

Summary
Biofilms are community architectures adopted by bacteria inclusive of a self-formed extracellular matrix that protects resident bacteria from diverse environmental stresses, and in many species, incorporates extracellular DNA (eDNA) and DNABII proteins for structural integrity throughout biofilm development. Here we present evidence that this eDNA-based architecture relies on the rare Z-form. Z-form DNA accumulates as biofilms mature and, through stabilization by the DNABII proteins, confers structural integrity to the biofilm matrix. Indeed, substances known to drive B-DNA into Z-DNA promoted biofilm formation while those that drive Z-DNA into B-DNA disrupted extant biofilms. Importantly, we demonstrated that the universal bacterial DNABII family of proteins stabilize both bacterial- and host-eDNA in the Z-form in situ. A model is proposed that incorporates the role of Z-DNA in biofilm pathogenesis, innate immune response and immune evasion.
Keywords: Z-DNA, extracellular DNA, biofilm matrix, DNase resistance, DNABII proteins
Graphical Abstract
In Brief:
The structural integrity of bacterial biofilms are strengthened by the action of bacterial DNABII proteins which drive the accumulation of extracellular, nuclease resistant Z-form DNA and inhibit host neutrophil extracellular trap function.
Introduction
Bacterial biofilms are comprised of a community of cells either aggregated or attached to a surface, which are embedded in a self-produced extracellular polymeric substance (EPS) matrix. This EPS contains extracellular DNA (eDNA), proteins, lipids, polysaccharides, biopolymers, and divalent cations; and provides a protective barrier against harsh environments, antimicrobials, and host immune effectors (Flemming and Wingender, 2010; Koo et al., 2017). Universally, the structure of eDNA is maintained by the two-member DNABII family of proteins (Devaraj et al., 2018), integration host factor (IHF) and histone-like protein (HU), of which at least one allele is present in the genomes of all eubacteria (Dey et al., 2017). The DNABII proteins bind to and bend DNA with high affinity (Swinger and Rice, 2004) and serve as accessory proteins in multiple nucleoprotein interactions within the bacterial cell (Grove, 2011; Mangan et al., 2006). We have previously shown that these proteins also serve a role extracellularly, wherein they act as linchpin proteins to stabilize the crossed-strand structure of eDNA, that these eDNA structures functionally resemble Holliday Junctions (HJ) (Devaraj et al., 2019) within the biofilm EPS, and further, are required for the stability of the biofilm matrix (Brockson et al., 2014; Devaraj et al., 2018). Targeted removal of DNABII proteins with specific antibodies results in rapid, significant biofilm collapse (Devaraj et al., 2015; Gunn et al., 2016; Novotny et al., 2013a; Rocco et al., 2018) with release of resident bacteria that are markedly more susceptible to antibiotics and host immune effectors (Brockson et al., 2014; Gunn et al., 2016; Mokrzan et al., 2020a; Novotny et al., 2013a).
eDNA is a ubiquitous component of the biofilm EPS and is compositionally similar to fragmented intracellular genomic DNA (Steinberger and Holden, 2005). Several studies have shown that nuclease-mediated degradation of eDNA will indeed prevent biofilm formation (Frederiksen et al., 2006; Gunn et al., 2016; Martins et al., 2012; Whitchurch et al., 2002). However, these nucleases typically have no obvious effect on mature in vitro (e.g.,>24h) biofilms, even though eDNA remains a dominant matrix component (Gunn et al., 2016; Hall-Stoodley et al., 2008; Kaplan et al., 2012; Koo et al., 2017; Novotny et al., 2013a; Saunders et al., 2020). The reason for this nuclease-resistant state of mature biofilms remains uncharacterized and is the focus of our work described here.
DNA in the B-form adopts a right-handed, low energy configuration that is sensitive to nuclease degradation and is the most common DNA configuration under physiologic conditions (Bezanilla et al., 1994; Suck and Oefner, 1986). Conversely, Z-DNA has a left-handed configuration with distinct nucleotide geometry but preserves Watson-Crick base pairing, is resistant to nuclease degradation (Ramesh and Brahmachari, 1989), and is not abundant intracellularly due to its high intrinsic energy state (Dumat et al., 2016; Ho et al., 1991; Kim et al., 2018). However, Z-DNA forming sequences are involved in multiple intracellular transactions (Zavarykina et al., 2019) (Shin et al., 2016; Zhou et al., 2009), (Ray et al., 2013), (Wang et al., 2006), (Ray et al., 2013; van der Vorst et al., 2018). Additionally, Z-DNA binding proteins, while rare, are involved in gene regulation (Oh et al., 2002), viral pathogenesis [e.g., E3L] (Kim et al., 2003; Kwon and Rich, 2005), innate immune sensing [e.g. ZBP1] (Kuriakose and Kanneganti, 2018; Newton et al., 2016), DNA recognition [e.g. ADAR-1] (Kim et al., 2000), and inflammation (Szczesny et al., 2018). There is also ample experimental evidence of Z-DNA formation in the presence of high ionic strength (Ali and Ali, 1997; Peck et al., 1982), Z-DNA binding proteins (Bae et al., 2011), negative supercoiling (Nordheim et al., 1982; Nordheim and Rich, 1983; Wittig et al., 1991), as well as induction of Z-DNA via nucleotide modification (e.g. methylation and or bromination) that can reduce the high energy activation barrier (Temiz et al., 2012). Although there are Z-prone DNA sequences [e.g. alternating dGdC (Moller et al., 1982)], all sequences of DNA are capable of conversion to the Z-form (Kypr et al., 2009). Despite recent advances in Z-DNA research, no extracellular role had yet been discovered.
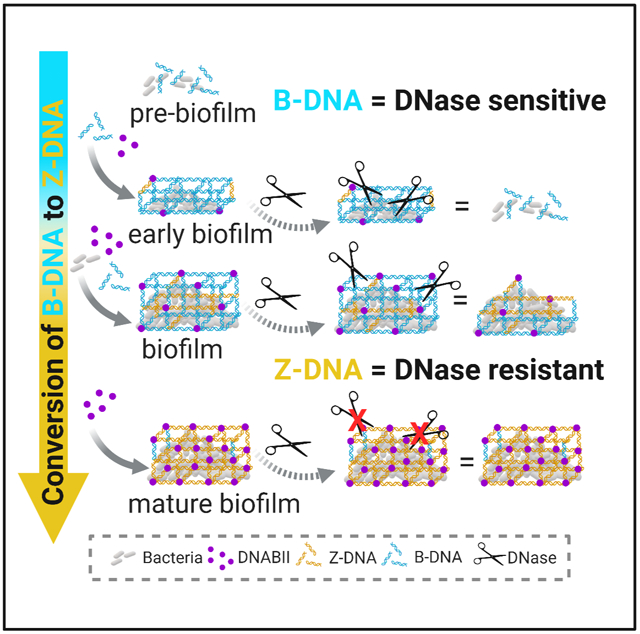
While no studies have provided evidence of non B-form DNA structures involved with biofilm stability (Kassinger and van Hoek, 2020), a recent publication does show the presence of B-form G-quadraplex structures that contribute to the structure of Pseudomonas aeruginosa biofilms (Seviour et al., 2021), thus highlighting the complex architectural eDNA structures of bacterial biofilms. Z-DNA is the most common conformation of DNA that is nuclease-resistant (Ramesh and Brahmachari, 1989; Thomas et al., 1985; Zhang et al., 2019). We thereby hypothesized that in mature bacterial biofilms, eDNA was in the Z-form. To examine this premise, we showed abundant Z-DNA within biofilms formed by multiple human pathogens in vitro, as well as within biofilm fragments present in clinical specimens. We used DNase treatment to distinguish the contribution of B-DNA versus Z-DNA to biofilm stability, and further, used compounds known to induce or revert Z-DNA to the B-form (and vice versa), to determine whether conversion of eDNA to B- or Z-forms affected biofilm architecture and/or mechanical properties. Our study provided compelling evidence of a stable presence of Z-DNA as a structural component of mature biofilms. Furthermore, Z-DNA confers the observed nuclease-resistance to these structures and thus, contributes significantly to the recalcitrance of eDNA-dependent bacterial biofilms. Importantly, we demonstrated that bacterial DNABII proteins inactivated NET-mediated bacterial killing function of polymorphonuclear leukocytes (PMNs) through conversion of host-derived B-DNA to Z-DNA.
Results
Z-DNA was present in all tested biofilms
DNase added at biofilm initiation, but not when mature, significantly inhibits biofilm formation (Frederiksen et al., 2006; Martins et al., 2012; Whitchurch et al., 2002). (Gunn et al., 2016; Hall-Stoodley et al., 2008; Kaplan et al., 2012). We recapitulated this phenomenon here with three human pathogens: Uropathogenic E. coli (UPEC), Klebsiella pneumoniae (Kp), and nontypeable Haemophilus influenzae (NTHI). Whereas Pulmozyme®, a DNase commonly used to debulk mucus and resident bacteria within the lungs of people with cystic fibrosis (CF) (Frederiksen et al., 2006) prevented biofilm formation, mature biofilms were resistant (Fig. S1A&B).
We previously showed that eDNA is both present in mature biofilms and an integral component of the EPS structure (Devaraj et al., 2015; Novotny et al., 2013a; Novotny et al., 2013b; Rocco et al., 2017). DNase readily degrades double-stranded DNA (ds-DNA), and to a lesser degree single-stranded DNA (ss-DNA) (Baranovskii et al., 2004), but not Z-DNA (Ramesh and Brahmachari, 1989). While ss-DNA is important for the initial phase of Neisseria gonorrhea biofilm formation, only ds-DNA was important for its biofilm structure (Zweig et al., 2014). We thereby hypothesized that the inability of DNase to affect mature biofilms was likely due to a nuclease-resistant DNA conformation, e.g., Z-DNA. To determine the presence of Z-DNA, we performed Immunofluorescence (IF) microscopy using monoclonal antibodies (Mabs). We first validated the specificity of these Mabs against a Z-DNA substrate [brominated poly(dGdC)] (Edgington and Stollar, 1992) (Fig. S2A) by ELISA (Fig.S2B & C), and confirmed no cross-reactivity to whole cell lysates of NTHI by Western blot (Fig. S2D)]. We then showed that NTHI genomic (gDNA) could be converted to Z-DNA in a non-sequence specific manner by bromination in the presence of 3.6 M NaCl (Fig. S2E). Lastly, we confirmed the resistance of brominated poly(dGdC) [Z-DNA] to degradation by DNase (Fig. S2F).
Via use of these highly specific Mabs directed against B-DNA (Heegaard et al., 1996) or Z-DNA (Moller et al., 1982), we first assessed whether Z-DNA was present in biofilms formed by several well-known biofilm-forming pathogens: NTHI, UPEC, Kp, P. aeruginosa, and Streptococcus mutans using IF. Strikingly, abundant levels of both B-DNA and Z-DNA were present within the EPS of the mature biofilms (40h) formed by all pathogens tested (Fig. 1A). Furthermore, we showed that while both Z-DNA and B-DNA increased within the EPS of biofilms formed over time (24, 40, 90h) by NTHI, Kp, and UPEC, after one week, eDNA in biofilms formed by NTHI was so strongly skewed to the Z-DNA form that the B-DNA signal was below the level of detection (Fig. S3A &B). Additionally, we confirmed that there are no secondary antibody cross-reactivity (e.g., nonspecific fluorescence) within NTHI biofilms (Fig. S3C). This finding demonstrates that, at least in vitro, as biofilms mature, Z-DNA continues to accumulate.
Figure 1. Z-DNA was present in biofilms formed by multiple bacterial species in vitro and in vivo.

(A) Both B and Z-DNA were present in biofilms. NTHI, UPEC, P. aeruginosa, S. mutans, and K. pneumoniae biofilms were established for 40h. Unfixed biofilms were incubated with rabbit monoclonal antibody against Z-DNA[Z22] [5 μg/ml] a murine monoclonal antibody against B-DNA[3519] [5 μg/ml], murine isotype IgG2a, and IgG purified from unimmunized rabbit serum [5μg/ml]. Then, biofilms were incubated with goat α-mouse IgG conjugated Alexa Fluor® 405 [cyan] and goat α-rabbit IgG conjugated Alexa Fluor® 488 [yellow], and counterstained with bacterial membrane stain FM™4–64 [white]. NTHI, n=3; UPEC, n=3; Pa, n=4; S.mu, n=3; Kp, n=4. (B) NTHI biofilms could be readily established on human airway epithelial cells (HAEs) NTHI [pMDC-P1, a GFP reporter isolate, (fuchsia)] biofilms were established for 16h on primary HAEs [gray], which were incubated with murine IgG1, IgG purified from unimmunized rabbit serum as negative controls. Differential interference contrast (DIC) microscopy was used to visualize HAEs. (C) B and Z DNA could be visualized on HAEs but only when NTHI biofilms were present. No fluorescent signal was detected when HAEs only (no NTHI inoculated) were labeled with B- and Z-DNA specific monoclonal antibodies (as described in A) followed by goat-α-rabbit IgG conjugated Alexa Fluor® [yellow], and goat-α-mouse IgG conjugated Alexa Fluor®594 [cyan]. (D) NTHI biofilms formed on HAEs immunolabeled for B-DNA and Z-DNA, as described above. Individual and merged fluorescent channel images shown. B-D, representative images shown, scale bars 5μM (n=6). (E) Representative images of immunohistochemical labeling of B-DNA with murine α-B-DNA[3519] and Z-DNA with rabbit αZ-DNA[Z22] in specimens collected during experimental and clinical diseases. Labeling was revealed with goat anti-rabbit IgG conjugated to Alexa Fluor® 647, goat anti-mouse IgG conjugated to Alexa Fluor® 488. Left: Sputum from an individual with cystic fibrosis (n=7). Right: NTHI biofilm formed for 14 days within the middle ear of a chinchilla with experimental otitis media (n=2). Insets, neg. control murine monoclonal antibody isotype IgG2a and IgG from unimmunized rabbit serum. Scale bar 10μm. (e.g., see also Figs. S2&S3)
Next, we examined the DNA content of biofilms formed on primary, polarized human airway epithelial cells (HAEs), as a bridge to in vivo experiments. We allowed the airway pathogen, NTHI (Mokrzan et al., 2016) to form a 16h biofilm on polarized HAEs (Fig. 1B) (Mokrzan et al., 2020b; Zhang et al., 2002), then probed for both B-DNA and Z-DNA by IF. In the absence of NTHI, no visible eDNA strands were observed (Fig 1C). In contrast, in HAEs with an apically formed NTHI biofilm eDNA strands of both B- and Z-DNA were observed (Fig. 1D). This result further confirmed that in all cases tested, Z-form DNA was present during biofilm formation.
We then determined by IF whether Z-DNA was also present in ex vivo biofilms recovered from the middle ear of a chinchilla with experimental otitis media due to NTHI, and examined sputum expectorated from a person with CF that was culture-positive for both methicillin-susceptible Staphylococcus aureus (MSSA) and Burkholderia cenocepacia. As shown in Fig. 1E, Z-DNA (yellow) was abundant within these single- and mixed-species biofilm fragments. Z-DNA was visualized as thick, fiber-like strands, which is a characteristic of Z-DNA (Chaires and Norcum, 1988) and was similar to that observed within biofilms formed on HAEs in vitro (Fig. 1D). Collectively, these data demonstrated Z-DNA as a component of the EPS of biofilms formed as part of the bacterial disease course.
Z-DNA was the primary structural form of eDNA within mature biofilms
We expanded our hypothesis to consider that in addition to DNase-resistance, Z-DNA and not B-DNA, served as an important structural form of eDNA. To test this hypothesis, we allowed NTHI, Kp, and UPEC to form mature biofilms in vitro for 24h, followed by incubation with DNase for 16h. We then quantified the relative B-DNA and Z-DNA content by IF microscopy, wherein fluorescence intensity was normalized against fluorescently labeled biofilm cells. As shown in Fig. 2A, whereas B-DNA levels were significantly reduced after incubation with DNase, Z-DNA levels were maintained. A comparison of the ratio of B-DNA and Z-DNA fluorescence in DNase-treated biofilms to their respective controls (untreated biofilms), demonstrated a notable shift towards a biofilm wherein Z-DNA was the predominant form (Fig. 2B). Additionally, NTHI biofilms were incubated for 40h in the presence of DNase throughout biofilm development and the relative B-DNA and Z-DNA content was quantified as described above. These data showed a statistically significant decrease in average thickness, a marked reduction in Z-DNA, and near abolishment of B-DNA, with a Z/B-DNA ratio that strongly favored Z-DNA (Fig. S4A–D). This finding suggests that the addition of DNase degrades B-DNA during biofilm development which lessens the production of Z-DNA. However once formed, Z-DNA was not vulnerable to DNase. These data also support the concept that Z-DNA could provide a mode of DNase resistance that was not due to protease inactivation (Saunders et al., 2020; Whitchurch et al., 2002). Collectively, these data suggested that B-DNA served as a reservoir of eDNA for Z-DNA formation, and that nuclease-resistant Z-DNA, not B-DNA, was critical to maintain the stability of bacterial biofilms.
Figure 2. Z-DNA remained intact after DNase treatment.
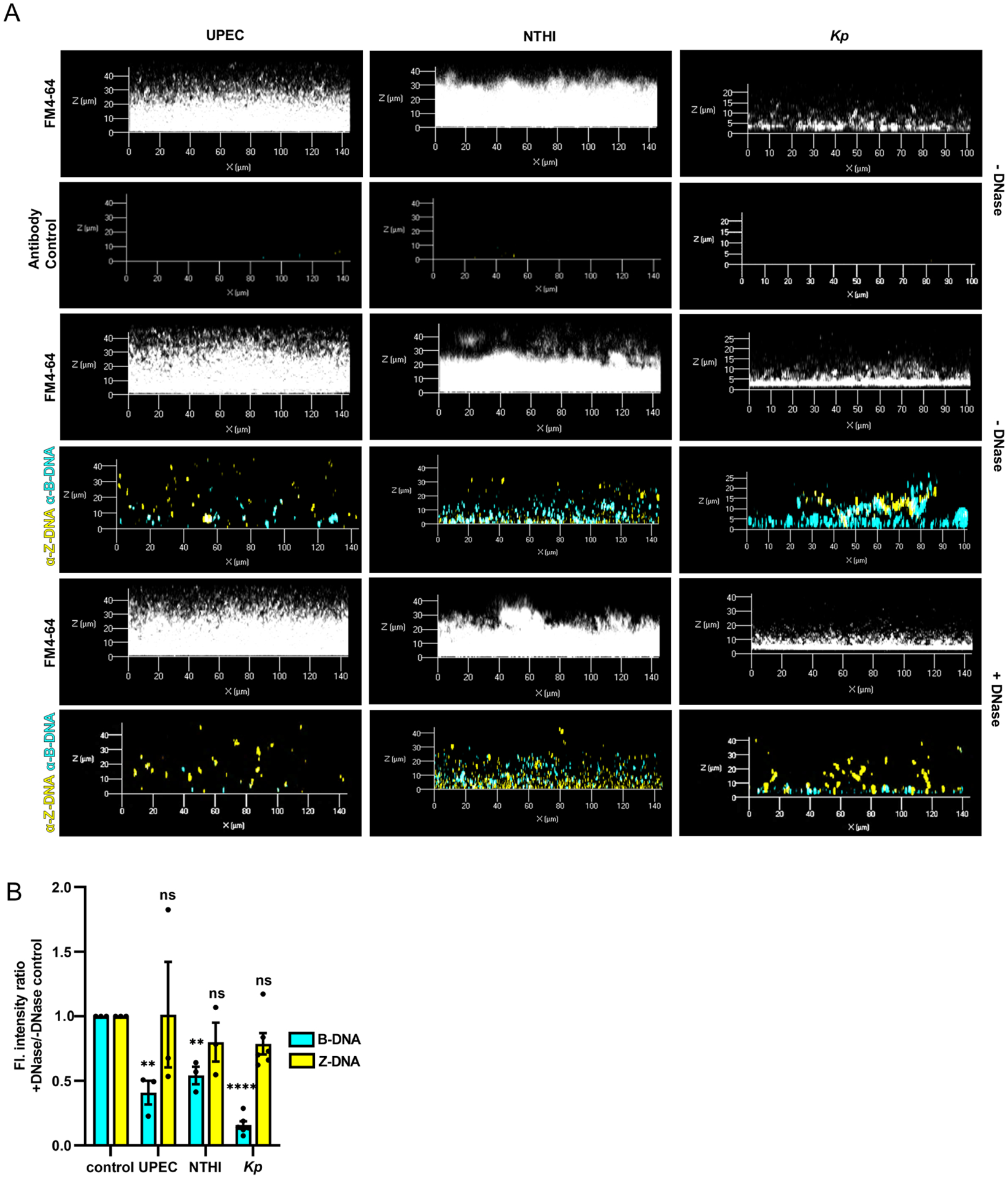
(A) UPEC, NTHI, and K. pneumoniae were allowed to form biofilms for 24h then incubated with DNase (Pulmozyme®; 40 U/ml in media) for 16h. Biofilms were then incubated with murine monoclonal raised against B-DNA[3519] [5 μg/ml] and rabbit monoclonal antibody raised against Z-DNA[Z22] [5 μg/ml] or IgG2a and IgG purified from unimmunized rabbit serum, then counterstained with FM™4–64. (B) Changes in abundance of B-DNA or Z-DNA after DNase treatment were quantified as the ratio of fluorescence intensity (F.I.) of B-DNA or Z-DNA compared to untreated biofilms (- DNase) using ImageJ software. F.I. was normalized to total biomass (FM™4–64 fluorescent signal). Error bars represent the SEM. UPEC, n=3; NTHI, n=3; Kp, n=6. Statistical significance compared to control (- DNase) was assessed by paired t-tests, **P<0.01; ****P<0.0001. (e.g., see also Figs. S1&S4).
Z-DNA-specific antibodies stimulated biofilm formation
Z-DNA-binding proteins (Bae et al., 2011; Kim et al., 2018), which include Z-DNA-specific antibodies, bind to and/or stabilize Z-form DNA structure (Lafer et al., 1985; Moinuddin et al., 1998) and thus also shift the B/Z DNA equilibrium towards the Z-DNA conformation (Lafer et al., 1986; Lee et al., 2018). We thereby hypothesized that the addition of Z-DNA-specific antibodies would stabilize and shift the equilibrium of eDNA to favor the Z-conformation and facilitate biofilm formation. To this end, we added purified IgG isolated from Z-DNA-specific polyclonal (Safina et al., 2017) antibodies (pAB), as well as a specific Z-DNA Mab (Thomas et al., 1991) to cultures of NTHI and/or UPEC at the time of biofilm initiation. As seen in Fig. S5A and 5B, addition of either polyclonal or monoclonal Z-DNA-specific antibodies induced a significant, dose-dependent increase in biofilm average thickness.
A shift in B/Z equilibrium of eDNA modulated biofilm structure
The equilibrium of B-DNA to Z-DNA conversion is also facilitated in the presence of multi-valent cations or high ionic strength (Pohl and Jovin, 1972), whereas the binding of intercalating agents tend to favor the B-DNA conformation (Mirau and Kearns, 1983). We used these principles to attempt to drive eDNA into either the B- or Z-forms to determine if either state was preferentially associated with biofilm formation. To drive B-DNA into Z-DNA, we utilized the ability of cerium (III) chloride (CeCl3) to form self-assembled Z-DNA aggregate structures at low concentrations (<1 mM). CeCl3 binds to the phosphate backbone of DNA, which thus neutralizes the electrostatic repulsion, allowing the Z-DNA form to be energetically favored (Bhanjadeo et al., 2017). We first confirmed that CeCl3 induced conversion of gDNA to Z-form (Bhanjadeo et al., 2017) in a dose-dependent manner by spectroscopic A260/295 absorbance ratio assay (Thomas and Messner, 1988) (Fig. S5C). To now determine whether CeCl3 altered the Z-DNA content of biofilms, mature NTHI biofilms were incubated with increasing concentrations of CeCl3, then probed for the presence of Z-DNA by IF microscopy. As shown in Fig. 3A&B, CeCl3 induced an increase in Z-DNA signal and a dose-dependent increase in overall fluorescent intensity of bacterial biomass (Fig. 3C). We confirmed that CeCl3 did not negatively affect NTHI planktonic growth (Fig.S5D) but resulted in a greater proportion of bacteria that favored the biofilm state (Fig S5E). These results suggested that an equilibrium shift from B-DNA to Z-DNA enhanced biofilm formation by promoting a biofilm dominant state over planktonic growth.
Figure 3. B/Z eDNA ratio modulates the physical properties of biofilms.

NTHI biofilms formed for 24h were incubated with CeCl3 (500μM) for 16h, then incubated with unimmunized rabbit IgG (5 μg/ml) or rabbit α-Z-DNA[Z22] [5 μg/ml], then with goat α-rabbit IgG conjugated with Alexa Fluor® 488 and imaged by CLSM. (A) Representative image of an NTHI biofilm with 500μM CeCl3. (B&C) ImageJ quantifications of Z-DNA (Alexa Fluor® 488) and bacterial cells (FM™4–64) signal intensities. Statistical significance compared to control was assessed by paired t-tests, *P<0.05, **P<0.01, n=7. To determine the mechanical properties of biofilms, NTHI biofilms were formed for 24h on glass fluorodishes, then incubated with media or 500μM CeCl3 for 16h at 37°C. Additionally, 24h biofilms incubated with media or CeCl3 for 16h, then treated with Pulmozyme® (50μg/ml) for 1h.(D) Stress-strain curves of NTHI biofilms were determined from axial mechanical indentation analysis. (E) Young’s Modulus was determined from the lower linear region of the stress-strain curve depicted in (D). Statistical significance determined by One-way ANOVA. * p <0.05, ns; not significant compared to media only control. NTHI biofilms formed for 24h were incubated with chloroquine (5μM) for 16h then incubated with unimmunized rabbit IgG (5 μg/ml) or rabbit α-Z-DNA[Z22] (5 μg/ml) and then with goat α-rabbit IgG conjugated with Alexa Fluor® 488 and imaged by CLSM. (F) Representative image of an NTHI biofilm with 5μM chloroquine. (G&H) ImageJ quantifications of Z-DNA (Alexa Fluor® 488) and bacterial cells (FM™4–64) in the presence of chloroquine. Statistical significance compared to control was assessed by paired t-tests, *P<0.05, **P<0.01, n=3. Mechanical properties of 40h NTHI biofilms formed on glass fluorodishes and incubated with media or chloroquine (5μM) for 1h at 37°C. (I) Stress-strain curves of NTHI biofilms treated with chloroquine were determined as described previously (D). (J) Young’s Modulus was determined as described in (E), from stress-strain curve in (G). Statistical significance determined by One-way ANOVA or unpaired t-test * p <0.05, ns; not significant compared to media only control, n=3 [2 biofilms for each replicate was analyzed for a total of 6 biofilms per group] (K, L & M) Modulation of B/Z eDNA increased the susceptibility of biofilms to DNase after treatment with chloroquine. (K) NTHI biofilms were formed for 40h and incubated with chloroquine, DNase, or both, then stained with LIVE/DEAD®, fixed, visualized via CLSM, and images analyzed by COMSTAT. Average thickness as a percent of media control, n=7. (L) Images of the sputum solids disruption assay (n=1) where clinical sputum exudates were incubated with DPBS (control), chloroquine (100μM), DNase (30U), or in combination for 1h at 37°C, then analyzed for change in OD600 over time, n=4. (M) Fold change in OD600 relative to t=0. Higher OD600 relative to t=0 indicates biofilm disruption. Scale bar 10μm. Error bars represent the SEM. Statistical significance compared to control was assessed by unpaired t-tests, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, n=4. (e.g., see also Figs. S5, S6 & Table S1).
We then used rheological analysis to determine if DNase (removal of B-DNA) and/or CeCl3 (induction of Z-DNA) altered the bulk mechanical properties of a biofilm, using NTHI as a model. We performed axial mechanical indentation on untreated (media) biofilms and those incubated with DNase, CeCl3, or a sequential addition of both (e.g, induction of Z-DNA followed by removal of B-DNA) wherein an 8mm geometry was lowered onto the biofilm, using an approach rate of 1μm/s, and the force required to compress the biofilm was determined (Devaraj et al., 2019). From the stress-strain curves, there was a significant difference in the mechanical properties between all three treatments compared to untreated biofilms (Fig. 3D). The Young’s Modulus, an indication of how stiff a material is in response to a normal force (Timoshenko and Goodier, 1970), was determined at the lower linear portion of the force-displacement curve, per Eq. 1 (STAR Methods, in section “axial mechanical indentations of NTHI biofilms”) as demonstrated in Fig. S5F. In support of our observations from the stress-strain curves, there was a significant statistical difference between the Young’s Modulus and all three conditions compared to control biofilms. (Fig. 3E). These data indicated that DNase, CeCl3, CeCl3+DNase resulted in an increased Young’s Modulus which is indicative of a more rigid biofilm. This outcome suggested that reduction of the proportion of B-DNA (e.g., via incubation with Pulmozyme®), without affecting the steady state levels of Z-DNA, increased the stiffness and thus, the stability of the biofilm EPS. Additionally, incubation of DNase after conversion to Z-DNA (e.g., CeCl3 treated) demonstrated no change in rigidity, which suggested that the mode of action of CeCl3 treatment on biofilms was directed toward eDNA. Taken together, we concluded that the stiffness of the biofilm could be attributed to eDNA being solely in the Z-form.
To drive Z-DNA into B-DNA, we utilized the DNA-intercalating agent chloroquine, which prevents the formation of Z-DNA (Kwakye-Berko and Meshnick, 1990). We hypothesized that chloroquine, an immunosuppressant drug used for the prevention and treatment of malaria, as well as for disorders such as Systemic Lupus erythematosus[SLE](Al-Bari, 2015), would shift the B-Z equilibrium of mature biofilms to primarily the B-form, and thus reduce biofilm structural integrity. We first confirmed that chloroquine prevented NaCl-induced Z-DNA conversion by spectroscopic absorbance ratio assay (Fig. S5G) and further, showed there was no effect on NTHI growth (Fig. S5H). We allowed NTHI to establish a biofilm (24h) followed by incubation with increased concentrations of chloroquine (at concentrations that had no effect on NTHI growth), then probed for the presence of Z-DNA by IF. Chloroquine caused a significant reduction in overall Z-DNA signal and a dose-dependent decrease of NTHI in the biofilm state, which suggested that Z-DNA was critical for biofilm stabilization (Fig. 3F–H). Moreover, we observed that chloroquine partitioned biofilm-resident bacteria toward the planktonic state (Fig. S5I), which suggested that the DNA-intercalating effects of chloroquine drove Z-DNA into B-DNA, thus likely disrupting the biofilm EPS which lead to the release of bacteria from biofilm residence, without cell killing. We also performed rheological analysis to determine how the conversion of Z-DNA to B-DNA affected biofilm mechanics. Axial mechanical indentation was performed on untreated NTHI biofilms (media) or those incubated with chloroquine, as described above. Stress-strain analysis indicated that the mechanics of chloroquine-incubated biofilms were altered, relative to the control biofilm (Fig. 3I). The derived Young’s Modulus of chloroquine-treated NTHI biofilms was significantly lower, compared to control biofilms (Fig. 3J), which indicated that conversion of Z-DNA to B-DNA, destabilized the mechanical integrity of the biofilm. In contrast, NTHI biofilms incubated with DNase had a significantly higher Young’s Modulus, compared to control biofilms (Fig. 3E). This latter result suggested that reduction of the proportion of B-DNA (e.g., via incubation with Pulmozyme®), without affecting the steady state levels of Z-DNA, increased the stiffness of the biofilm EPS.
Thus, according to our hypothesis, as Z-form DNA returns to B-form in the presence of chloroquine, this transformation should restore DNase susceptibility to the biofilm. To this end, NTHI biofilms (40h), were incubated with chloroquine and/or DNase for 1h. As shown in Figs. 3K & S6A, there was a significant decrease in biofilm average thickness, which indicated that an equilibrium shift towards a B-DNA-dependent EPS transitioned the biofilm from a DNase-resistant to a DNase-sensitive state (Fig. 3K). To determine whether this approach could be applied to clinical specimens, we obtained expectorated sputum samples from individuals with CF, which contain both a lattice of e-DNA and DNABII proteins (Novotny et al., 2013a). Sputa were culture-positive for P. aeruginosa (mucoid and non-mucoid) and S. aureus, and all were resistant to specific antibiotics (Table S1). We employed a previously established sputum solids disruption assay (Gustave et al., 2013) to determine whether conversion of Z- to B-DNA increased susceptibility of CF sputum to DNase and/or chloroquine. To test this, we incubated sputum for 1h with buffer, chloroquine (100μM), DNase (30U) or a combination of both, followed by optical density measurements to quantify the extent of disruption (Gustave et al., 2013). A representative image of the sputum solids disruption assay (Fig. 3L) and the fold change of optical density (Fig. 3M) demonstrated complete disruption of CF sputum solids when treated with both chloroquine and DNase. IF microscopy demonstrated that each clinical specimen contained both B- and Z-DNA (Fig. S6B), organized within a distinct eDNA-rich lattice. Collectively, these data suggested that stable Z-DNA is the mode of DNase-resistance in the eDNA-dependent biofilm.
Resolution of extracellular HJs prevents Z-DNA formation in biofilms
We previously demonstrated, via the use of Holliday Junction (HJ)-specific resolvases (RuvABC and RusA), that HJs are critical for the mechanical and structural stability of the eDNA lattice of bacterial biofilms (Devaraj et al., 2019). Here, we asked if the presence of HJs was also critical for Z-DNA stability. To test this, we allowed NTHI biofilms to form in the presence of RusA (10μg/ml) for 16h, then quantified the steady state levels of both B-DNA and Z-DNA, as described above. The resolution of HJs by RusA markedly reduced the overall fluorescent intensity of bacterial biomass of the biofilm, with a concomitant reduction in the steady state level of B-DNA, but nearly eliminated Z-DNA formation (Fig. 4A–D). These data suggested that maintenance of the HJ-like structures was critical for Z-DNA stability.
Figure 4. Resolution of HJs prevent, whereas DNABII proteins facilitate Z-DNA content within the biofilm.

NTHI biofilms were initiated in the presence or absence of RusA (10 μg/ml) for 16h at 37°C, then washed and incubated with rabbit monoclonal antibody against Z-DNA[Z22] [5 μg/ml] and a murine monoclonal antibody against B-DNA α-B-DNA[3519] [5 μg/ml], murine isotype IgG2a (Invitrogen, 02-6200), or IgG purified from unimmunized rabbit [5μg/ml], then with goat α-rabbit IgG conjugated Alexa Fluor® 405 (yellow) and goat α-mouse IgG conjugated Alexa Fluor® 488 (cyan). (A) Representative image of RusA prevention of Z-DNA. Signal intensities were quantified by ImageJ: (B) biomass (FM™4–64 fluorescent signal), (C) B-DNA (Alexa Fluor® 594) or (D) Z-DNA intensity (Alexa Fluor® 488), n=3. (E) Representative image of NTHI biofilms that were initiated and maintained in the presence or absence of exogenous HUNTHI (2 μg/ml) for 40h at 37°C. Biofilms were then washed and incubated with rabbit monoclonal antibody against Z-DNA[Z22] [5μg/ml] or IgG purified from unimmunized rabbit [5μg/ml], then with goat α-rabbit IgG conjugated Alexa Fluor® 488 (yellow). (F) Z-DNA intensity (Alexa Fluor® 488) and (G) changes in biomass (FM™4–64) were quantified by ImageJ, n=3. To determine the mechanical properties of biofilms, NTHI biofilms were formed on fluorodishes and incubated with RusA or HUNTHI as described above. Control experiments were performed, where HUNTHI was added as described above, but was treated with Pulmozyme® (50μg/ml) or RusA (10μg/ml) + Pulmozyme® (50μg/ml) for 1h. Additionally, NTHI biofilms were grown for 24h, incubated with CeCl3 (500μM) for 16h, then treated with RusA (10μg/ml) for 1h at 37°C (H) Stress-strain curves were determined from axial mechanical indentation analysis. (I) Young’s Modulus was determined from the lower linear region of the stress-strain curve(H), n=3 [2 biofilms for each replicate were analyzed for a total of 6 biofilms per group]. Statistical significance determined by One-way ANOVA and Dunnett’s multiple comparison test. * p <0.05, a p-value ≤ 0.01 is indicated by**, a p-value ≤ 0.001 is indicated by ***, and a p-value <0.0001 is indicated by ****, ns; not significant compared to control. Error bars represent the SEM.
Exogenous DNABII proteins facilitate Z-DNA within biofilms
We reasoned that facilitation of biofilm development would have a specific kinetic threshold to which exogenous addition of HU would enhance Z-DNA accumulation. To test this, NTHI was incubated (and maintained) in the presence of HUNTHI (100nM) throughout biofilm development for 40h. We performed IF for Z-DNA and found that an increased accumulation of Z-DNA was evident compared to the untreated control (Fig. 4E–G), which indicated that for NTHI, DNABII proteins are not limiting for biomass but continue to be required to modulate the equilibrium of the B-to-Z transition. Despite the fact that both CeCl3 and DNABII require B-DNA as a reservoir to create stable Z-DNA, these agents likely achieve this outcome via different mechanisms.
Z-DNA contributes to biofilm mechanical properties via HJs.
To test whether HJ constraint stabilized Z-DNA in the EPS of bacterial biofilms, axial mechanical indentation was performed on control NTHI biofilms or those incubated with either a DNABII protein (HUNTHI) or RusA, as described above. In addition, we added DNase for 1h to biofilms incubated with HUNTHI (throughout development), RusA (16h), or a combination of both. Stress-strain analysis demonstrated that treatment with either protein altered the mechanical properties of the biofilm, compared to the control (Fig. 4H). The Young’s Modulus for HUNTHI-incubated biofilms was significantly greater than controls (Fig. 4I). This indicated that increased Z-DNA in the biofilm, by interactions with a DNABII protein, led to significantly stiffer biofilms, similar to what was observed for CeCl3 incubated biofilms (Fig. 3E). Conversely, the Young’s Modulus of RusA-incubated biofilms was significantly less than controls (Fig. 4I), which indicated that decreased Z-DNA destabilizes biofilm mechanical properties, similar to chloroquine treatment (Fig. 3J). DNase did not alter the Young’s Modulus of either HU- or RusA- treated biofilms nor did HU alter the effect of RusA-treated biofilms. These results suggested that DNase has no additive or reductive effect on biofilm architecture once Z-DNA is either induced (via HJ stabilization by DNABII proteins) or resolved (via RusA). We demonstrated previously that HJs pre-bound with DNABII proteins are cleaved by RusA (Devaraj et al., 2019), thereby the failure of exogenous HU to mitigate RusA activity is expected. To provide further evidence that conversion of B to Z-DNA by CeCl3 was dependent on eDNA, we incubated NTHI biofilms with CeCl3 for 16h, followed by incubation with RusA for 1h. As shown, the Young’s Modulus was similar to RusA alone, but reduced as compared to CeCl3 alone (Fig. 3E) i.e., the increased rigidity of CeCl3-converted biofilms was completely abrogated by RusA (Fig. 4I), which suggested that the increased rigidity by CeCl3 was reliant on eDNA, and not due to other factors within the biofilm. Together, these results confirmed that extracellular HJs, mediated by DNABII proteins, contributed to the mechanical rigidity and structure of the biofilm via stabilization of Z-DNA.
DNABII proteins drive B-DNA dominant NETs to Z-DNA during PMA-induced NETosis
eDNA is ubiquitous in nature and a crucial structural component of multi-cellular communities (Ibanez de Aldecoa et al., 2017). While eubacteria release DNA in vitro (Ibanez de Aldecoa et al., 2017) (Jurcisek et al., 2017), biofilms within a host likely incorporate both bacterial and host-derived DNA (Alhede et al., 2020). Indeed, immune cells such as neutrophils release eDNA and associated antimicrobial compounds (e.g., histones) by NETosis (Dubois et al., 2012; Dwyer et al., 2014; Papayannopoulos et al., 2011) to control pathogens. This release of eukaryotic eDNA provides biofilm-resident bacteria an opportunity to incorporate both eukaryotic and bacterial-derived eDNA into their EPS. In this regard, there is a continuum of bacterial- and host-derived eDNA present at the site of infection (Alhede et al., 2020). While it is expected that the interface between bacterial and host eDNA would vary in nucleoprotein complexes, we wondered whether DNABII proteins could, in and of themselves, also drive eukaryotic eDNA from its native B-form into the Z-form. First, we confirmed that neutrophils induced by phorbol-myristate-acetate (PMA) produced neutrophil extracellular traps (NETs) (Fig. 5A, Fig. S7A&B) and determined that the primary form of eDNA within PMA-induced NETs is B-DNA (Fig. 5B & Fig. S7B). To recapitulate exposure to bacterial biofilms and their associated DNABII proteins, we added HUNTHI to neutrophils at the time of PMA-induction. As demonstrated in Fig. 5B, HU not only condensed the PMA-induced NETs, but converted a proportion of eDNA from B-form to Z-DNA. Conversely, CbpA, another minor groove DNA-binding protein (Chintakayala et al., 2015) also released by NTHI (Devaraj et al., 2018), failed to alter the appearance or proportion of B-form of the DNA (Fig. S7B). Inactivation of NETs by DNase is well characterized and contributes to a loss of bacterial killing activity (Brinkmann et al., 2004; Mohanty et al., 2019), but whether a bacterial protein could mitigate bacterial killing is unknown. To test this hypothesis, we incubated neutrophils with DNase, HUNTHI, CbpA, or buffer for 4h with NTHI (16h after seeding where the bacteria are in equilibrium between residence in the young biofilm and the planktonic state due to natural biofilm remodeling). Triton-X-100 was added to lyse neutrophils to enable recovery of viable intracellular bacteria and total CFU NTHI were enumerated for each condition to measure the relative percent bacterial killing within the system attributable to PMN NETosis (Robledo-Avila et al., 2020). As shown in Fig. 5C, addition of either DNase or HU resulted in a marked reduction of measurable bacterial killing by NETs as compared to media control or CbpA. These collective results suggested that the proximity of DNABII proteins to host-derived eDNA not only could affect the form of eDNA, but also the ability of host cells to kill bacterial pathogens and/or control bacterial biofilm proliferation. We showed recently in a biofilm recovered from the middle ear of a chinchilla 11 days after challenge with NTHI (Devaraj et al., 2021) that host proteins (e.g., HMGB1) are localized near PMNs and PMN-derived eDNA, while DNABII proteins are primarily localized near the bacterial biofilm. However, at this host-pathogen interface, both proteins were visualized by IF. The ability of HMGB1 to disrupt bacterial biofilms (Devaraj et al., 2021), and the inactivation of NET-mediated bacterial killing function by DNABII proteins suggest a delicate balance at this interface. To now examine these regions within the same cryosections (Devaraj et al. 2021) for spatial distribution of B and Z-DNA, we observed a dense and extensive PMN-rich region located on and above the bacterial biofilm that is adhered to the middle ear mucosa (Fig. 6A&B). The PMN-dense region contained both B-form and Z-form DNA within the eDNA lattice, however the adherent NTHI biofilm was predominantly Z-DNA rich (Fig. 6C–E). Higher magnification insets of Panel E (Fig. 6) provide further characterization and additional evidence of the localization of B-form and Z-form DNA within NETs. The uppermost portion above the biofilm within the middle ear lumen (e.g., recently migrated PMNs), contained predominantly B-DNA (Inset F). The top third “newer” region of the PMN-rich area (Inset G) contained both forms of DNA, while the bottom third “older” PMN region closest to the bacterial biofilm (Inset H) contained primarily Z-DNA. Z-DNA was the predominant form of eDNA within the bacterial biofilm adherent to the middle ear mucosa (Inset I). Thus, as bacteria-released DNABII proteins accumulate not only within bacterial eDNA, but also appeared to have migrated to NET-derived eDNA where we hypothesize that conversion of NETs eDNA to Z-DNA could mitigate NET functions (e.g. bacterial killing), either through changes in NET structure and/or their capacity to maintain a high concentration gradient of antimicrobial molecules [e.g. histones which fail to bind Z-DNA (Burton et al., 1978; Fishel et al., 1991; Nickol et al., 1982)].
Figure 5. DNABII proteins inactivated NET function through the conversion of B-form NET eDNA to Z-form DNA.
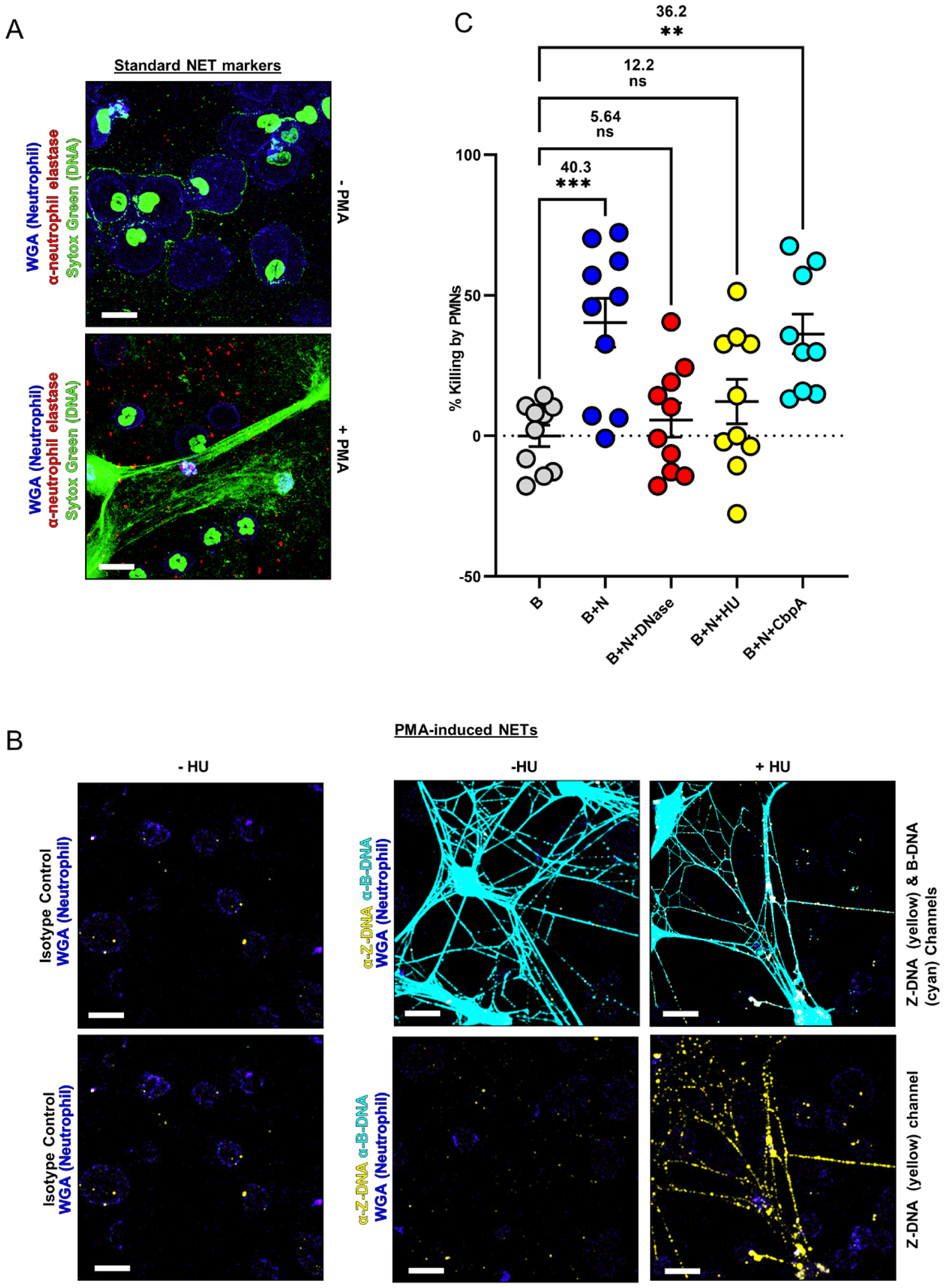
Neutrophils (1×105) were allowed to attach to an 8 well glass chamberslide and induced to NET with 200nM PMA (phorbol-12-myristate-13-acetate) in the presence or absence of HUNTHI (500nM) for 3.5h at 37°C. NETs were then fixed, washed, blocked with 10% goat serum followed by incubation with rabbit polyclonal antibody against human neutrophil elastase (1:100), rabbit monoclonal antibody against Z-DNA[Z22] (5μg/ml), and a murine monoclonal antibody against B-DNA (5μg/ml). Then NETs were incubated with goat α-rabbit IgG conjugated Alexa Fluor® 594 and goat α-mouse IgG conjugated Alexa Fluor® 488, wheat germ agglutinin (WGA 350), and SYTOX™ nucleic acid stain. NETs were imaged by CLSM. (A) Representative images of NETs stained with the traditional NET markers, SYTOX™ (nucleic acid stain, green), neutrophil elastase (active NETs, red), and WGA (membrane stain, blue), n=3. (B) Representative images of NETs formed in the presence or absence of HUNTHI demonstrated an increased Z-DNA signal only when exogenous DNABII proteins were added. Note: Merged Z-DNA (yellow) and B-DNA (cyan) co-localize and appear white, n=3. (C) NETs killing is inactivated by DNABII proteins. NTHI biofilms were challenged with human neutrophils (B+N) and treated with 10 units/ml of Pulmozyme® (B+N+Pulmo), 1μM HU (B+N+HU) or 1 μM CbpA (B+N+CbpA) for 4h, NTHI biofilm without neutrophils was used as a control (B). The bacteria challenged with neutrophils (B+N) [mean= 40.3%] and the group treated with CbpA (B+N+CbpA) [mean=36.2%] showed a reduction in the relative % killing as compared to the biofilm control group (B). The bacteria treated with Pulmozyme® (B+N+Pulmo) [mean= 5.64%] or with HU (B+N+HU) [mean= 12.2%] did not show differences in the relative % killing of bacteria by PMNs. The results suggest that Pulmozyme® or HU treatment inactivated the NET function of bacterial killing. The graph plot replicates were derived from 4 healthy donors ± SEM. The statistical analysis was performed with One-way ANOVA and Dunett’s multiple comparison test. (**=p<0.01, ***=p<0.001). (e.g., see also Fig. S7)
Figure 6. Tiled CSLM images of an immunolabeled biofilm recovered from the middle ear of a chinchilla 11 days after challenge with NTHI.

[lu – lumen of the middle ear; PMNs – dense extensive PMN-rich region above the bacterial biofilm; b – NTHI biofilm adherent to the middle ear mucosa; e – epithelium that lines the middle ear space]. The cryosection was labeled with: (A) isotype control rabbit serum and isotype control mouse serum + Alexa Fluor® 594-conjugated goat α-rabbit serum and Alexa Fluor® 488-conjugated goat α-mouse serum respectively; (B) DAPI to identify ds-DNA; (C) rabbit α-Z-DNA + Alexa Fluor® 594-conjugated goat α-rabbit serum (pseudocolored yellow); (D) murine anti-B-DNA + Alexa Fluor® 488-conjugated goat α-mouse serum (pseudocolored cyan); (E) composite image of overlayed panels B-D. Insets F-I are higher magnification 3D reconstructions of z-stack images taken from the portions of the biomass as indicated by the boxed regions in Panel E. Inset F: the uppermost portion of the biofilm closest to the lumen of the middle ear where the most recently migrated PMNs are located; Inset G: top third of the PMN-rich region of this cryosection; Inset H: bottom third of the PMN-rich region; Inset I: bacterial biofilm. Note that ds-DNA is present throughout this cryosection as evidenced by DAPI labeling in Panel B, however it is only in B-form in the PMN-rich region and not within the bacterial biofilm, as evidence by Panel D. Conversely, DNA in the bacterial biofilm is in Z-form as evidenced by labeling visible in Panel C but absent in Panel D. Labeling within insets provide additional evidence for the B-form DNA that predominates in the NETs formed by the most recently migrated PMNs (F); strands of DNA labeled for both B- and Z-form DNA in PMN-rich regions that are ‘older’ (e.g. within the middle region of the cryosection) (G); exclusively Z-form DNA labeling of NETs formed by PMNs closest to the bacterial biofilm (H); and exclusively Z-form of the eDNA within the bacterial biofilm that is adherent to the middle ear mucosa (I). Scale bar in panels A-E = 200 μm; in panels F-I = 5 μm. [Note: to best resolve deep blue DAPI labeling against the black background in panels A & B, the brightness of these two panels only was increased].
Discussion
Since its discovery almost 50 years ago, Z-DNA has been exclusively characterized for its intracellular functions. In our present study, we revealed that extracellular Z-DNA provided a critical structural contribution and DNase-resistance to the eDNA-dependent EPS of bacterial biofilms.
Z-DNA provides multiple advantages over B-DNA as a biofilm matrix material. First, Z-DNA has a 3-fold greater persistence length compared to B-DNA, and thereby can more effectively contribute to the structural integrity of the EPS. Second, we have argued that an underlying EPS for all bacteria that enter a multispecies biofilm would have great value to foster inclusion of diverse bacterial genera at large but exclude elements of potentially hazardous environments (Goodman et al., 2011). Since all eubacteria express DNA and DNABII proteins, all eubacteria produce the materials requisite for an inclusive EPS. The fact that it appears that a biofilm structure inclusive of Z-DNA is also conserved, provides additional evidence in support of this model. Third, an underlying Z-DNA-dependent EPS is highly resistant to DNA nucleases. It is well known that various pathogens secrete nucleases to release themselves from the B form-eDNA tendrils released by NETs, likely a critical step in pathogen dispersal and propagation from the biofilm reservoir (Storisteanu et al., 2017; Tran et al., 2016). With the eDNA of bacterial biofilms in the Z-form, and the NETs eDNA initially in the B-form, secreted nucleases would allow bacteria to leave biofilm residence without the danger of NET-mediated killing, as evidenced by the likelihood of DNABII-mediated inactivation. Finally, nearly all DNA-binding proteins bind to B-form DNA but not to Z-DNA. Indeed, the potent antimicrobial activity of NET-released histone is reliant upon histones being in the bound state which effectively concentrates their antimicrobial activity within this B-form eDNA NET (Brinkmann et al., 2004). Given that DNABII proteins are likely the rate limiting effector to drive NET eDNA into the Z-form, it has not escaped our attention that any eDNA (e.g., that deployed by NETs) within the effective diffusion radius of the bacterial biofilm EPS could be subject to conversion to the Z-form. When active on NET-deployed eDNA, this would induce histones into an unbound state, and thus a loss of their concentration gradient and effectiveness against targeted biofilms. The effective failure of localized histone-mediated antimicrobial activity would be a strong immune evasion mechanism for biofilm-resident bacteria. Moreover, histones are cytotoxic (Singh et al., 2010), and thus histones released from NETs by conversion to Z-form have the potential to inadvertently cause collateral damage to host tissues.
How does Z-DNA form within the biofilm EPS? B- and Z-form DNA [regardless of sequence (Kypr et al., 2009)] are in equilibrium (Guéron et al., 2000; Lee et al., 2018; Zacharias et al., 1988), with the B-form dominating under physiologic conditions. While the equilibrium shift from B to Z can readily be induced (Ali and Ali, 1997; Bhanjadeo et al., 2017; Thomas and Messner, 1988; Zhang et al., 2019), these conditions do not typically occur in native systems due to the high intrinsic energy requirement of Z-DNA (Dumat et al., 2016; Ho et al., 1991; Kim et al., 2018). Our results show a system with, and dependent on, development of a stable Z-DNA state that increases in magnitude with biofilm maturity. Indeed CeCl3, a known catalyst of Z-DNA formation, increased both the magnitude of the biomass and the Z-DNA content of the biofilm. In addition, DNase treatment of mature biofilms which reduces the proportion of B-DNA but does not affect the extant steady state levels of Z-DNA, also stiffened biofilm structure. This outcome indicated that the presence of some B-DNA makes biofilms less stiff and hence more flexible, a property that may be important during the life cycle of the biofilm. In contrast, chloroquine, a known intercalator of B-DNA that drives Z-DNA back into the B-DNA state, reduced both the magnitude of the biomass and Z-DNA content of the biofilm. Interestingly, addition of DNABII proteins has an effect that is similar, but not identical, to CeCl3 addition. According to our model, under native conditions the equilibrium state of DNA is predominantly in the B-form. The presence of endogenous extracellular DNABII proteins constrains more of the eDNA in the Z-form, which stabilizes the eDNA-dependent EPS (Top panel: Fig. 7A–C). When CeCl3 is added, the equilibrium shift toward production of Z-DNA is increased, which drives a greater proportion of eDNA into the Z-form (Top panel: Fig. 7D). In contrast, when chloroquine is added the equilibrium shift which drives DNA into the B-form is increased, which favors disassembly of the eDNA-dependent EPS (Top panel: Fig. 7E). When exogenous DNABII is added, the equilibrium shift favors more eDNA in the Z-form (Top panel: Fig. 7F). While DNABII proteins bind various DNA substrates (Bonnefoy and Rouviere-Yaniv, 1991; Kamashev, 2000; Kamashev and Rouviere-Yaniv, 2000; Rice et al., 1996; Swinger and Rice, 2004; Swinger and Rice, 2007), there is no direct evidence that DNABII proteins bind Z-DNA, thus we posit that instead, they constrain and stabilize the Z-DNA (Bae et al., 2011) configuration via HJ-like structures. Indeed, in the solved IHF co-crystal with DNA, the terminal nucleotides in the complex are in the anti-syn conformation indicative of Z-DNA (Rice et al., 1996; Zhou et al., 2019). Thus, in this context, HJs are conduits for both B and Z-form DNA. Per our proposed model, B-form predominates when linear ds-DNA has free ends, and the DNABII proteins are in equilibrium between HJ-bound and linear DNA-bound (Bottom panel: Fig. 7A) wherein the ds-DNA is wrapped or twisted around DNABII proteins (a known property of at least HU proteins). As the DNABII proteins migrate to stabilize HJs and a closed loop is formed (Bottom panel: Fig. 7B), the ends of DNA could be locked in a high energy state (e.g. where the twist vacated by the DNABII proteins is converted to supercoiling) conducive for Z-DNA formation (Nordheim and Rich, 1983; Thomas and Messner, 1988; Wittig et al., 1991), with as few as 3 HJs that serve to constrain three strands of ds-DNA (Bottom panel: Fig. 7C) and only utilize 2 of 4 arms of each HJ. The remaining arms would act as conduits to the B-DNA reservoir. In agreement with this model, if there is a sufficient steady state level of DNABII proteins to stabilize the HJs, the configuration is locked, and as a consequence, the B-form DNA-binding DNABII proteins can no longer bind the intervening sequences as they are in a Z-configuration (Bottom panel: Fig. 7D&E).
Figure 7. Models of the B/Z-DNA equilibrium and the HJ constrained eDNA of the biofilm.

Top panel: (A) In the absence of DNABII proteins, there is no eDNA lattice structure. (B) In the presence of DNABII proteins, native biofilm the equilibrium shifts between B-form and Z-form eDNA. (C) Antibodies directed against DNABII proteins collapse bacterial biofilms as this favors B-form eDNA. (D) CeCl3 favors Z-form eDNA, which stabilizes the biofilm matrix and promotes biofilm residence. (E) Chloroquine favors B-form eDNA, which destabilizes the biofilm matrix and releases bacteria from biofilm residence. (F) Exogenous DNABII proteins shift the equilibrium to favor Z-DNA via Holliday Junction-like stabilization. Bottom panel: (A) B-form predominates when linear ds-DNA has free ends, and the DNABII proteins are in equilibrium between HJ-bound and linear DNA-bound. (B) DNABII proteins migrate to stabilize HJs and a closed loop is formed. (C) The ends of DNA could be locked in a high energy state [>3 HJs]. The transition from (B) to (C) is inhibited by Chloroquine, whereas the CeCl3 stimulates the transition from (C) to (D) [e.g., supercoiling “flips” to Z-form. (D) Z-DNA is constrained. (E) Z-DNA is stabilized within the ds-DNA matrix [4HJs].
This model is also consistent with several of our observations. First, when chloroquine binds ds-DNA, it unwinds the DNA, thus absorbing the writhe that DNABII proteins create when migrated to the HJs, thereby reverting Z-DNA back into B-form. Second, since CeCl3 promotes Z-DNA formation, it would act as an additional stimulatory factor in the transition from supercoiling (Bottom panel: Fig. 7C) to “flipping” into Z-DNA (Bottom panel: Fig. 7D). Indeed, this may indicate that other naturally occurring cations (e.g., polyamines) could also facilitate the stabilization of Z-DNA (Thomas and Messner, 1988),(Thomas et al., 1991). Third, cleavage of the HJs would release the ends of the ds-DNA allowing free rotation of the DNA ends with an equilibrium shift back to B-form DNA and concomitant collapse of the biofilm matrix eDNA-rich structure. Interestingly, others have shown the reverse phenomenon with similar, yet synthetic structures. When B-form DNA is locked into a 2-D array of HJ’s [DNA origami (Shrestha et al., 2016)] such that the ends are not free to rotate, conditions that otherwise favor Z-DNA cannot facilitate the transition from B-DNA (Rajendran et al., 2013). While the exact mechanism of lattice formation awaits future studies, only when the HJ-like structures within the biofilm EPS are present is Z-DNA observed.
Whether the host immune system directly reconciles an abundance of Z-DNA in the EPS of bacterial biofilms has yet to be shown, though there is ample evidence for the involvement of Z-DNA-binding proteins in the innate immune response and host-pathogen interactions (Feng et al., 2011; Kim et al., 2003; Kwon and Rich, 2005; Oh et al., 2002; Ray et al., 2013; Shin et al., 2016; Szczesny et al., 2018; van der Vorst et al., 2018; Wang et al., 2006; Zavarykina et al., 2019). We propose that bacterial derived Z-DNA within biofilms is a major contributor to the reservoir of Z-DNA within the host. Z-DNA is more antigenic than B-DNA (Moller et al., 1982; Rich and Zhang, 2003), and antibodies specific to Z-DNA are more abundant in autoimmune diseases, in particular SLE (Rich and Zhang, 2003) and rheumatoid arthritis (RA) (Sibley et al., 1984). Individuals with SLE or RA also have a higher prevalence of bacterial infections (Bouza et al., 2001). This clinical outcome may be the result of host-derived Z-DNA-binding proteins, or the induction of Z-DNA specific antibodies during the course of these chronic diseases that inadvertently stabilizes the bacterial biofilm EPS. We also propose that bacterial DNABII proteins facilitate B-to Z-DNA conversion of NET-deployed eDNA at the site of bacterial biofilm-mediated disease, which mitigates the action of host immune effectors. Based on our model, we propose that DNABII interaction with host eDNA results in a three-pronged effect. First, it would permit bacteria to utilize the reservoir of NET eDNA for biofilm development. Second, any antimicrobial B-form DNA-binding proteins (e.g., histones) would lose their affinity for the NET-derived eDNA as it is converted into the Z-form. Finally, it is also possible that bacterial infections are part of a cycle, where the adaptive immune response produces Z-DNA specific antibodies in response to biofilm infections, which inadvertently results in increased initiation and durability of bacterial biofilm formation as mediated by stabilization of a Z-DNA-rich structure (Moinuddin et al., 1998), reduces the threshold of conditions needed for Z-DNA induction (Lafer et al., 1985), and ultimately promotes chronic infection. Taken together, the role of Z-DNA in the structural stability of bacterial biofilms could be part of a much larger signal recognition response by the host innate immune system to detect foreign DNA, a hierarchical transition from rare DNA structure with an uncharacterized cellular role to a central component in the innate immune response. The development and use of therapeutic agents designed to drive biofilm eDNA back into its native B-form could provide additional approaches for clinical resolution, or prevention, of biofilm-mediated diseases.
Limitations of the Study
Despite strong evidence that supports a structural role of Z-DNA in the eDNA-dependent EPS of bacterial biofilms, there are two limitations to this study. First, while the antibodies we used that were directed against B or Z DNA are highly specific, their overall access to each form of DNA substrate within the biofilm matrix could be variable and dependent upon the overall structure and/or constituents of the biofilm. If true, this result would not affect our overall conclusions, and instead would speak to the relative contributions of B-DNA and Z-DNA to biofilm structure. Second, while our data strongly supports DNA, DNABII proteins and Z-DNA as necessary components of the eDNA dependent matrix, we cannot rule out that these components are insufficient to recapitulate the native biofilm structure; other universally available biofilm components may be required. Future experiments will focus on determining the fine structure of this extracellular nucleoprotein matrix to further dissect the apparent underlying architecture common to bacterial biofilms.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Steven D. Goodman (Steven.Goodman@NationwideChildrens.org).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The published article includes all data sets generated or analyzed during this study except for additional original replicate data [Figures 1 and 5; Supplemental Figures 2, 3, and 7] which is deposited within Mendeley Data [DOI: 10.17632/fpwbypb4kd.1].
Experimental Model and Subject Details
Bacterial strains
NTHI strain 86–028NP (Harrison et al., 2005), NTHI 86–028NP/pMDC-P1(Mokrzan et al., 2016), S. mutans strain UA159 (Mashburn-Warren et al., 2010), UPEC strain UTI89 (Mulvey et al., 2001), P. aeruginosa strain ATCC 27853, and a clinical isolate of K. pneumoniae (Rosen et al., 2008) were used.
Human derived samples
Well-differentiated primary human airway epithelial cells (HAEs) from healthy donors were obtained from the C3 Epithelial Cell Core at Nationwide Children’s Hospital. Human sputum from individuals with a diagnosis of cystic fibrosis were obtained from the Cure CF Columbus Translational Core CF sputum samples through IRB11-00790. Human neutrophils were isolated from blood donated from healthy adult control subjects that span the demographic spectrum of central Ohio to the Abigail Wexner Research Institute Blood Donor service after obtaining informed consent and under approved IRB protocol (#00002860) at Nationwide Children’s Hospital.
Chinchilla model
Adult (501–859 g) outbred, mixed sex chinchillas (Chinchilla langigera) are procured from a single vendor, Raushcher’s Chinchilla Ranch, LLC., located in La Rue, Ohio (USDA license #31A0040). To induce experimental otitis media (OM), chinchillas are injected transbullarly with a small inoculum of nontypeable Haemophilus influenzae (NTHI; strain #86-028NP) to deliver ~1000 CFU. In this model, NTHI with form a mucosal biofilm that fill >50% of the middle ear within 4 days of challenge (Novotny et al., 2011). On either day 11 or 14 after challenge, animals were sacrificed per approved IACUC protocol, and mucosal biofilms were collected, frozen in OCT at the time of collection and processed for imaging as we have described earlier (Devaraj et al., 2021; Novotny et al., 2011). Chinchilla Otitis media samples were collected in accordance with the NIH Guide for the Care and Use of Laboratory Animals and under protocol 01304AR at Nationwide Children’s Hospital.
Method Details
Bacterial growth
NTHI was grown in Brain Heart Infusion (sBHI) broth supplemented with hemin (2 μg/ml) and β-NAD (2 μg/ml). UPEC and K. pneumoniae were grown in Luria Broth (LB), P. aeruginosa was grown in Tryptic Soy Broth (TSB), and S. mutans was grown in Todd Hewett Broth (THB). Cultures were grown at 37°C in a 5% CO2 humidified incubator.
In vitro biofilms
Biofilms were allowed to form in a LabTek n 8-well glass chambered slide (Fisher Scientific 155411) and maintained for 24–40h with replenishment of the respective media at 16 and 24h post biofilm initiation. S. mutans biofilms were maintained for 40h, with replenishment of media at 24h. NTHI was grown as indicated above then diluted to 2.5 × 105 CFU/ml in 200μl sBHI. UPEC and K. pneumoniae were cultured on LB agar overnight at 37°C in 5% CO2 humidified atmosphere then suspended in LB broth to an OD490 of 0.65, diluted (1:12 in LB), and incubated statically for 2.5h. The cultures were then diluted to 2.5 × 105 CFU/ml in 200μl LB and inoculated into an 8-well glass chamber slide. P. aeruginosa biofilms were grown as above but were not incubated statically for the additional 2.5h. After 40h, the pellicle at the air-liquid interface was collected onto a glass slide. For S. mutans, an overnight culture was diluted 1:25 in THB broth then incubated at 37°C in 5% CO2 to an OD490 of 0.65. The culture was then diluted to OD490 0.05 and seeded into an 8-well glass chamber slide.
Antibodies
The following commercial antibodies were used: murine monoclonal antibody against ds-DNA [3519] (B-DNA) (Abcam, ab27156), murine monoclonal isotype IgG2a (Invitrogen, 02–6200) and 2b (Invitrogen, 02-6300)), rabbit monoclonal antibody against α-Z-DNA[Z22] (Absolute Antibodies, Ab00783-23.0), murine monoclonal antibody against Z-DNA [Z22] (Absolute Antibodies, Ab00783-3.0), rabbit monoclonal IgG isotype control (Abcam, Ab172730), naive sheep serum (Novus, 5-001-A), sheep polyclonal antibody against Z-DNA (Abcam, Ab2079), rabbit polyclonal α-neutrophil elastase (Abcam, Ab68672). IgG was purified from unimmunized rabbit whole serum as previously described (Devaraj et al., 2018).
Nuclease resistance of biofilms
NTHI 86–028NP and UPEC strains were grown as described. Biofilm growth was initiated in the presence or absence of Pulmozyme® (Genentech, dornase alfa, NDC Code 50242-100-40) [50 or 125 μg/ml] for 16h for prevention assays or added at 24h for disruption assays. Biofilms were then washed twice with 0.9% (w/v) saline, stained with LIVE/DEAD® (Invitrogen, L7007) per manufacturer’s instructions, fixed, visualized by confocal laser scanning microscope (CLSM) [Zeiss LSM 800 with Zen 2.6 Software, and images analyzed with COMSTAT software (Heydorn et al., 2000).
Antibody specificity of B and Z-DNA antibodies
Induction of Z-form DNA from genomic DNA (gDNA) [purified from NTHI using the MasterPure™ Gram Positive DNA Purification Kit (Epicentre, MGP04100) and poly(dGdC) (InvivoGen, tlrl-pgcn)] was as previously described by bromination of DNA substrates in the presence of 4M NaCl (Edgington and Stollar, 1992) with a few modifications as follows. Poly(dGdC) DNA or gDNA substrates (200 μg) were incubated overnight in 10.5ml 20mM Na citrate buffer with 4M NaCl supplemented with 500μl bromine water (RICCA, 1195–16) at room temperature, followed by buffer exchange with QIAamp Midi spin column (Qiagen) and quantification with Biotek Epoch Take3 plate reader. Z-DNA formation was determined by the absorbance ratio of 260 to 295nm as described previously (B.Chaires, 1983; Pohl and Jovin, 1972) for non-brominated or brominated poly(dGdC) (1 μg) substrates in a 100μl volume of a10mM Na-Cacodylate, 10mM NaCl, and 2mM EDTA buffer of within a Nunc 96 well UV transparent plate (ThermoFisher Scientific, 8404). Absorbance values were obtained by a Biotek Synergy H1 spectrophotometer at 260 and 295nm wavelengths. Buffer without DNA substrates served as blanks. Specificity of monoclonal antibodies was confirmed by ELISA using a Nunc MaxiSorp C-bottom 96 well plate (ThermoFisher Scientific, 44-2404-21) coated with 1 μg of poly dGdC (B-DNA) or brominated poly dGdC (Z-DNA102), followed by blocking with 0.5% (w/v) BSA in PBS for 1h at 37°C. Wells were then probed with murine IgG1(ThermoFisher Scientific, 02–6100) [neg. control], murine monoclonal antibody against either B-DNA[3519] (Abcam, ab27156) or Z-DNA[Z22] (Absolute Antibodies, Ab00783–3.0) for 1h at 37°C, and detected with goat α-mouse IgG-conjugated to HRP (Invitrogen, 31430) at 1:1000. TMB (3,3’,5,5’-tetramethylbenzidine) (MP Biomedicals, Inc., 0215234650) was the colorimetric substrate used for HRP detection. The stop solution was 2M H2SO4 (Bethyl Laboratories, E115). Absorbance values at 450nm were obtained on a BMG Labtech Fluostar Omega plate reader. An image of a representative microtiter plate was captured on a Protein Simple FluorChemM instrument.
Pulmozyme® protection assay
Poly-dGdC (2 ug) and br-poly-dGdC (2 ug), prepared as described above were incubated in a buffer containing 10 mM cacodylate pH 7.0, 10 mM NaCl and 100 μM CaCl2, with and without 0.5 units Pulmozyme® (Genentech, dornase alfa, NDC Code 50242-100-40) for 10 minutes at 37°C. EDTA at a final concentration of 5 mM was then added to each tube to quench the reactions. A 20μl aliquot of each was electrophoresed on a 1% agarose gel containing ethidium bromide at120V for 30 minutes. The resulting gel was imaged on a BioRad Gel Doc instrument, using ImageLab software.
Verification of antibody cross-reactivity by Western Blot
Bacterial cell lysates were prepared by collection of a NTHI biofilms formed for 40h in chambered coverglass slides as described (Devaraj et al., 2018). Lysates were heated to 100°C for 5 min in 4X Laemmli Sample buffer (BioRad) and 20ug protein applied per well of 12% sodium dodecyl sulfate–polyacrylamide gel. Proteins were electrophoretically separated in Tris-glycine-SDS buffer (BioRad), transferred to PVDF membrane (BioRad) and blocked with 2% bovine serum albumin (Millipore-Sigma) in Tris-buffered saline. Membranes were probed with murine monoclonal antibody to B-DNA (as determined in “antibody specificity of B- and Z-DNA antibodies”), murine monoclonal antibody to Z-DNA or murine IgG2a isotype antibody control. Additionally, reactivity of IgG isolated from sheep anti-Z DNA or unimmunized sheep serum was assessed. As a positive antibody control, chinchilla antiserum raised against NTHI 86-028NP whole outer membrane protein preparation was incorporated (Novotny et al., 2000). Murine antibodies were revealed with goat anti-mouse IgG-HRP (Invitrogen, 31430), sheep antibodies were detected with rabbit anti-sheep IgG-HRP (Abcam, Ab6747) and chinchilla antibodies shown with Protein A-HRP (Invitrogen, 101023) and developed with Pierce 1-Step Ultra TMB Blotting Solution (ThermoFisher Scientific, 34018. Images were captured on a Protein Simple FluorChemM instrument.
B-DNA and Z-DNA increases during biofilm maturation
Biofilms of strains NTHI 86-029 NP, UPEC UTI89, and Klebsiella pneumoniae were allowed to form in an 8-well glass chamber slide as described above for 24, 40, 90h, and 1 week. At each indicated time-point, biofilms were washed once with phosphate buffered saline (PBS) and probed with either murine monoclonal antibody against B-DNA[3519] (Abcam, ab27156) [5 μg] or murine monoclonal antibody against Z-DNA[Z22] (Absolute Antibodies, Ab00783-3.0) [5 μg] and their respective murine isotype IgG2a (Invitrogen, 02–6200) or IgG2b (Invitrogen, 02-6300) [5 μg] controls in 1ml of a diluent that contained 5% (bovine serum albumin) [Fisher Scientific, BP9703100] BSA (w/v) in PBS for 1h at room temperature. The biofilms were then washed once with PBS and incubated in PBS that contained 5% (w/v) BSA with 1:200 dilution of the respective goat-α mouse IgG conjugated Alexa Fluor® 488 (Abcam, A11001) for 1hr at room temperature, and stained with 1:400 dilution of FM™4–64 (Invitrogen, T3166). The biofilms were then washed once with PBS and imaged using a 63x oil objective on a Zeiss LSM 800 confocal microscope (Carl Zeiss Inc.).
Visualization of B-DNA and Z-DNA in biofilms formed in vitro
To probe for B-DNA and Z-DNA, biofilms were allowed to form as described above, then washed once with PBS and incubated with rabbit monoclonal antibody raised against Z-DNA[Z22] (Absolute Antibodies, Ab00783–23.0) (5 μg), IgG purified from unimmunized rabbit serum (5 μg), with murine monoclonal antibody raised against B-DNA[3519] (Abcam, ab27156) (5 μg), or murine isotype IgG2a (Invitrogen, 02–6200) (5 μg) in 1 ml of PBS that contained 5% (w/v) BSA for 2 hours at room temperature. Biofilms were washed with PBS and incubated with either a 1:200 dilution of goat α-rabbit IgG conjugated to Alexa Fluor® 488 (Invitrogen, A11008) or goat α-mouse IgG conjugated to Alexa Fluor® 405 (Invitrogen, A31553), and counterstained with 5 μg FM™4–64 (Invitrogen, T3166) /ml PBS that contained 5% BSA. Biofilms were then washed once with PBS and imaged using a 63x oil objective on a Zeiss LSM 800 microscope.
Visualization of B-DNA and Z-DNA in biofilms formed on HAEs
Well-differentiated primary human airway epithelial cells (HAEs) from one healthy donor were obtained from the C3 Epithelial Cell Core at Nationwide Children’s Hospital. Cells were added to 6.5mm2 diameter Costar Transwell permeable inserts (Corning Inc., 3470) with a pore size of 0.4μm then incubated as described to permit polarization(Mokrzan et al., 2016; Zhang et al., 2002) and differentiation. Prior to inoculation with NTHI, the apical surface of the polarized HAEs was first gently washed with PBS to remove excess accumulated mucus. NTHI 86-028NP/pMDC-P1, a reporter construct wherein the promoter for NTHI ompP5 drives expression of green fluorescent protein (Mokrzan et al., 2016) was inoculated on to HAE cultures at a MOI of 100 bacteria per epithelial cell, and incubated for 16 h. Non-adherent NTHI were removed by aspiration and the apical surface of the HAE cultures blocked with 10% normal goat serum (Bethyl Laboratories, Inc., IHC-GS50)- 0.05M Tris-0.15M NaCl, pH 7.4 buffer. Cultures were incubated with antibody cocktails that contained murine monoclonal antibody to B-form DNA [3519] (Abcam, ab27156) and rabbit monoclonal antibody to Z-form DNA[Z22] (Absolute Antibodies, Ab00783-23.0) (5 μg each antibody/ml buffer). Negative antibody controls consisted of murine IgG1 isotype antibody (ThermoFisher Scientific, 02-6100) and IgG enriched from unimmunized rabbit serum (5 μg each antibody/ml buffer). Labeling was revealed with goat anti-rabbit IgG conjugated to Alexa Fluor® 647 (Invitrogen, A21245) and goat anti-mouse IgG conjugated to Alexa Fluor® 594 (Invitrogen, A11032) [5 μg each antibody /ml]. One set of HAE cultures was not inoculated with NTHI; however, it was probed with antibodies to B-form and Z-form DNA to validate that any DNA-specific signal observed was due to DNA release from NTHI and not from epithelial cells. Transwell membranes were excised, mounted on to SuperFrost™ Plus slides (Fisher Scientific, 22-037-246) with ProLong™ Diamond (Invitrogen, P36961) and coverslipped prior to visualization on a Zeiss LSM800 confocal scanning laser microscope.
Visualization of B-DNA and Z-DNA within clinical specimens
To examine for the presence of B- or Z-forms of DNA within biofilms formed during disease, archived specimens frozen in OCT at the time of collection were examined: (1) a 14-day old NTHI biofilm formed in the middle ear of a chinchilla during experimental otitis media and (2) sputum from an individual with CF wherein the specimen was culture positive for MSSA and Burkholderia cenocepacia complex. Ten μm serial sections were cut and processed via standard protocol (Jurcisek and Bakaletz, 2007). Briefly, slides were air dried, fixed in cold acetone, then equilibrated in 0.05M Tris-HCl, 0.15M NaCl-, 0.05% Tween 20 buffer, pH 7.4. Sections were blocked with image-iT FX signal enhancer (Invitrogen, I36933)) followed by Super Block (ScyTec Laboratories, Inc., AAA999). Specimens were incubated with primary antibody cocktail comprised of murine monoclonal antibody against B-DNA [3519] (Abcam, ab27156) (5.0 μg/ml), rabbit monoclonal antibody against Z-DNA (5.0 μg/ml) diluted in 5% (w/v) BSA in PBS. IgG purified from serum from unimmunized rabbit or murine IgG1antibody (ThermoFisher Scientific, 02-6100) served as the respective negative antibody controls for non-specific binding. Labeling was revealed with goat anti-rabbit IgG conjugated to Alexa Fluor® 647 (Invitrogen, A21245), goat anti-mouse IgG conjugated to Alexa Fluor® 488 (Invitrogen, A11001) [each used at 5 μg/ml] in PBS. Prolong Gold™ (Invitrogen, P36930) or Prolong Gold™ with NucBlue™ Live (Invitrogen, R37605) was applied prior to placement of a coverslip. Sections were viewed using a Zeiss LSM 800 confocal microscope and images rendered with Zeiss Zen 2.6 software.
Quantification of Z-DNA and B-DNA within nuclease-treated biofilms
NTHI 86-029 NP, UPEC, and K. pneumoniae, were allowed to form biofilms, then maintained for 40 hours as described above in “in vitro biofilms”. At 24 hours, biofilms were incubated in the presence or absence of Pulmozyme® (Genentech, dornase alfa) [40U/ml] for 16h at 37°C. Alternatively, to determine whether B-DNA was a reservoir for Z-DNA development, Pulmozyme® (Genentech, dornase alfa, NDC Code 50242-100-40) was added at initiation followed by subsequent additions at 16 and 24h for a total of 40h. The biofilms were then washed with PBS and incubated with 200 μl of PBS that contained 5% (w/v) BSA and either murine monoclonal antibody raised against B-DNA [3519] (Abcam, ab27156) [5 μg/ml] or rabbit monoclonal antibody raised against Z-DNA [Z22] (Absolute Antibodies, Ab00783-23.0) [5 μg/ml] or the respective naive murine IgG isotype 2aIgG2a (Invitrogen, 02-6200) (5μg/ml) or IgG purified from unimmunized rabbit [5μg/ml] controls for 1h at room temperature. The biofilms were then washed once with PBS and incubated with the respective goat-α mouse IgG conjugated Alexa Fluor® 405 (Invitrogen, A31553) and goat-α rabbit IgG conjugated Alexa Fluor® 488 (Invitrogen, A11008) in PBS that contained 5% (w/v) BSA for 1h at room temperature, and counterstained with 5 μg of FM™4–64(Invitrogen, T3166) /ml in PBS. The biofilms were then washed with PBS and imaged using a 63x oil objective on a Zeiss LSM 800 confocal microscope (Carl Zeiss Inc.). The fluorescence intensity of B-DNA (Alexa Fluor® 405 signal) and Z-DNA (Alexa Fluor® 488 signal) were normalized to the fluorescence intensity of FM™4–64. Changes in fluorescence intensity was determined by ImageJ software by determining the mean intensity of Z-stacks. NTHI 86-028NP was incubated as described previously. Biofilm growth was initiated in the presence or absence of Pulmozyme® (Genentech, dornase alfa, NDC Code 50242-100-40) [100 μg/ml], followed by subsequent additions at 16 and 24h for a total of 40h. The biofilms were washed, incubated with the appropriate monoclonal antibodies, and visualized as described in “Quantification of Z-DNA and B-DNA within nuclease treated biofilms”.
Stimulation of biofilm formation by anti-Z-DNA
NTHI and UPEC biofilms were initiated in the presence of 0, 1, or 5 μg/ml polyclonal α-Z DNA antibody IgG (Abcam, ab2079) or unimmunized sheep IgG (Novus, 5-001-A) and grown for 16h. Additionally, UPEC biofilms were initiated in the presence of 0 or 5 μg/ml murine monoclonal α-Z-DNA[Z22] (Absolute Antibodies, Ab00783-3.0) or murine isotype 2b (Invitrogen, 02-6300). Biofilms were then analyzed by LIVE/DEAD® (Invitrogen, L7007) as described above in “nuclease resistance in biofilms”.
Confirmation of B/Z-DNA conversion by OD 260/295
Genomic DNA (400ng) was incubated in a reaction buffer as described previously “Antibody specificity of B- and Z-DNA antibodies” and supplemented with 0, 0.25, 0.5, and 1mM CeCl3 for 2h at 37°C. Poly(dGdC) (1μg) was incubated with either NaCl (3.6M), chloroquine (100μM), or a combination of both for 2h at 37°C. The absorbance values were obtained as described above in “Antibody specificity of B and Z-DNA antibodies”. Reaction buffers that contained chloroquine or NaCl (absent DNA) served as blanks. The values were plotted as the ratio of A260/295.
Growth curves of planktonic NTHI
Planktonic growth curves of NTHI were initiated at OD490 of 0.02 in sBHI supplemented with 0, 250, 500, or 1000 μM CelCl3 or with 0, 10, 50, 100, or 200 μM chloroquine in a 96-well plate and grown for 16h at 37°C using a Biotek Synergy H1 multi-mode plate reader.
Modulation of B/Z eDNA of bacterial biofilms
Cultures of NTHI were incubated as described above. At 24h, CeCl3 (Alpha Aesar, CAS 7790-86-5), a compound known to induce Z-DNA (Bhanjadeo et al., 2017), was added to a final concentration of 0.25, 0.5, or 1mM in sBHI to convert B- to Z-DNA. Chloroquine (Sigma, C6628–100G), a known inhibitor of Z-DNA formation (Kwakye-Berko and Meshnick, 1990) was added to a final concentration of 1μM or 5 μM in sBHI to convert Z-DNA to B-DNA. The effect of DNABII proteins on Z-DNA within the biofilm EPS was analyzed by the exogenous addition of HUNTHI (Novotny et al., 2016) [2 μg/ml] at initiation and throughout biofilm development as previously described(Devaraj et al., 2018). Biofilms were probed for Z-DNA with IgG purified from unimmunized rabbit serum (5 μg/ml) or rabbit α-Z-DNA [Z22] (Absolute Antibodies, Ab00783-23.0) [5 μg/ml] in 5% (w/v) BSA in PBS, then goat α-rabbit conjugated with Alexa Fluor® 488 (Invitrogen, A11008), and stained with FM™4–64 (Invitrogen, T3166) before being analyzed by immunofluorescence microscopy with calculation of partition coefficients determined as described previously (Devaraj et al., 2015).
Axial mechanical indentation of NTHI biofilms
NTHI biofilms were grown in 35 mm FluoroDishes (World Precision Instruments, FD35-100) for 40 h at 37°C with 5% CO2 as described above. After 24h, biofilms were treated with CeCl3 (500μM) for 16h, then incubated with media or Pulmozyme® (50μg/ml) for 1h at 37°C with 5% CO2. Media only treatment was used as a control. To test the effect of chloroquine, NTHI biofilms were grown for 40 h and treated with cholorquine (5μM) for 1 h at 37°C with 5% CO2. NTHI biofilms were initiated in the presence of RusA (10μg/ml) or HUNTHI (2μg/ml) and supplemented at 16 and 24h for a total of 40h. Additionally, HUNTHI was added as throughout biofilm development as described in “Modulation of B/Z eDNA of bacterial biofilms” for 40h then were treated with Pulmozyme® (50μg/ml) or RusA (10μg/ml) + Pulmozyme® (50μg/ml) for 1h. Additionally, NTHI biofilms were grown for 24h, incubated with CeCl3 (500μM) for 16h, then treated with RusA (10μg/ml) for 1h at 37°C with 5% CO2. Biofilms were then washed twice with PBS and maintained in 600 μl mL PBS until rheological analysis.
Axial mechanical indention was performed on treated NTHI biofilms as previously described (Devaraj et al., 2019). Briefly, the FluoroDish containing treated or control biofilms was transferred to the Pelteir plate of a Discovery Hybrid Rheometer-2 (DHR-2; TA instruments) fitted with an 8 mm sand-blasted Smart Swap parallel plate geometry. The geometry was lowered onto the submerged biofilm at an approach rate of 1 μm/s. Three biological replicates were performed, each with duplicate replicates. Two to three measurements were taken for each biofilm. TRIOS v5 (TA instruments) software was used.
For data interpretation, force-displacement curves were converted to stress-strain curves as previously described (Gloag et al., 2018). The Young’s modulus (E) was determined using the previously described force-displacement relationship, according to equation 1 (Timoshenko and Goodier, 1970),
| (1) |
where r is the radius of the geometry (0.004m), and v is the assumed Poisson’s ratio for a biofilm (v = 0.5) (Rmaile et al., 2013). The slope is of the force-displacement curve (N/m), taken at the lower “linear” portion corresponding to 0–40% strain.
The susceptibility of biofilms to DNase after chloroquine
NTHI biofilms were established as described above. At 40h, chloroquine (Sigma, C6628-100G) [5 μM], Pulmozyme®(Genentech, dornase alfa, NDC Code 50242-100-40) [50 μg/ml] or a combination of both in sBHI were added to mature biofilms and incubated for 1h at 37°C. Biofilms were then washed twice with 0.9% (w/v) saline, stained with LIVE/DEAD® (Invitrogen, L7007) per manufacturer’s instructions, fixed, visualized by confocal laser scanning microscope (CLSM), and images analyzed by COMSTAT (Heydorn et al., 2000).
The susceptibility of clinical specimens to DNase and chloroquine
Fresh human sputum from 4 individuals with a diagnosis of cystic fibrosis were obtained from the Cure CF Columbus Translational Core, divided into approximately equal volumes and placed into wells of a 24-well plate as described (Gustave et al., 2013). The remainder of each specimen was snap-frozen in OCT and processed for immunofluorescent detection of B-DNA and Z-DNA as described above. To evaluate the susceptibility of each fresh sputum specimen to disruption, the following treatments were applied: 10μM chloroquine, 100μM chloroquine, 10U Pulmozyme® (Genentech, dornase alfa, NDC Code 50242-100-40), 30U Pulmozyme®, 10μM chloroquine+10U Pulmozyme®, 100μM chloroquine+10U Pulmozyme®, 10μM chloroquine+30U Pulmozyme® or 100μM chloroquine+30U Pulmozyme® or diluent (DPBS) only. Sputa were incubated static for 1h at 37°C and changes in the optical density in the solution that surrounded the sputum solids were determined at 600nm in a 6×6 matrix 14mm in diameter with a BMG Labtech Fluorostar Omega plate reader. Images of sputum specimens were captured using a Zeiss SV6 microscope and Zeiss Zen software. As part of routine clinical analysis, an aliquot of each sputum specimen was also submitted for speciation to the Laboratory Services at Nationwide Children’s Hospital.
Prevention of Z-DNA formation by a HJ resolvase
NTHI 86-028NP was incubated as described previously. RusA was purified as described (Devaraj et al., 2019). Biofilm growth was initiated in the presence or absence of RusA (10μg/ml) for 16h. The biofilms were washed, incubated with murine monoclonal antibody raised against B-DNA [3519] (Abcam, ab27156) [5 μg/ml] and rabbit monoclonal antibody raised against Z-DNA [Z22] (Absolute Antibodies, Ab00783-23.0) [5 μg/ml] or the respective murine IgG isotype IgG2a (Invitrogen, 02-6200) [5μg/ml] or IgG purified from unimmunized rabbit [5μg/ml] controls for 1h at room temperature. The biofilms were then washed once with PBS and incubated with the respective goat-α mouse IgG conjugated Alexa Fluor® 405 (Invitrogen, A31553) and goat-α rabbit IgG conjugated Alexa Fluor® 488 (Invitrogen, A11008) in PBS that contained 5% (w/v) BSA for 1h at room temperature, and counterstained with 5 μg of FM™4–64 (Invitrogen, T3166) /ml in PBS. The biofilms were then washed with PBS and imaged using a 63x oil objective on a Zeiss LSM 800 confocal microscope (Carl Zeiss Inc.). The fluorescence intensity of B-DNA (Alexa Fluor® 405 signal) and Z-DNA (Alexa Fluor® 488 signal) were normalized to the fluorescence intensity of FM™4–64. Changes in fluorescence intensity was determined by ImageJ software by determining the mean intensity of Z-stacks.
DNABII proteins drive B-DNA dominant NETs to Z-DNA during PMA-induced NETosis
Human neutrophils were isolated from blood using the EasySep™ Human neutrophil isolation kit (Stemcell Technologies Inc., 17957). Neutrophils (1×105) were incubated and allowed to attach to 8-well glass chamberslides followed by induction of NETs with 200nM PMA (phorbol-12-myristate-13-acetate) for 3.5h in the presence or absence of 500nM HUNTHI (Novotny et al., 2016) or CbpA [purified as described (Devaraj et al., 2018)] and served as a negative control. NETs were formalin fixed, blocked with 10% normal goat serum (Life Technologies, 50062Z), and incubated with rabbit monoclonal antibody raised against Z-DNA [Z22] (Absolute Antibodies, Ab00783–23.0) (5 μg), from rabbit isotype control (Abcam, ab172730) [5 μg], with murine monoclonal antibody raised against B-DNA [3519] (Abcam, ab27156) [5 μg], or murine isotype IgG2a (Invitrogen, 02-6200) [5 μg] in 1 ml of PBS for 16h hours at 4°C. NETs were washed with PBS and incubated for 1h with 1:200 dilution of goat α-rabbit IgG conjugated to Alexa Fluor® 594 (Invitrogen, A11037) or goat α-mouse IgG conjugated to Alexa Fluor® 488 (Invitrogen, A11001) and 1:1000 dilution wheat germ agglutinin conjugated to Alexa Fluor® 350 (Invitrogen, W11263) [PMN membrane stain]. As a control, common NETs markers were used to verify proper activation. NETs were incubated similarly with 1:100 rabbit polyclonal antibody raised against human neutrophil elastase (Abcam, ab68672) to label elastase which is indicative of active NETs DNA structure. 1:5000 SYTOX™ green nucleic acid stain (Invitrogen, S7020), and 1:1000 wheat germ agglutinin conjugated to Alexa Fluor® 350 (Invitrogen, W11263) to counterstain the plasma membrane of the neutrophils. NETs were imaged with a Zeiss LSM 800 confocal microscope (Carl Zeiss Inc.) and rendered with Zeiss Zen 2.6 software.
NET inactivation of Bacterial Killing
NTHI biofilms were allowed to establish for 16h as described above. Briefly, 2.5×105 bacteria were incubated for 16h in a 24 well glass-plates prior to the addition of neutrophils. Peripheral blood neutrophils were purified from healthy donors by magnetic negative selection (Stemcell Technologies Inc., 17957) and adjusted to be able to deliver 1×106 PMNs/well. Prior to the addition of PMNs, the young biofilms were washed carefully with PBS to remove non-adherent bacteria, followed by the addition of 300 μl of RPMI supplemented with 10% FBS. Neutrophils were then added to the wells and treated with either: 1μM HUNTHI, 1 μM CbpA, or 10 units Pulmozyme® (Genentech, dornase alfa, NDC Code 50242-100-40)/ml for 4h. After incubation, during which time biofilm remodeling occurs, 0.1% Triton X-100 was added to the cultures to release any intracellular bacteria from the PMNs after which all bacteria were recovered by homogenized via pipetting, followed by serial dilutions and plating (Robledo-Avila et al., 2020). The percentage of reduction of the number of NTHI (total CFU) and % relative killing was calculated as indicated in the following formulas: [(CFU of each sample) ÷ (average CFU of NTHI only controls) × 100 = % viable]: [% Killing = 100% − (% viable)]. The relative % killing was normalized so that the NTHI only control represented 0% killing as follows: [100% (e.g., average of NTHI control) – (% killing of each sample) = relative % killing]. The graph plot replicates were derived from 4 healthy donors ± SEM. The statistical analysis was performed with One-way ANOVA and Dunett multiple comparison test. (**=p<0.01, ***=p<0.001).
Image of 11 day challenged chinchilla sections
Biofilms that had been allowed to form for eleven days within the middle ear of a chinchilla in an experimental model of otitis media induced by NTHI were recovered, as described in detail previously were used (Devaraj et al., 2021). Briefly, bullae which contained the NTHI biofilm adherent to the middle ear mucosa and associated PMNs that had migrated to the middle ear in response to the bacterial challenge were embedded in OCT (Fisher Scientific, Waltham, MA), cut into ten micron sections and labeled for IF imaging using rabbit anti-Z-DNA and/or mouse anti-B-DNA or their associated isotype control sera, then incubated with goat anti-rabbit conjugated to Alexa Fluor® 594 and/or goat anti-mouse conjugated to Alexa Fluor® 488. Sections were then coverslipped with Prolong Glass containing NucBlue as the mounting medium (Molecular Probes, Eugene, OR). Images were captured using a Zeiss LSM 800 with Airyscan.
Quantification and Statistical Analysis
Quantification of average thickness, fluorescence intensity, optical density, and absorbance values were determined as described in STAR Methods. Statistical significance was determined by either unpaired, paired t-test, Tukey’s multiple comparisons test, Dunnett’s multiple comparison test, or One-way ANOVA with GraphPad Prism (version 8.0 and 9.1) and is described as appropriate, within the respective figure legends. A p-value ≤ 0.05 is indicated by *, a p-value ≤ 0.01 is indicated by**, a p-value ≤ 0.001 is indicated by ***, and a p-value <0.0001 is indicated by ****. Error values represent the SEM.
Supplementary Material
Figure S1. Mature biofilms were resistant to DNase, related to Figure 2 (A) Representative images of UPEC biofilms incubated with DNase. (B) Increasing concentrations of DNase were added at initial biofilm seeding of NTHI, Kp and UPEC (prevention of biofilm development) or 24h after seeding (disruption of mature biofilms). After DNase treatment, biofilms were incubated for an additional 16h, then stained with LIVE/DEAD® viability stain, fixed, visualized via CLSM, and images analyzed by COMSTAT. Error bars represent the standard error of the mean (SEM). Statistical significance of DNase treatment compared to control (- DNase) was assessed by unpaired t-tests, **, P<0.01 and ***, P<0.001. NTHI prevention, n=4 and disruption, n=3; Kp prevention and disruption, n=5; UPEC prevention and disruption, n=3. DNase inhibited the formation of biofilms by UPEC, Kp and NTHI, but did not disrupt these structures once formed.
Figure S2. Verification of the specificity of B- and Z-DNA specific antibodies, related to Figure 1 and S5 (A) Brominated poly(dGdC) (10 μg/ml) or non-brominated poly(dGdC) (10 μg/ml) were incubated in buffer, the absorbance values at 260nm and 295nm were measured, then the A260/295 ratio was calculated. Poly(dGdC) had a ratio of 10.51 ± 1.33 indicative of B-DNA, while Br-poly(dGdC) had a ratio of 2.73 ± 0 which is indicative of Z-DNA. Statistical significance compared to control (non-brominated poly(dGdC) was assessed by unpaired t-tests, ***P<0.001, n=3. (B) Representative ELISA plate image demonstrated the specificity of monoclonal antibodies to B- or Z-form DNA. An ELISA plate was coated with 1 μg of poly dGdC (B-DNA) or brominated poly dGdC (Z-DNA102), blocked with 0.5% (w/v) BSA, then probed with murine IgG1 (ms isotype) and naive rabbit serum (NRS) (neg. controls), or murine monoclonal antibody directed against B-DNA or Z-DNA (ms anti-B or -Z, respectively), followed by detection with a secondary goat α-mouse IgG-HRP. TMB (3,3’,5,5’-tetramethylbenzidine) was the colorimetric substrate used for HRP detection (dark gray wells). (C) Quantification of the ELISA for specificity of the B- and Z-DNA monoclonal antibodies. Statistical significance of murine α-Z-DNA and α-B-DNA were compared to control (no dGdC buffer) and assessed by Tukey’s multiple comparisons test, **P<0.01, ***P<0.001, and P<0.0001, n=4. (D) Western Blot analysis of bacterial biofilm whole-cell lysates (L) (20 μg) probed with the indicated antibodies (M – molecular mass standards), n=4. The monoclonal antibodies used in this study which are directed to B-DNA and Z-DNA are specific and do not cross react with bacterial lysates. (E) Brominated gDNA (4 μg/ml) or non-brominated gDNA (4 μg/ml) were incubated in buffer or 3.6M NaCl, absorbance values at 260nm and 295nm were measured, then the A260/295 ratio was calculated. Brominated Poly(dGdC) and non-brominated poly(dGdC) were used as internal controls. gDNA had a ratio of 10.69 ± 3.36 indicative of B-DNA. Conversely, Br-gDNA had a ratio of 2.19 ± 0.06 and gDNA 3.6M NaCl had a ratio of 2.76 ± 0.53 which are both indicative of Z-DNA. Statistical significance compared to control (non-brominated) was assessed by unpaired t-tests, n=3. (F) Poly(dGdC) and Br-poly(dGdC) were incubated in the presence or absence of Pulmozyme® (5U/ml) for 10 minutes at 37°C. The DNA degradation was assessed by gel electrophoresis which showed that Br-poly(dGdC) was protected from the activity of DNase, n=3. ***P<0.001. Error bars represent the SEM.
Figure S3. B-DNA and Z-DNA increased within the EPS of biofilms, related to Figure 1 (A) 24, 40, 90h, or (B) 1 week old NTHI, Kp, and UPEC biofilms were examined. Unfixed biofilms were incubated with a murine monoclonal antibody against either B-DNA (5 μg/ml) or Z-DNA (5 μg/ml), then revealed via incubation with goat α-mouse IgG conjugated to Alexa Fluor® 488. Biofilms were visualized by CLSM. B-DNA (cyan) and Z-DNA (yellow) accumulated as the biofilms matured, n=3. (C) NTHI biofilms (40h) were incubated with the same primary, control antibodies, and buffer (no primary antibody), then visualized with the same secondary antibodies as described above, n=3. No cross-reactivity of the secondary conjugated antibodies was observed.
Figure S4. Removal of B-DNA throughout biofilm development reduced Z-DNA, related to Figure 2 NTHI biofilms were initiated in the presence or absence of DNase (80 U/ml) for 40h. Unfixed biofilms were incubated with a murine monoclonal antibody against either B-DNA[3519] (5 μg/ml) or Z-DNA (5 μg/ml), then revealed via incubation with goat α-mouse IgG conjugated to Alexa Fluor® 405 and goat α-rabbit IgG conjugated to Alexa Fluor® 488. Changes in either: (A) biomass (FM™4–64 fluorescent signal); (B) Z-DNA (Alexa Fluor® 488) or (C) B-DNA (Alexa Fluor® 405) were quantified by image J software. (D) The ratio of relative B-DNA to Z-DNA of IF intensity of DNase incubated biofilms were compared to the intensity of the media control. Error bars represent the SEM. Statistical significance compared to control (no DNase) was assessed by unpaired t-tests, *P>0.05 **P<0.01, n=4.
Figure S5. Z-DNA-specific antibodies stimulated biofilm formation, CeCl3 induced Z-DNA, whereas chloroquine converted Z-DNA to B-DNA and partitioned planktonic NTHI differentially, related to Figure 3, 4 &S2 (A) NTHI and UPEC biofilms were initiated in the presence of increased amounts (1 or 5 μg/ml) of either IgG from unimmunized sheep serum (pAB sheep), sheep polyclonal α-Z-DNA (pAB-Z-DNA) or media for 16h, n=4. (B) UPEC biofilms were initiated in the presence of rabbit monoclonal α-Z-DNA (mAB Z-DNA), rabbit monoclonal isotype 2b (isotype control) or media for 16h. Biofilm average thickness was analyzed by LIVE/DEAD® staining with COMSTAT image analysis. Error bars represent the SEM. Statistical significance compared to control was assessed by paired t-tests, *P<0.05, **P<0.01, n=6. (C) Genomic DNA isolated from NTHI (4 μg/ml) was incubated with increased concentrations of CeCl3 (0, 0.25, 0.5, and 1 mM) in buffer, and analyzed by spectroscopic absorbance ratio assay. The ratio determined for samples that contained either 0 or 0.25mM CeCl3 were 14.17 ± 6.79 or 15.61 ± 4.75 respectively, which indicated B-DNA. In contrast, ratios determined for 0.5 and 1mM CeCl3 were 3.90 ± 0.76 or 2.90 ± 1.51, respectively, which indicated Z-DNA. Error bars represent the SEM. Statistical significance compared to control was assessed by unpaired t-tests, *P<0.05, n=3. (D) Growth curves of NTHI grown planktonically in the presence of increased concentrations of CeCl3 (0.25, 0.5, and 1 mM) indicated a minimal effect on growth (n=3, averaged curves). (E) NTHI was allowed to form a biofilm for 24h in the presence of CeCl3. At 40h, relative CFU of the planktonic and biofilm populations were assessed to calculate the relative percent of planktonic versus biofilm resident NTHI which represents the ‘partition coefficient’, n=3. (F) To determine the Young’s modulus of treated and untreated NTHi biofilms, the slope of the lower linear portion of the force-displacement curve that corresponded to 0 – 40% strain (Figs. 3 and 4), where R2 ≥ 0.9, was determined. Using the slope, the Young’s modulus was calculated according to equation 1. Depicted is the force-displacement region of a representative untreated control NTHi biofilm that corresponded to 0 – 40% strain. The linear fit of the curve is indicated in red; the dashed lines indicate the 95% confidence interval. The R2 value of the linear fit is indicated. (G) Poly(dGdC) DNA (10 μg/ml) was incubated with 0.1 mM chloroquine, 3.6M NaCl or a combination of both, followed by analysis of the spectroscopic absorbance ratio assay. The absorbance 260/295nm ratios calculation for poly(dGdC) and 0.1mM chloroquine were 11.36 ± 1.72 and 8.25 ± 0.90 respectively which indicated B-DNA, while the ratio value calculated for 3.6M NaCl was 3.62 ± 0.37, which confirmed Z-DNA, n=3. Chloroquine prevented NaCl- induced Z-DNA formation as determined by the ratio calculation of 6.61 ± 0.32. (H) Planktonic NTHI growth curves with increased concentrations of chloroquine (0.01, 0.05, 0.1, and 0.2 mM) indicated no effect on growth (n=3, averaged curves). (I) NTHI was allowed to form a biofilm for 24h followed by the addition of increasing concentrations of chloroquine. At 40h, the partition coefficients were calculated, as above. Error bars represent the SEM. Statistical significance compared to control was assessed by paired t-tests, *P<0.05, n=7.
Figure S6. Modulation of B/Z eDNA increased the susceptibility of biofilms to DNase, related to Figure 3 and Table S1 NTHI biofilms were formed for 40h and incubated with chloroquine, DNase, or both, then stained with LIVE/DEAD®, fixed, visualized via CLSM, and images analyzed by COMSTAT, quantified in Fig 3K, n=7. (A) Representative images of NTHI biofilms incubated with DNase and/or chloroquine. (B) Representative images of IF labeling of B-DNA with murine α-B-DNA[3519] and Z-DNA with rabbit αZ-DNA[Z22] in 4 unique specimens collected during exacerbation of clinical disease and assayed in Fig 3M, n=1. Labeling was revealed with goat anti-rabbit IgG conjugated to Alexa Fluor® 647, goat anti-mouse IgG conjugated to Alexa Fluor® 488. Scale bar 10μm.
Figure S7. PMA-induced NET eDNA was predominantly comprised of B-DNA, related to Figure 5 Neutrophils (1×105) were allowed to attach to an 8 well glass chamberslide and incubated in the presence or absence of CbpA (500nM) upon induction with PMA (200nM) for 3.5h at 37°C. NETs were then fixed, washed, blocked with 10% goat serum followed by incubation with rabbit polyclonal antibody against human neutrophil elastase (1:100), rabbit monoclonal antibody against Z-DNA (5 μg/ml), and a murine monoclonal antibody against B-DNA (5 μg/ml). Then NETs were incubated with goat α-rabbit IgG conjugated Alexa Fluor® 594 and goat α-mouse IgG conjugated Alexa Fluor® 488, wheat germ agglutinin (WGA 350), and SYTOX™ nucleic acid stain. NETs were imaged by CLSM. (A) Representative image of no PMA control, n=3. (B) Representative images of NETs formed in the presence or absence of CbpA (no Z-DNA induction), n=3.
Z-DNA is stable and abundant extracellularly
Z-DNA provides structure and DNase-resistance to the biofilm matrix
Bacterial DNABII proteins mitigate NET function
Acknowledgments
This work was supported by NIH grants R01DC011818 and R01AI155501 (to S.D.G. and L.O.B.) and R01GM124436 (to P.S.). We thank Jeffrey Melvin Ph.D. for thoughtful discussions. We also thank the C3 Epithelial Cell Core at Nationwide Children’s Hospital which is supported by a Research Development Grant from the Cystic Fibrosis Foundation. Finally, we extend our gratitude to those individuals who donated CF sputum samples through IRB11-00790 which was approved by the Nationwide Children’s Hospital Institutional Review Board.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
S.D.G., and L.O.B., are founders, shareholders and members of the scientific advisory board of Clarametyx Biosciences, Inc.
References
- Al-Bari MA (2015). Chloroquine analogues in drug discovery: new directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J Antimicrob Chemother 70, 1608–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhede M, Alhede M, Qvortrup K, Kragh KN, Jensen PO, Stewart PS, and Bjarnsholt T (2020). The origin of extracellular DNA in bacterial biofilm infections in vivo. Pathog Dis 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali N, and Ali R (1997). High salt and solvent induced Z-conformation in native calf thymus DNA. Biochem Mol Biol Int 41, 1227–1235. [DOI] [PubMed] [Google Scholar]
- B.Chaires J (1983). Daunomycin nhibits the B- Z transition in poly d(G-). Nucleic Acids Research 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Kim D, Kim KK, Kim YG, and Hohng S (2011). Intrinsic Z-DNA is stabilized by the conformational selection mechanism of Z-DNA-binding proteins. J Am Chem Soc 133, 668–671. [DOI] [PubMed] [Google Scholar]
- Baranovskii AG, Buneva VN, and Nevinsky GA (2004). Human deoxyribonucleases. Biochemistry (Mosc) 69, 587–601. [DOI] [PubMed] [Google Scholar]
- Bezanilla M, Drake B, Nudler E, Kashlev M, Hansma PK, and Hansma HG (1994). Motion and enzymatic degradation of DNA in the atomic force microscope. Biophysical Journal 67, 2454–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhanjadeo MM, Nayak AK, and Subudhi U (2017). Cerium chloride stimulated controlled conversion of B-to-Z DNA in self-assembled nanostructures. Biochem Biophys Res Commun 482, 916–921. [DOI] [PubMed] [Google Scholar]
- Bonnefoy E, and Rouviere-Yaniv J (1991). HU and IHF, two homologous histone-like proteins of Escherichia coli, form different protein-DNA complexes with short DNA fragments. EMBO J 10, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouza E, Moya JG, and Munoz P (2001). Infections in systemic lupus erythematosus and rheumatoid arthritis. Infect Dis Clin North Am 15, 335–361, vii. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, and Zychlinsky A (2004). Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535. [DOI] [PubMed] [Google Scholar]
- Brockson ME, Novotny LA, Mokrzan EM, Malhotra S, Jurcisek JA, Akbar R, Devaraj A, Goodman SD, and Bakaletz LO (2014). Evaluation of the kinetics and mechanism of action of anti-integration host factor-mediated disruption of bacterial biofilms. Mol Microbiol 93, 1246–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton DR, Butler MJ, Hyde JE, Phillips D, Skidmore CJ, and Walker IO (1978). The interaction of core histones with DNA: equilibrium binding studies. Nucleic Acids Res 5, 3643–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaires JB, and Norcum MT (1988). Structure and stability of Z* DNA. J Biomol Struct Dyn 5, 1187–1207. [DOI] [PubMed] [Google Scholar]
- Chintakayala K, Sellars LE, Singh SS, Shahapure R, Westerlaken I, Meyer AS, Dame RT, and Grainger DC (2015). DNA recognition by Escherichia coli CbpA protein requires a conserved arginine-minor-groove interaction. Nucleic Acids Res 43, 2282–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj A, Buzzo J, Rocco CJ, Bakaletz LO, and Goodman SD (2018). The DNABII family of proteins is comprised of the only nucleoid associated proteins required for nontypeable Haemophilus influenzae biofilm structure. Microbiologyopen 7, e00563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj A, Buzzo JR, Mashburn-Warren L, Gloag ES, Novotny LA, Stoodley P, Bakaletz LO, and Goodman SD (2019). The extracellular DNA lattice of bacterial biofilms is structurally related to Holliday junction recombination intermediates. Proc Natl Acad Sci U S A 116, 25068–25077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj A, Justice SS, Bakaletz LO, and Goodman SD (2015). DNABII proteins play a central role in UPEC biofilm structure. Mol Microbiol 96, 1119–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj A, Novotny LA, Robledo-Avila FH, Buzzo JR, Mashburn-Warren L, Jurcisek JA, Tjokro NO, Partida-Sanchez S, Bakaletz LO, and Goodman SD (2021). The extracellular innate-immune effector HMGB1 limits pathogenic bacterial biofilm proliferation. J Clin Invest 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey D, Nagaraja V, and Ramakumar S (2017). Structural and evolutionary analyses reveal determinants of DNA binding specificities of nucleoid-associated proteins HU and IHF. Molecular Phylogenetics and Evolution 107, 356–366. [DOI] [PubMed] [Google Scholar]
- Dubois AV, Gauthier A, Brea D, Varaigne F, Diot P, Gauthier F, and Attucci S (2012). Influence of DNA on the activities and inhibition of neutrophil serine proteases in cystic fibrosis sputum. Am J Respir Cell Mol Biol 47, 80–86. [DOI] [PubMed] [Google Scholar]
- Dumat B, Larsen AF, and Wilhelmsson LM (2016). Studying Z-DNA and B- to Z-DNA transitions using a cytosine analogue FRET-pair. Nucleic Acids Res 44, e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer M, Shan Q, D’Ortona S, Maurer R, Mitchell R, Olesen H, Thiel S, Huebner J, and Gadjeva M (2014). Cystic fibrosis sputum DNA has NETosis characteristics and neutrophil extracellular trap release is regulated by macrophage migration-inhibitory factor. J Innate Immun 6, 765–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgington SM, and Stollar BD (1992). Immunogenicity of Z-DNA Depends on the Size of Polynucleotide Presented in Complexes with Methylated Bsa. Molecular Immunology 29, 609–617. [DOI] [PubMed] [Google Scholar]
- Feng S, Li H, Zhao J, Pervushin K, Lowenhaupt K, Schwartz TU, and Droge P (2011). Alternate rRNA secondary structures as regulators of translation. Nat Struct Mol Biol 18, 169–176. [DOI] [PubMed] [Google Scholar]
- Fishel R, Derbyshire MK, Moore SP, and Young CSH (1991). Biochemical studies of homologous and nonhomologous recombination in human cells. Biochimie 73, 257–267. [DOI] [PubMed] [Google Scholar]
- Flemming HC, and Wingender J (2010). The biofilm matrix. Nature Reviews Microbiology 8, 623–633. [DOI] [PubMed] [Google Scholar]
- Frederiksen B, Pressler T, Hansen A, Koch C, and Hoiby N (2006). Effect of aerosolized rhDNase (Pulmozyme) on pulmonary colonization in patients with cystic fibrosis. Acta Paediatr 95, 1070–1074. [DOI] [PubMed] [Google Scholar]
- Gloag ES, German GK, Stoodley P, and Wozniak DJ (2018). Viscoelastic properties of Pseudomonas aeruginosa variant biofilms. Scientific reports 8, 9691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman SD, Obergfell KP, Jurcisek JA, Novotny LA, Downey JS, Ayala EA, Tjokro N, Li B, Justice SS, and Bakaletz LO (2011). Biofilms can be dispersed by focusing the immune system on a common family of bacterial nucleoid-associated proteins. Mucosal Immunol 4, 625–637. [DOI] [PubMed] [Google Scholar]
- Grove A (2011). Functional Evolution of Bacterial Histone-Like HU Proteins. Curr Issues Mol Biol 13, 1–12. [PubMed] [Google Scholar]
- Guéron M, Demaret JP, and Filoche M (2000). A Unified Theory of the B-Z Transition of DNA in High and Low Concentrations of Multivalent Ions. Biophysical Journal 78, 1070–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn JS, Bakaletz LO, and Wozniak DJ (2016). J Biol Chem 291, 12538–12546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustave JE, Jurcisek JA, McCoy KS, Goodman SD, and Bakaletz LO (2013). Targeting bacterial integration host factor to disrupt biofilms associated with cystic fibrosis. J Cyst Fibros 12, 384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall-Stoodley L, Nistico L, Sambanthamoorthy K, Dice B, Nguyen D, Mershon WJ, Johnson C, Hu FZ, Stoodley P, Ehrlich GD, et al. (2008). Characterization of biofilm matrix, degradation by DNase treatment and evidence of capsule downregulation in Streptococcus pneumoniae clinical isolates. BMC Microbiol 8, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison A, Dyer DW, Gillaspy A, Ray WC, Mungur R, Carson MB, Zhong H, Gipson J, Gipson M, Johnson LS, et al. (2005). Genomic sequence of an otitis media isolate of nontypeable Haemophilus influenzae: comparative study with H. influenzae serotype d, strain KW20. J Bacteriol 187, 4627–4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heegaard NHH, Olsen DT, and Larsen K-LP (1996). Immuno-capillary electrophoresis for the characterization of a monoclonal antibody against DNA. Journal of Chromatography A 744, 285–294. [DOI] [PubMed] [Google Scholar]
- Heydorn A, Nielsen AT, Hentzer M, Sternberg C, Givskov M, Ersboll BK, and Molin S (2000). Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology 146 (Pt 10), 2395–2407. [DOI] [PubMed] [Google Scholar]
- Ho PS, Kagawa TF, Tseng KH, Schroth GP, and Zhou GW (1991). Prediction of a crystallization pathway for Z-DNA hexanucleotides. Science 254, 1003–1006. [DOI] [PubMed] [Google Scholar]
- Ibanez de Aldecoa AL, Zafra O, and Gonzalez-Pastor JE (2017). Mechanisms and Regulation of Extracellular DNA Release and Its Biological Roles in Microbial Communities. Front Microbiol 8, 1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurcisek JA, and Bakaletz LO (2007). Biofilms formed by nontypeable Haemophilus influenzae in vivo contain both double-stranded DNA and type IV pilin protein. J Bacteriol 189, 3868–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurcisek JA, Brockman KL, Novotny LA, Goodman SD, and Bakaletz LO (2017). Nontypeable Haemophilus influenzae releases DNA and DNABII proteins via a T4SS-like complex and ComE of the type IV pilus machinery. Proc Natl Acad Sci U S A 114, E6632–E6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamashev D (2000). The histone-like protein HU binds specifically to DNA recombination and repair intermediates. The EMBO Journal 19, 6527–6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamashev D, and Rouviere-Yaniv J (2000). The histone-like protein HU binds specifically to DNA recombination and repair intermediates. EMBO J 19, 6527–6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan JB, LoVetri K, Cardona ST, Madhyastha S, Sadovskaya I, Jabbouri S, and Izano EA (2012). Recombinant human DNase I decreases biofilm and increases antimicrobial susceptibility in staphylococci. J Antibiot (Tokyo) 65, 73–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassinger SJ, and van Hoek ML (2020). Biofilm architecture: An emerging synthetic biology target. Synth Syst Biotechnol 5, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Lim SH, Lee AR, Kwon DH, Song HK, Lee JH, Cho M, Johner A, Lee NK, and Hong SC (2018). Unveiling the pathway to Z-DNA in the protein-induced B-Z transition. Nucleic Acids Res 46, 4129–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YG, Lowenhaupt K, Maas S, Herbert A, Schwartz T, and Rich A (2000). The zab domain of the human RNA editing enzyme ADAR1 recognizes Z-DNA when surrounded by B-DNA. J Biol Chem 275, 26828–26833. [DOI] [PubMed] [Google Scholar]
- Kim YG, Muralinath M, Brandt T, Pearcy M, Hauns K, Lowenhaupt K, Jacobs BL, and Rich A (2003). A role for Z-DNA binding in vaccinia virus pathogenesis. Proc Natl Acad Sci U S A 100, 6974–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo H, Allan RN, Howlin RP, Stoodley P, and Hall-Stoodley L (2017). Targeting microbial biofilms: current and prospective therapeutic strategies. Nature Reviews Microbiology 15, 740–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriakose T, and Kanneganti TD (2018). ZBP1: Innate Sensor Regulating Cell Death and Inflammation. Trends Immunol 39, 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwakye-Berko F, and Meshnick S (1990). Sequence preference of chloroquine binding to DNA and prevention of Z-DNA formation. Mol Biochem Parasitol 39, 275–278. [DOI] [PubMed] [Google Scholar]
- Kwon JA, and Rich A (2005). Biological function of the vaccinia virus Z-DNA-binding protein E3L: gene transactivation and antiapoptotic activity in HeLa cells. Proc Natl Acad Sci U S A 102, 12759–12764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kypr J, Kejnovska I, Renciuk D, and Vorlickova M (2009). Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res 37, 1713–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafer EM, Sousa R, Ali R, Rich A, and Stollar BD (1986). The effect of anti-Z-DNA antibodies on the B-DNA-Z-DNA equilibrium. J Biol Chem 261, 6438–6443. [PubMed] [Google Scholar]
- Lafer EM, Sousa R, and Rich A (1985). Anti-Z-DNA Antibody-Binding Can Stabilize Z-DNA in Relaxed and Linear Plasmids under Physiological Conditions. Embo Journal 4, 3655–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AR, Kim NH, Seo YJ, Choi SR, and Lee JH (2018). Thermodynamic Model for B-Z Transition of DNA Induced by Z-DNA Binding Proteins. Molecules 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan MW, Lucchini S, Danino V, Croinin TO, Hinton JC, and Dorman CJ (2006). The integration host factor (IHF) integrates stationary-phase and virulence gene expression in Salmonella enterica serovar Typhimurium. Mol Microbiol 59, 1831–1847. [DOI] [PubMed] [Google Scholar]
- Martins M, Henriques M, Lopez-Ribot JL, and Oliveira R (2012). Addition of DNase improves the in vitro activity of antifungal drugs against Candida albicans biofilms. Mycoses 55, 80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashburn-Warren L, Morrison DA, and Federle MJ (2010). A novel double-tryptophan peptide pheromone controls competence in Streptococcus spp. via an Rgg regulator. Mol Microbiol 78, 589–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirau PA, and Kearns DR (1983). The effect of intercalating drugs on the kinetics of the B to Z transition of poly(dG-dC). Nucleic Acids Res 11, 1931–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty T, Fisher J, Bakochi A, Neumann A, Cardoso JFP, Karlsson CAQ, Pavan C, Lundgaard I, Nilson B, Reinstrup P, et al. (2019). Neutrophil extracellular traps in the central nervous system hinder bacterial clearance during pneumococcal meningitis. Nat Commun 10, 1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moinuddin, Arjumand S, and Ali A (1998). SLE autoantibodies binding to native calf thymus DNA brominated in high salt. Lupus 7, 524–529. [DOI] [PubMed] [Google Scholar]
- Mokrzan EM, Ahearn CP, Buzzo JR, Novotny LA, Zhang Y, Goodman SD, and Bakaletz LO (2020a). Nontypeable Haemophilus influenzae newly released (NRel) from biofilms by antibody-mediated dispersal versus antibody-mediated disruption are phenotypically distinct. Biofilm 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokrzan EM, Dairo KA, Novotny LA, and Bakaletz LO (2020b). Nontypeable Haemophilus influenzae Responds to Virus-Infected Cells with a Significant Increase in Type IV Pilus Expression. mSphere 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokrzan EM, Ward MO, and Bakaletz LO (2016). Type IV Pilus Expression Is Upregulated in Nontypeable Haemophilus influenzae Biofilms Formed at the Temperature of the Human Nasopharynx. J Bacteriol 198, 2619–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller A, Gabriels JE, Lafer EM, Nordheim A, Rich A, and Stollar BD (1982). Monoclonal antibodies recognize different parts of Z-DNA. J Biol Chem 257, 12081–12085. [PubMed] [Google Scholar]
- Mulvey MA, Schilling JD, and Hultgren SJ (2001). Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect Immun 69, 4572–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, Lill JR, Roose-Girma M, Warming S, Solon M, et al. (2016). RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 540, 129–133. [DOI] [PubMed] [Google Scholar]
- Nickol J, Behe M, and Felsenfeld G (1982). Effect of the B--Z transition in poly(dG-m5dC). poly(dG-m5dC) on nucleosome formation. Proc Natl Acad Sci U S A 79, 1771–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordheim A, Lafer EM, Peck LJ, Wang JC, David Stollar B, and Rich A (1982). Negatively supercoiled plasmids contain left-handed Z-DNA segments as detected by specific antibody binding. Cell 31, 309–318. [DOI] [PubMed] [Google Scholar]
- Nordheim A, and Rich A (1983). The sequence (dC-dA)n X (dG-dT)n forms left-handed Z-DNA in negatively supercoiled plasmids. Proc Natl Acad Sci U S A 80, 1821–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotny LA, Amer AO, Brockson ME, Goodman SD, and Bakaletz LO (2013a). Structural stability of Burkholderia cenocepacia biofilms is reliant on eDNA structure and presence of a bacterial nucleic acid binding protein. PLoS One 8, e67629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotny LA, Clements JD, and Bakaletz LO (2011). Transcutaneous immunization as preventative and therapeutic regimens to protect against experimental otitis media due to nontypeable Haemophilus influenzae. Mucosal Immunol 4, 456–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotny LA, Clements JD, and Bakaletz LO (2013b). Kinetic analysis and evaluation of the mechanisms involved in the resolution of experimental nontypeable Haemophilus influenzae-induced otitis media after transcutaneous immunization. Vaccine 31, 3417–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotny LA, Jurcisek JA, Goodman SD, and Bakaletz LO (2016). Monoclonal antibodies against DNA-binding tips of DNABII proteins disrupt biofilms in vitro and induce bacterial clearance in vivo. EBioMedicine 10, 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotny LA, Jurcisek JA, Pichichero ME, and Bakaletz LO (2000). Epitope mapping of the outer membrane protein P5-homologous fimbrin adhesin of nontypeable Haemophilus influenzae. Infect Immun 68, 2119–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DB, Kim YG, and Rich A (2002). Z-DNA-binding proteins can act as potent effectors of gene expression in vivo. Proc Natl Acad Sci U S A 99, 16666–16671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papayannopoulos V, Staab D, and Zychlinsky A (2011). Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PLoS One 6, e28526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck LJ, Nordheim A, Rich A, and Wang JC (1982). Flipping of cloned d(pCpG)n.d(pCpG)n DNA sequences from right- to left-handed helical structure by salt, Co(III), or negative supercoiling. Proc Natl Acad Sci U S A 79, 4560–4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl FM, and Jovin TM (1972). Salt-induced Co-operative Conformational Change of a Synthetic DNA: Equilibrium and Kinetic Studies with Poly(dG-dC) J Mol Biol 67, 375–396. [DOI] [PubMed] [Google Scholar]
- Rajendran A, Endo M, Hidaka K, and Sugiyama H (2013). Direct and real-time observation of rotary movement of a DNA nanomechanical device. J Am Chem Soc 135, 1117–1123. [DOI] [PubMed] [Google Scholar]
- Ramesh N, and Brahmachari SK (1989). Structural alteration from non-B to B-form could reflect DNase I hypersensitivity. J Biomol Struct Dyn 6, 899–906. [DOI] [PubMed] [Google Scholar]
- Ray BK, Dhar S, Henry C, Rich A, and Ray A (2013). Epigenetic regulation by Z-DNA silencer function controls cancer-associated ADAM-12 expression in breast cancer: cross-talk between MeCP2 and NF1 transcription factor family. Cancer Res 73, 736–744. [DOI] [PubMed] [Google Scholar]
- Rice PA, Yang S, Mizuuchi K, and Nash HA (1996). Crystal structure of an IHF-DNA complex: a protein-induced DNA U-turn. Cell 87, 1295–1306. [DOI] [PubMed] [Google Scholar]
- Rich A, and Zhang S (2003). Z-DNA: the long road to biological function. Nat Rev Genet 4, 566–572. [DOI] [PubMed] [Google Scholar]
- Rmaile A, Carugo D, Capretto L, Zhang X, Wharton JA, Thurner PJ, Aspiras M, Ward M, and Stoodley P (2013). Microbial tribology and disruption of dental plaque bacterial biofilms. Wear 306, 276–284. [Google Scholar]
- Robledo-Avila FH, Ruiz-Rosado JD, Brockman KL, and Partida-Sanchez S (2020). The TRPM2 Ion Channel Regulates Inflammatory Functions of Neutrophils During Listeria monocytogenes Infection. Front Immunol 11, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocco CJ, Bakaletz LO, and Goodman SD (2018). Targeting the HUbeta Protein Prevents Porphyromonas gingivalis from Entering into Preexisting Biofilms. J Bacteriol 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocco CJ, Davey ME, Bakaletz LO, and Goodman SD (2017). Natural antigenic differences in the functionally equivalent extracellular DNABII proteins of bacterial biofilms provide a means for targeted biofilm therapeutics. Mol Oral Microbiol 32, 118–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DA, Pinkner JS, Jones JM, Walker JN, Clegg S, and Hultgren SJ (2008). Utilization of an intracellular bacterial community pathway in Klebsiella pneumoniae urinary tract infection and the effects of FimK on type 1 pilus expression. Infect Immun 76, 3337–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safina A, Cheney P, Pal M, Brodsky L, Ivanov A, Kirsanov K, Lesovaya E, Naberezhnov D, Nesher E, Koman I, et al. (2017). FACT is a sensor of DNA torsional stress in eukaryotic cells. Nucleic Acids Res 45, 1925–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders SH, Tse ECM, Yates MD, Otero FJ, Trammell SA, Stemp EDA, Barton JK, Tender LM, and Newman DK (2020). Extracellular DNA Promotes Efficient Extracellular Electron Transfer by Pyocyanin in Pseudomonas aeruginosa Biofilms. Cell 182, 919–932 e919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seviour T, Winnerdy FR, Wong LL, Shi X, Mugunthan S, Foo YH, Castaing R, Adav SS, Subramoni S, Kohli GS, et al. (2021). The biofilm matrix scaffold of Pseudomonas aeruginosa contains G-quadruplex extracellular DNA structures. NPJ Biofilms Microbiomes 7, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SI, Ham S, Park J, Seo SH, Lim CH, Jeon H, Huh J, and Roh TY (2016). Z-DNA-forming sites identified by ChIP-Seq are associated with actively transcribed regions in the human genome. DNA Res 23, 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha P, Emura T, Koirala D, Cui Y, Hidaka K, Maximuck WJ, Endo M, Sugiyama H, and Mao H (2016). Mechanical properties of DNA origami nanoassemblies are determined by Holliday junction mechanophores. Nucleic Acids Res 44, 6574–6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley JT, Lee JS, and Decoteau WE (1984). Left-handed “Z” DNA antibodies in rheumatoid arthritis and systemic lupus erythematosus. J Rheumatol 11, 633–637. [PubMed] [Google Scholar]
- Singh RK, Liang D, Gajjalaiahvari UR, Kabbaj MH, Paik J, and Gunjan A (2010). Excess histone levels mediate cytotoxicity via multiple mechanisms. Cell Cycle 9, 4236–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberger RE, and Holden PA (2005). Extracellular DNA in single- and multiple-species unsaturated biofilms. Appl Environ Microbiol 71, 5404–5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storisteanu DM, Pocock JM, Cowburn AS, Juss JK, Nadesalingam A, Nizet V, and Chilvers ER (2017). Evasion of Neutrophil Extracellular Traps by Respiratory Pathogens. Am J Respir Cell Mol Biol 56, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suck D, and Oefner C (1986). Structure of DNase I at 2.0 A resolution suggests a mechanism for binding to and cutting DNA. Nature 321, 620–625. [DOI] [PubMed] [Google Scholar]
- Swinger KK, and Rice PA (2004). IHF and HU: flexible architects of bent DNA. Current Opinion in Structural Biology 14, 28–35. [DOI] [PubMed] [Google Scholar]
- Swinger KK, and Rice PA (2007). Structure-based analysis of HU-DNA binding. J Mol Biol 365, 1005–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesny B, Marcatti M, Ahmad A, Montalbano M, Brunyanszki A, Bibli SI, Papapetropoulos A, and Szabo C (2018). Mitochondrial DNA damage and subsequent activation of Z-DNA binding protein 1 links oxidative stress to inflammation in epithelial cells. Sci Rep 8, 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temiz NA, Donohue DE, Bacolla A, Luke BT, and Collins JR (2012). The role of methylation in the intrinsic dynamics of B- and Z-DNA. PLoS One 7, e35558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JR, Bolla RI, Rumbyrt JS, and Schlessinger D (1985). DNase I-resistant nontranscribed spacer segments of mouse ribosomal DNA contain poly(dG-dT).poly(dA-dC). Proc Natl Acad Sci U S A 82, 7595–7598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas TJ, Gunnia UB, and Thomas T (1991). Polyamine-induced B-DNA to Z-DNA conformational transition of a plasmid DNA with (dG-dC)n insert. J Biol Chem 266, 6137–6141. [PubMed] [Google Scholar]
- Thomas TJ, and Messner RP (1988). Structural Specificity of Polyamines in Left-handed Z-DNA Formation J Mol Biol 201, 463–467. [DOI] [PubMed] [Google Scholar]
- Timoshenko S, and Goodier J (1970). Theory of Elasticity, third edn (McGraw Hill Higher Education; ). [Google Scholar]
- Tran TM, MacIntyre A, Hawes M, and Allen C (2016). Escaping Underground Nets: Extracellular DNases Degrade Plant Extracellular Traps and Contribute to Virulence of the Plant Pathogenic Bacterium Ralstonia solanacearum. PLoS Pathog 12, e1005686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vorst EPC, Weber C, and Donners M (2018). A Disintegrin and Metalloproteases (ADAMs) in Cardiovascular, Metabolic and Inflammatory Diseases: Aspects for Theranostic Approaches. Thromb Haemost 118, 1167–1175. [DOI] [PubMed] [Google Scholar]
- Wang G, Christensen LA, and Vasquez KM (2006). Z-DNA-forming sequences generate large-scale deletions in mammalian cells. Proc Natl Acad Sci U S A 103, 2677–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitchurch CB, Tolker-Nielsen T, Ragas PC, and Mattick JS (2002). Extracellular DNA required for bacterial biofilm formation. Science 295, 1487. [DOI] [PubMed] [Google Scholar]
- Wittig B, Dorbic T, and Rich A (1991). Transcription is associated with Z-DNA formation in metabolically active permeabilized mammalian cell nuclei. Proc Natl Acad Sci U S A 88, 2259–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias W, Jaworski A, Larson JE, and Wells RD (1988). The B- to Z-DNA equilibrium in vivo is perturbed by biological processes. Proc Natl Acad Sci U S A 85, 7069–7073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavarykina TM, Atkarskaya MV, and Zhizhina GP (2019). The Structural and Functional Properties of Z-DNA. Biophysics 64, 671–682. [Google Scholar]
- Zhang L, Peeples ME, Boucher RC, Collins PL, and Pickles RJ (2002). Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J Virol 76, 5654–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Cui Y, An R, Liang X, Li Q, Wang H, Wang H, Fan Y, Dong P, Li J, et al. (2019). Topologically Constrained Formation of Stable Z-DNA from Normal Sequence under Physiological Conditions. J Am Chem Soc 141, 7758–7764. [DOI] [PubMed] [Google Scholar]
- Zhou C, Zhou F, and Xu Y (2009). Comparative analyses of distributions and functions of Z-DNA in Arabidopsis and rice. Genomics 93, 383–391. [DOI] [PubMed] [Google Scholar]
- Zhou H, Sathyamoorthy B, Stelling A, Xu Y, Xue Y, Pigli YZ, Case DA, Rice PA, and Al-Hashimi HM (2019). Characterizing Watson-Crick versus Hoogsteen Base Pairing in a DNA-Protein Complex Using Nuclear Magnetic Resonance and Site-Specifically (13)C- and (15)N-Labeled DNA. Biochemistry 58, 1963–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweig M, Schork S, Koerdt A, Siewering K, Sternberg C, Thormann K, Albers SV, Molin S, and van der Does C (2014). Secreted single-stranded DNA is involved in the initial phase of biofilm formation by Neisseria gonorrhoeae. Environ Microbiol 16, 1040–1052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Mature biofilms were resistant to DNase, related to Figure 2 (A) Representative images of UPEC biofilms incubated with DNase. (B) Increasing concentrations of DNase were added at initial biofilm seeding of NTHI, Kp and UPEC (prevention of biofilm development) or 24h after seeding (disruption of mature biofilms). After DNase treatment, biofilms were incubated for an additional 16h, then stained with LIVE/DEAD® viability stain, fixed, visualized via CLSM, and images analyzed by COMSTAT. Error bars represent the standard error of the mean (SEM). Statistical significance of DNase treatment compared to control (- DNase) was assessed by unpaired t-tests, **, P<0.01 and ***, P<0.001. NTHI prevention, n=4 and disruption, n=3; Kp prevention and disruption, n=5; UPEC prevention and disruption, n=3. DNase inhibited the formation of biofilms by UPEC, Kp and NTHI, but did not disrupt these structures once formed.
Figure S2. Verification of the specificity of B- and Z-DNA specific antibodies, related to Figure 1 and S5 (A) Brominated poly(dGdC) (10 μg/ml) or non-brominated poly(dGdC) (10 μg/ml) were incubated in buffer, the absorbance values at 260nm and 295nm were measured, then the A260/295 ratio was calculated. Poly(dGdC) had a ratio of 10.51 ± 1.33 indicative of B-DNA, while Br-poly(dGdC) had a ratio of 2.73 ± 0 which is indicative of Z-DNA. Statistical significance compared to control (non-brominated poly(dGdC) was assessed by unpaired t-tests, ***P<0.001, n=3. (B) Representative ELISA plate image demonstrated the specificity of monoclonal antibodies to B- or Z-form DNA. An ELISA plate was coated with 1 μg of poly dGdC (B-DNA) or brominated poly dGdC (Z-DNA102), blocked with 0.5% (w/v) BSA, then probed with murine IgG1 (ms isotype) and naive rabbit serum (NRS) (neg. controls), or murine monoclonal antibody directed against B-DNA or Z-DNA (ms anti-B or -Z, respectively), followed by detection with a secondary goat α-mouse IgG-HRP. TMB (3,3’,5,5’-tetramethylbenzidine) was the colorimetric substrate used for HRP detection (dark gray wells). (C) Quantification of the ELISA for specificity of the B- and Z-DNA monoclonal antibodies. Statistical significance of murine α-Z-DNA and α-B-DNA were compared to control (no dGdC buffer) and assessed by Tukey’s multiple comparisons test, **P<0.01, ***P<0.001, and P<0.0001, n=4. (D) Western Blot analysis of bacterial biofilm whole-cell lysates (L) (20 μg) probed with the indicated antibodies (M – molecular mass standards), n=4. The monoclonal antibodies used in this study which are directed to B-DNA and Z-DNA are specific and do not cross react with bacterial lysates. (E) Brominated gDNA (4 μg/ml) or non-brominated gDNA (4 μg/ml) were incubated in buffer or 3.6M NaCl, absorbance values at 260nm and 295nm were measured, then the A260/295 ratio was calculated. Brominated Poly(dGdC) and non-brominated poly(dGdC) were used as internal controls. gDNA had a ratio of 10.69 ± 3.36 indicative of B-DNA. Conversely, Br-gDNA had a ratio of 2.19 ± 0.06 and gDNA 3.6M NaCl had a ratio of 2.76 ± 0.53 which are both indicative of Z-DNA. Statistical significance compared to control (non-brominated) was assessed by unpaired t-tests, n=3. (F) Poly(dGdC) and Br-poly(dGdC) were incubated in the presence or absence of Pulmozyme® (5U/ml) for 10 minutes at 37°C. The DNA degradation was assessed by gel electrophoresis which showed that Br-poly(dGdC) was protected from the activity of DNase, n=3. ***P<0.001. Error bars represent the SEM.
Figure S3. B-DNA and Z-DNA increased within the EPS of biofilms, related to Figure 1 (A) 24, 40, 90h, or (B) 1 week old NTHI, Kp, and UPEC biofilms were examined. Unfixed biofilms were incubated with a murine monoclonal antibody against either B-DNA (5 μg/ml) or Z-DNA (5 μg/ml), then revealed via incubation with goat α-mouse IgG conjugated to Alexa Fluor® 488. Biofilms were visualized by CLSM. B-DNA (cyan) and Z-DNA (yellow) accumulated as the biofilms matured, n=3. (C) NTHI biofilms (40h) were incubated with the same primary, control antibodies, and buffer (no primary antibody), then visualized with the same secondary antibodies as described above, n=3. No cross-reactivity of the secondary conjugated antibodies was observed.
Figure S4. Removal of B-DNA throughout biofilm development reduced Z-DNA, related to Figure 2 NTHI biofilms were initiated in the presence or absence of DNase (80 U/ml) for 40h. Unfixed biofilms were incubated with a murine monoclonal antibody against either B-DNA[3519] (5 μg/ml) or Z-DNA (5 μg/ml), then revealed via incubation with goat α-mouse IgG conjugated to Alexa Fluor® 405 and goat α-rabbit IgG conjugated to Alexa Fluor® 488. Changes in either: (A) biomass (FM™4–64 fluorescent signal); (B) Z-DNA (Alexa Fluor® 488) or (C) B-DNA (Alexa Fluor® 405) were quantified by image J software. (D) The ratio of relative B-DNA to Z-DNA of IF intensity of DNase incubated biofilms were compared to the intensity of the media control. Error bars represent the SEM. Statistical significance compared to control (no DNase) was assessed by unpaired t-tests, *P>0.05 **P<0.01, n=4.
Figure S5. Z-DNA-specific antibodies stimulated biofilm formation, CeCl3 induced Z-DNA, whereas chloroquine converted Z-DNA to B-DNA and partitioned planktonic NTHI differentially, related to Figure 3, 4 &S2 (A) NTHI and UPEC biofilms were initiated in the presence of increased amounts (1 or 5 μg/ml) of either IgG from unimmunized sheep serum (pAB sheep), sheep polyclonal α-Z-DNA (pAB-Z-DNA) or media for 16h, n=4. (B) UPEC biofilms were initiated in the presence of rabbit monoclonal α-Z-DNA (mAB Z-DNA), rabbit monoclonal isotype 2b (isotype control) or media for 16h. Biofilm average thickness was analyzed by LIVE/DEAD® staining with COMSTAT image analysis. Error bars represent the SEM. Statistical significance compared to control was assessed by paired t-tests, *P<0.05, **P<0.01, n=6. (C) Genomic DNA isolated from NTHI (4 μg/ml) was incubated with increased concentrations of CeCl3 (0, 0.25, 0.5, and 1 mM) in buffer, and analyzed by spectroscopic absorbance ratio assay. The ratio determined for samples that contained either 0 or 0.25mM CeCl3 were 14.17 ± 6.79 or 15.61 ± 4.75 respectively, which indicated B-DNA. In contrast, ratios determined for 0.5 and 1mM CeCl3 were 3.90 ± 0.76 or 2.90 ± 1.51, respectively, which indicated Z-DNA. Error bars represent the SEM. Statistical significance compared to control was assessed by unpaired t-tests, *P<0.05, n=3. (D) Growth curves of NTHI grown planktonically in the presence of increased concentrations of CeCl3 (0.25, 0.5, and 1 mM) indicated a minimal effect on growth (n=3, averaged curves). (E) NTHI was allowed to form a biofilm for 24h in the presence of CeCl3. At 40h, relative CFU of the planktonic and biofilm populations were assessed to calculate the relative percent of planktonic versus biofilm resident NTHI which represents the ‘partition coefficient’, n=3. (F) To determine the Young’s modulus of treated and untreated NTHi biofilms, the slope of the lower linear portion of the force-displacement curve that corresponded to 0 – 40% strain (Figs. 3 and 4), where R2 ≥ 0.9, was determined. Using the slope, the Young’s modulus was calculated according to equation 1. Depicted is the force-displacement region of a representative untreated control NTHi biofilm that corresponded to 0 – 40% strain. The linear fit of the curve is indicated in red; the dashed lines indicate the 95% confidence interval. The R2 value of the linear fit is indicated. (G) Poly(dGdC) DNA (10 μg/ml) was incubated with 0.1 mM chloroquine, 3.6M NaCl or a combination of both, followed by analysis of the spectroscopic absorbance ratio assay. The absorbance 260/295nm ratios calculation for poly(dGdC) and 0.1mM chloroquine were 11.36 ± 1.72 and 8.25 ± 0.90 respectively which indicated B-DNA, while the ratio value calculated for 3.6M NaCl was 3.62 ± 0.37, which confirmed Z-DNA, n=3. Chloroquine prevented NaCl- induced Z-DNA formation as determined by the ratio calculation of 6.61 ± 0.32. (H) Planktonic NTHI growth curves with increased concentrations of chloroquine (0.01, 0.05, 0.1, and 0.2 mM) indicated no effect on growth (n=3, averaged curves). (I) NTHI was allowed to form a biofilm for 24h followed by the addition of increasing concentrations of chloroquine. At 40h, the partition coefficients were calculated, as above. Error bars represent the SEM. Statistical significance compared to control was assessed by paired t-tests, *P<0.05, n=7.
Figure S6. Modulation of B/Z eDNA increased the susceptibility of biofilms to DNase, related to Figure 3 and Table S1 NTHI biofilms were formed for 40h and incubated with chloroquine, DNase, or both, then stained with LIVE/DEAD®, fixed, visualized via CLSM, and images analyzed by COMSTAT, quantified in Fig 3K, n=7. (A) Representative images of NTHI biofilms incubated with DNase and/or chloroquine. (B) Representative images of IF labeling of B-DNA with murine α-B-DNA[3519] and Z-DNA with rabbit αZ-DNA[Z22] in 4 unique specimens collected during exacerbation of clinical disease and assayed in Fig 3M, n=1. Labeling was revealed with goat anti-rabbit IgG conjugated to Alexa Fluor® 647, goat anti-mouse IgG conjugated to Alexa Fluor® 488. Scale bar 10μm.
Figure S7. PMA-induced NET eDNA was predominantly comprised of B-DNA, related to Figure 5 Neutrophils (1×105) were allowed to attach to an 8 well glass chamberslide and incubated in the presence or absence of CbpA (500nM) upon induction with PMA (200nM) for 3.5h at 37°C. NETs were then fixed, washed, blocked with 10% goat serum followed by incubation with rabbit polyclonal antibody against human neutrophil elastase (1:100), rabbit monoclonal antibody against Z-DNA (5 μg/ml), and a murine monoclonal antibody against B-DNA (5 μg/ml). Then NETs were incubated with goat α-rabbit IgG conjugated Alexa Fluor® 594 and goat α-mouse IgG conjugated Alexa Fluor® 488, wheat germ agglutinin (WGA 350), and SYTOX™ nucleic acid stain. NETs were imaged by CLSM. (A) Representative image of no PMA control, n=3. (B) Representative images of NETs formed in the presence or absence of CbpA (no Z-DNA induction), n=3.
Data Availability Statement
The published article includes all data sets generated or analyzed during this study except for additional original replicate data [Figures 1 and 5; Supplemental Figures 2, 3, and 7] which is deposited within Mendeley Data [DOI: 10.17632/fpwbypb4kd.1].