

Abstract
Objective:
To identify correlates of incident HIV infection in rapidly growing HIV molecular clusters.
Design:
Phylogenetic analysis of HIV public health surveillance data.
Methods:
High-priority HIV genetic transmission clusters with evidence of rapid growth in 2012 (i.e., clusters with a pairwise genetic distance ≤0.005 substitutions/site and at least 3 cases diagnosed in 2012) were identified using HIV-TRACE. Then, we investigated cluster growth, defined as HIV cases diagnosed in the following 5 years that were genetically linked to these clusters. For clusters that grew during the follow-up period, Bayesian molecular clock phylogenetic inference was performed to identify clusters with evidence of incident HIV infection (as opposed to diagnosis of previously infected cases) during this follow-up period.
Results:
Of the 116 rapidly growing clusters identified, 73 (63%) had phylogenetic evidence for an incident HIV case during the 5-year follow-up period. Correlates of an incident HIV case arising in clusters included a greater number of diagnosed but virally unsuppressed cases in 2012, a greater number of inferred undiagnosed cases in the cluster in 2012, and a younger time of most recent common ancestor for the cluster.
Conclusions:
These findings suggest that incident infections in rapidly growing clusters originate equally from diagnosed but unsuppressed cases and undiagnosed infections. These results highlight the importance of promoting retention in care and viral suppression as well as partner notification and other case-finding activities when investigating and intervening on high-priority molecular transmission clusters.
Keywords: HIV, transmission cluster, molecular epidemiology, suppression, diagnosis, phylogenetics
INTRODUCTION
Reducing the incidence of HIV is an important goal of HIV public health surveillance and prevention activities [1]. Ensuring that all people with HIV infection receive a diagnosis and that they all achieve sustained viral suppression is critical to realizing this reduction in incidence. Individuals who sustain suppression of viral replication through antiretroviral therapy do not transmit HIV through sexual activity [2–4].
Molecular transmission network analysis of HIV genetic sequence data can identify clusters of HIV cases with direct and indirect epidemiological connections [5–9]. Those clusters that have experienced the most recent growth (i.e., addition of cases with genetically highly similar viruses) are the most likely to give rise to newly diagnosed, genetically linked HIV cases in the near future [10, 11]. Further, these rapidly growing clusters have been documented to have a mean transmission rate 8-times the national average in the United States [12]. These clusters represent attractive targets for public health intervention, because they comprise individuals more likely to be associated with future HIV cases.
Growth in molecular transmission clusters can be due to both the diagnosis of previously undiagnosed infections (i.e., HIV transmitted prior to cluster identification, but diagnosed afterwards) and incident infection (i.e., HIV transmitted after cluster identification) [11, 13]. These incident infections can arise from transmission from (i) diagnosed, but virally unsuppressed cases in the transmission cluster and (ii) cases that would have been linked to the cluster but were undiagnosed at the time of cluster identification.
We conducted a retrospective analysis of rapidly growing HIV transmission clusters using data from United States National HIV Surveillance System (NHSS). We identified clusters that experienced rapid growth in 2012 and determined the correlates of subsequent incident infection in the following 5 years [12, 14].
METHODS
Data source.
We analyzed data reported to NHSS for HIV-1 cases with an associated HIV partial polymerase (pol) sequence for diagnoses occurring in 2010 through 2017 using data reported to CDC through December 31st, 2017. Our analysis was restricted to the six states with genotype reporting completeness above 50% across these years: Connecticut, Michigan, New York, South Carolina, Texas, and Washington. Perinatal cases and people under the age of 13 with an HIV diagnosis were not included.
Baseline network analysis.
We analyzed the first reported pol genotype (protease and reverse transcriptase, sites 2253–3749 in the HXB2 reference sequence) at least 500 nucleotides in length for each case. A genetic transmission network was constructed using HIV-TRACE [15] from 19,511 cases diagnosed between 2010 through 2012. To identify rapidly growing clusters, we applied a conservative genetic distance threshold of 0.005 substitutions/site, which is more likely to be associated with recent transmission partners [8, 16]. We identified molecular transmission clusters that would have been considered priority for public health action as of December 31st, 2012. High-priority clusters were defined according to criteria for recent and rapid growth outlined in Oster et al. [12]: genetic partners linked using a 0.005 substitutions/site threshold and at least 3 cases diagnosed in the most recent year (i.e., 2012). Genetic distance was calculated using an ambiguity fraction threshold of 0.015 in HIV-TRACE. For each individual in these priority clusters, we analyzed reported viral load data to determine whether they were virally unsuppressed at the end of 2012 (i.e., viral load ≥200 copies/ml or no reported viral load result in the previous 12 months).
Follow-up molecular transmission network analysis.
We then characterized the growth of these priority clusters over a 5-year period between January 1st, 2013 through December 31st, 2017. We used HIV-TRACE to identify cases newly diagnosed during this follow-up period that were genetically linked to the original priority clusters at 0.005 substitutions/site.
Phylogenetic analysis.
To distinguish cluster growth due to incident HIV infection from delayed diagnosis of prevalent HIV cases, we inferred tip-dated phylogenies for each priority cluster using BEAST v1.8.4 [17]. A TN93 substitution model [18] was applied and a Bayesian Skyline Plot was used as a coalescent prior [19]. As in previous investigations of clusters identified in NHSS [12, 20], we applied a strict molecular clock with a normal prior on the substitution rate with mean of 1.2201 × 10−3 substitutions/site/year and a narrow standard deviation of 1 × 10−6.
Markov chain Monte Carlo (MCMC) runs were performed in duplicate and had a length of 10 million generations, sampling every 1000 generations; the first 10% of generations were removed as burn-in. Convergence and mixing were assessed in Tracer [21] such that all relevant parameters had an estimated sample size >200. Maximum clade credibility (MCC) trees were constructed in TreeAnnotator.
For each cluster, we performed two rounds of MCMC analyses. The first MCMC round was run for the clusters at the end of 2012 to determine their time to most recent common ancestor (TMRCA) at the time of prioritization. We included cases diagnosed between 2010 through 2012, even when the date of genotype sampling for this case post-dated 2012, due to delayed genotyping.
The second round of MCMC analyses was performed on these same clusters at the end of 2017, to identify instances of incident cluster growth, and included subsequently diagnosed linked cases. The number of subsequent incident cases were inferred from the median number of post-2012 internal nodes in posterior distribution of phylogenies (Figure 1). Each post-2012 internal node is evidence of a viral lineage diversification after initial prioritization, which implies a transmission event. When counting the number of post-2012 internal nodes, we ignored internal nodes arising from the delayed genotyping of a cluster member who was diagnosed between 2010 through 2012 (see Figure S1 for an example).
Figure 1. Example inference of incident infection of growing clusters characterized by internal node post-dating the identification of the high-priority cluster.
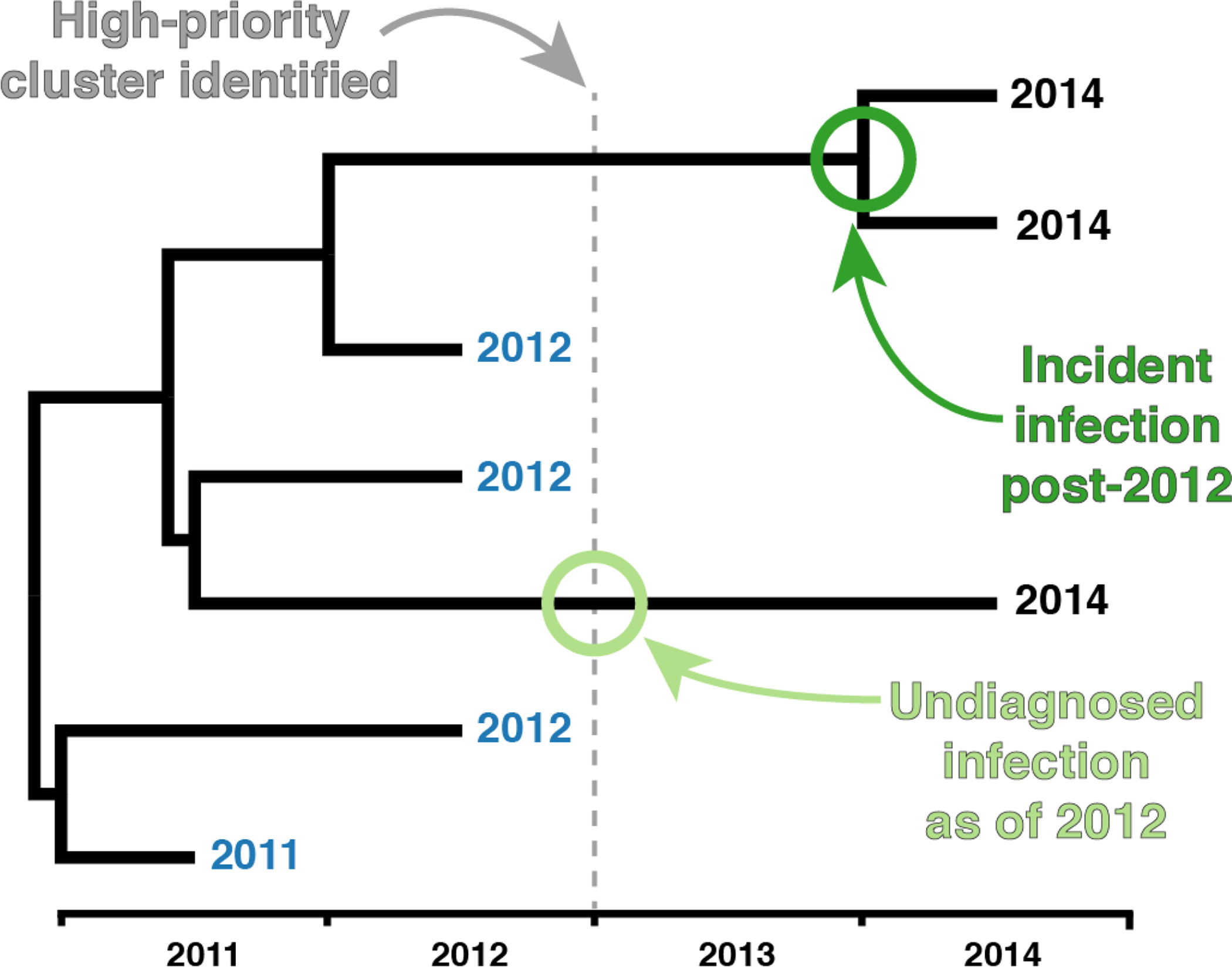
Tip labels represent date of diagnosis. Tip dates colored in blue were identified as cases in the high-priority cluster as of identification in 2012. In this example tree, we can infer an incident infection post-2012 (dark green circle); however, we cannot determine which, if either descendant is the source or recipient partner with incident HIV. Further, we can infer an undiagnosed infection as of 2012 (light green circle).
We then estimated the number of undiagnosed previously infected cases in these clusters as the total number of post-2012 cases less the number of inferred incident cases. We acknowledge that this number is likely overestimated using our approach, because at least two closely related cases diagnosed post-2012 are required to identify a transmission event using this phylogenetic approach. To partially control for this bias in the regression models (described below), we subtracted the number of post-2012 clades (i.e., dark green circles in Figure 1) from the estimated of number of newly diagnosed, previously infected cases in each cluster (i.e., light green circles in Figure 1).
Logistic regression analysis.
We performed univariate and multivariate logistic regression analyses to identify correlates of a priori interest between incident cluster growth and the characteristics of the cluster as it was observed at the end of 2012. The binary outcome for each cluster was whether or not the MCMC analysis inferred a post-2012 internal node. The cluster-level covariates as of the end of 2012 were the number of diagnosed but virally unsuppressed cases (VL ≥200 copies/ml), the number of inferred undiagnosed cases, the number of total cases in the cluster, the TMRCA, the median log10 viral load, whether the majority of the cluster was Black/African American, and whether the majority of the cluster was men who have sex with men (MSM). We selected majority Black/African American and majority MSM because these groups were the most common in the priority clusters.
We also identified correlates of growth due to the diagnosis of previously infected case in a priority cluster. We performed univariate and multivariate logistic regression analyses in which evidence for at least one inferred undiagnosed case was the binary outcome.
Data availability statement.
The data analyzed in this article were collected and analyzed as part of CDC routine surveillance activities. CDC is not permitted to share or distribute any surveillance data due to an assurance of confidentiality authorized under Section 308(d) of the Public Health Service Act (USA). Each of the six states has primary authority for determining whether their laws and regulations permit the submission to GenBank or other open databases. Therefore, these data cannot be made publicly available by the authors.
RESULTS
Characterizing priority molecular transmission clusters.
From the 19,511 HIV cases included, we inferred 116 priority clusters in 2012 comprising 759 cases. The median size for these priority clusters was 5 cases, ranging between 3 and 33 cases (Figure 2). Only 16% of these clusters comprised 10 or more cases in 2012. Regarding race/ethnicity composition, 36 clusters (31%) were majority Black/African American, 29 (25%) were majority Hispanic/Latino, and 31 (27%) were majority White. Regarding transmission risk, 100 priority clusters (86%) were majority MSM, two were majority heterosexual (2%), and three were majority people who infected drugs (PWID) (3%). Clusters had a mean TMRCA of 2.9 years (95% range between 0.4 and 7.1 years).
Figure 2. Priority clusters and growth due to incident and previously undiagnosed cases, sorted by cluster size.
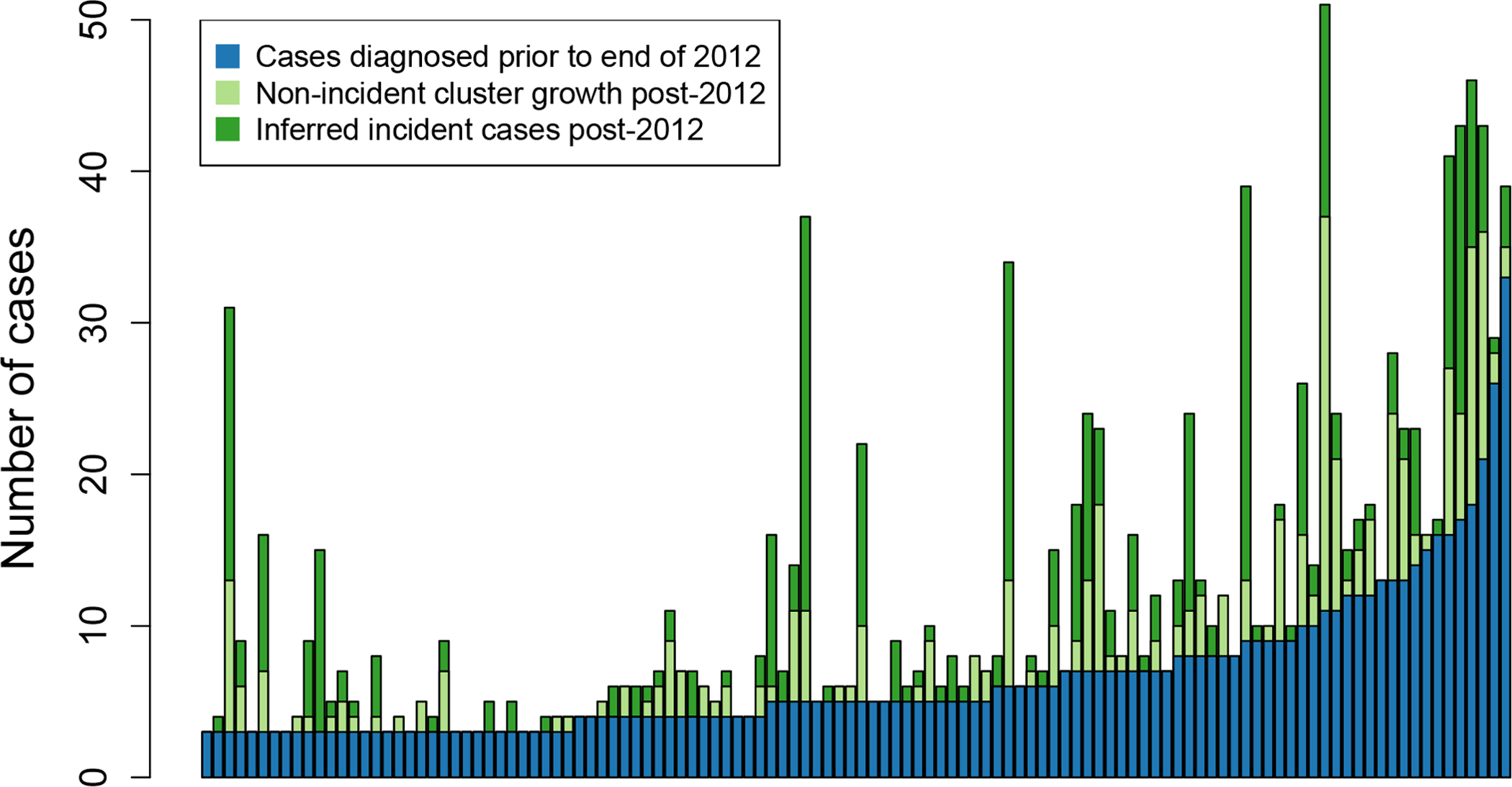
Each column represents a single high-priority cluster (in 2012 in blue) and its subsequent growth (in light and dark green).
Growth of priority clusters.
Eighty two (71%) high-priority clusters experienced growth during 2013–2017 (Figure 2). There were 641 cases diagnosed post-2012 that were genetically linked to these priority clusters. Nine clusters added more than 20 cases in the follow-up period, with one cluster growing from 3 cases in 2012 to 31 cases in 2017. We inferred evidence for growth due to incident cases in 73 (63%) clusters and growth due to undiagnosed cases in 68 (59%) clusters.
Predictors of cluster growth due to incident HIV.
The number of virally unsuppressed cases and the inferred number of undiagnosed cases in priority clusters at the end of 2012 were significantly associated with future incident growth in these clusters (p < 0.05; logistic regression; Table 1). These associations were similar in magnitude and robust to the inclusion of cluster size, cluster TMRCA, median log10 viral load, and cluster composition by race/ethnicity and transmission risk in the multivariate logistic regression model.
Table 1.
Predictors of incident cluster growth in priority clusters using univariate and multivariate logistic regression models.
| Cluster attributea | OR | 95% CI | p-value | AOR | 95% CI | p-value |
|---|---|---|---|---|---|---|
| # diagnosed, unsuppressed cases | 1.78 | 1.34–2.51 | <0.001 | 1.65 | 1.17–2.47 | 0.028 |
| # undiagnosed cases | 1.71 | 1.29–2.48 | 0.001 | 1.62 | 0.90–1.51 | 0.011 |
| Cluster size | 1.24 | 1.09–1.46 | 0.005 | 1.14 | 0.55–0.96 | 0.304 |
| TMRCA | 0.99 | 0.84–1.17 | 0.930 | 0.75 | 0.84–1.42 | 0.041 |
| Median log10 viral load | 1.23 | 0.97–1.57 | 0.093 | 1.09 | 0.37–3.15 | 0.535 |
| Majority Black/African American | 1.27 | 0.56–2.96 | 0.577 | 1.07 | 0.04–0.91 | 0.897 |
| Majority MSM | 0.35 | 0.08–1.16 | 0.115 | 0.23 | 1.17–2.47 | 0.049 |
Attributes of the cluster comprising cases diagnosed between 2010 through 2012
OR, odds ratio; AOR, adjusted odds ratio; CI, confidence interval; TMRCA
The size of the priority cluster in 2012 was associated with incident growth in the univariate model (Table 1); however, this association is not significant in the multivariate model. On its own, the cluster TMRCA, representing the upper bound on the timing of the first transmission event within the cluster, is not associated with future incident growth (p=0.930). However, upon adjusting for cluster size, clusters with older TMRCAs were associated with significantly fewer future incident cases (p=0.041). The only other cluster attribute included in our model significantly associated with incident growth in priority clusters was transmission risk, whereby clusters comprised of majority MSM were less likely to have incident growth.
Predictors of cluster growth due to subsequent diagnosis of previously infected case.
When we examined correlates of subsequent diagnosis of a genetically linked, previously infected HIV case, the number of unsuppressed cases in priority clusters at the end of 2012 was significantly associated in the univariate regression model (p=0.004) but not in the multivariate model (p=0.064) (Table 2). The same pattern was seen to a greater extent with the 2012 cluster size in the univariate and multivariate models.
Table 2.
Predictors of cluster growth due to the subsequent diagnosis of a previously infected case in priority clusters using univariate and multivariate logistic regression models.
| Cluster attributea | OR | 95% CI | p-value | AOR | 95% CI | p-value |
|---|---|---|---|---|---|---|
| # diagnosed, unsuppressed cases | 1.36 | 1.12–1.71 | 0.004 | 1.35 | 1.00–1.89 | 0.064 |
| Cluster size | 1.13 | 1.03–1.27 | 0.026 | 1.02 | 0.86–1.22 | 0.839 |
| TMRCA | 1.11 | 0.94–1.33 | 0.214 | 0.99 | 0.80–1.22 | 0.919 |
| Median log10 viral load | 1.02 | 0.80–1.31 | 0.864 | 0.93 | 0.71–1.22 | 0.591 |
| Majority Black/African American | 1.19 | 0.51–2.79 | 0.683 | 1.00 | 0.39–2.53 | 0.996 |
| Majority MSM | 1.09 | 0.36–2.36 | 0.875 | 0.96 | 0.29–3.26 | 0.949 |
Attributes of the cluster comprising cases diagnosed between 2010 through 2012
OR, odds ratio; AOR, adjusted odds ratio; CI, confidence interval; TMRCA, time of most recent common ancestor; MSM, men who have sex with men
DISCUSSION
We performed a retrospective study to characterize the correlates of incident HIV infection in HIV molecular transmission clusters that had recently exhibited rapid cluster growth. We characterized these high-priority clusters at the end of 2012 and used a phylodynamic approach to characterize incident HIV infection. We report that the number of previously diagnosed, virally unsuppressed cases in these clusters at the end of 2012 were predictive of subsequent incident infection. The inferred number of previously infected HIV cases undiagnosed as of 2012 in these clusters was equally associated with subsequent incident HIV infection in these clusters. Further, recently established clusters, with younger TMRCAs, were more likely to give rise to incident HIV infection. These findings highlight the importance of promoting both retention in care and viral suppression among people with diagnosed HIV infection as well as partner notification and other case-finding activities when investigating and intervening on high-priority molecular transmission clusters [22].
Translating the identification of high-priority clusters into a reduction in HIV incidence is a major public health goal. Standard public health intervention can be marshalled in real-time in response to clusters identified through molecular surveillance [6]. Individuals in these clusters who have unsuppressed viral load or are out-of-care can be prioritized for adherence support, relinkage to care, or other appropriate public health services. In addition, identifying HIV-infected people who are unaware of their HIV status or HIV-uninfected people at risk of HIV acquisition can be facilitated by case-finding activities. Case-finding via eliciting named sexual and injection drug-using partners tends to have high yields in outbreak settings [23, 24]. Therefore, prioritizing partner elicitation services for people in rapidly growing clusters may be an effective use of public health resources. Further, exploration of the social contact networks connected to these clusters represents an attractive method of identifying people living with HIV without a diagnosis, people with an HIV diagnosis who are out-of-care, or HIV-uninfected people at higher risk of HIV acquisition[25]. In addition, clusters with shared venues or geography could identify priorities for increased HIV testing. Although it is still untested which of these approaches will yield the greatest decrease in future HIV incidence, the results presented here suggest that rapidly growing clusters can help identify individuals to whom these services can be prioritized.
Individual-level HIV incidence testing data is not available in NHSS; therefore, we used a phylodynamic approach to identify clusters with incident infections. However, our phylodynamic approach precludes us from identifying any single individual in these high-priority clusters as an incident or prevalent HIV case. Rather, we identified clusters with phylogenetic evidence of incident infection based on the presence of internal node ages representing an HIV transmission event. For an internal node with two descendants (dark green circle in Figure 1), we cannot distinguish the source partner from the recipient partner with incident HIV. Further, both descendants could be incident cases, with an original cluster member or person whose infection has remained undiagnosed or does not have a sequence available serving as the source partner. In addition, our approach does not consider cluster members with older diagnosis (i.e., before 2010), who could be contributing to ongoing transmission. Also, there are likely incident cases remaining undiagnosed. Therefore, our approach necessarily undercounts the number of incident cases in these clusters. It is for this reason that we used a bivariate outcome, rather than a continuous outcome, in our regression analyses.
The findings reported here are limited to high-priority clusters identified in high-completeness jurisdictions in the United States. Further, growth in these clusters could only be determined for incident and prevalent cases with a reported viral genotype. Notably, traditional correlates of cluster growth such as race/ethnicity and transmission risk (i.e., MSM) [10, 11] were no longer associated with incident infection in our analysis. This counter-intuitive finding likely stems from the observation that of the 17 non-majority MSM priority clusters, all but the two of these clusters experienced incident growth. This apparent discrepancy between previous studies of HIV transmission clusters and the results reported here are likely due to our focus on high-priority clusters. By focusing our analysis on individuals in clusters recently experiencing rapid growth, we restrict our findings to individuals within each of these race/ethnicity and transmission risk sub-populations who were already associated with rapid cluster growth.
The stated goal of the planned Ending the HIV Epidemic initiative is to reduce the incidence of HIV in the United States by 90% by 2030 [1]. This initiative anticipates using HIV molecular epidemiology to detect HIV clusters, allowing public health response to deliver prevention and care services, including testing, HIV care, and pre-exposure prophylaxis (PrEP), to reach people that need them. Our results support this framework. This combination of approaches may help us achieve the Ending the HIV Epidemic goals.
Supplementary Material
Funding.
JOW was funded in part by the CDC, an NIH-NIAID K01 Career Development Award (K01AI110181), and an NIH-NIAID R01 (AI135992). JOW was also funded by a research grant to his institution by Gilead Sciences; this grant was unrelated to the work presented here.
Footnotes
Publisher's Disclaimer: Disclaimer. The findings and conclusions of this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
REFERENCES
- 1.Frieden TR, Foti KE, Mermin J. Applying Public Health Principles to the HIV Epidemic--How Are We Doing? N Engl J Med 2015; 373(23):2281–2287. [DOI] [PubMed] [Google Scholar]
- 2.Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, Kumarasamy N, et al. Prevention of HIV-1 infection with early antiretroviral therapy. N Engl J Med 2011; 365(6):493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fideli US, Allen SA, Musonda R, Trask S, Hahn BH, Weiss H, et al. Virologic and immunologic determinants of heterosexual transmission of human immunodeficiency virus type 1 in Africa. AIDS Res Hum Retroviruses 2001; 17(10):901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quinn TC, Wawer MJ, Sewankambo N, Serwadda D, Li C, Wabwire-Mangen F, et al. Viral load and heterosexual transmission of human immunodeficiency virus type 1. Rakai Project Study Group. N Engl J Med 2000; 342(13):921–929. [DOI] [PubMed] [Google Scholar]
- 5.Oster AM, France AM, Mermin J. Molecular Epidemiology and the Transformation of HIV Prevention. JAMA 2018. [DOI] [PubMed] [Google Scholar]
- 6.Poon AF, Joy JB, Woods CK, Shurgold S, Colley G, Brumme CJ, et al. The impact of clinical, demographic and risk factors on rates of HIV transmission: a population-based phylogenetic analysis in British Columbia, Canada. J Infect Dis 2015; 211(6):926–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith DM, May SJ, Tweeten S, Drumright L, Pacold ME, Kosakovsky Pond SL, et al. A public health model for the molecular surveillance of HIV transmission in San Diego, California. AIDS 2009; 23(2):225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wertheim JO, Kosakovsky Pond SL, Forgione LA, Mehta SR, Murrell B, Shah S, et al. Social and Genetic Networks of HIV-1 Transmission in New York City. PLoS Pathog 2017; 13(1):e1006000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewis F, Hughes GJ, Rambaut A, Pozniak A, Leigh Brown AJ. Episodic sexual transmission of HIV revealed by molecular phylodynamics. PLoS medicine 2008; 5(3):e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dennis AM, Hue S, Billock R, Levintow S, Sebastian J, Miller WC, et al. HIV-1 Phylodynamics to Detect and Characterize Active Transmission Clusters in North Carolina. J Infect Dis 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wertheim JO, Murrell B, Mehta SR, Forgione LA, Kosakovsky Pond SL, Smith DM, et al. Growth of HIV-1 Molecular Transmission Clusters in New York City. J Infect Dis 2018; 218(12):1943–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oster AM, France AM, Panneer N, Banez Ocfemia MC, Campbell E, Dasgupta S, et al. Identifying Clusters of Recent and Rapid HIV Transmission Through Analysis of Molecular Surveillance Data. J Acquir Immune Defic Syndr 2018; 79(5):543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Little SJ, Kosakovsky Pond SL, Anderson CM, Young JA, Wertheim JO, Mehta SR, et al. Using HIV networks to inform real time prevention interventions. PLoS One 2014; 9(6):e98443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Centers for Disease Control and Prevention Division of HIV/AIDS Prevention. Detecting, investigating, and responding to HIV transmission clusters, Version 1.0. In; 2017.
- 15.Kosakovsky Pond SL, Weaver S, Leigh Brown AJ, Wertheim JO. HIV-TRACE (Transmission Cluster Engine): a tool for large scale molecular epidemiology of HIV-1 and other rapidly evolving pathogens. Mol Biol Evol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le Vu S, Ratmann O, Delpech V, Brown AE, Gill ON, Tostevin A, et al. Comparison of cluster-based and source-attribution methods for estimating transmission risk using large HIV sequence databases. Epidemics 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 2012; 29(8):1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 1993; 10(3):512–526. [DOI] [PubMed] [Google Scholar]
- 19.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol 2005; 22(5):1185–1192. [DOI] [PubMed] [Google Scholar]
- 20.Wertheim JO, Oster AM, Johnson JA, Switzer WM, Saduvala N, Hernandez AL, et al. Transmission fitness of drug-resistant HIV revealed in a surveillance system transmission network. Virus Evolution 2017:In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst Biol 2018; 67(5):901–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wertheim JO, Chato C, Poon AFY. Comparative analysis of HIV sequences in real time for public health. Curr Opin HIV AIDS 2019; 14(3):213–220. [DOI] [PubMed] [Google Scholar]
- 23.Cranston K, Alpren C, John B, Dawson E, Roosevelt K, Burrage A, et al. Notes from the Field: HIV Diagnoses Among Persons Who Inject Drugs - Northeastern Massachusetts, 2015–2018. MMWR Morb Mortal Wkly Rep 2019; 68(10):253–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peters PJ, Pontones P, Hoover KW, Patel MR, Galang RR, Shields J, et al. HIV Infection Linked to Injection Use of Oxymorphone in Indiana, 2014–2015. N Engl J Med 2016; 375(3):229–239. [DOI] [PubMed] [Google Scholar]
- 25.Wohl AR, Ludwig-Barron N, Dierst-Davies R, Kulkarni S, Bendetson J, Jordan W, et al. Project Engage: Snowball Sampling and Direct Recruitment to Identify and Link Hard-to-Reach HIV-Infected Persons Who Are Out of Care. J Acquir Immune Defic Syndr 2017; 75(2):190–197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data analyzed in this article were collected and analyzed as part of CDC routine surveillance activities. CDC is not permitted to share or distribute any surveillance data due to an assurance of confidentiality authorized under Section 308(d) of the Public Health Service Act (USA). Each of the six states has primary authority for determining whether their laws and regulations permit the submission to GenBank or other open databases. Therefore, these data cannot be made publicly available by the authors.