1020–1030. [DOI] [PubMed] [Google Scholar]
- Benveniste H, Liu X, Koundal S, Sanggaard S, Lee H, Wardlaw J. 2019. The glymphatic system and waste clearance with brain aging: A review. Gerontology. 65:106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz BA, Nandadasa S, Takeuchi M, Grady RC, Takeuchi H, LoPilato RK, Kakuda S, Somerville RPT, Apte SS, Haltiwanger RS, et al. 2016. Genetic and biochemical evidence that gastrulation defects in Pofut2 mutants result from defects in ADAMTS9 secretion. Dev Biol. 416:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon M, Wallmeier J, Ma L, Loges NT, Jaspers M, Olbrich H, Dougherty GW, Raidt J, Werner C, Amirav I, et al. 2014. MCIDAS mutations result in a mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Commun. 5:4418. [DOI] [PubMed] [Google Scholar]
- Bradshaw NJ, Trossbach SV, Kober S, Walter S, Prikulis I, Weggen S, Korth C. 2020. Disrupted in schizophrenia 1 regulates the processing of reelin in the perinatal cortex. Schizophr Res. 215:506–513. [DOI] [PubMed] [Google Scholar]
- Canda MT, Doganay Caglayan L, Demir AB, Demir N. 2018. Prenatal detection of Peters plus-like syndrome. Turk J Obstet Gynecol. 15:273–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantaut-Belarif Y, Sternberg JR, Thouvenin O, Wyart C, Bardet PL. 2018. The Reissner Fiber in the cerebrospinal fluid controls morphogenesis of the body axis. Curr Biol. 28:2479–2486.e2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona-Calero EM, Gonzalez-Marrero I, Gonzalez-Toledo JM, Castaneyra-Ruiz A, Paz-Carmona H, Castaneyra-Ruiz L, Fernandez-Rodriguez P, Ruiz-Mayor ML, Castaneyra-Perdomo A. 2009. Reissner's fibre proteins and p73 variations in the cerebrospinal fluid and subcommissural organ of hydrocephalic rat. Anat Histol Embryol. 38:282–285. [DOI] [PubMed] [Google Scholar]
- Del Bigio MR. 1995. The ependyma A protective barrier between brain and cerebrospinal fluid. Glia. 14:1–13. [DOI] [PubMed] [Google Scholar]
- Dietrich P, Shanmugasundaram R, Shuyu E, Dragatsis I. 2009. Congenital hydrocephalus associated with abnormal subcommissural organ in mice lacking huntingtin in Wnt1 cell lineages. Hum Mol Genet. 18:142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet A, Overall CM. 2011. Broad coverage identification of multiple proteolytic cleavage site sequences in complex high molecular weight proteins using quantitative proteomics as a complement to edman sequencing. Mol Cell Proteomics. 10:M110 003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Takeuchi H, Leonhard-Melief C, Shroyer KR, Dlugosz M, Haltiwanger RS, Holdener BC. 2010. O-fucosylation of thrombospondin type 1 repeats restricts epithelial to mesenchymal transition (EMT) and maintains epiblast pluripotency during mouse gastrulation. Dev Biol. 346:25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y, Wang SH, Song J, Mironova Y, Ming GL, Kolodkin AL, Giger RJ. 2014. Semaphorin 5A inhibits synaptogenesis in early postnatal- and adult-born hippocampal dentate granule cells. Elife. 3:e04390 doi: 10.7554/eLife.04390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubail J, Vasudevan D, Wang LW, Earp SE, Jenkins MW, Haltiwanger RS, Apte SS. 2016. Impaired ADAMTS9 secretion: A potential mechanism for eye defects in Peters plus syndrome. Sci Rep. 6:33974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichele G, Bodenschatz E, Ditte Z, Gunther AK, Kapoor S, Wang Y, Westendorf C. 2020. Cilia-driven flows in the brain third ventricle. Philos Trans R Soc Lond B Biol Sci. 375:20190154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto H, Nelson CM, Somerville RP, Mielke K, Dixon LJ, Powell K, Apte SS. 2010. Cooperation of two ADAMTS metalloproteases in closure of the mouse palate identifies a requirement for versican proteolysis in regulating palatal mesenchyme proliferation. Development. 137:4029–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Llebrez P, Ggrondona JM, Perez J, Lopez-Aranda MF, Estivill-Torrus G, Llebrez-Zayas PF, Soriano E, Ramos C, Lallemand Y, Bach A, et al. 2004. Msx1-deficient mice fail to form prosomere 1 derivatives, subcommissural organ, and posterior commissure and develop hydrocephalus. J Neuropathol Exp Neurol. 63:574–586. [DOI] [PubMed] [Google Scholar]
- Gavet O, Pines J. 2010. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell. 18:533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves-Mendes N, Simon-Chazottes D, Creveaux I, Meiniel A, Guenet JL, Meiniel R. 2003. Mouse SCO-spondin, a gene of the thrombospondin type 1 repeat (TSR) superfamily expressed in the brain. Gene. 312:263–270. [DOI] [PubMed] [Google Scholar]
- Gray RS, Gonzalez R, Ackerman SD, Minowa R, Griest JF, Bayrak MN, Troutwine B, Canter S, Monk KR, Sepich DS, et al. 2021. Postembryonic screen for mutations affecting spine development in zebrafish. Dev Biol. 471:18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota Y, Meunier A, Huang S, Shimozawa T, Yamada O, Kida YS, Inoue M, Ito T, Kato H, Sakaguchi M, et al. 2010. Planar polarity of multiciliated ependymal cells involves the anterior migration of basal bodies regulated by non-muscle myosin II. Development. 137:3037–3046. [DOI] [PubMed] [Google Scholar]
- Hisanaga A, Morishita S, Suzuki K, Sasaki K, Koie M, Kohno T, Hattori M. 2012. A disintegrin and metalloproteinase with thrombospondin motifs 4 (ADAMTS-4) cleaves Reelin in an isoform-dependent manner. FEBS Lett. 586:3349–3353. [DOI] [PubMed] [Google Scholar]
- Holdener BC, Haltiwanger RS. 2019. Protein O-fucosylation: Structure and function. Curr Opin Struct Biol. 56:78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdener BC, Percival CJ, Grady RC, Cameron DC, Berardinelli SJ, Zhang A, Neupane S, Takeuchi M, Jimenez-Vega JC, Uddin SMZ, et al. 2019. ADAMTS9 and ADAMTS20 are differentially affected by loss of B3GLCT in mouse model of Peters plus syndrome. Hum Mol Genet. 28:4053–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubmacher D, Schneider M, Berardinelli SJ, Takeuchi H, Willard B, Reinhardt DP, Haltiwanger RS, Apte SS. 2017. Unusual life cycle and impact on microfibril assembly of ADAMTS17, a secreted metalloprotease mutated in genetic eye disease. Sci Rep. 7:41871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh MS, Todd MA, Picketts DJ. 2009. SCO-ping out the mechanisms underlying the etiology of hydrocephalus. Physiology (Bethesda). 24:117–126. [DOI] [PubMed] [Google Scholar]
- Ibanez-Tallon I, Pagenstecher A, Fliegauf M, Olbrich H, Kispert A, Ketelsen UP, North A, Heintz N, Omran H. 2004. Dysfunction of axonemal dynein heavy chain Mdnah5 inhibits ependymal flow and reveals a novel mechanism for hydrocephalus formation. Hum Mol Genet. 13:2133–2141. [DOI] [PubMed] [Google Scholar]
- Janssen L, Dupont L, Bekhouche M, Noel A, Leduc C, Voz M, Peers B, Cataldo D, Apte SS, Dubail J, et al. 2016. ADAMTS3 activity is mandatory for embryonic lymphangiogenesis and regulates placental angiogenesis. Angiogenesis. 19:53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Zhou J, Qin X, Zheng H, Gao B, Liu X, Jin G, Zhou Z. 2020. MT1-MMP deficiency leads to defective ependymal cell maturation, impaired ciliogenesis, and hydrocephalus. JCI Insight. 5:e132782. doi: 10.1172/jci.insight.132782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Kulkarni AV, Limbrick DD, Warf BC. 2016. Hydrocephalus in children. The Lancet. 387:788–799. [DOI] [PubMed] [Google Scholar]
- Kakuda S, Haltiwanger RS. 2014. Analyzing the posttranslational modification status of Notch using mass spectrometry. Methods Mol Biol. 1187:209–221. [DOI] [PubMed] [Google Scholar]
- Kaur C, Rathnasamy G, Ling EA. 2016. The choroid plexus in healthy and diseased brain. J Neuropathol Exp Neurol. 75:198–213. [DOI] [PubMed] [Google Scholar]
- Khatri D, Gosal JS, Das KK, Bhaisora KS. 2019. Peter plus syndrome: A Neurosurgeon's perspective. J Pediatr Neurosci. 14:148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma K, Keusch JJ, Hegemann B, Luther KB, Klein D, Hess D, Haltiwanger RS, Hofsteenge J. 2006. Identification and characterization of abeta1,3-glucosyltransferase that synthesizes the Glc-beta1,3-Fuc disaccharide on thrombospondin type 1 repeats. J Biol Chem. 281:36742–36751. [DOI] [PubMed] [Google Scholar]
- Lang B, Song B, Davidson W, MacKenzie A, Smith N, McCaig CD, Harmar AJ, Shen S. 2006. Expression of the human PAC1 receptor leads to dose-dependent hydrocephalus-related abnormalities in mice. J Clin Invest. 116:1924–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goff C, Somerville RP, Kesteloot F, Powell K, Birk DE, Colige AC, Apte SS. 2006. Regulation of procollagen amino-propeptide processing during mouse embryogenesis by specialization of homologous ADAMTS proteases: Insights on collagen biosynthesis and dermatosparaxis. Development. 133:1587–1596. [DOI] [PubMed] [Google Scholar]
- Lesnik Oberstein SAJ, Kriek M, White SJ, Kalf ME, Szuhai K, den Dunnen JT, Breuning MH, Hennekam RC. 2006. Peters plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am J Hum Genet. 79:562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnik Oberstein SAJ, Ruivenkamp CAL, Hennakam RC. 2007[Updated 2017 Aug 24]. Peters plus syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, LJH B, Stephens A, Amemiya A, editors. Gene Reviews (R) [Internet]. Seattle (WA): University of Washington, Seattle. [PubMed] [Google Scholar]
- Lett RL, Wang W, O'Connor TP. 2009. Semaphorin 5B is a novel inhibitory cue for corticofugal axons. Cereb Cortex. 19:1408–1421. [DOI] [PubMed] [Google Scholar]
- Luo Y, Koles K, Vorndam W, Haltiwanger RS, Panin VM. 2006a. Protein O-fucosyltransferase 2 adds O-fucose to thrombospondin type 1 repeats. J Biol Chem. 281:9393–9399. [DOI] [PubMed] [Google Scholar]
- Luo Y, Nita-Lazar A, Haltiwanger RS. 2006b. Two distinct pathways for O-fucosylation of epidermal growth factor-like or thrombospondin type 1 repeats. J Biol Chem. 281:9385–9392. [DOI] [PubMed] [Google Scholar]
- Martínez-Peña y Valenzuela I, Carmona-Calero EM, Perez-Gonzalez H, Ormazabal-Ramos C, Fernandez-Rodriguez P, Gonzalez-Marrero I, Castaneyra-Perdomo A, Ferres-Torres R. 2006. Alterations of the cerebrospinal fluid proteins and subcommissural organ secretion in the arterial hypertension and ventricular dilatation. A study in SHR rats. Histol Histopathol. 21:179–185. [DOI] [PubMed] [Google Scholar]
- McClenahan FK, Sharma H, Shan X, Eyermann C, Colognato H. 2016. Dystroglycan suppresses notch to regulate stem cell niche structure and function in the developing postnatal subventricular zone. Dev Cell. 38:548–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch DR, Nelson CM, Dixon LJ, Silver DL, Wylie JD, Lindner V, Sasaki T, Cooley MA, Argraves WS, Apte SS. 2009. ADAMTS metalloproteases generate active versican fragments that regulate interdigital web regression. Dev Cell. 17:687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead TJ, Apte SS. 2018. ADAMTS proteins in human disorders. Matrix Biol. 71–72:225–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiniel A. 2007. The secretory ependymal cells of the subcommissural organ: Which role in hydrocephalus? Int J Biochem Cell Biol. 39:463–468. [DOI] [PubMed] [Google Scholar]
- Meiniel O, Meiniel A. 2007. The complex multidomain organization of SCO-spondin protein is highly conserved in mammals. Brain Res Rev. 53:321–327. [DOI] [PubMed] [Google Scholar]
- Mirzadeh Z, Han YG, Soriano-Navarro M, Garcia-Verdugo JM, Alvarez-Buylla A. 2010. Cilia organize ependymal planar polarity. J Neurosci. 30:2600–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz RI, Kahne T, Herrera H, Rodriguez S, Guerra MM, Vio K, Hennig R, Rapp E, Rodriguez E. 2019. The subcommissural organ and the Reissner fiber: Old friends revisited. Cell Tissue Res. 375:507–529. [DOI] [PubMed] [Google Scholar]
- Nakajima M, Matsuda K, Miyauchi N, Fukunaga Y, Watanabe S, Okuyama S, Perez J, Fernandez-Llebrez P, Shen J, Furukawa Y. 2011. Hydrocephalus and abnormal subcommissural organ in mice lacking presenilin-1 in Wnt1 cell lineages. Brain Res. 1382:275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandadasa S, Kraft CM, Wang LW, O'Donnell A, Patel R, Gee HY, Grobe K, Cox TC, Hildebrandt F, Apte SS. 2019. Secreted metalloproteases ADAMTS9 and ADAMTS20 have a non-canonical role in ciliary vesicle growth during ciliogenesis. Nat Commun. 10:953. 10.1038/s41467-019-08520-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandadasa S, Nelson CM, Apte SS. 2015. ADAMTS9-mediated extracellular matrix dynamics regulates umbilical cord vascular smooth muscle differentiation and rotation. Cell Rep. 11:1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandadasa S, Szafron JM, Pathak V, Murtada SI, Kraft CM, O'Donnell A, Norvik C, Hughes C, Caterson B, Domowicz MS, et al. 2020. Vascular dimorphism ensured by regulated proteoglycan dynamics favors rapid umbilical artery closure at birth. Elife. 9:e60683. doi: 10.7554/eLife.60683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nualart F, Hein S, Rodriguez EM, Oksche A. 1991. Identification and partial characterization of the secretory glycoproteins of the bovine subcommissural organ-Reissner's fiber complex. Evidence for the existence of two precursor forms. Mol Brain Res. 11:227–238. [DOI] [PubMed] [Google Scholar]
- Ogino H, Hisanaga A, Kohno T, Kondo Y, Okumura K, Kamei T, Sato T, Asahara H, Tsuiji H, Fukata M, et al. 2017. Secreted metalloproteinase ADAMTS-3 inactivates Reelin. J Neurosci. 37:3181–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohata S, Alvarez-Buylla A. 2016. Planar organization of multiciliated ependymal (E1) cells in the brain ventricular epithelium. Trends Neurosci. 39:543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohata S, Nakatani J, Herranz-Perez V, Cheng J, Belinson H, Inubushi T, Snider WD, Garcia-Verdugo JM, Wynshaw-Boris A, Alvarez-Buylla A. 2014. Loss of Dishevelleds disrupts planar polarity in ependymal motile cilia and results in hydrocephalus. Neuron. 83:558–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olstad EW, Ringers C, Hansen JN, Wens A, Brandt C, Wachten D, Yaksi E, Jurisch-Yaksi N. 2019. Ciliary beating compartmentalizes cerebrospinal fluid flow in the brain and regulates ventricular development. Curr Biol. 29:229–241 e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orts-Del'Immagine A, Cantaut-Belarif Y, Thouvenin O, Roussel J, Baskaran A, Langui D, Koeth F, Bivas P, Lejeune FX, Bardet PL, et al. 2020. Sensory neurons contacting the cerebrospinal fluid require the Reissner Fiber to detect spinal curvature in vivo. Curr Biol. 30:827–839 e824. [DOI] [PubMed] [Google Scholar]
- Perez-Figares JM, Jimenez AJ, Rodriguez EM. 2001. Subcommissural organ, cerebrospinal fluid circulation, and hydrocephalus. Microsc Res Tech. 52:591–607. [DOI] [PubMed] [Google Scholar]
- Ramos C, Fernandez-Llebrez P, Bach A, Robert B, Soriano E. 2004. Msx1 disruption leads to diencephalon defects and hydrocephalus. Dev Dyn. 230:446–460. [DOI] [PubMed] [Google Scholar]
- Redmond SA, Figueres-Onate M, Obernier K, Nascimento MA, Parraguez JI, Lopez-Mascaraque L, Fuentealba LC, Alvarez-Buylla A. 2019. Development of ependymal and postnatal neural stem cells and their origin from a common embryonic progenitor. Cell Rep. 27:429–441 e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis LM, Tyler RC, Abdul-Rahman O, Trapane P, Wallerstein R, Broome D, Hoffman J, Khan A, Paradiso C, Ron N, et al. 2008. Mutation analysis of B3GALTL in Peters plus syndrome. Am J Med Genet A. 146A:2603–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Manzaneque JC, Fernandez-Rodriguez R, Rodriguez-Baena FJ, Iruela-Arispe ML. 2015. ADAMTS proteases in vascular biology. Matrix Biol. 44–46:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez EM, Oksche A, Hein S, Rodríguez S, Yulis R. 1984. Comparative immunocytochemical study of the subcommissural organ. Cell Tissue Res. 237:427–441. [DOI] [PubMed] [Google Scholar]
- Rodríguez EM, Oksche A, Hein S, Yulis CR. 1992. Cell biology of the subcommissural organ. Int Rev Cytol. 135:39–121. [DOI] [PubMed] [Google Scholar]
- Rodriguez EM, Oksche A, Montecinos H. 2001. Human subcommissural organ, with particular emphasis on its secretory activity during the fetal life. Microsc Res Tech. 52:573–590. [DOI] [PubMed] [Google Scholar]
- Rodriguez EM, Rodriguez S, Hein S. 1998. The subcommissural organ. Microsc Res Tech. 41:98–123. [DOI] [PubMed] [Google Scholar]
- Sato T, Sato M, Kiyohara K, Sogabe M, Shikanai T, Kikuchi N, Togayachi A, Ishida H, Ito H, Kameyama A, et al. 2006. Molecular cloning and characterization of a novel human beta1,3-glucosyltransferase, which is localized at the endoplasmic reticulum and glucosylates O-linked fucosylglycan on thrombospondin type 1 repeat domain. Glycobiology. 16:1194–1206. [DOI] [PubMed] [Google Scholar]
- Schneider M, Al-Shareffi E, Haltiwanger RS. 2017. Biological functions of fucose in mammals. Glycobiology. 27:601–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M, Kumar V, Nordstrom LU, Feng L, Takeuchi H, Hao H, Luca VC, Garcia KC, Stanley P, Wu P, et al. 2018. Inhibition of delta-induced Notch signaling using fucose analogs. Nat Chem Biol. 14:65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoner K, Kohlhase J, Muller AM, Schramm T, Plassmann M, Schmitz R, Neesen J, Wieacker P, Rehder H. 2013. Hydrocephalus, agenesis of the corpus callosum, and cleft lip/palate represent frequent associations in fetuses with Peters' plus syndrome and B3GALTL mutations. Fetal PPS phenotypes, expanded by Dandy Walker cyst and encephalocele. Prenat Diagn. 33:75–80. [DOI] [PubMed] [Google Scholar]
- Schwartz NB, Domowicz M. 2004. Proteoglycans in brain development. Glycoconj J. 21:329–341. [DOI] [PubMed] [Google Scholar]
- Shan X, Tomlinson L, Yang Q, Colognato H. 2018. Distinct requirements for extracellular and intracellular MMP12 in the development of the adult V-SVZ neural stem cell niche. Stem Cell Reports. 10:984–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova A, Preller M, Taft MH, Pujols J, Ventura S, Tiemann B, Buettner FF, Bakker H. 2019. C-mannosylation supports folding and enhances stability of thrombospondin repeats. Elife. 8:e52978 doi: 10.7554/eLife.52978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F, Sottile J. 2011. MT1-MMP regulates the turnover and endocytosis of extracellular matrix fibronectin. J Cell Sci. 124:4039–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somera KC, Jones HC. 2004. Reduced subcommissural organ glycoprotein immunoreactivity precedes aqueduct closure and ventricular dilatation in H-Tx rat hydrocephalus. Cell Tissue Res. 315:361–373. [DOI] [PubMed] [Google Scholar]
- Takagishi M, Sawada M, Ohata S, Asai N, Enomoto A, Takahashi K, Weng L, Ushida K, Ara H, Matsui S, et al. 2017. Daple coordinates planar polarized microtubule dynamics in ependymal cells and contributes to hydrocephalus. Cell Rep. 20:960–972. [DOI] [PubMed] [Google Scholar]
- Thomas M, Lu JJ, Ge Q, Zhang C, Chen J, Klibanov AM. 2005. Full deacylation of polyethylenimine dramatically boosts its gene delivery efficiency and specificity to mouse lung. Proc Natl Acad Sci USA. 102:5679–5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troutwine BR, Gontarz P, Konjikusic MJ, Minowa R, Monstad-Rios A, Sepich DS, Kwon RY, Solnica-Krezel L, Gray RS. 2020. The Reissner Fiber is highly dynamic in vivo and controls morphogenesis of the spine. Curr Biol. 30:2352–2362.e3. 10.1016/j.cub.2020.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Schooneveld MJ, Delleman JW, Beemer FA, Bleeker-Wagemakerp EM. 1984. Peters-plus a new syndrome. Ophthalmic Paediatr Genet. 4:141–146. [DOI] [PubMed] [Google Scholar]
- Vasudevan D, Haltiwanger RS. 2014. Novel roles for O-linked glycans in protein folding. Glycoconj J. 31:417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan D, Takeuchi H, Johar SS, Majerus E, Haltiwanger RS. 2015. Peters plus syndrome mutations disrupt a noncanonical ER quality-control mechanism. Curr Biol. 25:286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vio K, Rodriguez S, Navarrete EH, Perez-Figares JM, Jimenez AJ, Rodriguez EM. 2000. Hydrocephalus induced by immunological blockage of the subcommissural organ-Reissner's fiber (RF) complex by maternal transfer of anti-RF antibodies. Exp Brain Res. 135:41–52. [DOI] [PubMed] [Google Scholar]
- Vio K, Rodríguez S, Yulis CR, Oliver C, Rodríguez EM. 2008. The subcommissural organ of the rat secretes Reissner's fiber glycoproteins and CSF-soluble proteins reaching the internal and external CSF compartments. Cerebrospinal Fluid Res. 5:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallingford JB. 2010. Planar cell polarity signaling, cilia and polarized ciliary beating. Curr Opin Cell Biol. 22:597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LW, Nandadasa S, Annis DS, Dubail J, Mosher DF, Willard BB, Apte SS. 2019. A disintegrin-like and metalloproteinase domain with thrombospondin type 1 motif 9 (ADAMTS9) regulates fibronectin fibrillogenesis and turnover. J Biol Chem. 294:9924–9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Wey S, Zhang Y, Ye R, Lee AS. 2009. Role of the unfolded protein response regulator GRP78=BiP in development, cancer, and neurological disorders. Antioxid Redox Signal. 11:2307–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weh E, Reis LM, Tyler RC, Bick D, Rhead WJ, Wallace S, McGregor TL, Dills SK, Chao MC, Murray JC, et al. 2014. Novel B3GALTL mutations in classic Peters plus syndrome and lack of mutations in a large cohort of patients with similar phenotypes. Clin Genet. 86:142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weh E, Takeuchi H, Muheisen S, Haltiwanger RS, Semina EV. 2017. Functional characterization of zebrafish orthologs of the human Beta 3-glucosyltransferase B3GLCT gene mutated in Peters plus syndrome. PLoS One. 12:e0184903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Chen L, Zheng PS, Yang BB. 2002. Beta 1-integrin-mediated glioma cell adhesion and free radical-induced apoptosis are regulated by binding to a C-terminal domain of PG-M/versican. J Biol Chem. 277:12294–12301. [DOI] [PubMed] [Google Scholar]
- Xu C, Bailly-Maitre B, Reed JC. 2005. Endoplasmic reticulum stress: Cell life and death decisions. J Clin Invest. 115:2656–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Simonneau C, Kilker R, Oakley L, Byrne MD, Nichtova Z, Stefanescu I, Pardeep-Kumar F, Tripathi S, Londin E, et al. 2019. Murine MPDZ-linked hydrocephalus is caused by hyperpermeability of the choroid plexus. EMBO Mol Med. 11:e9540. doi: 10.15252/emmm.201809540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A, Berardinelli SJ, Leonhard-Melief C, Vasudevan D, Liu TW, Taibi A, Giannone S, Apte SS, Holdener BC, Haltiwanger RS. 2020. O-Fucosylation of ADAMTSL2 is required for secretion and is impacted by geleophysic dysplasia-causing mutations. J Biol Chem. 295:15742–15753. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.