

Abstract
Background:
Deficiency of adenosine deaminase 2 (DADA2) is an autoinflammatory disease caused by deleterious ADA2 variants. The frequency of these variants in the general population, and hence the expected disease prevalence, remain unknown.
Objective:
We aim to characterize the functional impact and carrier frequency of ADA2 variants.
Methods:
We performed functional studies and in silico analysis on 163 ADA2 variants, including DADA2-associated variants and population variants identified in the Genome Aggregation Database (gnomAD). We estimated the carrier rate using the aggregate frequency of deleterious variants.
Results:
Functional studies of ADA2 variants revealed that 77/85 (91%) of DADA2-associated variants reduced ADA2 enzymatic function by > 75%. Analysis of 100 ADA2 variants in gnomAD showed a full spectrum of impact on ADA2 function, rather than a dichotomy of benign versus deleterious variants. We found several in silico algorithms that effectively predicted the impact of ADA2 variants with high sensitivity and specificity, and confirmed a correlation between the residual function of ADA2 variants in vitro and the plasma ADA2 activity of individuals carrying these variants (n = 45; r = 0.649; p < 0.0001). Using < 25% residual enzymatic activity as the cut-off to define potential pathogenicity, integration of our results with gnomAD population data revealed an estimated carrier frequency of at least 1 in 236 individuals, corresponding to an expected DADA2 disease prevalence of ~1 in 222,000 individuals.
Conclusion:
Functional annotation guides the interpretation of ADA2 variants to create a framework that enables estimation of DADA2 carrier frequency and disease prevalence.
Keywords: adenosine deaminase 2, DADA2, ADA2 variants, carrier frequency, disease prevalence
Graphical Abstract
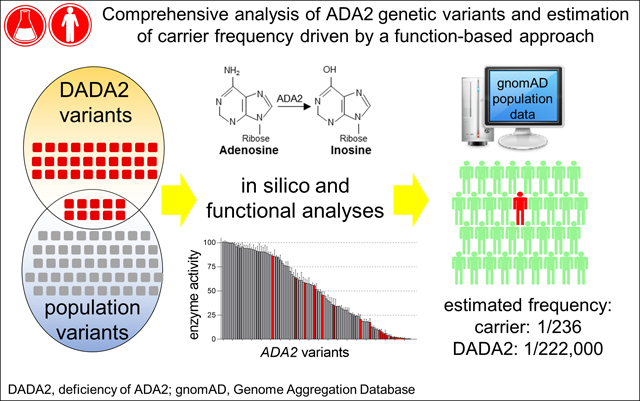
Capsule Summary:
We analyzed the impact of disease-associated variants and population variants on the function of adenosine deaminase 2 (ADA2). This study provides a framework to analyze ADA2 genetic variants and calculate carrier frequency and disease prevalence.
Introduction
Advances in immunology and genetics have accelerated the discovery of monogenic immune defects. Depending on the function of the affected gene, these monogenic diseases may be characterized by immunodeficiency, autoinflammation, autoimmunity or a hybrid of these features.1 Rapid diagnosis and prompt initiation of targeted therapy are critical to control disease severity and mitigate organ damage.2
Genetic panels that evaluate a large number of monogenic immune defects are increasingly used by immunologists and rheumatologists. However, these panels often uncover variants in multiple genes including variants of uncertain significance and thus careful interpretation is required.3 Guidelines to evaluate the pathogenicity of genetic variants have been established by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology.4 Pathogenicity of a genetic variant is strongly supported by prior disease-associated variants affecting the same amino acid or established functional studies confirming a detrimental effect on protein function.5
Adenosine deaminase 2 (ADA2) is a myeloid cell-derived plasma protein that catalyzes the deamination of extracellular adenosine to inosine.6, 7 Biallelic deleterious variants in the encoding gene ADA2 (formerly known as CECR1) on chromosome 22q11 cause a monogenic disease of systemic vasculitis, early-onset stroke, bone marrow failure and immunodeficiency known as deficiency of ADA2 (DADA2).8, 9 Although DADA2 is considered a rare disease, genetic variation in ADA2 is not uncommon. Data from Genome Aggregation Database (gnomAD) revealed more than 200 missense and predicted loss-of-function (pLoF) variants in >140,000 people from the general population.10
In this study, we performed functional analysis of disease-associated ADA2 variants as well as a large number of population variants in gnomAD. Our work constructs a roadmap to interpret ADA2 variants and allows estimation of carrier frequency and disease prevalence for DADA2.
Methods
Patients and samples:
These studies were approved by the Institutional Review Boards at Boston Children’s Hospital and the National Institutes of Health. Informed consent was obtained from healthy individuals, carriers of ADA2 variants and patients with DADA2 prior to serum / plasma collection.
Identification of ADA2 variants.
Single nucleotide variants of ADA2 were identified in gnomAD v2.1.1 (Ensembl gene ID: ENSG00000093072.11) and sorted by allelic frequency. The 101 missense variants with the highest allelic frequency and allele count > 1 in gnomAD were selected for functional analysis. Expression plasmids were created for 100 variants because two of the variants resulted in the same amino acid change (p.G47R). DADA2-associated variants were identified from a review of publications between February 2014 and October 2020. A literature search was performed in PubMed, using the search terms: “DADA2” and “adenosine deaminase 2.” Large genomic deletions and splice site variants were not included due to the lack of patient materials to study the genetic rearrangements. Eighty-five unique DADA2-associated variants were identified for further analysis. A complete list of DADA2-associated variants analyzed by functional studies is provided in Online Repository Table E1. Plasmids for 53 of these variants were generated from our previous studies.11, 12
Plasmid construction and site-directed mutagenesis.
Construction of pcDNA3.1-Myc/His(−) plasmid that encodes wildtype ADA2 was described.11 Site-directed mutagenesis was performed using the Q5 Mutagenesis Kit (New England Biolabs, Ipswich, MA). Plasmids were purified using Purelink Quick Plasmid Miniprep kit (Thermo Fisher Scientific, Waltham, MA). Variants were confirmed by sequencing using T7 and BHG-R primers (Genewiz, Cambridge, MA). Primer sequences for plasmid construction are available upon request.
Quantification of ADA2 enzymatic activity.
HEK 293T cells were seeded in 24-well plates (1×105 cells / well) and transfected with wildtype or mutant ADA2 plasmids using Fugene 6 (Promega, Madison, WI) according to manufacturer’s instruction. Medium was collected after 72 hours of incubation at 37°C. ADA2 activity in culture supernatant and human plasma was measured using an established spectrophotometry assay in BioTek’s Synergy HTX Multi-Mode Reader (Winooski, VT).9, 13 This assay quantifies the adenosine-dependent generation of ammonia in the presence of EHNA, a selective inhibitor of ADA1. The final concentrations of adenosine and EHNA were adjusted to 12 mM (saturating for ADA2) and 0.1 mM, respectively. This assay was fully validated (i.e., for linearity, range, precision, accuracy, specificity, robustness, and ruggedness), and results closely correlate with ADA2 activity measured with an independent high performance liquid chromatography assay that directly quantifies the conversion of adenosine to inosine (MS Hershfield, unpublished). The activity of each sample was normalized to wildtype ADA2 activity measured on the same run. Each variant was analyzed by at least 3 independent experiments.
In silico analysis of ADA2 variants.
Description and source of prediction algorithms assessed in this study are available in Online Repository Table E2. Prediction metrics from some of the algorithms were generated in bulk through wANNOVAR software14 and subsequently verified by analysis from the original sources.
Calculation of carrier frequency and disease prevalence.
Previously reported DADA2-associated variants, the 100 most common missense variants and pLOF variants in gnomAD (with duplicates removed) were included in the analysis. ACMG curation was not applied in this analysis, rather a disruption of ADA2 enzymatic activity by > 75% (i.e., residual ADA2 enzymatic activity of <25%) was used to define deleterious variants for these analyses. The allelic frequencies of missense variants and pLoF variants were combined to determine the aggregate allelic frequency (q). Predicted carrier frequency (2pq) and disease prevalence (q2) were determined using Hardy-Weinberg equilibrium (p2 + 2pq + q2 = 1).
Statistical analysis.
Differences between two groups were analyzed using the Mann-Whitney U test. Correlation between two variables was assessed using Spearman’s rank-order correlation. Receiver operator characteristic (ROC) curves and area under the curve (AUC) calculations were used to analyze the performance of prediction algorithms. All tests were 2-sided, and p < 0.05 was considered significant. Statistical analyses were performed using Prism 8.0 software (GraphPad Software, La Jolla, Calif).
Results
Analysis of disease-associated ADA2 variants
To date, approximately 300 patients and 100 unique disease-associated ADA2 variants have been described in the literature and the Infevers database (https://infevers.umai-montpellier.fr).15 Using a functional assay to quantify ADA2 enzymatic activity, we first analyzed 85 disease-associated variants including 63 missense variants, 3 single amino acid deletions, and 19 pLoF variants (12 insertions / deletions with frameshift and 7 nonsense variants; Online Repository Table E1). Large genomic deletions and splice site variants were not included in the analysis.
Ectopic expression of each mutant construct in 293T cells revealed reduced enzymatic activity for all disease-associated variants compared to wildtype ADA2 (Figure 1A). pLoF variants resulted in complete absence of ADA2 activity while missense variants and single amino acid deletions showed a wide range of residual enzymatic activity (0.5 to 86%). Overall, 94% of all disease-associated variants displayed a reduction of ADA2 activity by >50% while 91% showed <25% residual enzymatic activity (Figure 1B). Missense variants with residual ADA2 activity were distributed among the various functional domains (Figure 1C).
Figure 1. Functional and in silico analysis of DADA2-associated variants.
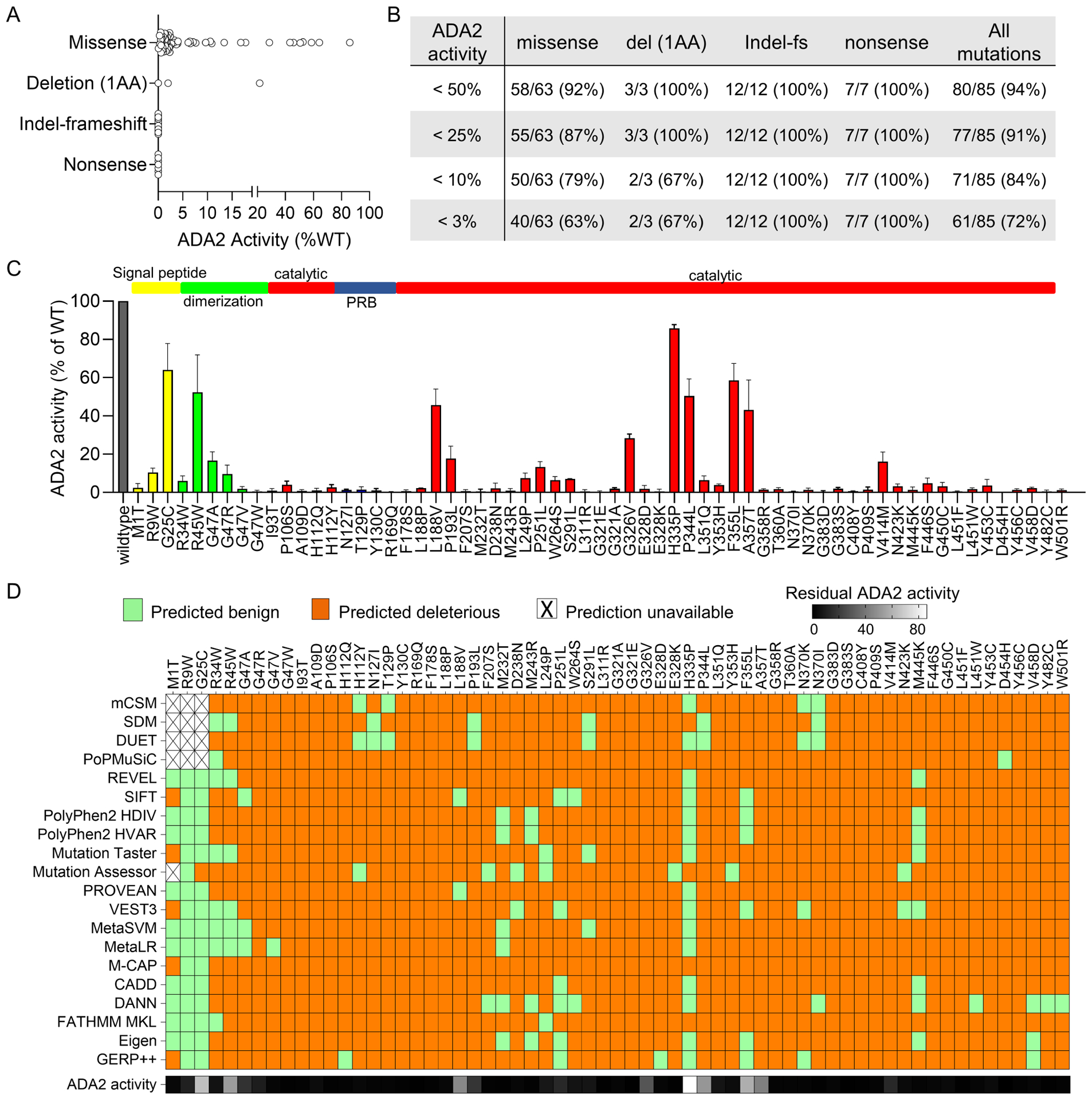
A) ADA2 enzymatic activity of known DADA2-associated variants stratified by variant type. B) Evaluation of DADA2-association variants by various cut-off values of residual ADA2 enzyme activity. C) Residual ADA2 activity of missense variants displayed by amino acid positions and functional domains. D) In silico analysis of DADA2-associated variants by 20 prediction algorithms. For all panels, ADA2 enzymatic activity of mutant constructs is normalized as percentage of residual activity relative to wildtype ADA2.
The potential pathogenicity of genetic variants is often evaluated by in silico prediction algorithms. We utilized results from the functional assay to benchmark the performance of 20 established prediction algorithms for the analysis of ADA2 missense variants. A brief description of these algorithms is provided in Online Repository Table E2. These algorithms predicted most of the disease-associated variants as deleterious with sensitivity ranging from 76 – 97% (Figure 1D and Online Repository Figure E1A). Many of the algorithms incorrectly predicted several N-terminal variants as benign changes, likely due to the divergent secretory peptide sequence among species (Online Repository Figure E1B). Interestingly, two disease-associated variants with the highest residual activity (H335P [86%] and G25C [65%]) were predicted benign variants by most algorithms. H335P was found in trans with Y130C (1% residual activity) in one case while homozygous G25C was described in two patients.16, 17 Taken together, these findings illustrated that the prediction algorithms are generally effective in determining pathogenic ADA2 variants and that many disease-associated variants are hypomorphic variants with residual enzymatic function.
Evaluation of ADA2 variants in gnomAD
To study ADA2 genetic variants in the general population, we explored the genomic data from >140,000 individuals available in gnomAD.10 Among the missense and pLoF ADA2 variants found in gnomAD, only 18 variants were classified as “pathogenic” or “likely pathogenic” based on ACMG criteria. Overall, R169Q was the most common disease-associated variant with a MAF of 4.74 × 10−4 (carrier frequency: 1 in 1054 individuals; Online Repository Table E3). Seven missense variants without prior association with DADA2 had higher allelic frequencies compared to R169Q. To better capture the impact of ADA2 variants on protein function, we analyzed 100 missense variants with the highest MAF in gnomAD. This list covered >99% of all missense ADA2 variants based on allele counts and 93% of variants in gnomAD (3,375 / 3,623 allele counts) after excluding the common polymorphism H335R (MAF 0.34; >99% enzymatic activity relative to wildtype ADA2).
Functional analysis of these 100 variants showed a full spectrum of ADA2 activity ranging from 100% to < 0.5% of the levels found in wildtype ADA2 control (Figure 2A). Twenty-one of these variants were previously identified in patients with DADA2, mostly with the vasculitis phenotype (red bars; also see Online Repository Table E3). Contrary to the notion that rare variants are more likely to be pathogenic, residual ADA2 enzymatic activity measurements did not correlate with allelic frequencies (Figure 2B), though analysis was restricted to mostly rare variants (MAF <1%). Similarly, the distribution of allelic frequencies was comparable between the subset of DADA2-associated variants and all other variants (p = 0.602; data not shown).
Figure 2. Functional and in silico analysis of missense ADA2 variants in gnomAD.
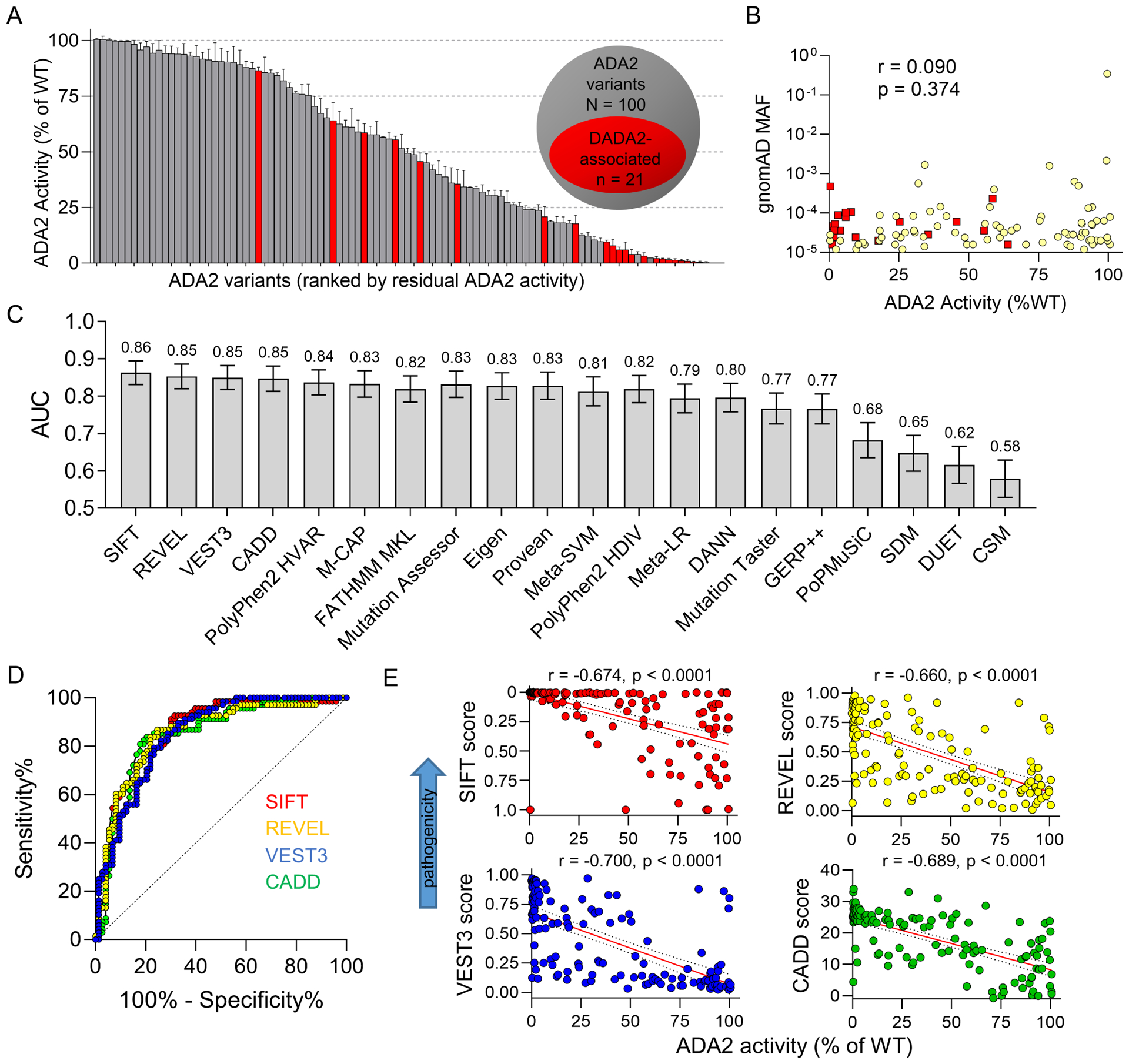
A) Enzymatic activity of 100 most frequent missense ADA2 variants in gnomAD. Red bars indicate DADA2-associated variants. B) Correlation between minor allelic frequency and residual enzymatic activity of ADA2 variants. Red squares indicate DADA2-associated variants. C and D) Performance of prediction algorithms in predicting the pathogenicity of ADA2 variants assessed by ROC curve analysis. Area under the curve (AUC) from ROC analysis of all algorithms are displayed in panel C and ROC curves of SIFT, REVEL, CADD and VEST3 are illustrated in panel D. E) Correlation plots of raw scores from SIFT (range 1 – 0), REVEL (range 0 – 1), CADD (range 1 – 40) and VEST3 algorithms (range 0 – 1) with residual enzymatic activity of ADA2 variants (n = 100 for all panels).
This dataset allowed a more comprehensive assessment of the prediction algorithms. We combined the data from DADA2-associated variants (Figure 1A) and analyzed a total of 141 unique ADA2 missense variants. We used <25% residual enzymatic activity as a conservative cut-off to define deleterious variants because >90% of DADA2-associated variants have activity under this threshold. A correlation matrix was constructed for ADA2 activity and prediction scores generated from the 20 algorithms (Online Repository Figure E2A). Receiver operator characteristic (ROC) analysis showed that SIFT, REVEL, VEST3 and CADD were among the algorithms with the best performance in predicting benign vs. deleterious variants (Figure 2C, D). Raw scores generated from these prediction algorithms correlated well with the enzymatic activity of ADA2 variants (Figure 2E). Because a few DADA2-associated variants had residual activity greater than 25%, we performed additional ROC analyses of the algorithms using 50% and 75% residual enzymatic activity as the cut-off to evaluate another definition for including deleterious (possibly hypomorphic) variants (Online Repository Figure E2B). Taken together, these findings provide a guide to interpret the functional significance of ADA2 variants in the gnomAD database.
Plasma ADA2 activity in carriers of ADA2 variants
To assess the physiologic relevance of these in vitro data, we measured plasma ADA2 levels in 45 carriers of ADA2 variants. The majority of these individuals (n = 36) were family members of patients with DADA2. The remaining individuals (n = 9) were found to have a monoallelic ADA2 variant during genetic evaluation for recurrent febrile illness of unknown etiology and none of them received treatment that is likely to alter ADA2 levels. Fourteen unique missense variants analyzed above and 9 pLoF variants (indel-frameshift or nonsense) were found among these carriers. DADA2 patients with biallelic variants were not further compared as they possessed very low plasma ADA2 activity and therefore it would not be feasible to distinguish the impact of each allele (Figure 3A).
Figure 3. Plasma ADA2 activity in carriers of ADA2 variants.
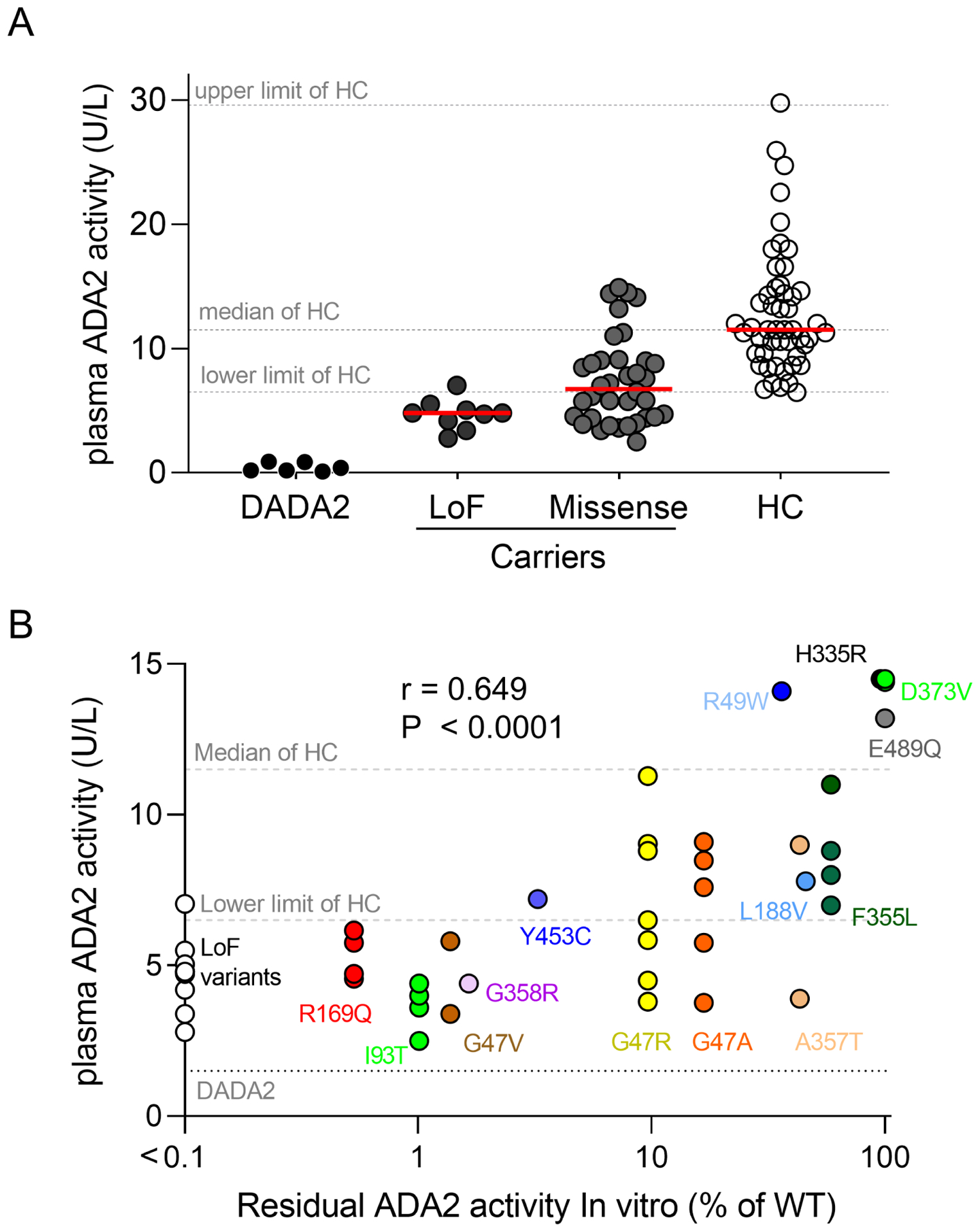
A) Comparison of plasma ADA2 activity levels in patients with DADA2 (n = 6), carriers of predicted pLoF variants (n = 9), carriers of missense variants (n = 36) and age-matched healthy controls (HC; n = 50). B) Correlation between plasma ADA2 activity in carriers (n = 45) and residual enzymatic activity of specific variants determined by in vitro studies. Individual variant and label are color coded. Dotted lines indicate reference range for healthy controls and patients with DADA2. Upper and lower limit of healthy controls were defined as the 98th and 2nd percentile, respectively.
We segregated the carrier group by the type of ADA2 variants and found that individuals that possess a pLoF variant all had plasma ADA2 levels below the lower limit of healthy controls. In contrast, carriers of missense variants displayed a wider range of plasma ADA2 activity that overlaps with levels found in age-matched healthy controls (Figure 3A). Correlation studies revealed that carriers of variants that most profoundly alter ADA2 function (R169Q, G47V, I93T and G358R) had the lowest plasma ADA2 activity (Figure 3B). Carriers of hypomorphic variants that display greater residual function including G47R and G47A tended to show variable plasma ADA2 activity and some were within the range of healthy controls. Accordingly, carriers of variants with minimal impact on ADA2 activity in vitro (H335R, D373V, E489Q) displayed normal plasma enzymatic activity (Figure 3B). Overall, plasma ADA2 activity in carriers generally correlated with the expected functional impact of the ADA2 variants but the variability was greater for hypomorphic variants (Spearman r = 0.649; p < 0.0001).
Estimation of carrier frequency and disease prevalence
The prevalence of a monogenic disease can be estimated by the reported incidence in a population or by aggregate allelic frequency of deleterious variants.18–20 We explored the prevalence and carrier rate of variants causing the DADA2 phenotype by integrating the allelic frequency of ADA2 variants in gnomAD and the curated functional data on these variants (Figure 4A). Because these calculations are heavily dependent on the cut-off of residual ADA2 activity used to determine pathogenic variants, we examined carrier frequency at cut-off values ranging from 3% to 75% (Figure 4B).
Figure 4. Carrier frequency analysis of pathogenic ADA2 variants in gnomAD.
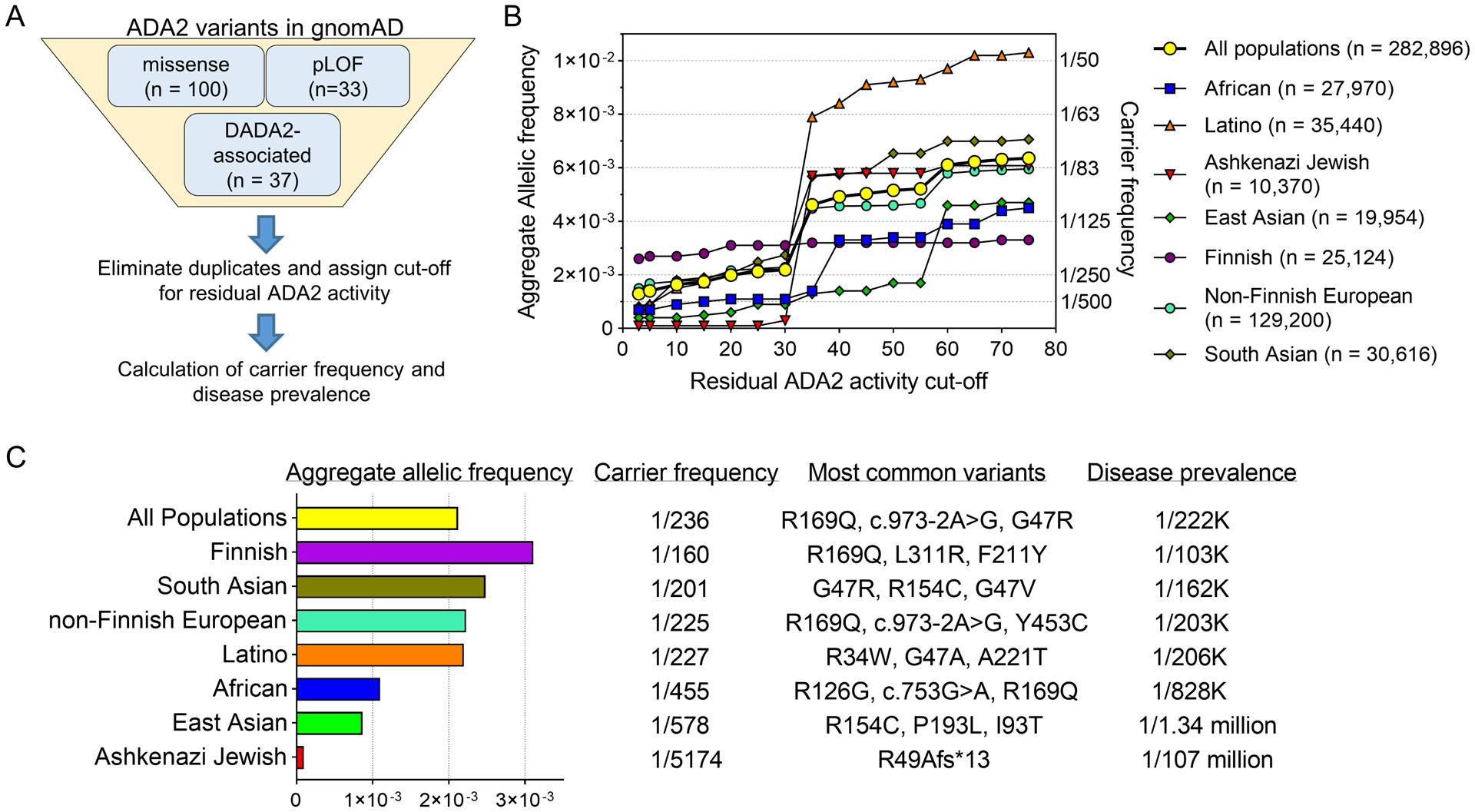
A) Schematic for selection of ADA2 variants to compute carrier frequency. B) Aggregate allelic frequency (left axis) and carrier frequency (right axis) of deleterious ADA2 variants at various cut-off values of residual ADA2 activity. n represents the total number of alleles for each population in gnomAD. C) Calculation of aggregate allelic frequency, carrier frequency of deleterious ADA2 variants (defined by the cut-off of <25% residual ADA2 activity) and DADA2 disease prevalence.
We elected to use 25% residual enzymatic activity as the cut-off to determine deleterious variants because >90% of DADA2-associated variants displayed residual activity under this threshold (see Figure 1B). Combining all gnomAD populations, the aggregate allelic frequency of pathogenic missense variant and pLoF variants was 0.00212, yielding a carrier frequency of 1 in 236 individuals and an estimated DADA2 disease prevalence of ~1 in 222,164 individuals (Figure 4C). Based on the Hardy-Weinberg equilibrium, these calculations predict over 30,000 DADA2 cases among the world population of 7.7 billion individuals. If a strict cut-off of 10% was applied to determine deleterious variants (i.e., >90% reduction of ADA2 activity), the overall carrier rate was ~1 in 305 individuals. The carrier rate sharply increased to 1 in 108 individuals at the cut-off of 35% (i.e., >65% reduction of ADA2 activity), largely due to the prevalence of two hypomorphic variants, M309I and P435A (Online Repository Table E4).
The aggregate allelic frequency, carrier rate and the most common pathogenic variants for subpopulations in gnomAD (based on the 25% residual activity cut-off) are detailed in Figure 4C. The Finnish population exhibited the highest carrier rate (1 in 160) and estimated disease prevalence (~1 in 103,217) primarily due to the high allelic frequency of the well-established DADA2-associated variant R169Q (MAF 0.0019). South Asians exhibited the next highest carrier frequency with G47R as the most common variant (MAF 0.00075). In contrast, African and Ashkenazi Jewish populations displayed the lowest frequency of deleterious ADA2 variants in gnomAD. Taken together, the combination of gnomAD data and functional analysis of ADA2 variants provides an informed, data-driven approach to estimate carrier frequency in different populations.
Discussion
The availability of next-generation sequencing panels and exome / genome sequencing has improved our ability to detect monogenic immune disorders. Genetic variants identified from these studies require careful interpretation; functional studies can provide complementary information in determining the pathogenicity of specific variants.3, 5 Our study constitutes the most comprehensive analysis to date on variants in the ADA2 gene. We performed in silico and functional studies on 163 ADA2 variants encompassing disease-associated variants and population variants in gnomAD. These data provide a guide to interpret ADA2 variants and create a framework to estimate the prevalence of DADA2.
Analysis of the 100 most common missense ADA2 variants in gnomAD illustrates that it is likely an oversimplification to assign variants dichotomously as “benign” versus “pathogenic”. Instead, the impact of these variants on ADA2 function exists as a spectrum without a discrete cut-off, and cannot be predicted by allelic frequencies. While our study is focused on ADA2, these concepts are likely applicable to other monogenic disease-associated genes. To benchmark the analysis of in silico prediction algorithms, we established < 25% residual enzymatic activity as a conservative cut-off to determine pathogenic ADA2 variants based on levels observed in the >90% of disease associated variants. However, it is clear that several hypomorphic variants with residual activity above this cut-off have been causally linked to cases of DADA2, typically as a part of compound heterozygous variants.17, 21–23 It is also possible that these variants may impact protein production and/or function in ways not reflected by our in vitro system.
How much residual ADA2 activity is needed to protect against the development of autoinflammation is an important question to consider. For example, the F355L variant possesses >50% residual activity and carriers of this variant generally display plasma ADA2 activity in the low normal range. Despite the relatively high allelic frequency of this variant (0.0028) in the East Asian population, F355L has been described in only one DADA2 patient with the vasculitis phenotype as part of compound heterozygous variants (in trans with E328K).22 The M309I variant displayed a reduction of ADA2 enzymatic activity by 65% in our functional assay and is present in 1/300 individuals in the general population (MAF 0.00167). Similarly, P435A is a common variant in the Latino population (MAF 0.0043; 1 in 116 individuals) and exhibited residual ADA2 activity of 32%. Interestingly, two cases of DADA2 with M309I as part of compound heterozygous variants (in trans with R169Q) were described while we were preparing the current manuscript.24 Inheritance of a more pathogenic allele in trans may be necessary for these hypomorphic variants to cause DADA2. Alternatively, other epigenetic factors and environmental triggers may contribute to disease development in susceptible individuals with hypomorphic variants.
The pathophysiology of DADA2 is an area of intense investigation. Recent studies have suggested that disruption of adenosine homeostasis in the absence of ADA2 triggers the formation of neutrophil extracellular traps and activation of type I interferon signaling.25, 26 ADA2 may also have additional functions as a lysosomal nuclease.27 While the physiologic function of ADA2 awaits further clarification, measurement of deaminase activity remains the best approach to study ADA2 variants at this time. Parallel studies using plasma from carriers of ADA2 variants further support the utility of our in vitro analysis in predicting in vivo ADA2 activity. Loss of enzymatic activity is a common feature of DADA2-associated variants and correspondingly, near-absent plasma ADA2 enzymatic activity is confirmatory for the diagnosis of DADA2 in patients. It is notable that plasma ADA2 activity in carriers with the same hypomorphic variant can also vary considerably, suggesting that additional factors may regulate protein expression by the mutant and/or wild-type allele.
In our experience, carriers of disease-associated variants are typically healthy without the hallmark manifestations of DADA2. These individuals are usually found by targeted screening due to the diagnosis of DADA2 in a family member. Interestingly, carrier status for several isolated cases in our study were identified by genetic evaluation for periodic fever syndromes. A systematic study of carriers of deleterious ADA2 variants is needed to address the impact of reduced plasma ADA2 levels on health.
The estimated carrier frequency of pathogenic ADA2 variants and disease prevalence of DADA2 may seem surprisingly high given that approximately 300 cases have been reported in the literature to date. However, DADA2 was described only 6 years ago, and timely diagnosis may still be limited by awareness of this condition and availability of genetic or enzymatic activity testing. Moreover, recognition of DADA2 may be further hindered by incomplete penetrance, variable age of disease onset, and pleiotropic manifestations of the disease, which may include bone marrow failure or immunodeficiency, with or without the classic features of systemic inflammation and vasculitis.28, 29
The high frequency of R169Q in Finnish and Northern European populations and G47R in South Asians are supported by data from DADA2 patients in those regions.30, 31 The actual prevalence of DADA2 may be greater than our conservative estimate because a number of DADA2-associated variants with residual activity > 25% are not captured in the calculations. Some disease-associated variants are not found in gnomAD, and very rare missense variants (MAF < 1×10−6) in gnomAD were also not included in the analysis, although their impact on disease prevalence is likely minimal.
Relying on the assumption of Hardy-Weinberg equilibrium, these data also cannot account for consanguinity or enrichment of specific ADA2 variants in certain populations, such as the high carrier frequency of G47R in the Georgian Jewish population.8 An additional limitation of our approach is incomplete representation of populations in gnomAD. For example, genomic data from several hotspots for DADA2 including Turkey, Pakistan and India are not well represented in the database. A significant fraction of reported DADA2 cases, including many with homozygous G47R variants, has been described in these regions.30, 32 Furthermore, individuals with severe pediatric diseases and their first degree relatives were excluded from the gnomAD curation process.10 Taking these factors into account, the actual carrier frequency of deleterious ADA2 variants is likely higher than the current estimate.
Finally, our study is limited by the focus on genotype and does not specify the various phenotypes of DADA2. Like many other monogenic syndromes, genotype does not always translate to phenotype and cases of incomplete penetrance have been described in individuals with DADA2.29, 30, 33 The precise disease penetrance for DADA2 is currently unknown and may be difficult to estimate without a natural history study of symptomatic and asymptomatic DADA2 cases.
Our work demonstrates the utility of functional studies to annotate genetic data for monogenic immune disorders. The combination of in silico analysis and functional studies described here provides a roadmap to interpret ADA2 variants. Interrogation of variants in gnomAD further revealed high carrier frequency of ADA2 variants and provided the first estimation of DADA2 prevalence in various populations. These findings underscore the need to improve access to diagnostic testing for this monogenic disease with pleiotropic presentations.
Supplementary Material
Clinical Implications:
The combination of functional analysis and population data on ADA2 variants illustrates a data-driven approach to study the genetics of monogenic immune diseases.
Acknowledgements
We thank Drs. Raif Geha, Craig Platt and members of the Nigrovic laboratory for helpful discussions. We thank NG and SK in the Hershfield Laboratory for assistance with developing the spectrophotometric assay for quantitation of ADA2 activity. We thank the gnomAD investigators for creation of this resource and Michael Wilson for preparing gnomAD sites vcf data files used in this study.
Funding Sources:
This work was supported by the National Institute of Health / National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) R01-AR065538, R01-AR073201, R01-AR075906 and P30-AR070253 (P.A.N.), K08-AR074562 (P.Y.L.), the Rheumatology Research Foundation Investigator Award (P.Y.L.), Boston Children’s Hospital Faculty Career Development Award (P.Y.L) and by the Chan Zuckerberg Initiative to the Rare Genomes Projects (S.B. and A.O.L).
Abbreviations:
- ADA2
Adenosine Deaminase 2
- CADD
Combined Annotation Dependent Depletion
- CECR1
Cat Eye Syndrome Critical Region 1
- DADA2
Deficiency of ADA2
- gnomAD
Genome Aggregate Database
- pLoF
predicted loss of function
- MAF
Minor Allelic Frequency
- REVEL
Rare Exome Variant Ensemble Learner
- ROC
Receiver operating characteristic
- SIFT
Sorting Intolerant from Tolerant
- VEST
Variant Effect Scoring Tool
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest disclosure statement: The authors declare no conflict of interest relevant to the study.
References
- 1.Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol 2017; 18:832–42. [DOI] [PubMed] [Google Scholar]
- 2.Nigrovic PA, Lee PY, Hoffman HM. Monogenic autoinflammatory disorders: Conceptual overview, phenotype, and clinical approach. J Allergy Clin Immunol 2020; 146:925–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sullivan KE. The scary world of variants of uncertain significance (VUS): A hitchhiker’s guide to interpretation. J Allergy Clin Immunol 2020. [DOI] [PubMed] [Google Scholar]
- 4.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med 2019; 12:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schrader WP, Pollara B, Meuwissen HJ. Characterization of the residual adenosine deaminating activity in the spleen of a patient with combined immunodeficiency disease and adenosine deaminase deficiency. Proc Natl Acad Sci U S A 1978; 75:446–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zavialov AV, Yu X, Spillmann D, Lauvau G. Structural basis for the growth factor activity of human adenosine deaminase ADA2. J Biol Chem 2010; 285:12367–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med 2014; 370:921–31. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Stone DL, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med 2014; 370:911–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020; 581:434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee PY, Huang Y, Zhou Q, Schnappauf O, Hershfield MS, Li Y, et al. Disrupted N-linked glycosylation as a disease mechanism in deficiency of ADA2. J Allergy Clin Immunol 2018; 142:1363–5 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee PY, Kellner ES, Huang Y, Furutani E, Huang Z, Bainter W, et al. Genotype and functional correlates of disease phenotype in deficiency of adenosine deaminase 2 (DADA2). J Allergy Clin Immunol 2020; 145:1664–72 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee PY, Schulert GS, Canna SW, Huang Y, Sundel J, Li Y, et al. Adenosine deaminase 2 as a biomarker of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Ann Rheum Dis 2020; 79:225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang X, Wang K. wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet 2012; 49:433–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarrauste de Menthiere C, Terriere S, Pugnere D, Ruiz M, Demaille J, Touitou I. INFEVERS: the Registry for FMF and hereditary inflammatory disorders mutations. Nucleic Acids Res 2003; 31:282–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rama M, Duflos C, Melki I, Bessis D, Bonhomme A, Martin H, et al. A decision tree for the genetic diagnosis of deficiency of adenosine deaminase 2 (DADA2): a French reference centres experience. Eur J Hum Genet 2018; 26:960–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang W, Yu Z, Gou L, Zhong L, Li J, Ma M, et al. Single-Center Overview of Pediatric Monogenic Autoinflammatory Diseases in the Past Decade: A Summary and Beyond. Front Immunol 2020; 11:565099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu W, Pajusalu S, Lake NJ, Zhou G, Ioannidis N, Mittal P, et al. Estimating prevalence for limb-girdle muscular dystrophy based on public sequencing databases. Genet Med 2019; 21:2512–20. [DOI] [PubMed] [Google Scholar]
- 19.Hanany M, Rivolta C, Sharon D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc Natl Acad Sci U S A 2020; 117:2710–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schrodi SJ, DeBarber A, He M, Ye Z, Peissig P, Van Wormer JJ, et al. Prevalence estimation for monogenic autosomal recessive diseases using population-based genetic data. Hum Genet 2015; 134:659–69. [DOI] [PubMed] [Google Scholar]
- 21.Caorsi R, Penco F, Grossi A, Insalaco A, Omenetti A, Alessio M, et al. ADA2 deficiency (DADA2) as an unrecognised cause of early onset polyarteritis nodosa and stroke: a multicentre national study. Ann Rheum Dis 2017; 76:1648–56. [DOI] [PubMed] [Google Scholar]
- 22.Keer N, Hershfield M, Caskey T, Unizony S. Novel compound heterozygous variants in CECR1 gene associated with childhood onset polyarteritis nodosa and deficiency of ADA2. Rheumatology (Oxford) 2016; 55:1145–7. [DOI] [PubMed] [Google Scholar]
- 23.Petty RE, Cabral DA. Vasculitis and Its Classification. In: Petty RE, Laxer RM, Lindsley C, Wedderburn LR, editors. Textbook of Pediatric Rheumatology. 7th ed: Elsevier; 2016. p. 448–532. [Google Scholar]
- 24.Cooray S, Omyinmi E, Hong Y, Papadopoulou C, Harper L, Al-Abadi E, et al. Anti-tumour necrosis factor treatment for the prevention of ischaemic events in patients with deficiency of adenosine deaminase 2 (DADA2). Rheumatology (Oxford) 2021. [DOI] [PubMed] [Google Scholar]
- 25.Carmona-Rivera C, Khaznadar SS, Shwin KW, Irizarry-Caro JA, O’Neil LJ, Liu Y, et al. Deficiency of adenosine deaminase 2 triggers adenosine-mediated NETosis and TNF production in patients with DADA2. Blood 2019; 134:395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dhanwani R, Takahashi M, Mathews IT, Lenzi C, Romanov A, Watrous JD, et al. Cellular sensing of extracellular purine nucleosides triggers an innate IFN-beta response. Sci Adv 2020; 6:eaba3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greiner-Tollersrud OK, Boehler V, Bartok E, Krausz M, Polyzou A, Schepp J, et al. ADA2 is a lysosomal DNase regulating the type-I interferon response. bioRxiv 2020:2020.06.21.162990. [Google Scholar]
- 28.Lee PY. Vasculopathy, Immunodeficiency, and Bone Marrow Failure: The Intriguing Syndrome Caused by Deficiency of Adenosine Deaminase 2. Front Pediatr 2018; 6:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyts I, Aksentijevich I. Deficiency of Adenosine Deaminase 2 (DADA2): Updates on the Phenotype, Genetics, Pathogenesis, and Treatment. J Clin Immunol 2018; 38:569–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma A, Naidu G, Sharma V, Jha S, Dhooria A, Dhir V, et al. Deficiency of adenosine deaminase 2 (DADA2) in Adults and Children: Experience from India. Arthritis Rheumatol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trotta L, Martelius T, Siitonen T, Hautala T, Hamalainen S, Juntti H, et al. ADA2 deficiency: Clonal lymphoproliferation in a subset of patients. J Allergy Clin Immunol 2018; 141:1534–7 e8. [DOI] [PubMed] [Google Scholar]
- 32.Ozen S, Batu ED, Taskiran EZ, Ozkara HA, Unal S, Guleray N, et al. A Monogenic Disease with a Variety of Phenotypes: Deficiency of Adenosine Deaminase 2. J Rheumatol 2019; 47:117–25. [DOI] [PubMed] [Google Scholar]
- 33.Nanthapisal S, Murphy C, Omoyinmi E, Hong Y, Standing A, Berg S, et al. Deficiency of Adenosine Deaminase Type 2: A Description of Phenotype and Genotype in Fifteen Cases. Arthritis Rheumatol 2016; 68:2314–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.