

Abstract
Zhu-Tokita-Takenouchi-Kim (ZTTK) syndrome is caused by de novo loss-of-function variants in the SON gene (MIM #617140). This multi-systemic disorder is characterized by intellectual disability, seizures, abnormal brain imaging, variable dysmorphic features and various congenital anomalies. The wide application and increasing accessibility of whole exome sequencing (WES) has helped to identify new cases of ZTTK syndrome over the last few years. To date, there have been approximately 45 cases reported in the literature. Here, we describe 15 additional individuals with variants in the SON gene, including those with missense variants bringing the total number of known cases to 60. We have reviewed the clinical and molecular data of these new cases and all previously reported cases to further delineate the most common as well as emerging clinical findings related to this syndrome. Furthermore, we aim to delineate any genotype-phenotype correlations specifically for a recurring pathogenic four base pair deletion (c.5753_5756del) along with discussing the impact of missense variants seen in the SON gene.
Keywords: SON, whole exome sequencing, multi-systemic disorder, genotype-phenotype correlation
Introduction
Loss-of-function variants in the SON gene (MIM #182465) have been recognized as causative of Zhu-Tokita-Takenouchi-Kim (ZTTK) syndrome (MIM #617140). Common features include intellectual disability, dysmorphic craniofacial features, hypotonia, and abnormalities of the brain. Many individuals also have musculoskeletal abnormalities and additional clinical features including congenital heart and genitourinary system defects (Kim, Shinde, et al., 2016; Y. Yang, Xu, Yu, Huang, & Yang, 2019). The phenotype associated with variants in the SON gene was initially suggested in 2015 when a de novo frameshift variant was identified in a 5-year-old female with developmental delay, seizure disorder, minor dysmorphic features, macrocephaly, brain white matter abnormalities, intestinal atresia, and a ventricular septal defect (VSD) (Zhu et al., 2015). The same variant was identified in an individual with postnatal growth retardation, infantile hypotonia, intellectual disability, and cardiac abnormalities (Takenouchi, Miura, Uehara, Mizuno, & Kosaki, 2016). SON was then implicated as a possible candidate gene for the neurological phenotype seen in Braddock-Carey 21q22 microdeletion syndrome encompassing SON (Takenouchi et al., 2016).
To date, the largest published cohort of disease-causing SON variants includes 20 individuals. (Kim, Shinde, et al., 2016). The reported phenotype in these individuals include intellectual disability (20 of 20), facial dysmorphisms (20 of 20), brain malformation (17 of 19), neurological features (17 of 20), musculoskeletal abnormalities (17 of 20), and eye/vision abnormalities (15 of 20). Other features were short stature, heart defects, gastrointestinal malformations, and urogenital malformations (Kim, Shinde, et al., 2016). Following this publication, there have been additional case reports with loss-of-function variants in the SON gene associated with a similar multi-system phenotype (Quintana Castanedo et al., 2020; Slezak et al., 2020; Tan et al., 2020; Y. Yang et al., 2019). Tokita et al. (2016) was the first to describe an individual with two de novo missense mutations in cis configuration in the SON gene. It is unclear whether these variants were a complex allele or whether an individual variant contributes to the disease phenotype (Tokita et al., 2016). Recently, dental and nail abnormalities have been suggested to be a part of the ZTTK syndrome phenotype after an individual with a loss-of-function variant in SON was reported to have enamel hypoplasia, microdontia, and retronychia (Quintana Castanedo et al., 2020). We have included an updated phenotype for this reported individual in this study.
Here, we describe 15 additional previously unreported cases carrying rare, pathogenic variants in SON, bringing the total number of known cases to 60. Moreover, we have reviewed the clinical and molecular data of these new cases and compared them to the previously reported cases, with the goal of further delineating the phenotypic spectrum of ZTTK syndrome and to establish potential genotype-phenotype correlations.
Methods
Individuals 1, 2, and 3 were identified by research whole exome sequencing at the Institute for Genomic Medicine at Columbia University Irving Medical Center (CUIMC). Individual 4 was identified by reanalysis of clinical whole exome sequencing at the Laboratory of Personalized Genomic Medicine (PGM) at CUIMC. DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources) was utilized to contact physicians of individuals 5–12, and 16. Individuals 13,14, and 51 were ascertained via direct contact by clinicians. For this publication, we were able to obtain additional information on individual 51 who was previously reported by Castanedo et al. (2020).
Whole exome sequencing (WES) was performed for 12 individuals to identify disease-causing variants using standard methods. Whole genome sequencing was performed for one individual while three individuals underwent gene panel testing related to either epilepsy or intellectual disability. Informed consent was obtained from each family prior to testing and approval of the ethics board was obtained in each institution. Detailed methodology for testing procedures can be found in the supplementary material.
For the literature review, relevant papers describing individuals with SON variants were identified through PubMed (https://www.ncbi.nlm.nih.gov/pubmed), OMIM (Online Mendelian Inheritance of Man, http://www.omim.org/), HGMD (Human Gene Mutation Database, http://www.hgmd.cf.ac.uk/ac/index.php), and DECIPHER. A total of 45 cases were ascertained from the literature: twenty cases from Kim et al. (2016), seven from Tokita et al. (2016), one from Takenouchi et al. (2016), one from Y.Yang, et al. (2019), two from Kim et al. (2019), two from Slezak et al. (2020), one from Yang et al. (2020), one from Tan et al. (2020), one from Castanedo et al. (2020) and nine individuals from DECIPHER. Of note, limited information was available for the nine individuals from DECIPHER. Attempts were made to obtain more phenotypic information on these cases but were unsuccessful.
Results
Molecular Findings
In the 15 newly described individuals, rare, heterozygous variants in the SON gene (GenBank: NM_138927.3) were detected. Fourteen of the 15 individuals were heterozygous for a loss-of-function SON variant and in one case, a rare missense variant was identified. In the majority of the cases, the SON variants were de novo (10 of 15) while in the remaining cases (5 of 15), inheritance was unknown. All loss-of-function variants were absent in the Genome Aggregation Database (gnomAD) v2.1.1 (Karczewski et al., 2020) while one missense variant (Individual 9) was observed one time. Detailed variant information is included in Table 1. The distribution of SON variants observed in our cohort as well as previously reported cases is shown in Figure 1.
Table 1:
Review of systems in present cohort
| Symptoms | Individual 1 | Individual 2 | Individual 3 | Individual 4 | Individual 5 |
|---|---|---|---|---|---|
| Variant ID | 21-34921860-CAAAG-C | 21-34924667-GCCTACGAGCGCTCTATGATGTCC-G | 21-34924945-C-A | 21-34927287-CAGTTAG-CAG | 21-34927298-C-T |
| NM_138927.3 (c.) | c.326_329del | c.3131_3153del | c.3408C>A | c.5753_5756del | c.5761C>T |
| NP_620305 (p.) | p.Lys109SerfsTer39 | p.Glu1046GlyfsTer2 | p.Tyr1136Ter | p.Val1918GlufsTer87 | p.Arg1921Ter |
| Inheritance | De novo | De novo | Unknown | Unknown | Unknown |
| Sanger confirmation | Yes | Yes | Yes | Yes | Yes |
| Classification | Pathogenic | Pathogenic | Likely Pathogenic | Pathogenic | Likely Pathogenic |
| Sex and Age | Male, 9 years | Male, 6 years | Female, 4 years | Male, 22 years | Female, 4.5 years |
| Height at last evaluation: cm (percentile, SD) | 113 (10–25th) | 84 (<5th) | 64 (<3rd) | (<1st, −2.74 SD) | 90.6 (−3 SD) |
| Weight at last evaluation: kg (percentile, SD) | 18 (3–10th) | 9.7 (<5th) | 6.45 (<3rd) | 71.4 (72nd, +0.58 SD) | 12 (−3 SD) |
| Head Circumference: cm (percentile, SD) | 53.5 (90th) | 84 (75th) | 46 (90–95th) | 56 cm (73rd, +0.63 SD) | 46.7 (−3 SD) |
| Investigation | WES | WES | WES | WES | WES |
| Prior Genetic Testing | Karyotype, microarray, metabolic profile, AFP skin biopsy and fibroblast studies, Fragile X, Angelman/Prader Willi testing | ||||
| DD/ID | + | + | + | + | + |
| NA | Severe | Moderate | Mild | Moderate | |
| Seizures | + | - | + | - | - |
| Status epilepticus | Status epilepticus, febrile seizures, focal epilepsy | - | - | ||
| Abnormal Brain MRI | + | + | + | + | - |
| T2 Flair hyperintensity within the dorsal brainsteam, and within the bilateral periatrial white matter (mild periventricular leukomalacia), arachnoid cyst | ventricular enlargement, agenesis of the corpus callosum, polymicrogyria | subependymal nodule, subtle increased T2 signal greater left periatrial white with mild volume loss | retention cysts in paranasal sinuses with thickening of the mucosal membranes | - | |
| Hypotonia | + | + | + | + | + |
| Eye Anomalies | NA | NA | + | NA | - |
| NA | NA | Exotropia, dissociated vertical up gaze | NA | ||
| Cardiac defect | NA | NA | - | NA | - |
| NA | NA | - | NA | - | |
| GI abnormalities | NA | + | - | - | + |
| NA | Feeding difficulties | - | - | Intestinal Malrotation | |
| GU abnormalities | NA | NA | + | - | NA |
| NA | NA | Echogenic medullary pyraminds | - | NA | |
| Musculoskeletal abnormalities | + | + | + | + | + |
| Unsteady gait, slim habitus | Mild pectus excavatum | Disproportionate short trunk,shortened height of the thoracic vertebrae right sided weakness, pes planus, genu varus bilaterally | Small feet, high arched left foot w/ absent nails on 3rd and 4th toes | Hypermobility and joint pain | |
| Short stature | - | - | + | + | + |
| Dysmorphic features | NA | + | + | + | + |
| NA | broad forehead, macrocephalic appearance | upslanting palpebral fissures, upturned nose | small whorl at the anterior hairline, large ears with earlobe creases | prominent nasal bridge, R preauricular ear tag, sparse hair all over, scalp particularly | |
| Other | Happy affect, constant drooling | - | Hearing loss, recurrent otitis media | Constant drooling, migraines | - |
| Individual 6 | Individual 7 | Individual 8 | Individual 9 | Individual 10 | |
| Variant ID | 21-34931675-CA-C | 21-34926312-CTCTA-C | 21-34927287-CAGTTAG-CAG | 21-34924751-C-T | 21-34924300-C-A |
| NM_138927.3 (c.) | c.6461del | c.4776_4779del | c.5753_5756del | c.3214C>T | c.2763C >A |
| NP_620305 (p.) | p.Asn2154IlefsTer2 | p.Ser1594LeufsTer28 | p.Val1918GlufsTer87 | p.Arg1072Cys | p.Tyr921Ter |
| Inheritance | De novo | De novo | De novo | Unknown | De novo |
| Sanger confirmation | Yes | NA | Yes | Yes | No |
| Classification | Pathogenic | Pathogenic | Pathogenic | VUS | Likely Pathogenic |
| Sex and Age | Male 10, years | Male, 5 years | Female, 13 years | Male, 12 years | Female, 6 years |
| Height at last evaluation: cm (percentile, SD) | 130.4 (9th) | 77.5 (<0.4th, −3.1) | 149.3 (5th, −1.63 SD) | 109.4 (50th, −0.18 SD) | 92 (<1st, −5.7 SD) |
| Weight at last evaluation: cm (percentile, SD) | 24.7 (2–9th) | 9.2 (1st, −2.3) | 37.7 (6th, −1.57 SD) | 18.4 (50th, −0.17 SD) | 12 (50th) |
| Head Circumference: cm (percentile, SD) | 53 (9–25th) | 50.3 (94th, +1.6 SD) | 53 (75th) | 48.9 (<0.4th, −2.64 SD) | 48 (50th) |
| Investigation | WES | WES and WGS | WES | WES | WES |
| Prior Genetic Testing | Microarray (1q21 gain-VOUS) | Prader Willi/Angelman syndrome, SMN1 testing | Karyotype, microarray, Single gene testing (PTPN11, SOS1, BRAF, MAP2K1, MAP2K2, HRAS, RAF1, SCHOC2, KANSL) | Microarray (7q34 dup, Yp11.32 dup, paternally inherited) | Karyotype, microarray, Coffin-Siris panel, ARID1B, Sensenbrenner panel |
| DD/ID | + | + | + | + | + |
| Moderate | Moderate | Moderate to Severe | Moderate to severe | Severe | |
| Seizures | + | - | + | + | + |
| 1 febrile seizure | - | Focal epilepsy | Status epilepticus (5x) | Febrile seizures | |
| Abnormal Brain MRI | + | + | + | - | + |
| Hypoplastic corpus callosum | Plagiocephaly, enlarged ventricles, thin septum pellucidum., a congenital fusion of the right C5–6 facet joint and probably also the disc space, butterfly vertebrae at T11 | Asymmetrical insular cortex, left side | Normal MRI | Enlarged ventricles, agenesis of the corpus callosum | |
| Hypotonia | + | + | + (with spasticity) | - | + |
| Eye Anomalies | - | - | + | - | + |
| - | - | Strabismus, heterotopia | - | Cerebral vision impairment with severely reduced vision | |
| Cardiac defect | - | - | + | - | + |
| - | - | Large ASD (spontaneous closing) | - | ASD | |
| GI abnormalities | + | + | - | - | + |
| Persistent chronic diarrhea | Early feeding difficulties | - | - | Severe feeding difficulties | |
| GU abnormalities | - | + | - | - | + |
| - | Pylectasis, vesicoureteral reflux | - | - | Vesicoureteral reflux | |
| Musculoskeletal abnormalities | + | + | - | - | + |
| Fifth finger clinodactyly | Delayed walked and hypermobility | - | - | Mild scoliosis, hip dysplasia, short fifth toes | |
| Short stature | - | NA | + | - | + |
| Dysmorphic features | + | + | + | - | + |
| deep set eyes, broad nasal bridge, tubular nose, prominent ears, widely spaced teeth, hypoplastic nails | plagiocephaly, almond shaped eyes, short toes | Frontal bossing, low set posterior rotated ears, downslanting palpebral fissures, ptosis, flat mid face, high palate, full lower lip, prognathism of the mandibule, high hairline, hypoplastic nails | - | abnormal head shape with frontal bossing, low-set ears, sparse scalp hair | |
| Other | Absent adult teeth, eczema, hand flapping, sleep issues, ADHD | Underactive thyroid | - | - | Hearing loss, craniosynostosis, recurrent infections (ear, UT). Anus somewhat posteriorly placed |
| Individual 11 | Individual 12 | Individual 13 | Individual 14 | Individual 15 | |
| Variant ID | 21-34926311-ACT-A | 21-34925981-CATGTT-C | 21-34923695-AC-A | 21-34926199-TA-T | 21-34927287-CAGTTAG-CAG |
| NM_138927.3 (c.) | c.4777_4778del | c.4448_4452del | c.2160delC | c.4663delA | c.5753_5756del |
| NP_620305 (p.) | p.Leu1593IlefsTer11 | p.Val1483GlufsTer4 | p.Met721TrpfsTer6 | p.1555fsTer68 | p.Val1918GlufsTer87 |
| Inheritance | De novo | Unknown | De novo | De novo | De novo |
| Sanger confirmation | Yes | In Progress | Yes | Yes | Yes |
| Classification | Likely Pathogenic | Likely Pathogenic | Pathogenic | Likely Pathogenic | Pathogenic |
| Sex and Age | Male, 3 years | Female, 6 years | Male, 7 years | Male, 6.5 months | Male, 10 years |
| Height at last evaluation: cm (percentile, SD) | 87.5 (<5th, −2.7 SD) | 104.6 (<0.4th −2.7 SD) | 103 (−2.65 SD) | 67 (25th) | 138.5 (<25th) |
| Weight at last evaluation: kg (percentile, SD) | 9.98 (<5th, −3.2 SD) | 13.7 (<0.4th, −3.8 SD) | 16.1 (17th) | 8.19 (50th) | 36.5 (50–75th) |
| Head Circumference: cm (percentile, SD) | 47.6 (7th, −1.45 SD) | 50.4 (4th, −1.7 SD) | 48.5 (−2.69 SD) | 48 cm (25th) | 52.8 (9th) |
| Investigation | Epilepsy Panel | WGS | WES | WES | WES |
| Prior Genetic Testing | Microarray, Angelman/Prader willi syndrome, Metabolic screening | Microarray, Cornelia de Lange panel, testing for Wiedemann Steiner, KBG, and Rubinstein Taybi | Microarray, Noonan syndrome panel | Microarray, Fragile X testing, epilepsy gene panel, Smith-Lemli-Opitz testing | Karyotype, microarray, Fragile X |
| DD/ID | + | + | + | + | + |
| Mild to Moderate | Moderate | Severe (nonverbal) | Moderate | Severe (nonverbal) | |
| Seizure | + | - | + | + | - |
| Febrile seizures | - | Generalized tonic clonic seizures | Febrile seizures, generalized tonic-clonic seizures | - | |
| Abnormal Brain MRI | + | + | + | + | - |
| Delayed myelination, enlarged lateral ventricles, thin corpus callosum | Signal changes in the periventricular regions and temporal poles bilaterall, mild elongation of the temporal horns, thinning of the corpus callosum and mild crowding of the foramen of magnum. | Right fronto-parietal-temporal polymicrogiri, mild deep white matter reduction, dysmorphisms and dilatation of lateral ventricles, incomplete hippocampal inversion, corpus callosum hypoplasia, enlargement of frontotemporal subarachnoid spaces | Prominence of the ventricular system and extra-acial spaces, particularly in the frontal region and vertex | Normal brain MRI | |
| Hypotonia | + | - | + | + | + |
| Eye Anomalies | - | - | + | + | + |
| - | - | Hypermetropia, Strabismus | Convergent squint, visual impairment, hypermetropia, astigmatism | Refractive error and astigmatism | |
| Cardiac defect | - | - | - | - | - |
| - | - | - | - | - | |
| GI abnormalities | - | + | + | + | + |
| - | Early feeding difficulties | Recurrent vomiting until 3 years | Feeding difficulties | Early feeding difficulties | |
| GU abnormalities | - | - | - | + | - |
| - | - | - | Recurrent UTIs | - | |
| Musculoskeletal abnormalities | + | - | + | + | + |
| Pes planovalgus | - | Talipes equinovarus, clinodactyly of the 2nd toe, drumstick appearance of terminal phalanges of hands, spasticity of lower limbs | hypermobility,mild 2–3 toe syndactyly | 2–3 toe syndactyly | |
| Short stature | + | + | + | - | - |
| Dysmorphic features | + | + | + | + | + |
| Frontal bossing, bulbous and depressed nasal tip, low hanging columella, short philtrum, thin upper lip, prominent chin | down slanting palpebral fissures with lateral eyebrow flare | sparse eyebrows laterally, overhanging nasal tip, down slanting palpebral fissures, short nose, smooth philtrum, everted lower lip, mild prognathism | low set ears with upturned lobe, wide spaced front teeth | small palpebral fissures, tubular nose, down-turned corners of mouth, inverted nipples, slithly tapering fingers, Sandal gap, thin ridged nails | |
| Other | - | recurrent serious lower respiratory chest infections in early years with ICU admission | IgA and IgG deficit, cryptorchidisms (surgically corrected), hypothyroidism | - | Larynogmalacia, torticollis, plaigocephaly, poor sleep, hand flapping, two episodes of acute, self-resolving hemiparesis |
Fig 1:
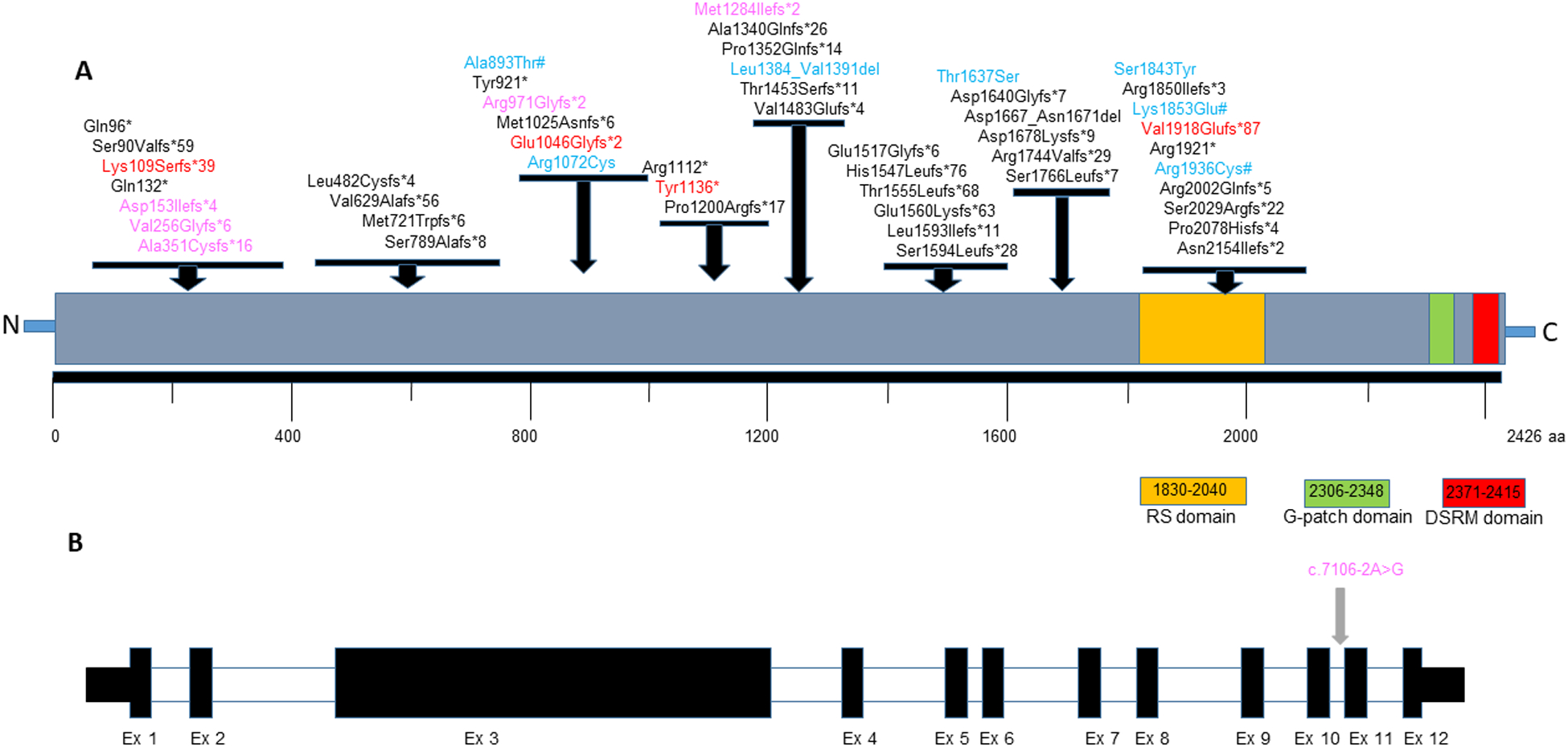
A. Distribution of variants seen in affected individuals on the SON protein. The LoF variants identified in this study are shown in red, missense & in-frame deletion variants are shown in blue. Predicted loss of function variants from DECIPHER cases are shown in pink and missense changes from DECIPHER cases are shown in blue followed by a #. Other variants reported in literature are shown in black color (NP_620305.3). The location of the functional domains in SON protein were adapted from Hickey et al 2013. B. Schematic showing the exons and the splice variant reported in the SON gene (NM_138927.4).
Clinical Findings
Neurodevelopment
All individuals in our cohort presented with developmental and/or intellectual disability (15 of 15, 100%). The degree of intellectual disability ranged from mild to severe, although the majority were noted to be moderate or severe (12 of 14). Most individuals achieved independent ambulation between the ages of two years and three years but two were not able to ambulate independently at their current age of six years. Two individuals were noted to have regression with one individual having regression following an encephalitic episode with intractable seizures. Speech delay was seen in all cases, with three individuals being non-verbal. Most individuals had better receptive language than expressive language. Abnormal muscle tone was seen in all individuals except for two (13 of 15, 87%), making it the most common neurodevelopmental symptom observed in this cohort.
The majority of individuals (12 of 15, 80%) had abnormal brain imaging. The most common findings were hypoplasia or agenesis of the corpus callosum (6 of 12, 50%) and ventricular enlargement (5 of 12, 42%). In addition, most (8 of 12, 67%) had multiple findings on brain MRI. Of those with abnormal brain imaging, nine presented with seizures. Of the three individuals with normal brain imaging, one had a diagnosis of focal epilepsy. Nine individuals total (60%) had a history of seizures with various epilepsy types. Five of 9 individuals were noted to have seizures associated with febrile illness while three were noted to have at least one episode of status epilepticus. In general, seizures were successfully managed with medication including levetiracetam, carbamazepine or clobazam.
Growth
Seven of 15 individuals (47%) had persistent growth delay (defined as both weight and height at least two standard deviations below the mean at the time of their last visit or below the third centile). There was no consistency in trends related to head circumference in our cohort. Eight of 14 individuals (57%) had short stature, defined as two standard deviations below the mean height for age.
Vision
Seven of 12 individuals (58%) had abnormal vision. The most common visual abnormality was strabismus and hypermetropia, each seen in three individuals.
Cardiovascular
Congenital heart defects were seen in two of 11 individuals (18%). Both individuals had an atrial septal defect (ASD) at birth. Cardiac information was not available for three individuals.
Gastrointestinal
Nine of 14 individuals (64%) in our cohort had gastrointestinal symptoms, of which six were reported with feeding difficulties. Other gastrointestinal features noted were recurrent vomiting, chronic diarrhea, and intestinal malrotation.
Genitourinary
Genitourinary anomalies were observed in four of 13 individuals (31%). Two individuals had vesicoureteral reflux, one had recurrent urinary tract infections, one had pyelectasis detected prenatally, and one had echogenic medullary pyramids that were thought to be related to nephrocalcinosis.
Musculoskeletal
The majority (12 of 15, 80%) of individuals in our cohort had musculoskeletal abnormalities. Four of twelve individuals had either clinodactyly or syndactyly of the fingers or toes while three were noted to have joint hypermobility. Gait abnormalities were noted in two individuals but it is unknown whether the abnormalities were related to a neurological or musculoskeletal deficit. Foot anomalies were present in three individuals including pes planovalgus (1 of 3), high arched feet (1 of 4) and talipes equinovarus (1 of 4).
Discussion
SON, located on chromosome region 21q22.11, encodes an RNA and DNA binding protein that acts as an mRNA splicing cofactor by promoting efficient RNA splicing of transcripts with weak and alternative splice sites and also plays a role in gene repression (Ahn et al., 2011; Kim, Baddoo, et al., 2016). The full length SON protein (total aa= 2426) contains three C-terminal conserved domains –an arginine/serine (RS)-rich domain, a G-patch domain, and a double-stranded RNA-binding motif (Kim, Shinde, et al., 2016; Ueda et al., 2020) (Figure 1). This gene is constrained for loss-of-function variants as seen in the gnomAD population database v2.1.1 (pLI=1, o/e ratio 0.05). Kim et al. (2016) showed a significant down regulation in the expression of SON transcript and protein in individuals with ZTTK syndrome when compared to unaffected parents and unrelated controls (Kim, Shinde, et al., 2016). SON knockdown in human cell lines resulted in global changes in gene expression including significant reduced expression in genes involved in neuronal cell migration, embryonic survival, metabolism, and mitochondrial function, which was confirmed in peripheral blood mononuclear cells (PBMCs) from affected individuals. Additionally, these PBMCs showed defective RNA splicing and accumulation of mis-spliced products. A morpholino mediated knockdown of a well-conserved human SON homolog in a zebra fish model, exhibited wide range of developmental defects mimicking features observed in affected individuals with ZTTK syndrome (Kim, Shinde, et al., 2016). More recently, shRNA mediated knockdown of SON in the developing mouse brain, using in utero electroporation, has shown neuronal migration abnormalities during corticogenesis and reduced spine density on mature cortical neurons (Ueda et al., 2020).
ZTTK syndrome is a multi-systemic condition that has been previously described in 45 individuals (Kim et al., 2019; Kim, Shinde, et al., 2016; Quintana Castanedo et al., 2020; Slezak et al., 2020; Takenouchi et al., 2016; Tan et al., 2020; Tokita et al., 2016; L. Yang & Yang, 2020; Y. Yang et al., 2019). In this study, we describe 15 previously unreported individuals with SON variants, and obtained additional clinical information on the individual recently described by Castanedo et al. (2020). Detailed clinical and molecular findings were compared to the cases reported in the literature (Table 2, Supplementary Table 1).
Table 2:
Comparison of symptoms across all patients
| Symptoms | Individual 1 | Individual 2 | Individual 3 | Individual 4 | Individual 5 | Individual 6 | Individual 7 | Individual 8 | Individual 9 | Individual 10 | Individual 11 | Individual 12 | Individual 13 | Individual 14 | Individual 15 | Present Cohort |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DD/ID | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | 15/15 |
| Abnormal Muscle Tone | + | + | + | + | + | + | + | + | - | + | + | - | + | + | + | 13/15 |
| Seizures | + | - | + | - | - | + | - | + | + | + | + | - | + | + | - | 9/15 |
| Abnormal Brain MRI | + | + | + | + | - | + | + | + | - | + | + | + | + | + | - | 12/15 |
| Eye anomalies | NA | NA | + | NA | + | - | - | + | - | + | - | - | + | + | + | 7/12 |
| Cardiac Defect | NA | NA | - | NA | - | - | - | + | - | + | - | - | - | - | NA | 2/11 |
| GI abnormalities | NA | + | - | - | + | + | + | - | - | + | - | + | + | + | + | 9/14 |
| GU abnormalities | NA | _ | + | - | NA | - | + | - | - | + | - | - | - | + | - | 4/13 |
| Musculoskeletal abnormalities | + | + | + | + | + | + | + | - | - | + | + | - | + | + | + | 12/15 |
| Short Stature | - | - | + | + | + | - | NA | + | - | + | + | + | + | - | - | 8/14 |
| Dysmorphic features | NA | + | + | + | + | + | + | + | - | + | + | + | + | + | + | 13/14 |
| Symptoms | Kim et al. (2016) | Tokita et al. (2016) | Takenouchi et al. (2016) | Yang et al. (2019) | Kim et al. (2019) | Slezak et al. (2020) | Yang et al. (2020) | Tan et al. (2020) | Castanedo et al. (2020) | DECIPHER | Total | Percentage |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DD/ID | 20/20 | 7/7 | 1/1 | 1/1 | NA | 2/2 | 1/1 | 1/1 | 1/1 | 9/9 | 58/58 | 100% |
| Abnormal Muscle Tone | 15/20 | 5/6 | 0/1 | N/A | NA | 2/2 | 0/1 | 1/1 | 1/1 | 1/1 | 38/48 | 79.2% |
| Seizures | 11/20 | 4/6 | 0/1 | N/A | NA | 1/1 | 1/1 | 0/1 | 1/1 | 4/4 | 31/50 | 58% |
| Abnormal Brain MRI | 17/19 | 5/6 | 0/1 | 1/1 | NA | 1/1 | 0/1 | 1/1 | 1/1 | 2/2 | 40/48 | 83.3% |
| Eye anomalies | 15/20 | 5/7 | 0/1 | N/A | NA | 2/2 | 0/1 | 0/1 | 1/1 | 3/3 | 33/48 | 68.8% |
| Cardiac Defect | 5/20 | 3/6 | 1/1 | 0/1 | NA | 1/2 | 0/1 | 0/1 | 1/1 | 1/1 | 14/45 | 31.1% |
| GI abnormalities | 3/20 | 6/7 | 0/1 | 0/1 | NA | 2/2 | 1/1 | 1/1 | 0/1 | 3/3 | 25/51 | 47.1% |
| GU abnormalities | 6/20 | 3/6 | 1/1 | NA | 2/2 | 1/2 | 0/1 | 1/1 | 0/1 | 2/2 | 20/48 | 39.5% |
| Musculoskeletal abnormalities | 17/20 | 6/7 | 1/1 | 0/1 | NA | 1/1 | 0/1 | 0/1 | 1/1 | 2/2 | 40/50 | 80% |
| Short Stature | 10/20 | 1/7 | 1/1 | 1/1 | NA | 0/2 | 1/1 | 1/1 | 1/1 | 1/1 | 25/49 | 51% |
| Dysmorphic features | 20/20 | 7/7 | 1/1 | 1/1 | NA | 2/2 | 0/1 | 1/1 | 1/1 | 8/8 | 54/56 | 96.4% |
Please refer to Table 2 in the Supplementary Materials Section for a list of all SON variants (in this cohort and previously reported), organized by publication. The distribution of the known 48 unique SON variants is shown in Figure 1. Among these, 33 (69%) were frameshift variants, six (13%) were nonsense variants, one (2%) was a splice site variant, six (13%) were missense variants, and two (4%) were in-frame deletions. Three variants (p.Val1918GlufsTer87, p.Met1284IlefsTer2, and p.Arg1112Ter), were seen in more than one affected individual. Ten of 60 individuals (17%), including three individuals in our cohort, were found to have the recurrent 4-bp deletion (c.5753_5756del) predicted to result in a frameshift-p.Val1918Glufs*87 making this variant the most prevalent pathogenic alteration seen in ZTTK syndrome. This recurrent hotspot variant maps to the Arginine/Serine (RS) -rich domain in the coding exon 3 of 12 and is predicted to undergo nonsense-mediated decay resulting in reduced mRNA levels (Kim, Shinde, et al., 2016). Interestingly, cardiac defects were more common among individuals who carried this variant compared to those who did not [5 of 8 (63%) vs. 19 of 37 (24%); Table 3], suggesting a possible genotype-phenotype correlation. Intragenic deletions have not been reported in affected individuals, except for a SON whole gene deletion, which has been reported once (Kim, Shinde, et al., 2016). Variants were de novo in all cases in which both parents were available for testing.
Table 3:
Comparison of symptoms between different types of SON variants
| Symptoms | All individuals | Recurring frameshift variant (n=10) | Missense/in-frame variants (n=6) | Loss of function variants (n=52) | ||||
|---|---|---|---|---|---|---|---|---|
| DD/ID | 58/58 | 100% | 10/10 | 100% | 6/6 | 100% | 52/52 | 100% |
| Abnormal Muscle Tone | 38/48 | 79.2% | 6/10 | 60% | 3/4 | 75% | 35/44 | 79.6% |
| Seizures | 31/50 | 58% | 5/9 | 55.6% | 2/4 | 50% | 29/46 | 63% |
| Abnormal Brain MRI | 40/48 | 81.3% | 7/10 | 70% | 0/1 | 0% | 40/47 | 86.4% |
| Eye anomalies | 33/48 | 68.8% | 7/9 | 77.8% | 2/5 | 40% | 31/43 | 72.1% |
| Cardiac Defect | 14/45 | 31.1% | 5/8 | 62.5% | 1/3 | 33.3% | 13/42 | 31% |
| GI abnormalities | 25/51 | 47.1% | 4/10 | 40% | 1/3 | 33.3% | 24/48 | 50% |
| GU abnormalities | 20/48 | 41.7% | 4/10 | 40% | 0/3 | 0% | 20/45 | 44.4% |
| Musculoskeletal abnormalities | 40/50 | 81.6% | 8/10 | 80% | 3/4 | 75% | 37/46 | 80.4% |
| Short Stature | 25/49 | 51% | 5/10 | 50% | 1/3 | 33.3% | 24/46 | 52.2% |
| Dysmorphic features | 54/57 54/56 | 94.7% | 10/10 | 100% | 5/6 | 83.3% | 49/50 | 98% |
n= number of individuals
In our cohort, the missense variant seen in Individual 9 (p.Arg1072Cys) was classified as a variant of uncertain significance based on criteria established by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (Richards et al., 2015). This individual had normal development until an encephalitic episode with intractable seizures at the age of 2 years and 9 months. It is unknown whether this episode caused his neurodevelopmental deficits, although his mother became concerned with his development after the episode. Apart from the neurodevelopmental deficits, he had no abnormalities in other systems. Parental testing has not been done for the variant and it was present as a single heterozygous allele in gnomAD, making this a weak candidate for the phenotype seen in this individual.
Three additional individuals with missense variants were identified through the DECIPHER study, two with de novo variants and one with an inherited variant. Family history information on the individual with the inherited variant was not available for review. Limited phenotype information was available for these individuals despite attempts of obtaining more information, but all three were noted to have intellectual disability. Tokita et al. (2016) reported an individual (Subject 7) who harboured two de novo missense variants in the SON gene (p.Thr1637Ser, p.Ser1843Tyr). It is unclear whether one or both variants (complex allele) contributed to the disease phenotype. This individual presented with multi-system anomalies including intellectual disability, hypotonia, feeding difficulties, gall bladder agenesis, left lung agenesis, cervical right chondroma, and multiple cardiac defects. Interestingly, gall bladder agenesis and lung agenesis have not been reported in any other individuals with pathogenic SON variants and may suggest a phenotype expansion or may be explained by an additional, unrelated genetic etiology yet to be identified. Supplementary Figure 1 shows an MTR landscape of all missense variants discussed.
A comparison of clinical features where cases (n=number of individuals) with a missense or in-frame variant (n=6) to those with loss-of-function variants (n=54) showed increased incidence of multi-system involvement including abnormal brain imaging, genitourinary anomalies, and growth deficiency in the latter group (Table 3), though larger datasets are needed for assessing genotype–phenotype correlation. Overall, currently, the clinical significance of the missense variants in SON gene is unclear. It is important to note that although there are six individuals with missense or in-frame variants, there are a total of seven variants given one individual harbours two variants. Of the seven missense or in-frame deletion SON variants, two were localized in the conserved RS domain (p.Ser1843Tyr, p.Arg1936Cys). It is speculated that variants in the DNA binding region may impact the binding of SON to other proteins and thus modulate transcriptional repression (Ueda et al., 2020).
Genetic testing prior to next generation sequencing was done for all individuals in our cohort, including karyotype, microarray and Angelman/Prader-Willi methylation testing (Table 1). Interestingly, Individuals 8 and 14 were clinically suspected to have a Rasopathy, one individual (Individual 3) suspected to have Tuberous Sclerosis, while two other individuals (Individuals 10 & 12) were highly suspected to have Coffin-Siris syndrome prior to identifying a pathogenic variant in SON. This highlights the phenotypic variability associated with ZTTK syndrome and further promotes the utility of genome wide testing (WES, WGS) for the diagnosis of rare disorders (Slezak et al., 2020; L. Yang & Yang, 2020)
Table 2 provides a summary of clinical findings of individuals in our cohort and previously reported individuals (n=60). All individuals who were evaluated had developmental delay (58 of 58, or 100%). The majority of individuals were noted to have moderate or severe intellectual disability (29 of 37, or 78%).
The majority of individuals exhibited both motor and speech delays although exact ages at which developmental milestones were attained was not available for all the cases. The earliest independent ambulation and speech was achieved at 12 months. This was in an individual in our cohort (individual 9). He exhibited regression at age 2, following an encephalitic episode resulting in the loss of speech. Given this history, it is unclear whether his deficits are related to the identified SON variant. Regression was seen in four other individuals, three in the cohort reported by Tokita et al. 2016 and two individuals in this cohort. In individuals for which this skill was documented, independent ambulation was achieved between 24 months and 9 years. Language development was documented in 24 individuals across studies. In individuals for which this skill was documented, 6 of 24 (25%) are nonverbal and 18 of 24 (75%) had speech delay. Speech was achieved between 24 months and 9 years of age among these individuals, with expressive language being more severely affected than receptive language. Of the six individuals who are nonverbal, three are currently non-ambulatory. Consistent with previous studies, individuals with ZTTK syndrome are delayed in achieving developmental milestones and the age at which they attain these milestones has an extremely wide range. We did not find any genotype-phenotype correlations between variant type and severity of neurodevelopmental deficits.
Seizures were noted for 31 of 50 individuals (62%). Onset of seizures in all individuals ranged from 6 months of age to 8 years. Among those 31 individuals with a history of seizures, five individuals (16%) were reported to have at least one status epilepticus episode. Eight of 31 (26%) reported to have at least one febrile seizure. Abnormal brain imaging is commonly noted both in our present cohort and across all individuals in the literature (both at about 80%). Hypotonia was a prominent feature of ZTTK syndrome, present in about 80% of individuals (38 of 48 individuals). Hypotonia progressed to spasticity in three individuals, two from our cohort (Individual 8 and 13) and one (Subject 3) reported in Tokita et al. (2016).
Apart from neurodevelopmental deficits, most individuals exhibit other multi-system symptoms with the most common being the musculoskeletal abnormalities. These were present in 40 of 50 (80%) individuals with a range of many different presentations. This was consistent within each individual study as well. Most often individuals present with minor anomalies of the hand and feet, hypermobility, and spine anomalies including three (8%) with hemi vertebrae, three (8%) with scoliosis, and one with exaggerated lumbar lordosis (3%) (Kim, Shinde, et al., 2016; Tokita et al., 2016).
Vision abnormalities were seen in 69% (33 of 48). Of these, more than half (19 of 33, or 58%) presented with strabismus. Other eye abnormalities were hypermetropia, cortical visual impairment, and astigmatism. Gastrointestinal symptoms were seen in 25 of 51 (47%) individuals. Feeding difficulties in the neonatal period was common, present in 18 of 25 (72%) individuals. Intestinal malrotation has been reported in three individuals: Individual 5 from the present cohort, an individual by Kim et al. (2016), and an individual reported by Slezak et al. (2020). Three individuals were noted to have duodenal malformations including atresia in two of the three individual (Kim, Shinde, et al., 2016; Slezak et al., 2020). Growth delay and short stature are present in about half (25 of 49, 51%) of all individuals. Yang et al. (2019) reported a case of a 13-year-old female individual with ZTTK syndrome who had a growth hormone provocation test revealing a peak level of 7.37 ng/ml, while Yang et al. 2020 reported an 11-year-old female individual who had a similar peak value of 8.1 ng/mL (reference value: >15 ng/mL). To our knowledge, only one individual (reported in Castanedo et al. (2020)) received growth hormone treatment due to her short stature. This individual had an adequate response to growth hormone treatment. However, additional studies are required to determine the effectiveness of growth hormone treatment in individuals with ZTTK syndrome.
Genitourinary anomalies were seen in 20 of 48 individuals (42%). Three of 18 individuals (17%) had vesicoureteral reflux, four (22%) noted a history of recurrent urinary tract infections, and three (17%) were noted to have horseshoe kidneys on ultrasound. Unilateral and/or bilateral renal dysplasia was noted in three individuals as well. Renal ultrasounds should be considered for all individuals with ZTTK syndrome. Interestingly, Kim et al. (2019) showed that SON haploinsufficiency led to aberrant pre-mRNA splicing and subsequent downregulation of genes crucial for kidney development and function, which may explain the renal phenotype seen in individuals with ZTTK syndrome.
Cardiac defects were noted in 14 of 45 individuals (31%), with ventricular and atrial septal defects being the most common type of cardiac finding reported. Additionally, aortic valve abnormalities were seen in three individuals and abnormal placement of the carotid arteries was seen in one individual. Of these fourteen individuals, five harboured the recurrent 4bp deletion.
Hearing loss was also noted in a few individuals. Two individuals in our cohort had hearing loss from a young age (Individuals 3 and 10). Individual 3 was noted to have bilateral hearing loss at age 4 while individual 10 was noted to have congenital bilateral hearing loss. In addition, three others with hearing loss have been noted in the literature (Kim, Shinde, et al., 2016; Tokita et al., 2016) although specific details on the type of hearing loss was not available for review. Hearing loss has not been previously established as a part of this syndrome, but given this data, it may be an emerging feature of this condition and an audiology referral after diagnosis may be warranted.
Conclusion
We report 15 individuals with variants in the SON gene, raising the number of potentially affected individuals with ZTTK syndrome to 60. We provide further evidence for neurodevelopmental deficits caused by haploinsufficiency of this gene. The most common features seen in ZTTK syndrome include developmental delay/intellectual disability (100%), abnormal brain imaging (83%), hypotonia (79%) and musculoskeletal abnormalities (80%). Less frequent findings include congenital heart defects (31%), gastrointestinal abnormalities (47%) and/or genitourinary abnormalities (42%). Hearing loss also may be an emerging phenotype seen in these individuals, making an audiology referral warranted for individuals with ZTTK syndrome. In addition, there might be some a genotype-phenotype correlation with a higher tendency of cardiac malformations in individuals with the recurrent frameshift variant in SON (p.Val1918Glufs*87), although further studies on a larger dataset are needed. Although we describe six individuals with missense variants and one individual with an in-frame deletion in SON in this report, further research including functional studies is needed to establish the pathogenicity of these variants.
Supplementary Material
Acknowledgements
We acknowledge the contribution of the DECIPHER Consortium. The DECIPHER study makes use of data generated by the DECIPHER community. A full list of centers who contributed to the generation of the data is available from decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. Funding for the project was provided by the Wellcome Trust. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund [grant number HICF-1009-003], a parallel funding partnership between Wellcome and the Department of Health, and the Wellcome Sanger Institute [grant number WT098051]. The views expressed in this publication are those of the author(s) and not necessarily those of Wellcome or the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network. In addition, this publication was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant Number UL1TR001873. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Conflicts of Interest
There are no conflicts of interest to declare for any of the authors listed in this publication.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable requests
References:
- Ahn EY, DeKelver RC, Lo MC, Nguyen TA, Matsuura S, Boyapati A, … Zhang DE (2011). SON controls cell-cycle progression by coordinated regulation of RNA splicing. Mol Cell, 42(2), 185–198. doi: 10.1016/j.molcel.2011.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, … MacArthur DG (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Baddoo MC, Park EY, Stone JK, Park H, Butler TW, … Ahn EY (2016). SON and Its Alternatively Spliced Isoforms Control MLL Complex-Mediated H3K4me3 and Transcription of Leukemia-Associated Genes. Mol Cell, 61(6), 859–873. doi: 10.1016/j.molcel.2016.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Park EY, Chitayat D, Stachura DL, Schaper J, Lindstrom K, … Ahn EE (2019). SON haploinsufficiency causes impaired pre-mRNA splicing of CAKUT genes and heterogeneous renal phenotypes. Kidney Int, 95(6), 1494–1504. doi: 10.1016/j.kint.2019.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Shinde DN, Reijnders MRF, Hauser NS, Belmonte RL, Wilson GR, … Ahn EYE (2016). De Novo Mutations in SON Disrupt RNA Splicing of Genes Essential for Brain Development and Metabolism, Causing an Intellectual-Disability Syndrome. Am J Hum Genet, 99(3), 711–719. doi: 10.1016/j.ajhg.2016.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana Castanedo L, Sánchez Orta A, Maseda Pedrero R, Santos Simarro F, Palomares Bralo M, Feito Rodríguez M, & de Lucas Laguna R (2020). Skin and nails abnormalities in a patient with ZTTK syndrome and a de novo mutation in SON. Pediatr Dermatol, 37(3), 517–519. doi: 10.1111/pde.14113 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … Rehm HL (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 17(5), 405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slezak R, Smigiel R, Rydzanicz M, Pollak A, Kosinska J, Stawinski P, … Ploski R (2020). Phenotypic expansion in Zhu-Tokita-Takenouchi-Kim syndrome caused by de novo variants in the SON gene. Mol Genet Genomic Med, e1432. doi: 10.1002/mgg3.1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenouchi T, Miura K, Uehara T, Mizuno S, & Kosaki K (2016). Establishing SON in 21q22.11 as a cause a new syndromic form of intellectual disability: Possible contribution to Braddock-Carey syndrome phenotype. Am J Med Genet A, 170(10), 2587–2590. doi: 10.1002/ajmg.a.37761 [DOI] [PubMed] [Google Scholar]
- Tan Y, Duan L, Yang K, Liu Q, Wang J, Dong Z, … Lin L (2020). A novel frameshift variant in SON causes Zhu-Tokita-Takenouchi-Kim Syndrome. J Clin Lab Anal, 34(8), e23326. doi: 10.1002/jcla.23326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokita MJ, Braxton AA, Shao Y, Lewis AM, Vincent M, Kury S, … Walkiewicz MA (2016). De Novo Truncating Variants in SON Cause Intellectual Disability, Congenital Malformations, and Failure to Thrive. Am J Hum Genet, 99(3), 720–727. doi: 10.1016/j.ajhg.2016.06.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda M, Matsuki T, Fukada M, Eda S, Toya A, Iio A, … Nakayama A (2020). Knockdown of Son, a mouse homologue of the ZTTK syndrome gene, causes neuronal migration defects and dendritic spine abnormalities. Mol Brain, 13(1), 80. doi: 10.1186/s13041-020-00622-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, & Yang F (2020). A de novo heterozygous variant in the SON gene is associated with Zhu-Tokita-Takenouchi-Kim syndrome. Mol Genet Genomic Med, e1496. doi: 10.1002/mgg3.1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Xu L, Yu Z, Huang H, & Yang L (2019). Clinical and genetic analysis of ZTTK syndrome caused by SON heterozygous mutation c.394C>T. Mol Genet Genomic Med, 7(11), e953. doi: 10.1002/mgg3.953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu YF, McSweeney KM, … Goldstein DB (2015). Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med, 17(10), 774–781. doi: 10.1038/gim.2014.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.