

Summary
Cilia are microtubule-based structures that either transmit information into the cell or move fluid outside of the cell. There are many human diseases that arise from malfunctioning cilia. Though mammalian models provide vital insights into the underlying pathology of these diseases, aquatic organisms such as Xenopus and zebrafish provide valuable tools to help screen and dissect out the underlying causes of these diseases. In this review we focus on recent studies that identify or describe different types of human ciliopathies and outline how aquatic organisms have aided our understanding of these diseases.
Keywords: Xenopus, Zebrafish, Cilia, Kidney, Nasal, Node, Ciliopathy, cystic kidney
Introduction
Cilia are tubulin based structures that protrude from the cell. True cilia (in contrast to stereocilia) have a similar structure. At the base of each cilium is a basal body made up of a centriole (Dahl 1963; Preble, Giddings, and Dutcher 1999; Reese 1965). The basal body, which is a microtubule organizing center, is thought to be the platform by which the rest of the cilium is assembled. At the apical end of the basal body is the transition zone (Diener, Lupetti, and Rosenbaum 2015). This zone likely contains hundreds of proteins that anchor the cilia and regulate trafficking into and out of the cilia (Diener et al. 2015). The structure that protrudes from the cells is the axoneme, which is supported by a ring of microtubules (Sun et al. 2019). Two primary characteristics are used to classify types of cilia, monocilia versus multicilia and motile versus primary (sensory) cilia. With a few exceptions, in mammals primary cilia are typically present as a single sensory cilium that extends from the cell with an axoneme that contains nine microtubule pairs arranged in a ring (9+0 arrangement) (Nikai, Rose, and Cattoni 1970) (Fig. 1). These sensory cilia allow cells to detect and respond to fluid flow, hormones, or other sensory stimuli from their extracellular environment. In contrast, motile cilia can be present as either monocilia (one per cell) or multicilia (many per cell) and function to move fluid in their environment. Typically in motile cilia, the outer ring of tubules contain arms made up of the microtubule motor protein, dynein, and an additional two inner microtubules which aid in ciliary movement (9+2 arrangement) (Rhodin and Dalhamn 1956). Because the development of motile cilia requires much of the same machinery as primary cilia, mutations that affect one type of cilia can affect other types within the organism.
Figure 1:
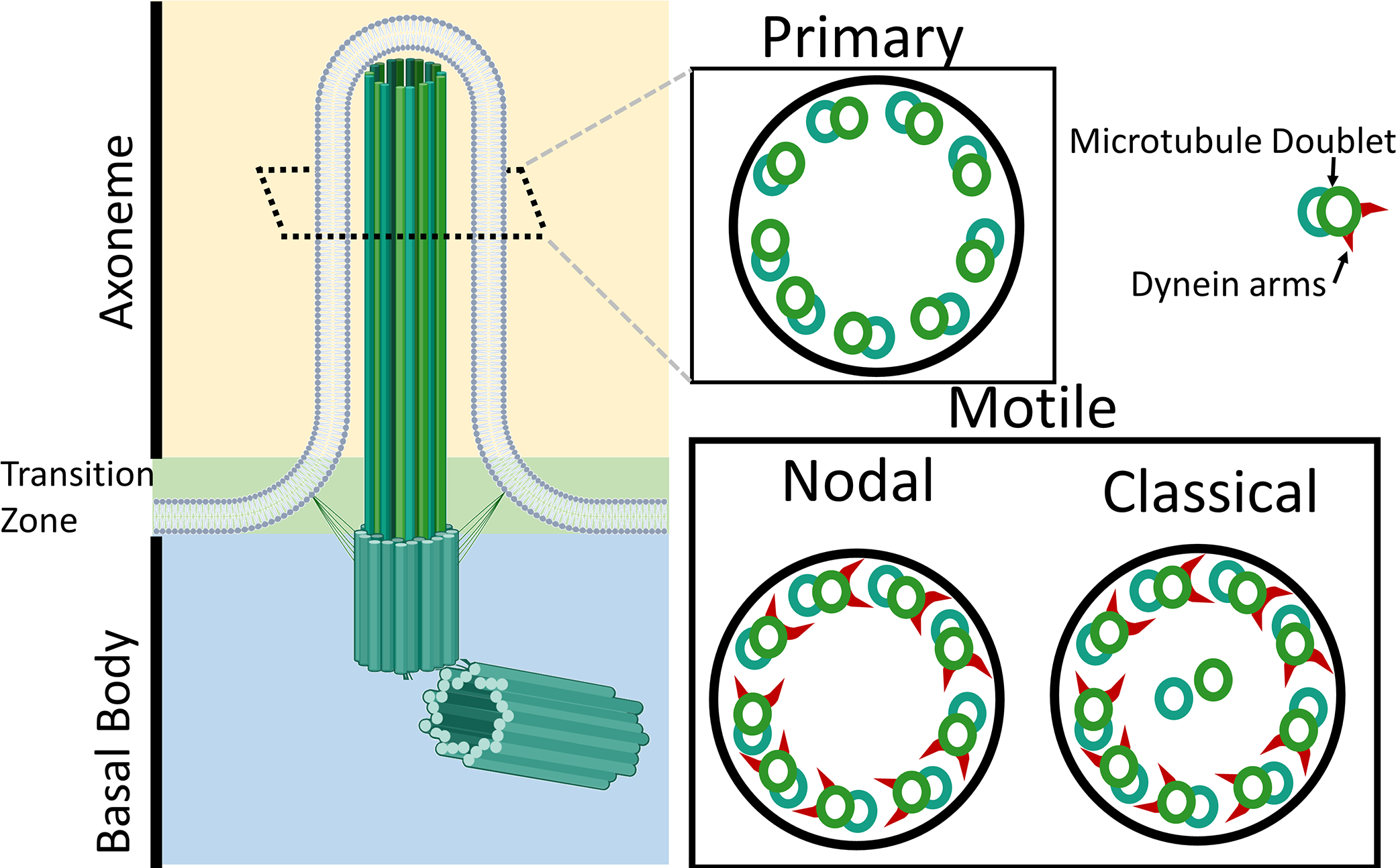
Structure of cilia. The cilia are composed of three main sections, the axoneme which performs the sensory or movement function, the transition zone which likely contains over 100 proteins which function to anchor the cilia and regulate transport to and from the cilia, and the basal body which is a centriole that functions as a tubulin organizing center to form the cilia. Diagram showing the cross section of the axoneme of common types of motile and primary cilia in vertebrates.
In humans, there are many different syndromic diseases that are caused by malformed or dysfunctional cilia. Diseases in this category are called ciliopathies. There are many different ciliopathies, each give rise to specific phenotypes depending on the gene that is mutated. Some examples include Polycystic kidney disease, Nephronophthisis, Bardet–Biedl syndrome, Joubert syndrome, Oral-facial-digital syndrome I, and situs inversus. Though these diseases are characterized by malformed or malfunctioning cilia, the role cilia play in a number of ciliopathies is largely unknown or heavily debated. Additionally, with the advent of whole exome sequencing and the increased accessibility of genome wide association studies, researchers are finding new candidate genes underlying human many diseases. Given that generating mouse lines is both costly and time consuming, other model organisms that do not have these limitations such as Xenopus and zebrafish are being used to identify and study candidate genes (Grove, Eckardt, and McLaughlin 2016).
Aquatic organisms have been used for years to study the function of ciliary components. In 1940s researchers were describing in Xenopus how motile cilia aid in the oocyte movement within the upper part of the oviduct (Waring, Landgrebe, and Neill 1941). Shortly after this, motile cilia were described as an adaptation in embryos to aid in the transport of small food particles to the stomach (Dodd 1950). Though zebrafish is a newer model organism it has also played a pivotal role in the study of cilia. In recent years, both ZFIN (the zebrafish genome database) and Xenbase (the Xenopus genome database) have put forth a unified effort to update their databases to aid in the modeling human diseases, including ciliopathies (Bradford et al. 2017; Nenni et al. 2019).
Aquatic organisms such as zebrafish (Danio rerio), Xenopus laevis, and Xenopus tropicalis provide many technological advantages over other vertebrate systems to study cilia function and development. The short developmental times and many offspring per clutch allow for experiments to be performed using hundreds of embryos in a matter of days. Ciliogenesis in these organisms can be seen within 24 hours of fertilization, and most of the ciliated organs are functional within a few days to a few weeks of development. Additionally, zebrafish have a short generation time, reaching maturity in three to four months, which allows for quick generation of mutant and transgenic lines (Lawrence et al. 2012). Also, the ability to easily isolate and culture stem cells from the Xenopus models allows for the generation of tissue organoids that develop in a few days. Aquatic models develop externally and are therefore easier to manipulate through several different methods.
Genetic manipulation can be easily accomplished by microinjection in many aquatic vertebrate species. Injection of morpholinos, or antisense RNA, lead to a quick means of gene knockdown, and injection of CRISPR guide pools are an alternate technique to generate genetic mutants (Bhattacharya et al. 2015; Chang et al. 2013; Clements et al. 2017; Delay et al. 2018). Given that many of these techniques are highly efficient and phenotypes can be validated through rescue experiments, analysis can be done in the F0 generation without the need to generate lines. It is now common to validate a morpholino experiment with CRISPR knockouts to recapitulate phenotypes (DeLay, Baldwin, and Miller 2019). Also, since these organisms are aquatic, drug treatments can be administrated through the water in which the embryos are growing in order to identify treatments, perform rescues, or to validate phenotypes seen from another technique. Furthermore, transgenic animals can be created through the aid of TOL2 transposons, ISceI meganuclease, or CRISPR guided homologous recombination (Aslan et al. 2017; Corkins et al. 2018; Fisher et al. 2006; Miller, Lee, and McCrea 2014; Ogino, McConnell, and Grainger 2006). Some aquatic species also have advantages over their mammalian counterparts. Because Xenopus is fate-mapped, targeted injections can be carried out at the two cell stage to affect only half of the embryo, leaving the other half as an internal control. Alternatively, injecting at later cell stages allows for targeting specific subsets of tissues, avoiding lethal or compounding phenotypes (Moody 1987b, 1987a). With accelerated genome sequencing in humans leading to identification of an abundance of putative disease genes, modeling novel mutations in aquatic organism greatly streamlines the identification of disease-causing genes, underlying pathways they act through, and potential treatments for diseases.
Primary cilia
Primary cilia, also known as sensory cilia, are typically monocilia that protrude from the cell. These sensory cilia allow the cells to respond to fluid flow, hormones, or other sensory stimuli from their extracellular environment. Most cell types have primary cilia at some point in their development, and loss of these cilia in humans leads to a wide variety of problems, including loss of senses such as vision, smell or hearing (Beales and Kenny 2014). For example, the BBSome is part of the cilia transport machinery, and loss of BBSome components are associated with renal abnormalities and loss of smell (Laurence and Moon 1995; Uytingco et al. 2019; Veleri et al. 2012) Loss of primary cilia also leads to developmental abnormalities such as craniofacial deformities, vision problems, cystic liver and kidneys, and intellectual disabilities (Brugmann, Cordero, and Helms 2010; Noda et al. 2016; Zhao and Malicki 2007). As an example, in humans, loss of the Joubert syndrome protein CEP290 leads to severe neurological disorders, cystic kidney disease, and vision loss (Srivastava et al. 2017). A number of these phenotypes are also seen in zebrafish and mice (Baye et al. 2011; Rachel et al. 2015).
Primary cilia are sensory organelles that affect several genetic signaling pathways. The two main pathways known to be regulated by cilia are Wnt and hedgehog signaling (HH), though other pathways such as PDGFRA are affected by the loss of cilia (Huangfu et al. 2003; Schmid et al. 2018; Wheway, Nazlamova, and Hancock 2018). How cilia regulate Wnt signaling, however, is heavily debated. Loss of primary cilia typically results in hypersensitivity to canonical Wnt signals (Ajima and Hamada 2011; Lancaster, Schroth, and Gleeson 2011; Ocbina, Tuson, and Anderson 2009). There are a few theories as to how this occurs. Components of the Wnt signaling pathway including GSK3, a β-catenin inhibitor, are found around the basal bodies of the cilia. Activation of these components may lead to β-catenin’s degradation (Corbit et al. 2008). As β-catenin is required for canonical Wnt signaling, degradation of β-catenin should lead to decreased Wnt signaling. Other theories involve calcium signaling, given that calcium ions can also inhibit Wnt signaling via the Wnt/Ca2+ pathway and the cilia contain mechanoresponsive calcium channels called polycystins (Kühl et al. 2000; Li et al. 2018). Alternatively, it is also possible that the polycystin complex more directly targets Wnt signaling through direct binding of β-catenin (Lal et al. 2008). Any or all of these theories potentially play a role in ciliary Wnt signaling.
Kidney cilia
Wnt signaling plays a pivotal role in kidney development and tubule formation, and with a few exceptions, many mutations that affect primary cilia result in kidneys abnormalities. The mammalian kidney develops in three stages: the pronephros, mesonephros, and the metanephros. All three forms of the kidney use many of the same genetic pathways with each successive remodeling of the kidney (Blackburn and Miller 2019; Brändli 1999). The basic unit of filtration, the nephron, is present in all three successive forms of the kidney. These nephrons are tubules that are primarily made up of primary ciliated epithelial cells (Fig. 2) (Carlier 1900). However, the pronephros of both zebrafish and Xenopus use motile multiciliated cells to drive fluid flow through the kidney (Dressler 2006; Kramer-Zucker et al. 2005; Serluca et al. 2009). Ciliopathies of the kidney are the result of dysfunctional primary cilia, as motile cilia are not found in mammalian kidneys. Therefore, researchers tend to use other systems such as epidermal and nasal cilia when studying motile cilia, while sensory cilia tend to be the primary focus in kidney ciliopathies.
Figure 2:
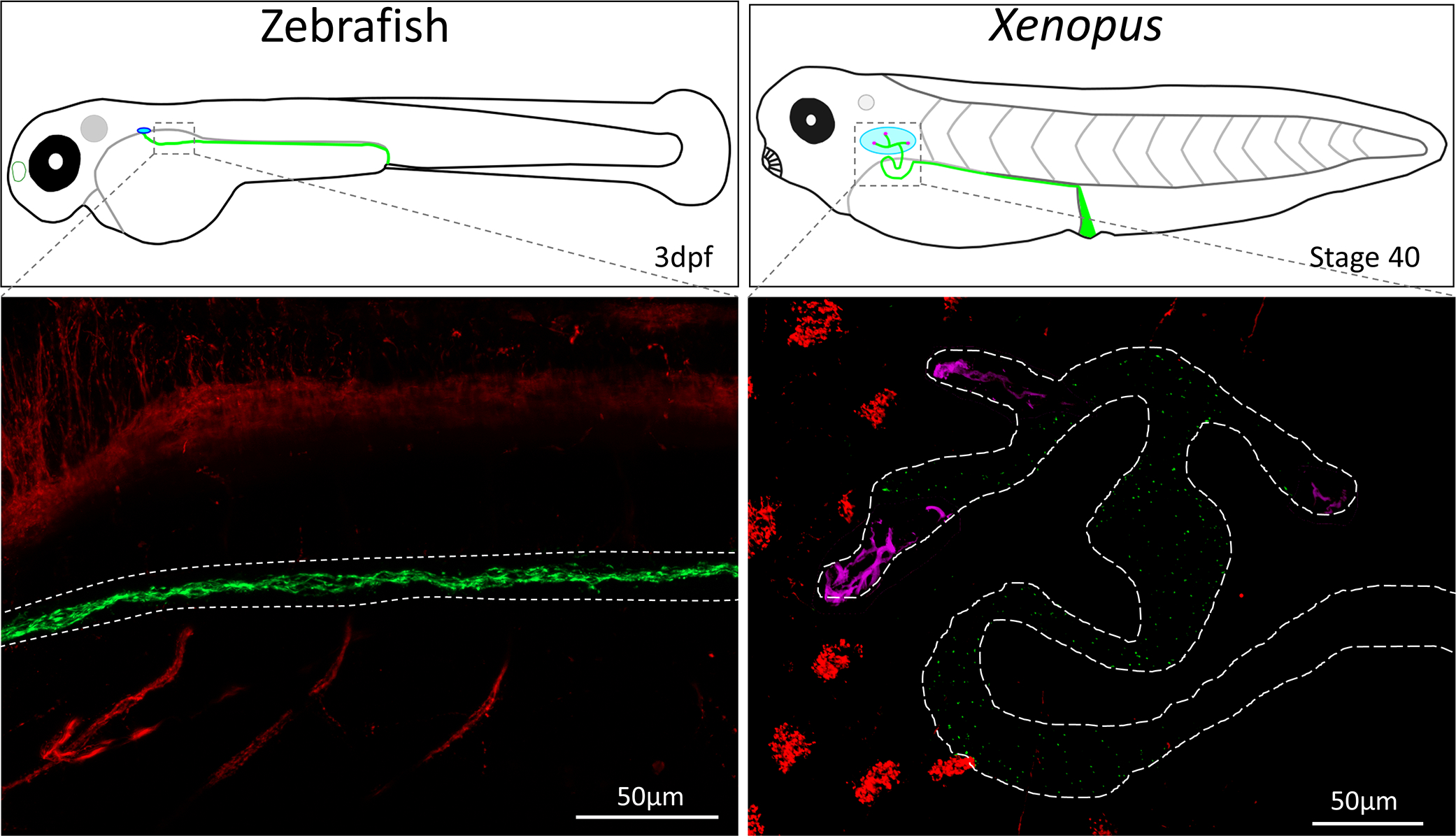
Confocal images of wholemount zebrafish (3dpf) and Xenopus laevis (Stage 37) kidney cilia. Cilia were stained using an acetylated alpha-tubulin antibody (Sigma T6793) which labels the neurons and cilia. Kidney cilia are pseudocolored in green while neurons and epithelial cilia are pseudocolored in red. The zebrafish and Xenopus kidney are outlined in white dashed lines, and motile multiciliated cells in the kidney are pseudocolored in magenta. Images were taken on a Zeiss LSM800 confocal microscope.
The predominant kidney phenotype seen in ciliopathies is cystic kidney diseases (CKDs) which occur in ~1/800 births, making them one of the most common life-threatening hereditary disorders (Belibi and Edelstein 2010; Wilson and Goilav 2007). Specific manifestations of each CDK depend on the gene affected. However, a common cause is malformation or dysfunction of primary cilia (Gascue, Katsanis, and Badano 2011). The most common cystic kidney disease arises from heritable mutations in the polycystin proteins PKD1 or PKD2 (cilia localized Ca2+ transporter complex), resulting in adult onset autosomal dominant polycystic kidney disease (ADPKD) (Harris and Torres 2009; Peters and Sandkuijl 1992). The polycystin complex transports calcium into the cilium in response to fluid movement (Chen et al. 1999; Huang et al. 2007; Zhu et al. 1996). Treatment of ADPKD primarily focuses on treating the symptoms with the eventual requirement of a kidney transplant or dialysis (Gascue et al. 2011; Patel, Chowdhury, and Igarashi 2009; Rizk and Chapman 2008). PKD is the cause of ~5% of all kidney failures requiring transplant (Lowrie and Hampers 1981).
Zebrafish PKD models of either pkd1 and pkd2 mutations have been established (Mangos et al. 2010; Obara et al. 2006). In addition, Pkd1 and Pkd2 morpholino knockdown results in cystic kidneys in both zebrafish and Xenopus (Zhang, Tran, and Wessely 2018). One of the underlying symptoms of polycystic kidney patients is the development of fibrosis. Fibrosis is the inappropriate extracellular matrix deposition, which normally occurs in response to injury. Morpholino knockdown of either Pkd1 or Pkd2 in zebrafish leads to inappropriate expression of collagen, which leads tail curvature defects. Knockdown of the collagen Col2a1 partially rescued this phenotype (Mangos et al. 2010). Given this tail curvature phenotype, pharmaceutical screens were undertaken to identify treatments for this disease (Metzner et al. 2020). From this screen, two novel pathways were identified alk5 kinase and non-canonical androgen receptors. Inhibition of these pathways not only rescues the curvature phenotype but also rescues the cystic kidney phenotypes seen in Pkd1 morphants. Additional work done in zebrafish has also found that the drug Metformin reduces the severity of cyst formation in pkd2 mutant models (Chang et al. 2017). Metformin is an AMPK activating drug. AMPK can directly phosphorylate β-catenin (Zhao et al. 2010), and Metformin has been found to inhibit Wnt signaling in mouse models of colin cancer (Park, Kim, and Kee 2019).
Forward genetic screens are possible in aquatic organisms, allowing for the identification of novel genes/pathways involved in human diseases. A mutagenesis was performed to identify novel genes that cause cystic kidney disease. For this screen they infected zebrafish with a virus that semi-randomly inserts a genetic element in the genome (Golling et al. 2002). This inserted element allows for quick identification of the affected genes. Approximately 400 unique genes were mutated and screened for cystic kidney diseases. As zebrafish are transparent, large kidney cysts are directly observable under a dissection scope without the need for staining. From this screen, 12 genes were pulled with six novel genes that have no identified biochemical function and are conserved to humans (Sun et al. 2004). Most of these genes showed similar phenotypes outside of the kidney which mimicked that of other known ciliopathies. Though not a novel pathway, one of the biggest gene families pulled was that of the IFT complexes. One of the first human ciliopathies identified was the result of a mutation in the gene ift88 (aka Polaris, or ORPK) (Cano et al. 2004). This study identified a new member of this complex involved in cystic kidney development in vertebrates. It also identified novel genes in cilia biogenesis that also result in cystic kidneys. Not only does zebrafish provide a good model to identify new genes involved in the development of cystic kidney disease, but it has also led to the identification of novel genes involved in ciliogenesis.
Neural cilia
There are a number of ciliopathies that lead to intellectual disabilities, including Joubert syndrome, Meckel syndrome, Bardet–Biedl syndrome, and Hydrolethalus syndrome (Valente et al. 2014). How loss of a cilia gene leads to mental impairment is largely unknown. As with other symptoms that are caused by malformed or dysfunctional cilia, the hedgehog (HH) and WNT signaling pathways are likely the underlying cause.
In humans, Joubert Syndrome is the result of a ciliopathy characterized by the absence or maldevelopment of a specific brain structure called the cerebellar vermis. This disease is associated with approximately 30 genes, and the majority of these genes either localize to or are involved in the assembly of the transition zones of the cilia (Shi et al. 2017). One of the more commonly associated genes with this disease is AHI1 (Jouberin or JBTS3). AHI1 is a ciliary transition zone protein of unknown function. The human AHI1 is structurally more similar to the zebrafish Ahi1 than the mouse Ahi1 (Zhu et al. 2019). Therefore, experiments were undertaken in zebrafish to understand the underlying defects upon loss of Ahi1. Joubert Syndrome patients not only have structural problems within the brain, but they also have vision problems (Parisi et al. 2006). During development of the visual system, the axons from each eye extend to the back of the brain crossing the midline and connecting to the opposite side of the brain (Joukal 2017). Given that zebrafish are optically clear, the neural retinal projections can be directly visualized by the injection of lipophilic dyes into the eye (Baier et al. 1996). This allows for easy tracking of axon migration. Either mutations in or loss of Ahi1 in zebrafish lead to problems with either crossing the midline or axonal elongation. Similar experiments have been done with other genes associated with Joubert syndrome, such as ARL13B (Zhu et al. 2020) and INPP5E (Luo, Lu, and Sun 2012). A novel causative gene Pibf1 was identified by exome sequencing of human Joubert syndrome patients, and the cilia phenotypes were verified in Xenopus (Ott et al. 2019).
A novel ciliopathy recently identified in Xenopus involves the protein Dyrk1a [dual specificity tyrosine-(Y)-phosphorylation-regulated kinase 1 A]. dryk1a is a gene that is associated with both Down syndrome and DYRK1A related intellectual disability syndrome. In humans, Down syndrome is the result of an extra copy of chromosome 21 which results in an extra copy of DYRK1A. On the opposite end of the spectrum DYRK1A related intellectual disability syndrome is the result of a mutated copy of DYRK1A leading to haploinsufficiency (Blackburn et al. 2019). Dyrk1a is not classically thought of as a ciliopathy gene, but recent work in Xenopus has found that dyrk1a is localized to puncta along ciliary axonemes and that loss of dyrk1a leads to loss of cilia in the epidermis (Willsey et al. 2020). RNAseq data indicate that cell cycle control genes are upregulated in Dyrk1a CRISPants. Since the cilia are thought to stall cell division, loss of cilia in DYRK1A related intellectual disability syndrome patients may lead to inappropriate cell division (Plotnikova, Pugacheva, and Golemis 2009; Tucker, Pardee, and Fujiwara 1979). Like many other ciliopathies loss of dyrk1a or many other cilia related genes involved in neural development are also associated with kidney abnormalities (Blackburn et al. 2019; Parisi et al. 2006).
Motile cilia.
Motile cilia form using similar machinery as primary cilia, but they are unlikely to have a sensory function. Motile cilia move fluid by oscillating back and forth in a wave like motion pushing against the fluid (Mov 1,2). Through electron microscopy, motile cilia can be identified through their characteristic central pair of microtubule filaments and dynein arms that extend from the 9 microtubule doublets within the axoneme (Fig. 1) (Rhodin 1959). Dynein is a microtubule motor protein that functions to move the cilia (King 2012). In mammals, motile cilia are found in the respiratory epithelium, fallopian tubes, sperm, and parts of the nervous system. Therefore, problems with motile cilia result in the inability to clear mucus from the lungs, leading to chronic infections and breathing difficulties (Austin-Tse et al. 2013). Additionally, infertility is seen in both genders, (Inaba and Mizuno 2016; Milla 2016; Raidt et al. 2015; Schneider et al. 2005) and hydrocephalus results from insufficient movement of cerebral fluid by motile cilia (Lee 2013). Though multiciliated tissues can be made from pluripotent stem cells, the process is laborious and takes days to form. Therefore there are currently no efficient cell culture models of either motile cilia or multiciliated cells, indicating the need for an animal model (Firth et al. 2014).
Epidermal cilia are one of the most studied cilia models in Xenopus. Motile multiciliated cells cover much of the epidermis (Fig. 3) and start to differentiate around 10 hours after fertilization (Nieuwkoop and Faber stage 11.5), and the motile cilia are fully formed and properly orientated within 35 hours (Stage 28) (Collins, Ventrella, and Mitchell 2020; Werner and Mitchell 2013). The likely function of these cells is to maintain fluid flow over the embryo to prevent bacteria or fungi from colonizing the skin, similar to their role in the mammalian lung. Given that the orientation of these cilia is essential to maintain directional fluid flow, pathways such as the planar cell polarity pathway aligns the cilia (Mitchell et al. 2009; Park et al. 2008; Yasunaga et al. 2015). It is easy observe the function of these cilia, as Xenopus sitting in a dish will slowly move anteriorly due to the fluid flow from these ciliary movements. The ciliary flow can also be demonstrated by placing dyes or beads are near the head of the embryo and observing their progression towards the posterior end of the embryo (Mov 3). These cilia are also easy to visualize outside of the embryo with the injection of a number of ciliary, or membrane markers (Werner and Mitchell 2013; Woolner, Miller, and Bement 2010). This ease of visualization and manipulation has lead to a better understanding of the mechanisms that is involved in both motile and primary cilia development (Kim et al. 2018; Marra et al. 2019).
Figure 3:
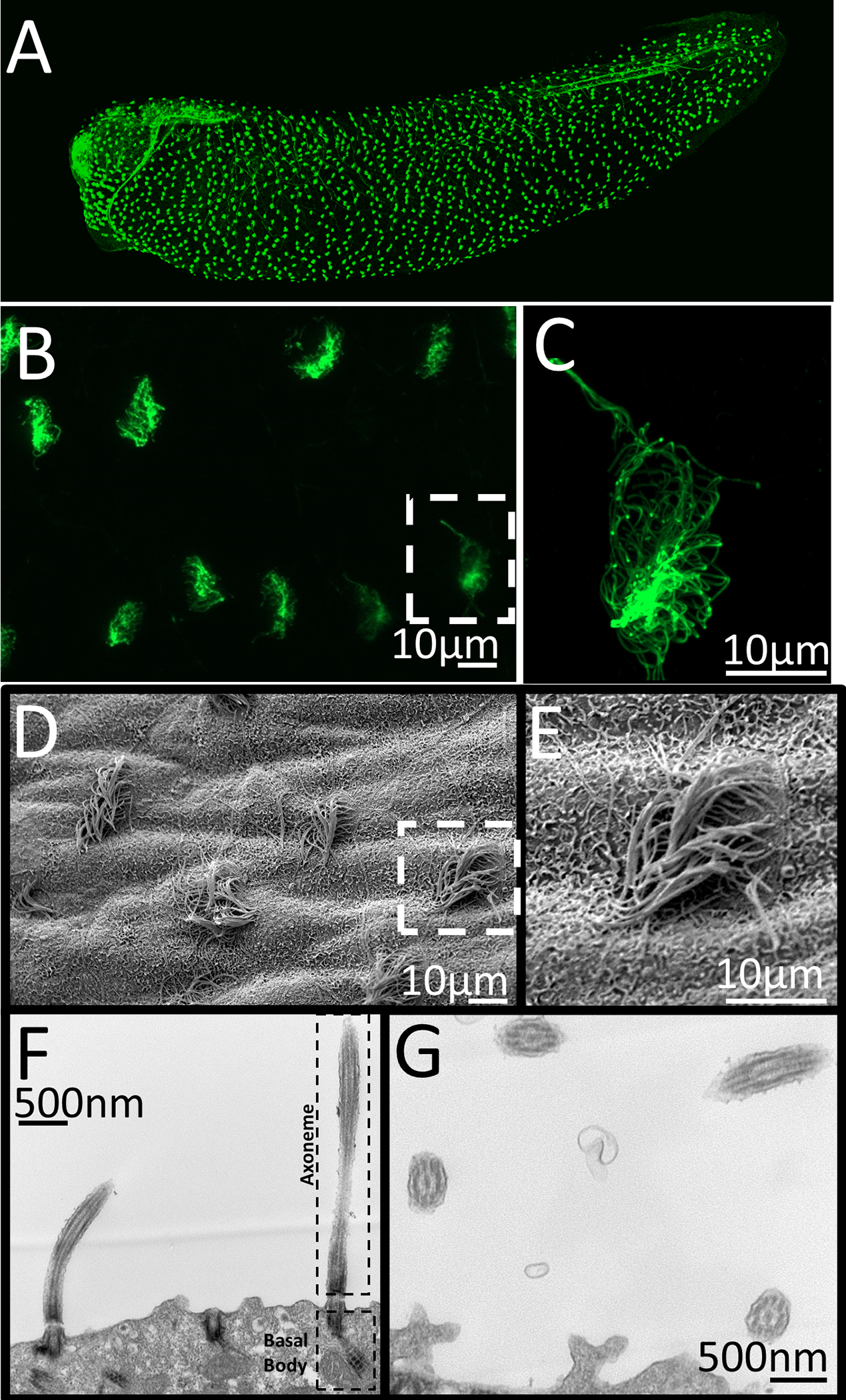
Images of Xenopus laevis motile epidermal cilia. A-C) Confocal imaging of acetylated alpha-tubulin stained whole mount Xenopus embryo. D-E) Scanning electron micrograph of the skin of whole mount Xenopus embryo. E-F) Transmission electron microscopy showing sections through cilia. F) Image showing basal body and axoneme of motile cilia G.) Image showing cross-section and the 9+2 microtubule structure of motile cilia. C,E) Zoomed in image of white dashed box in B and D.
The Xenopus embryonic epidermis is a mucociliary organ that is much like that of the upper respiratory tract in terrestrial vertebrates (Whitsett 2018). In fact, it contains many of the same cell types as the mucociliary epithelium of the lung, making it a good model of this tissue (Haas et al. 2019; Walentek 2018). Also, many of the genetic pathways involved in differentiating multiciliated cells, such as Notch and Wnt, are conserved in both the mammalian respiratory system and the Xenopus epidermis (Haas et al. 2019; Marcet et al. 2011; Rock et al. 2011; Schmid et al. 2017). In addition, lethal genes can be studied in Xenopus using organoids. Injection of mRNA, morpholino or CRISPR constructs followed by Isolation of pluripotent stem cells from blastula stage is and accessible technique. At the blastula stage, the cells sitting on top of the blastocoel, called the animal cap, can be explanted and differentiated ex vivo into many different tissue types, including kidney, neuronal and mucociliary epidermal tissues (Kim et al. 2020; Li et al. 2008; Sater, Steinhardt, and Keller 1993; Uochi and Asashima 1996). Given that each of these cells contain yolk, they will stay viable in a saline solution at room temperature for many days. The animal cap contains a pigmented epidermis. The removal of this epidermis stimulates the underlying cells to form a transparent mucociliary epidermis in under 24 hours, allowing for direct visualization of the developing mucociliary tissue. These advantages make it an attractive model that can overcome the technological challenges of mammalian systems.
Xenopus and zebrafish have been used to identify the pathways that facilitate the development of multiciliated cells. Just like in mouse, in Xenopus and zebrafish the transcription factor FoxJ1 appears to be a master regulator of motile cilia (Chen et al. 1998; Stubbs et al. 2008; Yu et al. 2008). In Xenopus and zebrafish FoxJ1 expression is largely restricted to motile ciliated cells such as multiciliated epidermal cells, multiciliated cells of the kidney and motile cilia of the node (Pohl and Knöchel 2004). Loss of FoxJ1 leads to loss of motile cilia and its misexpression can lead to ectopic cilia formation. Though FoxJ1 regulates the formation of motile cilia, it does not appear to influence the development or function of primary sensory cilia. The Rtx family of transcription factors, like FoxJ1, transcriptionally activates genes required for the development of motile cilia (Lemeille et al. 2020). Rtx proteins (Rtx1, Rtx2, Rtx3) potentially regulate a large number of gene targets. Approximately 350 genes have been identified as potential Rtx targets in Xenopus. To identify novel genes involved in the formation of cilia, injections of 259 unique plasmids from the human orfeome library encoding Rtx2 target proteins labeled with GFP were carried out. Each protein was coinjected with an RFP cilia marker and assayed for its localization within the cilia (Tu et al. 2018). 40 of these genes localize to ciliary structures, and 28 of these 40 have not previously been reported to have ciliary function or localization. Though injection of 259 constructs is still a significant amount of work, there are very few vertebrates in which this task is even feasible.
Validation of potential genes involved in motile ciliopathies is feasible in both Xenopus and zebrafish. In zebrafish and Xenopus the nasal pit is surrounded by peripheral motile cilia (Fig. 4) (Rachev et al. 2020; Reiten et al. 2017). These cilia are the easiest cilia in the zebrafish embryo to visualize. These cilia function to direct the flow of fluid over the sensory cells within the pit, including cells with primary cilia. This flow increases sensitivity and temporal resolution of the animal’s ability to detect odors in their environment. In humans, primary ciliary dyskinesia is a syndrome defined by nonfunctional motile cilia leading to chronic respiratory infections. Although, many of these patients also suffer from the other problems associated with motile ciliopathies (Bush et al. 1998). A genome wide association study on patients suffering from primary ciliary dyskinesia identified ten candidate genes to be a possible cause of this disfunction. (Austin-Tse et al. 2013). These patients suffered from a motile ciliopathy in which dynein arms fail to form. This results in the development of cilia that are unable to move. To test these candidate genes, morpholinos were injected then assessed for ciliary movement by brightfield microscopy of the olfactory pit. Of these ten genes, three showed strong motile ciliopathy phenotypes (c21orf59, ccdc65, and c15orf26). Injection of mRNAs allows for the expression of the exogenous human protein. This expression rescues the phenotypes observed upon knockdown of these genes in zebrafish. This not only confirms the phenotypes are due to knockdown of their respective gene, but it also demonstrates that the knockdowns affect the orthologs of the human genes and the function of these genes are evolutionarily conserved from human to zebrafish. Furthermore, multiple mutations in C21ORF59 were identified in patients (Austin-Tse et al. 2013). Severity of these mutations were measured in zebrafish by knocking down the endogenous protein and then expressing a copy of the human mutation to see if any of the human mutations rescued the morphant phenotypes. In this study, not only were multiple gene targets knocked down to identify causative genes, patient mutations were modeled in zebrafish, suggesting that these mutations likely play a causative role in the development of primary ciliary dyskinesia.
Figure 4:
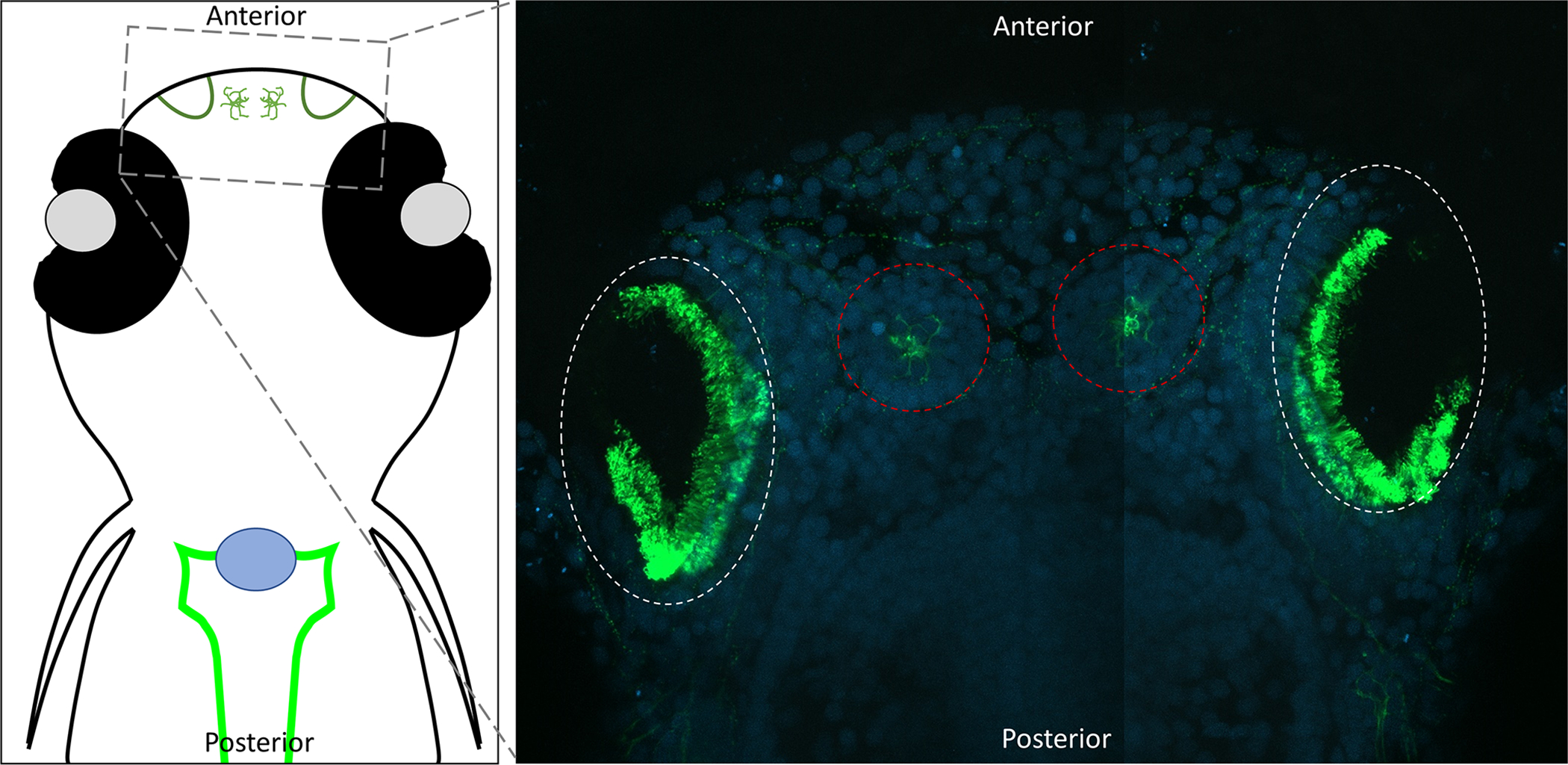
Confocal images of the motile cilia lining the zebrafish nasal (olfactory) pit. Dorsal view of 8dpf zebrafish embryos with head towards the top of the image. Embryos were fixed and stained with acetylated alpha-tubulin (Green) (Sigma T6793) and DAPI (Blue). Acetylated tubulin labels both the cilia and neurons. Nasal pits are circled in white, and neural mast cells are circled in red.
Complex and abnormal systems.
There are many biological systems that require a combination of motile cilia and sensory cilia. In the example given above, the zebrafish olfactory system uses motile cilia to maintain the flow of odors over primary cilia in the olfactory cleft. Both of the zebrafish and Xenopus pronephros use motile cilia to move fluid through the kidney tubules, which are lined with primary cilia. Another complex system is the left-right organizer.
Nodal cilia
The development of a left-right axis is an evolutionarily conserved process in vertebrates. Late in gastrulation near the onset of neurulation, a concavity called the node (aka gastrocoel roof plate, or Kupffer’s vesicle) is formed. In most vertebrates, the node contains sensory cilia on the edge of this depression, and motile monociliated cells within the cleft. These are abnormal motile cilia in that each cell contains a single cilia that do not have central pair of microtubules, but they do have the dynein arms required for movement (Huang, Hirota, and Sawamoto 2009). By currently unknown mechanisms, the establishment of planer cell polarity causes a posterior tilt of the motile cilia (Antic et al. 2010; Chien et al. 2018). This results in a leftward flow of fluid and activates the cilia on the left side of the node (Duncan and Khokha 2016; Okabe, Xu, and Burdine 2008; Schweickert et al. 2007). Activation of the cilia of the left half of the embryo results in opening of polycystin calcium channels. This influx of calcium represses calcium sensitive factors such as Coco (aka. Dand5) resulting in the activation of factors such as Nodal and Xnr1 on the left half of the embryo (Fig. 5) (Kamura et al. 2011; Schweickert et al. 2010). When pathways that affect the node are disrupted, development organs such as the heart and intestine are likewise disrupted. In some circumstances, a complete reversal of left right patterning occurs which is known as situs inversus. In this case, the intestine curves in the opposite direction, the heart is flipped, and the liver is on the right side. Approximately 1 in 10,000 individuals have some reversal of their left-right axis (Sharma 2012). A complete reversal normally does not cause any problems (Duncan and Khokha 2016). However, if the axis only partially inverts, then a serious situation called Heterotaxy syndrome occurs, which results coronary and digestive problems potentially requiring surgical correction (Hynes, Gau, and Titus 1973; Stamm et al. 2002; Yu et al. 2009).
Figure 5:
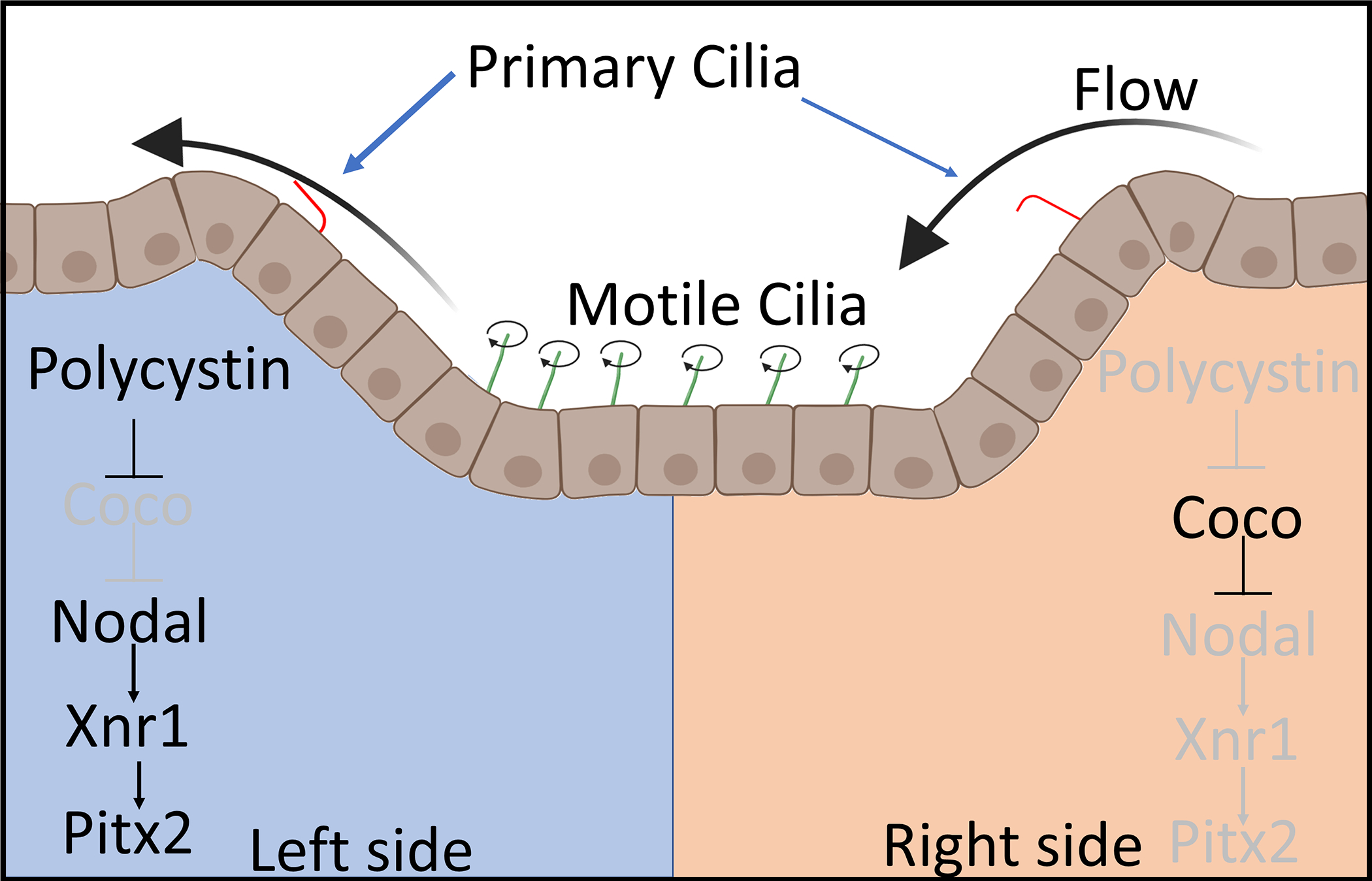
Diagram of a posterior view of the Left-Right organizer and its functions. Motile cilia (green) create a leftward flow of fluid over the cleft. This leftward flow activated primary cilia (red) on the left half of the cleft resulting in the opening of polycystin calcium channels. Calcium influx inhibits a protein Coco leading to activation of Nodal signaling.
In vertebrates, mutation of the Polycystin genes can not only lead to polycystic kidney disease but also left-right axis defects. Mutations in pkd1l1 (Pkd1 like #1) and pkd2 in vertebrates, lead to left-right axis problems (Bataille et al. 2011; Vetrini et al. 2016; Vick et al. 2018). Zebrafish studies have shown that Pkd1l1 forms a heterotetramer with Pkd2, which is important in regulating calcium signaling through the cilia within the node. In zebrafish, defects to nodal signaling can result in a distinctive curvature to the tail fin (Bisgrove et al. 2005). This obvious phenotype has allowed for high throughput screens using drug libraries to find treatments for Pkd2 caused by nodal ciliopathies (Metzner et al. 2020). From this screen, drugs that inhibited Alk5, a TGFβ receptor, were shown to rescue pkd2 tail curvature and cystic kidney phenotypes. Other potential drugs were identified as potential candidates for further study such, as COX-2 and HDAC inhibitors.
Genome sequencing of human patients has revealed many new putative disease genes. Recently, 61 candidate genes were identified as possibly causing axis problems in patients with situs inversus (Fakhro et al. 2011). Xenopus was used to test these genes as possible candidates in left-right axis formation. Seven genes showed promising expression within the ciliated cell of the node, and loss of five of these genes (rock2, galnt11, nek2, nup118, and tgfb2) resulted in axis defects, such as cardiac and intestinal looping anomalies. Loss of these genes also leads to misexpression of pitx2, which is a gene involved in axis formation and is largely only expressed on the left side of the embryo. Since these genes are expressed in other ciliary tissues, such as the kidney, it is likely that many of they play a role in either the development of or the signaling from cilia. Other genes have been identified from similar screens, starting from sequencing of human genome and utilizing Xenopus to identify factors involved in axis formation including Fgf4r (Sempou et al. 2018), Shroom3 (Tariq et al. 2011), and Pfkp (Cowan et al. 2016).
Auditory and vestibular cilia
Ear cilia are made up of two types of cilia known as kinocilia and stereocilia. Kinocilia are true cilia made up of tubulin. In mice, the kinocilia are predominantly the classical 9+0 form seen in sensory cilia; however, in mice 3–14% of the cilia have a 9+2 conformation associated with motile cilia (Sobkowicz, Slapnick, and August 1995). Stereocilia, however, are not true cilia, as they are made of actin and have more in common with microvilli than of cilia. The function of stereocilia is to attach to the kinocilia via cadherins and act as a directional signal (Müller 2008). One of these cadherins, Cdh23, was identified from a zebrafish mutant line called sputnik (Söllner et al. 2004). Kinocilia transmit sound by opening potassium channels in response to pulling from the attached stereocilia. Movement away from the stereocilia depolarizes the membrane by influx of potassium and movement towards the stereo cilia hyperpolarizes the membrane. Depolarization of the membrane leads to neurotransmitter release. Though few studies have focused on cilia in the Xenopus or zebrafish ear, we know that the zebrafish otic cilia have a similar structure and function as mouse cilia (Kindt, Finch, and Nicolson 2012; Whitfield 2020). However, unlike the adult human ear, regeneration of the ciliated sensory cells occurs in zebrafish (Monroe, Rajadinakaran, and Smith 2015). How this regeneration occurs is currently unknown, but it may help with understanding of hearing loss in humans.
Conclusion
Aquatic organisms, such as zebrafish and Xenopus, provide powerful tools to identify and dissect underlying causes of human diseases. Cilia are found in many tissues of the human and play a role in a multitude of cellular processes. Many of these disease manifestations, such as cystic diseases of the kidney, neurological malformations and the disrupted node function are conserved in zebrafish and Xenopus. The ability to perform mutagenesis and morpholino knockdowns in F0 organisms has provided the ability to rapidly screen many candidate genes from human genome sequencing studies. Expression of mRNAs allows for validation of knockdown/knockouts and characterization of the human mutations. This also allows for large scale protein expression analysis of a few hundred genes, leading to the possibility of performing large scale genetic screens to identify novel pathways to study. Their aquatic nature allows for efficient large scale drug screens to identify possible treatments. Coupling these features with their transparent epidermis and the multiple functional assays that have been developed to study cilia allows for efficient analysis of human ciliopathies.
Supplementary Material
Movie 1: Ciliary movement of multiciliated cells. Stage 40 X. laevis kidneys were dissected and dissociated into a single cell suspension. Cells were washed into PBS and DIC Movie was taken using an Olympus IX81 microscope. As the cilia beat it pushes against the fluid causing the cell to rotate.
Movie 2: Beating of Xenopus epidermal cilia. X. laevis embryos were injected with mRNA encoding for eGFP-alpha-tubulin (Woolner et al. 2010). Live stage 30 embryos were mounted on well slides and cilia were imaged on a 3i spinning disk confocal microscope.
Movie 3: In Xenopus motile cilia on their epidermis move fluid over their skin. Unanesthetized X. laevis embryos (Stage 30) were placed in a round bottom dish to prevent movement. On drop of 2mg/ml Rhodamine-dextran (Molecular probes D1817) placed on the posterior end of the tadpole. The embryo was imaged using an Olympus SZX16 dissection microscope. The Rhodamine diffuses around to the anterior before being pulled over the embryo.
Acknowledgements
We appreciate the helpful suggestions and advice throughout this project from the members of the laboratories of R. K. Miller, P. D. McCrea, J. Park as well as M. Kloc. A special thanks Yoshihiro Komatsu for the use of equipment in this project. We thank the animal care technicians and veterinarians, including J.C. Whitney and T.H. Gomez who took care of the animals, even during Hurricane Harvey. We appreciate training through the Cell & Developmental Biology of Xenopus: Gene Discovery & Disease course at Cold Spring Harbor, with a special thanks to Dr. Karen Liu, Dr. Mustafa Khokha and Dr. Emily Mis for preparation of RNA and embryos. Figures 1 and 5 were made with the help of Biorender.com.
National Institutes of Health (NIH) grants (R03DK118771 and R01DK115655 to R.K.M.), and startup funding from the Department of Pediatrics, Pediatric Research Center at UTHealth McGovern Medical School (to R.K.M.).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Ajima Rieko, and Hamada Hiroshi. 2011. “Wnt Signalling Escapes to Cilia.” Nature Cell Biology. [DOI] [PubMed] [Google Scholar]
- Antic Dragana, Stubbs Jennifer L., Suyama Kaye, Kintner Chris, Scott Matthew P., and Axelrod Jeffrey D.. 2010. “Planar Cell Polarity Enables Posterior Localization of Nodal Cilia and Left-Right Axis Determination during Mouse and Xenopus Embryogenesis.” PLoS ONE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslan Yetki, Tadjuidje Emmanuel, Zorn Aaron M., and Cha Sang Wook. 2017. “High-Efficiency Non-Mosaic CRISPR-Mediated Knock-in and Indel Mutation in F0 Xenopus.” Development (Cambridge). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin-Tse Christina, Halbritter Jan, Zariwala Maimoona A., Gilberti Renée M., Gee Heon Yung, Hellman Nathan, Pathak Narendra, Liu Yan, Panizzi Jennifer R., Patel-King Ramila S., Tritschler Douglas, Bower Raqual, O’Toole Eileen, Porath Jonathan D., Hurd Toby W., Chaki Moumita, Diaz Katrina A., Kohl Stefan, Lovric Svjetlana, Daw Yang Hwang Daniela A. Braun, Schueler Markus, Airik Rannar, Otto Edgar A., Leigh Margaret W., Noone Peadar G., Carson Johnny L., Davis Stephanie D., Pittman Jessica E., Ferkol Thomas W., Atkinson Jeffry J., Olivier Kenneth N., Sagel Scott D., Dell Sharon D., Rosenfeld Margaret, Milla Carlos E., Loges Niki T., Omran Heymut, Porter Mary E., King Stephen M., Knowles Michael R., Drummond Iain A., and Hildebrandt Friedhelm. 2013. “Zebrafish Ciliopathy Screen plus Human Mutational Analysis Identifies C21orf59 and CCDC65 Defects as Causing Primary Ciliary Dyskinesia.” American Journal of Human Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baier Herwig, Klostermann Stefan, Trowe Torsten, Karlstrom Rolf O., Nüsslein-Volhard Christiane, and Bonhoeffer Friedrich. 1996. “Genetic Dissection of the Retinotectal Projection.” Development. [DOI] [PubMed] [Google Scholar]
- Bataille Stanislas, Demoulin Nathalie, Devuyst Olivier, Audrézet Marie Pierre, Dahan Karin, Godin Michel, Fonts Michel, Pirson Yves, and Burtey Stéphane. 2011. “Association of PKD2 (Polycystin 2) Mutations with Left-Right Laterality Defects.” American Journal of Kidney Diseases. [DOI] [PubMed] [Google Scholar]
- Baye Lisa M., Patrinostro Xiaobai, Swaminathan Svetha, Beck John S., Zhang Yan, Stone Edwin M., Sheffield Val C., and Slusarski Diane C.. 2011. “The N-Terminal Region of Centrosomal Protein 290 (CEP290) Restores Vision in a Zebrafish Model of Human Blindness.” Human Molecular Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beales Philip L., and Kenny Thomas D.. 2014. “Towards the Diagnosis of a Ciliopathy.” in Ciliopathies. [Google Scholar]
- Belibi FA, and Edelstein CL. 2010. “Novel Targets for the Treatment of Autosomal Dominant Polycystic Kidney Disease.” Expert Opin Investig Drugs 19(3):315–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya Dipankan, Marfo Chris A., Li Davis, Lane Maura, and Khokha Mustafa K.. 2015. “CRISPR/Cas9: An Inexpensive, Efficient Loss of Function Tool to Screen Human Disease Genes in Xenopus.” Developmental Biology 408(2):196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgrove Brent W., Snarr Brian S., Emrazian Anoush, and Yost H. Joseph. 2005. “Polaris and Polycystin-2 in Dorsal Forerunner Cells and Kupffer’s Vesicle Are Required for Specification of the Zebrafish Left-Right Axis.” Developmental Biology. [DOI] [PubMed] [Google Scholar]
- Blackburn Alexandria T. M., Bekheirnia Nasim, Uma Vanessa C., Corkins Mark E., Xu Yuxiao, Rosenfeld Jill A., Bainbridge Matthew N., Yang Yaping, Liu Pengfei, Madan-Khetarpal Suneeta, Delgado Mauricio R., Hudgins Louanne, Krantz Ian, Rodriguez-Buritica David, Wheeler Patricia G., Gazali Lihadh Al, Mohamed Al Shamsi Aisha Mohamed Saeed, Gomez-Ospina Natalia, Chao Hsiao-Tuan, Mirzaa Ghayda M., Scheuerle Angela E., Kukolich Mary K., Scaglia Fernando, Eng Christine, Helen Rankin Willsey, Braun Michael C., Lamb Dolores J., Miller Rachel K., and Bekheirnia Mir Reza. 2019. “DYRK1A-Related Intellectual Disability: A Syndrome Associated with Congenital Anomalies of the Kidney and Urinary Tract.” Genetics in Medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn Alexandria T. M., and Miller Rachel K.. 2019. “Modeling Congenital Kidney Diseases in Xenopus Laevis.” DMM Disease Models and Mechanisms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford Yvonne M., Toro Sabrina, Ramachandran Sridhar, Ruzicka Leyla, Howe Douglas G., Eagle Anne, Kalita Patrick, Martin Ryan, Taylo. Moxon Sierra A., Schaper Kevin, and Westerfield Monte. 2017. “Zebrafish Models of Human Disease: Gaining Insight into Human Disease at ZFIN.” ILAR Journal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brändli André W. 1999. “Towards a Molecular Anatomy of the Xenopus Pronephric Kidney.” International Journal of Developmental Biology. [PubMed] [Google Scholar]
- Brugmann Samantha A., Cordero Dwight R., and Helms Jill A.. 2010. “Craniofacial Ciliopathies: A New Classification for Craniofacial Disorders.” American Journal of Medical Genetics, Part A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush A, Cole P, Hariri M, Mackay I, Phillips G, O’Callaghan C, Wilson R, and Warner JO. 1998. “Primary Ciliary Dyskinesia: Diagnosis and Standards of Care.” European Respiratory Journal. [DOI] [PubMed] [Google Scholar]
- Cano David A., Murcia Noel S., Pazour Gregory J., and Hebrok Matthias. 2004. “Orpk Mouse Model of Polycystic Kidney Disease Reveals Essential Role of Primary Cilia in Pancreatic Tissue Organization.” Development. [DOI] [PubMed] [Google Scholar]
- Carlier EW 1900. “Note on the Presence of Ciliated Cells in the Human Adult Kidney.” Journal of Anatomy. [PMC free article] [PubMed] [Google Scholar]
- Chang Ming Yang, Ma Tsu Lin, Hung Cheng Chieh, Tian Ya Chung, Chen Yung Chang, Yang Chih Wei, and Cheng Yi Chuan. 2017. “Metformin Inhibits Cyst Formation in a Zebrafish Model of Polycystin-2 Deficiency.” Scientific Reports. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Nannan, Sun Changhong, Gao Lu, Zhu Dan, Xu Xiufei, Zhu Xiaojun, Xiong Jing Wei, and Jeff Xi Jianzhong. 2013. “Genome Editing with RNA-Guided Cas9 Nuclease in Zebrafish Embryos.” Cell Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Jianchun, Knowles Heather J., Hebert Jennifer L., and Hackett Brian P.. 1998. “Mutation of the Mouse Hepatocyte Nuclear Factor/Forkhead Homologue 4 Gene Results in an Absence of Cilia and Random Left-Right Asymmetry.” Journal of Clinical Investigation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Xing Zhen, Vassilev Peter M., Basora Nuria, Ji Bin Peng Hideki Nomura, Segal Yoav, Brown Edward M., Reeders Stephen T., Hediger Matthias A., and Zhou Jing. 1999. “Polycystin-L Is a Calcium-Regulated Cation Channel Permeable to Calcium Ions.” Nature. [DOI] [PubMed] [Google Scholar]
- Chien Yuan Hung, Srinivasan Shyam, Keller Ray, and Kintner Chris. 2018. “Mechanical Strain Determines Cilia Length, Motility, and Planar Position in the Left-Right Organizer.” Developmental Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements Thomas P., Tandon Bhavna, Lintel Hendrik A., McCarty Joseph H., and Wagner Daniel S.. 2017. “RICE CRISPR: Rapidly Increased Cut Ends by an Exonuclease Cas9 Fusion in Zebrafish.” Genesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins Caitlin, Ventrella Rosa, and Mitchell Brian J.. 2020. “Building a Ciliated Epithelium: Transcriptional Regulation and Radial Intercalation of Multiciliated Cells.” in Current Topics in Developmental Biology. [DOI] [PubMed] [Google Scholar]
- Corbit Kevin C., Shyer Amy E., Dowdle William E., Gaulden Julie, Singla Veena, and Reiter Jeremy F.. 2008. “Kif3a Constrains β-Catenin-Dependent Wnt Signalling through Dual Ciliary and Non-Ciliary Mechanisms.” Nature Cell Biology. [DOI] [PubMed] [Google Scholar]
- Corkins Mark E., Hanania Hannah L., Krneta-Stankic Vanja, Delay Bridget D., Pearl Esther J., Lee Moonsup, Ji Hong, Davidson Alan J., Horb Marko E., and Miller Rachel K.. 2018. “Transgenic Xenopus Laevis Line for in Vivo Labeling of Nephrons within the Kidney.” Genes 9(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan Jason R., Tariq Muhammad, Shaw Chad, Rao Mitchell, Belmont John W., Lalani Seema R., Smolarek Teresa A., and Ware Stephanie M.. 2016. “Copy Number Variation as a Genetic Basis for Heterotaxy and Heterotaxy-Spectrum Congenital Heart Defects.” Philosophical Transactions of the Royal Society B: Biological Sciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl Hans A. 1963. “Fine Structure of Cilia in Rat Cerebral Cortex.” Zeitschrift Für Zellforschung Und Mikroskopische Anatomie. [DOI] [PubMed] [Google Scholar]
- DeLay Bridget D., Baldwin Tanya A., and Miller Rachel K.. 2019. “Dynamin Binding Protein Is Required for Xenopus Laevis Kidney Development.” Frontiers in Physiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delay Bridget D., Corkins Mark E., Hanania Hannah L., Salanga Matthew, Deng Jian Min, Sudou Norihiro, Taira Masanori, Horb Marko E., and Miller Rachel K.. 2018. “Tissue-Specific Gene Inactivation in Xenopus Laevis: Knockout of Lhx1 in the Kidney with CRISPR/Cas9.” Genetics 208(2):673–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diener Dennis R., Lupetti Pietro, and Rosenbaum Joel L.. 2015. “Proteomic Analysis of Isolated Ciliary Transition Zones Reveals the Presence of ESCRT Proteins.” Current Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd JM 1950. “Ciliary Feeding Mechanisms in Anuran Larvæ.” Nature (165):238. [DOI] [PubMed] [Google Scholar]
- Dressler Gregory R. 2006. “The Cellular Basis of Kidney Development.” Annual Review of Cell and Developmental Biology. [DOI] [PubMed] [Google Scholar]
- Duncan Anna R., and Khokha Mustafa K.. 2016. “Xenopus as a Model Organism for Birth Defects-Congenital Heart Disease and Heterotaxy.” Seminars in Cell and Developmental Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhro Khalid A., Choi Murim, Ware Stephanie M., Belmont John W., Towbin Jeffrey A., Lifton Richard P., Khokha Mustafa K., and Brueckner Martina. 2011. “Rare Copy Number Variations in Congenital Heart Disease Patients Identify Unique Genes in Left-Right Patterning.” Proceedings of the National Academy of Sciences of the United States of America. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth Amy L., Dargitz Carl T., Qualls Susan J., Menon Tushar, Wright Rebecca, Singer Oded, Gage Fred H., Khanna Ajai, and Verma Inder M.. 2014. “Generation of Multiciliated Cells in Functional Airway Epithelia from Human Induced Pluripotent Stem Cells.” Proceedings of the National Academy of Sciences of the United States of America. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher Shannon, Grice Elizabeth A., Vinton Ryan M., Bessling Seneca L., Urasaki Akihiro, Kawakami Koichi, and McCallion Andrew S.. 2006. “Evaluating the Biological Relevance of Putative Enhancers Using Tol2 Transposon-Mediated Transgenesis in Zebrafish.” Nature Protocols. [DOI] [PubMed] [Google Scholar]
- Gascue Cecilia, Katsanis Nicholas, and Badano Jose L.. 2011. “Cystic Diseases of the Kidney: Ciliary Dysfunction and Cystogenic Mechanisms.” Pediatric Nephrology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golling Gregory, Amsterdam Adam, Sun Zhaoxia, Antonelli Marcelo, Maldonado Ernesto, Chen Wenbiao, Burgess Shawn, Haldi Maryann, Artzt Karen, Farrington Sarah, Lin Shuh Yow, Nissen Robert M., and Hopkins Nancy. 2002. “Insertional Mutagenesis in Zebrafish Rapidly Identifies Genes Essential for Early Vertebrate Development.” Nature Genetics. [DOI] [PubMed] [Google Scholar]
- Grove Erin, Eckardt Sigrid, and McLaughlin K. John. 2016. “High-Speed Mouse Backcrossing through the Female Germ Line.” PLoS ONE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas Maximilian, Vázquez José Luis Gómez, Sun Dingyuan Iris, Tran Hong Thi, Brislinger Magdalena, Tasca Alexia, Shomroni Orr, Vleminckx Kris, and Walentek Peter. 2019. “ΔN-Tp63 Mediates Wnt/β-Catenin-Induced Inhibition of Differentiation in Basal Stem Cells of Mucociliary Epithelia.” Cell Reports. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris Peter C., and Torres Vicente E.. 2009. “Polycystic Kidney Disease.” Annual Review of Medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Kaiyao, Diener Dennis R., Mitchell Aaron, Pazour Gregory J., Witman George B., and Rosenbaum Joel L.. 2007. “Function and Dynamics of PKD2 in Chlamydomonas Reinhardtii Flagella.” Journal of Cell Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Shihhui, Hirota Yuki, and Sawamoto Kazunobu. 2009. “Various Facets of Vertebrate Cilia: Motility, Signaling, and Role in Adult Neurogenesis.” Proceedings of the Japan Academy Series B: Physical and Biological Sciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu Danwei, Liu Aimin, Rakeman Andrew S., Murcia Noel S., Niswander Lee, and Anderson Kathryn V.. 2003. “Hedgehog Signalling in the Mouse Requires Intraflagellar Transport Proteins.” Nature. [DOI] [PubMed] [Google Scholar]
- Hynes Kieran M., Gau Gerald T., and Titus Jack L.. 1973. “Coronary Heart Disease in Situs Inversus Totalis.” The American Journal of Cardiology. [DOI] [PubMed] [Google Scholar]
- Inaba Kazuo, and Mizuno Katsutoshi. 2016. “Sperm Dysfunction and Ciliopathy.” Reproductive Medicine and Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joukal Marek. 2017. “Anatomy of the Human Visual Pathway.” in Homonymous Visual Field Defects. [Google Scholar]
- Kamura Keiichiro, Kobayashi Daisuke, Uehara Yuka, Koshida Sumito, Iijima Norio, Kudo Akira, Yokoyama Takahiko, and Takeda Hiroyuki. 2011. “Pkd1l1 Complexes with Pkd2 on Motile Cilia and Functions to Establish the Left-Right Axis.” Development. [DOI] [PubMed] [Google Scholar]
- Kim Hye Young, Jackson Timothy R., Stuckenholz Carsten, and Davidson Lance A.. 2020. “Tissue Mechanics Drives Regeneration of a Mucociliated Epidermis on the Surface of Xenopus Embryonic Aggregates.” Nature Communications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Seongjae, Ma Lina, Shokhirev Maxim N., Quigley Ian, and Kintner Chris. 2018. “Multicilin and Activated E2f4 Induce Multiciliated Cell Differentiation in Primary Fibroblasts.” Scientific Reports. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindt Katie S., Finch Gabriel, and Nicolson Teresa. 2012. “Kinocilia Mediate Mechanosensitivity in Developing Zebrafish Hair Cells.” Developmental Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King Stephen M. 2012. “Integrated Control of Axonemal Dynein AAA+ Motors.” Journal of Structural Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer-Zucker Albrecht G., Olale Felix, Haycraft Courtney J., Yoder Bradley K., Schier Alexander F., and Drummond Iain A.. 2005. “Cilia-Driven Fluid Flow in the Zebrafish Pronephros, Brain and Kupffer’s Vesicle Is Required for Normal Organogenesis.” Development. [DOI] [PubMed] [Google Scholar]
- Kühl Michael, Sheldahl Laird C., Malbon Craig C., and Moon Randall T.. 2000. “Ca2+/Calmodulin-Dependent Protein Kinase II Is Stimulated by Wnt and Frizzled Homologs and Promotes Ventral Cell Fates in Xenopus.” Journal of Biological Chemistry. [DOI] [PubMed] [Google Scholar]
- Lal Mark, Song Xuewen, Pluznick Jennifer L., Giovanni Valeria Di, Merrick David M., Rosenblum Norman D., Chauvet Veronique, Gottardi Cara J., Pei York, and Caplan Michael J.. 2008. “Polycystin-1 C-Terminal Tail Associates with β-Catenin and Inhibits Canonical Wnt Signaling.” Human Molecular Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Schroth J, and Gleeson JG. 2011. “Subcellular Spatial Regulation of Canonical Wnt Signalling at the Primary Cilium.” Nat Cell Biol 13(6):700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurence JZ, and Moon RC. 1995. “Four Cases of ‘Retinitis Pigmentosa’ Occurring in the Same Family, and Accompanied by General Imperfections of Development. 1866.” Obesity Research. [DOI] [PubMed] [Google Scholar]
- Lawrence Christian, Adatto Isaac, Best Jason, James Althea, and Maloney Kara. 2012. “Generation Time of Zebrafish (Danio Rerio) and Medakas (Oryzias Latipes) Housed in the Same Aquaculture Facility.” Lab Animal. [DOI] [PubMed] [Google Scholar]
- Lee Lance. 2013. “Riding the Wave of Ependymal Cilia: Genetic Susceptibility to Hydrocephalus in Primary Ciliary Dyskinesia.” Journal of Neuroscience Research. [DOI] [PubMed] [Google Scholar]
- Lemeille Sylvain, Paschaki Marie, Baas Dominique, Laurette Morlé Jean Luc Duteyrat, Aouatef Ait-Lounis Emmanuèle Barras, Soulavie Fabien, Jerber Julie, Thomas Joëlle, Zhang Yong, Holtzman Michael J., Kistler W. Stephen, Reith Walter, and Durand Bénédicte. 2020. “Interplay of RFX Transcription Factors 1, 2 and 3 in Motile Ciliogenesis.” Nucleic Acids Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Ao, Xu Yuchen, Fan Song, Meng Jialin, Shen Xufeng, Xiao Qian, Li Yuan, Zhang Li, Zhang Xiansheng, Wu Guanqing, Liang Chaozhao, and Wu Dianqing. 2018. “Canonical Wnt Inhibitors Ameliorate Cystogenesis in a Mouse Ortholog of Human ADPKD.” JCI Insight. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FQ, Mofunanya A, Harris K, and Takemaru K. 2008. “Chibby Cooperates with 14–3-3 to Regulate Beta-Catenin Subcellular Distribution and Signaling Activity.” J Cell Biol 181(7):1141–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrie Edmund G., and Hampers CL. 1981. “The Success of Medicare’s End-Stage Renal-Disease Program.” New England Journal of Medicine. [DOI] [PubMed] [Google Scholar]
- Luo Na, Lu Jingping, and Sun Yang. 2012. “Evidence of a Role of Inositol Polyphosphate 5-Phosphatase INPP5E in Cilia Formation in Zebrafish.” Vision Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangos Steve, Pui Ying Lam Angela Zhao, Liu Yan, Mudumana Sudha, Vasilyev Aleksandr, Liu Aiping, and Drummond Iain A.. 2010. “The ADPKD Genes Pkd1a/b and Pkd2 Regulate Extracellular Matrix Formation.” DMM Disease Models and Mechanisms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcet Brice, Chevalier Benoît, Luxardi Guillaume, Coraux Christelle, Laure Emmanuelle Zaragosi Marie Cibois, Karine Robbe-Sermesant Thomas Jolly, Cardinaud Bruno, Moreilhon Chimène, Giovannini-Chami Lisa, Nawrocki-Raby Béatrice, Birembaut Philippe, Waldmann Rainer, Kodjabachian Laurent, and Barbry Pascal. 2011. “Control of Vertebrate Multiciliogenesis by MiR-449 through Direct Repression of the Delta/Notch Pathway.” Nature Cell Biology. [DOI] [PubMed] [Google Scholar]
- Marra Amanda N., Adeeb Basma D., Chambers Brooke E., Drummond Bridgette E., Ulrich Marisa, Addiego Amanda, Springer Meghan, Poureetezadi Shahram J., Chambers Joseph M., Ronshaugen Matthew, and Wingert Rebecca A.. 2019. “Prostaglandin Signaling Regulates Renal Multiciliated Cell Specification and Maturation.” Proceedings of the National Academy of Sciences of the United States of America. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzner A, Griffiths JD, Streets AJ, Markham E, Philippou T, Van Eeden FJM, and Ong ACM. 2020. “A High Throughput Zebrafish Chemical Screen Reveals ALK5 and Non-Canonical Androgen Signalling as Modulators of the Pkd2 −/− Phenotype.” Scientific Reports. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milla Carlos E. 2016. “The Evolving Spectrum of Ciliopathies and Respiratory Disease.” Current Opinion in Pediatrics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller Rachel K., Lee Moonsup, and McCrea Pierre D.. 2014. “ The Xenopus Pronephros.” in Xenopus Development. [Google Scholar]
- Mitchell Brian, Stubbs Jennifer L., Huisman Fawn, Taborek Peter, Yu Clare, and Kintner Chris. 2009. “The PCP Pathway Instructs the Planar Orientation of Ciliated Cells in the Xenopus Larval Skin.” Current Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe Jerry D., Rajadinakaran Gopinath, and Smith Michael E.. 2015. “Sensory Hair Cell Death and Regeneration in Fishes.” Frontiers in Cellular Neuroscience. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody Sally A. 1987a. “Fates of the Blastomeres of the 16-Cell Stage Xenopus Embryo.” Developmental Biology. [DOI] [PubMed] [Google Scholar]
- Moody Sally A. 1987b. “Fates of the Blastomeres of the 32-Cell-Stage Xenopus Embryo.” Developmental Biology 122(2):300–319. [DOI] [PubMed] [Google Scholar]
- Müller Ulrich. 2008. “Cadherins and Mechanotransduction by Hair Cells.” Current Opinion in Cell Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nenni Mardi J., Fisher Malcolm E., Christina James-Zorn Troy J. Pells, Ponferrada Virgilio, Chu Stanley, Fortriede Joshua D., Burns Kevin A., Wang Ying, Lotay Vaneet S., Wang Dong Zhou, Segerdell Erik, Chaturvedi Praneet, Karimi Kamran, Vize Peter D., and Zorn Aaron M.. 2019. “XenBase: Facilitating the Use of Xenopus to Model Human Disease.” Frontiers in Physiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikai H, Rose GG, and Cattoni M. 1970. “Electron Microscopy of Solitary Cilia in Human Gingiva and Rat Oral Mucosa.” Journal of Dental Research. [DOI] [PubMed] [Google Scholar]
- Noda Kazuo, Kitami Megumi, Kitami Kohei, Kaku Masaru, and Komatsu Yoshihiro. 2016. “Canonical and Noncanonical Intraflagellar Transport Regulates Craniofacial Skeletal Development.” Proceedings of the National Academy of Sciences of the United States of America. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara Tomoko, Mangos Steven, Liu Yan, Zhao Jinhua, Wiessner Stephanie, Kramer-Zucker Albrecht G., Olale Felix, Schier Alexander F., and Drummond Iain A.. 2006. “Polycystin-2 Immunolocalization and Function in Zebrafish.” Journal of the American Society of Nephrology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocbina Polloneal Jymmiel R., Tuson Miquel, and Anderson Kathryn V.. 2009. “Primary Cilia Are Not Required for Normal Canonical Wnt Signaling in the Mouse Embryo.” PLoS ONE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino Hajime, McConnell William B., and Grainger Robert M.. 2006. “High-Throughput Transgenesis in Xenopus Using I-SceI Meganuclease.” Nature Protocols. [DOI] [PubMed] [Google Scholar]
- Okabe Noriko, Xu Bo, and Burdine Rebecca D.. 2008. “Fluid Dynamics in Zebrafish Kupffer’s Vesicle.” Developmental Dynamics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott Tim, Kaufmann Lilian, Granzow Martin, Hinderhofer Katrin, Bartram Claus R., Susanne Theiß Angelika Seitz, Paramasivam Nagarajan, Schulz Angela, Moog Ute, Blum Martin, and Evers Christina M.. 2019. “The Frog Xenopus as a Model to Study Joubert Syndrome: The Case of a Human Patient with Compound Heterozygous Variants in PIBF1.” Frontiers in Physiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi Melissa A., Doherty D, Eckert ML, Shaw DWW, Ozyurek H, Aysun S, Giray O, Al Swaid A, Al Shahwan S, Dohayan N, Bakhsh E, Indridason OS, Dobyns WB, Bennett CL, Chance PF, and Glass IA. 2006. “AHI1 Mutations Cause Both Retinal Dystrophy and Renal Cystic Disease in Joubert Syndrome.” Journal of Medical Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Song Yi, Kim Dasarang, and Kee Sun Ho. 2019. “Metformin-Activated AMPK Regulates β-Catenin to Reduce Cell Proliferation in Colon Carcinoma RKO Cells.” Oncology Letters. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Tae Joo, Mitchell Brian J., Abitua Philip B., Kintner Chris, and Wallingford John B.. 2008. “Dishevelled Controls Apical Docking and Planar Polarization of Basal Bodies in Ciliated Epithelial Cells.” Nature Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel Vishal, Chowdhury Renuka, and Igarashi Peter. 2009. “Advances in the Pathogenesis and Treatment of Polycystic Kidney Disease.” Current Opinion in Nephrology and Hypertension. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters DJ, and Sandkuijl LA. 1992. “Genetic Heterogeneity of Polycystic Kidney Disease in Europe.” Contributions to Nephrology. [DOI] [PubMed] [Google Scholar]
- Plotnikova Olga V., Pugacheva Elena N., and Golemis Erica A.. 2009. “Primary Cilia and the Cell Cycle.” Methods in Cell Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl Barbara S., and Knöchel Walter. 2004. “Isolation and Developmental Expression of Xenopus FoxJ1 and FoxK1.” Development Genes and Evolution. [DOI] [PubMed] [Google Scholar]
- Preble Andrea M., Giddings Thomas M. Jr., and Dutcher Susan K.. 1999. “Basal Bodies and Centrioles: Their Function and Structure.” Current Topics in Developmental Biology 49:207–33. [DOI] [PubMed] [Google Scholar]
- Rachel Rivka A., Yamamoto Erin A., Dewanjee Mrinal K., May-Simera Helen L., Sergeev Yuri V., Hackett Alice N., Pohida Katherine, Munasinghe Jeeva, Gotoh Norimoto, Wickstead Bill, Fariss Robert N., Dong Lijin, Li Tiansen, and Swaroop Anand. 2015. “CEP290 Alleles in Mice Disrupt Tissue-Specific Cilia Biogenesis and Recapitulate Features of Syndromic Ciliopathies.” Human Molecular Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachev Ev, Schuster-Gossler Karin, Fuhl Franziska, Ott Tim, Tveriakhina Lena, Beckers Anja, Hegermann Jan, Boldt Karsten, Mai Michaela, Kremmer Elisabeth, Ueffing Marius, Blum Martin, and Gossler Achim. 2020. “CFAP43 Modulates Ciliary Beating in Mouse and Xenopus.” Developmental Biology. [DOI] [PubMed] [Google Scholar]
- Raidt Johanna, Werner Claudius, Menchen Tabea, Dougherty Gerard W., Olbrich Heike, Loges Niki T., Schmitz Ralf, Pennekamp Petra, and Omran Heymut. 2015. “Ciliary Function and Motor Protein Composition of Human Fallopian Tubes.” Human Reproduction. [DOI] [PubMed] [Google Scholar]
- Reese TS 1965. “Olfactory Cilia in the Frog.” Journal of Cell Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiten Ingrid, Uslu Fazil Emre, Fore Stephanie, Pelgrims Robbrecht, Ringers Christa, Verdugo Carmen Diaz, Hoffman Maximillian, Lal Pradeep, Kawakami Koichi, Pekkan Kerem, Yaksi Emre, and Jurisch-Yaksi Nathalie. 2017. “Motile-Cilia-Mediated Flow Improves Sensitivity and Temporal Resolution of Olfactory Computations.” Current Biology. [DOI] [PubMed] [Google Scholar]
- Rhodin J 1959. “LXVII Ultrastructure of the Tracheal Ciliated Mucosa in Rat and Man.” Annals of Otology, Rhinology … Laryngology. [Google Scholar]
- Rhodin Johannes, and Dalhamn Tore. 1956. “Electron Microscopy of the Tracheal Ciliated Mucosa in Rat.” Zeitschrift Für Zellforschung Und Mikroskopische Anatomie. [DOI] [PubMed] [Google Scholar]
- Rizk Dana, and Chapman Arlene. 2008. “Treatment of Autosomal Dominant Polycystic Kidney Disease (ADPKD): The New Horizon for Children with ADPKD.” Pediatric Nephrology. [DOI] [PubMed] [Google Scholar]
- Rock Jason R., Gao Xia, Xue Yan, Randell Scott H., Kong Young Yun, and Hogan Brigid L. M.. 2011. “Notch-Dependent Differentiation of Adult Airway Basal Stem Cells.” Cell Stem Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sater Amy K., Steinhardt Richard A., and Keller Ray. 1993. “Induction of Neuronal Differentiation by Planar Signals in Xenopus Embryos.” Developmental Dynamics. [DOI] [PubMed] [Google Scholar]
- Schmid Andreas, Sailland Juliette, Novak Lisa, Baumlin Nathalie, Fregien Nevis, and Salathe Matthias. 2017. “Modulation of Wnt Signaling Is Essential for the Differentiation of Ciliated Epithelial Cells in Human Airways.” FEBS Letters. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid Fabian Marc, Schou Kenneth Bødtker, Vilhelm Martin Juel, Holm Maria Schrøder, Breslin Loretta, Farinelli Pietro, Larsen Lars Allan, Andersen Jens Skorstengaard, Pedersen Lotte Bang, and Christensen Søren Tvorup. 2018. “IFT20 Modulates Ciliary PDGFRα Signaling by Regulating the Stability of Cbl E3 Ubiquitin Ligases.” Journal of Cell Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, and Christensen ST. 2005. “PDGFRalphaalpha Signaling Is Regulated through the Primary Cilium in Fibroblasts.” Curr Biol 15(20):1861–66. [DOI] [PubMed] [Google Scholar]
- Schweickert Axel, Vick Philipp, Getwan Maike, Weber Thomas, Schneider Isabelle, Eberhardt Melanie, Beyer Tina, Pachur Anke, and Blum Martin. 2010. “The Nodal Inhibitor Coco Is a Critical Target of Leftward Flow in Xenopus.” Current Biology. [DOI] [PubMed] [Google Scholar]
- Schweickert Axel, Weber Thomas, Beyer Tina, Vick Philipp, Bogusch Susanne, Feistel Kerstin, and Blum Martin. 2007. “Cilia-Driven Leftward Flow Determines Laterality in Xenopus.” Current Biology. [DOI] [PubMed] [Google Scholar]
- Sempou Emily, Lakhani Osaamah Ali, Amalraj Sarah, and Khokha Mustafa K.. 2018. “Candidate Heterotaxy Gene FGFR4 Is Essential for Patterning of the Left-Right Organizer in Xenopus.” Frontiers in Physiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serluca Fabrizio C., Xu Bo, Okabe Noriko, Baker Kari, Lin Shin Yi, Sullivan-Brown Jessica, Konieczkowski David J., Jaffe Kimberly M., Bradner Joshua M., Fishman Mark C., and Burdine Rebecca D.. 2009. “Mutations in Zebrafish Leucine-Rich Repeat-Containing Six-like Affect Cilia Motility and Result in Pronephric Cysts, but Have Variable Effects on Left-Right Patterning.” Development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma Sharada. 2012. “Situs Inversus Totalis (Dextroversion) - An Anatomical Study.” Anatomy & Physiology. [Google Scholar]
- Shi Xiaoyu, Garcia Galo, Van De Weghe Julie C., McGorty Ryan, Pazour Gregory J., Doherty Dan, Huang Bo, and Reiter Jeremy F.. 2017. “Super-Resolution Microscopy Reveals That Disruption of Ciliary Transition-Zone Architecture Causes Joubert Syndrome.” Nature Cell Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobkowicz HM, Slapnick SM, and August BK. 1995. “The Kinocilium of Auditory Hair Cells and Evidence for Its Morphogenetic Role during the Regeneration of Stereocilia and Cuticular Plates.” Journal of Neurocytology. [DOI] [PubMed] [Google Scholar]
- Söllner Christian, Nicolson Teresa, Rauch Gerd Jörg, Geisler Robert, Schuster Stephan C., Siemens Jan, Müller Ulrich, Nicolson Teresa, Van Bebber F, Busch-Nentwich E, Dahm R, Frank O, Frohnhöfer HG, Geiger H, Gilmour D, Holley S, Hooge J, Jülich D, Knaut H, Maderspacher F, Maischein HM, Neumann C, Nüsslein-Volhard C, Roehl H, Schönberger U, Seiler C, Sidi S, Sonawane M, Wehner A, Erker P, Habeck H, Hagner U, Hennen Kaps CE, Kirchner A, Koblizek T, Langheinrich U, Loeschke C, Metzger C, Nordin R, Odenthal J, Pezzuti M, Schlombs K, de Santana-Stamm J, Trowe T, Vacun G, Walderich B, Walker A, and Weiler C. 2004. “Mutations in Cadherin 23 Affect Tip Links in Zebrafish Sensory Hair Cells.” Nature. [DOI] [PubMed] [Google Scholar]
- Srivastava Shalabh, Ramsbottom Simon A., Molinari Elisa, Alkanderi Sumaya, Filby Andrew, White Kathryn, Henry Charline, Saunier Sophie, Miles Colin G., and Sayer John A.. 2017. “A Human Patient-Derived Cellular Model of Joubert Syndrome Reveals Ciliary Defects Which Can Be Rescued with Targeted Therapies.” Human Molecular Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm Christof, Friehs Ingeborg, Duebener Lennart F., Zurakowski David, Mayer John E., Jonas Richard A., and Del Nido Pedro J.. 2002. “Improving Results of the Modified Fontan Operation in Patients with Heterotaxy Syndrome.” Annals of Thoracic Surgery. [DOI] [PubMed] [Google Scholar]
- Stubbs JL, Oishi I, Izpisua Belmonte JC, and Kintner C. 2008. “The Forkhead Protein Foxj1 Specifies Node-like Cilia in Xenopus and Zebrafish Embryos.” Nat Genet 40(12):1454–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Shufeng, Fisher Rebecca L., Bowser Samuel S., Pentecost Brian T., and Sui Haixin. 2019. “Three-Dimensional Architecture of Epithelial Primary Cilia.” Proceedings of the National Academy of Sciences of the United States of America. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Amsterdam A, Pazour GJ, Cole DG, Miller MS, and Hopkins N. 2004. “A Genetic Screen in Zebrafish Identifies Cilia Genes as a Principal Cause of Cystic Kidney.” Development 131(16):4085–93. [DOI] [PubMed] [Google Scholar]
- Tariq Muhammad, Belmont John W., Lalani Seema, Smolarek Teresa, and Ware Stephanie M.. 2011. “SHROOM3 Is a Novel Candidate for Heterotaxy Identified by Whole Exome Sequencing.” Genome Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Fan, Sedzinski Jakub, Ma Yun, Marcotte Edward M., and Wallingford John B.. 2018. “Protein Localization Screening in Vivo Reveals Novel Regulators of Multiciliated Cell Development and Function.” Journal of Cell Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker Robert W., Pardee Arthur B., and Fujiwara Keigi. 1979. “Centriole Ciliation Is Related to Quiescence and DNA Synthesis in 3T3 Cells.” Cell. [DOI] [PubMed] [Google Scholar]
- Uochi Takaaki, and Asashima Makoto. 1996. “Sequential Gene Expression during Pronephric Tubule Formation in Vitro in Xenopus Ectoderm.” Development Growth and Differentiation. [DOI] [PubMed] [Google Scholar]
- Uytingco Cedric R., Williams Corey L., Xie Chao, Shively Dana T., Green Warren W., Ukhanov Kirill, Zhang Lian, Nishimura Darryl Y., Sheffield Val C., and Martens Jeffrey R.. 2019. “BBS4 Is Required for Intraflagellar Transport Coordination and Basal Body Number in Mammalian Olfactory Cilia.” Journal of Cell Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente Enza Maria, Rosti Rasim O., Gibbs Elizabeth, and Gleeson Joseph G.. 2014. “Primary Cilia in Neurodevelopmental Disorders.” Nature Reviews Neurology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veleri Shobi, Bishop Kevin, Dall. Nogare Damian E., English Milton A., Foskett Trevor J., Chitnis Ajay, Sood Raman, Liu Paul, and Swaroop Anand. 2012. “Knockdown of Bardet-Biedl Syndrome Gene BBS9/PTHB1 Leads to Cilia Defects.” PLoS ONE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrini Francesco, D’Alessandro Lisa C. A., Akdemir Zeynep C., Braxton Alicia, Azamian Mahshid S., Eldomery Mohammad K., Miller Kathryn, Kois Chelsea, Sack Virginia, Shur Natasha, Rijhsinghani Asha, Chandarana Jignesh, Ding Yan, Holtzman Judy, Jhangiani Shalini N., Muzny Donna M., Gibbs Richard A., Eng Christine M., Hanchard Neil A., Harel Tamar, Rosenfeld Jill A., Belmont John W., Lupski James R., and Yang Yaping. 2016. “Bi-Allelic Mutations in PKD1L1 Are Associated with Laterality Defects in Humans.” American Journal of Human Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vick Philipp, Kreis Jennifer, Schneider Isabelle, Tingler Melanie, Getwan Maike, Thumberger Thomas, Beyer Tina, Schweickert Axel, and Blum Martin. 2018. “An Early Function of Polycystin-2 for Left-Right Organizer Induction in Xenopus.” IScience. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walentek Peter. 2018. “Manipulating and Analyzing Cell Type Composition of the Xenopus Mucociliary Epidermis.” in Methods in Molecular Biology. [DOI] [PubMed] [Google Scholar]
- Waring H, Landgrebe FW, and Neill RM. 1941. “Ovulation and Oviposition in Anura.” Journal of Experimental Biology. [Google Scholar]
- Werner Michael E., and Mitchell Brian J.. 2013. “Using Xenopus Skin to Study Cilia Development and Function.” in Methods in Enzymology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheway Gabrielle, Nazlamova Liliya, and Hancock John T.. 2018. “Signaling through the Primary Cilium.” Frontiers in Cell and Developmental Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield Tanya T. 2020. “Cilia in the Developing Zebrafish Ear.” Philosophical Transactions of the Royal Society B: Biological Sciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitsett Jeffrey A. 2018. “Airway Epithelial Differentiation and Mucociliary Clearance.” in Annals of the American Thoracic Society. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey Helen Rankin, Xu Yuxiao, Everitt Amanda, Dea Jeanselle, Exner Cameron R. T., Jeremy Willsey A, State Matthew W., and Harland Richard M.. 2020. “Neurodevelopmental Disorder Risk Gene Dyrk1a Is Required for Ciliogenesis and Brain Size in Xenopus Embryos.” Development (Cambridge). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson PD, and Goilav B. 2007. “Cystic Disease of the Kidney.” Annu Rev Pathol 2:341–68. [DOI] [PubMed] [Google Scholar]
- Woolner Sarah, Miller Ann L., and Bement William M.. 2010. “Imaging the Cytoskeleton in Live Xenopus Laevis Embryos. Cytoskeleton Methods and Protocols.” Business. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga Takayuki, Hoff Sylvia, Schell Christoph, Martin Helmstädter Oliver Kretz, Kuechlin Sebastian, Yakulov Toma A., Engel Christina, Barbara Müller Robert Bensch, Ronneberger Olaf, Huber Tobias B., Lienkamp Soeren S., and Walz Gerd. 2015. “The Polarity Protein Inturned Links NPHP4 to Daam1 to Control the Subapical Actin Network in Multiciliated Cells.” Journal of Cell Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu David C., Thiagarajan Ravi R., Laussen Peter C., Laussen James P., Jaksic Tom, and Weldon Christopher B.. 2009. “Outcomes after the Ladd Procedure in Patients with Heterotaxy Syndrome, Congenital Heart Disease, and Intestinal Malrotation.” Journal of Pediatric Surgery. [DOI] [PubMed] [Google Scholar]
- Yu Xianwen, Ng Chee Peng, Habacher Hermann, and Roy Sudipto. 2008. “Foxj1 Transcription Factors Are Master Regulators of the Motile Ciliogenic Program.” Nature Genetics. [DOI] [PubMed] [Google Scholar]
- Zhang Bo, Tran Uyen, and Wessely Oliver. 2018. “Polycystin 1 Loss of Function Is Directly Linked to an Imbalance in G-Protein Signaling in the Kidney.” Development (Cambridge, England). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Chengtian, and Malicki Jarema. 2007. “Genetic Defects of Pronephric Cilia in Zebrafish.” Mechanisms of Development. [DOI] [PubMed] [Google Scholar]
- Zhao Junxing, Yue Wanfu, Zhu Mei J., Sreejayan Nair, and Du Min. 2010. “AMP-Activated Protein Kinase (AMPK) Cross-Talks with Canonical Wnt Signaling via Phosphorylation of β-Catenin at Ser 552.” Biochemical and Biophysical Research Communications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Jian, Wang Han Tsing, Chen Yu Rong, Yan Ling Ya, Han Ying Ying, Liu Ling Yan, Cao Ying, Liu Zhi Zhi, and Xu Hong A.. 2020. “The Joubert Syndrome Gene Arl13b Is Critical for Early Cerebellar Development in Zebrafish.” Neuroscience Bulletin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Louyin, Chen Laiqiang, Yan Lingya, Perkins Brian D., Li Shihua, Li Baoming, Xu Hong A., and Li Xiao Jiang. 2019. “Mutant Ahi1 Affects Retinal Axon Projection in Zebrafish Via Toxic Gain of Function.” Frontiers in Cellular Neuroscience. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Xi, Jiang Meisheng, Peyton Michael, Boulay Guylain, Hurst Raymond, Stefani Enrico, and Birnbaumer Lutz. 1996. “Trp, a Novel Mammalian Gene Family Essential for Agonist-Activated Capacitative Ca2+ Entry.” Cell. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie 1: Ciliary movement of multiciliated cells. Stage 40 X. laevis kidneys were dissected and dissociated into a single cell suspension. Cells were washed into PBS and DIC Movie was taken using an Olympus IX81 microscope. As the cilia beat it pushes against the fluid causing the cell to rotate.
Movie 2: Beating of Xenopus epidermal cilia. X. laevis embryos were injected with mRNA encoding for eGFP-alpha-tubulin (Woolner et al. 2010). Live stage 30 embryos were mounted on well slides and cilia were imaged on a 3i spinning disk confocal microscope.
Movie 3: In Xenopus motile cilia on their epidermis move fluid over their skin. Unanesthetized X. laevis embryos (Stage 30) were placed in a round bottom dish to prevent movement. On drop of 2mg/ml Rhodamine-dextran (Molecular probes D1817) placed on the posterior end of the tadpole. The embryo was imaged using an Olympus SZX16 dissection microscope. The Rhodamine diffuses around to the anterior before being pulled over the embryo.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.