

Abstract
Here we report the synthesis of interesting 3-alkyl-4-hydroxy-1-aryl-4-(propa-1,2-dienyl)1H-pyrazol-5(4H)-ones and 9-alkyl-7-aryl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-ones, starting from 1,2-diaza-1,3-dienes (DDs) and propargyl alcohol. The reaction proceeds through a sequence Michael-type nucleophilic attack/cyclization/[2,3]-Wittig rearrangement. In the same way, the reaction between the aforementioned DDs and allyl alcohol furnished 4-allyl-4-hydroxy-3-alkyl-1-aryl-1H-pyrazol-5(4H)-ones. A DFT study was also carried out, in order to have decisive clarifications about the mechanism.
Keywords: [2,3]-Wittig rearrangement; pyrazolones; propargyl alcohol; allyl alcohol; 1,2-diaza-,1,3-dienes; DFT study
1. Introduction
Heterocycles, in particular those that contain one or more nitrogen atoms, are an extremely important class of compounds widely distributed in nature, as they represent the majority of active molecules employed in biological, pharmacological, and industrial chemistry [1,2,3,4,5].
Among them, pyrazolones play a crucial role as they have a wide range of applications, in particular in the pharmaceutical field [6,7,8]. In fact, they are known to have, along with many others, antimicrobial [9], antitubercular [10], antifungal [7,11,12,13], and antibacterial [7,11,12,13] properties, and also antitumor [7,14], gastric secretion stimulatory [15], anticonvulsant [16], and antimalarial [17] activities. Besides, they are employed as starting materials for the preparation of dyes. [18].
It is for this cause that organic chemists [19,20] as well as our group [21] are actively engaged in the development of new synthetic strategies for their synthesis.
In particular, spirocyclic motifs [19,20,21] containing pyrazolone core are widely represented in many synthetic bioactive and natural products [22,23,24,25,26,27,28,29]. So, several methods for the preparation of such complex three-dimensional structures have been developed starting from simple and readily accessible precursors [22,23,24,25,26,27,28,29].
On the other hand, the α-hydroxyallenes are also a valuable class of derivatives that constitute both versatile synthetic intermediates as well as the core of different compounds of synthetic or natural origin, which manifest biological activities [30,31,32,33,34,35,36].
Among the several approaches that have been employed for their synthesis [37,38,39,40,41,42,43,44,45], the [2,3]-Wittig rearrangement of propargylic ethers represents an efficient way to assemble them [46,47]. The major limitation of this versatile bond reorganization process, which has many other applications in organic synthesis, is mainly correlated to the use of strong bases and very low temperatures together with the laborious, complicated, and expensive procedures required [48,49,50].
Quite recently, we have published a preliminary paper, in which, by exploiting the ability of 1,2-diaza-1,3-dienes (DDs) as Michael-acceptors [51,52,53,54,55,56,57,58], we have synthesized α-(prop-2-yn-1-yloxy)hydrazones 3 (Scheme 1) [59]. These compounds containing a propargylic ether function have been demonstrated to be able to give easily and in very mild conditions the [2,3]-Wittig rearrangements. In detail, the synthesis involves 1-methyloxycarbonyl-DDs 1a–c and propargyl alcohol 2a [60,61] and it is conducted at room temperature, in dichloromethane in the presence of a catalytic amount of a base such as the 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU). In these conditions, the reaction has furnished the corresponding α-(prop-2-yn-1-yloxy)hydrazones 3a–c (41–61%), in 24.0–72.0 h (Scheme 1) [59], by means of the nucleophilic Michael-type attack of the oxygen atom of propargyl alcohol 2a to the terminal carbon atom of the DDs 1.
Scheme 1.

Synthesis of α-(prop-2-yn-1-yloxy)hydrazones 3a–c and of 9-alkyl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-ones 5a–c [59].
The subsequent treatment of the so-obtained hydrazonic compounds 3a–c with 4 equiv. of a weak base, as K2CO3, in methanol has furnished new and unexpected 9-alkyl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-ones 5a–c, in good yields (67–73%) in 1.0–1.5 h whose structure was confirmed by X-ray spectroscopy (Scheme 1) [59].
For the formation of products 5a–c, we hypothesized a preliminary base-promoted nucleophilic attack of the hydrazonic nitrogen at the ester group of Michael adducts 3, with the loss of an alcohol molecule, to give the pyrazolone ring, followed by a further cyclization process passing through a [2,3]-Wittig-type rearrangement [59]. In order to better clarify the proposed mechanism and to have some variability on the substituents of DDs as well as to increase the scope of the reaction, we have decided to extend our studies to investigate the reactivity of differently substituted DDs such as 1-aryl-DDs 1d–g with propargyl and allyl alcohols 2a,b, respectively. The results of this study are the object of the present work.
2. Results and Discussion
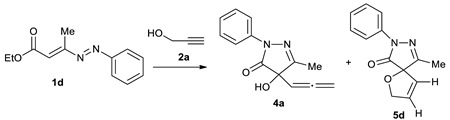
To conduct the optimization process, we chose as a representative model the 1-phenyl-DD 1d and the same propargyl alcohol 2a (Table 1). Initially, we have verified whether the conditions previously employed with the 1-methyloxycarbonyl-DDs 1a–c and 2a to obtain the adducts 3, that is CH2Cl2, DBU (0.1 equiv.) (Scheme 1) [59], can also be adapted to the DDs otherwise substituted such as 1d (Table 1, entry 1). Unfortunately, in this case, the reaction did not work at all. This occurrence is probably due to the lower electrophilicity of 1-aryl-DDs 1d–g (Scheme 2) compared to that of 1-methoxycarbonyl-DDs 1a–c (Scheme 1), as evidenced by kinetics studies previously published by some of us [62].
Table 1.
Screening of different conditions in the reaction between 1-phenyl-DD 1d and propargyl alcohol 2a to obtain 4-hydroxy-3-methyl-1-phenyl-4-(propa-1,2-dienyl)-1H-pyrazol-5(4H)-one 4a and 9-methyl-7-phenyl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-one 5d a.
| |||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | Equivalents | T | 4a; 5d Yield (%) b |
| 1 | CH2Cl2 | DBU | 0.1 | rt | no reaction |
| 2 | CH2Cl2 | DBU | 1 | rt | complicated mixture c |
| 3 | MeCN | DBU | 0.1 | rt | no reaction |
| 4 | MeCN | DBU | 0.1 | 60 °C | complicated mixture c |
| 5 | MeCN | DBU | 1 | rt | complicated mixture c |
| 6 | THF | DBU | 0.1 | rt | no reaction |
| 7 | THF | DBU | 1 | rt | complicated mixture c |
| 8 | 2a as sr d | DBU | 1 | rt | complicated mixture c |
| 9 | MeCN | DIPEA | 2 | rt | no reaction |
| 10 | MeCN | DIPEA | 4 | rt | no reaction |
| 11 | THF | DIPEA | 4 | rt | no reaction |
| 12 | CH2Cl2 | DIPEA | 4 | rt | no reaction |
| 13 | 2a as sr d | DIPEA | 4 | rt | no reaction |
| 14 | 2a as sr d | DIPEA | 4 | 60 °C | no reaction |
| 15 | MeCN | NaH | 0.1 | rt | complicated mixture c |
| 16 | THF | NaH | 0.1 | rt | complicated mixture c |
| 17 | THF | NaH | 0.1 | −20 °C | complicated mixture c |
| 18 | THF | NaH | 0.1 | −78 °C | complicated mixture c |
| 19 | CH2Cl2 | NaH | 0.1 | rt | complicated mixture c |
| 20 | 2a as sr d | NaH | 0.1 | rt | complicated mixture c |
| 21 | MeCN | MeONa | 0.1 | rt | complicated mixture c |
| 22 | THF | MeONa | 0.1 | rt | complicated mixture c |
| 23 | THF | MeONa | 0.1 | −20 °C | complicated mixture c |
| 24 | CH2Cl2 | MeONa | 0.1 | rt | complicated mixture c |
| 25 | 2a as sr d | MeONa | 0.1 | rt | complicated mixture c |
| 26 | THF | t-BuONa | 0.1 | rt | complicated mixture c |
| 27 | THF | t-BuONa | 0.1 | −20 °C | complicated mixture c |
| 28 | MeCN | K2CO3 | 4 | rt | complicated mixture c |
| 29 | THF | K2CO3 | 4 | rt | complicated mixture c |
| 30 | CH2Cl2 | K2CO3 | 4 | rt | complicated mixture c |
| 31 | 2a as sr d | K2CO3 | 4 | rt | complicated mixture c |
| 32 | 2a as sr d | K2CO3 | 4 | 60 °C | 4a: 11%; 5d: 29% |
| 33 | 2a as sr d | Cs2CO3 | 4 | 60 °C | 4a: 9%; 5d: 19% |
| 34 | 2a as sr d | Na2CO3 | 4 | 60 °C | 4a: 6%; 5d: 11% |
a The reactions were performed at 0.5 mmol scale of 1-phenyl-DD 1d in 3 mL of solvent, using 1.2 equiv. of alcohol 2a (except when 2a was employed as solvent/reagent). b Yields of isolated 4a and 5d, based on 1-phenyl-DD 1d. c TLC analysis revealed traces of products 4a and 5d. d Propargyl alcohol was used as solvent/reagent (3 mL of 2a were employed).
Scheme 2.

Synthesis and plausible mechanism for the base-promoted synthesis of 3-alkyl-4-hydroxy-1-aryl-4-(propa-1,2-dienyl)1H-pyrazol-5(4H)-ones 4a–d and of 9-alkyl-7-aryl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-ones 5d–g.
So, we have tested several solvents, such as dichloromethane, acetonitrile, and tetrahydrofuran, and also the same propargyl alcohol used both as solvent and reagent (Table 1). Furthermore, a series of organic and inorganic bases as promoters have been used, such as DBU, DIPEA, NaH, MeONa, and K2CO3 (Table 1).
The increment of the DBU to 1.0 equivalent in CH2Cl2, MeCN, THF, or using 2a as solvent/reagent (entries 2, 5, 7, 8), as well as the use of a catalytic amount of the DBU at 60 °C in MeCN (entry 4), have produced complicated reaction mixtures, in which a TLC analysis revealed the presence of 3-methyl-4-hydroxy-1-phenyl-4-(propa-1,2-dienyl)1H-pyrazol-5(4H)-one 4a and 9-methyl-7-phenyl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-one 5d (Table 1), but only in traces. The reactions carried out with DIPEA (entries 9–14) or a catalytic amount of DBU, both at r.t. and by heating (entries 1, 3, 4, and 6) did not work at all, while with NaH (entries 15–20) or MeONa (entries 21–25), they gave complicated mixtures, regardless of the solvent conditions employed.
Additionally, the attempts to carry out the reaction using strong bases at lower temperatures such as NaH at –20 °C (entry 17) or –78 °C (entry 18), as well as MeONa at –20 °C (entry 23) or t-BuONa at room temperature (entry 26) or at −20 °C (entry 27) have also failed, giving complicated reaction mixtures.
Additionally in the case of the use of 4 equiv. of K2CO3 in any solvent at room temperature, only traces of 4a and 5d were obtained (entries 23–26).
The trend of the reaction improves by using four equiv. of K2CO3 using 3 mL of propargy alcohol 2a as solvent/reagent at 60 °C, giving 4a and 5d in 11% and 29% yields, respectively, and these are the best conditions found in our screening (Table 1, entry 32).
At this point, to tentatively improve the yields, we have tried some different carbonates, such as Cs2CO3 (entry 33) or Na2CO3 (entry 34), in the same best conditions found with K2CO3 but we have obtained lower yields of 4a and 5d.
So, with these most optimal conditions possible in hand, we performed the reactions between 1-aryl-DDs 1d–g and propargyl alcohol 2a used as solvent-reagent, at 60 °C in the presence of 4 equiv. of K2CO3.
After 8.0–11.5 h, at the disappearance of the red color of the DDs, a TLC monitoring revealed a complicated reaction mixture in which there are two main spots, very close to each other. Their separation was very difficult having requested a first purification by column chromatography on silica gel and then by preparative thin-layer chromatography with more successive elutions. The so-obtained products were identified as 3-alkyl-4-hydroxy-1-aryl-4-(propa-1,2-dienyl)1H-pyrazol-5(4H)-ones 4a–d and 9-alkyl-7-aryl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-ones 5d–g (Scheme 2, Table 2). It is impossible to isolate other significant products as the reaction appears as a dirty complicated mixture of degradation compounds.
Table 2.
Yields and reaction times for the synthesis of 3-alkyl-4-hydroxy-1-aryl-4-(propa-1,2-dienyl)1H-pyrazol-5(4H)-ones 4a–d a and of 9-alkyl-7-aryl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-ones 5d–g.
| Entry | 1 | R1 | R2 | R3 | 4 | Yield (%) b | 5 | Yield (%) c | Time (h) d |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1d | Et | Me | H | 4a | 11 | 5d | 29 | 11.5 |
| 2 | 1e | Me | Me | Cl | 4b | 10 | 5e | 17 | 10.0 |
| 3 | 1f | Me | Me | OMe | 4c | 14 | 5f | 21 | 9.0 |
| 4 | 1g | Me | Et | H | 4d | 18 | 5g | 18 | 8.0 |
a Reaction conditions: DDs 1d–g (1 mmol), propargyl alcohol 2a (3 mL), K2CO3 (4 mmol), at 60 °C (oil bath). b Yield of pure isolated 3-alkyl-4-hydroxy-1-aryl-4-(propa-1,2-dienyl)1H-pyrazol-5(4H)-ones 4a–d referred to 1. c Yield of pure isolated 9-alkyl-7-aryl-1-oxa-7,8- diazaspiro[4.4]nona-3,8-dien-6-ones 5d–g referred to 1. d Time of disappearance of 1.
As the first step of the reaction, we hypothesized the formation of the non-isolable hydrazone intermediates 3, by means of the nucleophilic Michael-type attack of the oxygen atom of propargyl alcohol 2a to the terminal carbon atom of the azoene system of the DDs 1. The base-promoted nucleophilic attack of the hydrazonic nitrogen at the ester group of hydrazones 3, with the loss of an alcohol molecule, gives the corresponding non-isolable pyrazolone derivative I (Scheme 2).
In order to obtain decisive information to explain the subsequent Wittig rearrangement involved in the formation of the following final products 4a–d and 5d–g, a DFT study was conducted.
The base-promoted deprotonation of the intermediate I can occur at the α or α′ positions, leading to the formation of anion species II or II′, respectively (Scheme 3). Intermediate II can theoretically undergo both [1,2]- or [2,3]-Wittig rearrangements, furnishing products III or IV, respectively, while intermediate II′ can give the [1,2]- or [1,4]-rearrangement, giving products III′ or III″, respectively (Scheme 3) [40,47,49,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81].
Scheme 3.

Possible Wittig rearrangements of 4-(prop-2-yn-1-yloxy)-1H-pyrazol-5(4H)-ones I.
It is noteworthy that the process here described is highly regioselective, since the deprotonation occurs only in the α position of the intermediate I to produce II, as this proton is more acid than the one in the α′ position, being activated both from an amidic carboxylic group as well as from an imino function. As a confirmation of this event, the two products of deprotonation II and II′ have been optimized by DFT methods. As supposed, the former II resulted in being more stable than the latter by 33.8 kcal/mol (see Supplementary Materials).
Then, in the formation of the 3-alkyl-4-hydroxy-1-aryl-4-(propa-1,2-dienyl)1H-pyrazol-5(4H)-ones 4a–d, the Wittig rearrangement observed is of the [2,3]-type, as the spectroscopic data of the products 4 clearly support (Scheme 2 and Scheme 3). To exclude that, in the concomitant formation of the 9-alkyl-7-aryl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-ones 5d–g, the [1,2]-Wittig rearrangement was involved by means of cyclization of intermediate III (Scheme 3), a DFT study has been carried out.
From intermediate II (Scheme 3), the two different possibilities, that is [1,2]- and [2,3]-rearrangement, have been explored. In the former, a single transition state (TS1) is necessary to obtain the final product, but the activation free energy is quite high (ΔGǂ = 49 kcal/mol) due to the strain of the incipient three-members ring in TS1 (Figure 1). On the other hand, for the [2,3]-rearrangement, two different TSs (TS2 and TS3) are necessary. In the former, the carbanion of II attacks the terminal propargylic carbon giving the conjugated base of 9-alkyl-7-aryl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-one IV. Successively, in TS3, the bond between the oxygen and the carbon 1 breaks, giving V, which after reprotonation, will lead to 4a. The free energy of TS2 and TS3 are quite similar (11.1 and 10.4 kcal/mol, respectively).
Figure 1.

Energy profile for the [1,2]- and [2,3]-rearrangement for the propargylic derivative.
The energy profile for the two mechanisms (Figure 1) shows that the [2,3]-rearrangement is largely favored over the other one, both thermodynamically and kinetically, de facto excluding that a [1,2]-Wittig rearrangement may be involved in the concomitant formation of 5. So, the formation of the 9-alkyl-7-aryl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-ones 5d–g happens by means of the protonation of intermediate IV (Scheme 2, Figure 1).
However, the high activation energy determined by the DFT study for the formation of final products 4 and 5 can explain why their yields are so low. On the other hand, it is reported in the literature that the yields of Wittig rearrangement products are commonly quite low [63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84].
It is noteworthy that, commonly to trigger the Wittig rearrangements, the use of strong Brønsted bases, such as BuLi or t-BuLi [63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84], is required, and usually, the reactions happen at very low temperatures. In our system instead, much milder conditions, such as a weak base as K2CO3 and a temperature of 60 °C, are able to efficiently promote the α-deprotonation and the consequent [2,3]-Wittig rearrangement. This aspect together with the possibility to conduct the synthesis under solvent-free conditions makes it eco-friendly and less harmful to the environment.
It is well known that allyl alcohol is a valuable building block in organic syntheses, due to its versatile reactivity as alkylating agent [82,83,84,85].
For this fact and also with the intent to verify if the mild conditions employed for [2,3]-Wittig rearrangement in the reaction of the DDs 1 with propargyl alcohol 2a could be extended to other substrates, we have planned to conduct the reaction between DDs 1d–h and allyl/crotyl alcohol 2b,c (Scheme 4). To our great pleasure, the reactions conducted using 2b, as solvent and reagent, at 60 °C and in the presence of 4 equiv. of K2CO3, have actually provided the corresponding 4-allyl-4-hydroxy-3-alkyl-1aryl-1H-pyrazol-5(4H)-ones 6a–e (25–70%) (Scheme 4, Path A, Table 3).
Scheme 4.

Synthesis of hydrazonic adducts 3d,e, 4-allyl-4-hydroxy-3-alkyl-1-aryl-1H-pyrazol-5(4H)-ones 6a–d and tert-butyl 4-allyl-4-hydroxy-3-methyl-5-oxo-4,5-dihydro-1H-pyrazole-1-carboxylate 6e.
Table 3.
Yields and reaction times for the synthesis of adducts 3d,e, and 1H-pyrazol-5(4H)-ones 6a–e.
| Entry | 1 | R1 | R2 | R3 | 2 | R4 | 3 a | Yield (%) b |
Time (h) |
6 | Yield (%) |
Time (h) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1d | Et | Me | Ph | 2b | H | 6a c | 25 e | 8.0 | |||
| 2 | 1e | Me | Me | 4-Cl-Ph | 2b | H | 6b c | 33 e | 10.0 | |||
| 3 | 1f | Me | Me | 4-OMe-Ph | 2b | H | 6c c | 33 e | 6.0 | |||
| 4 | 1g | Me | Et | Ph | 2b | H | 6d c | 28 e | 16.0 | |||
| 5 | 1h | Et | Me | COOBut | 2b | H | 3d | 37 | 0.1 | 6e d | 70 f | 4.0 |
| 6 | 1h | Et | Me | COOBut | 2c | Me | 3e | 22 | 0.1 |
a Reaction conditions: DD 1h (1 mmol), allyl (2b) or crotyl (2c) alcohol (3 mL), K2CO3 (1 mmol), rt. b Yields of 3d,e referred to 1. c Reaction conditions: DDs 1d–g (1 mmol), allyl alcohol 2b (3 mL), K2CO3 (4 mmol), at 60 °C (oil bath). d Reaction conditions: adduct 3d (1 mmol) in ethanol (3 mL), K2CO3 (1 mmol), rt. e Yields of 4-allyl-4-hydroxy-3-alkyl-1-aryl-1H-pyrazol-5(4H)-ones 6a–d referred to 1. f Yield of tert-butyl 4-allyl-4-hydroxy-3-methyl-5-oxo-4,5-dihydro-1H-pyrazole-1-carboxylate 6e referred to 3d.
Also in this case, the reaction proceeds through the preliminary formation of a hydrazonic adduct intermediate 3, by means of the nucleophilic attack of the oxygen of allyl alcohol 2 to the terminal carbon atom of the azoene system of the DD 1, followed by a base-promoted cyclization due to a nucleophilic attack of the hydrazonic nitrogen at the ester group of hydrazone 3, with the loss of an alcohol molecule, to give the corresponding non-isolable pyrazolone VI (Scheme 5). Now, the base-promoted loss of the hydrogen in the 4 position of the pyrazolone ring can theoretically promote both [1,2]- and [2,3]-Wittig rearrangements, however, furnishing in both cases the same final products 6a–d (Scheme 4 and Scheme 5) [17,18].
Scheme 5.

Possible [1,2]- or [2,3]-Wittig rearrangements of 4-(allyloxy)-1H-pyrazol-5(4H)-ones III.
So, to clarify which of the two mechanisms was involved, we tried to introduce a methyl substituent on the terminal carbon atom of the double bond of the alcohol and therefore we tested the crotyl alcohol 2c (R4 = Me, Scheme 4 and Scheme 5) in the reaction with 1-aryl-DDs 1d–g. To our large disappointment, in all cases, the reaction was unsuccessful, despite using various conditions of solvent, base, and temperature (for the conditions tested, see Table S1 in the Supplementary Materials).
We have then investigated the behavior of t-butoxycarbonyl-DD 1h, chosen as an example, by virtue of its incremented electrophilic character due to the replacement of the aryl on the N1 of the azo-ene system with the BOC moiety [62].
As supposed, both allyl (2b) and crotyl (2c) alcohol reacted with 1h, at room temperature, with only one equivalent of K2CO3, under solvent-free conditions, furnishing the hydrazonic adduct intermediates 3d,e (Scheme 4, Path B and Scheme 5, Table 3). Unfortunately, while compound 3d, if treated with 1 equiv. of K2CO3 in ethanol, was converted into the corresponding 4-allyl-4-hydroxy-3-alkyl-1-alcoxycarbonyl-1H-pyrazol-5(4H)-one 6e, 3e did not furnish the corresponding pyrazol-5(4H)-one under any of the countless conditions tested (Scheme 4, Path B and Scheme 5, Table 3. For the conditions tested, see Table S2 in the Supplementary Materials).
Then, a DFT study was carried out for the reaction between the allylic moiety and DDs, for which, given the lack of reactivity of substituted allyl alcohol, it is difficult to obtain experimental information about the mechanism. Also, in this case, the strain in the TS of the [1,2]-rearrangement (TS1A) makes this path high in energy (ΔGǂ = 57.1 kcal/mol) and not a viable option. Differently than before, the [2,3]-rearrangement is not a two-step mechanism, as the formation of the Cα-C1 bond and the O-C1 bond cleavage are concerted (TS2A). The activation barrier of the [2,3]-rearrangement is 16.2 kcal/mol, which is higher than in the case of the propargylic moiety but still viable, in principle (Figure 2).
Figure 2.

Energy profile for the [1,2]- and [2,3]-rearrangement for the allylic derivative.
Both the mechanisms lead to the same product, VII, but the latter is less stable than the starting material, VI, by 1.4 kcal/mol. This difference is positive and almost constant with all the dielectric values used in the calculations, ranging from toluene to water.
Using the crotyl alcohol, two different products, 7 or 8, are possible, depending on the reaction mechanism (Scheme 5). Both the [1,2]- and [2,3]-rearrangement would lead to a thermodynamically unfavored product (ΔG° = 2.8 and 7.3 kcal/mol).
Finally, if using 1-t-butoxycarbonyl-DD 1h instead of the 1-aryl-ones, the framework is slightly different. In this case, the use of allyl alcohol leads to a product that is thermodynamically favored (9, ΔG° = −1.1 kcal/mol) and it is the same with both the [1,2]- and [2,3]-rearrangements. Given the previous results (Figure 2), it is most likely that only the [2,3]-rearrangement is active. The use of crotyl alcohol leads to a slightly different scenario: the [1,2]-rearrangement would lead to a substantially thermoneutral product (10, ΔG° = −0.09 kcal/mol), but, as seen before, this way is kinetically forbidden, whereas the [2,3]-rearrangement, which would be kinetically viable, leads to a product that is thermodynamically forbidden (11, ΔG° = 6.7 kcal/mol) (see Supp. Information).
3. Experimental Section
3.1. General
All chemicals and solvents were purchased from commercial suppliers and used as received. 1,2-Diaza-1,3-dienes were prepared as reported [86,87,88] and used as EE/EZ isomer mixtures. Melting points were determined in open capillary tubes and are uncorrected. FTIR spectra were obtained as Nujol mulls. All 1H NMR and 13C NMR spectra were recorded at 400 and 100 MHz, respectively. Proton and carbon spectra were referenced internally to solvent signals, using values of δ = 7.27 ppm for proton and δ = 77.00 ppm for carbon (middle peak) in CDCl3. All coupling constants (J) are given in Hz. All the NH and OH exchanged with D2O. Precoated silica gel plates of 0.25 mm were employed for analytical thin-layer chromatography. All new compounds showed satisfactory elemental analysis. Mass spectra were recorded in the ESI-MS mode. The nomenclature was generated using ACD/IUPAC Name (version 3.50, 5 April 1998), Advanced Chemistry Development Inc., Toronto, ON, Canada.
3.2. General Procedure for the Synthesis of 3-Alkyl-4-hydroxy-1-aryl-4-(propa-1,2-dienyl) 1H-Pyrazol-5(4H)-ones 4a–d and of 9-Alkyl-7-aryl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-ones 5d–g, Starting from 1,2-Diaza-1,3-dienes 1d–g and Propargyl Alcohol 2a
To a magnetically stirred mixture of 1,2-diaza-1,3-diene 1d–g (1 mmol) and propargyl alcohol 2a (3 mL) at 60 °C (oil bath), K2CO3 (4 mmol) was added and the suspension was left to stand under these conditions for the appropriate time (8.0–11.5 h) until the disappearance of the reagent 1 (TLC monitoring). The crude mixture was then purified by column chromatography on silica gel and successively on thin-layer chromatography to afford 3-alkyl-4-hydroxy-1-aryl-4-(propa-1,2-dienyl)1H-pyrazol-5(4H)-ones 4a–d and 9-alkyl-7-aryl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-ones 5d–g. Compounds 4b,d and 5e,g were crystallized from EtOAc-light petroleum ether, while 4a,c and 5d,f were found to be oils.
4-Hydroxy-3-methyl-1-phenyl-4-(propa-1,2-dienyl)-1H-pyrazol-5(4H)-one (4a).
4a was isolated by column chromatography (acetate/cyclohexane 20:80) and then by thin-layer chromatography (three elutions in acetate/cyclohexane 20:80 mixture) in 11% yield as brown oil; 1H-NMR (400 MHz, CDCl3, 25 °C): δ 2.21 (s, 3H), 4.09 (brs, 1H), 5.09 (d, J = 6.4, 2H), 5.40 (t, J = 6.4 Hz, 1H), 7.20 (t, J = 7.6, 1Har), 7.40 (t, J = 7.2 Hz, 2Har), 7.86 (d, J = 8.8 Hz, 2Har) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ 13.3 (q), 78.0 (s), 80.7 (t), 89.7 (d), 118.8 (d), 125.3 (d), 128.8 (d), 137.6 (s), 161.2 (s), 172.4 (s), 207.6 (s) ppm; IR (Nujol, cm−1): νmax 3302, 1953, 1719; MS (ESI): m/z 229.25 [M + H+]; anal. calcd. for C13H12N2O2 (228.25): C 68.41, H 5.30, N 12.27; found: C 68.58, H 5.35, N 12.18.
1-(4-Chlorophenyl)-4-hydroxy-3-methyl-4-(propa-1,2-dienyl)-1H-pyrazol-5(4H)-one (4b).
4b was isolated by column chromatography (acetate/cyclohexane 20:80) and then by thin-layer chromatography (three elutions in acetate/cyclohexane 20:80 mixture) in 10% yield as a brown solid; mp: 85–87 °C; 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 2.20 (s, 3H), 4.28–4.30 (m, 1H), 5.13 (d, J = 6.4 Hz, 2H), 5.36 (t, J = 6.4 Hz, 1H), 7.36 (d, J = 8.8 Hz, 2Har), 7.83 (d, J = 9.2 Hz, 2Har) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ 13.3 (q), 77.6 (s), 81.3 (t), 89.6 (d), 119.8 (d), 128.9 (d), 130.4 (s), 136.2 (s), 160.9 (s), 171.8 (s), 207.6 (s) ppm; IR (Nujol, cm−1): νmax 3302, 1953, 1719; MS (ESI): m/z 261.22 [M − H+]; anal. calcd. for C13H11N2O2Cl (262.69): C 59.44, H 4.22, N 10.66; found: C 59.21, H 4.28, N 10.60.
4-Hydroxy-1-(4-methoxyphenyl)-3-methyl-4-(propa-1,2-dienyl)-1H-pyrazol-5(4H)-one (4c).
4c was isolated by column chromatography (acetate/cyclohexane 20:80) and then by thin-layer chromatography (three elutions in acetate/cyclohexane 20:80 mixture) in 14% yield as brown oil; 1H-NMR (400 MHz, CDCl3, 25 °C): δ 2.20 (s, 3H), 3.83 (s, 3H), 4.29 (d, J = 2.4 Hz, 1H), 5.15 (d, J = 6.8 Hz, 2H), 5.35 (t, J = 6.8 Hz, 1H), 6.94 (d, J = 9.2 Hz, 2Har), 7.74 (d, J = 9.2 Hz, 2Har) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ 13.2 (q), 55.5 (q), 77.2 (s), 81.2 (t), 89.9 (d), 114.1 (d), 120.6 (d), 130.6 (s), 157.2 (s), 160.4 (s), 171.4 (s), 207.6 (s) ppm; IR (Nujol, cm−1): νmax 3242, 1956, 1712, 1678; MS (ESI): m/z 259.12 [M + H+]; anal. calcd. for C14H14N2O3 (258.27): C 65.11, H 5.46, N 10.85; found: C 65.22, H 5.40, N 10.90.
3-Ethyl-4-hydroxy-1-phenyl-4-(propa-1,2-dienyl)-1H-pyrazol-5(4H)-one (4d).
4d was isolated by column chromatography (acetate/cyclohexane 20:80) and then by thin-layer chromatography (three elutions in acetate/cyclohexane 20:80 mixture) in 18% yield as an orange solid; mp: 101–103; 1H-NMR (400 MHz, CDCl3, 25 °C): δ 1.32 (t, J = 7.2 Hz, 3H), 2.55–2.66 (m, 2H), 4.27–4.29 (m, 1H), 5.11 (d, J = 6.8 Hz, 2H), 5.37 (t, J = 6.4 Hz, 1H), 7.21 (t, J = 7.2 Hz, 2Har), 7.41 (t, J = 7.6 Hz, 2Har), 7.89 (d, J = 8. Hz, 1Har) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 9.3 (q), 21.0 (t), 78.1 (s), 81.1 (t), 90.1 (d), 118.8 (d), 125.3 (d), 128.8 (d), 137.7 (s), 164.6 (s), 172.4 (s), 207.5 (s) ppm; IR (Nujol, cm−1): νmax 3279, 1956, 1691; MS (ESI): m/z 243.12 [M + H+]; anal. calcd. for C14H14N2O2 (242.27): C 69.41, H 5.82, N 11.56; found: C 69.32, H 5.89, N 11.65.
9-Methyl-7-phenyl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-one (5d).
5d was isolated by column chromatography (acetate/cyclohexane 20:80) and then by thin-layer chromatography (three elutions in acetate/cyclohexane 20:80 mixture) in 29% yield as orange oil; 1H-NMR (400 MHz, CDCl3, 25 °C): δ 2.09 (s, 3H), 4.94–4.99 and 5.12–5.16 (2m, 2H), 5.62–5.65 (m, 1H), 6.46–6.48 (m, 1H), 7.19 (t, J = 7.6 Hz, 1Har), 7.40 (t, J = 7.6 Hz, 2Har), 7.89 (dd, J = 8.4 Hz, J = 1.2, 2Har) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 13.1 (q), 78.4 (t), 93.7 (s), 118.5 (d), 123.8 (d), 125.1 (d), 128.8 (d), 133.5 (d), 138.0 (s), 159.8 (s), 170.8 (s) ppm; IR (Nujol, cm−1): νmax 3314, 3081, 1762, 1597; MS (ESI): m/z 227.20 [M − H+]; anal. calcd. for C13H12N2O2 (228.25): C 68.41, H 5.30, N 12.27; found: C 68.31, H 5.38, N 12.39.
7-(4-Chlorophenyl)-9-methyl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-one (5e).
5e was isolated by column chromatography (acetate/cyclohexane 20:80) and then by thin-layer chromatography (three elutions in acetate/cyclohexane 20:80 mixture) in 17% yield as an orange solid; mp: 108–109 °C; 1H-NMR (400 MHz, CDCl3, 25 °C): δ 2.08 (s, 3H), 4.94–4.98 and 5.11–5.15 (2m, 2H), 5.62–5.64 (m, 1H), 6.47–6.49 (m, 1H), 7.36 (d, J = 9.2 Hz, 2Har), 7.86 (d, J = 9.2 Hz, 2Har) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ 13.1 (q), 78.4 (t), 93.6 (s), 119.6 (d), 123.6 (d), 128.9 (d), 130.1 (s), 133.7 (d), 136.6 (s), 160.1 (s), 170.7 (s) ppm; IR (Nujol, cm−1): νmax = 3278, 3093, 1722, 1595; MS (ESI): m/z 261.22 [M − H+]; anal. calcd. for C13H11N2O2Cl (262.69): C 59.44, H 4.22, N 10.66; found: C 59.56, H 4.28, N 10.74.
7-(4-Methoxyphenyl)-9-methyl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-one (5f).
5f was isolated by column chromatography (acetate/cyclohexane 20:80) and then by thin-layer chromatography (three elutions in acetate/cyclohexane 20:80 mixture) in 21% yield as brown oil; 1H-NMR (400 MHz, CDCl3, 25 °C): δ 2.08 (s, 3H), 3.82 (s, 3H), 4.94–4.98 and 5.12–5.16 (2m, 2H), 5.62–5.65 (m, 1H), 6.46–6.48 (m, 1H), 6.93 (d, J = 9.2 Hz, 2Har), 7.77 (d, J = 9.2 Hz, 2Har) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ 13.1 (q), 55.5 (q), 78.4 (t), 93.6 (s), 114.0 (d), 120.4 (d), 123.8 (d), 131.4 (s), 133.4 (d), 157.0 (s), 159.7 (s), 170.4 (s) ppm; IR (Nujol, cm−1): νmax 3279, 3086, 1731, 1717, 1610; MS (ESI): m/z 257.13 [M − H+]; anal. calcd. for C14H14N2O3 (258.27): C 65.11, H 5.46, N 10.85; found: C 64.99, H 5.54, N 10.76.
9-Ethyl-7-phenyl-1-oxa-7,8-diazaspiro[4.4]nona-3,8-dien-6-one (5g).
5g was isolated by column chromatography (acetate/cyclohexane 20:80) and then by thin-layer chromatography (three elutions in acetate/cyclohexane 20:80 mixture) in 18% yield as a yellow solid; mp: 121–123 °C; 1H-NMR (400 MHz, CDCl3, 25 °C): δ 1.27 (t, J = 7.2 Hz, 3H), 2.34–2.54 (m, 2H), 4.93–4.97 and 5.11–5.15 (2m, 2H), 5.63–5.66 (m, 1H), 6.44–6.46 (m, 1H), 7.19 (t, J = 7.6 Hz, 1Har), 7.41 (t, J = 7.6 Hz, 2Har), 7.92 (dd, J = 8.8 Hz, J = 1.2, 2Har) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ 9.7 (q), 21.1 (t), 78.2 (t), 93.7 (s), 118.5 (d), 124.1 (d), 125.0 (d), 128.8 (d), 133.1 (d), 138.0 (s), 163.5 (s), 170.9 (s) ppm; IR (Nujol, cm−1): νmax 3096, 1714, 1632, 1597; MS (ESI): m/z 241.25 [M − H+]; anal. calcd. for C14H14N2O2 (242.27): C 69.41, H 5.82, N 11.56; found: C 69.31, H 5.77, N 11.62.
3.3. General Procedure for the Synthesis of Tert-Butyl 2-(3-(Allyloxy)-4-ethoxy-4-oxobutan-2-ylidene) Hydrazinecarboxylate (3d) or Tert-Butyl 2-(3-(but-2-en-1-yloxy)-4-ethoxy-4-oxobutan-2-ylidene) Hydrazinecarboxylate (3e), Starting from 1,2-Diaza-1,3-diene 1h and Allyl (2b) or Crotyl (2c) Alcohol
To a magnetically stirred mixture of 1,2-diaza-1,3-diene 1h (1 mmol) and allyl (2b) or crotyl (2c) alcohol (10 equiv.) at room temperature, K2CO3 (1 mmol) was added and the suspension was left to stand under these conditions for 0.1 h until the disappearance of the reagent 1 (TLC monitoring). The crude mixture was quickly filtered and then purified by column chromatography on silica gel to afford compounds 3d,e, that were crystallized from EtOAc.
Tert-butyl 2-(3-(allyloxy)-4-ethoxy-4-oxobutan-2-ylidene)hydrazinecarboxylate (3d).
3d was isolated by column chromatography (acetate/cyclohexane 40:60) in 37% yield as a white solid; mp: 119–120 °C; 1H-NMR (400 MHz, CDCl3, 25 °C): δ 1.27 (t, J = 7.2 Hz, 3H), 1.52 (s, 9H), 1.83 (s, 3H), 4.02–4.12 (m, 2H), 4.20–4.28 (m, 2H), 4.63 (s, 1H), 5.21 (d, J = 10.0 Hz, 1H), 5.28–5.35 (m, 1H), 5.86–5.95 (m, 1H), 7.60 (s, 1H) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ 10.8 (q), 14.1 (q), 28.2 (q), 61.4 (t), 70.8 (t), 81.6 (s), 81.8 (d), 118.2 (t), 133.5 (d), 146.8 (s), 152.3 (s), 169.1 (s) ppm; IR (Nujol, cm−1): νmax 3235, 1760, 1725, 1704; MS (ESI): m/z 303.22 [M + H+]; anal. calcd. for C14H24N2O5 (302.17): C 55.98, H 8.05, N 9.33; found: C 56.16, H 8.40, N 9.18.
Tert-butyl 2-(3-(but-2-en-1-yloxy)-4-ethoxy-4-oxobutan-2-ylidene)hydrazinecarboxylate (3e).
3e was isolated by column chromatography (acetate/cyclohexane 40:60) in 22% yield as a white solid; mp: 158–160 °C; 1H-NMR (400 MHz, CDCl3, 25 °C): δ 1.27 (t, J = 7.2 Hz, 3H), 1.52 (s, 9H), 1.71 (dd, J = 6.4 Hz, J = 0.8 Hz, 3H), 1.82 (s, 3H), 3.96–4.01 (m, 2H), 4.22 (q, J = 7.2 Hz, 2H), 4.62 (s, 1H), 5.53–5.61 (m, 1H), 5.70–5.79 (m, 1H), 7.57 (s, 1H) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ 10.8 (q), 14.2 (q), 17.8 (q), 28.2 (q), 61.4 (t), 70.6 (t), 81.4 (d), 81.5 (s), 126.3 (d), 131.1 (d), 146.9 (s), 152.2 (s), 169.3 (s) ppm; IR (Nujol, cm−1): νmax 3372, 3269, 1715, 1627; MS (ESI): m/z 313.09 [M − H+]; anal. calcd. for C15H26N2O5 (314.18): C 57.31, H 8.34, N 8.91; found: C 57.16, H 8.41, N 8.98.
3.4. General Procedure for the Synthesis of 4-Allyl-4-hydroxy-3-alkyl-1-aryl-1H-pyrazol-5(4H)-ones 6a–d, Starting from 1,2-Diaza-1,3-dienes 1d–g and Allyl Alcohol 2b
To a magnetically stirred mixture of 1,2-diaza-1,3-dienes 1d–g (1 mmol) and allyl alcohol 2b (3 mL) at 60 °C (oil bath), K2CO3 (4 mmol) was added and the suspension was left to stand under these conditions for the appropriate time (8.0–16.0 h), until the disappearance of the reagent 1 (TLC monitoring). The crude mixture was then purified by column chromatography on silica gel to afford 4-allyl-4-hydroxy-3-alkyl-1-aryl-1H-pyrazol-5(4H)-ones 6a–d as oils, in the case of 6a,c,d or solid that was crystallized from EtOAc-light petroleum ether in the case of 6b.
3.5. General Procedure for the Synthesis of 4-Allyl-4-hydroxy-3-methyl-1-alcoxycarbonyl-1H-pyrazol-5(4H)-one 6e, Starting from 3d
To a magnetically stirred solution of tert-butyl 2-(3-(allyloxy)-4-ethoxy-4-oxobutan -2-ylidene)hydrazinecarboxylate 3d (1 mmol) in ethanol (3 mL), K2CO3 (1 mmol) was added and the suspension was left to stand under these conditions for 2.0 h, until the disappearance of the reagent 3 (TLC monitoring). The crude mixture was then filtered and purified by column chromatography on silica gel to afford 6e as oil.
4-Allyl-4-hydroxy-3-methyl-1-phenyl-1H-pyrazol-5(4H)-one (6a).
6a was isolated by column chromatography (acetate/cyclohexane 20:80) in 25% yield as brown oil; 1H-NMR (400 MHz, CDCl3, 25 °C): δ 2.18 (s, 3H), 2.59–2.73 (m, 2H), 3.90 (brs, 1H), 5.15–5.24 (m, 2H), 5.55–5.65 (m, 1H), 7.20 (t, J = 7.6 Hz, 1Har), 7.39 (t, J = 8.8 Hz, 2Har), 7.83 (d, J = 7.6 Hz, 2Har) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ 13.2 (q), 40.6 (t), 79.6 (s), 118.9 (d), 121.4 (t), 125.4 (d), 128.5 (d), 128.8 (d), 137.4 (s), 161.5 (s), 173.4 (s) ppm; IR (Nujol, cm−1): νmax 3263, 1683, 1596; MS (ESI): m/z 231.30 [M + H+]; anal. calcd. for C13H14N2O2 (230.26): C 67.81, H 6.13, N 12.17; found: C 67.70, H 6.19, N 12.22.
4-Allyl-1-(4-chlorophenyl)-4-hydroxy-3-methyl-1H-pyrazol-5(4H)-one (6b).
6b was isolated by column chromatography (acetate/cyclohexane 20:80) in 33% yield as a yellow solid crystallized from EtOAc-light petroleum ether; mp: 131–133 °C; 1H-NMR (400 MHz, CDCl3, 25 °C): δ 2.18 (s, 3H), 2.58–2.71 (m, 2H), 4.19 (brs, 1H), 5.15–5.24 (m, 2H), 5.52–5.62 (m, 1H), 7.33 (d, J = 8.8 Hz, 2Har), 7.79 (d, J = 8.8 Hz, 2Har) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ 13.2 (q), 40.5 (t), 79.6 (s), 119.9 (d), 121.5 (t), 128.3 (d), 128.9 (d), 130.5 (s), 135.9 (s), 161.9 (s), 173.5 (s) ppm; IR (Nujol, cm−1): νmax 3302, 1685, 1625; MS (ESI): m/z 265.09 [M + H+]; anal. calcd. for C13H13N2O2Cl (264.71): C 58.99, H 4.95, N 10.58; found: C 59.11, H 4.89, N 10.66.
4-Allyl-4-hydroxy-1-(4-methoxyphenyl)-3-methyl-1H-pyrazol-5(4H)-one (6c).
6c was isolated by column chromatography (acetate/cyclohexane 20:80) in 33% yield as brown oil; 1H-NMR (400 MHz, CDCl3, 25 °C): δ 2.17 (s, 3H), 2.60–2.64 (m, 2H), 3.82 (s, 3H), 4.10 (brs, 1H), 5.20–5.27 (m, 2H), 5.60–5.69 (m, 1H), 6.93 (d, J = 9.2 Hz, 2Har), 7.72 (d, J = 9.2 Hz, 2Har) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ 13.3 (q), 40.7 (t), 55.5 (q), 79.0 (s), 114.0 (d), 120.7 (d), 121.5 (t), 128.5 (d), 130.8 (s), 157.2 (s), 160.8 (s), 172.5 (s) ppm; IR (Nujol, cm−1): νmax 3357, 1693, 1608; MS (ESI): m/z 259.23 [M − H+]; anal. calcd. for C14H16N2O3 (260.29): C 64.60, H 6.20, N 10.76; found: C 64.51, H 6.24, N 10.67.
4-Allyl-3-ethyl-4-hydroxy-1-phenyl-1H-pyrazol-5(4H)-one (6d).
6d was isolated by column chromatography (acetate/cyclohexane 20:80) in 28% yield as red oil; 1H-NMR (400 MHz, CDCl3, 25 °C): δ 1.32 (t, J = 7.2 Hz, 3H), 2.44–2.70 (m, 4H), 3.65 (brs, 1H), 5.16–5.25 (m, 2H), 5.56–5.66 (m, 1H), 7.20 (t, J = 7.2 Hz, 1Har), 7.40 (t, J = 8.4 Hz, 2Har), 7.87 (dd, J = 8.8 Hz, J = 1.2 Hz, 2Har) ppm; 13C NMR (100 MHz, CDCl3, 25 °C): δ 9.1 (q), 21.0 (t), 40.9 (t), 79.7 (s), 118.8 (d), 121.4 (t), 125.2 (d), 128.6 (d), 128.8 (d), 137.6 (s), 164.9 (s), 173.5 (s) ppm; IR (Nujol, cm−1): νmax 3313, 1689, 1625, 1597; MS (ESI): m/z 245.14 [M + H+]; anal. calcd. for C14H16N2O2 (244.29): C 67.83, H 6.60, N 11.47; found: C 67.71, H 6.65, N 11.54.
Tert-butyl 4-allyl-4-hydroxy-3-methyl-5-oxo-4,5-dihydro-1H-pyrazole-1-carboxylate (6e).
6e was isolated by column chromatography (acetate/cyclohexane 20:80) in 70% yield as pale yellow oil; 1H-NMR (400 MHz, CDCl3, 25 °C): δ 1.59 (s, 9H), 2.13 (s, 3H), 2.55 (d, J = 7.2 Hz, 2H), 3.10 (brs, 1H), 5.20–5.30 (m, 2H), 5.61–5.71 (m, 1H) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ 13.4 (q), 28.0 (q), 40.3 (t), 78.1 (s), 84.9 (s), 122.2 (t), 127.9 (d), 147.5 (s), 161.5 (s), 173.2 (s) ppm; IR (Nujol, cm−1): νmax 3268, 1678, 1619; MS (ESI): m/z 253.22 [M − H+]; anal. calcd. for C12H18N2O4 (254.28): C 56.68, H 7.13, N 11.02; found: C 56.84, H 7.02, N 10.86.
4. DFT Calculations
All the geometries have been optimized with ORCA 4.1.0 [89,90], using the BP86 functional in conjunction with a def2-TZVP basis set for all the atoms. Dispersion forces were taken into account using the D3 correction with Becke−Johnson damping [91]. The effect of the solvent has been simulated through the Continuum-like Polarizable Continuum Model (C-PCM, dichloromethane if not otherwise specified). All the geometries have been confirmed to be stationary points, with zero (intermediates) or one (transition states) imaginary frequency.
5. Conclusions
In conclusion, we have established a protocol to synthesize new and appealing 4-hydroxy-4-(propa-1,2-dienyl) or 4-hydroxy-4-allyl-1H-pyrazol-5(4H)-ones and spiro dihydrofuran-pyrazolones, starting from 1,2-diaza-1,3-dienes and propargyl or allyl alcohols under very mild conditions. The transformation shows attractive features since it excludes the employment of strong bases and low temperatures.
The obtained products have potentially interesting properties. In fact, often, the biological activities of molecules that incorporate more scaffolds are not simply attributable to the sum of the characteristics shown by the individual functionalities, but synergistic effects can increase its effectiveness, or induce the manifestation of new characteristics.
Finally, a DFT study carried out on these reactions allowed us to obtain definitive elucidations on the mechanism which involves a [2,3]-Wittig rearrangement.
Acknowledgments
The authors thank Anna Maria Gioacchini who competently performed the MS spectra.
Supplementary Materials
The following are available online: experimental procedures and spectral data of all compounds, copies of 1H-NMR and 13C-NMR spectra of compounds 3 d,e, 4a–d, 5d–g, 6a–d, Tables S1 and S2 refer to the screening of conditions in the reaction between DDs 1d–g and crotyl alcohol 2b and to tentatively convert hydrazone 3e into the corresponding pyrazolone, respectively. DFT optimized geometries and energies.
Author Contributions
Conceptualization: F.M.; investigation and methodology: G.M.; data curation: G.F. and S.S.; DFT calculation: G.C. and M.C.; project administration and writing—review and editing: L.D.C. All authors have read and agreed to the published version of the manuscript.
Funding
The authors declare no competing financial interest.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data can be consulted by contacting the correspong authors.
Conflicts of Interest
There are no conflict to declare.
Sample Availability
Samples of the compounds are not available from the authors.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Khan H., Mubarak M.S., Amin S. Antifungal Potential of Alkaloids as an Emerging Therapeutic Target. Curr. Drug Targets. 2017;18:1825–1835. doi: 10.2174/1389450117666160719095517. [DOI] [PubMed] [Google Scholar]
- 2.Ain Q.-U., Khan H., Mubarak M.S., Pervaiz A. Plant Alkaloids Antiplatelet Agent: Drugs of the Future in the Light of Recent Developments. Front. Pharmacol. 2016;7:292. doi: 10.3389/fphar.2016.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rahman A., Basha A. Indole Alkaloids. Harwood Academic Publishers; Amsterdam, The Netherlands: 1997. [Google Scholar]
- 4.Cordell G.A., editor. The Alkaloids: Chemistry and Biology. Academic Press; San Diego, CA, USA: 2000. [Google Scholar]
- 5.Marti C., Carreire E.M. Construction of Spiro[pyrrolidine-3,3′-oxindoles]—Recent Applications to the Synthesis of Oxindole Alkaloids. Eur. J. Org. Chem. 2003;2003:2209–2219. doi: 10.1002/ejoc.200300050. [DOI] [Google Scholar]
- 6.Varvounis G. Pyrazol-3-ones. Part IV: Synthesis and Applications. In: Katritzky A.R., editor. Advances in Heterocyclic Chemistry. Vol. 98. Academic Press; New York, NY, USA: 2009. pp. 143–223. [Google Scholar]
- 7.Dhawan S., Narang R., Khatik G.L., Chopra H.K., Nayak S.K. Strategies for chemical synthesis of pyrazolone derivatives and their bio-significance. J. Chem. Pharm. Res. 2016;8:969–981. [Google Scholar]
- 8.Horton D.A., Bourne G.T., Smythe M.L. The Combinatorial Synthesis of Bicyclic Privileged Structures or Privileged Substructures. Chem. Rev. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- 9.Hassan A.E., Moustafa A.H., Tolbah M.M., Zohdy H.F., Haikal A.Z. Synthesis and Antimicrobial Evaluation of Novel Pyrazolones and Pyrazolone Nucleosides. Nucleosides Nucleotides Nucleic Acids. 2012;31:783–800. doi: 10.1080/15257770.2012.732250. [DOI] [PubMed] [Google Scholar]
- 10.Castagnolo D., Manetti F., Radi M., Bechi B., Pagano M., De Logu A., Meleddu R., Saddi M., Botta M. Synthesis, biological evaluation, and SAR study of novel pyrazole analogues as inhibitors of Mycobacterium tuberculosis: Part 2. Synthesis of rigid pyrazolones. Bioorg. Med. Chem. 2009;17:5716–5721. doi: 10.1016/j.bmc.2009.05.058. [DOI] [PubMed] [Google Scholar]
- 11.Saidachary G., Veera Prasad K., Divya D., Sing A., Ramesh U., Sridhar B., China Raju B. Convenient one-pot synthesis, anti-mycobacterial and anticancer activities of novel benzoxepinoisoxazolones and pyrazolones. Eur. J. Med. Chem. 2014;76:460–469. doi: 10.1016/j.ejmech.2014.02.042. [DOI] [PubMed] [Google Scholar]
- 12.Sujatha K., Shanthi G., Selvam N.P., Manoharan S., Perumal P.T., Rajendran M. Synthesis and antiviral activity of 4,4′-(arylmethylene)bis(1H-pyrazol-5-ols) against peste des petits ruminant virus (PPRV) Bioorg. Med. Chem. Lett. 2009;19:4501–4503. doi: 10.1016/j.bmcl.2009.02.113. [DOI] [PubMed] [Google Scholar]
- 13.Bondock S., Rabie R., Etman H.A., Fadda A.A. Synthesis and antimicrobial activity of some new heterocycles incorporating antipyrine moiety. Eur. J. Med. Chem. 2008;43:2122–2129. doi: 10.1016/j.ejmech.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 14.Casas J.S., Castellano E.E., Ellena J., Garcia-Tasende M.S., Peres-Paralle M.L., Sanchez A., Sanchez-Gonzales A., Sordo J., Touceda A. New Pd(II) and Pt(II) complexes with N,S-chelated pyrazolonate ligands: Molecular and supramolecular structure and preliminary study of their in vitro antitumoral activity. J. Inorg. Biochem. 2008;102:33–45. doi: 10.1016/j.jinorgbio.2007.06.032. [DOI] [PubMed] [Google Scholar]
- 15.Rosiere C.E., Grossman M.I. An Analog of Histamine that Stimulates Gastric Acid Secretion without other Actions of Histamine. Science. 1951;113:651–653. doi: 10.1126/science.113.2945.651.a. [DOI] [PubMed] [Google Scholar]
- 16.Sngh B.K., Sachan N., Chawla P. Synthesis and Pharmacological Screening of Novel 1,3-Disubstituted 5-Pyrazolones as Anticonvulsant Agents. Curr. Bioact. Compd. 2013;9:279–287. doi: 10.2174/1573407210666140307011414. [DOI] [Google Scholar]
- 17.Harmon R.E., Geller B.L., Gupta S.I., Herbert H.M., Chitharanjan D. Antimalarial properties of a variety of substituted p-sulfamoylphenylazo compounds. J. Pharm. Sci. 1970;59:1031–1033. doi: 10.1002/jps.2600590729. [DOI] [PubMed] [Google Scholar]
- 18.Basaif S.A., Hassan M.A., Gobouri A.A. AlCl3-Catalyzed diazocoupling of 1-(aryl/hetaryl)-3-phenyl-1H-pyrazol-2-in-5-ones in aqueous medium. Synthesis of hetaryl-azopyrazolones and their application as disperse dyes. Dyes Pigm. 2007;72:387–391. doi: 10.1016/j.dyepig.2005.09.025. [DOI] [Google Scholar]
- 19.Gribble G.W., Joule J.A., editors. Progress in Heterocyclic Chemistry. Vol. 98. Elsevier; Oxford, UK: 2014. pp. 143–223. [Google Scholar]
- 20.Katritzky A.R., Ramsden C.A., Scriven E.F.V., Taylor R.J.K., editors. Comprehensive Heterocyclic Chemistry III. Vol. 98. Elsevier; Oxford, UK: 2008. pp. 143–223. [Google Scholar]
- 21.Attanasi O.A., De Crescentini L., Favi G., Filippone P., Mantellini F., Perrulli F.R., Santeusanio S. Cultivating the Passion to Build Heterocycles from 1,2-Diaza-1,3-dienes: The Force of Imagination. Eur. J. Org. Chem. 2009:3109–3127. doi: 10.1002/ejoc.200900243. [DOI] [Google Scholar]
- 22.Li Y.-F., Chen Z.-J., Jiao W.-Y., Chen Z.-J., Chen I. Syntheses of Spiro(2-oxopyrrolidinyl)-5,4′-pyrazolones via Organocatalyzed Michael/Ammonolysis Cascade Reaction of 4-Aminopyrazolones and α,β-Unsaturated Acyl Phosphates. Synlett. 2021;32:923–929. doi: 10.1055/a1362-0296. [DOI] [Google Scholar]
- 23.Wang W., Bao X., Wei S., Nawaz S., Qu J., Wang B. Asymmetric sequential annulation/aldol process of 4-isothiocyanato pyrazolones and allenones: Access to novel spiro[pyrrole–pyrazolones] and spiro[thiopyranopyrrole–pyrazolones] Chem. Commun. 2021;57:363–366. doi: 10.1039/D0CC07113G. [DOI] [PubMed] [Google Scholar]
- 24.Zhou J., Huang W.-J., Jiang G.-F. Synthesis of Chiral Pyrazolone and Spiropyrazolone Derivatives through Squaramide-Catalyzed Reaction of Pyrazolin-5-ones with o-Quinone Methides. Org. Lett. 2018;20:1158–1161. doi: 10.1021/acs.orglett.8b00025. [DOI] [PubMed] [Google Scholar]
- 25.Zheng J., Wang S.-B., Zheng C., You S.-L. Asymmetric Synthesis of Spiropyrazolones by Rhodium-Catalyzed C(sp2)−H Functionalization/Annulation Reactions. Angew. Chemie, Int. Ed. 2017;56:4540–4544. doi: 10.1002/anie.201700021. [DOI] [PubMed] [Google Scholar]
- 26.Wang L., Li S., Chauhan P., Hack D., Philipps A.-R., Puttreddy R., Rissanen K., Raabe G., Enders D. Asymmetric, Three-Component, One-Pot Synthesis of Spiropyrazolones and 2,5-Chromenediones from Aldol Condensation/NHC-Catalyzed Annulation Reactions. Chem. Eur. J. 2016;22:5123–5127. doi: 10.1002/chem.201600515. [DOI] [PubMed] [Google Scholar]
- 27.Hack D., Dürr A.B., Deckers K., Chauhan P., Seling N., Rübenach L., Mertens L., Raabe G., Shoenebeck F., Enders D. Asymmetric Synthesis of Spiropyrazolones by Sequential Organo-and Silver Catalysis. Angew. Chemie Int. Ed. 2016;55:1797–1800. doi: 10.1002/anie.201510602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molvi K.I., Haque N., Awen B.Z.S., Zameeruddian M. Synthesis of Spiro Compounds as Medicinal Agents; New Opportunities for Drug. Design and Discovery. Part I: A Review. World J. Pharm. Pharm. Sci. 2014;3:536–563. doi: 10.1002/chin.201525271. [DOI] [Google Scholar]
- 29.Rios R. Enantioselective methodologies for the synthesis of spiro compounds. Chem. Soc. Rev. 2012;41:1060–1074. doi: 10.1039/C1CS15156H. [DOI] [PubMed] [Google Scholar]
- 30.Chu W.-D., Zhang Y., Wang J.B. Recent advances in catalytic asymmetric synthesis of allenes. Catal. Sci. Technol. 2017;7:4570–4579. doi: 10.1039/C7CY01319A. [DOI] [Google Scholar]
- 31.Ye J.T., Ma S.M. Palladium-Catalyzed Cyclization Reactions of Allenes in the Presence of Unsaturated Carbon–Carbon Bonds. Acc. Chem. Res. 2014;47:989–1000. doi: 10.1021/ar4002069. [DOI] [PubMed] [Google Scholar]
- 32.Ye J.T., Ma S.M. Conquering three-carbon axial chirality of allenes. Org. Chem. Front. 2014;1:1210–1224. doi: 10.1039/C4QO00208C. [DOI] [Google Scholar]
- 33.Neff R.K., Frantz D.E. Recent Advances in the Catalytic Syntheses of Allenes: A Critical Assessment. ACS Catal. 2014;4:519–528. doi: 10.1021/cs401007m. [DOI] [Google Scholar]
- 34.Yu S.H., Ma S.M. How easy are the syntheses of allenes? Chem. Commun. 2011;47:5384–5418. doi: 10.1039/C0CC05640E. [DOI] [PubMed] [Google Scholar]
- 35.Ma S.M. Some Typical Advances in the Synthetic Applications of Allenes. Chem. Rev. 2005;105:2829–2871. doi: 10.1021/cr020024j. [DOI] [PubMed] [Google Scholar]
- 36.Hoffmann-Röder A., Krause N. Synthesis and Properties of Allenic Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed. 2004;43:1196–1216. doi: 10.1002/anie.200300628. [DOI] [PubMed] [Google Scholar]
- 37.Liao Y., Yin X.M., Wang X.H., Yu W.Z., Fang D.M., Hu L.R., Wang M., Liao J. Enantioselective Synthesis of Multisubstituted Allenes by Cooperative Cu/Pd-Catalyzed 1,4-Arylboration of 1,3-Enynes. Angew. Chem. Int. Ed. 2020;59:1176–1180. doi: 10.1002/anie.201912703. [DOI] [PubMed] [Google Scholar]
- 38.Li S., Tang Z.W., Wang Y., Wang D., Wang Z.L., Yu C.X., Li T.J., Wei D.H., Yao C.S. NHC-Catalyzed Aldol-Like Reactions of Allenoates with Isatins: Regiospecific Syntheses of γ-Functionalized Allenoates. Org. Lett. 2019;21:1306–1310. doi: 10.1021/acs.orglett.8b04082. [DOI] [PubMed] [Google Scholar]
- 39.Tang Y., Xu J., Yang J., Lin L.L., Feng X.M., Liu X.H. Asymmetric three-component reaction for the synthesis of tetrasubstituted allenoates via allenoate-copper intermediates. Chem. 2018;4:1658–1672. doi: 10.1016/j.chempr.2018.04.012. [DOI] [Google Scholar]
- 40.Xu X., Zhang J.L., Dong S.X., Lin L.L., Lin X.B., Liu X.H., Feng X.M. Nickel(II)-Catalyzed Asymmetric Propargyl [2,3] Wittig Rearrangement of Oxindole Derivatives: A Chiral Amplification Effect. Angew. Chem. Int. Ed. 2018;57:8734–8738. doi: 10.1002/anie.201804080. [DOI] [PubMed] [Google Scholar]
- 41.Jiang Y., Diagne A.B., Thomson R.J., Schaus S.E. Enantioselective Synthesis of Allenes by Catalytic Traceless Petasis Reactions. J. Am. Chem. Soc. 2017;139:1998–2005. doi: 10.1021/jacs.6b11937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tap A., Blond A., Wakchaure V.N., List B. Chiral Allenes via Alkynylogous Mukaiyama Aldol Reaction. Angew. Chem. Int. Ed. 2016;55:8962–8965. doi: 10.1002/anie.201603649. [DOI] [PubMed] [Google Scholar]
- 43.Wang G., Liu X.H., Chen Y.S., Yang J., Li J., Lin L.L., Feng X.M. Diastereoselective and Enantioselective Alleno-aldol Reaction of Allenoates with Isatins to Synthesis of Carbinol Allenoates Catalyzed by Gold. ACS Catal. 2016;6:2482–2486. doi: 10.1021/acscatal.6b00294. [DOI] [Google Scholar]
- 44.Li Z.J., Boyarskikh V., Hansen J.H., Autschbach J., Musaev D.G., Davies H.M.L. Scope and Mechanistic Analysis of the Enantioselective Synthesis of Allenes by Rhodium-Catalyzed Tandem Ylide Formation/[2,3]-Sigmatropic Rearrangement between Donor/Acceptor Carbenoids and Propargylic Alcohols. J. Am. Chem. Soc. 2012;134:15497–15504. doi: 10.1021/ja3061529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moniz G.A., Wood J.L. Catalyst-Based Control of [2,3]- and [3,3]-Rearrangement in α-Diazoketone-Derived Propargyloxy Enols. J. Am. Chem. Soc. 2001;123:5095–5097. doi: 10.1021/ja015727h. [DOI] [PubMed] [Google Scholar]
- 46.Isobe M., Ploysuk C. The [2,3]-Wittig Rearrangement. In: Rojas C.M., editor. Molecular Rearrangements in Organic Synthesis. Wiley; Hoboken, NJ, USA: 2015. pp. 539–567. [Google Scholar]
- 47.Nakai T., Mikami K. [2,3]-Wittig sigmatropic rearrangements in organic synthesis. Chem. Rev. 1986;86:885–902. doi: 10.1021/cr00075a011. [DOI] [Google Scholar]
- 48.Perali R.S., Boddu U.R., Sankar D.C. [1,2]- vs [2,3]-Wittig Rearrangement in Carbohydrate Derived Alkenyl Systems. Org. Lett. 2021;23:3850–3853. doi: 10.1021/acs.orglett.1c00988. [DOI] [PubMed] [Google Scholar]
- 49.Xu X., Dong S., Feng L., Wang S., Liu X., Feng X. Kinetic Resolution of Propargylic Ethers via [2,3]-Wittig Rearrangement to Synthesize Chiral α-Hydroxyallenes. Org. Lett. 2020;22:2692–2696. doi: 10.1021/acs.orglett.0c00649. [DOI] [PubMed] [Google Scholar]
- 50.Rodríguez R.I., Ramírez E., Fernández-Salas J.A., Sánchez-Obregón R., Yuste F., Alemán J. Asymmetric [2,3]-Wittig Rearrangement: Synthesis of Homoallylic, Allenylic, and Enynyl α-Benzyl Alcohols. Org. Lett. 2018;20:8047–8051. doi: 10.1021/acs.orglett.8b03659. [DOI] [PubMed] [Google Scholar]
- 51.Lopes S.M.M., Cardoso A.L., Lemos A., Pinho e Melo T.M.V.D. Recent Advances in the Chemistry of Conjugated Nitrosoalkenes and Azoalkenes. Chem. Rev. 2018;118:11324–11352. doi: 10.1021/acs.chemrev.8b00375. [DOI] [PubMed] [Google Scholar]
- 52.Ciccolini C., Gatti F.G., Gatti G., Giorgi G., Mari G., Mantellini F., Favi G. Synthesis of Polycyclic Fused Indoline Scaffolds through a Substrate-Guided Reactivity Switch. J. Org. Chem. 2020;85:11409–11425. doi: 10.1021/acs.joc.0c01489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mari G., De Crescentini L., Favi G., Santeusanio S., Mantellini F. Metal and Oxidant Free Construction of Substituted-and/or Polycyclic Indoles: A Useful Alternative to Bischler and Related Syntheses. Eur. J. Org. Chem. 2020:5411–5424. doi: 10.1002/ejoc.202000845. [DOI] [Google Scholar]
- 54.Mari G., Favi G., Santeusanio S., Mantellini F., De Crescentini L. A practical and effective method for the N–N bond cleavage of N-amino-heterocycles. Org. Chem. Front. 2019;6:3408–3414. doi: 10.1039/C9QO00895K. [DOI] [Google Scholar]
- 55.Mari G., Ciccolini C., De Crescentini L., Favi G., Santeusanio S., Mancinelli M., Mantellini F. Metal and Oxidant-Free Brønsted Acid-Mediated Cascade Reaction to Substituted Benzofurans. J. Org. Chem. 2019;84:10814–10824. doi: 10.1021/acs.joc.9b01363. [DOI] [PubMed] [Google Scholar]
- 56.Mari G., Verboni M., De Crescentini L., Favi G., Santeusanio S., Mantellini F. Assembly of fully substituted 2, 5-dihydrothiophenes via a novel sequential multicomponent reaction. Org. Chem. Front. 2018;5:2108–2114. doi: 10.1039/C8QO00343B. [DOI] [Google Scholar]
- 57.Mari G., De Crescentini L., Mantellini F., Santeusanio S., Favi G. 5-Methylene N-acyl dihydropyridazinium ions as novel Mannich-type acceptors in 1, 4 additions of nucleophiles. Org. Chem. Front. 2018;5:1308–1311. doi: 10.1039/C8QO00032H. [DOI] [Google Scholar]
- 58.Lemos A. Cycloaddition Reactions of Conjugated Azoalkenes. In: Attanasi O.A., Spinelli D., editors. Targets in Heterocyclic Systems—Chemistry and Properties. Vol. 14. Società Chimica Italiana; Rome, Italy: 2010. pp. 1–18. [Google Scholar]
- 59.De Crescentini L., Perrulli F.R., Favi G., Santeusanio S., Giorgi G., Attanasi O.A., Mantellini F. Reactions of 1, 2-diaza-1, 3-butadienes with propargyl alcohol as an approach to novel bi-heterocyclic systems. Org. Biomol. Chem. 2016;14:8674–8678. doi: 10.1039/C6OB01595F. [DOI] [PubMed] [Google Scholar]
- 60.Ayers B.J., Chan P.W.H. Harnessing the Versatile Reactivity of Propargyl Alcohols and their Derivatives for Sustainable Complex Molecule Synthesis. Synlett. 2015;26:1305–1339. doi: 10.1002/chin.201533282. [DOI] [Google Scholar]
- 61.Egi M., Akai S. Transition Metal-Catalyzed Intramolecular Cyclization of Propargyl Alcohols and Their Derivatives for the Synthesis of Highly Substituted Five-Membered Oxygen Heterocycles. Heterocycles. 2015;91:931–958. doi: 10.3987/REV-15-818. [DOI] [Google Scholar]
- 62.Kanzian T., Nicolini S., De Crescentini L., Attanasi O.A., Ofial A.R., Mayr H. Electrophilic Reactivities of 1, 2-Diaza-1, 3-dienes. Chem. Eur. J. 2010;16:12008–12016. doi: 10.1002/chem.201000828. [DOI] [PubMed] [Google Scholar]
- 63.Wang F., Wang J., Zhang Y., Yang J. The [1,2]-and [1,4]-Wittig rearrangement. Tetrahedron. 2020;76:130857. doi: 10.1016/j.tet.2019.130857. [DOI] [Google Scholar]
- 64.Kimm M., Ošeka M., Kaabel S., Metsala A., Järving I., Kanger T. [2,3]-Wittig Rearrangement as a Formal Asymmetric Alkylation of α-Branched Ketones. Org. Lett. 2019;21:4976–4980. doi: 10.1021/acs.orglett.9b01495. [DOI] [PubMed] [Google Scholar]
- 65.Peňaška T., Mejer Mojzes M., Filo J., Jurdáková H., Mečiarová M., Šebesta R. Organocatalysts Effect on the Stereoselectivity of [2,3]-Wittig Rearrangement. Eur. J. Org. Chem. 2019;2019:605–610. doi: 10.1002/ejoc.201801697. [DOI] [Google Scholar]
- 66.Kitabayashi Y., Fukuyama T., Yokoshima S. Synthesis of the [7-5-5] tricyclic core of Daphniphyllum alkaloids. Org. Biomol. Chem. 2018;16:3556–3559. doi: 10.1039/C8OB00859K. [DOI] [PubMed] [Google Scholar]
- 67.Nath S.R., Joshi K.A. Mechanistic investigation in the [1, 4] and [1, 2] Wittig rearrangement reactions: A DFT study. Phys. Chem. Chem. Phys. 2018;20:21457–21473. doi: 10.1039/C8CP01045E. [DOI] [PubMed] [Google Scholar]
- 68.Tsubuki M., Kamata T., Okita H., Arai M., Shigihara A., Honda T. Wittig rearrangement of allyl and propargyl furfuryl ethers leading to 2-furylmethanol derivatives. Chem. Commun. 1999:2263–2264. doi: 10.1039/a907312d. [DOI] [Google Scholar]
- 69.Marshall J.-A., Robinson E.D., Zapata A. [2,3] Wittig rearrangement of nonracemic propargyl ethers leading to allenes of high stereochemical integrity. J. Org. Chem. 1989;54:5854–5855. doi: 10.1021/jo00286a013. [DOI] [Google Scholar]
- 70.Mikami K., Azuma K.-I., Nakai T. [2,3]-Wittig sigmatropic rearrangement of crotyl propargyl ether system. An emerging tool for control of acyclic stereochemistry. Tetrahedron. 1984;40:2303–2308. doi: 10.1016/0040-4020(84)80013-7. [DOI] [Google Scholar]
- 71.Everett R.K., Wolfe J.P. Aza-Wittig Rearrangements of N-Benzyl and N-Allyl Glycine Methyl Esters. Discovery of a Surprising Cascade Aza-Wittig Rearrangement/Hydroboration Reaction. J. Org. Chem. 2015;80:9041–9056. doi: 10.1021/acs.joc.5b01286. [DOI] [PubMed] [Google Scholar]
- 72.Anderson J.C., Davies E.A. Diastereoselective synthesis of substituted prolines via 5-endo-trig cyclisations of aza-[2,3]-Wittig sigmatropic rearrangement products. Tetrahedron. 2010;66:6300–6308. doi: 10.1016/j.tet.2010.04.095. [DOI] [Google Scholar]
- 73.Vogel K. The aza-Wittig rearrangement. Synthesis. 1997;5:497–505. doi: 10.1055/s-1997-1231. [DOI] [Google Scholar]
- 74.Ošeka M., Kimm M., Kaabel S., Järving I., Rissanen K., Kanger T. Asymmetric Organocatalytic Wittig [2,3]-Rearrangement of Oxindoles. Org. Lett. 2016;18:1358–1361. doi: 10.1021/acs.orglett.6b00291. [DOI] [PubMed] [Google Scholar]
- 75.McGowan G., Thomas E.J. Synthesis of macrocyclic precursors of phomactins using [2,3]-Wittig rearrangements. Org. Biomol. Chem. 2009;7:2576–2590. doi: 10.1039/b903256h. [DOI] [PubMed] [Google Scholar]
- 76.Barbazanges M., Meyer C., Cossy J. Stereoselective synthesis of 1, 2-aminoalcohols by [2, 3]-Wittig rearrangements. Org. Lett. 2007;9:3245–3248. doi: 10.1021/ol0711725. [DOI] [PubMed] [Google Scholar]
- 77.Barluenga J., Fañanás F.J., Sanz R., Marcos C., Trabada M. On the Reactivity of o-Lithioaryl Ethers: Tandem Anion Translocation and Wittig Rearrangement. Org. Lett. 2002;4:1587–1590. doi: 10.1021/ol0258029. [DOI] [PubMed] [Google Scholar]
- 78.Liang J., Hoard D.W., Van Khau V., Martinelli M.J., Moher E.D., Moore R.E., Tius M.A. Synthesis of unit A of cryptophycin via a [2, 3]-Wittig rearrangement. J. Org. Chem. 1999;64:1459–1463. doi: 10.1021/jo9815958. [DOI] [PubMed] [Google Scholar]
- 79.Ahman J., Somfai P. Aza-[2, 3]-Wittig rearrangements of vinylaziridines. J. Am. Chem. Soc. 1994;116:9781–9782. doi: 10.1021/ja00100a066. [DOI] [PubMed] [Google Scholar]
- 80.Anderson J.C., Roberts C.A. The Tri-n-butyltin Group as a Novel Stereocontrol Element and Synthetic Handle in the Aza-[2,3]-Wittig Sigmatropic Rearrangement. Tetrahedron Lett. 1998;39:159–162. doi: 10.1016/S0040-4039(97)10477-4. [DOI] [Google Scholar]
- 81.Marshall J.A., Wang X. Synthesis of enantioenriched. alpha-hydroxy-alpha-allenylacetic acids by [2, 3] Wittig rearrangement of. alpha-(propargyloxy) acetates. J. Org. Chem. 1991;56:4913–4918. doi: 10.1021/jo00016a020. [DOI] [Google Scholar]
- 82.Lumbroso A., Cooke M.L., Breit B. Catalytic asymmetric synthesis of allylic alcohols and derivatives and their applications in organic synthesis. Angew. Chem. Int. Ed. 2013;52:1890–1932. doi: 10.1002/anie.201204579. [DOI] [PubMed] [Google Scholar]
- 83.Sundararaju B., Achard M., Bruneau C. Transition metal catalyzed nucleophilic allylic substitution: Activation of allylic alcohols via π-allylic species. Chem. Soc. Rev. 2012;41:4467–4483. doi: 10.1039/c2cs35024f. [DOI] [PubMed] [Google Scholar]
- 84.Bandini M. Allylic alcohols: Sustainable sources for catalytic enantioselective alkylation reactions. Angew. Chem. Int. Ed. 2011;50:994–995. doi: 10.1002/anie.201006522. [DOI] [PubMed] [Google Scholar]
- 85.Tamaru Y. Activation of allyl alcohols as allyl cations, allyl anions, and amphiphilic allylic species by palladium. Eur. J. Org. Chem. 2005:2647–2656. doi: 10.1002/ejoc.200500076. [DOI] [Google Scholar]
- 86.Attanasi O.A., Filippone P., Mei A., Santeusanio S. Effect of Metal Ions in Organic Synthesis; Part XXIII. Easy and High-Yield Direct Synthesis of 3-Aminocarbonyl-1-ureidopyrroles by the Copper(II) Chloride-Catalyzed Reaction of Aminocarbonylazoalkenes with 3-Oxoalkanamides. Synthesis. 1984:671–672. doi: 10.1055/s-1984-30928. [DOI] [Google Scholar]
- 87.Attanasi O.A., Filippone P., Mei A., Santeusanio S. Effect of metal ions in organic synthesis. XXIV: Facile one-flask synthesis of 2-alkoxycarbonylamino-3-aminocarbonylpyrroles by reaction of alkoxycarbonylazoalkenes with 3-oxoalkanamides under copper(II) chloride catalysis. Synthesis. 1984:873–874. doi: 10.1055/s-1984-31005. [DOI] [Google Scholar]
- 88.Preti L., Attanasi O.A., Caselli E., Favi G., Ori C., Davoli P., Felluga F., Prati F. One-Pot Synthesis of Imidazole-4-Carboxylates by Microwave-Assisted 1,5-Electrocyclization of Azavinyl Azomethine Ylides. Eur. J. Org. Chem. 2010:4312–4320. doi: 10.1002/ejoc.201000434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Neese F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012;2:73–78. doi: 10.1002/wcms.81. [DOI] [Google Scholar]
- 90.Neese F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017;8:e1327. doi: 10.1002/wcms.1327. [DOI] [Google Scholar]
- 91.Grimme S., Antony J., Ehrlich S., Krieg H.A. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data can be consulted by contacting the correspong authors.