

Summary
Dragonflies and damselflies are among the earliest flying insects with extant representatives. However, unraveling details of their long evolutionary history, such as egg laying (oviposition) strategies, is impeded by unresolved phylogenetic relationships, particularly in damselflies. Here we present a transcriptome-based phylogenetic reconstruction of Odonata, analyzing 2,980 protein-coding genes in 105 species representing nearly all the order’s families. All damselfly and most dragonfly families are recovered as monophyletic. Our data suggest a sister relationship between dragonfly families of Gomphidae and Petaluridae. According to our divergence time estimates, both crown-Zygoptera and -Anisoptera arose during the late Triassic. Egg-laying with a reduced ovipositor apparently evolved in dragonflies during the late Jurassic/early Cretaceous. Lastly, we also test the impact of fossil choice and placement, particularly, of the extinct fossil species, †Triassolestodes asiaticus, and †Proterogomphus renateae on divergence time estimates. We find placement of †Proterogomphus renateae to be much more impactful than †Triassolestodes asiaticus.
Subject areas: Evolutionary biology, Phylogenetics
Graphical abstract
Highlights
-
•
Relationships of dragonflies and damselflies are unraveled using transcriptomes
-
•
Earliest flying insects – Odonata and extinct relatives – date back to Permian period
-
•
Both extant dragonflies and damselflies started diverging in the Triassic period
Evolutionary biology; Phylogenetics
Introduction
Few insects elicit as positive a human response as dragonflies and damselflies. Along with butterflies, ladybird beetles, and perhaps fireflies, dragonflies and damselflies are generally beloved across cultures. Odonata (the order containing damselflies and dragonflies) are sister to the order Ephemeroptera — the mayflies — and together, they are considered the sister to the rest of Pterygota (Misof et al., 2014). There are 6,426 described extant species within Odonata, grouped in three suborders: Anisozygoptera, Anisoptera (dragonflies), and Zygoptera (damselflies) (https://www.pugetsound.edu/academics/academic-resources/slater-museum/biodiversity-resources/dragonflies/world-odonata-list2/- accessed Nov 12th, 2020). There are 2,967 recorded species of dragonflies and 3,332 recorded species of damselflies. The suborder Anisozygoptera is monogeneric and contains three species, all belonging to the genus Epiophlebia. Ubiquitous in lentic (flowing) and lotic (still water) habitats (Letsch et al., 2016), dragonflies and damselflies (Odonata) are globally distributed freshwater insects with aquatic nymphs and terrestrial adults. All odonates are predatory, both as adults and nymphs, and consume other insects, small fishes, and tadpoles (Corbet, 1999). Dragonflies and damselflies can be found in a variety of ecosystems, from arid desert (Suhling et al., 2003) to the arctic tundra (Kohli et al., 2018).
To date, relatively few of the extant species of Odonata have been analyzed in a phylogenetic context. Historically, phylogenetics and taxonomy of Odonata has largely been based on morphological characteristics and most studies focused on characters based on wing venation, genitalic, and more recently, larval traits (e.g., Tillyard, 1917; Fraser and Tillyard, 1957; Trueman, 1996; Rehn, 2003; Trueman, 2007). During the last two decades, odonate phylogenies have been inferred based on a few molecular markers using Sanger sequence data and focusing on the relationships among families and genera. Ballare and Ware (2011) summarize the results of past molecular studies on Odonata. While Sanger sequence data have been useful for resolving parts of the odonate tree of life (e.g., Ware et al., 2017), they have failed to resolve inter- and intra-familial relationships within Anisoptera and Zygoptera (e.g., Ware et al., 2007; Pilgrim and von Dohlen, 2008; Dijkstra et al., 2014). For example, within Anisoptera, the relative phylogenetic positions of Cavilabiata, Gomphidae, and Petaluridae are still debated. Previous morphological investigations have hypothesized Gomphidae and Petaluridae to be sister to all other dragonflies (for example, Blanke et al., 2013). However, multiple recent molecular studies have firmly placed Aeshnidae (and Austropetaliade when included) as sister to all other anisopteran families (e.g., Letsch et al., 2016; Bybee et al., 2021; Suvorov et al., 2021). Gomphidae and Petaluridae most likely diverged after Aeshnidae and Austropetalidae diverged, according to recent studies. However, the timing and interfamilial relationships of Gomphidae and the other anisopteran families remained unresolved and raise pressing questions: What is the sister to Gomphidae? Are Gomphidae sister to Petaluridae or, instead, are they more closely related to the group Cavilabiata owing to their similar oviposition style? The phylogenetic relationships of the groups Cavilabiata, Gomphidae, and Petaluridae to one another have major implications for our understanding of the evolution of dragonfly oviposition strategies in Odonata.
A robustly resolved and reliably inferred phylogeny of Odonata is a prerequisite for reliable divergence time estimates. There are only a few studies that have estimated divergence times within Odonata by analyzing molecular data. Most of these studies were focused on estimating the ages of familial or super-familial groups (Dumont et al., 2005; Sánchez-Herrera et al., 2020; Ware et al., 2007, Ware et al., 2009, 2012, 2014; Letsch et al., 2016; Thomas et al., 2013; Davis et al., 2011). In the context of a study covering all Palaeoptera (Ephemeroptera and Odonata), Thomas et al. (2013) estimated ages across the order Odonata. They found that the crown damselflies diverged in the late Triassic, approximately 230 million years ago (Zygoptera; 216.2 my (202.7–241.2 My)) while crown Anisoptera were estimated to have diverged around the late Jurassic (Anisoptera; 163.4 my (145.8–192.7 My)). Misof et al. (2014), in the context of a study covering all major lineages of insects, again recovered a Triassic age for the origin of crown Odonata (with confidence intervals ranging from late Permian to early Triassic). The fossil record of crown Odonata consists of 828 fossil species distributed across 517 genera (www.palaeodb.org; compression and amber fossils). Of these, several fossils have already been suggested as suitable calibrations for an odonate time tree (Kohli et al., 2016). The oldest putative crown odonate fossil species belongs to the 250-million-year-old extinct family Triassolestidae (see references cited by Kohli et al. (2016)). However, this placement is debated and it is alternatively considered a stem lineage close to the crown (Tierney et al., 2020). Whether fossils are treated as crown- or stem-members of a particular group can dramatically influence the age of a particular node (example: Ruane et al., 2011), but the extent of this impact in other parts of the tree has not been well studied.
Here, we present one of the first phylogenomic studies using transcriptome data to reliably infer the phylogeny of Odonata. It includes 103 odonate species covering most extant families (except Austropetaliidae and Neopetaliidae, see Table S1). Odonate transcriptomic data have been incorporated in studies of vision (e.g., Bybee et al., 2012; Futahashi et al., 2015) and embryology (Simon et al., 2017), but to date a large-scale phylogenomic treatment of Odonata is lacking. Our study has two main goals. First is toaddress difficult to resolve nodes within Anisoptera—particularly relationship between families Gomphidae and Petaluridae—by utilizing transcriptomes to reconstruct a reliable phylogeny of Odonata. We do so by applying both concatenation and gene tree based coalescent approaches and looking at various measures for branch support. Our second goal is to provide the most comprehensive divergence time estimation in Odonata by using the most inclusive set of fossils assembled for calibration and to discuss the impact of the specific odonate fossils on the recovered node ages.
Results
Orthology assignment, protein domain and gene identification and alignments
We concatenated the aligned amino acid sequences into a supermatrix comprising 116 species and 1,070,111 alignment sites. We partitioned our data using protein domain identification as suggested by Misof et al. (2014) to improve the biological realism by modelling sequence evolution based on protein domains rather than gene partitions. Analysis of our dataset with the alignment masking software MARE version 0.1.2-rc reduced the data set by excluding the species Acanthaeschna victoria and a total of 245,328 alignment sites. The final amino acid (AA) matrix had a total of 2,890 protein domain partitions with 824,783 aligned amino acid positions and 115 species (supermatrix A) (See STAR Methods and Tables S1 and S2). The corresponding nucleotide alignments including only second codon positions and all three codon positions comprised of 824,783 (supermatrix B) and 2,474,349 nucleotide positions (supermatrix C), respectively. The protein domain partitioning was used for the maximum likelihood (ML) tree reconstruction of the concatenated data set (See STAR Methods for details on ML analysis parameters).
We also performed multispecies coalescent (MSC) analysis based on gene partitions. After performing alignment masking with MARE version 0.1.2-rc, our original dataset with 2,980 gene partitions and 116 species was reduced to 2,413 genes and 115 species after removing Acanthaeschna victoria.
For all the dating analyses, we used a further reduced and unpartitioned amino acid data set that contained only positions that were presented in at least 99% of the species. The resulting supermatrix D comprised 53,091 amino acid positions and this matrix was used for the divergence time estimation analysis.
Topology estimation and congruence
Topology
The tree topologies recovered using the concatenation approach and protein domain partitions for amino acid (supermatrix A) and the two corresponding nucleotide datasets (NT123, NT2 shown in Figure S1) are congruent with respect to suborder monophyly and interfamilial relationships. The ML tree obtained using RaxML and supermatrix A comprising of amino acids is show in Figure 1.
Figure 1.

Phylogenetic relationships in Odonata recovered using the amino acid (AA) dataset
All branches are recovered with 100% bootstrap support except the two branches leading to the nodes in Anisoptera highlighted with white circles. Results of the four-cluster likelihood mapping (FcLM) for relationships among Petaluridae, Gomphidae, and Cavilabiata are also represented.
Damselflies (Zygoptera) are recovered as sister to Epiprocta, the group that comprises Anisozygoptera and Anisoptera (dragonflies) (Figure 1). Within the scope of our sampling, Lestoidea are sister to the remaining Zygoptera. In line with prior studies (e.g., Rehn, 2003; Bybee et al., 2008) Hemiphlebia, the only representative in the family Hemiphlebiidae, is recovered as sister to Lestidae. Calopterygoidea, damselflies with often brightly colored wings and comprising of Chlorocyphidae and Calopterygidae, is recovered as sister to Coenagrionoidea, the bluets (a family which includes Megaloprepus caerulatus, the odonate with the largest known wingspan; Fincke (1984)).
Anisozygoptera, represented here by the Japanese endemic species Epiophlebia superstes, is recovered as sister to the dragonflies (Anisoptera). As in previous studies (e.g., Letsch et al., 2016), we recover Aeshnidae as sister to the remaining dragonfly families (Figure 1). The families Gomphidae and Petaluridae are recovered as sister taxa. This clade is in turn recovered as sister to the superfamily Cavilabiata (Figure 1). Within Cavilabiata, Chlorogomphidae + Cordulegastridae are recovered as sister to Libelluloidea. Cordulephya pygmea and Eusynthemis guttata, two species of family Synthemistidae endemic to Australia, are recovered in a paraphyletic assemblage. Therefore, the monophyly of Synthemistidae cannot be determined with this limited taxon sampling. Lastly, Macromiidae is recovered as sister to the families (Cordullidae + Libellulidae).
The phylogenetic tree recovered from the MSC analysis (Figure S2) based on all the genes is effectively similar to the concatenation tree (Figure 1) with respect to suborder monophyly and interfamilial relationships. However, there is one key exception: The families Gomphidae and Petaluriade are no longer recovered as sister taxa as in the ML concatenation tree (Figure 1). Instead, Gomphidae is recovered as sister to a clade containing Cavilabiata and Petaluridae (Figures 2 and S2). In fact, we found that the relationship between groups Gomphidae, Petaluridae, and Cavilabata changed based on speed of gene evolution. When reconstructing a species tree based on the fastest 25%, fastest 50% and slowest 75% genes, we recovered the same topology as in the case of using all the genes (Figure 2). When we used the slowest 50% genes for species tree reconstruction, the groups Gomphidae, Petaluridae, and Cavilabiata are recovered with a support of 37% which can essentially be interpreted as a polytomy. However, when using the 25% slowest genes for the species reconstruction, families Gomphidae and Petaluridae switch to being sisters and the recovered topology is the same as that recovered in the ML analysis (Figure 2).
Figure 2.

Different relationship between Gomphidae, Petaluridae, and Cavilabiata recovered based on the rate of evolution of genes
Rate of gene evolution decreases from left to right. On the extreme left is the relationship recovered using a subset of fastest 50% of the genes while on the extreme right is the relationship recovered from a subset of slowest 25% of the genes. Numbers on the nodes represent the quartet support for the branch leading upto the nodes recovered in an MSC analysis.
Lastly, we found no difference in the MSC topology when branches with low bootstrap values (cut off values of 10%, 20%, 30%, 40%, and 50% bootstrap support were tested) in the gene trees were collapsed before species tree reconstruction.
Congruence and support
All branches were recovered with maximal statistical non-parametric bootstrap support except for two clades of Anisoptera (Figure 1, highlighted in white circles). Branch support for Macromiidae + (Corduliidae + Libellulidae) is 98%, while the sister group relationship of Petaluridae and Gomphidae is recovered with 99% support. Previous studies recovered Gomphidae and Petaluridae as sister groups as we have here (e.g., Bybee et al., 2008 but with low support); however, Four-cluster Likelihood Mapping (FcLM) did not provide support for this sister group relationship (Table S2). In FcLM analyses Gomphidae + Petaluridae grouping was only supported by 11.7% of the quartets, whereas a clade comprising Cavilabiata and Gomphidae received high quartet support (78% of the quartets). This indicates that the ML tree reconstruction results are likely biased and that the signal comes from non-randomly distributed data, not necessarily from violating assumptions of stationarity (nc/aa frequencies constant over time), reversibility (substitution rates equal in both directions), and homogeneity (substitution rates constant along the tree). We conclude this from a slightly higher support for Gomphidae + Petaluridae in the analyses of permutation scheme I and a decreasing support for this topology in the analyses of permutation scheme II. Interestingly, support for Cavilabiata + Gomphidae remains highest in all permutation schemes, that is, even after removing all phylogenetic signal, suggesting a bias towards this topology in the FcLM analyses (see STAR Methods and Table S2 for more information).
We also used FcLM to test a sister group relationship between: i) Cordulegastroidea and Libelluloidea, ii) Aeshnidae and all remaining Anisoptera, iii) Corduliidae and Libellulidae, and iv) the monophyly of Synthemistidaes.l.. For all these relationships, the FcLM results were in congruence with the ML tree results (Figure 1). We therefore considered the results of the ML tree to not be severely biased.
We also assessed support for all the branches in the MSC tree by calculating gene and site concordance factors (gCF and sCF) (Minh et al., 2020a) (Figure S2) as implemented in IQTREE version 2.0-rc2 (Minh et al., 2020b). We obtained low gCF and sCF values for some deep and most shallow branches in our tree. Concordance factor values are fundamentally different from the traditional bootstrap support values and, hence, are interpreted differently. gCF and sCF values tend to be generally lower than bootstrap values. gCF estimates the proportions of genes that support a branch in a recovered topology, while sCF estimates the proportion of sites (in each gene partition) out of the total number of decisive sites that support the branch of interest (Minh et al., 2020a). Concordance factor values enable us to understand the underlying signal variation for a branch unlike bootstrap values with their statistical support. Lower gCF values in our data could be a result of the quality of information in the genes, the overall length of the genes, or they simply indicate a lot of underlying variation in the inferred phylogenetic relationships, which might be improved with additional taxa in the dataset.
Of particular interest in our case are the branches that lead to families Gomphidae and Petaluridae. The split between Gomphidae and the clade Petaluridae + Cavilabiata, as recovered with MSC analyses, has a gCF value of 19.7% (the lowest gCF possible value being 0%) and a sCF value of 35.8% (the lowest sCF possible value being 0%, but sCF are rarely lower than 33% for larger data sets, see Minh et al., 2020a), indicating that there is a lot of variation in signal for this branch, which is in agreement with the results from other analyses (i.e., uncertainty in the relationships).
Divergence time estimation
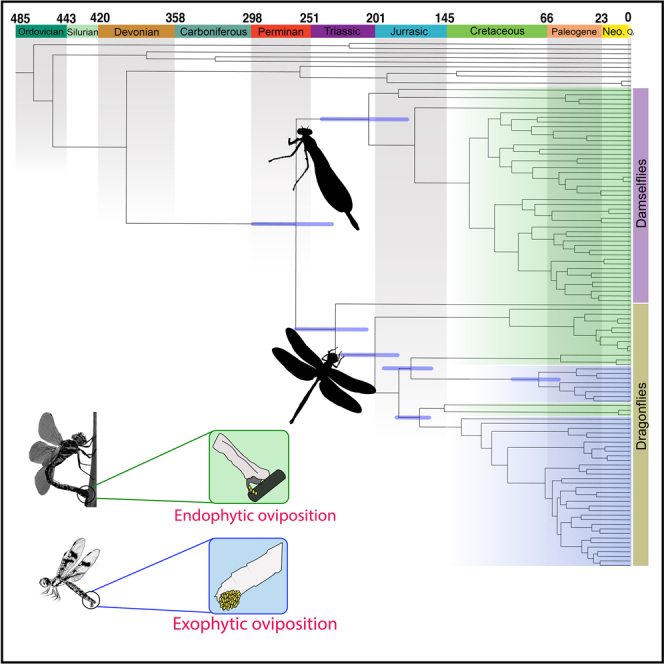
We used a comprehensive fossil dataset using newly assessed fossils in combination with those used by Kohli et al. (2016) (see Figure S3 and Table S4) to infer a well-resolved time calibrated phylogeny of Odonata. We estimated divergence time for Odonata time tree under three different scenarios: “Primary”, “Minus Triassolestidae”, and “Stem placement”, by changing one fossil calibration each in “Minus Triassolestidae”, and “Stem placement” in comparison to the “Primary” scenario (see manuscript section “Effect of fossil choice and placement” and STAR Methods for more details). We believe that the fossil calibration set used in the “Stem placement” scenario, where the fossil †Proterogomphusrenateaeis placed on the node Gomphidae + Petaluridae is the most accurate calibration scheme based on suggestions by Parham et al. (2012). Therefore, we present results and discuss chronology of Odonata evolution from this scenario (Figure 3; also see Figures 4 and S4 for divergence estimates from other scenarios as well).
Figure 3.

Evolution of egg-laying behavior in Odonata are shown in accordance with the recovered phylogenetic relationships
Lineages with endophytic oviposition, those that lay their eggs in plant material, are shaded in green. Lineages with exophytic oviposition (usually laying eggs on the surface of water) are shaded in blue. Origin of the predators that may have influenced the evolution of egg-laying strategies in dragonflies and damselflies are represented along the geological time scale. Silhouettes of putative predators that were present at various geological times (left to right): fish, frogs, pterosaurs, and birds. All the ages along the geological time scale are in millions of years.
Figure 4.

Divergence times estimates comparisons between crown or stem placement of the fossil †Proterogomphus renateae (indicated with brown line)
Six different nodes of interest are indicated with different colors (cyan = Odonata, red = Anisoptera, orange = Zygoptera, green = Gomphidae +Petaluridae, pink = Gomphidae and yellow = Cavilabiata). These nodes are joined across the two scenarios with a dashed line to indicate the difference in the recovered divergence time estimates. We recover older ages under the †Proterogomphusrenateae crown placement scenario compared to the †Proterogomphus renateae stem placement scenario. Hollowed circles on a node indicate a slower substitution rate on the branch leading to this node compared to the same node in the other scenario. Substitution rates leading to the nodes Odonata, Anisoptera, Zygopter, and Gomphidae were slower in the stem placement scenario compared to the crown placement scenario. For these branches the substitution rates were faster in the crown scenario compared to the Stem scenario. By contrast, for branches leading to Zygoptera and Gomphidae + Petaluridae (Petaluroidea), rates in the stem scenario were faster compared to the crown scenario; this difference, however, was minor.
Our results show that the age of crown damselflies (Zygoptera) is 206.3 My (176–243.2 My). Calopterygoidea is recovered as 112 My (93.8–133.8 My) old. Coenagrionoidea, recovered as sister to Calopterygoidea, are estimated to be 107 My (90–127.4 My) old. Epiophlebia superstes (Anisozygoptera), sister to all dragonflies, represents the crown-Epiophlebiidae family and is 232.1 My (235.1–297.3 My) old. Crown-Anisoptera, i.e., dragonflies as recognized today, split from Anisozygoptera around 201.0 My (207.7–263.3 My). Within Anisoptera, the first splitting lineage Aeshnidae, starts diversifying around 95 My (73.0–131.3 My). Gomphidae + Petaluridae split form the remaining dragonfly lineages around 172 My (156.68–194 My). The age of crown-Gomphidae is Cretaceous, around 72 My (56.5–92.85 My) while the age of crown-Petaluridae is 55.14 My (31.95–93.65 My). Crown-Cavilabiata has a late Jurassic age, around 165 My (158.3–183.87 My). The most speciose family of dragonflies, Libellulidae, start diversifying around K/T boundary, with an age for crown-Libellulidae found to be 67 My (58.1–77.3 My).
Effect of fossil choice and placement
We tested the effect of two contested fossils: †Triassolestodes asiaticusand †Proterogomphus renateae. †Triassolestodes asiaticus has been suggested for calibrating crown-Odonata (Kohli et al., 2016), but this suggestion has been debated (see STAR Methods for further details). Therefore, we tested the effect of presence or absence of this fossil on divergence estimates in our “Minus Triassolestidae” scenario. Our results (see Figure S4 and Table S4) show that the fossil †Triassolestodes asiaticus does not dramatically affect the outcome of our divergence time estimation analysis. Throughout the chromogram, the only noticeable affects seen were on the age estimates of the nodes Anisoptera, Epiprocta, Odonata, and Zygoptera (Figure 4). Out of these, crown-Odonata changed by 6.23 Mya (a 2.09% change), the largest shift in node age caused by this fossil throughout the tree. The age of the node Epiprocta, the node from which the Triasssolestidae fossil was removed, only changed by 1.92%. Removal of †Triassolestodes asiaticus led to an increased precision, that is a smaller confidence interval (CI) of node age estimates. However, just like node ages, this change was very small. The most noticeable impact of †Triassolestodes asiaticus on the CI length was on the nodes Odonata (CI was reduced by 34.23%) and Epiprocta (CI reduced by 19.8%). Lastly, removal of †Triassolestodes asiaticus did not lead to dramatic changes in recovered substitution rates on branches leading to the nodes of interest (Table S4).
Additionally, we tested the impact of the placement of †Proterogomphus renateae in our “Stem placement” scenario. This fossil was assigned a crown Gomphidae placement by (Kohli et al., 2016), but given our current taxon sampling, which contains more recently diverged Gomphidae lineages, it was deemed an inappropriate calibration at the crown Gomphidae node in our case. Our sampling for Gomphidae in this study was largely opportunistic since at the time no Gomphidae molecular phylogeny had been reconstructed. Ware et al. (2017) published a Gomphidae phylogeny after our sequencing was completed, and this study revealed that the earliest branching lineages of Gomphidae were in the Lindeniinae (which we have not included here). Because of the absence of Lindeniinae from our phylogenetic reconstruction, †Proterogomphus renateae should not be placed on crown Gomphidae. Therefore, a stem placement, i.e, the Gomphidae + Petaluridae node in our current topology, was considered a more appropriate placement for †Proterogomphus renateae rather than crown-Gomphidae as suggested by Kohli et al. (2016).
Unlike the impact of †Triassolestodes asiaticus, a stem placement of †Proterogomphus renateae led to strongly altered and younger ages on nodes throughout the topology (Figure 4). The largest age difference was seen on the crown-Gomphidae node, which was recovered 81 million years younger (a 53% decrease in age) under the “Stem placement” scenario (Figure 4). Deeper nodes, such as those of Anisoptera, Cavilabiata, Epiprocta, Odonata, and Zygoptera, were all recovered on average to be 30 million years younger (a 11–15% decrease in age) when †Proterogomphus renateae was used as a stem rather than crown calibration. CI lengths either increased or decreased depending on the nodes. However, the absolute change in lengths of confidence intervals was high in several cases: On the Gomphidae node, the CI changed by 165% with the range increasing from 13.69 Mya to 36.35 Mya in breadth. Lastly, the distribution of the posterior probabilities for the nodes in this scenario was strikingly different from the priors for the nodes.
Discussion
We inferred a robust phylogeny of the order Odonata using transcriptomic data. We employed rigorous testing of the fossils to present the first comprehensive divergence time estimation analysis for the dragonflies and damselflies. We first discuss our results from the phylogenetic analysis and the implications of the inferred divergence times on our understanding of Odonata evolution.
Relationships and evolution of oviposition in Odonata
Phylogenetic relationships in Zygoptera have previously been considered chaotic, and a fully resolved phylogeny of Zygoptera does not yet exist. Past studies have only resolved parts of the Zygoptera family tree (Dumont et al., 2005; Sánchez-Herrera et al., 2020; Vega-Sánchez et al., 2019). Many of the traditionally recognized families, such as Amphipterygidae and Megapodagrionidae, as well as genera such as Agriocnemis and Pseudagrion have been shown to be polyphyletic (Dijkstra et al., 2014), which clearly hampers the compilation of a reliable taxon sampling to target deep phylogenetic relationships within Zygoptera. Our transcriptomic data support Lestoidea as the earliest branching lineage in Zygoptera (c.f. Dijkstra et al., 2014; Dumont et al., 2010; Bybee et al., 2008; Carle et al., 2008); Lestoidea comprises Hemiphlebiidae, Lestidae, Perilestidae, and Synlestidae. However, we lacked the latter two families in this analysis. Our analyses further support the position of Platystictidae as sister group to all remaining Zygoptera (c.f. Dijkstra et al., 2014; Dumont et al., 2010; Bybee et al., 2008). The latter are further subdivided into the superfamilies Calopterygoidea and Coenagrionoidea. A clade Platycnemidae + Coenagrionidae has been recovered in other analyses (e.g., Dijkstra et al., 2014), which is not surprising based on morphological characters that support this grouping. Calopterygidae is recovered as sister to Chlorocyphidae; both taxa are known for their striking wing coloration patterns used in communication. The taxonomy of Zygoptera cannot be simplified with the taxon sampling here, and we note that future sampling should be expanded to Megapodagrionidae and other families that have been traditionally considered “junk drawer” taxa.
Within Anisoptera, the following families are recovered as monophyletic with strong support; Aeshnidae, Cordullidae, Gomphidae, Libellulidae, Macromiidae, and Petaluridae. Yet, despite the vast amount of data used here, we still find a lack of resolution in relationships within Libelluloidea (which comprises Corduliidae, Libellulidae, Macromiidae, and Synthemistidae) and among Cavilabiata, Gomphidae, and Petaluridae. Our results recovered two different relationships based on the method and genes used for the analysis. Concatenation based maximum likelihood phylogenetic tree recovered Gomphidae and Petaluridae as sister taxa, while the multispecies coalescence topology recovers Gomphidae as sister to Cavilabiata + Petaluriade. However, when restricted to slower genes, MSC analyses also recover the group Gomphidae + Petaluridae instead of Cavilabiata + Petaluridae (Figure 2). Gene filtering, based on several factors such as phylogenetic signal, incomplete fragments, and evolutionary rate variation among species, has been shown to dramatically affect the resultant species tree (Xi et al., 2015; Sayyari et al., 2017; Vankan et al., 2021). In our case as we go from fast evolving genes to slow evolving genes the recovered topology changes (Figure 2). Fast evolving genes suffer from saturation and hence can add high levels of noise to the phylogenetic signal to noise ratio. In contract, slow genes that tend to be less saturated can be helpful in resolving deep relationships like that between the families Gomphidae and Petaluraide. The slowest evolving genes in our case recover the same topology as the concatenation-based ML tree. Further, this relationship recently has also been recovered by Bybee et al. (2021) and Suvorov et al. (2021) using next-generation data (Anchor Hybrid enrichment and Transcriptomic data respectively), suggesting that the relationship Gomphidae + Petaluridae might reflect a relationship in the actual odonata tree of life.
Yet, neither of these two relationships—Gomphidae + Petaluridae and Cavilabiata + Petaluridae—are supported by FcLM analysis. Rather we find support for a third relationship: Petaluridae as sister to Cavilabiata + Gomphidae, a grouping that was also recovered by Letsch et al. (2016). Surprisingly, this relationship is supported by FcLM analysis even in the slow gene datasets.
The relationship among Gomphidae, Petaluridae, and Cavilabiata is conceivably difficult to resolve because of a short internode and/or a rapid radiation or ancestral introgression. Perhaps resolution of this is also confounded by the fact that Gomphidae seem to have much faster substitution rates in the likelihood analyses than nearby groups. Suvorov et al. (2021) suggest incomplete lineage sorting or introgression to be possibly responsible for the low resolution in this group. Perhaps, additional evidence to unravel relationship between Cavilabiata, Gomphidae, and Petaluridae can be found in morphology.
Anisozygoptera, Zygoptera, and some Anisoptera lay their eggs within plant material using an ovipositor, made up of a grouping of structures called gonapophyses (scleritized genital structures; see Matushkina (2011) for review). This process of egg-laying is termed endophytic oviposition. The ovipositor of Zygoptera, Aeshnoidea, and Petaluridae comprises anterior and posterior gonapophyses enclosed by gonoplacs for endophytic oviposition. By contrast, Gomphidae and Libelluloidea (comprising Corduliidae, Libellulidae, Macromiidae, and Synthemistidae) have a reduced or vestigal ovipositor, instead laying their eggs by tapping their abdomens on the surface of water to disperse their eggs (Tillyard, 1917; Corbet, 1999; Carle, 1995). This strategy, called “exophytic oviposition”, takes less time than endophytic oviposition (Corbet, 1999), allows greater egg deposition (∼1,500 eggs per clutch are possible through exophytic oviposition compared to ∼300 eggs in endophytic oviposition), and expands suitable larval niche space to include temporary and transient bodies of water that may not host plant life. In the dragonflies that employ exophytic oviposition, the gonoplacs are vestigial and in some of these taxa the anterior and posterior gonapophyses are also vestigial (see figures in Matushkina (2011)), perhaps suggesting this reduction was shared by the ancestor to those which possess this reduction. However, (Matushkina, 2011) showed that some Libelluloidea retain ovipositor associated muscles and rudiments of the apparatus on the 9th abdominal segment, but that such muscles and rudiments are absent in the Gomphidae she examined. Since a reduction in the ovipositor seems to be different in the two exophytically ovipositing groups, this could suggest independent reductions. All in all, morphological data from the ovipositor system does not clearly support a particular phylogenetic relationship but simply does not lend support for a Gomphidae and Libelluloidea sister group relationship. What does this mean for our understanding of the evolution of egg laying? Our results and the literature seem to show conflicting support for either (1) convergent reduction in the ovipositor in groups Cavilabiata and Gomphoidea (FcLM) or (2) independent reduction in the ovipositor (tree topology, morphology).
Gomphidae is estimated to have diverged in the Cretaceous closer to the K/T boundary while Cavilabiata appear during the late Jurassic. Based on these origin times Cavilabiata and Gomphidae are likely to have experienced different predation pressures (Figure 3). Several predators impose risks to odonates while undertaking oviposition: Birds, fish, and frogs frequently consume individuals that are copulating or ovipositing (Corbet, 1999). Frogs have likely been a strong source of predation pressure for a large portion of odonate history as there are records of early Triassic frogs (Triadobatrachus; Ascarrunz et al. (2016)). Although the oldest crown birds emerged in the Cretaceous (Asteriornis, (Field et al., 2020)) there were likely stem birds that emerged before them (e.g. Archaeopteryx from late Jurassic), hence it is unclear how long they have been a source of predation on odonates. Bony fish have been around far longer, certainly predating the rise of Odonata, and presumably acting as a predation threat over the course of odonate evolution.
Chronological history of Odonata
Odonata are recovered as an ancient lineage, which probably originated during the Carboniferous. The fossil record suggests that during the Pennsylvanian (323–298 My) and the Permian (298–251 My), stem-odonates both diversified and were ubiquitously distributed, including often famously large griffinflies and the more gracile damselfly-like †Archizygoptera (Li et al., 2013; Nel and Huang, 2009; Nel et al., 2012). Our results suggest that crown-Odonata, dragonflies, and damselflies as we recognize them today, diverged from their ancient relatives during the Permian (Figure 4). While some of the observed temporal gaps among clades can be due to our taxon sample of extant species (i.e., due to undersampling early diverging genera in families like Gomphidae), most ages are congruent with conclusions based on the fossil record. Moreover, several temporal gaps compose a genuine documentation of the extinction of once flourishing lineages. Crown-Odonata and crown-Aeshnidae can be regarded as a small subset of the historical diversity of their respective lineages. Other groups, such as Epiophlebiidae and Petaluridae, seem to have remained rare throughout their history. It must be emphasized that the upper boundaries of the observed gaps should not be understood as representing extinction events. A vivid example is the stem-odonatan †Protomyrmeleontoidea, which originated in the Permian and have their latest representative in the Early Cretaceous (Deregnaucourt et al., 2019; Nel et al., 2005), that is, more than 100 My after the origin of crown-Odonata.
The timing of appearance of the main extant lineages of Odonata does not clearly relate to global events which shaped present organismic life. Notably, the group does not seem to have experienced important radiation events or severe replacement of its constituents. A possible exception is a putative mid-Cretaceous diversification of Zygoptera, suggested by both our analysis and the content of Myanmar amber (Zheng and Jarzembowski, 2020), which would then be concomitant with the latest record of the †Protomyrmeleontoidea, which, judging from their wing morphology, must have occupied a flight mode niche similar to that of non-Calopterygidae Zygoptera.
On a more general note, since Odonata, which are generalist top predators, inhabit freshwater areas and are capable of extensive dispersal, their broad niche space might have rendered the group relatively immune to historical global changes. One of the causes of extinction of particular lineages may have been a strong preference for oviposition host-plants which themselves may have declined, such as Sphenophytes, possibly used by griffinflies to host their eggs (Béthoux et al., 2004). Also, given the critical role of flight performances for foraging, predator avoidance, and reproductive success, their obsolescence might have become detrimental for some lineages, but this remains difficult to evaluate yet. Regardless of the assumed historical resilience of odonates, the fast, human-induced rarefaction and homogenization of habitats suitable to them (Sánchez-Bayo and Wyckhuys, 2019) represents a threat that has no ancient counterpart.
Effect of fossil choice and placement
Fossil choice and placement have been shown to influence recovered age estimates (Parham et al., 2012; Inoue et al., 2010; Warnock et al., 2012; Dos Reis and Yang, 2013). Here, we have tested this premise in an ancient group, well represented in the fossil record, for a topology that had several challenging nodes to recover. We expected the inclusion/exclusion of the debated †Triassolestodes asiaticus (Tierney et al., 2020) to have the greatest effect on the node age estimates due to its placement on an ancient node, crown-Epiprocata, on the Odonata tree. However, we find that removal of this fossil neither changes the recovered ages nor does it affect the precision of ages (Figure S4 and Table S4). The only observable differences, albeit small, are seen on the Epiprocata node (where the fossil calibration is placed) and the three surrounding nodes: Anisoptera, Odonata, and Zygoptera. Our results lead to the conclusion that †Triassolestodes asiaticus had a very localized effect. A more global impact of this fossil is perhaps prevented by the fact that all the nodes surrounding it also have fossil calibrations associated with them. Therefore, in a multi-calibration scenario where the surrounding nodes are not calibrated, the impact of †Triassolestodes asiaticus would perhaps be more profound. Our findings, however, do not imply that fossil choice is not pertinent to divergence time estimation. Rather, fossil choice cannot be treated separately from fossil placement as we discuss below.
Unlike the impact of †Triassolestodes asiaticus, a stem placement of †Proterogomphus renateae led to strongly altered and younger ages of nodes throughout the topology (Figure 4). An alarming aspect of the dramatic influence of this fossil is the extremely tight confidence interval recovered for crown-Gomphidae in the “primary” scenario, which should be treated with extreme caution given the contrasting stem placement results. In general, a short CI length has often been thought to imply a more accurate node age. Hence, an extremely small confidence interval, like the one on crown-Gomphidae, could mistakenly be interpreted as an extremely reliable result. However, the only reason we recover such a small CI for this node is because the age on that node is being pushed towards its upper limit with the older fossil calibration on it, while molecular data suggests a younger age on that node. Lastly, as with node ages and CIs, we see dramatic differences in substitution rates on certain branches when †Proterogomphus renateae was included or excluded (Table S4). However, unlike node age and CI, the greatest impact of using †Proterogomphus renateae as a stem calibration was seen for Odonata, which made the substitution rate 24% lower compared with the primary scenario.
The impact of the gomphid calibration underscores the importance of carefully considering taxon sampling during the experimental setup phase rather than sampling species opportunistically, if divergence estimation of a group is an end goal. This is particularly important in highly speciose groups like the insects where it is not always feasible to have sampled every lineage. Our iterative analyses using varying fossil calibration sets strongly highlight the relatively local rather than global impact of fossil calibrations. These analyses underscore the importance of considering stem- and crown-placement of fossils when conducting divergence time estimation analyses.
Limitations of the study
This study uses one of the most comprehensive transcriptomic dataset generated for Odonata till date. However, just like any other study undertaking molecular phylogenetics, we envision that the phylogenetic hypotheses recovered in this study are likely to change upon addition of more taxon and additional transcriptomes. Similarly, conclusions on the timing of evolution of Odonata may also perhaps change upon addition of more taxa and different fossil calibrations.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact: Manpreet Kohli, mkohli@amnh.org.
Materials availability
This study did not generate new reagents.
Experimental models and subject details
No experimental model organism was used in this study.
Method details
Taxa selection and transcriptomics
Note on taxon names
In several NCBI files and the 1KITE wiki page (http://www.1kite.org) some species names may be different compared to the text here and the main paper due to identification updates and corrections. Valid names are found in the main text and Table S1. It concerns the following names: Uropetala carovei was analyzed as Uropetala carovei chiltoni, but we have removed the subspecies name; Libellula fulva was changed to Ladona fulva, correcting the genus name; Sarasaeschna pryeri was changed to reflect the synonymization of Sarasaeschna and Oligoaseshna; Aeshna isosceles was changed to the correct genus Anaciaeschna; the species name was corrected from Gomphaeschna furculata to Gomphaeschna furcillata; the species name for Dorocordulia was confirmed to be lepida; the species Psolodesmus kuroiwae was corrected to Psolodesmus mandarinus kuroiwae; the species Cericion plagiosum was corrected to Paracercion plagiosum; for the species Austroargiolestes icteromelas the family was corrected to Megapodagrionidae; the species Baetis pumilis was not confirmed, so we changed the name to Baetis s.p.; the genus name for Chlorocnemis nigripes was corrected to Allocnemis, the species name of Crocothemis erythraeawas corrected to Crocothemis erythrea; the species name Diplacodes lefebvrei was corrected to Diplacodes lefebvrii; the species Cordulephya pygmaea was corrected to Cordulephya pygmea.
Specimen collection, sequencing, and taxon sampling
Specimens were either hand-collected or collected with insect nets. Upon capture, alive specimens were immediately immersed and ground in RNAlater (Qiagen, Hilden, Germany) and then stored at 4°C until further processing. Detailed information (e.g., sex, developmental stage, collection site) are provided in Table S1. Whenever possible, we preserved a voucher specimen (collected from the same collection locality, same morphotype as the sample used for transcriptome sequencing) in >95% ethanol, which are deposited in the Biobank at the Zoological Research Museum Alexander Koenig (Bonn, Germany). The extraction of RNA, generation of cDNA libraries, transcriptome sequencing and de novo assembly of transcriptome data were conducted at the Beijing Genomics Institute (BGI) Shenzhen following the procedure described in Misof et al. (2014) and Peters et al. (2017). Transcriptome sequencing for 102 odonate species and two mayfly species (Ephemeroptera) was performed on an Illumina HiSeq 2000platform: Except for seven samples, paired-end sequencing was done with a read-length of 150 base pairs (Illumina, San Diego, CA, USA, generating ca. 2.5 Gigabases for each sample (Table S1). For seven samples we used the TruSeq mRNA library Pre Kit (Illumina, Ca. USA) due to limited RNA inputs (<1 μg) which were sequenced with 90 bp PE reads (Table S1). We assembled raw reads using the software tool SOAPdenovo-Trans-31kmer (Xi et al., 2015) as described in Peters et al. (2017). Quality assessment, including check and removal of contaminants of the de novo assembled transcriptomes, as well as submission to the NCBI Sequence Read Archive (SRA) and the Transcriptome Shotgun Assembly (TSA) database were conducted as described in Peters et al. (2017). For details on the number of contigs before and after contamination check see Table S1. Transcriptome data published with this study are deposited in GenBank NCBI under the 1KITE umbrella project (Table S1). We further complemented our data by including three odonate species as well as two mayflies, three Zygentoma species, and two bristle tails (Archaeognatha) that had been previously published Misof et al. (2014) using the current assembly version (Pauli et al., 2016). In our transcriptome dataset, we also used nine outgroup species (Ephemeroptera, Zygentoma and Archaeognatha). The ortholog set from genome data of Ladona fulva and Ephemera danica (see section below) were additionally included in the final dataset (Assembly GCA_000376725.1 downloaded from ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/376/725/GCA_000376725.1_Lful_1.0/, and Assembly GCA_000507165.1 downloaded from ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/507/165/GCA_000507165.1_Edan_1.0/ using the portal at https://i5k.nal.usda.gov/content/data-downloads).
Orthology assignment
We compiled an ortholog set comprising 2,986 clusters of single-copy protein encoding sequences (ortholog groups, OGs) using the OrthoDB v8 database (Kriventseva et al., 2015). Setting the hierarchical level to “Arthropoda”, we included the official gene sets (OGS) of four species with well sequenced and annotated genomes species as reference species in the ortholog set: Daphnia pulex OGS v1.22 (Colbourne et al., 2011), Zootermopsis nevadensis OGS v2.2 (Terrapon et al., 2014), Ephemera danica OGS v0.5.3 (i5K-Consortium, 2013), Ladona fulva OGS v0.5.3 (i5K-Consortium, 2013). The OGS were screened for the occurrence of isoforms (and alternative transcripts on the coding sequence level), and we only kept one isoform per OG with helper scripts from the Orthograph package. Selenocysteine (“U”) was replaced by “X” to avoid problems in downstream analyses. A list of 2,986 OGs and the OGS on protein coding and CDS level were used as input to search for contigs of putative single-copy orthologous genes in our newly sequenced and previously published transcriptome assemblies.
We used the software package Orthograph v0.5.7 (Petersen et al., 2017) to search putative single copy OGs in our transcriptomes with the following dependencies: BLAST + v2.2.28 (Camacho et al., 2009), Exonerate v2.2 (Slater and Birney, 2005), and MAFFT v7.123 (Katoh and Standley, 2014). We applied on putative ortholog transcripts the reciprocal best hit search in relaxed mode: we considered the best-reciprocal hit (BRH) criterion as fulfilled if the first reciprocal hit matched either Ladona fulva or Ephemera danica. The maximum number of candidate hits to consider and the maximum number of reciprocal best hits were both set to 50. We further allowed the open reading frame (ORF) to be extended while the orthologous region was retained with a default overlap of 50%. For all other Orthograph parameters default settings were applied. When summarizing Orthograph results, terminal stop codons were removed and internal stop codons (∗) and selenocysteine (U) were replaced by X and NNN, respectively, in all amino acid and corresponding nucleotide sequences. In total, 2,980 of the 2,986 target genes were represented in the 116 transcriptomes. Note that for two species (Cordulephya pygmea and Tyriobapta torrida) we had sequenced two samples, but kept, after orthology assignment, only the samples with the higher number of orthologous hits which left transcriptome data of altogether 114 species.
Multiple sequence alignments
We constructed multiple sequence alignments (MSAs) on the translational level with MAFFT, using the L-INSi algorithm. Resulting alignments were checked for misaligned amino acid sequences (hereafter referred to as outliers) as outlined by Misof et al. (2014). Outliers spread across 464 OGs and were subsequently refined using a profile alignment approach as described in Misof et al. (2014). Alignment refinement of the 1,897 outlier sequences succeeded in 1,469 instances (i.e., sequences). The remaining 428 sequences, referring to a total of 251 OGs, remained outliers and were consequently removed from the MSAs. Furthermore, we removed all sequences for the reference species Daphnia pulex and Zootermopsis nevadensis as they were used only for orthology prediction, and not as outgroups; instead we used mayflies and basal hexapods as our outgroups (see above). Corresponding nucleotide sequences were removed from the nucleotide sequence files as well. We then inferred the MSA of each ortholog group on the transcriptional (nucleotide) level corresponding to the amino acid MSAs using a modified version (see Misof et al. (2014) [S2] for details on the software modification) of the software Pal2Nal v14 (Suyama et al., 2006) using the amino acid MSAs as a blueprint. Finally, we discarded all gap-only sites identified on the amino acid level from amino acid and nucleotide MSAs.
Protein domain identification, alignment masking, optimizing datasets
For the phylogenetic analyses we used a domain-based partitioning scheme to improve the biological realism of evolutionary models. It has been argued in Misof et al. (2014) that protein domains rather than genes are the evolutionary units that should be used as data blocks in partitioned phylogenetic analyses. Therefore, in our manuscript we focus preliminarily on the data sets partitioned according to protein domains. Following the strategy outlined in Misof et al. (2014), we used the Domain-identification-v1.3 and Domain-parser-v1.4.1 programs, which in turn invoked the software PFAM_SCAN.PL v1.5, HMMSCAN from the HMMER to identify protein clans, families and single domains as well as non-annotated regions (so called voids) in each gene MSA on the amino acid level. In this way, we identified Pfam-A domains with clan annotation, and Pfam-A domains without clan annotation. Pfam-A domains with clan annotation were pooled into 263 data blocks. Pfam-A domains belonging to the same clan and Pfam-A domains without clan annotation but with the same name were pooled into Pfam-A data block domains (820 data blocks). The void regions were pooled for each gene into 1807 data blocks.
Parallel to the protein domain identification on amino acid and nucleotide level, we used a modified version of Aliscore v1.2 (Misof and Misof, 2009) to identify sections of putative alignment ambiguities in the amino acid MSAs of each OG. Aliscore v1.2 was applied using the default sliding window size, the option –r 1∗1027 specifying the maximum number of pairwise sequence comparisons, and option –e (i. e., indicating gappy amino acid sequence data, see Meusemann et al. (Meusemann et al., 2010)). Corresponding lists for nucleotide MSAs were generated with custom-made Perl scripts (see supplementary material from Misof et al. (2014)). Blocks of putative alignment ambiguities or alignment sections indistinguishable from randomized data were subsequently removed from each data block at the amino acid and nucleotide level using custom-made Perl-scripts. Trailing ends (gap symbols) at the beginning and at the end of each resulting MSAs were replaced with 'X's (translational level) and/or 'NNN's (transcriptional level), respectively. OG MSAs were finally concatenated into supermatrices. Additional details on this procedure are described in Peters et al. (2017).
Dataset diagnostics and dataset optimization
We assessed the putative signal of each data block at the translational level with the software MARE v0.1.2-rc (Meyer and Misof, 2011) using default settings. To account and reduce the amount of data that violates stationary, (time) reversible and homogenous (SRH) conditions in the nucleotide dataset with all three codon positions, we generated a second nucleotide supermatrix, but only with the second codon positions (115 taxa, 824,783 aligned sites). We tested whether the evolution of our datasets was under globally stationary, reversible and homogeneous (SRH) conditions, which includes a test for possible compositional heterogeneity of amino acids using SymTest v2.0.47 (https://github.com/ottmi/symtest) on all three supermatrices (“amino acid = AA”, “nucleotide = NT123” and “nucleotide second codon positions only = NT2” datasets). The analysis in SymTest used matched-pairs tests of homogeneity as in Misof et al. (2014). Heatmaps generated from these tests (based on p values of Bowker’s matched-pairs test of symmetry) were used to determine which sequence pairs matched SRH conditions. We further applied Alistat v1.6 (Wong et al., 2020) without partition information and defaults on all three supermatrices. The overall completeness score for the alignment (Ca), the minimum C-score for individual sequences (Cr_min), and the maximum C-score for individual sequences (Cr_max) are shown in Table S2.
Selecting optimal partition schemes and model selection
We selected the most appropriate number of partitions (i.e., merged data blocks) with PartitionFinder v2.0.0 (Lanfear et al., 2014, 2016) in combination with the provided RaxML version. For the SOS amino acid supermatrix AA (2890 initial data blocks, 115 taxa, 824,783 amino acid positions) we used the following rcluster algorithm: --rcluster --rcluster-max 11,560 --rcluster-percent 100 --weights 1,1,0,1 -q -p 20 --all-states --min-subset-size 100. We used a maximum likelihood start tree and had branch lengths linked. We restricted models due to computational restrictions to the following options: LG + G (Yang and Temin, 1994; Le and Gascuel, 2008; Gu et al., 1995; Soubrier et al., 2012), WAG + G (Whelan and Goldman, 2001), DCMUT + G (Kosiol and Goldman, 2005), JTT + G (Jones et al., 1992), BLOSUM62 + G (Henikoff and Henikoff, 1992), LG + G + F, WAG + G + F, DCMUT + G + F, JTT + G + F, BLOSUM62 + G + F, LG4X (Le et al., 2012). This resulted in 1217 merged data blocks, i. e. partitions. For both nucleotide supermatrices, the one with only the second codon positions and the one including all three codon positions, we applied the k-means algorithm (Frandsen et al., 2015) that starts with a single block to find an optimal partition scheme. We restricted the models to GTR only (GTR, GTR + G, GTR + I + G) due to a restriction of RaxML v.8.2.8 (Stamatakis, 2014) and otherwise chose again the AICc as selecting criterion of the best schemes and the best fitting model. (Note that k-means is not available anymore in current PartitionFinder versions). While the dataset with 2nd codon positions only resulted in 123 partitions, the dataset with all codon positions resulted in 704 partitions. We consider the AA dataset as the main data set which is used also for the FCLM, MSC and divergence time estimates, see below.
Rogue taxon analyses
We tested all three datasets for rogue taxa using all inferred bootstrap trees with RogueNaRok v1.0 (Aberer et al., 2013) providing the best inferred ML tree and otherwise using default settings.
Maximum likelihood tree inference and statistical support
Phylogenetic relationships were inferred for all three supermatrices under the maximum likelihood (ML) optimality criterion as implemented in IQ-TREE v1.5.1 (Nguyen et al., 2015; Chernomor et al., 2016) using the best partition scheme and selected models. We applied the edge-proportional partition model allowing different evolutionary rates across partitions (option –ssp). We performed 50 independent tree searches with random starting trees, using the median approximation for Gamma rate heterogeneity (option –gmedian) (i.e., 50 independent IQ-Tree jobs). We ran this with an increased number of unsuccessful iterations before stopping (–numstop 300), and otherwise used the default settings. The resulting number of unique tree topologies was assessed with Unique Tree (v.1.9; provided by Thomas Wong, available upon request). For each of the three datasets, the tree topology of the 50 ML trees varied only in the arrangement of a few species; all familial, super-familial and sub-ordinal relationships were identical. Statistical support was estimated using a non-parametric bootstrap analysis with 100 replicates for each dataset; the settings for the bootstrap analyses are as described above. We ensured bootstrap convergence with the a posteriori bootstrap criterion (Pattengale et al., 2010) as implemented in RaxML (options:”-I autoMRE”, -B 0.01, --bootstop-perms = 10,000, performing the test 10 times with different random seeds). Bootstrap convergence was met for all three datasets in all analyses. Bootstrap support was mapped onto the ML tree with the best log likelihood. Full ML tree for AA dataset is shown in Figure 1 and for NT2 and NT123 datasets are shown in Figure S1. Additionally, we calculated an alternative support measure described by Guindon et al. (2010) with 10,000 replicates for each dataset and as above, we mapped the support onto the best inferred ML tree for each dataset (Figures 1 and S1).
Multispecies coalescence
Apart from using the supermatrix approach using protein domain partitions we conducted gene tree/species tree estimation to investigate the evolutionary history of our group. We were primarily interested in examining if and how incomplete lineage sorting, and different gene histories might affect our conclusions. In a supermatrix approach, even though each partition has its own rate of evolution, partitions are linked by the same tree topology. Multispecies coalescence methods treat each gene as a separate evolutionary unit and allow each of these to evolve under a different topology. Such methods can provide a more robust view of the evolutionary history of the target group and help identify instances of incomplete lineage sorting.
As the MSC methodology entails, we conducted our analysis using genes as partitions, unlike our supermatrix approach which was conducted using the protein domains. Data preparation for the genes was similar to as described above except that the alignments prepared in MAFFT were partitioned into genes rather than protein domains after using Aliscore and Alicut. We used the software package MARE v0.1.2-rc to identify informative subsets of genes and taxa. We conducted MARE analysis using default settings except taxa sets were restricted using the option -c to match the final taxa set to that of the supermatrix topology. Using this approach, we obtained a total number of 2413 gene partitions (reduced from 116 taxa and 2980 partitions). Our MSC analysis was only conducted for the AA dataset, the main dataset used for results and conclusions in our manuscript. The final reduced alignment contained 115 taxa and 871,847 amino acid characters.
We estimated gene trees for each of the gene loci using the software package IQTREE v2.1.2 (multicore version for Linux). Evolutionary model for each of the gene fragment was chosen using ModelFinder as implemented in IQTREE v2.1.2 by conducting a full IQTREE tree reconstruction analysis on the gene partitioned dataset and extracting the model information from the log file. IQTREE crashed on one gene dataset (EOG82FVZC), leaving us with 2412 gene trees in the MSC analyses. We then estimated a species tree, using the multispecies coalescent method in software package ASTRAL III v5.15.2 (Zhang et al., 2018). We choose this method since it was shown to be statistically consistent under instances of incomplete lineage sorting (Mirarab et al., 2014). The recovered MSC tree is shown in Figure S2. We tested the robustness of our species tree by collapsing branches in gene trees that had less than 10%, 20%, 30%, 40% and 50% bootstrap support.
Lastly, we also investigated the effect of rate of evolution of the genes on the recovered species tree. Genes were categorized as fastest or slowest genes based on their speed recovered in the full IQTREE analysis used to determine models for individual genes. More precisely, the relative “speeds” printed to the iqtree file of the full analysis were used to subdivide the set of genes into speed classes: slowest 25%, 50%, and 75% containing respectively 603, 1206, 1809 genes, and the fastest 50% and 75% of the genes containing 1206 and 1809 genes. Species trees were then reconstructed in ASTRAL III v5.15.2 for all five subsets of corresponding gene trees. Relative rates (“speed” values) as determined by IQTREE differed considerably among the genes. In the four quartiles of the genes sorted by their relative rates, the rates ranged from 0.018–0.469 (slowest 25%), 0.471–0.865 (second speed quartile), 0.865–1.427 (third speed quartile), 1.429–6.180 (fastest 25%), showing that the fastest gene evolved about 343 times faster than the slowest gene.
Gene and site concordance factor
We estimated the gene and site concordance factors for our supermatrix and species coalescent tree using the software package IQTREE v2.0-rc2. We used the option --gcf and –scf 100 to calculate GCF and the SCF values for our trees. These were estimated only for trees generated using the AA dataset, including the MSC tree. Figure S2 shows the results of the concordance factor analysis.
Hypothesis testing with four-cluster likelihood mapping (FcLM) for alternative and putatively confounding signal
We assessed the support for specific phylogenetic relationships within Odonata using the four-cluster likelihood mapping (FcLM) method (Strimmer and von Haeseler, 1997), as implemented in IQ-Tree v1.6.1x. We tested altogether five different relationships which are described below. For each hypothesis, we defined four groups of interest (three monophyletic taxa plus an outgroup). For each test, we generated specific datasets for which only those partitions were retained, for which at least one representative species was present. For all possible quartets, the log likelihood scores for the three possible topologies were calculated and depicted in a 2D-simplex graph. To receive sufficient coverage for each hypothesis, all possible quartets were considered. Permutation tests were used to infer the potential impact of confounding signal. The datasets were permuted in three ways as in Misof et al. (2014), i.e., by either (1) destroying phylogenetic signal but keeping the among-lineage heterogeneity originating from amino acid frequencies and the distribution of missing data; by (2) destroying phylogenetic signal, making the dataset homogeneous among lineages by drawing amino acids randomly from the LG model frequencies, but keeping the distribution of missing data; or by (3) modifying (2) to also permute the positions of missing data. For a more detailed description of this approach, and a rationale, see Misof et al. (2014). Defined groups, the number of partitions, amino acid sites and the number of amino acid sites are listed below for each hypothesis. Lastly, hypothesis 1 was also tested with slow and fast gene partitions as well (described in section “Multispecies Coalescence”).
For all FcLM analyses the following topologies and groups were defined:
T1: G1, G2 – G3, G4 (relationship recovered in the ML tree reconstruction)
T2: G1, G3 – G2, G4
T3: G1, G4 – G2, G3
Hypothesis 1: Sister group relationship between Petaluridae and Gomphidae
Data set: 1209 partitions, 822214 aa sites
Unique quartets: 9504
Groups: G1: Petaluridae (4 Taxa) G2: Gomphidae (8 Taxa), G3 Cavilabiata (41 Taxa), G4 Aeshnidae + Epiophlebia (11 Taxa)
See Table S3 for the results
Hypothesis 2: Sister group relationship between Cordulegastroidea and Libelluloidea
Data set: 1212 partitions, 823385 aa sites
Unique quartets: 10890
Groups: G1: Petaluridae + Gomphidae (12 Taxa) G2: Aeshnidae (10 Taxa) G3: Cordulegastroidea (i.e., Cordulegastridae + Chlorogomphidae, 3 Taxa), G3 Libelluloidea (37 Taxa)
See Table S3 for the results.
Hypothesis 3: Aeshnidae is sister group to all remaining Anisoptera
Data set: 1203 partitions, 820607 aa sites
Unique quartets: 3960
Groups: G1: Epiophlebia (1 Taxa), G2: Aeshnidae (10 Taxa), G3: Petaluridae + Gomphidae (12 Taxa), G4: Cavilabiata (41 Taxa)
See Table S3 for the results.
Hypothesis 4: Corduliidae is sister group to Libellulidae
Data set: 1203 partitions, 820907 aa sites
Unique quartets : 1750
Groups: G1: Libellulidae (25 Taxa), G2: Corduliidae (5 Taxa), G3 Macromiidae (3 taxa), G4: remaining Cavilabiata (5 Taxa)
See Table S3 for the results.
Hypothesis 5: Status of Synthemistidae s.l. (sensu Letsch et al., 2016, i.e., GSI-Complex sensu Ware et al., 2007)
Data set: 1077 partitions, 774,160 aa sites
Unique quartets : 93
Groups: G1: Eusynthemis, G2: Cordulephya, G3: MCL-Complex (33 Taxa), G4: Cordulegastroidea (3 Taxa)
See Table S3 for the results.
Fossil selection and divergence time estimation
Fossil selection
The fossil record of crown Odonata consists of 828 species distributed across 517 genera (http://www.palaeodb.org; including both compression and amber fossils). Analysis of odonate fossils relies heavily on wing venation, as compression fossils rarely preserve details of genitalic, thoracic, penile or larval traits, while wing venation is usually well-preserved. Additionally, odonate wing venation provides a plethora of characters, owing to its rich and complex evolutionary history (Fraser and Tillyard, 1957). Indeed, most previous morphological studies have relied heavily on wing venation, yet often acknowledging the occurrence of homoplasies (e.g., Fraser and Tillyard, 1957; Gloyd, 1959; Geijskes, 1970). Many wing venation characters may support convergent relationships and should therefore be used in conjunction with others (Hennig, 1981; Fleck et al., 2008). Kohli et al. (2016) published a list of appropriate fossil calibrations for Odonata, as part of the Fossil Calibration Database (https://fossilcalibrations.org/). Thus, we used Kohli et al. (2016) as a starting point for our fossil selection. This paper describes a curated list of ten odonate fossils, with extensive phylogenetic and age justification to be used for divergence time estimation. It primarily focuses on Anisoptera and suggests fossil calibrations for the crown nodes of Odonata, Zygoptera, Epiprocta, Anisoptera, Aeshnidae, Gomphidae, Cavilabiata, Macromiidae, Cordullidae and Libellulidae. In the current study, we established a set of additional fossils to extend our fossil sampling to cover several other branches or clades, including splits in Zygoptera. In total we used 17 fossils as calibration for our molecular clock analysis. Phylogenetic placement and age for these fossils can be found in Table S4 and Figure S3. Phylogenetic and age justification for the fossils chosen in this study can be found below. Lastly, since publication of Kohli et al. (2016) ages for the fossil species †Triassolestodes asiaticus, †Mersituria ludmilae, †Sinacymatophlebia mongolica, †Gomphaeschna inferna and †Juralibellulaningchengensis have changed due to stratigraphic age updates of several fossil beds. Minimum age for all fossils considered by Kohli et al. (2016) were updated according to recent data (Wolfe et al., 2016; and reference therein). We conducted all our divergence time analysis using these updated ages (Table S4).
Divergence times estimation analysis
A recent study (Tierney et al., 2020) suggests that †Triassolestodes asiaticus is perhaps not an appropriate fossil for dating the crown Odonata node (see section “Updated Kohli et al. selection”). Additionally, we also determined that perhaps †Proterogomphus renateae is more appropriately placed at the stem Gomphidae node rather than Crown Gomphidae node based on our current taxon sampling (see section “Updated Kohli et al. selection”). Since fossil choice and fossil placement are known to heavily influence the outcome of a divergence time analysis, we decided to test the effect of †Triassolestodes asiaticus and †Proterogomphus renateae on our divergence time analysis. Therefore, we ran divergence time estimation under three different scenarios. In the first scenario, designated as “Primary” Scenario, we used all 17 fossils. In the second scenario, designated as “Minus Triassolestidae” scenario, we tested the effect of fossil choice. We ran this scenario with all the fossils minus †Triassolestodes asiaticus and hence in this scenario a total of 16 fossils were used for the time calibration of nodes. In the third scenario, designated as “Stem placement” scenario we tested the effect of fossil placement. We used all the 17 fossils as in the first scenario, however, we placed †Proterogomphus renateae on the stem Gomphidae node (or crown Gomphidae + Petaluridae node) instead of crown Gomphidae like in the “Primary” Scenario.
All divergence time analyses were conducted on the amino acid data set in MCMCtree as implemented in the software package PAML, version 4.9 g (Yang, 2007) with the inferred ML tree (which is also the same topology recovered in the MSC analysis using slowest 25% gene dataset) provided as fixed input topology. Due to computational restrictions, we generated a modified version of the amino acid dataset by condensing the amino acid supermatrix so that it contained only genes that were found across all included species. Furthermore, we applied AliStat on this condensed data set to retain only those positions, which were present in at least 99 % of the species. The resulting supermatrix comprised 53,091 amino acid positions. For all dating analyses, we used this reduced and unpartitioned amino acid data set. Fossils were specified as point calibrations, using uniform distributions with hard upper and lower bounds (Table S4). The root age of the tree was set to 484.5 million years, based on the age of the earliest putative aquatic-terrestrial transition trackways (Macnaughton et al., 2002; Dunlop et al., 2013). All parameters for defining prior distribution, including the offset (p) and scaling (c) parameters were set to default. We used the JTT + I + G4 amino acid substitution model for calculating the Hessian matrix for our dataset. For each of the scenarios, we performed two independent MCMC runs (chains) with the independent rates clock model and uniform prior distributions. We examined the joint prior distributions to ensure reasonable fossil choices and placement on the tree (see Warnock et al., 2012 for a discussion of the effects of prior distributions in dating analyses). Using the software tool Tracer v1.6 (Rambaut et al., 2018), we ensured an effective sample size (ESS) of >200 for each run and checked for convergence through pairwise comparison of the posteriors. Divergence time estimates from all the three scenarios are shown in Table S4 (See also Figures 4 and S4).
Fossil validation
We surveyed several fossils for use as calibrations to extend our sampling beyond Kohli et al. (2016). In general, as in Misof et al. (2014), we used the five principles set forth by Parham et al. (2012) and Ksepka et al. (2015), as best fossil calibration practices; criteria are listed below as C1 (accession number for fossil), C2 (apomorphy-based or phylogenetic analysis), C3 (agreement of morphology and molecular phylogenetic data), C4 (detailed locality and stratigraphy data for fossil provided), and C5 (radioisotopic age or numeric age references for fossil given). Further, these selections were conducted using a rationale previously elaborated by Evangelista et al. (2019). In short, a fossil is selected if (a) it can be assigned to a particular node (‘ultimate level’) on the basis of a strict apomorphy (‘Class 1’ character state), or if (b) it can be assigned to a particular node (e.g. dolphin) on the basis of a homoplastic state (‘Class 2’; e.g. dorsal fin), but which, within a context is supported by a strict apomorphy (e.g. placenta), such that it becomes itself a strict apomorphy (e.g. dolphins). A fossil can be assigned to the node dolphin on the basis of a homoplastic characters (or Class 2 character), like dorsal fin. That is because within Placentalia, possessing a dorsal fin indicates a dolphin, but without context, ‘dorsal fin’ is not a decisive character.) Two exceptions to this rationale are Sinocalopteryx shangyongensis and Palaeolibellula zherikhini, placement for which is supported by a sequence of homoplastic character states only.
Crown-Ephemeroptera
Stem-[Eurylophella sp. + Ephemera danica]/crown-[(Eurylophella sp. + Ephemera danica) + (Epeorus assimilis + Ecdyonurus insignis)]
Fossil: Australiphemera revelataMcCafferty, 1990
Original description:Mccafferty (1990).
Further descriptive accounts: None.
Locality: Crato (112.6 Ma; see localities ages section).
CR1 (single/multiple OTUs with museum numbers): Yes (see original description).
CR2 (apomorphy-based or phylogenetic analysis): See below.
CR3 (agreement of morphology and molecular data): See below.
CR4 (detailed locality and stratigraphy data provided): Yes (original description).
CR5 (radioisotopic age or numeric age references given): Yes (see localities section).
Phylogenetic justification& remarks:
The placement of Australiphemera revelata as stem- [Eurylophella sp. + Ephemera danica] / crown-[(Eurylophellasp. + Ephemera danica) + (Epeorus assimilis + Ecdyonurus insignis)] is based on the following character states:
Ultimate level:
Class 1: (1) in fore-wing proximal part, MP2 diverging obliquely from MP, approaching CuA before diverging from it; concurrently, CuA approaching CuP before diverging from it.
Class 2: none found/considered.
Contextual level:
Class 1: none found/considered.
Class 2: none found/considered.
The character state (1), very conspicuous, is listed by (Kluge, 2004) as apomorphic of the taxon Fimbriatotergaliae, including the selected fossil species and the genera Eurylophella and Ephemera. The Fimbriatotergaliae is recovered monophyletic in our analysis. It must be mentioned that two other species from the same locality and also described by Mccafferty (1990), namely Microphemera neotropica and Pristiplocia rupestris, are equally suitable for calibration of the corresponding node.
Stem-Odonata/crown-(Odonata + Ephemeroptera)
Preliminary remarks: Stem representatives of both Odonata and Ephemeroptera are relevant to calibrate the corresponding node. A stem-Odonata is favored because all putative stem-Ephemeroptera are younger. Moreover, the actual systematic position of the earliest putative stem-Ephemeroptera is not well ascertained.
Fossil: Erasipteroides valentiniBrauckmann, Koch & Kemper, 1985
Original description:Brauckmann et al. (1985).
Further descriptive accounts:Bechly et al. (2001) redescribed the holotype of the species and described an additional specimen, they regarded as probably belonging to Erasipteroides valentini; this specimen was further positively assigned to the species (Brauckmann and Herd, 2002).
Locality: Hagen-Vorhalle (319.9 My; see localities ages section).
CR1 (single/multiple OTUs with museum numbers): Yes (see original description further descriptive accounts).
CR2 (apomorphy-based or phylogenetic analysis): Yes (see below).
CR3 (agreement of morphology and molecular data): Yes (see below).
CR4 (detailed locality and stratigraphy data provided): Yes (see further descriptive account).
CR5 (radioisotopic age or numeric age references given): Yes (see localities ages section).
Phylogenetic justification & remarks:
The placement of Erasipteroides valentini as a stem-Odonata/crown-(Odonata + Ephemeroptera) is based on the following character states:
Ultimate level:
Class 1: (1) in both wing pairs, slightly distal of wing base, CuP fused with AA for some distance, and CuA fused with CuP (or CuP + AA) for some distance; (2) in both wing pairs, fusion of MP with Cu near or at the wing base.
Class 2: none found/considered.
Contextual level:
Class 1: (2) none found/considered.
Class 2: (3) none found/considered.
In the Hagen-Vorhalle locality, stem-Odonata are represented by several species in addition to Erasipteroides valentini, including Zessinellasiope and Namurotypussippeli (Brauckmann et al., 1985, Brauckmann et al., 2003; Brauckmann, 1988; Brauckmann and Zessin, 1989; Bechly et al., 2001). All of them display the character states whose relevance is discussed below.
The wing venation of total-Odonata remained debated until Riek and Kukalová-Peck (1984) described the Eugeropteridae, in which vein fusions that are not immediately patent in crown-Odonata and their closer stem-relatives could be clearly evidenced. Note, however, that a debate remains on whether the Media (Riek and Kukalová-Peck, 1984) or the Radius (see Béthoux, 2015: Fig. 8) forms a single stem at the wing base of Eugeropteridae. This does not affect the relevance of the character states allowing the placement of Erasipteroides valentini as stem-Odonata.
All known total-Odonata possess, slightly distal to the wing base, a sharp inflexion of CuA and CuP (the ‘kink’ of Riek and Kukalová-Peck (1984)) when they approximate each other, and AA. In Eugeropteridae these veins do not fuse together; instead, they are connected by a series of aligned cross-veins. In all known total-Odonata except the Eugeropteridae these cross-veins are captured by AA and CuP. As a result, CuP fuses with AA, and CuA with CuP (or CuP + AA, if AA remains fused with CuP for some distance). This organization is herein regarded as representing a single character state. It is unique to total-Odonata but Eugeropteridae (notably, it is absent in Ephemeroptera (notably, it is absent in Ephemeroptera Béthoux, 2015)).
A further relevant character state is the fusion of MP with Cu, near or at the wing base (2). This fusion is not present in Eugeropteridae. Indeed, its occurrence is evidence by the wing morphology of Erasipteroides valentini and of related species, in which MP is substantiated by a short, free portion, parallel to Cu, visible near the wing base before it fuses with Cu (Nel and Huang, 2009; Li et al., 2013). The occurrence of a full fusion (i.e., without basal free portion of MP) in extant species (Riek and Kukalová-Peck, 1984) is well-evidenced by the elevation of the respective veins in their distal portion and is largely admitted. This character is unique to total-Odonata but Eugeropteridae (it is absent in Ephemeroptera; Béthoux, 2015). To our knowledge, the affinities of Erasipteroides valentini and of related and sub-contemporaneous species with crown-Odonata have never been challenged, even under topological homology conjectures elaborated prior to Riek and Kukalová-Peck (1984) essential account.
Crown-Zygoptera
Sister-group of Sapho gloriosa/crown-[Sapho gloriosa + [(Calopteryx splendens + (Mnais pruinosa + Psolodesmus kuroiwae)) + (Vestalis amoena + Vestalis amethystina]]
Fossil: ‘Sapho’ sannoisiensisMeunier, 1914
Original description:Meunier (1914).
Further descriptive accounts:Nel and Papazian (1992) re-described the material of this species and discussed its phylogenetic affinities.
Locality: Aix-en-Provence (23.0 My; see localities ages section).
CR1 (single/multiple OTUs with museum numbers): Yes (see further descriptive account).
CR2 (apomorphy-based or phylogenetic analysis): Yes (see below).
CR3 (agreement of morphology and molecular data): Yes (see below).
CR4 (detailed locality and stratigraphy data provided): Yes (see further descriptive account).
CR5 (radioisotopic age or numeric age references given): Yes (see localities ages section).
Phylogenetic justification & remarks:
The placement of ‘Sapho’ sannoisiensis as a close relative of Sapho spp. (and of Umma spp.) is based on the following character states:
Ultimate level:
Class 1: (1) in both wing pairs, area between CuP + AA and the posterior wing margin with a secondary vein branching at the level of the quadrangle.
Class 2: none found/considered.
Contextual level:
Class 1: (2) in both wing pairs, between the arculus and the nodus, RP1+2 + IR2 fused with RA.
Class 2: (3) in both wing pairs, between the arculus and the nodus, RP1+2 + IR2 directed towards RA (can be considered a state of previous character); (4) in both wing pairs, posterior wing margin without sharp angle near opposite to, or basal to, the quadrangle (i.e., petiole absent); (4) discoidal cell crossed by more than 2 cross-veins.
The character state (1) is unique to Sapho spp.,Umma spp. and Matrona spp. (Bridges, 1994; Hämäläinen et al., 2011 among others). As pointed out by Nel and Papazian (1992) ‘Sapho’ sannoisiensis can be assigned to any of these two genera. Several additional character states unambiguously indicate that the fossil is a Calopterygidae, some of which are listed above. Given our species sample, it is placed as sister-group to Sapho gloriosa.
Stem-Calopterygidae / crown-(Calopterygidae + Chlorocyphidae)
Fossil: SinocalopteryxshangyongensisLin, Petrulevičius, Huang, Nel & Engel, 2010
Original description:Lin et al. (2010).
Further descriptive accounts: None.
Locality: Shangyong (47.8 My; see localities ages section).
CR1 (single/multiple OTUs with museum numbers): Yes (see further descriptive account).
CR2 (apomorphy-based or phylogenetic analysis): Yes (see below).
CR3 (agreement of morphology and molecular data): Yes (see below).
CR4 (detailed locality and stratigraphy data provided): Yes (see original description).
CR5 (radioisotopic age or numeric age references given): Yes (see localities ages section).
Phylogenetic justification & remarks:
Note that the holotype of the species is a fore-wing base. Another specimen preserving the median and apical parts of a hindwing (only the median part is documented by a drawing) was also recovered and considered conspecific with the holotype Lin et al. (2010). This is very likely.
The placement of Sinocalopteryx shangyongensis as a stem-Calopterygidae/crown-(Calopterygidae + Chlorocyphidae) is based on a series of character states which distribution, as indicated by the obtained molecular-based topology, indicates some degree of homoplasy with respect to other families and within Calopterygidae. However, the placement of Sinocalopteryx shangyongensis at any other node would generate additional homoplasies. These states are:
Ultimate level
Class 1: none found/considered.
Class 2: (1) pterostigma absent; (2) in fore-wing, posterior wing margin without sharp angle near opposite to, or basal to, the quadrangle (i.e., petiole absent).
Contextual level
Class 1: none found/considered.
Class 2: (3) discoidal cell very long; (4) discoidal cell crossed by more than 3 cross-veins;
A very long discoidal cell (3) occurs in Calopterygidae, Polythoridae and some Pseudostigmatidae. The occurrence of more than 3 cross-veins in this cell (4), a derived condition according to the obtained topology, excludes the possibility of affinities with the latter family. Note that the corresponding cells, in Anisoptera (i.e., the triangle and hypertriangle) are crossed by numerous cross-veins in Aeshnidae, Chlorogomphidae, and some large Libellulidae. Note also that the presence of numerous cross-veins in the discoidal cell (or triangle and hypertriangle) can co-occur with the presence of numerous cross-veins in the area between R + MA and MP + Cu, basal to the arculus (Calopterygidae, Polythoridae, Chlorogomphidae, some Aeshnidae, and the fossil Aeshnidiidae). These two conditions might therefore represent states of a single character.
In this context, the lack of pterostigma (1) indicates affinities with the Calopterydidae, even though this trait is not present in all members of the family. Notably, several species of Hetaerina spp. possess a pterostigma. The lack of petiole (2) further ascertains the placement of Sinocalopteryxshangyongensis as a stem- or crown-Calopterygidae.
The lack of the curvature of RP1+2 (+ IR2) towards RA (and the fusion of RP1+2 + IR2 with RA) challenges this statement. The trait is present in most Calopterygidae (most of which additionally possess a fusion of RP1+2 + IR2 with RA, absent in Sinocalopteryx shangyongensis). It was regarded as a possible reversal (Lin et al., 2010). This is plausible, given that in the calopterygid Matrona spp. the curvature can be moderate (Hämäläinen et al., 2011).
A character state of possible importance, but whose occurrence is difficult to evaluate based on published data, is the fusion of IR2 with RP1+2. In Sinocalopteryx shangyongensis it is evidenced by the elevation of RP1+2 (+ IR2) basal to the divergence of IR2: the portion of RP1+2 basal to the point of divergence of IR2 is convex, while it is expected to be concave; it demonstrates that the convex IR2 is fused with it. This state is possibly present in Dicteriadidae, Calopterygidae, Euphaeidae, and Polythoridae. It might have relevance at the contextual level. Several other character states were originally considered (Lin et al., 2010) but are not decisive for a positive assignment of Sinocalopteryx shangyongensis to the Calopterygidae. For example, Lin et al. (2010) excluded possible affinities of Sinocalopteryx shangyongensis with the Polythoridae based on the lack of ‘polythorid type of arculus’ (i.e., the merging of the points of divergence of RA, RP and MA: i.e., arculus reduced to a single point). However, possessing this state clearly is a derived condition;hence, its absence does not preclude Sinocalopteryx shangyongensis from being a stem-Polythoridae. Note that Euphaeidae also possess a very short arculus.
The placement of the species with respect to other known Calopterygidae is not evident. The posteriorly pectinate CuA sharply contrasts with the condition displayed in Dicteriadidae, Polythoridae and most Calopterygidae, with the notable exception of Baydera spp. and, to a lower extant, Vestalis spp. (both Calopterygidae; Hämäläinen, 2004). The RP1/RP2 split located basal to the nodus (documented in the hind-wing specimen) is also an unusual feature for a Calopterygidae, but it occurs in Iridictyon spp. (the corresponding species lack cross-veins in the area between R + MA and MP + Cu, basal to the arculus, while they occur in Sinocalopteryxshangyongensis).
In summary, the placement of Sinocalopteryx shangyongensis as stem-Calopterygidae is only based on a set of ‘Class 2’ character states. However, provided that the placement of the species at any other node within Odonata would imply further homoplasies, the species is selected as stem-Calopterygidae.
Crown-Anisoptera
Stem-Anisoptera / crown-[Anisoptera+Epiophlebiidae]
Preliminary remarks: Stem representatives of both Anisoptera and Zygoptera are relevant to calibrate the corresponding node. A stem-Anisoptera is favored because all suitable stem-Zygoptera, including Congqingiarhora (Zhang, 1992), are younger. See section 'Updated Kohli et al.'s (2016) selection, the case of Triassolestodes asiaticus.
Stem-[Hemianax ephippiger + (Planaeschna sp. + Anax imperator)] / crown-[Aeshna isoceles + (Hemianax ephippiger + (Planaeschna sp. + Anax imperator))]
Fossil: Merlax bohemicusProkop & Nel, 2000
Original description:Prokop and Nel (2000).
Further descriptive accounts: None.
Locality: Bílina mine (18.6 M; see localities ages section).
CR1 (single/multiple OTUs with museum numbers): Yes (original description).
CR2 (apomorphy-based or phylogenetic analysis): See below.
CR3 (agreement of morphology and molecular data): See below.
CR4 (detailed locality and stratigraphy data provided): Yes (original description).
CR5 (radioisotopic age or numeric age references given): Yes (see localities section).
Phylogenetic justification & remarks:
The placement of Merlax bohemicus as crown-[Aeshna isoceles + (Hemianax ephippiger + (Planaeschna sp. + Anax imperator)) ] / stem-[Hemianax ephippiger + (Planaeschna sp. + Anax imperator)] is based on the following character states:
Ultimate level:
Class 1: (1) convex bending of RP2 located opposite, or distal to, the distal edge of the pterostigma.
Class 2: none considered.
Contextual level:
Class 1: (2) area between RP3+4 and MA gradually tapering until MA is sharply bent posteriorly, this resulting into a broadening of the RP3+4 / MA area (often filled with at least two rows of cells); (3) IR2 forked.
Class 2: none considered.
Among extant species represented in our analysis, the character state (1) is present in Anax imperator and Hemianax ephippiger (note that it is absent in Planaeschna sp. and Aeshna isoceles). The occurrence of a bending of RP2 was noticed by Prokop and Nel (2000), who relied on this trait, among others, to place the fossil within the Aeshnidae. Note that the specific location of this bending allows a placement of this fossil within Aeschnidae.
The character state (2) is present in some Aeshna spp., Anax, Anaciaeschna, Andeaschna, etc. (note that, however, the state is absent in Planaeschna), and further indicates affinities within the Aeschnidae. A distal narrowing of the corresponding area is often present, but its occurrence (or lack thereof) cannot be assessed in the fossil. Among Anisoptera, the character state (3) occurs only in Aeschnidae.
Stem-Libellulidae / crown-(Libellulidae + Corduliidae)
Fossil: Palaeolibellulazherikhini Fleck, Nel & Martí;nez-Delclòs, 1999
Original description:Fleck et al. (1999).
Further descriptive accounts: None.
Locality: Kzyl-Zhar locality (89.5 Ma; see localities ages section).
CR1 (single/multiple OTUs with museum numbers): Yes (original description).
CR2 (apomorphy-based or phylogenetic analysis): See below.
CR3 (agreement of morphology and molecular data): See below.
CR4 (detailed locality and stratigraphy data provided): Yes (original description).
CR5 (radioisotopic age or numeric age references given): Yes (see localities ages section).
Phylogenetic justification & remarks:
The placement of Palaeolibellulazherikhini as stem-Libellulidae / crown-(Libellulidae + Corduliidae) is based on the following character states:
Ultimate level:
Class 1: none found/considered.
Class 2: (1) in fore-wing, triangle at least twice longer than broad.
Contextual level:
Class 1: none found/considered.
Class 2: (2) origin of RP1+2 oblique; (3) first cell distal to the subnodus long (‘libellulid gap’ sensu Bechly, 1996); (4) occurrence of Mspl; (5) occurrence of Rspl; (6) presence of a strong concave ‘supplementary sector’ developed between RP1 and RP2 (‘posterior pseudo-branch of RP1’).
The character state (1) is present in Libellulidae and in some members of the fossil family Aeschnidiidae, regarded as related to Libellulidae (Fleck and Nel, 2003). However, the numerous additional states listed above provide a strong support to an assignment to the Libellulidae.
Stem-Chlorogomphidae / crown-(Chlorogomphidae + Cordulegastridae)
Fossil: AraripechlorogomphusmurataiBechly & Ueda 2002
Original description: Bechly and Ueda (2002)
Further descriptive accounts: None.
Locality: Crato (112.6 My; see localities ages section).
CR1 (single/multiple OTUs with museum numbers): Yes (original description).
CR2 (apomorphy-based or phylogenetic analysis): See below.
CR3 (agreement of morphology and molecular data): See below.
CR4 (detailed locality and stratigraphy data provided): Yes (original description).
CR5 (radioisotopic age or numeric age references given): Yes (see localities ages section).
Phylogenetic justification & remarks:
The placement of Araripechlorogomphus muratai as stem-Chlorogomphidae / crown-(Chlorogomphidae + Cordulegastridae) is based on the following character states:
Ultimate level:
Class 1: in hindwing, one of the CuA anterior furcal angles (distal to either the connection of CuA with the closure of the anal loop, or to the first CuA fork) is low (less than approximately 120°)
Class 2: none found/considered.
Contextual level:
Class 1: none found/considered.
Class 2: none found/considered.
In the original description the fossil is placed in the fossil family Araripechlorogomphidae, considered sister-group of Chlorogomphidae (Bechly and Ueda, 2002). In other words, the family is composed of stem-Chlorogomphidae. However, the relevance of the listed character states is not evident. Instead, we focused on the angle formed by CuA at one of its forks. We define the (a) anterior, (b) inner, and (c) posterior furcal angles of a vein as the angles formed at the fork, by (a) the initial stem and the anterior branch resulting from the fork, (b) the anterior and posterior branches resulting from the fork, and (c) the stem and the posterior branch resulting from the fork. In Chlorogomphidae, CuA has its ultimate or penultimate fork with a low anterior furcal angle (i.e., has its anterior branch arising very obliquely). Among extant species, the character state is unique to Chlorogomphidae.
Complementary selection, localities age
Hagen-Vorhalle (319.9 Ma)
Geographic location: Hagen-Vorhalle, Germany.
Geological settings:Ziegelschiefer Formation.
Stratigraphic data:‘Namurian B’, Late Carboniferous.
Age justification: See Wolfe et al. (2016) and references therein.
Bílina mine (18.6 Ma)
Geographic location: Bohemia, Czech Republic.
Geological settings: Most Formation.
Stratigraphic data: Burdigalian, Early Miocene.
Age justification: The concerned fossiliferous horizon at Bílina mine is the ‘Clayey Superseam Horizon’, from which the holotype of Merlax bohemicus was recovered (Prokop andNel et al., 2002). This horizon belongs to the Holešice Member, which is overlain by the Libkovice Member (Grygar and Mach, 2013; and references therein). Temporal constraints based on fossil mammals at the base of the Holešice Member, coupled with paleomagnetism and cylcostratigraphy, allowed an age of 18.6 Ma for the boundary between Holešice and Libkovice Members to be inferred (Grygar et al., 2014). This age is consistent with palaeofloristic correlations (Kvaček et al., 2004).
Kzyl-Zhar (89.5 Ma)
Geographic location:Chili district, Kazakhstan.
Geological settings: No information.
Stratigraphic data: Turonian, Early Cretaceous.
Age justification: The age of the locality is derived from its content in fossil plants (Shilin, 1986) and was widely adopted. The upper boundary of the Turonian is dated to 89.8Ma±0.3Myr (Cohen et al., 2013), providing a minimum age of 89.5 Ma.
Crato (112.6 Ma)
Geographic location: Brazil.
Geological settings: Crato Formation.
Stratigraphic data: Aptian, Lower Cretaceous.
Age justification: See Wolfe et al. (2016) and references therein.
Aix-en-Provence (23.0 Ma)
Geographic location: Bouches du Rhone, France.
Geological settings: Aix-en-Provence Formation.
Stratigraphic data:Chattian, Oligocene.
Age justification: Investigations on the age of this locality was summarized by (Gaudant et al., 2018). Various biostratigraphic markers converge towards a Late Chattian age. The upper boundary of the Chattian is 23.03 Ma (Cohen et al., 2013).
Shangyong (47.8 Ma)
Geographic location: Mengla County, Yunnan Province, China.
Geological settings: Undefined stratum in the vicinity of Mengyejing Formation.
Stratigraphic data: Undetermined stage, Eocene.
Age justification: The age justification for this locality was discussed by (Lin et al., 2010). Referring to Chen and Shen (1980) and Shen et al. (2006), these authors adopted an Ypresian (Early Eocene) age based on the occurrence of the conchostracan Paraleptestheria menglaensis and the associated fauna occurring in the Lofochai Group (also referred to as Lofuozhai; specifically, in the Zhuguikeng Member of the Nongshan Formation). However, Chen and Shen (1980) admitted that, although an early Eocene age is ‘probable’, this fauna is ‘generally considered as Eocene’ (p. 188). Note that an Ypresian age for Paraleptestheria menglaensis is given by Shen et al. (2006), but without justification. It should be noted that the occurrence of the Peckichara longa - Obtusochara elliptica charophyte complex together with Paraleptestheria menglaensis provides a useful, indirect evidence on the age of the latter, namely Early Eocene (Yang et al., 1979).
Lin et al. (2010) reported that the precise strata from which fossil insects were recovered is ‘undefined’, in the vicinity of the Mengyejing Formation, which is supposedly of Late Cretaceous age. Apparently, this age incongruity relates with inconsistent naming of geological formations. According to (Wang et al., 2015), the Denghei Formation that they regard as Eocene to Oligocene occurs in the vicinity of Mengla, nearby Shangyong. Alternatively, according to Tong et al. (2013), referring to the same area, the Mengyejing Formation encompasses Paleocene strata only, while the Xiaoyaku Formation is composed of Early Eocene strata. In conclusion, regardless of the formation they have been assigned to, Paleocene and Eocene strata occur in the vicinity of the Shangyong village. In conclusion an Ypresian age for the Shangyong locality is plausible but might prove under-estimated.
Updated Kohli et al.’s (2016) selection
Below we comment on the fossil selection by Kohli et al. (2016) carried out using a rationale commonly applied in fossil odonate systematics: in order to cope with homoplastic characters, a systematic assignment is usually based on a set of states whose relevance is assessed on a case-by-case basis. In one case the proposed phylogenetic placement is now contentious, and in the other it should be reconsidered given our sample of extant species.
Fossil: Triassolestodes asiaticusPritykina, 1981
Original description: Pritykina (1981)
Further descriptive accounts: Nel et al., (2002)
Locality: Madygen (228.4 Ma).
The species was selected by Kohli et al. (2016) as crown-Odonata resting on published accounts in which the species is assigned to the Triassolestidae, but without firmly establishing the placement of the species as crown-Odonata. Conversely, Tierney et al., (2020) argued that the Triassolestidae represent a paraphyletic assemblage of stem-Odonata, in which Triassolestodes asiaticus lacks the posterior lobe typical of the Lobodonata, including crown-Odonata and their closer stem-relatives. The latter includes the Mesophlebiidae and a number of ‘fossil anisozygopterans’ which all possess an open discoidal cell, thus precluding their assignment to crown-Odonata. In summary, the placement of Triassolestodes asiaticus is contentious. Analyses were therefore run with and without this particular fossil.
Fossil: ProterogomphusrenateaeBechly, Nel, Martínez-Delclòs & Fleck, 1998
Original description: Bechly et al., (1998)
Further descriptive accounts: None.
Locality:Solnhofen (150 Ma; Kohli et al., 2016).
The species was enlisted as crown-Gomphidae in Kohli et al. (2016). Character states supporting this placement are: (1) in hind-wing, distal side of triangle angulate; (2) in the MA–MP area, basal-most intercalary forming a rectilinear vein-like element’ (original wording: ‘in hind-wing, trigonal planate(postrigonal sector)’; also called ‘supplementary trigonal branch’); (3) in hind-wing, less than 5 antefurcal cross-veins; (4) branching of RP at midfork symmetrical.
The character state (1) is also present in Petaluridae (the sister-group of Gomphidae according to the obtained topology), and absent in some Gomphidae, such as Aphylla, Stylogomphus and Ophiogomphus (the two latter genera included in our analysis; Belle, 1964; Garrison et al., 2006) and should therefore be considered with caution. The character state (2) occurs in Petaluridae (including stem-representatives; (Coram and Nel, 2009), is homoplastic within extant Gomphidae (e.g., is absent in Stylogomphus and Ophiogomphus) and can prove somewhat difficult to evaluate in some cases. Moreover, its occurrence in Proterogomphusrenateae is not evident. As for character state (4), it is difficult to evaluate (it can be considered present in Petaluridae) and is, also, homoplastic within extant Gomphidae.
Kohli et al. (2016) refer to the phylogenetic analysis by Vernoux et al. (2010), performed with an ingroup composed of eight terminal units, with three extant Gomphidae genera and five extinct genera (including Proterogomphus). Note that while the tree presented by these authors indicate a clade (Progomphus+(Gomphus+ fossils)), there is no character state supporting it (see p. 98). According to Vernoux et al. (2010), four fossil genera, including Proterogomphus, form a monophyletic group which sister-group is Gomphus (representing the Brevicubitalia). This group is supported by the character state (3).(Character 30, state 1, in Vernoux et al., 2010). Note that Progomphus was miscoded for this character (see Riek and Kukalová-Peck, 1984). Caution is needed when relying on it, as it is homoplastic across Anisoptera, but can even vary at the intra-individual level in fossil relatives of Gomphidae (See Bechly, 2010: figs 74,75). Nevertheless, the state is indeed present in all extant species selected in our analysis. All possess less than 4 (and usually less than 3) antefurcal cross-veins. Therefore, according to this character, Proterogomphusrenateae is a sister-group to our selection of Gomphidae. In other words, given our sample, it is not ideal to calibrate the first split of (our selection of) Gomphidae but the Gomphidae/Petaluridae split instead. The latter option is consistent with the age of the earliest stem-Petaluridae (useful to calibrate the same split), a fossil recovered from the same locality as Proterogomphusrenateae (see Nel et al., 1998).
Acknowledgments
We thank Daniela Bartel for making the ortholog set, and Robert Waterhouse for analytics support. We thank Ondrej Hlinka for bioinformatics assistance and Karen Meusemann for help in running pre and main analyses. Thank you to Alexander Blanke for leading the initial Transodonata 1KITE-working group. We thank all who provided specimens: Alex Blanke, Akira Nishida, Chris Manchester, Daichi Kato, Guenther Theischinger, Jeanne Wilbrandt, John Trueman, Julian Glos, Kai Schütte, KaoruSekiya, Karen Meusemann, Manfred Niehuis, Martin Kubiak, Matthew Gimmel, Melissa Sanchez-Herrera, Miai Sakai, Mika Sugimura, MitsutoshiSugimura, Ola Fincke, Osamu Tanabe, Ralph S. Peters, Reiner Richter, Richard A. B. Leschen, Rudolf Meier, Hiroshi Yokota, Sae Nomura, Tanja Ziesmann, Thomas Buckley, ToshiyukiTeramoto, William Kuhn, Xiao-Li Tong, X.Y., X.Z., Yoko Watanabe, YutaMashimo, Dominic Evangelista, and Erik Svensson. We are grateful to M. Hämäläinen for assistance with access to the relevant literature and the UK Geological Survey. ON thanks Netta Dorchin for help preparing field trips to Israel, and Mike Nelson and David Trently for help finding odonates in Tennessee (USA). ON furthermore acknowledges the Israeli Nature and National Parks Protection Authority for granting permission to collect samples. JLW would like to acknowledge NSF grants #1564386 and 1453147. ON acknowledges the German Research Foundation (DFG) for supporting field trips within Germany and to Israel (NI 1387/1-1). Transcriptome sequencing and data curation under 1KITE were supported by funds from BGI-Shenzhen through XZ.
Author contributions
X.Z, R.M., O.N., X.Y., B.M., and J.W., collected specimen. S.L., X.Z., A.D., L.P., and B.M. performed transcriptome sequencing, contamination checks, and assembly. S.L., X.Z., A.D., and B.M. performed transcriptome data curation and A.D., submitted the transcriptomes to NCBI. A.D., C.M., L.P., and S.G. provided tools and script for transcriptome analysis. M.K., H.L., C.G., and C.M., performed phylogenetic estimation analyses. M.K., and C.M. conducted the multispecies coalescence analysis. O.B., I.D., J.R., and T.W. collected the fossil calibration data. M.K., H.L., O.B., and J.W., conducted divergence time estimation analysis. M.K., H.L., O.B., and J.W., wrote the original draft of the manuscript. M.K., H.L., and O.B., worked on the figures and tables for the manuscript. M.K., H.L., C.G., X.Z., A.D., C.M., O.N., T.W., and J.W. reviewed and edited the manuscript. B.M. secured the funding for the project.
Declaration of interests
Authors declare no competing interests.
Published: November 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103324.
Supplemental information
Data and code availability
-
•
Data - The sequence datasets generated during the present study are available in NCBI (accession numbers in Table S1).
-
•
Code - We have no unreported custom computer code or algorithms.
-
•
Other - No “All other items” to report.
References
- Aberer A.J., Krompass D., Stamatakis A. Pruning rogue taxa improves phylogenetic accuracy: an efficient algorithm and webservice. Syst. Biol. 2013;62:162–166. doi: 10.1093/sysbio/sys078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascarrunz E., Rage J.-C., Legreneur P., Laurin M. Triadobatrachusmassinoti, the earliest known lissamphibian (Vertebrata: Tetrapoda) re-examined by μCT scan, and the evolution of trunk length in batrachians. Contrib. Zoolog. 2016;85:201–234. [Google Scholar]
- Ballare E.F., Ware J.L. Dragons fly, biologists classify: an overview of molecular odonate studies, and our evolutionary understanding of dragonfly and damselfly (Insecta: odonata) behavior. Int. J. Odonatology. 2011;14:137–147. [Google Scholar]
- Bechly G. Morphologische Untersuchungen am Flügelgeäder der rezenten Libellen und deren Stammgruppenvertreter (Insecta; Pterygota; Odonata) unter besonderer Berücksichtigung der Phylogenetischen Systematik und des Grundplanes der ∗Odonata - Petalura, spec. 1996;2:402. [Google Scholar]
- Bechly G. Additions to the fossil dragonfly fauna from the lower cretaceous crato formation of Brazil (Insecta: odonata) Palaeodiversity. 2010;3:11–77. [Google Scholar]
- Bechly G., Brauckmann C., Zessin W., Gröning E. New results concerning the morphology of the most ancient dragonflies (Insecta: Odonatoptera) from the Namurian of Hagen-Vorhalle (Germany) J. Zoolog. Syst. Evol. Res. 2001;39:209–226. [Google Scholar]
- Bechly G., Nel A., Martínez-Delclòs X., Fleck G. Four new dragonflies from the upper jurassic of Germany and the lower cretaceous of Mongolia (Anisoptera: Hemeroscopidae, Sonidae, and Proterogomphidae fam. nov.) Odonatologica. 1998;27:149–187. [Google Scholar]
- Bechly G., Ueda K. The first fossil record and first new world record for the dragonfly clade chlorogomphida (Insecta: odonata: Anisoptera: Araripechlorogomphidae n. fam.) from the crato limestone (lower cretaceous, Brazil) Stutt.Beit.Naturk. (B) 2002;328:1–11. [Google Scholar]
- Belle J. Surinam dragonflies of the genus Aphylla, with a description of a new species. Stud. Fauna Suriname other Guyanas. 1964;7:22–35. [Google Scholar]
- Béthoux O. The late Carboniferous Triplosoba pulchella is not a fly in the ointment but a stem-mayfly. Syst. Entomol. 2015;40:342–356. [Google Scholar]
- Béthoux O., Galtier J., Nel A. Earliest evidence of insect endophytic oviposition. Palaios. 2004;19:408–413. [Google Scholar]
- Blanke A., Greve C., Mokso R., Beckmann F., Misof B. An updated phylogeny of Anisoptera including formal convergence analysis of morphological characters. Syst. Entomol. 2013;38:474–490. [Google Scholar]
- Brauckmann C. Zwei neue Insekten (Odonata, Megasecoptera) aus dem Namurium von Hagen-Vorhalle (West-Deutschland) Dortmunder Beiträge zur Landeskunde, Naturwissenschaftliche Mitteilungen. 1988;22:91–101. [Google Scholar]
- Brauckmann C., Herd K.J. Insekten-Funde aus dem Westfalium D (Ober-Karbon) des Piesberges bei Osnabrück (Deutschland) Teil 1: Palaeoptera. Osnabrücker Naturwissenschaftliche Mitteilungen. 2002;28:27–69. [Google Scholar]
- Brauckmann C., Koch L., Kemper M. Spinnentiere (Arachnida) und Insekten aus den Vorhalle-Schichten (Namurium B; Ober-Karbon) von Hagen-Vorhalle (West-Deutschland) Geologie und Paläontologie in Westfalen. 1985;3:1–132. [Google Scholar]
- Brauckmann C., Schöllmann L., Sippel W. Die fossilen insekten, spinntiere und eurypteriden von Hagen-Vorhalle. Geologie und Paläontologie in Westfalen. 2003;59:1–89. [Google Scholar]
- Brauckmann C., Zessin W. Neue meganeuridae aus dem Namurium von Hagen-Vorhalle (BRD) und die phylogenie der meganisoptera (Insecta, Odonata) Deutsche Entomologische Z. 1989;36:177–215. [Google Scholar]
- Bridges C.A. Charles A. Bridges; 1994. Catalogue of the Family-Group, Genus-Group, and Species-Group Names of the Odonata of the World. [Google Scholar]
- Bybee S.M., Johnson K.K., Gering E.J., Whiting M.F., Crandall K.A. All the better to see you with: a review of odonate color vision with transcriptomic insight into the odonate eye. Organisms Divers.Evol. 2012;12:241–250. [Google Scholar]
- Bybee S.M., Kalkman V.J., Erickson R.J., Frandsen P.B., Breinholt J.W., Suvorov A., Dijkstra K.-D.B., Cordero-Rivera A., Skevington J.H., Abbott J.C. Phylogeny and classification of Odonata using targeted genomics. Mol. Phylogenet. Evol. 2021;160:107115. doi: 10.1016/j.ympev.2021.107115. [DOI] [PubMed] [Google Scholar]
- Bybee S.M., Ogden T.H., Branham M.A., Whiting M.F. Molecules, morphology and fossils: a comprehensive approach to odonate phylogeny and the evolution of the odonate wing. Cladistics. 2008;24:477–514. doi: 10.1111/j.1096-0031.2007.00191.x. [DOI] [PubMed] [Google Scholar]
- Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carle F. Evolution, taxonomy, and biogeography of ancient Gondwanian libelluloides, with comments on anisopteroid evolution and phylogenetic systematics (Anisoptera: Libelluloidea) Odonatologica. 1995;24:383–424. [Google Scholar]
- Carle F.L., Kjer K.M., May M.L. Evolution of odonata, with special reference to Coenagrionoidea (Zygoptera) Arthropod Syst. Phylogeny. 2008;66:37–44. [Google Scholar]
- Chen P.-J., Shen Y.-B. On the Paraleptestheria menglaensis fauna with reference to the age of the Lofochai Group. Acta Palaeontologica Sinica. 1980;19:182–189. [Google Scholar]
- Chernomor O., von Haeseler A., Minh B.Q. Terrace aware data structure for phylogenomic inference from supermatrices. Syst. Biol. 2016;65:997–1008. doi: 10.1093/sysbio/syw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen K.M., Finney S.C., Gibbard P.L., Fan J.-X. The ICS international chronostratigraphic chart. Episodes. 2013;36:199–204. [Google Scholar]
- Colbourne J.K., Pfrender M.E., Gilbert D., Thomas W.K., Tucker A., Oakley T.H., Tokishita S., Aerts A., Arnold G.J., Basu M.K. The ecoresponsive genome of Daphnia pulex. Science. 2011;331:555–561. doi: 10.1126/science.1197761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coram R.A., Nel A. A new petalurid dragonfly from the lower cretaceous of southern England (odonata: Petalurida: ?Cretapetaluridae) Palaeodiversity. 2009;2:205–208. [Google Scholar]
- Corbet P.S. 1999. Dragonflies: behaviour and ecology of Odonata, Harley books. https://www.amazon.com/Dragonflies-Behavior-Ecology-Odonata-Comstock/dp/0801425921. [Google Scholar]
- Davis R.B., Nicholson D.B., Saunders E.L., Mayhew P.J. Fossil gaps inferred from phylogenies alter the apparent nature of diversification in dragonflies and their relatives. BMC Evol.Biol. 2011;11:252. doi: 10.1186/1471-2148-11-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deregnaucourt I., Wappler T., Anderson J.M., Béthoux O. The wing venation of the Protomyrmeleontidae (Insecta: Odonatoptera) reconsidered thanks to a new specimen from Molteno (Triassic; South Africa) Hist. Biol. 2019:1–7. [Google Scholar]
- Dijkstra K.-D.B., Kalkman V.J., Dow R.A., Stokvis F.R., van Tol J. Redefining the damselfly families: a comprehensive molecular phylogeny of Z ygoptera (O donata) Syst. Entomol. 2014;39:68–96. [Google Scholar]
- Dos Reis M., Yang Z. The unbearable uncertainty of Bayesian divergence time estimation. J. Syst. Evol. 2013;51:30–43. [Google Scholar]
- Dumont H.J., Vanfleteren J.R., De Jonckheere J.F., Weekers P.H. Phylogenetic relationships, divergence time estimation, and global biogeographic patterns of calopterygoid damselflies (Odonata, Zygoptera) inferred from ribosomal DNA sequences. Syst. Biol. 2005;54:347–362. doi: 10.1080/10635150590949869. [DOI] [PubMed] [Google Scholar]
- Dumont H.J., Vierstraete A., Vanfleteren J.R. A molecular phylogeny of the Odonata (Insecta) Syst. Entomol. 2010;35:6–18. [Google Scholar]
- Dunlop J.A., Scholtz G., Selden P.A. In: Arthropod Biology and Evolution: Molecules, Development, Morphology. Minelli A., Boxshall G., Fusco G., editors. Springer; 2013. Water-to-Land transitions; pp. 417–439. [Google Scholar]
- Evangelista D.A., Wipfler B., Béthoux O., Donath A., Fujita M., Kohli M.K., Legendre F., Liu S., Machida R., Misof B. An integrative phylogenomic approach illuminates the evolutionary history of cockroaches and termites (Blattodea) Proc. R. Soc. B. 2019;286:20182076. doi: 10.1098/rspb.2018.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field D.J., Benito J., Chen A., Jagt J.W., Ksepka D.T. Late Cretaceous neornithine from Europe illuminates the origins of crown birds. Nature. 2020;579:397–401. doi: 10.1038/s41586-020-2096-0. [DOI] [PubMed] [Google Scholar]
- Fincke O.M. Giant damselflies in a tropical forest: reproductive biology of Megaloprepus coerulatus with notes on Mecistogaster (Zygoptera: Pseudostigmatidae) Adv. Odonatology. 1984;2:13–27. [Google Scholar]
- Finn R.D., Clements J., Eddy S.R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R.D., Coggill P., Eberhardt R.Y., Eddy S.R., Mistry J., Mitchell A.L., Potter S.C., Punta M., Qureshi M., Sangrador-Vegas A., et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–D285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleck G., Nel A. Revision of the mesozoic family Aeschnidiidae (Odonata: Anisoptera) Zoologica. 2003;153:1–180. [Google Scholar]
- Fleck G., Nel A., Martínez-Delclòs X. The oldest record of libellulid dragonflies from the upper cretaceous of Kazakhstan (Insecta: odonata, Anisoptera) Cretaceous Res. 1999;20:655–658. [Google Scholar]
- Fleck G., Ullrich B., Brenk M., Wallnisch C., Orland M., Bleidissel S., Misof B. A phylogeny of anisopterous dragonflies (Insecta, Odonata) using mtRNA genes and mixed nucleotide/doublet models. J. Zoolog. Syst. Evol. Res. 2008;46:310–322. [Google Scholar]
- Frandsen P.B., Calcott B., Mayer C., Lanfear R. Automatic selection of partitioning schemes for phylogenetic analyses using iterative k-means clustering of site rates. BMC Evol.Biol. 2015;15:13. doi: 10.1186/s12862-015-0283-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser F.C., Tillyard R.J. Royal Zoological Society of New South Wales; 1957. Reclassification of the Order Odonata. [Google Scholar]
- Futahashi R., Kawahara-Miki R., Kinoshita M., Yoshitake K., Yajima S., Arikawa K., Fukatsu T. Extraordinary diversity of visual opsin genes in dragonflies. Proc. Natl. Acad. Sci. U S A. 2015;112:E1247–E1256. doi: 10.1073/pnas.1424670112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison R.W., Von Ellenrieder N., Louton J.A. JHU Press; 2006. Dragonfly Genera of the New World: An Illustrated and Annotated Key to the Anisoptera, Baltimore, Maryland, USA. [Google Scholar]
- Gaudant J., Nel A., Nury D., Véran M., Carnevale G. The uppermost Oligocene of Aix-en-Provence (Bouches-du-Rhône, Southern France): a Cenozoic brackish subtropical Konservat-Lagerstätte, with fishes, insects and plants. Comptes Rendus Palevol. 2018;17:460–478. [Google Scholar]
- Geijskes D. Generic characters of the South American Corduliidae, with descriptions of the species found in the Guyanas. Stud. Fauna Suriname other Guyanas. 1970;12:1–42. [Google Scholar]
- Gloyd L.K. Elevation of the Macromia group to family status (Odonata) Entomol. News. 1959;70:197–205. [Google Scholar]
- Grygar T.M., Mach K. Regional chemostratigraphic key horizons in the macrofossil-barren siliciclastic lower Miocene lacustrine sediments (Most Basin, Eger Graben, Czech Republic) Bull. Geosciences. 2013;88:557–571. [Google Scholar]
- Grygar T.M., Mach K., Schnabl P., Pruner P., Laurin J., Martinez M. A lacustrine record of the early stage of the Miocene climatic optimum in Central Europe from the most basin, Ohře (eger) Graben, Czech Republic. Geol. Mag. 2014;151:1013–1033. [Google Scholar]
- Gu X., Fu Y.-X., Li W.-H. Maximum likelihood estimation of the heterogeneity of substitution rate among nucleotide sites. Mol. Biol. Evol. 1995;12:546–557. doi: 10.1093/oxfordjournals.molbev.a040235. [DOI] [PubMed] [Google Scholar]
- Guindon S., Dufayard J.-F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Hämäläinen M. Caloptera damselflies from Fujian (China), with description of a new species and taxonomic notes (Zygoptera: Calopterygoidea) Odonatologica. 2004;33:371–398. [Google Scholar]
- Hämäläinen M., Yu X., Zhang H. Descriptions of Matrona oreades spec. nov. and Matrona corephaea spec. nov. from China (Odonata: Calopterygidae) Zootaxa. 2011;2830:20–28. [Google Scholar]
- Henikoff S., Henikoff J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. U S A. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig W. John Wiley & Sons Ltd; 1981. Insect Phylogeny. [Google Scholar]
- i5K-Consortium The i5K Initiative: advancing arthropod genomics for knowledge, human health, agriculture, and the environment. J. Hered. 2013;104:595–600. doi: 10.1093/jhered/est050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue J., Donoghue P.C., Yang Z. The impact of the representation of fossil calibrations on Bayesian estimation of species divergence times. Syst. Biol. 2010;59:74–89. doi: 10.1093/sysbio/syp078. [DOI] [PubMed] [Google Scholar]
- Jermiin L, Ott M. SymTest v. 2.0.47 2017. https://github.com/ottmi/symtest.
- Jones D.T., Taylor W.R., Thornton J.M. The rapid generation of mutation data matrices from protein sequences. Bioinformatics. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. Humana Press; Totowa, NJ: 2014. MAFFT: iterative refinement and additional methods. Multiple sequence alignment methods; pp. 131–146. [DOI] [PubMed] [Google Scholar]
- Kluge N.J. Kluwer Academic Publishers; 2004. The phylogenetic system of Ephemeroptera; p. 456. [Google Scholar]
- Kohli M.K., Sahlén G., Kuhn W.R., Ware J.L. Extremely low genetic diversity in a circumpolar dragonfly species, Somatochlora sahlbergi (Insecta: odonata: Anisoptera) Scientific Rep. 2018;8:1–10. doi: 10.1038/s41598-018-32365-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli M.K., Ware J.L., Bechly G. How to date a dragonfly: fossil calibrations for odonates. Palaeontol. Electronica. 2016;19:1–14. [Google Scholar]
- Kosiol C., Goldman N. Different versions of the Dayhoff rate matrix. Mol. Biol. Evol. 2005;22:193–199. doi: 10.1093/molbev/msi005. [DOI] [PubMed] [Google Scholar]
- Kriventseva E.V., Tegenfeldt F., Petty T.J., Waterhouse R.M., Simao F.A., Pozdnyakov I.A., Ioannidis P., Zdobnov E.M. OrthoDB v8: update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res. 2015;43:D250–D256. doi: 10.1093/nar/gku1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksepka D.T., Parham J.F., Allman J.F., Benton M.J., Carrano M.T., Cranston K.A., Donoghue P.C., Head J.J., Hermsen E.J., Irmis R.B. The fossil calibration database—a new resource for divergence dating. Syst. Biol. 2015;64:853–859. doi: 10.1093/sysbio/syv025. [DOI] [PubMed] [Google Scholar]
- Kück P., Meusemann K., Dambach J., Thormann B., von Reumont B.M., Wägele J.W., Misof B. Parametric and non-parametric masking of randomness in sequence alignments can be improved and leads to better resolved trees. Front Zool. 2010;7:10. doi: 10.1186/1742-9994-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvaček Z., Dvořák Z., Mach K., Sakala J. Severočeské doly a.s., Chomutov & Granit; 2004. Třetihorní rostliny severočeské hnědouhelné pánve. [Google Scholar]
- Lanfear R., Calcott B., Kainer D., Mayer C., Stamatakis A. Selecting optimal partitioning schemes for phylogenomic datasets. BMC Evol.Biol. 2014;14:82. doi: 10.1186/1471-2148-14-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear R., Frandsen P.B., Wright A.M., Senfeld T., Calcott B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2016;34:772–773. doi: 10.1093/molbev/msw260. [DOI] [PubMed] [Google Scholar]
- Le S.Q., Dang C.C., Gascuel O. Modeling protein evolution with several amino acid replacement matrices depending on site rates. Mol. Biol. Evol. 2012;29:2921–2936. doi: 10.1093/molbev/mss112. [DOI] [PubMed] [Google Scholar]
- Le S.Q., Gascuel O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008;25:1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- Letsch H., Gottsberger B., Ware J.L. Not going with the flow: a comprehensive time-calibrated phylogeny of dragonflies (Anisoptera: odonata: Insecta) provides evidence for the role of lentic habitats on diversification. Mol. Ecol. 2016;25:1340–1353. doi: 10.1111/mec.13562. [DOI] [PubMed] [Google Scholar]
- Li Y., Béthoux O., Pang H., Ren D. Early Pennsylvanian Odonatoptera from the Xiaheyan locality (Ningxia, China): new material, taxa, and perspectives. Fossil Rec. 2013;16:117–139. [Google Scholar]
- Lin Q.-B., Petrulevičius J.F., Huang D.-Y., Nel A., Engel M.S. First fossil Calopterygoidea (odonata: Zygoptera) from southeastern Asia: a new genus and species from the Paleogene of China. Geobios. 2010;43:349–353. [Google Scholar]
- Lin Q.-B., Szwedo J., Huang D.-Y., Stroiński A. Weiwoboidae fam. Nov. Of ‘higher’Fulgoroidea (Hemiptera: Fulgoromorpha) from the Eocene deposits of Yunnan, China. Acta Geologica Sinica-English Edition. 2010;84:751–755. [Google Scholar]
- Macnaughton R.B., Cole J.M., Dalrymple R.W., Braddy S.J., Briggs D.E., Lukie T.D. First steps on land: arthropod trackways in Cambrian-Ordovician eolian sandstone, southeastern Ontario, Canada. Geology. 2002;30:391–394. [Google Scholar]
- Matushkina N.A. Morphology of exophytic ovipositors in dragonflies (Odonata: Gomphidae, Corduliidae, Libellulidae), with particular reference to ovipositor muscles and sensilla. Int. J. Odonatology. 2011;14:233–248. [Google Scholar]
- Mccafferty W.P. In: Insects from the Santana Formation, Lower Cretaceous, of Brazil. Grimaldi D.A., editor. Vol. 195. American Museum of Natural History; 1990. Ephemeroptera; pp. 20–50. [Google Scholar]
- Meunier F. Nouvelles recherches sur quelques insectes du Sannoisien d’Aix-en-Provence. Bulletin de la Société Géologique de France. 1914;4:187–198. + pls186–187. [Google Scholar]
- Meusemann K., von Reumont B.M., Simon S., Roeding F., Strauss S., Kück P., Ebersberger I., Walzl M., Pass G., Breuers S. A phylogenomic approach to resolve the arthropod tree of life. Mol. Biol. Evol. 2010;27:2451–2464. doi: 10.1093/molbev/msq130. [DOI] [PubMed] [Google Scholar]
- Meyer B., Misof B. 2011. MARE v0.1.2-rc: MAtrix REduction - a Tool to Select Optimized Data Subsets from Supermatrices for Phylogenetic Inference.http://mare.zfmk.de [Google Scholar]
- Minh B.Q., Hahn M.W., Lanfear R. New methods to calculate concordance factors for phylogenomic datasets. Mol. Biol. Evol. 2020;37:2727–2733. doi: 10.1093/molbev/msaa106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh B.Q., Schmidt H.A., Chernomor O., Schrempf D., Woodhams M.D., von Haeseler A., Lanfear R. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020;37:1530–1534. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab S., Reaz R., Bayzid M.S., Zimmermann T., Swenson M.S., Warnow T. ASTRAL: genome-scale coalescent-based species tree estimation. Bioinformatics. 2014;30:i541–i548. doi: 10.1093/bioinformatics/btu462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misof B., Liu S., Meusemann K., Peters R.S., Donath A., Mayer C., Frandsen P.B., Ware J., Flouri T., Beutel R.G. Phylogenomics resolves the timing and pattern of insect evolution. Science. 2014;346:763–767. doi: 10.1126/science.1257570. [DOI] [PubMed] [Google Scholar]
- Misof B., Misof K. A Monte Carlo approach successfully identifies randomness in multiple sequence alignments: a more objective means of data exclusion. Syst. Biol. 2009;58:21–34. doi: 10.1093/sysbio/syp006. [DOI] [PubMed] [Google Scholar]
- Nel A., Bechly G., Jarzembowski E., Delclòs X. A revision of the fossil petalurid dragonflies (Insecta: odonata: Anisoptera: Petalurida) Paleontologica Lombarda. 1998;10:1–69. [Google Scholar]
- Nel A., Bechly G., Prokop J., Béthoux O., Fleck G. Systematics and evolution of Paleozoic and mesozoic damselfly-like Odonatoptera of the ‘Protozygopteran’grade. J. Paleontol. 2012;86:81–104. [Google Scholar]
- Nel A., Huang D. First Chinese cymatophlebiidaefrom the middle jurassic of inner Mongolia (Odonata: Anisoptera: Aeshnoptera) Palaeodiversity. 2009;2:e204. [Google Scholar]
- Nel A., Marie V., Schmeissne S. Revision of the lower mesozoicdragonfly family Triassolestidae (Odonata: Epiproctophora) Ann. de Paléontologie. 2002;2002:189–214. [Google Scholar]
- Nel A., Papazian M. Redescription de calopteryx sannoisiensis Meunier, 1914, Odonata fossile de l'Oligocène terminal d'Aix-en-Provence (Odonata, Calopterygidae) Entomologica Gallica. 1992;3:10–12. [Google Scholar]
- Nel A., Petrulevičius J.F., Martínez-Delclós X. New mesozoic protomyrmeleontidae (Insecta: Odonatoptera: Archizygoptera) from Asia with a new phylogenetic analysis. J. Syst. Palaeontology. 2005;3:187–201. [Google Scholar]
- Nguyen L.-T., Schmidt H.A., von Haeseler A., Minh B.Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham J.F., Donoghue P.C., Bell C.J., Calway T.D., Head J.J., Holroyd P.A., Inoue J.G., Irmis R.B., Joyce W.G., Ksepka D.T. Best practices for justifying fossil calibrations. Syst. Biol. 2012;61:346–359. doi: 10.1093/sysbio/syr107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattengale N.D., Alipour M., Bininda-Emonds O.R., Moret B.M., Stamatakis A. How many bootstrap replicates are necessary? J. Comput. Biol. 2010;17:337–354. doi: 10.1089/cmb.2009.0179. [DOI] [PubMed] [Google Scholar]
- Pauli T., Vedder L., Dowling D., Petersen M., Meusemann K., Donath A., Peters R.S., Podsiadlowski L., Mayer C., Liu S. Transcriptomic data from panarthropods shed new light on the evolution of insulator binding proteins in insects. BMC Genomics. 2016;17:861. doi: 10.1186/s12864-016-3205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters R.S., Krogmann L., Mayer C., Donath A., Gunkel S., Meusemann K., Kozlov A., Podsiadlowski L., Petersen M., Lanfear R. Evolutionary history of the Hymenoptera. Curr. Biol. 2017;27:1013–1018. doi: 10.1016/j.cub.2017.01.027. [DOI] [PubMed] [Google Scholar]
- Petersen M., Meusemann K., Donath A., Dowling D., Liu S., Peters R.S., Podsiadlowski L., Vasilikopoulos A., Zhou X., Misof B. Orthograph: a versatile tool for mapping coding nucleotide sequences to clusters of orthologous genes. BMC Bioinformatics. 2017;18:1–10. doi: 10.1186/s12859-017-1529-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilgrim E.M., von Dohlen C.D. Phylogeny of the Sympetrinae (Odonata: Libellulidae): further evidence of the homoplasious nature of wing venation. Syst. Entomol. 2008;33:159–174. [Google Scholar]
- Pritykina L.N. In: Novye Iskopaemye Nasekomye S Terrotorii SSSR. Vishniakova V.N., Dlussky G.M., Pritykina L.N., editors. Vol. 183. Trudy Paleontologicheskogo instituta, Akademiya Nauk SSSR.; 1981. NovyetriasovyestrekozysrednejAzii; pp. 5–42. [Google Scholar]
- Prokop J., Nel A. Merlax bohemicus gen. n., sp. n., a new fossil dragonfly from the Lower Miocene of northern Bohemia (Odonata: Aeshnidae) Eur. J. Entomol. 2000;97:427–431. [Google Scholar]
- Punta M., Coggill P.C., Eberhardt R.Y., Mistry J., Tate J., Boursnell C., Pang N., Forslund K., Ceric G., Clements J., et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A., Drummond A.J., Xie D., Baele G., Suchard M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018;67:901. doi: 10.1093/sysbio/syy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehn A.C. Phylogenetic analysis of higher-level relationships of Odonata. Syst. Entomol. 2003;28:181–240. [Google Scholar]
- Riek E.F., Kukalová-Peck J. A new interpretation of dragonfly wing venation based upon Early Upper Carboniferous fossils from Argentina (Insecta: Odonatoidea) and basic character states in pterygote wings. Can. J. Zoolog. 1984;62:1150–1166. [Google Scholar]
- Ruane S., Pyron R., Burbrink F. Phylogenetic relationships of the Cretaceous frog Beelzebufo from Madagascar and the placement of fossil constraints based on temporal and phylogenetic evidence. J. Evol. Biol. 2011;24:274–285. doi: 10.1111/j.1420-9101.2010.02164.x. [DOI] [PubMed] [Google Scholar]
- Sánchez-Bayo F., Wyckhuys K.A. Worldwide decline of the entomofauna: a review of its drivers. Biol. Conservation. 2019;232:8–27. [Google Scholar]
- Sánchez-Herrera M., Beatty C.D., Nunes R., Salazar C., Ware J.L. An exploration of the complex biogeographical history of the Neotropical banner-wing damselflies (Odonata: Polythoridae) BMC Evol. Biol. 2020;20:1–14. doi: 10.1186/s12862-020-01638-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayyari E., Whitfield J.B., Mirarab S. Fragmentary gene sequences negatively impact gene tree and species tree reconstruction. Mol. Biol. Evol. 2017;34:3279–3291. doi: 10.1093/molbev/msx261. [DOI] [PubMed] [Google Scholar]
- Shen Y.-B., Gallego O.F., Buchheim H.P., Biaggi R.E. Eocene conchostracans from the Laney member of the green river formation, Wyoming, USA. J. Paleontol. 2006;80:447–454. [Google Scholar]
- Shilin P.V. SSR; 1986. Pozdnemelovoy Flory Kazakhstana [Late Cretaceous Floras of Kazakhstan], [Google Scholar]
- Simon S., Sagasser S., Saccenti E., Brugler M.R., Schranz M.E., Hadrys H., Amato G., Desalle R. Comparative transcriptomics reveal developmental turning points during embryogenesis of a hemimetabolous insect, the damselfly Ischnura elegans. Sci. Rep. 2017;7:1–14. doi: 10.1038/s41598-017-13176-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater G.S.C., Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubrier J., Steel M., Lee M.S., Der Sarkissian C., Guindon S., Ho S.Y., Cooper A. The influence of rate heterogeneity among sites on the time dependence of molecular rates. Mol. Biol. Evol. 2012;29:3345–3358. doi: 10.1093/molbev/mss140. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer K., von Haeseler A. Likelihood-mapping: a simple method to visualize phylogenetic content of a sequence alignment. Proc. Natl. Acad. Sci. U S A. 1997;94:6815–6819. doi: 10.1073/pnas.94.13.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhling F., Jödicke R., Schneider W. Odonata of African arid regions–are there desert species. Cimbebasia. 2003;18:207–224. [Google Scholar]
- Suvorov A., Scornavacca C., Fujimoto M.S., Bodily P., Clement M., Crandall K.A., Whiting M.F., Schrider D.R., Bybee S.M. Deep ancestral introgression shapes evolutionary history of dragonflies and damselflies. Syst. Biol. 2021:syab063. doi: 10.1093/sysbio/syab063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama M., Torrents D., Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrapon N., Li C., Robertson H.M., Ji L., Meng X., Booth W., Chen Z., Childers C.P., Glastad K.M., Gokhale K., et al. Molecular traces of alternative social organization in a termite genome. Nat. Commun. 2014;5:3636. doi: 10.1038/ncomms4636. [DOI] [PubMed] [Google Scholar]
- Thomas J.A., Trueman J.W., Rambaut A., Welch J.J. Relaxed phylogenetics and the Palaeoptera problem: resolving deep ancestral splits in the insect phylogeny. Syst. Biol. 2013;62:285–297. doi: 10.1093/sysbio/sys093. [DOI] [PubMed] [Google Scholar]
- Tierney A., Deregnaucourt I., Anderson J.M., Tierney P., Wappler T., Béthoux O. The Triassic Mesophlebiidae, a little closer to the crown of the Odonata (Insecta) than other ‘triassolestids’ Alcheringa: Australas. J. Palaeontology. 2020;44:279–285. [Google Scholar]
- Tillyard R.J. CUP Archive; 1917. The Biology of dragonflies:(Odonata or Paraneuroptera) [Google Scholar]
- Tong Y.-B., Yang Z., Zheng L.-D., Xu Y.-L., Wang H., Gao L., Hu X.-Z. Internal crustal deformation in the northern part of Shan-Thai Block: new evidence from paleomagnetic results of Cretaceous and Paleogene redbeds. Tectonophysics. 2013;608:1138–1158. [Google Scholar]
- Trueman J. A preliminary cladistic analysis of odonate wing venation. Odonatologica. 1996;25:59–72. [Google Scholar]
- Trueman J.W. A brief history of the classification and nomenclature of Odonata. Zootaxa. 2007;1668:381–394. [Google Scholar]
- Vankan M., Ho S.Y., Duchêne D.A. Evolutionary rate variation among lineages in gene trees has a negative impact on species-tree inference. Syst. Biol. 2021 doi: 10.1093/sysbio/syab051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Sánchez Y.M., Mendoza-Cuenca L.F., González-Rodríguez A. Complex evolutionary history of the American Rubyspot damselfly, Hetaerina americana (Odonata): evidence of cryptic speciation. Mol. Phylogenet. Evol. 2019;139:106536. doi: 10.1016/j.ympev.2019.106536. [DOI] [PubMed] [Google Scholar]
- Vernoux J., Huang D.-Y., Jarzembowski E.A., Nel A. The Proterogomphidae: a worldwide mesozoic family of gomphid dragonflies (odonata: Anisoptera: Gomphides) Cretaceous Res. 2010;31:94–100. [Google Scholar]
- Wang L., Liu C., Fei M., Shen L., Zhang H., Zhao Y. First SHRIMP U–Pb zircon ages of the potash-bearing Mengyejing formation, Simao Basin, southwestern Yunnan, China. Cretaceous Res. 2015;52:238–250. [Google Scholar]
- Ware J., Karlsson M., Sahlén G., Koch K. Evolution of reproductive strategies in libellulid dragonflies (Odonata: Anisoptera) Organisms Divers.Evol. 2012;12:313–323. [Google Scholar]
- Ware J., May M., Kjer K. Phylogeny of the higher Libelluloidea (Anisoptera: odonata): an exploration of the most speciose superfamily of dragonflies. Mol. Phylogenet. Evol. 2007;45:289–310. doi: 10.1016/j.ympev.2007.05.027. [DOI] [PubMed] [Google Scholar]
- Ware J.L., Beatty C.D., Sánchez Herrera M., Valley S., Johnson J., Kerst C., May M.L., Theischinger G. The petaltail dragonflies (Odonata: Petaluridae): mesozoic habitat specialists that survive to the modern day. J. Biogeogr. 2014;41:1291–1300. [Google Scholar]
- Ware J.L., Pilgrim E., May M.L., Donnelly T.W., Tennessen K. Phylogenetic relationships of North American Gomphidae and their close relatives. Syst. Entomol. 2017;42:347–358. doi: 10.1111/syen.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware J.L., Simaika J.P., Samways M.J. Biogeography and divergence time estimation of the relict Cape dragonfly genus Syncordulia: global significance and implications for conservation. Zootaxa. 2009;2216:22–36. [Google Scholar]
- Warnock R.C., Yang Z., Donoghue P.C. Exploring uncertainty in the calibration of the molecular clock. Biol. Lett. 2012;8:156–159. doi: 10.1098/rsbl.2011.0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan S., Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001;18:691–699. doi: 10.1093/oxfordjournals.molbev.a003851. [DOI] [PubMed] [Google Scholar]
- Wolfe J.M., Daley A.C., Legg D.A., Edgecombe G.D. Fossil calibrations for the arthropod tree of life. Earth-Science Rev. 2016;160:43–110. [Google Scholar]
- Wong T.K., Kalyaanamoorthy S., Meusemann K., Yeates D.K., Misof B., Jermiin L.S. A minimum reporting standard for multiple sequence alignments. NAR Genomics and Bioinformatics. 2020;2:lqaa024. doi: 10.1093/nargab/lqaa024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Z., Liu L., Davis C.C. Genes with minimal phylogenetic information are problematic for coalescent analyses when gene tree estimation is biased. Mol. Phylogenet. Evol. 2015;92:63–71. doi: 10.1016/j.ympev.2015.06.009. [DOI] [PubMed] [Google Scholar]
- Yang H., Wang Z., Li M., Huang B. Nanjing Institute of Geology and Paleontology; 1979. Stratigraphic Subdivision, Correlation, Paleofaunas, and Floras of South China Mesozoic to Early Tertiary Red Beds. Mesozoic and Cenozoic Red Beds of South China (Nanjing, China) [Google Scholar]
- Yang S., Temin H.M. A double hairpin structure is necessary for the efficient encapsidation of spleen necrosis virus retroviral RNA. EMBO J. 1994;13:713–726. doi: 10.1002/j.1460-2075.1994.tb06311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Paml 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Zhang C., Rabiee M., Sayyari E., Mirarab S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics. 2018;19:15–30. doi: 10.1186/s12859-018-2129-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. Congqingia rhora gen. nov., spec. nov.–a new dragonfly from the Upper Jurassic of eastern China (Anisozygoptera: Congqingiidae fam. nov.) Odonatologica. 1992;21:375–383. [Google Scholar]
- Zheng D., Jarzembowski E.A. A brief review of Odonata in mid-Cretaceous Burmese amber. Int. J. Odonatology. 2020;23:13–21. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data - The sequence datasets generated during the present study are available in NCBI (accession numbers in Table S1).
-
•
Code - We have no unreported custom computer code or algorithms.
-
•
Other - No “All other items” to report.