

Abstract
The biomarker p16 plays a role in aging and is upregulated in aged organs and cells, including bone marrow mesenchymal stem cells (BM-MSCs), which play a leading role in fracture healing. Several studies have reported delayed fracture healing in geriatric mice. However, the relationship between p16 expression and fracture healing in geriatric mice remains poorly understood. In this study, we found that fracture healing was accelerated in p16 deletion (p16-/-) mice, and the number of migrated BM-MSCs from p16-/- mice increased. The expressions of SDF-1 and CXCR4 were also upregulated in p16-/- mice. Increased cell percentage at S phase in cell cycle, enhanced expressions of CDK4/6, pRB, and E2F1, decreased expression of RB, and elevated expressions of SOX9, PCNA, and COL2A1 were detected in p16-/- mice. The expressions of COL10A1, MMP13, OSTERIX, and COL1A1 were also high in p16-/- mice. Moreover, the expressions of p-AKT, p-mTOR, HIF-1α, and VEGF-A in BM-MSCs and expression of VEGF-A in callus were upregulated in p16-/- mice. The expression of VEGF in the serum of p16-/- mice was also higher than that of wild type mice. Thus, deletion of p16 enhances migration, division, and differentiation of BM-MSCs, promotes proliferation and maturation of chondrocytes, activates osteoblastogenesis, and facilitates vascularization to accelerate fracture healing, providing a novel strategy to treat fracture in the elderly.
Keywords: p16, geriatric mice, fracture healing, endochondral ossification, vascularization
Introduction
Fracture healing is considered to be a specialized postnatal regenerative process that recapitulates embryonic skeletal development [1]. Numerous physiologic and environmental factors negatively influence bone regeneration after fracture [2], of which aging is an important one [3-6]. In a prospective observational research involving 1133 patients who suffered from femoral neck fractures, the incidence of nonunion was elevated from 5.9% in patients younger than 40 years to 24.9% in patients older than 70 years [7]. In a study to detect changes that occur during fracture healing, delayed chondrocyte proliferation and maturation, vascular invasion, and less bone formation at the tibial fracture site were observed in elderly mice [8]. With increasing age, age-related changes, including a decrease in the amount and division potential of mesenchymal stem cells (MSCs), lower responsiveness of MSCs to signal molecules, reduced blood vessel formation, and changes in signaling molecules levels, are responsible for delayed healing in the elderly [9]. It seems that enhancing cell recruitment, proliferation and differentiation, promoting osteoblastogenesis, and facilitating vascular invasion may be key targets for accelerating fracture healing in the elderly [6].
A biomarker of aging, p16, is upregulated in aged organs and tissues, including the bone marrow [10], and acts as a cyclin-dependent kinase (CDK) inhibitor through repressing cell cycle progress to prohibit cell proliferation and trigger aging in mammals [11]. Che et al. found that p16 deficiency enhances nucleus pulposus (NP) cell proliferation [12]. Moreover, p16 expression suppresses angiogenesis in breast cancer cells [13]. When p16 was deleted, pro-angiogenic factors were overexpressed to promote vascularization in pancreatic carcinoma [14]. Few studies have focused on the relationship between p16 and fracture healing in the elderly, and the effects and mechanisms of p16 on fracture healing remain poorly understood.
In this study, we deleted the p16 gene in geriatric mice and found that bone healing in p16-deficient mice was accelerated after femoral shaft fracture. After p16 deletion, the migration, division, and differentiation of bone marrow MSCs (BM-MSCs) were enhanced, chondrocyte proliferation and maturation were strengthened, osteoblastogenesis was activated, and blood vessel invasion was facilitated, indicating that inactivation of p16 might be a novel strategy for treating fractures in the elderly.
Material and methods
Mouse BM-MSCs isolation and culture
p16 deficient (p16-/-) mice and their wild-type (WT) littermates were prepared as previously described by Che et al. [12]. Animal use was approved by the Institutional Animal Care and Use Committee of Nanjing Medical University (approval number: IACUC-1904037). Mice were housed in the Experimental Animal Center in compliance with guidelines of the Institute for Laboratory Animal Research of Nanjing Medical University. Mouse BM-MSCs were isolated as previously described [15] and cultured in α-MEM (modified Eagle’s medium alpha) supplemented with 10% fetal bovine serum, penicillin (100 units/ml) and streptomycin (100 mg/ml) at 37°C and 5% CO2. BM-MSCs were characterized and confirmed using a FACS system.
Transwell assay
To determine the effects of p16 on cell migration, a transwell assay was performed.
In brief, 2 × 105 BM-MSCs derived from WT or p16-/- mice were added to the upper wells separated by a 0.8 μm pore size polyethylene terephthalate membrane. After 48 h, the non-migrated cells were carefully removed from the top of the membrane. The membranes were then fixed with 10% methanol and stained with crystal violet staining solution (Boyetime, Shanghai, China). After air-drying, the membranes were detected under a light microscope (Olympus, Japan) in five randomly selected fields (original magnification, × 100).
Immunofluorescence (IF)
Cultured BM-MSCs were induced to differentiate into chondrocytes for 14 days and confirmed by toluidine blue staining. IF staining of proliferating cell nuclear antigen (PCNA) and COL2A1 in chondrocytes was carried out as previously described [12].
Cell counting kit-8 (CCK-8) cell viability assay
The CCK-8 assay (KeyGen, Nanjing, China) was employed to evaluate cell proliferation. Cells were cultured in 96-well plates (5000 cells/well) and allowed to grow for 24, 48, and 72 h. 10 ml CCK reagent was added to each well and incubated for 3-4 h. The absorbance at 450 nm was measured with a spectrophotometer reader (Thermo Electron, Massachusetts, USA).
Alkaline phosphatase (ALP) staining
Cultured BM-MSCs were induced to differentiate into osteoblasts for 14 days and rinsed three times using PBS. Cells were fixed in 100% ethanol for 15 min and stained with ALP staining solution for 15 min.
Alizarin red staining
Cultured BM-MSCs were induced to differentiate into osteoblasts for 21 days and rinsed three times using PBS. Cells were fixed in 95% ethanol for 10 minutes and stained with alizarin red staining solution for 30 min.
Flow cytometry analyses
Chondrocytes were rinsed three times with PBS and fixed in 70% ethanol. The specimen was then examined by flow cytometry using a FACSCalibur flow cytometer.
Human umbilical vein endothelial cells (HUVECs) tube formation assay
Each well in 96-well plates was coated with 50 μl ice-cold Matrigel matrix (BD Biosciences, USA), and the matrix was solidified at 37°C for 30 min. HUVECs were suspended at a density of 2 × 105/ml with medium supernatant harvested from BM-MSCs derived from p16-/- and WT mice. HUVECs (1 × 104) were seeded onto the matrix and incubated at 37°C and 5% CO2. After 4 h of incubation, tubule meshes in five randomly selected fields (original magnification, × 200) were photographed, and the number of meshes was counted.
Animal handling
Femoral shaft fracture and intramedullary fixation were performed in eighty 12-month-old p16-/- and WT mice. The mice were carefully handled and kept warm during and after surgery. None of the surgeries failed, and no complications were detected during the experiments. On postoperative days 1, 7, 10, 14, 21, and 28, the mice were humanely sacrificed. Blood, clot at the fracture site, or operated femurs were collected for subsequent examinations.
Specimen processing
Some of the harvested femurs were fixed in periodate-lysine-paraformaldehyde fixation solution at pH 7.2 to 7.4 for 24 h and then immersed in PBS in a refrigerator at 4°C for radiologic and histologic examinations. The remaining femurs were used for RNA extraction.
Radiologic examinations
The fixed femurs were analyzed by X-ray and micro-computed tomography (micro-CT) scans. The region of interest was the callus. The microstructural parameters, including bone mineral density (BMD) and bone volume to total volume ratio (BV/TV) were calculated.
Histologic staining
After radiological examinations, the femurs were embedded in paraffin, and serially sectioned at 5 μm intervals. Hematoxylin and eosin (H&E) staining and toluidine blue staining were performed according to standard protocols.
Immunohistochemistry (IHC)
Immunohistochemical staining of SOX9, COL1A1, COL2A1, COL10A1, PCNA, matrix metalloproteinase 13 (MMP13), OSTERIX and vascular endothelial growth factor-A (VEGFA) were performed as previously described [16]. The following primary antibodies were used: Anti-SOX9 (ab185966, abcam, UK), Collagen Type I (14695-1-AP, Proteintech, USA), Collagen Type II (28459-1-AP, Proteintech, USA), Anti-Collagen X (ab58632, Abcam, UK), PCNA (10205-2-AP, Proteintech, USA), Anti-MMP13 (ab39012, Abcam, UK), Anti-Sp7/Osterix (ab22552, Abcam, UK) and VEGFA (66828-1-Ig, Proteintech, USA).
RNA extraction and real time quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was collected from BM-MSCs, clots, chondrocytes, or calluses using TRIzol reagent (Beyotime, Shanghai, China), and RNA concentration was measured with a Nanodrop spectrophotometer (Thermo Scientific, USA). Subsequently, RNA was reverse transcribed to cDNA using HiScript III Reverse Transcriptase (Vazyme, Nanjing, China). Using the ABI 7500 Fast Real-Time PCR System (Applied Biosystems), qRT-PCR was performed. Table 1 tabulates the qRT-PCR primer sequences. The expression levels of target genes were normalized to GAPDH using the comparative 2-ΔΔCt method.
Table 1.
PCR primer sequences
| Name | Sequence (5’-3’) |
|---|---|
| SDF-1-F | TGCATCAGTGACGGTAAACCA |
| SDF-1-R | TTCTTCAGCCGTGCAACAATC |
| CXCR4-F | GAAGTGGGGTCTGGAGACTAT |
| CXCR4-R | TTGCCGACTATGCCAGTCAAG |
| CDK4-F | ATGGCTGCCACTCGATATGAA |
| CDK4-R | TCCTCCATTAGGAACTCTCACAC |
| CDK6-F | GGCGTACCCACAGAAACCATA |
| CDK6-R | AGGTAAGGGCCATCTGAAAACT |
| RB-F | TGCATCTTTATCGCAGCAGTT |
| RB-R | GTTCACACGTCCGTTCTAATTTG |
| E2F1-F | CAGAACCTATGGCTAGGGAGT |
| E2F1-R | GATCCAGCCTCCGTTTCACC |
| COL1A1-F | GCTCCTCTTAGGGGCCACT |
| COL1A1-R | CCACGTCTCACCATTGGGG |
| COL2A1-F | GGGAATGTCCTCTGCGATGAC |
| COL2A1-R | GAAGGGGATCTCGGGGTTG |
| COL10A1-F | TTCTGCTGCTAATGTTCTTGACC |
| COL10A1-R | GGGATGAAGTATTGTGTCTTGGG |
| MMP13-F | CTTCTTCTTGTTGAGCTGGACTC |
| MMP13-R | CTGTGGAGGTCACTGTAGACT |
| OSTERIX-F | ATGGCGTCCTCTCTGCTTG |
| OSTERIX-R | TGAAAGGTCAGCGTATGGCTT |
| SOX9-F | GAGCCGGATCTGAAGAGGGA |
| SOX9-R | GCTTGACGTGTGGCTTGTTC |
| PCNA-F | TTTGAGGCACGCCTGATCC |
| PCNA-R | GGAGACGTGAGACGAGTCCAT |
| VEGFA-F | CTGCCGTCCGATTGAGACC |
| VEGFA-R | CCCCTCCTTGTACCACTGTC |
| GADPH-F | ATGATTCTACCCACGGCAAG |
| GADPH-R | CTGGAAGATGGTGATGGGTT |
Protein extraction and western blot analysis
Proteins were extracted from BM-MCSs, chondrocytes, or clots at the fracture site and primary antibodies against CXCL12/SDF-1 (10205-2-AP, Proteintech, USA), CXCR4 (11073-2-AP, Proteintech), CDK4 (11026-1-AP, Proteintech, USA), CDK6 (14052-1-AP, Proteintech, USA), Rb (17218-1-AP, Proteintech, USA), Anti-Rb (phospho S807) (ab184796, abcam, UK), E2F1 (66515-1-Ig, Proteintech, USA), Collagen Type I (14695-1-AP, Proteintech, USA), Anti-Sp7/Osterix (ab22552, Abcam, UK), Anti-pan-AKT (ab8805, Abcam, UK), Anti-AKT (phospho T308) (ab38449, Abcam, UK), Anti-mTOR (ab32028, Abcam, UK), Anti-mTOR (phospho S2448) (ab109268, Abcam, UK), Anti-HIF-1 alpha (ab16066, Abcam, USA), VEGFA (66828-1-Ig, Proteintech, USA), and HRP-Conjugated Beta Actin (HRP-60008, Proteintech, USA) were employed. Immunoreactive bands were visualized with ECL (Beyotime, Shanghai, China) and quantified by Scion Image Beta 4.02.
Enzyme-linked immunosorbent assay (ELISA)
Serum samples were obtained from blood collected from the eyeballs of mice on postoperative days 7, 10, 14, 21, and 28 in each group. Serum VEGF levels were determined using an ELISA kit (KeyGen, Nanjing, China).
Statistical analyses
Numerical data were presented as mean ± standard deviation (SD). Student’s t-test was performed to evaluate differences between groups. The SPSS software (Version 25.0) and GraphPad Prism (Version 8.0) were used for statistical analyses, and differences in results were considered significant at P<0.05.
Results
Deletion of p16 accelerated fracture healing in geriatric mice
To explore the effect of p16 deletion on fracture healing, we performed femoral shaft fracture in 12-month-old WT and p16-/- mice. Radiologic and histologic examinations were performed to assess the fracture healing. The results showed that the percentage of soft callus area peaked on postoperative day 7 and declined gradually, while the percentage of bony callus increased from postoperative day 7 in p16-/- mice. In WT mice, the percentage of soft callus area peaked on postoperative day 10, and the percentage of bony callus increased from postoperative day 10 (Figure 1A-C). On postoperative days 21 and 28, a higher BMD was observed in the callus of p16-/- mice. BV/TV in callus in p16-/- mice was higher and lower than that in WT mice on postoperative days 21 and 28, respectively (Figure 1D-F). These results indicated that p16 deletion accelerated fracture healing in geriatric mice.
Figure 1.

Deletion of p16 accelerated fracture healing in geriatric mice. Representative micrographs of (A) H&E staining and (B) toluidine blue staining and (C) percentages of soft and bony callus at different postoperative timepoints. (D) Representative X-ray radiographs and (E) coronal reconstruction images of micro-CT scans of callus. (F) Quantitative analysis of callus microstructural parameters, including bone mineral density (BMD) and bone volume to total volume ratio (BV/TV). n=4, *P<0.05, **P<0.01, ***P<0.001, compared with WT mice.
Deletion of p16 upregulated SDF-1 and CXCR4 expression levels and led to BM-MSCs migration
BM-MSCs play a vital role in bone regeneration. BM-MSCs migrate to injured sites upon stimulation with relevant chemokines, such as stromal cell-derived factor-1 (SDF-1) and its receptor CXCR4 [17]. BM-MSC migration was evaluated by migration assay, and SDF-1 and CXCR4 expression levels were assessed by western blot analysis and qRT-PCR. The results showed that the number of migrated BM-MSCs from p16-/- mice was higher than that from WT mice (Figure 2A). CXCR4 expression was upregulated in p16-/- mice-derived BM-MSCs (Figure 2B, 2D, 2E). SDF-1 expression in clots at the injury site in p16-/- mice was also significantly higher than that in WT mice on postoperative day 1 (Figure 2C, 2F, 2G). These results showed that p16 deletion upregulated SDF-1 and CXCR4 expression levels and led to BM-MSC migration.
Figure 2.
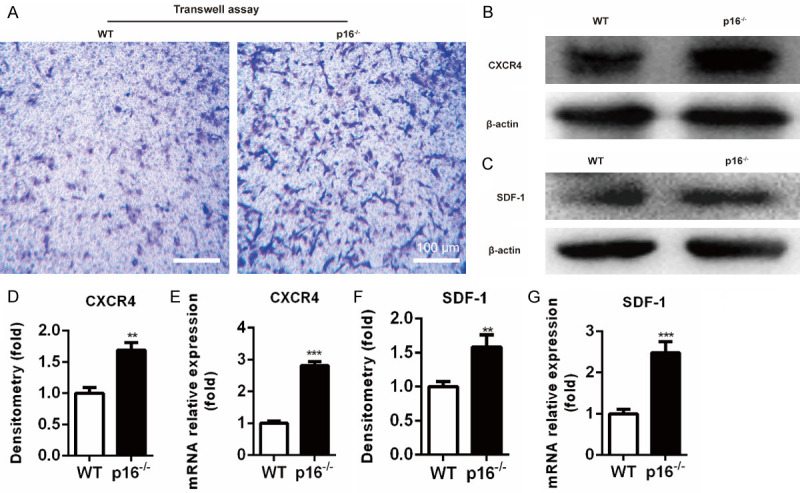
Deletion of p16 promoted BM-MSCs migration. A. Transwell assay was performed to assess the migration of BM-MSCs. B, C. Western blot analysis was performed to determine the protein expression levels of SDF-1 in clots at the injury site on postoperative day 1 and CXCR4 in BM-MSCs. β-actin was used as loading control. D, F. The quantified protein levels of SDF-1 and CXCR4 were evaluated by densitometric analysis. E, G. The mRNA levels of SDF-1 and CXCR4 were measured by qRT-PCR, calculated as ratio relative to GAPDH mRNA, and expressed relative to WT. n=4, **P<0.01, ***P<0.001, compared with WT mice.
Deletion of p16 encouraged BM-MSCs to proliferate and differentiate into chondrocytes
Migrated BM-MSCs proliferate and then differentiate into chondrocytes, and SOX9 is required for chondrogenesis [18]. Flow cytometry and western blot analysis were employed to explore the effect of p16 on BM-MSC proliferation. SOX9 expression in callus was investigated with IHC and qRT-PCR on postoperative days 7, 10 and 14. Results showed that the ratios of cells in the G0/G1 and S phases in p16-/- mice were lower and higher, respectively, than those in WT mice (Figure 3A). The expression levels of CDK4, CDK6, pRB, and E2F1 were upregulated while RB expression level was downregulated in BM-MSCs from p16-/- mice (Figure 3B-D). The SOX9 expression in callus was increased on postoperative day 7 while decreased on postoperative days 10 and 14 in p16-/- mice (Figure 3E-G). These results demonstrated that deletion of p16 could encourage BM-MSCs to proliferate via activating the CDK4/6-pRB-E2F pathway and to differentiate into chondrocytes.
Figure 3.

Deletion of p16 encouraged BM-MSCs to proliferate and differentiate into chondrocytes. A. Cell-cycle distribution of BM-MSCs was determined by flow cytometry. B. Western blot analysis was performed to assess the protein expression levels of members of the CDK4/6-pRB-E2F pathway, including CDK4, CDK6, RB, pRB, and E2F1 in BM-MSCs. β-actin was used as loading control. C. The quantified protein levels of CDK4, CDK6, RB, pRB, and E2F1 in BM-MSCs were evaluated by densitometric analysis. D. The mRNA levels of CDK4, CDK6, RB, pRB and E2F1 in BM-MSCs were measured by qRT-PCR, calculated as ratio relative to GAPDH mRNA and expressed relative to WT. E. Representative immunohistochemical micrographs of SOX9, a key regulator of chondrocyte differentiation. F. The percentages of SOX9-positive cells in callus on postoperative days 7, 10, and 14. G. The mRNA levels of SOX9 in callus on postoperative days 7, 10, and 14 were measured by qRT-PCR, calculated as ratio relative to GAPDH mRNA and expressed relative to WT. n=4, *P<0.05, **P<0.01, ***P<0.001, compared with WT mice.
Deletion of p16 promoted chondrocyte proliferation
The differentiated chondrocytes then proliferate to form cartilage callus to connect the fracture ends. IF, CCK-8 cell viability assay, IHC, and qRT-PCR were employed to detect alterations of indicators of chondrocyte proliferation, including PCNA and COL2A1. We found that p16 deletion significantly elevated the percentage of PCNA-positive cells (Figure 4A, 4B) and COL2A1-positive areas (Figure 4C, 4D), and the absorbance at 450 nm (Figure 4E). Similar results were observed in the callus. PCNA (Figure 4F-H) and COL2A1 (Figure 4I-K) expression levels were upregulated in p16-/- mice on postoperative day 7 while downregulated on postoperative days 10 and 14. These results demonstrated that p16 deletion promoted chondrocytes to proliferate in the early stage of fracture healing.
Figure 4.

Deletion of p16 promoted chondrocyte proliferation. Representative immunofluorescent micrographs of (A) PCNA, an indicator of proliferation, and (C) COL2A1, a specific marker of chondrocytes in chondrocytes differentiated from BM-MSCs. The percentages of (B) PCNA-positive cells and (D) COL2A1-positive areas in chondrocytes differentiated from BM-MSCs. (E) CCK-8 assay was performed to measure the absorbance at 450 nm to assess chondrocyte proliferation in chondrocytes differentiated from BM-MSCs. Representative immunohistochemical micrographs of (F) PCNA and (I) COL2A1 in callus on postoperative days 7, 10 and 14. The percentages of (G) PCNA-positive cells and (J) COL2A1-positive areas in callus on postoperative days 7, 10, and 14. (G) The mRNA levels of (H) PCNA and (K) COL2A1 in callus on postoperative days 7, 10, and 14 were measured by qRT-PCR, calculated as ratio relative to GAPDH mRNA and expressed relative to WT. n=4, *P<0.05, **P<0.01, ***P<0.001, compared with WT mice.
Deletion of p16 promoted chondrocyte maturation
To uncover the effect of p16 deletion on chondrocyte maturation, we employed IHC, and qRT-PCR to detect alterations in indicators of chondrocyte maturation, including COL10A1 and MMP13 in callus. The results showed that the expression levels of COL10A1 and MMP13 were elevated and declined in p16-/- mice on postoperative days 10 and 14, respectively (Figure 5A-F). These results suggested that deletion of p16 advanced chondrocyte maturation.
Figure 5.
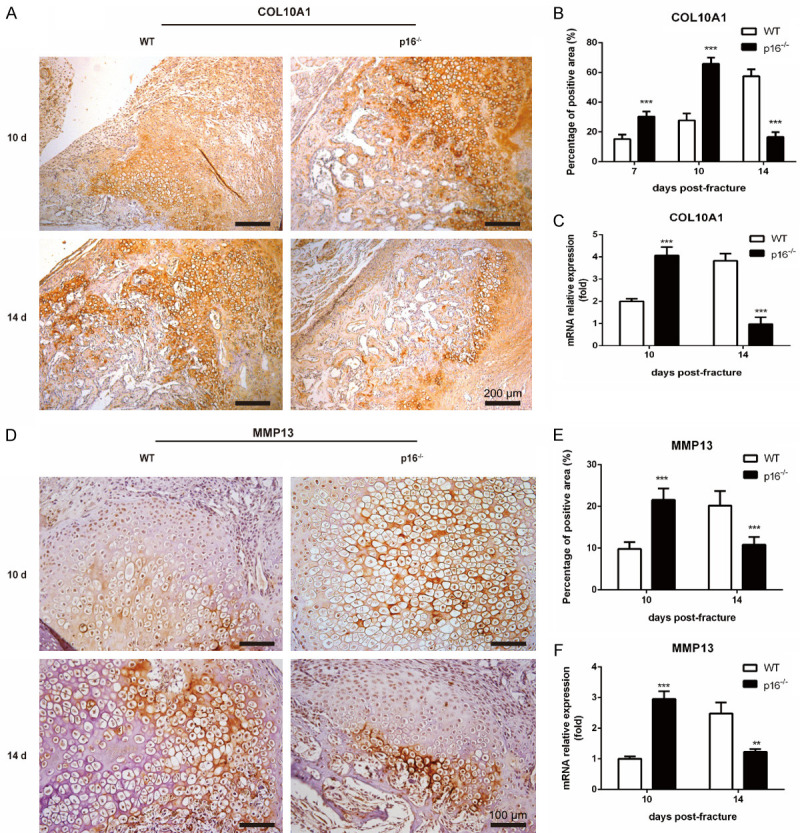
Deletion of p16 promoted chondrocyte maturation. Representative immunohistochemical micrographs of (A) COL10A1, a specific marker of hypertrophic chondrocytes and (D) MMP13 in callus on postoperative days 10 and 14. The percentages of (B) COL10A1-positive and (E) MMP13-positive areas in callus on postoperative days 10 and 14. The mRNA levels of (C) COL10A1 and (F) MMP13 in callus on postoperative days 10 and 14 were measured by qRT-PCR, calculated as ratio relative to GAPDH mRNA and expressed relative to WT. n=4, **P<0.01, ***P<0.001, compared with WT mice.
Deletion of p16 stimulated osteoblastogenesis
To study the effect of p16 deletion on osteoblastogenesis, western blot analysis, qRT-PCR, ALP staining, alizarin red staining and IHC were employed to detect alterations of osteoblastic differentiation markers, including OSTERIX, COL1A1 and ALP. We found that the expression levels of OSTERIX and COL1A1 were upregulated in BM-MSCs from p16-/- mice (Figure 6A-C). ALP activity and calcium deposition were enhanced in BM-MSCs from p16-/- mice (Figure 6D). The OSTERIX expression in callus in p16-/- mice was increased on postoperative days 7, 10 and 14 (Figure 6E-G). The COL1A1 expression in callus in p16-/- mice was increased on postoperative days 10 and 14 while decreased on postoperative day 21 (Figure 6H-J). These results indicated that deletion of p16 stimulated osteoblastogenesis.
Figure 6.

Deletion of p16 facilitated osteoblastogenesis. (A) Western blot analysis was performed to determine the protein expression levels of osteoblastic differentiation markers, including OSTERIX and COL1A1 in induced BM-MSCs. β-actin was used as loading control. (B) The quantified protein levels of OSTERIX and COL1A1 in induced BM-MSCs were evaluated by densitometric analysis. (C) The mRNA levels of OSTERIX and COL1A1 in induced BM-MSCs were measured by qRT-PCR, calculated as ratio relative to GAPDH mRNA and expressed relative to WT. (D) ALP staining and alizarin red staining images of induced BM-MSCs. Representative immunohistochemical micrographs of (E) OSTERIX in callus on postoperative days 7, 10 and 14, and (H) COL1A1 in callus on postoperative days 10, 14 and 21. The percentages of (F) OSTERIX-positive cells in callus on postoperative days 7, 10, and 14, and (I) COL1A1-positive areas in callus on postoperative days 10, 14, and 21. The mRNA levels of (G) OSTERIX in callus on postoperative days 7, 10, and 14, and (J) COL1A1 in callus on postoperative days 10 and 14 were measured by qRT-PCR, calculated as ratio relative to GAPDH mRNA and expressed relative to WT. n=4, *P<0.05, **P<0.01, ***P<0.001, compared with WT mice.
Deletion of p16 facilitated vascularization by activating AKT/mTOR/HIF-α pathway
Vascularization is essential for bone regeneration, and VEGF is a key regulator of vascularization [19]. To determine the effect of p16 deletion on vascularization in fracture healing, HUVECs tube formation assay, IHC, and ELISA were employed to detect VEGF expression and endothelial tube formation. The results showed that the number of closed mesh constructions bounded by differentiated HUVECs was significantly increased when incubated in the serum-free conditioned media collected from p16-/- mice-derived BM-MSCs for 4 h (Figure 7A). The expression levels of VEGF-A in callus (Figure 7B, 7C) and VEGF in serum (Figure 7D) were also increased before postoperative day 21 in p16-/- mice. To further investigate whether p16 deletion upregulated VEGF expression through activation of the AKT/mTOR/HIF-1α pathway, western blot analysis was performed. The expression levels of AKT and mTOR were downregulated, while the expression levels of p-AKT, p-mTOR, HIF-1α, and VEGF-A were upregulated in BM-MSCs from p16-/- mice (Figure 7E, 7F). These results showed that p16 deletion facilitated vascularization by activating the AKT/mTOR/HIF-1α pathway.
Figure 7.

Deletion of p16 stimulated vascularization by activating the AKT/mTOR/HIF-1α pathway. HUVEC tube formation assay was performed to determine the differentiation of HUVECs into capillary-like structures. A. Representative micrographs and quantitation of numbers of HUVEC meshes. B. Representative immunohistochemical micrographs of VEGF-A in callus on postoperative days 7, 10, and 14. C. The percentages of VEGF-A-positive cells in callus on postoperative days 7, 10, and 14. D. The serum VEGF level detected with ELISA. E. Western blot analysis was employed to assess the protein expression levels of members of the AKT/mTOR/HIF-1α pathway, including p-AKT, AKT, p-mTOR, mTOR, HIF-1α, and VEGF-A. β-actin was used as loading control. F. The quantified protein levels of CDK4, CDK6, RB, pRB, and E2F1 were evaluated by densitometric analysis. n=4, **P<0.01, ***P<0.001, compared with WT mice.
Discussion
Bone is one of the few tissues that can perfectly regenerate without scar formation. After initial injury, fractures heal through either primary or secondary healing, which is composed of intramembranous and endochondral ossification [20,21]. Secondary fracture healing is the most common form of fracture repair, involving cartilaginous callus formation, mineralization, resorption, and conversion to bony callus [22].
To initiate the fracture repair program, MSCs from surrounding tissues or remote hematopoietic sites must be recruited and migrated to the fracture site under regulation by relevant cytokines, particularly SDF-1. SDF-1 is induced in the periosteum of injured bone and binds to its receptor CXCR4 to recruit MSCs to the injury site [17]. SDF-1-mediated signaling drives MSCs migration and significantly enhances cartilage regeneration in osteochondral defects in rats [23]. Disruption of SDF-1 and/or CXCR-4 impairs MSC chemotaxis and decreases bone formation [17,24]. In the present study, SDF-1 expression in clots and CXCR-4 expression in BM-MSCs were both elevated, and migration of BM-MSCs was also increased in p16-/- mice.
Following migrating to the injury site, MSCs undergo proliferation and differentiation. p16 is a CDK inhibitor, which restrains the cell cycle [25]. It binds to and inactivates CDK4/6, suppresses phosphorylation of RB (pRB) and activation of E2F, and prevents the cell cycle from passing through the G1/S checkpoint, thus inhibiting cell proliferation [11]. Cell cycle and western blot analysis in this study illustrated that p16 deletion could accelerate the progression of BM-MSCs from G1 to S phase to enhance proliferation by activating the CDK4/6-pRB-E2F pathway. Similar results were obtained in NP cells in a study conducted by Che et al. [12].
SOX9 plays an essential role in chondrogenesis and in maintaining the cartilaginous phenotype and hypertrophic maturation [26-28]. It triggers the differentiation of MSCs into chondrocytes and regulates the expression of canonical extracellular matrix (ECM) proteins of chondrocytes [29,30]. Chondrocytes proliferate to form cartilage callus to bridge the fracture gap and provide immediate stabilization. In our study, increased expression levels of SOX9, PCNA and CLO2A1 were observed in p16-/- mice, which was consistent with the histologic and radiologic findings.
Conversion of chondrocytes from a proliferative to hypertrophic state and osteoblastogenesis are required for the replacement of the cartilage callus by bony callus [31]. CLO10A1 is a specific marker of hypertrophic chondrocytes. Hypertrophic chondrocytes also secrete MMP13, which degrades ECM and plays critical roles in development of growth plate cartilage and in endochondral ossification [32]. Hypertrophic chondrocytes restore the expression of osteogenic promoters RUNX2 after losing SOX9 expression [26]. RUNX2 transcriptionally regulates osteoblastogenesis through the transcription factor OSTERIX. Afterwards, OSTERIX induces COL1A1 expression through binding to specificity protein 1 sites in the bone enhancer and proximal promoter regions [33]. Osteoblasts express canonical extracellular markers of bone, including ALP, and osteocalcin to form hard callus [34]. The hard callus formation usually peaks on postoperative day 14 in animal models [35]. As the hard callus formation progresses and the calcified cartilage converts to woven bone, the callus becomes more solid and mechanically rigid [22]. Interestingly, in p16-/- mice, the expressions of OSTERIX and COL1A1 coincidentally reached a peak on postoperative day 14 in this study.
Bone is a highly vascularized organ, and revascularization has been recognized as an essential component of successful bone regeneration [19,36]. Disrupted vascularization results in a high risk of fracture nonunion [37,38]. VEGF is a key driver of vascular regeneration. It binds to its receptors to enhance proliferation and sprouting of endothelial cells, recruit endothelial progenitor cells to the fracture site, promote blood vessel invasion and transform the avascular cartilaginous callus into a vascularized osseous tissue [19,20]. Suppression of the activities of VEGF or its receptors leads to delayed callus vascularization, decreased vessel volume, and reduced callus formation [39,40].
In addition to inhibiting cell cycle progression, p16 plays a crucial role in regulating VEGF expression and revascularization at the fracture site. HIF-1α is a positive regulator of VEGF and is under the control of the AKT/mTOR pathway [41]. Active AKT activates mTOR and its major target, HIF-1α, to induce angiogenesis [42,43]. p16 suppresses the AKT/mTOR/HIF-1α signaling pathway to inhibit proangiogenic effects in breast cancer [44]. Similar results were obtained in human gliomas, where restoration of p16 downregulated VEGF expression and prevented angiogenesis [45]. We found that VEGF expression was upregulated and the AKT/mTOR/HIF-1α pathway was activated in p16-/- mice. It has been suggested that p16 deletion facilitates vascularization by activating the AKT/mTOR/HIF-1α pathway.
Thus, from the data in the current study, we conclude that p16 deletion accelerated fracture healing in geriatric mice by enhancing the migration, division, and differentiation of BM-MSCs, promoting proliferation and maturation of chondrocytes, activating osteoblastogenesis, and facilitating vascularization. This is a novel strategy to treat fractures in the elderly.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81572149).
Disclosure of conflict of interest
None.
References
- 1.Gerstenfeld LC, Cullinane DM, Barnes GL, Graves DT, Einhorn TA. Fracture healing as a post-natal developmental process: molecular, spatial, and temporal aspects of its regulation. J Cell Biochem. 2003;88:873–884. doi: 10.1002/jcb.10435. [DOI] [PubMed] [Google Scholar]
- 2.Borrelli J Jr, Pape C, Hak D, Hsu J, Lin S, Giannoudis P, Lane J. Physiological challenges of bone repair. J Orthop Trauma. 2012;26:708–711. doi: 10.1097/BOT.0b013e318274da8b. [DOI] [PubMed] [Google Scholar]
- 3.Meyer RA Jr, Tsahakis PJ, Martin DF, Banks DM, Harrow ME, Kiebzak GM. Age and ovariectomy impair both the normalization of mechanical properties and the accretion of mineral by the fracture callus in rats. J Orthop Res. 2001;19:428–435. doi: 10.1016/S0736-0266(00)90034-2. [DOI] [PubMed] [Google Scholar]
- 4.Meyer RA Jr, Meyer MH, Tenholder M, Wondracek S, Wasserman R, Garges P. Gene expression in older rats with delayed union of femoral fractures. J Bone Joint Surg Am. 2003;85:1243–1254. doi: 10.2106/00004623-200307000-00010. [DOI] [PubMed] [Google Scholar]
- 5.Bak B, Andreassen TT. The effect of aging on fracture healing in the rat. Calcif Tissue Int. 1989;45:292–297. doi: 10.1007/BF02556022. [DOI] [PubMed] [Google Scholar]
- 6.Foulke BA, Kendal AR, Murray DW, Pandit H. Fracture healing in the elderly: a review. Maturitas. 2016;92:49–55. doi: 10.1016/j.maturitas.2016.07.014. [DOI] [PubMed] [Google Scholar]
- 7.Meinberg EG, Clark D, Miclau KR, Marcucio R, Miclau T. Fracture repair in the elderly: clinical and experimental considerations. Injury. 2019;50(Suppl 1):S62–S65. doi: 10.1016/j.injury.2019.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu C, Miclau T, Hu D, Hansen E, Tsui K, Puttlitz C, Marcucio RS. Cellular basis for age-related changes in fracture repair. J Orthop Res. 2005;23:1300–1307. doi: 10.1016/j.orthres.2005.04.003.1100230610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gruber R, Koch H, Doll BA, Tegtmeier F, Einhorn TA, Hollinger JO. Fracture healing in the elderly patient. Exp Gerontol. 2006;41:1080–1093. doi: 10.1016/j.exger.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boquoi A, Arora S, Chen T, Litwin S, Koh J, Enders GH. Reversible cell cycle inhibition and premature aging features imposed by conditional expression of p16Ink4a. Aging Cell. 2015;14:139–147. doi: 10.1111/acel.12279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Che H, Li J, Li Y, Ma C, Liu H, Qin J, Dong J, Zhang Z, Xian CJ, Miao D, Wang L, Ren Y. p16 deficiency attenuates intervertebral disc degeneration by adjusting oxidative stress and nucleus pulposus cell cycle. Elife. 2020;9:e52570. doi: 10.7554/eLife.52570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li M, Zhang Z, Hill DL, Wang H, Zhang R. Curcumin, a dietary component, has anticancer, chemosensitization, and radiosensitization effects by down-regulating the MDM2 oncogene through the PI3K/mTOR/ETS2 pathway. Cancer Res. 2007;67:1988–1996. doi: 10.1158/0008-5472.CAN-06-3066. [DOI] [PubMed] [Google Scholar]
- 14.Buscail L, Bournet B, Dufresne M, Torrisani J, Cordelier P. Advance in the biology of pancreatic of cancer. Bull Cancer. 2015;102:S53–61. doi: 10.1016/S0007-4551(15)31218-2. [DOI] [PubMed] [Google Scholar]
- 15.Khan M, Mohsin S, Khan SN, Riazuddin S. Repair of senescent myocardium by mesenchymal stem cells is dependent on the age of donor mice. J Cell Mol Med. 2011;15:1515–1527. doi: 10.1111/j.1582-4934.2009.00998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu X, Li J, Zhang H, Wang H, Yin G, Miao D. Pyrroloquinoline quinone prevents testosterone deficiency-induced osteoporosis by stimulating osteoblastic bone formation and inhibiting osteoclastic bone resorption. Am J Transl Res. 2017;9:1230–1242. [PMC free article] [PubMed] [Google Scholar]
- 17.Kitaori T, Ito H, Schwarz EM, Tsutsumi R, Yoshitomi H, Oishi S, Nakano M, Fujii N, Nagasawa T, Nakamura T. Stromal cell-derived factor 1/CXCR4 signaling is critical for the recruitment of mesenchymal stem cells to the fracture site during skeletal repair in a mouse model. Arthritis Rheum. 2009;60:813–823. doi: 10.1002/art.24330. [DOI] [PubMed] [Google Scholar]
- 18.Akiyama H, Kim JE, Nakashima K, Balmes G, Iwai N, Deng JM, Zhang Z, Martin JF, Behringer RR, Nakamura T, de Crombrugghe B. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc Natl Acad Sci U S A. 2005;102:14665–14670. doi: 10.1073/pnas.0504750102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keramaris NC, Calori GM, Nikolaou VS, Schemitsch EH, Giannoudis PV. Fracture vascularity and bone healing: a systematic review of the role of VEGF. Injury. 2008;39(Suppl 2):S45–57. doi: 10.1016/S0020-1383(08)70015-9. [DOI] [PubMed] [Google Scholar]
- 20.Bahney CS, Zondervan RL, Allison P, Theologis A, Ashley JW, Ahn J, Miclau T, Marcucio RS, Hankenson KD. Cellular biology of fracture healing. J Orthop Res. 2019;37:35–50. doi: 10.1002/jor.24170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marsell R, Einhorn TA. The biology of fracture healing. Injury. 2011;42:551–555. doi: 10.1016/j.injury.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerstenfeld LC, Alkhiary YM, Krall EA, Nicholls FH, Stapleton SN, Fitch JL, Bauer M, Kayal R, Graves DT, Jepsen KJ, Einhorn TA. Three-dimensional reconstruction of fracture callus morphogenesis. J Histochem Cytochem. 2006;54:1215–1228. doi: 10.1369/jhc.6A6959.2006. [DOI] [PubMed] [Google Scholar]
- 23.Mustapich T, Schwartz J, Palacios P, Liang H, Sgaglione N, Grande DA. A novel strategy to enhance microfracture treatment with stromal cell-derived factor-1 in a rat model. Front Cell Dev Biol. 2020;8:595932. doi: 10.3389/fcell.2020.595932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Granero-Molto F, Weis JA, Miga MI, Landis B, Myers TJ, O’Rear L, Longobardi L, Jansen ED, Mortlock DP, Spagnoli A. Regenerative effects of transplanted mesenchymal stem cells in fracture healing. Stem Cells. 2009;27:1887–1898. doi: 10.1002/stem.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 26.Dy P, Wang W, Bhattaram P, Wang Q, Wang L, Ballock RT, Lefebvre V. Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev Cell. 2012;22:597–609. doi: 10.1016/j.devcel.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikegami D, Akiyama H, Suzuki A, Nakamura T, Nakano T, Yoshikawa H, Tsumaki N. Sox9 sustains chondrocyte survival and hypertrophy in part through Pik3ca-Akt pathways. Development. 2011;138:1507–1519. doi: 10.1242/dev.057802. [DOI] [PubMed] [Google Scholar]
- 28.Leung VY, Gao B, Leung KK, Melhado IG, Wynn SL, Au TY, Dung NW, Lau JY, Mak AC, Chan D, Cheah KS. SOX9 governs differentiation stage-specific gene expression in growth plate chondrocytes via direct concomitant transactivation and repression. PLoS Genet. 2011;7:e1002356. doi: 10.1371/journal.pgen.1002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bell DM, Leung KK, Wheatley SC, Ng LJ, Zhou S, Ling KW, Sham MH, Koopman P, Tam PP, Cheah KS. SOX9 directly regulates the type-II collagen gene. Nat Genet. 1997;16:174–178. doi: 10.1038/ng0697-174. [DOI] [PubMed] [Google Scholar]
- 30.Lefebvre V, Huang W, Harley VR, Goodfellow PN, de Crombrugghe B. SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol Cell Biol. 1997;17:2336–2346. doi: 10.1128/mcb.17.4.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu DP, Ferro F, Yang F, Taylor AJ, Chang W, Miclau T, Marcucio RS, Bahney CS. Cartilage to bone transformation during fracture healing is coordinated by the invading vasculature and induction of the core pluripotency genes. Development. 2017;144:221–234. doi: 10.1242/dev.130807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inada M, Wang Y, Byrne MH, Rahman MU, Miyaura C, Lopez-Otin C, Krane SM. Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc Natl Acad Sci U S A. 2004;101:17192–17197. doi: 10.1073/pnas.0407788101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ortuno MJ, Susperregui AR, Artigas N, Rosa JL, Ventura F. Osterix induces Col1a1 gene expression through binding to Sp1 sites in the bone enhancer and proximal promoter regions. Bone. 2013;52:548–556. doi: 10.1016/j.bone.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 34.Gerstenfeld LC, Shapiro FD. Expression of bone-specific genes by hypertrophic chondrocytes: implication of the complex functions of the hypertrophic chondrocyte during endochondral bone development. J Cell Biochem. 1996;62:1–9. doi: 10.1002/(SICI)1097-4644(199607)62:1%3C1::AID-JCB1%3E3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 35.Einhorn TA. The cell and molecular biology of fracture healing. Clin Orthop Relat Res. 1998;335:S7–21. doi: 10.1097/00003086-199810001-00003. [DOI] [PubMed] [Google Scholar]
- 36.Hu K, Olsen BR. The roles of vascular endothelial growth factor in bone repair and regeneration. Bone. 2016;91:30–38. doi: 10.1016/j.bone.2016.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dickson KF, Katzman S, Paiement G. The importance of the blood supply in the healing of tibial fractures. Contemp Orthop. 1995;30:489–493. [PubMed] [Google Scholar]
- 38.Brownlow HC, Reed A, Simpson AH. The vascularity of atrophic non-unions. Injury. 2002;33:145–150. doi: 10.1016/s0020-1383(01)00153-x. [DOI] [PubMed] [Google Scholar]
- 39.Jacobsen KA, Al-Aql ZS, Wan C, Fitch JL, Stapleton SN, Mason ZD, Cole RM, Gilbert SR, Clemens TL, Morgan EF, Einhorn TA, Gerstenfeld LC. Bone formation during distraction osteogenesis is dependent on both VEGFR1 and VEGFR2 signaling. J Bone Miner Res. 2008;23:596–609. doi: 10.1359/JBMR.080103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Street J, Bao M, deGuzman L, Bunting S, Peale FV Jr, Ferrara N, Steinmetz H, Hoeffel J, Cleland JL, Daugherty A, van Bruggen N, Redmond HP, Carano RA, Filvaroff EH. Vascular endothelial growth factor stimulates bone repair by promoting angiogenesis and bone turnover. Proc Natl Acad Sci U S A. 2002;99:9656–9661. doi: 10.1073/pnas.152324099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skinner HD, Zheng JZ, Fang J, Agani F, Jiang BH. Vascular endothelial growth factor transcriptional activation is mediated by hypoxia-inducible factor 1alpha, HDM2, and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT signaling. J Biol Chem. 2004;279:45643–45651. doi: 10.1074/jbc.M404097200. [DOI] [PubMed] [Google Scholar]
- 43.Ma Z, Wang LZ, Cheng JT, Lam WST, Ma X, Xiang X, Wong AL, Goh BC, Gong Q, Sethi G, Wang L. Targeting HIF-1-mediated metastasis for cancer therapy. Antioxid Redox Signal. 2020 doi: 10.1089/ars.2019.7935. [DOI] [PubMed] [Google Scholar]
- 44.Al-Ansari MM, Hendrayani SF, Tulbah A, Al-Tweigeri T, Shehata AI, Aboussekhra A. p16INK4A represses breast stromal fibroblasts migration/invasion and their VEGF-A-dependent promotion of angiogenesis through Akt inhibition. Neoplasia. 2012;14:1269–1277. doi: 10.1593/neo.121632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harada H, Nakagawa K, Iwata S, Saito M, Kumon Y, Sakaki S, Sato K, Hamada K. Restoration of wild-type p16 down-regulates vascular endothelial growth factor expression and inhibits angiogenesis in human gliomas. Cancer Res. 1999;59:3783–3789. [PubMed] [Google Scholar]