

Abstract
The ESKAPE pathogens comprise a group of multidrug-resistant bacteria that are the leading cause of nosocomial infections worldwide. The prevalence of antibiotic resistant strains and the relative ease by which bacteria acquire resistance genes highlight the continual need for the development of novel antibiotics against new drug targets. The methylerythritol phosphate (MEP) pathway is an attractive target for the development of new antibiotics. The MEP pathway governs the synthesis of isoprenoids, which are key lipid precursors for vital cell components such as ubiquinone and bacterial hopanoids. Additionally, the MEP pathway is entirely distinct from the corresponding mammalian pathway, the mevalonic acid (MVA) pathway, making the first committed enzyme of the MEP pathway, 1-deoxy-d-xylulose 5-phosphate reductoisomerase (IspC), an attractive target for antibiotic development. To facilitate drug development against two of the ESKAPE pathogens, Acinetobacter baumannii and Klebsiella pneumoniae, we cloned, expressed, purified, and characterized IspC from these two Gram-negative bacteria. Enzyme inhibition assays using IspC from these two pathogens, and compounds fosmidomycin and FR900098, indicate IC50 values ranging from 19.5–45.5 nM. Antimicrobial susceptibility tests with these inhibitors reveal that A. baumannii is susceptible to FR900098, whereas K. pneumoniae is susceptible to both compounds. Finally, to facilitate structure-based drug design of inhibitors targeting A. baumannii IspC, we determined the 2.5 Å crystal structure of IspC from A. baumannii in complex with inhibitor FR900098, and cofactors NADPH and magnesium.
Keywords: drug discovery, antibiotics, multidrug-resistance, bacteria, Acinetobacter baumannii, Klebsiella pneumoniae
Acinetobacter baumannii and Klebsiella pneumoniae are Gram-negative, opportunistic pathogens frequently encountered in the healthcare and military sectors. They comprise two of the ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species), a group of multidrug-resistant bacteria that are currently the leading cause of nosocomial infections worldwide.1 The first record of an A. baumannii nosocomial infection was during the Korean War, and its pervasiveness has propagated throughout military history.2−5 During the wars in Iraq and Afghanistan, A. baumannii was a dominant cause of complicated wound healing in injured military personnel,6,7 earning itself the name “Iraqibacter”.8 In addition to bodily wounds, A. baumannii infections commonly occur in the blood (septicemia), urinary tract, and lungs (pneumonia).9A. baumannii remains a threat to civilians and veterans due to the spread of multidrug-resistant (MDR) strains to civilian hospitals via infected soldiers.10,11A. baumannii is particularly successful in the hospital environment due to its persistence on inanimate surfaces (e.g., it can survive for up to 5 months on plasticware via biofilm formation12,13) and due to its highly plastic genome which readily accepts antibiotic resistance genes.11,14 Consequently, patients whose care requires the use of ventilators or catheters, or a prolonged hospital stay, are at especially high risk of infection.9 Generally, carbapenems are the main course of treatment for Acinetobacter infections, and thus, resistance to carbapenems is concerning.15,16 According to the 2019 Antibiotic Resistance Threats Report (AR Threats Report), issued by the Centers for Disease Control (CDC), carbapenem-resistant Acinetobacter caused an estimated 8500 infections and 700 deaths in hospitalized patients in 2017.17
K. pneumoniae is a bacterium that belongs to a large family of Gram-negative bacteria known as Enterobacteriaceae.18K. pneumoniae is a naturally occurring member of human body flora that typically colonizes the intestines, where it does not cause disease.19 Like A. baumannii infections, most K. pneumoniae infections occur in healthcare settings, where patients are exposed via ventilators, catheters, or bodily wounds.19 Consequently, K. pneumoniae is a common cause of bacterial pneumonia and has been identified as the second most common source of urinary tract infections (UTIs) following Escherichia coli.20−23K. pneumoniae infections are largely attributed to the surface pili of the bacteria, which aids in its adherence to the respiratory and urinary epithelium.24 Additionally, Klebsiella species are the most antibiotic resistant of all identified Enterobacteriaceae.24 In 1983, K. pneumoniae was first shown to exhibit resistance to various β-lactam antibiotics, via production of extended-spectrum β-lactamases (ESBLs).25,26 As indicated in the 2019 CDC AR Threats Report, ESBL-producing Enterobacteriaceae caused an estimated 197400 infections and 9100 deaths in hospitalized patients in 2017.17 Typically, carbapenems are regarded as the antibiotic class of choice for treatment of ESBL-producing infections;27 however, in addition to β-lactam antibiotic resistance, many ESBL-producing K. pneumoniae have also acquired resistance to quinolones, aminoglycosides, and carbapenems, further complicating treatment.28 Per the CDC 2019 AR Threats Report, carbapenem-resistant Enterobacteriaceae caused an estimated 13100 infections and 1100 deaths in hospitalized patients in 2017.17
In February 2017, the World Health Organization published its first ever list of antibiotic-resistant “priority pathogens”, which included both carbapenem-resistant A. baumannii and K. pneumoniae and ESBL-producing K. pneumoniae as “Priority 1: Critical” pathogens.29 The rapid increase of MDR Gram-negative pathogens, such as A. baumannii and K. pneumoniae, has led to increased use of the “last-resort drug” colistin, despite its serious nephrotoxicity and neurotoxicity issues.26,30,31 With the elevated usage of colistin to treat MDR Gram-negative infections, the emergence of colistin-resistant pathogens now poses a dire health threat and an urgent need for developing a novel class of antibiotics.26 Underscoring this need, in September 2016, a patient died in Reno, Nevada due to K. pneumoniae induced sepsis.32 The specific strain of K. pneumoniae isolated from her infection was found to be resistant to all 26 antibiotics available in the United States, including colistin.32
Increasing antimicrobial resistance, in conjunction with the shrinking arsenal of effective antibiotics to treat MDR A. baumannii and K. pneumoniae infections, have prompted an urgent need for developing new antibiotics with novel targets. The methylerythritol phosphate (MEP) pathway of isoprenoid biosynthesis (Figure 1) is an attractive target for the development of novel antimicrobial drugs. Isoprenoids comprise a large and diverse group of over 30000 known products with vital biological functions, such as electron transport and peptidoglycan biosynthesis in bacteria.33−36 Bacteria synthesize isoprenoids via the methylerythritol phosphate (MEP) pathway, whereas mammals synthesize isoprenoids via the mevalonic acid (MVA) pathway.37,38 The MEP pathway is entirely distinct from the MVA pathway making it an attractive target for antibiotic development.37,38
Figure 1.

MEP Pathway. The MEP pathway is used by many eubacteria, as well as plants and apicomplexan protozoa. Pyruvate is condensed with glyceraldehyde 3-phosphate to yield 1-deoxy-d-xylulose 5-phosphate, DXP.39 1-deoxy-d-xylulose 5-phosphate reductoisomerase (IspC) catalyzes the reduction and isomerization of DXP to yield 2-C-methylerythritol 4-phosphate (MEP).40 Isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) are produced after an additional five enzymatic steps.37,41−46 IPP and DMAPP are precursors for isoprenoids, such as bacterial hopanoids, bactoprenols, ubiquinone, and menaquinone.33,36
Two potent phosphonate inhibitors of the first committed MEP pathway enzyme, IspC, include fosmidomycin (a) and its acetyl derivative FR900098 (b) (Figure 2), which are naturally produced by the filamentous bacteria Streptomyces lavendulae and Streptomyces rubellomurinus, respectively.47−49 Fosmidomycin, in conjunction with clindamycin, piperaquine, or azithromycin has shown promise in the treatment of malaria.50−56 In addition to demonstrating growth inhibition of a causative agent of malaria, Plasmodium falciparum, fosmidomycin and/or FR900098 have also shown in vitro growth inhibition of the bacteria Yersinia pestis, Escherichia coli, K. pneumoniae, and Francisella tularensis.35,57−62
Figure 2.
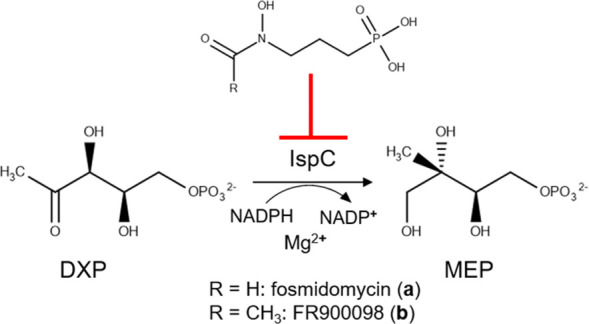
Inhibition of IspC. IspC is inhibited by two potent phosphonate inhibitors, fosmidomycin (a) and its acetyl derivative FR900098 (b).
Herein, we describe the cloning, expression, purification, and kinetic characterization of the first committed MEP pathway enzyme, IspC, from A. baumannii (AbIspC) and K. pneumoniae (KpIspC). Additionally, we characterize enzyme and bacterial growth inhibition using two known IspC inhibitors, fosmidomycin and FR900098. Furthermore, to facilitate structure-based drug design of inhibitors targeting AbIspC, we determined the 2.5 Å crystal structure of AbIspC in complex with inhibitor, FR900098, and cofactors NADPH and magnesium.
Results and Discussion
MEP Pathway as an Antimicrobial Target forA. baumannii and K. pneumoniae
To evaluate the effectiveness of fosmidomycin and FR900098 in inhibiting the growth of K. pneumoniae and A. baumannii, we performed antimicrobial susceptibility tests. The determined minimum inhibitory concentrations (MICs) for fosmidomycin and FR900098 for each strain of A. baumannii and K. pneumoniae are summarized in Table 1. Each strain is indicated as being either resistant (R), or susceptible (S), to each compound.
Table 1. MICs of Fosmidomycin and FR900098 against A. baumannii (Ab) and K. pneumoniae (Kp) Strains with Rifampicin as the Controla.
| Strains | Fosmidomycin | FR900098 | Rifampicin |
|---|---|---|---|
| MIC (μg/mL) | MIC (μg/mL) | MIC (μg/mL) | |
| Ab5075 | >512 (R) | 256 (S) | 4 (S) |
| Ab5711 | >512 (R) | >512 (R) | 4 (S) |
| Ab19606 | >512 (R) | 128 (S) | 2 (S) |
| KpBAA-1705 | 128 (S) | 256 (S) | 64 (S) |
| KpNSC-277 | 64 (S) | 256 (S) | 64 (S) |
Susceptibility to compounds is represented as “S” and resistance as “R”. Each experiment was performed in triplicate.
Ab5075, which was isolated from a patient at Walter Reed Army Medical Center (WRAMC), is a highly virulent MDR strain,63 and Ab5711 is a substantial biofilm-forming MDR clinical strain.64 Virulence has been linked to antibiotic resistance in multiple animal infection models,65 and biofilm-forming pathogens generally exhibit decreased susceptibility to antibacterial agents.66 Accordingly, higher MIC values were expected for Ab5075 and Ab5711. However, Ab19606 is a less virulent and antibiotic susceptible strain obtained from ATCC; therefore, lower MIC values were expected. KpBAA-1705, obtained from ATCC, is a MDR carbapenem-resistant K. pneumoniae strain, and KpNSC-277 is a MDR carbapenem-sensitive clinical strain. Because the MEP pathway is present in many Gram-negative bacteria, Fosmidomycin and FR900098 were generally expected to have inhibition effects on the whole cells.
As depicted in Table 1, all three A. baumannii strains (Ab5075, Ab5711, and Ab19606) were resistant to fosmidomycin at the tested drug concentration range (1–512 μg/mL). However, A. baumannii strains Ab5075 and Ab19606 were susceptible to FR900098 with MICs of 256 and 128 μg/mL, respectively; strain Ab5711 was resistant to FR900098. Conversely, the K. pneumoniae strains (KpBAA-1705 and KpNSC-277) were more susceptible to fosmidomycin than the A. baumannii strains, with MICs of 128 and 64 μg/mL, respectively. Both K. pneumoniae strains were susceptible to FR900098 with an MIC of 256 μg/mL.
To enter bacterial cells and inhibit the MEP pathway, fosmidomycin is dependent on the glycerol-3-phosphate transporter (GlpT).48,67,68 Some pathogens which are lacking the GlpT transporter, such as Mycobacterium tuberculosis, are impermeable to fosmidomycin.69 However, lipophilic phosphonate prodrugs have proven effective MEP pathway inhibitors against bacteria lacking the GlpT transporter.48,69−72 A BLAST search with the E. coli K-12 GlpT protein sequence (accession no. P08194) identifies a homologous transport protein in the K. pneumoniae KpBAA-1705 proteome (accession no. EMR29960; 93.06% identity). A BLAST search with the E. coli K-12 GlpT protein sequence and the A. baumannii Ab5075 proteome did not identify a GlpT transporter. The BLAST searches were performed with the National Center for Biotechnology Information (NCBI) BLAST (blastp) suite. Sequenced proteomes for KpNSC-277, Ab5711, and Ab19606 were not available for searching. A. baumannii resistance to fosmidomycin may therefore be due to poor cellular uptake. Additionally, previous studies have shown that uptake of FR900098 is only partially dependent on GlpT, which may explain the growth inhibition of A. baumannii strains Ab5075 and Ab19606.48 Furthermore, resistance of Ab5711 to fosmidomycin and FR900098 could be due to its substantial biofilm formation characteristics in vitro.64 Fosmidomycin and FR900098 are hydrophilic compounds which may not be able to effectively penetrate the A. baumannii and K. pneumoniae cell. Alternatively, or in combination, resistance to Fosmidomycin and FR900098 may be due to efflux. However, additional studies are warranted to validate the mechanism of cellular uptake and/or efflux in A. baumannii and K. pneumoniae species conclusively. Because fosmidomycin and/or FR900098 clearly inhibited A. baumannii and K. pneumoniae growth, we cloned and characterized both the A. baumannii and K. pneumoniae IspC enzyme to further assess IspC as an antibiotic target for A. baumannii and K. pneumoniae and to provide a framework for developing more potent derivatives.
Characterization of A. baumannii and K. pneumoniae IspC Enzymes
To enable the enzymatic characterization of A. baumannii IspC, the A. baumannii ispC gene was PCR amplified from genomic DNA, cloned into a pBG1861 vector, and transformed into chemically competent E. coli BL21(DE3) codon plus RIL cells for protein expression. The resulting recombinant AbIspC enzyme was affinity purified to near homogeneity via an N-terminal histidine tag. In parallel, the K. pneumoniae ispC gene was fully synthesized (GenScript USA Inc., Piscataway, NJ), cloned into a pMCSG28 vector, and transformed into chemically competent E. coli BL21(DE3) codon plus RIL cells. The resulting recombinant KpIspC enzyme was affinity purified to near homogeneity via a C-terminal histidine tag.
The catalytic activity of each purified recombinant enzyme was determined by a spectrophotometric assay monitoring the substrate dependent oxidation of NADPH. Nonlinear regression fitting of enzyme velocity versus substrate concentration was used to determine the apparent kinetic constants (Figure 3). The KMapp for DXP was obtained using assays performed with a saturating concentration of NADPH (150 μM), whereas the KMapp for NADPH was obtained using assays performed with 400 μM DXP. Generally, the recombinant A. baumannii IspC and K. pneumoniae IspC have KMapp, DXP, KMapp, NADPH, kcatDXP, and kcatNADPH values that are comparable to those reported for homologous enzymes from other organisms (Table 2). The apparent specificity constant (KcatDXP/KMDXP) of both A. baumannii and K. pneumoniae IspC are approximately 2-fold higher than that reported for Yersinia pestis IspC, approximately 30-fold lower than that reported for the E. coli enzyme, and approximately 2-fold lower than those reported for the Francisella tularensis and Mycobacterium tuberculosis enzymes (Table 2).
Figure 3.

Substrate dependent catalytic activity of A. baumannii and K. pneumoniae IspC. Shown are Michaelis–Menten plots of AbIspC reaction velocity as a function of (A) DXP concentration and (B) NADPH concentration. Also shown are the Michaelis–Menten plots of KpIspC reaction velocity as a function of (C) DXP concentration and (D) NADPH concentration. KMapp values were obtained using GraphPad Prism 5.0. Least-squares best fit of the data to the Michaelis–Menten equation produces the kinetic parameters depicted here and listed in Table 2. The R2 value for each plot is indicated. All assays were performed in duplicate. The error bars indicate the standard deviation for each data point.
Table 2. IspC Apparent Kinetic Parameters.
| AbIspC | KpIspC | EcIspCa | FtIspCa | MtIspCa | YpIspCa | |
|---|---|---|---|---|---|---|
| KMapp (DXP) (μM)b | 127.6 ± 7.2 | 155.5 ± 10.8 | 81–250 | 103.7 ± 12.1 | 47 | 221.5 ± 34.3 |
| KMapp(NADPH)(μM) | 19.53 ± 0.53 | 14.51 ± 0.83 | 0.5–18 | 13.3 ± 1.5 | 29.7 | 12.7 ± 1.5 |
| kcatDXP (s–1)b | 1.92 ± 0.03 | 2.21 ± 0.06 | 33 | 2.0 ± 0.09 | 1.2 | 1.7 |
| kcatNADPH (s–1) | 1.64 ± 0.01 | 1.96 ± 0.03 | 1.3 ± 0.04 | 1.0 | ||
| kcatDXP/KMDXP (M–1 min–1) | 9.0 × 105 ± 5.3 × 104 | 8.5 × 105 ± 6.3 × 104 | 2.4 × 107 | 1.2 × 106 | 1.5 × 106 | 4.6 × 105 |
| IC50fosmidomycin (nM)c | 46.8 (38.2–57.2) | 20.2 (15.9–25.6) | 35 | 247 | 80 | 710 |
| IC50FR900098 (nM) | 23.9 (21.4–26.7) | 23.1 (18.3–29.2) | 35 | 230 | 160 | 231 |
| ref | This study | This study | (40,74−76) | (35,72) | (71,77) | (57) |
EcIspC, FtIspC, MtIspC, YpIspC = recombinant IspC from Escherichia coli, Francisella tularensis, Mycobacterium tuberculosis, and Yersinia pestis, respectively.
KMapp and kcat values were obtained using GraphPad Prism 5.0.
The 95% confidence intervals of the IC50 values are indicated parenthetically. All assays were performed in duplicate. The standard errors for the KMapp and kcat values for DXP and NADPH, and kcatDXP/KMDXP are indicated for AbIspC and KpIspC.
IspC requires a divalent cation–generally Mg2+ or Mn2+ – for catalysis.35,57,73 Like many other IspC enzymes,35,57,73 evaluation of enzyme activity in the presence of various divalent cations reveals that recombinant AbIspC and KpIspC preferentially use Mg2+ (Figure 4), although approximately 10% and 6% enzyme activity (relative to Mg2+) is retained for AbIspC and KpIspC, respectively, when Mn2+ is used.
Figure 4.
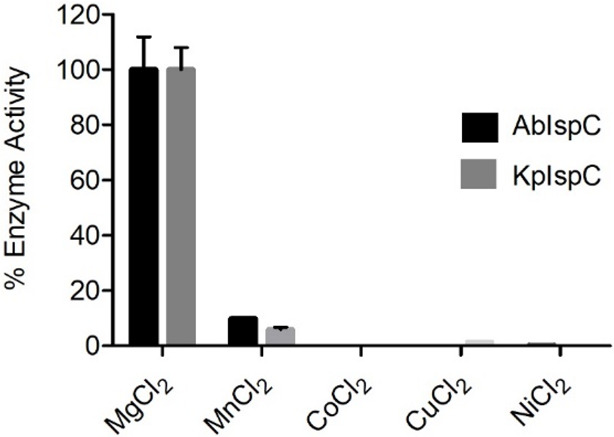
Cation specificity of A. baumannii and K. pneumoniae IspC. Enzyme assays were performed with fixed concentrations of NADPH (150 μM), DXP (400 μM), and divalent cation (25 mM). AbIspC and KpIspC prefer Mg2+. Assays were performed in duplicate. The error bars indicate the standard deviation.
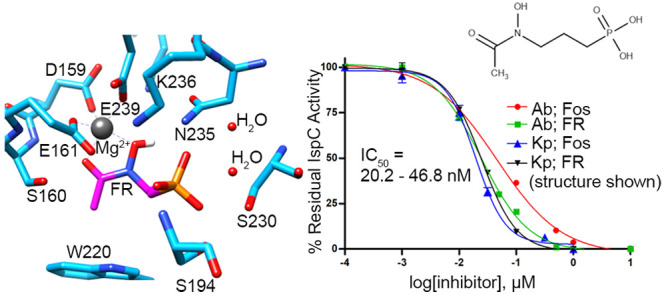
Next, we determined the half-maximal inhibitory (IC50) concentrations of the phosphonate inhibitors fosmidomycin and FR900098, with AbIspC and KpIspC (Figure 5). While AbIspC was more potently inhibited by FR900098 than fosmidomycin, KpIspC was inhibited by fosmidomycin and FR900098 at similar potencies. The improved potency of FR900098 over fosmidomycin against AbIspC agrees with the MIC values wherein all three A. baumannii strains were resistant to fosmidomycin at 1–512 μg/mL. It is noteworthy that, in general, fosmidomycin and FR900098 are more potent against AbIspC and KpIspC than the homologous recombinant enzymes from F. tularensis, M. tuberculosis, and Y. pestis (Table 2), thus highlighting their promise as targets for therapeutics against A. baumannii and K. pneumoniae.
Figure 5.

Dose-dependent inhibition of AbIspC and KpIspC. IC50 values for AbIspC were determined using (A) fosmidomycin or (B) FR900098. IC50 values for KpIspC were also determined using (C) fosmidomycin or (D) FR900098. The R2 value for each plot is indicated. IC50 values were obtained using GraphPad Prism 5.0. Assays were performed in duplicate. The error bars indicate the standard deviation for each data point.
Overall Structure of AbIspC in Complex with FR900098, NADPH, and Mg2+
The structure of AbIspC in complex with FR900098, NADPH, and Mg2+ was refined at 2.5 Å resolution with Rfree and Rwork values of 24% and 23%, respectively (Figure 6). AbIspC crystallized in space group P21212 and the unit cell parameters were a = 66.572, b = 118.251, c = 53.911, and α = β = γ = 90. The refinement and statistics are summarized in Table 3. AbIspC crystallized as a monomer in the asymmetric unit, with the homodimer formed by crystallographic symmetry. The biological assembly of IspC is known to be a homodimer.78 The overall structure of the AbIspC monomer (Figure 6A) is similar to those that have been previously reported, in which each monomer consists of three domains: a central catalytic domain, an N-terminal NADPH-binding domain, and a C-terminal α-helical domain.79,80 The AbIspC N-terminal NADPH binding domain is comprised of a seven-stranded parallel β-sheet and six α-helices joined by a Rossman or adenosine diphosphate (ADP)-binding βαβ fold which binds NADPH in a classical manner.81 The C-terminal α-helical domain is comprised of a four-helix bundle. The central catalytic domain, which binds the divalent magnesium ion and FR900098, is located within a deep cleft at the center of the monomer, and is covered by an active site loop (Figure 7) which is known to close over the active site upon substrate binding.80,82 The four-stranded β-sheet which sits below the catalytic domain contains one parallel and two antiparallel alignments and is known to comprise part of the dimer interaction with the second IspC monomer.79
Figure 6.

Structure of the AbIspC complex. (A) The overall crystal structure of AbIspC (blue) in complex with NADPH (yellow), FR900098 (magenta), and Mg2+ (gray). The N- and C-termini are indicated. (B) Focused view of the AbIspC FR900098 and Mg2+ binding site. The Fo-Fc difference map around FR900098 and Mg2+ is shown in green mesh, contoured at a radius of 3.0 Å around the ligand. Residues 191–193 and 258 and NADPH were deleted for visualization of the binding site. Bonded waters are shown. AbIspC structure (PDB ID: 4ZN6) was used as a search model for phasing.
Table 3. Crystallography Data Collection and Refinement Statistics.
| Recombinant AbIspCa | |
|---|---|
| Wavelength (Å) | 1.54 |
| Resolution range (Å) | 33.917–2.490 (2.533–2.490) |
| Space group | P21212 |
| Unit cell | 66.572 118.251 53.911 90 90 90 |
| Unique reflections | 28399 (15339) |
| Multiplicity | 7.23 |
| Completeness (%) | 98.57 (98.57) |
| Mean I/sigma(I) | 23.09 |
| Wilson B-factor | 23.33 |
| R-merge | 0.2494 |
| R-work | 0.2307 |
| R-free | 0.2442 |
| RMS(bonds) | 0.008 |
| Number of H2O | 147 |
| Ramachandran favored (%) | 97.46 |
| Ramachandran outliers (%) | 0 |
| Clashscore | 0 |
| Average B-factor | 23.65 |
| macromolecules | 23.78 |
| ligands | 19.34 |
Statistics for the highest-resolution shell are shown in parentheses.
Figure 7.

The active site loop. (A) The AbIspC quaternary complex (blue) superimposed with apo AbIspC (PDB ID: 4ZN6, orange). The N- and C-termini are indicated. The active site loop (residues 216–223, violet) is closed over the active site in the quaternary complex. The conformation of both the N-terminal NADPH binding domain and the central catalytic domain are not greatly shifted by ligand binding. However, there is a distinct shift in the position of the four-helix bundle comprising the C-terminal α-helical domain. (B) Zoomed in view of the active site loop. The Fo-Fc electron density difference map, contoured at a radius of 2.8 Å around the loop region (residues 216–223), is shown in green mesh. Electron density can be observed for residues 216, and 220–223.
Active Site and Active Site Loop
FR900098 is bound to the active site via hydrogen bonding interactions with the oxygens of its phosphonate and hydroxamate groups, coordination with the divalent magnesium ion, and hydrophobic interactions with the propyl backbone of FR900098 (Figure 6B). The phosphonate oxygens of FR900098 form hydrogen bonds with Asn235, two water molecules, and highly conserved residues, Ser230, Ser194, and Lys236.74,79 Both the side chain and backbone nitrogen of Ser194 are hydrogen bonded to the phosphonate oxygen. The divalent magnesium cation is coordinated to highly conserved residues Asp159, Glu161, and Glu239 and the carbonyl and hydroxyl oxygens of the FR900098 hydroxamate group in a distorted trigonal bipyramidal geometry.74,79 The metal interactions are similar to those observed in other quaternary IspC complexes with FR900098, NADPH, and a divalent cation. In the quaternary structure of IspC from M. tuberculosis in complex with FR900098, NADPH, and Mn2+, only five oxygen atoms coordinate each metal ion, with an approximate octahedral geometry.83 Similarly, in the quaternary structure of IspC from Plasmodium falciparum in complex with FR900098, Mg2+, and NADPH, the Mg2+ ion binds to three protein ligands and two inhibitor atoms, resulting in a distorted trigonal bipyramidal geometry.84 The Ser160 side chain and its backbone nitrogen also form hydrogen bonds with the hydroxyl oxygen of the hydroxamate group. The active site loop is closed over the FR900098 and Mg2+ binding site, and the propyl backbone of FR900098 interacts with the adjacent Trp220 and Met222 (of the active site loop) at approximate distances of 4.138 and 4.150 Å, respectively.
Closed Conformation of AbIspC in Complex with FR900098, NADPH, and Mg2+
The primary difference between the quaternary AbIspC structure (AbIspC in complex with FR900098, NADPH, and Mg2+) and the apo AbIspC structure (deposited by the Seattle Structural Genomics Center for Infectious Disease, PDB ID: 4ZN6) is the overall conformation of the enzyme, wherein the apo structure adopts a more open conformation and the quaternary structure adopts a closed conformation. This can be observed by superimposing the two structures (Figures 7 and 8). In the quaternary structure, the flexible loop region (residues 216–223) is closed over the active site, whereas, in the apo AbIspC structure (PDB ID: 4ZN6), the flexible loop region is disordered. The loop region in the quaternary structure is well-ordered, and residues 216–223 were built into the Fo-Fc difference electron density map (Figure 7B). As observed from the superimposed structures, the conformation of both the N-terminal NADPH binding domain and the central catalytic domain are not greatly shifted by the binding of FR900098, NADPH, and Mg2+ in the quaternary structure (Figure 7A). However, there is a distinct shift in the position of the four-helix bundle comprising the C-terminal α-helical domain (Figure 7 and 8).
Figure 8.

The C-terminal α-helical domain. The AbIspC quaternary complex (blue) superimposed with apo AbIspC (PDB ID: 4ZN6, orange). The N- and C-termini are indicated where visible. The C-terminal α-helices adopt a more open conformation in the apo structure (1αa, 2αa, 3αa, 4αa), and a closed conformation in the quaternary complex (1αb, 2αb, 3αb, 4αb). (A) C-terminal α-helices 2 (residues 339–355) and 4 (residues 383–404) in the quaternary complex (2αb and 4αb) and in the apo conformation (2αa and 4αa). 2αb is shifted approximately 4.585 Å toward the active site relative to 2αa. 4αb is shifted approximately 5.880 Å toward the active site relative to 4αa. (B) C-terminal α-helices 1 (residues 325–335) and 3 (residues 361–374) in the quaternary complex (1αb and 3αb) and in the apo conformation (1αa and 3αa). 1αb is shifted approximately 2.163 Å toward the active site relative to 1αa. 3αb is shifted approximately 5.053 Å toward the active site relative to 3αa.
Conclusion
In summary, we have shown that A. baumannii and K. pneumoniae IspC are valid targets for the development of novel antibiotics. Although all A. baumannii strains were resistant to fosmidomycin at 512 μg/mL and Ab5711 was resistant to both inhibitors at 512 μg/mL, A. baumannii strains Ab5075 and Ab19606 were susceptible to FR900098 at MICs of 256 and 128 μg/mL, respectively. A. baumannii resistance to fosmidomycin may be due to lack of GlpT uptake and/or impermeability. Ab5711’s resistance to both fosmidomycin and FR900098 could likewise be attributed to impermeability due to the substantial biofilm formation characteristics of Ab5711. Conversely, K. pneumoniae was more susceptible to fosmidomycin than FR900098. Strains KpBAA-1705 and KpNSC-277 were susceptible to fosmidomycin at MICs of 128 and 64 μg/mL, respectively, whereas, both strains were susceptible to FR900098 at an MIC of 256 μg/mL. As mentioned, fosmidomycin uptake is GlpT-dependent, whereas FR900098 uptake is partially GlpT-dependent. K. pneumoniae’s improved susceptibility to fosmidomycin over FR900098 may be attributed to uptake via GlpT. Nonetheless, Fosmidomycin and FR900098 are highly hydrophilic compounds; therefore, resistance may be generally attributed to impermeability regardless of GlpT uptake. Future studies are warranted to assess the method of uptake, or efflux, of these compounds in A. baumannii and K. pneumoniae.
Unlike the bacterial growth inhibition assays, both A. baumannii and K. pneumoniae IspC were potently inhibited by fosmidomycin and FR900098 in vitro. Additionally, both A. baumannii and K. pneumoniae IspC were generally more susceptible to the inhibitors than other homologous enzymes. The in vitro potency of fosmidomycin and FR900098 against A. baumannii and K. pneumoniae IspC highlights their promise as targets for therapeutics against A. baumannii and K. pneumoniae. We resolved the quaternary structure of AbIspC in complex with FR900098, NADPH, and Mg2+. This structure can guide additional structure–based drug design, which may yield derivatives with more favorable bacterial growth inhibition. As mentioned previously, where cellular penetration has posed a challenge for bacterial growth inhibition, lipophilic phosphonate prodrugs have demonstrated potential for effective bacterial growth inhibition. Accordingly, lipophilic prodrugs of fosmidomycin and FR900098 analogs likely represent the best prospects for A. baumannii and K. pneumoniae, as well as other ESKAPE pathogens.
Methods
Bacterial Strains and Growth Conditions
Clinical isolates of A. baumannii strains Ab5075 and Ab5711 and K. pneumoniae strain KpNSC-277 were obtained from the Multidrug-resistant organism Repository and Surveillance Network (MRSN) at Walter Reed Army Institute of Research. Ab5075 is the most virulent and multidrug-resistant strain. Ab5711 is also an MDR strain and is known to form substantial biofilms in vitro. A. baumannii strain Ab19606, K. pneumoniae strain KpBAA-1705, and Escherichia coli strain Ec25922 were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). The bacterial isolates were cultured on blood agar plates (Tryptic Soy Agar with 5% sheep blood) at 37 °C for 16–18 h prior to antimicrobial susceptibility tests. Recombinant proteins were expressed in Escherichia coli BL21 CodonPlus (DE3)-RIL cells obtained from Stratagene (La Jolla, CA). E. coli was cultured at 37 °C in Luria–Bertani (LB) media supplemented with 100 μg/mL ampicillin and 50 μg/mL chloramphenicol with constant shaking at 250 rpm. Agar (1.5 wt %/vol) was added to prepare solid media.
Antimicrobial Susceptibility Assays
The minimum inhibitory concentration (MIC) of each compound was evaluated using the broth microdilution method according to the guidelines described in the Clinical and Laboratory Standards Institute (CLSI).85 Fosmidomycin and FR900098 were purchased from Sigma-Aldrich (St. Louis, MO). Inhibitors stock solution were prepared in phosphate buffered saline (PBS) and stored at −80 °C. The compounds were 2-fold diluted in cation-adjusted Mueller-Hinton Broth (CAMHB) in 96-well round-bottom, polystyrene microtiter plates to final concentrations ranging from 1 to 512 μg/mL. 100 μL of the drug containing broth was dispensed into the wells of the 96-well plate in triplicate. Wells containing broth only served as growth and sterility controls. Prior to antimicrobial susceptibility testing, A. baumannii strains were cultured on blood agar plates at 37 °C for 16–18 h, and K. pneumoniae strains were cultured in CAMHB media at 37 °C for 5–6 h.
Fresh colonies and suspension cultures in PBS were adjusted spectrophotometrically to an optical density of 0.1, at wavelength of 600 nm, to yield approximately 1 × 108 colony-forming units (CFU)/mL. The bacterial suspension was further diluted in CAMHB, and 5 μL of the suspension was inoculated into respective wells to yield a starting inoculum of around 5 × 105 CFU/mL. The 96-well plates were incubated at 37 °C for 18 to 20 h. The MIC was recorded as the lowest concentration of antimicrobial agent that inhibits the visible growth of the bacterial isolate after 18–20 h incubation.85,86E-coli strain ATCC 25922 was used as the quality control strain with ampicillin as the antibiotic Rifampicin was used as the control against A. baumannii and K. pneumoniae strains. A minimum of three replicates were performed for each compound.
Cloning, Expression, and Purification of A. baumannii and K. pneumoniae IspC
The A. baumannii and K. pneumoniae IspC genes (ispC) were identified in the complete genome sequence using primary sequence homology with orthologs from other organisms. Sequence alignment was performed with the National Center for Biotechnology Information (NCBI) BLAST (blastp) suite with the following NCBI reference sequences: E. coli (U00096.2, AAC73284.1), F. tularensis (AJ749949.2, CAG46207.1), M. tuberculosis (NC_000962.3, NP_217386.2), Y. pestis (NC_003143.1, YP_002346091.1), A. baumannii (NZ_AFCZ00000000.2, EJP43779.1), and K. pneumoniae (KK036887.1, EWF09955.1).
The A. baumannii IspC clone was generously provided by the Seattle Structural Genomics Center for Infectious Disease (SSGCID). To prepare the clone, the A. baumannii ispC gene was PCR amplified from genomic DNA and cloned into a pBG1861 vector to yield pAbIspC, facilitating the expression of A. baumannii IspC with an N-terminal His6-tag. The K. pneumoniae ispC gene was fully synthesized (GenScript USA Inc., Piscataway, NJ) and cloned into a pMCSG28 vector to yield pKpIspC, facilitating the expression of K. pneumoniae IspC with a C-terminal His6-tag.
Each expression plasmid (pAbIspC and pKpIspC) was separately transformed into chemically competent E. coli BL21 CodonPlus (DE3)-RIL cells for protein expression. To express the His-tagged protein, a 10 mL overnight seed culture was added to 1 L of LB media supplemented with 100 μg/mL ampicillin and 50 μg/mL chloramphenicol and then incubated with shaking at 37 °C and 250 rpm. At an OD600 of 0.8, protein expression was induced with addition of isopropyl b-d-thiogalactopyranoside (IPTG) to 0.5 mM and the culture was further incubated with shaking at 37 °C and 250 rpm for an additional 18 h. Cells were harvested via centrifugation (4648g, 20 min, 4 °C) and stored at −80 °C. Protein was subsequently isolated and purified from the cells via chemical lysis and affinity chromatography.
Cells were lysed with lysis buffer A (100 mM Tris pH 8.0, 0.032% lysozyme, 3 mL per gram cell pellet), followed by lysis buffer B (0.1 M CaCl2, 0.1 M MgCl2, 0.1 M NaCl, 0.020% DNase, 0.3 mL per gram cell pellet). Clarified cell lysate was collected after centrifugation (48,000g, 20 min, 4 °C) and passed through a TALON immobilized metal affinity column (Clontech Laboratories, Mountain View, CA).
The column was washed with 20 column volumes of equilibrium buffer (50 mM HEPES pH 7.5, 300 mM NaCl), 10 column volumes of wash buffer A (50 mM HEPES pH 7.5, 300 mM NaCl, 10 mM imidazole), and 15 column volumes of wash buffer B (100 mM HEPES pH 7.5, 600 mM NaCl, 20 mM imidazole). The protein was eluted with 5 column volumes of elution buffer (150 mM imidazole pH 7.0, 300 mM NaCl). Buffer was exchanged with protein storage buffer (100 mM Tris pH 7.5, 1 mM NaCl, 5 mM DTT) during concentration by ultrafiltration. Protein concentration was determined using Advanced Protein Assay Reagent (Cytoskeleton, Denver CO) with γ-globulins (Sigma-Aldrich) as the standard and by measuring the absorbance at 280 nm (A280). Purified protein was visualized via Coomassie stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The yield of AbIspC averages 20 mg per 1 L shake flask, whereas the yield of KpIspC averages 10 mg per 1 L shake flask.
Crystallization
Crystallization experiments were set up with AbIspC according to the conditions published by the Seattle Structural Genomics Center for Infectious Disease (SSGCID) in the RCSB Protein Data Bank (PDB ID: 4ZN6). Crystals were obtained via sitting drop vapor diffusion at 16 °C from drops containing 100 mM sodium citrate:HCl pH 5.60, 250 mM ammonium sulfate, and 24% PEG 4000. Crystals were transferred to 100 mM sodium citrate:HCl pH 5.60, 250 mM ammonium chloride, and 24% PEG 4000 at 16 °C. Subsequently, the crystals were soaked with 50 mM magnesium chloride, 1 mM NADPH, and 50 mM FR900098.
Data Collection and Processing
The crystals were cryoprotected in mother liquor containing 10% ethylene glycol, followed by mother liquor containing 20% ethylene glycol, and then flash frozen in a N2 gas stream. Data was collected at 100 K to 2.4 Å resolution using a Bruker Microstar Rotating Anode X-ray generator with a Pt 135 CCD detector in-house at the Walter Reed Army Institute of Research X-ray Crystallography Center with integration and scaling using Proteum (Bruker).
Molecular Replacement and Refinement
The structure was determined by molecular replacement using the AbIspC structure (PDB ID 4ZN6) as a search model for phasing. Molecular replacement was performed using the program Phaser within the PHENIX suite. The NADPH, FR900098, and Mg2+ molecules were built within the mFo-Fc difference density map using Coot and Phenix. Refinement of the AbIspC:NADPH:FR900098:Mg tetra-complex was performed using the phenix.refine within the Phenix software suite and Coot.
Enzyme Assays
Data was analyzed using GraphPad Prism 5.0 for Windows (GraphPad Software Inc., San Diego, CA). A. baumannii and K. pneumoniae IspC activity was assayed at 37 °C by spectrophotometrically monitoring the enzyme catalyzed oxidation of NADPH upon addition of 1-deoxy-d-xylulose 5-phosphate (DXP; Echelon Biosciences, Salt Lake City, UT) to the assay mixture, as described previously.87 The oxidation of NADPH was monitored at 340 nm using an Agilent 8453 UV–vis spectrophotometer equipped with a temperature regulated cuvette holder. All assays were performed in duplicate.
To determine the apparent KM for DXP, 120 μL assay solutions contained 100 mM Tris pH 7.8, 25 mM MgCl2, 150 μM NADPH, 0.18 μM IspC, and variable concentrations of DXP. The assay solution was incubated at 37 °C for 10 min, prior to addition of DXP, to facilitate the association of NADPH with the enzyme. To determine the apparent KM for NADPH, assays were performed with a fixed DXP concentration (400 μM) and variable concentrations of NADPH. The kinetic constants were determined by nonlinear regression to the Michaelis–Menten equation using GraphPad PRISM version 5.00 for Windows (GraphPad Software Inc., San Diego, CA). IC50 values were determined by nonlinear regression to a dose–response curve: Y= (100)/(1 + 10∧((LogIC50-X)) using GraphPad PRISM version 5.00 for Windows.
To determine cation specificity, assays were performed with either MgCl2, CoCl2, CuCl2, MnCl2, or NiCl2 at 25 mM final concentration. Half-maximal inhibition (IC50) of enzyme activity by fosmidomycin and FR900098 was determined using a plot of fractional enzyme activity as a function of inhibitor concentration using GraphPad Prism 5.0 for Windows. As both inhibitors are slow, tight binding inhibitors,40 fosmidomycin and FR900098 were preincubated with the enzyme at 37 °C for 10 min prior to the addition of the substrate, DXP.
Acknowledgments
This work was generously supported by the George Mason University Department of Chemistry and Biochemistry, the U.S. Army MRDC (W81XWH-17-C-0066), The Military Infectious Disease Research Program (W0161_15_WR to S.M.N), the NIH (5R01AI123433-04), and by an appointment to the Student Research Participation Program at the Walter Reed Army Institute of Research administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and USAMRDC. We would like to thank the Seattle Structural Genomics Center for Infectious Disease (SSGCID) for generously providing the A. baumannii IspC clone and their protein crystallization experimental conditions. The Seattle Structural Genomics Center for Infectious Disease (www.SSGCID.org) is supported by Federal Contract No. HHSN272201700059C from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services.
Glossary
Abbreviations Used
- FR
- Fos
Fosmidomycin
- Ab
Acinetobacter baumannii
- Kp
Klebsiella pneumoniae
Accession Codes
7S04
The authors declare no competing financial interest.
Notes
This work has been reviewed by the Walter Reed Army Institute of Research (WRAIR). There is no objection to its presentation and/or publication. The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or reflecting the views of the Department of Defense, the Department of the Army, or any other agency of the U.S. Government.
References
- Santajit S.; Indrawattana N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. BioMed Res. Int. 2016, 2016, 1. 10.1155/2016/2475067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg R. B.; Wetzler T. F.; Newton A.; Howard J. M.; Davis J. H.; Strawitz J. The Bacterial Flora of the Blood Stream in the Korean Battle Casualty. Ann. Surg. 1955, 141 (3), 366–368. 10.1097/00000658-195503000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong M. J. Septic Complications of War Wounds. JAMA, J. Am. Med. Assoc. 1972, 219 (8), 1044–1047. 10.1001/jama.219.8.1044. [DOI] [PubMed] [Google Scholar]
- Petersen K.; Riddle M. S.; Danko J. R.; Blazes D. L.; Hayden R.; Tasker S. A.; Dunne J. R. Trauma-Related Infections in Battlefield Casualties from Iraq. Ann. Surg. 2007, 245 (5), 803–811. 10.1097/01.sla.0000251707.32332.c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun J. H.; Murray C. K.; Manring M. M. Multidrug-Resistant Organisms in Military Wounds from Iraq and Afghanistan. Clin. Orthop. Relat. Res. 2008, 466 (6), 1356–1362. 10.1007/s11999-008-0212-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootz T. D.; Marra A. Acinetobacter Baumannii: An Emerging Multidrug-Resistant Threat. Expert Rev. Anti-Infect. Ther. 2008, 6 (3), 309–325. 10.1586/14787210.6.3.309. [DOI] [PubMed] [Google Scholar]
- Dallo S. F.; Weitao T. Insights into Acinetobacter War-Wound Infections, Biofilms, and Control. Adv. Skin Wound Care 2010, 23 (4), 169–174. 10.1097/01.ASW.0000363527.08501.a3. [DOI] [PubMed] [Google Scholar]
- Richards A. M.; Kwaik Y. A.; Lamont R. J. Code Blue: Acinetobacter Baumannii, a Nosocomial Pathogen with a Role in the Oral Cavity. Mol. Oral Microbiol. 2015, 30 (1), 2–15. 10.1111/omi.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acinetobacter in Healthcare Settings | HAI | CDC https://www.cdc.gov/hai/organisms/acinetobacter.html (accessed 2018 −10 −16).
- McQueary C. N.; Kirkup B. C.; Si Y.; Barlow M.; Actis L. A.; Craft D. W.; Zurawski D. V. Extracellular Stress and Lipopolysaccharide Modulate Acinetobacter Baumannii Surface-Associated Motility. J. Microbiol. 2012, 50 (3), 434–443. 10.1007/s12275-012-1555-1. [DOI] [PubMed] [Google Scholar]
- Christoff R. M.; Murray G. L.; Kostoulias X. P.; Peleg A. Y.; Abbott B. M. Synthesis of Novel 1,2,5-Oxadiazoles and Evaluation of Action against Acinetobacter Baumannii. Bioorg. Med. Chem. 2017, 25 (24), 6267–6272. 10.1016/j.bmc.2017.08.015. [DOI] [PubMed] [Google Scholar]
- Kramer A.; Schwebke I.; Kampf G. How Long Do Nosocomial Pathogens Persist on Inanimate Surfaces? A Systematic Review. BMC Infect. Dis. 2006, 6, 130. 10.1186/1471-2334-6-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaras A. P.; Dorsey C. W.; Edelmann R. E.; Actis L. A. Attachment to and Biofilm Formation on Abiotic Surfaces by Acinetobacter Baumannii: Involvement of a Novel Chaperone-Usher Pili Assembly System. Microbiology 2003, 149 (12), 3473–3484. 10.1099/mic.0.26541-0. [DOI] [PubMed] [Google Scholar]
- Doi Y.; Murray G. L.; Peleg A. Y. Acinetobacter Baumannii: Evolution of Antimicrobial Resistance-Treatment Options. Semin Respir Crit Care Med. 2015, 36 (1), 85–98. 10.1055/s-0034-1398388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalopoulos A.; Falagas M. E. Treatment of Acinetobacter Infections. Expert Opin. Pharmacother. 2010, 11 (5), 779–788. 10.1517/14656561003596350. [DOI] [PubMed] [Google Scholar]
- Hakyemez I. N.; Kucukbayrak A.; Tas T.; Yikilgan A. B.; Akkaya A.; Yasayacak A.; Akdeniz H. Nosocomial Acinetobacter Baumannii Infections and Changing Antibiotic Resistance. Pak J. Med. Sci. 2013, 29 (5), 1245–1248. 10.12669/pjms.295.3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC . The biggest antibiotic-resistant threats in the U.S. https://www.cdc.gov/drugresistance/biggest-threats.html (accessed 2019 −12 −31).
- Tzouvelekis L. S.; Markogiannakis A.; Psichogiou M.; Tassios P. T.; Daikos G. L. Carbapenemases in Klebsiella Pneumoniae and Other Enterobacteriaceae: An Evolving Crisis of Global Dimensions. Clin. Microbiol. Rev. 2012, 25 (4), 682–707. 10.1128/CMR.05035-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebsiella pneumoniae in Healthcare Settings | HAI | CDC https://www.cdc.gov/hai/organisms/klebsiella/klebsiella.html (accessed 2020 −01 −03).
- Behzadi P.; Behzadi E.; Yazdanbod H.; Aghapour R.; Akbari Cheshmeh M.; Salehian Omran D. A Survey on Urinary Tract Infections Associated with the Three Most Common Uropathogenic Bacteria. Maedica (Buchar) 2010, 5 (2), 111–115. [PMC free article] [PubMed] [Google Scholar]
- Farajnia S.; Alikhani M. Y.; Ghotaslou R.; Naghili B.; Nakhlband A. Causative Agents and Antimicrobial Susceptibilities of Urinary Tract Infections in the Northwest of Iran. Int. J. Infect. Dis. 2009, 13 (2), 140–144. 10.1016/j.ijid.2008.04.014. [DOI] [PubMed] [Google Scholar]
- Spahiu L.; Hasbahta V. Most Frequent Causes of Urinary Tract Infections in Children. Med. Arh 2010, 64 (2), 88–90. [PubMed] [Google Scholar]
- Martin R. M.; Bachman M. A. Colonization, Infection, and the Accessory Genome of Klebsiella Pneumoniae. Front. Cell. Infect. Microbiol. 2018, 8, 4. 10.3389/fcimb.2018.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K.Chapter 21: Enterobacteriaceae. In Sherris Medical Microbiology: An Introduction to Infectious Diseases; McGraw-Hill: 2004; pp 343–372. [Google Scholar]
- Knothe H.; Shah P.; Krcmery V.; Antal M.; Mitsuhashi S. Transferable Resistance to Cefotaxime, Cefoxitin, Cefamandole and Cefuroxime in Clinical Isolates of Klebsiella Pneumoniae and Serratia MarcescensUbertragbare Resistenz Gegenüber Cefotaxim, Cefoxitin, Cefamandol Und Cefuroxim in Klinischen Isolaten von Klebsiella Pneumoniae Und Serratia Marcescens. Infection 1983, 11 (6), 315–317. 10.1007/BF01641355. [DOI] [PubMed] [Google Scholar]
- Ah Y.-M.; Kim A.-J.; Lee J.-Y. Colistin Resistance in Klebsiella Pneumoniae. Int. J. Antimicrob. Agents 2014, 44 (1), 8–15. 10.1016/j.ijantimicag.2014.02.016. [DOI] [PubMed] [Google Scholar]
- Tamma P. D.; Rodriguez-Bano J. The Use of Noncarbapenem β-Lactams for the Treatment of Extended-Spectrum β-Lactamase Infections. Clin. Infect. Dis. 2017, 64 (7), 972–980. 10.1093/cid/cix034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugnaioli C.; Luzzaro F.; Luca F. D.; Brigante G.; Perilli M.; Amicosante G.; Stefani S.; Toniolo A.; Rossolini G. M. CTX-M-Type Extended-Spectrum β-Lactamases in Italy: Molecular Epidemiology of an Emerging Countrywide Problem. Antimicrob. Agents Chemother. 2006, 50 (8), 2700–2706. 10.1128/AAC.00068-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO | WHO publishes list of bacteria for which new antibiotics are urgently needed https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed 2017 −11 −30).
- Beringer P. The Clinical Use of Colistin in Patients with Cystic Fibrosis. Curr. Opin. Pulm. Med. 2001, 7 (6), 434–440. 10.1097/00063198-200111000-00013. [DOI] [PubMed] [Google Scholar]
- Landman D.; Georgescu C.; Martin D. A.; Quale J. Polymyxins Revisited. Clin. Microbiol. Rev. 2008, 21 (3), 449–465. 10.1128/CMR.00006-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.Notes from the Field: Pan-Resistant New Delhi Metallo-Beta-Lactamase-Producing Klebsiella Pneumoniae — Washoe County, Nevada, 2016. MMWR Morb Mortal Wkly Rep 2017, 66. 33. 10.15585/mmwr.mm6601a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange B. M.; Rujan T.; Martin W.; Croteau R. Isoprenoid Biosynthesis: The Evolution of Two Ancient and Distinct Pathways across Genomes. Proc. Natl. Acad. Sci. U. S. A. 2000, 97 (24), 13172–13177. 10.1073/pnas.240454797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard J.; Moreira D. Origins and Early Evolution of the Mevalonate Pathway of Isoprenoid Biosynthesis in the Three Domains of Life. Mol. Biol. Evol. 2011, 28 (1), 87–99. 10.1093/molbev/msq177. [DOI] [PubMed] [Google Scholar]
- Jawaid S.; Seidle H.; Zhou W.; Abdirahman H.; Abadeer M.; Hix J. H.; van Hoek M. L.; Couch R. D. Kinetic Characterization and Phosphoregulation of the Francisella Tularensis 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase (MEP Synthase). PLoS One 2009, 4 (12), e8288. 10.1371/journal.pone.0008288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuston S.; Begley M.; Gahan C. G. M.; Hill C. Isoprenoid Biosynthesis in Bacterial Pathogens. Microbiology 2012, 158 (6), 1389–1401. 10.1099/mic.0.051599-0. [DOI] [PubMed] [Google Scholar]
- Rohdich F.; Bacher A.; Eisenreich W. Isoprenoid Biosynthetic Pathways as Anti-Infective Drug Targets. Biochem. Soc. Trans. 2005, 33 (4), 785–791. 10.1042/BST0330785. [DOI] [PubMed] [Google Scholar]
- Singh N.; Chevé G.; Avery M. A.; McCurdy C. R. Targeting the Methyl Erythritol Phosphate (MEP) Pathway for Novel Antimalarial, Antibacterial and Herbicidal Drug Discovery: Inhibition of 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase (DXR) Enzyme. Curr. Pharm. Des. 2007, 13 (11), 1161–1177. 10.2174/138161207780618939. [DOI] [PubMed] [Google Scholar]
- Lange B. M.; Wildung M. R.; McCaskill D.; Croteau R. A Family of Transketolases That Directs Isoprenoid Biosynthesis via a Mevalonate-Independent Pathway. Proc. Natl. Acad. Sci. U. S. A. 1998, 95 (5), 2100–2104. 10.1073/pnas.95.5.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppisch A. T.; Fox D. T.; Blagg B. S. J.; Poulter C. D. E.; Coli M. E. P. Synthase: Steady-State Kinetic Analysis and Substrate Binding. Biochemistry 2002, 41 (1), 236–243. 10.1021/bi0118207. [DOI] [PubMed] [Google Scholar]
- Altincicek B.; Kollas A. K.; Sanderbrand S.; Wiesner J.; Hintz M.; Beck E.; Jomaa H. GcpE Is Involved in the 2-C-Methyl-D-Erythritol 4-Phosphate Pathway of Isoprenoid Biosynthesis in Escherichia Coli. J. Bacteriol. 2001, 183 (8), 2411–2416. 10.1128/JB.183.8.2411-2416.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham F. X.; Lafond T. P.; Gantt E. Evidence of a Role for LytB in the Nonmevalonate Pathway of Isoprenoid Biosynthesis. J. Bacteriol. 2000, 182 (20), 5841–5848. 10.1128/JB.182.20.5841-5848.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altincicek B.; Duin E. C.; Reichenberg A.; Hedderich R.; Kollas A.-K.; Hintz M.; Wagner S.; Wiesner J.; Beck E.; Jomaa H. LytB Protein Catalyzes the Terminal Step of the 2-C-Methyl-D-Erythritol-4-Phosphate Pathway of Isoprenoid Biosynthesis. FEBS Lett. 2002, 532 (3), 437–440. 10.1016/S0014-5793(02)03726-2. [DOI] [PubMed] [Google Scholar]
- McAteer S.; Coulson A.; McLennan N.; Masters M. The LytB Gene of Escherichia Coli Is Essential and Specifies a Product Needed for Isoprenoid Biosynthesis. J. Bacteriol. 2001, 183 (24), 7403–7407. 10.1128/JB.183.24.7403-7407.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohdich F.; Hecht S.; Gärtner K.; Adam P.; Krieger C.; Amslinger S.; Arigoni D.; Bacher A.; Eisenreich W. Studies on the Nonmevalonate Terpene Biosynthetic Pathway: Metabolic Role of IspH (LytB) Protein. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (3), 1158–1163. 10.1073/pnas.032658999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam P.; Hecht S.; Eisenreich W.; Kaiser J.; Grawert T.; Arigoni D.; Bacher A.; Rohdich F. Biosynthesis of Terpenes: Studies on 1-Hydroxy-2-Methyl-2-(E)-Butenyl 4-Diphosphate Reductase. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (19), 12108–12113. 10.1073/pnas.182412599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuhara M.; Kuroda Y.; Goto T.; Okamoto M.; Terano H.; Kohsaka M.; Aoki H.; Imanaka H. Studies on New Phosphonic Acid Antibiotics. III. Isolation and Characterization of FR-31564, FR-32863 and FR-33289. J. Antibiot. 1980, 33 (1), 24–28. 10.7164/antibiotics.33.24. [DOI] [PubMed] [Google Scholar]
- McKenney E. S.; Sargent M.; Khan H.; Uh E.; Jackson E. R.; Jose G. S.; Couch R. D.; Dowd C. S.; van Hoek M. L. Lipophilic Prodrugs of FR900098 Are Antimicrobial against Francisella Novicida In Vivo and In Vitro and Show GlpT Independent Efficacy. PLoS One 2012, 7 (10), e38167. 10.1371/journal.pone.0038167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuhara M.; Kuroda Y.; Goto T.; Okamoto M.; Terano H.; Kohsaka M.; Aoki H.; Imanaka H. Studies on New Phosphonic Acid Antibiotics. I. FR-900098, Isolation and Characterization. J. Antibiot. 1980, 33 (1), 13–17. 10.7164/antibiotics.33.13. [DOI] [PubMed] [Google Scholar]
- Evaluation of Fosmidomycin and Clindamycin in the Treatment of Acute Uncomplicated P.Falciparum Malaria in Children - Full Text View - https://clinicaltrials.gov/ct2/show/NCT01464138 (accessed 2017 −11 −16).
- Evaluation of Fosmidomycin and Clindamycin in the Treatment of Acute Uncomplicated Plasmodium Falciparum Malaria - Full Text View - https://clinicaltrials.gov/ct2/show/NCT01361269 (accessed 2017 −11 −16).
- Fosmidomycin With Clindamycin or With Clindamycin Plus Artesunate - Full Text View - https://clinicaltrials.gov/ct2/show/NCT01002183 (accessed 2017 −11 −16).
- Evaluation of Fosmidomycin and Piperaquine in the Treatment of Acute Falciparum Malaria - Full Text View - https://clinicaltrials.gov/ct2/show/NCT02198807 (accessed 2017 −11 −16).
- Fosmidomycin and Azithromycin for Acute Uncomplicated Plasmodium Falciparum Malaria (P. Malaria) in Adults - Full Text View - https://clinicaltrials.gov/ct2/show/NCT01464125 (accessed 2017 −11 −16).
- Treatment of Malaria in Gabon With Fosmidomycin-Clindamycin - Full Text View - https://clinicaltrials.gov/ct2/show/NCT00217451 (accessed 2017 −11 −16).
- Efficacy of Fosmidomycin-Clindamycin for Treating Malaria in Gabonese Children - Full Text View - https://clinicaltrials.gov/ct2/show/NCT00214643 (accessed 2017 −11 −16).
- Haymond A.; Johny C.; Dowdy T.; Schweibenz B.; Villarroel K.; Young R.; Mantooth C. J.; Patel T.; Bases J.; Jose G. S.; Jackson E. R.; Dowd C. S.; Couch R. D. Kinetic Characterization and Allosteric Inhibition of the Yersinia Pestis 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase (MEP Synthase). PLoS One 2014, 9 (8), e106243. 10.1371/journal.pone.0106243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jomaa H.; Wiesner J.; Sanderbrand S.; Altincicek B.; Weidemeyer C.; Hintz M.; Türbachova I.; Eberl M.; Zeidler J.; Lichtenthaler H. K.; Soldati D.; Beck E. Inhibitors of the Nonmevalonate Pathway of Isoprenoid Biosynthesis as Antimalarial Drugs. Science 1999, 285 (5433), 1573–1576. 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- Davey M. S.; Tyrrell J. M.; Howe R. A.; Walsh T. R.; Moser B.; Toleman M. A.; Eberl M. A Promising Target for Treatment of Multidrug-Resistant Bacterial Infections ▽. Antimicrob. Agents Chemother. 2011, 55 (7), 3635–3636. 10.1128/AAC.00382-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu H. C.; Kamimura T. In Vitro and in Vivo Antibacterial Activity of FR-31564, a Phosphonic Acid Antimicrobial Agent. Antimicrob. Agents Chemother. 1981, 19 (6), 1013–1023. 10.1128/AAC.19.6.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mine Y.; Kamimura T.; Nonoyama S.; Nishida M.; Goto S.; Kuwahara S. In Vitro and in Vivo Antibacterial Activities of FR-31564, a New Phosphonic Acid Antibiotic. J. Antibiot. 1980, 33 (1), 36–43. 10.7164/antibiotics.33.36. [DOI] [PubMed] [Google Scholar]
- Yokota Y.; Murakawa T.; Nishida M. In Vitro Synergism of FR-31564, a New Phosphonic Acid Antibiotic. J. Antibiot. 1981, 34 (7), 876–883. 10.7164/antibiotics.34.876. [DOI] [PubMed] [Google Scholar]
- Jacobs A. C.; Thompson M. G.; Black C. C.; Kessler J. L.; Clark L. P.; McQueary C. N.; Gancz H. Y.; Corey B. W.; Moon J. K.; Si Y.; Owen M. T.; Hallock J. D.; Kwak Y. I.; Summers A.; Li C. Z.; Rasko D. A.; Penwell W. F.; Honnold C. L.; Wise M. C.; Waterman P. E.; Lesho E. P.; Stewart R. L.; Actis L. A.; Palys T. J.; Craft D. W.; Zurawski D. V. AB5075, a Highly Virulent Isolate of Acinetobacter Baumannii, as a Model Strain for the Evaluation of Pathogenesis and Antimicrobial Treatments. mBio 2014, 5 (3), e01076–01014. 10.1128/mBio.01076-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C. L.; Neu H. M.; Gilbreath J. J.; Michel S. L. J.; Zurawski D. V.; Merrell D. S. Copper Resistance of the Emerging Pathogen Acinetobacter Baumannii. Appl. Environ. Microbiol. 2016, 82 (20), 6174–6188. 10.1128/AEM.01813-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder M.; Brooks B. D.; Brooks A. E. The Complex Relationship between Virulence and Antibiotic Resistance. Genes 2017, 8 (1), 39. 10.3390/genes8010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.-H.; Su P.-W.; Moi S.-H.; Chuang L.-Y. Biofilm Formation in Acinetobacter Baumannii: Genotype-Phenotype Correlation. Molecules 2019, 24 (10), 1849. 10.3390/molecules24101849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto Y.; Furukawa S.; Ogihara H.; Yamasaki M. Fosmidomycin Resistance in Adenylate Cyclase Deficient (Cya) Mutants of Escherichia Coli. Biosci., Biotechnol., Biochem. 2003, 67 (9), 2030–2033. 10.1271/bbb.67.2030. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Watts K. M.; Hodge D.; Kemp L. M.; Hunstad D. A.; Hicks L. M.; Odom A. R. A Second Target of the Antimalarial and Antibacterial Agent Fosmidomycin Revealed by Cellular Metabolic Profiling. Biochemistry 2011, 50 (17), 3570–3577. 10.1021/bi200113y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. C.; Parish T. Dxr Is Essential in Mycobacterium Tuberculosis and Fosmidomycin Resistance Is Due to a Lack of Uptake. BMC Microbiol. 2008, 8, 78. 10.1186/1471-2180-8-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Jose G.; Jackson E. R.; Uh E.; Johny C.; Haymond A.; Lundberg L.; Pinkham C.; Kehn-Hall K.; Boshoff H. I.; Couch R. D.; Dowd C. S. Design of Potential Bisubstrate Inhibitors against Mycobacterium Tuberculosis (Mtb) 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase (Dxr)-Evidence of a Novel Binding Mode. MedChemComm 2013, 4 (7), 1099–1104. 10.1039/c3md00085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhiman R. K.; Schaeffer M. L.; Bailey A. M.; Testa C. A.; Scherman H.; Crick D. C. 1-Deoxy-d-Xylulose 5-Phosphate Reductoisomerase (IspC) from Mycobacterium Tuberculosis: Towards Understanding Mycobacterial Resistance to Fosmidomycin. J. Bacteriol. 2005, 187 (24), 8395–8402. 10.1128/JB.187.24.8395-8402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uh E.; Jackson E. R.; San Jose G.; Maddox M.; Lee R. E.; Lee R. E.; Boshoff H. I.; Dowd C. S. Antibacterial and Antitubercular Activity of Fosmidomycin, FR900098, and Their Lipophilic Analogs. Bioorg. Med. Chem. Lett. 2011, 21 (23), 6973–6976. 10.1016/j.bmcl.2011.09.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyrou A.; Blanchard J. S. Kinetic and Chemical Mechanism of Mycobacterium Tuberculosis 1-Deoxy-d-Xylulose-5-Phosphate Isomeroreductase. Biochemistry 2004, 43 (14), 4375–4384. 10.1021/bi049974k. [DOI] [PubMed] [Google Scholar]
- Kuzuyama T.; Takahashi S.; Takagi M.; Seto H. Characterization of 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase, an Enzyme Involved in Isopentenyl Diphosphate Biosynthesis, and Identification of Its Catalytic Amino Acid Residues. J. Biol. Chem. 2000, 275 (26), 19928–19932. 10.1074/jbc.M001820200. [DOI] [PubMed] [Google Scholar]
- Kuzuyama T.; Shimizu T.; Takahashi S.; Seto H. Fosmidomycin, a Specific Inhibitor of 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase in the Nonmevalonate Pathway for Terpenoid Biosynthesis. Tetrahedron Lett. 1998, 39, 7913–7916. 10.1016/S0040-4039(98)01755-9. [DOI] [Google Scholar]
- Walker J. R.; Poulter C. D. Synthesis and Evaluation of 1-Deoxy-d-Xylulose 5-Phosphate Analogues as Chelation-Based Inhibitors of Methylerythritol Phosphate Synthase. J. Org. Chem. 2005, 70 (24), 9955–9959. 10.1021/jo0516786. [DOI] [PubMed] [Google Scholar]
- Andaloussi M.; Henriksson L. M.; Wiȩckowska A.; Lindh M.; Björkelid C.; Larsson A. M.; Suresh S.; Iyer H.; Srinivasa B. R.; Bergfors T.; Unge T.; Mowbray S. L.; Larhed M.; Jones T. A.; Karlén A. Design, Synthesis, and X-Ray Crystallographic Studies of α-Aryl Substituted Fosmidomycin Analogues as Inhibitors of Mycobacterium Tuberculosis 1-Deoxy-d-Xylulose 5-Phosphate Reductoisomerase. J. Med. Chem. 2011, 54 (14), 4964–4976. 10.1021/jm2000085. [DOI] [PubMed] [Google Scholar]
- Henriksson L. M.; Unge T.; Carlsson J.; Aqvist J.; Mowbray S. L.; Jones T. A. Structures of Mycobacterium Tuberculosis 1-Deoxy-D-Xylulose-5-Phosphate Reductoisomerase Provide New Insights into Catalysis. J. Biol. Chem. 2007, 282 (27), 19905–19916. 10.1074/jbc.M701935200. [DOI] [PubMed] [Google Scholar]
- Yajima S.; Hara K.; Iino D.; Sasaki Y.; Kuzuyama T.; Ohsawa K.; Seto H. Structure of 1-Deoxy-d-Xylulose 5-Phosphate Reductoisomerase in a Quaternary Complex with a Magnesium Ion, NADPH and the Antimalarial Drug Fosmidomycin. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2007, 63 (6), 466–470. 10.1107/S1744309107024475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mac Sweeney A.; Lange R.; Fernandes R. P. M.; Schulz H.; Dale G. E.; Douangamath A.; Proteau P. J.; Oefner C. The Crystal Structure of E.Coli 1-Deoxy-d-Xylulose-5-Phosphate Reductoisomerase in a Ternary Complex with the Antimalarial Compound Fosmidomycin and NADPH Reveals a Tight-Binding Closed Enzyme Conformation. J. Mol. Biol. 2005, 345 (1), 115–127. 10.1016/j.jmb.2004.10.030. [DOI] [PubMed] [Google Scholar]
- Hanukoglu I. Proteopedia: Rossmann Fold: A Beta-Alpha-Beta Fold at Dinucleotide Binding Sites. Biochem. Mol. Biol. Educ. 2015, 43 (3), 206–209. 10.1002/bmb.20849. [DOI] [PubMed] [Google Scholar]
- Steinbacher S.; Kaiser J.; Eisenreich W.; Huber R.; Bacher A.; Rohdich F. Structural Basis of Fosmidomycin Action Revealed by the Complex with 2-C-Methyl-d-Erythritol 4-Phosphate Synthase (IspC) Implications for the Catalytic Mechanism and Anti-Malaria Drug Development. J. Biol. Chem. 2003, 278 (20), 18401–18407. 10.1074/jbc.M300993200. [DOI] [PubMed] [Google Scholar]
- Björkelid C.; Bergfors T.; Unge T.; Mowbray S. L.; Jones T. A. Structural Studies on Mycobacterium Tuberculosis DXR in Complex with the Antibiotic FR-900098. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68 (2), 134–143. 10.1107/S0907444911052231. [DOI] [PubMed] [Google Scholar]
- Umeda T.; Tanaka N.; Kusakabe Y.; Nakanishi M.; Kitade Y.; Nakamura K. T.. Molecular Basis of Fosmidomycin’s Action on the Human Malaria Parasite Plasmodium Falciparum. Sci. Rep. 2011, 1. 10.1038/srep00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLSI . Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Eighth ed.; Clinical and Laboratory Standards Institute, Wayne, PA, 2009; Vol. 8th ed. M07–A8. [Google Scholar]
- Wiegand I.; Hilpert K.; Hancock R. E. W. Agar and Broth Dilution Methods to Determine the Minimal Inhibitory Concentration (MIC) of Antimicrobial Substances. Nat. Protoc. 2008, 3 (2), 163–175. 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- Takahashi S.; Kuzuyama T.; Watanabe H.; Seto H. A 1-Deoxy-D-Xylulose 5-Phosphate Reductoisomerase Catalyzing the Formation of 2-C-Methyl-D-Erythritol 4-Phosphate in an Alternative Nonmevalonate Pathway for Terpenoid Biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 1998, 95 (17), 9879–9884. 10.1073/pnas.95.17.9879. [DOI] [PMC free article] [PubMed] [Google Scholar]