

Abstract
BACKGROUND AND AIMS:
The molecular mechanisms underlying successful fecal microbiota transplantation (FMT) for recurrent Clostridioides difficile infection (rCDI) remain poorly understood. The primary objective of this study was to characterize alterations in microRNAs (miRs) following FMT for rCDI.
METHODS:
Sera from 2 prospective multicenter randomized controlled trials were analyzed for miRNA levels with the use of the Nanostring nCounter platform and quantitative reverse-transcription (RT) polymerase chain reaction (PCR). In addition, rCDI-FMT and toxin-treated animals and ex vivo human colonoids were used to compare intestinal tissue and circulating miRs. miR inflammatory gene targets in colonic epithelial and peripheral blood mononuclear cells were evaluated by quantitative PCR (qPCR) and 3′UTR reporter assays. Colonic epithelial cells were used for mechanistic, cytoskeleton, cell growth, and apoptosis studies.
RESULTS:
miRNA profiling revealed up-regulation of 64 circulating miRs 4 and 12 weeks after FMT compared with screening, of which the top 6 were validated in the discovery cohort by means of RT-qPCR. In a murine model of relapsing-CDI, RT-qPCR analyses of sera and cecal RNA extracts demonstrated suppression of these miRs, an effect reversed by FMT. In mouse colon and human colonoids, C difficile toxin B (TcdB) mediated the suppressive effects of CDI on miRs. CDI dysregulated DROSHA, an effect reversed by FMT. Correlation analyses, qPCR ,and 3′UTR reporter assays revealed that miR-23a, miR-150, miR-26b, and miR-28 target directly the 3′UTRs of IL12B, IL18, FGF21, and TNFRSF9, respectively. miR-23a and miR-150 demonstrated cytoprotective effects against TcdB.
CONCLUSIONS:
These results provide novel and provocative evidence that modulation of the gut microbiome via FMT induces alterations in circulating and intestinal tissue miRs. These findings contribute to a greater understanding of the molecular mechanisms underlying FMT and identify new potential targets for therapeutic intervention in rCDI.
Keywords: Fecal Transplantation, C difficile, microRNA, DROSHA
Graphical Abstract

Fecal microbiota transplantation (FMT) is a well established treatment for recurrent Clostridioides difficile infection (rCDI). Accumulating evidence also supports FMT as a potential treatment for other disorders associated with intestinal dysbiosis, including inflammatory bowel diseases, cancer, metabolic syndrome, and neuropsychiatric disorders.1
Despite the effectiveness of FMT in rCDI, its mechanisms of action remain poorly explored. Current evidence suggests the success of FMT may be attributed in part to the reconstitution of intestinal microbiota, restoration of secondary bile acid metabolism, and modulation of immune-mediated inflammatory responses.2,3 We have previously reported that effective FMT for rCDI is associated with activation of the bile acid–farnesoid X receptor–fibroblast growth factor (FGF) pathway and decreased serum C-X-C motif chemokine 11 (CXCL11), interleukin-18 (IL18), tumor necrosis factor (TNF)–related activation–induced cytokine, IL12B, CXCL6, and tnf receptor superfamily member 9 (TNFRSF9).4 The gut microbiota can modify host cell responses to stimuli (eg, metabolites) through alterations in the host epigenome and, ultimately, gene expression.5 MicroRNAs (miRNAs) are thought to be one way in which the gut microbiota communicates with the human host. These short noncoding RNA molecules (containing ~22 nucleotides) are expressed as individual genes or as parts of longer transcripts and are processed by machinery involving DROSHA and DICER nucleases, which generate the mature miRNAs. Mature miRNAs are loaded on Argonaute AGO–containing complexes and bind to complementary sequences in the 3′-untranslated region (3′UTR) of messenger RNAs (mRNAs), resulting in transcript degradation and translational suppression of target genes.6 Bacterial pathogens clearly alter host miRNA expression,7 but less is known regarding the effect of commensal bacteria on the host miRNAome. A recent study has demonstrated how host fecal miRNAs, normal components of feces, can enter certain bacteria (eg, Fusobacterium nucleatum and Escherichia coli) and regulate bacterial gene transcription and growth.8 However, the alterations in circulating miRNAs of rCDI patients undergoing FMT and the functional effects they may exert on downstream targets remain unknown.
Herein, we characterize the impact of FMT on circulating miRNA signatures to better understand immunological mechanisms relevant to FMT in the treatment of rCDI. Our findings suggest a conserved mechanism involved in regulating host miRNAs by C difficile and identify new miRNA inflammatory targets in response to FMT.
Materials and Methods
Participants’ Clinical Data, Sample Collection, and Storage
Randomly selected rCDI subjects participating in 2 clinical trials (capsule- vs colonoscopy-delivered FMT9 and fresh vs frozen enema-delivered FMT10) comprised the discovery and replication cohorts, respectively. Blood samples were collected from October 2014 to December 2016 (discovery cohort) and from July 2012 to September 2014 (replication cohort) and stored at −80°C. Only sera with sufficient volume were selected for miRNA analysis. Healthy control subjects (n = 42; mean ± SD age 53.3 ± 20.7 years; 30 women [71.4%]) were defined as asymptomatic adults undergoing screening colonoscopy recruited in Edmonton, Canada. Clinical and demographic information were collected from medical records. Participant baseline characteristics are presented in Table 1. All participants provided written informed consent under the approvals granted by the Research Ethics Boards of the University of Alberta (Pro 1994 and 49006), St. Joseph’s Healthcare (11-3622), and Hamilton Health Sciences (12-505) in Canada.
Table 1.
Participant Baseline Characteristics for the Discovery and Replication Cohorts
| Discovery cohort—NCT02254811 |
Replication cohort—NCT01398969 |
|||||
|---|---|---|---|---|---|---|
| Capsule (n = 25) |
Colonoscopy (n = 17) |
P value | Fresh (n = 11) |
Frozen (n = 13) |
Pvalue | |
| Age, y | 59.0 ± 19.7) | 56.4 ± 8.7 | .6624 | 74.3 ± 14.1 | 71.6 ± 18.6 | .7363 |
|
| ||||||
| Female | 21 (84.0%) | 9 (52.9%) | .0659 | 4 (36.4%) | 9 (69.2%) | .2305 |
|
| ||||||
| Charlson comorbodity index | 3 (2–5) | 3 (0–4) | .4353 | |||
|
| ||||||
| Immunosuppressed patients | 2 (8.0%) | 3 (17.6%) | .6439 | 3 (27.3%) | 1 (7.7%) | .4637 |
|
| ||||||
| Use of immune modulator | ||||||
|
| ||||||
| Costicosteroid | 0 (0%) | 1 (5.9%) | .8443 | |||
|
| ||||||
| Immunosuppresant | 1 (4.0%) | 1 (5.9%) | 1 | 4 (36.4%) | 3 (23.1%) | .7926 |
|
| ||||||
| Biologic | 2 (8.0%) | 1 (5.9%) | 1 | 5 (45.5%) | 4 (30.8%) | .751 |
|
| ||||||
| Body mass index, kg/m2 | 25.3 ± 6.6 | 27.0 ± 4.1 | .3113 | |||
|
| ||||||
| Inpatient status at screening | 3 (12%) | 0 (0%) | .3833 | 7 (63.6%) | 6 (46.2%) | .6561 |
|
| ||||||
| PPI use prior to FMT | 4 (16%) | 1 (5.9%) | .6111 | 5 45.5%) | 5 (38.5%) | 1 |
|
| ||||||
| No. of RCDI episodes before FMT; median | 3 (3–4) | 3 (3–4) | .3893 | 3 (2.5–4.0) | 3 (3–4) | .4641 |
|
| ||||||
| Duration of rCDI before FMT | 65 (49–96) | 70 (57–135) | .2648 | |||
|
| ||||||
| No. of CDI-related hospital admissions | 1 (0–2) | 0 (0–0) | .0057 | |||
|
| ||||||
| Inflammatory bowel disease | ||||||
|
| ||||||
| Ulcerative colitis | 2 (8.3%) | 3 (1.8%) | .6439 | |||
|
| ||||||
| Crohn’s disease | 2 (8.3%) | 1 (5.9%) | 1 | 2 (18.2%) | 2 (15.4%) | 1 |
|
| ||||||
| Hemoglobin, g/dL | 137 (129–144) | 138 (132–144) | .6079 | |||
|
| ||||||
| WBC, 109/L | 8 (6.8–8.6) | 6.5 (5.1–6.9) | .0143 | 15.30 (8.35–21.70) | 8.9 (6.7–12.6) | .2212 |
|
| ||||||
| Albumin, g/L | 40.5 (38.8–43.3) | 40 (38–42) | .5857 | 31 (28–34) | 30 (24–34) | .8557 |
|
| ||||||
| CRP, mg/L | 1.9 (0.9–5.8) | 6.2 (1.3–10.2) | .244 | |||
|
| ||||||
| Creatinine (mg/dL) | 70 (58.75–76) | 70 (59–84) | .7404 | 88 (69.5–127) | 85 (64–128) | .6849 |
Values are mean ± SD, n (%), or median (range).
CDI, Clostridioides difficile infection; CRP, C-reactive protein; FMT, fecal microbiota transplantation; PPI, proton pump inhibitor; rCDI, recurrent Clostridioides difficile infection; WBC, white blood count.
Mouse Model of rCDI
A mouse model of rCDI was used to assess miRNA, mRNA, and protein levels following infection and treatment with FMT, as described previously.11 Animal work was approved by the Clemson University Institutional Animal Care and Use Committee (IACUC). Additional details are available in the Supplementary Methods. Briefly, 6–8-week-old C57BL/6 mice were given 0.5 mg/mL cefoperazone (MP Biochemicals; cat. no. 199695) in sterile drinking water (Gibco Laboratories; cat. no. 15230) ad libitum for 5 days (n = 42). Two days after cessation of antibiotics, the mice received 103 C difficile strain 630 spores resuspended in 1 mL sterile water, prepped as described previously (day 0; n = 36).12 A subset of mice were killed 4 days after infection (dpi) to assess miRNA during acute infection (n = 9). The remainder of the mice received 0.4 mg/mL vancomycin (Sigma; cat no. V2002) starting at 4 dpi for 5 days (4-9 dpi) ad libitum in sterile drinking water (n = 27). At 11 dpi, FMT prepped from untreated (n = 8) healthy age-matched mice (mFMT) was administered via oral gavage to a group of mice. Each mouse received 100 μL FMT material diluted in phosphate-buffered saline solution (PBS) (~0.2 g fresh fecal material in 1.5 mL prereduced PBS, homogenized via mixing and gravity filtering). One group of mice received all antibiotics and mFMT but no C difficile inoculum (handling and experimental control; n = 6). The remainder of infected mice (n = 12) did not receive FMT (noFMT). Fecal sampling was conducted throughout the experiment, and end point cecal sampling to assess C difficile load was determined by plating 20 μL of content from individual samples in 1:10 PBS and serially diluted on taurocholate cycloserine cefoxitin fructose agar under anaerobic conditions (Coy Laboratory Products, Grass Lake, MI, USA). The colony-forming units (CFU)/mL content was determined after overnight incubation at 37°C. Mice were killed at 21 dpi, and cecal contents, tissue, and serum were flash frozen in liquid nitrogen and kept at −80°C for downstream analyses.
Mouse Toxin Challenge Model
The animal protocol was approved by the Vanderbilt University Medical Center IACUC; 6–8-week-old C57BL/6 mice (Jackson Labs) were observed from arrival to ensure normal health. Mice were separated into 3 groups (n = 6 per group) to receive intrarectal instillations of either purified recombinant whole C difficile toxin B(TcdB), TcdA and TcdB, or Hank’s Balanced Salt Solution (HBSS) vehicle control, as described elsewhere.13 Further details on the purification of the toxins are described in the Supplementary methods. Toxins were derived from the VPI 10463 C difficile reference strain and prepared as 15 μg in a total volume of 100 μL per instillation. Mice were anesthetized with isofluorane and confirmed to be sedated by toe pinch. One mL HBSS was instilled intrarectally to evacuate stools with a flexible plastic gavage applicator (20 G × 30 mm; gavageneedle.com). Instillation was performed over 30 seconds while lightly pinching closed the anus, which was held for an additional 30 seconds as previously described.13 Mice were returned to cages to recover. After 2–5 hours, mice were killed by CO2 inhalation and cervical dislocation. Whole blood was extracted via cardiac puncture and allowed to clot in RNAsefree microcentrifuge tubes for 15 minutes at room temperature before centrifugation for 15 minutes at 1500g at 4°C. Serum was transferred to a fresh tube and flash frozen in liquid nitrogen. The colon was isolated and dissected from surrounding visceral tissue. The whole colon was washed in chilled sterile 1× PBS before portions of the middle and distal colon were combined and flash frozen for protein and miRNA analysis.
Treatment of Human Colonoids With C difficile Toxins
Deidentified human colon tissue was obtained through the Cooperative Human Tissue Network. On the day of colon resection surgery, normal marginal colon mucosa was resected and placed into Dulbecco’s Modified Eagle Medium (DMEM) at 4°C. Within hours, the tissue was prepared into normal human colonoids with Intesticult Organoid Growth Medium (Stem Cell Technologies) according to the manufacturer’s protocol (https://cdn.stemcell.com/media/files/pis/DX21423-PIS_1_0_0.pdf). Colonoids were suspended in Matrigel matrix (Corning) with standard growth factor concentration for maintenance and passage. Matrigel with reduced growth factors was used for suspending colonoids in the final passage for the experiment. Eight hours before toxin exposure, organoids were serum starved with DMEM and no growth serum. Colonoids were exposed to TcdA (10 pmol/L), TcdB (10 pmol/L), or DMEM vehicle negative control for either 30 minutes or 6 hours. Colonoid-containing Matrigel was washed once with 1× PBS and flash frozen in liquid nitrogen for protein analysis. For miRNA analysis, colonoids were removed from Matrigel with the use of Gentle Cell Dissociation Reagent (Stem Cell Technologies) and resuspended in RNAlater (Invitrogen). Colonoids in RNAlater were stored at 4°C overnight before freezing at −80°C.
Serum miRNA Isolation and High-Content Analysis
Human serum was isolated by centrifugation (2200g for 10 minutes at room temperature) from whole blood and snapfrozen. RNA was isolated from human and animal serum samples (200 μL) with the use of the miRNeasy Serum/Plasma Kit (Qiagen) according to the manufacturer’s instructions. Eluted RNA from serum samples was further purified and concentrated with the use of Amicon Ultra YM-3 columns (3000 kDa MWCO; Millipore).
For high-content miRNA analysis, RNAs after hybridization reactions were processed with the use of the nCounter Prep Station and nCounter Digital Analyzer. miRNA levels (n = 800) were analyzed with the use of the nSolver software v3.0 (Nanostring Technologies, Seattle, WA, USA). Normalization was performed with the use of all miRNAs (n = 110) with coefficients of variation <70%.14
Additional information is available in the Supplementary Methods.
Statistical Analysis
All statistical analyses were performed in SPSS v24 and R v3.5.1. Descriptive statistics for participant characteristics at baseline were reported using mean ± SD and percentages. All data are expressed as mean and SEM. Systematic within-week changes for each miRNA were examined using a nonparametric longitudinal method (nparLD in R package) followed by Wilcoxon signed rank test for pairwise comparisons. The association between the metavariables and miRNAs was assessed by means of Spearman correlation. Heatmaps for capsule and colonoscopy combined and separately for capsule and colonoscopy delivered FMT were generated in the R package ComplexHeatmap2.15 Normality was checked by means of the Kolmogorov-Smirnov test. Graph generation, fold changes, and statistical significance in levels of circulating miRNAs were assessed with the use of quantitative polymerase chain reaction (qPCR) by means of OriginPro and Wilcoxon matched-pairs signed rank test. Significance differences were considered when *P < 0.05; **P < 0.01; ***P < 0.001.
Results
FMT Modifies Circulating miRNAs in rCDI Patients
For miRNA profiling, we first analyzed serum samples derived from the discovery cohort.9 Table 1 describes patient baseline characteristics. Sera obtained at screening and 4 and 12 weeks after FMT from 42 participants who achieved a clinical cure after FMT were subjected to miRNA analysis using the Nanostring nCounter platform. miRNA profiling of 126 samples revealed the significant up-regulation of 64 circulating miRNAs 4 and 12 weeks after FMT compared with screening (Figure 1A). The miRNAs with the highest levels detected are depicted in Figure 1B. Similar changes in miRNA levels were detected in recipients of either by-capsule or colonoscopic FMT (Supplementary Figure 1). Pathway enrichment analysis identified overlaps between the top 3 miRNA-regulated pathways, B-cell lymphoma 2, Runt-related transcription factor 2 (linked to nuclear factor κB inhibitor β), and phosphoinositide 3-kinase (regulatory class 1A p55-γ), linking inflammatory signaling to immune cell survival and differentiation (Figure 1C). Pathway analysis also uncovered commonalities with other diseases (eg, inflammatory bowel disease and multiple sclerosis) and cell functions such as apoptosis (Supplementary Figures 2 and 3).
Figure 1.
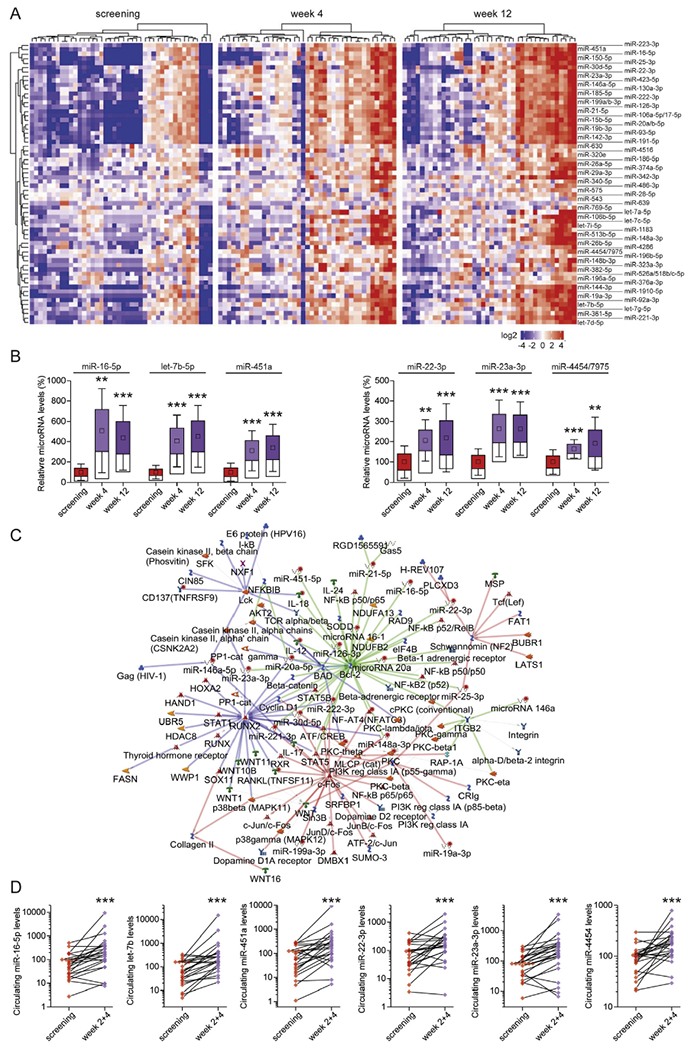
Fecal microbiota transplantation (FMT) in patients with Clostridioides difficile infection regulates the levels of circulating microRNAs (miRNAs). (A) Heatmap representation of the significantly up-regulated circulating miRNAs 4 and 12 weeks after FMT treatment compared with the screening time point (n = 42), as assessed with the nCounter Nanostring platform. (B) Representative box plots depicting the miRNAs with highest levels of detection. Box plots denote mean % change ± SEM, inner boxes represent mean, and error bars represent 95% confidence intervals (CIs). Statistical significance of FMT effect on circulating miRNAs was determined by nonparametric longitudinal method followed by Wilcoxon signed-rank test for pairwise comparisons. **P < 0.01; ***P < 0.001. (C) The overlapping top 3 miRNA-regulated pathways, as assessed with the Metacore network analysis software. (D) Validation of the top 6 up-regulated miRNAs in the replication cohort 2 and 4 weeks after FMT as assessed with reverse-transcription quantitative polymerase chain reaction (RT-qPCR). Fold changes and statistical significance in circulating levels of miRNAs were determined with OriginPro and Wilcoxon matched-pairs signed rank test. ***P < 0.001.
We next sought to validate our discovery cohort results. For the replication cohort, we assessed 24 patients at 3 time points: screening and 2 and 4 weeks after FMT.10 Table 1 describes patient baseline characteristics. The top 6 up-regulated miRNAs from the discovery cohort analysis (Figure 1B) were selected for reverse-transcription (RT) qPCR validation in our replication cohort. Individual miRNAs were found to be up-regulated in 78%–94% of samples analyzed, with the average changes ranging from 3- to 12-fold (Figure 1D).
FMT Modifies Intestinal Tissue miRNAs in a Mouse Model of rCDI
The concerted increase of circulating miRNAs by FMT suggests that C difficile–associated dysbiosis may regulate miRNAs through a conserved mechanism. We tested this hypothesis with the use of a mouse model of rCDI.8 Animals pretreated with cefoperazone received 103 C difficile spores, and at 4 dpi were exposed to vancomycin. At 11 dpi, a group of mice received fresh FMT derived from healthy mice. Sera collected 21 dpi were subjected to RT-qPCR. Parallel to what we found in rCDI patients, the same top 5 miRNAs (miR-4454 has not been characterized in mice) and in addition 2 potentially functional miRNAs were up-regulated after FMT. C difficile recurrence (Supplementary Figure 4) resulted in down-regulation of the tested circulating miRNAs in mice (Figure 2A) concomitant with a decrease in animal body weight (Supplementary Figure 5). To examine the time dependence of miRNA regulation by CDI, we assessed miRNA levels during the acute phase of infection (4 dpi). RT-qPCR analysis revealed that the suppression of circulating host miRNAs may be an early sign of CDI (Supplementary Figure 6). Importantly, the inhibitory effects of rCDI on miRNA levels was reversed 10 days after FMT (Figure 2A).
Figure 2.

FMT reverses the effects of C difficile on circulating and tissue miRNAs in a mouse model of recurrent C difficile infection (CDI). Box plots depicting the changes in miRNA levels in (A) sera and (B) ceca from animals treated with FMT, infected with C difficile (CDI), and infected with C difficile and treated with FMT (CDI+FMT) compared with FMT donors. Box plots denote mean % change ± SEM, inner boxes represent mean, and error bars represent 95% CIs. miRNA levels were assessed with RT-qPCR and normalized against (A) RNU1A1 and cel-miR-39 (spike-in) or (B) RNU5G and 5S rRNA, and compared with control (donor) samples set as 100%. Statistical significance was determined by Student t test. Compared with donor: *P < 0.05; **P < 0.01; ***P < 0.001. Compared with CDI: #P < 0.05; ##P < 0.01; ###P < 0.001. Other abbreviations as in Figure 1.
We next tested whether the circulating miRNA changes reflect the effects of rCDI on colonic tissue. RNA extracts from the ceca were analyzed by means of RT-qPCR. Our results showed that tissue miRNA levels altered similarly to circulating miRNAs (Figure 2B). Comparisons of the changes of individual miRNA levels in matched tissue and serum samples, from all animals, showed significant and positive correlation between circulating and ceca-expressed miRNAs (Supplementary Figure 7). This supports the notion that alterations in circulating miRNAs may be originating from colonic tissues. Furthermore, we found even stronger positive correlation between matched colonic and circulating miRNAs derived from an 84-year-old male patient with fulminant CDI treated with FMT by colonoscopy (Supplementary Figure 8). In mice, tissue miRNA levels down-regulated by rCDI reached statistical significance in the early phase of infection (Supplementary Figure 9) and FMT up-regulated the tissue miRNAs, coinciding with the reduction of C difficile load (Supplementary Figure 4) and the recovery of animal body weight (Supplementary Figure 5).
Toxin B Suppresses miRNAs in the Intestinal Mucosa
C difficile pathogenicity is primarily mediated by exotoxins that induce cell death. We investigated the effects of purified TcdA and TcdB on host miRNA regulation. Mice were treated by intrarectal instillation with a combination of TcdB and TcdA, TcdB alone (15 μg), or vehicle (HBSS), and colonic tissues and sera were collected 2–5 hours after instillation. RT-qPCR analysis of serum RNA extracts showed that the toxins had no effect on circulating miRNAs (Supplementary Figure 10). However, expression of the same miRNAs was suppressed in colonic tissues of C difficile–infected mice. Notably, TcdB alone suppressed miRNA levels, an effect enhanced by its combination with TcdA (Figure 3A). The discrepancy between circulating and tissue miRNAs in this model suggests that the impact of the toxins is luminally confined and may not lead to changes in circulating miRNA levels owing to local and time-restricted exposure of animals to toxins at early time points (2–5 hours).
Figure 3.
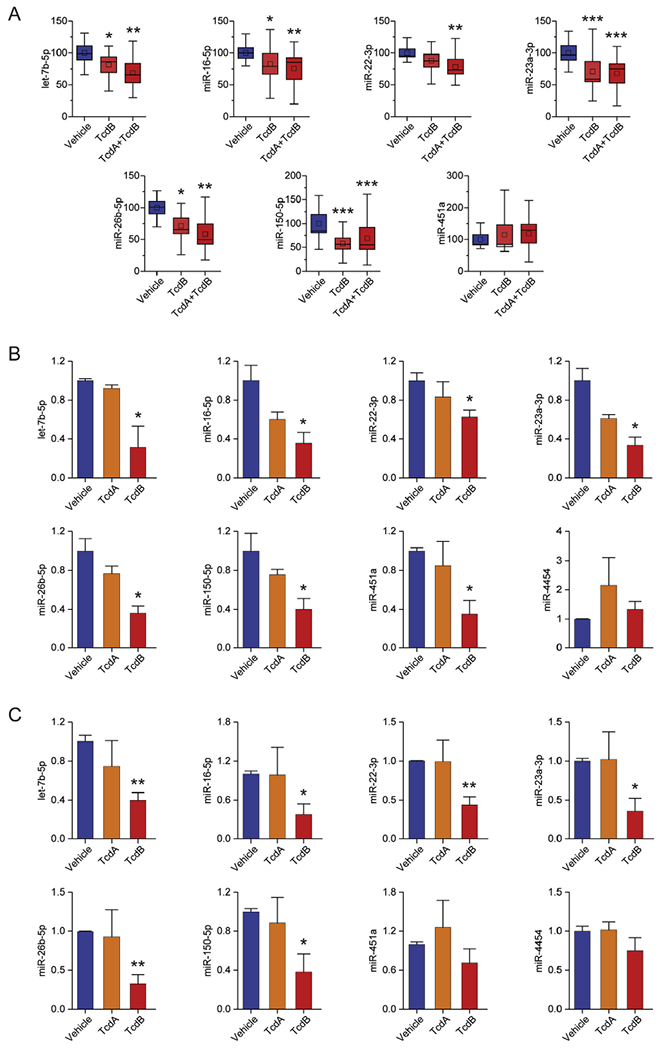
Down-regulation of miRNAs in mouse colonic tissues and human colonoids treated with C difficile toxins (Tcds). (A) Box plots depicting the changes in miRNA levels in colonic tissues from animals treated with TcdB and combination of TcdA with TcdB, compared with HBSS-treated control animals (Vehicle). Box plots denote mean % change ± SEM, inner boxes represent mean, and error bars represent 95% CIs. miRNA levels were assessed with RT-qPCR, normalized against RNU5G and 5S rRNA, and compared with control samples (Vehicle) set as 100%. Changes in miRNA levels after (B) 30 minutes and (C) 6 hours in colonoids treated with TcdA or TcdB, compared with DMEM-treated control samples (Vehicle). miRNA levels assessed with RT-qPCR were normalized against RNU1A1 and 5S rRNA and expressed as mean ± SEM compared with control samples set as 1. Statistical significance was determined by Student t test. *P < 0.05; **P < 0.01; ***P < 0.001. HBSS, Hank’s Balanced Salt Solution; other abbreviations as in Figure 1.
The above findings suggest the effect of CDI on miRNA regulation is mediated by the direct effect of TcdA and/or TcdB on colonic epithelial cells and the observed alterations in circulating miRNAs in FMT-treated patients may result from changes at the tissue level. To test this hypothesis, we exposed normal human colon organoids (colonoids) to TcdA or TcdB and analyzed miRNA expression. Colonoids from freshly isolated human colon mucosa were treated with TcdA (10 pmol/L), TcdB (10 pmol/L), or vehicle (DMEM), and cell extracts were obtained 30 minutes and 6 hours later. RT-qPCR analysis showed that 30 minutes after exposure to TcdB, all miRNAs studied were reduced (Figure 3B), an effect sustained for 6 hours (Figure 3C). These results show that miRNA suppression in CDI is attributed to the activity of TcdB and further support the hypothesis that FMT-mediated up-regulation of circulating miRNAs is driven, at least in part, at the epithelial level.
FMT Counteracts C difficile Effects on miRNA Biogenesis
The evidence collectively show that the miRNAs studied adhere to a common mode of regulation, although they are encoded by different genes and controlled by different mechanisms at the level of transcription. In fact, the miRNAs studied are located in different chromosomes, are intragenic (exonic or intronic) or intergenic, and their expression is under the control of distinct regulatory elements. Therefore, their regulation may be the result of a universal mechanism. Following transcription, miRNA maturation is a process shared by most miRNAs, and their concerted up-regulation by FMT suggests that miRNA processing may be affected by C difficile–associated dysbiosis. Therefore, we analyzed the effects of C difficile and FMT on the expression of enzymes playing a central role in miRNA biogenesis (DROSHA, DICER1, AGO2). RT-qPCR analysis of cecal RNA extracts from rCDI mice suggested that CDI suppresses Drosha expression by 50% (Figure 4A, left), with minor changes on Dicer1 and Ago2 mRNA levels (Supplementary Figure 11). A more pronounced effect (>80% decrease) on Drosha expression was evident at the protein level (Figure 4A, right). Importantly, the effects on both Drosha mRNA and protein were reversed by FMT (Figure 4A). Similarly, in colonic tissues of mice treated with exotoxins, Drosha mRNA levels dropped by 40% (Figure 4B). In human colonoids, TcdB showed small but significant effects on DROSHA mRNA (Figure 4C, left), while Western blot analysis revealed a robust decrease (>60%) in protein levels (Figure 4C, right).
Figure 4.

C difficile infection suppresses DROSHA expression and miRNA processing. (A) Drosha mRNA and protein levels in ceca from animals treated with FMT, infected with C difficile (CDI), infected with C difficile and treated with FMT (CDI+FMT), compared with FMT donors. (B) Drosha mRNA levels in colonic tissues from animals treated with TcdB or combination of TcdA with TcdB, compared with control samples (Vehicle). (C) Drosha mRNA and protein levels in colonoids treated with TcdA or TcdB, compared with DMEM-treated control subjects (Vehicle). (D) DROSHA protein levels in NCM356 colonic epithelial cells on knockdown of DROSHA by means of short interfering (si) RNA (left) and treated with TcdB (right), compared with nontargeting siRNA (siControl) and DMEM-treated control samples (Vehicle), respectively. (E) Primary (pri) miRNA transcript levels in NCM356 cells on knockdown of DROSHA (top) and in colonoids treated with TcdA or TcdB bottom), compared with nontargeting siRNA (siControl) and DMEM-treated control samples (Vehicle), respectively. mRNA and pri-miRNA levels assessed with RT-qPCR were normalized against β-Actin and GAPDH and expressed as mean ± SEM compared with control samples set as 1. DROSHA protein levels were assessed with immunoblot analysis. alpha-Tubulin was used as the loading control. Bottom right: schematic representation of the proposed model of miRNA regulation by CDI. Statistical significance was determined by Student t test. *P < 0.05; **P < 0.01; compared with CDI: #P < 0.01. DMEM, Dulbecco’s Modified Eagle Medium; other abbreviations as in Figure 1.
To investigate the role of drosha suppression in mediating the CDI effects on the regulation of miRNA levels, we knocked down drosha in colonic epithelial cells (NCM356) by means of small interfering RNA. We verified that the knockdown of drosha mimics the effects of TcdB on drosha protein levels in these cells (Figure 4D). RNA extracts from these cells were subjected to RT-qPCR for the levels of the primary transcripts (pri-miRNAs) and the mature forms of 3 different miRNAs, known to be transcriptionally regulated by distinct mechanisms.16–18 The results showed that on DROSHA inhibition, the levels of the pri-miRNAs (Figure 4E, top) are increased with the concurrent and significant decrease in mature miRNA levels (Supplementary Figure 12). We then measured the levels of these pri-miRNAs in colonoids treated with TcdB. In the same line, we found that although the mature miRNAs are suppressed by TcdB (Figure 3), the levels of the respective pri-miRNAs are increased (Figure 4E, bottom).
Combined, these data suggest that drosha expression is decreased in response to CDI, a phenomenon regulated at both the transcriptional and post-transcriptional level. Furthermore, they attribute the concerted changes in miRNA levels to the dysregulation of miRNA biogenesis machinery (proposed model shown in Figure 4E) by rCDI and its recovery by FMT treatment.
FMT-Regulated miRNAs Possess Functional Properties
We next investigated the downstream effects of FMT-regulated circulating miRNAs in our rCDI patient cohorts compared with healthy control subjects. We assessed miRNAs predicted to target specific chemokines and cytokines, which we previously found to be down-regulated by FMT.4 Based on TargetScan prediction analyses we found that the levels of 6 miRNAs in FMT-treated rCDI patients inversely correlate with the serum levels of FGF21, IL12B, IL18, and TNFRSF9 proteins (Supplementary Figure 13). miR-23a, miR-26b and miR-130a are predicted to target the 3′UTR of FGF21 mRNA, miR-23a is predicted to target the 3′UTR of IL12B, and miR-150 the 3′UTR of IL18, whereas miR-20a and miR-28 are the 3′UTR of TNFRSF9. Overexpression of these miRNAs in colonic epithelial cells showed that miR-26b, miR-23a, miR-150, and miR-130a suppress the mRNA levels of FGF21, IL12B, IL18, and TNFRSF9, respectively (Figure 5A). These findings were further validated by 3′UTR reporter assays, where mutations in their target sequences within the 3′UTRs reversed their suppressive effects (Figure 5B), suggesting that the 4 miRNAs directly target these mRNAs.
Figure 5.

Effects of FMT-regulated miRNAs on circulating proteins in patients with CDI. (A) Effects of miR-26b-5p, miR-23a-3p, miR-150-5p, and miR-28-5p overexpression on the levels of FGF21, IL12B, IL18, and TNFRSF9 mRNAs, respectively, in colonic epithelial cells. Gene expression data normalized against β-Actin and GAPDH are expressed as mean ± SEM compared with miR-C–transfected cells set as 1. (B) Effects of miR-26b-5p, miR-23a-3p, miR-150-5p, and miR-28-5p on the activity of FGF21, IL12B, IL18, and TNFRSF9 mRNA 3′UTRs, as assessed with luciferase reporter assays. 3′UTR sequences were cloned in a reporter vector downstream of the Renilla Luciferase gene. The reporter vector was transfected in colonic epithelial cells and luciferase activity was measured 24 hours after the overexpression of the respective miRNAs in the same cells. Direct targeting of the 3′ UTR by the miRNA was validated with assays using deletion mutants (DMs) of the respective miRNA target sequences. Renilla Luciferase activity was normalized against the activity of the Firefly Luciferase gene expressed by the same vector. miR-C (cel-miR-39–p), a nontargeting miRNA, was used as negative control. (C) CDI effect on the levels of circulating miR-26b-5p, miR-23a-3p, miR-150-5p, and miR-28-5p in patients. The levels of serum miRNAs in C difficile patients compared with healthy control subjects (n = 42), as assessed with the nCounter Nanostring platform. Box plots denote mean % change ± SEM, inner boxes represent mean, and error bars represent 95% CIs. (D) Effects of IL18 on toxin-mediated cell growth inhibition. NCM356 cell growth was monitored in real time as % of confluence (IncuCyte). (E) Effects of il18 on TcdB-mediated cell growth inhibition. NCM460 cell survival was assessed by measuring metabolically active cells (Cell-Titer Glo) and expressed as mean ± SEM compared with untreated cells set as 100%. Statistical significance was determined by Student t test. Compared with miR-C (A and B), healthy control samples (C), and TcdB alone (D and E): *P < 0.05; **P < 0.01; ***P < 0.001. Compared with wild-type 3′UTRs: ##P < 0.01; ###P < 0.001. UTR, untranslated region; other abbreviations as in Figures 1 and 2.
In addition to the changes observed for these miRNAs in the mouse model of rCDI human colonoids (Figure 2), in mouse colonic tissues treated with TcdB (Figure 3), and in FMT-treated rCDI patients (Figure 1A), we found that rCDI decreases the levels of these 4 miRNAs in sera compared with healthy control samples (Figure 5C). The use of miRNA inhibitors against miR-26b and miR-150, in peripheral blood mononuclear cells derived 4 weeks after FMT treatment, partly (FGF21) or completely (IL18) reversed the effect of FMT on their expression, suggesting a functional role for these 2 miRNAs in FMT therapy (Supplementary Figure 14). Pretreatment of colonic epithelial cells with these cytokines revealed that though FGF21, IL12B, and TNFRSF9 had no effects (Supplementary Figure 15), IL18 sensitizes cells to TcdB but not TcdA (Figure 5D), an effect validated in a second mucosal cell line (Figure 5E). The identified miRNA-target interactions provide novel links between inflammation and metabolism (Supplementary Figure 16).
FMT-Regulated miRNAs Modulate Susceptibility to C difficile Toxin Effects
We next examined whether miRNAs up-regulated by FMT can affect the colonic epithelial response to C difficile toxins. First, we tested the effect of miRNA up-regulation, alone or in combination with TcdB, on cell survival. The results showed that individual miRNAs had minor effects on TcdB cytotoxicity. However, combination of miR-23a-3p and miR-150-5p significantly increased cell survival (Figure 6A). A live-cell analysis assay verified the above findings (Supplementary Figure 17) and suggested that miR-23a-3p and miR-150-5p alone (Supplementary Figure 18) or in combination confer a growth advantage to cells treated with TcdB (Figure 6B). In accordance, the viability of a second mucosal cell line exposed to TcdB was significantly increased by the combined overexpression of miR-23a-3p and miR-150-5p (Figure 6C).
Figure 6.
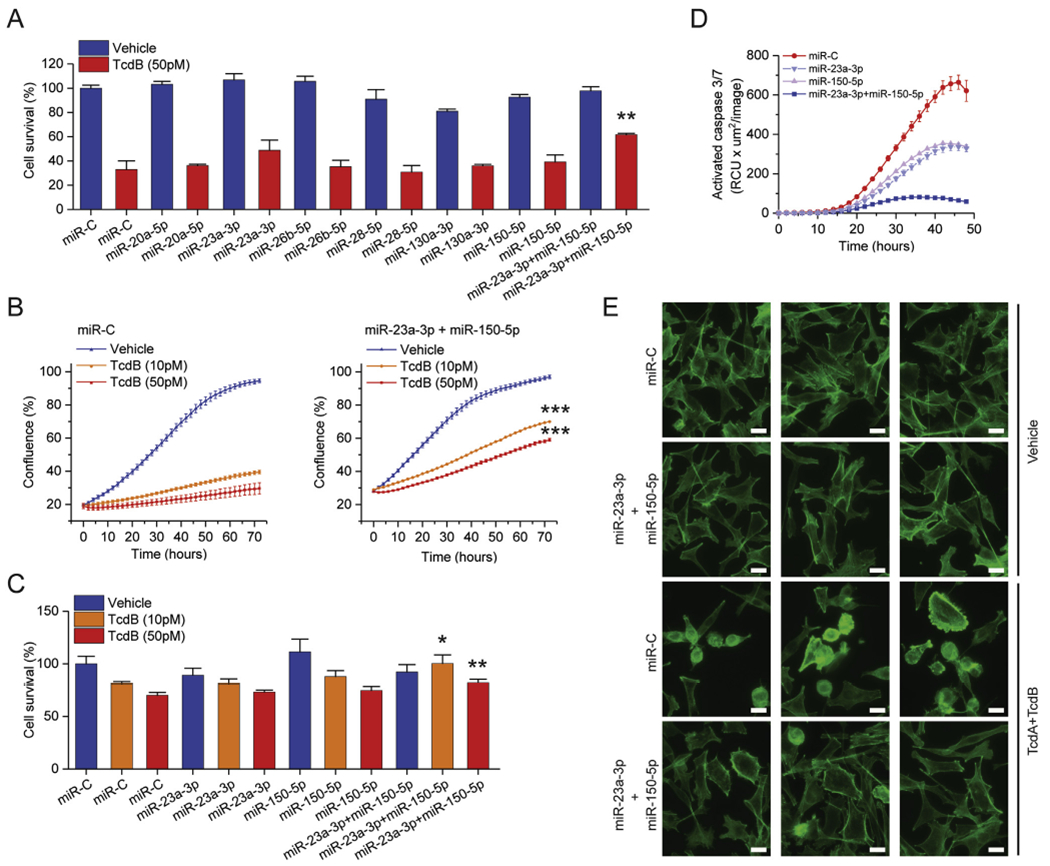
Functional effects of FMT-regulated miRNAs on colonic epithelial cells. (A) Effects of FMT-regulated miRNA overexpression on TcdB-mediated cell growth inhibition. NCM356 cell survival was assessed by measuring metabolically active cells (Cell-Titer Glo) and expressed as mean ± SEM compared with miR-C transfected cells set as 100%. (B) Effects of miR-23a-3p and miR-150-5p overexpression on TcdB-mediated NCM356 cell growth inhibition. Cells were monitored in real time as % of confluence (IncuCyte). (C) Effects of miR-23a-3p and miR-150-5p overexpression on TcdB-mediated NCM460 cell growth inhibition. Cell survival was assessed by measuring metabolically active cells (Cell-Titer Glo) and expressed as mean ± SEM compared with miR-C transfected cells set as 100%. (D) Effects of miR-23a-3p and miR-150-5p overexpression on TcdB-induced cell apoptosis. NCM356 cell apoptosis was monitored in real time as activated caspase3/7 fluorescence (IncuCyte). (E) Effects of miR-23a-3p and miR-150-5p overexpression on TcdB-mediated cytoskeleton rearrangements. NCM356 cytoskeleton organization was studied by fluorescence microscopy after phalloidin staining. miR-C (cel-miR-39–3p), a nontargeting miRNA, was used as negative control. Statistical significance was determined by Student t test. *P < 0.05; **P < 0.01. Abbreviations as in Figures 1 and 2
Although miR-23a-3p and miR-150-5p had minor effect on the early TcdB-mediated cytotoxicity, they promoted cell survival (Supplementary Figure 19). Using a real-time caspase-3/7 detection assay, we found that TcdB induces cell apoptosis in a dose-dependent manner (Supplementary Figure 20); miR-23a-3p and miR-150-5p had additive effects in reducing susceptibility to TcdB-induced apoptosis over time (Figure 6D). A major cytopathic effect attributed to TcdB is the induction of cell morphologic and cytoskeleton changes, which can be visualized by staining colonic epithelial cells with fluorescence-conjugated phalloidin. Using this method, we observed major morphologic changes to epithelial cells, such as loss of actin stress fibers, spindle-like formation, and cell rounding, when exposed to both TcdA and TcdB. Quantification of the ratio of rounded and spindle-like cells revealed that TcdB-induced cytopathic effects were significantly counteracted by the overexpression of miR-23a-3p and miR-150-5p (Figure 6E and Supplementary Figure 21).
Discussion
This is the first study to examine the effects of FMT for rCDI on miRNA signatures. Our findings in a clinical rCDI cohort demonstrate the concerted regulation of miRNA expression by FMT. Validating these observations in a mouse model of rCDI and organoids, we provide evidence that miRNA processing in colonic epithelial cells is directly altered by C difficile toxins and may be affected by C difficile–associated dysbiosis. Conditional knockout of the miRNA-processing enzyme Dicer in murine intestinal epithelial cells, which secrete fecal miRNAs, has been shown to modulate the gut microbiota and exacerbate colitis. This phenotype can be rescued via wild-type fecal transplantation.8 From this observation, we hypothesized that colitis due to CDI can suppress circulating miRNAs, which can be restored by FMT. In support of our hypothesis, predominant suppression of miRNAs was observed in 2 independent cohorts of rCDI patients but was differentiated from observed miRNA suppression in other colitis patient cohorts, such as in patients with chronic inflammatory bowel diseases (ulcerative colitis14 and Crohn’s disease19) and associated colorectal cancer.20 For example, miR-30 family members are up-regulated in both ulcerative colitis and Crohn’s disease, but suppressed in rCDI, although miR-23a-3p is up-regulated only in ulcerative colitis and down-regulated in rCDI. These differences may be leveraged for future diagnostic purposes and could help identify patients with inflammatory bowel disease who can benefit from biotherapeutic products such as FMT, which currently demonstrate variable efficacy compared with patients with CDI.
The miRNA signature in the mouse models suggest that changes in circulating miRNAs reflect alterations in the intestinal epithelial cells. Importantly, the effects of C difficile infection on miRNA levels are reversed in both sera and tissues after FMT, paralleling the post-FMT serum miRNA changes observed in patients with CDI after FMT. TcdA and TcdB have been causally linked to the pathogenetic mechanisms of CDI.21 At the cellular level, toxins induce cytoskeleton reorganization and tight junction disruption resulting in cell rounding and cell death.22 Here, we show that TcdB suppresses key inflammation-related miRNAs in murine intestinal tissues and human colonoids. Moreover, we identify 2 miRNAs up-regulated by FMT, miR-23a and miR-150, which exert cytoprotective effects against TcdB. Interestingly, we found that IL18, previously shown to contribute to mucosal damage,23 sensitizes colonic epithelial cells to TcdB. In addition, we show that IL18 is a direct target of miR-150, suggesting a new mechanism by which FMT may counteract C difficile–induced epithelial disruption. Additional miRNA-regulated cytokines may be involved in the regulation of anti-inflammatory effects of FMT. We found that miR-23a targets IL12B, an essential activator of TH1-cell development, associated with CDI recurrence.24 Collectively, these data support a role for miRNAs suppressed by toxins as a new pathogenic mechanism for CDI. We propose that the restoration of these miRNAs by FMT contributes to the protection of epithelial barrier integrity. Changes in circulating miRNAs may also contribute to extracolonic manifestations of CDI including cardiac, renal, and neurologic impairment.22
Our analyses of the miRNA biogenesis machinery illustrate that down-regulation of miRNAs is likely through the suppression of DROSHA by CDI/TcdB and is restored following FMT. Inhibition of DROSHA results in defective miRNA processing with accumulation of pri-miRNAs. Our findings suggest the biphasic regulation of the microprocessor by CDI. The temporal effect of TcdB suggests that early miRNA regulation is attributed to a nontranscriptional mechanism. In fact, DROSHA protein is reduced in response to TcdB before its mRNA levels are suppressed. Different types of stress have been associated with the stability of DROSHA protein. Under oxidative stress, DROSHA is phosphorylated by p38 MAPK at the N-terminus. This results in disruption of its interaction with DGSCR8, relocation to the cytoplasm, and protein degradation.25 Indeed, the cytotoxic effect of TcdB has been shown to depend on assembly of the host epithelial cell NADPH oxidase complex and the production of reactive oxygen species.26 Other metabolic inputs may be involved. The mammalian Target of Rapamycin nutrient/amino acid sensor activates MDM2, which catalyzes DROSHA ubiquitination, marking it for degradation.27 The long-term suppression of both DROSHA mRNA and protein may involve a gene transcription regulatory mechanism. The transcription factor c-MYC activates DROSHA gene and up-regulates DROSHA protein,28 and is under the control of the WNT/β-catenin pathway, which is suppressed by both toxins.29,30
The regulation of miRNA levels by C difficile may not rely solely on the effects of toxins on mucosal cells. miRNA suppression, mediated by surface layer proteins of specific C difficile ribotypes, has been proposed to attenuate the host’s immune response, resulting in a more persistent infection in mice.31 Interestingly, the suppression of circulating miRNAs on depletion of regulatory T cells (Tregs), reported in mouse models of autoimmune diseases,32 suggests potential links between Treg function, CDI, miRNAs, and FMT. Recent experimental evidence has linked the effectiveness of FMT for colitis with the induction of IL10 and Transforming Growth Factor β, cytokines critical for Treg accumulation in the intestine.33 Whether FMT-directed immunosuppression aids in the recovery from C difficile requires further investigation.3
Intriguingly, in C difficile-naïve animals treated with FMT we observed a trend toward higher Drosha and Dicer mRNA levels and alterations in the miRNA profiles. These control animals were conditioned with cefoperazone (an antibiotic against both gram-positive cocci and gramnegative bacteria) to facilitate C difficile colonization. This observation proposes that host-microbe interactions regulate the host miRNA biogenesis machinery, which in turn may affect directly or indirectly, through the immune response, gut microbiota composition.
Conclusion
We here reported changes in circulating and colonic miRNAs in the context of FMT for rCDI, validating our observations across 2 independent randomized trials. Using 2 different animal models and human colonoids and colonic epithelial cells, we substantiated that C difficile highjacks miRNA biogenesis and showed how miRNA restoration contributes to the therapeutic effects of FMT. Our findings strongly support the need for further mapping of the epitranscriptomic changes associated with FMT. While our data in rCDI mice suggest that TcdB affects Drosha rather than Dicer1 or Ago2 expression, studies with conditional knockout mice of the miRNA-processing machinery, such as Dicer or Drosha, may reveal additional molecular mechanisms or metabolic pathways affected by CDI-induced colitis.
Together, these results provide novel and provocative evidence that modulation of the gut microbiome via FMT induces changes in circulating miRNAs, and that a subset of these miRNAs down-regulates inflammatory protein expression and protects epithelial cells. These findings add new insight into the molecular mechanisms underlying C difficile pathogenesis and FMT and identify new potential targets for therapeutic intervention.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
Fecal microbiota transplantation (FMT) is highly effective at preventing recurrent Clostridioides difficile infection (rCDI). However, the mechanisms of action remain largely unknown. MicroRNAs (miRNAs), short noncoding RNA sequences that bind to complementary sequences of mRNA and can regulate gene expression, may be a potential mechanism by which commensal microbiota communicate with the human host.
NEW FINDINGS
We identified several significant alterations in circulating miRNAs following successful FMT treatment in 2 independent rCDI patient cohorts. miRNA signatures were validated in animal models and human colonoids. We further demonstrated that FMT-regulated miRNAs regulate cell properties and target IL12B, IL18, FGF21, and TNFRSF9, integral in pathways linking to inflammation, autoimmunity, and cancer.
LIMITATIONS
Deeper characterization of the epitranscriptome in FMT is required.
IMPACT
These results describe a new mechanism of action of FMT against rCDI and provide potential new therapeutic targets for conditions associated with intestinal dysbiosis.
Acknowledgments
The authors are grateful to the participants that have made this research possible. The authors are grateful to Matt Emberg, Melanie Lingaya, and Yirga Falcone for their technical assistance in sample preparation. Authors Anna M. Seekatz, Nicholas O. Markham, Tung On Yau, and Maria Hatziapostolou contributed equally.
Funding
This work was supported by a University of Nottingham Research Priority Area grant to Tanya M. Monaghan and supplemented by the National Institute for Health Research Nottingham Digestive Diseases Biomedical Research Centre, based at Nottingham University Hospitals NHS Trust and University of Nottingham, Litwin Initiative at the Crohn’s and Colitis Foundation to Christos Polytarchou, Nottingham Trent University Quality Research funds to Christos Polytarchou and Maria Hatziapostolou, Micheal Smith Health Research Foundation to Christine Lee, National Institute for Diabetes and Digestive and Kidney Diseases grant K01-DK111794 to Anna M. Seekatz, and research funding provided by Alberta Health Services and University of Alberta Hospital Foundation to Dina Kao. The funders were not involved in study design, writing the report or decision for publication.
Abbreviations used in this paper:
- AGO
argonaute
- FGF
fibroblast growth factor
- FMT
fecal microbiota transplantation
- IL
interleukin
- pri-miRNA
primary microRNA transcript
- rCDI
recurrent Clostridioides difficile infection
- Tcd
C difficile toxin
- TNF
tumor necrosis factor
- TNFRSF9
TNF receptor superfamily member 9
Footnotes
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2021.03.050.
Conflicts of interest
Tanya M. Monaghan is a consultant advisor for Takeda. The authors declare no conflicts.
References
- 1.d’Haens GR, Jobin C. Fecal microbial transplantation for diseases beyond recurrent Clostridium difficile infection. Gastroenterology 2019;157(3):624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khoruts A, Sadowsky MJ. Understanding the mechanisms of action of faecal microbiota transplantation. Nat Rev Gastroenterol Hepatol 2016;13(9):508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petri WA Jr, Frisbee AL. Considering the immune system during fecal microbiota transplantation for Clostridioides difficile infection. Gastroenterology 2020;26(5):496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monaghan T, Mullish BH, Patterson J, et al. Effective fecal microbiota transplantation for recurrent Clostridioides difficile infection in humans is associated with increased signalling in the bile acid–farnesoid X receptor–fibroblast growth factor pathway. Gut Microbes 2019;10(2):142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qin Y, Wade PA. Crosstalk between the microbiome and epigenome: messages from bugs. J Biochem 2018; 163(2):105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol 2019;20:21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aguilar C, Mano M, Eulalio A. MicroRNAs at the hostbacteria interface: host defense or bacterial offense. Trends Microbiol 2019;27(3):206–218. [DOI] [PubMed] [Google Scholar]
- 8.Liu S, da Cunha AP, Rezende RM, et al. The host shapes the gut microbiota via fecal microRNA. Cell Host Microbe 2016;19(1):32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kao D, Roach B, Silva M, et al. Effect of oral capsule-vs colonoscopy-delivered fecal microbiota transplantation on recurrent Clostridium difficile infection: a randomised clinical trial. JAMA 2017;318(20):1985–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee C, Steiner T, Petrof EO, et al. Frozen vs fresh fecal microbiota transplantation and clinical resolution of diarrhea in patients with recurrent Clostridium difficile infection: a randomized clinical trial. JAMA 2016; 315(2):142–149. [DOI] [PubMed] [Google Scholar]
- 11.Seekatz AM, Theriot CM, Molloy CT, et al. Fecal microbiota transplantation eliminates Clostridium difficile in a murine model of relapsing disease. Infect Immun 2015; 83(10):3838–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Theriot CM, Koumpouras CC, Carlson PE, et al. Cefoperazone-treated mice as an experimental platform to assess differential virulence of Clostridium difficile strains. Gut Microbes 2011;2(6):326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirota SA, lablokov V, Tulk SE, et al. Intrarectal instillation of Clostridium difficile toxin A triggers colonic inflammation and tissue damage: development of a novel and efficient mouse model of Clostridium difficile toxin exposure. Infect Immun 2012;80(12):4474–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Polytarchou C, Oikonomopoulos A, Mahurkar S, et al. Assessment of circulating microRNAs for the diagnosis and disease activity evaluation in patients with ulcerative colitis by using the nanostring technology. Inflamm Bowel Dis 2015;21(11):2533–9253. [DOI] [PubMed] [Google Scholar]
- 15.Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016;32(18):2847–2849. [DOI] [PubMed] [Google Scholar]
- 16.Ofir M, Hacohen D, Ginsberg D. miR-15 and miR-16 are direct transcriptional targets of E2F1 that limit E2F-induced proliferation by targeting cyclin E. Mol Cancer Res 2011;9(4):440–447. [DOI] [PubMed] [Google Scholar]
- 17.Hassan MQ, Gordon JA, Beloti MM, et al. A network connecting Runx2, SATB2, and the miR-23a~27a~24–2 cluster regulates the osteoblast differentiation program. Proc Natl Acad Sci U S A 2010;107(46):19879– 19884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu T, Chong Y, Cai B, et al. DNA methyltransferase 1–mediated CpG methylation of the miR-150-5p promoter contributes to fibroblast growth factor receptor 1–driven leukemogenesis. J Biol Chem 2019;294(48):18122– 18130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oikonomopoulos A, Polytarchou C, Joshi S, et al. Identification of circulating microRNA signatures in Crohn’s disease using the nanostring ncounter technology. Inflamm Bowel Dis 2016;22(9):2063–2069. [DOI] [PubMed] [Google Scholar]
- 20.Chen G, Feng Y, Li X, et al. Post-transcriptional gene regulation in colitis associated cancer. Front Genet 2019; 10:585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chandrasekaran R, Lacy DB. The role of toxins in Clostridium difficile infection. FEMS Microbiol Rev 2017; 41(6):723–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Bella S, Ascenzi P, Siarakas S, et al. Clostridium difficile toxins A and B: insights into pathogenic properties and extraintestinal effects. Toxins (Basel) 2016; 8(5):134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nowarski R, Jackson R, Gagliani N, et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell 2015; 163:1444–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yacyshyn MB, Reddy TN, Plageman LR, et al. Clostridium difficile recurrence is characterized by pro-inflammatory peripheral blood mononuclear cell (PBMC) phenotype. J Med Microbiol 2014. October;63(Pt 10):1260–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Q, Li W, She H, et al. Stress induces p38 MAPK-mediated phosphorylation and inhibition of drosha-dependent cell survival. Mol Cell 2015;57(4):721–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farrow Ma, Chumbler NM Lapierre LA, et al. Clostridium difficile toxin B–induced necrosis is mediated by the host epithleial cell NADPH oxidase complex. Proc Natl Acad Sci U S A 2013;110(46):18674–18679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ye P, Liu Y, Chen C, et al. An mTORC1-Mdm2-drosha axis for miRNA biogenesis in response to glucose- and amino acid-deprivation. Mol Cell 2015;57(4):708–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Zhao X, Gao P, et al. c-Myc modulates microRNA processing via the transcriptional regulation of drosha. Sci Rep 2013;3:1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bezerra Lima B, Faria Fonseca B, da Graça Amado N, Moreira Lima D, Albuquerque Ribeiro R, Garcia Abreu J, de Castro Brito GA. Clostridium difficile toxin A attenuates Wnt/β-catenin signaling in intestinal epithelial cells. Infect Immun 2014;82(7):2680–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen P, Tao L, Wang T, et al. Structural basis for recognition of frizzled proteins by Clostridium difficile toxin B. Science 2018;360(6389):664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kennedy KF. The effects of surface layer proteins isolated from Clostridium difficile on TLR4 signalling. PhD thesis. Dublin City University, March 2016. [Google Scholar]
- 32.Jin F, Hu H, Xu M, et al. Serum microRNA profiles serve as novel biomarkers for autoimmune diseases. Front Immunol 2018;9:2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burrell C, Garavaglia F, Cribiu FM, Ercoli G, et al. Therapeutic faecal microbiota transplantation controls intestinal inflammation through IL10 secretion by immune cells. Nat Comm 2018;9:5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.