

Abstract
Dyskerin and its homologs are ancient and conserved enzymes that catalyze the most common post-transcriptional modification found in cells, pseudouridylation. The resulting pseudouridines provide stability to RNA molecules and regulate ribosome biogenesis and splicing events. Dyskerin does not act independently—it is the core component of a protein heterotetramer, which associates with RNAs that contain the H/ACA motif. The variety of H/ACA RNAs that guide the function of this ribonucleoprotein (RNP) complex highlights the diversity of cellular processes in which dyskerin participates. When associated with small nucleolar (sno) RNAs, it regulates ribosomal (r) RNAs and ribosome biogenesis. By interacting with small Cajal body (sca) RNAs, it targets small nuclear (sn) RNAs to regulate pre-mRNA splicing. As a component of the telomerase holoenzyme, dyskerin binds to the telomerase RNA to modulate telomere maintenance. In a disease context, dyskerin malfunction can result in multiple detrimental phenotypes. Mutations in DKC1, the gene that encodes dyskerin, cause the premature aging syndrome X-linked dyskeratosis congenita (X-DC), a still incurable disorder that typically leads to bone marrow failure. In this review, we present the classical and most recent findings on this essential protein, discussing the evolutionary, structural, and functional aspects of dyskerin and the H/ACA RNP. The latest research underscores the role that dyskerin plays in the regulation of gene expression, translation efficiency, and telomere maintenance, along with the impacts that defective dyskerin has on aging, cell proliferation, haematopoietic potential, and cancer.
Keywords: H/ACA ribonucleoprotein, DKC1, dyskeratosis congenita, dyskerin, pseudouridylation, telomerase
INTRODUCTION
Premature aging syndromes, bone-marrow failure and several types of cancer are detrimental—or even fatal—health conditions that may be caused by defects in the expression and function of a protein known as dyskerin (Kirwan and Dokal 2008; Angrisani et al. 2014; Townsley et al. 2014; Grill and Nandakumar 2020). Dyskerin, encoded by the gene DKC1, was discovered in 1998 after the screening of patients harboring mutations associated with the premature aging disease, X-linked recessive dyskeratosis congenita (X-DC) (Heiss et al. 1998). Dyskerin is highly conserved evolutionarily with orthologs in multiple organisms, such as Cbf5 in yeast, Nap57 in rats, and Nop60B (minifly) in Drosophila. Conserved counterparts (also known as Cbf5) in Archaea further support the importance of this protein throughout evolution (Czekay and Kothe 2021).
Prior to dyskerin identification in humans, homologs from yeast and rats were discovered while studying nucleolar proteins involved in ribosome biogenesis, transport of molecules between the nucleus and the cytoplasm, and centromere and kinetochore binding (Meier and Blobel 1990, 1992, 1994; Jiang et al. 1993; Yu and Meier 2014). A few years later, these nucleolar proteins were identified as homologous to the E. coli pseudouridine synthase TruB (Koonin 1996; Cadwell et al. 1997). Moreover, the yeast homolog was demonstrated to interact with small nucleolar (sno) RNAs that display a secondary structure known as the H/ACA motif (Lafontaine et al. 1998) (more details below). These discoveries led to the conclusion that the dyskerin homologs effectively function in ribosomal biogenesis through ribosomal RNA (rRNA) pseudouridylation. Pseudouridylation is the most common RNA post-transcriptional modification. Besides the modification of rRNA, dyskerin also pseudouridylates spliceosomal small nuclear (sn) RNAs, participating in premessenger RNA (mRNA) splicing events (Yu and Meier 2014). Furthermore, dyskerin associates with a particular H/ACA RNA, the telomerase RNA component (TERC, or hTR in humans) (Mitchell et al. 1999), as part of the telomerase holoenzyme. Dyskerin is essential for regulating the accumulation of hTR and, consequently, for the activity of telomerase in elongating telomeres, the ends of linear chromosomes (MacNeil et al. 2016).
In this review, we provide an overview of the main concepts and discoveries related to the evolution of dyskerin and its homologs, focusing on its main structural features, function and regulation in multiple cellular processes, as well as its role in human disease.
AN ANCIENT PROTEIN
Dyskerin is a member of a group of enzymes known as pseudouridine synthases (Koonin 1996). In general, pseudouridine synthases share greater structural than sequence similarity (Fig. 1A,B). These enzymes are grouped in different families (TruA, TruB, TruD, RsuA, and RluA). Dyskerin is part of the TruB family, which is also the name given to its catalytic domain. Substrate specificity varies widely among pseudouridine synthases, some acting on one specific RNA only, while others act on many RNAs containing similar structures. Pseudouridine synthase substrate RNA binding is mediated by different motifs such as the thumb motif, also known as the β7–10 loop (Fig. 1B). An RNA binding region is also located in the carboxy-terminal portion of the TruB pseudouridine synthase family members (Hamma and Ferré-D'Amaré 2006).
FIGURE 1.

Dyskerin sequence and structural conservation and H/ACA RNAs general secondary structure. (A) Simplified schematic of the main domains found in dyskerin. The regions delimited by the red shade are conserved among eukaryotes and by the yellow shade are conserved from Archaea to eukaryotes. The numbers indicate the amino acid sequence in human dyskerin. TRUB: pseudouridine synthase domain, PUA: pseudouridine synthase and archaeosine transglycosylase domain, NLS/NoLS: nuclear and nucleolar localization signals (in blue), NTE and CTE: amino- and carboxy-terminal extensions. (B) Superimposition of human dyskerin (red—PDB: 7bgb), archaeal and yeast homologs (shades of blue and purple—PDB: 3hjy, 3hay, and 3u28). The thumb motif in varied conformations is marked with an arrow in the structures—this motif (corresponding to residues 182 to 194 in humans), however, is disordered and not modeled in the human and in some homolog structures. (C) Simplified general structure of eukaryotic H/ACA RNAs, displaying the two hairpins linked by the H box. The ACA box is located three nucleotides before the 3′ end of the molecule. The number of bulges and stems, as well as their size, vary widely among different H/ACA RNAs. The corresponding P1 and P2 stems mentioned in structural studies of archaeal H/ACA RNAs are indicated in the figure. The substrate RNA (violet) sits in the pseudouridylation pocket, the bulge highlighted in orange, base-pairing with the guide H/ACA RNA except for the target uridine (U)/pseudouridine (Ψ) and an extra nucleotide 3′ of the target U/Ψ (N, in this case). The target Ψ sits 14 to 16 nt from the H or ACA box. Figures 1B, 2, and 5B were prepared using UCSF ChimeraX (Goddard et al. 2018; Pettersen et al. 2021).
RNA-binding proteins are found in all domains of life and participate in a wide variety of cellular processes involving RNA. In dyskerin, the RNA-binding motif is known as the pseudouridine synthase and archaeosine transglycosylase domain (PUA) (Fig. 1A). The size of the PUA domain varies among different species but generally displays highly conserved glycine and hydrophobic residues in bacteria, archaea and eukaryotes (Cerrudo et al. 2014, 2015). Features found only in eukaryotic dyskerin homologs are the amino-terminal extension (NTE) and the carboxy-terminal extension (CTE). The amino- and the carboxy-terminal sequences are the most variable regions of this protein (Cerrudo et al. 2015).
Unlike most pseudouridine synthases, dyskerin and its homologs require auxiliary factors for RNA substrate recognition and modification. Archaeal and eukaryotic dyskerin homologs form ribonucleoprotein (RNP) complexes with conserved protein components and RNAs. The core protein components of the mature RNP are the nucleolar protein 10 (NOP10), nonhistone protein 2 (NHP2) and the glycine–arginine-rich protein 1 (GAR1) in humans, with homologs conserved from archaea to eukaryotes (Kiss et al. 2010; Czekay and Kothe 2021). The variable components of these RNPs are snoRNAs and small Cajal body (sca) RNAs, both of which are H/ACA RNAs that contain an H box (represented by the consensus sequence ANANNA, with N being any nucleotide) and the ACA box motif. H/ACA snoRNAs display a secondary structure with two hairpins separated by an H box single-stranded hinge, in addition to the ACA box, which is located three nucleotides from the 3′ end (Fig. 1C). H/ACA RNAs have a median sequence length of 133 residues, and each hairpin commonly displays other structural motifs, including internal loops and bulges (Fig. 1C; Balakin et al. 1996; Ganot et al. 1997). Because of the H/ACA RNA component, the ribonucleoprotein complex is known as an H/ACA RNP.
Apart from the conserved H and ACA boxes and the bulges in the hairpin structure that correspond to the pseudouridylation pocket required for the catalytic activity of dyskerin (Fig. 1C), H/ACA RNAs display some particularities that diverged in evolution. One of them is related to the general structure of the H/ACA RNAs: whereas most eukaryotic H/ACA RNAs contain two hairpins, archaeal H/ACA RNAs typically consist of a single hairpin (Rozhdestvensky et al. 2003). Another difference is the archaeal specific K-turn motif in the upper, turn region of the hairpin, which promotes a conformational kink in an RNA helix (Czekay and Kothe 2021). This K-turn motif is involved in the binding of L7ae (the archaeal homolog of eukaryotic NHP2) to the H/ACA RNA. The K-turn is, therefore, of special importance for the archaeal H/ACA RNP structure, and highlights the differences in binding specificity of L7ae and NHP2 to H/ACA RNAs and other components of the RNP (Wang and Meier 2004; Li and Ye 2006; Li et al. 2011b). Another feature found in the H/ACA RNA hTR is the biogenesis-promoting (BIO) box (Egan and Collins 2012a,b) located in the 3′ hairpin and that interacts with NHP2 (Ghanim et al. 2021).
Overall, the sequence and structural conservation observed in dyskerin and its homologs is a clear indication of the functional relevance of pseudouridine synthases in regulating cellular processes and physiology.
STRUCTURE, LOCALIZATION, AND NUCLEAR COMPLEXES
Dyskerin is an L-shaped protein of 514 residues with a molecular weight of ∼58 kDa (Fig. 2A). It is largely hydrophilic, and its homologs display similar hydrophilicity plots (Cerrudo et al. 2015). A common fold and core comprise a β-sheet bisected by a catalytic cleft, which in turn is surrounded by different loop and helix motifs. An aspartic acid residue, considered critical for enzymatic activity, is found in a loop in the catalytic cleft of all pseudouridine synthases (Fig. 2A; Hamma and Ferré-D'Amaré 2006). The amino- and carboxy-terminal regions contain sequences associated with subcellular localization of the protein: the nuclear and nucleolar localization signals (NLS and NoLS, respectively) (Fig. 1A), which are usually rich in lysines (Cerrudo et al. 2015). Most of the structural data regarding human dyskerin are derived from either homology models based on archaeal or yeast Cbf5 or on studies of the human telomerase holoenzyme (Hamma et al. 2005; Li and Ye 2006; Rashid et al. 2006; Liang et al. 2007, 2009a; Duan et al. 2009; Li et al. 2011a,b; Nguyen et al. 2018; Ghanim et al. 2021). In all cases, the structure of dyskerin has been elucidated as part of H/ACA RNPs.
FIGURE 2.

Structure of dyskerin and the H/ACA RNP. (A) Cartoon of human dyskerin (PDB: 7bgb), highlighting the TruB catalytic domain in orange, with the catalytic aspartate D125 represented by spheres in magenta, and the PUA RNA binding domain in light green. The catalytic core of dyskerin is the upper, horizontal portion of the structure consisting of multiple helices and strands. (B) Including NOP10 (pink), NHP2 (yellow), and GAR1 (dark green) to dyskerin (red), the structure of the human H/ACA heterotetramer is shown in a space-filling representation (PDB: 7bgb). (C) Archaeal H/ACA RNP, with the corresponding homolog components shown in the same colors as in B, in addition to a single-hairpin H/ACA RNA (PDB: 2hvy). The triangular shape of the RNP is indicated by the dotted gray lines.
Dyskerin is a component of H/ACA ribonucleoprotein complexes
Dyskerin is the core protein and catalytic subunit of the H/ACA RNP (Fig. 2B). The other components include proteins that stabilize the RNP structure, folding and interactions with other molecules. High-resolution structures of H/ACA RNPs were initially revealed for unicellular organisms, such as archaea and yeast (Li and Ye 2006; Rashid et al. 2006; Li et al. 2011b). Data from a 2.1 Å crystal structure of the archaeal homolog of dyskerin, Cbf5, in association with Nop10 and Gar1, demonstrates the structural similarity between Cbf5 and E. coli TruB, including the superimposition of the catalytic aspartic acid residue when comparing both pseudouridine synthase structures (Rashid et al. 2006). Gar1 binds to Cbf5, stabilizing the dyskerin homolog for RNA substrate binding, without necessarily interacting with the RNA. The amino-terminal portion of Cbf5 was suggested to structurally stabilize the RNA binding region of the protein by providing an extra β-strand to the PUA domain β-sheet. Nop10 and Gar1 independently bind to Cbf5 adjacent to the catalytic domain of the enzyme. Nop10 undergoes conformational changes upon binding to Cbf5, possibly favoring stable binding to Cbf5 as well as a stable positioning of the target uridine for pseudouridylation (Rashid et al. 2006).
Another high-resolution crystal structure of an archaeal RNP included L7ae (an NHP2 homolog) (Li and Ye 2006). This structure showed that the mature RNP complex forms a T-shaped or a triangular-like structure composed of Cbf5, Nop10, L7ae, and Gar1 (Fig. 2C). Cbf5 forms the center of the RNP with its catalytic site and a vertex of the structure consists of its PUA domain. Gar1 associates with Cbf5 near the center of the structure, forming another vertex. The last of the vertices represents L7ae, which is bound to Cbf5 in the center of the structure via interactions with Nop10. The archaeal H/ACA RNA binds to one side of the tetramer in an axis formed by Cbf5, Nop10, and L7ae, not interacting with Gar1 (Fig. 2C), which is instead involved in regulating substrate turnover. The pseudouridylation pocket of the H/ACA RNA, required for dyskerin's catalytic activity, binds near the active site of Cbf5 (Li and Ye 2006). The thumb motif (Fig. 1B) is located in proximity of the catalytic core. The thumb changes conformation to interact with the substrate RNA and other Cbf5 residues or with Gar1 in the absence of substrate and is therefore important for proper binding of the RNP to the substrate RNA as well as for substrate turnover (Duan et al. 2009). Another archaeal H/ACA RNP structure (lacking Gar1) bound to substrate RNA revealed that Nop10, L7ae, and a portion of the guide RNA hold this same guide H/ACA RNA more stably by shifting it closer to the catalytic core of Cbf5, while the protein–protein interactions within the RNP do not exhibit significant changes. Thus, the same core protein components of the H/ACA RNP, with minor conformational rearrangements among them, are able to adapt the complex to the multiple RNAs with which the RNP interacts (Liang et al. 2009a).
Subsequent studies of the Saccharomyces cerevisiae H/ACA RNP revealed that the eukaryotic complex displays considerable structural similarity to archaeal RNPs (Li et al. 2011a,b). Complementary chemical crosslinking data show that the location where Nhp2 binds to the eukaryotic complex corresponds to the binding location of L7Ae in the archaeal RNP. However, Nhp2 is not essential for the catalytic activity of the yeast Cbf5, differing from the essential role of L7Ae for the catalytic function of the archaeal counterpart. This may be explained by the reduced sequence and structural conservation in the eukaryotic upper-stem region of H/ACA RNAs (Li et al. 2011b).
The telomerase holoenzyme
The architecture and stoichiometry of the human telomerase holoenzyme components were initially elucidated via tandem affinity purification assays, as well as by measurements of hTR accumulation and enzymatic activity of the holoenzyme reconstituted in vivo (Egan and Collins 2010). Two sets of H/ACA core proteins (dyskerin, NOP10, NHP2, and GAR1) but only one copy of the telomerase reverse transcriptase (TERT)—which is the catalytic subunit responsible for repeatedly reverse transcribing the hTR template region—and a single copy of the telomerase Cajal body protein 1 (TCAB1) associate with hTR to form a functional telomerase holoenzyme. TCAB1 associates specifically with the Cajal body (CAB) box region of hTR (Fig. 3; Tycowski et al. 2009; Venteicher et al. 2009).
FIGURE 3.
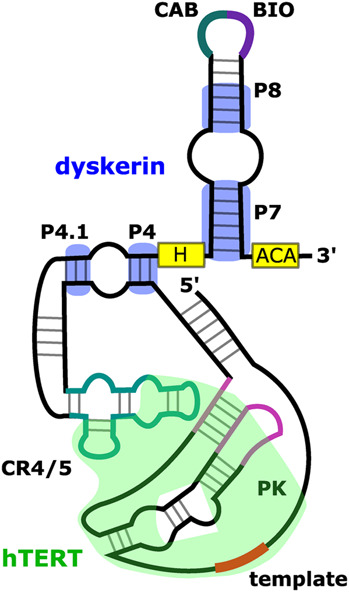
The human telomerase RNA (hTR). This secondary structure is based on recent high-resolution data of the telomerase holoenzyme (Nguyen et al. 2018; Ghanim et al. 2021). Some regions of especial importance for the holoenzyme are highlighted: CR4/5 and t/PK (template/pseudoknot) are important for telomerase function and are located in the catalytic lobe (TERT associates with hTR approximately in the region highlighted in green). H/ACA RNP proteins (including dyskerin) associate with stems P7 and P8 in the 3′ hairpin and dyskerin associates with P4, located in the 5′ hairpin (approximate regions highlighted in blue). H and ACA represent the H box and the ACA box. CAB is the consensus sequence that directs the H/ACA complex to the Cajal bodies and the binding location of TCAB1 in the hTR molecule. BIO indicates the location of the biogenesis-promoting box, both on the apical 3′ hairpin loop.
A considerably diminished 5′ hairpin structure in hTR proved to be sufficient for the assembly of the two sets of H/ACA RNP core proteins, without negatively impacting hTR accumulation or the catalytic activity of the holoenzyme (Egan and Collins 2010). For this reason, it was suggested that cross-hairpin interactions favor the sequential or concomitant binding of the two sets of the H/ACA RNP with fewer protein–RNA interactions in the 5′ hairpin, a possible contribution of dyskerin motifs not present in the archaeal Cbf5 (Egan and Collins 2010).
A high-resolution structure of a complete H/ACA RNP of a multicellular species has only recently been elucidated (Ghanim et al. 2021). Initially, in a cryo-electron microscopy (cryo-EM) model based on pre-existent structures or homology models fitted to the holoenzyme densities map, it was demonstrated that the human telomerase holoenzyme consists of two lobes (Nguyen et al. 2018). One is the catalytic core formed by TERT bound to the template/pseudoknot (t/PK) and conserved regions 4 and 5 (CR4/5) domains of hTR (Fig. 3). The other lobe comprises two sets of the H/ACA heterotetramers (dyskerin, NOP10, NHP2, and GAR1) and one TCAB1 molecule. Each heterotetramer binds to a different hairpin of hTR in the 3′ terminal half of the molecule. One of the H/ACA heterotetramers associates with the 5′ hairpin exclusively via the binding of dyskerin to the P4 stem of hTR, whereas the other heterotetramer associates with the 3′ hairpin through more extensive interactions between the protein components and the P7 stem and P8 stem–loop of hTR (Fig. 3; Nguyen et al. 2018).
This structure was improved in a subsequent high-resolution cryo-EM reconstruction, in which the H/ACA lobe was resolved at 3.4 Å resolution, allowing the telomerase holoenzyme to be built de novo (Ghanim et al. 2021). In this recent model, a histone heterodimer consisting of H2A and H2B was found bound to the catalytic core of telomerase, representing unexpected and novel components of the holoenzyme.
Also, the carboxy- and amino-terminal extensions of dyskerin were modeled for the first time. Dyskerin contains a hydrophobic pocket formed by its amino terminus and the PUA domain. The two dyskerin subunits that associate with the 5′ and 3′ hairpins of hTR are respectively referred to as the 5′ dyskerin and 3′ dyskerin. A portion of the amino-terminal region of the 5′ dyskerin sits in the hydrophobic pocket of the 3′ dyskerin. Conversely, a carboxy-terminal helix of the 3′ dyskerin sits in the hydrophobic pocket of the 5′ dyskerin. Cross-hairpin interactions involving the H and ACA boxes as well as dyskerin were observed, a particularity of eukaryotic H/ACA RNPs (Ghanim et al. 2021). The association of the H/ACA RNP to the 5′ hairpin of hTR exclusively via interactions between dyskerin and hTR, in addition to the cross-hairpin interactions mediated by dyskerin, support the earlier explanation for the plasticity displayed by the complex to modifications in the hTR 5′ hairpin. From these observations, considering the two dyskerin copies are each able to bind to one hairpin and maintain the cross-hairpin interactions, the holoenzyme should tolerate changes in the 5′ hairpin.
Despite the lack of a canonical H/ACA motif in telomerase RNA from plants (Fajkus et al. 2019; Song et al. 2019), the dyskerin homolog in Arabidopsis nonetheless participates in the plant telomerase complex via interactions with the P1a stem that is formed by base-pairing between the 5′ and 3′ ends of the telomerase RNA (Song et al. 2021). Moreover, these interactions influence telomerase activity (Song et al. 2021). However, how dyskerin binds to an RNA molecule lacking a canonical H/ACA motif to modulate telomerase activity in plants remains unknown.
H/ACA RNP biogenesis and cellular localization
H/ACA RNP biogenesis proceeds via a complex, and not fully understood, mechanism of association of the core RNP proteins and the transient association of multiple assembly factors, with varying intranuclear localizations, depending on the stage of the biogenesis process and on the function of the mature RNP (Fig. 4). Early functional characterizations of human dyskerin revealed that it translocates to the nucleus, more specifically to the nucleolus and the Cajal bodies, similarly to its eukaryotic homologs. This localization is achieved by the cooperative effect of its lysine- and arginine-rich amino- and carboxy-terminal nuclear localization signals (Heiss et al. 1999). A few years later, the initial assembly model for the H/ACA RNP—based on the interactions identified for the RNP components translated in vitro—suggested that NOP10 binds to dyskerin (or, in this case, to the rat homolog NAP57), followed by the association of this dimer with NHP2, resulting in the core RNP protein trimer (Wang and Meier 2004). NHP2 binds nonspecifically to RNA and only demonstrates specific binding to H/ACA RNAs when in association with NOP10 and dyskerin, a possible evolutionary adaptation to the absence of the archaeal K-turn in eukaryotic H/ACA RNAs, as suggested by Wang and Meier (2004). It was first proposed that the RNP protein tetramer forms prior to the association with H/ACA RNAs and, since only dyskerin contains a classical NLS in its sequence, that the protein tetramer forms in the cytoplasm before being translocated into the nucleus (Wang and Meier 2004).
FIGURE 4.

H/ACA RNP biogenesis and function. In a general model of H/ACA RNP biogenesis, the assembly process initiates by the binding of the assembly factor SHQ1 to dyskerin, in the cytoplasm. This initial binding is likely necessary for the stabilization of an otherwise unstable unbound dyskerin. These two proteins then translocate into the nucleus, guided by dyskerin's NLS. In the nucleoplasm, NOP10, NHP2, and the assembly factor NAF1 associate with dyskerin, while SHQ1 dissociates from the enzyme. This newly formed, immature H/ACA complex then localizes to nascent H/ACA RNAs, likely by the interaction between NAF1 and RNA Polymerase II and its transcription factors. Pontin and reptin, as part of the general assembly factor R2TP complex, also interact with the immature H/ACA RNP at the H/ACA RNA transcription site, likely contributing to the dissociation of SHQ1 from dyskerin. GAR1 displaces NAF1 in the late stages of RNP biogenesis, forming the mature protein tetramer consisting of dyskerin, NOP10, NHP2 and GAR1 in association with an H/ACA RNA. Other additional factors may participate in the assembly process of H/ACA RNPs: SMN, Hsp90 and the adaptor Nufip. The assembly factors NAF1 and SHQ1 accumulate in the nucleoplasm and the cytoplasm but are absent from the sites of the mature H/ACA RNP localization: the nucleolus and the Cajal bodies. Depending on the H/ACA RNA component, the H/ACA RNP will localize to different subcellular compartments and have a different function. In the nucleolus, H/ACA snoRNAs are mostly involved in guiding the pseudouridylation of rRNAs, while some of them are directly involved in the processing of rRNAs, both participating in ribosome biogenesis. Some snoRNAs, however, may be exported to the cytoplasm and processed in a Dicer-dependent manner to act as micro RNA-like small RNAs in gene silencing. H/ACA scaRNAs, on the other hand, guide the H/ACA RNP to the Cajal bodies and target snRNAs, thus being involved in pre-mRNA splicing events. When associated with hTR, also a scaRNA, the H/ACA RNP has a more structural role in the formation of the telomerase holoenzyme. The functions of some H/ACA RNAs, called orphan RNAs are unknown.
Subsequently, NAF1 was identified as a new essential factor involved in the H/ACA RNP assembly process (Darzacq et al. 2006). It was proposed that NAF1 stabilizes the recently synthesized dyskerin (or NAP57) in the cytoplasm, either alone or in a core complex with NOP10 and NHP2. Then, all four proteins jointly translocate into the nucleus. By observing a nascent H/ACA RNA in a single-cell system, it was suggested that NAF1 is recruited to the H/ACA RNA transcription site along with dyskerin, NOP10 and NHP2, maintaining the RNP catalytically inactive while in this immature state (Darzacq et al. 2006). It is possible that the immature RNP is recruited to nascent H/ACA RNAs by a prior interaction between NAF1 and RNA Polymerase II or the immature RNP, already containing NAF1, is recruited by RNA Polymerase II to the transcription site (Fig. 4; Ballarino et al. 2005).
Another protein, SHQ1, was then uncovered as an assembly factor that acts in the initial stages of H/ACA RNP formation. Of the four core proteins of the immature RNP, SHQ1 associates exclusively with the dyskerin homolog, NAP57, both in vitro and in vivo. This occurs before NAP57 binds to NHP2, NOP10, or NAF1 (Grozdanov et al. 2009). SHQ1 localizes to the nucleoplasm but is excluded from the nucleolus and the Cajal bodies, which confirms its involvement only in the early stages of RNP assembly. Additionally, SHQ1 does not contain a classical NLS, and translocates from the cytoplasm to the nucleus possibly by its association with dyskerin in the cytoplasm, where it may act as a chaperone. Furthermore, the accumulation of H/ACA RNAs is dependent on SHQ1 (Grozdanov et al. 2009). It was proposed that SHQ1 stabilizes free dyskerin to prevent its promiscuous binding to RNAs and pseudouridylation activity because of its RNA mimicry mechanism, observed in the crystal structure of yeast Cbf5 interacting with yeast Shq1 (Walbott et al. 2011). The carboxy-terminal SHQ1-specific domain (SSD) binds to the PUA domain and to the CTE of Cbf5, simulating the contacts that Cbf5 has with RNA, thus preventing the interaction with RNAs and the other proteins that compose the RNP (Walbott et al. 2011).
An alternative assembly model was also proposed in which Shq1 is able to bind to Cbf5 in a pre-RNP complex consisting of Nop10 and Gar1 (Li et al. 2011a). The presence of a pre-RNP complex does not impede the binding of Shq1 to Cbf5 in this case, revealing a possible difference between the RNP assembly process in yeast and mammalian cells. The competition between RNA and SHQ1 for binding to the dyskerin homologs, however, was confirmed for both yeast and mammalian cells. This competition may lead to the displacement of SHQ1 from dyskerin by an H/ACA RNA (Li et al. 2011a). SHQ1 also contains an amino-terminal HSP90 cochaperone p23-like CHORD-containing proteins and Sgt1 (CS) domain. Based on the interaction between dyskerin or Cbf5 peptides and the CS domain, a model structure of the yeast SHQ1 CS domain bound to Cbf5 in the pre-H/ACA RNP was generated, in which the CS domain interacts with the amino-terminal portion of Cbf5 (Singh et al. 2015).
Following the discovery of SHQ1's role in stabilizing newly translated dyskerin and preventing its promiscuous and premature association with RNAs, the role of two other assembly factors in H/ACA RNP biogenesis was also uncovered. H/ACA RNP accumulation in vivo requires the presence of pontin and reptin. Pontin and reptin are two highly conserved proteins of the ATPases associated with diverse cellular activities (AAA+) family of ATPases, homologous to bacterial helicases (Neuwald et al. 1999; Hanson and Whiteheart 2005; Mao and Houry 2017). These two proteins form a heterohexameric ring in association with the proteins PIH1D1 and RPAP3, forming the R2TP complex (Kakihara and Houry 2012). This complex is likely recruited to the dyskerin-SHQ1 dimer via interaction between PIH1D1 and dyskerin, after it is translocated from the cytoplasm into the nucleoplasm (Machado-Pinilla et al. 2012), although pontin is also able to associate directly with dyskerin (Venteicher et al. 2008). R2TP probably interacts with dyskerin-SHQ1 at the time dyskerin is required to exchange SHQ1 for the nascent H/ACA RNAs at the site of transcription. In a proposed model, the pontin-reptin hexameric ring associated with PIH1D1 releases SHQ1 from dyskerin with the help of the carboxy-terminal tail of dyskerin, in an apparent ATP-independent manner. This allows dyskerin to bind to nascent H/ACA RNAs and the other proteins from the immature H/ACA RNP. The role of the other R2TP component, RPAP3, is not yet clear (Machado-Pinilla et al. 2012).
The transition between an immature or pre-RNP and a mature complex is marked by the displacement of NAF1 by GAR1 (Fig. 4). NAF1 and GAR1 compete to bind to dyskerin. Once GAR1 associates with the now mature H/ACA RNP, the complex localizes to sites of function, namely the nucleolus or the Cajal bodies (Darzacq et al. 2006). For this reason, H/ACA RNPs can be classified as either snoRNP or scaRNP, depending on the H/ACA RNA with which the RNP protein complex associates and the localization of the complexes. Whereas dyskerin already contains a NoLS, the localization of scaRNPs to the Cajal bodies is guided by the conserved CAB box consensus sequence (ugAG) found in the apical region of the scaRNA hairpins. The addition of this sequence is able to localize snoRNAs to Cajal bodies, and its mutation leads to the localization of scaRNAs in the nucleolus (Richard et al. 2003).
Post-translational modifications also contribute to the regulation of dyskerin and H/ACA RNP biogenesis. Through the process of poly ADP-ribosylation (PARylation) (Langelier et al. 2018), poly (ADP-ribose) polymerase 1 (PARP-1) was proposed to influence the RNA-binding function of dyskerin and GAR1, affecting telomerase RNP assembly (Savelyev et al. 2021). Dyskerin nuclear and subnuclear localization is also regulated by the small ubiquitin-like modifier (SUMO). The covalent post-translational modification by SUMO, known as SUMOylation, is involved in regulating cellular transport, activity, and stability of target proteins (Cappadocia and Lima 2018). SUMOylation sites were identified in dyskerin (Becker et al. 2013; Hendriks et al. 2014, 2018; Hendriks and Vertegaal 2016; Lamoliatte et al. 2017), of which several are located in its lysine-rich carboxy-terminal NLS. Dyskerin lacking the carboxy-terminal residues 446–514 accumulates mostly in the cytoplasm (MacNeil et al. 2021). The fusion of this cytoplasmic localized truncation variant with SUMO3 restores nuclear accumulation, but not nucleolar localization, possibly due to the loss of NoLS in this carboxy-terminal region along with important residues that might regulate the nucleolar localization of dyskerin, such as K467. SUMOylation of K467 was found to be essential for the interaction between dyskerin and GAR1, as well as the subnuclear localization of dyskerin in the nucleolus. These studies also identified a short noncovalent SUMO-interaction motif in GAR1. Thus, SUMO plays an important role in the H/ACA RNP biogenesis and its localization to the nucleolus (MacNeil et al. 2021). In addition to SUMO, the single-strand-selective monofunctional uracil DNA glycosylase 1 (SMUG1) is involved in the localization of dyskerin. The nucleolar distribution of dyskerin is influenced by the interaction between SMUG1 and dyskerin and Smug1–/– mouse embryonic fibroblasts exhibit dyskerin mislocalization (Kroustallaki et al. 2019).
Other than the components of the immature and mature H/ACA RNP and the assembly factors mentioned above, other molecules may participate in the H/ACA RNP assembly process, including the heat shock protein 90 (Hsp90) (Schopf et al. 2017) and the adaptor protein Nufip (Boulon et al. 2008), the survival of motor neurons (SMN) complex (Matera and Wang 2014) and the nucleolar and Cajal body phosphoprotein (Nopp140) (Fig. 4; Meier 2005). The discovery that SMN directly binds to GAR1 in vivo raised the possibility that it is an assembly factor for H/ACA RNPs (Pellizzoni et al. 2001). Depletion of Nopp140 in human cell lines results in structural changes in the Cajal bodies, reducing their size, along with the loss of scaRNPs, including the telomerase holoenzyme, in these membraneless compartments. Nopp140 thus plays an important function in Cajal body formation and localization of scaRNPs (Bizarro et al. 2019).
Once the mature H/ACA RNP is assembled, dyskerin remains bound to the newly transcribed H/ACA RNA, possibly requiring de novo synthesis of both molecules to form a new RNP complex. The other proteins, however, demonstrate a dynamic exchange between different RNPs (Kittur et al. 2006). Compared to yeast and archaeal H/ACA RNPs, there are few structural studies of human H/ACA RNPs, with the only high-resolution model available being the telomerase holoenzyme (Ghanim et al. 2021). Thus, it is still not possible to confirm whether all predicted structural rearrangements during assembly and function are conserved from archaeal and yeast homologs to humans. The variable amino- and carboxy-terminal regions of dyskerin are examples of motifs that are not conserved in archaeal structures. Furthermore, these regions harbor disease mutations and adopt a specific conformation when interacting with a second set of H/ACA RNPs. Also, there are still divergences in the proposed mechanisms of RNP assembly between different organisms. For instance, the suggested timing of and subcellular location where the core RNP proteins and assembly factors bind to dyskerin are different for mammalian and yeast cells (Grozdanov et al. 2009; Li et al. 2011a). Whereas yeast Shq1 is able to bind to Cbf5, Nop10, Nhp2, Naf1, and Gar1, the mammalian SHQ1 may participate only in the initial stages of RNP assembly, before dyskerin interacts with all the other RNP core proteins and NAF1 (Grozdanov et al. 2009; Li et al. 2011a). Further investigation may help to elucidate whether these different mechanisms are specific to particular organisms or whether a common mechanism for the H/ACA RNP biogenesis can be established for eukaryotes.
REGULATION AND FUNCTION OF DYSKERIN IN CELLULAR PROCESSES
H/ACA RNPs are essential for many processes that ensure cell viability, depending on the H/ACA RNA with which the core protein tetramer associates (Fig. 4). It is also important to highlight that some H/ACA RNAs fulfill functions that are independent of their traditional role in guiding the H/ACA RNP. In addition to guiding the pseudouridylation of the U2 snRNA, ACA45, for instance, can be further processed into a micro RNA-like small RNA in the cytoplasm in a Dicer dependent manner (Fig. 4). This novel small RNA may then associate with Argonaute proteins and participate in gene silencing events (Ender et al. 2008). Moreover, some H/ACA RNAs have no clear target RNA or processing and structural function and thus are considered orphan H/ACA RNAs. Pseudouridine synthases may also perform a pseudouridylation-independent function as a chaperone. Viability of Cbf5 null yeast can be partially rescued by expressing a catalytically inactive Cbf5 variant (Zebarjadian et al. 1999). Similarly, the growth defects of a TruB deletion in E. coli are rescued by the expression of catalytically inactive TruB (Gutgsell et al. 2000), which was proposed to be critical for bacterial fitness as a chaperone, and not necessarily pseudouridylation activity (Keffer-Wilkes et al. 2016). Below we will discuss the most common and some noncanonical functions of dyskerin in cellular processes.
Pseudouridylation
Pseudouridine, or 5-ribosyluracil, represented by Ψ, is a C5-glycoside isomer of uridine. Ψ is the most common modified nucleotide found in cells (Yu and Meier 2014). Pseudouridylation is dependent on an aspartic acid residue found in the active site of all pseudouridine synthases (Fig. 2A; Huang et al. 1998; Hamma and Ferré-D'Amaré 2006). The high-resolution crystal structure of the bacterial TruB bound to the substrate tRNA revealed that TruB is able to interact with the target uridine via base flipping. Flipping out the target nucleotide disrupts interactions within the unbound tRNA structure and allows TruB to access the nucleotide that will be pseudouridylated (Hoang and Ferré-D'Amaré 2001). The proposed mechanism for this reaction involves the nucleophilic attack of the target uridine by the conserved aspartate in the active site of pseudouridine synthases, such as dyskerin (Huang et al. 1998; Gu et al. 1999). The reaction results in a 180° rotation of the uracil across the N3–C6 axis, resulting in a glycosidic bond via a carbon atom instead of a nitrogen (Fig. 5A).
FIGURE 5.

Pseudouridylation. (A) The pseudouridylation reaction results in the conversion of uridine to pseudouridine by rotating the molecule in 180° across its N3–C6 axis (represented by the diagonal line). (B) Cartoon representation displaying the stacked helices and three-way junctions (J1 and J2) formed by the interaction between guide H/ACA RNA and substrate RNA. The target uridine is marked in the figure by an arrow. (PDB: 2P89 [Wu and Feigon 2007].)
This modification provides an extra hydrogen bond donor to the base and a more stable glycosidic bond, enhancing the stability of the molecule (Kierzek et al. 2014; Yu and Meier 2014). Moreover, quantitatively mapping pseudouridines in human and yeast transcriptomes has revealed an increasing number of pseudouridines and the extent to which different types of RNAs are pseudouridylated. By using a technique known as Ψ-seq and the knockout of different pseudouridine synthase genes, novel pseudouridylation sites were identified and validated for each of the enzymes tested. Pseudouridylation sites have been found at many residues important for substrate pairing in both H/ACA and C/D snoRNAs (Schwartz et al. 2014), the latter being a class of RNAs implicated in 2′-O-methylation of target RNAs (Balakin et al. 1996).
Models for the RNA duplex formed by the binding of substrate RNA to the pseudouridylation pocket of the guide H/ACA RNA were proposed based on two solution structures elucidated by NMR spectroscopy (Jin et al. 2007; Wu and Feigon 2007). Two coaxially stacked helices formed by the interaction between the pseudouridylation pocket of the guide H/ACA RNA and the substrate RNA molecule—one helix 5′ and the other 3′ of the target uridine—were identified (Fig. 5B). Moreover, two unusual three-way junctions are created by the binding of the substrate RNA to the H/ACA RNA (Fig. 5B), stabilizing the otherwise flexible pseudouridylation pocket and placing the uridine that will be modified in a favorable position in the active site of dyskerin. In this RNA duplex, the unpaired target uridine, along with its unpaired 3′ adjacent nucleotide, promote a sharp turn giving the substrate RNA a U-shaped structure when it is bound to one face of the guide H/ACA RNA. No H/ACA RNP protein components are required for the formation of this RNA duplex. The stacked helices that are formed in this RNA duplex are also responsible for determining the distance of 14 to 16 nt between the target uridine and the ACA box (Wu and Feigon 2007). However, the pseudouridylation pocket structure changes considerably in the presence of H/ACA RNP proteins thus facilitating the modification of the target uridine by dyskerin (Hamma and Ferré-D'Amaré 2010). RNA-guided pseudouridylation requires the formation of at least eight base pairs between the guide H/ACA RNA and the substrate RNA (De Zoysa et al. 2018). However, pseudouridylation still occurs when more than two unpaired nucleotides are added to the substrate RNA. These unpaired nucleotides, located near the base of the upper stem of the guide H/ACA RNA (Fig. 1C), reveal the flexibility of the pseudouridylation pocket to accommodate a variety of substrate RNAs without perfectly matching residues near the pseudouridine (De Zoysa et al. 2018).
Studies on the kinetics of association between guide and substrate RNAs have furthered our understanding of the mechanisms of pseudouridylation. In a recent study, the pseudouridylation of wild-type and modified substrate RNAs by the yeast H/ACA guide RNA snR34 was tested (Kelly et al. 2019). Modified substrate RNAs were designed to contain different types of base pairs and different numbers of mismatches with the guide RNA. The number of base pairs, when respecting a minimum of 3 bp on each side of the target uridine, reduces the initial velocity of pseudouridylation but does not influence the binding of these noncognate RNAs. The pseudouridylation activity, however, diminished drastically on noncognate substrate RNAs that exhibit reduced binding to the guide RNA, in comparison with the wild-type substrates. Nucleotide changes near the target uridine are the most detrimental to pseudouridylation. This observation suggests that mismatches in base pairing do not significantly reduce the affinity of substrate RNAs for guide RNAs and the H/ACA RNP, and binding might be transient, since pseudouridylation levels of wild-type substrate RNAs are maintained in the presence of competitor noncognate RNAs. The observed transient binding could be explained by a search mechanism for the actual target of the H/ACA RNP, or even connected to a chaperone function that the RNP may perform during ribosome biogenesis (Kelly et al. 2019). Thus, the flexibility of the pseudouridylation pocket in accommodating varied target RNAs would occur at the cost of reduced pseudouridylation activity, depending on the number of mismatches, especially near the target uridine. The precise reason for this flexibility remains to be elucidated.
Further details on the mechanisms involved in pseudouridylation were obtained from crystal structures of the archaeal H/ACA RNP bound to substrate RNA (Duan et al. 2009; Liang et al. 2009a). For pseudouridylation to occur, the target RNA is recruited via base-pairing with the guide H/ACA RNA—as discussed above—and also through interactions with the RNP proteins. The core RNP maintain a generally rigid structure, except for the PUA domain of Cbf5 and the P1 stem of the guide H/ACA RNA, which significantly shift positions when binding to substrate RNA. The substrate RNA associates with Cbf5 only via nonspecific interactions, which also demonstrates how dyskerin homologs are adaptable to different types of RNAs. The two ordered helices formed upon substrate RNA binding to the complementary guide sequences in the pseudouridylation pocket were also observed in this crystal structure. These helices maintain the RNA duplex in an appropriate conformation for catalysis. Another important conformational change that is observed involves the thumb region of Cbf5. The thumb interacts with GAR1 when no substrate is present in the catalytic core, and it interacts with the substrate RNA in the absence of GAR1. As the substrate RNA binds to the H/ACA RNP, the thumb changes from its open conformation when bound to GAR1, to a closed conformation, with multiple hydrogen-bonds, van der Waals and electrostatic interactions with the substrate RNA, without contacting the H/ACA guide RNA. This conformational change stabilizes the substrate in its U-shaped structure and effective pseudouridylation depends on proper interaction with the thumb (Duan et al. 2009; Liang et al. 2009a). However, despite the advances in understanding the structure and function of dyskerin and its homologs, the detailed mechanism of the pseudouridylation reaction is still not fully understood.
Ribosome and spliceosome biogenesis
H/ACA RNPs participate in ribosome biogenesis by pseudouridylating pre-rRNAs (Fig. 4). Pseudouridylation is important for rRNA folding and the modification is maintained in mature rRNAs. In addition to providing structural stability, rRNA modifications have been suggested to modulate the interaction between distant regions of the ribosome, either by influencing rRNA folding or via interactions with ribosomal proteins (Sloan et al. 2017; Ojha et al. 2020). Remarkably, pseudouridines cluster in regions that are important for ribosome function, such as the decoding center, the tRNA binding sites and the interface between the ribosomal subunits. This localized distribution of pseudouridines implies a regulatory role in ribosome function. In fact, the presence of pseudouridines affects translation efficiency and fidelity (Watkins and Bohnsack 2012; Sloan et al. 2017). Although the loss of single or a few pseudouridines does not generally cause detrimental effects on ribosome function, some specific pseudouridines have a critical role in ribosome biogenesis. The loss of a hypermodified pseudouridine (that includes a methylation at N1 and a 3-amino-3-carboxypropyl group) impedes proper ribosome biogenesis by delaying 18S rRNA processing and maturation (Liang et al. 2009b). Pseudouridylation of this particular base is especially important, since the methylation that occurs on this hypermodified pseudouridine is dependent on prior pseudouridylation at the same base (Liang et al. 2009b).
Also relevant for rRNA quality control in cells is the protein SMUG1. This base-excision repair enzyme binds to rRNA and removes base damage involving uracil and pyrimidine oxidation products. Found in the nucleolus and in Cajal bodies, SMUG1 interacts with dyskerin and possibly targets specific rRNAs in these subcellular locations (Jobert et al. 2013).
It is worth mentioning that additional noncanonical functions of H/ACA RNAs have been documented in ribosome biogenesis. The 3′ hairpin of snR30 (also known as U17) contains sequences that act as conserved 18S rRNA recognition elements in the bulge of the hairpin, which pair with pre-rRNA. In the upper stem of the 3′ hairpin, there is a putative binding site for a protein that might be involved in the nucleolytic processing of the pre-rRNA 35S into the 18S rRNA. Because of this observed function, snR30 may act as a guide RNA for 18S rRNA processing that is not related to the more commonly known function of H/ACA RNPs in pseudouridylating rRNAs (Fayet-Lebaron et al. 2009).
Lastly, pseudouridines are very common and conserved in spliceosomal snRNAs—most snRNAs contain at least one pseudouridine, which is located in regions that may alter the RNA duplex structure, facilitating the snRNA function in splicing events (Meier 2005). Ψ35 in the U2 snRNA, a component of the spliceosome complex, for instance, is important to maintain an appropriate conformation for a nucleophilic attack that occurs in the initial stages of splicing, allowing an increased formation of the pre-mRNA splicing product. However, the precise mechanisms of how pseudouridines affect spliceosome biogenesis and splicing events have not been fully elucidated (Wu et al. 2011).
Regulation of telomerase
The mechanisms controlling telomerase activity and telomere maintenance are complex, and regulate multiple cellular processes in health and disease (MacNeil et al. 2016). The telomerase RNA component, hTR, is essential for proper telomerase function, and hTR regulation and processing have been recently extensively reviewed (Qin and Autexier 2021). Dyskerin plays a critical role in the stabilization and maintenance of hTR levels—in the absence of dyskerin, hTR levels and telomerase activity decrease. Dyskerin, however, does not act alone in the regulation and maintenance of hTR. SMUG1, for instance, has been reported to act in base excision repair at the 3′ end of hTR. This function is essential for dyskerin to bind to nascent hTR. hTR from SMUG1 knockout cells maintains base modifications that impair dyskerin binding and results in accumulation of hTR intermediates and augmented hTR degradation in an exosome component 10 enzyme (EXOSC10)-independent manner. The base modifications to which SMUG1 binds are located near the binding site for dyskerin, between CR4/5 and the H box, though the precise hTR bases and the types of modifications that impede the binding of dyskerin have not yet been identified (Kroustallaki et al. 2019)
The role of dyskerin in hTR quality control was further explored in knockdown experiments of cellular nucleases. It was suggested that the poly(A) polymerase 5 (PAPD5) and poly(A) RNase (PARN) have opposing functions of adenylating and deadenylating the 3′ end of hTR, respectively, regulating the degradation of hTR by exosome nucleases (Shukla et al. 2016). In the absence of dyskerin or PARN, or when dyskerin binding to hTR is compromised, hTR levels decrease, due to decapping by the cytoplasmic enzyme DCP2 followed by 5′–3′ exoribonuclease 1 (XRN1) degradation and 3′–5′ degradation by EXOSC10. Consequently, hTR does not localize to Cajal bodies efficiently, and cytoplasmic hTR bodies are also observed. EXOSC10 activity is enhanced by PAPD5-catalyzed oligoadenylation. Depletion of EXOSC10 and DCP2, in cells also subjected to dyskerin or PARN knockdown, restores telomerase activity. Depletion of EXOSC10 is also able to restore hTR localization to Cajal bodies (Shukla et al. 2016). In this context, dyskerin may be dispensable for telomerase function, conflicting with it being a component of the telomerase holoenzyme. Dyskerin may be dispensable for hTR maturation when the aforementioned nucleases are alternatively regulated such that hTR localization and accumulation can be reestablished. Further studies are required to determine whether dyskerin directly regulates telomere extension or indirectly by controlling the localization and stability of the telomerase RNP.
Additionally, dyskerin may also be involved in the actual modification of hTR. By mapping pseudouridines in the human transcriptome, two putative pseudouridines were identified in hTR. One of these possible pseudouridylation sites is located in the P6.1 hairpin of hTR, a region that is part of the catalytic core of telomerase (Schwartz et al. 2014). This may be associated with a noncanonical role of dyskerin in telomerase stability and function, however the enzyme catalyzing these modifications has not yet been identified.
Regulation of dyskerin expression in stem cells
In the context of hematopoietic stem and progenitor cells (HSPC), dyskerin was demonstrated to have an essential role in regulating HSPC differentiation into primary erythroblasts (Richards et al. 2020). The rapid multiplication of erythroid cells—but not their differentiation—is correlated with increased dyskerin expression. The increased expression is associated with GATA1, a transcription factor specific to the erythroid cell lineage. Binding of GATA1 to the DKC1 promoter is connected to higher levels of dyskerin expression, which in turn results in increased levels of active telomerase in proliferating primary erythroblasts. Once HSPC commit to erythroid lineage, increased levels of telomerase activity are regulated mostly by DKC1 expression, instead of being modulated by hTERT expression. Higher levels of dyskerin required for erythroblasts multiplication might be related to the role of dyskerin in ribosome biogenesis, which is substantially increased during erythropoiesis (Richards et al. 2020). The relationship between dyskerin and the regulation of stem cells, however, goes beyond the correlation between cell differentiation and DKC1 expression. When harboring an X-DC mutation associated with bone-marrow hypoplasia, CD34+ hematopoietic progenitor cells display considerably decreased differentiation into myeloid and erythroid cells (Bellodi et al. 2013). This defective phenotype is only rescued by the expression of wild-type dyskerin, but not by catalytically inactive dyskerin, which implies a possible role of pseudouridylation activity as a requirement for cell differentiation (Bellodi et al. 2013). Dyskerin and the H/ACA RNP are also involved in the self-renewal and differentiation of mesenchymal stem cells (Zhang et al. 2017). More specifically, the small nucleolar RNA 7A (SNORA7A) promotes the proliferation and the self-renewal capability of mesenchymal stem cells derived from umbilical cord blood, while impeding osteogenic and adipogenic differentiation of these cells. SNORA7A requires the core protein components of the H/ACA RNP—especially dyskerin—for this regulatory function (Zhang et al. 2017).
DYSKERIN AND HUMAN DISEASE
Premature aging syndromes, commonly represented by telomere biology disorders, or telomeropathies (Townsley et al. 2014; Martinez and Blasco 2017), are among the most common conditions caused or affected by dyskerin malfunction. Moreover, ribosome disorders (Farley-Barnes et al. 2019) can also be influenced by defective dyskerin expression or function. Both of these types of conditions are also connected to the development or progression of cancer (Barbieri and Kouzarides 2020). We highlight and discuss below some of the main findings concerning the impact of dyskerin on human health.
Premature aging syndromes
Dyskeratosis congenita (DC) is the most typical example among the premature aging syndromes associated with dyskerin malfunction. The estimate is that this disease affects one in every one million people (Dokal et al. 2015). Though first described at the beginning of the 20th century, it was only in 1998 that the genetic causes of the disease were identified. The classical triad of symptoms used to diagnose DC involve mucocutaneous features: reticular skin pigmentation, oral leukoplakia and nail dystrophy. Over the years, different symptoms and causal mutations have been identified. We now know that X-linked DC and its severe variant, Hoyeraal–Hreidarsson syndrome (Knight et al. 1999a; Glousker et al. 2015), are caused by mutations in the DKC1 gene (Heiss et al. 1998; Knight et al. 1999b). The autosomal variants involve mutations in other genes, associated with the components of the H/ACA RNP (NOP10, NHP2), the telomerase holoenzyme (TERC, TERT), the shelterin complex (de Lange 2005) that binds telomeres (TINF2) (Walne and Dokal 2009), besides PARN, WRAP53 (TCAB1), the helicase RTEL1 and CTC1, a member of the CST complex, which participates in telomere elongation (Grill and Nandakumar 2020). Defects in hTR processing and in the regulation of the telomerase holoenzyme impacting telomere biology syndromes have recently been extensively reviewed (Grill and Nandakumar 2020; Nagpal and Agarwal 2020; Roake and Artandi 2020).
Structurally, multiple X-DC mutations are grouped in the PUA domain of the archaeal dyskerin homolog, suggesting that X-DC may be linked to defective dyskerin-RNA interaction (Rashid et al. 2006). In the human H/ACA RNP structure, a cluster of DC mutations map to the interface of both dyskerin subunits as well as to the dyskerin-hTR interface (Nguyen et al. 2018). Furthermore, the amino-terminal portion of the 5′ dyskerin which sits in the hydrophobic pocket of the 3′ dyskerin contains a cluster of DC and Hoyeraal–Hreidarsson mutations (Ghanim et al. 2021). X-DC mutations can also perturb naturally occurring SUMOylation sites in human dyskerin. The amino-terminal SUMOylated residues K39 and K43, when mutated, are associated with decreased levels of dyskerin, deficient hTR accumulation and telomerase activity, and shorter telomeres, even when SUMOylation is only mildly diminished (Brault et al. 2013). These same mutated residues, along with A353V in the PUA, lead to reduced interaction between dyskerin and H/ACA RNAs. Nucleolar localization of these dyskerin variants and protein–protein interactions within the RNP, however, are not impaired. Dyskerin mutated at the amino-terminal residues demonstrates a more pronounced reduction in association with hTR in comparison with other H/ACA RNAs, indicating a potential telomerase-centric role for these amino-terminal residues. All three variants lead to a decline of 3′ extended precursors, polyadenylated and total hTR accumulation. This observation led to a model suggesting that hTR precursors are degraded instead of processed due to their inability to properly associate with these dyskerin variants (MacNeil et al. 2019).
An unusual manifestation of defects in dyskerin was observed in one case of idiopathic pulmonary fibrosis, a disease commonly associated with telomere shortening. The investigation of this case of familial pulmonary fibrosis revealed that a silent mutation in DKC1 is responsible for the observed phenotype (Gaysinskaya et al. 2020). The causal silent mutation generates a splicing enhancer that results in missplicing events and degradation of the misspliced DKC1 mRNA product via nonsense-mediated decay. This leads to lower levels of dyskerin, which in turn significantly reduces the accumulation of hTR. This splicing defect, associated with the onset of idiopathic pulmonary fibrosis later in life, differs from missense mutations in DKC1, which normally manifest as DC earlier in life (Gaysinskaya et al. 2020).
Defects in ribosome biogenesis and cell proliferation
Some mutations in DKC1 and in NOP10 cause symptoms that considerably differ from the classical X-DC mucocutaneous phenotype. Cases of two families, one harboring a mutation in NOP10 and the other harboring an E206K mutation in DKC1, present symptoms of nephrotic syndrome and cataracts, previously unreported for mutations related to the H/ACA RNP (Balogh et al. 2020). They also exhibit auditory deficiency, enterocolitis, telomere attrition and premature death. Although telomere attrition was reported, symptoms implicate a defective cellular function independent of telomerase. The causal mutations are located at the interface between dyskerin and NOP10 near the catalytic site of the RNP. The absence of classical symptoms, the location of the mutation sites in the H/ACA RNP structure, and the reduced rRNA pseudouridine levels in these patients indicate that this disorder is related to impaired ribosomal biogenesis, caused by catalytic defects in rRNA pseudouridylation. The ribosomal dysfunction observed in humans was recapitulated in zebrafish with the equivalent DKC1 mutation (Balogh et al. 2020).
Altered pseudouridylation associated with DC has been further clarified by high-throughput experiments. Following a study on the human 80S ribosome based on a high-resolution cryo-EM structure (Natchiar et al. 2017), the chemical modifications found in the human 80S ribosome were determined by quantitative mass spectrometry (Taoka et al. 2018). In this latter study, the 28S rRNA component of the 80S ribosome derived from cells of X-DC patients had reduced levels of pseudouridylation at two specific nucleotides located on the external surface of the large ribosomal subunit. This reduced pseudouridylation may impact the interaction of the 28S rRNA with the ribosomal proteins and destabilize the ribosome, potentially leading to impaired translation (Taoka et al. 2018). Slightly lower pseudouridylation levels were also detected in rRNA and hTR by applying the Ψ-seq technique transcriptome-wide in DC patient-derived cells (Schwartz et al. 2014).
Mutations in the catalytic site of dyskerin can lead to especially detrimental effects in ribosome function. Yeast, murine and human cells display similar ribosomal ligand defects due to impaired pseudouridylation activity when the catalytic aspartic acid residue in dyskerin or its homologs is mutated (Jack et al. 2011). Ribosome defects are manifested by impaired ribosomal binding to the cricket paralysis virus (CrPV) internal ribosome entry site (IRES) element, and consequent defective IRES-dependent translation initiation. Furthermore, tRNAs, which are structurally similar to the CrPV IRES element, also had reduced affinity to ribosomes lacking pseudouridylated rRNA in its A (tRNA entry site), P (peptidyl-tRNA site) and decoding sites. Cells with reduced pseudouridylation levels demonstrate distinctive sensitivity to compounds that inhibit translation in comparison to cells expressing wild-type dyskerin. Observed growth defects may arise from impaired translation fidelity represented by increased frameshifting and stop codon readthrough during translation, due to decreased pseudouridylation in rRNA. This is a conserved mechanism among yeast and mammalian cells, and it promotes alterations in gene expression that may be associated with the phenotypes observed in DC (Jack et al. 2011).
A role for impaired pseudouridylation in premature aging syndromes caused by mutations in DKC1 is also supported by studies in mice. Hypomorphic DCK1 mice recapitulate the human phenotype of DC, including bone-marrow failure and high predisposition to tumor formation (Ruggero et al. 2003). The first generations of the mutant mice display decreased levels of pseudouridylation in rRNAs, higher amounts of immature rRNA molecules and hypersensitivity to drugs that inhibit translation. Telomere shortening manifests as multiple cell divisions occur, requiring more time to elicit symptoms in comparison to defective translation due to impaired ribosome biogenesis. Since mice telomeres are many times longer than human telomeres (Zijlmans et al. 1997; Calado and Dumitriu 2013), impaired pseudouridylation activity may be predominant in these hypomorphic DCK1 mice compared to human DC patients, especially in the early stage of the disease, when the mouse telomeres are not yet sufficiently short to elicit cellular phenotypes. Lastly, it is important to highlight that in addition to dyskerin malfunction, other defective components of the H/ACA RNP can also lead to similar disease phenotypes. NHP2 mutations, for instance, have been identified as the genetic cause of some cases of Høyeraal–Hreidarsson syndrome and pulmonary fibrosis (Benyelles et al. 2020).
The role of dyskerin in cancer
Altered dyskerin expression has been linked with tumor formation and multiple types of cancer (Alawi and Lee 2007; O'Brien et al. 2016; Ko et al. 2018; Elsharawy et al. 2020; Hou et al. 2020). The correlation between reduced levels of dyskerin and increased rates of tumor formation led to the suggestion that dyskerin may be a tumor suppressor (Ruggero et al. 2003). Dyskerin overexpression, on the other hand, has been associated with poor prognosis in breast cancer (Elsharawy et al. 2020) and higher prevalence of nodal metastasis in patients. This phenomenon may be linked to the increased proliferation and invasion capability observed in immortalized untransformed mammary epithelial cells overexpressing dyskerin. This aggressive phenotype could be caused by the improved translational efficiency observed in these cells, as a result of increases in specific rRNA modifications when dyskerin is overexpressed (Guerrieri et al. 2020).
Colorectal cancer tissues display increased expression levels of dyskerin, and this is associated with poor disease progression and survival. This correlation can be explained by dyskerin directly binding to the promoter of the transcription factor Hypoxia-inducible factor 1 (HIF-1) gene and increasing its expression (Hou et al. 2020). HIF-1 is normally activated by hypoxia in rapidly proliferating solid tumors. Under low oxygen concentration, the HIF-1α subunit accumulates and is translocated to the nucleus, where it interacts with the HIF-1β subunit. The resulting complex can activate the transcription of genes such as the vascular endothelial growth factor (VEGF), which in turn induces angiogenesis and metastasis in tumors (Weidemann and Johnson 2008). In this case, dyskerin expression levels can be used as a prognostic factor for colorectal cancer patients (Hou et al. 2020).
The expression of the DKC1 gene is also altered by the Protein-Disulfide Isomerase-Associated 3 (PDIA3). PDIA3, also known as Erp57, one of the members of the family of enzymes that catalyze the formation of native disulfide bonds (Ellgaard and Ruddock 2005). In humans, PDIA3 binds a G-quadruplex secondary structure (G4) motif in the promoter of the DKC1 gene, repressing its transcription (Ko et al. 2018). In the liver, where reactive oxygen species form at relatively high levels, PDIA3 is modified by oxidation leading to its cytoplasmic localization in hepatic cells. The release of PDIA3 from the G4 DNA in the DKC1 promoter allows the recruitment of the transcription complex resulting in increased dyskerin mRNA levels. The up-regulation of dyskerin expression mediated by PDIA3 oxidation and consequent removal from the DKC1 promoter is thus a common feature in hepatocellular carcinoma, in which an increased oxidative environment can be observed. In this case, dyskerin expression could be used as a factor for poor prognosis of disease progression. In fact, hepatocellular carcinoma cell survival is dependent on increased levels of dyskerin. Either the knockdown of dyskerin or the overexpression of PDIA3 (and subsequent increased suppression of DKC1 expression) are correlated with decreased invasion ability of hepatocellular carcinoma cells (Ko et al. 2018).
Hematological defects
Specific mutations in DKC1 may cause hematological defects by influencing hTR processing. Human embryonic stem cells (hESC) expressing dyskerin containing the X-DC mutation A353V display defects in the hematopoietic potential similar to those observed in X-DC patients. hTR degradation by the exosome complex is regulated by adenylation of its 3′ end. While silencing of either the exosome component gene 3 (EXOSC3) or PAPD5 results in diminished hTR degradation, restores telomerase activity, and reduces the levels of DNA double strand breaks signaling, only the silencing of PAPD5 is able to re-establish the hematopoietic potential of cells harboring this X-DC dyskerin variant (Fok et al. 2019). From these findings, the inhibition of PAPD5 was suggested as a potential treatment for DC. The compound RG7834, a PAPD5/7 inhibitor, caused a decline in the 3′ adenylation and increased cellular accumulation of hTR, as well as restored telomere lengthening and hematopoietic potential in cells displaying the X-DC mutation A353V. Overall, RG7834 elicited similar effects on cell function to those previously observed when the expression of PAPD5 was genetically silenced (Shukla et al. 2020).
CONCLUSIONS AND OUTLOOK
The presence of homologs in all domains of life substantiate the importance of dyskerin for cell function. The loss of dyskerin homologs is lethal or causes serious cell proliferation defects in multiple organisms, including mice, fruit flies, plants, and yeast (Jiang et al. 1993; Giordano et al. 1999; He et al. 2002; Kannan et al. 2008; Ge et al. 2010). During evolution, dyskerin developed close associations with other proteins and H/ACA RNAs. Many details on the H/ACA RNP biogenesis have been elucidated in the last two decades. However, there is no consensus on the suggested order of assembly of each component or factor in different organisms. The mechanism by which GAR1 displaces NAF1 to form the mature H/ACA RNP remains unknown. Furthermore, proposed mechanisms and supporting evidence for the role of some factors, such as Hsp90, the SMN complex and Nopp140, in the assembly process are lacking. Despite the relevance of dyskerin for fundamental biological processes, precise molecular mechanisms on how dyskerin and the H/ACA RNP act in pseudouridylation, rRNA processing, pre-mRNA splicing, and telomere elongation also remain incompletely characterized. It is remarkable how the same core protein components, with minimum conformational changes, and potentially independent of an external energy source, are able to associate with numerous H/ACA RNAs and then catalyze a nucleotide base change in so many target RNAs.
The role of dyskerin in health and disease is considerably complex and multifaceted. Point mutations in DKC1 and the diverse disease phenotypes displayed by X-DC patients confirm the essential role of this protein for human health. Still, differences in the biology of animal models and humans make the translation of discoveries associated with the causes of DC complicated. Mice, for instance, display significantly longer telomeres than humans, and this difference may account for a phenotypic disparity between the two species upon dyskerin dysfunction. Dyskerin mutations and a perturbed regulation of dyskerin levels seems to be more intimately connected with telomerase defects in humans whereas, in mice, ribosomal defects play a significant role. As it can be inferred from recent studies (Shukla et al. 2016), the part that dyskerin plays in telomerase function may be connected more predominantly with maintenance of hTR levels than with its presence in the holoenzyme structure. Nevertheless, some manifestations of DKC1 mutations in humans do implicate defective ribosomal biogenesis as the cause of the disease phenotype (Balogh et al. 2020). Moreover, the connection between dyskerin and cancer also requires further clarification, since the protein was proposed to act as a tumor suppressor in some instances and as a factor associated with poor prognosis in cases of breast cancer, neuroblastoma, and hepatocellular carcinoma.
DC, typically associated with dyskerin, is still an incurable disease. Novel treatments could be developed by targeting enzymes that participate in hTR processing, such as the suggested inhibition of PAPD5/7 (Shukla et al. 2020), or by the depletion of the exonucleases EXOSC10 and XRN1. Targeting dyskerin could be a tricky strategy though, since both lower and higher cellular levels of dyskerin are associated with disease. While disease phenotypes caused by point mutations in DKC1 can be rescued by expression of wild-type dyskerin, a CRISPR-based approach (Doudna and Charpentier 2014) could be studied as an alternative option for treating DC. Clinical trials using CRISPR to treat genetic conditions have already been conducted for transthyretin amyloidosis, sickle cell disease and β-thalassemia (Frangoul et al. 2021; Gillmore et al. 2021). However, these trials were based on silencing the faulty gene expression. Since dyskerin is important for multiple cellular processes, single base editing to change the mutated residue should be used instead. This is a very recent strategy and the limitations of the technique, including possible off-target effects, immune response to the foreign molecules and the delivery system as well as the high cost of experimental clinical trials, should be considered.
In conclusion, the incompletely elucidated mechanisms of the H/ACA RNP assembly, in addition to the many noncanonical functions reported in recent years, add to the complexity and significance of dyskerin for cellular processes—an opportunity for discoveries in many research fields for years to come.
ACKNOWLEDGMENTS
The authors thank Kelly Nguyen, Mark Bayfield, Jian Qin, and Adrian Young for helpful comments on the manuscript. Work on dyskerin in the laboratory of C.A. is supported by a Canadian Institutes of Health Research (CIHR) Project grant PJT-166130, and a McGill University Faculty of Medicine Internal Studentship to A.G.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.078953.121.
Freely available online through the RNA Open Access option.
REFERENCES
- Alawi F, Lee MN. 2007. DKC1 is a direct and conserved transcriptional target of c-MYC. Biochem Biophys Res Commun 362: 893–898. 10.1016/j.bbrc.2007.08.071 [DOI] [PubMed] [Google Scholar]
- Angrisani A, Vicidomini R, Turano M, Furia M. 2014. Human dyskerin: beyond telomeres. Biol Chem 395: 593–610. 10.1515/hsz-2013-0287 [DOI] [PubMed] [Google Scholar]
- Balakin AG, Smith L, Fournier MJ. 1996. The RNA world of the nucleolus: two major families of small RNAs defined by different box elements with related functions. Cell 86: 823–834. 10.1016/S0092-8674(00)80156-7 [DOI] [PubMed] [Google Scholar]
- Ballarino M, Morlando M, Pagano F, Fatica A, Bozzoni I. 2005. The cotranscriptional assembly of snoRNPs controls the biosynthesis of H/ACA snoRNAs in Saccharomyces cerevisiae. Mol Cell Biol 25: 5396–5403. 10.1128/MCB.25.13.5396-5403.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balogh E, Chandler JC, Varga M, Tahoun M, Menyhárd DK, Schay G, Goncalves T, Hamar R, Légrádi R, Szekeres Á, et al. 2020. Pseudouridylation defect due to DKC1 and NOP10 mutations causes nephrotic syndrome with cataracts, hearing impairment, and enterocolitis. Proc Natl Acad Sci 117: 15137–15147. 10.1073/pnas.2002328117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri I, Kouzarides T. 2020. Role of RNA modifications in cancer. Nat Rev Cancer 20: 303–322. 10.1038/s41568-020-0253-2 [DOI] [PubMed] [Google Scholar]
- Becker J, Barysch SV, Karaca S, Dittner C, Hsiao H-H, Diaz MB, Herzig S, Urlaub H, Melchior F. 2013. Detecting endogenous SUMO targets in mammalian cells and tissues. Nat Struct Mol Biol 20: 525–531. 10.1038/nsmb.2526 [DOI] [PubMed] [Google Scholar]
- Bellodi C, McMahon M, Contreras A, Juliano D, Kopmar N, Nakamura T, Maltby D, Burlingame A, Savage SA, Shimamura A, et al. 2013. H/ACA small RNA dysfunctions in disease reveal key roles for noncoding RNA modifications in hematopoietic stem cell differentiation. Cell Rep 3: 1493–1502. 10.1016/j.celrep.2013.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyelles M, O'Donohue M-F, Kermasson L, Lainey E, Borie R, Lagresle-Peyrou C, Nunes H, Cazelles C, Fourrage C, Ollivier E, et al. 2020. NHP2 deficiency impairs rRNA biogenesis and causes pulmonary fibrosis and Høyeraal-Hreidarsson syndrome. Hum Mol Genet 29: 907–922. 10.1093/hmg/ddaa011 [DOI] [PubMed] [Google Scholar]
- Bizarro J, Bhardwaj A, Smith S, Meier UT. 2019. Nopp140-mediated concentration of telomerase in Cajal bodies regulates telomere length. Mol Biol Cell 30: 3136–3150. 10.1091/mbc.E19-08-0429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulon S, Marmier-Gourrier N, Pradet-Balade B, Wurth L, Verheggen C, Jády BE, Rothé B, Pescia C, Robert M-C, Kiss T, et al. 2008. The Hsp90 chaperone controls the biogenesis of L7Ae RNPs through conserved machinery. J Cell Biol 180: 579–595. 10.1083/jcb.200708110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault ME, Lauzon C, Autexier C. 2013. Dyskeratosis congenita mutations in dyskerin SUMOylation consensus sites lead to impaired telomerase RNA accumulation and telomere defects. Hum Mol Genet 22: 3498–3507. 10.1093/hmg/ddt204 [DOI] [PubMed] [Google Scholar]
- Cadwell C, Yoon HJ, Zebarjadian Y, Carbon J. 1997. The yeast nucleolar protein Cbf5p is involved in rRNA biosynthesis and interacts genetically with the RNA polymerase I transcription factor RRN3. Mol Cell Biol 17: 6175–6183. 10.1128/MCB.17.10.6175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calado RT, Dumitriu B. 2013. Telomere dynamics in mice and humans. Semin Hematol 50: 165–174. 10.1053/j.seminhematol.2013.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappadocia L, Lima CD. 2018. Ubiquitin-like protein conjugation: structures, chemistry, and mechanism. Chem Rev 118: 889–918. 10.1021/acs.chemrev.6b00737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerrudo CS, Ghiringhelli PD, Gomez DE. 2014. Protein universe containing a PUA RNA-binding domain. FEBS J 281: 74–87. 10.1111/febs.12602 [DOI] [PubMed] [Google Scholar]
- Cerrudo CS, Mengual Gómez DL, Gómez DE, Ghiringhelli PD. 2015. Novel insights into the evolution and structural characterization of dyskerin using comprehensive bioinformatics analysis. J Proteome Res 14: 874–887. 10.1021/pr500956k [DOI] [PubMed] [Google Scholar]
- Czekay DP, Kothe U. 2021. H/ACA small ribonucleoproteins: structural and functional comparison between archaea and eukaryotes. Front Microbiol 12: 654370. 10.3389/fmicb.2021.654370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzacq X, Kittur N, Roy S, Shav-Tal Y, Singer RH, Meier UT. 2006. Stepwise RNP assembly at the site of H/ACA RNA transcription in human cells. J Cell Biol 173: 207–218. 10.1083/jcb.200601105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. 2005. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 19: 2100–2110. 10.1101/gad.1346005 [DOI] [PubMed] [Google Scholar]
- De Zoysa MD, Wu G, Katz R, Yu Y-T. 2018. Guide-substrate base-pairing requirement for box H/ACA RNA-guided RNA pseudouridylation. RNA 24: 1106–1117. 10.1261/rna.066837.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokal I, Vulliamy T, Mason P, Bessler M. 2015. Clinical utility gene card for: dyskeratosis congenita—update 2015. Eur J Hum Genet 23: 4. 10.1038/ejhg.2014.170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E. 2014. The new frontier of genome engineering with CRISPR-Cas9. Science 346: 1258096. 10.1126/science.1258096 [DOI] [PubMed] [Google Scholar]
- Duan J, Li L, Lu J, Wang W, Ye K. 2009. Structural mechanism of substrate RNA recruitment in H/ACA RNA-guided pseudouridine synthase. Mol Cell 34: 427–439. 10.1016/j.molcel.2009.05.005 [DOI] [PubMed] [Google Scholar]
- Egan ED, Collins K. 2010. Specificity and stoichiometry of subunit interactions in the human telomerase holoenzyme assembled in vivo. Mol Cell Biol 30: 2775–2786. 10.1128/MCB.00151-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan ED, Collins K. 2012a. An enhanced H/ACA RNP assembly mechanism for human telomerase RNA. Mol Cell Biol 32: 2428–2439. 10.1128/MCB.00286-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan ED, Collins K. 2012b. Biogenesis of telomerase ribonucleoproteins. RNA 18: 1747–1759. 10.1261/rna.034629.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L, Ruddock LW. 2005. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep 6: 28–32. 10.1038/sj.embor.7400311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsharawy KA, Mohammed OJ, Aleskandarany MA, Hyder A, El-Gammal HL, Abou-Dobara MI, Green AR, Dalton LW, Rakha EA. 2020. The nucleolar-related protein Dyskerin pseudouridine synthase 1 (DKC1) predicts poor prognosis in breast cancer. Br J Cancer 123: 1543–1552. 10.1038/s41416-020-01045-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ender C, Krek A, Friedländer MR, Beitzinger M, Weinmann L, Chen W, Pfeffer S, Rajewsky N, Meister G. 2008. A human snoRNA with microRNA-like functions. Mol Cell 32: 519–528. 10.1016/j.molcel.2008.10.017 [DOI] [PubMed] [Google Scholar]
- Fajkus P, Peška V, Závodník M, Fojtová M, Fulnečková J, Dobias Š, Kilar A, Dvořáčková M, Zachová D, Nečasová I, et al. 2019. Telomerase RNAs in land plants. Nucleic Acids Res 47: 9842–9856. 10.1093/nar/gkz695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley-Barnes KI, Ogawa LM, Baserga SJ. 2019. Ribosomopathies: old concepts, new controversies. Trends Genet 35: 754–767. 10.1016/j.tig.2019.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayet-Lebaron E, Atzorn V, Henry Y, Kiss T. 2009. 18S rRNA processing requires base pairings of snR30 H/ACA snoRNA to eukaryote-specific 18S sequences. EMBO J 28: 1260–1270. 10.1038/emboj.2009.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fok WC, Shukla S, Vessoni AT, Brenner KA, Parker R, Sturgeon CM, Batista LFZ. 2019. Posttranscriptional modulation of TERC by PAPD5 inhibition rescues hematopoietic development in dyskeratosis congenita. Blood 133: 1308–1312. 10.1182/blood-2018-11-885368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangoul H, Altshuler D, Cappellini MD, Chen Y-S, Domm J, Eustace BK, Foell J, de la Fuente J, Grupp S, Handgretinger R, et al. 2021. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N Engl J Med 384: 252–260. 10.1056/NEJMoa2031054 [DOI] [PubMed] [Google Scholar]
- Ganot P, Caizergues-Ferrer M, Kiss T. 1997. The family of box ACA small nucleolar RNAs is defined by an evolutionarily conserved secondary structure and ubiquitous sequence elements essential for RNA accumulation. Genes Dev 11: 941–956. 10.1101/gad.11.7.941 [DOI] [PubMed] [Google Scholar]
- Gaysinskaya V, Stanley SE, Adam S, Armanios M. 2020. Synonymous mutation in DKC1 causes telomerase RNA insufficiency manifesting as familial pulmonary fibrosis. Chest 158: 2449–2457. 10.1016/j.chest.2020.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Rudnick DA, He J, Crimmins DL, Ladenson JH, Bessler M, Mason PJ. 2010. Dyskerin ablation in mouse liver inhibits rRNA processing and cell division. Mol Cell Biol 30: 413–422. 10.1128/MCB.01128-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanim GE, Fountain AJ, van Roon A-MM, Rangan R, Das R, Collins K, Nguyen THD. 2021. Structure of human telomerase holoenzyme with bound telomeric DNA. Nature 593: 449–453. 10.1038/s41586-021-03415-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, Seitzer J, O'Connell D, Walsh KR, Wood K, et al. 2021. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med 385: 493–502. 10.1056/NEJMoa2107454 [DOI] [PubMed] [Google Scholar]
- Giordano E, Peluso I, Senger S, Furia M. 1999. minifly, a Drosophila gene required for ribosome biogenesis. J Cell Biol 144: 1123–1133. 10.1083/jcb.144.6.1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glousker G, Touzot F, Revy P, Tzfati Y, Savage SA. 2015. Unraveling the pathogenesis of Hoyeraal–Hreidarsson syndrome, a complex telomere biology disorder. Br J Haematol 170: 457–471. 10.1111/bjh.13442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE. 2018. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci 27: 14–25. 10.1002/pro.3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill S, Nandakumar J. 2020. Molecular mechanisms of telomere biology disorders. J Biol Chem 296: 100064. 10.1074/jbc.REV120.014017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozdanov PN, Roy S, Kittur N, Meier UT. 2009. SHQ1 is required prior to NAF1 for assembly of H/ACA small nucleolar and telomerase RNPs. RNA 15: 1188–1197. 10.1261/rna.1532109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu XR, Liu YQ, Santi DV. 1999. The mechanism of pseudouridine synthase I as deduced from its interaction with 5-fluorouracil-tRNA. Proc Natl Acad Sci 96: 14270–14275. 10.1073/pnas.96.25.14270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrieri AN, Zacchini F, Onofrillo C, Di Viggiano S, Penzo M, Ansuini A, Gandin I, Nobe Y, Taoka M, Isobe T, et al. 2020. DKC1 overexpression induces a more aggressive cellular behavior and increases intrinsic ribosomal activity in immortalized mammary gland cells. Cancers (Basel) 12: 3512. 10.3390/cancers12123512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutgsell N, Englund N, Niu L, Kaya Y, Lane BG, Ofengand J. 2000. Deletion of the Escherichia coli pseudouridine synthase gene truB blocks formation of pseudouridine 55 in tRNA in vivo, does not affect exponential growth, but confers a strong selective disadvantage in competition with wild-type cells. RNA 6: 1870–1881. 10.1017/S1355838200001588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamma T, Ferré-D'Amaré AR. 2006. Pseudouridine synthases. Chem Biol 13: 1125–1135. 10.1016/j.chembiol.2006.09.009 [DOI] [PubMed] [Google Scholar]
- Hamma T, Ferré-D'Amaré AR. 2010. The box H/ACA ribonucleoprotein complex: interplay of RNA and protein structures in post-transcriptional RNA modification. J Biol Chem 285: 805–809. 10.1074/jbc.R109.076893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamma T, Reichow SL, Varani G, Ferre-D'Amare AR. 2005. The Cbf5-Nop10 complex is a molecular bracket that organizes box H/ACA RNPs. Nat Struct Mol Biol 12: 1101–1107. 10.1038/nsmb1036 [DOI] [PubMed] [Google Scholar]
- Hanson PI, Whiteheart SW. 2005. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol 6: 519–529. 10.1038/nrm1684 [DOI] [PubMed] [Google Scholar]
- He J, Navarrete S, Jasinski M, Vulliamy T, Dokal I, Bessler M, Mason PJ. 2002. Targeted disruption of Dkc1, the gene mutated in X-linked dyskeratosis congenita, causes embryonic lethality in mice. Oncogene 21: 7740–7744. 10.1038/sj.onc.1205969 [DOI] [PubMed] [Google Scholar]
- Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, Poustka A, Dokal I. 1998. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet 19: 32–38. 10.1038/ng0598-32 [DOI] [PubMed] [Google Scholar]
- Heiss NS, Girod A, Salowsky R, Wiemann S, Pepperkok R, Poustka A. 1999. Dyskerin localizes to the nucleolus and its mislocalization is unlikely to play a role in the pathogenesis of dyskeratosis congenita. Hum Mol Genet 8: 2515–2524. 10.1093/hmg/8.13.2515 [DOI] [PubMed] [Google Scholar]
- Hendriks IA, Vertegaal ACO. 2016. A comprehensive compilation of SUMO proteomics. Nat Rev Mol Cell Biol 17: 581–595. 10.1038/nrm.2016.81 [DOI] [PubMed] [Google Scholar]
- Hendriks IA, D'Souza RCJ, Yang B, Verlaan-de Vries M, Mann M, Vertegaal ACO. 2014. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat Struct Mol Biol 21: 927–936. 10.1038/nsmb.2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks IA, Lyon D, Su D, Skotte NH, Daniel JA, Jensen LJ, Nielsen ML. 2018. Site-specific characterization of endogenous SUMOylation across species and organs. Nat Commun 9: 2456. 10.1038/s41467-018-04957-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang C, Ferré-D'Amaré AR. 2001. Cocrystal structure of a tRNA Ψ55 pseudouridine synthase: nucleotide flipping by an RNA-modifying enzyme. Cell 107: 929–939. 10.1016/S0092-8674(01)00618-3 [DOI] [PubMed] [Google Scholar]
- Hou P, Shi P, Jiang T, Yin H, Chu S, Shi M, Bai J, Song J. 2020. DKC1 enhances angiogenesis by promoting HIF-1α transcription and facilitates metastasis in colorectal cancer. Br J Cancer 122: 668–679. 10.1038/s41416-019-0695-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Pookanjanatavip M, Gu X, Santi DV. 1998. A conserved aspartate of tRNA pseudouridine synthase is essential for activity and a probable nucleophilic catalyst. Biochemistry 37: 344–351. 10.1021/bi971874+ [DOI] [PubMed] [Google Scholar]
- Jack K, Bellodi C, Landry DM, Niederer RO, Meskauskas A, Musalgaonkar S, Kopmar N, Krasnykh O, Dean AM, Thompson SR, et al. 2011. rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol Cell 44: 660–666. 10.1016/j.molcel.2011.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Middleton K, Yoon H, Fouquet C, Carbon J. 1993. An essential yeast protein, Cbf5p, binds in-vitro to centromeres and microtubules. Mol Cell Biol 13: 4884–4893. 10.1128/MCB.13.8.4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Loria JP, Moore PB. 2007. Solution structure of an rRNA substrate bound to the pseudouridylation pocket of a box H/ACA snoRNA. Mol Cell 26: 205–215. 10.1016/j.molcel.2007.03.014 [DOI] [PubMed] [Google Scholar]
- Jobert L, Skjeldam HK, Dalhus B, Galashevskaya A, Vågbø CB, Bjørås M, Nilsen H. 2013. The human base excision repair enzyme SMUG1 directly interacts with DKC1 and contributes to RNA quality control. Mol Cell 49: 339–345. 10.1016/j.molcel.2012.11.010 [DOI] [PubMed] [Google Scholar]
- Kakihara Y, Houry WA. 2012. The R2TP complex: discovery and functions. Biochim Biophys Acta Mol Cell Res 1823: 101–107. 10.1016/j.bbamcr.2011.08.016 [DOI] [PubMed] [Google Scholar]
- Kannan K, Nelson ADL, Shippen DE. 2008. Dyskerin is a component of the Arabidopsis telomerase RNP required for telomere maintenance. Mol Cell Biol 28: 2332–2341. 10.1128/MCB.01490-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keffer-Wilkes LC, Veerareddygari GR, Kothe U. 2016. RNA modification enzyme TruB is a tRNA chaperone. Proc Natl Acad Sci 113: 14306–14311. 10.1073/pnas.1607512113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly EK, Czekay DP, Kothe U. 2019. Base-pairing interactions between substrate RNA and H/ACA guide RNA modulate the kinetics of pseudouridylation, but not the affinity of substrate binding by H/ACA small nucleolar ribonucleoproteins. RNA 25: 1393–1404. 10.1261/rna.071043.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierzek E, Malgowska M, Lisowiec J, Turner DH, Gdaniec Z, Kierzek R. 2014. The contribution of pseudouridine to stabilities and structure of RNAs. Nucleic Acids Res 42: 3492–3501. 10.1093/nar/gkt1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirwan M, Dokal I. 2008. Dyskeratosis congenita: a genetic disorder of many faces. Clin Genet 73: 103–112. 10.1111/j.1399-0004.2007.00923.x [DOI] [PubMed] [Google Scholar]
- Kiss T, Fayet-Lebaron E, Jady BE. 2010. Box H/ACA small ribonucleoproteins. Mol Cell 37: 597–606. 10.1016/j.molcel.2010.01.032 [DOI] [PubMed] [Google Scholar]
- Kittur N, Darzacq X, Roy S, Singer RH, Meier UT. 2006. Dynamic association and localization of human H/ACA RNP proteins. RNA 12: 2057–2062. 10.1261/rna.249306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight SW, Heiss NS, Vulliamy TJ, Aalfs CM, McMahon C, Richmond P, Jones A, Hennekam RC, Poustka A, Mason PJ, et al. 1999a. Unexplained aplastic anaemia, immunodeficiency, and cerebellar hypoplasia (Hoyeraal-Hreidarsson syndrome) due to mutations in the dyskeratosis congenita gene, DKC1. Br J Haematol 107: 335–339. 10.1046/j.1365-2141.1999.01690.x [DOI] [PubMed] [Google Scholar]
- Knight SW, Heiss NS, Vulliamy TJ, Greschner S, Stavrides G, Pai GS, Lestringant G, Varma N, Mason PJ, Dokal I, et al. 1999b. X-linked dyskeratosis congenita is predominantly caused by missense mutations in the DKC1 gene. Am J Hum Genet 65: 50–58. 10.1086/302446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko E, Kim J-S, Ju S, Seo H-W, Chang Y, Kang J-A, Park S-G, Jung G. 2018. Oxidatively modified protein-disulfide isomerase-associated 3 promotes dyskerin pseudouridine synthase 1-mediated malignancy and survival of hepatocellular carcinoma cells. Hepatology 68: 1851–1864. 10.1002/hep.30039 [DOI] [PubMed] [Google Scholar]
- Koonin EV. 1996. Pseudouridine synthases: four families of enzymes containing a putative uridine-binding motif also conserved in dUTPases and dCTP deaminases. Nucleic Acids Res 24: 2411–2415. 10.1093/nar/24.12.2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroustallaki P, Lirussi L, Carracedo S, You P, Esbensen QY, Götz A, Jobert L, Alsøe L, Sætrom P, Gagos S, et al. 2019. SMUG1 promotes telomere maintenance through telomerase RNA processing. Cell Rep 28: 1690–1702. 10.1016/j.celrep.2019.07.040 [DOI] [PubMed] [Google Scholar]
- Lafontaine DL, Bousquet-Antonelli C, Henry Y, Caizergues-Ferrer M, Tollervey D. 1998. The box H+ACA snoRNAs carry Cbf5p, the putative rRNA pseudouridine synthase. Genes Dev 12: 527–537. 10.1101/gad.12.4.527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamoliatte F, McManus FP, Maarifi G, Chelbi-Alix MK, Thibault P. 2017. Uncovering the SUMOylation and ubiquitylation crosstalk in human cells using sequential peptide immunopurification. Nat Commun 8: 14109. 10.1038/ncomms14109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langelier M-F, Eisemann T, Riccio AA, Pascal JM. 2018. PARP family enzymes: regulation and catalysis of the poly(ADP-ribose) posttranslational modification. Curr Opin Struct Biol 53: 187–198. 10.1016/j.sbi.2018.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Ye K. 2006. Crystal structure of an H/ACA box ribonucleoprotein particle. Nature 443: 302–307. 10.1038/nature05151 [DOI] [PubMed] [Google Scholar]
- Li S, Duan J, Li D, Ma S, Ye K. 2011a. Structure of the Shq1–Cbf5–Nop10–Gar1 complex and implications for H/ACA RNP biogenesis and dyskeratosis congenita. EMBO J 30: 5010–5020. 10.1038/emboj.2011.427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Duan J, Li D, Yang B, Dong M, Ye K. 2011b. Reconstitution and structural analysis of the yeast box H/ACA RNA-guided pseudouridine synthase. Genes Dev 25: 2409–2421. 10.1101/gad.175299.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang B, Xue S, Terns RM, Terns MP, Li H. 2007. Substrate RNA positioning in the archaeal H/ACA ribonucleoprotein complex. Nat Struct Mol Biol 14: 1189–1195. 10.1038/nsmb1336 [DOI] [PubMed] [Google Scholar]
- Liang B, Zhou J, Kahen E, Terns RM, Terns MP, Li H. 2009a. Structure of a functional ribonucleoprotein pseudouridine synthase bound to a substrate RNA. Nat Struct Mol Biol 16: 740–U85. 10.1038/nsmb.1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X-H, Liu Q, Fournier MJ. 2009b. Loss of rRNA modifications in the decoding center of the ribosome impairs translation and strongly delays pre-rRNA processing. RNA 15: 1716–1728. 10.1261/rna.1724409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado-Pinilla R, Liger D, Leulliot N, Meier UT. 2012. Mechanism of the AAA+ ATPases pontin and reptin in the biogenesis of H/ACA RNPs. RNA 18: 1833–1845. 10.1261/rna.034942.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNeil DE, Bensoussan HJ, Autexier C. 2016. Telomerase regulation from beginning to the end. Genes (Basel) 7: 64. 10.3390/genes7090064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNeil DE, Lambert-Lanteigne P, Autexier C. 2019. N-terminal residues of human dyskerin are required for interactions with telomerase RNA that prevent RNA degradation. Nucleic Acids Res 47: 5368–5380. 10.1093/nar/gkz233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNeil DE, Lambert-Lanteigne P, Qin J, McManus F, Bonneil E, Thibault P, Autexier C. 2021. SUMOylation- and GAR1-dependent regulation of dyskerin nuclear and subnuclear localization. Mol Cell Biol 41: e00464–20. 10.1128/MCB.00464-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y-Q, Houry WA. 2017. The role of pontin and reptin in cellular physiology and cancer etiology. Front Mol Biosci 4: 58. 10.3389/fmolb.2017.00058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez P, Blasco MA. 2017. Telomere-driven diseases and telomere-targeting therapies. J Cell Biol 216: 875–887. 10.1083/jcb.201610111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera AG, Wang Z. 2014. A day in the life of the spliceosome. Nat Rev Mol Cell Biol 15: 108–121. 10.1038/nrm3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier UT. 2005. The many facets of H/ACA ribonucleoproteins. Chromosoma 114: 1–14. 10.1007/s00412-005-0333-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier UT, Blobel G. 1990. A nuclear localization signal binding protein in the nucleolus. J Cell Biol 111: 2235–2245. 10.1083/jcb.111.6.2235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier U, Blobel G. 1992. Nopp140 shuttles on tracks between nucleolus and cytoplasm. Cell 70: 127–138. 10.1016/0092-8674(92)90539-O [DOI] [PubMed] [Google Scholar]
- Meier U, Blobel G. 1994. Nap57, a mammalian nucleolar protein with a putative homolog in yeast and bacteria. J Cell Biol 127: 1505–1514. 10.1083/jcb.127.6.1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JR, Wood E, Collins K. 1999. A telomerase component is defective in the human disease dyskeratosis congenita. Nature 402: 551–555. 10.1038/990141 [DOI] [PubMed] [Google Scholar]
- Nagpal N, Agarwal S. 2020. Telomerase RNA processing: implications for human health and disease. Stem Cells 38: 1532–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natchiar SK, Myasnikov AG, Kratzat H, Hazemann I, Klaholz BP. 2017. Visualization of chemical modifications in the human 80S ribosome structure. Nature 551: 472–477. 10.1038/nature24482 [DOI] [PubMed] [Google Scholar]
- Neuwald AF, Aravind L, Spouge JL, Koonin EV. 1999. AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res 9: 27–43. 10.1101/gr.9.1.27 [DOI] [PubMed] [Google Scholar]
- Nguyen THD, Tam J, Wu RA, Greber BJ, Toso D, Nogales E, Collins K. 2018. Cryo-EM structure of substrate-bound human telomerase holoenzyme. Nature 557: 190–195. 10.1038/s41586-018-0062-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien R, Tran SL, Maritz MF, Liu B, Kong CF, Purgato S, Yang C, Murray J, Russell AJ, Flemming CL, et al. 2016. MYC-driven neuroblastomas are addicted to a telomerase-independent function of dyskerin. Cancer Res 76: 3604–3617. 10.1158/0008-5472.CAN-15-0879 [DOI] [PubMed] [Google Scholar]
- Ojha S, Malla S, Lyons SM. 2020. snoRNPs: functions in ribosome biogenesis. Biomolecules 10: 783. 10.3390/biom10050783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellizzoni L, Baccon J, Charroux B, Dreyfuss G. 2001. The survival of motor neurons (SMN) protein interacts with the snoRNP proteins fibrillarin and GAR1. Curr Biol CB 11: 1079–1088. 10.1016/S0960-9822(01)00316-5 [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE. 2021. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci 30: 70–82. 10.1002/pro.3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Autexier C. 2021. Regulation of human telomerase RNA biogenesis and localization. RNA Biol 18: 305–315. 10.1080/15476286.2020.1809196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid R, Liang B, Baker DL, Youssef OA, He Y, Phipps K, Terns RM, Terns MP, Li H. 2006. Crystal structure of a Cbf5-Nop10-Gar1 complex and implications in RNA-guided pseudouridylation and dyskeratosis congenita. Mol Cell 21: 249–260. 10.1016/j.molcel.2005.11.017 [DOI] [PubMed] [Google Scholar]
- Richard P, Darzacq X, Bertrand E, Jády BE, Verheggen C, Kiss T. 2003. A common sequence motif determines the Cajal body-specific localization of box H/ACA scaRNAs. EMBO J 22: 4283–4293. 10.1093/emboj/cdg394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards LA, Kumari A, Knezevic K, Thoms JA, von Jonquieres G, Napier CE, Ali Z, O'Brien R, Marks-Bluth J, Maritz MF, et al. 2020. DKC1 is a transcriptional target of GATA1 and drives upregulation of telomerase activity in normal human erythroblasts. Haematologica 105: 1517–1526. 10.3324/haematol.2018.215699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roake CM, Artandi SE. 2020. Regulation of human telomerase in homeostasis and disease. Nat Rev Mol Cell Biol 21: 384–397. 10.1038/s41580-020-0234-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozhdestvensky TS, Tang TH, Tchirkova IV, Brosius J, Bachellerie J-P, Hüttenhofer A. 2003. Binding of L7Ae protein to the K-turn of archaeal snoRNAs: a shared RNA binding motif for C/D and H/ACA box snoRNAs in Archaea. Nucleic Acids Res 31: 869–877. 10.1093/nar/gkg175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggero D, Grisendi S, Piazza F, Rego E, Mari F, Rao PH, Cordon-Cardo C, Pandolfi PP. 2003. Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science 299: 259–262. 10.1126/science.1079447 [DOI] [PubMed] [Google Scholar]
- Savelyev NV, Shepelev NM, Lavrik OI, Rubtsova MP, Dontsova OA. 2021. PARP1 regulates the biogenesis and activity of telomerase complex through modification of H/ACA-proteins. Front Cell Dev Biol 9: 621134. 10.3389/fcell.2021.621134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopf FH, Biebl MM, Buchner J. 2017. The HSP90 chaperone machinery. Nat Rev Mol Cell Biol 18: 345–360. 10.1038/nrm.2017.20 [DOI] [PubMed] [Google Scholar]
- Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, León-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES, et al. 2014. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159: 148–162. 10.1016/j.cell.2014.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Schmidt JC, Goldfarb KC, Cech TR, Parker R. 2016. Inhibition of telomerase RNA decay rescues telomerase deficiency caused by dyskerin or PARN defects. Nat Struct Mol Biol 23: 286–292. 10.1038/nsmb.3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Jeong H-C, Sturgeon CM, Parker R, Batista LFZ. 2020. Chemical inhibition of PAPD5/7 rescues telomerase function and hematopoiesis in dyskeratosis congenita. Blood Adv 4: 2717–2722. 10.1182/bloodadvances.2020001848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Wang Z, Cascio D, Feigon J. 2015. Structure and interactions of the CS domain of human H/ACA RNP assembly protein Shq1. J Mol Biol 427: 807–823. 10.1016/j.jmb.2014.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan KE, Warda AS, Sharma S, Entian K-D, Lafontaine DLJ, Bohnsack MT. 2017. Tuning the ribosome: the influence of rRNA modification on eukaryotic ribosome biogenesis and function. RNA Biol 14: 1138–1152. 10.1080/15476286.2016.1259781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Logeswaran D, Castillo-González C, Li Y, Bose S, Aklilu BB, Ma Z, Polkhovskiy A, Chen JJ-L, Shippen DE. 2019. The conserved structure of plant telomerase RNA provides the missing link for an evolutionary pathway from ciliates to humans. Proc Natl Acad Sci 116: 24542–24550. 10.1073/pnas.1915312116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Castillo-González C, Ma Z, Shippen DE. 2021. Arabidopsis retains vertebrate-type telomerase accessory proteins via a plant-specific assembly. Nucleic Acids Res 49: 9496–9507. 10.1093/nar/gkab699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taoka M, Nobe Y, Yamaki Y, Sato K, Ishikawa H, Izumikawa K, Yamauchi Y, Hirota K, Nakayama H, Takahashi N, et al. 2018. Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res 46: 9289–9298. 10.1093/nar/gky811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley DM, Dumitriu B, Young NS. 2014. Bone marrow failure and the telomeropathies. Blood 124: 2775–2783. 10.1182/blood-2014-05-526285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycowski KT, Shu M-D, Kukoyi A, Steitz JA. 2009. A conserved WD40 protein binds the Cajal body localization signal of scaRNP particles. Mol Cell 34: 47–57. 10.1016/j.molcel.2009.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher AS, Meng Z, Mason PJ, Veenstra TD, Artandi SE. 2008. Identification of ATPases pontin and reptin as telomerase components essential for holoenzyme assembly. Cell 132: 945–957. 10.1016/j.cell.2008.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher AS, Abreu EB, Meng Z, McCann KE, Terns RM, Veenstra TD, Terns MP, Artandi SE. 2009. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 323: 644–648. 10.1126/science.1165357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walbott H, Machado-Pinilla R, Liger D, Blaud M, Réty S, Grozdanov PN, Godin K, van Tilbeurgh H, Varani G, Meier UT, et al. 2011. The H/ACA RNP assembly factor SHQ1 functions as an RNA mimic. Genes Dev 25: 2398–2408. 10.1101/gad.176834.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walne AJ, Dokal I. 2009. Advances in the understanding of dyskeratosis congenita. Br J Haematol 145: 164–172. 10.1111/j.1365-2141.2009.07598.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Meier UT. 2004. Architecture and assembly of mammalian H/ACA small nucleolar and telomerase ribonucleoproteins. EMBO J 23: 1857–1867. 10.1038/sj.emboj.7600181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins NJ, Bohnsack MT. 2012. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip Rev RNA 3: 397–414. 10.1002/wrna.117 [DOI] [PubMed] [Google Scholar]
- Weidemann A, Johnson RS. 2008. Biology of HIF-1 α. Cell Death Differ 15: 621–627. 10.1038/cdd.2008.12 [DOI] [PubMed] [Google Scholar]
- Wu H, Feigon J. 2007. H/ACA small nucleolar RNA pseudouridylation pockets bind substrate RNA to form three-way junctions that position the target U for modification. Proc Natl Acad Sci 104: 6655–6660. 10.1073/pnas.0701534104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Yu AT, Kantartzis A, Yu Y-T. 2011. Functions and mechanisms of spliceosomal small nuclear RNA pseudouridylation. Wiley Interdiscip Rev RNA 2: 571–581. 10.1002/wrna.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y-T, Meier UT. 2014. RNA-guided isomerization of uridine to pseudouridine—pseudouridylation. RNA Biol 11: 1483–1494. 10.4161/15476286.2014.972855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebarjadian Y, King T, Fournier MJ, Clarke L, Carbon J. 1999. Point mutations in yeast CBF5 can abolish in vivo pseudouridylation of rRNA. Mol Cell Biol 19: 7461–7472. 10.1128/MCB.19.11.7461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xu C, Gu D, Wu M, Yan B, Xu Z, Wang Y, Liu H. 2017. H/ACA box small nucleolar RNA 7A promotes the self-renewal of human umbilical cord mesenchymal stem cells. Stem Cells 35: 222–235. 10.1002/stem.2490 [DOI] [PubMed] [Google Scholar]
- Zijlmans J, Martens UM, Poon SSS, Raap AK, Tanke HJ, Ward RK, Lansdorp PM. 1997. Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proc Natl Acad Sci 94: 7423–7428. 10.1073/pnas.94.14.7423 [DOI] [PMC free article] [PubMed] [Google Scholar]