

ABSTRACT
Parathyroid hormone (PTH) is produced by the parathyroid glands in response to low serum calcium concentrations where it targets bones, kidneys, and indirectly, intestines. The N‐terminus of PTH has been investigated for decades for its ability to stimulate bone formation when administered intermittently (iPTH) and is used clinically as an effective anabolic agent for the treatment of osteoporosis. Despite great interest in iPTH and its clinical use, the mechanisms of PTH action remain complicated and not fully defined. More than 70 gene targets in more than 90 murine models have been utilized to better understand PTH anabolic actions. Because murine studies utilized wild‐type mice as positive controls, a variety of variables were analyzed to better understand the optimal conditions under which iPTH functions. The greatest responses to iPTH were in male mice, with treatment starting later than 12 weeks of age, a treatment duration lasting 5–6 weeks, and a PTH dose of 30–60 μg/kg/day. This comprehensive study also evaluated these genetic models relative to the bone formative actions with a primary focus on the trabecular compartment revealing trends in critical genes and gene families relevant for PTH anabolic actions. The summation of these data revealed the gene deletions with the greatest increase in trabecular bone volume in response to iPTH. These included PTH and 1‐α‐hydroxylase (Pth;1α(OH)ase, 62‐fold), amphiregulin (Areg, 15.8‐fold), and PTH related protein (Pthrp, 10.2‐fold). The deletions with the greatest inhibition of the anabolic response include deletions of: proteoglycan 4 (Prg4, −9.7‐fold), low‐density lipoprotein receptor‐related protein 6 (Lrp6, 1.3‐fold), and low‐density lipoprotein receptor‐related protein 5 (Lrp5, −1.0‐fold). Anabolic actions of iPTH were broadly affected via multiple and diverse genes. This data provides critical insight for future research and development, as well as application to human therapeutics. © 2021 The Authors. Journal of Bone and Mineral Research published by Wiley Periodicals LLC on behalf of American Society for Bone and Mineral Research (ASBMR).
Keywords: ANABOLIC, BONE ANABOLISM, GENETIC ANIMAL MODELS, PARATHYROID‐RELATED DISORDERS, PTH
INTRODUCTION
Parathyroid hormone (PTH) has been approved by the US Food and Drug Administration (FDA) since 2002, when teriparatide, a 34–amino acid analog of PTH, was accepted for the treatment of osteoporosis. More recently a PTH related protein (PTHrP) analog was also approved for the treatment of osteoporosis under the name abaloparatide.( 1 ) It is well accepted that intermittent PTH (iPTH) therapy is anabolic for bone, whereas continuous PTH exposure is catabolic. The anabolic actions of iPTH in bone have been observed in animal models since 1929 using cats and rats.( 2 , 3 , 4 , 5 ) These results were recapitulated in human patients,( 6 , 7 ) which led to the approval of this anabolic agent for therapeutic purposes. However, the anabolic mechanism of iPTH is not fully understood, and this study aimed to reveal trends in critical genes and gene families relevant for iPTH anabolic actions.
As an endogenous endocrine mediator, PTH is released when the parathyroid gland detects a decrease in serum calcium concentration. Circulating PTH then targets the kidney and bone to increase serum calcium levels.( 5 ) The effects of PTH and PTHrP in bone are achieved by binding to its type 1 receptor (PTH1R, a G‐protein coupled receptor with seven transmembrane domains) on osteoblasts.( 8 , 9 ) This stimulates the production of receptor activator of nuclear factor κB ligand (RANKL) in osteoblasts and subsequent osteoclastogenesis.( 10 ) Indirectly, there is an increase in osteoblast numbers and bone formation.( 11 )
PTH is essential for fetal development, with newborn PTH‐deficient mice exhibiting reduced cartilage matrix mineralization and trabecular bone, due to fewer metaphyseal osteoblasts.( 12 ) Adult PTH‐null mice exhibit decreased serum calcium, decreased 1,25‐dihydroxyvitamin D3, and increased serum phosphate.( 13 ) Trabecular bone volume is increased in the femurs, tibias, and vertebrae of mutant mice, and the number and size of tibial osteoclasts are reduced. Furthermore, there is a decreased mineral apposition rate.
PTHrP‐null mice exhibit an osteoporotic phenotype that can be recapitulated in mice with targeted deletion in osteoblasts (Pthrp f/f ;cre colI ).( 14 ) This model is more specific to the local bone environment, in which iPTH treatment increased mineral apposition rate, bone volume, trabecular number, trabecular thickness, trabecular connectivity, and cortical thickness in long bones. This could be attributed to increased receptor availability without endogenous PTHrP or changes in receptor desensitization (i.e., increased number of receptors because there is not desensitization from PTHrP). In either case, it is likely that PTHrP can modulate the response to PTH via the PTH1R receptor.( 14 )
MATERIALS AND METHODS
Data for this study was collected from publications that have administered anabolic doses of iPTH from 2001 to 2020 (Figure 1). Papers were accessed by searching scholarly search engines, such as PubMed, through December 2020. A highly relevant and consistent outcome of trabecular bone volume per total volume was used as a key and focused measure to compare the anabolic response in experimental gene targeted mice to wild‐type controls in published studies. The PTH‐induced bone volume response was derived for both gene targeted and wild‐type mice (Table 1) separately [(PTH – Vehicle)/PTH]. Then, the relative response was calculated as a fold change by dividing the gene targeted response by the wild‐type response. A fold change of 1.0 indicates that there was no change in the anabolic response between wild‐type and gene‐targeted mice. If the fold change was greater than 1.0, the mutant mice had a greater anabolic response than wild‐type mice, whereas between 0 and 1.0 the mutant mice had a less anabolic response. A negative fold change indicates that the mutant response to iPTH was not anabolic. In some studies, actual numerical data was provided, whereas in others, data was derived from graphic representation. When bone volume was only depicted graphically, values were estimated by measurement with a ruler to derive the gene‐targeted response relative to wild type. Studies that showed an anabolic response to PTH in wild‐type controls were included whereas those that did not demonstrate an anabolic response in controls were excluded (there were very few studies that did not display an anabolic response).
FIGURE 1.
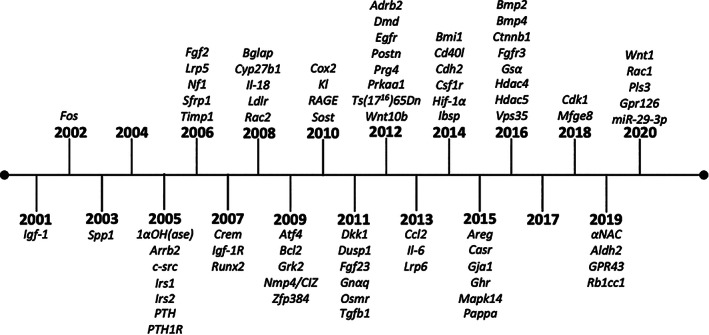
Timeline of gene targeted mouse models of PTH anabolic actions in bone. Abbreviation: PTH, parathyroid hormone.
TABLE 1.
Statistical analysis of the trabecular bone response in wild‐type mice
| Pearson's correlation | Linear regression of the slope | |||
|---|---|---|---|---|
| Category | r 2 | p | Slope | 95% CI |
| Gender | ||||
| Female (n = 44) | 0.8990 | <0.0001 | 1.031 | 0.8746 to 1.1870 |
| Male (n = 40) | 0.7698 | <0.0001 | 1.808 | 1.3160 to 2.3010 |
| Both (n = 11) | 0.3470 | 0.2957 | 0.748 | −0.7763 to 2.2720 |
| Bone site | ||||
| Tibia (n = 15) | 0.8631 | <0.0001 | 1.194 | 0.9090 to 1.4790 |
| Femur (n = 63) | 0.5204 | <0.0001 | 1.690 | 1.2750 to 2.1050 |
| Vertebrae (n = 21) | 0.1462 | 0.0872 | 0.620 | −0.0996 to 1.3400 |
| Age at start of treatment | ||||
| 0–2 weeks (n = 12) | 0.4261 | 0.0214 | 0.988 | 0.1802 to 1.7970 |
| 4–8 weeks (n = 22) | 0.3150 | 0.0066 | 0.752 | 0.2348 to 1.2690 |
| 9–10 weeks (n = 23) | 0.6942 | <0.0001 | 0.950 | 0.6640 to 1.2360 |
| 11–12 weeks (n = 25) | 0.7071 | <0.0001 | 1.530 | 1.1050 to 1.9540 |
| >12 weeks (n = 22) | 0.6239 | <0.0001 | 2.031 | 1.2950 to 2.7670 |
| Days per week of treatment | ||||
| 5–5.5 (n = 35) | 0.8758 | <0.0001 | 1.250 | 1.0060 to 1.4940 |
| 7 (n = 66) | 0.6487 | <0.0001 | 1.3178 | 0.9320 to 1.7010 |
| Treatment duration | ||||
| <4 weeks (n = 23) | 0.6880 | <0.0001 | 1.347 | 0.9357 to 1.7590 |
| 4 weeks (n = 48) | 0.3858 | <0.0001 | 0.885 | 0.5335 to 1.2016 |
| 5–6 weeks (n = 22) | 0.6749 | <0.0001 | 2.459 | 1.6630 to 3.2550 |
| 7–12 weeks (n = 12) | 0.6503 | 0.0015 | 0.790 | 0.3814 to 1.1970 |
| Treatment dose (μg/kg/day) | ||||
| ≦30 (n = 19) | 0.6201 | <0.0001 | 2.176 | 1.3050 to 3.0480 |
| 40 (n = 19) | 0.6799 | <0.0001 | 1.565 | 1.0150 to 2.1140 |
| 50–60 (n = 13) | 0.3717 | 0.0269 | 1.135 | 0.1559 to 2.1150 |
| 80 (n = 44) | 0.4488 | <0.0001 | 0.919 | 0.6021 to 1.2370 |
| 90–160 (n = 10) | 0.9454 | <0.0001 | 1.001 | 0.8050 to 1.1970 |
Notes: Data was pooled to analyze Pearson's correlation of the trabecular response of wild‐type mice to vehicle or iPTH. The r 2 and p value are reported from this analysis. The slope and 95% CI of the linear regression of the slope is also reported.
Abbreviations: CI, confidence interval; iPTH, intermittent parathyroid hormone.
Most commonly, human PTH(1‐34) (hPTH(1‐34)) was administered, although there were a few studies as indicated when the PTH differed (i.e., hPTH(1‐84) or derived from a different source). Doses ranged from 20 to 160 μg/kg/day, but was typically between 40 and 100 μg/kg/day, as specified in Table 2. PTH was administered by injection daily, 7 days/week, unless noted differently. Treatment time was typically 2 to 6 weeks of iPTH. The models are grouped under categories largely according to functional analyses in the Supplemental Material, alphabetically in Table 2. By assimilating the literature that has used anabolic PTH in genetic mouse models, we gain a better understanding of key genetic pathways as well as the overall complexity of PTH actions in bone.
TABLE 2.
Genetic models treated with iPTH
| Target gene | Genotype | Gender | PTH regimen | Age of mice during treatment | Bone site | FC in trabecular BV/TV | N.Ob/BS | N.Oc/BS | Strain | Year | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1α(OH)ase | 1α(OH)ase −/− | ♂ | 40 μg/kg/day hPTH(1‐34) | 12–16 weeks | Tibia | ~1.101 | No change | No change | C57BL/6J; BALB/c | 2008 | ( 15 ) |
| Ampkα1 | Ampkα1 −/− | NI | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 12–16 weeks | Tibia | ~4.250 | ND | ND | C57BL/6129/Sv | 2012 | ( 16 ) |
| Areg | Areg −/− | ♀ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 12–16 weeks | Femur | ~15.75 | ND | Decreased | 129/C57BL/6 | 2015 | ( 17 ) |
| Atf4 | Atf4 −/− | NI | 60 μg/kg/day hPTH(1‐34) | 5–33 days | Femur | ~0.468 | ND | ND | Swiss Black | 2009 | ( 18 ) |
| Atf4 | Atf4 −/− | NI | 60 μg/kg/day hPTH(1‐34) | 5–33 days | Vertebrae | ~0.353 | ND | ND | Swiss Black | 2009 | ( 18 ) |
| Bcl2 | Bcl2 −/− | NI | 50 μg/kg/day hPTH(1‐34) | 4–13 days | Tibia | 1.054 | ND | ND | 129/C57BL/6 | 2009 | ( 19 ) |
| Bcl2 | Bcl2 −/− Bim +/− | ♂ | 80 μg/kg/day hPTH(1‐34) | 16–20 weeks | Tibia | ND | ND | No change | C57BL/6 (10th generation) | 2010 | ( 20 ) |
| β‐arr2 | β‐arr2 −/− | ♂ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 12–16 weeks | Femur | ND | Increased | Increased | C57Bl/6 | 2005 | ( 21 ) |
| β‐arr2 | β‐arr2 −/− | ♂ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 12–16 weeks | Vertebrae | ~0.000 | ND | ND | C57Bl/6 | 2005 | ( 21 ) |
| β‐arr2 | β‐arr2 −/− | ♂ | 40 μg/kg/day hPTH(1‐34) | 9–17 weeks | Vertebrae | ~0.428 | Decreased | Decreased | C57Bl/6 | 2009 | ( 22 ) |
| β‐arr2 | β‐arr2 −/− | ♂ | 40 μg/kg/day hPTH(1‐34) | 9–17 weeks | Tibia | ~0.179 | ND | ND | C57Bl/6 | 2009 | ( 22 ) |
| β‐cat | Dmp1‐CreERt2;β‐cat f/f | ♂ | 30 μg/kg/day rhPTH(1‐34) | 12.5–17.5 weeks | Femur | ~2.115 | ND | ND | C57Bl/6 129 | 2016 | ( 23 ) |
| β‐cat | Dmp1‐CreERt2;β‐cat f/f | ♂ | 30 μg/kg/day rhPTH(1‐34) | 12.5–17.5 weeks | Vertebrae | ~2.571 | ND | ND | C57Bl/6 129 | 2016 | ( 23 ) |
| β‐cat | Osx‐Cre;β‐cat f/f | ♂ | 80 μg/kg/day rhPTH(1‐34) | 7–11 weeks | Femur | ~1.120 | ND | ND | C57Bl/6 (6th generation) | 2018 | ( 24 ) |
| β‐cat | Osx‐Cre;β‐cat f/f | ♂ | 80 μg/kg/day rhPTH(1‐34) | 7–11 weeks | Vertebrae | ~3.350 | ND | ND | C57Bl/6 (6th generation) | 2018 | ( 24 ) |
| β2AR | Adbr −/− | ♀ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 10–14 weeks | Femur | ~ −0.081 | ND | Decreased | C57Bl/6 | 2012 | ( 25 ) |
| β2AR | Adbr −/− | ♀ | 80 μg/kg/day hPTH(1‐34)(5 days/week) | 10–14 weeks | Vertebrae | ~ −0.131 | ND | ND | C57Bl/6 | 2012 | ( 25 ) |
| β2AR | Adbr −/− | ♀ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 54–58 weeks | Femur | ~ −0.113 | ND | No change | C57Bl/6 | 2012 | ( 25 ) |
| BMI1 | Bmi1 −/− | ♀♂ | 80 μg/kg/day hPTH(1‐34) | 1–4 weeks | Femur | Cannot determine (missing necessary controls) | Cannot determine (missing necessary controls) | ND | 129Ola FVB/N hybrid | 2014 | ( 26 ) |
| Bmp2, Bmp4 | R26CreER/R26CreER and Bmp2 C/C . Bmp2 C/C ; Bmp4 C/C ; R26Cre ER/+ (Bmp2/4 DCKO); OVX | ♀ | 40 μg/kg/day hPTH(1‐34) (5 days/week) | 10–12 to 16–18 weeks | Femur | Cannot determine (missing necessary controls) | ND | ND | NI | 2016 | ( 27 ) |
| BSP | Bsp −/− | ♂ | 0.8 μg/μl PTH 1‐84 (local injection) | 12–14 weeks | Calvaria | ~0.985 (BV reported) | ND | ND | 129/CD‐1 | 2015 | ( 28 ) |
| C‐FMS | MAFIA | ♀ | 50 μg/kg/day hPTH(1‐34) | 16–22 weeks | Tibia | ~0.127 | ND | Decreased | C57Bl/6J | 2014 | ( 29 ) |
| C‐FOS | c‐fos −/− | NI | 50 μg/kg/day hPTH(1‐34) | 4–21 days | Vertebrae | ~0.316 | ND | ND | C57Bl/6 (5th generation) | 2002 | ( 30 ) |
| CaSR | Col‐Bone CaSR Δflox/Δflox | NI | 50 μg/kg/day hPTH(1‐34) | 4–17 days | Tibia | ~0.893 | ND | ND | C57Bl/6 CD‐1 | 2015 | ( 31 ) |
| CD40L | CD40L −/− | ♀ | 80 μg/kg/day hPTH(1‐34) | 12–16 weeks | Femur | 0.135 | ND | Decreased | C57Bl/6 | 2014 | ( 32 ) |
| Cdh2 | Osx‐Cre::Cdh2 f/f | ♂ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 4 weeks of iPTH starting 12–16 weeks | Tibia | 3.815 | No change | Decreased | C57Bl/6 | 2014 | ( 33 ) |
| Cdh2 | Dmp1‐cre;Cdh2 f/f | ♂ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 8–12 weeks | Femur | 3.393 | Increased | Increased | C57Bl/6 | 2016 | ( 34 ) |
| Cdk1 | Osx‐Cre;Cdk1 f/f | ♀ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 12–16 weeks | Vertebrae | ~2.018 | Increased | No change | C57Bl/6129S6/SvEvTac | 2018 | ( 35 ) |
| Cox2 | Cox2 −/− | ♂ | 80 μg/kg/day hPTH(1‐34) | 20–23 weeks | Femur | 1.669 | Increased | No change | CD‐1 (9th generation) | 2010 | ( 36 ) |
| Cox2 | Cox2 −/− | ♂ | 80 μg/kg/day hPTH(1‐34) | 20–23 weeks | Vertebrae | 5.688 | ND | ND | CD‐1 (9th generation) | 2010 | ( 36 ) |
| Crem | Crem −/− | ♂ | 160 μg/kg/day hPTH(1‐34) | 10 days of iPTH from 11–12 weeks | Femur | ~0.312 | No change | Increased | 129Sv; C57BL/6 | 2007 | ( 37 ) |
| Cx43 | Cx43 ΔCT/fl ;DMP1‐8kb‐Cre | ♀ | 100 μg/kg/day hPTH(1‐34) | 16–18 weeks | Femur | 1.154 | ND | ND | C57Bl/6 | 2015 | ( 38 ) |
| Dkk1 | Dkk1 TG; 2.3‐kb rat collagen type Ia promoter | NI | 95 μg/kg/day hPTH(1‐34) | 34 days of iPTH from 12–14 weeks | Tibia | ND | Decreased | Decreased | C57Bl/6 CD‐1 | 2011 | ( 39 ) |
| Egdr | Egfr Wa5 (impaired EGFR signaling) | ♀ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 12–16 weeks | Femur | ~0.704** | ND | Decreased | C57Bl/6 | 2012 | ( 40 ) |
| Fgf2 | Fgf2 −/− | ♂ | 80 μg/kg/day hPTH(1‐34) | 8–12 weeks | Femur | 0.647 | Decreased | No change | Black Swiss 129Sv | 2006 | ( 41 ) |
| Fgf2 | Fgf2 −/− | ♀ | 80 μg/kg/day hPTH(1‐34) | 60–64 weeks | Femur | 0.139 | ND | ND | Black Swiss 129Sv | 2006 | ( 41 ) |
| Fgf2 | 3.6Col1GFPsaph tg/tg ;Fgf2 −/− | ♀ | 20 μg/kg/day PTH(1‐34) | 12 weeks (8 h) | Tibia | ND | ND | ND | Black Swiss 129Sv; FVB/N | 2018 | ( 42 ) |
| Fgf23 | Fgf23 −/− | NI | 100 μg/kg/day hPTH(1‐34) | 8–22 days | Femur | ~1.077 | No change | ND | C57Bl/6 129Sv | 2011 | ( 43 ) |
| Fgfr3 | Fgfr3 −/− | ♂ | 80 μg/kg/day hPTH(1‐34) | 16–20 weeks | Femur | ~2.533 | Decreased | Increased | C3H | 2016 | ( 44 ) |
| Fgfr3 | FGFR3 G369C/+ | NI | 80 μg/kg/day hPTH(1‐34) | 8–12 weeks | Femur | ~2.814 | ND | ND | C57Bl/6 | 2017 | ( 45 ) |
| Ghr | DMP1‐Cre;GHR f/f | ♀ | 80 μg/kg/day hPTH(1‐34) | 4–8 weeks | Femur | 0.234 | Decreased | No change | C57Bl/6 | 2015 | ( 46 ) |
| GPR126 | Osx‐cre;Gpr126 f/f | ♀♂ | 80 μg/kg/day hPTH(1‐34) | 5–30 days | Femur | ~1.975 | ND | ND | C57Bl/6 | 2020 | ( 47 ) |
| GRK2 | GRK1 TG ;1.3kb fragment of OG2 promoter | ♀♂ | 40 μg/kg/day hPTH(1‐34) | 36–40 weeks | Vertebrae | Cannot determine (missing necessary controls) | Increased | No change | B6SJLF1/J | 2009 | ( 48 ) |
| Gαs | Gα s Osx‐KO | ♀♂ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 8–12 weeks | Femur | 0.223 | Increased | Increased | C57Bl/6 CD1 | 2016 | ( 49 ) |
| HDAC4 | HDAC4 fl/fl ; DMP1‐cre | ♀ | 100 μg/kg/day hPTH(1‐34) (5 days/week) | 8–12 weeks | NI | ~0.971 | ND | ND | C57Bl/6 | 2016 | ( 50 ) |
| HDAC4; HDAC5 | HDAC5 −/− ; HDAC4 fl/fl ; DMP1‐cre | ♀ | 100 μg/kg/day hPTH(1‐34) (5 days/week) | 8–12 weeks | NI | ~2.111 | ND | ND | C57Bl/6 | 2016 | ( 50 ) |
| HDAC5 | HDAC5 −/− | ♀ | 100 μg/kg/day hPTH(1‐34) (5 days/week) | 8–12 weeks | NI | ~2.111 | ND | ND | C57Bl/6 | 2016 | ( 50 ) |
| Hif‐1α | Ocn‐Cre;Hif‐1αf/f | ♀ | 20 μg/kg/day hPTH(1‐34) | 10–16 weeks | Femur | ~1.511 | ND | ND | C57Bl/6 | 2014 | ( 51 ) |
| Hif‐1α | Ocn‐Cre;Hif‐1αf/f | ♀ | 40 μg/kg/day hPTH(1‐34) | 10–16 weeks | Femur | ~1.223 | ND | ND | C57Bl/6 | 2014 | ( 51 ) |
| Igf‐1 | Igf‐1 −/− | NI | 160 μg/kg/day hPTH(1‐34) | 5–6.5 weeks | Femur | ND | ND | ND | NI | 2001 | ( 52 ) |
| Igf‐1 | B6.C3H‐6T | ♀ | 50 μg/kg/day hPTH(1‐34) | 16–20 weeks | Femur | 0.704 | ND | ND | C57Bl/6 (10th generation) | 2005 | ( 53 ) |
| Igf‐1 | Igf1 fl/fl ; Albumin‐Cre | ♂ | 50 μg/kg/day hPTH(1‐34) (5 days/week) | 12–16 weeks | Vertebrae | ~2.150 | ND | ND | FVB/N, C57BL, and 129Sv | 2006 | ( 54 ) |
| Igf‐1 | ALS −/− | ♂ | 50 μg/kg/day hPTH(1‐34) (5 days/week) | 12–16 weeks | Vertebrae | ~ −0.300 | ND | ND | C57Bl/6 (6th generation) | 2006 | ( 54 ) |
| Igf‐1 | Igf1 fl/fl ; Albumin‐Cre; ALS −/− | ♂ | 50 μg/kg/day hPTH(1‐34) (5 days/week) | 12–16 weeks | Vertebrae | ~ −0.350 | ND | ND | FVB/N; C57BL 129Sv | 2006 | ( 54 ) |
| Igf‐1 | HIT (hepatic IGF‐1 transgene) | ♂ | 50 μg/kg/day hPTH(1‐34) | 12–16 weeks | Femur | ~1.622 | ND | ND | FVB/N | 2010 | ( 55 ) |
| Igf‐1 | HIT KO | ♂ | 50 μg/kg/day hPTH(1‐34) | 12–16 weeks | Femur | ~2.069 | ND | ND | FVB/N | 2010 | ( 55 ) |
| IGF‐IR | Ocn‐Cre;Igf‐IR f/f | NI | 80 μg/kg/day rat PTH(1‐34) | 12–14 weeks | Tibia and femur | ND | ND | ND | FVB/N | 2014 | ( 56 ) |
| IL18 | IL18 −/− | ♀ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 4 weeks of iPTH starting at 7–8 weeks | Tibia and femur | ND | ND | ND | DBA/1 | 2008 | ( 57 ) |
| IL6 | IL6 −/− | ♀♂ | 50 μg/kg/day hPTH(1‐34) | 3–24 days | Femur | ~0.596 | ND | Decreased | C57Bl/6 | 2013 | ( 58 ) |
| IL6 | IL6 −/− | ♀♂ | 50 μg/kg/day hPTH(1‐34) | 16–22 weeks | Femur | ~3.333 | ND | ND | C57Bl/6 | 2013 | ( 58 ) |
| Irs‐1 | Irs‐1 −/− | ♂ | 80 μg/kg/day hPTH(1‐34) | 10–14 weeks | Tibia and femur | 0.090 | No change | Decreased | C57Bl6 CBA | 2005 | ( 59 ) |
| Irs‐2 | Irs‐2 −/− | ♂ | 80 μg/kg/day hPTH(1‐34) | 10–14 weeks | Tibia and femur | 2.499 | Decreased | Decreased | C57Bl6 CBA | 2005 | ( 59 ) |
| Kl | Kl −/− | NI | 100 μg/kg/day hPTH(1‐34) | 8–22 days | Femur | ~1.077 | No change | ND | C57Bl/6 129Sv | 2010 | ( 43 ) |
| Ldlr | Ldlr −/− | ♀ | 40 μg/kg/day hPTH(1‐34) (5 days/week) | 20–25 weeks | Femur | 0.624 | Increased | Increased | C57Bl/6 | 2009 | ( 60 ) |
| Ldlr | Ldlr −/− ; pOBCol3.6GFPtpz and pOBCol2.3GFPCyan | ♀ | 40 μg/kg/day hPTH(1‐34) (5 days/week) | 5 weeks of iPTH starting at 8–12 weeks | Calvaria | ND | Decreased | ND | C57Bl/6 | 2013 | ( 61 ) |
| Ldlr | Ldlr −/− ; pOBCol3.6GFPtpz and pOBCol2.3GFPCyan | ♀ | 40 μg/kg/day hPTH(1‐34) (5 days/week) | 5 weeks of iPTH starting at 8–12 weeks | Femur | ND | Decreased | ND | C57Bl/6 | 2013 | ( 61 ) |
| Lrp5 | Lrp5 −/− | ♀♂ | 40 μg/kg/day hPTH(1‐34) (5 days/week) | 12–16 weeks | Hindlimb | ND | ND | ND | 129S/J | 2006 | ( 62 ) |
| Lrp5 | Lrp5 −/− | ♀ | 80 μg/kg/day hPTH(1‐34) (every other day) | 20–26 weeks | Femur | ~0.435 | ND | ND | C57Bl/6 | 2009 | ( 63 ) |
| Lrp5 | Lrp5 −/− | ♂ | 80 μg/kg/day hPTH(1‐34) (every other day) | 20–26 weeks | Femur | ~ −1.294 | ND | ND | C57Bl/6 | 2009 | ( 63 ) |
| Lrp5 | Lrp5 −/− | ♀ | 80 μg/kg/day hPTH(1‐34) (every other day) | 20–26 weeks | Vertebrae | ~10.000 | No change | No change | C57Bl/6 | 2009 | ( 63 ) |
| Lrp5 | Lrp5 −/− | ♂ | 80 μg/kg/day hPTH(1‐34) (every other day) | 20–26 weeks | Vertebrae | ~ −1.028 | No change | No change | C57Bl/6 | 2009 | ( 63 ) |
| Lrp6 | Ocn‐cre;Lrp6 f/f | ♂ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 8–12 weeks | Femur | ~ −1.255 | Decreased | No change | C57Bl/6J; 129 FVB/N | 2013 | ( 64 ) |
| Lrp6 | Ocn‐Cre;Lrp6 f/f | ♂ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 8–12 weeks | Femur | ND | ND | ND | C57Bl/6J; 129 FVB/N | 2015 | ( 65 ) |
| MCP‐1 | Mcp‐1 −/− | ♂ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 16–22 weeks | Tibia | ~0.084 | ND | Decreased | C57Bl/6 | 2013 | ( 66 ) |
| MCP‐1 | Mcp‐1 −/− | ♀♂ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 20–26 weeks | ND | ND | ND | ND | C57Bl/6 | 2013 | ( 66 ) |
| Mdx | C57BL/10ScSn/DMD‐mdx | ♂ | 30 μg/kg/day black bear PTH(1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 63, 64, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87) (5 days/week) | 4–10 weeks | Femur | ~5.833 | No change | Decreased | C57BL/610ScSn | 2012 | ( 88 ) |
| Mfge8 | Mfge8 −/− | ♀♂ | 50 μg/kg/day hPTH(1‐34) | 16–22 weeks | Tibia | ~2.000 (reported as FC) | ND | Decreased | C57Bl/6 | 2018 | ( 89 ) |
| MHC I | MHC I −/− | NI | 80 μg/kg/day hPTH(1‐34) | 5–9 weeks | Femur | ~0.173 | ND | ND | C57Bl/6 | 2009 | ( 72 ) |
| MHC I; MHC II | MHC I −/− ; MHC II −/− | NI | 80 μg/kg/day hPTH(1‐34) | 5–9 weeks | Femur | ~0.058 | ND | ND | C57Bl/6 | 2009 | ( 72 ) |
| MHC II | MHC II −/− | NI | 80 μg/kg/day hPTH(1‐34) | 5–9 weeks | Femur | ~1.038 | ND | ND | C57Bl/6 | 2009 | ( 72 ) |
| miR‐29‐3p | miR‐29‐3p decoy | ♀ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 12–16 weeks | Femur | ~8.858 | Increased | No change | C57Bl/6 | 2020 | ( 90 ) |
| Mkp1 | Mkp1 −/− | ♀ | 50 μg/kg/day hPTH(1‐34) (5–6 days/week) | 3–24 days | Femur | ~1.250 (reported as FC) | ND | ND | C57Bl/6 129 | 2011 | ( 91 ) |
| Nf1 | Nf1 +/− | ♂ | 80 μg/kg/day hPTH(1‐34) | 28 days of iPTH starting 8–12 weeks | Tibia | ~0.963 | ND | Increased | C57Bl/6 | 2006 | ( 92 ) |
| Nmp4 | Nmp4 −/− | ♀ | 30 μg/kg/day hPTH(1‐34) | 10–17 weeks | Femur | ~2.906 | ND | ND | C57Bl/6 (6th generation) | 2009 | ( 93 ) |
| Nmp4 | Nmp4 −/− | ♀ | 30 μg/kg/day hPTH(1‐34) | 10–12 weeks | Tibia | ~1.500 | ND | ND | C57Bl/6 (6th generation) | 2011 | ( 94 ) |
| Nmp4 | Nmp4 −/− | ♀ | 30 μg/kg/day hPTH(1‐34) | 10–17 weeks | Tibia | ~0.800 | ND | ND | C57Bl/6 (6th generation) | 2011 | ( 94 ) |
| Nmp4 | Nmp4 −/− | ♀ | 30 μg/kg/day hPTH(1‐34) | 10–12 weeks | Vertebrae | ~1.467 | ND | ND | C57Bl/6 (6th generation) | 2011 | ( 94 ) |
| Nmp4 | Nmp4 −/− | ♀ | 30 μg/kg/day hPTH(1‐34) | 10–17 weeks | Vertebrae | ~4.206 | ND | ND | C57Bl/6 (6th generation) | 2011 | ( 94 ) |
| Nmp4 | Nmp4 −/− | ♀ | 30 μg/kg/day hPTH(1‐34) | 10–13 weeks | Femur | ~2.523 | ND | ND | C57Bl/6 (6th‐7th generation) | 2012 | ( 95 ) |
| Ocn | Ocn −/− | ♀ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 10–14 weeks | Vertebrae | 1.266 | ND | ND | C57Bl/6 | 2008 | ( 96 ) |
| Ocn | Ocn −/− | ♀ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 10–14 weeks | Femur | 1.174 | No change | Increased | C57Bl/6 | 2008 | ( 96 ) |
| Opn | Opn −/− | ♀ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 7–11 weeks | Tibia and femur | ~1.362** | ND | Decreased | 129 | 2003 | ( 97 ) |
| OSMR | Osmr −/− | ♂ | 30 μg/kg/day hPTH(1‐34) (5 days/week) | 6–9 weeks | Tibia | ~ −0.518 | Decreased | Increased | C57Bl/6 | 2011 | ( 98 ) |
| p38α | Ocn‐Cre;p38α f/f | ♂ | 40 μg/kg/day hPTH(1‐34) | 12–16 weeks | Femur | ~0.415 | Decreased | Decreased | C57Bl/6 | 2015 | ( 99 ) |
| Pappa | Pappa −/− | ♀ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 12–18 weeks | Femur | ~0.277** | ND | ND | C57Bl/6; 129 | 2015 | ( 100 ) |
| PLS3 | Pls3 −/0 | ♂ | 80 μg/kg/day hPTH(1‐34) | 10–12 weeks | Vertebrae | ND | No change | ND | C57Bl/6 | 2020 | ( 101 ) |
| Postn | Postn−/− | ♀ | 40 μg/kg/day hPTH(1‐34) | 12–17 weeks | Femur | 1.106 | ND | Increased | C57Bl/6 | 2012 | ( 102 ) |
| Postn | Postn −/− | ♀ | 40 μg/kg/day hPTH(1‐34) | 12–17 weeks | Vertebrae | 1.762 | ND | ND | C57Bl/6 | 2012 | ( 102 ) |
| Prg4 | Prg4 −/− | ♀♂ | 50 μg/kg/day hPTH(1‐34) | 4–21 days | Femur | 1.239 | ND | ND | C57Bl/6 | 2012 | ( 71 ) |
| Prg4 | Prg4 −/− | ♀♂ | 50 μg/kg/day hPTH(1‐34) | 16–22 weeks | Femur | −9.692 | No change | Decreased | C57Bl/6 | 2012 | ( 71 ) |
| PTH and 1α(OH)ase | PTH −/− ;1α(OH)ase −/− | NI | 0.2 μg/kg/day rat PTH(1‐34)/day | 4–14 days | Femur | ~62.000 | Cannot determine (no reported WT+PTH) | Cannot determine (no reported WT+PTH) | C57BL/6J and BALB/c | 2005 | ( 103 ) |
| PTH1R | Lck‐Cre;PTH1R f/f | ♀ | 80 μg/kg/day hPTH(1‐34) | 2–6 weeks | Femur | 0.409 | Decreased | No change | C57Bl/6 | 2012 | ( 104 ) |
| PTH1R | Lck‐Cre;PTH1R f/f | ♀ | 80 μg/kg/day hPTH(1‐34) | 13–17 weeks | Femur | −0.314 | ND | ND | C57Bl/6 | 2012 | ( 104 ) |
| PTH1R | pdPTH1R | ♀ | 40 μg/kg/day hPTH(1‐34) (5 days/week) | 12–22 weeks | Vertebrae | ~0.837 | ND | ND | C57Bl/6 | 2012 | ( 105 ) |
| PTH1R | pdPTH1R | ♂ | 40 μg/kg/day hPTH(1‐34) (5 days/week) | 12–22 weeks | Vertebrae | ~0.890 | ND | ND | C57Bl/6 | 2012 | ( 105 ) |
| PTH1R | pdPTH1R | ♀ | 40 μg/kg/day hPTH(1‐34) (5 days/week) | 12–22 weeks | Femur | ~0.822 | ND | ND | C57Bl/6 | 2012 | ( 105 ) |
| PTH1R | pdPTH1R | ♂ | 40 μg/kg/day hPTH(1‐34) (5 days/week) | 12–22 weeks | Femur | ~1.000 | ND | ND | C57Bl/6 | 2012 | ( 105 ) |
| PTH1R | DMP1‐Cre;PTH1R f/f | ♀ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 4 weeks of iPTH (start age NI) | Femur | ND | ND | ND | C57Bl/6 dominant (mixed background) | 2013 | ( 106 ) |
| PTH1R | DMP1‐Cre;PTH1R f/f | ♀ | 80 μg/kg/day hPTH(1‐34) (5 days/week) | 4 weeks of iPTH (start age NI) | Vertebrae | 0.339 | ND | ND | C57Bl/6 dominant (mixed background) | 2013 | ( 106 ) |
| PTH1R | Dmp1‐Cre;PTH1R f/f | ♀ | 100 ng/g/day PTH(1‐34) | 16–20 weeks | Femur | ~0.739 | ND | ND | C57BL/6Nhsd | 2016 | ( 107 ) |
| PTH1R | Dmp1‐Cre;PTH1R f/f | ♂ | 100 ng/g/day PTH(1‐34) | 16–20 weeks | Femur | ~ −0.081 | ND | ND | C57BL/6Nhsd | 2016 | ( 107 ) |
| PTHRP | Pthrp +/− | ♂ | 40 μg/kg/day hPTH(1‐34) | 12–24 weeks | Femur | ~10.230 | ND | ND | FVB/N CD‐1 | 2005 | ( 14 ) |
| Rac1 | Osx‐Cre;Rac1 −/− | NI | 80 μg/kg/day hPTH(1‐34) | 4–8 weeks | Femur | ND | NI | NI | NI | 2020 | ( 108 ) |
| Rac2 | Rac2 −/− | NI | 80 μg/kg/day hPTH(1‐34) | 12–16 weeks | Tibia | ND | Increased | Increased | C57Bl/6 (used as control) | 2008 | ( 109 ) |
| Rag2 | Rag2 −/− | NI | 80 μg/kg/day hPTH(1‐34) | 5–9 weeks | Femur | ~0.406 | ND | ND | C57Bl6/J | 2009 | ( 72 ) |
| RAGE | RAGE −/− | ♀ | 30 μg/kg/day hPTH(1‐34) | 10–12 weeks | Femur | ~0.00 | ND | ND | C57Bl/6 | 2010 | ( 110 ) |
| RAGE | RAGE −/− | ♀ | 30 μg/kg/day hPTH(1‐34) | 10–17 weeks | Femur | ~0.495 | ND | ND | C57Bl/6 | 2010 | ( 110 ) |
| RAGE | RAGE −/− | ♀ | 30 μg/kg/day hPTH(1‐34) | 10–12 weeks | Vertebrae | ~1.857 | ND | ND | C57Bl/6 | 2010 | ( 110 ) |
| Runx2 | Runx2 Tg | ♀ | 100 μg/kg/day hPTH(1‐34) | 4–10 weeks | Femur | ~0.637 | ND | Increased | C57Bl/6 | 2007 | ( 111 ) |
| sFRP1 | sFRP −/− | ♀ | 100 μg/kg/day hPTH(1‐34) | 8–12 weeks | Femur | ~0.711 (reported as FC) | ND | ND | C57BL/6 (albino)‐129SvEv (LEX‐1) | 2006 | ( 112 ) |
| sFRP1 | sFRP −/− | ♀ | 100 μg/kg/day hPTH(1‐34) | 24–28 weeks | Femur | ~0.627 (reported as FC) | ND | ND | C57BL/6 (albino)‐129SvEv (LEX‐1) | 2006 | ( 112 ) |
| sFRP1 | sFRP −/− | ♀ | 100 μg/kg/day hPTH(1‐34) | 36–40 weeks | Femur | ~0.332 (reported as FC) | ND | ND | C57BL/6 (albino)‐129SvEv (LEX‐1) | 2006 | ( 112 ) |
| sFRP1 | sFRP1 Tg | ♀ | 40 μg/kg/day hPTH(1‐34) (5 days/week) | 12–14 weeks | Femur | 0.103 | ND | No change | FVB/N‐Swiss Webster hybrid | 2010 | ( 113 ) |
| sFRP1 | sFRP1 Tg | ♂ | 40 μg/kg/day hPTH(1‐34) (5 days/week) | 12–14 weeks | Femur | 0.120 | ND | No change | FVB/N‐Swiss Webster hybrid | 2010 | ( 113 ) |
| sFRP1 | sFRP1 Tg | ♀ | 40 μg/kg/day hPTH(1‐34) (5 days/week) | 12–14 weeks | Vertebrae | 0.099 | ND | ND | FVB/N‐Swiss Webster hybrid | 2010 | ( 113 ) |
| sFRP1 | sFRP1 Tg | ♂ | 40 μg/kg/day hPTH(1‐34) (5 days/week) | 12–14 weeks | Vertebrae | 0.402 | ND | ND | FVB/N‐Swiss Webster hybrid | 2010 | ( 113 ) |
| Sost | Sost TG | ♂ | 100 μg/kg/day hPTH(1‐34) (5–6 days/week) | 24–33 weeks | Femur | 0.391 | ND | No change | FVB, C57BL/6 | 2010 | ( 114 ) |
| Sost | Sost −/− | ♂ | 30 μg/kg/day hPTH(1‐34) | 10–16 weeks | Femur | ~0.779 | ND | ND | 129/SvJ and Black Swiss | 2011 | ( 115 ) |
| Sost | Sost −/− | ♂ | 90 μg/kg/day hPTH(1‐34) | 10–16 weeks | Femur | ~0.877 | ND | ND | 129/SvJ and Black Swiss | 2011 | ( 115 ) |
| TCRβ | TCRβ −/− | NI | 80 μg/kg/day hPTH(1‐34) | 5–9 weeks | Femur | 0.503 | Decreased | Increased | C57Bl/6 | 2009 | ( 72 ) |
| TGFβ1 | TGFβ1 −/− ,Rag2 −/− | ♂ | 40 μg/kg/day hPTH(1‐34) (5 days/week) | 8–12 weeks | Tibia | ~ −0.388 | Decreased | No change | C57Bl/6 | 2011 | ( 116 ) |
| TGIF1 | Tgif1 fl/fl ; DMP1‐cre | ♂ | 100 μg/kg/day hPTH(1‐34) (5 days/week) | 8–12 weeks | Tibia | ~0.103 | Decreased | No change | C57Bl/6 | 2019 | ( 117 ) |
| TGIF1 | Tgif1 −/− | ♂ | 100 μg/kg/day hPTH(1‐34) (5 days/week) | 8–12 weeks | Tibia | ~ −0.126 | Decreased | Decreased | C57Bl/6 | 2019 | ( 117 ) |
| Timp1 | Timp1 TG by type‐I collagen promoter | ♀ | 40 μg/kg/day hPTH(1‐34) | 10–16 weeks | Femur | 1.964 | ND | Decreased | C57BL/6 CBA | 2006 | ( 118 ) |
| Ts65Dn | Mosel for trisomy 21 | ♂ | 30 μg/kg/day hPTH(1‐34) | 12–16 weeks | Tibia | ~1.450 | No change | No change | C57BL/6; C3H/HeJ | 2012 | ( 119 ) |
| Ts65Dn | Mosel for trisomy 21 | ♂ | 80 μg/kg/day hPTH(1‐34) | 12–16 weeks | Tibia | ~1.450 | No change | No change | C57BL/6; C3H/HeJ | 2012 | ( 119 ) |
| Vps35 | Ocn‐Cre;Vps35 f/f | ♂ | 50 μg/kg/day hPTH(1‐34) (5 days/week) | 7–12 weeks | Femur | ~7.690 | ND | ND | C57Bl/6 | 2016 | ( 120 ) |
| Wnt1 | Wnt1 +/R235W | ♀ | 80 μg/kg/day hPTH(1‐34) | 52–56 weeks | Femur | ND | ND | ND | C57Bl/6 129 | 2020 | ( 121 ) |
Notes: A summary of each publication using iPTH in a genetic model is alphabetized by target gene. The genotype, gender, PTH regimen, age of mice during treatment, bone site, fold change in BV/TV comparing targeted gene versus WT (target gene/WT), N.Ob/BS, N.Oc/BS, strain, and year are listed.
Abbreviations: ♂, male; ♀, female; ~, values estimated from a graph; **, bone area reported; BV/TV, trabecular bone volume per total volume; FC, fold change; hPTH, human parathyroid hormone; iPTH, intermittent parathyroid hormone; ND, not determined; NI, not indicated; N.Ob/BS, number of osteoblasts per bone surface; N.Oc/BS, number of osteoclasts per bone surface; PTH, parathyroid hormone; WT, wild type.
RESULTS
Actions of iPTH in wild‐type mice
Because gene‐targeted murine studies utilized wild‐type mice as positive controls, a variety of variables were analyzed to better understand the optimal conditions under which iPTH functions. Trabecular bone volume was compiled and organized by different categories (Figure 2, Table 1). The groups were stratified by: sex, bone site, days per week of treatment, age at start of treatment, duration of treatment, and dose of iPTH. Strain was also considered and is listed in Table 2; however, the only strain that had a large enough sample size for consideration was C57BL/6. Because the interest of this section is in comparing different categories, we did not include strain in the analysis. Most of these groups had a significant, positive correlation between the control trabecular bone volume and the iPTH‐treated bone volume (Table 1). Using both sexes was an exception. Although this does not suggest that those indices should not be used in future studies, caution should be taken if drawing conclusions based only on trabecular bone volume.
FIGURE 2.
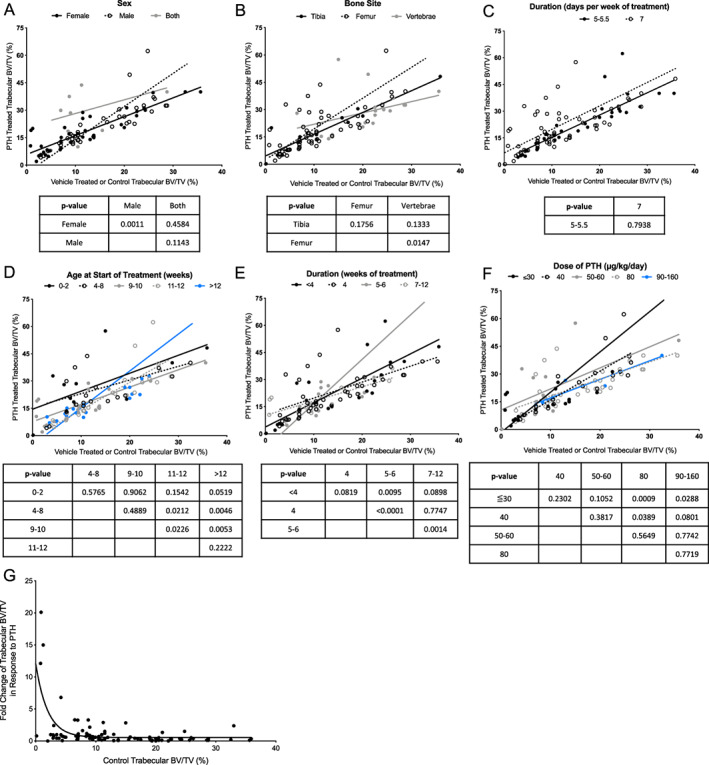
Trabecular bone response in WT mice. (A–F) Trabecular bone volume is graphed for vehicle‐treated (x axis) and PTH‐treated (y axis) WT mice. Each plot stratifies a different variable, including (A) sex, (B) bone site analyzed, (C) duration (days per week of treatment), (D) age at the start of treatment, (E) duration (weeks of treatment), or (F) dose of treatment. Linear regression of the slope was analyzed for each group and compared within a variable. The p values are reported in the charts under each graph, and correspond to the analysis between the column and row headers (i.e., in (A), the slope of the line for male and female has a p‐value of 0.0011). (G) Control trabecular bone volume in WT mice and the FC of trabecular bone volume in response to PTH in WT mice is plotted. The AIC is a statistical predictor of error between two models, and was used to confirm an inverse exponential relationship between control bone volume and the FC in bone volume with PTH in WT mice. Abbreviations: AIC, Akaike Information Criterion; PTH, parathyroid hormone; WT, wild‐type; FC, fold change.
Correlation graphs of the reported trabecular bone volume in control versus iPTH mice are shown in Figure 2 and are separated by the categories mentioned. In order to understand how the variables relate within a category, the data was modeled with a linear regression and the slopes and corresponding 95% confidence interval were compared. Groups that had a significant correlation are discussed in the Supplemental Material, but all of the data is presented. This data can be used to inform future study design and interpretation.
We hypothesized that if a mouse has a high baseline bone volume, there is less capacity to mount an anabolic response to iPTH. Similarly, if an animal has a low baseline bone volume, they would show a greater response to iPTH. Analysis of the graph in Figure 2G supports this, with the control bone volume plotted against the fold change response to PTH. Although biases exist because only studies that showed an anabolic response in wild‐type mice were included, statistics support an inverse exponential relationship between these variables. To confirm that the data had an exponential relationship, and not a linear one, we calculated the Akaike Information Criterion (AIC), a statistical predictor of error between two models. The AIC for the exponential model is 36.44 lower than the linear model, indicating that the exponential equation more precisely describes the relationship between the two variables.
Analysis of PTH anabolic actions in bone using gene‐targeted mice
The mechanism of anabolic iPTH and its effect on the bone microenvironment has been studied for decades, and numerous mechanisms have been proposed based on in vitro and in vivo models.( 67 , 68 , 69 ) A wide variety of genetic mouse models have been employed to elucidate the actions of PTH in bone over the past 20 years (Figures 1, 3, Table 2). With modern technology facilitating unprecedented genetic manipulation, this comprehensive study compiles the evidence of iPTH actions in gene‐targeted murine models. Of note, an important limitation is that although some mutations are global, many are focused on a subset of cells, and dependent on effective cre drivers and appropriate promoter selection. Hence the anabolic actions of PTH may reflect the effectiveness of the model as well as the targeted gene. Specific genotypes are indicated in Table 2, and are discussed in detail in the Supplemental Materials.
FIGURE 3.
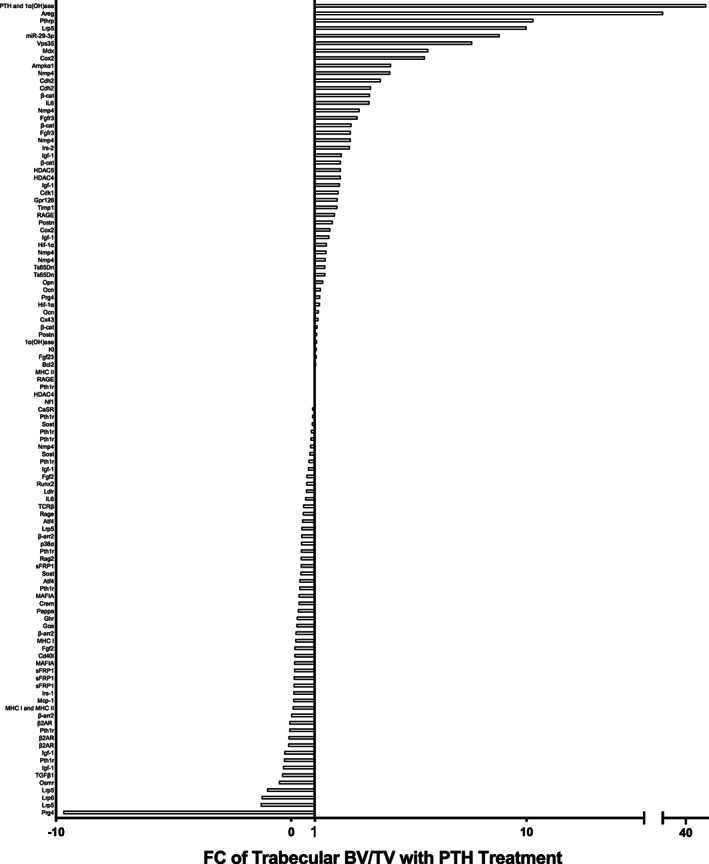
FC of PTH‐/control‐treated trabecular bone volume per total volume per targeted gene model. The response to PTH treatment in gene targeted murine models was calculated using the bone volume FC in mutant mice relative to the FC of control treated mice. The x axis lists the targeted gene. Some genes are listed multiple times, each of which represents a different study or cohort of animals listed in Table 2. If there was no change between control and genetically modified treated animals, the FC is 1, indicated by the marked line. Abbreviations: FC, fold change; PTH, parathyroid hormone.
The Supplemental Materials include detailed text descriptions of the literature using iPTH in gene‐targeted mice, which are summarized alphabetically by gene in Table 2. The models studied can be stratified by the function of the gene, including receptor activation and signaling pathways; downstream mediators in the fibroblast growth factor (FGF) family, wingless‐related integration site (Wnt) family, bone morphogenetic protein (BMP) family, insulin‐like growth factor (IGF), and growth hormone (GH), epidermal growth factor (EGF) family; and cell regulatory factors including apoptotic, immunity, extracellular matrix (ECM), cytoskeletal, and calcium regulation. The summation of these data demonstrated the gene deletions with the greatest increase in response to iPTH. These included PTH and 1‐α‐hydroxylase (Pth;1α(OH)ase, 62‐fold)( 70 ), amphiregulin (Areg, 15.8‐fold),( 17 ) and PTH‐related protein (Pthrp, 10.2‐fold).( 14 ) (Table 2). The deletions with the greatest inhibition of the anabolic response include deletions of: proteoglycan 4 (Prg4, −9.7‐fold),( 71 ) low‐density lipoprotein receptor‐related protein 6 (Lrp6, 1.3‐fold),( 64 ) and low‐density lipoprotein receptor‐related protein 5 (Lrp5, −1.0‐fold)( 63 ) (Table 2). Several notable genes demonstrated no alteration of the anabolic action of PTH, including major histocompatibility complex II knockout mice (Mhc II),( 72 ) bone sialoprotein (Bsp),( 28 ) and histone deacetylase 4 (Hdac4).( 50 ) The models with the most study were insulin‐like growth factor‐1 (Igf‐1).( 52 , 53 , 54 , 55 )
By detailing comparisons between reported iPTH studies, we are able to assimilate the role of different genes in the anabolic response. For example, Table 2 shows that mice with mutations in Igf‐1 can range in their response to iPTH, with bone volume fold changes relative to control mice from −0.3 ‐fold to 2.1‐fold.( 52 , 53 , 54 , 55 , 56 ) There has been long‐standing interest in this gene; it was the first genetic model to be studied with iPTH in 2001 because of the increase in IGF‐1 production from osteoblasts in response to PTH.( 52 ) A detailed analysis in the Supplemental Material compares the study design, mouse genetics, and conclusions of each report. These studies support a necessary role of IGF‐1 in the anabolic response, as well as downstream targets, such as insulin receptor substrate‐1 (IRS‐1).( 59 )
DISCUSSION
When mice are administered anabolic doses of PTH, signaling cascades affect proliferation and development of osteoblasts. There are many protein interactions and regulatory factors involved in this process, and it is unsurprising that when they are disrupted, the anabolic response does not achieve its full potential. The purpose of this study was to further elucidate PTH mechanisms by collectively analyzing the extensive work performed using mouse models.
The anabolic response in wild‐type mice was analyzed to understand baseline differences and influences. Of the variables analyzed, the greatest responses to iPTH were in male mice, with treatment starting later than 12 weeks of age, a treatment duration lasting 5 to 6 weeks, and a PTH dose of 30 to 60 μg/kg/day. This data should be used to inform future study design for efficient use of resources. For example, based on the correlation data, male and female mice should be analyzed separately when treated with iPTH.
Collectively, the data suggests that starting treatment at greater than 12 weeks of age yields the highest response to iPTH. Mice are considered mature adults at this stage, but peak bone mass is closer to 16 to 18 weeks. The murine skeleton continues to grow past sexual maturity (about 7 weeks), whereas the human skeleton does not. PTH is commonly prescribed in postmenopausal women, and this population would be more comparative to mice that are at least 12 months old. Of the more than 130 cohorts of mice studied, only one was in this age range.( 25 )
Administering PTH for at least 5 days per week is sufficient to yield an anabolic response. Although it is well documented that whereas continuous PTH is catabolic, iPTH is anabolic,( 73 ) this analysis has focused on the anabolic studies. Frolik et al.( 74 ) used a rat model to determine that the pharmacokinetics of PTH(1‐34) varies with differing treatment regimens. They found giving the same 80 μg/kg of PTH in a single injection or via six injections over 1 h resulted in an anabolic response. However, administering the same 80 μg/kg of PTH over 6 or 8 h produced a catabolic response. They associated the anabolic iPTH in a temporal manner with the rapid increase in serum calcium, followed by tapering.
Analyses for this examination focused on the tibias, femurs, and vertebrae. Although studies analyzing calvariae are reported in Table 2, there were not enough to include in the correlation analysis. In humans, bone mineral density in postmenopausal women that were randomly assigned to PTH or placebo showed a larger percent change in the lumbar spine than femoral neck.( 7 ) Of note, this is comparing different outcomes (bone volume for murine studies and bone mineral density for human), measured by different variables, and in a quadrupedal versus a bipedal species.
Relative to specific genetic aberrations that may inform PTH mechanisms, several trends are apparent from this analysis of more than 90 gene‐targeted studies. Bone health and energy metabolism are linked formulating a vital area of research interest. Many clinical conditions are also linked to altered energy expenditure, as reviewed by Motyl et al.( 75 ) Among these targeted murine models with the largest increases in anabolic response to iPTH were AMP‐activated protein kinase α1 (Ampkα1), hypoxia‐inducible factor 1‐alpha (Hif‐1α), and cyclooxygenase‐2 (Cox2). Ampkα1 regulates energy consumption in the cell, working to promote adenosine triphosphate (ATP) conservation or expenditure depending on current conditions.( 76 ) Mice lacking Ampkα1 have a low bone mass with an increased anabolic response to iPTH.( 16 ) Hif‐1α is referred to as the master regulator of hypoxia because it is an oxygen‐sensitive subunit of the Hif‐1 complex (with Hif‐1β). When oxygen is not present, Hif‐1α is stabilized and translocated to the nucleus to bind to hypoxia‐response elements.( 77 ) Cox2 has been identified as a hypoxia responsive gene in colorectal cancer.( 78 ) Authors of the work with Cox2 and iPTH were interested in its role regulating prostaglandin production, but it is possible that part of the effect of deleting this gene is affected by changes in energy metabolism. When these genes are deleted, the responsiveness to iPTH in bone is enhanced. Because these genes are activated when the cell is under metabolic stress and their actions limit the PTH response, it is conceivable that they allow the cell to work at the capacity allowed by current energy conditions, limited by oxygen concentrations.
Ampkα1 and Hif‐1α both regulate autophagy.( 79 , 80 ) PTH prevents osteoblast apoptosis, prolonging the life of these cells.( 81 ) It is also possible that in the absence of these genes, cell survival is further enhanced, leading to an increased response to iPTH. A presentation at the American Society for Bone and Mineral Research Annual Meeting in 2019 further connected autophagy and PTH mechanisms.( 82 ) Using mice that had autophagy‐deficient osteoblasts (Fip200 flox/flox; Osterix–cyclic recombinase [Osx‐cre]), Qi et al.( 82 ) showed a blunted anabolic response. Taken together, the evidence supports a relationship between autophagy and iPTH.
Canonical Wnt signaling promotes osteoblast expansion and function. Soluble ligands bind to the receptors (including LRPs) that induce stabilization of β catenin (β‐cat), allowing it to translocate to the nucleus and alter gene expression.( 83 ) In mice with mutations in Lrp6 and β‐cat, there were similar anabolic responses to PTH (vertebrae and femur when β‐cat deletion was under control of dentin matrix acidic phosphoprotein 1 [DMP1], and in the vertebrae when under control of Osx). Other Wnt family member proteins have been studied with iPTH, and it is clear that this pathway is critical for its anabolic effects in bone. N‐cadherin restrains Wnt signaling and bone formation in osteoblasts.( 84 ) Interestingly, when the gene for N‐cadherin, Cdh2, is disrupted, the anabolic response to iPTH is increased. When both positive and negative regulators of Wnts are affected, the response to iPTH increases, suggesting anabolic PTH is sensitive to slight changes in Wnts.
N‐cadherin may affect PTH responsiveness through other mechanisms as well. Expression of Cdh2 is increased with maturity of osteoblasts and decreased expression is associated with osteosarcoma.( 85 , 86 ) N‐cadherin mediates cell‐to‐cell adhesion, highlighting the effect of interaction with the microenvironment on osteoblasts. Mdx mice have a mutation in dystrophin, a protein that also helps osteoblasts interact with their environment by connecting the cytoplasm to the extracellular matrix in a complex. Disruption in dystrophin function increases the anabolic response to iPTH. Both N‐cadherin and dystrophin are affected by calcium. N‐cadherin is a calcium dependent glycoprotein, whereas Mdx mice exhibit increased intracellular calcium levels.( 87 ) It is possible that these changes in calcium regulation alter responsiveness to iPTH.
This work summarizes decades of work aimed to outline the mechanisms of anabolic iPTH, with more studies surely forthcoming. The reports described highlight the importance of many cell types in the bone microenvironment. Signaling starts in the osteoblast, depends on intracellular second messengers, and is then affected by/affects microenvironmental cues and other organ systems, formulating a complex and dynamic process that results in bone formation and bone accrual. The insights from the analysis of the pooled data provide better direction for future experiments and appropriate interpretation.
DISCLOSURES
Laura E. Zweifler, Amy J. Koh, and Stephanie Daignault‐Newton have no disclosures. Laurie K. McCauley owns Amgen Stock.
AUTHOR CONTRIBUTIONS
Study design: Laura E. Zweifler and Laurie K. McCauley. Data collection: Laura E. Zweifler and Amy J. Koh. Data analysis: Laura E. Zweifler, Amy J. Koh, and Stephanie Daignault‐Newton. Data interpretation: Laura E. Zweifler, Stephanie Daignault‐Newton, Laurie K. McCauley. Drafting manuscript: Laura E. Zweifler. Revising manuscript content: Laura E. Zweifler and Laurie K. McCauley. Approving final version of manuscript: Laura E. Zweifler, Amy J. Koh, Stephanie Daignault‐Newton, and Laurie K. McCauley. Laurie K. McCauley takes responsibility for the integrity of the data analysis.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/jbmr.4389.
Supporting information
Appendix S1 Supplemental Material
ACKNOWLEDGMENTS
We acknowledge the National Institute of Dental and Craniofacial Research (NIDCR, DE028455 to Laura E. Zweifler, DE022327 to Laurie K. McCauley), the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK, DK053904 to Laurie K. McCauley), and the National Cancer Institute (NCI, CA093900 to Stephanie Daignault‐Newton), of the National Institutes of Health (NIH) for funding support. We thank the Consulting for Statistics, Computing, and Analytics Research (CSCAR) team at the University of Michigan for their advice.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Shirley M. Abaloparatide: first global approval. Drugs. 2017;77(12):1363‐1368. [DOI] [PubMed] [Google Scholar]
- 2. Burrows RB. Variations produced in bones of growing rats by parathyroid extracts. Am J Anat. 1938;62(2):237‐290. [Google Scholar]
- 3. Pugsley LI. The effect of parathyroid hormone and of irradiated ergosterol on calcium and phosphorous metabolism in the rat. J Physiol. 1932;76(3):315‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pugsley LI, Selye H. The histological changes in the bone responsible for the action of parathyroid hormone on the calcium metabolism of the rat. J Physiol. 1933;79(1):113‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bauer W, Aub JC, Albright F. Studies of calcium and phosphorus metabolism: V. A study of the bone trabeculae as a readily available reserve supply of calcium. J Exp Med. 1929;49(1):145‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lindsay R, Nieves J, Formica C, et al. Randomised controlled study of effect of parathyroid hormone on vertebral‐bone mass and fracture incidence among postmenopausal women on oestrogen with osteoporosis. Lancet. 1997;350(9077):550‐555. [DOI] [PubMed] [Google Scholar]
- 7. Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1‐34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344(19):1434‐1441. [DOI] [PubMed] [Google Scholar]
- 8. Juppner H, Abou‐Samra AB, Freeman M, et al. A G protein‐linked receptor for parathyroid hormone and parathyroid hormone‐related peptide. Science. 1991;254(5034):1024‐1026. [DOI] [PubMed] [Google Scholar]
- 9. Datta NS, Abou‐Samra AB. PTH and PTHrP signaling in osteoblasts. Cell Signal. 2009;21(8):1245‐1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McSheehy PM, Chambers TJ. Osteoblast‐like cells in the presence of parathyroid hormone release soluble factor that stimulates osteoclastic bone resorption. Endocrinology. 1986;119(4):1654‐1659. [DOI] [PubMed] [Google Scholar]
- 11. Jilka RL, O'Brien CA, Bartell SM, Weinstein RS, Manolagas SC. Continuous elevation of PTH increases the number of osteoblasts via both osteoclast‐dependent and ‐independent mechanisms. J Bone Miner Res. 2010;25(11):2427‐2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miao D, He B, Karaplis AC, Goltzman D. Parathyroid hormone is essential for normal fetal bone formation. J Clin Invest. 2002;109(9):1173‐1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miao D, Li J, Xue Y, Su H, Karaplis AC, Goltzman D. Parathyroid hormone‐related peptide is required for increased trabecular bone volume in parathyroid hormone‐null mice. Endocrinology. 2004;145(8):3554‐3562. [DOI] [PubMed] [Google Scholar]
- 14. Miao D, He B, Jiang Y, et al. Osteoblast‐derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1‐34. J Clin Invest. 2005;115(9):2402‐2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Samadfam R, Xia Q, Miao D, Hendy GN, Goltzman D. Exogenous PTH and endogenous 1,25‐dihydroxyvitamin D are complementary in inducing an anabolic effect on bone. J Bone Miner Res. 2008;23(8):1257‐1266. [DOI] [PubMed] [Google Scholar]
- 16. Jeyabalan J, Shah M, Viollet B, et al. Mice lacking AMP‐activated protein kinase α1 catalytic subunit have increased bone remodelling and modified skeletal responses to hormonal challenges induced by ovariectomy and intermittent PTH treatment. J Endocrinol. 2012;214(3):349‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jay FF, Vaidya M, Porada SM, Andrukhova O, Schneider MR, Erben RG. Amphiregulin lacks an essential role for the bone anabolic action of parathyroid hormone. Mol Cell Endocrinol. 2015;417:158‐165. [DOI] [PubMed] [Google Scholar]
- 18. Yu S, Franceschi RT, Luo M, et al. Critical role of activating transcription factor 4 in the anabolic actions of parathyroid hormone in bone. PLoS One. 2009;4(10):e7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yamashita J, Datta NS, Chun YH, et al. Role of Bcl2 in osteoclastogenesis and PTH anabolic actions in bone. J Bone Miner Res. 2009;23(5):621‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nagase Y, Iwasawa M, Akiyama T, et al. Antiapoptotic molecule Bcl‐2 is essential for the anabolic activity of parathyroid hormone in bone. Ann N Y Acad Sci. 2010;1192:330‐337. [DOI] [PubMed] [Google Scholar]
- 21. Ferrari SL, Pierroz DD, Glatt V, et al. Bone response to intermittent parathyroid hormone is altered in mice null for β‐arrestin2. Endocrinology. 2005;146(4):1854‐1862. [DOI] [PubMed] [Google Scholar]
- 22. Gesty‐Palmer D, Flannery P, Yuan L, et al. A β‐arrestin‐biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci Transl Med. 2009;1(1):1ra. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kedlaya R, Kang KS, Hong JM, et al. Adult‐onset deletion of β‐catenin in (10kb) Dmp1‐expressing cells prevents intermittent PTH‐induced bone gain. Endocrinology. 2016;157(8):3047‐3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yu C, Xuan M, Zhang M, et al. Postnatal deletion of β‐catenin in osterix‐expressing cells is necessary for bone growth and intermittent PTH‐induced bone gain. J Bone Miner Metab. 2018;36(5):560‐572. [DOI] [PubMed] [Google Scholar]
- 25. Hanyu R, Wehbi VL, Hayata T, et al. Anabolic action of parathyroid hormone regulated by the β2‐adrenergic receptor. Proc Natl Acad Sci U S A. 2012;109(19):7433‐7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu R, Wang Q, Han Y, Li J, Yang XJ, Miao D. Parathyroid hormone administration improves bone marrow microenvironment and partially rescues haematopoietic defects in Bmi1‐null mice. PLoS One. 2014;9(4):e93864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Khan MP, Khan K, Yadav PS, et al. BMP signaling is required for adult skeletal homeostasis and mediates bone anabolic action of parathyroid hormone. Bone. 2016;92:132‐144. [DOI] [PubMed] [Google Scholar]
- 28. Bouleftour W, Bouet G, Granito RN, et al. Blocking the expression of both bone sialoprotein (BSP) and osteopontin (OPN) impairs the anabolic action of PTH in mouse calvaria bone. J Cell Physiol. 2015;230(3):568‐577. [DOI] [PubMed] [Google Scholar]
- 29. Cho SW, Soki FN, Koh AJ, et al. Osteal macrophages support physiologic skeletal remodeling and anabolic actions of parathyroid hormone in bone. Proc Natl Acad Sci U S A. 2014;111(4):1545‐1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Demiralp B, Chen HL, Koh AJ, Keller ET, McCauley LK. Anabolic actions of parathyroid hormone during bone growth are dependent on c‐fos. Endocrinology. 2002;143(10):4038‐4047. [DOI] [PubMed] [Google Scholar]
- 31. Al‐Dujaili SA, Koh AJ, Dang M, et al. Calcium sensing receptor function supports osteoblast survival and acts as a co‐factor in PTH anabolic actions in bone. J Cell Biochem. 2016;117(7):1556‐1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robinson JW, Li JY, Walker LD, et al. T cell‐expressed CD40L potentiates the bone anabolic activity of intermittent PTH treatment. J Bone Miner Res. 2015;30(4):695‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Revollo L, Kading J, Jeong SY, et al. N‐cadherin restrains PTH activation of Lrp6/ β‐catenin signaling and osteoanabolic action. J Bone Miner Res. 2015;30(2):274‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang H, Dong J, Xiong W, Fang Z, Guan H, Li F. N‐cadherin restrains PTH repressive effects on sclerostin/SOST by regulating LRP6‐PTH1R interaction. Ann N Y Acad Sci. 2016;1385(1):41‐52. [DOI] [PubMed] [Google Scholar]
- 35. Takahashi A, Mulati M, Saito M, et al. Loss of cyclin‐dependent kinase 1 impairs bone formation, but does not affect the bone‐anabolic effects of parathyroid hormone. J Biol Chem. 2018;293(50):19387‐19399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu M, Choudhary S, Voznesensky O, et al. Basal bone phenotype and increased anabolic responses to intermittent parathyroid hormone in healthy male COX‐2 knockout mice. Bone. 2010;47(2):341‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu F, Lee SK, Adams DJ, Gronowicz GA, Kream BE. CREM deficiency in mice alters the response of bone to intermittent parathyroid hormone treatment. Bone. 2007;40(4):1135‐1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pacheco‐Costa R, Davis HM, Sorenson C, et al. Defective cancellous bone structure and abnormal response to PTH in cortical bone of mice lacking Cx43 cytoplasmic C‐terminus domain. Bone. 2015;81:632‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yao GQ, Wu JJ, Troiano N, Insogna K. Targeted overexpression of Dkk1 in osteoblasts reduces bone mass but does not impair the anabolic response to intermittent PTH treatment in mice. J Bone Miner Metab. 2011;29(2):141‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schneider MR, Dahlhoff M, Andrukhova O, et al. Normal epidermal growth factor receptor signaling is dispensable for bone anabolic effects of parathyroid hormone. Bone. 2012;50(1):237‐244. [DOI] [PubMed] [Google Scholar]
- 41. Hurley MM, Okada Y, Xiao L, et al. Impaired bone anabolic response to parathyroid hormone in Fgf2−/− and Fgf2+/− mice. Biochem Biophys Res Commun. 2006;341(4):989‐994. [DOI] [PubMed] [Google Scholar]
- 42. Xiao L, Fei Y, Hurley MM. FGF2 crosstalk with Wnt signaling in mediating the anabolic action of PTH on bone formation. Bone Rep. 2018;9:136‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yuan Q, Sato T, Densmore M, et al. FGF‐23/Klotho signaling is not essential for the phosphaturic and anabolic functions of PTH. J Bone Miner Res. 2011;26(9):2026‐2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xie Y, Yi L, Weng T, et al. Fibroblast growth factor receptor 3 deficiency does not impair the osteoanabolic action of parathyroid hormone on mice. Int J Biol Sci. 2016;12(8):990‐999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen H, Sun X, Yin L, et al. PTH 1‐34 ameliorates the osteopenia and delayed healing of stabilized tibia fracture in mice with achondroplasia resulting from gain‐of‐function mutation of FGFR3. Int J Biol Sci. 2017;13(10):1254‐1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu Z, Kennedy OD, Cardoso L, et al. DMP‐1‐mediated Ghr gene recombination compromises skeletal development and impairs skeletal response to intermittent PTH. FASEB J. 2016;30(2):635‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sun P, He L, Jia K, et al. Regulation of body length and bone mass by Gpr126/Adgrg6. Sci Adv. 2020;6(12):eaaz0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang L, Quarles LD, Spurney RF. Unmasking the osteoinductive effects of a G‐protein‐coupled receptor (GPCR) kinase (GRK) inhibitor by treatment with PTH(1‐34). J Bone Miner Res. 2004;19(10):1661‐1670. [DOI] [PubMed] [Google Scholar]
- 49. Sinha P, Aarnisalo P, Chubb R, et al. Loss of Gsα in the postnatal skeleton leads to low bone mass and a blunted response to anabolic parathyroid hormone therapy. J Biol Chem. 2016;291(4):1631‐1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wein MN, Liang Y, Goransson O, et al. SIKs control osteocyte responses to parathyroid hormone. Nat Commun. 2016;7:13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Frey JL, Stonko DP, Faugere MC, Riddle RC. Hypoxia‐inducible factor‐1α restricts the anabolic actions of parathyroid hormone. Bone Res. 2014;2:14005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Miyakoshi N, Kasukawa Y, Linkhart TA, Baylink DJ, Mohan S. Evidence that anabolic effects of PTH on bone require IGF‐I in growing mice. Endocrinology. 2001;142(10):4349‐4356. [DOI] [PubMed] [Google Scholar]
- 53. Rosen CJ, Ackert‐Bicknell C, Beamer WG, et al. Allelic differences in a quantitative trait locus affecting insulin‐like growth factor‐I impact skeletal acquisition and body composition. Pediatr Nephrol. 2005;20(3):255‐260. [DOI] [PubMed] [Google Scholar]
- 54. Yakar S, Bouxsein ML, Canalis E, et al. The ternary IGF complex influences postnatal bone acquisition and the skeletal response to intermittent parathyroid hormone. J Endocrinol. 2006;189(2):289‐299. [DOI] [PubMed] [Google Scholar]
- 55. Elis S, Courtland HW, Wu Y, et al. Elevated serum IGF‐1 levels synergize PTH action on the skeleton only when the tissue IGF‐1 axis is intact. J Bone Miner Res. 2010;25(9):2051‐2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang Y, Menendez A, Fong C, ElAlieh HZ, Chang W, Bikle DD. Ephrin B2/EphB4 mediates the actions of IGF‐I signaling in regulating endochondral bone formation. J Bone Miner Res. 2014;29(8):1900‐1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Raggatt LJ, Qin L, Tamasi J, et al. Interleukin‐18 is regulated by parathyroid hormone and is required for its bone anabolic actions. J Biol Chem. 2008;283(11):6790‐6798. [DOI] [PubMed] [Google Scholar]
- 58. Cho SW, Pirih FQ, Koh AJ, et al. The soluble interleukin‐6 receptor is a mediator of hematopoietic and skeletal actions of parathyroid hormone. J Biol Chem. 2013;288(10):6814‐6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yamaguchi M, Ogata N, Shinoda Y, et al. Insulin receptor substrate‐1 is required for bone anabolic function of parathyroid hormone in mice. Endocrinology. 2005;146(6):2620‐2628. [DOI] [PubMed] [Google Scholar]
- 60. Huang MS, Lu J, Ivanov Y, et al. Hyperlipidemia impairs osteoanabolic effects of PTH. J Bone Miner Res. 2008;23(10):1672‐1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li X, Garcia J, Lu J, et al. Roles of parathyroid hormone (PTH) receptor and reactive oxygen species in hyperlipidemia‐induced PTH resistance in preosteoblasts. J Cell Biochem. 2014;115(1):179‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sawakami K, Robling AG, Ai M, et al. The Wnt co‐receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J Biol Chem. 2006;281(33):23698‐23711. [DOI] [PubMed] [Google Scholar]
- 63. Iwaniec UT, Wronski TJ, Liu J, et al. PTH stimulates bone formation in mice deficient in Lrp5. J Bone Miner Res. 2007;22(3):394‐402. [DOI] [PubMed] [Google Scholar]
- 64. Li C, Xing Q, Yu B, et al. Disruption of LRP6 in osteoblasts blunts the bone anabolic activity of PTH. J Bone Miner Res. 2013;28(10):2094‐2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li C, Wang W, Xie L, Luo X, Cao X, Wan M. Lipoprotein receptor‐related protein 6 is required for parathyroid hormone‐induced Sost suppression. Ann N Y Acad Sci. 2016;1364:62‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tamasi JA, Vasilov A, Shimizu E, et al. Monocyte chemoattractant protein‐1 is a mediator of the anabolic action of parathyroid hormone on bone. J Bone Miner Res. 2013;28(9):1975‐1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rodan GA, Martin TJ. Role of osteoblasts in hormonal control of bone resorption—a hypothesis. Calcif Tissue Int. 1981;33(4):349‐351. [DOI] [PubMed] [Google Scholar]
- 68. Jones SJ, Boyde A. Experimental study of changes in osteoblastic shape induced by calcitonin and parathyroid extract in an organ culture system. Cell Tissue Res. 1976;169(4):449‐465. [DOI] [PubMed] [Google Scholar]
- 69. Parfitt AM. The actions of parathyroid hormone on bone: relation to bone remodeling and turnover, calcium homeostasis, and metabolic bone diseases. Part II. PTH and bone cells: bone turnover and plasma calcium regulation. Metabolism. 1976;25(8):909‐955. [DOI] [PubMed] [Google Scholar]
- 70. Xue Y, Karaplis AC, Hendy GN, Goltzman D, Miao D. Genetic models show that parathyroid hormone and 1,25‐dihydroxyvitamin D3 play distinct and synergistic roles in postnatal mineral ion homeostasis and skeletal development. Hum Mol Genet. 2005;14(11):1515‐1528. [DOI] [PubMed] [Google Scholar]
- 71. Novince CM, Michalski MN, Koh AJ, et al. Proteoglycan 4: a dynamic regulator of skeletogenesis and parathyroid hormone skeletal anabolism. J Bone Miner Res. 2012;27(1):11‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Terauchi M, Li JY, Bedi B, et al. T lymphocytes amplify the anabolic activity of parathyroid hormone through Wnt10b signaling. Cell Metab. 2009;10(3):229‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dobnig H, Turner RT. The effects of programmed administration of human parathyroid hormone fragment (1‐34) on bone histomorphometry and serum chemistry in rats. Endocrinology. 1997;138(11):4607‐4612. [DOI] [PubMed] [Google Scholar]
- 74. Frolik CA, Black EC, Cain RL, et al. Anabolic and catabolic bone effects of human parathyroid hormone (1‐34) are predicted by duration of hormone exposure. Bone. 2003;33(3):372‐379. [DOI] [PubMed] [Google Scholar]
- 75. Motyl KJ, Guntur AR, Carvalho AL, Rosen CJ. Energy metabolism of bone. Toxicol Pathol. 2017;45(7):887‐893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hardie DG. AMP‐activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25(18):1895‐1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jiang BH, Zheng JZ, Leung SW, Roe R, Semenza GL. Transactivation and inhibitory domains of hypoxia‐inducible factor 1α. Modulation of transcriptional activity by oxygen tension. J Biol Chem. 1997;272(31):19253‐19260. [DOI] [PubMed] [Google Scholar]
- 78. Kaidi A, Qualtrough D, Williams AC, Paraskeva C. Direct transcriptional up‐regulation of cyclooxygenase‐2 by hypoxia‐inducible factor (HIF)‐1 promotes colorectal tumor cell survival and enhances HIF‐1 transcriptional activity during hypoxia. Cancer Res. 2006;66(13):6683‐6691. [DOI] [PubMed] [Google Scholar]
- 79. Zhang H, Bosch‐Marce M, Shimoda LA, et al. Mitochondrial autophagy is an HIF‐1‐dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283(16):10892‐10903. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 80. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest. 1999;104(4):439‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Qi SQ, Wang L, Choi HK, et al. Fip200, an essential autophagy gene, mediates the anabolic action of PTH in bone. J Bone Miner Res. 2019;34(32):22‐23.30536424 [Google Scholar]
- 83. Westendorf JJ, Kahler RA, Schroeder TM. Wnt signaling in osteoblasts and bone diseases. Gene. 2004;341:19‐39. [DOI] [PubMed] [Google Scholar]
- 84. Hay E, Laplantine E, Geoffroy V, et al. N‐cadherin interacts with axin and LRP5 to negatively regulate Wnt/β‐catenin signaling, osteoblast function, and bone formation. Mol Cell Biol. 2009;29(4):953‐964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ferrari SL, Traianedes K, Thorne M, et al. A role for N‐cadherin in the development of the differentiated osteoblastic phenotype. J Bone Miner Res. 2000;15(2):198‐208. [DOI] [PubMed] [Google Scholar]
- 86. Marie PJ. Role of N‐cadherin in bone formation. J Cell Physiol. 2002;190(3):297‐305. [DOI] [PubMed] [Google Scholar]
- 87. Morgenroth VH, Hache LP, Clemens PR. Insights into bone health in Duchenne muscular dystrophy. Bonekey Rep. 2012;1:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gray SK, McGee‐Lawrence ME, Sanders JL, Condon KW, Tsai CJ, Donahue SW. Black bear parathyroid hormone has greater anabolic effects on trabecular bone in dystrophin‐deficient mice than in wild type mice. Bone. 2012;51(3):578‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Michalski MN, Seydel AL, Siismets EM, et al. Inflammatory bone loss associated with MFG‐E8 deficiency is rescued by teriparatide. FASEB J. 2018;32(7):3730‐3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hrdlicka HC, Pereira RC, Shin B, et al. Inhibition of miR‐29‐3p isoforms via tough decoy suppresses osteoblast function in homeostasis but promotes intermittent parathyroid hormone‐induced bone anabolism. Bone. 2021;143:115779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mahalingam CD, Datta T, Patil RV, et al. Mitogen‐activated protein kinase phosphatase 1 regulates bone mass, osteoblast gene expression, and responsiveness to parathyroid hormone. J Endocrinol. 2011;211(2):145‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yu X, Milas J, Watanabe N, et al. Neurofibromatosis type 1 gene haploinsufficiency reduces AP‐1 gene expression without abrogating the anabolic effect of parathyroid hormone. Calcif Tissue Int. 2006;78(3):162‐170. [DOI] [PubMed] [Google Scholar]
- 93. Robling AG, Childress P, Yu J, et al. Nmp4/CIZ suppresses parathyroid hormone‐induced increases in trabecular bone. J Cell Physiol. 2009;219(3):734‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Childress P, Philip BK, Robling AG, et al. Nmp4/CIZ suppresses the response of bone to anabolic parathyroid hormone by regulating both osteoblasts and osteoclasts. Calcif Tissue Int. 2011;89(1):74‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. He Y, Childress P, Hood M Jr, et al. Nmp4/CIZ suppresses the parathyroid hormone anabolic window by restricting mesenchymal stem cell and osteoprogenitor frequency. Stem Cells Dev. 2013;22(3):492‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Machado do Reis L, Kessler CB, Adams DJ, Lorenzo J, Jorgetti V, Delany AM. Accentuated osteoclastic response to parathyroid hormone undermines bone mass acquisition in osteonectin‐null mice. Bone. 2008;43(2):264‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kitahara K, Ishijima M, Rittling SR, et al. Osteopontin deficiency induces parathyroid hormone enhancement of cortical bone formation. Endocrinology. 2003;144(5):2132‐2140. [DOI] [PubMed] [Google Scholar]
- 98. Walker EC, Poulton IJ, McGregor NE, et al. Sustained RANKL response to parathyroid hormone in oncostatin M receptor‐deficient osteoblasts converts anabolic treatment to a catabolic effect in vivo. J Bone Miner Res. 2012;27(4):902‐912. [DOI] [PubMed] [Google Scholar]
- 99. Thouverey C, Caverzasio J. Suppression of p38α MAPK signaling in osteoblast lineage cells impairs bone anabolic action of parathyroid hormone. J Bone Miner Res. 2016;31(5):985‐993. [DOI] [PubMed] [Google Scholar]
- 100. Clifton KB, Conover CA. Pregnancy‐associated plasma protein‐A modulates the anabolic effects of parathyroid hormone in mouse bone. Bone. 2015;81:413‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Yorgan TA, Sari H, Rolvien T, et al. Mice lacking plastin‐3 display a specific defect of cortical bone acquisition. Bone. 2020;130:115062. [DOI] [PubMed] [Google Scholar]
- 102. Bonnet N, Conway SJ, Ferrari SL. Regulation of β catenin signaling and parathyroid hormone anabolic effects in bone by the matricellular protein periostin. Proc Natl Acad Sci U S A. 2012;109(37):15048‐15053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Xue Y, Zhang Z, Karaplis AC, Hendy GN, Goltzman D, Miao D. Exogenous PTH‐related protein and PTH improve mineral and skeletal status in 25‐hydroxyvitamin D‐1α‐hydroxylase and PTH double knockout mice. J Bone Miner Res. 2005;20(10):1766‐1777. [DOI] [PubMed] [Google Scholar]
- 104. Bedi B, Li JY, Tawfeek H, et al. Silencing of parathyroid hormone (PTH) receptor 1 in T cells blunts the bone anabolic activity of PTH. Proc Natl Acad Sci U S A. 2012;109(12):E725‐E733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Datta NS, Samra TA, Abou‐Samra AB. Parathyroid hormone induces bone formation in phosphorylation‐deficient PTHR1 knockin mice. Am J Physiol Endocrinol Metab. 2012;302(10):E1183‐E1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Saini V, Marengi DA, Barry KJ, et al. Parathyroid hormone (PTH)/PTH‐related peptide type 1 receptor (PPR) signaling in osteocytes regulates anabolic and catabolic skeletal responses to PTH. J Biol Chem. 2013;288(28):20122‐20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Delgado‐Calle J, Tu X, Pacheco‐Costa R, et al. Control of bone anabolism in response to mechanical loading and PTH by distinct mechanisms downstream of the PTH receptor. J Bone Miner Res. 2017;32(3):522‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Huck K, Sens C, Wuerfel C, Zoeller C, Nakchbandi IA. The Rho GTPase RAC1 in osteoblasts controls their function. Int J Mol Sci. 2020;21(2):385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kawano T, Troiano N, Adams DJ, Wu JJ, Sun BH, Insogna K. The anabolic response to parathyroid hormone is augmented in Rac2 knockout mice. Endocrinology. 2008;149(8):4009‐4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Philip BK, Childress PJ, Robling AG, et al. RAGE supports parathyroid hormone‐induced gains in femoral trabecular bone. Am J Physiol Endocrinol Metab. 2010;298(3):E714‐E725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Merciris D, Marty C, Collet C, de Vernejoul MC, Geoffroy V. Overexpression of the transcriptional factor Runx2 in osteoblasts abolishes the anabolic effect of parathyroid hormone in vivo. Am J Pathol. 2007;170(5):1676‐1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Bodine PV, Seestaller‐Wehr L, Kharode YP, Bex FJ, Komm BS. Bone anabolic effects of parathyroid hormone are blunted by deletion of the Wnt antagonist secreted frizzled‐related protein‐1. J Cell Physiol. 2007;210(2):352‐357. [DOI] [PubMed] [Google Scholar]
- 113. Yao W, Cheng Z, Shahnazari M, Dai W, Johnson ML, Lane NE. Overexpression of secreted frizzled‐related protein 1 inhibits bone formation and attenuates parathyroid hormone bone anabolic effects. J Bone Miner Res. 2010;25(2):190‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kramer I, Loots GG, Studer A, Keller H, Kneissel M. Parathyroid hormone (PTH)‐induced bone gain is blunted in SOST overexpressing and deficient mice. J Bone Miner Res. 2010;25(2):178‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Robling AG, Kedlaya R, Ellis SN, et al. Anabolic and catabolic regimens of human parathyroid hormone 1‐34 elicit bone‐ and envelope‐specific attenuation of skeletal effects in Sost‐deficient mice. Endocrinology. 2011;152(8):2963‐2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wu X, Pang L, Lei W, et al. Inhibition of Sca‐1‐positive skeletal stem cell recruitment by alendronate blunts the anabolic effects of parathyroid hormone on bone remodeling. Cell Stem Cell. 2010;7(5):571‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Saito H, Gasser A, Bolamperti S, et al. TG‐interacting factor 1 (Tgif1)‐deficiency attenuates bone remodeling and blunts the anabolic response to parathyroid hormone. Nat Commun. 2019;10(1):1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Merciris D, Schiltz C, Legoupil N, Marty‐Morieux C, de Vernejoul MC, Geoffroy V. Over‐expression of TIMP‐1 in osteoblasts increases the anabolic response to PTH. Bone. 2007;40(1):75‐83. [DOI] [PubMed] [Google Scholar]
- 119. Fowler TW, McKelvey KD, Akel NS, et al. Low bone turnover and low BMD in Down syndrome: effect of intermittent PTH treatment. PLoS One. 2012;7(8):e42967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Xiong L, Xia WF, Tang FL, Pan JX, Mei L, Xiong WC. Retromer in osteoblasts interacts with protein phosphatase 1 regulator subunit 14C, terminates parathyroid hormone's signaling, and promotes its catabolic response. EBioMedicine. 2016;9:45‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Yorgan TA, Rolvien T, Sturznickel J, et al. Mice carrying a ubiquitous R235W mutation of Wnt1 display a bone‐specific phenotype. J Bone Miner Res. 2020;35(9):1726‐1737. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supplemental Material
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.