

Abstract
Optimising the function of a protein of length N amino acids by directed evolution involves navigating a ‘search space’ of possible sequences of some 20N. Optimising the expression levels of P proteins that materially affect host performance, each of which might also take 20 (logarithmically spaced) values, implies a similar search space of 20P. In this combinatorial sense, then, the problems of directed protein evolution and of host engineering are broadly equivalent. In practice, however, they have different means for avoiding the inevitable difficulties of implementation. The spare capacity exhibited in metabolic networks implies that host engineering may admit substantial increases in flux to targets of interest. Thus, we rehearse the relevant issues for those wishing to understand and exploit those modern genome-wide host engineering tools and thinking that have been designed and developed to optimise fluxes towards desirable products in biotechnological processes, with a focus on microbial systems. The aim throughput is ‘making such biology predictable’. Strategies have been aimed at both transcription and translation, especially for regulatory processes that can affect multiple targets. However, because there is a limit on how much protein a cell can produce, increasing kcat in selected targets may be a better strategy than increasing protein expression levels for optimal host engineering.
Keywords: flux, host, metabolomics, optimisation, proteomics, transcriptomics
Introduction
Much of microbial biotechnology consists conceptually of two main optimisation problems [1]: (i) deciding which proteins whose levels should be changed, and (ii) by which amounts. The former is ostensibly somewhat simpler, e.g. when a specific enzyme is the target for overproduction, since the assumption is then that the aim is simply the maximal production of the active target (whether intracellularly or in a secreted form). Where the overproduction of a small molecule is the target the optimal levels of individual metabolic network enzyme proteins depend on their specific kinetic properties and the consequent distribution of flux control (e.g. [2–8]). Since both circumstances ultimately seek to maximise the flux to the product of interest, we shall discuss them both, albeit mostly at a high level. Recognising that many pathways are poorly expressed in their natural hosts we shall be somewhat organism-agnostic [9,10], (though we largely ignore cell-free systems) since we are more interested in the principles (whether microscopic [11] or macroscopic [12]) than the minutiae.
The possible number of discrete manipulations one can perform on a given system is referred to as the ‘search space’. The overriding issue is that the number of changes one might make scales exponentially with the number of those considered, and is simply astronomical; the trick is to navigate the search space intelligently [13]. Modern methods, especially those recognising the potential of synthetic biology and host engineering to make ‘anything’ (e.g. [14–23]), are improving both computational [24] and experimental approaches. The main means of making such navigation more effective is by seeking to recognise those areas that are most ‘important’ or ‘difficult’ for the problem of interest, and focusing on them; this is generally true of combinatorial search problems (and to illustrate this, a nice example is given by the means by which the Eternity puzzle https://en.wikipedia.org/wiki/Eternity_puzzle was solved).
Forward and inverse problems, and how the latter are now within range
We find it useful to classify problems into ‘forward’ and ‘inverse’ problems, because (Figure 1) this is in fact how they are commonly presented [13]. In areas such as drug discovery, a typical forward problem might be represented by starting with a set of structures and paired quantitative properties or activities (QSAR/QSPR) with which one can set up a model or a nonlinear mapping in which the molecular structures are the inputs and the activities are the outputs. A good model will be able to ‘generalise’, in the sense that it can give accurate predictions for novel molecular structure. On a good day (such as here [25]), it will even be able to extrapolate, to make predictions of activities larger than it ever saw when it was being trained. This can be seen as a ‘forward’ problem (‘have molecule, want to predict properties’), nowadays known as a ‘discriminative’ problem [26–28]. However, what we really wish to solve [29,30] is the normally far harder inverse problem (‘have desired properties, want molecules’), nowadays referred to as a ‘generative’ problem [26,27,31–35] since the output is (the generation of) the solution of interest. Such generative methods are now well known in the image and natural language processing, and are becoming available in all kinds of related areas of present interest such as drug discovery [36] and protein sequence generation (e.g. [37–40]). Thus, while we have (and can more or less easily create [41]) reasonable whole-genome models of all kinds of microbes (and see below), what we effectively need to solve here again is the ‘inverse problem’ [30,42]. For organism optimisation this is mainly ‘have desired flux, need to optimise the gene sequences and expression profiles of my producer organism to create it’.
Figure 1. The cycle of knowledge in directed protein evolution.
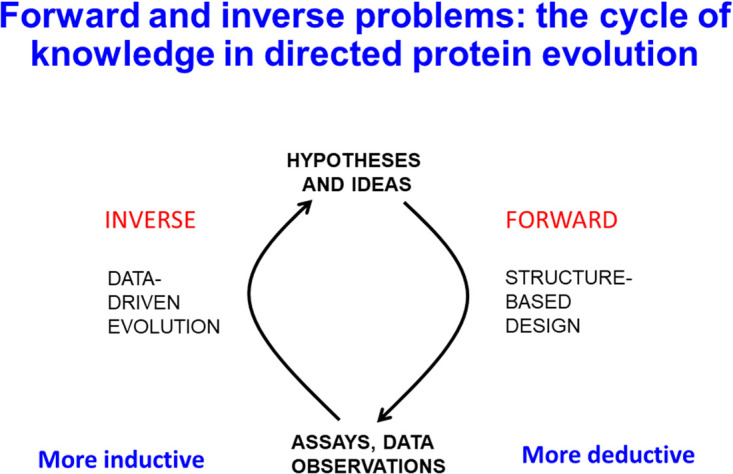
It is useful to contrast the worlds of (i) mental constructs including ideas and hypotheses from (ii) more physical worlds that include data and ‘observations’. Their interrelations are iterative but their nature depends on their directionality. In the post-genomic era, there has rightly been a trend away from the primacy in the biology of hypothesis-dependent deductive reasoning towards data-driven biology in which the best explanations are induced from available data.
Combinatorial problems of genetic sequences
The genetic search space in biology is enormous; even considering just a 30 mer of the standard nucleic acid bases can produce 430 (∼1018) different sequences. The enormity of this number can be illustrated by the fact that if each such sequence was arrayed as a 5 μm spot the array would take up an area of ∼29 km2 [43]. Obviously, the number of bases in just a smallish bacterial genome such as that of Escherichia coli MG1655 is some 105 times greater than 30, and it remains the case that we still know next to nothing about approximately one-third of the genes encoded therein [44], the so-called y-genes [45]. The expression levels of the identical protein sequence can vary several 100-fold just by changing the codons used [46], mainly because of the expression levels [47,48] and the stability of mRNA [49,50], as well as because of codon bias [51] and for other reasons [52]. Also, note that obtaining the best expression of the active protein is not simply a question of using the commonest codons [53], since (i) over-usage of an individual codon will necessarily deplete its tRNA, and (ii) sometimes it is necessary to slow down protein expression so as to avoid inclusion body formation [54]. Consequently, the problem is not made easier by substituting the term ‘nucleic acid bases’ in the above reasoning by the words ‘amino acids’ or ‘codons’. Indeed, the control of gene expression is distributed over the whole genome [55].
Combinatorial problems of protein engineering
Considering just the 20 ‘common’ amino acids, the number of sequence variants for M substitutions in a given protein of N amino acids is [56]. For a protein of 300 amino acids with random changes in just 1, 2, or 3 amino acids in the whole protein this evaluates to 5700, ca 16 million, and ca 30 billion, while even for a small protein of N = 165 amino acids (smaller than half that of the average protein length in Uniprot), the number of variants exceeds 1015 when M = 8. If we wish to include insertions and deletions, they can be considered as simply increasing the length of N and the number of variants to 21 (with a ‘gap’ being coded as a 21st amino acid). Obviously, if we just consider a fixed number of positions N the number of possibilities scales as 20N if any amino acid substitution is allowed. At all events, the dimensionality of the problem is equal to the number of things that can be varied, and it is the exponent in a relationship where the base (in the mathematical sense) is the number of values that it can take. In contrast, for a 165-, 350-, or 700-residue protein, although the number of ways of finding ‘the best’ five amino acids to vary is, respectively ∼108, ∼2.1010, and 1012, exhaustive search of those five amino acids always involves ‘just’ 205 = 3.2 million variants. Thus strategies (such as ProSAR [57–60]) that seek the best elements to mutate at all, even in the necessary absence of epistatic analyses (see below), have considerable merit.
Directed protein evolution
Overall, the solution to such a combinatorial search problem, as is used by the biology of course, is not to try to make these massive numbers of changes ‘in one go’ but to build on earlier successes in a generational or evolutionary manner, known in protein engineering as the design–build–test–learn (DBTL) cycle (Figure 2).
Figure 2. The design–build–test–learn (DBTL) paradigm for engineering biology.

Although usually considered solely at the level of protein directed evolution (and on which this diagram is based [13]), the DBTL strategy applies equally to host engineering.
Algorithmic strategies for doing so in general (including in physical sciences and engineering) are known variously as ‘genetic algorithms’ or ‘evolutionary computing’, and come in a variety of flavours (e.g. [61–69]). They have become well known for individual proteins in the form of directed evolution, as popularised in particular by Frances Arnold (e.g. [70–73]). Some recent reviews include [74–81]. Increasingly, the use of ‘deep mutational scanning’ [82–90], sometimes coupled to FACS-based sorting [91] (‘sort-seq’ [52,82,83,92]), is making available large amounts of sequence-activity pairs [85]. (We ourselves made available one million paired aptamer activity sequences in 2010 [93].) The general structure of an evolutionary algorithm is outlined in Figure 3. As (to some degree [94]) with natural evolution and organism breeding, it is up to the experimenter to select individuals to mutate or to recombine, specifically as one seeks combinations of traits that overall provide the desired phenotype.
Figure 3. Generalized evolutionary algorithms.

The elements of an evolutionary algorithm, in which a population of candidate solutions are mutated and recombined, iteratively with selection, to develop improved variants. Based in part on [95].
The problems of landscape ruggedness and epistasis
Notwithstanding the numerical combinatorial problems, the biggest problem in natural evolution involves that of epistasis, i.e. the very common circumstances in which the ‘best’ amino acid at a certain location depends on the precise nature of the amino acid at one or more other residues. The commonest way to think of these problems is in terms of the fitness landscape metaphor [96], as illustrated in Figure 4. In this representation, ‘where’ one is in the multidimensional search space is encoded via the X- and Y- co-ordinates, while the value of the (composite desired) property of interest, or the fitness, is represented as the height. Epistasis manifests as a sort of ruggedness in the landscape, and is more-or-less inevitable when residues that are ‘distant’ in the primary sequence are in contact; indeed their covariance provides an importance strategy for detecting such contacts from sequences alone (e.g. [97–100]). In particular, ‘sign’ epistasis occurs when A is better than B at location one when C is at location 2, but B is better than A at location one when D is at location two. It is easy to understand this in simple biophysical terms with respect to the likelihood of contact formation, in this case via ion pairs, if A, B, C, and D are, respectively, glutamate, lysine, arginine and aspartate. Indeed, the covariation of residues in protein families is widely used as a means of predicting 3D structure from sequence alone [97,98,101–103].
Figure 4. The landscape metaphor for understanding genotype–phenotype relationships in directed evolution and similar protein optimisation experiments.

The X–Y co-ordinates indicate where one is in the sequence space, while the height indicates the value of the desired objective function(s). Reproduced from an open-access publication at [13].
The ‘ruggedness’ of the fitness landscape is widely taken to reflect the ease with which it may be searched, and which kinds of search algorithms may be optimal [62,93,104]. Biological landscapes tend to be somewhat rugged, but not pathologically so [105,106]. The so-called NK landscapes (e.g. [107–110]) are convenient models, and are completely non-rugged when K = 0; experimentally, we found K ∼ 1 for protein binding to DNA sequences [93]. Ruggedness necessarily increases as protein length L increases, and reasonable routes joining everything upwards or neutrally so as to escape local minima decrease [111], though they do exist in high dimensions [112].
Such sign epistasis is both common and highly important [113–116], and is especially responsible for ruggedness and local isolation under selection. From what we know (e.g. [117]), while pairwise epistasis of this type is indeed very common [118], including in adjacent residues [119], higher-order epistasis is somewhat less so (plausibly for steric reasons). Armed with paired sequence and activity values, all one can do is to seek to interpolate between the few positions with known values and those without. However, if one simply keeps climbing locally one is inevitably likely to be trapped in a local minimum (or maximum in the landscape metaphor) from which it is very hard to escape by mutation alone. Weak mutation and strong selection are commonplace in natural evolution [120–127]) and consequently tend to disfavour lower fitnesses [128], exacerbating the problem of being trapped in a local minimum. This largely constrains natural evolution, and means that we can anticipate great improvements in organisms by seeking previously unknown sequences distant from known ‘peaks’.
Overall, then, the concept of epistasis implies that there is no monotonic ordering of the utility or performance of individual residues within a complete fitness landscape, and that depending on what else is going on there is some kind of a bell-shaped curve relating the utility of a given amino acid in the performance of a protein, to the rest of the protein landscape when that is allowed to be varied. As we shall see, and it is in fact inevitable, this is commonly mirrored more generally.
Combinatorial problems of host engineering
As presaged, the basic combinatorial problem of host engineering [129] is largely equivalent to that of protein engineering. We have P enzymes, each of which might be expressed at Q levels (to make life more reasonable in practice we would let these levels vary logarithmically, so 20 levels of a twofold increment gives a range of just over 106 (220 = 1 048 576)). Our problem comes (this is easily checked using the ‘COMBIN’ function in a spreadsheet programme such as MS-Excel, or online) because again the number of combinations NC explodes as P increases (for Q = 20, NC ∼ 1014 for P = 50, NC > 1020 for P = 100, and NC > 1030 for P = 300, which is lower than one-tenth of the number of gene products in E. coli). However, for P = 100, NC is only 100 and 4950 when just one or two variant levels are introduced that differ from those of the wild type (WT), respectively. As with any combinatorial search problem, appropriate application of modern Bayesian, machine learning, and design of experiment principles can assist with finding optimal combinations (e.g. [130–133].
The optimisation of flux in metabolic networks; metabolic control analysis
Much evidence exists (modulo ‘bet hedging’ [134–138]), that the majority of organisms in a population seek to maximise their instantaneous growth rate (the flux to biomass) [12,139], and thus understanding the control of flux is a core issue, whatever the flux of interest. The typical relationship between the flux through a metabolic pathway or network and the concentration of an individual enzyme typically follows some kind of curve like a rectangular hyperbola (similar to that relating activity to substrate concentration in the simplest Michaelis–Menten equation). An illustration is given in Figure 5.
Figure 5. Some potential relationships between the activity of an individual enzyme in a metabolic ‘pathway’ and a flux of interest.
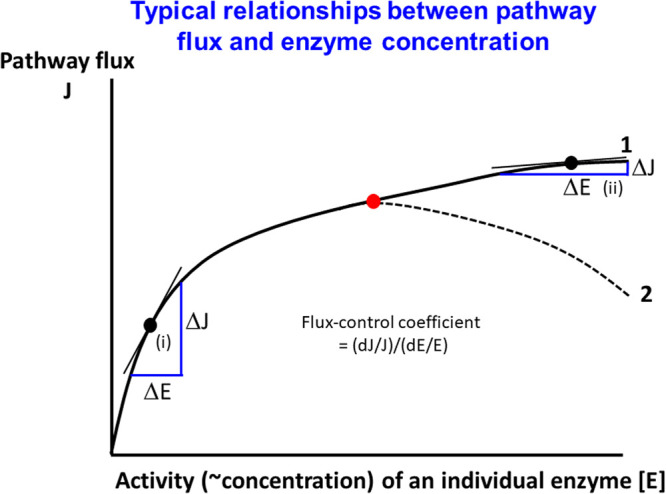
For these purposes, we consider that enzyme concentration and activity are proportionate. The broadly expected result is similar to that of curve 1, where there is a monotonic increase in flux as the enzyme's activity is increased. At lower enzyme activities (i) the slope of the tangent is reasonably high, while at higher activities (ii) a further increase in enzyme activity has little effect on pathway flux and the slope is correspondingly low. In some circumstances (curve 2), whether because of pleiotropic effects or because of the negative effects of an increased protein burden (see text), increases in enzyme activity beyond an optimum (marked in red) lead to decreases in pathway flux. The flux-control coefficient is the normalised slope relating pathway flux to enzyme activity at the operating point of interest.
Understanding why this is so is the territory of metabolic control analysis (MCA), which originated in the work of Kacser and Burns [140–142] and of Heinrich and Rapoport [143,144]. We refer readers to some reviews (e.g. [2,3,5–8,145–147]) and an online tutorial http://dbkgroup.org/metabolic-control-analysis/. MCA can be seen as a kind of local sensitivity analysis [148] (cf. [149]), in which the sensitivities (known as control coefficients, illustrated for a flux-control coefficient in Figure 5) add up either to zero or to one. The chief points of MCA for our purposes are that (i) every enzyme can contribute to the control of flux, but because their flux-control coefficients add up to zero the contribution of individual enzymes is mostly small, (ii) the distribution of control varies as the activity of an individual enzyme is increased (this is somewhat equivalent to epistasis), (iii) because it is activities that matter and because enzyme concentrations cannot be increased without limit, it is better to increase them directly (through increasing individual kcat values) rather than by increasing enzyme expression levels (in terms of kinetics this might only differ during transients, not in the steady state [42]), (iv) the best way to increase fluxes is to modulate multiple enzyme activities simultaneously [5,150], (v) because the sum of concentration-control coefficients is zero, individual steps can and do have substantial effects on the concentrations of metabolic intermediates (this is precisely why the metabolome can serve to amplify comparatively changes in the transcriptome or the proteome [151–153]). However, normally it is fluxes to the product in which one is interested for biotechnology, and in terms of increasing metabolic fluxes, one has to make choices from a combinatorial space [154], because there are necessarily fairly strong limitations on the total amount of protein that can be made by a given organism.
The concept of ‘spare capacity’
The fact that most enzymes have small flux-control coefficients (because they must add up to one) necessarily means that they must tend to have ‘spare capacity’; this is simply another way of saying that increasing or lowering their activity has relatively little effect of a pathway flux. Such spare capacity also allows for rapid responses in the face of changes in the environment [155–159]. This spare capacity of itself implies that there is plenty of ‘room for manoeuvre’ in host engineering. Indeed, the ‘spare capacity’ has been identified explicitly in a variety of systems [160], for instance in mitochondrial respiration (e.g. [161,162]) and others discussed below. This said, some other respiratory systems are barely able to keep pace with the need to oxidise reducing equivalents that can be produced at high rates (e.g. [12,163–166]). Depending on one's point of view of the desirability of forming the relevant products, a failure of spare capacity in some pathways might also be seen as contributing to so-called overflow metabolism [167], as may be evidenced by ‘metabolic footprinting’ [168,169] or ‘exometabolomics’ [170–173]. Ultimately, of course, the ‘adaptability’ of an organism typically depends on the environments in which it has naturally evolved [174,175].
Transporters are both massively important for biotechnology (e.g. [176–178]) and variably promiscuous (and see later for transporter engineering). In one recent approach [179,180], we have used flow cytometry to assess the ability of single-gene knockouts of some 530 genes E. coli (mostly transporter genes; the full list and datsets are in [179,180]) to take up a variety of fluorescent dyes. Some of the data, for SYBR Green uptake (whose fluorescence is massively enhanced upon binding to DNA), are replotted in Figure 6. The range is some 70-fold, reflecting the ability of multiple transporters to influence the uptake and efflux of the dye. Shown are the value for the WT and lines representing one half of and twofold that uptake (encompassing 361 of the 531 knockouts, ca 68%, studied). As expected, most manipulations have comparatively little effect, but there is a ‘long tail’ [181] (here in either direction) of a few that do. This is quite typical of biology, where it is also worth noting that the flow cytometric analysis of clonal cultures indicates a massive heterogeneity therein, presumably as a result of the differential expression of many hundreds of different enzymes. In some ways, the only surprise is that this variation is so small, and that is likely a result of evolution's necessary selection for robustness (e.g. [182–191]).
Figure 6. The modal extent of uptake of SYBR Green in single-gene knockouts of E. coli.

The data reflect flow cytometric analysis of the uptake of the fluorescent/ fluorogenic dye SYBR Green into 530 different single-gene knockouts of E. coli from the Keio collection. Data are replotted from the supplementary table given in [179]. Red symbols indicate y-genes (genes of ‘unknown’ function). The wild type (WT) is marked in green. The horizontal lines indicated uptakes of one half or double that of the WT.
Basic limitations of protein expression: how we cannot just make more of every protein to increase flux
As is well known, microbes adjust their ribosomal content to match their growth rate [12,192,193]. Thus, when the opportunity arises, they can funnel excess amino acids into ribosomal biosynthesis [194]. Indeed, increasing amino acid availability, as in the ‘terrific broth’ [195], does indeed assist protein production. Equally, it has been known for many years that, although there is considerable flexibility [196], cells do, as they must, have limitations on the total amount of protein that they can make [12,197], both as a flux to protein biosynthesis ‘as a whole’ and as a percentage of total biomass. To this extent, then, especially in laboratory cultures in rich media, the ability to biosynthesise protein is essentially a zero-sum game [198,199]: increasing the amount (concentration) of some proteins necessarily means decreasing the concentration of others. Since consensus metabolic networks have become established in standard organisms such as E. coli [200] and yeast [201] (and indeed humans [202,203]), attention has thus begun to shift to the wider proteome [204–207]. Experimentally, while the cell may ‘wish’ to retain some spare capacity, sometimes it is simply not possible. Thus, an early study [208] showed that increases in the expression of a variety of glycolytic enzymes in Zymomonas mobilis actually decreased the glycolytic flux; it was as though the cells were already at an optimal point (as marked in Figure 5 with a red symbol). Other studies [155,209,210] are reviewed by Bruggeman et al. [12]. A recent analysis in baker's yeast [211] by Nielsen and colleagues covers many of the issues. In this work [211], it was found that under nitrogen-limiting conditions, 75% of the total transcriptome and 50% of the proteome were produced in excess of what is necessary to maintain growth. This necessarily implies that there is scope for improving the host for the purposes of the biotechnologist.
The potential for host proteome optimisation
Given both the spare capacity, and the fact that many proteins are commonly expressed that are not essential either for cell growth or for assisting fluxes to the product of interest [212,213], it is obvious that attention might usefully be applied to proteome engineering [214–216] as part of host engineering. This becomes especially obvious when it is recognised that the expression levels of even the commonest proteins span some 5 orders of magnitude (in a roughly log-normal distribution) when assessed in baker's yeast using methods that provide absolute numbers [217]. While transcript levels were an important contributor to this variation, the number of protein molecules per transcript in the same study also showed an impressive range, from 40 to 180 000 proteins per transcript [217], implying considerable post-transcriptional control. A more recent study in the same organism detailed absolute protein abundances under some 42 conditions [218], with data replotted therefrom in Figure 7. This serves to show the relatively limited range available in both the total transcriptome and the total proteome in growing cell cultures. Such kinds of datasets (and others such as those in [219] with simple growth as the output) will be of massive value in the future for the purposes of host engineering, as they at once allow one to understand the conditions under which genes are expressed, the strength of the relevant promoters, as well as other features such as genes whose expression varies little and might thus be used for purposes of normalisation [220,221].
Figure 7. Relationship between absolute proteome and absolute transcriptome in baker's yeast grown under different conditions.

Data are replotted from supplementary table S1a of [218]. The dilution rate from 0.1 to 0.35 h−1 is encoded by the style of symbol, and the nitrogen source by the colour indicated (optimised for colour-blind legibility via the palettes at http://colorbrewer.org/). Numbers refer to the sample numbers in that table.
‘Minimal’ genomes
It is sometimes considered that because biotechnologists often aim to grow cells in rich media, one might usefully delete a lot of the biosynthetic capacity of a cell to ‘streamline’ a genome to make a ‘minimal’ genome. Actually, because of redundancy (A or B is required but not both), the number of redundant pairs n scales exponentially (as 2n) so the concept of the minimal genome is quite inaccurate. Anyway, any real ‘burden’ comes from the expression, not the possession, of a particular gene, so we focus on strategies that optimise expression. More interesting are essential genes, and a very nice genome-wide study provided a clever method for assessing them [222].
Transcription vs translation engineering
To vary the amount of a particular protein one can act at the transcriptional or translational levels (or of course both). The former might involve truncations, knockouts, transcription factor engineering (see later), mRNA stability, RNA polymerase engineering, transcription factor binding sites, and direct promoter engineering. A suite of such approaches has been referred to as global transcription machinery engineering (gTME) [223–227]. Translation engineering will tend to have a focus on translation initiation, elongation, and codon optimisation [228,229]. Table 1 gives some examples. We are not aware of any studies that would point experimenters towards a general preference for one or the other (from the MCA analysis above, more likely both), implying that such studies would be of value; it may be, of course, that every problem is more or less bespoke. What is certain, however, is that very little of the search space has ever been covered by natural evolution. A nice example of this is given by the work of Wu et al. [230] who used a transformer model (see [39,231–240]) to assess the ability of various signal peptide sequences (SPs) to induce protein secretion. They found that successful, model-generated SPs were diverse in sequence, sharing as little as 58% sequence identity with the closest known native signal peptide and possessed just 73 ± 9% on average. Unsurprisingly, given the tiny population of sequence space accessed during natural evolution, this is more generally true [241]. Given the many recent advances in deep learning for solving a variety of biological problems (e.g. [31,37–39,242–255]), it is clear that these data-driven [29] strategies will be in the vanguard of the ‘learn’ part of the DBTL cycle.
Table 1. Some examples of yield improvement by transcription or translation engineering.
| Focus | Details/comments | Selected references |
|---|---|---|
| Transcription | ||
| mRNA stability | Contributes as much to transcription as does codon usage | [46,47] |
| Promoter engineering | Guided, empirical strategies | [265–268] |
| Inducible Trp-T7 for serine production | [269] | |
| Novel tet-based use of machine learning | [270] | |
| Random variation on a trc promoter, allowing 60-fold variation in expression levels | [263] | |
| Promoter library module combinatorics for use in threonine production | [271] | |
| Reviews | [215,272–279] | |
| Sigma-factor-specific promoters | [280] | |
| Transcription factor engineering | See below | |
| DNA/RNA polymerase engineering | [281–284] | |
| Chromosomal integration site | Increased isobutanol production from E. coli, involving chromosomal integration at random sites, selection by cell sorting | [285] |
| β-carotene synthesis in Yarrowia lipolytica by simultaneous integration of a 3-module biosynthetic pathway plus selection by colony colour. | [286] | |
| Riboswitches | Can provide effective control | [287] |
| σ-factor engineering | Major transcriptional control point | [288] |
| Improved antibody production in E. coli | [289] | |
| Extracytoplasmic σ factors | [290] | |
| Use in cyanobacteria | [291] | |
| Terminator engineering (acts both transcriptionally and translationally) | Increased protein expression through reduced read-through, including Itaconic acid and betaxathin production. | [292–297] |
| Translation | ||
| Codon usage | Strong selection in S. cerevisiae leads to plasmid copy variation | [298,299] |
| Role in regulating protein folding | [300] | |
| Review of codon usage tables | [301] | |
| Ribosome binding sites (RBS) | RBS calculator | [302] |
| Machine learning in E. coli | [303] | |
| Multiprotein RBS optimisation in various bacteria | [304] | |
| Review of RBS calculator | [305] | |
| Phenotypic recording with deep learning, using more than 2.7 M sequence-function pairs | [306] | |
| Translation initiation optimisation | Reviews | [228,307,308] |
| Significant increase in serine overproduction | [309] | |
| 5-Methylpyrazine-2-carboxylic acid production | [310] | |
| 2,5-furandicarboxylic acid production | [311] | |
| tRNA engineering | Admits novel codons, of which some can encode non-canonical amino acids | [312] |
| tRNA synthetase engineering | [313] | |
It is generally agreed that while the use of extrachromosomal plasmids is useful for high-throughput screening applications, integration of pathways into the chromosomal DNA of the host organism is ultimately preferable in most production strains due to the well-established instability of plasmids during continuous growth [256,257]. Recently advances in CRISPR and other molecular biology techniques have allowed the integration of reporter genes into a high number of defined genomic sites. Significant variations in expression levels of reporter proteins by the site of genomic integration have been demonstrated in Saccharomyces [258], E. coli [259,260], Bacillus subtilis [261], Pseudomonas putida [262], and Acinetobacter baylyi [263]. Generally higher levels of expression are seen for integration at sites closer to the origin of replication, as during replication there is in essence a higher copy number of genes that are on the DNA strands replicated first [264]. As discussed in detail above it is rarely a sensible goal to maximise the expression level of all proteins in a relevant pathway, so the genomic integration site of heterologous proteins is an axis on which optimisation can be performed.
Systems modelling for host engineering
Much of the systems biology agenda (e.g. [30,145,146,314–318]) has recognised that to understand complex, nonlinear systems such as biochemical networks it is wise to model them in parallel with analysing them experimentally. This allows the performance in silico of ‘what if?’ kinds of experiments in a manner far less costly than doing them all, allowing one to choose a subset of the most promising. This is also sometimes referred to as e-science [319–321], or having a ‘digital twin’ [322,323] of the process of interest. It is, of course, very well established in fields such as chemical or electronic engineering, where it would be inconceivable to design a process plant or a new chip without modelling it in parallel.
In part, the success of those fields is because we know (because we have designed them) both the wiring diagram of how components or modules interact, and in addition, we know, quantitatively, the input–output characteristics of each module. This allows one to produce what amounts to a series of ordinary differential equations that, given a starting set of conditions, can model the time evolution of the system (by integrating the ordinary differential equations). Such models can be set up in biochemistry-friendly systems such as Copasi [324,325] (http://copasi.org/), CellDesigner [326–329] (http://www.celldesigner.org/), and Cytoscape [330–332] (https://cytoscape.org/). However, prerequisite to this being done accurately is that one has knowledge of the expression levels, kinetic rate equations, and rate constants for each of the steps. This is only rarely achieved (e.g. [333]), even when such details are not known and generalised equations that cover a wide range of force–flux conditions are used [334–336]. Consequently, so-called constraint-based methods have come to the fore. Chief among these is flux balance analysis (FBA) [146,315,317,337–341].
FBA recognises and exploits the massively important ‘stoichiometric’ constraints engendered by the fact that mass must be conserved [342–344], leading to atomic and molecular constraints reflected in reaction stoichiometries, and that consequently only certain kinds of fluxes and flux rations are possible in a known metabolic network. This simple but exceptionally powerful idea, equivalent to Kirchoff's laws in electrical circuit theory, comes into its own when one seeks to optimise fluxes to the desired end [345–348] (as in host engineering).
Software for performing FBA is also more or less widely available [340,349], the generic COBRA toolboxes [347,350–353] being especially popular. Such software is much aided by the development of various kinds of linguistic standards for describing systems biology models, such as BioPAX [354–357] (http://www.biopax.org/) and SBML [358] (http://sbml.org/Main_Page).
An especially potent implementation comes from the recognition that if the expression level of a given enzyme is treated as a surrogate for (or an approximation to) the actual flux through that step, then methods that maximise the correlation between predicted and real fluxes, while still admitting mass conservation, can, in fact, predict real fluxes astonishingly well (e.g. [359,360]). Given such a base model, it is then just a question of navigating the space of expression profiles to see those (combinations of) changes that have the greatest effect on the flux of interest. Note, however, that FBA (i) is blind to regulatory effects and (ii) cannot predict metabolite concentrations (only fluxes). Finally, here, it is worth remarking that advanced analyses based on molecular dynamics simulations are beginning to allow the calculation of enzymatic activities and epistatic interactions de novo (or at least to account for them) (e.g. [116,361–363]); as with other areas [364–366], the increasing availability of cheap computing will continue to make such methods both more potent and more accurate.
Genome-wide engineering to improve host performance
As seen in early work in E. coli [367], promoter engineering allows one to vary the amount of target enzymes both smoothy and extensively. Of course, nowadays this can be done on a genome-wide scale using methods such as CRISPR–Cas [368,369]. Thus Alper and colleagues [370] assessed the effects of the expression level of all 969 genes that comprise the ‘ito977’ model of Saccharomyces cerevisiae metabolism, with overproduction of betaxanthins as one of the objective functions. A particularly important finding was that in a good many cases knockdown rather than complete knockout was preferable, and that there was almost always an optimal level (as per Figure 5) in the range considered. This optimality has been widely reported (e.g. [371–375]), and interestingly (presumably for evolutionary reasons) typically corresponds to the expression level seen in the WT [12]! RNAi engineering can also be used to modulate expression levels [376].
Transformation engineering
Of the various means of genetic manipulation widely available (transformation, transduction, mating, etc.), transformation by exogenous DNA remains the most popular. This said, transformation using libraries of DNA is far less efficient than one would like [377,378], and it varies considerably with the organism of interest. Some cells [379] such as certain bacilli [380], streptococci [381], acinetobacters [382], and Vibrio spp. [383–386] are more-or-less ‘naturally’ competent, which others require considerable optimisation to achieve acceptable rates [387]. A veritable witches’ brew of cocktail components have been considered; at this stage, it seems that an empirical approach is needed for every organism (e.g. [388–393]). There is also the question of whether the vector to be used is intended to be or remain episomal or to integrate by recombination into the host chromosome. These are areas that will require especial attention for improved host engineering.
CRISPR–Cas-based genome engineering
The arrival of CRISPR–Cas9 and related genome editing tools [394,395] is well enough known as not to need detailed review (and many are available, e.g. [369,396–410]).
A recent advance incorporates the ability to incorporate a simple (barcoded) coupling between the gRNA that might have had an effect and its nature as encoded via a barcode. This is the CRISPR-enabled trackable genome engineering (CREATE) technology developed by Gill and colleagues (e.g. [396,404,411–413]). CREATE uses array-based oligos to synthesise and clone 100s of 1000s of cassettes containing a genome-targeting gRNA covalently linked to a dsDNA repair cassette encoding a designed mutation. After CRISPR/Cas9 genome editing, the frequency of each designed mutant can be tracked by high-throughput sequencing using the CREATE plasmid as a barcode. (A commercial version of this approach is now available as the Onyx™ instrument (https://www.inscripta.com/technology).)
A biotechnological example of the CREATE technology is that for lysine production (a mature, multi-billion $US market [414]) in E. coli [412]. Here the authors [412] designed over 16 000 mutations to perturb lysine production, and mapped their contributions toward resistance to a lysine antimetabolite (toxic amino acid analogue). They thereby identified a variety of different routes that can alter pathway function and flux, uncovering mechanisms that would have been difficult to design rationally — many were, in fact, unknown! In the event, mutations in genes linked to transport, biosynthesis, regulation, and degradation were uncovered, with some being as expected (showing the virtue of the strategy) and others—especially in DapF acting as a regulator—being entirely novel. Overall, this strategy provides an exceptionally potent, efficient and effective approach to the principled discovery of ‘novel’ genes involved in any bioprocess of interest that can be run at different ‘levels’ or in different ‘states’.
Transcription factor engineering
It is a curious fact that much of the community that studies plants has focused on the control of flux via transcription factors (TFs, e.g. for pigment production [415–418]), while microbiologists have tended historically to focus more directly on metabolic networks per se. This is starting to change.
The transcription factor-based regulatory network of E. coli is probably the best studied (e.g. [419–421], with over 200 TFs [422–424] organised into some 150 regulons [423,425,426]. Independent components analysis (ICA) is a useful, convenient, multivariate linear, and well-established technique for separating mixed signals into orthogonal contributions; it has been used to group these differential gene expression changes into over 300 iModulons [427,428]. One may suppose that semi-supervised methods of deep learning [31] will prove even more rewarding in terms of understanding coregulation.
A related study in yeast manipulated some 47 TFs (via a library containing over 83 000 mutations) affecting over 3000 genes, leading to a substantial improvement in both isopropanol and n-butanol tolerance. An analysis of the relevant gene expression changes showed that genes related to glycolysis played a role in the tolerance to isobutanol, while changes in mitochondrial respiration and oxidative phosphorylation were significant for tolerance to both isobutanol and isopropanol.
The number and nature of the genes regulated by TFs can vary considerably, and in a nice strategy Lastiri-Pancardo et al. [429] worked out those whose removal would provide maximal flexibility for the reorganisation of allocation of the rest of the proteome. For instance, feast/famine regulatory proteins/transcription factors [430,431] are common to both archaea and eubacteria; Lrp, in particular, is especially responsive to the concentration of leucine as an indicator of the cell's nutritional status. Overall, TFs seem a particularly useful target for intelligent host engineering (e.g. [432–434], including in biosensors [435–444]).
In addition to changing the expression levels of target genes, we will also wish to change their activities, and one obvious means is via mutation. The kinds of diversity creation that can effect mutation are summarised in Figure 8.
Figure 8. Types of diversity creation and genome engineering.

Different strategies for creating strain diversity as part of host engineering, set out as a ‘Boston matrix’ reflecting the variation between difference strategies in terms of the number of variants created and the number of genomic locations tested. Based on the material at https://www.youtube.com/watch?v=tb97SghfL_8&t=256s.
Methods for genome-wide introduction of mutations
Many of the genetic variations that improve the performance of microbial cell factories are not currently possible to design rationally, despite the large degree of genetic knowledge around many platform strains [378]. This is in large part due to the high degree of epistasis and the combinatorial problems discussed in detail above. While advances in AI are rapidly changing this (see above), improvements in microbial cell factories are presently still in many cases being found by wet laboratory techniques (Table 2) that introduce more-or-less random mutations across the genome and then select for strains with desired properties. These strains can be used directly, or with the plummeting costs of next-generation sequencing, beneficial mutations can be identified revealing new mechanisms and targets for further rational design. A further advantage of random mutagenesis relevant to some applications is that strains generated through random mutagenesis are considered ‘GMO free’, which allows one to avoid legal regulations that have been set up around some kinds of so-called genetically modified organisms [445,446].
Table 2. Example applications of techniques to introduce genome-wide mutations.
| Technique | Species | Purpose | Notes | References |
|---|---|---|---|---|
| UV | Kluyveromyces marxianus | Improved ethanol production | Used an automated platform incorporating UV mutagenesis. | [480] |
| Yarrowia lipolytica | Improved oil production | [481] | ||
| Chemical mutagenesis | Chlorella vulgaris | Light tolerance | [482] | |
| Brettanomyces bruxellensis | Reduced production of 4-ethylphenol, an undesirable by-product in wine fermentation | [483] | ||
| Yarrowia lipolytica | Increased lipid production | [484] | ||
| Lipomyces starkeyi | Increased production of triacylglycerol | [485] | ||
| Atmospheric and room temperature Plasma mutagenesis | Zymomonas mobilis | Acetic acid tolerance | [486] | |
| Spirulina platensis | Astaxanthin production | [487] | ||
| Escherichia coli | L-lysine production | Incorporated a biosensor for cell sorting | [488] | |
| Actinosynnema pretiosum | Production of the antibiotic Ansamitocin | Used in combination with genome shuffling | [489] | |
| Streptomyces mobaraensis | Production of the enzyme transglutaminase | |||
| epWGA | S. cerevisiae | Ethanol tolerance | [451] | |
| Lactobacillus pentosus | Lactic acid production | [490] | ||
| Zymomonas mobilis | Furfural tolerance | [491] | ||
| E. coli | Butanol tolerance. | [492] | ||
| Serialised ALE | Saccharomyces cerevisiae | β-caryophyllene production | [493] | |
| Corynebacterium glutamicum | Glutarate production | [494] | ||
| E. coli | Ionic liquid tolerance | [454] | ||
| Continuous ALE | Methylobacterium extorquens | Methanol tolerance | [495] | |
| E. coli | Conversion to generate all its biomass from CO2 | [496] | ||
| GREACE | E. coli | Lysine production | [478] | |
| E. coli | Butanol tolerance | [476] | ||
| E. coli | Cadmium resistance | [497] | ||
| S. cerevisiae | Acetic acid tolerance, reduced acetaldehyde production | [479] |
UV and chemical mutagenesis
The ability of UV radiation [447] and certain chemicals [448] to cause mutation has been established since the 1930s and 1940s, respectively. While there have been massive advances in the tools available for metabolic engineering and strain generation in the subsequent decades (some of which are outlined below), several recent papers illustrate that there is still utility in using UV radiation and mutagenic chemicals to introduce genetic diversity. These techniques are especially relevant when working with novel or poorly characterised strains for which other tools to introduce variation are lacking, since UV and chemical mutagens cause mutations efficiently in nearly all species.
Atmospheric and room temperature plasma mutagenesis
Atmospheric and room temperature plasma mutagenesis (ARTM) is a novel technique for introducing random mutagenesis. The application of plasma as a mutagenic agent was first described by Li and colleagues in a 2012 paper [449], in which it was used to generate a mutant library of Methylosinus trichosporium. In ARTM, a jet of helium, ionised by an electric field, is blown onto a sample, which (through a yet to be fully elucidated mechanism) causes DNA damage and mutations.
The ARTM technique has, according to a recent review [445], been applied to industrially relevant improvements in over 20 species including both Gram-positive and -negative bacteria, filamentous fungi, yeasts, algae, and cyanobacteria [445]. It has been shown in the umu test on Salmonella typhimurium that ARTM generates a higher rate of surviving mutated cells than do UV and chemical mutagenesis methods [450]. Despite the apparent advantages, the commercial unit is thus far only available in China, and the publications using ARTM appear to be exclusively from Chinese institutions.
Error-prone whole-genome amplification
Another technique to introduce mutations across the genome is error-prone whole-genome amplification (epWGA). In this, genomic DNA from the strain of interest is extracted and subjected to error-prone PCR, then retransformed into the initial strain [451]. The transformed cells are subjected to relevant selective pressure, for instance, to isolate strains that have improved property such as a tolerance to an inhibitor or increased product titre. This process can be performed iteratively, and with full genome sequencing beneficial mutations can be identified and isolated to quantify their effects.
Serialised adaptive laboratory evolution
One of the most widely used and well-established techniques to introduce (i.e. select) beneficial mutations is adaptive laboratory evolution (ALE) [452,453]. ALE is in principle a very simple technique in which cells are cultured under some form of selective pressure, such as the presence of a toxic substance. Cultures are generally serially propagated into media with incremental increases in selective pressure. During this, mutations that confer a fitness advantage accumulate and become fixed in the population. These mutations can then be discovered by sequencing and reintroduced explicitly into a strain of interest, or the evolved strain can be used directly as a platform in downstream applications (e.g. [454,455]).
In the most straightforward use case, tolerance ALE (TALE), cells are propagated in increasing concentrations of some compound that normally inhibits growth in order to improve tolerance (‘tolerance engineering’, Figure 9). Tolerance to toxic environments is still a major limiting factor in achievable yields from microbial cell factories [456,457]. This may be for example toxicity of the desired product (as is the case in fermentative butanol production [458]) or toxic inhibitors present in feed stocks (which is a major challenge in attempts to process lignocellulose hydrolysates [459–461]. ALE may also be used to improve utilisation of a preferred energy source, or to increase product titre directly (although the latter usually requires more advanced experimental design in order to couple production to a fitness advantage [462,463]). A very extensive recent review covering the applications of ALE in more detail is found in [464].
Figure 9. Adaptive laboratory evolution (ALE), illustrated here for tolerance engineering.

Cultures are grown in batch mode under conditions in which a stress leads their overall growth rate or yield to be suboptimal. As mutants that are more tolerant to the stress emerge they are selected for and take over the culture, with concomitant increases in growth rate or yield. The magnitude of the stress can then be increased and the process repeated as often as desired.
Continuous adaptive laboratory evolution
While ALE in its simplest form involves serial propagation of cells, continuous evolution techniques utilise variations on what are commonly referred to as ostat bioreactors. These use some form of detection from a growth chamber (commonly OD but such x-stats may also detect pH, dissolved oxygen and many other parameters). Cultures are maintained in a constant state of growth under steady conditions by dilution through automated addition of fresh media along with other supplements or inhibitors. In this way, a constant growth rate and smooth evolution curve can be achieved, compared with the more ‘punctuated equilibrium’ that is the hallmark of serialised ALE [465]. Traditionally cost has been something of a barrier in the use of turbidostats (albeit far from insurmountable [198,199,466–468]), which unlike serialised ALE require specialised detection probes and feedback systems [469]. Recently, however, several open source and low-cost chemostats have become available, reducing the financial barriers to entry at the cost of a requirement for significantly greater hands-on expertise [470–472].
Genome replication engineering assisted continuous evolution
The adaptive mutations that appear in both serialised and continuous ALE occur through the natural mutations occurring during DNA replication in growing cell populations. Although DNA replication in microbes is generally of very high fidelity (estimated to be on the order of 10−10 errors per base pair per generation [473] in wild-type strains), the high density of cells during cultivation (108–1010 per ml) still means that enough mutations will occur to generate strains with a fitness advantage. A higher mutation rate may be desirable, however [474], to increase the rate of adaptation or to allow adaptations towards more specialised phenotypes. Indeed, the mutation rate is itself adaptive [475].
The mutation rate in E. coli has been increased in a principled way through a technique called genome replication engineering assisted continuous evolution (GREACE) [476–478]. In this approach, a plasmid carrying a modified DNA proofreading element (the dnaQ gene) is transformed into the initial strain of interest, and then the transformed cells are subject to continuous ALE. Cells carrying the modified PE plasmid have deficiencies in proofreading ability and, therefore, accumulate mutations at a higher rate than do untransformed cells. Under strong selective pressure, higher mutation rates themselves confer a fitness advantage and the cells carrying the plasmid outcompete those that lose the plasmid. As the deficient proofreading machinery is present on a plasmid as opposed to the genome, once this is removed a strain with the accumulated mutations but a native DNA proofreading system can be recovered, allowing direct use in downstream industrial applications.
Since the initial demonstration of GREACE to generate tolerance to butanol [476], the GREACE methodology has also been extended to S. cerevisiae, substituting the dnaQ gene with an error-prone DNA polymerase from S. cerevisiae. Here it was successfully used to increase the tolerance to acetic acid and reduce the production of acetaldehyde in an ethanol-producing strain [479].
Vmax vs kcat
The activity of an enzyme, as expressed in the term Vmax, is the product of two terms, viz. the concentration of the enzyme E and its catalytic turnover rate kcat. Consequently, there are, broadly, two ways to speed up an individual step in a metabolic network: (i) increase the amount of catalyst (Vmax) or increase the activity of each catalyst molecule (kcat). While the former is the more common via well-established promoter engineering methods, we have long taken the view that the latter should be more effective. The reason is simple, i.e. to increase an enzyme concentration 10-fold requires the production of 10-fold more protein, and this is not always possible (see above). Indeed, especially for membrane proteins, the available real estate may be especially limited [498,499]. In contrast, an increase in kcat of a 100-fold, which is often easily obtainable in directed evolution programmes, means that one could increase the rate of an individual step by 10-fold while using even ten times less of the relevant protein. One example where massive overexpression of a target protein has been used in the overexpression of the efflux transporter for serine [499].
Membrane transporter engineering
Although we are aiming not to focus excessively on specific areas, we mention transporter engineering because (i) transporters normally exhibit considerable flux control for both substrate influx and product efflux, and (ii) they illustrate more generally how an often-neglected scientific area may benefit from the significant study [500]. In addition, it is (somewhat astonishingly [501]) widely still believed (or at least assumed) that all kinds of substrates simply cross biological membrane via passage through any bilayer that may be present. The facts are otherwise [176,502–509]. Those references rehearse the fact that even tiny molecules like water [510,511] do not pass unhindered through phospholipid bilayers in real biological membranes (whose protein : lipid ratio by mass is often 3 : 1), but require transporters. Recent examples of transporter engineering for biotechnological purposes include glycolipid surfactants [512] and fatty acids [513]. Flow cytometry can provide a convenient means of assessing the activities of certain transporters [179,180].
Molecular breeding
Classically, the predominance of diploidy in organisms such as penicillia has been seen as a significant disadvantage, as it prevents the emergence of traits that rely on similar activities in both genomes for expression. Indeed, MCA serves to explain the molecular basis of genetic dominance [141], and the use of haploid cells can provide a much great signal : noise in genetic competition experiments [199,514–521]. Yeasts such as those of the genus Saccharomyces are of special interest here, since they can sporulate as haploid forms of different mating types that can then interbreed, including interspecifically [522]. Perhaps surprisingly, the effects of this on transcription can be quite modest [523].
Growth rate engineering
As noted above, natural evolution tends to select for growth rate rather than growth yield [139]. However, typically if the product is not directly growth-associated (e.g. as with ‘secondary’ metabolites in idiophase [10,524]) or with two-stage fed-batch regimes where a growth phase is followed by a production phase, one is wanting cells not to grow at the expense of making product [525,526]. Certainly, ‘dormant’ (non-replicating) cells can be quite active metabolically [527–530]. Consequently, although not usually a focus of biotechnology, it remains the case that the more time cells spend in a fermentor non-productively the less good the process. This has led to the consideration of hosts such as Vibrio natriegens [50,531–541], whose optimal doubling time can be as little as 7 min, some threefold quicker than the widely quoted 20 min for E. coli in rich media. Whether or not organisms such as V. natriegens turn out to be valuable production hosts, there is no doubt that understanding how to make cell growth quicker might help enhance the rates of recombinant protein production. Turbidostats [542,543] could be seen as a ‘revved-up’ version of ALE in that they too select for (and demonstrate the levels of any) growth rate enhancement. However, they remain a surprisingly under-utilized system for manipulating microbial physiology, despite many advantages [466,467,544]. Continuing the theme of comparative ‘growth-omics’, the growth rates of yeasts are significantly slower than those of bacteria, the record (in terms of rate of biomass doubling) apparently being Kluyveromyces marxianus with a doubling time of some 52 min [498] (a growth rate approximately twice that of S. cerevisiae [541,545–547]). This was achieved [498] via a different kind of growth rate selection in a kind of ‘turbidostat’ called a pHauxostat [548–550]).
Consequently, selection for faster growth rates and the concomitant analysis of gene expression changes [546,551–555] would seem to be a powerful means of understanding how to improve cellular performance.
Medium optimisation and engineering
At one level, the fact that growth rates [219] and expression profiles [198,556–558] differ as different enzymes are expressed at different levels in different growth media is trivial, and essentially describes the whole of microbial physiology. At another level, it is far from trivial because medium optimisation represents yet another combinatorial search problem [559]. If the optimal concentration is considered to be within a known two orders of magnitude and to sit adequately therein within a twofold concentration range, each constituent could take at least 6 values (simply because 100 lies between 26 and 27). With 20 medium constituents, there is then a ‘search space’ of some 620 (∼4.1015) recipes to find the optimum. Even just taking metal ions, and noting that approximately half of all enzymes are metalloenzymes [560–562], it is clear that organisms have significant preferences for particular levels of metal ions [563]. In an early example, Weuster-Botz and Wandrey [564] used a genetic algorithm to increase the productivity of formate dehydrogenase in an established fermentation by more than 50%, finding that Ca++, Mn++, Zn++, Cu++, and Co++ had all been used at excessive levels previously. Obviously, the optimum can also change with the host genotype, so is not fixed even for a given species. Consequently, we feel that automated medium optimisation algorithms should also be at the heart of any host engineering programme. As a classical combinatorial optimisation problem [1,95], this is arguably best attacked by evolutionary algorithms (e.g. [61,62,565–567]); Link and Weuster-Botz [559] give an excellent summary of their applications in medium optimisation, including the rather infrequent cases (e.g. [568]) in which multiple objectives are to be optimised.
Looking to the future
The variation in expression of individual proteins, even within a nominally homogeneous or axenic culture of an isogenic organism, can vary considerably, leave alone those explicitly differentiated (e.g. [569–572]). This is also becoming ever clearer in differentiated organism via the emerging cell map projects (e.g. [573]) As single-cell transcriptomics, proteomics, and metabolomics become possible, and individual cells are easily sorted in a fluorescence-activated cell sorter, one can contemplate studies in which the expression profiles even of large numbers of nominally isogenic cells are compared with their productivity simultaneously. This may even include understanding of the spatial distribution of proteins within individual cells [574]. One can also imagine a far greater use of the methods of chemical genomics in affecting and understanding cellular behaviour; in this regard, the strategy of chemically induced selective protein degradation [575–580] seems likely to be of significant value.
We have purposely avoided focus on the production of any specific target molecules, since our aim is to help develop the BioEconomy generally. This said, the growth of ‘AI’ and deep learning alluded to above has already shown profound benefits in identifying chemical (e.g. [581–585]) and biosynthetic pathways (e.g. [586,587]), while our own work has developed deep learning methods for molecular generation [588] and molecular similarity [589], for navigating chemical space in a principled way [238], and in particular for predicting the structure of small molecules from their high-resolution mass spectra [239]. In this latter work, we developed a deep neural network with some 400 million interconnections [239], a number that just 3 years ago (writing in July 2021) would have been the largest published. Such has been the growth of large networks (approaching 1% of the interconnections in the human brain) that that number is now too low by a factor of more than 1000-fold [234], necessitating the development of specialist hardware and software to deal with it. Innovations in such kinds of computer engineering, including e.g. in optical computing, will be of considerable benefit. With these large networks has come the question of interpreting precisely how they are doing what they do so well (so-called ‘explainable AI’ or XAI [590–594]). XAI will of necessity lead both to better understanding and to sparser networks, and is an important part of the automation [595–598] (not covered here) that will help to speed up the DBTL cycle enormously.
Classically, electronic circuits were and are predictable because the input/output characteristics of the components are known, and because their wiring diagrams are expertly and precisely controlled by their designers. None of these facts is presently true of biology [599,600], and much of the future in both ‘pure’ organismal bioscience and in biotechnology will thus be about ‘making biology predictable’ [30].
Concluding remarks
This has been a purposely high-level overview of some of the possibilities in host engineering predicated on genome-wide analyses. Our main aim has been to draw attention to these developments, and to some of the means by which readers who are only loosely acquainted with them can incorporate these methods into their own work.
Take-home messages include
Host engineering, like directed protein evolution, is a combinatorial search problem.
Every enzyme potentially has an optimal expression level for every process.
This is not normally its maximal level, since the maximum amount of protein a cell can produce is fixed, including for a given growth rate; protein synthesis is largely a zero-sum game.
Changes in the individual concentrations of most enzymes at their operating point necessarily have little effect on fluxes.
Some areas of transcription and translation effect a more global control and thus can have greater effects and hence serve as better targets for host engineering.
kcat is a much better target for host and protein engineering than is Vmax.
Modern methods of modelling, including deep learning, are beginning to provide the ability to assess desirable changes in silico, as a prelude to developing a fully predictive biology.
The success of these messages will be judged by the rapidity with which the strategies they contain are adopted.
Abbreviations
- ALE
adaptive laboratory evolution
- ARTM
atmospheric and room temperature plasma mutagenesis
- DBTL
design–build–test–learn
- epWGA
error-prone whole-genome amplification
- FBA
flux balance analysis
- MCA
metabolic control analysis
- SPs
signal peptide sequences
- TFs
transcription factors
- WT
wild type
Open Access
Open access for this article was enabled by the participation of University of Liverpool in an all-inclusive Read & Publish pilot with Portland Press and the Biochemical Society under a transformative agreement with JISC.
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
Funding
L.J.M. and D.B.K. are funded by the Novo Nordisk Foundation (grant NNF NNF20CC0035580). Present funding also includes the UK BBSRC projects BB/R014744/1 (with GSK) and BB/T017481/1. We apologise to authors whose contributions were not included due to lack of space.
References
- 1.Kell, D.B. (2012) Scientific discovery as a combinatorial optimisation problem: how best to navigate the landscape of possible experiments? Bioessays 34, 236–244 10.1002/bies.201100144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kell, D.B. and Westerhoff, H.V. (1986) Metabolic control theory: its role in microbiology and biotechnology. FEMS Microbiol. Rev. 39, 305–320 10.1111/j.1574-6968.1986.tb01863.x [DOI] [Google Scholar]
- 3.Kell, D.B. and Westerhoff, H.V. (1986) Towards a rational approach to the optimization of flux in microbial biotransformations. Trends Biotechnol. 4, 137–142 10.1016/0167-7799(86)90163-0 [DOI] [Google Scholar]
- 4.Brown, G.C. (1991) Total cell protein concentration as an evolutionary constraint on the metabolic control distribution in cells. J. Theor. Biol. 153, 195–203 10.1016/S0022-5193(05)80422-9 [DOI] [PubMed] [Google Scholar]
- 5.Cornish-Bowden, A., Hofmeyr, J.-H.S. and Cárdenas, M.L. (1995) Strategies for manipulating metabolic fluxes in biotechnology. Bioorg. Chem. 23, 439–449 10.1006/bioo.1995.1030 [DOI] [Google Scholar]
- 6.Heinrich, R., Schuster, S. and Holzhütter, H.G. (1991) Mathematical analysis of enzymatic reaction systems using optimization principles. Eur. J. Biochem. 201, 1–21 10.1111/j.1432-1033.1991.tb16251.x [DOI] [PubMed] [Google Scholar]
- 7.Heinrich, R. and Schuster, S. (1996) The Regulation of Cellular Systems, Chapman & Hall, New York, NY [Google Scholar]
- 8.Fell, D.A. (1998) Increasing the flux in metabolic pathways: a metabolic control analysis perspective. Biotechnol. Bioeng. 58, 121–124 [DOI] [PubMed] [Google Scholar]
- 9.Zhang, M.M., Wang, Y., Anga, E.L. and Zhao, H. (2015) Engineering microbial hosts for production of bacterial natural products. Nat. Prod. Rep. 33, 963 10.1039/C6NP00017G [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang, G., Kell, D.B. and Borodina, I. (2021) Harnessing the yeast Saccharomyces cerevisiae for the production of fungal secondary metabolites. Essays Biochem. 65, 277–291 10.1042/EBC20200137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jun, S., Si, F., Pugatch, R. and Scott, M. (2018) Fundamental principles in bacterial physiology-history, recent progress, and the future with focus on cell size control: a review. Rep. Prog. Phys. 81, 056601 10.1088/1361-6633/aaa628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bruggeman, F.J., Planqué, R., Molenaar, D. and Teusink, B. (2020) Searching for principles of microbial physiology. FEMS Microbiol. Rev. 44, 821–844 10.1093/femsre/fuaa034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Currin, A., Swainston, N., Day, P.J. and Kell, D.B. (2015) Synthetic biology for the directed evolution of protein biocatalysts: navigating sequence space intelligently. Chem. Soc. Rev. 44, 1172–1239 10.1039/C4CS00351A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jensen, M.K. and Keasling, J.D. (2018) Synthetic Metabolic Pathways: Methods and Protocols, Humana Press, New York, NY [Google Scholar]
- 15.Nielsen, J. and Keasling, J.D. (2011) Synergies between synthetic biology and metabolic engineering. Nat. Biotechnol. 29, 693–695 10.1038/nbt.1937 [DOI] [PubMed] [Google Scholar]
- 16.Way, J.C., Collins, J.J., Keasling, J.D. and Silver, P.A. (2014) Integrating biological redesign: where synthetic biology came from and where it needs to go. Cell 157, 151–161 10.1016/j.cell.2014.02.039 [DOI] [PubMed] [Google Scholar]
- 17.Redden, H., Morse, N. and Alper, H.S. (2015) The synthetic biology toolbox for tuning gene expression in yeast. FEMS Yeast Res. 15, 1–10 10.1093/femsyr/fou003 [DOI] [PubMed] [Google Scholar]
- 18.de Lorenzo, V., Prather, K.L., Chen, G.Q., O'Day, E., von Kameke, C., Oyarzun, D.A.et al. (2018) The power of synthetic biology for bioproduction, remediation and pollution control: the UN's sustainable development goals will inevitably require the application of molecular biology and biotechnology on a global scale. EMBO Rep. 19, e45658 10.15252/embr.201745658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katz, L., Chen, Y.Y., Gonzalez, R., Peterson, T.C., Zhao, H. and Baltz, R.H. (2018) Synthetic biology advances and applications in the biotechnology industry: a perspective. J. Ind. Microbiol. Biotechnol. 45, 449–461 10.1007/s10295-018-2056-y [DOI] [PubMed] [Google Scholar]
- 20.Freemont, P.S. (2019) Synthetic biology industry: data-driven design is creating new opportunities in biotechnology. Emerg. Top. Life Sci. 3, 651–657 10.1042/ETLS20190040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clarke, L. and Kitney, R. (2020) Developing synthetic biology for industrial biotechnology applications. Biochem. Soc. Trans. 48, 113–122 10.1042/BST20190349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang, Y., Ding, W., Wang, Z., Zhao, H. and Shi, S. (2021) Development of host-orthogonal genetic systems for synthetic biology. Adv. Biol. (Weinh) 5, e2000252 10.1002/adbi.202000252 [DOI] [PubMed] [Google Scholar]
- 23.Wang, T., Ma, X., Du, G. and Chen, J. (2012) Overview of regulatory strategies and molecular elements in metabolic engineering of bacteria. Mol. Biotechnol. 52, 300–308 10.1007/s12033-012-9514-y [DOI] [PubMed] [Google Scholar]
- 24.Zielinski, D.C., Patel, A. and Palsson, B.O. (2020) The expanding computational toolbox for engineering microbial phenotypes at the genome scale. Microorganisms 8, 2050 10.3390/microorganisms8122050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodacre, R., Trew, S., Wrigley-Jones, C., Saunders, G., Neal, M.J., Porter, N.et al. (1995) Rapid and quantitative analysis of metabolites in fermentor broths using pyrolysis mass spectrometry with supervised learning: application to the screening of Penicillium chryosgenum fermentations for the overproduction of penicillins. Anal. Chim. Acta 313, 25–43 10.1016/0003-2670(95)00170-5 [DOI] [Google Scholar]
- 26.Ng, A.Y. and Jordan, M. I. (2001) On discriminative vs. generative classifiers: a comparison of logistic regression and naive Bayes. Prc NIPS 14, 841–848 [Google Scholar]
- 27.Baggenstoss, P.M. (2020) The Projected Belief Network Classfier: both Generative and Discriminative. arXiv 2008.06434
- 28.Verma, V.K., Liang, K.J., Mehta, N., Rai, P. and Carin, L. (2021) Efficient feature transformations for discriminative and generative continual learning. arXiv, 2103.13558
- 29.Kell, D.B. and Oliver, S.G. (2004) Here is the evidence, now what is the hypothesis? The complementary roles of inductive and hypothesis-driven science in the post-genomic era. Bioessays 26, 99–105 10.1002/bies.10385 [DOI] [PubMed] [Google Scholar]
- 30.Kell, D.B. and Knowles, J.D. (2006) The role of modeling in systems biology. In System Modeling in Cellular Biology: From Concepts to Nuts and Bolts (Szallasi, Z., Stelling, J. and Periwal, V., eds), pp. 3–18, MIT Press, Cambridge, U.K. [Google Scholar]
- 31.Kell, D.B., Samanta, S. and Swainston, N. (2020) Deep learning and generative methods in cheminformatics and chemical biology: navigating small molecule space intelligently. Biochem. J. 477, 4559–4580 10.1042/BCJ20200781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abid, M.A., Hedhli, I. and Gagné, C. (2021) A generative model for hallucinating diverse versions of super resolution images. arXiv, 2102.06624
- 33.Dupont, E., Teh, Y.W. and Doucet, A. (2021) Generative models as distributions of functions. arXiv, 2102.04776
- 34.Lamb, A. (2021) A brief introduction to generative models. arXiv, 2103.00265
- 35.Ruthotto, L. and Haber, E. (2021) An introduction to deep generative modeling. arXiv, 2103.05180
- 36.Jiménez-Luna, J., Grisoni, F., Weskamp, N. and Schneider, G. (2021) Artificial intelligence in drug discovery: recent advances and future perspectives. Expert Opin. Drug Discov. 16, 949–959 10.1080/17460441.2021.1909567 [DOI] [PubMed] [Google Scholar]
- 37.Biswas, S., Khimulya, G., Alley, E.C., Esvelt, K.M. and Church, G.M. (2021) Low-N protein engineering with data-efficient deep learning. Nat. Methods 18, 389–396 10.1038/s41592-021-01100-y [DOI] [PubMed] [Google Scholar]
- 38.Wu, Z., Johnston, K.E., Arnold, F.H. and Yang, K.K. (2021) Protein sequence design with deep generative models. arXiv, 2104.04457
- 39.Hie, B.L. and Yang, K.K. (2021) Adaptive machine learning for protein engineering. arXiv, 2106.05466
- 40.Li, G., Qin, Y., Fontaine, N.T., Ng Fuk Chong, M., Maria-Solano, M.A., Feixas, F.et al. (2021) Machine learning enables selection of epistatic enzyme mutants for stability against unfolding and detrimental aggregation. Chembiochem 22, 904–914 10.1002/cbic.202000612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swainston, N., Smallbone, K., Mendes, P., Kell, D.B. and Paton, N.W. (2011) The SuBliMinaL Toolbox: automating steps in the reconstruction of metabolic networks. Integrative Bioinf. 8, 186, 10.2390/biecoll-jib-2011-186 PMID: [DOI] [PubMed] [Google Scholar]
- 42.Kell, D.B. (2006) Metabolomics, modelling and machine learning in systems biology: towards an understanding of the languages of cells. The 2005 Theodor Bücher lecture. FEBS J. 273, 873–894 10.1111/j.1742-4658.2006.05136.x [DOI] [PubMed] [Google Scholar]
- 43.Knight, C.G., Platt, M., Rowe, W., Wedge, D.C., Khan, F., Day, P.et al. (2009) Array-based evolution of DNA aptamers allows modelling of an explicit sequence-fitness landscape. Nucleic Acids Res. 37, e6 10.1093/nar/gkn899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghatak, S., King, Z.A., Sastry, A. and Palsson, B.O. (2019) The y-ome defines the 35% of Escherichia coli genes that lack experimental evidence of function. Nucleic Acids Res. 47, 2446–2454 10.1093/nar/gkz030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rudd, K.E. (1998) Linkage map of Escherichia coli K-12, edition 10: the physical map. Microbiol. Mol. Biol. Rev. 62, 985–1019 10.1128/MMBR.62.3.985-1019.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kudla, G., Murray, A.W., Tollervey, D. and Plotkin, J.B. (2009) Coding-sequence determinants of gene expression in Escherichia coli. Science 324, 255–258 10.1126/science.1170160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Plotkin, J.B. and Kudla, G. (2011) Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 12, 32–42 10.1038/nrg2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu, P., Vogel, C., Wang, R., Yao, X. and Marcotte, E.M. (2007) Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 25, 117–124 10.1038/nbt1270 [DOI] [PubMed] [Google Scholar]
- 49.Boël, G., Letso, R., Neely, H., Price, W.N., Wong, K.H., Su, M.et al. (2016) Codon influence on protein expression in E. coli correlates with mRNA levels. Nature 529, 358–363 10.1038/nature16509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eichmann, J., Oberpaul, M., Weidner, T., Gerlach, D. and Czermak, P. (2019) Selection of high producers from combinatorial libraries for the production of recombinant proteins in Escherichia coli and Vibrio natriegens. Front. Bioeng. Biotechnol. 7, 254 10.3389/fbioe.2019.00254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tuller, T., Waldman, Y.Y., Kupiec, M. and Ruppin, E. (2010) Translation efficiency is determined by both codon bias and folding energy. Proc. Natl Acad. Sci. U.S.A. 107, 3645–3650 10.1073/pnas.0909910107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmitz, A. and Zhang, F. (2021) Massively parallel gene expression variation measurement of a synonymous codon library. BMC Genom. 22, 149 10.1186/s12864-021-07462-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Swainston, N., Currin, A., Day, P.J. and Kell, D.B. (2014) Genegenie: optimised oligomer design for directed evolution. Nucleic Acids Res. 12, W395–W400 10.1093/nar/gku336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caspers, M., Brockmeier, U., Degering, C., Eggert, T. and Freudl, R. (2010) Improvement of Sec-dependent secretion of a heterologous model protein in Bacillus subtilis by saturation mutagenesis of the N-domain of the AmyE signal peptide. Appl. Microbiol. Biotechnol. 86, 1877–1885 10.1007/s00253-009-2405-x [DOI] [PubMed] [Google Scholar]
- 55.Zrimec, J., Börlin, C.S., Buric, F., Muhammad, A.S., Chen, R., Siewers, V.et al. (2020) Deep learning suggests that gene expression is encoded in all parts of a co-evolving interacting gene regulatory structure. Nat. Commun. 11, 6141 10.1038/s41467-020-19921-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moore, J.C., Jin, H.M., Kuchner, O. and Arnold, F.H. (1997) Strategies for the in vitro evolution of protein function: enzyme evolution by random recombination of improved sequences. J. Mol. Biol. 272, 336–347 10.1006/jmbi.1997.1252 [DOI] [PubMed] [Google Scholar]
- 57.Berland, M., Offmann, B., Andre, I., Remaud-Simeon, M. and Charton, P. (2014) A web-based tool for rational screening of mutants libraries using ProSAR. Protein Eng. Des. Sel. 27, 375–381 10.1093/protein/gzu035 [DOI] [PubMed] [Google Scholar]
- 58.Chen, H., Borjesson, U., Engkvist, O., Kogej, T., Svensson, M.A., Blomberg, N.et al. (2009) ProSAR: a new methodology for combinatorial library design. J. Chem. Inf. Model. 49, 603–614 10.1021/ci800231d [DOI] [PubMed] [Google Scholar]
- 59.Fox, R.J., Davis, S.C., Mundorff, E.C., Newman, L.M., Gavrilovic, V., Ma, S.K.et al. (2007) Improving catalytic function by ProSAR-driven enzyme evolution. Nat. Biotechnol. 25, 338–344 10.1038/nbt1286 [DOI] [PubMed] [Google Scholar]
- 60.Zaugg, J., Gumulya, Y., Gillam, E.M. and Boden, M. (2014) Computational tools for directed evolution: a comparison of prospective and retrospective strategies. Methods Mol. Biol. 1179, 315–333 10.1007/978-1-4939-1053-3_21 [DOI] [PubMed] [Google Scholar]
- 61.Bäck, T., Fogel, D.B. and Michalewicz, Z. (1997) Handbook of Evolutionary Computation, IOPPublishing/Oxford University Press, Oxford, U.K. [Google Scholar]
- 62.Reeves, C.R. and Rowe, J.E. (2002) Genetic Algorithms - Principles and Perspectives: A Guide to GA Theory, Kluwer Academic Publishers, Dordrecht, the Netherlands [Google Scholar]
- 63.Onwubolu, G.C. and Davendra, D. (2010) Differential Evolution: A Handbook for Global Permutation-Based Combinatorial Optimization, Springer, Berlin, Germany [Google Scholar]
- 64.Ashlock, D. (2006) Evolutionary Computation for Modeling and Optimization, Springer, New York, NY [Google Scholar]
- 65.Coello, C., van Veldhuizen, C.A., and Lamont, D.A. and B, G. (2002) Evolutionary Algorithms for Solving Multi-Objective Problems, Kluwer Academic Publishers, New York, NY [Google Scholar]
- 66.Fogel, D.B. (2000) Evolutionary Computation: Toward A new Philosophy of Machine Intelligence, IEEE Press, Piscataway, NJ [Google Scholar]
- 67.Iba, H. and Noman, N. (2020) Deep Neural Evolution: Deep Learning with Evolutionary Computation, Springer, Berlin, Germany [Google Scholar]
- 68.O'Hagan, S., Knowles, J. and Kell, D.B. (2012) Exploiting genomic knowledge in optimising molecular breeding programmes: algorithms from evolutionary computing. PLoS One 7, e48862 10.1371/journal.pone.0048862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ventura, S. and Luna, J.M. (2016) Pattern Mining with Evolutionary Algorithms, Springer, Berlin, Germany [Google Scholar]
- 70.Arnold, F.H. and Volkov, A.A. (1999) Directed evolution of biocatalysts. Curr. Opin. Chem. Biol. 3, 54–59 10.1016/S1367-5931(99)80010-6 [DOI] [PubMed] [Google Scholar]
- 71.Bloom, J. D. and Arnold, F. H. (2009) In the light of directed evolution: pathways of adaptive protein evolution. Proc. Natl Acad. Sci. U.S.A. 106 Suppl 1, 9995–10000 10.1073/pnas.0901522106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Renata, H., Wang, Z.J. and Arnold, F.H. (2015) Expanding the enzyme universe: accessing non-natural reactions by mechanism-guided directed evolution. Angew. Chem. Int. Ed. Engl. 54, 3351–3367 10.1002/anie.201409470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tracewell, C.A. and Arnold, F.H. (2009) Directed enzyme evolution: climbing fitness peaks one amino acid at a time. Curr. Opin. Chem. Biol. 13, 3–9 10.1016/j.cbpa.2009.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bunzel, H.A., Anderson, J.L.R. and Mulholland, A.J. (2021) Designing better enzymes: insights from directed evolution. Curr. Opin. Struct. Biol. 67, 212–218 10.1016/j.sbi.2020.12.015 [DOI] [PubMed] [Google Scholar]
- 75.Engqvist, M.K.M. and Rabe, K.S. (2019) Applications of protein engineering and directed evolution in plant research. Plant Physiol. 179, 907–917 10.1104/pp.18.01534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Frey, R., Hayashi, T. and Buller, R.M. (2019) Directed evolution of carbon-hydrogen bond activating enzymes. Curr. Opin. Biotechnol. 60, 29–38 10.1016/j.copbio.2018.12.004 [DOI] [PubMed] [Google Scholar]
- 77.Kan, A. and Joshi, N.S. (2019) Towards the directed evolution of protein materials. MRS Commun. 9, 441–455 10.1557/mrc.2019.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Morrison, M.S., Podracky, C.J. and Liu, D.R. (2020) The developing toolkit of continuous directed evolution. Nat. Chem. Biol. 16, 610–619 10.1038/s41589-020-0532-y [DOI] [PubMed] [Google Scholar]
- 79.Qu, G., Li, A., Acevedo-Rocha, C.G., Sun, Z. and Reetz, M.T. (2020) The crucial role of methodology development in directed evolution of selective enzymes. Angew. Chem. Int. Ed. Engl. 59, 13204–13231 10.1002/anie.201901491 [DOI] [PubMed] [Google Scholar]
- 80.Yang, K.K., Wu, Z. and Arnold, F.H. (2019) Machine-learning-guided directed evolution for protein engineering. Nat. Methods 16, 687–694 10.1038/s41592-019-0496-6 [DOI] [PubMed] [Google Scholar]
- 81.Zeymer, C. and Hilvert, D. (2018) Directed evolution of protein catalysts. Annu. Rev. Biochem. 87, 131–157 10.1146/annurev-biochem-062917-012034 [DOI] [PubMed] [Google Scholar]
- 82.Araya, C.L. and Fowler, D.M. (2011) Deep mutational scanning: assessing protein function on a massive scale. Trends Biotechnol. 29, 435–442 10.1016/j.tibtech.2011.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bloom, J.D. (2015) Software for the analysis and visualization of deep mutational scanning data. BMC Bioinform. 16, 168 10.1186/s12859-015-0590-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fowler, D.M., Stephany, J.J. and Fields, S. (2014) Measuring the activity of protein variants on a large scale using deep mutational scanning. Nat. Protoc. 9, 2267–2284 10.1038/nprot.2014.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fowler, D.M. and Fields, S. (2014) Deep mutational scanning: a new style of protein science. Nat. Methods 11, 801–807 10.1038/nmeth.3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Klesmith, J.R., Bacik, J.P., Wrenbeck, E.E., Michalczyk, R. and Whitehead, T.A. (2017) Trade-offs between enzyme fitness and solubility illuminated by deep mutational scanning. Proc. Natl Acad. Sci. U.S.A. 114, 2265–2270 10.1073/pnas.1614437114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Livesey, B.J. and Marsh, J.A. (2020) Using deep mutational scanning to benchmark variant effect predictors and identify disease mutations. Mol. Syst. Biol. 16, e9380 10.15252/msb.20199380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mehlhoff, J.D. and Ostermeier, M. (2020) Biological fitness landscapes by deep mutational scanning. Methods Enzymol. 643, 203–224 10.1016/bs.mie.2020.04.023 [DOI] [PubMed] [Google Scholar]
- 89.Munro, D. and Singh, M. (2020) Demask: a deep mutational scanning substitution matrix and its use for variant impact prediction. Bioinformatics 6, 5322–5329 10.1093/bioinformatics/btaa1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Reeb, J., Wirth, T. and Rost, B. (2020) Variant effect predictions capture some aspects of deep mutational scanning experiments. BMC Bioinform. 21, 107 10.1186/s12859-020-3439-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Behrendt, L., Stein, A., Shah, S.A., Zengler, K., Sørensen, S.J., Lindorff-Larsen, K.et al. (2018) Deep mutational scanning by FACS-sorting of encapsulated E. coli micro-colonies. bioRxiv, 274753
- 92.Katz, N., Tripto, E., Granik, N., Goldberg, S., Atar, O., Yakhini, Z.et al. (2021) Overcoming the design, build, test bottleneck for synthesis of nonrepetitive protein-RNA cassettes. Nat. Commun. 12, 1576 10.1038/s41467-021-21578-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rowe, W., Platt, M., Wedge, D., Day, P.J., Kell, D.B. and Knowles, J. (2010) Analysis of a complete DNA-protein affinity landscape. J. R. Soc. Interface 7, 397–408 10.1098/rsif.2009.0193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kell, D.B. (2017) Evolutionary algorithms and synthetic biology for directed evolution: commentary on ‘on the mapping of genotype to phenotype in evolutionary algorithms’ by Peter A. Whigham, Grant Dick, and James Maclaurin. Genet. Program. Evol. Mach. 18, 373–378 10.1007/s10710-017-9292-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kell, D.B. and Lurie-Luke, E. (2015) The virtue of innovation: innovation through the lenses of biological evolution. J. R. Soc. Interface 12, 20141183 10.1098/rsif.2014.1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wright, S. (1932) The roles of mutation, inbreeding, crossbreeding and selection in evolution. In Proceedings of the Sixth International Congress of Genetics (Jones, D.F., ed.), pp. 356–366, Genetics Society of America, Austin/Ithaca, TX/NY [Google Scholar]
- 97.Marks, D.S., Hopf, T.A. and Sander, C. (2012) Protein structure prediction from sequence variation. Nat. Biotechnol. 30, 1072–1080 10.1038/nbt.2419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kosciolek, T. and Jones, D.T. (2014) De novo structure prediction of globular proteins aided by sequence variation-derived contacts. PLoS One 9, e92197 10.1371/journal.pone.0092197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kosciolek, T. and Jones, D.T. (2016) Accurate contact predictions using covariation techniques and machine learning. Proteins 84 Suppl 1, 145–151 10.1002/prot.24863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hopf, T.A., Green, A.G., Schubert, B., Mersmann, S., Scharfe, C.P.I., Ingraham, J.B.et al. (2019) The EVcouplings python framework for coevolutionary sequence analysis. Bioinformatics 35, 1582–1584 10.1093/bioinformatics/bty862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jones, D.T., Buchan, D.W., Cozzetto, D. and Pontil, M. (2012) PSICOV: precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics 28, 184–190 10.1093/bioinformatics/btr638 [DOI] [PubMed] [Google Scholar]
- 102.Rawi, R., Mall, R., Kunji, K., El Anbari, M., Aupetit, M., Ullah, E.et al. (2016) COUSCOus: improved protein contact prediction using an empirical Bayes covariance estimator. BMC Bioinform. 17, 533 10.1186/s12859-016-1400-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O.et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Richter, H. and Engelbrecht, A.P. (2014) Recent Advances in the Theory and Application of Fitness Landscapes, Springer, Berlin, Germany [Google Scholar]
- 105.Aguilar-Rodríguez, J., Payne, J.L. and Wagner, A. (2017) A thousand empirical adaptive landscapes and their navigability. Nat. Ecol. Evol. 1, 45 10.1038/s41559-016-0045 [DOI] [PubMed] [Google Scholar]
- 106.Blanco, C., Janzen, E., Pressman, A., Saha, R. and Chen, I.A. (2019) Molecular fitness landscapes from high-coverage sequence profiling. Annu. Rev. Biophys. 48, 1–18 10.1146/annurev-biophys-052118-115333 [DOI] [PubMed] [Google Scholar]
- 107.Kauffman, S.A. (1993) The Origins of Order, Oxford University Press, Oxford, U.K [Google Scholar]
- 108.Kauffman, S.A. and Macready, W.G. (1995) Search strategies for applied molecular evolution. J. Theor. Biol. 173, 427–440 10.1006/jtbi.1995.0074 [DOI] [PubMed] [Google Scholar]
- 109.Aita, T., Hayashi, Y., Toyota, H., Husimi, Y., Urabe, I. and Yomo, T. (2007) Extracting characteristic properties of fitness landscape from in vitro molecular evolution: a case study on infectivity of fd phage to E. coli. J. Theor. Biol. 246, 538–550 10.1016/j.jtbi.2006.12.037 [DOI] [PubMed] [Google Scholar]
- 110.Mater, A.C., Sandhu, M. and Jackson, C. (2020) The NK landscape as a versatile benchmark for machine learning driven protein engineering. bioRxiv, 2020.2009.2030.319780
- 111.Hwang, S., Schmiegelt, B., Ferretti, L. and Krug, J. (2018) Universality classes of interaction structures for NK fitness landscapes. J. Stat. Phys. 172, 226–278 10.1007/s10955-018-1979-z [DOI] [Google Scholar]
- 112.Zagorski, M., Burda, Z. and Waclaw, B. (2016) Beyond the hypercube: evolutionary accessibility of fitness landscapes with realistic mutational networks. PLoS Comput. Biol. 12, e1005218 10.1371/journal.pcbi.1005218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weinreich, D.M., Watson, R.A. and Chao, L. (2005) Perspective: sign epistasis and genetic constraint on evolutionary trajectories. Evolution 59, 1165–1174, PMID: [PubMed] [Google Scholar]
- 114.de Visser, J.A.G.M., Cooper, T.F. and Elena, S.F. (2011) The causes of epistasis. Proc. Biol. Sci. 278, 3617–3624 10.1098/rspb.2011.1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.de Visser, J.A.G.M. and Krug, J. (2014) Empirical fitness landscapes and the predictability of evolution. Nat. Rev. Genet. 15, 480–490 10.1038/nrg3744 [DOI] [PubMed] [Google Scholar]
- 116.Acevedo-Rocha, C.G., Li, A., D'Amore, L., Hoebenreich, S., Sanchis, J., Lubrano, P.et al. (2021) Pervasive cooperative mutational effects on multiple catalytic enzyme traits emerge via long-range conformational dynamics. Nat. Commun. 12, 1621 10.1038/s41467-021-21833-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Weinreich, D.M., Lan, Y.H., Jaffe, J. and Heckendorn, R.B. (2018) The influence of higher-order epistasis on biological fitness landscape topography. J. Stat. Phys. 172, 208–225 10.1007/s10955-018-1975-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Adams, R.M., Kinney, J.B., Walczak, A.M. and Mora, T. (2019) Epistasis in a fitness landscape defined by antibody-antigen binding free energy. Cell Syst. 8, 86–93.e83 10.1016/j.cels.2018.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gonzalez, C.E. and Ostermeier, M. (2019) Pervasive pairwise intragenic epistasis among sequential mutations in TEM-1 beta-lactamase. J. Mol. Biol. 431, 1981–1992 10.1016/j.jmb.2019.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gillespie, J.H. (1983) A simple stochastic gene substitution model. Theor. Popul. Biol. 23, 202–215 10.1016/0040-5809(83)90014-X [DOI] [PubMed] [Google Scholar]
- 121.Gillespie, J.H. (1984) Molecular evolution over the mutational landscape. Evolution 38, 1116–1129 10.1111/j.1558-5646.1984.tb00380.x [DOI] [PubMed] [Google Scholar]
- 122.Orr, H.A. (2005) The genetic theory of adaptation: a brief history. Nat. Rev. Genet. 6, 119–127 10.1038/nrg1523 [DOI] [PubMed] [Google Scholar]
- 123.Orr, H.A. (2006) The population genetics of adaptation on correlated fitness landscapes: the block model. Evolution 60, 1113–1124 10.1111/j.0014-3820.2006.tb01191.x [DOI] [PubMed] [Google Scholar]
- 124.Orr, H.A. (2006) The distribution of fitness effects among beneficial mutations in Fisher's geometric model of adaptation. J. Theor. Biol. 238, 279–285 10.1016/j.jtbi.2005.05.001 [DOI] [PubMed] [Google Scholar]
- 125.Orr, H.A. (2009) Fitness and its role in evolutionary genetics. Nat. Rev. Genet. 10, 531–539 10.1038/nrg2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Unckless, R.L. and Orr, H.A. (2009) The population genetics of adaptation: multiple substitutions on a smooth fitness landscape. Genetics 183, 1079–1086 10.1534/genetics.109.106757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Szendro, I.G., Franke, J., de Visser, J.A.G.M. and Krug, J. (2013) Predictability of evolution depends nonmonotonically on population size. Proc. Natl Acad. Sci. U.S.A. 110, 571–576 10.1073/pnas.1213613110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Carneiro, M. and Hartl, D.L. (2011) Adaptive landscapes and protein evolution. Proc. Natl Acad. Sci. U.S.A. 107 Suppl 1, 1747–1751 10.1073/pnas.0906192106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Naseri, G. and Koffas, M.A.G. (2020) Application of combinatorial optimization strategies in synthetic biology. Nat. Commun. 11, 2446 10.1038/s41467-020-16175-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Swainston, N., Dunstan, M., Jervis, A.J., Robinson, C.J., Carbonell, P., Williams, A.R.et al. (2018) Partsgenie: an integrated tool for optimising and sharing synthetic biology parts. Bioinformatics 34, 2327–2329 10.1093/bioinformatics/bty105 [DOI] [PubMed] [Google Scholar]
- 131.HamediRad, M., Chao, R., Weisberg, S., Lian, J., Sinha, S. and Zhao, H. (2019) Towards a fully automated algorithm driven platform for biosystems design. Nat. Commun. 10, 5150 10.1038/s41467-019-13189-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Radivojević, T., Costello, Z., Workman, K. and Garcia Martin, H. (2020) A machine learning automated recommendation tool for synthetic biology. Nat. Commun. 11, 4879 10.1038/s41467-020-18008-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhang, J., Petersen, S.D., Radivojevic, T., Ramirez, A., Pérez-Manríquez, A., Abeliuk, E.et al. (2020) Combining mechanistic and machine learning models for predictive engineering and optimization of tryptophan metabolism. Nat. Commun. 11, 4880 10.1038/s41467-020-17910-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Beaumont, H.J.E., Gallie, J., Kost, C., Ferguson, G.C. and Rainey, P.B. (2009) Experimental evolution of bet hedging. Nature 462, 90–93 10.1038/nature08504 [DOI] [PubMed] [Google Scholar]
- 135.Kaldalu, N., Hauryliuk, V. and Tenson, T. (2016) Persisters-as elusive as ever. Appl. Microbiol. Biotechnol. 100, 6545–6553 10.1007/s00253-016-7648-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Levy, S.F., Ziv, N. and Siegal, M.L. (2012) Bet hedging in yeast by heterogeneous, age-correlated expression of a stress protectant. PLoS Biol. 10, e1001325 10.1371/journal.pbio.1001325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kell, D.B., Potgieter, M. and Pretorius, E. (2015) Individuality, phenotypic differentiation, dormancy and ‘persistence’ in culturable bacterial systems: commonalities shared by environmental, laboratory, and clinical microbiology. F1000Research 4, 179 10.12688/f1000research.6709.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Salcedo-Sora, J.E. and Kell, D.B. (2020) A quantitative survey of bacterial persistence in the presence of antibiotics: towards antipersister antimicrobial discovery. Antibiotics 9, 508 10.3390/antibiotics9080508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Westerhoff, H.V., Hellingwerf, K.J. and van Dam, K. (1983) Thermodynamic efficiency of microbial growth is low but optimal for maximal growth rate. Proc. Natl Acad. Sci. U.S.A. 80, 305–309 10.1073/pnas.80.1.305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kacser, H. and Burns, J.A. (1973) The control of flux. In Rate Control of Biological Processes. Symposium of the Society for Experimental Biology (Davies, D.D., ed.), vol. 27, pp. 65–104, Cambridge University Press, Cambridge, U.K: [PubMed] [Google Scholar]
- 141.Kacser, H. and Burns, J.A. (1981) The molecular basis of dominance. Genetics 97, 639–666 10.1093/genetics/97.3-4.639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kacser, H. (1983) The control of enzyme systems in vivo: elasticity analysis of the steady state. Biochem. Soc. Trans. 11, 35–40 10.1042/bst0110035 [DOI] [PubMed] [Google Scholar]
- 143.Heinrich, R. and Rapoport, T.A. (1973) Linear theory of enzymatic chains: its application for the analysis of the crossover theorem and of the glycolysis of human erythrocytes. Acta Biol. Med. Ger. 31, 479–494 [PubMed] [Google Scholar]
- 144.Heinrich, R. and Rapoport, T.A. (1974) A linear steady-state treatment of enzymatic chains. General properties, control and effector strength. Eur. J. Biochem. 42, 89–95 10.1111/j.1432-1033.1974.tb03318.x [DOI] [PubMed] [Google Scholar]
- 145.Klipp, E., Herwig, R., Kowald, A., Wierling, C. and Lehrach, H. (2005) Systems Biology in Practice: Concepts, Implementation and Clinical Application, Wiley/VCH, Berlin, Germany [Google Scholar]
- 146.Palsson, B.Ø. (2006) Systems Biology: Properties of Reconstructed Networks, Cambridge University Press, Cambridge, U.K [Google Scholar]
- 147.Moreno-Sánchez, R., Saavedra, E., Rodríguez-Enríquez, S. and Olin-Sandoval, V. (2008) Metabolic control analysis: a tool for designing strategies to manipulate metabolic pathways. J. Biomed. Biotechnol. 2008, 597913 10.1155/2008/597913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rand, D.A. (2008) Mapping global sensitivity of cellular network dynamics: sensitivity heat maps and a global summation law. J. R. Soc. Interface 5 Suppl 1, S59–S69 10.1098/rsif.2008.0084.focus [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Saltelli, A., Ratto, M., Andres, T., Campolongo, F., Cariboni, J., Gatelli, D.et al. (2008) Global Sensitivity Analysis: the Primer, WileyBlackwell, New York, NY [Google Scholar]
- 150.Fell, D.A. and Thomas, S. (1995) Physiological control of metabolic flux: the requirement for multisite modulation. Biochem. J. 311, 35–39 10.1042/bj3110035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Oliver, S.G., Winson, M.K., Kell, D.B. and Baganz, F. (1998) Systematic functional analysis of the yeast genome. Trends Biotechnol. 16, 373–378 10.1016/S0167-7799(98)01214-1 [DOI] [PubMed] [Google Scholar]
- 152.Raamsdonk, L.M., Teusink, B., Broadhurst, D., Zhang, N., Hayes, A., Walsh, M.et al. (2001) A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat. Biotechnol. 19, 45–50 10.1038/83496 [DOI] [PubMed] [Google Scholar]
- 153.Kell, D.B. and Oliver, S.G. (2016) The metabolome 18 years on: a concept comes of age. Metabolomics 12, 148 10.1007/s11306-016-1108-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Karim, A.S., Dudley, Q.M., Juminaga, A., Yuan, Y., Crowe, S.A., Heggestad, J.T.et al. (2020) In vitro prototyping and rapid optimization of biosynthetic enzymes for cell design. Nat. Chem. Biol. 16, 912–919 10.1038/s41589-020-0559-0 [DOI] [PubMed] [Google Scholar]
- 155.Scott, M., Gunderson, C.W., Mateescu, E.M., Zhang, Z. and Hwa, T. (2010) Interdependence of cell growth and gene expression: origins and consequences. Science 330, 1099–1102 10.1126/science.1192588 [DOI] [PubMed] [Google Scholar]
- 156.Shoval, O., Sheftel, H., Shinar, G., Hart, Y., Ramote, O., Mayo, A.et al. (2012) Evolutionary trade-offs, pareto optimality, and the geometry of phenotype space. Science 336, 1157–1160 10.1126/science.1217405 [DOI] [PubMed] [Google Scholar]
- 157.Schuetz, R., Zamboni, N., Zampieri, M., Heinemann, M. and Sauer, U. (2012) Multidimensional optimality of microbial metabolism. Science 336, 601–604 10.1126/science.1216882 [DOI] [PubMed] [Google Scholar]
- 158.Mori, M., Schink, S., Erickson, D.W., Gerland, U. and Hwa, T. (2017) Quantifying the benefit of a proteome reserve in fluctuating environments. Nat. Commun. 8, 1225 10.1038/s41467-017-01242-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Basan, M., Honda, T., Christodoulou, D., Horl, M., Chang, Y.F., Leoncini, E.et al. (2020) A universal trade-off between growth and lag in fluctuating environments. Nature 584, 470–474 10.1038/s41586-020-2505-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Peebo, K., Valgepea, K., Maser, A., Nahku, R., Adamberg, K. and Vilu, R. (2015) Proteome reallocation in Escherichia coli with increasing specific growth rate. Mol. Biosyst. 11, 1184–1193 10.1039/C4MB00721B [DOI] [PubMed] [Google Scholar]
- 161.Desler, C., Hansen, T.L., Frederiksen, J.B., Marcker, M.L., Singh, K.K. and Juel Rasmussen, L. (2012) Is there a link between mitochondrial reserve respiratory capacity and aging? J. Aging Res. 2012, 192503 10.1155/2012/192503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Marchetti, P., Fovez, Q., Germain, N., Khamari, R. and Kluza, J. (2020) Mitochondrial spare respiratory capacity: mechanisms, regulation, and significance in non-transformed and cancer cells. FASEB J. 34, 13106–13124 10.1096/fj.202000767R [DOI] [PubMed] [Google Scholar]
- 163.Rieger, M., Käppeli, O. and Fiechter, A. (1983) The role of limited respiration in the incomplete oxidation of glucose by Saccharomyces cerevisiae. J. Gen. Microbiol. 129, 653–661 10.1074/jbc.273.38.24529 [DOI] [Google Scholar]
- 164.Sonnleitner, B. and Käppeli, O. (1986) Growth of Saccharomyces cerevisiae is controlled by its limited respiratory capacity - formulation and verification of a hypothesis. Biotechnol. Bioeng. 28, 927–937 10.1002/bit.260280620 [DOI] [PubMed] [Google Scholar]
- 165.van Hoek, P., van Dijken, J.P. and Pronk, J.T. (1998) Effect of specific growth rate on fermentative capacity of baker's yeast. Appl. Environ. Microbiol. 64, 4226–4233 10.1128/AEM.64.11.4226-4233.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Basan, M., Hui, S., Okano, H., Zhang, Z., Shen, Y., Williamson, J.R.et al. (2015) Overflow metabolism in Escherichia coli results from efficient proteome allocation. Nature 528, 99–104 10.1038/nature15765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Neijssel, O.M. and Tempest, D.W. (1976) The role of energy-splitting reactions in the growth of Klebsiella aerogenes NCTC 418 in aerobic chemostat culture. Arch. Microbiol. 110, 305–311 10.1007/BF00690243 [DOI] [PubMed] [Google Scholar]
- 168.Allen, J.K., Davey, H.M., Broadhurst, D., Heald, J.K., Rowland, J.J., Oliver, S.G.et al. (2003) High-throughput characterisation of yeast mutants for functional genomics using metabolic footprinting. Nat. Biotechnol. 21, 692–696 10.1038/nbt823 [DOI] [PubMed] [Google Scholar]
- 169.Kell, D.B., Brown, M., Davey, H.M., Dunn, W.B., Spasic, I. and Oliver, S.G. (2005) Metabolic footprinting and systems biology: the medium is the message. Nat. Rev. Microbiol. 3, 557–565 10.1038/nrmicro1177 [DOI] [PubMed] [Google Scholar]
- 170.Dörries, K. and Lalk, M. (2013) Metabolic footprint analysis uncovers strain specific overflow metabolism and D-isoleucine production of Staphylococcus aureus COL and HG001. PLoS One 8, e81500 10.1371/journal.pone.0081500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Paczia, N., Nilgen, A., Lehmann, T., Gätgens, J., Wiechert, W. and Noack, S. (2012) Extensive exometabolome analysis reveals extended overflow metabolism in various microorganisms. Microb. Cell Fact. 11, 122 10.1186/1475-2859-11-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Schmitz, K., Peter, V., Meinert, S., Kornfeld, G., Hardiman, T., Wiechert, W.et al. (2013) Simultaneous utilization of glucose and gluconate in Penicillium chrysogenum during overflow metabolism. Biotechnol. Bioeng. 110, 3235–3243 10.1002/bit.24974 [DOI] [PubMed] [Google Scholar]
- 173.Szenk, M., Dill, K.A. and de Graff, A.M.R. (2017) Why do fast-growing bacteria enter overflow metabolism? Testing the membrane real estate hypothesis. Cell Syst. 5, 95–104 10.1016/j.cels.2017.06.005 [DOI] [PubMed] [Google Scholar]
- 174.Koch, A.L. (1971) The adaptive responses of Escherichia coli to a feast and famine existence. Adv. Microbial. Physiol. 6, 147–217 10.1016/S0065-2911(08)60069-7 [DOI] [PubMed] [Google Scholar]
- 175.Poindexter, J. (1981) Oligotrophy: fast and famine existence. Adv. Microbial. Ecol. 5, 63–89 10.1007/978-1-4615-8306-6_2 [DOI] [Google Scholar]
- 176.Kell, D.B., Swainston, N., Pir, P. and Oliver, S.G. (2015) Membrane transporter engineering in industrial biotechnology and whole-cell biocatalysis. Trends Biotechnol. 33, 237–246 10.1016/j.tibtech.2015.02.001 [DOI] [PubMed] [Google Scholar]
- 177.Kell, D.B. (2019) Control of metabolite efflux in microbial cell factories: current advances and future prospects. In Fermentation Microbiology and Biotechnology, 4th edn (El-Mansi, E.M.T., Nielsen, J., Mousdale, D., Allman, T. and Carlson, R., eds), pp. 117–138, CRC Press, Boca Raton, FL [Google Scholar]
- 178.Wang, G., Møller-Hansen, I., Babaei, M., D'Ambrosio, V., Christensen, H.B., Darbani, B.et al. (2021) Transportome-wide engineering of Saccharomyces cerevisiae. Metab. Eng. 64, 52–63 10.1016/j.ymben.2021.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jindal, S., Yang, L., Day, P.J. and Kell, D.B. (2019) Involvement of multiple influx and efflux transporters in the accumulation of cationic fluorescent dyes by Escherichia coli. BMC Microbiol. 19, 195. also bioRxiv 603688v603681 10.1186/s12866-019-1561-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Salcedo-Sora, J.E., Jindal, S., O'Hagan, S. and Kell, D.B. (2021) A palette of fluorophores that are differentially accumulated by wild-type and mutant strains of Escherichia coli: surrogate ligands for bacterial membrane transporters. Microbiology 167, 001016 10.1099/mic.0.001016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Anderson, C.M. (2006) The Long Tail: how Endless Choice is Creating Unlimited Demand, Random House, London, U.K. [Google Scholar]
- 182.Bornholdt, S. and Sneppen, K. (2000) Robustness as an evolutionary principle. Proc. R. Soc. B-Biol. Sci. 267, 2281–2286 10.1098/rspb.2000.1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Morohashi, M., Winn, A.E., Borisuk, M.T., Bolouri, H., Doyle, J. and Kitano, H. (2002) Robustness as a measure of plausibility in models of biochemical networks. J. Theor. Biol. 216, 19–30 10.1006/jtbi.2002.2537 [DOI] [PubMed] [Google Scholar]
- 184.Kitano, H. (2004) Biological robustness. Nat. Rev. Genet. 5, 826–837 10.1038/nrg1471 [DOI] [PubMed] [Google Scholar]
- 185.Stelling, J., Sauer, U., Szallasi, Z., Doyle, III, F.J. and Doyle, J. (2004) Robustness of cellular functions. Cell 118, 675–685 10.1016/j.cell.2004.09.008 [DOI] [PubMed] [Google Scholar]
- 186.Wagner, A. (2005) Robustness and Evolvability in Living Systems, Princeton University Press, Princeton, NJ [Google Scholar]
- 187.Ma, W., Lai, L., Ouyang, Q. and Tang, C. (2006) Robustness and modular design of the Drosophila segment polarity network. Mol. Syst. Biol. 2, 70 10.1038/msb4100111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lehár, J., Krueger, A., Zimmermann, G. and Borisy, A. (2008) High-order combination effects and biological robustness. Mol. Syst. Biol. 4, 215 10.1038/msb.2008.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Gong, Z., Nielsen, J. and Zhou, Y.J. (2017) Engineering robustness of microbial cell factories. Biotechnol. J. 12, 1700014. 10.1002/biot.201700014 [DOI] [PubMed] [Google Scholar]
- 190.Klug, A., Park, S.C. and Krug, J. (2019) Recombination and mutational robustness in neutral fitness landscapes. PLoS Comput. Biol. 15, e1006884 10.1371/journal.pcbi.1006884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Donati, S., Kuntz, M., Pahl, V., Farke, N., Beuter, D., Glatter, T.et al. (2021) Multi-omics analysis of CRISPRi-knockdowns identifies mechanisms that buffer decreases of enzymes in E. coli metabolism. Cell Syst. 12, 56–67 10.1016/j.cels.2020.10.011 [DOI] [PubMed] [Google Scholar]
- 192.Schaechter, M., Maaløe, O. and Kjeldgaard, N.O. (1958) Dependency on medium and temperature of cell size and chemical composition during balanced grown of Salmonella typhimurium. J. Gen. Microbiol. 19, 592–606 10.1099/00221287-19-3-592 [DOI] [PubMed] [Google Scholar]
- 193.Neidhart, F.C., Ingraham, J.L. and Schaechter, M. (1990) Physiology of the Bacterial Cell: A Molecular Approach, Sinauer Associates, Sunderland, MA [Google Scholar]
- 194.Björkeroth, J., Campbell, K., Malina, C., Yu, R., Di Bartolomeo, F. and Nielsen, J. (2020) Proteome reallocation from amino acid biosynthesis to ribosomes enables yeast to grow faster in rich media. Proc. Natl Acad. Sci. U.S.A. 117, 21804–21812 10.1073/pnas.1921890117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tartof, K.D. and Hobbs, C.A. (1987) Improved media for growing plasmid and cosmid clones. Bethseda Res. Labs Focus. 9, 12 [Google Scholar]
- 196.Hill, W.G. (2005) A century of corn selection. Science 307, 683–684 10.1126/science.1105459 [DOI] [PubMed] [Google Scholar]
- 197.de Groot, D.H., Hulshof, J., Teusink, B., Bruggeman, F.J. and Planqué, R. (2020) Elementary growth modes provide a molecular description of cellular self-fabrication. PLoS Comput. Biol. 16, e1007559 10.1371/journal.pcbi.1007559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Castrillo, J.I., Zeef, L.A., Hoyle, D.C., Zhang, N., Hayes, A., Gardner, D.C.J.et al. (2007) Growth control of the eukaryote cell: a systems biology study in yeast. J. Biol. 6, 4 10.1186/jbiol54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Delneri, D., Hoyle, D.C., Gkargkas, K., Cross, E.J., Rash, B., Zeef, L.et al. (2008) Identification and characterization of high-flux-control genes of yeast through competition analyses in continuous cultures. Nat. Genet. 40, 113–117 10.1038/ng.2007.49 [DOI] [PubMed] [Google Scholar]
- 200.Reed, J.L. and Palsson, B.Ø. (2003) Thirteen years of building constraint-based in silico models of Escherichia coli. J. Bacteriol. 185, 2692–2699 10.1128/JB.185.9.2692-2699.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Herrgård, M.J., Swainston, N., Dobson, P., Dunn, W.B., Arga, K.Y., Arvas, M.et al. (2008) A consensus yeast metabolic network obtained from a community approach to systems biology. Nat. Biotechnol. 26, 1155–1160 10.1038/nbt1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Thiele, I., Swainston, N., Fleming, R.M.T., Hoppe, A., Sahoo, S., Aurich, M.K.et al. (2013) A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 31, 419–425 10.1038/nbt.2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Thiele, I., Sahoo, S., Heinken, A., Hertel, J., Heirendt, L., Aurich, M.K.et al. (2020) Personalized whole-body models integrate metabolism, physiology, and the gut microbiome. Mol. Syst. Biol. 16, e8982 10.15252/msb.20198982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Covert, M.W., Xiao, N., Chen, T.J. and Karr, J.R. (2008) Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics 24, 2044–2050 10.1093/bioinformatics/btn352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hou, J., Tyo, K.E., Liu, Z., Petranovic, D. and Nielsen, J. (2012) Metabolic engineering of recombinant protein secretion by Saccharomyces cerevisiae. FEMS Yeast Res. 12, 491–510 10.1111/j.1567-1364.2012.00810.x [DOI] [PubMed] [Google Scholar]
- 206.Chen, Y. and Nielsen, J. (2019) Energy metabolism controls phenotypes by protein efficiency and allocation. Proc. Natl Acad. Sci. U.S.A. 116, 17592–17597 10.1073/pnas.1906569116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chen, K., Anand, A., Olson, C., Sandberg, T.E., Gao, Y., Mih, N.et al. (2021) Bacterial fitness landscapes stratify based on proteome allocation associated with discrete aero-types. PLoS Comput. Biol. 17, e1008596 10.1371/journal.pcbi.1008596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Snoep, J.L., Yomano, L.P., Westerhoff, H.V. and Ingram, L.O. (1995) Protein burden in Zymomonas mobilis - negative flux and growth- control due to overproduction of glycolytic enzymes. Microbiology 141, 2329–2337 10.1099/13500872-141-9-2329 [DOI] [Google Scholar]
- 209.Bentley, W.E., Mirjalili, N., Andersen, D.C., Davis, R.H. and Kompala, D.S. (1990) Plasmid-encoded protein: the principal factor in the ‘metabolic burden’ associated with recombinant bacteria. Biotechnol. Bioeng. 35, 668–681 10.1002/bit.260350704 [DOI] [PubMed] [Google Scholar]
- 210.Dong, H., Nilsson, L. and Kurland, C.G. (1995) Gratuitous overexpression of genes in Escherichia coli leads to growth inhibition and ribosome destruction. J. Bacteriol. 177, 1497–1504 10.1128/jb.177.6.1497-1504.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Yu, R., Campbell, K., Pereira, R., Bjorkeroth, J., Qi, Q., Vorontsov, E.et al. (2020) Nitrogen limitation reveals large reserves in metabolic and translational capacities of yeast. Nat. Commun. 11, 1881 10.1038/s41467-020-15749-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Holms, W.H., Hamilton, I.D. and Mousdale, D. (1991) Improvements to microbial productivity by analysis of metabolic fluxes. J. Chem. Technol. Biotechnol. 50, 139–141 10.1002/jctb.280500119 [DOI] [Google Scholar]
- 213.Holms, H. (1996) Flux analysis and control of the central metabolic pathways in Escherichia coli. FEMS Microbiol. Rev. 19, 85–116 10.1111/j.1574-6976.1996.tb00255.x [DOI] [PubMed] [Google Scholar]
- 214.Valgepea, K., Peebo, K., Adamberg, K. and Vilu, R. (2015) Lean-proteome strains - next step in metabolic engineering. Front. Bioeng. Biotechnol. 3, 11 10.3389/fbioe.2015.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Glasscock, C.J., Lucks, J.B. and DeLisa, M.P. (2016) Engineered protein machines: emergent tools for synthetic biology. Cell Chem. Biol. 23, 45–56 10.1016/j.chembiol.2015.12.004 [DOI] [PubMed] [Google Scholar]
- 216.Guirimand, G., Kulagina, N., Papon, N., Hasunuma, T. and Courdavault, V. (2021) Innovative tools and strategies for optimizing yeast cell factories. Trends Biotechnol. 39, 488–504 10.1016/j.tibtech.2020.08.010 [DOI] [PubMed] [Google Scholar]
- 217.Lawless, C., Holman, S.W., Brownridge, P., Lanthaler, K., Harman, V.M., Watkins, R.et al. (2016) Direct and absolute quantification of over 1800 yeast proteins via selected reaction monitoring. Mol. Cell. Proteom. 15, 1309–1322 10.1074/mcp.M115.054288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Yu, R., Vorontsov, E., Sihlbom, C. and Nielsen, J. (2021) Quantifying absolute gene expression profiles reveals distinct regulation of central carbon metabolism genes in yeast. eLife 10, e65722 10.7554/eLife.65722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Nichols, R.J., Sen, S., Choo, Y.J., Beltrao, P., Zietek, M., Chaba, R.et al. (2011) Phenotypic landscape of a bacterial cell. Cell 144, 143–156 10.1016/j.cell.2010.11.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.O'Hagan, S., Wright Muelas, M., Day, P.J., Lundberg, E. and Kell, D.B. (2018) Genegini: assessment via the Gini coefficient of reference ‘‘housekeeping’’ genes and diverse human transporter expression profiles. Cell Syst. 6, 230–244 10.1016/j.cels.2018.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Muelas, W., Mughal, M., O'Hagan, F., Day, S., and Kell, P.J. and B, D. (2019) The role and robustness of the Gini coefficient as an unbiased tool for the selection of Gini genes for normalising expression profiling data. Sci. Rep. 9, 17960 10.1038/s41598-019-54288-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Gallagher, L.A., Bailey, J. and Manoil, C. (2020) Ranking essential bacterial processes by speed of mutant death. Proc. Natl Acad. Sci. U.S.A. 117, 18010–18017 10.1073/pnas.2001507117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Alper, H., Moxley, J., Nevoigt, E., Fink, G.R. and Stephanopoulos, G. (2006) Engineering yeast transcription machinery for improved ethanol tolerance and production. Science 314, 1565–1568 10.1126/science.1131969 [DOI] [PubMed] [Google Scholar]
- 224.Alper, H. and Stephanopoulos, G. (2007) Global transcription machinery engineering: a new approach for improving cellular phenotype. Metab. Eng. 9, 258–267 10.1016/j.ymben.2006.12.002 [DOI] [PubMed] [Google Scholar]
- 225.Liu, H., Yan, M., Lai, C., Xu, L. and Ouyang, P. (2010) gTME for improved xylose fermentation of Saccharomyces cerevisiae. Appl. Biochem. Biotechnol. 160, 574–582 10.1007/s12010-008-8431-9 [DOI] [PubMed] [Google Scholar]
- 226.Tan, F., Wu, B., Dai, L., Qin, H., Shui, Z., Wang, J.et al. (2016) Using global transcription machinery engineering (gTME) to improve ethanol tolerance of Zymomonas mobilis. Microb. Cell Fact. 15, 4 10.1186/s12934-015-0398-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.El-Rotail, A.A.M.M., Zhang, L., Li, Y., Liu, S.P. and Shi, G.Y. (2017) A novel constructed SPT15 mutagenesis library of Saccharomyces cerevisiae by using gTME technique for enhanced ethanol production. AMB Express 7, 111 10.1186/s13568-017-0400-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Knight, J.R.P., Garland, G., Poyry, T., Mead, E., Vlahov, N., Sfakianos, A.et al. (2020) Control of translation elongation in health and disease. Dis. Model. Mech. 13, dmm043208 10.1242/dmm.043208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Masoudi, M., Teimoori, A., Tabaraei, A., Shahbazi, M., Divbandi, M., Lorestani, N.et al. (2021) Advanced sequence optimization for the high efficient yield of human group A rotavirus VP6 recombinant protein in Escherichia coli and its use as immunogen. J. Med. Virol. 93, 3549–3556 10.1002/jmv.26522 [DOI] [PubMed] [Google Scholar]
- 230.Wu, Z., Yang, K.K., Liszka, M., Lee, A., Batzilla, A., Wernick, D.et al. (2020) Signal peptides generated by attention-based neural networks. ACS Synth. Biol. 9, 2154–2161 10.1021/acssynbio.0c00219 [DOI] [PubMed] [Google Scholar]
- 231.Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A.N.et al. (2017) Attention is all you need. arXiv, 1706.03762
- 232.Devlin, J., Chang, M.-W., Lee, K. and Toutanova, K. (2018) BERT: pre-training of deep bidirectional transformers for language understanding. arXiv, 1810.04805
- 233.Bepler, T. and Berger, B. (2021) Learning the protein language: evolution, structure, and function. Cell Syst. 12, 654–669.e653 10.1016/j.cels.2021.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Hutson, M. (2021) The language machines. Nature 591, 22–25 10.1038/d41586-021-00530-0 [DOI] [PubMed] [Google Scholar]
- 235.Kreutter, D., Schwaller, P. and Reymond, J.-L. (2021) Predicting enzymatic reactions with a molecular transformer. Chem. Sci. 12, 8648–8659 10.1039/D1SC02362D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Lin, T., Wang, Y., Liu, X. and Qiu, X. (2021) A survey of transformers. arXiv, 2106.04554
- 237.Singh, S. and Mahmood, A. (2021) The NLP cookbook: modern recipes for transformer based deep learning architectures. arXiv, 2104.10640
- 238.Shrivastava, A.D. and Kell, D.B. (2021) Fragnet, a contrastive learning-based transformer model for clustering, interpreting, visualising and navigating chemical space. Molecules 26, 2065 10.3390/molecules26072065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Shrivastava, A.D., Swainston, N., Samanta, S., Roberts, I., Wright Muelas, M. and Kell, D.B. (2021) MassGenie: a transformer-based deep learning method for identifying small molecules from their mass spectra. bioRxiv, 2021.2006.2025.449969 [DOI] [PMC free article] [PubMed]
- 240.Wu, Z., Johnston, K.E., Arnold, F.H. and Yang, K.K. (2021) Protein sequence design with deep generative models. Curr. Opin. Chem. Biol. 65, 18–27 10.1016/j.cbpa.2021.04.004 [DOI] [PubMed] [Google Scholar]
- 241.Repecka, D., Jauniskis, V., Karpus, L., Rembeza, E., Rokaitis, I., Zrimec, J.et al. (2021) Expanding functional protein sequence space using generative adversarial networks. Nat. Mach. Intell. 3, 324–333 10.1038/s42256-021-00310-5 [DOI] [Google Scholar]
- 242.LeCun, Y., Bengio, Y. and Hinton, G. (2015) Deep learning. Nature 521, 436–444 10.1038/nature14539 [DOI] [PubMed] [Google Scholar]
- 243.Schmidhuber, J. (2015) Deep learning in neural networks: an overview. Neural Netw. 61, 85–117 10.1016/j.neunet.2014.09.003 [DOI] [PubMed] [Google Scholar]
- 244.Elton, D.C., Boukouvalas, Z., Fuge, M.D. and Chung, P.W. (2019) Deep learning for molecular design: a review of the state of the art. Mol. Syst. Des. Eng. 4, 828–849 10.1039/C9ME00039A [DOI] [Google Scholar]
- 245.Gupta, A., Harrison, P.J., Wieslander, H., Pielawski, N., Kartasalo, K., Partel, G.et al. (2019) Deep learning in image cytometry: a review. Cytometry A 95, 366–380 10.1002/cyto.a.23701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Islam, M.M., Karray, F., Alhajj, R. and Zeng, J. (2020) A review on deep learning techniques for the diagnosis of novel coronavirus (COVID-19). arXiv, 2008.04815 [DOI] [PMC free article] [PubMed]
- 247.Langkvist, M., Karlsson, L. and Loutfi, A. (2014) A review of unsupervised feature learning and deep learning for time-series modeling. Pattern Recgnit. Lett. 42, 11–24 10.1016/j.patrec.2014.01.008 [DOI] [Google Scholar]
- 248.Minaee, S., Kalchbrenner, N., Cambria, E., Nikzad, N., Chenaghlu, M. and Gao, J. (2020) Deep learning based text classification: a comprehensive review. arXiv, 2004.03705
- 249.Paliwal, K., Lyons, J. and Heffernan, R. (2015) A short review of deep learning neural networks in protein structure prediction problems. Adv. Tech. Biol. Med. 3, 3 10.4172/2379-1764.1000139 [DOI] [Google Scholar]
- 250.Tripathi, N., Goshisht, M.K., Sahu, S.K. and Arora, C. (2021) Applications of artificial intelligence to drug design and discovery in the big data era: a comprehensive review. Mol. Divers 25, 1643–1664 10.1007/s11030-021-10237-z [DOI] [PubMed] [Google Scholar]
- 251.Zhang, H.-M. and Dong, B. (2020) A review on deep learning in medical image reconstruction. J. Oper. Res. Soc. China 8, 311–340 10.1007/s40305-019-00287-4 [DOI] [Google Scholar]
- 252.Zhou, S. K., Greenspan, H., Davatzikos, C., Duncan, J.S., Ginneken, B.V., Madabhushi, A.et al. (2020) A review of deep learning in medical imaging: Image traits, technology trends, case studies with progress highlights, and future promises. arXiv, 2008.09104
- 253.Le, N.Q.K., Ho, Q.T., Nguyen, T.T. and Ou, Y.Y. (2021) A transformer architecture based on BERT and 2D convolutional neural network to identify DNA enhancers from sequence information. Brief. Bioinform. 22, bbab005 10.1093/bib/bbab005 [DOI] [PubMed] [Google Scholar]
- 254.Song, B., Li, Z., Lin, X., Wang, J., Wang, T. and Fu, X. (2021) Pretraining model for biological sequence data. Brief. Funct. Genom. 20, 181–195 10.1093/bfgp/elab025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Wittmann, B.J., Johnston, K.E., Wu, Z. and Arnold, F.H. (2021) Advances in machine learning for directed evolution. Curr. Opin. Struct. Biol. 69, 11–18 10.1016/j.sbi.2021.01.008 [DOI] [PubMed] [Google Scholar]
- 256.Imanaka, T. and Aiba, S. (1981) A perspective on the application of genetic engineering: stability of recombinant plasmid. Ann. N Y Acad. Sci. 369, 1–14 10.1111/j.1749-6632.1981.tb14172.x [DOI] [PubMed] [Google Scholar]
- 257.Friehs, K. (2004) Plasmid copy number and plasmid stability. Adv. Biochem. Eng. Biotechnol. 86, 47–82 10.1007/b12440 [DOI] [PubMed] [Google Scholar]
- 258.Flagfeldt, D.B., Siewers, V., Huang, L. and Nielsen, J. (2009) Characterization of chromosomal integration sites for heterologous gene expression in Saccharomyces cerevisiae. Yeast 26, 545–551 10.1002/yea.1705 [DOI] [PubMed] [Google Scholar]
- 259.Scholz, S.A., Diao, R., Wolfe, M.B., Fivenson, E.M., Lin, X.N. and Freddolino, P.L. (2019) High-resolution mapping of the Escherichia coli chromosome reveals positions of high and low transcription. Cell Syst. 8, 212–225.e219 10.1016/j.cels.2019.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Bryant, J.A., Sellars, L.E., Busby, S.J. and Lee, D.J. (2014) Chromosome position effects on gene expression in Escherichia coli K-12. Nucleic Acids Res. 42, 11383–11392 10.1093/nar/gku828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Sauer, C., Syvertsson, S., Bohorquez, L.C., Cruz, R., Harwood, C.R., van Rij, T.et al. (2016) Effect of genome position on heterologous gene expression in Bacillus subtilis: an unbiased analysis. ACS Synth. Biol. 5, 942–947 10.1021/acssynbio.6b00065 [DOI] [PubMed] [Google Scholar]
- 262.Chaves, J.E., Wilton, R., Gao, Y., Munoz, N.M., Burnet, M.C., Schmitz, Z.et al. (2020) Evaluation of chromosomal insertion loci in the Pseudomonas putida KT2440 genome for predictable biosystems design. Metab. Eng. Commun. 11, e00139 10.1016/j.mec.2020.e00139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Biggs, B.W., Bedore, S.R., Arvay, E., Huang, S., Subramanian, H., McIntyre, E.A.et al. (2020) Development of a genetic toolset for the highly engineerable and metabolically versatile Acinetobacter baylyi ADP1. Nucleic Acids Res. 48, 5169–5182 10.1093/nar/gkaa167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Block, D.H., Hussein, R., Liang, L.W. and Lim, H.N. (2012) Regulatory consequences of gene translocation in bacteria. Nucleic Acids Res. 40, 8979–8992 10.1093/nar/gks694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Feng, X. and Marchisio, M.A. (2021) Novel S. cerevisiae hybrid synthetic promoters based on foreign core promoter sequences. Int. J. Mol. Sci. 22, 5704 10.3390/ijms22115704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Han, L., Cui, W., Suo, F., Miao, S., Hao, W., Chen, Q.et al. (2019) Development of a novel strategy for robust synthetic bacterial promoters based on a stepwise evolution targeting the spacer region of the core promoter in Bacillus subtilis. Microb. Cell Fact. 18, 96 10.1186/s12934-019-1148-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Liu, D., Mao, Z., Guo, J., Wei, L., Ma, H., Tang, Y.et al. (2018) Construction, model-based analysis, and characterization of a promoter library for fine-tuned gene expression in Bacillus subtilis. ACS Synth. Biol. 7, 1785–1797 10.1021/acssynbio.8b00115 [DOI] [PubMed] [Google Scholar]
- 268.Han, L., Chen, Q., Lin, Q., Cheng, J., Zhou, L., Liu, Z.et al. (2020) Realization of robust and precise regulation of gene expression by multiple sigma recognizable artificial promoters. Front. Bioeng. Biotechnol. 8, 92 10.3389/fbioe.2020.00092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Landberg, J., Mundhada, H. and Nielsen, A.T. (2020) An autoinducible trp-T7 expression system for production of proteins and biochemicals in Escherichia coli. Biotechnol. Bioeng. 117, 1513–1524 10.1002/bit.27297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Liu, X., Gupta, S.T.P., Bhimsaria, D., Reed, J.L., Rodríguez-Martinez, J.A., Ansari, A.Z.et al. (2019) De novo design of programmable inducible promoters. Nucleic Acids Res. 47, 10452–10463 10.1093/nar/gkz772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Wei, L., Xu, N., Wang, Y., Zhou, W., Han, G., Ma, Y.et al. (2018) Promoter library-based module combination (PLMC) technology for optimization of threonine biosynthesis in Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 102, 4117–4130 10.1007/s00253-018-8911-y [DOI] [PubMed] [Google Scholar]
- 272.Blazeck, J. and Alper, H.S. (2013) Promoter engineering: recent advances in controlling transcription at the most fundamental level. Biotechnol. J. 8, 46–58 10.1002/biot.201200120 [DOI] [PubMed] [Google Scholar]
- 273.Dehli, T., Solem, C. and Jensen, P.R. (2012) Tunable promoters in synthetic and systems biology. Subcell Biochem. 64, 181–201 10.1007/978-94-007-5055-5_9 [DOI] [PubMed] [Google Scholar]
- 274.Jin, L.Q., Jin, W.R., Ma, Z.C., Shen, Q., Cai, X., Liu, Z.Q.et al. (2019) Promoter engineering strategies for the overproduction of valuable metabolites in microbes. Appl. Microbiol. Biotechnol. 103, 8725–8736 10.1007/s00253-019-10172-y [DOI] [PubMed] [Google Scholar]
- 275.Xu, N., Wei, L. and Liu, J. (2019) Recent advances in the applications of promoter engineering for the optimization of metabolite biosynthesis. World J. Microbiol. Biotechnol. 35, 33 10.1007/s11274-019-2606-0 [DOI] [PubMed] [Google Scholar]
- 276.Tang, H., Wu, Y., Deng, J., Chen, N., Zheng, Z., Wei, Y.et al. (2020) Promoter architecture and promoter engineering in Saccharomyces cerevisiae. Metabolites 10, 320 10.3390/metabo10080320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Feng, X. and Marchisio, M.A. (2021) Saccharomyces cerevisiae promoter engineering before and during the synthetic biology era. Biology (Basel) 10, 504 10.3390/biology10060504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Helmann, J.D. (2019) Where to begin? Sigma factors and the selectivity of transcription initiation in bacteria. Mol. Microbiol. 112, 335–347 10.1111/mmi.14309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Bervoets, I. and Charlier, D. (2019) Diversity, versatility and complexity of bacterial gene regulation mechanisms: opportunities and drawbacks for applications in synthetic biology. FEMS Microbiol. Rev. 43, 304–339 10.1093/femsre/fuz001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Van Brempt, M., Clauwaert, J., Mey, F., Stock, M., Maertens, J., Waegeman, W.et al. (2020) Predictive design of sigma factor-specific promoters. Nat. Commun. 11, 5822 10.1038/s41467-020-19446-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Nikoomanzar, A., Chim, N., Yik, E.J. and Chaput, J.C. (2020) Engineering polymerases for applications in synthetic biology. Q. Rev. Biophys. 53, e8 10.1017/S0033583520000050 [DOI] [PubMed] [Google Scholar]
- 282.Ouaray, Z., Singh, I., Georgiadis, M.M. and Richards, N.G.J. (2020) Building better enzymes: molecular basis of improved non-natural nucleobase incorporation by an evolved DNA polymerase. Protein Sci. 29, 455–468 10.1002/pro.3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Cravens, A., Jamil, O.K., Kong, D., Sockolosky, J.T. and Smolke, C.D. (2021) Polymerase-guided base editing enables in vivo mutagenesis and rapid protein engineering. Nat. Commun. 12, 1579 10.1038/s41467-021-21876-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.O'Connor, N.J., Bordoy, A.E. and Chatterjee, A. (2021) Engineering transcriptional interference through RNA polymerase processivity control. ACS Synth. Biol. 10, 737–748 10.1021/acssynbio.0c00534 [DOI] [PubMed] [Google Scholar]
- 285.Saleski, T.E., Chung, M.T., Carruthers, D.N., Khasbaatar, A., Kurabayashi, K. and Lin, X.N. (2021) Optimized gene expression from bacterial chromosome by high-throughput integration and screening. Sci. Adv. 7, eabe1767 10.1126/sciadv.abe1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Cui, Z., Jiang, X., Zheng, H., Qi, Q. and Hou, J. (2019) Homology-independent genome integration enables rapid library construction for enzyme expression and pathway optimization in Yarrowia lipolytica. Biotechnol. Bioeng. 116, 354–363 10.1002/bit.26863 [DOI] [PubMed] [Google Scholar]
- 287.Nshogozabahizi, J.C., Aubrey, K.L., Ross, J.A. and Thakor, N. (2019) Applications and limitations of regulatory RNA elements in synthetic biology and biotechnology. J. Appl. Microbiol. 127, 968–984 10.1111/jam.14270 [DOI] [PubMed] [Google Scholar]
- 288.Tripathi, L., Zhang, Y. and Lin, Z. (2014) Bacterial sigma factors as targets for engineered or synthetic transcriptional control. Front. Bioeng. Biotechnol. 2, 33 10.3389/fbioe.2014.00033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.McKenna, R., Lombana, T.N., Yamada, M., Mukhyala, K. and Veeravalli, K. (2019) Engineered sigma factors increase full-length antibody expression in Escherichia coli. Metab. Eng. 52, 315–323 10.1016/j.ymben.2018.12.009 [DOI] [PubMed] [Google Scholar]
- 290.Todor, H., Osadnik, H., Campbell, E.A., Myers, K.S., Li, H., Donohue, T.J.et al. (2020) Rewiring the specificity of extracytoplasmic function sigma factors. Proc. Natl Acad. Sci. U.S.A. 117, 33496–33506 10.1073/pnas.2020204117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Srivastava, A., Varshney, R.K. and Shukla, P. (2021) Sigma factor modulation for cyanobacterial metabolic engineering. Trends Microbiol. 29, 266–277 10.1016/j.tim.2020.10.012 [DOI] [PubMed] [Google Scholar]
- 292.Curran, K.A., Morse, N.J., Markham, K.A., Wagman, A.M., Gupta, A. and Alper, H.S. (2015) Short synthetic terminators for improved heterologous gene expression in yeast. ACS Synth. Biol. 4, 824–832 10.1021/sb5003357 [DOI] [PubMed] [Google Scholar]
- 293.Chen, Y.J., Liu, P., Nielsen, A.A., Brophy, J.A., Clancy, K., Peterson, T.et al. (2013) Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat. Methods 10, 659–664 10.1038/nmeth.2515 [DOI] [PubMed] [Google Scholar]
- 294.Ito, Y., Terai, G., Ishigami, M., Hashiba, N., Nakamura, Y., Bamba, T.et al. (2020) Exchange of endogenous and heterogeneous yeast terminators in Pichia pastoris to tune mRNA stability and gene expression. Nucleic Acids Res. 48, 13000–13012 10.1093/nar/gkaa1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Cui, W., Lin, Q., Hu, R., Han, L., Cheng, Z., Zhang, L.et al. (2021) Data-driven and in silico-assisted design of broad host-range minimal intrinsic terminators adapted for bacteria. ACS Synth. Biol. 10, 1438–1450 10.1021/acssynbio.1c00050 [DOI] [PubMed] [Google Scholar]
- 296.Deaner, M. and Alper, H.S. (2018) Promoter and terminator discovery and engineering. Adv. Biochem. Eng. Biotechnol. 162, 21–44 10.1007/10_2016_8 [DOI] [PubMed] [Google Scholar]
- 297.Amarelle, V., Sanches-Medeiros, A., Silva-Rocha, R. and Guazzaroni, M.E. (2019) Expanding the toolbox of broad host-range transcriptional terminators for Proteobacteria through metagenomics. ACS Synth. Biol. 8, 647–654 10.1021/acssynbio.8b00507 [DOI] [PubMed] [Google Scholar]
- 298.Chu, D., Thompson, J. and von der Haar, T. (2014) Charting the dynamics of translation. Biosystems 119, 1–9 10.1016/j.biosystems.2014.02.005 [DOI] [PubMed] [Google Scholar]
- 299.Jossé, L., Singh, T. and von der Haar, T. (2019) Experimental determination of codon usage-dependent selective pressure on high copy-number genes in Saccharomyces cerevisiae. Yeast 36, 43–51 10.1002/yea.3373 [DOI] [PubMed] [Google Scholar]
- 300.Liu, Y. (2020) A code within the genetic code: codon usage regulates co-translational protein folding. Cell Commun. Signal. 18, 145 10.1186/s12964-020-00642-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Athey, J., Alexaki, A., Osipova, E., Rostovtsev, A., Santana-Quintero, L.V., Katneni, U.et al. (2017) A new and updated resource for codon usage tables. BMC Bioinform. 18, 391 10.1186/s12859-017-1793-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Salis, H.M. (2011) The ribosome binding site calculator. Methods Enzymol. 498, 19–42 10.1016/B978-0-12-385120-8.00002-4 [DOI] [PubMed] [Google Scholar]
- 303.Jervis, A.J., Carbonell, P., Vinaixa, M., Dunstan, M.S., Hollywood, K.A., Robinson, C.J.et al. (2019) Machine learning of designed translational control allows predictive pathway optimization in Escherichia coli. ACS Synth. Biol. 8, 127–136 10.1021/acssynbio.8b00398 [DOI] [PubMed] [Google Scholar]
- 304.Farasat, I., Kushwaha, M., Collens, J., Easterbrook, M., Guido, M. and Salis, H.M. (2014) Efficient search, mapping, and optimization of multi-protein genetic systems in diverse bacteria. Mol. Syst. Biol. 10, 731 10.15252/msb.20134955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Halper, S.M., Cetnar, D.P. and Salis, H.M. (2018) An automated pipeline for engineering many-enzyme pathways: computational sequence design, pathway expression-flux mapping, and scalable pathway optimization. Methods Mol. Biol. 1671, 39–61 10.1007/978-1-4939-7295-1_4 [DOI] [PubMed] [Google Scholar]
- 306.Höllerer, S., Papaxanthos, L., Gumpinger, A.C., Fischer, K., Beisel, C., Borgwardt, K.et al. (2020) Large-scale DNA-based phenotypic recording and deep learning enable highly accurate sequence-function mapping. Nat. Commun. 11, 3551 10.1038/s41467-020-17222-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Tarrant, D. and von der Haar, T. (2014) Synonymous codons, ribosome speed, and eukaryotic gene expression regulation. Cell Mol. Life Sci. 71, 4195–4206 10.1007/s00018-014-1684-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Vigar, J.R.J. and Wieden, H.J. (2017) Engineering bacterial translation initiation - do we have all the tools we need? Biochim. Biophys. Acta Gen. Subj. 1861, 3060–3069 10.1016/j.bbagen.2017.03.008 [DOI] [PubMed] [Google Scholar]
- 309.Rennig, M., Mundhada, H., Wordofa, G.G., Gerngross, D., Wulff, T., Worberg, A.et al. (2019) Industrializing a bacterial strain for l-serine production through translation initiation optimization. ACS Synth. Biol. 8, 2347–2358 10.1021/acssynbio.9b00169 [DOI] [PubMed] [Google Scholar]
- 310.Gu, L., Yuan, H., Lv, X., Li, G., Cong, R., Li, J.et al. (2020) High-yield and plasmid-free biocatalytic production of 5-methylpyrazine-2-carboxylic acid by combinatorial genetic elements engineering and genome engineering of Escherichia coli. Enzyme Microb. Technol. 134, 109488 10.1016/j.enzmictec.2019.109488 [DOI] [PubMed] [Google Scholar]
- 311.Yuan, H., Liu, Y., Li, J., Shin, H.D., Du, G., Shi, Z.et al. (2018) Combinatorial synthetic pathway fine-tuning and comparative transcriptomics for metabolic engineering of Raoultella ornithinolytica BF60 to efficiently synthesize 2,5-furandicarboxylic acid. Biotechnol Bioeng. 115, 2148–2155 10.1002/bit.26725 [DOI] [PubMed] [Google Scholar]
- 312.Katoh, T., Iwane, Y. and Suga, H. (2018) tRNA engineering for manipulating genetic code. RNA Biol. 15, 453–460 10.1080/15476286.2017.1343227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Perona, J.J. and Hadd, A. (2012) Structural diversity and protein engineering of the aminoacyl-tRNA synthetases. Biochemistry 51, 8705–8729 10.1021/bi301180x [DOI] [PubMed] [Google Scholar]
- 314.Alon, U. (2006) An Introduction to Systems Biology: Design Principles of Biological Circuits, Chapman and Hall/CRC, London, U.K. [Google Scholar]
- 315.Palsson, B.Ø. (2011) Systems Biology: Simulation of Dynamics Network States, Cambridge University Press, Cambridge, U.K. [Google Scholar]
- 316.Lerman, J.A., Hyduke, D.R., Latif, H., Portnoy, V.A., Lewis, N.E., Orth, J.D.et al. (2012) In silico method for modelling metabolism and gene product expression at genome scale. Nat. Commun. 3, 929 10.1038/ncomms1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Palsson, B.Ø. (2015) Systems Biology: Constraint-Based Reconstruction and Analysis, Cambridge University Press, Cambridge, U.K. [Google Scholar]
- 318.Ellis, M.A., Dalwadi, M.P., Ellis, M.J., Byrne, H.M. and Waters, S.L. (2021) A systematically reduced mathematical model for organoid expansion. Front. Bioeng. Biotechnol. 9, 670186 10.3389/fbioe.2021.670186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Hey, T. and Trefethen, A.E. (2005) Cyberinfrastructure for e-Science. Science 308, 817–821 10.1126/science.1110410 [DOI] [PubMed] [Google Scholar]
- 320.Hey, T. and Trefethen, A. (2003) e-Science and its implications. Philos. Trans. A Math. Phys. Eng. Sci. 361, 1809–1825 10.1098/rsta.2003.1224 [DOI] [PubMed] [Google Scholar]
- 321.Taylor, I.J., Deelman, E., Gannon, D.B. and Shields, M. (2007) Workflows for e-Science: Scientific Workflows for Grids, Springer, Guildford, U.K [Google Scholar]
- 322.Neethirajan, S. and Kemp, B. (2021) Digital twins in livestock farming. Animals (Basel) 11, 1008 10.3390/ani11041008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Stark, R., Fresemann, C. and Lindow, K. (2019) Development and operation of Digital Twins for technical systems and services. CIRP Annals 68, 129–132 10.1016/j.cirp.2019.04.024 [DOI] [Google Scholar]
- 324.Bergmann, F.T., Hoops, S., Klahn, B., Kummer, U., Mendes, P., Pahle, J.et al. (2017) COPASI and its applications in biotechnology. J. Biotechnol. 261, 215–220 10.1016/j.jbiotec.2017.06.1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Hoops, S., Sahle, S., Gauges, R., Lee, C., Pahle, J., Simus, N.et al. (2006) COPASI: a COmplex PAthway SImulator. Bioinformatics 22, 3067–3074 10.1093/bioinformatics/btl485 [DOI] [PubMed] [Google Scholar]
- 326.Funahashi, A., Tanimura, N., Morohashi, M. and Kitano, H. (2003) Celldesigner: a process diagram editor for gene-regulatory and biochemical networks. Biosilico 1, 159–162 10.1016/S1478-5382(03)02370-9 [DOI] [Google Scholar]
- 327.Funahashi, A., Matsuoka, Y., Jouraku, A., Morohashi, M., Kikuchi, N. and Kitano, H. (2008) Celldesigner 3.5: a versatile modeling tool for biochemical networks. Proc. IEEE 96, 1254–1265 10.1109/JPROC.2008.925458 [DOI] [Google Scholar]
- 328.Matsuoka, Y., Funahashi, A., Ghosh, S. and Kitano, H. (2014) Modeling and simulation using CellDesigner. Methods Mol. Biol. 1164, 121–145 10.1007/978-1-4939-0805-9_11 [DOI] [PubMed] [Google Scholar]
- 329.Balaur, I., Roy, L., Mazein, A., Karaca, S.G., Dogrusoz, U., Barillot, E.et al. (2020) Cd2sbgnml: bidirectional conversion between CellDesigner and SBGN formats. Bioinformatics 36, 2620–2622 10.1093/bioinformatics/btz969 [DOI] [PubMed] [Google Scholar]
- 330.Shannon, P., Markiel, A., Ozier, O., Baliga, N.S., Wang, J.T., Ramage, D.et al. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Pico, A.R., Bader, G.D., Demchak, B., Guitart Pla, O., Hull, T., Longabaugh, W.et al. (2014) The Cytoscape app article collection. F1000Research 3, 138 10.12688/f1000research.4642.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Demchak, B., Otasek, D., Pico, A.R., Bader, G.D., Ono, K., Settle, B.et al. (2018) The cytoscape automation app article collection. F1000Research 7, 800 10.12688/f1000research.15355.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Smallbone, K., Messiha, H.L., Carroll, K.M., Winder, C.L., Malys, N., Dunn, W.B.et al. (2013) A model of yeast glycolysis based on a consistent kinetic characterization of all its enzymes. FEBS Lett. 587, 2832–2841 10.1016/j.febslet.2013.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Liebermeister, W. and Klipp, E. (2006) Bringing metabolic networks to life: convenience rate law and thermodynamic constraints. Theor. Biol. Med. Model. 3, 41 10.1186/1742-4682-3-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Smallbone, K., Simeonidis, E., Broomhead, D.S. and Kell, D.B. (2007) Something from nothing: bridging the gap between constraint-based and kinetic modelling. FEBS J. 274, 5576–5585 10.1111/j.1742-4658.2007.06076.x [DOI] [PubMed] [Google Scholar]
- 336.Khodayari, A. and Maranas, C.D. (2016) A genome-scale Escherichia coli kinetic metabolic model k-ecoli457 satisfying flux data for multiple mutant strains. Nat. Commun. 7, 13806 10.1038/ncomms13806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Raman, K. and Chandra, N. (2009) Flux balance analysis of biological systems: applications and challenges. Brief. Bioinform. 10, 435–449 10.1093/bib/bbp011 [DOI] [PubMed] [Google Scholar]
- 338.Orth, J.D., Thiele, I. and Palsson, B.Ø. (2010) What is flux balance analysis? Nat. Biotechnol. 28, 245–248 10.1038/nbt.1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Gianchandani, E.P., Chavali, A.K. and Papin, J.A. (2010) The application of flux balance analysis in systems biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2, 372–382 10.1002/wsbm.60 [DOI] [PubMed] [Google Scholar]
- 340.Anand, S., Mukherjee, K. and Padmanabhan, P. (2020) An insight to flux-balance analysis for biochemical networks. Biotechnol. Genet. Eng. Rev. 36, 32–55 10.1080/02648725.2020.1847440 [DOI] [PubMed] [Google Scholar]
- 341.Dai, Z. and Locasale, J.W. (2017) Understanding metabolism with flux analysis: from theory to application. Metab. Eng. 43, 94–102 10.1016/j.ymben.2016.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Vanna, A. and Patsson, B.O. (1994) Metabolic flux balancing: basic concepts, scientific and practical use. Bio/Technology 12, 994–998 10.1038/nbt1094-994 [DOI] [Google Scholar]
- 343.Edwards, J.S., Ibarra, R.U. and Palsson, B.Ø. (2001) In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat. Biotechnol. 19, 125–130 10.1038/84379 [DOI] [PubMed] [Google Scholar]
- 344.Schuetz, R., Kuepfer, L. and Sauer, U. (2007) Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli. Mol. Syst. Biol. 3, 119 10.1038/msb4100162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Curran, K.A., Crook, N.C. and Alper, H.S. (2012) Using flux balance analysis to guide microbial metabolic engineering. Methods Mol. Biol. 834, 197–216 10.1007/978-1-61779-483-4_13 [DOI] [PubMed] [Google Scholar]
- 346.Sánchez, G., E, C. and Torres Sáez, R.G. (2014) Comparison and analysis of objective functions in flux balance analysis. Biotechnol. Prog. 30, 985–991 10.1002/btpr.1949 [DOI] [PubMed] [Google Scholar]
- 347.Heirendt, L., Arreckx, S., Pfau, T., Mendoza, S.N., Richelle, A., Heinken, A.et al. (2019) Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 14, 639–702 10.1038/s41596-018-0098-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Wang, X., Yu, L. and Chen, S. (2017) UP finder: a COBRA toolbox extension for identifying gene overexpression strategies for targeted overproduction. Metab. Eng. Commun. 5, 54–59 10.1016/j.meteno.2017.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Lakshmanan, M., Koh, G., Chung, B.K.S. and Lee, D.Y. (2014) Software applications for flux balance analysis. Briefings Bioinf. 15, 108–122 10.1093/bib/bbs069 [DOI] [PubMed] [Google Scholar]
- 350.Becker, S.A., Feist, A.M., Mo, M.L., Hannum, G., Palsson, B.O. and Herrgard, M.J. (2007) Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox. Nat. Protoc. 2, 727–738 10.1038/nprot.2007.99 [DOI] [PubMed] [Google Scholar]
- 351.Ebrahim, A., Lerman, J.A., Palsson, B.O. and Hyduke, D.R. (2013) COBRApy: constraints-based reconstruction and analysis for python. BMC Syst. Biol. 7, 74 10.1186/1752-0509-7-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Lloyd, C.J., Ebrahim, A., Yang, L., King, Z.A., Catoiu, E., O'Brien, E.J.et al. (2018) COBRAme: a computational framework for genome-scale models of metabolism and gene expression. PLoS Comput. Biol. 14, e1006302 10.1371/journal.pcbi.1006302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Schellenberger, J., Que, R., Fleming, R.M., Thiele, I., Orth, J.D., Feist, A.M.et al. (2011) Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat. Protoc. 6, 1290–1307 10.1038/nprot.2011.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Demir, E., Cary, M.P., Paley, S., Fukuda, K., Lemer, C., Vastrik, I.et al. (2010) The BioPAX community standard for pathway data sharing. Nat. Biotechnol. 28, 935–942 10.1038/nbt.1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Wright, D.W., Angus, T., Enright, A.J. and Freeman, T.C. (2014) Visualisation of BioPAX networks using BioLayout express (3D). F1000Research 3, 246 10.12688/f1000research.5499.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Benis, N., Schokker, D., Kramer, F., Smits, M.A. and Suarez-Diez, M. (2016) Building pathway graphs from BioPAX data in R. F1000Research 5, 2414 10.12688/f1000research.9582.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Agapito, G., Pastrello, C., Guzzi, P.H., Jurisica, I. and Cannataro, M. (2020) BioPAX-Parser: parsing and enrichment analysis of BioPAX pathways. Bioinformatics 36, 4377–4378 10.1093/bioinformatics/btaa529 [DOI] [PubMed] [Google Scholar]
- 358.Keating, S.M., Waltemath, D., Konig, M., Zhang, F., Drager, A., Chaouiya, C.et al. (2020) SBML level 3: an extensible format for the exchange and reuse of biological models. Mol. Syst. Biol. 16, e9110 10.15252/msb.20199110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Lee, D., Smallbone, K., Dunn, W.B., Murabito, E., Winder, C.L., Kell, D.B.et al. (2012) Improving metabolic flux predictions using absolute gene expression data. BMC Syst. Biol. 6, 73 10.1186/1752-0509-6-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Machado, D. and Herrgård, M. (2014) Systematic evaluation of methods for integration of transcriptomic data into constraint-based models of metabolism. PLoS Comput. Biol. 10, e1003580 10.1371/journal.pcbi.1003580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Jiménez-Osés, G., Osuna, S., Gao, X., Sawaya, M.R., Gilson, L., Collier, S.J.et al. (2014) The role of distant mutations and allosteric regulation on LovD active site dynamics. Nat. Chem. Biol. 10, 431–436 10.1038/nchembio.1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Osuna, S. (2020) The challenge of predicting distal active site mutations in computational enzyme design. Wires Comput. Mol. Sci. 11, e1502 10.1002/wcms.1502 [DOI] [Google Scholar]
- 363.Romero-Rivera, A., Garcia-Borràs, M. and Osuna, S. (2016) Computational tools for the evaluation of laboratory-engineered biocatalysts. Chem. Commun. 53, 284–297 10.1039/C6CC06055B [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Hestness, J., Narang, S., Ardalani, N., Diamos, G., Jun, H., Kianinejad, H.et al. (2017) Deep learning scaling is predictable, empirically. arXiv, 1712.00409
- 365.Kaplan, J., McCandlish, S., Henighan, T., Brown, T.B., Chess, B., Child, R.et al. (2020) Scaling laws for neural language models. arXiv, 2001.08361
- 366.Hernandez, D., Kaplan, J., Henighan, T. and McCandlish, S. (2021) Scaling laws for transfer. arXiv, 2102.01293
- 367.Jensen, P.R., Michelsen, O. and Westerhoff, H.V. (1995) Experimental determination of control by the H+-ATPase in Escherichia coli. J. Bioenerg. Biomembr. 27, 543–554 10.1007/BF02111653 [DOI] [PubMed] [Google Scholar]
- 368.Doudna, J.A. and Charpentier, E. (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096 10.1126/science.1258096 [DOI] [PubMed] [Google Scholar]
- 369.Liu, R., Liang, L., Freed, E.F. and Gill, R.T. (2021) Directed evolution of CRISPR/Cas systems for precise gene editing. Trends Biotechnol. 39, 262–273 10.1016/j.tibtech.2020.07.005 [DOI] [PubMed] [Google Scholar]
- 370.Bowman, E.K., Deaner, M., Cheng, J.F., Evans, R., Oberortner, E., Yoshikuni, Y.et al. (2020) Bidirectional titration of yeast gene expression using a pooled CRISPR guide RNA approach. Proc. Natl Acad. Sci. U.S.A. 117, 18424–18430 10.1073/pnas.2007413117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Jensen, P.R., Michelsen, O. and Westerhoff, H.V. (1993) Control analysis of the dependence of Escherichia coli physiology on the H+-ATPase. Proc. Natl Acad. Sci. U.S.A. 90, 8068–8072 10.1073/pnas.90.17.8068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Koebmann, B., Solem, C. and Jensen, P.R. (2005) Control analysis as a tool to understand the formation of the las operon in Lactococcus lactis. FEBS J. 272, 2292–2303 10.1111/j.1742-4658.2005.04656.x [DOI] [PubMed] [Google Scholar]
- 373.Solem, C., Koebmann, B., Yang, F. and Jensen, P.R. (2007) The las enzymes control pyruvate metabolism in Lactococcus lactis during growth on maltose. J. Bacteriol. 189, 6727–6730 10.1128/JB.00902-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Berkhout, J., Bosdriesz, E., Nikerel, E., Molenaar, D., de Ridder, D., Teusink, B.et al. (2013) How biochemical constraints of cellular growth shape evolutionary adaptations in metabolism. Genetics 194, 505–512 10.1534/genetics.113.150631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Keren, L., Hausser, J., Lotan-Pompan, M., Vainberg Slutskin, I., Alisar, H., Kaminski, S.et al. (2016) Massively parallel interrogation of the effects of gene expression levels on fitness. Cell 166, 1282–1294.e1218 10.1016/j.cell.2016.07.024 [DOI] [PubMed] [Google Scholar]
- 376.Wang, G., Björk, S.M., Huang, M., Liu, Q., Campbell, K., Nielsen, J.et al. (2019) RNAi expression tuning, microfluidic screening, and genome recombineering for improved protein production in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. U.S.A. 116, 9324–9332 10.1073/pnas.1820561116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Coussement, P., Bauwens, D., Maertens, J. and De Mey, M. (2017) Direct combinatorial pathway optimization. ACS Synth. Biol. 6, 224–232 10.1021/acssynbio.6b00122 [DOI] [PubMed] [Google Scholar]
- 378.Currin, A., Parker, S., Robinson, C.J., Takano, E., Scrutton, N.S. and Breitling, R. (2021) The evolving art of creating genetic diversity: From directed evolution to synthetic biology. Biotechnol. Adv. 50, 107762 10.1016/j.biotechadv.2021.107762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Blokesch, M. (2017) In and out-contribution of natural transformation to the shuffling of large genomic regions. Curr. Opin. Microbiol. 38, 22–29 10.1016/j.mib.2017.04.001 [DOI] [PubMed] [Google Scholar]
- 380.Cui, W., Han, L., Suo, F., Liu, Z., Zhou, L. and Zhou, Z. (2018) Exploitation of Bacillus subtilis as a robust workhorse for production of heterologous proteins and beyond. World J. Microbiol. Biotechnol. 34, 145 10.1007/s11274-018-2531-7 [DOI] [PubMed] [Google Scholar]
- 381.Straume, D., Stamsas, G.A. and Håvarstein, L.S. (2015) Natural transformation and genome evolution in Streptococcus pneumoniae. Infect. Genet. Evol. 33, 371–380 10.1016/j.meegid.2014.10.020 [DOI] [PubMed] [Google Scholar]
- 382.Jiang, X., Palazzotto, E., Wybraniec, E., Munro, L.J., Zhang, H., Kell, D.B.et al. (2020) Automating cloning by natural transformation. ACS Synth. Biol. 9, 3228–3235 10.1021/acssynbio.0c00240 [DOI] [PubMed] [Google Scholar]
- 383.Dalia, A.B., McDonough, E. and Camilli, A. (2014) Multiplex genome editing by natural transformation. Proc. Natl Acad. Sci. U.S.A. 111, 8937–8942 10.1073/pnas.1406478111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Dalia, A.B. (2018) Natural cotransformation and multiplex genome editing by natural transformation (MuGENT) of Vibrio cholerae. Methods Mol. Biol. 1839, 53–64 10.1007/978-1-4939-8685-9_6 [DOI] [PubMed] [Google Scholar]
- 385.Dalia, T.N., Hayes, C.A., Stolyar, S., Marx, C.J., McKinlay, J.B. and Dalia, A.B. (2017) Multiplex genome editing by natural transformation (MuGENT) for synthetic biology in Vibrio natriegens. ACS Synth. Biol. 6, 1650–1655 10.1021/acssynbio.7b00116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Simpson, C.A., Podicheti, R., Rusch, D.B., Dalia, A.B. and van Kessel, J.C. (2019) Diversity in natural transformation frequencies and regulation across Vibrio species. mBio 10, e02788–19 10.1128/mBio.02788-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Ren, J., Na, D. and Yoo, S.M. (2018) Optimization of chemico-physical transformation methods for various bacterial species using diverse chemical compounds and nanomaterials. J. Biotechnol. 288, 55–60 10.1016/j.jbiotec.2018.11.003 [DOI] [PubMed] [Google Scholar]
- 388.Kawai, S., Hashimoto, W. and Murata, K. (2010) Transformation of Saccharomyces cerevisiae and other fungi: methods and possible underlying mechanism. Bioeng. Bugs. 1, 395–403 10.4161/bbug.1.6.13257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Konishi, T. and Harata, M. (2014) Improvement of the transformation efficiency of Saccharomyces cerevisiae by altering carbon sources in pre-culture. Biosci. Biotechnol. Biochem. 78, 1090–1093 10.1080/09168451.2014.915730 [DOI] [PubMed] [Google Scholar]
- 390.Leong, C.G., Boyd, C.M., Roush, K.S., Tenente, R., Lang, K.M. and Lostroh, C.P. (2017) Succinate, iron chelation, and monovalent cations affect the transformation efficiency of Acinetobacter baylyi ATCC 33305 during growth in complex media. Can. J. Microbiol. 63, 851–856 10.1139/cjm-2017-0393 [DOI] [PubMed] [Google Scholar]
- 391.Calero, P. and Nikel, P.I. (2019) Chasing bacterial chassis for metabolic engineering: a perspective review from classical to non-traditional microorganisms. Microb. Biotechnol. 12, 98–124 10.1111/1751-7915.13292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Yu, S.C., Dawson, A., Henderson, A.C., Lockyer, E.J., Read, E., Sritharan, G.et al. (2016) Nutrient supplements boost yeast transformation efficiency. Sci. Rep. 6, 35738 10.1038/srep35738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Yu, S.C., Kuemmel, F., Skoufou-Papoutsaki, M.N. and Spanu, P.D. (2019) Yeast transformation efficiency is enhanced by TORC1- and eisosome-dependent signaling. Microbiologyopen 8, e00730 10.1002/mbo3.730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J.A. and Charpentier, E. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Knott, G.J. and Doudna, J.A. (2018) CRISPR-Cas guides the future of genetic engineering. Science 361, 866–869 10.1126/science.aat5011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Bao, Z., HamediRad, M., Xue, P., Xiao, H., Tasan, I., Chao, R.et al. (2018) Genome-scale engineering of Saccharomyces cerevisiae with single-nucleotide precision. Nat. Biotechnol. 36, 505–508 10.1038/nbt.4132 [DOI] [PubMed] [Google Scholar]
- 397.Deaner, M. and Alper, H.S. (2019) Enhanced scale and scope of genome engineering and regulation using CRISPR/Cas in Saccharomyces cerevisiae. FEMS Yeast Res 19, foz076 10.1093/femsyr/foz076 [DOI] [PubMed] [Google Scholar]
- 398.Hanna, R.E. and Doench, J.G. (2020) Design and analysis of CRISPR-Cas experiments. Nat. Biotechnol. 38, 813–823 10.1038/s41587-020-0490-7 [DOI] [PubMed] [Google Scholar]
- 399.Jessop-Fabre, M.M., Jakociunas, T., Stovicek, V., Dai, Z.J., Jensen, M.K., Keasling, J.D.et al. (2016) EasyClone-MarkerFree: a vector toolkit for marker-less integration of genes into Saccharomyces cerevisiae via CRISPR-Cas9. Biotechnol. J. 11, 1110–1117 10.1002/biot.201600147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Milne, N., Tramontin, L.R.R. and Borodina, I. (2020) A teaching protocol demonstrating the use of easyClone and CRISPR/Cas9 for metabolic engineering of Saccharomyces cerevisiae and Yarrowia lipolytica. FEMS Yeast Res. 20, foz062 10.1093/femsyr/foz062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Ronda, C., Maury, J., Jakociunas, T., Jacobsen, S.A., Germann, S.M., Harrison, S.J.et al. (2015) Credit: CRISPR mediated multi-loci gene integration in Saccharomyces cerevisiae. Microb. Cell Fact. 14, 97 10.1186/s12934-015-0288-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Molla, K.A. and Yang, Y. (2019) CRISPR/Cas-mediated base editing: technical considerations and practical applications. Trends Biotechnol. 37, 1121–1142 10.1016/j.tibtech.2019.03.008 [DOI] [PubMed] [Google Scholar]
- 403.Stovicek, V., Holkenbrink, C. and Borodina, I. (2017) CRISPR/Cas system for yeast genome engineering: advances and applications. FEMS Yeast Res. 17, fox030 10.1093/femsyr/fox030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Tarasava, K., Oh, E.J., Eckert, C.A. and Gill, R.T. (2018) CRISPR-enabled tools for engineering microbial genomes and phenotypes. Biotechnol. J. 13, e1700586 10.1002/biot.201700586 [DOI] [PubMed] [Google Scholar]
- 405.Modrzejewski, D., Hartung, F., Lehnert, H., Sprink, T., Kohl, C., Keilwagen, J.et al. (2020) Which factors affect the occurrence of off-target effects caused by the use of CRISPR/Cas: a systematic review in plants. Front. Plant. Sci. 11, 574959 10.3389/fpls.2020.574959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Lee, H.K., Oh, Y., Hong, J., Lee, S.H. and Hur, J.K. (2021) Development of CRISPR technology for precise single-base genome editing: a brief review. BMB Rep. 54, 98–105 10.5483/BMBRep.2021.54.2.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.McGlincy, N.J., Meacham, Z.A., Reynaud, K.K., Muller, R., Baum, R. and Ingolia, N.T. (2021) A genome-scale CRISPR interference guide library enables comprehensive phenotypic profiling in yeast. BMC Genom. 22, 205 10.1186/s12864-021-07518-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Anzalone, A.V., Koblan, L.W. and Liu, D.R. (2020) Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844 10.1038/s41587-020-0561-9 [DOI] [PubMed] [Google Scholar]
- 409.Replogle, J.M., Norman, T.M., Xu, A., Hussmann, J.A., Chen, J., Cogan, J.Z.et al. (2020) Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nat. Biotechnol. 38, 954–961 10.1038/s41587-020-0470-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Jakočiūnas, T., Bonde, I., Herrgård, M., Harrison, S.J., Kristensen, M., Pedersen, L.E.et al. (2015) Multiplex metabolic pathway engineering using CRISPR/Cas9 in Saccharomyces cerevisiae. Metab. Eng. 28, 213–222 10.1016/j.ymben.2015.01.008 [DOI] [PubMed] [Google Scholar]
- 411.Garst, A.D., Bassalo, M.C., Pines, G., Lynch, S.A., Halweg-Edwards, A.L., Liu, R.et al. (2017) Genome-wide mapping of mutations at single-nucleotide resolution for protein, metabolic and genome engineering. Nat. Biotechnol. 35, 48–55 10.1038/nbt.3718 [DOI] [PubMed] [Google Scholar]
- 412.Bassalo, M.C., Garst, A.D., Choudhury, A., Grau, W.C., Oh, E.J., Spindler, E.et al. (2018) Deep scanning lysine metabolism in Escherichia coli. Mol. Syst. Biol. 14, e8371 10.15252/msb.20188371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Liu, R., Liang, L., Choudhury, A., Bassalo, M.C., Garst, A.D., Tarasava, K.et al. (2018) Iterative genome editing of Escherichia coli for 3-hydroxypropionic acid production. Metab. Eng. 47, 303–313 10.1016/j.ymben.2018.04.007 [DOI] [PubMed] [Google Scholar]
- 414.Yokota, A. and Ikeda, M. (2017) Amino Acid Fermentation, Springer, Tokyo, Japan [Google Scholar]
- 415.Butelli, E., Titta, L., Giorgio, M., Mock, H.P., Matros, A., Peterek, S.et al. (2008) Enrichment of tomato fruit with health-promoting anthocyanins by expression of select transcription factors. Nat. Biotechnol. 26, 1301–1308 10.1038/nbt.1506 [DOI] [PubMed] [Google Scholar]
- 416.Zhang, Y., Butelli, E., Alseekh, S., Tohge, T., Rallapalli, G., Luo, J.et al. (2015) Multi-level engineering facilitates the production of phenylpropanoid compounds in tomato. Nat. Commun. 6, 8635 10.1038/ncomms9635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Appelhagen, I., Wulff-Vester, A.K., Wendell, M., Hvoslef-Eide, A.K., Russell, J., Oertel, A.et al. (2018) Colour bio-factories: towards scale-up production of anthocyanins in plant cell cultures. Metab. Eng. 48, 218–232 10.1016/j.ymben.2018.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Butelli, E., Licciardello, C., Ramadugu, C., Durand-Hulak, M., Celant, A., Reforgiato Recupero, G.et al. (2019) Noemi controls production of flavonoid pigments and fruit acidity and illustrates the domestication routes of modern citrus varieties. Curr. Biol. 29, 158–164.e152 10.1016/j.cub.2018.11.040 [DOI] [PubMed] [Google Scholar]
- 419.Browning, D.F. and Busby, S.J.W. (2016) Local and global regulation of transcription initiation in bacteria. Nat. Rev. Microbiol. 14, 638–650 10.1038/nrmicro.2016.103 [DOI] [PubMed] [Google Scholar]
- 420.Browning, D.F., Butala, M. and Busby, S.J.W. (2019) Bacterial transcription factors: regulation by Pick ‘N’ Mix. J. Mol. Biol. 431, 4067–4077 10.1016/j.jmb.2019.04.011 [DOI] [PubMed] [Google Scholar]
- 421.Busby, S.J.W. (2019) Transcription activation in bacteria: ancient and modern. Microbiology (Reading) 165, 386–395 10.1099/mic.0.000783 [DOI] [PubMed] [Google Scholar]
- 422.Fang, X., Sastry, A., Mih, N., Kim, D., Tan, J., Yurkovich, J.T.et al. (2017) Global transcriptional regulatory network for Escherichia coli robustly connects gene expression to transcription factor activities. Proc. Natl Acad. Sci. U.S.A. 114, 10286–10291 10.1073/pnas.1702581114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Mejia-Almonte, C., Busby, S.J.W., Wade, J.T., van Helden, J., Arkin, A.P., Stormo, G.D.et al. (2020) Redefining fundamental concepts of transcription initiation in bacteria. Nat. Rev. Genet. 21, 699–714 10.1038/s41576-020-0254-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Lempp, M., Farke, N., Kuntz, M., Freibert, S.A., Lill, R. and Link, H. (2019) Systematic identification of metabolites controlling gene expression in E. coli. Nat. Commun. 10, 4463 10.1038/s41467-019-12474-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Santos-Zavaleta, A., Sanchez-Perez, M., Salgado, H., Velazquez-Ramirez, D.A., Gama-Castro, S., Tierrafria, V.H.et al. (2018) A unified resource for transcriptional regulation in Escherichia coli K-12 incorporating high-throughput-generated binding data into RegulonDB version 10.0. BMC Biol. 16, 91 10.1186/s12915-018-0555-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Santos-Zavaleta, A., Salgado, H., Gama-Castro, S., Sánchez-Pérez, M., Gómez-Romero, L., Ledezma-Tejeida, D.et al. (2019) RegulonDB v 10.5: tackling challenges to unify classic and high throughput knowledge of gene regulation in E. coli K-12. Nucleic Acids Res. 47, D212–D220 10.1093/nar/gky1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Sastry, A.V., Gao, Y., Szubin, R., Hefner, Y., Xu, S., Kim, D.et al. (2019) The Escherichia coli transcriptome mostly consists of independently regulated modules. Nat. Commun. 10, 5536 10.1038/s41467-019-13483-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Sastry, A.V., Hu, A., Heckmann, D., Poudel, S., Kavvas, E. and Palsson, B.O. (2021) Independent component analysis recovers consistent regulatory signals from disparate datasets. PLoS Comput. Biol. 17, e1008647 10.1371/journal.pcbi.1008647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Lastiri-Pancardo, G., Mercado-Hernandez, J.S., Kim, J., Jimenez, J.I. and Utrilla, J. (2020) A quantitative method for proteome reallocation using minimal regulatory interventions. Nat. Chem. Biol. 16, 1026–1033 10.1038/s41589-020-0593-y [DOI] [PubMed] [Google Scholar]
- 430.Yokoyama, K., Ishijima, S.A., Clowney, L., Koike, H., Aramaki, H., Tanaka, C.et al. (2006) Feast/famine regulatory proteins (FFRPs): Escherichia coli Lrp, AsnC and related archaeal transcription factors. FEMS Microbiol. Rev. 30, 89–108 10.1111/j.1574-6976.2005.00005.x [DOI] [PubMed] [Google Scholar]
- 431.Kawashima, T., Aramaki, H., Oyamada, T., Makino, K., Yamada, M., Okamura, H.et al. (2008) Transcription regulation by feast/famine regulatory proteins, FFRPs, in archaea and eubacteria. Biol. Pharm. Bull. 31, 173–186 10.1248/bpb.31.173 [DOI] [PubMed] [Google Scholar]
- 432.Dossani, Z.Y., Reider Apel, A., Szmidt-Middleton, H., Hillson, N.J., Deutsch, S., Keasling, J.D.et al. (2018) A combinatorial approach to synthetic transcription factor-promoter combinations for yeast strain engineering. Yeast 35, 273–280 10.1002/yea.3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Naseri, G., Behrend, J., Rieper, L. and Mueller-Roeber, B. (2019) COMPASS for rapid combinatorial optimization of biochemical pathways based on artificial transcription factors. Nat. Commun. 10, 2615 10.1038/s41467-019-10224-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Li, J.W., Zhang, X.Y., Wu, H. and Bai, Y.P. (2020) Transcription factor engineering for high-throughput strain evolution and organic acid bioproduction: a review. Front. Bioeng. Biotechnol. 8, 98 10.3389/fbioe.2020.00098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Mahr, R. and Frunzke, J. (2016) Transcription factor-based biosensors in biotechnology: current state and future prospects. Appl. Microbiol. Biotechnol. 100, 79–90 10.1007/s00253-015-7090-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Skjoedt, M.L., Snoek, T., Kildegaard, K.R., Arsovska, D., Eichenberger, M., Goedecke, T.J.et al. (2016) Engineering prokaryotic transcriptional activators as metabolite biosensors in yeast. Nat. Chem. Biol. 12, 951–958 10.1038/nchembio.2177 [DOI] [PubMed] [Google Scholar]
- 437.Lin, J.L., Wagner, J.M. and Alper, H.S. (2017) Enabling tools for high-throughput detection of metabolites: metabolic engineering and directed evolution applications. Biotechnol. Adv. 35, 950–970 10.1016/j.biotechadv.2017.07.005 [DOI] [PubMed] [Google Scholar]
- 438.Cheng, F., Tang, X.L. and Kardashliev, T. (2018) Transcription factor-based biosensors in high-throughput screening: advances and applications. Biotechnol. J. 13, e1700648 10.1002/biot.201700648 [DOI] [PubMed] [Google Scholar]
- 439.Kasey, C.M., Zerrad, M., Li, Y., Cropp, T.A. and Williams, G.J. (2018) Development of transcription factor-based designer macrolide biosensors for metabolic engineering and synthetic biology. ACS Synth. Biol. 7, 227–239 10.1021/acssynbio.7b00287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Boada, Y., Vignoni, A., Picó, J. and Carbonell, P. (2020) Extended metabolic biosensor design for dynamic pathway regulation of cell factories. iScience 23, 101305 10.1016/j.isci.2020.101305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Snoek, T., Chaberski, E.K., Ambri, F., Kol, S., Bjørn, S.P., Pang, B.et al. (2020) Evolution-guided engineering of small-molecule biosensors. Nucleic Acids Res. 48, e3 10.1093/nar/gkz954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Mitchler, M.M., Garcia, J.M., Montero, N.E. and Williams, G.J. (2021) Transcription factor-based biosensors: a molecular-guided approach for natural product engineering. Curr. Opin. Biotechnol. 69, 172–181 10.1016/j.copbio.2021.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Ding, N., Zhou, S. and Deng, Y. (2021) Transcription-factor-based biosensor engineering for applications in synthetic biology. ACS Synth. Biol. 5, 911–922 10.1021/acssynbio.0c00252 [DOI] [PubMed] [Google Scholar]
- 444.Sonntag, C.K., Flachbart, L.K., Maass, C., Vogt, M. and Marienhagen, J. (2020) A unified design allows fine-tuning of biosensor parameters and application across bacterial species. Metab. Eng. Commun. 11, e00150 10.1016/j.mec.2020.e00150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Ottenheim, C., Nawrath, M. and Wu, J.C. (2018) Microbial mutagenesis by atmospheric and room-temperature plasma (ARTP): the latest development. Bioresour. Bioprocess. 5, 12 10.1186/s40643-018-0200-1 [DOI] [Google Scholar]
- 446.Yu, Q., Li, Y., Wu, B., Hu, W., He, M. and Hu, G. (2020) Novel mutagenesis and screening technologies for food microorganisms: advances and prospects. Appl. Microbiol. Biotechnol. 104, 1517–1531 10.1007/s00253-019-10341-z [DOI] [PubMed] [Google Scholar]
- 447.Altenburg, E. (1933) The production of mutations by ultra-violet light. Science 78, 587 10.1126/science.78.2034.587 [DOI] [PubMed] [Google Scholar]
- 448.Auerbach, C. and Robson, J.M. (1946) Chemical production of mutations. Nature 157, 302 10.1038/157302a0 [DOI] [PubMed] [Google Scholar]
- 449.Li, H.-P., Wang, Z.-B., Ge, N., Le, P.-S., Wu, H., Lu, Y.et al. (2012) Studies on the physical characteristics of the radio-frequency atmospheric-pressure glow discharge plasmas for the genome mutation of Methylosinus trichosporium. IEEE Trans Plasma Sci. 40, 2853–2860 10.1109/TPS.2012.2213274 [DOI] [Google Scholar]
- 450.Zhang, X., Zhang, C., Zhou, Q.Q., Zhang, X.F., Wang, L.Y., Chang, H.B.et al. (2015) Quantitative evaluation of DNA damage and mutation rate by atmospheric and room-temperature plasma (ARTP) and conventional mutagenesis. Appl. Microbiol. Biotechnol. 99, 5639–5646 10.1007/s00253-015-6678-y [DOI] [PubMed] [Google Scholar]
- 451.Luhe, A.L., Tan, L., Wu, J. and Zhao, H. (2011) Increase of ethanol tolerance of Saccharomyces cerevisiae by error-prone whole genome amplification. Biotechnol. Lett. 33, 1007–1011 10.1007/s10529-011-0518-7 [DOI] [PubMed] [Google Scholar]
- 452.Van den Bergh, B., Swings, T., Fauvart, M. and Michiels, J. (2018) Experimental design, population dynamics, and diversity in microbial experimental evolution. Microbiol. Mol. Biol. Rev. 82, e00008-00018 10.1128/MMBR.00008-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Phaneuf, P.V., Gosting, D., Palsson, B.O. and Feist, A.M. (2019) ALEdb 1.0: a database of mutations from adaptive laboratory evolution experimentation. Nucleic Acids Res. 47, D1164–D1171 10.1093/nar/gky983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Mohamed, E.T., Wang, S., Lennen, R.M., Herrgard, M.J., Simmons, B.A., Singer, S.W.et al. (2017) Generation of a platform strain for ionic liquid tolerance using adaptive laboratory evolution. Microb. Cell Fact. 16, 204 10.1186/s12934-017-0819-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Mohamed, E.T., Mundhada, H., Landberg, J., Cann, I., Mackie, R.I., Nielsen, A.T.et al. (2019) Generation of an E. coli platform strain for improved sucrose utilization using adaptive laboratory evolution. Microb. Cell Fact. 18, 116 10.1186/s12934-019-1165-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Dunlop, M.J., Dossani, Z.Y., Szmidt, H.L., Chu, H.C., Lee, T.S., Keasling, J.D.et al. (2011) Engineering microbial biofuel tolerance and export using efflux pumps. Mol. Syst. Biol. 7, 487 10.1038/msb.2011.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Mukhopadhyay, A. (2015) Tolerance engineering in bacteria for the production of advanced biofuels and chemicals. Trends Microbiol. 23, 498–508 10.1016/j.tim.2015.04.008 [DOI] [PubMed] [Google Scholar]
- 458.Peabody, G.L. and Kao, K.C. (2016) Recent progress in biobutanol tolerance in microbial systems with an emphasis on Clostridium. FEMS Microbiol. Lett. 363, fnw017 10.1093/femsle/fnw017 [DOI] [PubMed] [Google Scholar]
- 459.Weber, C., Farwick, A., Benisch, F., Brat, D., Dietz, H., Subtil, T.et al. (2010) Trends and challenges in the microbial production of lignocellulosic bioalcohol fuels. Appl. Microbiol. Biotechnol. 87, 1303–1315 10.1007/s00253-010-2707-z [DOI] [PubMed] [Google Scholar]
- 460.Jönsson, L.J., Alriksson, B. and Nilvebrant, N.O. (2013) Bioconversion of lignocellulose: inhibitors and detoxification. Biotechnol. Biofuels 6, 16 10.1186/1754-6834-6-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Liu, Z., Radi, M., Mohamed, E.T.T., Feist, A.M., Dragone, G. and Mussatto, S.I. (2021) Adaptive laboratory evolution of Rhodosporidium toruloides to inhibitors derived from lignocellulosic biomass and genetic variations behind evolution. Bioresour. Technol. 333, 125171 10.1016/j.biortech.2021.125171 [DOI] [PubMed] [Google Scholar]
- 462.Luo, H., Hansen, A.S.L., Yang, L., Schneider, K., Kristensen, M., Christensen, U.et al. (2019) Coupling S-adenosylmethionine-dependent methylation to growth: design and uses. PLoS Biol. 17, e2007050 10.1371/journal.pbio.2007050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Pontrelli, S., Fricke, R.C.B., Sakurai, S.S.M., Putri, S.P., Fitz-Gibbon, S., Chung, M.et al. (2018) Directed strain evolution restructures metabolism for 1-butanol production in minimal media. Metab. Eng. 49, 153–163 10.1016/j.ymben.2018.08.004 [DOI] [PubMed] [Google Scholar]
- 464.Sandberg, T.E., Salazar, M.J., Weng, L.L., Palsson, B.O. and Feist, A.M. (2019) The emergence of adaptive laboratory evolution as an efficient tool for biological discovery and industrial biotechnology. Metab. Eng. 56, 1–16 10.1016/j.ymben.2019.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Tan, Z.L., Zheng, X., Wu, Y., Jian, X., Xing, X. and Zhang, C. (2019) In vivo continuous evolution of metabolic pathways for chemical production. Microb. Cell Fact. 18, 82 10.1186/s12934-019-1132-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Markx, G.H., Davey, C.L. and Kell, D.B. (1991) The permittistat: a novel type of turbidostat. J. Gen. Microbiol. 137, 735–743 10.1099/00221287-137-4-735 [DOI] [Google Scholar]
- 467.Davey, H.M., Davey, C.L., Woodward, A.M., Edmonds, A.N., Lee, A.W. and Kell, D.B. (1996) Oscillatory, stochastic and chaotic growth rate fluctuations in permittistatically-controlled yeast cultures. Biosystems 39, 43–61 10.1016/0303-2647(95)01577-9 [DOI] [PubMed] [Google Scholar]
- 468.Delneri, D., Leong, H.S., Hayes, A., Davey, H.M., Kell, D.B. and Oliver, S.G. (2003) Assessing contributions to fitness of individual genes via genome-wide competition analysis. Yeast 20, S337–S337 [Google Scholar]
- 469.Dragosits, M. and Mattanovich, D. (2013) Adaptive laboratory evolution – principles and applications for biotechnology. Microb. Cell Fact. 12, 64 10.1186/1475-2859-12-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Miller, A.W., Befort, C., Kerr, E.O. and Dunham, M.J. (2013) Design and use of multiplexed chemostat arrays. J. Vis. Exp. 72, e50262 10.3791/50262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Guarino, A., Shannon, B., Marucci, L., Grierson, C., Savery, N. and Bernardo, M. (2019) A low-cost, open-source turbidostat design for in-vivo control experiments in synthetic biology. IFAC Papers Online 52, 244–248 10.1016/j.ifacol.2019.12.265 [DOI] [Google Scholar]
- 472.Steel, H., Habgood, R., Kelly, C.L. and Papachristodoulou, A. (2020) In situ characterisation and manipulation of biological systems with Chi.Bio. PLoS Biol. 18, e3000794 10.1371/journal.pbio.3000794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Drake, J.W. (1991) A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl Acad. Sci. U.S.A. 88, 7160–7164 10.1073/pnas.88.16.7160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Cox, E.C. and Gibson, T.C. (1974) Selection for high mutation rates in chemostats. Genetics 77, 169–184 10.1093/genetics/77.2.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Giraud, A., Matic, I., Tenaillon, O., Clara, A., Radman, M., Fons, M.et al. (2001) Costs and benefits of high mutation rates: adaptive evolution of bacteria in the mouse gut. Science 291, 2606–2608 10.1126/science.1056421 [DOI] [PubMed] [Google Scholar]
- 476.Luan, G., Cai, Z., Li, Y. and Ma, Y. (2013) Genome replication engineering assisted continuous evolution (GREACE) to improve microbial tolerance for biofuels production. Biotechnol. Biofuels 6, 137 10.1186/1754-6834-6-137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Luan, G., Bao, G., Lin, Z., Li, Y., Chen, Z., Li, Y.et al. (2015) Comparative genome analysis of a thermotolerant Escherichia coli obtained by Genome Replication Engineering Assisted Continuous Evolution (GREACE) and its parent strain provides new understanding of microbial heat tolerance. N. Biotechnol. 32, 732–738 10.1016/j.nbt.2015.01.013 [DOI] [PubMed] [Google Scholar]
- 478.Wang, X., Li, Q., Sun, C., Cai, Z., Zheng, X., Guo, X.et al. (2019) GREACE-assisted adaptive laboratory evolution in endpoint fermentation broth enhances lysine production by Escherichia coli. Microb. Cell Fact. 18, 106 10.1186/s12934-019-1153-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Xu, X., Liu, C., Niu, C., Wang, J., Zheng, F., Li, Y.et al. (2018) Rationally designed perturbation factor drives evolution in Saccharomyces cerevisiae for industrial application. J. Ind. Microbiol. Biotechnol. 45, 869–880 10.1007/s10295-018-2057-x [DOI] [PubMed] [Google Scholar]
- 480.Hughes, S.R., Bang, S.S., Cox, E.J., Schoepke, A., Ochwat, K., Pinkelman, R.et al. (2013) Automated UV-C mutagenesis of Kluyveromyces marxianus NRRL Y-1109 and selection for microaerophilic growth and ethanol production at elevated temperature on biomass sugars. J. Lab. Autom. 18, 276–290 10.1177/2211068213480037 [DOI] [PubMed] [Google Scholar]
- 481.Lindquist, M.R., López-Núñez, J.C., Jones, M.A., Cox, E.J., Pinkelman, R.J., Bang, S.S.et al. (2015) Irradiation of Yarrowia lipolytica NRRL YB-567 creating novel strains with enhanced ammonia and oil production on protein and carbohydrate substrates. Appl. Microbiol. Biotechnol. 99, 9723–9743 10.1007/s00253-015-6852-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Guardini, Z., Dall'Osto, L., Barera, S., Jaberi, M., Cazzaniga, S., Vitulo, N.et al. (2021) High carotenoid mutants of Chlorella vulgaris show enhanced biomass yield under high irradiance. Plants (Basel) 10, 911 10.3390/plants10050911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Gaona, I.JÁ., Assof, M.V., Jofré, V.P., Combina, M. and Ciklic, I.F. (2021) Mutagenesis, screening and isolation of Brettanomyces bruxellensis mutants with reduced 4-ethylphenol production. World J. Microbiol. Biotechnol. 37, 6 10.1007/s11274-020-02981-5 [DOI] [PubMed] [Google Scholar]
- 484.Katre, G., Ajmera, N., Zinjarde, S. and RaviKumar, A. (2017) Mutants of Yarrowia lipolytica NCIM 3589 grown on waste cooking oil as a biofactory for biodiesel production. Microb. Cell Fact. 16, 176 10.1186/s12934-017-0790-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Yamazaki, H., Kobayashi, S., Ebina, S., Abe, S., Ara, S., Shida, Y.et al. (2019) Highly selective isolation and characterization of Lipomyces starkeyi mutants with increased production of triacylglycerol. Appl. Microbiol. Biotechnol. 103, 6297–6308 10.1007/s00253-019-09936-3 [DOI] [PubMed] [Google Scholar]
- 486.Wu, B., Qin, H., Yang, Y., Duan, G., Yang, S., Xin, F.et al. (2019) Engineered Zymomonas mobilis tolerant to acetic acid and low pH via multiplex atmospheric and room temperature plasma mutagenesis. Biotechnol. Biofuels 12, 10 10.1186/s13068-018-1348-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.An, J., Gao, F., Ma, Q., Xiang, Y., Ren, D. and Lu, J. (2017) Screening for enhanced astaxanthin accumulation among Spirulina platensis mutants generated by atmospheric and room temperature plasmas. Algal Res. 25, 464–472 10.1016/j.algal.2017.06.006 [DOI] [Google Scholar]
- 488.Wang, Y., Li, Q., Zheng, P., Guo, Y., Wang, L., Zhang, T.et al. (2016) Evolving the L-lysine high-producing strain of Escherichia coli using a newly developed high-throughput screening method. J. Ind. Microbiol. Biotechnol. 43, 1227–1235 10.1007/s10295-016-1803-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Li, J., Guo, S., Hua, Q. and Hu, F. (2021) Improved AP-3 production through combined ARTP mutagenesis, fermentation optimization, and subsequent genome shuffling. Biotechnol. Lett. 43, 1143–1154 10.1007/s10529-020-03034-5 [DOI] [PubMed] [Google Scholar]
- 490.Ye, L., Zhao, H., Li, Z. and Wu, J.C. (2013) Improved acid tolerance of Lactobacillus pentosus by error-prone whole genome amplification. Bioresour. Technol. 135, 459–463 10.1016/j.biortech.2012.10.042 [DOI] [PubMed] [Google Scholar]
- 491.Huang, S., Xue, T., Wang, Z., Ma, Y., He, X., Hong, J.et al. (2018) Furfural-tolerant Zymomonas mobilis derived from error-prone PCR-based whole genome shuffling and their tolerant mechanism. Appl. Microbiol. Biotechnol. 102, 3337–3347 10.1007/s00253-018-8817-8 [DOI] [PubMed] [Google Scholar]
- 492.He, X., Xue, T., Ma, Y., Zhang, J., Wang, Z., Hong, J.et al. (2019) Identification of functional butanol-tolerant genes from Escherichia coli mutants derived from error-prone PCR-based whole-genome shuffling. Biotechnol. Biofuels 12, 73 10.1186/s13068-019-1405-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Godara, A. and Kao, K.C. (2021) Adaptive laboratory evolution of beta-caryophyllene producing Saccharomyces cerevisiae. Microb. Cell Fact. 20, 106 10.1186/s12934-021-01598-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Prell, C., Busche, T., Rückert, C., Nolte, L., Brandenbusch, C. and Wendisch, V.F. (2021) Adaptive laboratory evolution accelerated glutarate production by Corynebacterium glutamicum. Microb. Cell Fact. 20, 97 10.1186/s12934-021-01586-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Belkhelfa, S., Roche, D., Dubois, I., Berger, A., Delmas, V.A., Cattolico, L.et al. (2019) Continuous culture adaptation of Methylobacterium extorquens AM1 and TK 0001 to very high methanol concentrations. Front. Microbiol. 10, 1313 10.3389/fmicb.2019.01313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Gleizer, S., Ben-Nissan, R., Bar-On, Y.M., Antonovsky, N., Noor, E., Zohar, Y.et al. (2019) Conversion of Escherichia coli to generate all biomass carbon from CO2. Cell 179, 1255–1263 10.1016/j.cell.2019.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Qin, W., Zhao, J., Yu, X., Liu, X., Chu, X., Tian, J.et al. (2019) Improving cadmium resistance in Escherichia coli through continuous genome evolution. Front. Microbiol. 10, 278 10.3389/fmicb.2019.00278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Groeneveld, P., Stouthamer, A.H. and Westerhoff, H.V. (2009) Super life: how and why ‘cell selection’ leads to the fastest-growing eukaryote. FEBS J. 276, 254–270 10.1111/j.1742-4658.2008.06778.x [DOI] [PubMed] [Google Scholar]
- 499.Mundhada, H., Schneider, K., Christensen, H.B. and Nielsen, A.T. (2016) Engineering of high yield production of L-serine in Escherichia coli. Biotechnol. Bioeng. 113, 807–816 10.1002/bit.25844 [DOI] [PubMed] [Google Scholar]
- 500.César-Razquin, A., Snijder, B., Frappier-Brinton, T., Isserlin, R., Gyimesi, G., Bai, X.et al. (2015) A call for systematic research on solute carriers. Cell 162, 478–487 10.1016/j.cell.2015.07.022 [DOI] [PubMed] [Google Scholar]
- 501.Kell, D.B. and Welch, G.R. (2018) Belief: the baggage behind our being. OSF preprints, pnxcs https://osf.io/pnxcs/
- 502.Dobson, P.D. and Kell, D.B. (2008) Carrier-mediated cellular uptake of pharmaceutical drugs: an exception or the rule? Nat. Rev. Drug Disc. 7, 205–220 10.1038/nrd2438 [DOI] [PubMed] [Google Scholar]
- 503.Kell, D.B., Dobson, P.D., Bilsland, E. and Oliver, S.G. (2013) The promiscuous binding of pharmaceutical drugs and their transporter-mediated uptake into cells: what we (need to) know and how we can do so. Drug Disc. Today. 18, 218–239 10.1016/j.drudis.2012.11.008 [DOI] [PubMed] [Google Scholar]
- 504.Kell, D.B. and Oliver, S.G. (2014) How drugs get into cells: tested and testable predictions to help discriminate between transporter-mediated uptake and lipoidal bilayer diffusion. Front. Pharmacol. 5, 231 10.3389/fphar.2014.00231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Jones, C.M., Hernández Lozada, N.J. and Pfleger, B.F. (2015) Efflux systems in bacteria and their metabolic engineering applications. Appl. Microbiol. Biotechnol. 99, 9381–9393 10.1007/s00253-015-6963-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Kell, D.B. (2016) How drugs pass through biological cell membranes – a paradigm shift in our understanding? Beilstein Mag. 2, 5 http://www.beilstein-institut.de/download/628/609_kell.pdf [Google Scholar]
- 507.Kell, D.B. (2018) Control of metabolite efflux in microbial cell factories: current advances and future prospects. OSF preprints, xg9jh, https://osf.io/7t8gm/#!
- 508.Kell, D.B. (2021) The transporter-mediated cellular uptake and efflux of pharmaceutical drugs and biotechnology products: how and why phospholipid bilayer transport is negligible in real biomembranes. Molecules 26, 5629 10.3390/molecules26185629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Salcedo-Sora, J.E., Robison, A.T.R., Zaengle-Barone, J., Franz, K.J. and Kell, D.B. (2021) Membrane transporters involved in the antimicrobial activities of pyrithione in Escherichia coli. Molecules 26, 5826 10.3390/molecules26195826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Benga, G. (2003) Birth of water channel proteins-the aquaporins. Cell Biol. Int. 27, 701–709 10.1016/S1065-6995(03)00171-9 [DOI] [PubMed] [Google Scholar]
- 511.Agre, P. (2004) Aquaporin water channels (Nobel lecture). Angew. Chem. Int. Ed. Engl. 43, 4278–4290 10.1002/anie.200460804 [DOI] [PubMed] [Google Scholar]
- 512.Claus, S., Jenkins Sánchez, L. and Van Bogaert, I.N.A. (2021) The role of transport proteins in the production of microbial glycolipid biosurfactants. Appl. Microbiol. Biotechnol. 105, 1779–1793 10.1007/s00253-021-11156-7 [DOI] [PubMed] [Google Scholar]
- 513.López, S., M, J. and Van Bogaert, I.N.A. (2021) Microbial fatty acid transport proteins and their biotechnological potential. Biotechnol. Bioeng. 118, 2184–2201 10.1002/bit.27735 [DOI] [PubMed] [Google Scholar]
- 514.Giaever, G., Shoemaker, D.D., Jones, T.W., Liang, H., Winzeler, E.A., Astromoff, A.et al. (1999) Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat. Genet. 21, 278–283 10.1038/6791 [DOI] [PubMed] [Google Scholar]
- 515.Cubillos, F.A., Louis, E.J. and Liti, G. (2009) Generation of a large set of genetically tractable haploid and diploid saccharomyces strains. FEMS Yeast Res. 9, 1217–1225 10.1111/j.1567-1364.2009.00583.x [DOI] [PubMed] [Google Scholar]
- 516.Davey, H.M., Cross, E.J.M., Davey, C.L., Gkargkas, K., Delneri, D., Hoyle, D.C.et al. (2012) Genome-wide analysis of longevity in nutrient-deprived Saccharomyces cerevisiae reveals the importance of recycling in maintaining cell viability. Environ. Microbiol. 14, 1249–1260 10.1111/j.1462-2920.2012.02705.x [DOI] [PubMed] [Google Scholar]
- 517.Norris, M., Lovell, S. and Delneri, D. (2013) Characterization and prediction of haploinsufficiency using systems-level gene properties in yeast. G3 3, 1965–1977 10.1534/g3.113.008144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Lanthaler, K., Bilsland, E., Dobson, P., Moss, H.J., Pir, P., Kell, D.B.et al. (2011) Genome-wide assessment of the carriers involved in the cellular uptake of drugs: a model system in yeast. BMC Biol. 9, 70 10.1186/1741-7007-9-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Bürckstümmer, T., Banning, C., Hainzl, P., Schobesberger, R., Kerzendorfer, C., Pauler, F.M.et al. (2013) A reversible gene trap collection empowers haploid genetics in human cells. Nat. Methods 10, 965–971 10.1038/nmeth.2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Winter, G.E., Radic, B., Mayor-Ruiz, C., Blomen, V.A., Trefzer, C., Kandasamy, R.K.et al. (2014) The solute carrier SLC35F2 enables YM155-mediated DNA damage toxicity. Nat. Chem. Biol. 10, 768–773 10.1038/nchembio.1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Blomen, V.A., Májek, P., Jae, L.T., Bigenzahn, J.W., Nieuwenhuis, J., Staring, J.et al. (2015) Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096 10.1126/science.aac7557 [DOI] [PubMed] [Google Scholar]
- 522.Gibson, B., Geertman, J.A., Hittinger, C.T., Krogerus, K., Libkind, D., Louis, E.J.et al. (2017) New yeasts-new brews: modern approaches to brewing yeast design and development. FEMS Yeast Res. 17, fox038 10.1093/femsyr/fox038 [DOI] [PubMed] [Google Scholar]
- 523.Hovhannisyan, H., Saus, E., Ksiezopolska, E., Hinks Roberts, A.J., Louis, E.J. and Gabaldon, T. (2020) Integrative omics analysis reveals a limited transcriptional shock after yeast interspecies hybridization. Front. Genet. 11, 404 10.3389/fgene.2020.00404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Kell, D.B., Kaprelyants, A.S. and Grafen, A. (1995) On pheromones, social behaviour and the functions of secondary metabolism in bacteria. Trends Ecol. Evol. 10, 126–129 10.1016/S0169-5347(00)89013-8 [DOI] [PubMed] [Google Scholar]
- 525.Li, S., Jendresen, C.B., Landberg, J., Pedersen, L.E., Sonnenschein, N., Jensen, S.I.et al. (2020) Genome-wide CRISPRi-based identification of targets for decoupling growth from production. ACS Synth. Biol. 9, 1030–1040 10.1021/acssynbio.9b00143 [DOI] [PubMed] [Google Scholar]
- 526.Schramm, T., Lempp, M., Beuter, D., Sierra, S.G., Glatter, T. and Link, H. (2020) High-throughput enrichment of temperature-sensitive argininosuccinate synthetase for two-stage citrulline production in E. coli. Metab. Eng. 60, 14–24 10.1016/j.ymben.2020.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Kaprelyants, A.S., Gottschal, J.C. and Kell, D.B. (1993) Dormancy in non-sporulating bacteria. FEMS Microbiol. Rev. 10, 271–286 10.1111/j.1574-6968.1993.tb05871.x [DOI] [PubMed] [Google Scholar]
- 528.Kell, D.B., Kaprelyants, A.S., Weichart, D.H., Harwood, C.L. and Barer, M.R. (1998) Viability and activity in readily culturable bacteria: a review and discussion of the practical issues. Antonie van Leeuwenhoek 73, 169–187 10.1023/A:1000664013047 [DOI] [PubMed] [Google Scholar]
- 529.Beuter, D., Gomes-Filho, J.V., Randau, L., Díaz-Pascual, F., Drescher, K. and Link, H. (2018) Selective enrichment of slow-growing bacteria in a metabolism-wide CRISPRi library with a TIMER protein. ACS Synth. Biol. 7, 2775–2782 10.1021/acssynbio.8b00379 [DOI] [PubMed] [Google Scholar]
- 530.Lempp, M., Lubrano, P., Bange, G. and Link, H. (2020) Metabolism of non-growing bacteria. Biol. Chem. 401, 1479–1485 10.1515/hsz-2020-0201 [DOI] [PubMed] [Google Scholar]
- 531.Lee, H.H., Ostrov, N., Wong, B.G., Gold, M.A., Khalil, A.S. and Church, G.M. (2016) Vibrio natriegens, a new genomic powerhouse. bioRxiv, 058487
- 532.Weinstock, M.T., Hesek, E.D., Wilson, C.M. and Gibson, D.G. (2016) Vibrio natriegens as a fast-growing host for molecular biology. Nat. Methods 13, 849–851 10.1038/nmeth.3970 [DOI] [PubMed] [Google Scholar]
- 533.Lee, H.H., Ostrov, N., Gold, M.A. and Church, G.M. (2017) Recombineering in Vibrio natriegens. bioRxiv, 130088
- 534.Maida, I., Bosi, E., Perrin, E., Papaleo, M.C., Orlandini, V., Fondi, M.et al. (2013) Draft genome sequence of the fast-growing bacterium Vibrio natriegens strain DSMZ 759. Genome Announc. 1, e00648–13 10.1128/genomeA.00648-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Wang, Z., Lin, B., Hervey, W.J.T. and Vora, G.J. (2013) Draft genome sequence of the fast-growing marine bacterium Vibrio natriegens strain ATCC 14048. Genome Announc. 1, e00589-13 10.1128/genomeA.00589-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Becker, W., Wimberger, F. and Zangger, K. (2019) Vibrio natriegens: an alternative expression system for the high-yield production of isotopically labeled proteins. Biochemistry 58, 2799–2803 10.1021/acs.biochem.9b00403 [DOI] [PubMed] [Google Scholar]
- 537.Lee, H.H., Ostrov, N., Wong, B.G., Gold, M.A., Khalil, A.S. and Church, G.M. (2019) Functional genomics of the rapidly replicating bacterium Vibrio natriegens by CRISPRi. Nat. Microbiol. 4, 1105–1113 10.1038/s41564-019-0423-8 [DOI] [PubMed] [Google Scholar]
- 538.Wang, Z., Tschirhart, T., Schultzhaus, Z., Kelly, E.E., Chen, A., Oh, E.et al. (2020) Melanin produced by the fast-growing marine bacterium Vibrio natriegens through heterologous biosynthesis: characterization and application. Appl. Environ. Microbiol. 86, e02749-19 10.1128/AEM.02749-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Wu, F., Chen, W., Peng, Y., Tu, R., Lin, Y., Xing, J.et al. (2020) Design and reconstruction of regulatory parts for fast-frowing (sic) Vibrio natriegens synthetic biology. ACS Synth. Biol. 9, 2399–2409 10.1021/acssynbio.0c00158 [DOI] [PubMed] [Google Scholar]
- 540.Xu, J., Dong, F., Wu, M., Tao, R., Yang, J., Wu, M.et al. (2021) Vibrio natriegens as a pET-compatible expression host complementary to Escherichia coli. Front. Microbiol. 12, 627181 10.3389/fmicb.2021.627181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Pei, L. and Schmidt, M. (2018) Fast-growing engineered microbes: new concerns for gain-of-function research? Front. Genet. 9, 207 10.3389/fgene.2018.00207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Munson, R.J. (1970) Turbidostats. In Methods in Microbiology (Norris, J. R. and Ribbons, D. W., eds), vol. 2, pp. 349–376, Academic Press, Cambridge, MA [Google Scholar]
- 543.Watson, T.G. (1972) The present status and future prospects of the turbidostat. J. Appl. Chem. Biotechnol. 22, 229–243 10.1002/jctb.5020220206 [DOI] [Google Scholar]
- 544.Hoffmann, S.A., Wohltat, C., Muller, K.M. and Arndt, K.M. (2017) A user-friendly, low-cost turbidostat with versatile growth rate estimation based on an extended Kalman filter. PLoS One 12, e0181923 10.1371/journal.pone.0181923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Lane, M.M. and Morrissey, J.P. (2010) Kluyveromyces marxianus: a yeast emerging from its sister's shadow. Fungal Biol. Rev. 24, 17–26 10.1016/j.fbr.2010.01.001 [DOI] [Google Scholar]
- 546.Pir, P., Gutteridge, A., Wu, J., Rash, B., Kell, D.B., Zhang, N.et al. (2012) The genetic control of growth rate: a systems biology study in yeast. BMC Syst. Biol. 6, 4 10.1186/1752-0509-6-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547.Mo, W., Wang, M., Zhan, R., Yu, Y., He, Y. and Lu, H. (2019) Kluyveromyces marxianus developing ethanol tolerance during adaptive evolution with significant improvements of multiple pathways. Biotechnol. Biofuels 12, 63 10.1186/s13068-019-1393-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Fraleigh, S.P., Bungay, H.R. and Clesceri, L.S. (1990) Aerobic formation of ethanol by Saccharomyces cerevisiae in a computerized pHauxostat. J. Biotechnol. 13, 61–72 10.1016/0168-1656(90)90131-T [DOI] [Google Scholar]
- 549.Oltmann, L.F., Schoenmaker, G.S., Reunders, W.N.M. and Southamer, A.H. (1978) Modification of the pHauxostat culture method for the mass cultivation of bacteria. Biotech. Bioeng. 20, 921–925 10.1002/bit.260200613 [DOI] [PubMed] [Google Scholar]
- 550.von Schulthess, R., Bungay, H.R. and Fraleigh, S.P. (1990) Competition in a pHauxostat. Biotech. Lett. 12, 93–98 10.1007/BF01022422 [DOI] [Google Scholar]
- 551.Dikicioglu, D., Pir, P. and Oliver, S.G. (2013) Predicting complex phenotype-genotype interactions to enable yeast engineering: Saccharomyces cerevisiae as a model organism and a cell factory. Biotechnol. J. 8, 1017–1034 10.1002/biot.201300138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Avrahami-Moyal, L., Engelberg, D., Wenger, J.W., Sherlock, G. and Braun, S. (2012) Turbidostat culture of Saccharomyces cerevisiae W303-1A under selective pressure elicited by ethanol selects for mutations in SSD1 and UTH1. FEMS Yeast Res. 12, 521–533 10.1111/j.1567-1364.2012.00803.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Ullman, G., Wallden, M., Marklund, E.G.. Mahmutovic, A., Razinkov, I. and Elf, J. (2013) High-throughput gene expression analysis at the level of single proteins using a microfluidic turbidostat and automated cell tracking. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20120025 10.1098/rstb.2012.0025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554.Hans, S., Gimpel, M., Glauche, F., Neubauer, P. and Cruz-Bournazou, M.N. (2018) Automated cell treatment for competence and transformation of Escherichia coli in a high-throughput quasi-turbidostat using microtiter plates. Microorganisms 6, 60 10.3390/microorganisms6030060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.McGeachy, A.M., Meacham, Z.A. and Ingolia, N.T. (2019) An accessible continuous-culture turbidostat for pooled analysis of complex libraries. ACS Synth. Biol. 8, 844–856 10.1021/acssynbio.8b00529 [DOI] [PubMed] [Google Scholar]
- 556.Regenberg, B., Grotkjaer, T., Winther, O., Fausboll, A., Akesson, M., Bro, C.et al. (2006) Growth-rate regulated genes have profound impact on interpretation of transcriptome profiling in Saccharomyces cerevisiae. Genome Biol. 7, R107 10.1186/gb-2006-7-11-r107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Ramanathan, A. and Schreiber, S.L. (2007) Multilevel regulation of growth rate in yeast revealed using systems biology. J. Biol. 6, 3 10.1186/jbiol56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558.Gutteridge, A., Pir, P., Castrillo, J.I., Charles, P.D., Lilley, K.S. and Oliver, S.G. (2010) Nutrient control of eukaryote cell growth: a systems biology study in yeast. BMC Biol. 8, 68 10.1186/1741-7007-8-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Link, H. and Weuster-Botz, D. (2011) Medium formulation and development. In Comprehensive Biotechnology (Moo-Young, M., ed.), pp. 119–134, Elsevier, Amsterdam, the Netherlands [Google Scholar]
- 560.Andreini, C., Bertini, I., Cavallaro, G., Holliday, G.L. and Thornton, J.M. (2008) Metal ions in biological catalysis: from enzyme databases to general principles. J. Biol. Inorg. Chem. 13, 1205–1218 10.1007/s00775-008-0404-5 [DOI] [PubMed] [Google Scholar]
- 561.Andreini, C., Bertini, I., Cavallaro, G., Holliday, G.L. and Thornton, J.M. (2009) Metal-MACiE: a database of metals involved in biological catalysis. Bioinformatics 25, 2088–2089 10.1093/bioinformatics/btp256 [DOI] [PubMed] [Google Scholar]
- 562.Putignano, V., Rosato, A., Banci, L. and Andreini, C. (2018) MetalPDB in 2018: a database of metal sites in biological macromolecular structures. Nucleic Acids Res. 46, D459–D464 10.1093/nar/gkx989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.Chen, Y., Li, F., Mao, J., Chen, Y. and Nielsen, J. (2021) Yeast optimizes metal utilization based on metabolic network and enzyme kinetics. Proc. Natl Acad. Sci. U.S.A. 118, e2020154118 10.1073/pnas.2020154118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564.Weuster-Botz, D. and Wandrey, C. (1995) Medium optimization by genetic algorithm for continuous production of formate dehydrogenase. Process Biochem. 30, 563–571 10.1016/0032-9592(94)00036-0 [DOI] [Google Scholar]
- 565.Zitzler, E. (1999) Evolutionary Algorithms for Multiobjective Optimization: Methods and Applications, Shaker Verlag, Aachen, Germany [Google Scholar]
- 566.Michalewicz, Z. and Fogel, D.B. (2000) How to Solve it: Modern Heuristics, Springer-Verlag, Heidelberg, Germany [Google Scholar]
- 567.Corne, D., Dorigo, M. and Glover, F. (1999) New Ideas in Optimization, McGraw Hill, London, U.K [Google Scholar]
- 568.Sarma, M.V.R.K., Sahai, V. and Bisaria, V.S. (2009) Genetic algorithm-based medium optimization for enhanced production of fluorescent pseudomonad R81 and siderophore. Biochem. Eng. J. 47, 100–108 10.1016/j.bej.2009.07.010 [DOI] [Google Scholar]
- 569.Davey, H.M. and Kell, D.B. (1996) Flow cytometry and cell sorting of heterogeneous microbial populations: the importance of single-cell analysis. Microbiol. Rev. 60, 641–696 10.1128/mr.60.4.641-696.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Elowitz, M.B., Levine, A.J., Siggia, E.D. and Swain, P.S. (2002) Stochastic gene expression in a single cell. Science 297, 1183–1186 10.1126/science.1070919 [DOI] [PubMed] [Google Scholar]
- 571.Nordholt, N., van Heerden, J., Kort, R. and Bruggeman, F.J. (2017) Effects of growth rate and promoter activity on single-cell protein expression. Sci. Rep. 7, 6299 10.1038/s41598-017-05871-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572.Perkel, J.M. (2021) Single-cell analysis enters the multiomics age. Nature 595, 614–616 10.1038/d41586-021-01994-w34282340 [DOI] [Google Scholar]
- 573.Elmentaite, R., Kumasaka, N., Roberts, K., Fleming, A., Dann, E., King, H.W.et al. (2021) Cells of the human intestinal tract mapped across space and time. Nature 597, 250–255 10.1038/s41586-021-03852-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Go, C.D., Knight, J.D.R., Rajasekharan, A., Rathod, B., Hesketh, G.G., Abe, K.T.et al. (2021) A proximity-dependent biotinylation map of a human cell. Nature 595, 120–124 10.1038/s41586-021-03592-2 [DOI] [PubMed] [Google Scholar]
- 575.Bondeson, D.P., Mares, A., Smith, I.E.. Ko, E., Campos, S., Miah, A.H.et al. (2015) Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol, 611–617 10.1038/nchembio.1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.Cecchini, C., Pannilunghi, S., Tardy, S. and Scapozza, L. (2021) From conception to development: investigating PROTACs features for improved cell permeability and successful protein degradation. Front. Chem. 9, 672267 10.3389/fchem.2021.672267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Maniaci, C., Hughes, S.J., Testa, A., Chen, W., Lamont, D.J., Rocha, S.et al. (2017) Homo-PROTACs: bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat. Commun. 8, 830 10.1038/s41467-017-00954-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Neklesa, T.K., Winkler, J.D. and Crews, C.M. (2017) Targeted protein degradation by PROTACs. Pharmacol. Ther. 174, 138–144 10.1016/j.pharmthera.2017.02.027 [DOI] [PubMed] [Google Scholar]
- 579.Nowak, R.P. and Jones, L.H. (2021) Target validation using PROTACs: applying the four pillars framework. SLAS Discov. 26, 474–483 10.1177/2472555220979584 [DOI] [PubMed] [Google Scholar]
- 580.Toure, M. and Crews, C.M. (2016) Small-molecule PROTACS: new approaches to protein degradation. Angew. Chem. Int. Ed. Engl. 55, 1966–1973 10.1002/anie.201507978 [DOI] [PubMed] [Google Scholar]
- 581.Szymkuć, S., Gajewska, E.P., Klucznik, T., Molga, K., Dittwald, P., Startek, M.et al. (2016) Computer-assisted synthetic planning: the end of the beginning. Angew. Chem. Int. Ed. Engl. 55, 5904–5937 10.1002/anie.201506101 [DOI] [PubMed] [Google Scholar]
- 582.Grzybowski, B.A., Szymkuć, S., Gajewska, E.P., Molga, K., Dittwald, P., Wolos, A.et al. (2018) Chematica: a story of computer code that started to think like a chemist. Chem-Us 4, 390–397 10.1016/j.chempr.2018.02.024 [DOI] [Google Scholar]
- 583.Mikulak-Klucznik, B., Golebiowska, P., Bayly, A.A., Popik, O., Klucznik, T., Szymkuc, S.et al. (2020) Computational planning of the synthesis of complex natural products. Nature 588, 83–88 10.1038/s41586-020-2855-y [DOI] [PubMed] [Google Scholar]
- 584.Pflüger, P.M. and Glorius, F. (2020) Molecular machine learning: the future of synthetic chemistry? Angew. Chem. Int. Ed. Engl. 59, 18860–18865 10.1002/anie.202008366 [DOI] [PubMed] [Google Scholar]
- 585.Wilbraham, L., Mehr, S.H.M. and Cronin, L. (2021) Digitizing chemistry using the chemical processing unit: from synthesis to discovery. Acc. Chem. Res. 54, 253–262 10.1021/acs.accounts.0c00674 [DOI] [PubMed] [Google Scholar]
- 586.Delépine, B., Duigou, T., Carbonell, P. and Faulon, J.L. (2018) Retropath2.0: a retrosynthesis workflow for metabolic engineers. Metab. Eng. 45, 158–170 10.1016/j.ymben.2017.12.002 [DOI] [PubMed] [Google Scholar]
- 587.Koch, M., Duigou, T. and Faulon, J.L. (2020) Reinforcement learning for bioretrosynthesis. ACS Synth. Biol. 9, 157–168 10.1021/acssynbio.9b00447 [DOI] [PubMed] [Google Scholar]
- 588.Khemchandani, Y., O'Hagan, S., Samanta, S., Swainston, N., Roberts, T.J., Bollegala, D.et al. (2020) Deepgraphmolgen, a multiobjective, computational strategy for generating molecules with desirable properties: a graph convolution and reinforcement learning approach. J. Cheminform. 12, 53 10.1186/s13321-020-00454-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.Samanta, S., O'Hagan, S., Swainston, N., Roberts, T.J. and Kell, D.B. (2020) VAE-Sim: a novel molecular similarity measure based on a variational autoencoder. Molecules 25, 3446 10.3390/molecules25153446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Holzinger, A., Biemann, C., Pattichis, C.S. and Kell, D.B. (2017) What do we need to build explainable AI systems for the medical domain? arXiv, 1712.09923v09921
- 591.Samek, W., Montavon, G., Vedaldi, A., Hansen, L.K. and Müller, K.-R. (2019) Explainable AI: Interpreting, Explaining and Visualizing Deep Learning, Springer, Berlin, Germany [Google Scholar]
- 592.Arrieta, A.B., Díaz-Rodríguez, N., Del Ser, J., Bennetot, A., Tabik, S., Barbado, A.et al. (2020) Explainable artificial intelligence (XAI): concepts, taxonomies, opportunities and challenges toward responsible AI. Inform. Fusion. 58, 82–115 10.1016/j.inffus.2019.12.012 [DOI] [Google Scholar]
- 593.Rothman, D. (2020) Hands-on Explainable AI (XAI) with Python, Packt, Birmingham, U.K [Google Scholar]
- 594.Singh, A., Sengupta, S. and Lakshminarayanan, V. (2020) Explainable deep learning models in medical image analysis. arXiv, 2005.13799
- 595.Porwol, L., Kowalski, D.J., Henson, A., Long, D.L., Bell, N.L. and Cronin, L. (2020) An autonomous chemical robot discovers the rules of inorganic coordination chemistry without prior knowledge. Angew. Chem. Int. Ed. Engl. 59, 11256–11261 10.1002/anie.202000329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Gromski, P.S., Granda, J.M. and Cronin, L. (2020) Universal chemical synthesis and discovery with ‘The Chemputer’. Trends Chem. 2, 4–12 10.1016/j.trechm.2019.07.004 [DOI] [Google Scholar]
- 597.Mehr, S.H.M., Craven, M., Leonov, A.I., Keenan, G. and Cronin, L. (2020) A universal system for digitization and automatic execution of the chemical synthesis literature. Science 370, 101–108 10.1126/science.abc2986 [DOI] [PubMed] [Google Scholar]
- 598.Burger, B., Maffettone, P.M., Gusev, V.V., Aitchison, C.M., Bai, Y., Wang, X.et al. (2020) A mobile robotic chemist. Nature 583, 237–241 10.1038/s41586-020-2442-2 [DOI] [PubMed] [Google Scholar]
- 599.Kwok, R. (2010) Five hard truths for synthetic biology. Nature 463, 288–290 10.1038/463288a [DOI] [PubMed] [Google Scholar]
- 600.Meng, F. and Ellis, T. (2020) The second decade of synthetic biology: 2010-2020. Nat. Commun. 11, 5174 10.1038/s41467-020-19092-2 [DOI] [PMC free article] [PubMed] [Google Scholar]