

Abstract
Alzheimer’s disease (AD) is the most common form of dementia in the elderly followed by vascular dementia. In addition to clinically diagnosed dementia, cognitive dysfunction has been reported in diabetic patients. Recent studies are now beginning to recognize type 2 diabetes mellitus (T2DM), characterized by chronic hyperglycemia and insulin resistance, as a risk factor for AD and other cognitive disorders. While studies on insulin action have remained traditionally in the domain of peripheral tissues, the detrimental effects of insulin resistance in the central nervous system on cognitive dysfunction are increasingly being reported in recent clinical and preclinical studies. Brain functions require continuous supply of glucose and oxygen and a tight regulation of metabolic processes. Loss of this metabolic regulation has been proposed to be a contributor to memory dysfunction associated with neurodegeneration. Within the above scenario, this review will focus on the interplay among oxidative stress (OS), insulin resistance and AMPK dysfunctions in the brain by highlighting how these neurotoxic events contribute to neurodegeneration. We provide an overview on the detrimental effects of OS on proteins regulating insulin signaling and how these alterations impact cell metabolic dysfunctions through AMPK dysregulation. Such processes, we assert, are critically involved in the molecular pathways that underlie AD.
Keywords: Alzheimer disease and type 2 diabetes, oxidative and nitrosative stress, insulin resistance, neurodegeneration, biliverdin reductase-A, AMP-activated protein kinase
1. Introduction
The ever-increasing life expectancy has led to a dramatic increase in the prevalence of age-associated disorders, including type 2 diabetes mellitus (T2DM) and age-related dementia and Alzheimer disease (AD). Notably, there is a growing body of epidemiological evidence suggesting that metabolic syndrome and T2DM increase the risk of developing age-related cognitive decline, mild cognitive impairment (MCI), vascular dementia, and AD (reviewed in [1]).
Although the brain only represents 2% of the body’s total weight, it has a high demand for energy compared to other tissues, largely attributed to the extremely active and complex processes involved in neuronal transmission [2]. Neurons are a paramount example of high energy expenditure for their function and survival. This situation is reflected in large metabolic rates in neurons and by the comparatively higher sensibility of brain tissues to oxygen and glucose deprivation. Reactions controlling the conversion of nutrients into available cytosolic levels of ATP are critical to generate the potential metabolic work that is available to a neuron at any given time [3, 4].
Approximately 70% of the total energy is consumed for regulation of neuronal signaling (resting and action potentials, postsynaptic receptor signaling, the glutamine cycle, and postsynaptic Ca2+), and approximately 30% is used for non-signaling conduction activities (proteins, phospholipids, etc.). Indeed, failure to maintain basal energy levels can result in synaptic loss and cognitive impairment within few minutes, thus rendering the brain highly vulnerable to energy deficit-mediated damage [5, 6].
Among energy fuels, glucose is an essential substrate for the adult brain, and at least 25% of consumed glucose is used to drive basal brain activities [7]. Glucose metabolism sustains the physiological functions of the brain through glycolysis and mitochondrial oxidative phosphorylation (OXOPHOS), and its product, ATP, is the electrochemical basis for the maintenance of neurons and non-neuronal cells. Therefore, glucose metabolism and mitochondrial functions are essential ATP sources crucial for neuronal homeostasis [7].
Neuronal glucose utilization includes mechanisms that control its uptake. This involves glucose specific transporters (GLUTs), insulin signaling pathways and the entry of glycolytic metabolites into mitochondria that are further oxidized into the Krebs cycle [7].
Glucose import into the brain from the circulation is primarily mediated through the insulin-insensitive GLUT1, which is expressed by endothelial cells and astrocytes at the blood brain barrier (BBB) [8]. Within the brain parenchyma, GLUT3 and GLUT1, both of which are considered insulin-insensitive, are expressed widely by neurons and glial cells, respectively [8].
There is also evidence to support some functions of insulin in regulating brain glucose uptake. Indeed, several studies have been focused to unravel the pathological mechanisms and clinical implications that result from aberrant insulin signaling- i.e insulin resistance- both in the brain and periphery [8] (Figure 1). While much is known about the molecular aspects underlying insulin resistance in metabolic disorders, including obesity and T2DM, the exact mechanisms through which aberrant insulin signaling leads to cognitive decline need to be further elucidated.
Figure 1. Schematic representation of insulin signaling.
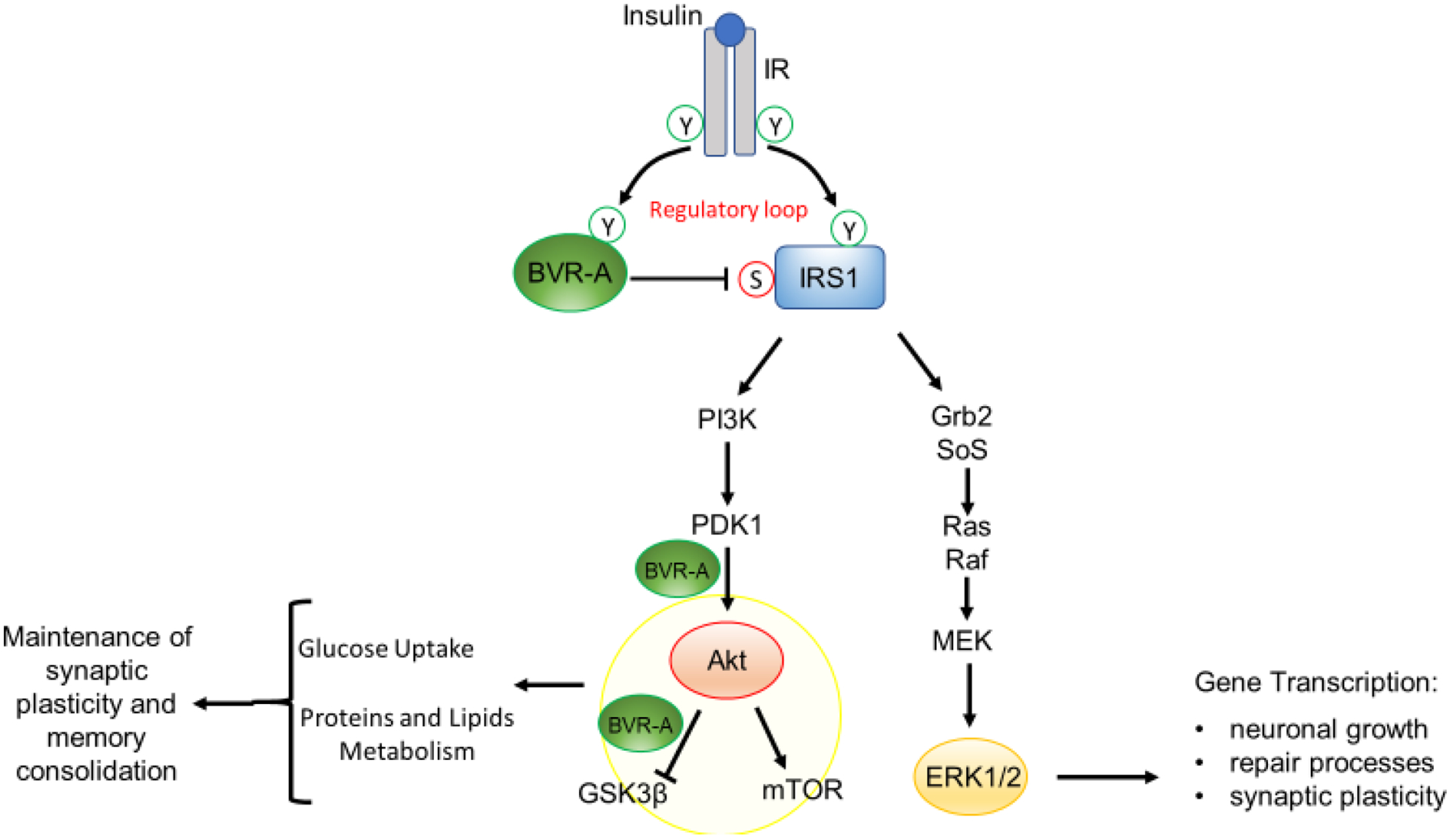
Under physiological conditions, the activation of insulin signaling requires the binding of insulin to the insulin receptor (IR), which auto-phosphorylates on Tyr residues (Y, e.g., Tyr1158/1162/1163) and promotes the receptor tyrosine kinase-mediated phosphorylation of its substrate (IRS1) on specific Tyr residues (e.g., 632). In parallel, IR phosphorylates BVR-A on specific Tyr residues and activates BVR-A to function as Ser/Thr/Tyr kinase. Then, as part of a regulatory loop, BVR-A phosphorylates IRS1 on inhibitory Ser residues (S, e.g., Ser307/312/616) to avoid IRS1 aberrant activation in response to IR. Once activated, IRS1 works as a scaffold protein, driving the activation of the two main arms of the insulin signaling: (1) the Ras/Raf/MAPK pathway (ERK1/2) mainly involved in gene transcription; and (2) the PI3K/Akt axis that is critical for glucose uptake as well as for protein and lipid metabolism. Moreover, Akt promotes the phosphorylation of several targets, among which are: (1) GSK3β (on Ser9, inhibitory site), which has a role in energy production; and (2) mTOR (on Ser2448, activating site), which regulates protein synthesis and autophagy. Within this axis, BVR-A works as scaffold protein facilitating the PDK1-mediated activation of Akt and the Akt-mediated inhibition of GSK3β. Arrows represent stimulation; lines represent inhibition. See text for more details.
In addition, living cells display multiple complementary mechanisms for regulation of energy homeostasis. Changes in the ATP/AMP gate the activity of multiple metabolic sensors which, in turn, induce a specific signaling cascade for short and long-term adaptations of neuronal functions. For example, all known eukaryotic cells, including neurons, harbor energy sensors, such as AMP-activated protein kinase (AMPK), which tend to restore ATP concentrations by decreasing anabolic and/or energy consuming processes, while increasing energy production through catabolism of post-energy challenges [9]. In model systems, sustained decreases in AMPK activity accompany insulin resistance, whereas AMPK activation increases insulin sensitivity. As well, activation of AMPK decreases endoplasmic reticulum (ER) stress and oxidative stress OS, and activates autophagy, all of which appear to be involved in the pathogenesis of insulin resistance [10].
In the sections of this review below, we discuss the interplay among OS, brain insulin resistance and AMPK dysfunctions to provide mechanistic insights into characterization of neuronal energy metabolism impairment, a key event for neurodegeneration both in T2DM and Alzheimer disease (AD) [11].
2. Alzheimer disease
Neurodegenerative disorders, including AD, share several pathological features, among which, bioenergetic defects are prominent characteristics [11]. AD is the predominant form of dementia affecting the elderly, and AD causes progressive degeneration of the brain. At the cellular level, AD is marked by a selective and progressive loss of nerve cells, dendritic spines and synapses, impaired neurotransmission, and progressive isolation of remaining nerve cells [12]. Neuropathologically, AD is characterized by deposition of extracellular β-amyloid (Aβ40 and Aβ42) in the brain parenchyma forming neuritic (senile) plaques (SPs), the presence of Aβ42 oligomers, β-amyloid angiopathy around cerebral blood vessels, and intraneuronal deposits of hyper-phosphorylated, abnormally conformed, and truncated tau protein in the form of neurofibrillary tangles (NFTs) [13].
Accumulating evidence indicates that in the development of AD, pathophysiological changes can occur up to 20–30 years before clinical symptoms manifest [14]. The limited amount of information regarding early stages of the neurodegenerative process, combined with to the fact that the disease’s clinical features that can lead to a diagnosis of AD occur at relatively advanced stages at which the degenerative processes have reached thresholds, emphasize the urgent need to identify the molecular aspects of the disease that are initiated in the pre-symptomatic stages.
Among putative candidates for such needs, emerging studies reveal that defects of energy metabolism, including glucose metabolism and mitochondrial activities, initiate many years before clinical manifestation of AD [15]. For example, with age, neuronal glucose uptake and proper mitochondrial function decline, reducing OXOPHOS activity, promoting increased oxidant species production [10]. These events reportedly occur decades before the onset of clinical symptoms of AD, exemplified in a study performed on young adults at high risk of developing AD [16].
Published reports indicate that global cerebral glucose metabolic rate is decreased by 20–25 % in AD patients, with the earliest change registered being in the hippocampus. In the 1980s, fluorodeoxyglucose positron emission tomography (FDG PET) studies showed brains from AD patients used less glucose than those from control subjects [17–21]. PET studies also showed decreased oxygen consumption by AD brains [22]. These studies elevated interest in a potential metabolic component for this disease. In addition, biochemical studies demonstrated activity deficiencies in bioenergetic-related enzymes such as Complex IV (cytochrome c oxidase), suggesting a mitochondrial defect [23].
While 18F-FDG PET studies have shown reduced brain glucose uptake in regions vulnerable to AD pathology [18–21], it is unclear whether an overall failure of regulation of brain glucose metabolism is a key etiopathogenic factor in AD and whether abnormalities of brain glucose homeostasis in AD are related to peripheral glucose concentration. Abnormalities in brain glucose homeostasis are intrinsic to AD pathogenesis and begin several years prior to clinical symptoms [14]. Interestingly, in a recent study by An and colleagues it has been reported that higher brain tissue glucose concentration, reduced glycolytic flux and lower levels of GLUT3 are related to severity of AD pathology and the expression of AD symptoms [24]. In addition, increasing fasting plasma glucose levels (but not the hyperglycemia as observed in T2DM individuals) are associated with higher brain tissue glucose concentrations [24]. Indeed, the longitudinal associations between fasting plasma glucose and brain tissue glucose concentrations are not driven by individuals with T2DM [24]. Hence, these observations suggest that it is not so much hyperglycemia as just reduced brain metabolism that favors higher brain glucose levels and related neurotoxicity in AD [24].
Furthermore, neuronal glucose hypometabolism negatively regulates the metabolic capacity of cells and leads to increased OS levels [17]. Moreover, modifications of key enzymes involved in glycolsis and in the Krebs cycle are probably major contributors to the hypometabolism observed in AD [11, 25]. Our group contributed this notion by demonstrating that aldolase, triosephosphate isomerase, glyceraldehyde-3-phosphate dehydrogenase, phosphoglycerate mutase 1 and α-enolase are oxidatively damaged in brain areas affected by AD pathology [11, 25]. Further, oxidatively modified aconitase, creatine kinase and ATP synthase are also present, which may contribute to the reduced glucose metabolism and ATP production in mitochondria [25]. Accordingly, several studies have shown that AD patients exhibit very early signs of metabolic changes in neurons and glial cells [11].
3. Type 2 diabetes mellitus
As well as growing older, a significant fraction of the populations of the developed economies also are obese and sedentary. Although this is most marked in the developed nations, it is truly an international trend. In addition, since many children in developed nations show dramatic increases in body mass index (BMI), this is a deeply concerning indicator of future health problems [26, 27]. For example, obesity increases risk of cancers, cardiovascular disease, and metabolic syndromes including T2DM [26, 27]. According to the International Diabetes Federation, T2DM affects 246 million people worldwide with an expected prevalence of 380 million by 2025, representing as much as 7.1% of the global adult population.
T2DM is a metabolic disease characterized by the relative resistance to peripheral insulin action, resulting in impaired glycemic control. Although genome-wide association studies have revealed that certain single nucleotide polymorphisms (SNPs) related to beta-cell function predispose one to the development of T2DM [28, 29], clinical characteristics such as obesity and lack of physical activity are regarded as the most important risk factors [30]. These latter predispose people to metabolic changes, which accumulate over the time and drive the inflammation and cell stress, leading to insulin resistance and ultimately to T2DM [30].
T2DM individuals show reduced glucose uptake/metabolism in the brain as measured by 18F-FDG PET scanning [31–35]. Interestingly, while non-diabetic subjects show a positive association between basal and glucose-stimulated insulin secretion and brain glucose uptake during euglycaemic-hyperinsulinaemic clamp, no such association was observed in T2DM individuals, thus suggesting that insulin resistance may contribute to reduced brain glucose uptake/metabolism in T2DM [31, 32]. On the other hand, as proposed by others, this phenomeon may represent an adaptative response to reduce the brain’s exposure to excessive circulating levels of glucose [35]. T2DM might be associated with defects in cerebral glucose metabolism stemming from decreased glucose transport across the BBB, a finding that might limit the brain’s ability to sense and respond appropriately to increments in peripheral glucose [31–35]. This defect may be driven by increasing insulin resistance, leading to further blunting of the brain glucose response to elevations in peripheral glucose [31–35]. In addition to that, Backestrom et al. reported that hyperglycemia affects neuronal and behavioural functions in participants with T2DM when their working memory is challenged, thus suggesting that working memory and the ability to perform complex activities during hyperglycaemic episodes are compromised [31].
In different clinical studies, an association of T2DM and neurodegenerative disorders as well as decline in memory has been described. A series of longitudinal studies reported that glucose intolerance and impairment of insulin secretion are associated with a higher risk to develop dementia or AD [36–38]. Indeed, it was shown that MCI subjects with normoglycemia at baseline had less functional and global cognitive decline, less whole-brain volume loss and lower conversion to AD than MCI subjects with impaired glycemia over 2 years of observations [39].
Vascular complications might only partially explain the increased incidence of neurodegeneration observed in patients with T2DM. Among others, the causes could be impaired β-amyloid (Aβ) clearance [40], up-regulation of amyloid precursor protein (APP) expression and Aβ deposition [41], or hyperinsulinemia that is present in T2DM. It is reported that such changes may play an important role in the formation of senile plaques [42]. On the other hand, patients with AD more frequently present with an impaired glucose metabolism or T2DM [43]. In addition, to further complicate this quite intricate puzzle, not all people developing AD show metabolic disorders as a comorbidity [1].
Hence, these observations raise questions thus far unanswered: whether T2DM is a cause, consequence, or compensatory counter-regulation to neurodegeneration, and whether neuronal insulin resistance indeed represents a risk factor for AD?
4. Insulin resistance links T2DM and AD
An important link between T2DM and the various forms of dementia lies is alterations of insulin signaling, particularly at the cerebral level. This process, named brain insulin resistance, negatively impacts memory functions by impairing the metabolic fueling of neurons that become dysfunctional [1]. Of note, the brain is an insulin-sensitive organ, in which insulin, beyond participating in glucose transport/metabolism, also regulates metabolic pathways required for the maintenance of memory and learning processes that are disrupted both in T2DM and in AD [44]. Consistent with this observation, systemic infusion of insulin under euglycemic hyper-insulinemic conditions in healthy humans demonstrated improvement with verbal memory and selective attention [45] suggested to be due to improved consolidation of information [46]. Likewise, intranasal insulin improves memory in both healthy humans and many patients with Alzheimer’s disease [47–49].
Conversely, insulin resistance is a condition characterized by a relative inability of target tissues to respond to the action of insulin. As a systemic disorder, insulin resistance locally affects several insulin-responsive tissues, such as adipose tissue (AT), liver and muscles, which are not able to correctly regulate glucose homeostasis [50]. AT inflammation and dysfunction is one of the major causes of disruption of the insulin signaling cascade in obesity and also represents a central trigger for development of insulin resistance in T2DM [51–54]. A similar inflammatory process is thought to occur also in the brain finally leading to brain insulin resistance [55]. In the brain, glial cells, especially astrocytes and microglia, undergo activation under pro-inflammatory conditions. Unchecked or chronic inflammation therefore becomes deleterious, leading to progressive neuronal damage [56].
Other than inflammation, increased OS levels were demonstrated to impair the activation of insulin signaling at the cellular level, thus representing an additional triggering mechanism in the development of insulin resistance both in T2DM and AD [57].
Because of these similarities, metabolic disorders (obesity and T2DM) and AD are pathological conditions that can provide insights into shared molecular mechanisms that lead to systemic and brain insulin resistance, as well as their interplay.
5. Molecular mechanisms driving development of brain insulin resistance
Most of the information collected until the present regard the development of systemic insulin resistance that is phenotypically characterized by: (i) an early phase during which hyperinsulinemia overcomes the initial reduction of glucose uptake and thus maintains an euglycemic state (pre-diabetes); and (ii) a late phase in which insulin resistance persists, hepatic glucose production rises, and endogenous insulin production falls, resulting in fasting and postprandial hyperglycemia [58]. Within this scenario, the epidemiological association of T2DM with cognitive dysfunction ranging from MCI to manifest dementia was shown [59]. This association already starts in midlife; and the longer the duration of diabetes, the higher the risk for developing slight cognitive impairment and eventually dementia [59]. Mostly importantly, cognitive decline seems to start well ahead of the manifestation of overt diabetes, in the prediabetic state [60]. During the MCI stage, peripheral insulin resistance was found to be associated with glucose dysmetabolism in different brain areas, with significant differences between individuals who progress to AD compared to those who did not [61]. In addition, genetic models of T2DM (including db/db mice), pharmacologically-induced T2DM models (such as streptozotocin-treated mice) and rodents fed a high-fat diet develop systemic insulin resistance, hyperglycemia and strong biochemical evidence of brain insulin resistance, coupled with memory deficits, synaptic abnormalities (structural, molecular and neurophysiological) and other brain abnormalities [62–65]. These lines of evidence support the hypothesis that peripheral insulin resistance might trigger the development of brain insulin resistance that is an early neuropathological mechanism leading to T2DM-associated cognitive decline.
Although peripheral alterations in metabolic disorders might drive development of brain insulin resistance and dementia, brain insulin resistance also can develop independently, while representing – similar to metabolic disorders – an early neuropathological aspect of neurodegenerative diseases [1]. Consonant with this observation, development of brain insulin resistance was reported to be evident during the early phases of AD in post-mortem brain from amnestic MCI (aMCI) [66, 67]. Markers of brain insulin resistance such as the levels of inhibited IRS1 (phosphorylated IRS1 at residue Ser636) were found to be significantly associated with cognitive decline and such association was even stronger than those observed for canonical hallmarks of AD, i.e., Aβ and Tau [66]. Furthermore, a recent study by Kapogiannis and colleagues reported that increased levels of inhibited IRS1 evaluated in neuronal-derived extracellular vesicles were among the strongest individual predictors for the development of AD in the general population [68]. For these reasons, understanding how brain insulin resistance develops represent a key challenge for future studies.
The precise molecular mechanisms underlying the development of insulin resistance have not been completely elucidated. Under physiological conditions, the activation of insulin signaling requires the binding of insulin to its receptor (IR), which auto-phosphorylates and promotes the phosphorylation of its substrate, insulin receptor substrate 1 (IRS1) on specific Tyr residues (e.g. 612 and 632). Once activated, IRS1 works as a scaffold protein driving the activation of the two main arms of the insulin signaling: 1) the PI3K/PDK1/Akt pathway; and 2) the MAPK pathway [69] (Figure 1). In peripheral tissues, the former is critical for linking IRS1 to the metabolic actions of insulin. Indeed, Akt activation induces downstream signaling, which leads to the regulation of a network of genes controlling: (i) glucose uptake and metabolism; (ii) lipid metabolism; and (iii) protein metabolism [69]. The latter, Ras–MEK–ERK1/2 signaling, predominantly controls cell growth and differentiation [69, 70]. In the brain, the activation of the PI3K/PDK1/Akt axis also is involved in the maintenance of synaptic plasticity and memory consolidation [71], synthesis of nitric oxide, which, in turn, plays a role in learning and memory processes [72]; in contrast, the MAPK cascade activation is responsible for the induction of several genes required for neuronal and synapse growth, maintenance and repair processes, as well as serving as a modulator of hippocampal synaptic plasticity that underlies learning and memory [73] (Figure 1). Both in metabolic disorders (obesity and T2DM) and AD, the uncoupling between IR and IRS1 occurs at the cellular level, leading to the inability of insulin to promote its cellular downstream effects (Figure 2). Thus, insulin resistance is characterized by the downregulation of IR expression and/or the inactivation of IRS proteins, the latter mediated by the phosphorylation of specific Ser residues (307, 312, 636). The aberrant activation of TNF-α receptor and stress-regulated kinases [i.e., JNK, IKkB, and PKR] as well as endoplasmic reticulum (ER) stress (PKR-mediated phosphorylation of eIF2α-P), are known mediators of IRS1 inactivation [56] (Figure 2).
Figure 2. Classical mechanisms favoring brain insulin resistance development.

The brain insulin resistance phenomenon is characterized by key events such as reduced IR protein levels and/or increased IRS1 inhibitory phosphorylation levels (e.g., Ser307, Ser636), that are responsible for the uncoupling between IR and IRS1. As result, despite insulin binding to IR, IR-mediated activation of IRS1 does not occur. As a consequence, pathways downstream from IRS1 are not activated. Within this scenario, the inhibitory phosphorylation of IRS1is mediated by the aberrant activation of stress-induced kinases (i.e., JNK, IKK, PKR). Arrows represent stimulation; lines represent inhibition. See text for more details.
However, all the above-mentioned proteins are down-stream effectors shared by several signaling pathways i.e., lipid metabolism and inflammation, and thus are not exclusively related to insulin signaling, although IRS1 is among their substrates [74]. Indeed, their involvement in insulin resistance requires the activation of other pathways. Furthermore, increased inflammation is a condition sufficient, but not necessary, during the development of insulin resistance, since disruption of inflammatory pathways by JNK deletion did not attenuate the pathological features of the early stage of systemic insulin resistance [75].
These fascinating aspects open new frontiers in the comprehension of the early mechanisms promoting the development of insulin resistance. Studies from our group showed that increased OS levels are antecedent to the rise of TNF-α in the brain of a mouse model of AD [76] and leads to the inactivation of IRS1 in vitro [76], thus suggesting a direct role in the development of brain insulin resistance. The increase in ROS levels inhibits the cellular production of ATP and decreases insulin secretion and sensitivity [77]. Similarly, oxidative damage affects a variety of signaling pathways related to the unfolded protein response and protein degradation, which could lead to insulin resistance [78, 79]. Moreover, previous studies have found that age-related mitochondrial disorders increase OS in individuals with T2DM, contributing to the development and progression of AD neuropathology [80–82].
6. Oxidative stress as an early event in the development of brain insulin resistance
Reactive species, especially ROS such as superoxide radical anion, hydrogen peroxide, and hydroxyl radical ions [83], are major contributors to OS. ROS produced endogenously have a physiological significance at low levels, especially in signaling pathways [84], while these mechanisms are not precisely understood because of the dual roles ROS play as both signaling and damaging agents.
With regard to the insulin signaling, previous studies suggested that a yet unknown intermediate stage during the IR activation process, the so-called redox priming step, exists in which oxidants like hydrogen peroxide (H2O2) facilitate, while some antioxidants such as butylated hydroxyanisole (BHA) or N-acetyl-cysteine (NAC) inhibit the insulin-induced IR autophosphorylation and thus activation [85]. An oxidative modification of cysteine residues within the IR was proposed as the structural basis of the “redox priming”, with Cys1138 in the proximity of catalytic aspartate 1132 being the most prominent candidate for the priming, since the non-oxidizable IR mutant Cys1138Ala was the only IR cysteine mutant that showed defective kinase activity in functional experiments [86]. The idea of redox priming was supported by the finding that insulin stimulation itself leads to generation of endogenous H2O2 in fat cells [87, 88]. Therefore, insulin-induced H2O2 could be the priming factor facilitating IR autophosphorylation in vivo. A subsequent study found that the role of H2O2 is not restricted to the redox priming of IR, but also includes inhibition of protein tyrosine phosphatase PTP1B, which inactivates the IR by dephosphorylating A-loop phosphotyrosine [89]. Collectively, insulin-induced H2O2 plays a role of a net positive regulator of IR activation through its concerted actions on the opposite activities of IR tyrosine kinase and PTP1B phosphatase. Similar results were also reported in neuronal cells in which insulin stimulation generated a spike in H2O2, while NAC, a Gpx1-dependent H2O2 scavenger, completely abrogated both the insulin-induced H2O2 and autophosphorylation of IR at Y1150/Y1151, thereby suggesting that the H2O2 signal is a critical requirement for the activation of the IR in neurons [90].
In addition to the mechanisms outlined above, age-associated increased OS levels along with reduced glutathione (GSH) levels were reported in both human [91] and animals, the latter including either AD-relevant [92] or aging-relevant models [92]. Similarly, insulin resistance and T2DM mice showed increased OS markers and reduced GSH levels in the brain [78, 93–97], thus suggesting a close link among OS, development of brain insulin resistance, and cognitive dysfunction. Consistent with this suggestion, brain plasticity, the capability of the brain to undergo structural and functional changes in response to environmental stimuli, is finely modulated by diet and nutrient-dependent hormones, including insulin [98]. Accordingly, alteration of insulin signaling into the central nervous system may accelerate brain aging, affect brain plasticity and promote synapse loss and neurodegeneration [99].
Intriguingly, synapse loss was found to correlate with increased OS levels, thus accounting for pathological changes observed during the clinically silent prodromal phases of AD [100]. OS has been widely recognized as a prodromal factor associated with AD neurodegeneration [11, 101, 102]. Multiple factors mainly related to mitochondrial dysfunction and energy metabolism deficit contribute to elevate cellular OS [103]. Brain structure, significant unsaturated lipid composition, high oxygen consumption, relatively high levels of redox-active transition metal ions (Fe2+ and Cu+), and rapid metabolic rate all contribute to brain susceptibility to toxic effects of OS. Moreover, the natural process of aging is associated with a physiological increase of OS over time [104–106]. As individuals age, ROS are integrated, to some extent, in the aging brain, disrupting redox-related communication and leading to cellular alterations, such as senescence and cell death, due to the inability to maintain redox homeostasis. This equilibrium in the cell is particularly important to keep the balanced microenvironment needed for multiple biological processes, from bioenergetics to vesicle transport and intracellular signaling. Further, oxidative damage is greater in aged brains than it is in younger brains [107–111]. Notably, this “physiological accumulation” of oxidative damage with aging is exacerbated during neurodegeneration and is associated with loss of cognitive functions [112]. At the neuronal level, oxidative damage related to aging, strongly impairs synaptic components involved in neuronal plasticity, cytoskeletal dynamics and cellular communication, among other alterations [111, 113, 114].
Such impairment also may occur at the expense of the insulin signaling cascade, thus triggering development of brain insulin resistance. In T2DM, consistent hyperglycemia, excessive lipid and high-level advanced glycation end products (AGEs) lead to ROS production [115]. In contrast, oxidative conditions-induced formation of AGEs and oxidation of other molecules [116], leading to cellular dysfunction, including impaired energy metabolism, altered cell signaling and cell cycle, impaired cell transport mechanisms and overall dysfunctional biological activity, immune system over-activation and inflammation [11, 116, 117]. Within this picture, it is interesting to note that levels of multiple DNA base oxidation products were found to be elevated in white blood cell DNA from patients with type II diabetes as compared with age-matched controls [118]. Similarly, oxidative stress-induced DNA fragmentation was observed among different brain regions in rats who developed diabetes [119]. Whether oxidative DNA damage plays a role by decreasing transcription of essential proteins involved in glucose metabolism in T2DM brain represent an intriguing hypothesis to be elucidated.
All these with cross-talk between AGEs and their receptors (RAGE) have been involved in the pathogenesis of diabetes, and a number of severe diabetic complications, including retinopathy, nephropathy, cardiovascular disease and nervous system dysfunction [120]. With the progression of T2DM, the increased OS and the reduced capacity of antioxidant defense can particularly damage both β-cells and neurons, resulting in progression of T2DM and related dementia [121].
Several reports highlight a close connection between insulin resistance and defects in energy metabolism driven by OS [57, 80, 122–125]. AD patients show reduced brain IR sensitivity [66, 126], hypophosphorylation of the IR, and downstream second messengers such as IRS1 [67, 80, 127, 128]. Increased OS levels in AD promote multiple effects, including the inhibition of cellular energy production, and the reduction of both insulin secretion and sensitivity [11, 77, 129]. In turn, defective insulin signaling-associated impairment in glucose uptake and utilization leads to a vicious cycle, in which reduced energy production parallel increased ROS and RNS levels that contribute to oxidative/nitrosative damage to mitochondria [80, 130]. In that regard, previous studies reported that brain insulin resistance, promotes an increase of mitochondrial ROS levels finally leading to neuronal apoptosis [131–134]. Furthermore, mitochondrial ROS overproduction enhances the accumulation of Aβ peptides and induces oxidative damage to proteins, lipids and nucleic acids in the brain under insulin resistance conditions [135–138]. In addition, brain is one of the most cholesterol-rich organs, and cholesterol oxidation products, i.e., oxysterols (e.g., 27-OHC, 7β-OHC, and 7-KC) were found elevated in AD brain and further exacerbate cell-damage by sustaining free-radical chain reactions [139–141] and insulin resistance [142]. Alterations of mitochondrial functions during brain insulin resistance also led to defects in synaptic plasticity mechanisms [143, 144]. Hence, brain insulin resistance, by favoring the imbalance in mitochondrial dynamics and functions within the brain, contribute to disturbances in neuronal apoptosis, as well as synaptic plasticity, cognitive decline, and cerebral degeneration.
7. Oxidative stress-induced damage in brain to proteins of the insulin signaling cascade
While a plethora of papers highlight the role for OS in favoring development of brain insulin resistance, whether increased ROS/RNS production also favor damage of proteins within the insulin signaling cascade is poorly explored. In our opinion, this represents an intriguing and novel aspect particularly in light of previous data from the Butterfield group showing OS-induced damage to proteins regulating cell metabolism in AD [25].
We identified two oxidatively modified proteins in AD brain or AD animal models highly relevant to insulin signaling: insulin degrading enzyme (IDE) [145–149]; and biliverdin reductase-A (BVR-A) [76, 150–154]. Moreover, both IDE and BVR-A were found to be dysregulated in T2DM/obesity peripheral tissues [155–160], thus suggesting their potential contribution to brain dysfunctions (Figure 3).
Figure 3. Oxidative stress-induced damage of IDE and BVR-A is associated with development of brain insulin resistance.
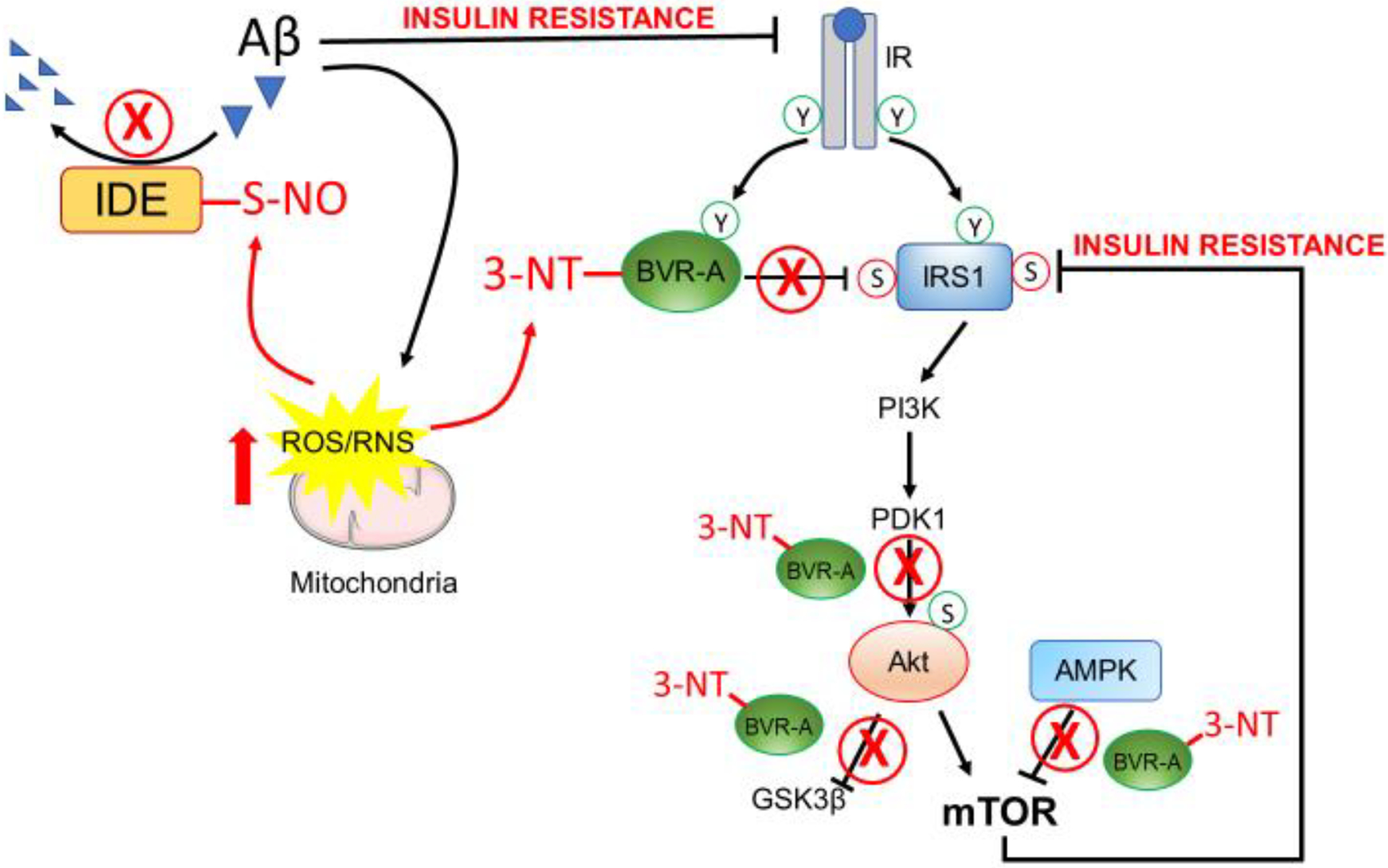
Mitochondria impairment results in increased levels of ROS/RNS that are able to damage proteins and lipids within cells. In particular, increased 3-NT modifications of BVR-A occur in AD brain and are responsible for reduced BVR-A activation and scaffold protein function. Such events result in the hyper-activation of IRS1 upstream in the pathway and in reduced activation of Akt downstream from IRS1. Moreover, the oxidative modification of BVR-A results in reduced ability for BVR-A to favor the AMPK-mediated inhibition of mTOR. As consequence, the hyper-activation of mTOR occurs and is responsible for the inhibition of IRS1 and thus the development of brain insulin resistance. Likewise, ROS/RNS elevation leads to the nitrosylation and consequent inhibition of IDE causing this enzyme not to contribute to Aβ degradation. In turn, increased Aβ levels lead to IR downregulation thereby reducing the pool of IR available at the plasma membrane to bind insulin. In parallel, increased Aβ levels further contribute to mitochondrial damage and increased ROS/RNS production. Arrows represent stimulation; lines represent inhibition. See text for more details.
In this picture oxidative-induced damage to IDE and BVR-A might represent early alterations triggering the development of brain insulin resistance.
We also acknowledge that other proteins belonging to the insulin signalling pathway, including IRS1, PTEN and Akt, were found oxidatively impaired by HNE-adducts in in vitro models [161–163]. However, the above-mentioned studies were conducted in adipocytes [161] and hepatic cells [162]. For that reason, in the next sections we focus on proteins found to be oxidatively impaired within the brain.
7.1. Insulin degrading enzyme oxidation
Insulin-degrading enzyme (IDE) is a neutral Zn2+−metallo-endopeptidase that is ubiquitously expressed in insulin-responsive and non-responsive cells [164]. IDE is evolutionarily ancient, with homologs present in phylogenetically diverse organisms of every kingdom [165]. As its name implies, IDE has a high affinity for insulin, but it can degrade a wide range of other peptide substrates, including glucagon, β-amyloid, and chemokine ligand 3 [164, 166]. IDE knockout mice are both glucose-intolerant and hyper-insulinemic, supporting the concept that IDE is important in the maintenance of normal blood glucose and insulin levels [167]. Human genetic studies have linked polymorphisms in the IDE gene to an increased risk for insulin resistance and T2DM [168–170]. Human genetic studies have also linked IDE to AD [171–173]. IDE hypofunction has been shown to contribute to the accumulation of Aβ plaques in animal models of AD [167]. In addition, a recent study from de la Monte and colleagues showed that IDE levels are altered both in mouse model of sporadic AD and in human AD brain suggesting that IDE plays a role in insulin deficiency and attendant insulin resistance in the early and intermediate stages of AD [174]. Consequently, factors that affect the activity of IDE could have significant impact on the progression of these diseases.
Indeed, a great interest in AD field toward IDE functions arose following the first identification of IDE as a physiologically relevant Aβ- degrading protease at multiple sites [175]. The original observation that IDE was able to hydrolyze Aβ in vitro [175] was extended by Selkoe and colleagues who demonstrated that a microglial cell line secreted a metalloprotease immunologically identical to IDE, which was able to regulate extracellular levels of Aβ [176]. IDE also protects mitochondria against Aβ accumulation and dysfunction [177]. Since that time, IDE has been regarded as a significant contributor to Aβ degradation both in vitro and in vivo.
A role for IDE in favoring brain insulin resistance mostly rely on its ability to degrade Aβ. In fact, as reported by De Felice and colleagues, Aβ oligomers are able to bind to IR causing a major loss of IRs from neuronal dendrites and thereby reduce the activation of insulin signaling in neuronal cells [178], i.e., a mechanism leading to brain insulin resistance. Conversely, insulin prevents Aβ oligomers-induced loss of surface IRs, neuronal oxidative stress, and synapse deterioration [179]. The mechanism of insulin protection involves IR signaling–dependent downregulation of oligomer binding to neurons. Thus, the protective action of insulin in rescuing the inhibition of IRS1 and thus recovering the activation of the insulin signaling cascade in neurons likely derives from its ability to block oligomer binding to neurons [179]. Correspondingly, the finding that Aβ plaque size inversely correlates with IDE expression and activity [180, 181] suggests that IDE deficiency could mediate plaque buildup and possibly cognitive impairment in AD. In this picture, IDE activity would favor Aβ degradation, thus preventing both IR loss and development of brain insulin resistance (Figure 3).
Linking the above to OS, previous reports demonstrated that IDE can be oxidatively modified, resulting in reduced activity both in T2DM and AD [145–148]. Hersh and colleagues first reported that IDE was inactivated by reaction with 4-hydroxy-2-nonenal (HNE) with the concomitant formation of protein adducts and that oxidation increased its susceptibility to proteolysis [149]. Later, Cordes and colleagues described an additional mechanism for IDE inactivation driven by nitric oxide (NO). NO donors caused S-nitrosylation of IDE that led to diminished insulin and Aβ degrading activities of IDE in vitro (Figure 3). Insulin-degrading activity appeared more sensitive to NO inhibition than Aβ degrading activity [145]. Similar results were collected in vivo by showing that the activity of IDE was lowered in APP/PS1 mice (a model for AD), but not in mice lacking the inducible form of nitric oxide synthase (NOS2) [APP/PS1/NOS2(−/−) mice]. These data suggest that NOS2 upregulation impaired amyloid-β degradation through negative regulation of IDE activity and, as a consequence, loss of NOS2 activity would positively influence Aβ clearance by IDE [146].
Results collected in AD were further extended to T2DM/obesity by the group of Lipton and co-workers [147]. In an elegant paper these authors reported in human and rodent tissues that elevated glucose, as found in T2DM, and oligomeric Aβ peptide, which as noted above is thought to be a key mediator of AD pathogenesis, coordinately increased neuronal Ca2+ and NO in an NMDA receptor-dependent manner [147]. The increase in NO results in S-nitrosylation of IDE, thus inhibiting insulin and Aβ catabolism as well as hyperactivating mitochondrial machinery. Consequent elevation in Aβ levels and compromised mitochondrial bioenergetics resulted in dysfunctional synaptic plasticity and synapse loss in cortical and hippocampal neurons. The NMDA receptor antagonist memantine attenuated these effects [147]. Thus, this study suggested that redox-mediated post-translational modification of IDE links Aβ and hyperglycemia to cognitive dysfunction in T2DM and AD [147] (Figure 3).
7.2. Biliverdin reductase-A (BVR-A) oxidation
The insulin signaling cascade contains several regulatory points, signal divergences, and crosstalk with other signaling pathways that represent critical nodes [182]; many steps are negatively regulated by phosphatases or inhibitory proteins, and the complexity of this signaling system is crucial for mediating the variety of insulin-related biological functions. Among them, the protein BVR-A emerged for its pleiotropic functions. BVR-A is mainly known for its canonical activity in the degradation pathway of heme, since BVR-A catalyzes the reduction of biliverdin (BV) to bilirubin (BR). However, previous studies in vitro showed that BVR-A arguably has a more diverse and expansive spectrum of functions because its numerous consensus regulatory motifs and its ability to fold into a protein–protein interactive structure [183]. Noteworthy, BVR-A is a unique serine/threonine/tyrosine (Ser/Thr/Tyr) kinase directly involved in the regulation of the insulin signaling [184, 185] at different levels. BVR-A is a direct target of the IR kinase activity, similar to IRS1. IR phosphorylates BVR-A on specific Tyr residues (Tyr198/228/291) and such phosphorylation is required for the activation of BVR-A kinase activity. In addition, as part of a regulatory loop, BVR-A phosphorylates IRS1 on inhibitory Ser residues (i.e. 307, 312, 616) to prevent IRS1 aberrant activation in response to IR [184] (Figure 1). In addition, BVR-A kinase activity is required for the activation of the extracellular signal-regulated kinases 1/2 (ERK1/2) [186]. Downstream from IRS1, BVR-A also functions as scaffold protein (Figure 1) favoring: (i) the PDK1-mediated activation of the aPKCζ [187], known to regulate either GLUT-4 translocation [188] or memory processes [189]; and (ii) the Akt-mediated inhibtion of GSK-3β [190]. Finally, BVR-A contains specific motifs in its sequence through which these motifs conceivably modulate IR kinase activity both negatively and positively [185].
Findings from animal models of obesity showed that loss of hepatic BVR-A is associated with altered glucose/insulin metabolism and increased fat accumulation [158]. Similarly, adipocyte-specific deletion of BVR-A caused increased expansion of visceral fat and adipocytes size, inflammation, reduced mitochondria number and hampered insulin signaling [157]. Evidence collected in vitro and animal models of metabolic disorders were strengthened by results from our group recently showing that reduced BVR-A levels were associated with the aberrant activation of insulin signaling, metabolic syndrome, non-alcoholic fatty liver disease (NAFLD) and visceral adipose tissue inflammation in obese individuals [155, 156, 191].
During the past several years, we have focused much research on BVR-A especially in AD [76, 150–153, 192–197]. In addition, we found that reduced levels or impaired BVR-A activation contribute to the development of insulin resistance and metabolic alterations in the brain that are known to drive neurodegenerative processes [76, 124, 154, 190, 195, 197, 198].
The Butterfield group was the first to demonstrate that BVR-A activation was reduced in post-mortem MCI and AD brain because of increased nitration [higher 3-nitrotyrosine levels (3NT) on BVR-A] [150, 151, 153, 195]. In addition, increased BVR-A nitration was associated with higher pIRS1 Ser307 levels in the same brain samples [67], thus suggesting that oxidatively induced damage of BVR-A likely contributes to the onset of brain insulin resistance in AD (Figure 3).
This hypothesis was further supported by data collected in a longitudinal study in which we found that the impairment of BVR-A is one of the earliest events observed during the development of brain insulin resistance in 3xTg-AD mice (a model to study AD) and that this phenomenon occurs before a consistent accumulation of Aβ and Tau as well as increased TNFα levels [76]. Of note, we proposed a role for OS in mediating BVR-A dysfunction: higher levels of OS markers paralleled the oxidative-induced damage at the expense of BVR-A, and both these events preceded the frank inhibition of IRS1 in the brain [76]. BVR-A oxidation reduced its kinase activity, which resulted first in the hyper-activation of IRS1 (in agreement with the regulatory role for BVR-A discussed above) and then in the inhibition of IRS1 mediated by the activation of inhibitory feedback mechanisms such as mTOR [76] (Figure 3).
The impairment of BVR-A also produces deleterious effects downstream from IRS1. Notwithstanding IRS1 hyper-activation, neither an increased activation of Akt nor an increased inhibition of GSK-3β were observed [190]. These data agree with the role for BVR-A in working as scaffold protein, favoring the PDK1-mediated activation of Akt as well as the Akt-mediated inhibition of GSK-3β in response to insulin [190, 199] (Figure 3). Therefore, OS-induced damage of BVR-A might turn-off the activation of insulin signaling, reducing its neuroprotective effects before a frank brain insulin resistance develops. These alterations produced detrimental effects on cognitive functions [198], while loss of BVR-A was found to impair long term potentiation (LTP), a key mechanism in learning, in cortical neurons [198].
As discussed above, brain insulin resistance has been proposed to have a role in the production and accumulation of Aβ in AD [66, 126, 127, 200]. Consequently, in a vicious cycle, increased Aβ oligomer generation further exacerbates brain insulin resistance through the down-regulation the of the IR, possibly because reduced IDE activity [179] (Figure 3). Within this picture, our group unraveled a novel mechanism linking BVR-A impairment, brain insulin resistance and increased Aβ production [154]. In particular, the impairment of BVR-A would favor the casein kinase I (CKI)-mediated phosphorylation of BACE1 and associated increased Aβ production along with the alteration of brain insulin signaling [154]. Therefore, these results support a role for BVR-A as molecular target whose dysfunction links brain insulin resistance with increased Aβ production in AD, which is associated with elevated oxidative stress [11].
Finally, increased 3-NT modifications of BVR-A observed both in human AD brain and in 3xTg-AD mice were found to be associated with mTOR hyper-activation [67, 76]. mTOR is a master regulator of autophagy [201, 202], and an hyper-activation of mTOR – which negatively regulates autophagy induction – along with the reduction of the autophagic flux was reported in AD [67, 203–205]. mTOR hyper-activation favors the accumulation of protein oxidative damage within the brain [79]. Moreover, mTOR hyper-activation is listed among the known feedback mechanisms responsible for IRS1 inhibition and the development of insulin resistance also in AD [67, 203–206]. Among the pathways responsible for mTOR hyperactivation in AD, our group identified the impairment of BVR-A as a crucial event responsible for the disruption of the AMPK/mTOR axis resulting in mTOR hyper-activation in the brain [76, 198, 207] (Figure 3).
This aspect is of interest because AMPK is considered a key cellular energy sensor and it has a crucial role in the control of several processes ranging from lipid biosynthesis and catabolism to glucose uptake and antioxidant defense and regulation of insulin signaling [208]. AMPK is dysregulated in major metabolic disorders such as diabetes, obesity, as well as in neurodegenerative diseases [209, 210]. A dysregulation of the BVR-A/AMPK axis fostered by increased OS levels in the brain may therefore contribute to the development of brain insulin resistance either through mTOR hyper-activation or other pathways as discussed below (Figure 3).
8. AMPK is a central hub between metabolic defects and neurodegeneration
Epidemiological studies demonstrated that metabolic diseases, such as obesity, diabetes and hypercholesterolemia, are common risk factors for cognitive impairment and sporadic AD [211, 212]. As supported by PET imaging analysis of brains from AD and MCI subjects, and of populations at-risk of dementia, reduced glucose uptake and utilization is an early sign of neurodegeneration [11, 213]. In addition, the reduction of glucose utilization was shown to positively correlate with cognitive impairment [214]. AMPK is a metabolic serine/threonine protein kinase that serves as an “energy receptor” by coordinating cell metabolism and energy needs [215]. Short-term effects of AMPK activation can rapidly regulate energy metabolism, while long-term effects can regulate gene transcription [216].
Several lines of evidence have demonstrated that dys-regulated AMPK is involved in the onset and progression of AD, serving as neuronal metabolic sensor [210, 217–219]. Also, in T2DM patients, aberrant AMPK activity alters the metabolism of glucose and lipids and affects the levels blood glucose as well as the profile of blood lipids [215, 220]. As noted above, the core pathological feature of T2DM is insulin resistance that results in decreased insulin sensitivity and glucose utilization rate, thus reducing energy production and promoting cognitive decline [1, 44, 221]. AMPK is closely related to the development of insulin resistance, thereby playing an important role in the reduction of cognitive performances associated with the pathogenesis of AD. Therefore, AMPK appears to link energy metabolism to neurodegeneration, and this, in turn, suggests that energy deficiency is associated with altered synaptic transmission and memory impairment [210, 218].
AMPK exists as a heterotrimeric complex composed of α-catalytic subunit and β- and γ-regulatory subunits. The α-, β-, and γ-subunits can also be found in different isoforms: γ1, γ2, or γ3 isoform for γ-subunit; β1 or β2 isoform for β-subunit; and α1 or α2 isoform for α-subunit [222, 223]. AMPK functions as an intracellular sensor of energy when activated in response to stress conditions, such as low glucose, hypoxia and ischemia, each of which depletes cellular ATP supplies and yields an increased AMP/ATP concentration ratio [224].
Regulation of AMPK activity involves both direct allosteric activation by AMP and reversible phosphorylation of AMPKα subunit on Thr172 by upstream kinases [222]. Phosphorylation (of Thr172) is quantitatively much more significant than the allosteric activation because it causes at least 50- to 100-fold increase in AMPK activation [210]. However, the activation of AMPK needs two conditions, including the γ-subunit conformational change, to expose the active phosphorylation site on the α-subunit and the subsequent phosphorylation of AMPK on its activating loop by upstream kinases [219]. The major mechanisms that lead to AMPK activation, through the phosphorylation of Thr172 are catalyzed by LKB1 complex (LKB1/STRAD/MO25) in response to the increased AMP/ATP ratio, and by calmodulin-dependent protein kinase kinase-beta (CaMKKβ) in response to elevated Ca2+ levels. Conversely, Thr172 is dephosphorylated by protein phosphatase-2C (PP2C) to turn active AMPK into the inactive form [215, 218, 223, 225]. The major function of AMPK is to switch on energy-producing processes and to switch off energy-consuming functions. Thus, AMPK promotes glucose uptake and glycolysis, fatty acid oxidation, and mitochondrial biogenesis, which ultimately increase ATP production. On the other hand, AMPK inhibits anabolic processes such as protein synthesis and decreases ATP consumption.
AMPK signaling also has a crucial role in regulating food intake in hypothalamic neurons [226]. AMPK negatively regulates several proteins central to ATP-consuming processes such as the transducer of regulated CREB activity 2 (TORC2), glycogen synthase, sterol regulatory element-binding proteins (SREBP), and tuberous sclerosis protein 2 (TSC2). Thus, AMPK activation results in the down-regulation or inhibition of gluconeogenesis, glycogen, lipid, and protein synthesis [218, 219].
With regard to the interaction between AMPK and insulin signaling, insulin resistance leads to repression of general AMPK activity through the over-activation of AKT. Further, by inhibiting AMPK activity, Aβ oligomers can decrease the surface levels of glucose transporters (GLUT) in hippocampal neurons, which results in insulin resistance [227]. In turn, insulin resistance can increase amyloidosis via mechanisms involved in the repression of insulin-PI3KAKT signaling [228], resulting in the upregulation of GSK3β activity. In agreement with the above, increased AMPK activation induces IRS phosphorylation at tyrosine residues, thereby promoting insulin signaling activities by inhibiting GSK3β [229, 230]. Furthermore, AMPK activation can promote the expression of GLUT4 or GLUT1, increasing the rate of glucose utilization and leading to the production of large amounts of ATP that improve neuronal activity and homeostasis [215].
As an energy sensor regulating all aspects of cellular functions, AMPK also has numerous downstream targets participating in biosynthetic pathways. Of particular interest is that AMPK can regulate protein homeostasis (synthesis/degradation rate), which is indispensable for long-lasting forms of synaptic plasticity and memory [219, 231].
8.1. Role of AMPK in Aβ and p-tau development
Given the participation of AMPK in the regulation of several mechanisms associated with memory and cognition, it is not surprising that abnormal AMPK activity has been reported in postmortem AD patients, as well as, in AD mouse models [215–217]. Intriguingly, AMPK dysregulation has been strongly associated with AD progression, however its role is still controversial, especially with regard to Aβ accumulation and tau phosphorylation. Studies reporting the positive or negative effects of AMPK on Aβ or tau demonstrated diverse mechanisms of action [226]. However, although there are conflicting results, there is no disagreement about the involvement of AMPK in AD pathogenesis. Several reports demonstrated that the activation of AMPK reduces the β-cleavage of APP in cultured rat cortical neurons, while knockout of AMPKα2 increases Aβ production [217, 232]. In agreement with these findings, the stimulation of AMPK by leptin was demonstrated to reduce Aβ deposition, whereas inhibitors of AMPK block the effects of leptin [233, 234]. This observation was corroborated by epidemiological studies, which showed lower leptin levels in AD patients compared to those in healthy individuals [226]. In addition, the direct stimulation of AMPK with 5-amino-imidazole-4-carboxamide ribonucleoside (AICAR), an AMPK agonist, replicated the effect of leptin and decreased Aβ production, whereas compound C, an AMPK inhibitor, increased Aβ production [235]. The pharmacological activation of AMPK by resveratrol in APP-transfected cells (APP-HEK293 and APP-N2a) or quercetin in aged mouse brain lowered the extracellular concentration of Aβ by reducing the expression of BACE1 and the formation of Aβ. Furthermore, metformin was shown to interact with BACE1 and AMPK; however, studies concerning the reduction of Aβ have been conflicting so far [236, 237]. Other studies, employing different AMPK agonist molecules, reported that the activation of AMPK decreases the expression of BACE1 at the transcription and translation levels [237]. Among the molecular mechanisms for reduction of BACE1 expression, a role for the attenuation of translation initiation factor eIF2α upon AMPK activation was proposed [238]. Moreover, the activation of AMPK is known to activate autophagy, and recent studies showed that upregulation of autophagy can reduce BACE1, therefore partially explaining why the levels of BACE1 were found to be reduced upon AMPK activation [215–217, 239].
Overall, the above-mentioned findings suggest that AMPK activation can moderate Aβ production by decreasing BACE1 expression; however, another reported mechanism by which AMPK control amyloidogenesis is by regulating the metabolism of cholesterol and sphingolipid [226]. AMPK regulates cholesterol and sphingolipid biosynthesis by controlling the gene expression and activity of associated enzymes. Recent pieces of evidence also show that reduction of cholesterol and sphingolipids decreases the generation of Aβ [240, 241]. However, the exact mechanisms linking AMPK activation with reduced Aβ production through sphingolipids and cholesterol metabolism is still under investigation.
In contrast, other studies supported the notion that the activation of AMPK can lead to the biogenesis of Aβ peptides and to the development of AD, of course being detrimental for the brain. As an example, metformin was shown to increase the biogenesis of Aβ through the activation of AMPK [242, 243]. In agreement, a population-based large-scale case-control study showed that the treatment of T2DM patients with metformin was associated with a slightly higher risk of AD development [244]. Additional studies demonstrated that metformin increased the generation of Aβ by different mechanisms involving the accumulation of autophagosomes and the resultant increase of β- and γ-secretases [245], as well as, the increase expression of BACE1 and APP [246]. Therefore, there is consistent evidence that the activation of AMPK with metformin might play a negative role in the pathogenesis of AD.
The role of AMPK in AD also is debatable with regard to tau phosphorylation [216]. Abnormally increased p-AMPK levels were found in the cytoplasm of cortical neurons in multiple tauopathies including AD. p-AMPK and p-tau protein demonstrated strong co-localization in pre-tangle-and- tangle-bearing neurons in most brain regions affected by tau pathology in AD patients [247]. Tau pathology is closely associated with mitochondrial failure due to its negative effects on mitochondrial transport, mitochondrial fission and fusion, and mitophagy [248]. Indeed, it has been proposed, that pathological forms of tau could disrupt axonal transport and cause synaptic damage by the increase of the pausing frequency of mitochondria movement the reduction of anterograde movement of mitochondrial [249]. Furthermore, tau play a significant role in the impairment of mitochondrial fission/fusion dynamics through the increase of the mitochondrial fission proteins, such as Drp1, and the decrease of fusion protein including OPA-1, Mfn1/2 [250]. Several in vitro studies demonstrated that AMPK is a tau kinase, which can phosphorylate tau protein at multiple sites within the microtubule binding domain and the flanking region [216, 251]. On the other hand, AMPK also may be involved in the reduction of tau levels and tau phosphorylation through various downstream mechanisms [215, 218]. For example, mounting evidence suggests that AMPK, by increasing the availability of NAD+, enhances SIRT1, a class III protein deacetylase, leading to decreased acetylation and increased degradation of tau protein [217]. Indeed, it was demonstrated that increased acetylation of tau inhibits the ubiquitinylation and the degradation processes of tau protein [252]. A further mechanism by which AMPK activation led to reduced tau phosphorylation is by activation of PP2A [253]. PP2A is one of the main dephosphorylating enzymes involved in the regulation of tau PTMs. Several studies have shown that the level and activity of PP2A are lowered in the brain from AD patients obtained by autopsy and in animal models of the disease, showing strong involvement of decreased PP2A in the generation of tauopathy [254, 255]. Furthermore, in T2DM animal models the significant increase of tau phosphorylation was associated with AMPK impairment as an effect of PP2A inhibition, decreased SIRT 1 activity, and GSK3β overactivity [256, 257]. Taken together, the activation of AMPK seems to interfere with tau phosphorylation by different mechanisms that include the decreased the activity of GSK3β, the increased the activity of PP2A and the increased proteasomal degradation of tau as an effect of SIRT1 activation [226]. However, the overall net effect of AMPK activation on tau phosphorylation remains to be fully elucidated. Indeed, as described above, AMPK has a direct negative effect on tauopathy and has an indirect protective effect through its downstream mediators. In addition, the indirect positive effects of AMPK are complex as they include the bidirectional regulation of different regulators of tau phosphorylation.
Therefore, up to the present, whether activation of AMPK activity would be either beneficial or detrimental for Aβ deposition and tau phosphorylation remains controversial.
8.2. AMPK regulates brain proteostasis and redox balance
In addition to participating in the regulation of insulin resistance, and of Aβ and p-tau formation, AMPK plays a crucial role in preventing AD by promoting autophagy and improving mitochondrial function [216, 217]. Reduced autophagy is observed during the development of AD [258]. The mammalian target of rapamycin complex 1 (mTORC1) inhibits the occurrence of autophagy, thus favoring the build-up of toxic proteins and the aggregation of Aβ and the formation of NFTs [206, 259]. mTORC1 is activated by insulin, growth factors and nutrients, and this complex participates in insulin signaling resulting in increased translation. Insulin induces mTORC1 activity by inhibiting the tumor suppressor complex (TSC1/TSC2), which is an endogenous mTORC1 repressor [260]. AMPK is widely recognized as an mTOR antagonist and prevents the mTORC1 from phosphorylating its substrates, thus affecting autophagy [215]. Indeed, AMPK promotes autophagy by directly activating Ulk1 through phosphorylation of Ser 317 and Ser 777, while mTOR activity prevents Ulk1 activation by phosphorylating Ulk1 Ser 757 and disrupting the interaction between Ulk1 and AMPK. Further, AMPK indirectly inhibits mTORC1 by the activation of TSC1 and TSC2 through the phosphorylation of both Thr1227 and Ser1345 on TSC2 (Figure 4). Moreover, AMPK is able to inhibit mTORC1 activity by directly phosphorylating RAPTOR, a component of the complex [243]. Recently, the activation of AMPK was shown to be important not only in autophagosome formation but also in autophagosome clearance and can improve the overall autophagic degradation of aberrant proteins [226]. Several studies have shown that AMPK is one of the major positive regulators of autophagy demonstrating that enhanced autophagy was able to promote the clearance of Aβ [261, 262]. Further evidence demonstrated that the increase in cytosolic Ca2+ can induce autophagy by inhibition of mTOR through the CaMKKβ-mediated activation of AMPK [263, 264].
Figure 4: Schematic representation of the “metabolic” regulations exerted by AMPK.
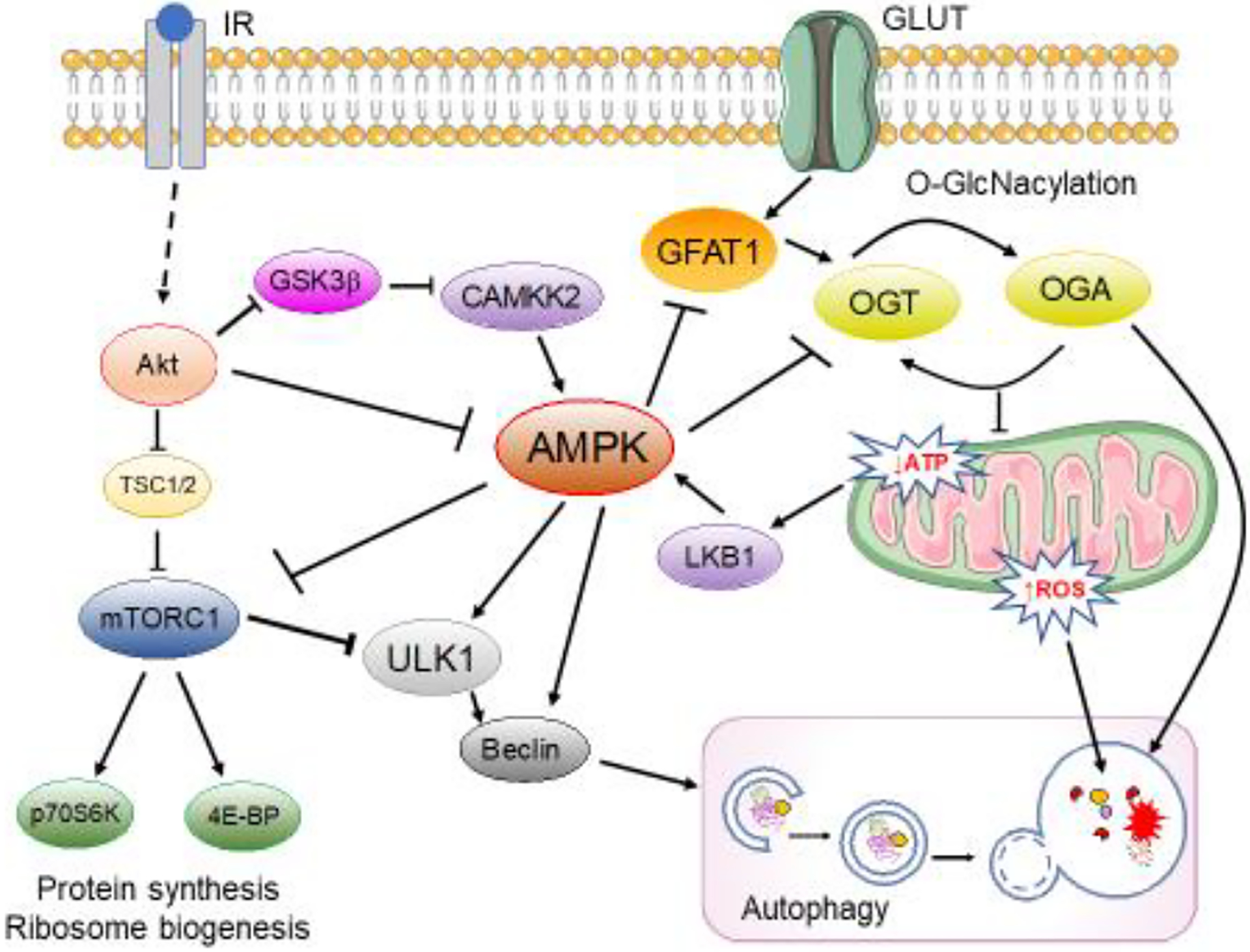
AMPK is activated by the LKB1 complex in response to the increase in the AMP/ATP ratio and by CaMKKβ in response to elevated Ca2+ levels, while insulin resistance leads to repression of general AMPK activity through the over-activation of Akt. AMPK promotes autophagy by directly activating Ulk1 through phosphorylation of Ser 317 and Ser 777, while mTOR activity prevents Ulk1 activation by phosphorylating Ulk1 Ser 757 and disrupting the interaction between Ulk1 and AMPK. Further, increased AMPK negatively regulates protein O-GlcNacylation by inhibiting GFAT1 and by regulating OGT localization. Arrows represent stimulation; lines represent inhibition. See text for more details.
Mitochondrial defects are also strong contributors to synaptic deficits, an early pathophysiological feature of AD [216]. Peroxisome proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1α) is a member of the transcription coactivator family, and it is the initiator of mitochondrial biosynthesis. Several studies indicated that the levels of PGC-1α are decreased in AD patients and low levels of PGC-1α can lead to mitochondrial dysfunction and oxidative stress [265, 266]. Thus, the correct regulation of PGC-1α may represent an important step for the maintenance of cognitive function in AD [267]. In agreement with the above discussion, the activation of AMPK promotes mitochondrial biogenesis, mitochondrial quality control and the regulation of mitochondrial shape through the phosphorylation of PGC-1α on Thr177 and Ser538 sites, which lead to its increased activity [268]. Upon activation, PGC-1α is then translocated into the nucleus and interacts with several transcription factors, including the nuclear factor erythroid 2-related factor 1 and 2 (Nrf1 and Nrf2) [269]. This interaction induces a series of mitochondrial protein gene sequences encoded by nuclear genes [270]. Moreover, PGC-1α is also activated by AMPK-driven deacetylation of SIRT1, thus promoting mitochondrial biogenesis [271].
The control of mitochondrial function and biogenesis exerted by AMPK also is strongly related with the regulation of redox homeostasis. As noted above, the main source of ROS is the electron transport chain (ETC) at the mitochondrial inner membrane. ROS generated by mitochondria can increase BACE1 activity and consequently promote amyloidogenic APP processing and Aβ overproduction [216]. Therefore, the regulation of ROS production might be crucial for protection against development of AD. Activation of AMPK can directly halt ROS overproduction via increasing the regulation of mitochondrial efficiency but also increase antioxidant protection through the activation of PGC-1α [271]. Therefore, AMPK functions as a redox-sensitive protein since it can be activated in response to excess ROS production, thereby coupling cellular ROS levels to mitochondrial metabolic homeostasis [272, 273]. Accordingly, cell lines lacking AMPKα expression displayed higher basal levels of mitochondrial ROS and, consistent with this finding, AMPKα knockout cells also rapidly underwent cell-death as a consequence of increased oxidative stress [273]. Different mechanisms are reported for the antioxidant function of AMPK. AMPK modulates the activity of several downstream proteins and processes involved in the redox balance including the enhancement expression of antioxidants, the activation of antioxidant enzymes, the decrease of ROS production and the stimulation of autophagic clearance [210, 215, 218]. Treatments with an AMPK agonist decreased intracellular and mitochondrial ROS burden, inhibited lipid peroxidation, and improved antioxidant contents by increasing the activity of endogenous antioxidant enzymes [274, 275]. In parallel, the administration of antioxidants was demonstrated to reduce the basal activation of AMPK [226].
Furthermore, as mentioned already, AMPK is an upstream activator of Nrf2, a master regulator of antioxidant gene programs [276, 277]. Once activated, Nrf2 translocates to the nucleus where it binds with the antioxidant response element and stimulates the expression of different antioxidant proteins such as heme oxygenase-1, NADPH quinone oxidoreductase-1, and Mn superoxide dismutase [278]. Nrf2 is reportedly reduced in AD human brain and in mouse models of the disease [279, 280]. Several in vivo and in vitro studies showed that the neuroprotective effect of AMPK activators was reversed by genetic deletion of both AMPK and/or Nrf2 genes [281, 282]. Therefore, the AMPK/Nrf2 pathway is one of the main antioxidant mechanisms of AMPK activation. In addition, PGC-1α, once activated by AMPK, translocates to the nucleus, and stimulates the expression of superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase-1 (GPX1) enzymes [283, 284]. The activation of AMPK enhances the expression of SIRT3, a NAD+ dependent protein deacetylase highly localized in the mitochondria [226]. SIRT3 in turn deacetylates SOD2 and decreases mitochondrial ROS [1, 220]. Thus, the induction of the AMPK-SIRT3-SOD2 pathway activates antioxidants already synthesized in the cell. In contrast, a recent study suggested that increased oxidative stress represents the underlaying mechanism leading to the concomitant activation and uncoupling of AMPK and mTOR, as observed in AD brain [285]. The induction of autophagy also is involved in the regulation of the redox status of the brain by degrading oxidized proteins and by activating antioxidant responses driven by the autophagy receptor p62/SQSTM1 [286–289]. Finally, AMPK also phosphorylates and decreases the transcription factor sterol regulatory element-binding protein-1c (SREBP-1c), thereby reducing the expression of lipogenic genes [210].
8.3. AMPK controls protein O-GlcNacylation
O-linked N-acetylglucosamine (O-GlcNAc) is a sugar post-translational modification participating in a diverse range of cell functions. A wide scope of cellular processes is controlled and fine-tuned by O-GlcNAc, including apoptosis, mitochondrial function, proliferation, and gene transcription [290–292]. The rate of protein O-GlcNAcylation is mediated by two enzymes: O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), and their alteration is involved in the disruption of O-GlcNAc cycling. O-GlcNAcylation is dependent on the intracellular concentration of UDP-GlcNAc from the hexosamine biosynthetic pathway (HBP) [293]. The HBP integrates the metabolic information of nutrients, including carbohydrates, amino acids, fatty acids, and nucleic acids, in the process of UPD-GlcNAc synthesis [294]. Further, HBP regulates energy homeostasis by controlling the production of both insulin and leptin [295]. O-GlcNAcylation is tightly coupled to insulin resistance since hyperglycemia- or hyperlipidemia-induced insulin resistance is closely related to altered HBP flux, and, in turn, the subsequent aberrant O-GlcNAcylation modifies insulin signaling, glucose uptake, gluconeogenesis, glycogen, and fatty acid synthesis [292, 296–298].
In this scenario, O-GlcNAcylation represents a key mechanism linking nutrient overload and insulin resistance, and its dysregulation might promote the transition from metabolic defects to chronic diseases such as T2DM and AD [299]. Indeed, obesity and peripheral hyperglycemia, by promoting insulin resistance and hypoglycemia in the brain, led to decreased O-GlcNAcylation of APP and Tau and to increased production of toxic Aβ and Tau aggregates, hallmarks of AD [290, 300]. HBP flux regulates multiple steps of the insulin signaling pathway and, in particular, IRS-1 has multiple O-GlcNAcylation sites, and its O-GlcNAcylation negatively correlates with changes in phosphorylation, which are a necessary in activating downstream pathways for glucose uptake [294, 296].
Investigations into the individual nutrient-sensitivities of the HBP, AMPK, and mTOR pathways revealed a complex regulatory dynamic, by which their unique responses to macromolecule levels coordinate cell behavior. Importantly, the crosstalk between these pathways fine-tunes the cellular response to nutrients (Figure 4). By the action of AMPK, GFAT1, the first rate-limiting enzyme of the HBP, undergoes negative regulation through increased phosphorylation on Ser243 [301, 302]. Studies on HFD mice demonstrated the concomitant occurrence of brain insulin resistance and reduced protein O-GlcNAcylation that converge in the development of AD markers [295]. Indeed, the brain from HFD mice exhibited increased inhibition of IRS-1 along with increased AMPK activation, which exerted a negative regulation on GFAT1 activity, by the direct phosphorylation of the Ser243 inhibitory residue, thus resulting in reduced protein O-GlcNacylation and increased phosphorylation of APP and tau [290, 295]. Different studies also showed that AMPK itself was O-GlcNAcylated on several residues, allowing one to posit further feedback mechanisms between these systems [299]. Thus, experimental evidence supports a regulatory role for OGT in controlling AMPK activity. On the other hand, AMPK phosphorylation of OGT on Thr444 increases OGT nuclear localization, increases nuclear O-GlcNAcylation and increases histone H3K9 acetylation, while O-GlcNAcylation of the H2B histone is lower, suggesting AMPK as a regulator of OGT activity and of O-GlcNAc mediated epigenetic modifications [299]. In addition, the rescue of O-GlcNAcylation brain levels was shown to stimulate autophagosome maturation and lysosomal degradation, suggesting a crosstalk between OGT/OGA cycling and autophagy that conceivably also might involve the AMPK/mTOR signaling [290].
9. Conclusions
AD and T2DM are two highly prevalent conditions affecting people worldwide. Clearly, a better understanding of the molecular bases of both conditions, and especially their intersection of molecular processes, we posit, will identify important therapeutic targets for both conditions.
It is toward this end that this review paper presents major underlying molecular processes in and the interplay among oxidative stress, glucose dysmetabolism, and the key protein AMPK in AD. Oxidative stress and glucose dysmetabolism are inextricably linked in the brain of AD individuals [11]. Insulin signaling, specifically the development of insulin resistance, also find commonality in both the role of oxidative stress and their origins and key molecular and clinically relevant characteristics of T2DM and AD.
Not only is oxidative stress involved in driving development of clinical and pathological aspects of diabetes and AD, but, in a vicious cycle, pathological aspects in T2DM and AD can lead to oxidative stress [11]. Insulin resistance results when the information inherent in the binding of insulin to the plasma membrane-resident insulin receptor cannot be transduced to intracellular biochemical changes (often phosphorylation of important cellular proteins). The IRS1 protein is intimately involved in insulin signaling mechanisms as we outline above. This protein is regulated by several processes, two of which are BVR-A and secondary to mTORC1 activation [67, 206, 207]. The latter leads to resultant phosphorylation of p70S6K and subsequent inhibition of IRS1 [206].
In the case of AD, sources of oxidative stress are numerous, with two of the most relevant ones being mitochondrial dysfunction [77, 103] and Aβ42 oligomer-associated lipid peroxidation and its downstream sequela of HNE modification of glucose metabolic proteins, ion-motive ATPases, intracellular Ca2+ accumulation, synaptic dysfunction, and neuronal death [11]. In the case of mitochondria, significant superoxide free radical leak from Complex I of the electron transport chain immediately is attacked by MnSOD in the matrix to produce H2O2. The latter compound, due to its trans-configuration, has a zero dipole moment and is highly soluble in the hydrophobic core of the mitochondrial lipid bilayer. Any adventitious reduced Fe2+ or Cu+ would immediately react with this H2O2 to produce ·OH radical which would wreak havoc on the mitochondria [11, 303]. In the case of lipid-resident Aβ42 oligomer-associated lipid peroxidation, much is now understood on how lipid peroxidation ensues [11]. A major product of lipid peroxidation, HNE is highly reactive and neurotoxic [303]. Brain-resident HNE-bound proteins are significantly elevated in AD and T2DM, meaning that key metabolic, regulatory, and structural proteins have altered structure and diminished function [303–311].
Insulin signaling is fundamental to metabolic pathways needed for processes related to learning and memory [1]. In both AD and T2DM, insulin signaling is disrupted due to insulin resistance [66, 67, 81]. Craft and co-workers demonstrated that intranasal insulin delivery directly to brain (which precluded peripheral effects of elevated insulin) helped individuals with MCI and AD to improve cognition [49]. This same group demonstrated that dose level, gender, and ApoE allele status had complex yielded complex relationships to cognition following intranasal insulin treatment [312]. More research is required to understand the molecular bases for these differences following intranasal insulin.
In addition to the above, other conclusions based on this review article are:
Hydrogen peroxide, in addition to being an important reactive oxygen species, is involved in the activation of the IR in neurons [89].
Oxidative stress is correlated to loss of synapses in AD and therefore in cognitive loss [100–104].
Insulin degrading enzyme normally cleaves Aβ, rendering it non-neurotoxic, but in brain of AD and T2DM persons and animal models thereof IDE is oxidatively modified by HNE [145–148], leading to increased levels of neurotoxic Aβ and insulin resistance decreased function of IDE in a destructive cycle. That is, more Aβ leads to more lipid peroxidation with consequent synaptic loss and neuronal death with consequent loss of cognition, a hallmark clinical characteristic of AD.
Defects in glucose metabolism are associated with insulin resistance in AD and MCI brains [11, 57, 80, 122] and important in the conversion of Down syndrome individuals to persons with Down syndrome and AD-like neuropathology and dementia [11, 128].
Biliverdin reductase-A and AMP-activated protein kinase are critical for the associations among obesity, T2DM, and development of AD. Moreover, AMPK is a central locus between metabolic defects and neurodegeneration [191, 210, 217–219].
Alzheimer disease is a devastating dementing disorder; T2DM and insulin resistance are significant risk factors for development of AD. The current review provides evidence of some of the underlying molecular processes by which this association exists and points to potential therapeutic targets to slow or, ideally, halt progression of AD. We predict that expanded research directed toward these underlying Aβ-associated dysfunctional metabolic mechanisms will ensue as the number of persons worldwide at risk of developing AD is ballooning. We trust that this review paper contributes to this effort.
Highlights.
Defects in energy metabolism are common features for T2DM and AD
Systemic insulin resistance is a risk factor for neurodegeneration
Neurodegeneration is associated with brain insulin resistance and oxidative stress
Oxidative stress contributes to brain insulin resistance by damaging BVR-A and IDE
AMPK dampens oxidative stress levels, thus regulating brain redox homeostasis
Acknowledgement
This work has been partially supported by a NIH grant [AG060056].
List of Abbreviations
- Aβ
β-amyloid protein
- AD
Alzheimer disease
- AGEs
advanced glycation end products
- AMPK
AMP-activated protein kinase
- APP
amyloid precursor protein
- ATP
Adenosine triphosphate
- AT
adipose tissue
- Akt
protein kinase B
- BVR-A
biliverdin reductase A
- BBB
blood brain barrier
- ER stress
endoplasmic reticulum stress
- ERK1/2
extracellular signal-regulated kinase 1/2
- GLUTs
glucose transporters
- GSK3β
Glycogen synthase kinase 3 beta
- HNE
4-hydroxy-2-nonenals
- IDE
insulin degrading enzyme
- IRS1
insulin receptor substrate1
- IR
insulin receptor
- IKkB
Inhibitor of nuclear factor kappa B kinase subunit neta
- JNK
c-Jun N-terminal kinase
- LTP
long term potentiation
- MAPK
mitogen-activated protein kinase
- MEK
mitogen-activated protein kinase kinase
- MCI
mild cognitive impairment
- mTOR
mammalian target of rapamycin
- NFTs
neurofibrillary tangles
- NMDA receptor
N-methyl-D-aspartate receptor
- Nrf2
nuclear factor erythroid 2–related factor 2
- O-GlcNAc
O-linked N-acetylglucosamine
- OGA
O-GlcNAcase
- OGT
O-GlcNAc transferase
- OXPHOS
oxidative phosphorylation
- OS
oxidative stress
- PDK1
phosphoinositide-dependent kinase 1
- PGC-1α
Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha
- PKCζ
protein kinase C zeta
- PPA2
inorganic pyrophosphatase 2
- PTEN
phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase
- PI3K
Phosphoinositide 3-kinases
- PKR
protein kinase R
- PTP1B
protein tyrosine phosphatase 1B
- 3-NT
3 -nitro-tyrosine
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- SOD
superoxide dismutase
- T2DM
type 2 diabetes mellitus
- TNF- α
tumor necrosis factor alpha
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no conflict of interest exists with any author.
References
- [1].Arnold SE, Arvanitakis Z, Macauley-Rambach SL, Koenig AM, Wang HY, Ahima RS, Craft S, Gandy S, Buettner C, Stoeckel LE, Holtzman DM, Nathan DM, Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums, Nat Rev Neurol 14(3) (2018) 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Howarth C, Gleeson P, Attwell D, Updated energy budgets for neural computation in the neocortex and cerebellum, J Cereb Blood Flow Metab 32(7) (2012) 1222–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Han R, Liang J, Zhou B, Glucose Metabolic Dysfunction in Neurodegenerative Diseases-New Mechanistic Insights and the Potential of Hypoxia as a Prospective Therapy Targeting Metabolic Reprogramming, Int J Mol Sci 22(11) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zhang X, Alshakhshir N, Zhao L, Glycolytic Metabolism, Brain Resilience, and Alzheimer’s Disease, Front Neurosci 15 (2021) 662242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Takata T, Okada Y, Effects of deprivation of oxygen or glucose on the neural activity in the guinea pig hippocampal slice--intracellular recording study of pyramidal neurons, Brain Res 683(1) (1995) 109–16. [DOI] [PubMed] [Google Scholar]
- [6].Yamane K, Yokono K, Okada Y, Anaerobic glycolysis is crucial for the maintenance of neural activity in guinea pig hippocampal slices, J Neurosci Methods 103(2) (2000) 163–71. [DOI] [PubMed] [Google Scholar]
- [7].Mergenthaler P, Lindauer U, Dienel GA, Meisel A, Sugar for the brain: the role of glucose in physiological and pathological brain function, Trends Neurosci 36(10) (2013) 587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Koepsell H, Glucose transporters in brain in health and disease, Pflugers Arch 472(9) (2020) 1299–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vergara RC, Jaramillo-Riveri S, Luarte A, Moenne-Loccoz C, Fuentes R, Couve A, Maldonado PE, The Energy Homeostasis Principle: Neuronal Energy Regulation Drives Local Network Dynamics Generating Behavior, Front Comput Neurosci 13 (2019) 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ruderman NB, Carling D, Prentki M, Cacicedo JM, AMPK, insulin resistance, and the metabolic syndrome, J Clin Invest 123(7) (2013) 2764–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Butterfield DA, Halliwell B, Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease, Nat Rev Neurosci 20(3) (2019) 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Holtzman DM, Morris JC, Goate AM, Alzheimer’s disease: the challenge of the second century, Sci Transl Med 3(77) (2011) 77sr1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT, Neuropathological alterations in Alzheimer disease, Cold Spring Harb Perspect Med 1(1) (2011) a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dubois B, Hampel H, Feldman HH, Scheltens P, Aisen P, Andrieu S, Bakardjian H, Benali H, Bertram L, Blennow K, Broich K, Cavedo E, Crutch S, Dartigues JF, Duyckaerts C, Epelbaum S, Frisoni GB, Gauthier S, Genthon R, Gouw AA, Habert MO, Holtzman DM, Kivipelto M, Lista S, Molinuevo JL, O’Bryant SE, Rabinovici GD, Rowe C, Salloway S, Schneider LS, Sperling R, Teichmann M, Carrillo MC, Cummings J, Jack CR Jr., G. Proceedings of the Meeting of the International Working, A.D. the American Alzheimer’s Association on “The Preclinical State of, July, U.S.A. Washington Dc, Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria, Alzheimers Dement 12(3) (2016) 292–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Swerdlow RH, Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease, J Alzheimers Dis 62(3) (2018) 1403–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jove M, Mota-Martorell N, Torres P, Ayala V, Portero-Otin M, Ferrer I, Pamplona R, The Causal Role of Lipoxidative Damage in Mitochondrial Bioenergetic Dysfunction Linked to Alzheimer’s Disease Pathology, Life (Basel) 11(5) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mosconi L, De Santi S, Li J, Tsui WH, Li Y, Boppana M, Laska E, Rusinek H, de Leon MJ, Hippocampal hypometabolism predicts cognitive decline from normal aging, Neurobiol Aging 29(5) (2008) 676–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Herholz K, Cerebral glucose metabolism in preclinical and prodromal Alzheimer’s disease, Expert Rev Neurother 10(11) (2010) 1667–73. [DOI] [PubMed] [Google Scholar]
- [19].Hunt A, Schonknecht P, Henze M, Seidl U, Haberkorn U, Schroder J, Reduced cerebral glucose metabolism in patients at risk for Alzheimer’s disease, Psychiatry Res 155(2) (2007) 147–54. [DOI] [PubMed] [Google Scholar]
- [20].Jagust WJ, Haan MN, Eberling JL, Wolfe N, Reed BR, Functional imaging predicts cognitive decline in Alzheimer’s disease, J Neuroimaging 6(3) (1996) 156–60. [DOI] [PubMed] [Google Scholar]
- [21].Mosconi L, Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD, Eur J Nucl Med Mol Imaging 32(4) (2005) 486–510. [DOI] [PubMed] [Google Scholar]
- [22].Magistretti PJ, Allaman I, A cellular perspective on brain energy metabolism and functional imaging, Neuron 86(4) (2015) 883–901. [DOI] [PubMed] [Google Scholar]
- [23].Marcus C, Mena E, Subramaniam RM, Brain PET in the diagnosis of Alzheimer’s disease, Clin Nucl Med 39(10) (2014) e413–22; quiz e423–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].An Y, Varma VR, Varma S, Casanova R, Dammer E, Pletnikova O, Chia CW, Egan JM, Ferrucci L, Troncoso J, Levey AI, Lah J, Seyfried NT, Legido-Quigley C, O’Brien R, Thambisetty M, Evidence for brain glucose dysregulation in Alzheimer’s disease, Alzheimers Dement 14(3) (2018) 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Butterfield DA, Di Domenico F, Swomley AM, Head E, Perluigi M, Redox proteomics analysis to decipher the neurobiology of Alzheimer-like neurodegeneration: overlaps in Down’s syndrome and Alzheimer’s disease brain, Biochem J 463(2) (2014) 177–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zimmet P, Globalization, coca-colonization and the chronic disease epidemic: can the Doomsday scenario be averted?, J Intern Med 247(3) (2000) 301–10. [DOI] [PubMed] [Google Scholar]
- [27].Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS, Koplan JP, The continuing epidemics of obesity and diabetes in the United States, JAMA 286(10) (2001) 1195–200. [DOI] [PubMed] [Google Scholar]
- [28].Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, Balkau B, Heude B, Charpentier G, Hudson TJ, Montpetit A, Pshezhetsky AV, Prentki M, Posner BI, Balding DJ, Meyre D, Polychronakos C, Froguel P, A genome-wide association study identifies novel risk loci for type 2 diabetes, Nature 445(7130) (2007) 881–5. [DOI] [PubMed] [Google Scholar]
- [29].Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, Hughes TE, Groop L, Altshuler D, Almgren P, Florez JC, Meyer J, Ardlie K, Bengtsson Bostrom K, Isomaa B, Lettre G, Lindblad U, Lyon HN, Melander O, Newton-Cheh C, Nilsson P, Orho-Melander M, Rastam L, Speliotes EK, Taskinen MR, Tuomi T, Guiducci C, Berglund A, Carlson J, Gianniny L, Hackett R, Hall L, Holmkvist J, Laurila E, Sjogren M, Sterner M, Surti A, Svensson M, Svensson M, Tewhey R, Blumenstiel B, Parkin M, Defelice M, Barry R, Brodeur W, Camarata J, Chia N, Fava M, Gibbons J, Handsaker B, Healy C, Nguyen K, Gates C, Sougnez C, Gage D, Nizzari M, Gabriel SB, Chirn GW, Ma Q, Parikh H, Richardson D, Ricke D, Purcell S, Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels, Science 316(5829) (2007) 1331–6. [DOI] [PubMed] [Google Scholar]
- [30].Sullivan PW, Morrato EH, Ghushchyan V, Wyatt HR, Hill JO, Obesity, inactivity, and the prevalence of diabetes and diabetes-related cardiovascular comorbidities in the U.S., 2000–2002, Diabetes Care 28(7) (2005) 1599–603. [DOI] [PubMed] [Google Scholar]
- [31].Backestrom A, Papadopoulos K, Eriksson S, Olsson T, Andersson M, Blennow K, Zetterberg H, Nyberg L, Rolandsson O, Acute hyperglycaemia leads to altered frontal lobe brain activity and reduced working memory in type 2 diabetes, PLoS One 16(3) (2021) e0247753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rebelos E, Mari A, Bucci M, Honka MJ, Hannukainen JC, Virtanen KA, Hirvonen J, Nummenmaa L, Heni M, Iozzo P, Ferrannini E, Nuutila P, Brain substrate metabolism and ss-cell function in humans: A positron emission tomography study, Endocrinol Diabetes Metab 3(3) (2020) e00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Roberts RO, Knopman DS, Cha RH, Mielke MM, Pankratz VS, Boeve BF, Kantarci K, Geda YE, Jack CR Jr., Petersen RC, Lowe VJ, Diabetes and elevated hemoglobin A1c levels are associated with brain hypometabolism but not amyloid accumulation, J Nucl Med 55(5) (2014) 759–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kepes Z, Aranyi C, Forgacs A, Nagy F, Kukuts K, Hascsi Z, Esze R, Somodi S, Kaplar M, Varga J, Emri M, Garai I, Glucose-level dependent brain hypometabolism in type 2 diabetes mellitus and obesity, Eur J Hybrid Imaging 5(1) (2021) 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hwang JJ, Jiang L, Hamza M, Sanchez Rangel E, Dai F, Belfort-DeAguiar R, Parikh L, Koo BB, Rothman DL, Mason G, Sherwin RS, Blunted rise in brain glucose levels during hyperglycemia in adults with obesity and T2DM, JCI Insight 2(20) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MM, Association of diabetes mellitus and dementia: the Rotterdam Study, Diabetologia 39(11) (1996) 1392–7. [DOI] [PubMed] [Google Scholar]
- [37].Luchsinger JA, Tang MX, Shea S, Mayeux R, Hyperinsulinemia and risk of Alzheimer disease, Neurology 63(7) (2004) 1187–92. [DOI] [PubMed] [Google Scholar]
- [38].Ronnemaa E, Zethelius B, Sundelof J, Sundstrom J, Degerman-Gunnarsson M, Berne C, Lannfelt L, Kilander L, Impaired insulin secretion increases the risk of Alzheimer disease, Neurology 71(14) (2008) 1065–71. [DOI] [PubMed] [Google Scholar]
- [39].Morris JK, Vidoni ED, Honea RA, Burns JM, Alzheimer’s Disease Neuroimaging I, Impaired glycemia increases disease progression in mild cognitive impairment, Neurobiology of aging 35(3) (2014) 585–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pluta R, Blood-brain barrier dysfunction and amyloid precursor protein accumulation in microvascular compartment following ischemia-reperfusion brain injury with 1-year survival, Acta Neurochir Suppl 86 (2003) 117–22. [DOI] [PubMed] [Google Scholar]
- [41].Sadowski M, Pankiewicz J, Scholtzova H, Li YS, Quartermain D, Duff K, Wisniewski T, Links between the pathology of Alzheimer’s disease and vascular dementia, Neurochem Res 29(6) (2004) 1257–66. [DOI] [PubMed] [Google Scholar]
- [42].de la Monte SM, Wands JR, Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer’s disease, Journal of Alzheimer’s disease : JAD 7(1) (2005) 45–61. [DOI] [PubMed] [Google Scholar]
- [43].Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC, Increased risk of type 2 diabetes in Alzheimer disease, Diabetes 53(2) (2004) 474–81. [DOI] [PubMed] [Google Scholar]
- [44].De Felice FG, Alzheimer’s disease and insulin resistance: translating basic science into clinical applications, J Clin Invest 123(2) (2013) 531–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kern W, Peters A, Fruehwald-Schultes B, Deininger E, Born J, Fehm HL, Improving influence of insulin on cognitive functions in humans, Neuroendocrinology 74(4) (2001) 270–80. [DOI] [PubMed] [Google Scholar]
- [46].Huang CC, Lee CC, Hsu KS, The role of insulin receptor signaling in synaptic plasticity and cognitive function, Chang Gung Med J 33(2) (2010) 115–25. [PubMed] [Google Scholar]
- [47].Reger MA, Watson GS, Frey WH 2nd, Baker LD, Cholerton B, Keeling ML, Belongia DA, Fishel MA, Plymate SR, Schellenberg GD, Cherrier MM, Craft S, Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype, Neurobiol Aging 27(3) (2006) 451–8. [DOI] [PubMed] [Google Scholar]
- [48].Zhang H, Hao Y, Manor B, Novak P, Milberg W, Zhang J, Fang J, Novak V, Intranasal insulin enhanced resting-state functional connectivity of hippocampal regions in type 2 diabetes, Diabetes 64(3) (2015) 1025–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B, Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial, Arch Neurol 69(1) (2012) 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Biddinger SB, Kahn CR, From mice to men: insights into the insulin resistance syndromes, Annu Rev Physiol 68 (2006) 123–58. [DOI] [PubMed] [Google Scholar]
- [51].Kahn SE, Hull RL, Utzschneider KM, Mechanisms linking obesity to insulin resistance and type 2 diabetes, Nature 444(7121) (2006) 840–6. [DOI] [PubMed] [Google Scholar]
- [52].Lazar MA, How obesity causes diabetes: not a tall tale, Science 307(5708) (2005) 373–5. [DOI] [PubMed] [Google Scholar]
- [53].Lin TH Chun, L. Kang, Adipose extracellular matrix remodelling in obesity and insulin resistance, Biochem Pharmacol 119 (2016) 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bluher M, Adipose tissue inflammation: a cause or consequence of obesity-related insulin resistance?, Clin Sci (Lond) 130(18) (2016) 1603–14. [DOI] [PubMed] [Google Scholar]
- [55].Ferreira ST, Clarke JR, Bomfim TR, De Felice FG, Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer’s disease, Alzheimers Dement 10(1 Suppl) (2014) S76–83. [DOI] [PubMed] [Google Scholar]
- [56].De Felice FG, Ferreira ST, Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease, Diabetes 63(7) (2014) 2262–72. [DOI] [PubMed] [Google Scholar]
- [57].Butterfield DA, Di Domenico F, Barone E, Elevated risk of type 2 diabetes for development of Alzheimer disease: a key role for oxidative stress in brain, Biochim Biophys Acta 1842(9) (2014) 1693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Watson GS, Craft S, Modulation of memory by insulin and glucose: neuropsychological observations in Alzheimer’s disease, Eur J Pharmacol 490(1–3) (2004) 97–113. [DOI] [PubMed] [Google Scholar]
- [59].Cheng G, Huang C, Deng H, Wang H, Diabetes as a risk factor for dementia and mild cognitive impairment: a meta-analysis of longitudinal studies, Intern Med J 42(5) (2012) 484–91. [DOI] [PubMed] [Google Scholar]
- [60].Yaffe K, Blackwell T, Whitmer RA, Krueger K, Barrett Connor E, Glycosylated hemoglobin level and development of mild cognitive impairment or dementia in older women, J Nutr Health Aging 10(4) (2006) 293–5. [PubMed] [Google Scholar]
- [61].Willette AA, Modanlo N, Kapogiannis D, Alzheimer’s Disease Neuroimaging I, Insulin resistance predicts medial temporal hypermetabolism in mild cognitive impairment conversion to Alzheimer disease, Diabetes 64(6) (2015) 1933–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Arnold SE, Lucki I, Brookshire BR, Carlson GC, Browne CA, Kazi H, Bang S, Choi BR, Chen Y, McMullen MF, Kim SF, High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice, Neurobiol Dis 67 (2014) 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Liu Z, Patil IY, Jiang T, Sancheti H, Walsh JP, Stiles BL, Yin F, Cadenas E, High-fat diet induces hepatic insulin resistance and impairment of synaptic plasticity, PLoS One 10(5) (2015) e0128274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Martins IV, Rivers-Auty J, Allan SM, Lawrence CB, Mitochondrial Abnormalities and Synaptic Loss Underlie Memory Deficits Seen in Mouse Models of Obesity and Alzheimer’s Disease, J Alzheimers Dis 55(3) (2017) 915–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ramos-Rodriguez JJ, Ortiz O, Jimenez-Palomares M, Kay KR, Berrocoso E, Murillo-Carretero MI, Perdomo G, Spires-Jones T, Cozar-Castellano I, Lechuga-Sancho AM, Garcia-Alloza M, Differential central pathology and cognitive impairment in pre-diabetic and diabetic mice, Psychoneuroendocrinology 38(11) (2013) 2462–75. [DOI] [PubMed] [Google Scholar]
- [66].Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE, Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline, J Clin Invest 122(4) (2012) 1316–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R, Perluigi M, Butterfield DA, Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD, J Neurochem 133(5) (2015) 739–49. [DOI] [PubMed] [Google Scholar]
- [68].Kapogiannis D, Mustapic M, Shardell MD, Berkowitz ST, Diehl TC, Spangler RD, Tran J, Lazaropoulos MP, Chawla S, Gulyani S, Eitan E, An Y, Huang CW, Oh ES, Lyketsos CG, Resnick SM, Goetzl EJ, Ferrucci L, Association of Extracellular Vesicle Biomarkers With Alzheimer Disease in the Baltimore Longitudinal Study of Aging, JAMA Neurol 76(11) (2019) 1340–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].White MF, Insulin signaling in health and disease, Science 302(5651) (2003) 1710–1. [DOI] [PubMed] [Google Scholar]
- [70].Gehart H, Kumpf S, Ittner A, Ricci R, MAPK signalling in cellular metabolism: stress or wellness?, EMBO Rep 11(11) (2010) 834–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Horwood JM, Dufour F, Laroche S, Davis S, Signalling mechanisms mediated by the phosphoinositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat, Eur J Neurosci 23(12) (2006) 3375–84. [DOI] [PubMed] [Google Scholar]
- [72].Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM, Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity, Nat Rev Neurosci 8(10) (2007) 766–75. [DOI] [PubMed] [Google Scholar]
- [73].Akter K, Lanza EA, Martin SA, Myronyuk N, Rua M, Raffa RB, Diabetes mellitus and Alzheimer’s disease: shared pathology and treatment?, Br J Clin Pharmacol 71(3) (2011) 365–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Copps KD, White MF, Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2, Diabetologia 55(10) (2012) 2565–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, Ham M, Talukdar S, Chen A, Lu WJ, Bandyopadhyay GK, Schwendener R, Olefsky J, Kim JB, Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance, Diabetes 60(10) (2011) 2474–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Barone E, Di Domenico F, Cassano T, Arena A, Tramutola A, Lavecchia MA, Coccia R, Butterfield DA, Perluigi M, Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm, Free Radic Biol Med 91 (2016) 127–42. [DOI] [PubMed] [Google Scholar]
- [77].Moreira PI, Santos MS, Seica R, Oliveira CR, Brain mitochondrial dysfunction as a link between Alzheimer’s disease and diabetes, J Neurol Sci 257(1–2) (2007) 206–14. [DOI] [PubMed] [Google Scholar]
- [78].Maiese K, Chong ZZ, Shang YC, Mechanistic insights into diabetes mellitus and oxidative stress, Curr Med Chem 14(16) (2007) 1729–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Tramutola A, Lanzillotta C, Arena A, Barone E, Perluigi M, Di Domenico F, Increased Mammalian Target of Rapamycin Signaling Contributes to the Accumulation of Protein Oxidative Damage in a Mouse Model of Down’s Syndrome, Neurodegener Dis 16(1–2) (2016) 62–8. [DOI] [PubMed] [Google Scholar]
- [80].de la Monte SM, Insulin resistance and Alzheimer’s disease, BMB Rep 42(8) (2009) 475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].de la Monte SM, Tong M, Brain metabolic dysfunction at the core of Alzheimer’s disease, Biochem Pharmacol 88(4) (2014) 548–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Orth M, Schapira AH, Mitochondria and degenerative disorders, Am J Med Genet 106(1) (2001) 27–36. [DOI] [PubMed] [Google Scholar]
- [83].Schieber M, Chandel NS, ROS function in redox signaling and oxidative stress, Curr Biol 24(10) (2014) R453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Brand MD, Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling, Free Radic Biol Med 100 (2016) 14–31. [DOI] [PubMed] [Google Scholar]
- [85].Schmid E, El Benna J, Galter D, Klein G, Droge W, Redox priming of the insulin receptor beta-chain associated with altered tyrosine kinase activity and insulin responsiveness in the absence of tyrosine autophosphorylation, FASEB J 12(10) (1998) 863–70. [DOI] [PubMed] [Google Scholar]
- [86].Schmid E, Hotz-Wagenblatt A, Hacj V, Droge W, Phosphorylation of the insulin receptor kinase by phosphocreatine in combination with hydrogen peroxide: the structural basis of redox priming, FASEB J 13(12) (1999) 1491–500. [DOI] [PubMed] [Google Scholar]
- [87].May JM, de Haen C, Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells, J Biol Chem 254(7) (1979) 2214–20. [PubMed] [Google Scholar]
- [88].Mukherjee SP, Lane RH, Lynn WS, Endogenous hydrogen peroxide and peroxidative metabolism in adipocytes in response to insulin and sulfhydryl reagents, Biochem Pharmacol 27(22) (1978) 2589–94. [DOI] [PubMed] [Google Scholar]
- [89].Goldstein BJ, Mahadev K, Wu X, Redox paradox: insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets, Diabetes 54(2) (2005) 311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Storozhevykh TP, Senilova YE, Persiyantseva NA, Pinelis VG, Pomytkin IA, Mitochondrial respiratory chain is involved in insulin-stimulated hydrogen peroxide production and plays an integral role in insulin receptor autophosphorylation in neurons, BMC Neurosci 8 (2007) 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Sultana R, Butterfield DA, Role of oxidative stress in the progression of Alzheimer’s disease, J Alzheimers Dis 19(1) (2010) 341–53. [DOI] [PubMed] [Google Scholar]
- [92].Sultana R, Perluigi M, Butterfield DA, Oxidatively modified proteins in Alzheimer’s disease (AD), mild cognitive impairment and animal models of AD: role of Abeta in pathogenesis, Acta Neuropathol 118(1) (2009) 131–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Garcia-Fernandez M, Delgado G, Puche JE, Gonzalez-Baron S, Castilla Cortazar I, Low doses of insulin-like growth factor I improve insulin resistance, lipid metabolism, and oxidative damage in aging rats, Endocrinology 149(5) (2008) 2433–42. [DOI] [PubMed] [Google Scholar]
- [94].Maiese K, Morhan SD, Chong ZZ, Oxidative stress biology and cell injury during type 1 and type 2 diabetes mellitus, Current neurovascular research 4(1) (2007) 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Pintana H, Apaijai N, Pratchayasakul W, Chattipakorn N, Chattipakorn SC, Effects of metformin on learning and memory behaviors and brain mitochondrial functions in high fat diet induced insulin resistant rats, Life Sci 91(11–12) (2012) 409–14. [DOI] [PubMed] [Google Scholar]
- [96].Liao YJ, Ueno M, Nakagawa T, Huang C, Kanenishi K, Onodera M, Sakamoto H, Oxidative damage in cerebral vessels of diabetic db/db mice, Diabetes Metab Res Rev 21(6) (2005) 554–9. [DOI] [PubMed] [Google Scholar]
- [97].Carvalho C, Cardoso S, Correia SC, Santos RX, Santos MS, Baldeiras I, Oliveira CR, Moreira PI, Metabolic alterations induced by sucrose intake and Alzheimer’s disease promote similar brain mitochondrial abnormalities, Diabetes 61(5) (2012) 1234–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Mainardi M, Fusco S, Grassi C, Modulation of hippocampal neural plasticity by glucose-related signaling, Neural Plast 2015 (2015) 657928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Kullmann S, Heni M, Hallschmid M, Fritsche A, Preissl H, Haring HU, Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans, Physiol Rev 96(4) (2016) 1169–209. [DOI] [PubMed] [Google Scholar]
- [100].Kamat PK, Kalani A, Rai S, Swarnkar S, Tota S, Nath C, Tyagi N, Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer’s Disease: Understanding the Therapeutics Strategies, Mol Neurobiol 53(1) (2016) 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA, Oxidative damage is the earliest event in Alzheimer disease, J Neuropathol Exp Neurol 60(8) (2001) 759–67. [DOI] [PubMed] [Google Scholar]
- [102].Butterfield DA, Drake J, Pocernich C, Castegna A, Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide, Trends Mol Med 7(12) (2001) 548–54. [DOI] [PubMed] [Google Scholar]
- [103].Lanzillotta C, Di Domenico F, Perluigi M, Butterfield DA, Targeting Mitochondria in Alzheimer Disease: Rationale and Perspectives, CNS Drugs 33(10) (2019) 957–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Halliwell B, Oxidative stress and neurodegeneration: where are we now?, J Neurochem 97(6) (2006) 1634–58. [DOI] [PubMed] [Google Scholar]
- [105].Di Domenico F, Pupo G, Giraldo E, Badia MC, Monllor P, Lloret A, Schinina ME, Giorgi A, Cini C, Tramutola A, Butterfield DA, Vina J, Perluigi M, Oxidative signature of cerebrospinal fluid from mild cognitive impairment and Alzheimer disease patients, Free Radic Biol Med 91 (2016) 1–9. [DOI] [PubMed] [Google Scholar]
- [106].Butterfield D.A.a.S., R. E, Protein Oxidation Processes in Aging Brain, Adv. Cell Aging Gerontol 2 (1997) 161–191. [Google Scholar]
- [107].Head E, Oxidative damage and cognitive dysfunction: antioxidant treatments to promote healthy brain aging, Neurochem Res 34(4) (2009) 670–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Sohal RS, Ku HH, Agarwal S, Forster MJ, Lal H, Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse, Mech Ageing Dev 74(1–2) (1994) 121–33. [DOI] [PubMed] [Google Scholar]
- [109].Liu R, Liu IY, Bi X, Thompson RF, Doctrow SR, Malfroy B, Baudry M, Reversal of age-related learning deficits and brain oxidative stress in mice with superoxide dismutase/catalase mimetics, Proc Natl Acad Sci U S A 100(14) (2003) 8526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Stadtman ER, Berlett BS, Reactive oxygen-mediated protein oxidation in aging and disease, Chem Res Toxicol 10(5) (1997) 485–94. [DOI] [PubMed] [Google Scholar]
- [111].Barone E, Mosser S, Fraering PC, Inactivation of brain Cofilin-1 by age, Alzheimer’s disease and gamma-secretase, Biochim Biophys Acta 1842(12 Pt A) (2014) 2500–9. [DOI] [PubMed] [Google Scholar]
- [112].Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, Bohr VA, Ageing as a risk factor for neurodegenerative disease, Nat Rev Neurol 15(10) (2019) 565–581. [DOI] [PubMed] [Google Scholar]
- [113].Mattson MP, Magnus T, Ageing and neuronal vulnerability, Nat Rev Neurosci 7(4) (2006) 278–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Perluigi M, Barone E, Di Domenico F, Butterfield DA, Aberrant protein phosphorylation in Alzheimer disease brain disturbs pro-survival and cell death pathways, Biochim Biophys Acta 1862(10) (2016) 1871–82. [DOI] [PubMed] [Google Scholar]
- [115].Ahmad W, Ijaz B, Shabbiri K, Ahmed F, Rehman S, Oxidative toxicity in diabetes and Alzheimer’s disease: mechanisms behind ROS/ RNS generation, J Biomed Sci 24(1) (2017) 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Rehman K, Akash MSH, Mechanism of Generation of Oxidative Stress and Pathophysiology of Type 2 Diabetes Mellitus: How Are They Interlinked?, J Cell Biochem 118(11) (2017) 3577–3585. [DOI] [PubMed] [Google Scholar]
- [117].Henriksen EJ, Diamond-Stanic MK, Marchionne EM, Oxidative stress and the etiology of insulin resistance and type 2 diabetes, Free Radic Biol Med 51(5) (2011) 993–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Rehman A, Nourooz-Zadeh J, Moller W, Tritschler H, Pereira P, Halliwell B, Increased oxidative damage to all DNA bases in patients with type II diabetes mellitus, FEBS Lett 448(1) (1999) 120–2. [DOI] [PubMed] [Google Scholar]
- [119].Singh P, Jain A, Kaur G, Impact of hypoglycemia and diabetes on CNS: correlation of mitochondrial oxidative stress with DNA damage, Mol Cell Biochem 260(1–2) (2004) 153–9. [DOI] [PubMed] [Google Scholar]
- [120].Nowotny K, Jung T, Hohn A, Weber D, Grune T, Advanced glycation end products and oxidative stress in type 2 diabetes mellitus, Biomolecules 5(1) (2015) 194–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Verdile G, Fuller SJ, Martins RN, The role of type 2 diabetes in neurodegeneration, Neurobiol Dis 84 (2015) 22–38. [DOI] [PubMed] [Google Scholar]
- [122].Craft S, Insulin resistance syndrome and Alzheimer’s disease: age- and obesity-related effects on memory, amyloid, and inflammation, Neurobiol Aging 26 Suppl 1 (2005) 65–9. [DOI] [PubMed] [Google Scholar]
- [123].Zhao WQ, Townsend M, Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease, Biochim Biophys Acta 1792(5) (2009) 482–96. [DOI] [PubMed] [Google Scholar]
- [124].Lanzillotta C, Tramutola A, Di Giacomo G, Marini F, Butterfield DA, Di Domenico F, Perluigi M, Barone E, Insulin resistance, oxidative stress and mitochondrial defects in Ts65dn mice brain: A harmful synergistic path in down syndrome, Free Radic Biol Med 165 (2021) 152–170. [DOI] [PubMed] [Google Scholar]
- [125].Di Domenico F, Sultana R, Ferree A, Smith K, Barone E, Perluigi M, Coccia R, Pierce W, Cai J, Mancuso C, Squillace R, Wiengele M, Dalle-Donne I, Wolozin B, Butterfield DA, Redox proteomics analyses of the influence of co-expression of wild-type or mutated LRRK2 and Tau on C. elegans protein expression and oxidative modification: relevance to Parkinson disease, Antioxid Redox Signal 17(11) (2012) 1490–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM, Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine, J Alzheimers Dis 8(3) (2005) 247–68. [DOI] [PubMed] [Google Scholar]
- [127].Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM, Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes?, J Alzheimers Dis 7(1) (2005) 63–80. [DOI] [PubMed] [Google Scholar]
- [128].Tramutola A, Lanzillotta C, Di Domenico F, Head E, Butterfield DA, Perluigi M, Barone E, Brain insulin resistance triggers early onset Alzheimer disease in Down syndrome, Neurobiol Dis 137 (2020) 104772. [DOI] [PubMed] [Google Scholar]
- [129].Gerbitz KD, Gempel K, Brdiczka D, Mitochondria and diabetes. Genetic, biochemical, and clinical implications of the cellular energy circuit, Diabetes 45(2) (1996) 113–26. [DOI] [PubMed] [Google Scholar]
- [130].Neumann KF, Rojo L, Navarrete LP, Farias G, Reyes P, Maccioni RB, Insulin resistance and Alzheimer’s disease: molecular links & clinical implications, Curr Alzheimer Res 5(5) (2008) 438–47. [DOI] [PubMed] [Google Scholar]
- [131].Sripetchwandee J, Chattipakorn N, Chattipakorn SC, Links Between Obesity-Induced Brain Insulin Resistance, Brain Mitochondrial Dysfunction, and Dementia, Front Endocrinol (Lausanne) 9 (2018) 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Sa-Nguanmoo P, Tanajak P, Kerdphoo S, Satjaritanun P, Wang X, Liang G, Li X, Jiang C, Pratchayasakul W, Chattipakorn N, Chattipakorn SC, FGF21 improves cognition by restored synaptic plasticity, dendritic spine density, brain mitochondrial function and cell apoptosis in obese-insulin resistant male rats, Horm Behav 85 (2016) 86–95. [DOI] [PubMed] [Google Scholar]
- [133].Pipatpiboon N, Pratchayasakul W, Chattipakorn N, Chattipakorn SC, PPARgamma agonist improves neuronal insulin receptor function in hippocampus and brain mitochondria function in rats with insulin resistance induced by long term high-fat diets, Endocrinology 153(1) (2012) 329–38. [DOI] [PubMed] [Google Scholar]
- [134].Koliaki C, Roden M, Alterations of Mitochondrial Function and Insulin Sensitivity in Human Obesity and Diabetes Mellitus, Annu Rev Nutr 36 (2016) 337–67. [DOI] [PubMed] [Google Scholar]
- [135].Tong M, de la Monte SM, Mechanisms of ceramide-mediated neurodegeneration, J Alzheimers Dis 16(4) (2009) 705–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Biessels GJ, Strachan MW, Visseren FL, Kappelle LJ, Whitmer RA, Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions, Lancet Diabetes Endocrinol 2(3) (2014) 246–55. [DOI] [PubMed] [Google Scholar]
- [137].Dragicevic N, Mamcarz M, Zhu Y, Buzzeo R, Tan J, Arendash GW, Bradshaw PC, Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice, J Alzheimers Dis 20 Suppl 2 (2010) S535–50. [DOI] [PubMed] [Google Scholar]
- [138].Baek SH, Park SJ, Jeong JI, Kim SH, Han J, Kyung JW, Baik SH, Choi Y, Choi BY, Park JS, Bahn G, Shin JH, Jo DS, Lee JY, Jang CG, Arumugam TV, Kim J, Han JW, Koh JY, Cho DH, Jo DG, Inhibition of Drp1 Ameliorates Synaptic Depression, Abeta Deposition, and Cognitive Impairment in an Alzheimer’s Disease Model, J Neurosci 37(20) (2017) 5099–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Niki E, Oxidant-specific biomarkers of oxidative stress. Association with atherosclerosis and implication for antioxidant effects, Free Radic Biol Med 120 (2018) 425–440. [DOI] [PubMed] [Google Scholar]
- [140].Palozza P, Barone E, Mancuso C, Picci N, The protective role of carotenoids against 7-keto-cholesterol formation in solution, Mol Cell Biochem 309(1–2) (2008) 61–8. [DOI] [PubMed] [Google Scholar]
- [141].Testa G, Staurenghi E, Zerbinati C, Gargiulo S, Iuliano L, Giaccone G, Fanto F, Poli G, Leonarduzzi G, Gamba P, Changes in brain oxysterols at different stages of Alzheimer’s disease: Their involvement in neuroinflammation, Redox Biol 10 (2016) 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Gamba P, Staurenghi E, Testa G, Giannelli S, Sottero B, Leonarduzzi G, A Crosstalk Between Brain Cholesterol Oxidation and Glucose Metabolism in Alzheimer’s Disease, Front Neurosci 13 (2019) 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Kothari V, Luo Y, Tornabene T, O’Neill AM, Greene MW, Geetha T, Babu JR, High fat diet induces brain insulin resistance and cognitive impairment in mice, Biochim Biophys Acta Mol Basis Dis 1863(2) (2017) 499–508. [DOI] [PubMed] [Google Scholar]
- [144].Spinelli M, Fusco S, Mainardi M, Scala F, Natale F, Lapenta R, Mattera A, Rinaudo M, Li Puma DD, Ripoli C, Grassi A, D’Ascenzo M, Grassi C, Brain insulin resistance impairs hippocampal synaptic plasticity and memory by increasing GluA1 palmitoylation through FoxO3a, Nat Commun 8(1) (2017) 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Cordes CM, Bennett RG, Siford GL, Hamel FG, Nitric oxide inhibits insulin-degrading enzyme activity and function through S-nitrosylation, Biochem Pharmacol 77(6) (2009) 1064–73. [DOI] [PubMed] [Google Scholar]
- [146].Kummer MP, Hulsmann C, Hermes M, Axt D, Heneka MT, Nitric oxide decreases the enzymatic activity of insulin degrading enzyme in APP/PS1 mice, J Neuroimmune Pharmacol 7(1) (2012) 165–72. [DOI] [PubMed] [Google Scholar]
- [147].Akhtar MW, Sanz-Blasco S, Dolatabadi N, Parker J, Chon K, Lee MS, Soussou W, McKercher SR, Ambasudhan R, Nakamura T, Lipton SA, Elevated glucose and oligomeric beta-amyloid disrupt synapses via a common pathway of aberrant protein S-nitrosylation, Nat Commun 7 (2016) 10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Cordes CM, Bennett RG, Siford GL, Hamel FG, Redox regulation of insulin degradation by insulin-degrading enzyme, PLoS One 6(3) (2011) e18138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Shinall H, Song ES, Hersh LB, Susceptibility of amyloid beta peptide degrading enzymes to oxidative damage: a potential Alzheimer’s disease spiral, Biochemistry 44(46) (2005) 15345–50. [DOI] [PubMed] [Google Scholar]
- [150].Barone E, Di Domenico F, Cenini G, Sultana R, Coccia R, Preziosi P, Perluigi M, Mancuso C, Butterfield DA, Oxidative and nitrosative modifications of biliverdin reductase-A in the brain of subjects with Alzheimer’s disease and amnestic mild cognitive impairment, J Alzheimers Dis 25(4) (2011) 623–33. [DOI] [PubMed] [Google Scholar]
- [151].Barone E, Di Domenico F, Cenini G, Sultana R, Cini C, Preziosi P, Perluigi M, Mancuso C, Butterfield DA, Biliverdin reductase--a protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment, Biochim Biophys Acta 1812(4) (2011) 480–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Barone E, Mancuso C, Di Domenico F, Sultana R, Murphy MP, Head E, Butterfield DA, Biliverdin reductase-A: a novel drug target for atorvastatin in a dog pre-clinical model of Alzheimer disease, J Neurochem 120(1) (2012) 135–46. [DOI] [PubMed] [Google Scholar]
- [153].Di Domenico F, Pupo G, Mancuso C, Barone E, Paolini F, Arena A, Blarzino C, Schmitt FA, Head E, Butterfield DA, Perluigi M, Bach1 overexpression in Down syndrome correlates with the alteration of the HO-1/BVR-a system: insights for transition to Alzheimer’s disease, J Alzheimers Dis 44(4) (2015) 1107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Triani F, Tramutola A, Di Domenico F, Sharma N, Butterfield DA, Head E, Perluigi M, Barone E, Biliverdin reductase-A impairment links brain insulin resistance with increased Abeta production in an animal model of aging: Implications for Alzheimer disease, Biochim Biophys Acta Mol Basis Dis 1864(10) (2018) 3181–3194. [DOI] [PubMed] [Google Scholar]
- [155].Ceccarelli V, Barchetta I, Cimini FA, Bertoccini L, Chiappetta C, Capoccia D, Carletti R, Di Cristofano C, Silecchia G, Fontana M, Leonetti F, Lenzi A, Baroni MG, Barone E, Cavallo MG, Reduced Biliverdin Reductase-A Expression in Visceral Adipose Tissue is Associated with Adipocyte Dysfunction and NAFLD in Human Obesity, Int J Mol Sci 21(23) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Cimini FA, Arena A, Barchetta I, Tramutola A, Ceccarelli V, Lanzillotta C, Fontana M, Bertoccini L, Leonetti F, Capoccia D, Silecchia G, Di Cristofano C, Chiappetta C, Di Domenico F, Baroni MG, Perluigi M, Cavallo MG, Barone E, Reduced biliverdin reductase-A levels are associated with early alterations of insulin signaling in obesity, Biochim Biophys Acta Mol Basis Dis 1865(6) (2019) 1490–1501. [DOI] [PubMed] [Google Scholar]
- [157].Stec DE, Gordon DM, Nestor-Kalinoski AL, Donald MC, Mitchell ZL, Creeden JF, Hinds TD Jr., Biliverdin Reductase A (BVRA) Knockout in Adipocytes Induces Hypertrophy and Reduces Mitochondria in White Fat of Obese Mice, Biomolecules 10(3) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Hinds TD Jr., Burns KA, Hosick PA, McBeth L, Nestor-Kalinoski A, Drummond HA, AlAmodi AA, Hankins MW, Vanden Heuvel JP, Stec DE, Biliverdin Reductase A Attenuates Hepatic Steatosis by Inhibition of Glycogen Synthase Kinase (GSK) 3beta Phosphorylation of Serine 73 of Peroxisome Proliferator-activated Receptor (PPAR) alpha, J Biol Chem 291(48) (2016) 25179–25191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Pivovarova O, Hohn A, Grune T, Pfeiffer AF, Rudovich N, Insulin-degrading enzyme: new therapeutic target for diabetes and Alzheimer’s disease?, Ann Med 48(8) (2016) 614–624. [DOI] [PubMed] [Google Scholar]
- [160].Leissring MA, Gonzalez-Casimiro CM, Merino B, Suire CN, Perdomo G, Targeting Insulin-Degrading Enzyme in Insulin Clearance, Int J Mol Sci 22(5) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Demozay D, Mas JC, Rocchi S, Van Obberghen E, FALDH reverses the deleterious action of oxidative stress induced by lipid peroxidation product 4-hydroxynonenal on insulin signaling in 3T3-L1 adipocytes, Diabetes 57(5) (2008) 1216–26. [DOI] [PubMed] [Google Scholar]
- [162].Shearn CT, Fritz KS, Reigan P, Petersen DR, Modification of Akt2 by 4-hydroxynonenal inhibits insulin-dependent Akt signaling in HepG2 cells, Biochemistry 50(19) (2011) 3984–96. [DOI] [PubMed] [Google Scholar]
- [163].Zhang H, Forman HJ, Signaling by 4-hydroxy-2-nonenal: Exposure protocols, target selectivity and degradation, Arch Biochem Biophys 617 (2017) 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Gonzalez-Casimiro CM, Merino B, Casanueva-Alvarez E, Postigo-Casado T, Camara-Torres P, Fernandez-Diaz CM, Leissring MA, Cozar-Castellano I, Perdomo G, Modulation of Insulin Sensitivity by Insulin-Degrading Enzyme, Biomedicines 9(1) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Makarova KS, Grishin NV, Thermolysin and mitochondrial processing peptidase: how far structure-functional convergence goes, Protein Sci 8(11) (1999) 2537–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Fernandez-Gamba A, Leal MC, Morelli L, Castano EM, Insulin-degrading enzyme: structure-function relationship and its possible roles in health and disease, Curr Pharm Des 15(31) (2009) 3644–55. [DOI] [PubMed] [Google Scholar]
- [167].Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S, Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo, Proc Natl Acad Sci U S A 100(7) (2003) 4162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Furukawa Y, Shimada T, Furuta H, Matsuno S, Kusuyama A, Doi A, Nishi M, Sasaki H, Sanke T, Nanjo K, Polymorphisms in the IDE-KIF11-HHEX gene locus are reproducibly associated with type 2 diabetes in a Japanese population, J Clin Endocrinol Metab 93(1) (2008) 310–4. [DOI] [PubMed] [Google Scholar]
- [169].Karamohamed S, Demissie S, Volcjak J, Liu C, Heard-Costa N, Liu J, Shoemaker CM, Panhuysen CI, Meigs JB, Wilson P, Atwood LD, Cupples LA, Herbert A, Study NFH, Polymorphisms in the insulin-degrading enzyme gene are associated with type 2 diabetes in men from the NHLBI Framingham Heart Study, Diabetes 52(6) (2003) 1562–7. [DOI] [PubMed] [Google Scholar]
- [170].Nordman S, Ostenson CG, Efendic S, Gu HF, Loci of TCF7L2, HHEX and IDE on chromosome 10q and the susceptibility of their genetic polymorphisms to type 2 diabetes, Exp Clin Endocrinol Diabetes 117(4) (2009) 186–90. [DOI] [PubMed] [Google Scholar]
- [171].Bertram L, Blacker D, Mullin K, Keeney D, Jones J, Basu S, Yhu S, McInnis MG, Go RC, Vekrellis K, Selkoe DJ, Saunders AJ, Tanzi RE, Evidence for genetic linkage of Alzheimer’s disease to chromosome 10q, Science 290(5500) (2000) 2302–3. [DOI] [PubMed] [Google Scholar]
- [172].Bjork BF, Katzov H, Kehoe P, Fratiglioni L, Winblad B, Prince JA, Graff C, Positive association between risk for late-onset Alzheimer disease and genetic variation in IDE, Neurobiol Aging 28(9) (2007) 1374–80. [DOI] [PubMed] [Google Scholar]
- [173].Blomqvist ME, Chalmers K, Andreasen N, Bogdanovic N, Wilcock GK, Cairns NJ, Feuk L, Brookes AJ, Love S, Blennow K, Kehoe PG, Prince JA, Sequence variants of IDE are associated with the extent of beta-amyloid deposition in the Alzheimer’s disease brain, Neurobiol Aging 26(6) (2005) 795–802. [DOI] [PubMed] [Google Scholar]
- [174].Delikkaya B, Moriel N, Tong M, Gallucci G, de la Monte SM, Altered expression of insulin-degrading enzyme and regulator of calcineurin in the rat intracerebral streptozotocin model and human apolipoprotein E-epsilon4-associated Alzheimer’s disease, Alzheimers Dement (Amst) 11 (2019) 392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Kurochkin IV, Goto S, Alzheimer’s beta-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme, FEBS Lett 345(1) (1994) 33–7. [DOI] [PubMed] [Google Scholar]
- [176].Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, Hersh LB, Selkoe DJ, Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation, J Biol Chem 273(49) (1998) 32730–8. [DOI] [PubMed] [Google Scholar]
- [177].Leal MC, Magnani N, Villordo S, Buslje CM, Evelson P, Castano EM, Morelli L, Transcriptional regulation of insulin-degrading enzyme modulates mitochondrial amyloid beta (Abeta) peptide catabolism and functionality, J Biol Chem 288(18) (2013) 12920–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Zhao WQ, De Felice FG, Fernandez S, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL, Amyloid beta oligomers induce impairment of neuronal insulin receptors, FASEB J 22(1) (2008) 246–60. [DOI] [PubMed] [Google Scholar]
- [179].De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, Viola KL, Zhao WQ, Ferreira ST, Klein WL, Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers, Proceedings of the National Academy of Sciences of the United States of America 106(6) (2009) 1971–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Vekrellis K, Ye Z, Qiu WQ, Walsh D, Hartley D, Chesneau V, Rosner MR, Selkoe DJ, Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme, J Neurosci 20(5) (2000) 1657–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Zhang J, Guo Y, Wang Y, Song L, Zhang R, Du Y, Long-term treadmill exercise attenuates Abeta burdens and astrocyte activation in APP/PS1 mouse model of Alzheimer’s disease, Neurosci Lett 666 (2018) 70–77. [DOI] [PubMed] [Google Scholar]
- [182].Taniguchi CM, Emanuelli B, Kahn CR, Critical nodes in signalling pathways: insights into insulin action, Nat Rev Mol Cell Biol 7(2) (2006) 85–96. [DOI] [PubMed] [Google Scholar]
- [183].Kapitulnik J, Maines MD, Pleiotropic functions of biliverdin reductase: cellular signaling and generation of cytoprotective and cytotoxic bilirubin, Trends Pharmacol Sci 30(3) (2009) 129–37. [DOI] [PubMed] [Google Scholar]
- [184].Lerner-Marmarosh N, Shen J, Torno MD, Kravets A, Hu Z, Maines MD, Human biliverdin reductase: a member of the insulin receptor substrate family with serine/threonine/tyrosine kinase activity, Proc Natl Acad Sci U S A 102(20) (2005) 7109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Gibbs PE, Lerner-Marmarosh N, Poulin A, Farah E, Maines MD, Human biliverdin reductase-based peptides activate and inhibit glucose uptake through direct interaction with the kinase domain of insulin receptor, FASEB J 28(6) (2014) 2478–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Lerner-Marmarosh N, Miralem T, Gibbs PE, Maines MD, Human biliverdin reductase is an ERK activator; hBVR is an ERK nuclear transporter and is required for MAPK signaling, Proc Natl Acad Sci U S A 105(19) (2008) 6870–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Lerner-Marmarosh N, Miralem T, Gibbs PE, Maines MD, Regulation of TNF-alpha-activated PKC-zeta signaling by the human biliverdin reductase: identification of activating and inhibitory domains of the reductase, FASEB J 21(14) (2007) 3949–62. [DOI] [PubMed] [Google Scholar]
- [188].Kim YB, Kotani K, Ciaraldi TP, Henry RR, Kahn BB, Insulin-stimulated protein kinase C lambda/zeta activity is reduced in skeletal muscle of humans with obesity and type 2 diabetes: reversal with weight reduction, Diabetes 52(8) (2003) 1935–42. [DOI] [PubMed] [Google Scholar]
- [189].Zhang Y, Zong W, Zhang L, Ma Y, Wang J, Protein kinase M zeta and the maintenance of long-term memory, Neurochem Int 99 (2016) 215–20. [DOI] [PubMed] [Google Scholar]
- [190].Sharma N, Tramutola A, Lanzillotta C, Arena A, Blarzino C, Cassano T, Butterfield DA, Di Domenico F, Perluigi M, Barone E, Loss of biliverdin reductase-A favors Tau hyperphosphorylation in Alzheimer’s disease, Neurobiol Dis 125 (2019) 176–189. [DOI] [PubMed] [Google Scholar]
- [191].Cimini FA, Barchetta I, Zuliani I, Pagnotta S, Bertoccini L, Dule S, Zampieri M, Reale A, Baroni MG, Cavallo MG, Barone E, Biliverdin reductase-A protein levels are reduced in type 2 diabetes and are associated with poor glycometabolic control, Life Sci 284 (2021) 119913. [DOI] [PubMed] [Google Scholar]
- [192].Barone E, Di Domenico F, Mancuso C, Butterfield DA, The Janus face of the heme oxygenase/biliverdin reductase system in Alzheimer disease: it’s time for reconciliation, Neurobiol Dis 62 (2014) 144–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Di Domenico F, Perluigi M, Barone E, Biliverdin Reductase-A correlates with inducible nitric oxide synthasein in atorvastatin treated aged canine brain, Neural Regen Res 8(21) (2013) 1925–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Barone E, Cenini G, Sultana R, Di Domenico F, Fiorini A, Perluigi M, Noel T, Wang C, Mancuso C, St Clair DK, Butterfield DA, Lack of p53 decreases basal oxidative stress levels in the brain through upregulation of thioredoxin-1, biliverdin reductase-A, manganese superoxide dismutase, and nuclear factor kappa-B, Antioxid Redox Signal 16(12) (2012) 1407–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Di Domenico F, Barone E, Mancuso C, Perluigi M, Cocciolo A, Mecocci P, Butterfield DA, Coccia R, HO-1/BVR-a system analysis in plasma from probable Alzheimer’s disease and mild cognitive impairment subjects: a potential biochemical marker for the prediction of the disease, J Alzheimers Dis 32(2) (2012) 277–89. [DOI] [PubMed] [Google Scholar]
- [196].Barone E, Cenini G, Di Domenico F, Noel T, Wang C, Perluigi M, St Clair DK, Butterfield DA, Basal brain oxidative and nitrative stress levels are finely regulated by the interplay between superoxide dismutase 2 and p53, J Neurosci Res 93(11) (2015) 1728–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Mancuso C, Siciliano R, Barone E, Curcumin and Alzheimer disease: this marriage is not to be performed, J Biol Chem 286(3) (2011) le3; author reply le4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Barone E, Tramutola A, Triani F, Calcagnini S, Di Domenico F, Ripoli C, Gaetani S, Grassi C, Butterfield DA, Cassano T, Perluigi M, Biliverdin Reductase-A Mediates the Beneficial Effects of Intranasal Insulin in Alzheimer Disease, Mol Neurobiol 56(4) (2019) 2922–2943. [DOI] [PubMed] [Google Scholar]
- [199].Miralem T, Lerner-Marmarosh N, Gibbs PE, Jenkins JL, Heimiller C, Maines MD, Interaction of human biliverdin reductase with Akt/protein kinase B and phosphatidylinositol-dependent kinase 1 regulates glycogen synthase kinase 3 activity: a novel mechanism of Akt activation, FASEB J 30(8) (2016) 2926–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].de la Monte SM, Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease, Drugs 72(1) (2012) 49–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Laplante M, Sabatini DM, mTOR Signaling, Cold Spring Harb Perspect Biol 4(2) (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Perluigi M, Di Domenico F, Butterfield DA, mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy, Neurobiol Dis 84 (2015) 39–49. [DOI] [PubMed] [Google Scholar]
- [203].O.N. C, PI3-kinase/Akt/mTOR signaling: impaired on/off switches in aging, cognitive decline and Alzheimer’s disease, Exp Gerontol 48(7) (2013) 647–53. [DOI] [PubMed] [Google Scholar]
- [204].Caccamo A, Majumder S, Richardson A, Strong R, Oddo S, Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments, J Biol Chem 285(17) (2010) 13107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Vartak RS, Rodin A, Oddo S, Differential activation of the mTOR/autophagy pathway predicts cognitive performance in APP/PS1 mice, Neurobiol Aging 83 (2019) 105–113. [DOI] [PubMed] [Google Scholar]
- [206].Perluigi M, Di Domenico F, Barone E, Butterfield DA, mTOR in Alzheimer disease and its earlier stages: Links to oxidative damage in the progression of this dementing disorder, Free Radic Biol Med 169 (2021) 382–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Lanzillotta C, Zuliani I, Vasavda C, Snyder SH, Paul BD, Perluigi M, Di Domenico F, Barone E, BVR-A Deficiency Leads to Autophagy Impairment through the Dysregulation of AMPK/mTOR Axis in the Brain-Implications for Neurodegeneration, Antioxidants (Basel) 9(8) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Jeon SM, Regulation and function of AMPK in physiology and diseases, Exp Mol Med 48(7) (2016) e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Carling D, AMPK signalling in health and disease, Curr Opin Cell Biol 45 (2017) 31–37. [DOI] [PubMed] [Google Scholar]
- [210].Peixoto CA, Oliveira WH, Araujo S, Nunes AKS, AMPK activation: Role in the signaling pathways of neuroinflammation and neurodegeneration, Exp Neurol 298(Pt A) (2017) 31–41. [DOI] [PubMed] [Google Scholar]
- [211].Frisardi V, Imbimbo BP, Metabolic-cognitive syndrome: metabolic approach for the management of Alzheimer’s disease risk, J Alzheimers Dis 30 Suppl 2 (2012) S1–4. [DOI] [PubMed] [Google Scholar]
- [212].Frisardi V, Solfrizzi V, Seripa D, Capurso C, Santamato A, Sancarlo D, Vendemiale G, Pilotto A, Panza F, Metabolic-cognitive syndrome: a cross-talk between metabolic syndrome and Alzheimer’s disease, Ageing Res Rev 9(4) (2010) 399–417. [DOI] [PubMed] [Google Scholar]
- [213].Counts SE, Ikonomovic MD, Mercado N, Vega IE, Mufson EJ, Biomarkers for the Early Detection and Progression of Alzheimer’s Disease, Neurotherapeutics 14(1) (2017) 35–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Mosconi L, Pupi A, De Leon MJ, Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease, Ann N Y Acad Sci 1147 (2008) 180–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Chen M, Huang N, Liu J, Huang J, Shi J, Jin F, AMPK: A bridge between diabetes mellitus and Alzheimer’s disease, Behav Brain Res 400 (2021) 113043. [DOI] [PubMed] [Google Scholar]
- [216].Yang L, Jiang Y, Shi L, Zhong D, Li Y, Li J, Jin R, AMPK: Potential Therapeutic Target for Alzheimer’s Disease, Curr Protein Pept Sci 21(1) (2020) 66–77. [DOI] [PubMed] [Google Scholar]
- [217].Wang X, Zimmermann HR, Ma T, Therapeutic Potential of AMP-Activated Protein Kinase in Alzheimer’s Disease, J Alzheimers Dis 68(1) (2019) 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Liu YJ, Chern Y, AMPK-mediated regulation of neuronal metabolism and function in brain diseases, J Neurogenet 29(2–3) (2015) 50–8. [DOI] [PubMed] [Google Scholar]
- [219].Cai Z, Yan LJ, Li K, Quazi SH, Zhao B, Roles of AMP-activated protein kinase in Alzheimer’s disease, Neuromolecular Med 14(1) (2012) 1–14. [DOI] [PubMed] [Google Scholar]
- [220].Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P, Rooyackers O, Zhou G, Williamson JM, Ljunqvist O, Efendic S, Moller DE, Thorell A, Goodyear LJ, Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes, Diabetes 51(7) (2002) 2074–81. [DOI] [PubMed] [Google Scholar]
- [221].van der Heide LP, Ramakers GM, Smidt MP, Insulin signaling in the central nervous system: learning to survive, Prog Neurobiol 79(4) (2006) 205–21. [DOI] [PubMed] [Google Scholar]
- [222].Viollet B, Athea Y, Mounier R, Guigas B, Zarrinpashneh E, Horman S, Lantier L, Hebrard S, Devin-Leclerc J, Beauloye C, Foretz M, Andreelli F, Ventura-Clapier R, Bertrand L, AMPK: Lessons from transgenic and knockout animals, Front Biosci (Landmark Ed) 14 (2009) 19–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Viollet B, Andreelli F, AMP-activated protein kinase and metabolic control, Handb Exp Pharmacol (203) (2011) 303–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Shackelford DB, Shaw RJ, The LKB1-AMPK pathway: metabolism and growth control in tumour suppression, Nat Rev Cancer 9(8) (2009) 563–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Viollet B, Lantier L, Devin-Leclerc J, Hebrard S, Amouyal C, Mounier R, Foretz M, Andreelli F, Targeting the AMPK pathway for the treatment of Type 2 diabetes, Front Biosci (Landmark Ed) 14 (2009) 3380–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Assefa BT, Tafere GG, Wondafrash DZ, Gidey MT, The Bewildering Effect of AMPK Activators in Alzheimer’s Disease: Review of the Current Evidence, Biomed Res Int 2020 (2020) 9895121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Seixas da Silva GS, Melo HM, Lourenco MV, Lyra ESNM, de Carvalho MB, Alves-Leon SV, de Souza JM, Klein WL, da-Silva WS, Ferreira ST, De Felice FG, Amyloid-beta oligomers transiently inhibit AMP-activated kinase and cause metabolic defects in hippocampal neurons, J Biol Chem 292(18) (2017) 7395–7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX, Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes, J Pathol 225(1) (2011) 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Anderson NJ, King MR, Delbruck L, Jolivalt CG, Role of insulin signaling impairment, adiponectin and dyslipidemia in peripheral and central neuropathy in mice, Dis Model Mech 7(6) (2014) 625–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Cheng KK, Lam KS, Wang B, Xu A, Signaling mechanisms underlying the insulin-sensitizing effects of adiponectin, Best Pract Res Clin Endocrinol Metab 28(1) (2014) 3–13. [DOI] [PubMed] [Google Scholar]
- [231].Su KH, Dai C, Metabolic control of the proteotoxic stress response: implications in diabetes mellitus and neurodegenerative disorders, Cell Mol Life Sci 73(22) (2016) 4231–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Won JS, Im YB, Kim J, Singh AK, Singh I, Involvement of AMP-activated-protein-kinase (AMPK) in neuronal amyloidogenesis, Biochem Biophys Res Commun 399(4) (2010) 487–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Greco SJ, Hamzelou A, Johnston JM, Smith MA, Ashford JW, Tezapsidis N, Leptin boosts cellular metabolism by activating AMPK and the sirtuins to reduce tau phosphorylation and beta-amyloid in neurons, Biochem Biophys Res Commun 414(1) (2011) 170–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Greco SJ, Sarkar S, Johnston JM, Tezapsidis N, Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells, Biochem Biophys Res Commun 380(1) (2009) 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Greco SJ, Sarkar S, Johnston JM, Zhu X, Su B, Casadesus G, Ashford JW, Smith MA, Tezapsidis N, Leptin reduces Alzheimer’s disease-related tau phosphorylation in neuronal cells, Biochem Biophys Res Commun 376(3) (2008) 536–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Hettich MM, Matthes F, Ryan DP, Griesche N, Schroder S, Dorn S, Kraubeta S, Ehninger D, The anti-diabetic drug metformin reduces BACE1 protein level by interfering with the MID1 complex, PLoS One 9(7) (2014) e102420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Zhang H, Zhao C, Cao G, Guo L, Zhang S, Liang Y, Qin C, Su P, Li H, Zhang W, Berberine modulates amyloid-beta peptide generation by activating AMP-activated protein kinase, Neuropharmacology 125 (2017) 408–417. [DOI] [PubMed] [Google Scholar]
- [238].Ma Y, Ma B, Shang Y, Yin Q, Hong Y, Xu S, Shen C, Hou X, Liu X, Flavonoid-rich ethanol extract from the leaves of Diospyros kaki attenuates cognitive deficits, amyloid-beta production, oxidative stress, and neuroinflammation in APP/PS1 transgenic mice, Brain Res 1678 (2018) 85–93. [DOI] [PubMed] [Google Scholar]
- [239].Chen RF, Zhang T, Sun YY, Sun YM, Chen WQ, Shi N, Shen F, Zhang Y, Liu KY, Sun XJ, Oxygen-glucose deprivation regulates BACE1 expression through induction of autophagy in Neuro-2a/APP695 cells, Neural Regen Res 10(9) (2015) 1433–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Kim Y, Kim C, Jang HY, Mook-Jung I, Inhibition of Cholesterol Biosynthesis Reduces gamma-Secretase Activity and Amyloid-beta Generation, J Alzheimers Dis 51(4) (2016) 1057–68. [DOI] [PubMed] [Google Scholar]
- [241].Cho YY, Kwon OH, Park MK, Kim TW, Chung S, Elevated cellular cholesterol in Familial Alzheimer’s presenilin 1 mutation is associated with lipid raft localization of beta-amyloid precursor protein, PLoS One 14(1) (2019) e0210535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Chen Y, Zhou K, Wang R, Liu Y, Kwak YD, Ma T, Thompson RC, Zhao Y, Smith L, Gasparini L, Luo Z, Xu H, Liao FF, Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription, Proc Natl Acad Sci U S A 106(10) (2009) 3907–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Markowicz-Piasecka M, Sikora J, Szydlowska A, Skupien A, Mikiciuk-Olasik E, Huttunen KM, Metformin - a Future Therapy for Neurodegenerative Diseases : Theme: Drug Discovery, Development and Delivery in Alzheimer’s Disease Guest Editor: Davide Brambilla, Pharm Res 34(12) (2017) 2614–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Imfeld P, Bodmer M, Jick SS, Meier CR, Metformin, other antidiabetic drugs, and risk of Alzheimer’s disease: a population-based case-control study, J Am Geriatr Soc 60(5) (2012) 916–21. [DOI] [PubMed] [Google Scholar]
- [245].Son SM, Shin HJ, Byun J, Kook SY, Moon M, Chang YJ, Mook-Jung I, Metformin Facilitates Amyloid-beta Generation by beta- and gamma-Secretases via Autophagy Activation, J Alzheimers Dis 51(4) (2016) 1197–208. [DOI] [PubMed] [Google Scholar]
- [246].Picone P, Vilasi S, Librizzi F, Contardi M, Nuzzo D, Caruana L, Baldassano S, Amato A, Mule F, San Biagio PL, Giacomazza D, Di Carlo M, Biological and biophysics aspects of metformin-induced effects: cortex mitochondrial dysfunction and promotion of toxic amyloid pre-fibrillar aggregates, Aging (Albany NY) 8(8) (2016) 1718–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Vingtdeux V, Davies P, Dickson DW, Marambaud P, AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies, Acta Neuropathol 121(3) (2011) 337–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Perez MJ, Jara C, Quintanilla RA, Contribution of Tau Pathology to Mitochondrial Impairment in Neurodegeneration, Front Neurosci 12 (2018) 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K, Toyoshima Y, Hasegawa M, Hisanaga S, Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease, J Neurosci 32(7) (2012) 2430–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Guha S, Johnson GVW, Nehrke K, The Crosstalk Between Pathological Tau Phosphorylation and Mitochondrial Dysfunction as a Key to Understanding and Treating Alzheimer’s Disease, Mol Neurobiol 57(12) (2020) 5103–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F, The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Abeta oligomers through Tau phosphorylation, Neuron 78(1) (2013) 94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, Gan L, Acetylation of tau inhibits its degradation and contributes to tauopathy, Neuron 67(6) (2010) 953–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Kim HS, Moon S, Paik JH, Shin DW, Kim LS, Park CS, Ha J, Kang JH, Activation of the 5’-AMP-Activated Protein Kinase in the Cerebral Cortex of Young Senescence-Accelerated P8 Mice and Association with GSK3beta- and PP2A-Dependent Inhibition of p-tau(3)(9)(6) Expression, J Alzheimers Dis 46(1) (2015) 249–59. [DOI] [PubMed] [Google Scholar]
- [254].Theendakara V, Bredesen DE, Rao RV, Downregulation of protein phosphatase 2A by apolipoprotein E: Implications for Alzheimer’s disease, Mol Cell Neurosci 83 (2017) 83–91. [DOI] [PubMed] [Google Scholar]
- [255].Gratuze M, Julien J, Petry FR, Morin F, Planel E, Insulin deprivation induces PP2A inhibition and tau hyperphosphorylation in hTau mice, a model of Alzheimer’s disease-like tau pathology, Sci Rep 7 (2017) 46359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Kim HS, Moon S, Kim S, Lee MJ, Suk MH, Park DH, Shin DW, Park CS, Kang JH, Chronological changes in the expression of phosphorylated tau and 5AMPactivated protein kinase in the brain of senescenceaccelerated P8 mice, Mol Med Rep 15(5) (2017) 3301–3309. [DOI] [PubMed] [Google Scholar]
- [257].Planel E, Tatebayashi Y, Miyasaka T, Liu L, Wang L, Herman M, Yu WH, Luchsinger JA, Wadzinski B, Duff KE, Takashima A, Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms, J Neurosci 27(50) (2007) 13635–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Colacurcio DJ, Pensalfini A, Jiang Y, Nixon RA, Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer’s Disease, Free Radic Biol Med 114 (2018) 40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Di Domenico F, Barone E, Perluigi M, Butterfield DA, The Triangle of Death in Alzheimer’s Disease Brain: The Aberrant Cross-Talk Among Energy Metabolism, Mammalian Target of Rapamycin Signaling, and Protein Homeostasis Revealed by Redox Proteomics, Antioxid Redox Signal 26(8) (2017) 364–387. [DOI] [PubMed] [Google Scholar]
- [260].Tramutola A, Lanzillotta C, Di Domenico F, Targeting mTOR to reduce Alzheimer-related cognitive decline: from current hits to future therapies, Expert Rev Neurother 17(1) (2017) 33–45. [DOI] [PubMed] [Google Scholar]
- [261].Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T, The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice, J Clin Invest 118(6) (2008) 2190–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Guo J, Chang L, Zhang X, Pei S, Yu M, Gao J, Ginsenoside compound K promotes beta-amyloid peptide clearance in primary astrocytes via autophagy enhancement, Exp Ther Med 8(4) (2014) 1271–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Grotemeier A, Alers S, Pfisterer SG, Paasch F, Daubrawa M, Dieterle A, Viollet B, Wesselborg S, Proikas-Cezanne T, Stork B, AMPK-independent induction of autophagy by cytosolic Ca2+ increase, Cell Signal 22(6) (2010) 914–25. [DOI] [PubMed] [Google Scholar]
- [264].Tong X, Smith KA, Pelling JC, Apigenin, a chemopreventive bioflavonoid, induces AMP-activated protein kinase activation in human keratinocytes, Mol Carcinog 51(3) (2012) 268–79. [DOI] [PubMed] [Google Scholar]
- [265].Handschin C, Kobayashi YM, Chin S, Seale P, Campbell KP, Spiegelman BM, PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy, Genes Dev 21(7) (2007) 770–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D, Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration, Cell 127(1) (2006) 59–69. [DOI] [PubMed] [Google Scholar]
- [267].Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, Pasinetti GM, PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia, Arch Neurol 66(3) (2009) 352–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Jager S, Handschin C, St-Pierre J, Spiegelman BM, AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha, Proc Natl Acad Sci U S A 104(29) (2007) 12017–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Lin J, Handschin C, Spiegelman BM, Metabolic control through the PGC-1 family of transcription coactivators, Cell Metab 1(6) (2005) 361–70. [DOI] [PubMed] [Google Scholar]
- [270].Hood DA, Tryon LD, Carter HN, Kim Y, Chen CC, Unravelling the mechanisms regulating muscle mitochondrial biogenesis, Biochem J 473(15) (2016) 2295–314. [DOI] [PubMed] [Google Scholar]
- [271].Gravandi MM, Fakhri S, Zarneshan SN, Yarmohammadi A, Khan H, Flavonoids modulate AMPK/PGC-1alpha and interconnected pathways toward potential neuroprotective activities, Metab Brain Dis (2021). [DOI] [PubMed] [Google Scholar]
- [272].Herzig S, Shaw RJ, AMPK: guardian of metabolism and mitochondrial homeostasis, Nat Rev Mol Cell Biol 19(2) (2018) 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel SP, Andrzejewski S, Raissi TC, Pause A, St-Pierre J, Jones RG, AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species, Cell Rep 21(1) (2017) 1–9. [DOI] [PubMed] [Google Scholar]
- [274].Colombo SL, Moncada S, AMPKalpha1 regulates the antioxidant status of vascular endothelial cells, Biochem J 421(2) (2009) 163–9. [DOI] [PubMed] [Google Scholar]
- [275].Cheng PW, Ho WY, Su YT, Lu PJ, Chen BZ, Cheng WH, Lu WH, Sun GC, Yeh TC, Hsiao M, Tseng CJ, Resveratrol decreases fructose-induced oxidative stress, mediated by NADPH oxidase via an AMPK-dependent mechanism, Br J Pharmacol 171(11) (2014) 2739–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Lim JL, Wilhelmus MM, de Vries HE, Drukarch B, Hoozemans JJ, van Horssen J, Antioxidative defense mechanisms controlled by Nrf2: state-of-the-art and clinical perspectives in neurodegenerative diseases, Arch Toxicol 88(10) (2014) 1773–86. [DOI] [PubMed] [Google Scholar]
- [277].Dinkova-Kostova AT, Kostov RV, Kazantsev AG, The role of Nrf2 signaling in counteracting neurodegenerative diseases, FEBS J 285(19) (2018) 3576–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Lanzillotta C, Di Domenico F, Stress Responses in Down Syndrome Neurodegeneration: State of the Art and Therapeutic Molecules, Biomolecules 11(2) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Lanzillotta C, Zuliani I, Tramutola A, Barone E, Blarzino C, Folgiero V, Caforio M, Valentini D, Villani A, Locatelli F, Butterfield DA, Head E, Perluigi M, Abisambra JF, Di Domenico F, Chronic PERK induction promotes Alzheimer-like neuropathology in Down syndrome: Insights for therapeutic intervention, Prog Neurobiol 196 (2021) 101892. [DOI] [PubMed] [Google Scholar]
- [280].Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, Hamilton RL, Chu CT, Jordan-Sciutto KL, Expression of Nrf2 in neurodegenerative diseases, J Neuropathol Exp Neurol 66(1) (2007) 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].Joo MS, Kim WD, Lee KY, Kim JH, Koo JH, Kim SG, AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550, Mol Cell Biol 36(14) (2016) 1931–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Duan J, Guan Y, Mu F, Guo C, Zhang E, Yin Y, Wei G, Zhu Y, Cui J, Cao J, Weng Y, Wang Y, Xi M, Wen A, Protective effect of butin against ischemia/reperfusion-induced myocardial injury in diabetic mice: involvement of the AMPK/GSK-3beta/Nrf2 signaling pathway, Sci Rep 7 (2017) 41491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM, Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators, Cell 127(2) (2006) 397–408. [DOI] [PubMed] [Google Scholar]
- [284].Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA, Masliah E, La Spada AR, PGC-1alpha rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function, Sci Transl Med 4(142) (2012) 142ra97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Majd S, Power JHT, Oxidative Stress and Decreased Mitochondrial Superoxide Dismutase 2 and Peroxiredoxins 1 and 4 Based Mechanism of Concurrent Activation of AMPK and mTOR in Alzheimer’s Disease, Curr Alzheimer Res 15(8) (2018) 764–776. [DOI] [PubMed] [Google Scholar]
- [286].Tramutola A, Lanzillotta C, Barone E, Arena A, Zuliani I, Mosca L, Blarzino C, Butterfield DA, Perluigi M, Di Domenico F, Intranasal rapamycin ameliorates Alzheimer-like cognitive decline in a mouse model of Down syndrome, Transl Neurodegener 7 (2018) 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [287].Li L, Tan J, Miao Y, Lei P, Zhang Q, ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms, Cell Mol Neurobiol 35(5) (2015) 615–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Di Domenico F, Tramutola A, Barone E, Lanzillotta C, Defever O, Arena A, Zuliani I, Foppoli C, Iavarone F, Vincenzoni F, Castagnola M, Butterfield DA, Perluigi M, Restoration of aberrant mTOR signaling by intranasal rapamycin reduces oxidative damage: Focus on HNE-modified proteins in a mouse model of down syndrome, Redox Biol 23 (2019) 101162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Di Domenico F, Zuliani I, Tramutola A, Shining a light on defective autophagy by proteomics approaches: implications for neurodegenerative illnesses, Expert Rev Proteomics 16(11–12) (2019) 951–964. [DOI] [PubMed] [Google Scholar]
- [290].Zuliani I, Lanzillotta C, Tramutola A, Francioso A, Pagnotta S, Barone E, Perluigi M, Di Domenico F, The Dysregulation of OGT/OGA Cycle Mediates Tau and APP Neuropathology in Down Syndrome, Neurotherapeutics 18(1) (2021) 340–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].Tramutola A, Sharma N, Barone E, Lanzillotta C, Castellani A, Iavarone F, Vincenzoni F, Castagnola M, Butterfield DA, Gaetani S, Cassano T, Perluigi M, Di Domenico F, Proteomic identification of altered protein O-GlcNAcylation in a triple transgenic mouse model of Alzheimer’s disease, Biochim Biophys Acta Mol Basis Dis 1864(10) (2018) 3309–3321. [DOI] [PubMed] [Google Scholar]
- [292].Akan I, Olivier-Van Stichelen S, Bond MR, Hanover JA, Nutrient-driven O-GlcNAc in proteostasis and neurodegeneration, J Neurochem 144(1) (2018) 7–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Di Domenico F, Lanzillotta C, Tramutola A, Therapeutic potential of rescuing protein O-GlcNAcylation in tau-related pathologies, Expert Rev Neurother 19(1) (2019) 1–3. [DOI] [PubMed] [Google Scholar]
- [294].Hart GW, Nutrient regulation of signaling and transcription, J Biol Chem 294(7) (2019) 2211–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [295].Zuliani I, Lanzillotta C, Tramutola A, Barone E, Perluigi M, Rinaldo S, Paone A, Cutruzzola F, Bellanti F, Spinelli M, Natale F, Fusco S, Grassi C, Di Domenico F, High-Fat Diet Leads to Reduced Protein O-GlcNAcylation and Mitochondrial Defects Promoting the Development of Alzheimer’s Disease Signatures, Int J Mol Sci 22(7) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [296].Ansari SA, Emerald BS, The Role of Insulin Resistance and Protein O-GlcNAcylation in Neurodegeneration, Front Neurosci 13 (2019) 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [297].Pinho TS, Verde DM, Correia SC, Cardoso SM, Moreira PI, O-GlcNAcylation and neuronal energy status: Implications for Alzheimer’s disease, Ageing Res Rev 46 (2018) 32–41. [DOI] [PubMed] [Google Scholar]
- [298].Di Domenico F, Owen JB, Sultana R, Sowell RA, Perluigi M, Cini C, Cai J, Pierce WM, Butterfield DA, The wheat germ agglutinin-fractionated proteome of subjects with Alzheimer’s disease and mild cognitive impairment hippocampus and inferior parietal lobule: Implications for disease pathogenesis and progression, J Neurosci Res 88(16) (2010) 3566–77. [DOI] [PubMed] [Google Scholar]
- [299].Cork GK, Thompson J, Slawson C, Real Talk: The Inter-play Between the mTOR, AMPK, and Hexosamine Biosynthetic Pathways in Cell Signaling, Front Endocrinol (Lausanne) 9 (2018) 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].Hastings NB, Wang XH, Song LX, Butts BD, Grotz D, Hargreaves R, Hess JF, Hong KLK, Huang CRR, Hyde L, Laverty M, Lee JL, Levitan D, Lu SX, Maguire M, Mahadomrongkul V, McEachern EJ, Ouyang XS, Rosahl TW, Selnick H, Stanton M, Terracina G, Vocadlo DJ, Wang GF, Duffy JL, Parker EM, Zhang LL, Inhibition of O-GlcNAcase leads to elevation of O-GlcNAc tau and reduction of tauopathy and cerebrospinal fluid tau in rTg4510 mice, Mol Neurodegener 12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Zibrova D, Vandermoere F, Goransson O, Peggie M, Marino KV, Knierim A, Spengler K, Weigert C, Viollet B, Morrice NA, Sakamoto K, Heller R, GFAT1 phosphorylation by AMPK promotes VEGF-induced angiogenesis, Biochem J 474(6) (2017) 983–1001. [DOI] [PubMed] [Google Scholar]
- [302].Scott JW, Oakhill JS, The sweet side of AMPK signaling: regulation of GFAT1, Biochem J 474(7) (2017) 1289–1292. [DOI] [PubMed] [Google Scholar]
- [303].Halliwell B, Gutteridge JM (), in: Free Radicals in Biology & Medicine, Oxford University Press; New York, 2015. [Google Scholar]
- [304].Butterfield DA, Lauderback CM, Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress, Free Radic Biol Med 32(11) (2002) 1050–60. [DOI] [PubMed] [Google Scholar]
- [305].Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA, The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: the role of Abeta1-42, J Neurochem 78(2) (2001) 413–6. [DOI] [PubMed] [Google Scholar]
- [306].Butterfield DA, Castegna A, Lauderback CM, Drake J, Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death, Neurobiol Aging 23(5) (2002) 655–64. [DOI] [PubMed] [Google Scholar]
- [307].Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, Coccia R, Markesbery WR, Butterfield DA, Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease, Neurobiol Dis 30(1) (2008) 107–20. [DOI] [PubMed] [Google Scholar]
- [308].Montine TJ, Neely MD, Quinn JF, Beal MF, Markesbery WR, Roberts LJ, Morrow JD, Lipid peroxidation in aging brain and Alzheimer’s disease, Free Radic Biol Med 33(5) (2002) 620–6. [DOI] [PubMed] [Google Scholar]
- [309].Perluigi M, Coccia R, Butterfield DA, 4-Hydroxy-2-nonenal, a reactive product of lipid peroxidation, and neurodegenerative diseases: a toxic combination illuminated by redox proteomics studies, Antioxid Redox Signal 17(11) (2012) 1590–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Sultana R, Perluigi M, Butterfield DA, Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer disease brain, Free Radic Biol Med 62 (2013) 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Butterfield DA, Brain lipid peroxidation and alzheimer disease: Synergy between the Butterfield and Mattson laboratories, Ageing Res Rev 64 (2020) 101049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [312].Claxton A, Baker LD, Wilkinson CW, Trittschuh EH, Chapman D, Watson GS, Cholerton B, Plymate SR, Arbuckle M, Craft S, Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or Alzheimer’s disease, J Alzheimers Dis 35(4) (2013) 789–97. [DOI] [PMC free article] [PubMed] [Google Scholar]