

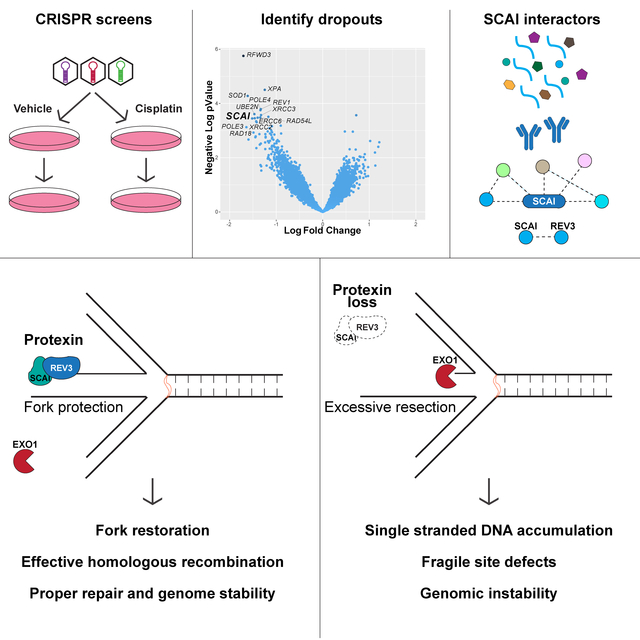
Summary
Protection of stalled replication forks is critical to genomic stability. Using genetic and proteomic analyses we discovered the Protexin complex containing the ssDNA binding protein SCAI and the DNA polymerase REV3. Protexin is required specifically for protecting forks stalled by nucleotide depletion, fork barriers, fragile sites and DNA interstrand crosslinks (ICLs) where it promotes homologous recombination and repair. Protexin loss leads to ssDNA accumulation and profound genomic instability in response to ICLs. Protexin interacts with RNA POL2 and both oppose EXO1’s resection of DNA on forks remodeled by the FANCM translocase activity. This pathway acts independently of BRCA/RAD51-mediated fork stabilization, and cells with BRCA2 mutations were dependent on SCAI for survival. These data suggest that Protexin and its associated factors establish a new fork protection pathway that counteracts fork resection in part through a REV3 polymerase-dependent resynthesis mechanism of excised DNA, particularly at ICL stalled forks.
Keywords: CRISPR, Inter-strand crosslinks, resection, homologous recombination, replication stress, Protexin, SCAI, REV3L, FANCM, EXO1
eTOC blurb
Adeyemi et al. performed genome-wide cisplatin sensitivity screens and identified SCAI, which they show to be important for maintaining genome stability following replication stress. They show SCAI is in complex with a polymerase, REV3, which they term Protexin. Protexin maintains DNA integrity after damage by protecting replication forks from nucleases.
Graphical Abstract
Introduction
Sensing and responding to DNA damage is critical to the maintenance of genomic stability and the prevention of numerous diseases including birth defects and cancer (Ciccia and Elledge, 2010). DNA polymerase blocks during replication (i.e. replication stress) can cause fork collapse and genomic instability (Zeman and Cimprich, 2014). Single-stranded DNA (ssDNA) accumulation at sites of replication stress acts as a trigger for the activation of the ATR kinase, which coordinates the replication stress response (Cimprich and Cortez, 2008).
DNA strand crosslinks are exceptionally toxic (Garaycoechea et al., 2018; Langevin et al., 2011), especially inter-strand crosslinks (ICLs) that covalently ligate both DNA strands, constituting a block to replication and transcription. Eukaryotes employ a distinct set of genes to repair ICLs, and germline mutations in this pathway leads to Fanconi anemia (FA), characterized by cancer predisposition, bone marrow and hematopoietic defects and other phenotypes (Ceccaldi et al., 2016; Kottemann and Smogorzewska, 2013). The FANCM complex recognizes the ICL and recruits the FA core complex (Xue et al., 2015) to set up the critical activating step for repair via ubiquitination of the FANCI-FANCD2 complex by FANCL. ATR is required for this activation step via phosphorylation of FANCI (Andreassen et al., 2004; Smogorzewska et al., 2007). Repair of ICLs involves multiple nucleases such as XPF-ERCC1, FAN1, MUS81 and SNM1A. How they are regulated and the exact steps in which they function is not fully clear, however, FANCI-D2 ubiquitination is important for these incisions events that occur prior to translesion synthesis (TLS) and homologous recombination (HR).
HR genes such as BRCA2 and RAD51 function in the FA pathway and are critical for maintenance of fork stability by preventing exonuclease-dependent fork degradation (Chen et al., 2018; Schlacher et al., 2011). HR at stalled forks is now believed to be regulated differently than HR at double strand breaks (DSBs) (Ray Chaudhuri et al., 2016; Willis et al., 2014). One difference is fork reversal at stalled forks. Fork reversal is mediated by remodeling enzymes like the DNA helicases (BLM, WRN, and others) and the DNA translocases FANCM, SMARCAL1, ZRANB3, RAD54 and HLTF (Quinet et al., 2017). Reversed forks can resemble one-ended DSBs and are thus susceptible to nuclease digestion or end-joining events that cause genomic instability. Thus, fork protection mechanisms promote genomic stability following PARP and cisplatin treatment (Ray Chaudhuri et al., 2016) by limiting NHEJ and resection.
In this study, through a genome-wide analysis of genes required for crosslink repair, we identified a previously uncharacterized mechanism of fork protection and fork restoration control mediated by the newly found Protexin complex. Protexin is critical for the replication stress response that protects stalled replication forks. Absence of Protexin causes ssDNA accumulation and profound genomic instability in response to agents that stall forks.
Results
A genome-wide cisplatin sensitivity screen identifies multiple novel genes involved in crosslink repair including SCAI.
We used CRISPR screening to identify genes required for ICL repair. The screen (Figure 1A) is described in detail in the experimental procedures. U2OS cells infected with a lentiviral sgRNA library targeting 18,148 genes, 5 gRNAs/gene. Selected cells were treated with cisplatin (hereafter cisp) or vehicle for 24 h before washout and passaged for 5 population doublings (PDs) before collection (PD5 arm). A second cisp treatment was performed at PD5, and the cells were allowed to grow for a further 5 PDs before collection (PD10 arm). NextGen sequencing coupled with bioinformatic analyses using MaGeCK (Li et al., 2014) and EdgeR (Robinson et al., 2010) identified gRNAs that change abundance following treatment (Tables S1–S3).
Figure 1. Genetic screen identifies genes important for cisp sensitivity including SCAI.

(A) Screen schematic. Cisp treatment was 1.5 μM at PD0 and was repeated at 1 μM at PD5 for the PD10 arm. PD - population doubling. (B) Pathway map showing the top scoring known DNA repair genes from our screens. Some DNA replication and RNA transcription genes were omitted for space constraints. (C) Genes with significant p-values at both PD5 and PD10 or false discovery rates (FDR) <= 0.05 were included. Venn diagram shows the hits identified by EdgeR and MAGeCK. (D) Mevalonate pathway map. Red - genes that scored in the screen. (E) U2OS cells were transduced with lentiviruses expressing shRNAs against the indicated genes, no shRNA, or a control shRNA, C. Cells in 96-well plates were treated with vehicle or 1 μM cisp for 24 h and analyzed by AlamarBlue viability assays at 72 h. Data is mean ± SEM of two independent experiments. *= p<0.05, **= p<0.01, ***=p<0.001. (F) Colony survival assays (CSA) showing survival of control or SCAI siRNA-treated U2OS cells treated with cisp. Data are normalized to untreated cells for each siRNA condition. Mean ± SEM survival of 2 independent experiments shown. Inset. Western blot showing depletion of SCAI. (G) CSA showing survival of WT and SCAI null with or without siRNA to FANCA. Mean ± SEM of two independent experiments shown. Inset. Western blots showing SCAI and FANCA depletion.
ICL lesions are complex and multiple pathways cooperate in their repair. Analysis of genes with significant dropout p-values at PD5 and PD10 revealed multiple pathways (Figure 1B). As anticipated, the screen identified many ICL repair genes including members of the FA pathway. Nineteen of the 21 known FA genes scored in the top 5% of hits in one or both arms (Tables S1-S2).
Almost all components of the nucleotide excision repair (NER) pathway scored, as did several RNA polymerase subunits. ICL repair often involves a DSB intermediate, and many HR genes scored including RAD51 paralogs, filament processors and stabilizers including RAD54L, FIGNL1, MMS22L and HELQ. Microhomology-mediated end joining (MMEJ) genes such as POLQ scored more strongly than their non-homologous end joining (NHEJ) counterparts, likely because any DSBs that arise are resected, and largely repaired by HR with MMEJ serving a backup role.
Analysis of both screen arms identified ~400 high confidence hits (Figure 1C, Table S4), many with no prior DNA repair link (Figures S1A and S1B). Chromatin remodelers (e.g. BPTF), anti-apoptotic genes, membrane trafficking proteins, and strikingly, several members of the mevalonate metabolism pathway (e.g. MVD, MVK and HMGCS1) scored strongly (Figure 1D). GO term enrichment analyses (Figure S2E) mirrored the findings from Figure 1B. Aside from repair-related themes, one prominent GO term was “RNA secondary structure unwinding” due to multiple RNA helicases scoring such DHX15, DDX46 and DDX47.
We validated several candidates (Figure 1E and Figures S1C - S1F) including POLE4, a histone fold protein that binds POLE3. ADSL is important for purine biosynthesis. Multiple shRNAs to ADSL showed significant ICL-sensitivity. OGDH catalyzes the conversion of alpha-ketoglutarate to succinyl-CoA in the Krebs’s cycle. Although present in mitochondria, it associated with stalled replication forks (Dungrawala et al., 2015). Several guides to OGDH scored (Figure S1C) and 2 of 3 OGDH shRNAs showed cisp-sensitivity (Figure 1E). Overall, our screens present a useful resource for identification and characterization of ICL repair genes.
SCAI promotes ICL repair
A top scoring gene, scoring with all 5 gRNAs, in both cisp-treated arms was SCAI (Figures S1A, S1B and S1G, Tables S2–S3). SCAI was originally identified as a regulator of invasive cell migration (Brandt et al., 2009), but recently was implicated in DSB repair (Hansen et al., 2016). The marked sensitivity to cisp in our screens suggested that it may play a distinct role in replication stress. To validate SCAI, we depleted it in HeLa and U2OS cells using siRNA pools. SCAI depletion resulted in reduced cell viability as determined by clonogenic survival assays (CSAs) following cisp treatment (Figures 1F and S2A).
Using Cas9-editing, we generated two independent SCAI null clones in U2OS cells (Figure S2B). SCAI nulls showed cisp-sensitivity equal to FANCA depletion (Figure 1G) and FANCA depletion in the SCAI null cells led only to a slight reduction in survival that was not statistically significant (Figure 1G). These data suggest that SCAI functions in ICL repair, possibly in the FA pathway.
SCAI protects against genomic instability, cell cycle defects and defective replication restart after replication stress
We characterized SCAI’s role in repair of IR, Cisp, camptothecin (CPT) and hydroxyurea (HU)-induced replication stress using multicolor competition assays (MCAs) (Figure 2A). SCAI depletion significantly increased sensitivity to CPT, HU, and marked sensitivity to cisp treatment but not IR (Figure 2A). Sensitivity to HU was also seen in CSAs in SCAI null cells (Figure 2B).
Figure 2. SCAI promotes repair during replication stress and forms a novel complex with REV3.

(A) Top. Schematic of the MCA assay. Middle. Relative sensitivity of SCAI and ATM depletion following treatment with 3 Gray (Gy) IR, 2 mM HU, 100 nM CPT or 1 μM cisp (Cisp) for 24 h. Data are normalized to untreated cells. Mean ± SEM values are plotted. Bottom panel: Western blots showing SCAI and ATM depletion. (B) CSA showing survival of WT and SCAI null U2OS cells following treatment with 0.5, 1 or 2 mM HU for 24 h. Mean ± SEM of 2 independent experiments shown. (C) Representative images showing increased genomic instability in MMC-treated, 293Ts following SCAI siRNA compared to control. (D, E) Quantification of aberrations (D) and radial percentages (E) from experiment shown in (C). Mean ± SEM of 2 independent experiments shown. (F) IP mass spec analyses showing normalized fold changes of IP’d peptides from FLAG-HA-SCAI vs vector-expressing 293-TREX cells following mock or cisp treatment. (G) 293Ts transfected with vector, GFP-SCAI and/or FLAG-REV3 as indicated for 60 h and treated or not with 2 μM cisp for 3 h. Pellets were IP’d with control or FLAG beads and processed for immunoblotting. (H) 293T cells were transfected with vector or FLAG-REV3 and processed as above.
Such pathologic responses to replication stress often lead to genome instability. To examine this, we performed chromosomal analysis of metaphase spreads from SCAI-depleted 293Ts. SCAI loss resulted in a significant increase in chromosomal aberrations (Figures 2C–2E), including chromatid breaks and gaps on par with FANCD2 loss (Figure 2D). Thus, SCAI is profoundly important for the maintenance of genome stability after replication stress.
SCAI loss caused a profound accumulation in S (24 h) and G2 (44 h) when treated with cisp but not in untreated cells (Figures S2B, S2C and S2G). To examine replication stress recovery, cells treated with HU or cisp for 18 h were released into fresh media for 3 to 6 h then pulsed with BrdU to determine the rate at which cells begin to restore DNA synthesis (Figure S2F). Cisp treated SCAI mutants showed markedly reduced BrdU incorporation compared to WT controls (Figure S2I). Similar but milder effects were seen following HU (Figure S2H). The defects in cell cycle recovery after replication stress in SCAI mutants indicates a key role for SCAI in repairing damage or restarting forks.
SCAI forms a complex with REV3.
To identify interactions that might mediate SCAI function, we inducibly expressed FLAG-HA-SCAI in TREX-293 cells. Mass spectrometry of SCAI pulldowns revealed multiple SCAI binding partners (Table S5) (Figure 2F) that include chromatin components KDM3B, DOT1L, 53BP1 and HELB, a DNA helicase with roles in end resection (Tkac et al., 2016). None of these scored as cisp sensitizers in our screen. Of interest, we found constitutive interactions with REV3L (REV3), MAD2L2 (REV7) and REV1 (Figure 2F). REV7 and REV3 together form the pol zeta polymerase complex, and function with REV1 in TLS (Prakash et al., 2005). All three genes scored very highly in our screen for ICL repair factors (Figure 1B and Tables S2–S5).
SCAI and REV3 association was validated by co-IPs in cells expressing GFP-SCAI and FLAG-REV3 (Figure 2G). SCAI also interacted with 53BP1 (Figure 2F) raising the possibility that SCAI exists in one large complex. However, FLAG-REV3 was able to pull down endogenous SCAI but not 53BP1 (Figure 2H) and REV3 was not detected in 53BP1 complexes (Gupta et al., 2018).
To visualize SCAI following replication stress we performed immunofluorescence (IF) for SCAI following cisp or MMC treatment. SCAI forms foci in response to DSBs and localizes to these foci partly by interaction with 53BP1 (Hansen et al., 2016). These SCAI nuclear foci co-localized with γH2AX (Figure S3A) and were detergent resistant (Figure S3B) suggesting chromatin association. Chromatin fractionation experiments revealed a cisp dose-dependent increase in SCAI accumulation on chromatin (Figure S3C).
Upon IR, SCAI formed fewer but larger foci compared to MMC (Figures 3A and 3B). Importantly, while 53BP1 depletion reduced SCAI foci after IR, it did not block SCAI foci following MMC (Figures 3B and 3C). REV3 depletion also had no effect on SCAI recruitment to foci (Figures 3B and 3C). Neither depletion of 53BP1 nor ATM (Figures 2A and S3D) caused ICL-sensitivity. These data suggest that SCAI exists in at least two separate complexes that respond differently to various kinds of DNA damage, and SCAI’s role during replication stress is independent of 53BP1. We named the SCAI-REV3 complex Protexin for reasons delineated below.
Figure 3. Protexin is important for genomic stability and repair at stalled forks.

(A) IF images showing foci of FLAG-SCAI and 53BP1 following 100 nM MMC (6h) or 4Gy IR (4h) treatment. (B) FLAG-SCAI U2OS transfected with the indicated siRNAs (48 h) then treated and processed for IF as in (A). (C) Quantification of the experiment shown in (B). At least 50 cells were counted per condition. (D) Top. Schematic showing the EGFP-based HR-Flex reporter described in experimental procedures. D-EGFP: donor EGFP. Bottom. Reporter cell lines were infected with the indicated shRNAs and assessed for GFP expression by flow after 14d. (E) Schematic of the Tus/6xTer HR reporter and HR repair products of Tus-Ter induced fork stalling. TrGFP is 5’ truncated GFP. The triangle shows the 6xTer site adjacent to I-SceI. Open ovals A and B: 5’ and 3’ artificial RFP exons. STGC, LTGC: short tract and long tract gene conversion HR repair outcomes. LTGC generates wtRFP through splicing (red filled ovals). (F) HR frequencies in mouse 6xTer/HR cells co-transfected with Tus (top left panel) or I-SceI (top right panel) and either control or two different mouse siRNAs against SCAI. Total HR represents combination of STGC and LTGC values. Data represent the mean ± SEM. Welch’s test, ***=p<0.001. (G) HR frequencies in 6xTer/HR cells co-transfected with Tus (top left panel) or I-SceI (top right panel) and either control or siRNAs against REV3. Data represent the mean ± SEM. Welch’s test, ****=p<0.0001.
Protexin prevents fork breakage and promotes repair at common fragile sites
Fragile sites are chromosomal regions that are susceptible to chromosome breaks and genomic instability, and are common sources of replication stress (Durkin and Glover, 2007). At some of these sites hairpins form ahead of and stall the replication fork requiring repair by fork reversal. Without FANCM, cells are unable to properly replicate through the synthetic common fragile site (CFS) hairpin structures, resulting in fork breakage, detected as increases in mitotic recombination and GFP levels using a modified DR-GFP reporter (Figure 3D) (Wang et al., 2018). We examined Protexin’s roles in maintaining fragile site integrity using this assay. SCAI or REV3 depletion led to increased fork breakage at CFSs (Figure 3D). Notably, the extent of mitotic recombination in the absence of Protexin was not as high as that seen following FANCM depletion, possibly because, as shown below, Protexin is also important for recombination at stalled forks, which is required for the assay readout. This data suggests that Protexin is required for stability at fragile sites.
Protexin is required for HR specifically at stalled forks
SCAI loss showed severe inviability following replication stress, thus we sought to determine whether Protexin regulates HR specifically at stalled forks. We employed a reporter in which six bacterial Ter sites interrupt the reading frame of an enhanced GFP (eGFP) gene inserted in the ROSA26 locus of a mouse embryonic stem cell line (Willis et al., 2014). Expression of the bacterial Tus protein and its binding to the 6X Ter array leads to site-specific bidirectional fork arrest and eventual fork breakage. Generation of WT GFP detectable by flow cytometry requires recombination of the stalled fork with a 5’ adjacent truncated GFP copy (Tr-GFP). The same flow assay allows for additional quantification of HR induced by a chromosomal DSB due to the presence of an I-SceI site next to the 6X Ter array (Figure 3E). In repeated experiments in mouse ES cells using two independent siRNAs, we observed minor effects of depletion of SCAI depletion (Figure 3F) or REV3 (Figure 3G) on I-SceI-induced HR repair that do not reach statistical significance.
In contrast, there was an ~65% and 60% reduction in Tus-induced total HR (Figure 3F, left panel) following SCAI (Figure S3E) or REV3 (Figure S3F) depletion, respectively. This was mostly attributable to reduced short tract gene conversion (STGC - an error free form of HR repair) at stalled forks following REV3 or SCAI loss. Their loss did not result in substantially reduced long tract gene conversion (LTGC – a rarer and more error prone form of repair). Thus, upon loss of Protexin, conventional DSB repair remains largely intact while HR repair by Tus/Ter induced fork stalling is substantially reduced.
Protexin prevents aberrant RPA phosphorylation and ssDNA accumulation after replication stress
Next, we assayed for DDR activation in response to ICLs in response to ICLs after Protexin loss. Chk1 phosphorylation increased following SCAI depletion (Figures 4A and S4B) only in the presence of ICLs. We also observed striking increases in RPA2 phosphorylation (pRPA) following SCAI depletion with multiple siRNAs or in SCAI null cells (Figures 4A and 4B) in a 53BP1 (Figure S4D) or HELB (Figure S5D)-independent manner. This was seen in multiple cell lines (RPE, Figure S4C; HeLa, Figure S3C), and could be rescued by expressing siRNA-resistant SCAI (Figure 4C). SCAI protein levels also appeared to increase following replication stress (Figure 4A). Kinetic analysis revealed that the increased pRPA first became detectible at 8 h after ICL treatment (Figure S4A), with similar kinetics to FANCD2 activation (Figure S6A). This pRPA occurs on chromatin (Figure S3C). Increased pRPA was also seen after HU upon SCAI depletion (Figure 4A), but was less pronounced because significant pRPA occurs in controls due to HU-induced replication fork uncoupling.
Figure 4. SCAI limits accumulation of nuclear ssDNA during replication stress.

(A) U2OS cells transfected with the indicated siRNAs for 72 h, then treated with 2 μM Cisp or 1 mM HU for 18 h, harvested and processed for western blotting with the indicated antibodies. ND-no drug. (B) WT U2OS and two independent SCAI null clones (7, 11) were treated with the indicated doses of cisp for 18 h before immunoblotted. (C) U2OS cells were transduced with vector or siRNA resistant FLAG-SCAI. Cells were then treated with indicated siRNAs, with or without 1.5 μM cisp for 18 h before western blotting. (D) WT and SCAI-null U2OS were treated with the indicated siRNAs for 72 h, then treated with vehicle (ND) or 1.5 μM cisp for 18 h before immunoblotting. (E) WT and REV7-null U2OS were transfected with the indicated siRNAs for 72 h, then treated 2 μM cisp for 18 h before immunoblotting. (F) HeLas were transfected with the indicated siRNA for 60 h before treatment with no drug or 0.5 μM Cisp for 6 h. Cells were processed for RPA2 IF. Intensity of nuclear RPA staining from was quantified in 50 cells using ImageJ and plotted. T-tests, **** = p<0.0001. (G) WT or SCAI null U2OS were BrdU labeled and then treated with vehicle (ND) or 1 μM MMC for 6 h and processed for BrdU IF. Average number of BrdU foci per cell was quantified and plotted on the right. T-tests, **** = p<0.0001.
Depletion of REV3 with multiple siRNAs phenocopied SCAI loss for pRPA with ICLs (Figures 4D, S4E–S4H) while REV1 depletion showed a small effect on pRPA. pRPA accumulation was the same in the individual and double depletions of REV3 and SCAI indicating epistasis. Like REV3, REV7 is essential for TLS as part of pol zeta, however, REV7 loss had no effect on pRPA levels and did not affect the phenotype of depletion of either SCAI or REV3 in response to ICLs (Figure 4E). This is consistent with a TLS-independent role for REV3 in preventing increased pRPA during ICL repair.
RPA on ssDNA in S phase is phosphorylated by ATR at replication blocks. To directly visualize whether SCAI loss led to ssDNA accumulation, we performed both IF and flow cytometry-based assays to quantify changes in BrdU content under non-denaturing conditions (a proxy for the extent of ssDNA content). SCAI loss led to significantly increased numbers of BrdU+ ssDNA foci per cell following MMC treatment (Figure 4G). Increased BrdU intensity was also observed in the absence of SCAI when only the nascent strand was labeled (Figure S4I). Taken together, our data demonstrates that SCAI and REV3 appear to function together in the Protexin complex to limit ssDNA formation following treatment replication stress agents.
Protexin protects against excessive fork degradation by EXO1
Protexin may limit ssDNA by protecting forks from nucleases, as suggested for BRCA2 (Schlacher et al., 2011). Using DNA combing techniques and sequential pulsing of CldU, then IdU followed by HU treatment, we examined fork lengths in the absence of SCAI. SCAI loss resulted in moderately reduced length of the second signal, diminishing the IdU:CldU ratio (Figure 5D). This is consistent with the reduced recovery of DNA synthesis seen upon HU- treatment of SCAI mutant cells (S2H). Thus, SCAI protects stalled forks.
Figure 5. SCAI protects stalled forks but not DSBs against resection by EXO1 and promotes resistance to PARP inhibitors.

(A) Cells treated with the indicated siRNA for 72 h were treated with vehicle or 2 mM cisp for 16 h and analyzed by immunoblotting. (B) IF analyses showing representative BrdU foci from WT and SCAI nulls following 60 h treatment with the indicated siRNAs. Cells were treated with MMC for 6 h prior to IF. (C) Quantification of B. (D) Top. Schematic showing DNA fiber assay protocol. Cells were pulsed with CldU followed by IdU for 30 m each after which forks were stalled by 4 mM HU treatment for 4.5 h. Bottom. WT and SCAI-nulls were treated with indicated siRNAs for 60 h before labeling as in schematic. DNA combing analyses was performed and approximately 100 fibers were quantified and plotted. Mann Whitney test, ns=p>0.05, **=p<0.01, ***=p<0.001. (E) WT and SCAI-null U2OS were treated with the indicated siRNAs for 72 h, then treated with vehicle (ND) or 2 μM cisp for 18 h before immunoblotting. The same samples were run in lanes 2–3 and lanes 10–11. Depletion of indicated proteins is shown in Figure S5F. (F) ER-AsiSi U2OS transfected with the indicated siRNAs for 60 h were treated with Tamoxifen to induce DSBs. Cells were harvested in low-melting agarose, proteinase-treated and genomic DNA was extracted. After restriction digest, qPCR was performed to determine resection efficiency. (G) BRCA1-null, TP53-null RPE1 cells expressing SCAI or vector control were treated with vehicle, or the indicated doses of olaparib or cisp for 2 weeks and allowed to form colonies. Mean ± SD two independent experiments. Representative images shown.
To determine the nucleases responsible for ssDNA generation, we depleted known resection enzymes. Neither CtIP (Figure S5A) or MRE11 (Figure S5B) depletion, or the MRE11 inhibitor mirin (Figure S5C) reduced pRPA in SCAI null cells following cisp treatment. However, we did observe a slight, but repeatable, reduction in pRPA following depletion of either DNA2 (Figure 5A) or WRN (Figure S5E), perhaps via replication slowing owing to DNA2’s role in Okazaki fragment maturation, resulting in fewer fork converging lesions. In contrast, EXO1 depletion completely abolished the increases in pRPA seen following cisp (Figure 5A) or MMC (Figure S6E) treatment, suggesting that SCAI activity is important to limit EXO1 function. EXO1 depletion also abolished pRPA seen following REV3 loss (Figure 6A).
Figure 6. Protexin protects FANCM-reversed forks from degradation.

(A) U2OS transfected with the indicated siRNAs 60 h were treated with vehicle (ND) or 2 μM cisp for 18 h before immunoblotting. (B) IF showing representative BrdU foci from WT and SCAI nulls 60 h post treatment with indicated siRNAs. Cells were treated with MMC for 6 h prior to IF analyses. Quantification shown on the right. At least 100 cells were counted per condition. (C) WT and SCAI-nulls transduced with vector (V), WT or K117R mutant (Mut) FANCM ORFs were then treated with control or siRNA to FANCM 3’ UTR for 72 h, then treated with vehicle or 1.5 μM cisp for 18 h before immunoblotting. (D) WT and SCAI-null U2OS were treated with control or the indicated siRNAs for 72 h. Cells were then treated with vehicle (ND) or 1.5 μM cisp for 18 h before immunoblotting. (E) U2OS cells expressing vector (VECTOR), WT-FANCM (FANCM-ORF) or K117R-FANCM ORFs (K117R-ORF) were transfected with control (siNeg) or siRNAs to FANCM 3’UTR (siFancm) for 48 h, treated with vehicle or the indicated MMC doses and analyzed by CSAs. Data are normalized to untreated cells for each siRNA condition. Mean ± SEM survival of two independent experiments shown. (F) WT and SCAI-null U2OS were treated with siRNAs to EXO1 or control for 48 h before 16 h treatment with vehicle or 2 μM cisp. CSAs showing increased survival upon depletion of EXO1 in SCAI-nulls. Viability is relative to vehicle-treated cells. Mean ± SEM of two independent experiments shown. Western blots shown on the right. (G) WT and SCAI-null U2OS were treated with 0.5 μM cisp for 18 h before staining for RAD51 foci by IF. Representative images shown. Quantified in right panel. At least 100 cells were counted in each condition. (H). WT and SCAI-nulls were treated with control or siRNA to BRCA2 for 48 h. Cells were seeded onto plates for CSA. Representative images shown on the right.
To examine ssDNA more directly, we stained for BrdU foci in WT and SCAI null cells following depletion of EXO1 and observed markedly fewer BrdU foci in SCAI null cells following EXO1 depletion (Figure 5B,C). Furthermore, DNA combing analyses showed that depletion of EXO1 suppressed the fork protection defect observed in SCAI mutants (Figure 5D).
Unlike its role during replication stress, SCAI promotes resection at DSBs and mediates resistance to PARP inhibitors.
Programmed DSBs that form during the unhooking step precede HR during ICL repair. This raises the question as to whether the increased ssDNA seen in the absence of Protexin was due to resection of those DSBs. If so, preventing DSB formation should abolish the increased pRPA seen after Protexin loss. We depleted enzymes that mediate the ‘unhooking’ step of ICL repair, FAN1, ERCC1, SLX4 and MUS81; none of these blocked pRPA accumulation in SCAI nulls (Figure 5E, S6F). Loss of ERCC1 caused moderate increases in pRPA in WT cells (Figure 5E). Although an unknown nuclease could be involved, taken together, these experiments suggest that increased pRPA was not due to DSB formation.
Since SCAI associated with several genes that control resection following DSBs, e.g. 53BP1, HELB, we examined Protexin roles in resection of DSBs. We used a well characterized system to quantitate ssDNA formation at enzyme-induced DSB sites (Zhou et al., 2014). Unlike members of the 53BP1-RIF1-REV7 pathway, neither SCAI nor REV3 loss increased resection at DSB sites (Figure 5F). SCAI loss appeared to, if anything, lead to reduced resection presumably through SCAI’s negative regulation of 53BP1 (Isobe et al., 2017).
Loss of 53BP1 pathway genes causes increased resistance to PARPi treatment (Lord and Ashworth, 2017). Since SCAI appears to play opposing roles to 53BP1 during DSB repair by promoting resection, we hypothesized that overexpression of SCAI might promote resistance to PARP inhibitors. Indeed, overexpression of SCAI in BRCA1 null RPE cells (Figure 5G–H) increased survival following PARPi or cisp treatment as assayed by CSAs. Thus, SCAI appears to differentially regulate DNA resection depending on the context of repair.
Protexin protects FANCM-reversed forks from degradation during ICL repair
To understand where in ICL repair SCAI acts, we examined several FA pathway steps. SCAI was not important for activation of FANCI foci or FANCD2 monoubiquitination after treatment with ICL agents (Figure S6A–C). Furthermore, depletion of the core FA gene FANCA (Figure S6F, lanes 5 & 6), FANCD2 (Figure S6F, lanes 7 & 8) or expression of two different dominant negative mutants in FANCI that block FANCD2 ubiquitination (Ishiai et al., 2008) (Figure S6H) had little or no effect on the generation of pRPA in SCAI mutants treated with cisp. Together, these data suggest that the core FA pathway per se was not important for ssDNA accumulation seen SCAI mutants.
Notably, FANCM depletion markedly reduced pRPA after cisp or MMC treatment in the absence of SCAI (S6E, F) or REV3 (Figure 6A) and reduced ssDNA generation as measured by BrdU foci formation (Figure 6B). FANCM forms a complex with FAAP24 and the histone fold complex proteins MHF1 and MHF2. Depletion of either FAAP24 or MHF2 also led to reduction in pRPA in SCAI null cells (Figure S6G). Thus, a FANCM-specific function that acts independently of the FA complex or FA pathway activation is essential for driving ssDNA accumulation in the absence of Protexin.
A study using Xenopus extracts showed that reversal of one of the converging forks takes place during the process of ICL repair (Abdullah et al., 2017; Amunugama et al., 2018). ssDNA accumulation during SCAI loss might occur via fork reversal and subsequent 5’ strand degradation by EXO1. FANCM is a highly conserved member of the XPF heterodimeric endonuclease 3’ flap family. Its fork remodeling roles are mediated by its ATP-dependent translocase domain, promoting branch point migration and fork reversal (Gari et al., 2008a; Gari et al., 2008b), a biochemical function conserved in S. cerevisiae (Mph1), S. pombe (Fml1) and archaebacteria (Hef) (Meng and Zhao, 2017). A single point mutation (K117R) specifically abolishes its translocase and fork reversal activities (Gari et al., 2008a; Gari et al., 2008b), acting as a separation of function mutant, since it retains proficiency for recruitment and activation of the FANC-ID complex (Xue et al., 2008). To determine whether the FANCM translocase activity is required for ssDNA accumulation following SCAI loss, we depleted FANCM in SCAI null cells. FANCM loss led to reduced pRPA, which was restored upon expression of WT FANCM but not the K117R mutant (Figure 6C). This phenotype is specific to FANCM, as depletion of fork remodelers ZRANB3 nor SMARCAL1 failed to impact pRPA after SCAI loss (Figure 6D).
The K117R mutant also failed to rescue the increased sensitivity of FANCM-depleted cells to MMC (Figure 6E) demonstrating that FANCM’s translocase activity was important for ICL repair. Also, whereas FANCM depletion led to reduced survival following cisp treatment both in WT and SCAI null cells (Figure S6I), depletion of EXO1 resulted in substantial increases in survival (3–4-fold) compared to control siRNA in the absence of SCAI (Figure 6F), suggesting that SCAI protects from cell death in response to ICLs at least in part by limiting excessive EXO1-dependent resection of FANCM-reversed forks. Unlike ICL treatment, in response to HU FANCM loss did not prevent the increased pRPA seen in SCAI null cells although it did lower overall pRPA (Figure S6J). Taken together, these findings suggest that the FANCM dependent fork reversal activity is required to generate the substrate for EXO1-dependent ssDNA accumulation seen in the absence of Protexin.
Protexin mediates a RAD51-independent mechanism of fork stabilization control
Several protection mechanisms have been identified that act via stabilization of RAD51 on the replication fork (Rickman and Smogorzewska, 2019) which is thought to be resistant to degradation by nucleases such as MRE11. To determine whether Protexin acts in a similar fashion, we examined RAD51 foci formation following replication stress. Remarkably, SCAI depletion had no effect on RAD51 foci formation (Figure 6G), suggesting Protexin prevents fork resection in a manner independent of RAD51 stabilization. BRCA2 plays important roles in fork stabilization via RAD51 filament regulation unlike SCAI. If they work in a parallel protection pathway, they should genetically interact. Consistent with distinct fork protection mechanisms, SCAI mutants were more sensitive to BRCA2 depletion than controls, even in the absence of damage (Figure 6H).
REV3 catalyzes DNA synthesis to restrain ssDNA accumulation.
While REV3’s TLS role was not important for Protexin function, we wished to know if REV3’s polymerase activity played a role in protecting stalled forks. Thus, we established cell lines stably expressing either vector alone, GFP-REV3(WT), or GFP fused to a previously published REV3 D2781A/D2783A active site mutation (MUT-REV3) that abrogates polymerase activity (Lange et al., 2016). We then depleted endogenous REV3 using siRNAs against the 3’ UTR which is absent in the transgenes. Interestingly, whereas the WT REV3 cDNA was able to reduce pRPA relative to control cells, polymerase-dead REV3 mutant did not (Figures S7A and S7B). Equivalent REV3 expression and foci formation was confirmed by IP-western blots (Figure S7A) and IF assays (Figure S7C). The failure of the polymerase defective mutant to complement Protexin’s prevention of pRPA formation was observed in further experiments done using three individual MUT-REV3 clones compared to WT-REV3 clones (Figure 7A). This data suggests that REV3 actively synthesizes DNA to prevent or control accumulation of RPA (and thus ssDNA) in the absence of Protexin, possibly through a replacement mechanism.
Figure 7. Protexin loss exposes a previously uncharacterized role for RNA polymerase activity and REV3 polymerase activity in fork protection.

(A) WT U2OS were stably transfected with linearized plasmids expressing tagged vector, WT REV3 or REV3 D2781A/D2783A (lacking polymerase activity) and clones were selected. WT U2OS or the above clones were treated with control or an siRNA to endogenous REV3’s 3’ UTR as indicated for 48 h then treated with cisp for 18 h before immunoblotting. (B) WT and SCAI-null U2OS cells were treated with 2 μM cisp or vehicle for 10 h then with vehicle control or 10 μg/ml α-amanitin (POL2i) for 6 h before immunoblotting. (C). WT and SCAI-null U2OS were treated with EdU 15 minutes prior to treatment with vehicle or 5 μM cisp for 3 h. Cells were pre-extracted and stained for RPA2 and EdU (via click-it reaction). Nuclear intensity of EdU positive cells was measured by imageJ. Representative images shown. (D) WT U2OS were treated with EdU and either vehicle, 5 μM cisp for 3 h, α-Amanitin for 2 h or both (α-Amanitin added after 1 h cisp treatment) and processed as in (C). Representative images shown. (E) Quantification of the experiments in (C). T-tests, **** = p<0.0001. (F) Quantification of the experiments in (D). T-tests, **** = p<0.0001. (G) WT U2OS were treated with control or siRNA to FANCM for 48 h then with EdU and 5 μM cisp for 1 hr followed by vehicle or 100 μM DRB for 2 h before processing as in (C). Quantification shown. T-tests, **** = p<0.0001. (H) WT U2OS were treated with EdU and 5 μM cisp for 1 hr. Vehicle, α-Amanitin (POL2i) or ML-60218 (POL3i) were then added for 2 h before processing as in (C). Quantification shown. T-tests, **** = p<0.0001 (I) WT and SCAI-null U2OS treated with 1 μM cisp for 8 h were processed for PLAs using the indicated antibodies. Quantification shown in bottom panel. Mann Whitney test, ***=p<0.001. (J) SCAI-null U2OS transfected with the indicated siRNAs for 60 h, were treated with 1 μM cisp for 8 h and processed for PLAs as in (I). Quantification shown in bottom panel. Mann Whitney test, ***=p<0.001. (K) WT and SCAI-null U2OS transfected with the indicated siRNAs for 60 h, were treated with 1 μM cisp for 8 h, pre-extracted, fixed and processed for PLAs using the indicated antibodies. Quantification shown in bottom panel. Mann Whitney test, ***=p<0.001.
Protexin loss exposes a previously uncharacterized role for RNA polymerase activity in stalled fork repair.
Strikingly, we detected substantial enrichment of every RNA polymerase subunit in SCAI IPs (Table S6). Reversed forks induced by crosslinks primarily leave a 3’ overhang (Amunugama et al., 2018). RNA polymerase II (POL2) can recognize ssDNA overhangs (Kadesch and Chamberlin, 1982), and recently, both POL2 (Pessina et al., 2019) and POL3 (Liu et al., 2021) have been shown to be recruited to and to catalyze RNA synthesis at DSBs to promote resection. The strong association of POL2 with SCAI prompted us to investigate whether POL2 was playing similar pro-resection functions at stalled forks induced by cisp treatment. If true, POL2 inhibition should prevent pRPA accumulation. Surprisingly, while blocking POL2 activity using α-amanitin (POL2i) slightly reduced pRPA in vehicle treated cells, doing so in cells pre-treated with cisp led instead to marked increases in pRPA (Figure 7B) that approached levels seen following Protexin loss. POL2i treatment of SCAI-null cells did not result in similar increases in (nor a reduction in) pRPA levels (Figure 7B). Similar increases were also seen following treatment with DRB, another POL2 inhibitor that acts via a different mechanism (Figure S7D). The increased pRPA seen upon POL2 inhibition was dependent on FANCM and EXO1 activity (Figure S7E), suggesting that it occurred similarly to Protexin loss.
To rule out potential indirect effects from slightly prolonged (6-hour) POL2 inhibition, we used an IF-based approach assaying for detergent-resistant RPA2 foci (used as a proxy for ssDNA accumulation) on chromatin in S phase cells tracked using EdU labeling. Following 3 h cisp treatment, SCAI null cells had increased RPA2 amounts on chromatin compared to untreated controls or cisp-treated WT (Figures 7C and 7E). Also, while treatment of WT cells with either POL2i or cisp alone had no effect on total RPA2 chromatin levels, inhibition of POL2 for 2 h in the presence of cisp led to marked increases RPA2 amounts on chromatin in these cells (Figures 7D and 7F) similar to what was seen upon SCAI loss. Treatment with DRB led to a similar, FANCM-dependent increase in RPA2 levels on chromatin in WT cells (Figure 7G). Recently, RNA POL3 was shown to be recruited to DSB sites where it regulates resection (Liu et al., 2021). Unlike that seen following POL2 inhibition, POL3 inhibition did not result in increased RPA on chromatin in the presence of cisp (Figure 7H). Taken together, our results demonstrate that POL2 activity has important roles in controlling FANCM-dependent RPA loading, and thus ssDNA accumulation, during ICL repair.
To directly visualize POL2 and its possible direct roles at stalled forks, we performed proximity ligation assays (PLAs). Since γH2AX foci form at damage sites upon cisp treatment, we used PLA to detect POL2 association with γH2AX. While we detected a few foci in WT cells (Figure 7I, bottom panel), we consistently observed increased amounts of POL2-H2AX foci in SCAI null cells upon cisp treatment (Figure 7I) that only occurred when both antibodies were present (Figure S7F and S7G). We also observed increased association of a 2nd distinct POL2 antibody with one to FANCI in the absence of SCAI (Figure S7H), ruling out any potential effects of SCAI loss on γH2AX levels, as SCAI loss does not affect FANCI recruitment to or foci formation at ICLs (Figure S6B and S6C). Thus, this data is consistent with the interpretation that RNA POL2 is present at ICL sites in both WT and SCAI null cells and is consistent with a role for POL2 at these sites. The increased antibody signal with POL2 and FANCI or H2AX at fork sites seen in the absence of SCAI could be due to persistence of the ssDNA substrate for POL2 recruitment and activity due to failure of either fork restoration and/or HR. Thus, Protexin loss likely exposed a transient role for POL2 at ICL sites.
Consistent with POL2 presence at ICL sites, depletion of FANCM or EXO1 led to reduction in the amounts of POL2 - γH2AX PLA signals in the absence of SCAI (Figure 7J), suggesting that, similar to ssDNA accumulation seen upon SCAI loss (Figure 4G), the association of POL2 and H2AX was dependent on FANCM and EXO1 activity. FANCM or EXO1 depletion did not affect γH2AX levels in the absence of Protexin (Figure 6A). To directly visualize POL2 products at ICL sites following cisp treatment, we performed PLAs against FANCI and RNA-DNA hybrids (using a known monoclonal antibody, S9.6). We detected increased numbers of S9.6/FANCI foci in the absence of SCAI (Figure 7K) although hybrid induction cannot be firmly concluded as these interpretations are dependent on S9.6 specificity for RNA-DNA hybrids (Crossley et al., 2021; Hartono et al., 2018). These experiments suggested the possibility that RNA-DNA hybrids might transiently form on the reversed fork, and that this structure may accumulate due to increases in ssDNA upon SCAI loss (Figure S7I).
Discussion
SCAI is a novel replication stress and crosslink repair factor
In this study we found many genes not previously implicated in ICL repair and focused on SCAI, which we show is important for ICL repair, fork protection under multiple circumstances and genomic stability. SCAI acts in the FA pathway and was epistatic with FANCA and genetically interacts with FANCM. Whether SCAI mutation in humans presents with an FA-related phenotype is unknown. However, loss of even one allele in humans is heavily selected against, pLi= 1 (https://gnomad.broadinstitute.org/). Thus, if SCAI mutants could result in a recessive FA phenotype, the homozygous mutants would be exceedingly rare as single LOF alleles are selected against. This is also true for REV3L and REV1, pLi=1.
SCAI forms a complex with REV3 that controls ssDNA accumulation
SCAI forms a complex, Protexin, with REV3. Protexin acts as a general regulator of stalled fork dynamics being required in response to HU, fragile sites and for HR repair at forks stalled by the Tus/Ter complex. A striking phenotype due to Protexin loss was accumulation of ssDNA in response to ICLs in S phase. That Protexin controls resection at stalled forks is supported by multiple observations. First, depletion of either component of Protexin led to increases in chromatin-bound pRPA and ssDNA following cisp, MMC and HU treatment. Secondly, DNA fiber assays showed increased fork resection following HU treatment in the absence of SCAI. Third, recovery of DNA synthesis is impaired following HU-treatment in SCAI mutant cells. Finally, RPA accumulation in mutants can be prevented by loss of exonucleases. Importantly, the role for REV3 in regulation of ssDNA accumulation upon replication stress was likely independent of its previously described role in TLS as loss of REV7 did not affect ssDNA accumulation.
The Protexin complex promotes reversed fork stability
ICL-induced ssDNA accumulation in Protexin deficient cells requires the FANCM-FAAP24-MHF2 complex but not members of the core FA complex or FANCI-D2. FANCM initiates repair at ICLs suggesting that the ssDNA accumulation occurs at ICL sites. Genetic evidence suggests the role of FANCM in the Protexin pathway involves fork reversal. In principle, this ssDNA could be generated by either resection on the lagging strand or after fork reversal. Fork reversal occurs during ICL repair both in Xenopus extracts (Amunugama et al., 2018) and in mammalian cells (Zellweger et al., 2015). FANCM possesses fork reversal activity in vitro (Gari et al., 2008a).
FANCM’s role in mediating pRPA accumulation in Protexin mutants could be separated from its role in FA pathway activation, as the defect in ssDNA accumulation in the absence of SCAI could not be rescued by the K117R FANCM mutant required for fork reversal in vitro. This mutant is severely defective in ICL repair in vivo but maintains the role of FANCM in recruiting core complex and FA signaling (Gari et al., 2008a; Meetei et al., 2005; Xue et al., 2008). Assuming that the K117R mutant is specific for fork reversal, these data suggest that Protexin acts to protect reversed forks from either extensive reversed strand resection and/or the inability to restore the resected DNA. Resection on the lagging strand pre-reversal is also possible but would be a minor component of overall resection. Resection of DSBs that form during ICL repair was ruled out by the lack of effect of mutants that prevent DSB formation in ICL repair e.g. FA core complex, FANCD2, MUS81 and SLX4.
How does Protexin act to prevent excessive ssDNA accumulation?
Protexin limits or counteracts EXO1 activity at stalled forks but must have additional activities as EXO1 loss only partly rescues ICL sensitivity in SCAI mutants and the HR defect at Tus/Ter stalled forks is not rescued by EXO1 depletion (data not shown). An alternative formal possibility is that ssDNA occurs constitutively upon fork reversal (e.g. leading strand advancement past the lagging strand leading to a 3’ overhang) and Protexin counteracts FANCM fork reversal activity. However, FANCM loss did not prevent increases in pRPA following HU in the absence of SCAI. It should be noted that while the TLS role of REV3 is likely not required for the function of Protexin at stalled forks, the converse is not true, i.e. that Protexin has no role in TLS, and remains to be examined. Furthermore, EXO1 and FANCM work coordinately because the phenotypes of EXO1 and FANCM loss on proliferative fitness across 600 cell lines is highly correlated [Cancer Dependency Map (https://depmap.org/portal/)] as FANCM is the most closely correlated gene with EXO1 in over 18,000 genes (Pearson Coefficient = 0.33).
Protexin promotes resynthesis of excised DNA.
While protecting DSBs or reversed forks from resection is thought to occur by prevention of exonuclease accessibility, the requirement for REV3 suggested the possibility of resynthesis of resected DNA. This model is supported by the observation that REV3 polymerase activity was required for preventing ssDNA accumulation analogous to what has been proposed for POL alpha and the CST complex at DSBs (Mirman et al., 2018). CST uses primase to prime and, although it is not fully clear how a primer for REV3 might be generated, it is possible that RNA synthesized by POL2 could be involved. RNA polymerase can recognize and initiate RNA synthesis on 3’ overhangs (Kadesch and Chamberlin, 1982) although this possible function will require future biochemical investigation.
Recently, RNA POL3 has been shown to be recruited to DSBs where it generates transient RNA that is required for HR, although there is conflicting data on whether POL2 or POL3 was required. Using genetic and imaging approaches we have shown that RNA POL2 is present at stalled forks. Inhibition of POL2 but not POL3 using multiple drugs led to FANCM-and EXO1-dependent increases in RPA2 at stalled forks. We also observed potential formation of RNA-DNA hybrids that persist in the absence of Protexin, although that is dependent on the specificity of the S9.6 antibody for RNA-DNA hybrids. In this role one hypothetical model suggests that POL2 recognizes ssDNA ends generated during fork reversal and ultimately plays a positive role in protecting these reversed forks. POL2 recognition of ssDNA may be a common predicament faced during many types of repair, and loss of Protexin might be uncovering a previously unrecognized role for RNA POL2 upon fork stalling and reversal.
Differential strategies for fork and DSB protection from resection and roles in HR.
For reversed stalled forks, limited resection is protective to prevent NHEJ, a role performed by DNA2 at HU-stalled forks (Thangavel et al., 2015). Excessive resection at stalled forks, however, could be deleterious. For example, RNA persistence in the absence of Protexin could block ssDNA from participating in HR or reversing fork reversal which could increase genomic instability. Thus, while loss of anti-resection factors at DSBs leads to increased HR, this is not the case at stalled forks. Importantly, our data suggests that SCAI has a pro-resection role with respect to the 53BP1 complex at DSBs as SCAI overproduction provided resistance to PARP inhibitors in BRCA1 mutants. Thus, SCAI could protect reversed forks by both countering NHEJ while also preventing excessive resection and promoting HR at forks.
In the absence of Protexin, excessive resection and inability to fill-in the resected DNA in response to ICLs leads to ssDNA accumulation. Inhibition of RNA POL2, which forms a complex with SCAI, phenocopies Protexin loss. How the POL2 and REV3 polymerases coordinate is not clear. One possibility is their interaction initiates REV3 activity after priming by POL2. Nevertheless, Protexin acts in concert with POL2, possibly analogously to the back-filling role proposed for the Shieldin-CST complex at DSBs to prevent pathological excessive resection. It is also possible that a multifaceted mechanism operates that also includes directly impeding EXOI activity. The fact that short-term POL2 inhibition phenocopies Protexin loss and that POL2 is present at the stalled fork suggests a direct role for POL2 at the fork. However, we cannot rule out the possibility that POL2 controls the expression of a short-lived RNA that acts in trans to protect forks.
Why allow resection in the first place if it can result in a need for a complex repair process? The answer is not known but one possibility is that it simply cannot be avoided kinetically during S phase and must be worked around. Alternatively, resection could help drive fork reversal kinetics in the case of an ICL. However, restoration of the reversed fork may require dsDNA resynthesis. ssDNA may be problematic for fork restoration energetically. Failure to properly restore fork function, particularly if it occurs on both sides of the ICL, could lead to partially unreplicated chromosomes and chromosomal breakage upon mitotic entry. While much remains to be known, it is clear that the Protexin complex and its associated proteins are playing a key role in addressing replication stress, and future elucidation of its mechanisms will illuminate a critical aspect of DNA repair.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
The lead contact is Stephen Elledge (selledge@genetics.med.harvard.edu)
Materials availability
Materials generated in this study are available upon request.
Data and code availability
All generated data is available in the main text or supplemental tables. Original images have been deposited in Mendeley and can be accessed using the following link: http://dx.doi.org/10.17632/35f2n4wsnv.1. Any additional information is available upon request.
This paper does not report original code
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human cell lines
U2OS cells were passaged in McCoys 5A media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. RPE1 cells were maintained in DMEM:F12 media supplemented with 10% FBS and 1% penicillin/streptomycin. HeLa cells and 293T cells were grown in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. U2OS expressing WT and Dom Neg FANCI were previously described (Ishiai et al., 2008). REV7 KO U2OS cells were previously described (Bluteau et al., 2017). ER-AsiSI cells were previously described (Zhou et al., 2014). All cells were maintained at 37°C in 5% CO2.
Mouse cell lines
Mouse ES cells were cultured in DMEM supplemented with 15% fetal bovine serum, 500 U/mL LIF, 2 mM glutamine, 1 mM sodium pyruvate, 20 mM Hepes, β-mercaptoethanol, non-essential amino acids, and penicillin/streptomycin.
METHOD DETAILS
SiRNA transfections
HeLa, RPE1, U2OS, or mouse 6xTer/HR ES cells were seeded onto 6-well plates and transfected with siRNAs the next day. In other instances, siRNAs were reverse transfected into cells at 20–40 nM using Lipofectamine RNAiMAX reagent (Invitrogen) or Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cells were harvested or treated with drugs 2–4 days later.
Plasmids, cloning and virus production/viral transduction
FLAG-SCAI, FLAG-HA-SCAI and GFP-tagged SCAI were generated by LR cloning. siRNA-resistant SCAI constructs were generated by site directed mutagenesis. FLAG-REV3 was generated by transferring the REV3 ORF via restriction digestion from JT113-pETDuet1-(R)-hREV3 (Addgene) into a modified FNpCNA3 (Addgene) vector. FANCM-WT and FANCM-K117R were previously described (Wang et al., 2018). shRNAs against RAD54, POLE4, ADSL, and OGDH were isolated from a previously described pGIPZ library (Silva et al., 2005). shRNAs against REV3 and SCAI were generated by cloning into pLKO lentiviral vectors.
Lentiviruses were packaged in 293T cells via transfection with PolyJet (SignaGen) or TransIt reagent (MiRus). At 36 and 60h after transfection, harvested supernatants were filtered through 0.45mm-pore low protein binding membranes, concentrated using Lenti-X concentrator (Clontech) according to the manufacturer’s instructions and treated with Benzonase (Millipore). Viral titers were calculated by clonogenic assays of serially diluted virus in the cell line of interest. All transductions were performed in the presence of polybrene. Infected U2OS cells were selected using puromycin (1 μg/ml; Clontech) for 3 days, Geneticin (Neomycin - 800 μg/ml; Invitrogen) for 7 days, or blasticidin (7.5 μg/ml; Invitrogen) for 4 days.
CRISPR library construction and CRISPR screen
The CRISPR library used in this screen was described previously (Martin et al., 2017). Briefly, a library of gRNAs targeting 18,166 genes with five gRNAs per gene for a total of ∼91,000 gRNAs was synthesized and cloned into the lentiCRISPR V2 puro vector. Pooled virus was prepared by transfecting 293T cells with the library plasmid pool with lentiviral packaging vectors. Viral supernatants were harvested after transfection and concentrated with lenti-X concentrator solution (Clontech) before being titred by colony formation assays in U2OS cells. Cisp doses for the screen were determined by titrating a series of drug concentrations in U2OS cells and examining cell survival after 3 days. For the screen, U2OS cells were infected at a low MOI (0.2) with a representation of 500 in duplicate. Cells were selected with puromycin (1 μg/mL) for 3 days until an uninfected control plate completely died. An initial cell pellet was taken as PD0 and cells in each replicate were split into two populations, vehicle treated or treated with 1.5 μM cisp. Vehicle treated cells were grown for 5 PDs and harvested. Drug treated cells were washed after 24 h and grown for 5 PDs before being harvested. For the second arm of the screen, cells were further treated with 1 μM cisp, washed and cultured for another 5 PDs. Control cells were treated with vehicle and cultured for another 5 PDs as well before a final cell pellet was collected at PD10. Genomic DNA was isolated by phenol/chloroform extraction and gRNAs were PCR amplified with barcoded primers for sequencing on an Illumina NextSeq 500. Sequencing reads were aligned to the initial library to obtain read counts for each PD and condition. MAGeCK, EdgeR and R were used to calculate p-values, FDRs, and log2 fold changes for comparison between the PD0, PD5 and PD10 cisp-treated and vehicle-treated samples. GO term analyses were performed using the GenePattern server (The Broad Institute).
Generation of CRISPR knockout SCAI cells
The following gRNA sequences 5’ CACCGTGCTGAAGATGATATCCCAC and 5’ AAACGTGGGATATCATCTTCAGCAC were annealed and cloned into LentiCrispr v2 (Addgene). U2OS cells were infected and selected in puromycin for 3 days. Single cells were cloned and SCAI knockout status was ascertained by western blotting.
Multicolor competition assays (MCAs)
MCAs were performed as described previously (Smogorzewska et al., 2007). Briefly, GFP-labeled U2OS cells were transfected with the indicated test siRNAs while RFP-labeled U2OS cells were transfected with control siRNAs. After 48 h, cells were mixed in equal quantities in six-well plates. Cells were then treated with the indicated dose of drug or vehicle control for 24 h. Fresh media was added and cells were maintained for 7 to 10 days after treatment. Subsequently, the percentage of GFP and RFP labeled cells were quantified by FACS analyses using a BD LSR II.
Colony formation assay
For colony formation assays, WT or CRISPR KO cells were transfected as above after which cells were exposed to the indicated doses of drugs for 16 to 24 h and adjusted for plating depending on dose of drug in six-well plates. Colonies were fixed and stained after 10 −14 days and scored with a colony counting pen (VWR).
96-well plate viability assays
Cell viability assays using AlamarBlue (ThermoFisher) and CellTiter-Glo 2 (Promega) were performed according to the manufacturers’ instructions. Briefly, following shRNA infections or siRNA treatment, cells were seeded at 500 to 1000 cells per well in triplicate in 96 well plates. Cells were then treated with the indicated drugs for 24 h, washed and left to recover for 48 to 72 h prior to being read on a VICTOR X5 Multilabel Plate Reader (PerkinElmer).
Western blotting
Western blotting was done as previously described (Elia et al., 2015). Briefly, cells were harvested and lysed in SDS lysis buffer or RIPA buffer on ice. Cells were sonicated, protein content was measured and analyzed using electrophoresis gels.
Quantitative reverse transcription PCR (RT-qPCR) analyses
Total RNA was isolated from U2OS cells or mouse 6xTer/HR ES cells using the RNeasy plus mini kit (Qiagen) and reverse transcribed into cDNA using SuperScript IV reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. RT-qPCR was performed in triplicate using the TaqMan Gene Expression Master Mix (Life Technologies) and the respective gene-specific TaqMan Gene expression Assays (Thermo Scientific) on an Applied Biosystems Fast 7500 machine. GAPDH served as an endogenous normalization control.
Cell cycle analyses
Cells were treated with siRNA as above and treated or not with drugs as indicated. Cells were then harvested and fixed in 4% formaldehyde for 15 m at room temperature. Alternatively, cells were fixed in 70% ethanol for 15 m on ice. Cells were then pelleted, washed in PBS and resuspended in 50 μg/ml propidium iodide solution containing 0.1 mg/ml RNase A as well as 0.05% Trition X-100 for 40 m at 37°C, resuspended in PBS and flow cytometry was performed using BD-LSR II (Becton Dickinson). Data were analyzed using Flowjo software (Tree Star, OR).
BrdU restart assay
Cells were treated with vehicle or drug as indicated in the figure legends. Cells were then labeled with 10 μM BrdU for 30 m, washed and harvested. BrdU incorporation was assayed using an APC BrdU flow kit (BD Pharmingen) according to the manufacturer’s instructions and analyzed by flow cytometry on a BD-LSRII Flow Cytometer (Becton Dickinson). Data was collected via BD FACS Diva software (Becton Dickinson). Analysis of cell cycle data was performed using FloJo software.
Chromatin fractionation assay
HeLa or U2OS cells were harvested and washed with cold PBS. Sedimented cells were resuspended in cold Solution 1 consisting of 10 mM Hepes (pH 7.9), 0.1% Triton X-100, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 1 mM DTT, protease and phosphatase inhibitor cocktails. After a 5 m incubation, samples were centrifuged at 1300g for 5 m, and the supernatant removed as the soluble fraction. Sedimented nuclei were washed once with Solution 1 and lysed in Solution 2 (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT) for 10 m. Samples were centrifuged at 1300 g for 5 m, and the chromatin-enriched pellets washed once with Solution 2 followed by resuspension in lysis buffer containing 50mM Tris ph 6.8, 100 mM NaCl, 1.7% SDS, 7% glycerol and protease and phosphatase inhibitors. After sonication, sample buffer was added to 1X and the samples were boiled for western blotting.
Immunofluorescence assay
For IF, cells were transfected or not with the indicated siRNAs, then 24 h later plated onto glass coverslips in 6-well plates. After 24–36 h, drug treatments were performed for the indicated durations. Cells were then washed with PBS, fixed with 4% paraformaldehyde for 15 m and extracted with 0.5% Triton X-100 in PBS for 10 m. In some cases cells were pre-extracted using cytoskeleton buffer (containing 10 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES), pH 6.8, 100 mM NaCl, 300 mM sucrose, 1 mM MgCl2, 1 mM EGTA as well as 0.5% Triton X-100, protease and phosphatase inhibitors) for 5 m on ice prior to the fixation and permeabilization steps. Cells were then blocked in 3% BSA in PBS and incubated with primary and secondary antibodies. The coverslips were mounted and nuclei visualized with DAPI Fluoromount-G (Southern Biotech) and images were acquired using an Olympus FV3000 confocal microscope.
Detection of ssDNA via BrdU staining
ssDNA generated by following treatment with ICL agents was detected as previously described (Huang et al., 2010). Briefly, WT or SCAI-null U2OS cells were reverse transfected or not with the indicated siRNAs and then grown for 36 h in media containing 20 μM BrdU. Afterwards cells were cultured without BrdU overnight prior to treatment with vehicle or 1 M MMC for 6–8 h. Cells were washed, fixed with cold methanol, rinsed with acetone, and then incubated with anti-BrdU antibody (BD Biosciences, 347580) without DNA denaturation. Total BrdU incorporation was determined under denatured condition via treatment with 2.5 M HCl for 80 minutes.
The native BrdU flow cytometry assay described in Figure S3H was performed as previously described (Tkac et al., 2016) with modifications. Briefly, control or SCAI siRNA treated cells were labeled with BrdU for 10 h followed by treatment with vehicle or indicated doses of cisplatin for 6 h. BrdU intensity was assayed using flow cytometry under non-denaturing conditions.
DSB resection assay
Measurement of resection at DSBs using the ER-AsiSI U2OS cell lines were performed as previously described (Zhou et al., 2014). DSBs were measured 4 h following addition of 4-hydroxytamoxifen to induce breaks.
Metaphase spreads
Cells were either transfected with SCAI specific siRNA or control nontargeted siRNA using Lipofectamine RNAiMax (Life Tech). The cells were then treated with 20ng/ml MMC for 48 h or left untreated. Following treatment, the cells were exposed to colcemid (100ng/ml) for 2 h, treated with a hypotonic solution (0.075M KCL) for 20 m and fixed with 3:1 methanol: acetic acid. Slides were stained with Wright’s stain and 50 metaphase spreads were scored for aberrations. Metaphase spreads were observed using a Zeiss Axio Imager microscope and captured using CytoVision software from Applied Imaging.
IP mass spectrometry analyses
IP mass spec analysis was done as previously described (Elia et al., 2015). 293T cells expressing FLAG-HA-tagged SCAI or vector control were treated with vehicle or 3 μM for 3 h and then lysed in low-salt lysis buffer (50 mM Tris, 150 mM NaCl, 10 mM NaF, 0.5% NP-40, pH 7.5) containing 2 mM N-Ethylmaleimide, protease inhibitor tablet, and phosphatase inhibitor cocktails. Lysates were clarified by centrifugation at 14,000g for 10 m. The insoluble pellet was then sonicated in low-salt lysis buffer and clarified a second time by centrifugation at 14,000g. Supernatants were combined and immunoprecipitated with monoclonal anti-HA agarose (Sigma) for 2 h at 4°C, washed 4 times in low-salt lysis buffer and eluted with 500 μg/mL HA peptide (Sigma). SCAI-interacting proteins were TCA precipitated, digested with trypsin, desalted using Stage tips, and analyzed by LC-MS/MS.
Co-immunoprecipitation (Co-IP) analyses
293T cells were transfected with the indicated plasmids for 60 h, treated with cisp or vehicle, harvested in cold PBS and lysed in RIPA buffer without SDS. Lysates were clarified by centrifugation and the supernatants were precleared with Protein G Dynabeads (Invitrogen) and immunoprecipitated using Anti-FLAG® M2 Magnetic Beads (Sigma) for 2 h at 4°C. Beads were washed four times and boiled in sample buffer prior to western blotting.
EdU Click-It assays
EdU click-it assays were performed according to the manufacturer’s recommendations (Sigma). EdU was added to cells at a final concentration of 10 μM. Cells were pre-extracted to remove non-chromatin bound RPA2 before staining.
Proximity ligation assays
For Proximity ligation assays (PLA), cells were fixed and permeabilized as with IF assays except in experiments detecting interaction of FANCI and S9.6 antibodies where cells were pre-extracted prior to fixation. Cells were then incubated with primary antibody combinations and PLAs were performed using Duolink in situ Mouse/Rabbit Kit (Sigma) according to the manufacturer’s instructions. Additional antibodies used for PLA include rabbit anti-RNA polymerase II (Bethyl, A300–653A), mouse monoclonal anti- Pol II (8WG16) (Santa Cruz, sc-56767) and anti-DNA-RNA Hybrid Antibody, clone S9.6 (Millipore, MABE1095).
DNA fiber assay
U20S or SCAI-null cells were transfected with control siRNAs or siRNAs to EXO1. After 60 h, cells were incubated with 25 mM CldU for 30 m, washed and subsequently treated with 250mM IdU for 30 mM. Cells were then treated 4.5 mM HU for 4.5 h. Cells were harvested and replication combing assay was performed using the FiberComb machine (Genomic Vision) according to the manufacturer’s protocol. Briefly, cells were counted and embedded in low melting point agarose plugs, then treated with proteinase K overnight. Agarose plugs were then washed and digested with agarose, then poured into FiberComb wells and combed onto silanized coverslips. Coverslips were stained as above and visualized by fluorescence microscopy using an Olympus FluoView FV3000 confocal microscope. Images were analyzed with ImageJ. More than 100 fibers were counted for each condition. In all figures, data represent the mean and SEM; p values were calculated using Mann-Whitney test.
CFS reporter assays
CFS assays were done as previously described (Wang et al., 2018). Cells infected with lentivirus vectors to express shRNAs to silence the indicated genes were cultured for two weeks. Mitotic recombination was measured by FACS.
Homologous recombination assay
1.6 × 105 mouse 6xTer/HR embryonic stem reporter cells were co-transfected in suspension with 0.35 μg pcDNA3β control vector, pcDNA3β-myc NLS-I-SceI, or pcDNA3β-myc NLS-Tus, or along with either 20 pmol siLUC, siSCAI or siREV3 siRNAs using Lipofectamine 2000 (Invitrogen) as previously described (Willis et al., 2014). For each independent experiment, HR frequencies from duplicate samples were scored 72 h after transfection by flow cytometry using a Becton Dickinson 5 Laser LSRII and values corrected for background events and for transfection efficiency (65–85%). Between 3–6 × 105 events were scored per duplicate samples. Transfection efficiency was measured by parallel transfection with 0.05 μg pcDNA3β-GFP expression vector, 0.30 μg pcDNA3β control vector, and 20 pmol siRNA. In all figures, data represents the mean and standard error of the mean (SEM) derived from independent experiments performed on different days (SEM = standard deviation/√n, where n= number of experiments). P-values were calculated using a two-tailed unpaired t-test.
Supplementary Material
Table S1. Read counts from cisp screens. Related to Figure 1.
Table S2. MAGeCK analyses of untreated and cisp treated screen arms at both population doublings. Related to Figure 1.
Table S3. EdgeR analyses of untreated and cisp treated screen arms at both population doublings. Related to Figure 1.
Table S4. List of hits from cisp screens. Related to Figure 1.
Table S5. SCAI IP mass spectrometry results. Related to Figure 5.
Table S7. List of oligonucleotides used in study. Related to Key Resources Table
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SCAI | Abcam | Ab124688 |
| Mouse anti-Vinculin | Sigma | V9131 |
| Mouse monoclonal anti-Flag | Sigma | F1804 |
| Rabbit monoclonal anti-Flag | Sigma | F7425 |
| Anti-BrdU antibody [BU1/75 (ICR1)] | Abcam | Ab6326 |
| GFP | Abcam | ab290 |
| ATM | Abcam | ab32420 |
| FANCA | Bethyl | A301-980 |
| FANCI | Bethyl | A301-254 |
| FANCD2 | Novus | NB100-182 |
| 53BP1 | Bethyl | A300-272A |
| BRCA2 | Bethyl | A300-005A |
| SMARCAL1 | Santa Cruz | sc-376377 |
| ZRANB3 | Bethyl | A303-033A |
| HELB | Abcam | ab202141 |
| SLX4 | Bethyl | A302-270A |
| ERCC1 | Cell Signaling | 3885S |
| SNM1A | Bethyl | A303-747A |
| FAAP24 | GeneTex | GTX117277 |
| RNAseH2A | Abcam | ab92876 |
| REV7 | Santa Cruz | sc-135977 |
| CtIP | Bethyl | A300-488A |
| MRE11 | Cell Signaling | 4895 |
| MUS81 | Santa Cruz | sc-53382 |
| ORC2 | Abcam | ab68348 |
| DNA2 | Abcam | ab96488 |
| EXO1 | Bethyl | A302-640A |
| RPA2-P-S4/8 | Bethyl | A300-245A |
| RPA32 | GeneTex | GTX70258 |
| CHK1-P-S345 | Cell Signaling | 2348S |
| CHK1 | Santa Cruz | Sc-8408 |
| γH2AX | Millipore | 05-636 |
| H2AX | Bethyl | A300-082A |
| Bacterial and virus strains | ||
| Lenti-Blast-3Flag FANCM WT | Wang et al, 2018 | N/A |
| Lenti-Blast-3Flag FANCM K117R | Wang et al, 2018 | N/A |
| LentiCrispr v2 | Sanjana et al, 2014 | Addgene #52961 |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| DMSO | Amresco | N182 |
| Hydroxyurea | Sigma | H8627-10G |
| Mitomycin C (MMC) | Santa Cruz Biotech | M4287-2MG |
| Cisplatin (cisp) | HMS Ludwig | N/A |
| Mirin | Sigma | M9948 |
| Olaparib | Selleck chemicals | S1060 |
| 4-hydroxytamoxifen | Sigma | H7904-5MGG |
| α-Amanitin | Abcam | ab144512 |
| 5,6-Dichlorobenzimidazole 1-β-D-ribofuranoside (DRB) | Sigma | D1916 |
| RNA Polymerase III inhibitor (ML-60218) | Sigma | 557403 |
| Superscript IV reverse transcriptase | Invitrogen | 18090050 |
| Lipofectamine RNAiMAX | Invitrogen | 13778075 |
| Lipofectamine 3000 | ThermoFisher | L3000008 |
| TransIT®-LT1 Transfection Reagent | Mirus | MIR2300 |
| 5-Chloro-2′-deoxyuridine (CIdU) | Sigma | C6891 |
| 5-Iodo-2′-deoxyuridine (IdU) | Sigma | I7125 |
| cOmplete, Mini, EDTA-free, Protease Inhibitor Cocktail Tablets | Roche | 11836170001 |
| Phosphatase Inhibitor Cocktail Set I | Sigma | 524624 |
| Phosphatase Inhibitor Cocktail Set II | Sigma | 524625 |
| Halt Protease and Phosphatase Inhibitor Cocktail (100X) | Fisher | 78440 |
| Critical commercial assays | ||
| RNeasy Plus mini kit | Qiagen | 74134 |
| Taqman Gene expression Mastermix | Life Technologies | 4369016 |
| Taqman Gene Expression Assay Human REV1 | ThermoFisher Scientific | Catalog # 4331182 Assay ID: Hs00249411_m1 |
| Taqman Gene Expression Assay Human REV3L | ThermoFisher Scientific | Catalog # 4331182 Assay ID: Hs00161301_m1 |
| Taqman Gene Expression Assay Human GAPDH | ThermoFisher Scientific | Catalog # 4331182, Assay ID:Hs99999905_m1 |
| AlamarBlue | ThermoFisher | DAL1025 |
| CellTitre-Glo 2 | Promega | G9242 |
| Click-iT™ Plus EdU Cell Proliferation Kit for Imaging, Alexa Fluor™ 594 dye | ThermoFisher | C10639 |
| APC BrdU Flow kit | BD biosciences | 552598 |
| Quickchange II XL Site Directed Mutagenesis Kit | Agilent | 200521 |
| Deposited data | ||
| http://dx.doi.org/10.17632/35f2n4wsnv.1 | ||
| Experimental models: Cell lines | ||
| Human: U2OS | ATCC | N/A |
| Human: U2OS SCAI KO | This paper | N/A |
| Human: U2OS WT FANCI | Ishiai et al, 2008 | N/A |
| Human: U2OS DN FANCI-1 | Ishiai et al, 2008 | N/A |
| Human: U2OS DN FANCI-2 | Ishiai et al, 2008 | N/A |
| Human: ER-AsiSi U2OS | Zhou et al, 2014 | N/A |
| Human: REV7 KO U2OS | Bluteau et al, 2017 | N/A |
| Human: HeLa | ATCC | N/A |
| Human: RPE | ATCC | N/A |
| Human: 293T | ATCC | N/A |
| Mouse: 6xTer/HR ES cells | Willis et al, 2014 | N/A |
| Experimental models: Organisms/strains | ||
| Oligonucleotides | ||
| Listed in Table S7 | ||
| Recombinant DNA | ||
| REV3L (NM_002912) Human Tagged ORF Clone | Origene | RG222943 |
| pETDuet1-(R)-hREV3 | Tomida et al, 2015 | Addgene #64872 |
| GFP-REV3 | This paper | N/A |
| Flag-SCAI | This paper | N/A |
| FLAG-HA-SCAI | This paper | N/A |
| GFP-SCAI | This paper | N/A |
| Software and algorithms | ||
| MaGeck | Li et al, 2014 | https://sourceforge.net/p/mageck/wiki/Home/ |
| EdgeR | Robinson et al, 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Flowjo | N/A | https://www.flowjo.com/ |
| ImageJ | N/A | https://imagej.nih.gov/ij/ |
| Graphpad Prism | N/A | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
Highlights.
Genome-wide cisplatin sensitivity screens show that SCAI promotes ICL repair
SCAI forms Protexin complexes with REV3, preventing ssDNA accumulation after damage
Protexin loss leads to excessive EXO1 resection of FANCM reversed forks at ICLs
RNA polymerase interacts with Protexin to restore forks and promote repair
Acknowledgements
We thank E. Wooten, Q. Xu, M. Li, J. Walter and T. de Lange for helpful discussions. We thank G. Legube and T. Paull for the ER-AsiSI U2OS cells. R.O.A. was an HHMI fellow of the Life Sciences Research Foundation. S.J.E. is supported by a grant from the National Cancer Institute of the National Institutes of Health under Award Number 1R01CA234600. S.J.E. is an investigator with the Howard Hughes Medical Institute.
Footnotes
Declaration of Interests
The authors declare no competing interests. S.J.E. is a member of the Molecular Cell advisory board.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdullah UB, McGouran JF, Brolih S, Ptchelkine D, El-Sagheer AH, Brown T, and McHugh PJ (2017). RPA activates the XPF-ERCC1 endonuclease to initiate processing of DNA interstrand crosslinks. EMBO J 36, 2047–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amunugama R, Willcox S, Wu RA, Abdullah UB, El-Sagheer AH, Brown T, McHugh PJ, Griffith JD, and Walter JC (2018). Replication Fork Reversal during DNA Interstrand Crosslink Repair Requires CMG Unloading. Cell Rep 23, 3419–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen PR, D’Andrea AD, and Taniguchi T (2004). ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev 18, 1958–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluteau D, Masliah-Planchon J, Clairmont C, Rousseau A, Ceccaldi R, d’Enghien CD, Bluteau O, Cuccuini W, Gachet S, de Latour RP, et al. (2017). Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest 127, 1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt DT, Baarlink C, Kitzing TM, Kremmer E, Ivaska J, Nollau P, and Grosse R (2009). SCAI acts as a suppressor of cancer cell invasion through the transcriptional control of beta1-integrin. Nat Cell Biol 11, 557–568. [DOI] [PubMed] [Google Scholar]
- Ceccaldi R, Sarangi P, and D’Andrea AD (2016). The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol 17, 337–349. [DOI] [PubMed] [Google Scholar]
- Chen CC, Feng W, Lim PX, Kass EM, and Jasin M (2018). Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu Rev Cancer Biol 2, 313–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, and Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Mol Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich KA, and Cortez D (2008). ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 9, 616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley MP, Brickner JR, Song C, Zar SMT, Maw SS, Chedin F, Tsai MS, and Cimprich KA (2021). Catalytically inactive, purified RNase H1: A specific and sensitive probe for RNA-DNA hybrid imaging. J Cell Biol 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, and Cortez D (2015). The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol Cell 59, 998–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkin SG, and Glover TW (2007). Chromosome fragile sites. Annu Rev Genet 41, 169–192. [DOI] [PubMed] [Google Scholar]
- Elia AE, Wang DC, Willis NA, Boardman AP, Hajdu I, Adeyemi RO, Lowry E, Gygi SP, Scully R, and Elledge SJ (2015). RFWD3-Dependent Ubiquitination of RPA Regulates Repair at Stalled Replication Forks. Mol Cell 60, 280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaycoechea JI, Crossan GP, Langevin F, Mulderrig L, Louzada S, Yang F, Guilbaud G, Park N, Roerink S, Nik-Zainal S, et al. (2018). Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 553, 171177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari K, Decaillet C, Delannoy M, Wu L, and Constantinou A (2008a). Remodeling of DNA replication structures by the branch point translocase FANCM. Proc Natl Acad Sci U S A 105, 16107–16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari K, Decaillet C, Stasiak AZ, Stasiak A, and Constantinou A (2008b). The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol Cell 29, 141–148. [DOI] [PubMed] [Google Scholar]
- Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, Typas D, Lammers M, Mailand N, Nussenzweig A, et al. (2018). DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 173, 972–988 e923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RK, Mund A, Poulsen SL, Sandoval M, Klement K, Tsouroula K, Tollenaere MA, Raschle M, Soria R, Offermanns S, et al. (2016). SCAI promotes DNA double-strand break repair in distinct chromosomal contexts. Nat Cell Biol 18, 1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartono SR, Malapert A, Legros P, Bernard P, Chedin F, and Vanoosthuyse V (2018). The Affinity of the S9.6 Antibody for Double-Stranded RNAs Impacts the Accurate Mapping of R-Loops in Fission Yeast. J Mol Biol 430, 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Kim JM, Shiotani B, Yang K, Zou L, and D’Andrea AD (2010). The FANCM/FAAP24 complex is required for the DNA interstrand crosslink-induced checkpoint response. Mol Cell 39, 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, Saberi A, Kinoshita E, Kinoshita-Kikuta E, Koike T, et al. (2008). FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat Struct Mol Biol 15, 1138–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isobe SY, Nagao K, Nozaki N, Kimura H, and Obuse C (2017). Inhibition of RIF1 by SCAI Allows BRCA1-Mediated Repair. Cell Rep 20, 297–307. [DOI] [PubMed] [Google Scholar]
- Kadesch TR, and Chamberlin MJ (1982). Studies of in vitro transcription by calf thymus RNA polymerase II using a novel duplex DNA template. J Biol Chem 257, 5286–5295. [PubMed] [Google Scholar]
- Kottemann MC, and Smogorzewska A (2013). Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493, 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange SS, Tomida J, Boulware KS, Bhetawal S, and Wood RD (2016). The Polymerase Activity of Mammalian DNA Pol zeta Is Specifically Required for Cell and Embryonic Viability. PLoS Genet 12, e1005759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langevin F, Crossan GP, Rosado IV, Arends MJ, and Patel KJ (2011). Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475, 53–58. [DOI] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Hua Y, Wang J, Li L, Yuan J, Zhang B, Wang Z, Ji J, and Kong D (2021). RNA polymerase III is required for the repair of DNA double-strand breaks by homologous recombination. Cell 184, 1314–1329 e1310. [DOI] [PubMed] [Google Scholar]
- Lord CJ, and Ashworth A (2017). PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TD, Cook DR, Choi MY, Li MZ, Haigis KM, and Elledge SJ (2017). A Role for Mitochondrial Translation in Promotion of Viability in K-Ras Mutant Cells. Cell Rep 20, 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, Steltenpool J, Stone S, Dokal I, Mathew CG, et al. (2005). A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet 37, 958–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, Bianchi A, Zimmermann M, Durocher D, and de Lange T (2018). 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 560, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessina F, Giavazzi F, Yin Y, Gioia U, Vitelli V, Galbiati A, Barozzi S, Garre M, Oldani A, Flaus A, et al. (2019). Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors. Nat Cell Biol 21, 1286–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash S, Johnson RE, and Prakash L (2005). Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem 74, 317–353. [DOI] [PubMed] [Google Scholar]
- Quinet A, Lemacon D, and Vindigni A (2017). Replication Fork Reversal: Players and Guardians. Mol Cell 68, 830–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman K, and Smogorzewska A (2019). Advances in understanding DNA processing and protection at stalled replication forks. J Cell Biol 218, 1096–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, and Jasin M (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JM, Li MZ, Chang K, Ge W, Golding MC, Rickles RJ, Siolas D, Hu G, Paddison PJ, Schlabach MR, et al. (2005). Second-generation shRNA libraries covering the mouse and human genomes. Nat Genet 37, 1281–1288. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D’Andrea AD, et al. (2007). Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129, 289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA, et al. (2015). DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 208, 545–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkac J, Xu G, Adhikary H, Young JTF, Gallo D, Escribano-Diaz C, Krietsch J, Orthwein A, Munro M, Sol W, et al. (2016). HELB Is a Feedback Inhibitor of DNA End Resection. Mol Cell 61, 405–418. [DOI] [PubMed] [Google Scholar]
- Tomida J, Takata K, Lange SS, Schibler AC, Yousefzadeh MJ, Bhetawal S, Dent SY, and Wood RD (2015). REV7 is essential for DNA damage tolerance via two REV3L binding sites in mammalian DNA polymerase zeta. Nucleic Acids Res 43, 1000–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Li S, Oaks J, Ren J, Li L, and Wu X (2018). The concerted roles of FANCM and Rad52 in the protection of common fragile sites. Nat Commun 9, 2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, and Scully R (2014). BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 510, 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Sung P, and Zhao X (2015). Functions and regulation of the multitasking FANCM family of DNA motor proteins. Genes Dev 29, 1777–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Li Y, Guo R, Ling C, and Wang W (2008). FANCM of the Fanconi anemia core complex is required for both monoubiquitination and DNA repair. Hum Mol Genet 17, 1641–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, and Lopes M (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208, 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, and Cimprich KA (2014). Causes and consequences of replication stress. Nat Cell Biol 16, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Caron P, Legube G, and Paull TT (2014). Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic Acids Res 42, e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Read counts from cisp screens. Related to Figure 1.
Table S2. MAGeCK analyses of untreated and cisp treated screen arms at both population doublings. Related to Figure 1.
Table S3. EdgeR analyses of untreated and cisp treated screen arms at both population doublings. Related to Figure 1.
Table S4. List of hits from cisp screens. Related to Figure 1.
Table S5. SCAI IP mass spectrometry results. Related to Figure 5.
Table S7. List of oligonucleotides used in study. Related to Key Resources Table
Data Availability Statement
All generated data is available in the main text or supplemental tables. Original images have been deposited in Mendeley and can be accessed using the following link: http://dx.doi.org/10.17632/35f2n4wsnv.1. Any additional information is available upon request.
This paper does not report original code
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.