

Abstract

Herein, we describe a study of the synthesis, characterization, and catalytic properties of a cis-dichlorido seleno-chelated Hoveyda–Grubbs type complex (Ru8). Such a complex has been obtained through a straightforward and high-yielding synthetic protocol in three steps from the commercially available 2-bromobenzaldehyde in good overall yield (54%). The catalytic profile, especially the latency of this complex, has been probed through selected olefin metathesis reactions such as ring-closing metathesis (RCM), self-cross-metathesis (self-CM) and ring-opening metathesis polymerization (ROMP). In addition to its high latency, the selenium Hoveyda-type complex Ru8 exhibits a switchable behavior upon thermal activation. Of interest, while the corresponding sulfur-chelated Hoveyda type catalyst is reported to be only activated by heat, the selenium analogue was found to be active upon both heat and light irradiation.
Introduction
Since carbon–carbon bond formation is a central aim in organic chemistry, olefin metathesis takes a crucial place in this field. The versatility and the wide application scope of metathesis allowed its use for compounds relevant as pharmaceuticals and the preparation of well-defined polymers as well as challenging target-oriented synthesis, on both the academic and industrial level.1 This methodology became widespread along with the successive enhancement of the catalyst of olefin metathesis, starting from the first well-defined ruthenium based Grubbs complex (PCy3)2(Cl)2Ru=CHPh (Gru-I, Figure 1)2 followed by the introduction of an N-heterocylic ligand (NHC) yielding to the second-generation Grubbs type catalyst (NHC)(PCy3)(Cl)2Ru=CHPh (Gru-II)3 to the phosphine-free Ru complex bearing a bidentate 2-alkoxybenzylidene ligand [(NHC)(2-isopropoxy-κ-O-benzylidene)RuCl2] (Hov-II, Figure 1).4 Furthermore, the structural flexibility of these complexes allowed a great adjustability of their properties5 such as an increased immunity toward unwanted C=C bond shifting,6E/Z selectivity,7 immobilization/heterogenization of molecular complexes,8 compatibility with green and aqueous media,5f latency,9 and light-activated olefin metathesis.10
Figure 1.
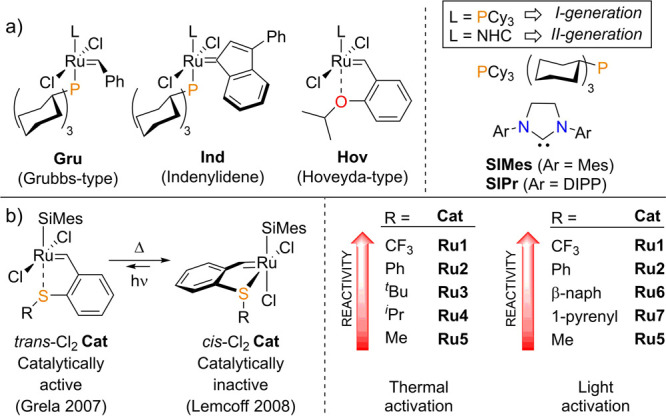
(a) Selected ruthenium-based catalysts for olefin metathesis and the neutral ligands. (b) trans- and cis-dichlorido S-chelated Ru benzylidene complexes and the influence of the S substituent on the reactivity. (Mes = mesityl (2,4,6-trimethylphenyl), DIPP = 2,6-diisopropylphenyl, NHC = N-heterocyclic carbene, SIMes =1,3-bis-mesityl-2-imidazolidin-2-ylidene, SIPr = 1,3-bis-(2,6-triisopropylphenyl)-2-imidazolidinylidene).
A new class of complexes emerged with the simple replacement of the chelating oxygen atom in the Hov-II structure by other main-group elements such as N,11 S,12−14 P,15 or halogen16 atoms (Figure 1b). These catalysts exist in two isomeric forms, trans- and cis- dichlorido ruthenium complexes, as depicted in Figure 1. Among all of these novel catalysts, the sulfur-chelated Ru-Hoveyda type complexes are some of the most extensively studied, notably by Lemcoff and co-workers.17 Of interest, this group demonstrated that the trans-Cl2 form of these complexes was the active precatalyst in olefin metathesis transformations but readily isomerized in solution to the thermodynamically more stable and inactive cis-Cl2 complexes.14a The very same group explained this stabilization of the cis form by the deleterious strong trans influence of the NHC ligand preventing the coordination/stabilization of a second σ-donor ligand in a trans position.15,18 Remarkably, while all of these cis-Cl2 catalysts can be activated by heat and exhibit a thermal-switchable behavior, the cis-Cl2 complex can easily isomerize to the catalytically active trans form upon irradiation with UV-A light. Such light-activated catalytic systems were found to be the most reactive in apolar solvents such as benzene due to a higher rate of isomerization and a significant stabilization of the trans-Cl2 form of the complexes.18
Lemcoff and co-workers demonstrated that the reactivity of such S-chelated complexes was strongly dependent on the steric14b,19 and the electronic properties14b of the S-substituent (denoted R in Figure 1) with the reactivity trend CF3 > Ph > tBu > iPr > Et > Me19 with thermal activation and CF3 > Ph > β-naphthyl > 1-pyrenyl > iPr11g,14b with 350 nm irradiation. On the basis of this observation, we assumed that the replacement of the S atom by the larger Se atom could afford some alteration of the reactivity of the corresponding Ru catalyst in comparison to Ru4 (SiPr).18,20 While the synthesis of such a seleno-chelated Ru complex has been already reported by the Lemcoff group,15 the reactivity profile of such a system has never been explored.15 Indeed, Lemcoff and co-workers synthesized and partially characterized the selenium Hoveyda–Grubbs complex to confirm the theoretical predictions of the favored cis conformation of such catalyst but did not study the preferred activation method or the cis–trans equilibrium of such a complex. In addition to not being the primary goal of the study, the absence of an in-depth catalytic examination was probably precipitated by the tedious low-yield synthesis of the required selenium-containing benzylidene ligand.
Herein, we report the improved synthesis of the selenium variant of the Hoveyda–Grubbs catalyst and its characterization and catalytic profile, including its latency and activity in selected olefin metathesis benchmark reactions.
Results and Discussion
The first synthesis of the selenium-bearing benzylidene ligand precursor was disclosed in 2009 using methyl 2-aminobenzoate as the starting material.15,21 In addition to being laborious and very low-yielding (3.4%), this preparation involved some hazardous compounds such as potassium selenocyanate in acidic media. Furthermore, this pathway included the selenation step at the very beginning of the synthesis and forced the handling of potentially toxic volatile organoselenium compounds.22 We thus considered to revisit the synthesis of such a molecule with two main aims in mind: (1) avoiding most of these aforementioned steps and hazardous reactants and also (2) introducing the selenium atom at a late stage of the synthesis. Therefore, we considered the synthesis of selenide 3 from the corresponding bromo derivative 2 via a simple one-step formation of a Grignard reagent and its subsequent reaction with metallic selenium as described by Tang23 (Scheme 1). To do so, bromostyrene 2 was treated with 1.1 equiv of Mg chips in freshly distilled THF at room temperature in the presence of a substoichiometric amount of lithium chloride (0.1 equiv).24 Once the organomagnesium derivative was formed, the gray selenium (1.0 equiv) was added in one portion followed by an excess of isopropyl iodide (3 equiv).25 The product 3 was then obtained in 74% yield with a simple extraction in DCM and could be used without further purification. Of note, the use of 1 equiv of LiCl for the first step of this reaction (instead of 0.1 equiv) proved to be deleterious for the formation of the selenolate intermediates Ar-Se-MgBr and prevented any further reaction. In comparison with the previous synthesis, such a methodology allowed the isolation of the selenide derivative 3 in a 67% yield from the commercially available 2-bromobenzaldehyde 1 and the selenium atom was introduced in the very last step. Of importance, such a late introduction of the heteroatom in the organic synthesis renders it safer than previous syntheses, since no further required step forces the handling and manipulation of potentially toxic volatile organoselenium compounds.
Scheme 1. Synthesis of the Selenide 3 and Corresponding Ru Complex Ru8 and ORTEP Diagram of Ru8 with 50% Probability Ellipsoids.
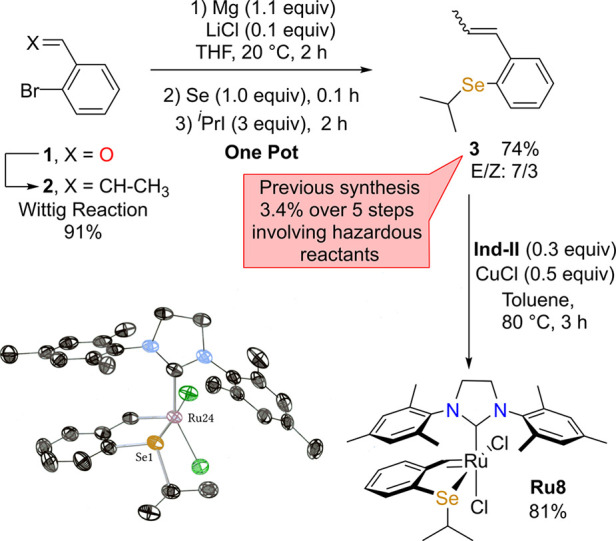
Hydrogen atoms are omitted for clarity.
A simple ligand exchange of 3 (1 equiv) with the second-generation ruthenium indenylidene complex Ind-II (0.3 equiv) in the presence of CuCl (0.5 equiv) as a phosphine scavenger allowed the isolation of the corresponding cis-dichlorido seleno-chelated complex Ru8 (depicted in Scheme 1) as unique isomer within 81% yield. 1H and 13C NMR as well as a single-crystal X-ray analysis of Ru8 confirmed the cis-dichlorido geometry. The selenium–ruthenium bond length was 2.4479(7) Å, in line with reported L-type ligand aryl/alkyl–Se–Ru(II) bonds in the Cambridge Crystallographic Database.15,26 Of note, 77Se NMR spectroscopy confirmed the coordination of the selenium atoms to the Ru(II) center, since the 77Se peaks of 3 (δ 368.8 and 379.5 ppm for the E and Z isomers, respectively) are shifted downfield to δ 518.9 ppm in Ru8 (Δδ = 150.1 and 139.3 ppm).
The activity and the stability of complex Ru8 were then probed through the RCM reaction of the benchmark substrate27 diethyl diallylmalonate (4). The first attempts of an RCM reaction of 4 were undertaken with 1 mol % of Ru8 (0.5 M in DCM or C6D6, Table 1, entries 1 and 4). Whether in DCM or in benzene-d6 (0.5 M), the complex Ru8 was found to be completely inactive at room temperature. Remarkably, such solutions of Ru8 in the presence of RCM substrates could be stored for more than 1 month without any sign of reaction or of decomposition of the complex, demonstrating the high latency as well as the exceptional stability of Ru8. A further study of this complex led us to consider a thermal activation of Ru8 as reported by Lemcoff for the sulfur-containing counterpart Ru4.14a,28 The reaction mixture of 4 in the presence of Ru8 was thus heated at 40 °C in DCM for 48 h, but no conversion of 4 could be observed under such conditions. When DCM was replaced with benzene, the reaction mixture could be heated to 80 °C and this temperature allowed the activation of Ru8, which promoted the RCM of 4 and led to a modest conversion of 29% within 7 h. Of high interest, while the activation of Ru8 is demonstrated by the conversion of 4, the trans-Cl2 species, the active form of this type of catalyst, has never been observed in 1H NMR of the crude mixture in such an activation mode (vide infra and in the Supporting Information).
Table 1. RCM of 4 Promoted by Complex Ru8 upon Thermal or Light Activation.

| entry | solvent | activation | time (h) | conversn (%) |
|---|---|---|---|---|
| 1 | CD2Cl2 | none, 20 °C | 720 | <5 |
| 2 | CD2Cl2 | heat, reflux | 48 | <5 |
| 3 | CD2Cl2 | hν, 20 °C | 24 | 25 |
| 4 | C6D6 | none, 20 °C | 720 | <5 |
| 5 | C6D6 | heat, 80 °C | 7 | 29a |
| 6 | C6D6 | heat, 80 °C | 24 | 63a |
| 7 | C6D5CD3 | heat, 80 °C | 24 | 79 |
| 8 | C6D5CD3 | heat, 110 °C | 5 | 78 |
| 9 | C6D6 | hν, 20 °C | 12 | 73 |
The formation of a side product, namely the product resulting from cycloisomerization, was observed.
A longer reaction time at 80 °C (up to 24 h) allowed an improved final conversion of 63% at the expense of the selectivity of the reaction (entry 6, Table 1). Indeed, in the case of prolonged heating of the Ru8 solution with 4, the product of cycloisomerization (ca. 7%) was observed in 1H NMR, which is probably due to the decomposition of the Ru species. Replacing benzene with deuterated toluene (C6D5CD3) within the same conditions of temperature (80 °C) and catalyst loading of Ru8 (1 mol %) offered a conversion rate of the reaction similar to that in benzene, leading to a conversion of 30% after 6 h of heating and an increased final conversion of 79% yield after 24 h at 80 °C (entry 8, Table 1). Gratifyingly, the rate of the reaction dramatically increased when the mixture was heated to 110 °C, offering a conversion of 78% within only 5 h (entry 8, Table 1).
Finally, light irradiation was then considered to activate Ru8 and promote the RCM of diene 4. To assess the feasibility of such an activation, a solution of Ru8 in DCM was first irradiated at 365 nm and the reaction progress was monitored by NMR spectroscopy. Gratifyingly, 1H NMR unveiled the formation of the trans-Cl2 isomer of Ru8 with an alkylidene proton signal at 17.72 ppm (vs 17.46 ppm for the cis isomer). Such a species has been detected in a small quantity of ca. 4% after 3 h of irradiation at 365 nm (Keq determined as 4.2 × 10–2). On the basis of this successful activation, a mixture of 4 in the presence of 1 mol % of Ru8 in DCM was placed in a UV reactor and irradiated at 365 nm. Such an activation method allowed the RCM of 4 to occur, leading to a conversion of 25% after 24 h of light irradiation. A longer irradiation time did not considerably improve the conversion of the reaction, since after 48 h only 32% of 4 was consumed. On the other hand, in benzene, RCM of 4 proceeded more quickly and was more productive on exposure to light in comparison to the same transformation in DCM or that activated by heat, allowing for 73% conversion within 12 h. Such a result is consistent with the higher rate of activation of Ru8 in benzene than in DCM under light irradiation with a Keq value determined to be 8.7 × 10–2. Of note, 1H NMR of the crude RCM reaction mixture in benzene unveiled a similar activation of Ru8 with an isomerization rate of up to 9% after 3 h of irradiation. After 12 h under such conditions (Table 1, entry 9), the catalyst Ru8 was completely decomposed and no alkylidene proton signal could be observed in 1H NMR, preventing an improvement in the yield upon a longer time of irradiation (24 h). To the best of our knowledge, such light activation has never been described with the S-chelated Hoveyda type complex Ru4 containing an isopropyl moiety as the sulfur substituent. Lemcoff and co-workers showed that Ru4, in contrast to the S-trifluoromethyl- or S-aromatic-functionalized Ru1–2 and Ru6–7, did not promote the RCM of diene 4 upon light activation regardless of the reaction conditions of time and solvent.14b,17 This successful light activation of Ru8 demonstrated that, as postulated earlier, the replacement of the sulfur with a selenium atom in the S-chelated Hoveyda type complexes afford some additional features, even with the simple iPr moiety as a substituent of the heteroatom.
Of interest, in the case of heat activation, complex Ru8 demonstrated a thermally switchable behavior similar to that of its sulfur-chelating counterpart Ru4: i.e., as soon as the reaction mixture was cooled to room temperature, the RCM of 4 was detained and no further evolution could be observed for days at 23 °C. Such a reaction could be resumed by heating the reaction mixture once again at 80 °C, and the complex Ru8 could sustain several heating–cooling cycles without any sign of decomposition.14a,28 Such behavior is consistent with the absence of the trans-Cl2 form of Ru8 observed under thermal conditions. In contrast, this switchable behavior of Ru8 was lost with the irradiation activation mode (Figure 2, top graph). Indeed, even after the end of the irradiation cycle, we could observe that the RCM of 4 was still evolving, as depicted in Figure 2 (top graph). This absence of switchable behavior upon light activation of Ru8 has been attributed to the persistence of the reactive trans-Cl2 isomer in benzene,1b,2,10 which exhibited a half-life time of trans-Ru8 of ca. 6 h.11c,11g,14b,29 The switchable behavior of Ru8 under the irradiation mode of activation could be reached only through a catalyst activation–deactivation protocol elegantly developed by Lemcoff and co-workers.14b The latter group proposed that heating the reaction mixture after the irradiation cycle with UV would “turn off” the RCM of 4 by forcing the isomerization of the trans-Cl2 catalyst to the thermodynamically more stable inactive cis-Cl2 species. To do so, the reaction mixture was simply heated at 80 °C for a short period of time of 5 min after each light activation cycle (Figure 2, bottom graph). Gratifyingly, in the case of the RCM of 4 catalyzed by Ru8, all trans species isomerized back to the metathesis-inactive cis-Cl2 complex and the reaction stopped. As in the case of the thermal-switchable reaction, the reaction conversion resumed with a new cycle of irradiation at 365 nm as, depicted in Figure 2.
Figure 2.
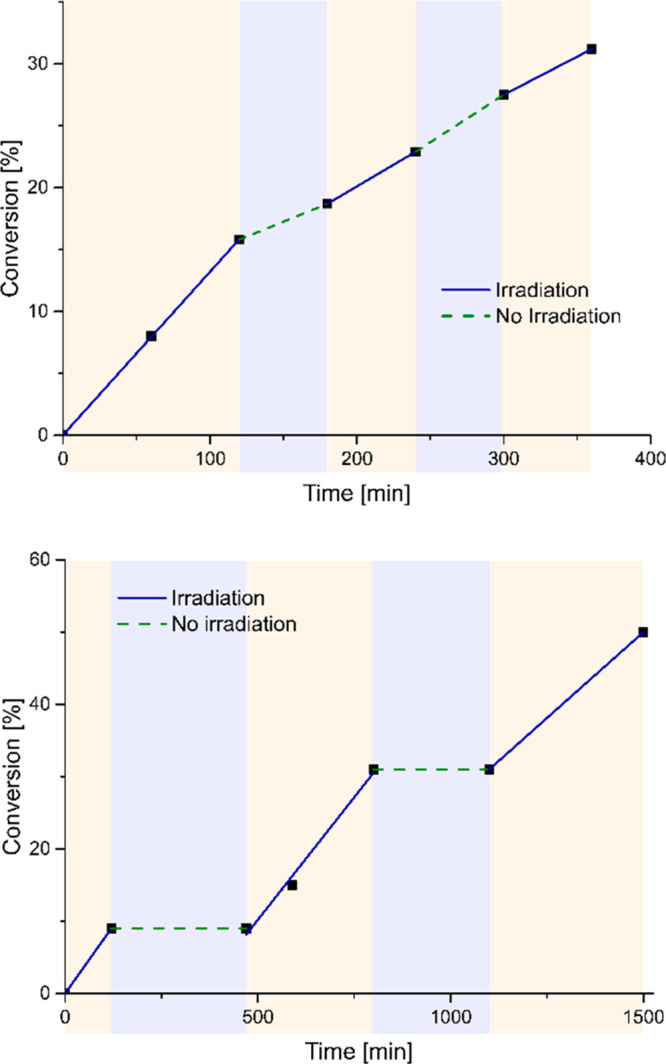
Evolution of the UV-activated RCM of 4 catalyzed by Ru8 (1 mol %). (top) Cycle of irradiation at 365 nm (blue line) and no irradiation (green dashed line). (bottom) Application of the activation–deactivation protocol: cycle of UV irradiation followed by 5 min of heating at 80 °C (blue line) and resting period at room temperature with no irradiation (green dashed line). The conversion was measured by 1H NMR. Lines are visual aids only.
The reactivity and the latency of the cis-dichlorido complex Ru8 were further probed through a series of ROMP reactions with different monomers offering variable ring strain, such as cyclooctene (COE, low ring strain) and dicyclopentadiene (DCPD, high ring strain). First, the ROMP of COE was carried out under several conditions of activation methods, solvents, and catalyst loadings, as reported in Table 2. The catalyst Ru8 demonstrated a perfect latency for the ROMP of COE with 150 ppm (0.015 mol %) catalyst loading in toluene (0.5 M), since no conversion of COE could be observed in 1H NMR or GC without irradiation (Table 2, entry 1). On the other hand, when the reaction mixture was exposed to UV, the ROMP of COE reached completion within 2 h, yielding the corresponding polymer in quantitative yield (E/Z ratio 3.5/1, Mn = 188.5 kDa, PDi = 2.21). When similar reactions were undertaken under neat conditions (150 ppm of Ru8), the catalyst conserved its property of latency (Table 2, entry 3) and the monomer was consumed only under light activation within 1 h (Table 2, entry 4). In both cases, ROMP of COE with Ru8 did not offer well-defined polymeric structures, since high polydispersity indexes from 1.74 to 2.21 were observed. Such a range of PDi can be explained by a high propagation rate and a slow initiation rate of the reaction, which is in line with the aforementioned results with regard to the very slow activation of the catalyst (vide supra).30
Table 2. Evaluation of the Performance of Ru8 in ROMP of COE.

| entry | activation | solvent | time (h) | conversna,b (%) | E/Za | theor Mn (kDa) | Mn (kDa) | PDi |
|---|---|---|---|---|---|---|---|---|
| 1 | dark | Tol | 72 | <5 | ||||
| 2 | hν | Tol | 2 | >99 | 3.5/1 | 726 | 188.4 | 2.21 |
| 3 | dark | neat | 72 | <5 | ||||
| 4 | hν | neat | 1 | >99 | 4/1 | 726 | 173.9 | 1.74 |
| 5 | hν | DCM | 20 | 37 | nd | 271 | 6445 | 2.26 |
| 6 | 80 °C | neat | 72 | >99 | nd | 726 | 127 | ≥7.66 |
| 7c | hν | neat | 24 | 82 | nd | 9900 | 21.5 | 2.64 |
Determined by 1H NMR spectroscopy. nd denotes not determined.
The conversion is the average value of at least two reactions. GPC measurements have been performed on one sample for each entry.
10 ppm of Ru8.
Replacing toluene with DCM as well as changing the activation source to heat (80 °C) dramatically affected the outcome of the reaction (Table 2, entries 5 and 6). When DCM was used as the solvent for the ROMP of COE (150 ppm of Ru8), the conversion of the starting material did not exceed 37%, which is reminiscent of the low conversion of 4 in DCM presented in Table 1. In contrast, heating a reaction mixture of COE and catalyst Ru8 under neat conditions at 80 °C did not prevent the reaction from reaching completion (99% conversion within 72 h). Nevertheless, this activation method dramatically affected the size distribution of the polymers, since the polydispersity index of the corresponding polymer was ≥7.66 (vs 1.74 in the case of light activation). Here again, this high value of PDi is in agreement with the very low activation rate of the catalyst, as previously observed for the RCM reaction of diallylmalonate 4 at 80 °C with catalyst Ru8, for which the trans-Cl2 form of Ru8 was not detectable in 1H NMR (see Table 1, entry 5). Furthermore, such a high PDi value could indicate the presence of a side reaction with a high kinetic rate, such as isomerization of the C=C bonds as previously described in the thermal activation of Ru8. It is noteworthy that the catalyst loading was decreased to as little as 10 ppm for the ROMP reaction of COE under neat conditions, and the reaction still offered a satisfactory yield of polyCOE of 82%.
Using Ru8 (150 ppm) under neat conditions in the presence of 1 equiv of mesitylene as an internal standard, we decided to study the scope of the ROMP reaction. First, cyclooctadiene (COD) was selected for this study. While COD is structurally similar to COE and exhibits a higher ring strain, the ROMP of COD (Scheme 2) with light activation proved to be less efficient than that of COE, leading to a maximum yield of 68% after 6 h of reaction. Moreover, while 1H NMR spectra confirmed the formation of the corresponding polymer, its size distribution was so broad that we were not able to determine the corresponding polydispersity index. On the other hand, monomers presenting a higher ring constraint in comparison to COE and COD were found to be very reactive under the conditions depicted in Scheme 2. Indeed, within only 1 h of irradiation and with 150 ppm of Ru8, the reaction of polymerization of both norbornene (6) and DCPD reached completion (vs 2 h for COE and 6 h for COD). In the case of the highly reactive monomer 6, the catalyst loading could be decreased to as little as 10 ppm without any deletion of the conversion (Scheme 2). Indeed, within 12 h of irradiation, the very low loading of Ru8 allowed the reaction to reach completion and the polydispersity was determined to be 2.04. Such PDi is higher than that of COE (1.74), in accordance with the respective ring constraints and consequently the respective reactivities of the monomers. Finally, the reaction of DCPD with 150 ppm of Ru8 led to the isolation of a rigid colorless material demonstrating the cross-linking of the corresponding polymers. When the catalyst loading was decreased to 10 ppm, the ROMP reaction of DCPD did not reach completion (84% conversion within 12 h) and led to the isolation of a soft solid.
Scheme 2. Scope of the ROMP with Ru8 with Light Activation.

N.D. denotes not detected.
On the basis of the observed stability and reactivity of the complex Ru8, the industrially relevant self-CM of methyl oleate (MO) was then considered. The group of Lemcoff demonstrated that the light activation used for self-CM was extremely beneficial to avoid a fast catalyst decomposition and removed the main limitation of the efficiency of these reactions: the isomerization of the double bonds (C=C bonds shifting, caused by Ru-decomposition products) leading to a complex mixture of products.14f Indeed, in a previous work, Lemcoff and co-workers used the trifluoromethyl-substituted sulfur-chelating Hoveyda type complex Ru1 to promote this reaction under light activation and achieved a perfect selectivity toward unwanted C=C bond shifting of the self-CM, yielding only the corresponding products: octadec-9-ene (C18) and the diester (DE) (no homologues of C18 as well as DE were observed). We wanted to compare the outcome of the reaction of MO catalyzed by Ru1 with the equilibrium obtained when Ru8 was used as a catalyst (Scheme 3).
Scheme 3. Self-CM of Methyl Oleate (MO) Promoted by Ru8 under Light Activation.
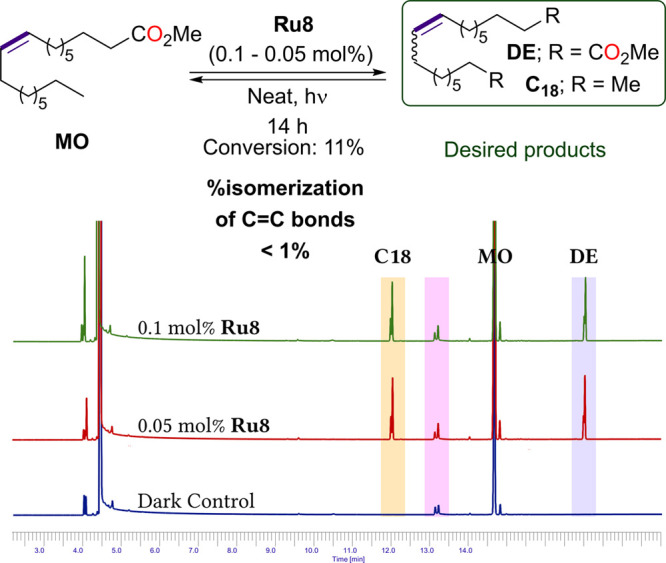
GC traces of the crude reaction. In pink, presence of contamination in the starting material.
To do so, the self-CM of MO was undertaken under conditions similar to those previously reported: 1000 ppm of catalyst under neat condtions and under irradiation for 14 h (Scheme 3). Satisfyingly, the GC traces unveiled a very high selectivity (>99%) toward the expected products C18 and DE, since no isomerization products of the C=C bond shifting could be observed (Scheme 3). Of note, when the catalyst loading was decreased to 500 ppm (0.05 mol %), the same selectivity was obtained, demonstrating the robustness of such an activation method for the self-CM of MO.
Conclusions
In summary, we reported herein a novel, straightforward synthesis of the selenium-containing styrene derivative 3 in an excellent overall yield of 67% starting from commercially available 2-bromobenzaldehyde. The synthesis of the corresponding complex Ru8 was successfully achieved (81% yield) and thus allowed us to study in detail its latency, activation, and reactivity through a set of benchmark RCM, ROMP, and self-CM reactions. Of interest, while the corresponding S-chelated Ru complex Ru4 was reported to be activated only by heat, complex Ru8 demonstrated its versatility upon both thermal and light activation. Similarly to Ru4, the seleno-chelated ruthenium catalyst exhibited a thermal-switchable behavior for the RCM of diethyl diallylmalonate. On the other hand, on activation by irradiation, Ru8 lost its switchable property due to the persistence of the active trans-Cl2 form of this selenium-containing catalyst in benzene. The latency of Ru8 was proven as well in the ROMP of COE and self-CM of methyl oleate. In all of these reactions, Ru8 was found to be very active and efficiently promoted polymerization reactions of norbornene and COE with a catalyst loading as low as 10 ppm. Furthermore, the self-CM of methyl oleate demonstrated an excellent selectivity of >99% toward C=C bond shifting.
We demonstrated that the replacement of the S atom with Se in the Hoveyda–Grubbs type structure endowed the resulting catalyst with an interesting behavior of light activation. Further development of such seleno-chelated complexes may follow the same successful strategy that was used by Lemcoff and co-workers for the sulfur-chelating Hoveyda–Grubbs family catalysts: variation of the selenoether fragment in the benzylidene ligand, such as the introduction of phenylselenolate or trifluoromethylselenolate in place of SeiPr in Ru8.
Experimental Section
General Remarks
Unless otherwise noted, all materials were purchased from commercial suppliers and used as received. All reactions requiring exclusion of oxygen and moisture were carried out in dry glassware with dry solvents (purified using the MBraun SPS) under a dry and oxygen-free atmosphere using Schlenk technique. The bottles with ruthenium catalysts were stored under argon atmosphere, but no special precautions were taken to avoid air or moisture exposure in the moment of extracting catalysts from the bottles. Nuclear magnetic resonance (NMR; 1H, 13C, and 77Se) spectra were recorded on Agilent Mercury 400 MHz spectrometer at ambient temperature with CDCl3 or CD2Cl2 as the solvent. Chemical shifts (δ) are given in parts per million (ppm) downfield from tetramethylsilane with a residual undeuterated solvent peak used as a reference: CDCl3 (δH 7.26 ppm, δC 77.16 ppm), CD2Cl2 (δH 5.32 ppm, δC 53.84 ppm). Coupling constants (J) are reported in hertz (Hz) and refer to H,H couplings. The following abbreviations are used in order to indicate the multiplicity of the signal: s (singlet), d (doublet), t (triplet), q (quartet), quin (quintet), sext (sextet), hept (heptet), dd (doublet of doublets), dt (doublet of triplets), ddd (doublet of doublets of doublets), etc., bs (broad signal), m (multiplet). The data obtained were processed with the software MestReNova. Gas chromatography (GC) measurements were carried out using a PerkinElmer Clarus 580 instrument employing an InertCap 5MS-Sil column and helium as a carrier gas. Infrared (IR) spectra were recorded using a JASCO FT/IR-6200 spectrometer. The substances were examined as a film or in solution. Wavenumbers (ν̃) are reported in cm–1. The data obtained were processed with the software Omni32. High-resolution mass spectrometry (HRMS) measurements were carried out using an AutoSpec Premier spectrometer. Column chromatography was performed using Merck Millipore silica gel (60, particle size 0.043–0.063 nm). Size exclusion chromatography was performed by a specialized laboratory at the University of Warsaw (Dr. Elżbieta Megiel, Faculty of Chemistry, Poland), using a Shimadzu Nexera high-pressure liquid chromatograph (Shimadzu Corporation) in THF at 40 °C with a flow rate of 1.0 mL min–1. Two columns placed in series were used: ReproGel 1000 and 10000 (Dr. Maisch), 5 μm, 300 mm × 8 mm. Samples were detected with a Shimadzu Model RID-20A refractive index detector.
1-Bromo-2-(prop-1-en-1-yl)benzene (2)
Ethyltriphenylphosphonium bromide (8.7 g, 23.1 mmol, 2.3 equiv) and potassium tert-butoxide (2.7 g, 23.1 mmol, 2.3 equiv) were placed in an oven-dried flask. A 20 mL portion of anhydrous THF was added. The reaction mixture was stirred at room temperature for 30 min. Then the mixture was cooled to −78 °C and the appropriate aldehyde 1 (1.9 g, 10.1 mmol, 1 equiv) was added dropwise. Stirring was continued over 2 h at rt. The reaction mixture was quenched with a saturated aqueous solution of NH4Cl (50 mL), and the THF was removed under reduced pressure. DCM (50 mL) was added, and the layers were separated. The aqueous phase was extracted with DCM (2 × 50 mL). The organic layer was dried over anhydrous MgSO4, and afterward, the solid was filtered off and the organic solution was concentrated by the evaporation of DCM. The crude product was purified using quick column chromatography (stationary phase SiO2, eluent DCM) in order to remove triphenylphosphine oxide from the alkene. The product was obtained as a yellow oil (1.81 g, 91%) and could be used without further purification. Further purification was performed by distillation using a Kugelrohr apparatus (p ≈ 10 mbar, T = 145 °C) to give pure product 2 (1.55 g, 78%, E/Z = 1.7:1). Major isomer (E): 1H NMR (400 MHz, CDCl3) δ 7.53 (dd, J = 8.0, 1.3 Hz, 1H, HAr), 7.47 (dd, J = 7.8, 1.7 Hz, 1H), 7.27–7.20 (m, 1H), 7.06 (ddd, J = 7.9, 7.3, 1.7 Hz, 1H), 6.74 (dq, J = 15.7, 1.8 Hz, 1H), 6.19 (dq, J = 15.6, 6.7 Hz, 1H), 1.93 (dd, J = 6.7, 1.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 137.8, 132.9, 130.1, 129.0, 128.2, 127.5, 126.9, 123.1, 18.8.
Isopropyl(2-(prop-1-en-1-yl)phenyl)selane (3)
In a glovebox filled with argon, a 25 mL round-bottom flask was charged with Mg (finely ground, 26 mg, 1.1 mmol, 1.1 equiv), lithium chloride (4.8 mg, 0.1 mmol, 0.1 equiv), and THF (3 mL). To the resulting suspension was added a solution of bromostyrene derivative 2 (200 mg, 1.0 mmol, 1.0 equiv) in THF (2 mL) dropwise. The reaction mixture was stirred at room temperature until the complete consumption of magnesium (leading to a clear colorless to pale yellow solution). A Grignard reagent was thus formed, and gray Se (80 mg, 1.0 mmol, 1.0 equiv) was added in one portion with vigorous stirring. The dark suspension of selenium in the reaction mixture turned rapidly (after 15 min) into a homogeneous clear orange solution, indicating the full conversion of selenium. The flask was taken outside of the glovebox, and 2-iodopropane (509 mg, 3 mmol, 3 equiv) was added to the reaction mixture. After the addition of alkylating agent, a precipitate appeared instantly, leading to a gray to yellow turbid suspension. The reaction mixture was stirred at room temperature overnight, and afterward water was added (5 mL). The product was extracted with DCM (3 × 10 mL). The organic phases were combined, washed with brine, and dried over anhydrous Na2SO4. The volatiles were removed under reduced pressure, and the yellow oil was dried overnight under high vacuum (1 × 10–3 mbar). The product 3 was obtained as a yellow oil (177.6 mg, 74%, E/Z = 1.7:1) and could be used without further purification. Major isomer (E): 1H NMR (400 MHz, CDCl3) δ 7.55 (dd, J = 7.7, 1.1 Hz, 1H), 7.47 (dd, J = 7.8, 1.2 Hz, 1H), 7.13 (m, 2H), 6.95 and 6.79 (dd, J = 15.6, 1.5 Hz, 1H), 6.22–6.04 (m, 1H), 3.41 (sept, J = 6.8 Hz, 1H), 1.91 (dd, J = 6.6, 1.7 Hz, 3H) 1.39 (d, J = 6.8 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 141.2, 135.8, 131.4, 127.9, 127.6, 127.15, 126.8, 33.9, 24.2, 18.8, 14.5, 14.4; 77Se NMR (76 MHz, CDCl3) δ 368.82. HRMS (ESI): calculated mass for C12H17Se [M + H]+, 241.0490; found, 241.0490(3).
Catalyst Ru8
A dry Schlenk vessel was charged with ruthenium complex Ind-II (100 mg, 0.105 mmol, 1 equiv) under an argon atmosphere. Then, 15 mL of anhydrous toluene was added, resulting in a deep red solution. Selenium-containing styrene 3 (62 mg, 0.315 mmol, 3 equiv) and anhydrous CuCl (15 mg, 0.158 mmol, 1.5 equiv) were added to the reaction mixture, and then it was stirred for 3 h at 80 °C. Complete conversion of the Ind-II catalyst was observed using TLC (stationary phase SiO2, eluent EtOAc/n-hexane 10/90 v/v, Rf,Ind-II = 0.39, Rf,Ru8 = 0) after that time. All of the volatiles were removed under reduced pressure, and the crude product was purified using column chromatography (stationary phase SiO2, eluent EtOAc/n-hexane from 20/80 to 80/20 v/v). The product complex Ru8 was obtained as a green solid (67 mg, 81%). 1H NMR (400 MHz, CD2Cl2): δ 17.47 (s, 1H), 7.52–7.41 (m, 2H), 7.20 (ddd, J = 7.7, 5.4, 3.0 Hz, 1H), 7.12 (s, 1H), 7.01 (s, 1H), 6.89 (s, 1H), 6.83 (d, J = 7.5 Hz, 1H), 5.94 (s, 1H), 4.25–3.72 (m, 5H), 2.67 (s, 1H), 2.53 (s, 1H), 2.47 (s, 1H), 2.36 (s, 1H), 2.15 (s, 1H), 1.54 (d, J = 4.4 Hz, 4H), 1.30 (t, J = 5.0 Hz, 2H), 1.02 (d, J = 6.8 Hz, 1H). 13C NMR (101 MHz, CD2Cl2): δ δ 284.6, 215.0, 157.5, 140.3, 140.2, 138.4, 137.8, 136.8, 135.4, 132.4, 131.8, 130.7, 129.8, 129.7, 129.6, 129.2, 128.7, 124.9, 51.6, 51.3, 36.4, 23.7, 21.9, 21.1, 20.9, 20.3, 20.1, 18.7, 17.8. 77Se NMR (76 MHz, CD2Cl2): δ 518.9.
General Procedure for the RCM Reaction of 4
In a glovebox filled with argon, diene 4 (48.5 mg, 50 μL, 0.2 mmol, 1 equiv) was added to a solution of Ru8 (1.38 mg, 0.002 mmol, 1 mol %) in the appropriate solvent (0.4 mL) in a screw-cap NMR tube. The NMR tube was taken outside of the glovebox, and the reaction mixture was stirred at 20 °C or heated or placed in a UV reactor for the required time. Data were recorded using 32 scans with a D1 delay time minimum of 5 s between each pulse. The progress of the reaction was monitored through the disappearance of the methylene signals of 4 (2.63 ppm) and the growth of the methylene proton signal of the product 5 (3.01 ppm).
General Procedure for the ROMP Reactions
A 15 mL vial equipped with a screw cap was charged with a cyclic substrate (1 equiv). Unless the reaction was carried out under neat conditions, an appropriate solvent (0.5 M) was added (DCM or toluene). To the resulting solution was added mesitylene (1 equiv) as an internal standard followed by the addition of a Ru8 catalyst stock solution in DCM (1 M, 0.0150 or 0.001 mol %). For the reactions carried out under neat conditions, the solvent (DCM) was removed under reduced pressure before photochemical activation. The vial was then placed in a UV reactor (365 nm) or in a heated oil bath (80 °C) and was left for the required time. The NMR spectrum of the crude solution was recorded, and it highlighted the presence of the polymer. The resulting polymer was precipitated out with acetone and methanol and then filtered off.
General Procedure for the Reaction of Self-CM of MO
Methyl oleate (MO) (341 μL, 1 mmol) was introduced into a 1.5 mL screw-capped vial. The air in the flask was replaced by argon. The catalyst (0.69 mg, 0.001 equiv, 1 μmol, 0.1 mol % or 0.35 mg, 0.5 μmol, 0.05 mol %) was added as a CH2Cl2 solution. CH2Cl2 was evaporated from the vessel by high vacuum followed by refilling the vial with argon before placing the vial in a UV reactor. Then, the reaction mixture was irradiated for 14 h. After this time, a solution of SnatchCat was added to the reaction mixture and diluted in toluene. The resulting solution was injected into the GC-MS.
Acknowledgments
The authors acknowledge research support from the National Science Centre grant UMO-2016/22/E/ST4/00573. The study was carried out at the Biological and Chemical Research Centre, University of Warsaw, established within the project cofinanced by the European Union from the European Regional Development Fund under the Operational Programme Innovative Economy, 2007–2013. The authors thank Dr. Elżbieta Megiel and Piotr Cieciórski from the Laboratory of Technology of Organic Functional Materials, Faculty of Chemistry, University of Warsaw, for a collaboration with regard to polymer analysis.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.1c00484.
Additional experimental details, crystal data for Ru8, and NMR spectra for compounds 3 and Ru8 (PDF)
Accession Codes
CCDC 2105961 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- a Grela K.Olefin Metathesis: Theory and Practice; Wiley: 2014. [Google Scholar]; b Grubbs R. H.; Wenzel A. G.; O’Leary D. J.; Khosravi E.. Handbook of Metathesis. 2nd ed.; Wiley-VCH: 2015. [Google Scholar]; c Cossy J.; Arseniyadis S.; Meyer C.. Metathesis in Natural Product Synthesis: Strategies, Substrates and Catalysts; Wiley-VCH: 2010. [Google Scholar]; d Phillips J. H. Latest Industrial Uses of Olefin Metathesis. Organometallic Chemistry in Industry 2020, 259–282. 10.1002/9783527819201.ch10. [DOI] [Google Scholar]
- a Trnka T. M.; Grubbs R. H. The Development of L2X2RuCHR Olefin Metathesis Catalysts: An Organometallic Success Story. Acc. Chem. Res. 2001, 34, 18–29. 10.1021/ar000114f. [DOI] [PubMed] [Google Scholar]; b Schwab P.; Grubbs R. H.; Ziller J. W. Synthesis and Applications of RuCl2(CHR′)(PR3)2: The Influence of the Alkylidene Moiety on Metathesis Activity. J. Am. Chem. Soc. 1996, 118, 100–110. 10.1021/ja952676d. [DOI] [Google Scholar]
- a Scholl M.; Trnka T. M.; Morgan J. P.; Grubbs R. H. Increased ring closing metathesis activity of ruthenium-based olefin metathesis catalysts coordinated with imidazolin-2-ylidene ligands. Tetrahedron Lett. 1999, 40, 2247–2250. 10.1016/S0040-4039(99)00217-8. [DOI] [Google Scholar]; b Scholl M.; Ding S.; Lee C. W.; Grubbs R. H. Synthesis and Activity of a New Generation of Ruthenium-Based Olefin Metathesis Catalysts Coordinated with 1,3-Dimesityl-4,5-dihydroimidazol-2-ylidene Ligands. Org. Lett. 1999, 1, 953–956. 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]; c Ackermann L.; Fürstner A.; Weskamp T.; Kohl F. J.; Herrmann W. A. Ruthenium carbene complexes with imidazolin-2-ylidene ligands allow the formation of tetrasubstituted cycloalkenes by RCM. Tetrahedron Lett. 1999, 40, 4787–4790. 10.1016/S0040-4039(99)00919-3. [DOI] [Google Scholar]
- a Garber S. B.; Kingsbury J. S.; Gray B. L.; Hoveyda A. H. Efficient and Recyclable Monomeric and Dendritic Ru-Based Metathesis Catalysts. J. Am. Chem. Soc. 2000, 122, 8168–8179. 10.1021/ja001179g. [DOI] [Google Scholar]; b Gessler S.; Randl S.; Blechert S. Synthesis and metathesis reactions of a phosphine-free dihydroimidazole carbene ruthenium complex. Tetrahedron Lett. 2000, 41, 9973–9976. 10.1016/S0040-4039(00)01808-6. [DOI] [Google Scholar]
- a Diesendruck C. E.; Tzur E.; Lemcoff N. G. The Versatile Alkylidene Moiety in Ruthenium Olefin Metathesis Catalysts. Eur. J. Inorg. Chem. 2009, 2009, 4185–4203. 10.1002/ejic.200900526. [DOI] [Google Scholar]; b Paradiso V.; Costabile C.; Grisi F. Ruthenium-based olefin metathesis catalysts with monodentate unsymmetrical NHC ligands. Beilstein J. Org. Chem. 2018, 14, 3122–3149. 10.3762/bjoc.14.292. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Monsigny L.; Kajetanowicz A.; Grela K. Ruthenium Complexes Featuring Unsymmetrical N-Heterocyclic Carbene Ligands–Useful Olefin Metathesis Catalysts for Special Tasks. Chem. Rec. 2021, 10.1002/tcr.202100126. [DOI] [PubMed] [Google Scholar]; d Kajetanowicz A.; Grela K. Nitro and Other Electron Withdrawing Group Activated Ruthenium Catalysts for Olefin Metathesis Reactions. Angew. Chem., Int. Ed. 2021, 60, 13738–13756. 10.1002/anie.202008150. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Masoud S. M.; Vorobyeva D. V.; Petropavlovskikh D. A.; Bruneau C.; Osipov S. N. Fluorine-containing ruthenium-based olefin metathesis catalysts. Russ. Chem. Rev. 2021, 90, 419–450. 10.1070/RCR4984. [DOI] [Google Scholar]; f Matsuo T. Functionalization of Ruthenium Olefin-Metathesis Catalysts for Interdisciplinary Studies in Chemistry and Biology. Catalysts 2021, 11, 359. 10.3390/catal11030359. [DOI] [Google Scholar]
- Rouen M.; Queval P.; Borré E.; Falivene L.; Poater A.; Berthod M.; Hugues F.; Cavallo L.; Baslé O.; Olivier-Bourbigou H.; Mauduit M. Selective Metathesis of α-Olefins from Bio-Sourced Fischer–Tropsch Feeds. ACS Catal. 2016, 6, 7970–7976. 10.1021/acscatal.6b01428. [DOI] [Google Scholar]
- a Dawood K. M.; Nomura K. Recent Developments in Z-Selective Olefin Metathesis Reactions by Molybdenum, Tungsten, Ruthenium, and Vanadium Catalysts. Adv. Synth. Catal. 2021, 363, 1970–1997. 10.1002/adsc.202001117. [DOI] [Google Scholar]; b Müller D. S.; Baslé O.; Mauduit M. A tutorial review of stereoretentive olefin metathesis based on ruthenium dithiolate catalysts. Beilstein J. Org. Chem. 2018, 14, 2999–3010. 10.3762/bjoc.14.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Szczepaniak G.; Kosiński K.; Grela K. Towards “cleaner” olefin metathesis: tailoring the NHC ligand of second generation ruthenium catalysts to afford auxiliary traits. Green Chem. 2014, 16, 4474–4492. 10.1039/C4GC00705K. [DOI] [Google Scholar]; b Buchmeiser M. R.; Grela K. Immobilization of Olefin Metathesis Catalysts. Olefin Metathesis: Theory and Practice 2014, 495–514. 10.1002/9781118711613.ch20. [DOI] [Google Scholar]
- Monsaert S.; Lozano Vila A.; Drozdzak R.; Van Der Voort P.; Verpoort F. Latent olefin metathesis catalysts. Chem. Soc. Rev. 2009, 38, 3360–3372. 10.1039/b902345n. [DOI] [PubMed] [Google Scholar]
- a Eivgi O.; Phatake R. S.; Nechmad N. B.; Lemcoff N. G. Light-Activated Olefin Metathesis: Catalyst Development, Synthesis, and Applications. Acc. Chem. Res. 2020, 53, 2456–2471. 10.1021/acs.accounts.0c00495. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Eivgi O.; Lemcoff N. G. Turning the Light On: Recent Developments in Photoinduced Olefin Metathesis. Synthesis 2018, 50, 49–63. 10.1055/s-0036-1589113. [DOI] [Google Scholar]
- a Ung T.; Hejl A.; Grubbs R. H.; Schrodi Y. Latent Ruthenium Olefin Metathesis Catalysts That Contain an N-Heterocyclic Carbene Ligand. Organometallics 2004, 23, 5399–5401. 10.1021/om0493210. [DOI] [Google Scholar]; b Slugovc C.; Burtscher D.; Stelzer F.; Mereiter K. Thermally Switchable Olefin Metathesis Initiators Bearing Chelating Carbenes: Influence of the Chelate’s Ring Size. Organometallics 2005, 24, 2255–2258. 10.1021/om050141f. [DOI] [Google Scholar]; c Barbasiewicz M.; Szadkowska A.; Bujok R.; Grela K. Structure and Activity Peculiarities of Ruthenium Quinoline and Quinoxaline Complexes: Novel Metathesis Catalysts. Organometallics 2006, 25, 3599–3604. 10.1021/om060091u. [DOI] [Google Scholar]; d Poater A.; Ragone F.; Correa A.; Szadkowska A.; Barbasiewicz M.; Grela K.; Cavallo L. Mechanistic Insights into the cis–trans Isomerization of Ruthenium Complexes Relevant to Catalysis of Olefin Metathesis. Chem. - Eur. J. 2010, 16, 14354–14364. 10.1002/chem.201001849. [DOI] [PubMed] [Google Scholar]; e Peeck L. H.; Savka R. D.; Plenio H. Fast Olefin Metathesis at Low Catalyst Loading. Chem. - Eur. J. 2012, 18, 12845–12853. 10.1002/chem.201201010. [DOI] [PubMed] [Google Scholar]; f Żukowska K.; Szadkowska A.; Pazio A. E.; Woźniak K.; Grela K. Thermal Switchability of N-Chelating Hoveyda-type Catalyst Containing a Secondary Amine Ligand. Organometallics 2012, 31, 462–469. 10.1021/om2011062. [DOI] [Google Scholar]; g Ginzburg Y.; Anaby A.; Vidavsky Y.; Diesendruck C. E.; Ben-Asuly A.; Goldberg I.; Lemcoff N. G. Widening the Latency Gap in Chelated Ruthenium Olefin Metathesis Catalysts. Organometallics 2011, 30, 3430–3437. 10.1021/om200323c. [DOI] [Google Scholar]; h Żukowska K.; Pump E.; Pazio A. E.; Woźniak K.; Cavallo L.; Slugovc C. Consequences of the electronic tuning of latent ruthenium-based olefin metathesis catalysts on their reactivity. Beilstein J. Org. Chem. 2015, 11, 1458–1468. 10.3762/bjoc.11.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grela K.; Szadkowska A.; Barbasiewicz M.; Kadyrov R.. Neuartige schwefelhaltige Metathese Katalysatoren. (Degussa AG) DE Patent Application DE102007020694.3, 2007.
- Kadyrov R.; Rosiak A.; Tarabocchia J.; Szadkowska A.; Bieniek M.; Grela K.. New Concepts in Designing Ruthenium Based Second Generation Olefin Metathesis Catalysts and their Application. In Catalysis of Organic Reactions: Twenty-second Conference; Prunier M. L., Ed.; CRC Press: 2008; Chemical Industries Series, Vol. 123, p 568. [Google Scholar]
- a Ben-Asuly A.; Tzur E.; Diesendruck C. E.; Sigalov M.; Goldberg I.; Lemcoff N. G. A Thermally Switchable Latent Ruthenium Olefin Metathesis Catalyst. Organometallics 2008, 27, 811–813. 10.1021/om701180z. [DOI] [Google Scholar]; b Ben-Asuly A.; Aharoni A.; Diesendruck C. E.; Vidavsky Y.; Goldberg I.; Straub B. F.; Lemcoff N. G. Photoactivation of Ruthenium Olefin Metathesis Initiators. Organometallics 2009, 28, 4652–4655. 10.1021/om9004302. [DOI] [Google Scholar]; c Tzur E.; Szadkowska A.; Ben-Asuly A.; Makal A.; Goldberg I.; Woźniak K.; Grela K.; Lemcoff N. G. Studies on Electronic Effects in O-, N- and S-Chelated Ruthenium Olefin-Metathesis Catalysts. Chem. - Eur. J. 2010, 16, 8726–8737. 10.1002/chem.200903457. [DOI] [PubMed] [Google Scholar]; d Levin E.; Mavila S.; Eivgi O.; Tzur E.; Lemcoff N. G. Regioselective Chromatic Orthogonality with Light-Activated Metathesis Catalysts. Angew. Chem., Int. Ed. 2015, 54, 12384–12388. 10.1002/anie.201500740. [DOI] [PubMed] [Google Scholar]; e Żukowska K.; Pączek Ł.; Grela K. Sulfoxide-Chelated Ruthenium Benzylidene Catalyst: a Synthetic Study on the Utility of Olefin Metathesis. ChemCatChem 2016, 8, 2817–2823. 10.1002/cctc.201600538. [DOI] [Google Scholar]; f Rozenberg I.; Eivgi O.; Frenklah A.; Butilkov D.; Kozuch S.; Goldberg I.; Lemcoff N. G. Synthesis and Catalytic Properties of Sulfur-Chelated Ruthenium Benzylidenes Bearing a Cyclic (Alkyl)(amino)carbene Ligand. ACS Catal. 2018, 8, 8182–8191. 10.1021/acscatal.8b02122. [DOI] [Google Scholar]
- Diesendruck C. E.; Tzur E.; Ben-Asuly A.; Goldberg I.; Straub B. F.; Lemcoff N. G. Predicting the Cis–Trans Dichloro Configuration of Group 15–16 Chelated Ruthenium Olefin Metathesis Complexes: A DFT and Experimental Study. Inorg. Chem. 2009, 48, 10819–10825. 10.1021/ic901444c. [DOI] [PubMed] [Google Scholar]
- Barbasiewicz M.; Michalak M.; Grela K. A New Family of Halogen-Chelated Hoveyda–Grubbs-Type Metathesis Catalysts. Chem. - Eur. J. 2012, 18, 14237–14241. 10.1002/chem.201202817. [DOI] [PubMed] [Google Scholar]
- Nechmad N. B.; Lemcoff N. G. Sulfur-Chelated Ruthenium Olefin Metathesis Catalysts. Synlett 2021, 32, 258–266. 10.1055/s-0040-1707231. [DOI] [Google Scholar]
- Pump E.; Cavallo L.; Slugovc C. A theoretical View on the Thermodynamic cis–trans Equilibrium of Dihalo Ruthenium Olefin Metathesis (pre-)Catalysts. Monatsh. Chem. 2015, 146, 1131–1141. 10.1007/s00706-015-1433-8. [DOI] [Google Scholar]
- Kost T.; Sigalov M.; Goldberg I.; Ben-Asuly A.; Lemcoff N. G. Latent sulfur chelated ruthenium catalysts: Steric acceleration effects on olefin metathesis. J. Organomet. Chem. 2008, 693, 2200–2203. 10.1016/j.jorganchem.2008.03.028. [DOI] [Google Scholar]
- Singh F. V.; Wirth T.. Selenium Compounds as Ligands and Catalysts. In Organoselenium Chemistry; Wiley-VCH: 2011; pp 321–360. [Google Scholar]
- The synthesis of the benzylidene precursor reported by Lemcoff and co-workers consists of a sequence of selenation via the formation of a diazonium salt followed by a reduction, an oxidation, selenium alkylation, and finally a Wittig olefination.
- a Nogueira C. W.; Barbosa N. V.; Rocha J. B. T. Toxicology and pharmacology of synthetic organoselenium compounds: an update. Arch. Toxicol. 2021, 95, 1179–1226. 10.1007/s00204-021-03003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nogueira C. W.; Zeni G.; Rocha J. B. T. Organoselenium and Organotellurium Compounds: Toxicology and Pharmacology. Chem. Rev. 2004, 104, 6255–6286. 10.1021/cr0406559. [DOI] [PubMed] [Google Scholar]
- Lin H. M.; Tang Y.; Li Z. H.; Liu K. D.; Yang J.; Zhang Y. M. A novel and efficient synthesis of selenides. ARKIVOC 2013, 2012, 146–156. 10.3998/ark.5550190.0013.814. [DOI] [Google Scholar]
- Li-Yuan Bao R.; Zhao R.; Shi L. Progress and developments in the turbo Grignard reagent i-PrMgCl·LiCl: a ten-year journey. Chem. Commun. 2015, 51, 6884–6900. 10.1039/C4CC10194D. [DOI] [PubMed] [Google Scholar]
- Iwaoka M.Nucleophilic Selenium. In Organoselenium Chemistry, Wirth T. Ed.; Wiley-VCH: 2011; pp 53–109. [Google Scholar]
- a Singh P.; Das D.; Singh M.; Singh A. K. Ligand Concentration And Reaction Time Controlled Successive Substitution Of Chloro And H6-Benzene Of Di-M-Chlorobis{H6-Benzene)Dichlororuthenium(II)} With Selenated Schiff Base: Formation Of Half Sandwich Ru(II) Complex Highly Active Catalyst For Oxidation Of Alcohols. Inorg. Chem. Commun. 2010, 13, 223–226. 10.1016/j.inoche.2009.11.011. [DOI] [Google Scholar]; b Das D.; Singh P.; Prakash O.; Singh A. K. Reactions of Benzene Based Half Sandwich Ruthenium(II) Complex with 2,6-bis((Phenylseleno)methyl)pyridine: Preferential Substitution of Ring Resulting in a Catalyst of High Activity for Oxidation of Alcohols. Inorg. Chem. Commun. 2010, 13, 1370–1373. 10.1016/j.inoche.2010.07.039. [DOI] [Google Scholar]; c Singh P.; Singh M.; Singh A. K. Half Sandwich Complexes of Ru(II) and Complexes of Pd(II) and Pt(II) with Seleno and Thio Derivatives of Pyrrolidine: Synthesis, Structure and Applications as Catalysts for Organic Reactions. J. Organomet. Chem. 2009, 694, 3872–3880. 10.1016/j.jorganchem.2009.08.004. [DOI] [Google Scholar]; d Singh P.; Singh A. K. Transfer Hydrogenation of Ketones and Catalytic Oxidation of Alcohols with Half-Sandwich Complexes of Ruthenium(II) Designed Using Benzene and Tridentate (S, N, E) Type Ligands (E = S, Se, Te). Organometallics 2010, 29, 6433–6442. 10.1021/om100807b. [DOI] [Google Scholar]; e Kumar R.; Singh V. V.; Jain N.; Singh A. K. Fast Transfer Hydrogenation(TH) in Aerobic Condition and Oxidation of Alcohols with N-Methylmorpholine-N-oxide Catalyzed by Ru(II) Ligated with Chalcogenated Pyridines and PPh3. ChemistrySelect 2020, 5, 9572–9578. 10.1002/slct.202002031. [DOI] [Google Scholar]; f Levason W.; Quirk J. J.; Reid G.; Smith S. M. Synthesis, Spectroscopic and Redox Properties of Ruthenium Complexes with Selenoether Macrocycles: Crystal Structures of cis-[RuCl2([16]aneSe4)] and trans-[RuCl(PPh3)([16]aneSe4)]PF6([16]aneSe4= 1,5,9,13-tetraselenacyclohexadecane). J. Chem. Soc., Dalton Trans. 1997, 3719–3724. 10.1039/a703667a. [DOI] [Google Scholar]; g Levason W.; Orchard S. D.; Reid G. Synthesis, Characterisation and Reactions of Ruthenium(II) Complexes Based Upon [RuL3]2+(L3 = tripodal Triseleno- or Tritelluro-ether) Fragments. Structures of [RuCl2(PPh3){MeC(CH2SeMe)3}] and [RuCl2(dmso){MeC(CH2SeMe)3}]. J. Chem. Soc., Dalton Trans. 2000, 4550–4554. 10.1039/b007487j. [DOI] [Google Scholar]; h Champness N. R.; Levason W.; Preece S. R.; Webster M. Synthesis and Spectroscopic Studies of Ruthenium(II) Selenoether Complexes: Structure of trans-[Ru&{PhSe(CH2)2SePh}2Cl2]. Polyhedron 1994, 13, 881–886. 10.1016/S0277-5387(00)83004-1. [DOI] [Google Scholar]
- Ritter T.; Hejl A.; Wenzel A. G.; Funk T. W.; Grubbs R. H. A Standard System of Characterization for Olefin Metathesis Catalysts. Organometallics 2006, 25, 5740–5745. 10.1021/om060520o. [DOI] [Google Scholar]
- Diesendruck C. E.; Vidavsky Y.; Ben-Asuly A.; Lemcoff N. G. A Latent S-Chelated Ruthenium Benzylidene Initiator for Ring-Opening Metathesis Polymerization. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 4209–4213. 10.1002/pola.23476. [DOI] [Google Scholar]
- Benitez D.; Goddard W. A. The Isomerization Equilibrium between Cis and Trans Chloride Ruthenium Olefin Metathesis Catalysts from Quantum Mechanics Calculations. J. Am. Chem. Soc. 2005, 127, 12218–12219. 10.1021/ja051796a. [DOI] [PubMed] [Google Scholar]
- These high polydispersity indexes combined with the significant differences between the theoretical and experimental values of the number-average molecular mass (Mn) suggest that competitive chain-transfer reactions took place during the polymerization.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.