

Abstract
Purpose
DNA methylation alterations have emerged as front-runners in cfDNA biomarker development. However, much effort to date has focused on single cancers. In this context, gastrointestinal (GI) cancers constitute the second leading cause of cancer-related deaths worldwide; yet there is no blood-based assay for the early detection and population screening of GI cancers.
Experimental Design
Herein, we performed a genome-wide DNA methylation analysis of multiple gastrointestinal (GI) cancers to develop a pan-GI diagnostic assay. By analyzing DNA methylation data from 1781 tumor and adjacent normal tissues, we first identified differentially methylated regions (DMRs) between individual GI cancers and adjacent normal, as well as across GI cancers. We next prioritized a list of 67,832 tissue DMRs by incorporating all significant DMRs across various GI cancers to design a custom, targeted bisulfite-sequencing platform. We subsequently validated these tissue-specific DMRs in 300 cell-free DNA-specimens and applied machine learning algorithms to develop three distinct categories of DMR-panels
Results
We identified three distinct DMR panels. 1) Cancer-specific biomarker panels with AUC values of 0.98 (colorectal cancer), 0.98 (hepatocellular carcinoma), 0.94 (esophageal squamous cell carcinoma), 0.90 (gastric cancer), 0.90 (esophageal adenocarcinoma), and 0.85 (pancreatic ductal adenocarcinoma); 2) A pan-GI panel that detected all GI cancers with an AUC of 0.88; and 3) A multi-cancer (tissue of origin) prediction panel, EpiPanGI Dx, with a prediction accuracy of 0.85–0.95 for most GI cancers.
Conclusions
Using a novel biomarker discovery approach, we provide the first evidence for a cfDNA methylation assay that offers robust diagnostic accuracy for GI cancers.
Introduction
Despite improved overall survival rates due to recent advancements in cancer therapies, cancer remains the second leading cause of mortality worldwide (1). At present in the United States, average-risk or asymptomatic population screening is recommended for only colorectal (CRC), breast, cervical, lung, and prostate cancers (2). Population screening for low prevalence cancers is challenging due to a lack of cost-effective diagnostic tools (3). Thus, to facilitate population screening and thereby eradicate the mortality associated with cancer, a universal cancer screening test that is non-invasive, simple, and robust is urgently needed.
Circulating tumor DNA released into the bloodstream by a tumor cell carries both a genetic and an epigenetic signature of the cell of origin, and is therefore becoming a key tool in developing liquid biopsy-based biomarkers for early detection and treatment monitoring (4). Unfortunately, the diversity of genetic mutations across cancers and the prevalence of these mutations across large genomic regions makes it challenging to develop mutation-based, pan-cancer diagnostic tests (5). In contrast, epigenetic DNA methylation changes occur in specific genomic regions called CpG islands and can be consistently measured using bisulfite sequencing in various biological fluids, including plasma, serum, urine, and saliva. Due to their high cancer specificity, and their appearance during the earliest phases of cancer development, aberrant DNA methylation alterations provide an excellent avenue by which to develop pan-cancer liquid biopsy-based diagnostic markers (6,7). However, most recent studies investigating plasma cell-free DNA (cfDNA) methylation patterns for biomarker development have focused on only individual cancers (8–10), whereas few investigated multiple cancers (11,12).
Gastrointestinal (GI) cancers, including CRC, hepatocellular carcinoma (HCC), esophageal squamous cell carcinoma (ESCC), gastric cancer (GC), esophageal adenocarcinoma (EAC), and pancreatic ductal adenocarcinoma (PDAC) constitute the second leading cause of cancer-related deaths worldwide, yet there is no blood-based assay for early detection and/or population screening of GI cancers. Due to their generally low prevalence and lack of cost-effective screening tools, except for CRC (13), most GI cancers present at a late stage, leading to a high mortality rate, and underscoring the need for improved screening tools. Most studies to date investigated genome-wide methylation patterns at the tissue level in individual cancers, subsequently selecting the most significant tissue markers for testing in the cfDNA of the corresponding cancer type. In this way, these single-cancer studies failed to analyze DNA methylation patterns in an unbiased and comprehensive manner, and thereby lack the ability to discover pan-cancer-specific markers. To address this challenge and to identify methylation markers across GI cancers, we performed a genome-wide DNA methylation analysis of multiple GI cancers, which we used to develop a novel cfDNA methylation biomarker panel for the early detection of individual GI cancers, a pan-GI diagnostic panel, and a multi-GI cancer prediction panel (EpiPanGI Dx).
Methods
Patients and clinicopathological data
Whole genome 450k tissue DNA methylation data across six GI cancers (CRC, HCC, ESCC, GC, EAC, PDAC) and adjacent normal tissues were obtained from the The Cancer Genome Atlas (TCGA) and GSE72872 dataset (14). Complete clinical, epidemiological, molecular, and histopathological data are available at the TCGA website: https://tcga-data.nci.nih.gov/tcga/. Retrospective plasma cfDNA specimens collected from 300 patients with the six GI cancers and healthy age-matched controls were collected from various institutes. Written informed consent was obtained from all patients and the study was approved by the institutional review boards of all participating institutions. The study adhered to Declaration of Helsinki ethical guidelines.
Specimen processing of patient plasma samples
Plasma samples were transferred to 2-mL microcentrifuge tubes and centrifuged at 16,000g for 10 minutes at 4°C to remove any cellular debris. Circulating cfDNA (10–100 ng) was extracted from 1–2 ml plasma using the QIAamp Circulating Nucleic Acid kit (Qiagen) and quantified using the Quant-iT high-sensitivity Picogreen double-stranded DNA Assay Kit (Invitrogen by Thermo Fisher Scientific). For targeted methylation sequencing, 10 ng plasma cfDNA was first bisulfite treated using the ZYMO Gold Kit. A Swift Bioscience Methyl-Seq library preparation kit was adapted to generate individual libraries incorporating 13 PCR cycles and overnight ligation. Custom targeted CpG methylation probes were designed using the Roche Nimblegen target capture kit, Custom SeqCap Epi Choice 30 MB. Libraries were quantified using the Quant-iT high-sensitivity Picogreen double-stranded DNA Assay Kit before equimolarly pooling 10 individual libraries per capture consisting of 2 μg total DNA. Hybridization and capture were performed using VK SeqCap Epi Reagent Kit Plus and SeqCap EZ hybridization/wash kit from Roche Nimblegen. For blocking, a universal blocker (IDT technologies) was used. Pooled libraries were sequenced on an Illumina NovaSeq S4 using paired-end, 100-base-pair reads, incorporating 150 individual libraries per lane. Sequencing matrices including the coverage distribution and methylation ratio distribution of gitBS in all plasma samples are included in Fig. S1 and S2.
Plasma targeted bisulfite data processing, DMR calling, and visualization
For each plasma sample, after trimming adaptor and low-quality bases, BSMAP (2.90) was used to align bisulfite sequencing reads to the hg19 human genome assembly. The methylation ratio of CpG sites was calculated using the methratio.py script (from BSMAP package). CpG methylation ratios supported by less than 4 reads were discarded before downstream analysis. Metilene (0.2–7) was used for calculating de novo DMRs between two conditions, e.g., normal vs. cancer. For each CpG site, at least three samples of each condition must have a non-missing value. Missing values were imputed using Metilene during DMR calling. Because the methylation difference between normal and cancer tissues is typically diluted in plasma, we selected DMRs based on a relatively loose cut-off (absolute methylation difference more than 0.1 and p-value less than 0.05) for downstream analysis. The methylation level of a DMR was represented as the mean methylation ratio of its CpG sites. The z-score of each DMR methylation level was used for heatmap visualization. Ward clustering and Euclidean distance were used for heatmap plotting.
Machine learning methods used for developing various GI cancer detection panels
Feature selection for individual GI cancer detection and pan-GI cancer detection.
For individual GI cancer prediction, normal and cancer plasma samples were randomly partitioned into a training set (70%) and a test set (30%). Within the training set only, DMR identification and feature selection (using the “Boruta” R package to select the top 200 informative DMRs) were performed in normal and cancer plasma samples for each GI cancer. For pan-GI cancer detection, samples from the training sets or testing sets for each GI cancer were pooled into a single pan-GI training set or testing set, respectively. Using the training set, DMRs identified from each GI cancer were also pooled, for a total of approximately 8000 DMRs for feature selection (using the Boruta R package to select the top 200 informative DMRs).
Feature selection for multi-GI cancer classification.
Plasma samples from six GI cancers and healthy people were used for classification analysis. ESCC and EAC were combined as one class, given their high similarity. Plasma samples from each class were randomly partitioned into a training set (70%) and a test set (30%) independently. Using the training set, class-specific DMRs were identified by one-versus-rest comparisons. Approximately 4000 DMRs identified from all classes were pooled together and the top 200 informative DMRs were selected (using Boruta R package with default parameters) for downstream GI cancer classification.
Feature selection using the Boruta R package.
After splitting the data into training and test sets, the Boruta R package was used to select the most informative DMRs from the training set for cancer detection. Given the randomness introduced by missing value imputation and random forest construction, we repeated the feature selection step 50 times and finally choose the top 200 DMRs that were most frequently selected by the Boruta algorithm for subsequent analyses.
Prediction model training and evaluation.
Training sets were used to train random forest (R package “ranger”) models for individual GI cancer prediction, pan-GI cancer prediction, and multi-GI cancer classification, respectively. The hyperparameters were tuned by 10-fold cross-validation. For model evaluation, the remaining 30% test sets were used to plot the ROC curve and calculate the AUC scores for each random forest model. The training-test set split, DMR calling, and feature selection were repeated 10 times to avoid overestimating model performance.
Independent cohort validation.
PDAC patient samples were from two independent cohorts (58 samples from University of Pittsburg and 16 samples from Medical College of Wisconsin (MCW)). The PDAC Pittsburg cohort, which has more patient samples, was used for DMR calling, feature selection (top 200 informative DMRs), and model training. The AUC scores for this model in detecting cancer were calculated using the PDAC MCW cohort.
Early stage cancer prediction.
For CRC, HCC, GC and PDAC, cancer stage information was available and therefore we looked at the early stage cancer prediction accuracy in these four cancers. For this, we took all late stage (stage IV) cancer samples, along with 70% of the normal plasma samples for DMR calling, feature selection (top 200 informative DMRs), and model training. The performance of the trained model was then evaluated using the early stage (stage I-III) cancer samples and the remaining 30% of normal samples.
Informative DMR validation using cancer tissue data.
Calculated beta values of 450K methylation array data for TCGA-COAD, TCGA-LIHC, TCGA-ESCA, TCGA-STAD and TCGA-PAAD were downloaded from the UCSC Xena database. Calculated beta values of 450K methylation array data for EAC was downloaded from GEO (GSE72872). The 450K CpG sites were mapped to the informative DMRs selected for individual GI cancer detection, pan-GI cancer detection, and multi-GI cancer classification. The methylation level of the informative DMRs for each cancer tissue sample was calculated by taking the mean of the mapped CpG site beta values. The normal and cancer tissue samples were randomly partitioned into a training (70%) and test set (30%) manner. We trained a random forest model with the training set and calculated the AUC scores of the model with the remaining test set.
Results
Development of a GI-targeted bisulfite sequencing panel (gitBS)
The study design describing tissue discovery, followed by plasma cfDNA validation, is illustrated in Fig. 1 and S1A. We first analyzed 450K methylation array data from 1781 tumor and adjacent normal tissues from six different GI cancers: CRC, HCC, ESCC, GC, EAC, and PDAC. By comparing data from tumor vs. normal tissues within each GI cancer, as well as across all GI cancers, we identified a total of 67,832 regions of interest (ROI), based on significant differentially methylated probes with a p-value <0.001 and an absolute delta beta of 0.20 across all the comparisons (Table S1–2). The covered regions were highly enriched for promoters as well as gene body regions (Fig. S3B), which are more susceptible to aberrant methylation during oncogenesis. We merged overlapping tissue-level ROIs from the various GI cancers to design a targeted SeqCap Epi-based bisulfite sequencing platform, which we termed the “GI-targeted bisulfite sequencing (gitBS)” panel (Table S3). Compared to a previously reported strategy (15), we used a meticulous analysis of every significant probe identified via a 450K tissue analysis across six GI cancers to build our gitBS panel, which included a much broader genomic region (~30 MB) covering approximately 1% of the human genome.
Fig. 1. Study design depicting tissue discovery and plasma validation of EpiPanGI Dx.
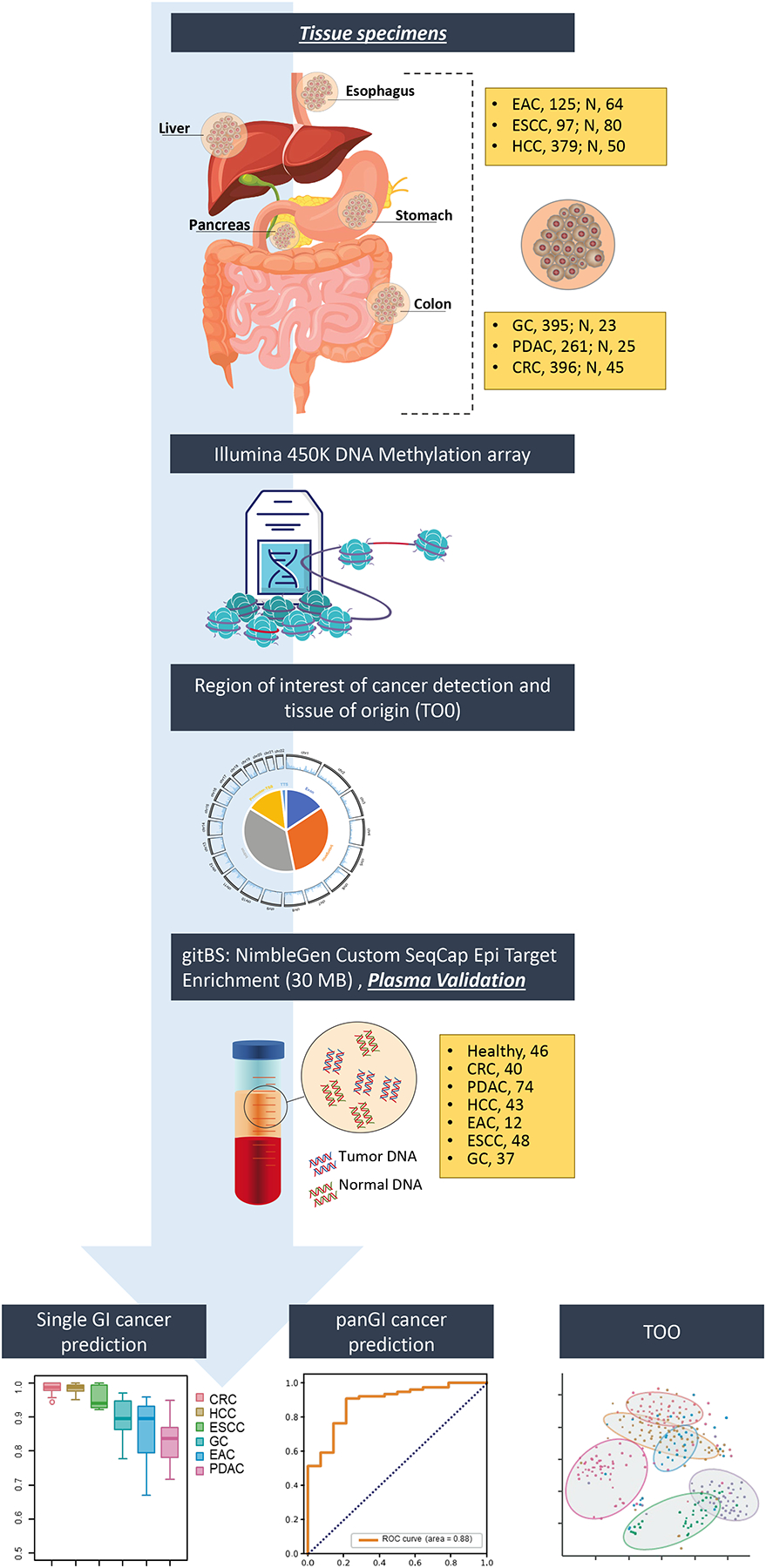
Genome-wide 450k DNA methylation analysis on individual GI cancers vs. adjacent normal tissues and across six GI cancers, resulted in the identification of 67,832 differentially methylated regions of interest. Subsequently, we developed custom plasma specific gitBS target enrichment panel to evaluate in plasma cfDNA (n=300). This resulted in the identification of plasma DMR panels for the detection of individual GI cancers, pan-GI cancers, and tissue of origin using machine learning algorithms.
Evaluation of gitBS in plasma cfDNA
To evaluate the comprehensive list of tissue-specific markers in plasma cfDNA, we performed gitBS on 300 total plasma samples collected from patients with CRC, HCC, ESCC, GC, EAC, or PDAC, and age-matched controls (Table S4). In comparing the individual GI cancers with controls, we identified a total of 216,887 differentially methylated CpGs consisting of 10,677 DMRs, in CRC (5689), HCC (1072), ESCC (1063), GC (949), EAC (1177), and PDAC (727) (Table S5). To confirm the diagnostic power of the identified DMR panels across each GI cancer, we performed hierarchical clustering based on the identified DMRs for each GI cancer type. For most GI cancers, we observed a clear separation of two clusters representing cancer vs. normal samples (Fig. S4–S8). For PDAC, although the boundary between cancer and normal clusters was less clear, most PDAC samples clustered together (Fig. S9). Overall, our results indicate that these DMRs could be used as potential biomarkers for GI cancer detection.
Development of cfDNA methylation panels for individual GI cancer detection
To develop plasma specific DMR panels for individual GI cancer detection, we used machine learning algorithms. Briefly, we have split plasma samples from GI cancer patients and healthy controls into training (70%) and test sets (30%). De novo DMRs between GI cancer and healthy controls were identified only with samples from training sets. Next, we performed feature selection based on the Boruta algorithm, which is known to be powerful for biological features (16). We then used the chosen DMRs to train a random forest model, which outperformed several other machine learning techniques for GI cancer detection, such as logistic regression model, support vector machine and K-nearest neighbor models (Fig. S10). We used PDAC for this comparison since PDAC plasma samples are not well separated from healthy controls based on the clustering result (Fig. S9). Finally, we evaluated prediction model performance by calculating Area Under the ROC Curve (AUC) scores using the test set samples. We repeated the entire process 10 times to prevent biases due to data set splitting. Our cancer prediction models achieved the best performance for CRC and HCC, with median AUC scores of 0.98; prediction models for the other GI cancers had median AUC scores of 0.94 (ESCC), 0.90, (GC), 0.90 (EAC), and 0.85 (PDAC), which is higher or comparable to previous reports (17,18) (Fig. 2A).
Fig. 2. Individual GI cancer detection accuracy using informative plasma DMRs identified from gitBS.
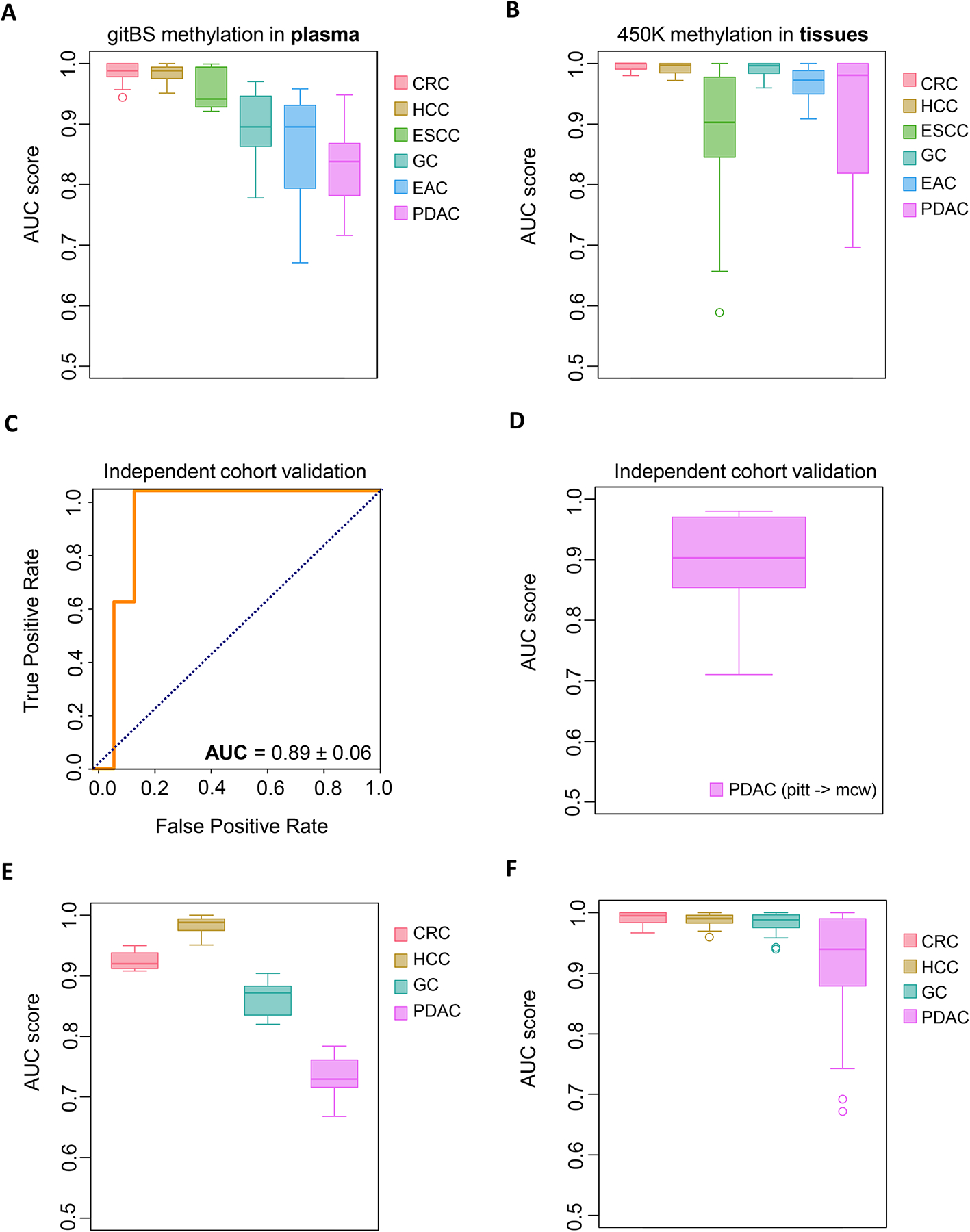
(A) Prediction accuracy of the machine learning model trained for each GI cancer. Samples (n=300) were randomly partitioned into a training set (70%) and a test set (30%) 10 times. DMR calling, feature selection, and model training were performed on the training sets. Boxplots show prediction model AUC scores calculated in test sets for each GI cancer. Sample size: CRC (40), PDAC (74), HCC (43), EAC (12), ESCC (48), GC (37), normal (46). (B) Use of the informative plasma DMRs from A to predict cancer in GI cancer tissues dataset (n=1781). Boxplots show AUC scores of 10 independent runs. (C, D) Representative ROC curve (left) and AUC score (right) for the PDAC independent validation set (10 runs). (E) Late stage (stage IV) plasma samples, along with randomly selected 70% normal plasma sample, were used for DMR calling, feature selection, and model training. This whole process was repeated for 10 times to avoid bias due to sample selection. Boxplots show AUC scores of prediction models on early stage (Stage I-III) plasma samples (CRC: 29, HCC:36, GC:16, PDAC:35). (F) Use of informative plasma DMRs from panel E to predict cancer in early stage GI cancer tissues (n=1257).
We subsequently applied the plasma derived DMR panels established using machine learning to distinguish GI cancer tissues from adjacent normal tissues. As expected, the median AUC scores of models for each of the GI cancers were 0.99 (CRC), 0.99 (HCC), 0.90 (ESCC), 1.00 (GC), 0.97 (EAC), and 0.94 (PDAC). Consistent with the performance of the PDAC model in plasma, the model performed relatively poorly at predicting PDAC in tissue (Fig. 2B). Therefore, we tested the PDAC DMRs in another independent plasma cohort. Interestingly, the machine learning model, trained and tested with PDAC plasma samples from the first cohort, achieved even higher prediction accuracy in the independent PDAC cohort, with an AUC of 0.89 (Fig. 2C and D).
Given that the ultimate goal of cancer screening is to identify cancer at an early stage, we evaluated the ability of the plasma DMRs to detect early stage GI cancers in CRC (29), HCC (36), GC (16) and PDAC (35). We did not have access to early stage EAC and ESCC and hence are not tested. Our models achieved median AUC scores of 0.92 (CRC), 0.99 (HCC), 0.87 (GC), and 0.73 (PDAC) for predicting early stage plasma samples in the test set (Fig. 2E). When applied to early stage tumor tissues in the same four GI cancers, the DMR panels achieved median AUC values of 0.99 (CRC), 0.99 (HCC), 0.99 (GC), and 0.94 (PDAC) (Fig. 2F). Altogether, these results indicate that DNA methylation aberrations we identified have great potential for detecting individual GI cancers along with early stage cancers.
Development of a Pan-GI cancer detection model
Having performed this study in individual GI cancers, we next used our DMR data to identify a pan-GI classifier. To do this, we pooled the training sets and test sets used for each individual GI cancer prediction model together as a pan-GI training set and test set, respectively. We also pooled the DMRs identified from each GI cancer for pan-GI cancer feature selection and model training. We achieved a median AUC of 0.88 for the pan-GI cancer prediction model in the test set plasma cohort (Fig. 3A). Similarly, the plasma DMRs achieved an excellent AUC of 0.98 in distinguishing pan-GI cancer tissues from normal tissues (Fig. 3B).
Fig. 3. Pan-GI cancer detection accuracy using informative plasma DMRs identified from gitBS.
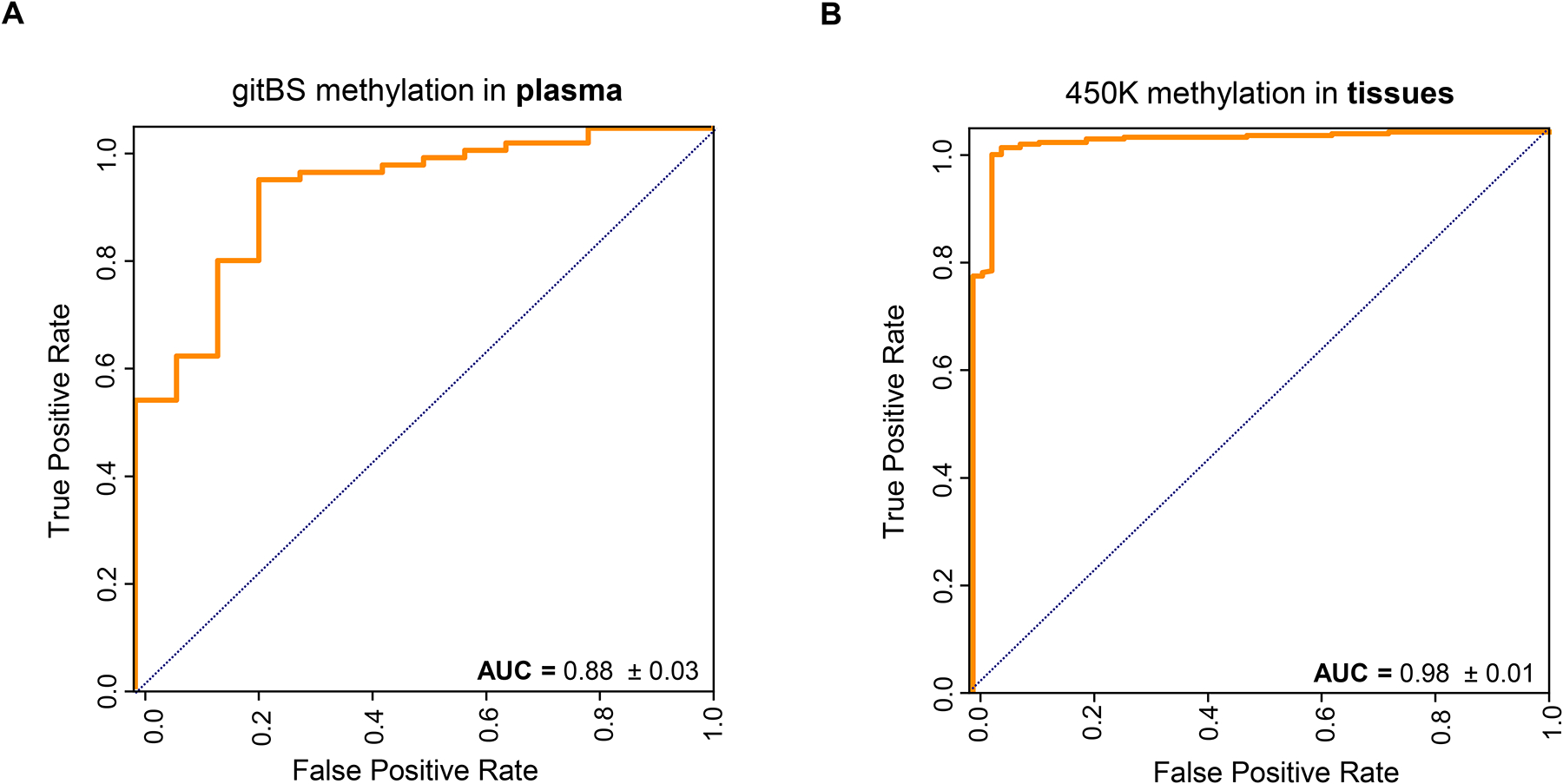
(A) Plasma samples of each GI cancer were randomly partioned into a training set (70%) and test set (30%) 10 times. Training sets of all GI cancers were pooled for training a pan-GI cancer prediction model. Representative ROC curve and AUC scores for the combined test sets are shown. (B) Use of informative plasma DMRs from A to predict pan-GI cancer in tissue samples.
Development of multi-GI cancer classification model EpiPanGIDx
Lastly, we have developed a plasma multi-GI cancer prediction model EpiPanGIDx using random forest that in addition to identify all GI cancers, also have the ability to reveal the tissue of origin. Given that ESCC and EAC both develop from the esophagus, we treated them as the same class in our model. For each class versus the other GI cancers, we identified class-specific plasma DMRs (Table S6), which we then pooled for feature selection and model training. In the test set, our models classified samples into normal plasma, CRC, PDAC, HCC and ESCC/EAC with higher accuracy than previous studies (17) (Fig. 4A). Clustering the data using a t-SNE plot also showed clear separation of most GI cancers from healthy samples and from one another (Fig. 4C). The class-specific plasma DMRs also successfully classified GI cancer and normal tissues with high accuracy (Fig. 4B and 4D). Collectively, these results prove the feasibility of utilizing cfDNA methylation markers for not only GI cancer detection, but also for identifying the tissue of origin of GI cancers.
Fig. 4. Multi-GI cancer tissue of origin classification using informative plasma DMRs identified from gitBS.
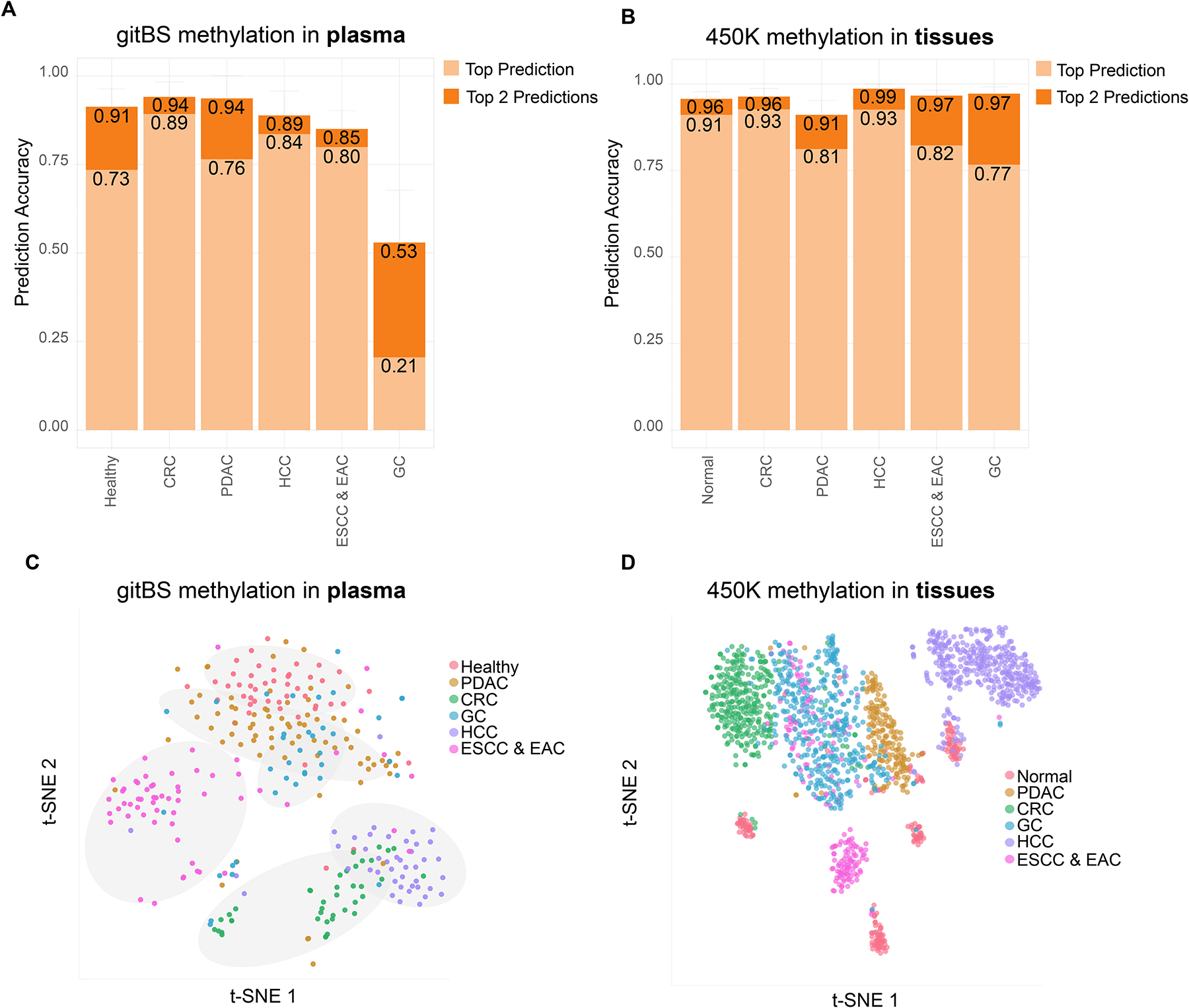
(A) Classification accuracy of the plasma samples from GI cancer patients. The number on the y-axis indicates the ratio of samples being correctly predicted. Light orange: sample labels were the same as the top prediction. Dark orange: sample labels were among the top 2 predictions. (B) Use of informative plasma DMRs from A to classify GI cancer tissues. (C, D) t-SNE plots for plasma samples (n=300) and GI cancer tissue samples (1781) generated using informative plasma DMRs.
Identification of minimum DMRs needed to achieve optimal accuracy across all GI cancers
Finally, to advance the development of powerful and cost-effective cfDNA methylation biomarker panels for GI cancer detection, we also evaluated the performance of our models when varying number of informative DMRs were selected for model training. For individual GI cancer prediction models, the top 50 DMRs were sufficient for achieving optimal accuracy for each GI cancer. Even with as few as 10 DMRs, models for HCC or CRC prediction still showed excellent performance, with AUC scores >0.95 (Fig. 5 and S11–S16 and Table S7). For both the pan-GI and multi-GI classification models, at least the top 150 informative DMRs were required to achieve the optimal performance (Fig. 5 and S17–S19 and Table S7).
Fig. 5. AUC plots with variable numbers of informative DMRs across GI cancers.
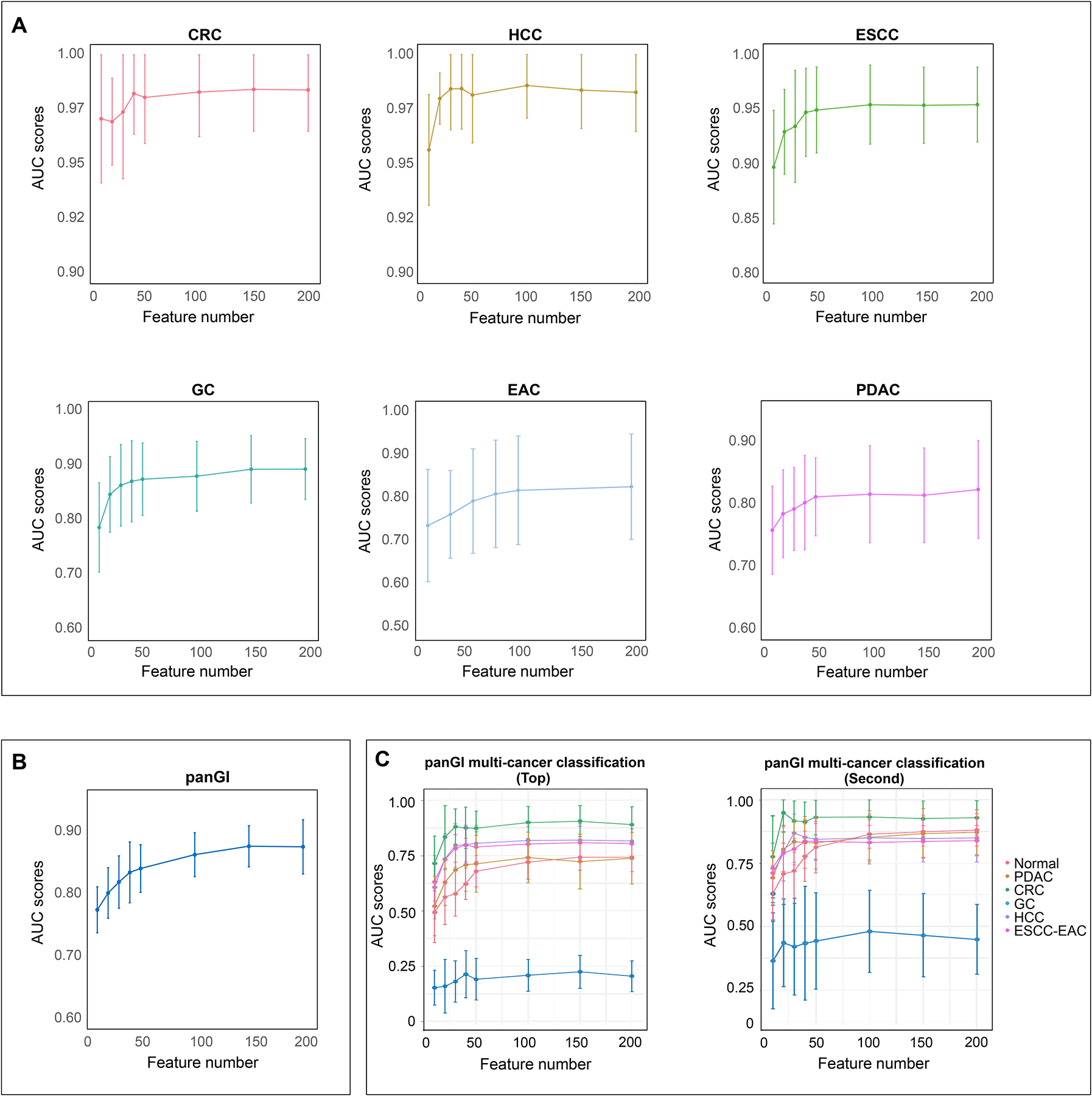
(A) Individual GI cancer prediction models. (B) Pan-GI cancer prediction model. (C) Multi-GI cancer tissue of origin classification model.
Discussion
The lack of population-based screening for all cancers is attributed to the low prevalence of many cancers in the general population (3,19). However, by developing sensitive multi-cancer or multi-organ diagnostic tests, population screening could be implemented, even for low-prevalence cancers. In this regard, GI cancers, which encompass a variety of cancer types, provide a unique opportunity for developing a pan-GI diagnostic assay. Alquist et al., showed that using a pan-GI diagnostic assay, only 83 patients need to be screened to diagnose one positive patient with GI cancer (3). Herein, we performed a comprehensive genome-wide DNA methylation study across six GI cancers to identify a non-invasive plasma DMR panel “EpiPanGI Dx” that predicted tissue-of-origin of all GI cancers with high accuracy.
Most previous studies either studied individual GI cancers (9,10,20) or selected a panel of significant tissue markers and subsequently validated them in cfDNA using PCR-based methods (21,22). Thus, cancer specificity was not well studied, and those studies failed to build multi-organ diagnostic assays to implement cost-effective population screening tests. In contrast, we first identified every tissue-significant CpG across six GI cancers, followed by development of plasma-specific diagnostic panels for the accurate detection of GI cancer tissues of origin using a single targeted methylation test, EpiPanGI Dx. Compared to previous studies (18), we selected fewer DMRs for prediction, which makes our model more feasible for large-scale validation studies and clinical practice (Fig. 5 and S11–S19). In addition, a low cost per sample, as well as a low (10 ng) input required for cfDNA, makes our targeted methylation assay very feasible for clinical use.
Recent plasma cfDNA methylation studies showed that targeted methylation sequencing is quite robust in discovering multi-tissue cfDNA methylation markers. Most notably, Liu et al (23). identified tissue of origin methylation markers across 50 different cancers. Another study used targeted methylation sequencing to identify plasma cfDNA markers using that differentiate between CRC, non-small-cell lung cancer, breast cancer, and melanoma (24). Shen et al. used a cfMeDIP–seq method to discover plasma DMRs that differentiate between multiple solid cancers including pancreatic, CRC, breast, lung, renal, and bladder cancers (18). However, ours is the first study in which organ-specific methylation markers were used to develop a multi-GI cancer cfDNA assay. Excitingly, the detection accuracy of our EpiPanGI Dx assay, with as few as 50 DMRs, was quite high across all GI cancers, considering it is a multi-cancer diagnostic test. Furthermore, our EpiPanGI Dx assay developed from plasma cfDNA showed excellent diagnostic accuracy (AUC 0.91–0.99) when applied back to GI cancer tissue cohorts. Thus, the markers we trained and validated in plasma cfDNA are highly cancer specific. PDAC showed somewhat lower accuracy and this could be attributed to the tumor purity and further validation of our markers can help us refining the signatures in PDAC.
The unique strengths of our study are 1) Comprehensive profiling of all GI cancer tissue methylation markers followed by the development of a targeted plasma cfDNA panel for the development of EpiPanGI Dx 2) Use of machine learning algorithms with training and validation sets, as well as using 10x cross-validation, to compute the accuracy of the EpiPanGI Dx assay across GI cancers 3) In addition, the assay is quite cost-effective as our models require fewer biomarkers than previously reported studies (18) and therefore will be more feasible for the development of diagnostic panels for large-scale clinical usage 4) Our assay can be performed using as little as 10 ng cfDNA 4) Although the plasma samples were collected from several different parts of the world, the detection accuracy of the EpiPanGI Dx assay in cfDNA, as well as the performance of the test in tissue data, shows the robustness of our markers.
Our study also has several limitations. First, the study is retrospective; therefore, we could not test the true population screening ability of our models. Second, although we showed our assay to be quite robust in identifying early stage cancers at both the tissue and plasma level, the number of samples used to represent each stage was limited. Third, although many previous studies showed the superiority of cfDNA methylation markers over genomic mutations for cancer detection, we did not have the mutation profiles of our cfDNA samples to be able to directly compare (or even combine) the diagnostic performance of our methylation assay relative to genomic mutations. However, in future studies, we expect a combination of epigenomic and genomic markers to further improve the accuracy and robustness of cfDNA-based early detection markers.
In summary, we developed a robust, sensitive, targeted methylation-based cfDNA test for multi-GI cancer detection. Our EpiPanGI Dx assay needs further validation in completely independent retrospective and prospective datasets for clinical translation as early diagnostic markers. Further large-scale prospective validation will pave a way to test the performance of our assay in population-level screening for GI cancers. Nevertheless, these findings underscore the potential utility of cfDNA methylation markers for non-invasive, cost-effective, and early detection of GI cancers and serve as a platform for future organ-specific methylation studies for multi-cancer detection.
Supplementary Material
Translational Relevance.
Gastrointestinal (GI) cancers constitute the second leading cause of cancer-related deaths worldwide, yet there is no blood-based assay for early detection and/or population screening of all GI cancers. Due to their generally low prevalence and lack of cost-effective screening tools, except for CRC, most GI cancers present at a late stage, leading to a high mortality rate, and underscoring the need for improved screening tools. Owing to their high cancer specificity, DNA methylation alterations have emerged as front-runners in cell-free DNA biomarker development. Herein, we performed a genome-wide DNA methylation analysis of multiple gastrointestinal (GI) cancer tissues, subsequently validated the tissue-specific DMRs in 300 cell-free DNA specimens by designing a custom, targeted bisulfite sequencing platform. In summary, we developed a robust, sensitive, targeted methylation-based assay for multi-GI cancer detection. Our EpiPanGI Dx assay needs further validation in completely independent retrospective and prospective datasets for clinical translation as early diagnostic markers.
Acknowledgements:
We thank Feng Gao for participating and sharing his expertise in analyzing the 450K tissue data.
Grant support:
This work was supported by R01 (CA72851, CA181572, CA184792, CA202797) and U01 (CA187956, CA214254) grants from the National Cancer Institute, National Institutes of Health; RP140784 from the Cancer Prevention Research Institute of Texas; grants from the Sammons Cancer Center and Baylor Foundation, as well as funds from the Baylor Scott & White Research Institute, Dallas, TX, USA awarded to Ajay Goel. This work was also supported by R01 (HG007538, CA193466, CA228140) grants awarded to Wei Li. M.I.P. was jointly supported by the SAMRC with funds received from the National Department of Health and the MRC UK with funds from the UK Government’s Newton Fund and GSK. S.J.M. was supported by the ACS Clinical Research Professorship, DK118250 and CA211457, and endowed professorship Myerberg/Hendrix Professor of Gastroenterology
Footnotes
Disclosures:
All authors have nothing to disclose
Data availability:
All data associated with this study are presented in the paper or Supplementary Materials. The raw plasma cfDNA gitBS sequencing data reported in this paper have been deposited into the Gene Expression Omnibus (GEO), under accession number GSE149438.
References
- 1.Torre LA, Siegel RL, Ward EM, Jemal A. Global Cancer Incidence and Mortality Rates and Trends--An Update. Cancer Epidemiol Biomarkers Prev 2016;25(1):16–27 doi 10.1158/1055-9965.EPI-15-0578. [DOI] [PubMed] [Google Scholar]
- 2.Smith RA, Andrews KS, Brooks D, Fedewa SA, Manassaram-Baptiste D, Saslow D, et al. Cancer screening in the United States, 2019: A review of current American Cancer Society guidelines and current issues in cancer screening. CA Cancer J Clin 2019;69(3):184–210 doi 10.3322/caac.21557. [DOI] [PubMed] [Google Scholar]
- 3.Ahlquist DA. Universal cancer screening: revolutionary, rational, and realizable. NPJ Precis Oncol 2018;2:23 doi 10.1038/s41698-018-0066-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Pol Y, Mouliere F. Toward the Early Detection of Cancer by Decoding the Epigenetic and Environmental Fingerprints of Cell-Free DNA. Cancer Cell 2019;36(4):350–68 doi 10.1016/j.ccell.2019.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Mroz EA, Rocco JW. The challenges of tumor genetic diversity. Cancer 2017;123(6):917–27 doi 10.1002/cncr.30430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lam K, Pan K, Linnekamp JF, Medema JP, Kandimalla R. DNA methylation based biomarkers in colorectal cancer: A systematic review. Biochim Biophys Acta 2016;1866(1):106–20 doi 10.1016/j.bbcan.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Kandimalla R, van Tilborg AA, Zwarthoff EC. DNA methylation-based biomarkers in bladder cancer. Nat Rev Urol 2013;10(6):327–35 doi 10.1038/nrurol.2013.89. [DOI] [PubMed] [Google Scholar]
- 8.Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun 2018;9(1):5068 doi 10.1038/s41467-018-07466-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu RH, Wei W, Krawczyk M, Wang W, Luo H, Flagg K, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater 2017;16(11):1155–61 doi 10.1038/nmat4997. [DOI] [PubMed] [Google Scholar]
- 10.Luo H, Zhao Q, Wei W, Zheng L, Yi S, Li G, et al. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Sci Transl Med 2020;12(524) doi 10.1126/scitranslmed.aax7533. [DOI] [PubMed] [Google Scholar]
- 11.Shen SY, Singhania R, Fehringer G, Chakravarthy A, Roehrl MHA, Chadwick D, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018;563(7732):579–83 doi 10.1038/s41586-018-0703-0. [DOI] [PubMed] [Google Scholar]
- 12.Guo S, Diep D, Plongthongkum N, Fung HL, Zhang K, Zhang K. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat Genet 2017;49(4):635–42 doi 10.1038/ng.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Provenzale D, Gupta S, Ahnen DJ, Markowitz AJ, Chung DC, Mayer RJ, et al. NCCN Guidelines Insights: Colorectal Cancer Screening, Version 1.2018. J Natl Compr Canc Netw 2018;16(8):939–49 doi 10.6004/jnccn.2018.0067. [DOI] [PubMed] [Google Scholar]
- 14.Krause L, Nones K, Loffler KA, Nancarrow D, Oey H, Tang YH, et al. Identification of the CIMP-like subtype and aberrant methylation of members of the chromosomal segregation and spindle assembly pathways in esophageal adenocarcinoma. Carcinogenesis 2016;37(4):356–65 doi 10.1093/carcin/bgw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo H, Zhao Q, Wei W, Zheng L, Yi S, Li G, et al. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Science Translational Medicine 2020;12(524). [DOI] [PubMed] [Google Scholar]
- 16.Degenhardt F, Seifert S, Szymczak S. Evaluation of variable selection methods for random forests and omics data sets. Briefings in bioinformatics 2017;20(2):492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018;359(6378):926–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen SY, Singhania R, Fehringer G, Chakravarthy A, Roehrl MH, Chadwick D, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018;563(7732):579. [DOI] [PubMed] [Google Scholar]
- 19.Cole P, Morrison AS. Basic issues in population screening for cancer. J Natl Cancer Inst 1980;64(5):1263–72. [PubMed] [Google Scholar]
- 20.Qin Y, Wu CW, Taylor WR, Sawas T, Burger KN, Mahoney DW, et al. Discovery, Validation, and Application of Novel Methylated DNA Markers for Detection of Esophageal Cancer in Plasma. Clin Cancer Res 2019;25(24):7396–404 doi 10.1158/1078-0432.CCR-19-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eissa MAL, Lerner L, Abdelfatah E, Shankar N, Canner JK, Hasan NM, et al. Promoter methylation of ADAMTS1 and BNC1 as potential biomarkers for early detection of pancreatic cancer in blood. Clin Epigenetics 2019;11(1):59 doi 10.1186/s13148-019-0650-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freitas M, Ferreira F, Carvalho S, Silva F, Lopes P, Antunes L, et al. A novel DNA methylation panel accurately detects colorectal cancer independently of molecular pathway. J Transl Med 2018;16(1):45 doi 10.1186/s12967-018-1415-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden M. TEMPORARY REMOVAL: Response to W.C. Taylor, and C. Fiala and E.P. Diamandis. Ann Oncol 2020. doi 10.1016/j.annonc.2020.06.008. [DOI] [PubMed] [Google Scholar]
- 24.Liu L, Toung JM, Jassowicz AF, Vijayaraghavan R, Kang H, Zhang R, et al. Targeted methylation sequencing of plasma cell-free DNA for cancer detection and classification. Ann Oncol 2018;29(6):1445–53 doi 10.1093/annonc/mdy119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are presented in the paper or Supplementary Materials. The raw plasma cfDNA gitBS sequencing data reported in this paper have been deposited into the Gene Expression Omnibus (GEO), under accession number GSE149438.