

Abstract
Purpose:
To report the association of procollagen-lysine 2-oxoglutarate 5-dioxygenase 2 (PLOD2) mutations with bilateral primary congenital glaucoma (PCG) in monozygotic twins and with nondominant juvenile-onset primary open-angle glaucoma (JOAG).
Methods:
We utilized family-based whole-exome sequencing to detect disease-causing mutations in a pair of monozygotic twins with de-novo PCG and compared its existence in 50 nonfamilial cases of JOAG and 30 healthy controls. To validate the identified mutations, direct Sanger sequencing was performed. For further evaluation of gene expression in the ocular tissues, we performed whole-mount in situ hybridization in zebrafish embryos.
Results:
We identified a novel missense mutation (c.1925A>G, p.Tyr642Cys) in the PLOD2 gene in the monozygotic twin pair with PCG and another missense mutation (c.1880G>A, p.Arg627Gln) in one JOAG patient. Both mutations identified were heterozygous. Neither the parents of the twins nor the parents of the JOAG patient harbored the mutation and it was probably a de-novo change. The zebrafish in situ hybridization revealed expression of the PLOD2 gene during embryogenesis of the eye.
Conclusion:
We observed an association of PLOD2 mutations with PCG and with nonfamilial JOAG. This new gene needs to be further investigated for its role in pathways associated with glaucoma pathogenesis.
Keywords: Congenital glaucoma, glaucoma, juvenile glaucoma, PLOD2
The first genetic site associated with primary congenital glaucoma (PCG) GLC3A was discovered at 2p21.[1] The CYP1B1 gene at this locus encoding cytochrome P4501B1 is found to be most commonly associated with PCG[2] and cases of juvenile-onset open-angle glaucoma (JOAG).[3,4,5] Akarsu et al.[6] identified the 1p36 locus (GLC3B) linked to PCG; later, two other loci (GLC3C and GLC3D) were discovered, including the LTBP2 gene.[7,8,9]
Among JOAG patients, the Myocilin (MYOC) gene is the most commonly known to be implicated with its prevalence varying from 5% to 35% among JOAG patients depending on the ethnic group studied.[4,10,11,12,13] MYOC mutations have also been shown to be associated with PCG,[14,15,16] emphasizing the genetic overlap between the two subtypes of glaucoma. Apart from MYOC, CYP1B1,[3,17] LTBP2[18,19] and CPAMD8[20] genes are also found to be associated with JOAG.
Among cases of congenital glaucoma, in addition to CYP1B1 and LTBP2, TEK, GPATCH, and PRSS56 mutations have also been associated;[21,22,23,24] however, together they explain only 10%–50% of those affected, depending on the population studied.[9,25,26] Genetic heterogeneity in nonsyndromic congenital and juvenile glaucoma[27,28] is the reason why efforts are being made to identify novel variants associated with them.
Next-generation sequencing enables exome-wide identification of novel variants and is currently the method of choice for identifying novel variants especially for diseases with genetic heterogeneity.[29] We undertook this study to analyze genetic variant(s) that could be the cause of PCG in a pair of monozygotic twins with the help of whole exome sequencing (WES).
Methods
Clinical evaluation
The parents of monozygotic twins presented to our hospital with complaints of the children having watering from their eyes since birth. Both the children were healthy males aged 8 months. The twins had similar facial features, body habitus, and blood group typing [Fig. 1]. They were of Indian ethnicity belonging to the Jat community in Haryana. This was a nonconsanguineous family and the parents had three children [Fig. 2].
Figure 1.

Monozygotic twins at 8 months of age
Figure 2.

(a) The pedigree of the monozygotic twins with PLOD2 (p.Tyr642Cys) mutation. Affected status shaded in black is for PCG. - /-indicates homozygous wild genotype. - /+ indicates heterozygous mutation for p.Tyr642Cys. Y indicates age in Years at the last follow-up. (b) DNA Sanger sequencing traces revealed wild allele at c. 1925 of PLOD2 in father (II.1) mother (II.2) and sibling (III.1) of the monozygotic twins and confirmed a heterozygous G allele [c.1925A>G(p.Tyr642Cys)] in the monozygotic twins (III.2, III.3)
The twins were examined under anesthesia. Child B had horizontal corneal diameters of 14.5 and 15 mm in the right eye (RE) and left eye (LE), respectively. Corneas were hazy with the presence of circumferential Haab striae. The anterior chamber was deep with normal iris pattern and clear lens. Fundus examination revealed a cup disc ratio of 0.9:1 with a thin neuroretinal rim in both eyes. His IOP with Perkins tonometer was 28 and 30 mm Hg on timolol 0.25% eye drop in the RE and LE, respectively. Trabeculotomy and trabeculectomy with the use of mitomycin-C were performed in both eyes. The other twin (Child C) also had horizontal corneal diameters of 14 and 14.5 mm in the RE and LE, respectively. His fundus examination also revealed a cup disc ratio of 0.9:1 with a thin neuroretinal rim in both eyes. The IOP was 24 mm Hg on timolol 0.25% eye drop in both eyes. Both eyes of this child were also operated for glaucoma. The postoperative course in both children remained uneventful. Follow-up over 10 years post-surgery showed an IOP of <18 mm Hg in both eyes of both twins with a single anti-glaucoma drug.
The parents and the sister of the twins were carefully examined to rule out raised IOP or glaucoma over the 10-year follow-up and were found to have a normal anterior segment and no evidence of glaucoma.
Genetic evaluation
The study was carried out in accordance with the tenets of the Declaration of Helsinki. Ethical approval was obtained from our institute’s ethics committee and written informed consent was provided by the parents for blood collection and genetic evaluation.
Exome sequencing was carried out in the family with the monozygotic twin pair concordant for PCG. For comparison, we selected from our cohort 50 other patients with non-familial JOAG and 30 healthy controls. Two ml of blood, drawn from the antecubital vein of cases and controls were collected in ethylene diamine tetraacetate (EDTA) vial, and DNA was isolated by salting out method. Exome capture was achieved using the Sure Select Human All Exon V5 (50.4 Mb) (Agilent Technologies, Santa Clara, California, USA) by following the manufacturer’s instructions. Briefly, 1 mg of genomic DNA was fragmented (150–200 bp) and ligated to adapter primers and then PCR amplified. Further, biotinylated RNA capture probes (~120 bp) were used for hybridization of amplified DNA-fragment libraries. Hybridized DNA was recovered by streptavidin-coated magnetic bead separation (Dynal, Invitrogen, Carlsbad, CA). HiSeq2000System (Illumina, San Diego, CA). Captured DNA was eluted and then subject to flow-cell massively-parallel sequencing on a HiSeq2000System (Illumina, San Diego, CA). Hybrid-capture libraries were amplified to add the sequencing primers and identifying tags and then subjected to paired-end (2 × 101 bp read length) multiplex sequencing. All samples were sequenced with a minimum of 100× coverage.
Exome variant analysis
All the FastQ files were assessed for the quality of raw sequence data. Raw reads with poor base quality and sequence adapters were trimmed by trimmomatic. The clean reads with Phred score >20 were mapped to the human reference genome build hg19/GRCh37 using Burrows–Wheeler Aligner (BWA). SAM tools were used for sorting of reads and duplicate reads were marked using Picard tools (http://picard.sourceforge.net/). Genome analysis tool kit (GATK) v2.7.2)[30] was used for calling variants. Further, the variant calling format files (VCF) were annotated using Golden Helix VarSeq Software v. 1.2.1 (Bozeman, MT). Target coverage and read depth were reviewed by the Integrated Genomics Viewer (IGV, http://www. broadinstitute.org/igv/). Variant filtering was based on (a) read depth (DP) of >10, (b) genotype quality (GQ) score of >20, and (c) predicted missense or loss-of-function mutation. All exonic single nucleotide polymorphisms (SNPs) from the respective VCF files of the monozygotic twins were compared to determine the concordant and discordant variants; the variants were considered concordant if the genotypes were identical in both VCF files.
We prioritized the variants by following steps. (1) Rare or novel variants (having <1% minor allele frequency) including frameshift, splice, stop gain, stop loss, or missense predicted to be damaging by at least one of the pathogenic prediction software were filtered. Minor allele frequency (MAF) was accessed from public databases including 1000 Genome Project phase 3 (www. 1000genomes.org), dbSNP Common 151 (http://ncbi.nlm.nih.gov/SNP/), ExomeVariantServer (EVS, http://evs.gs.washington.edu/EVS/); (2) potential disease-causing variants were further selected by including variants shared by affected twins and excluded variants shared by unaffected sibling; (3) included de-novo variants; and (4) only those variants that were potentially pathogenic and had an allele frequency <0.001 in the South Asian population. We then chose our genes based on other patients with JOAG that had a similar gene with an associated pathogenic or a variant with uncertain significance. From this analysis, we got the procollagen-lysine 2-oxoglutarate 5-dioxygenase 2 (PLOD2) gene (also called LH2) with a de novo mutation present in both twins as well as in a JOAG patient. Further, the prioritized variants were validated via PCR amplification and by Sanger sequencing using an ABI 3730XL sequencer (Applied Biosystems/Life Technologies, Carlsbad, CA, USA).
Whole-mount in situ hybridization
For functional characterization of the gene, we looked for its expression in ocular tissues of the zebrafish embryo using whole-mount in situ hybridization (WISH). The zebrafish has advantages for studying the gene expression in ocular development because of its fast development, the optical clarity during embryogenesis, and the readily available genomic sequence. The details of the methodology for WISH are provided in the supplementary file.
Results
No pathogenic variants (including copy number variations) were detected that cosegregated with the disease in the PCG twins at any of the known loci for Mendelian forms of POAG (GLC1A, GLC1B, GLC1D-Q) or PCG (GLC3A-D), including the known causative genes, namely MYOC (GLC1A), CYP1B1 (GLC3A), WDR36 (GLC1G), ASB10 (GLC1F), OPTN (GLC1E), NTF4 (GLC1O), TBK1 (GLC1P), LTBP2 (GLC3C), PAX6, TEK, ANGPT1, and FOXC1.
We identified a heterozygous, nonsynonymous, missense deleterious mutation in the PLOD2 gene on exon 17 (chr3:145789071T>CGRch37;NM_000935.2:c.1925A>G; p.Tyr642Cys) in the twins. The allele frequency of this variant is not reported in the dbSNP database nor in the 1000 Genomes database. This de-novo heterozygous mutation was confirmed in both the twins as well as its absence in both the parents and sister by Sanger sequencing [Fig. 2]. The mutation was predicted to be deleterious (0.807) by the Condel program.[31] One JOAG patient was found to harbor another heterozygous nonsynonymous, missense mutation in the PLOD2 gene on exon 17 (chr3: 145789116 C>TGRch37; NM_000935.2:c.1880G>A; p.Arg627Gln) (rs748652746). The allele frequency of this variant in the South Asian population is reported to be 0.000556 [Table 1]. The SIFT and Polyphen score of this variant were 0.46 and 0.59, respectively. The patient had been diagnosed with JOAG since the age of 31 years with an untreated IOP of 28 mm Hg and a 0.7:1 cupping in BE. His anterior segment was within normal limits (normal corneal size). Gonioscopy revealed a featureless angle that was probably due to the compaction of trabecular beams, suggesting an immature trabecular meshwork. The variation was validated by Sanger sequencing [Fig. 3]. The parents of this JOAG patient were unaffected and did not harbor the mutation. His siblings were unavailable for examination. Both these PLOD2 variants are highly conserved in the evolutionary process with a genomic evolutionary rate profiling (GERP) value of >5 [Fig. 4]. Table 2 gives the variants that segregated and those that did not segregate with the disease, including those variants associated with glaucoma, for both the monozygotic twins as well as the JOAG patient.
Table 1.
The characteristics of the two variants detected in the PCG and JOAG patients
| Diagnosis | Chr. Position | Exon | Codon | Protein Change | gnomAD variant count | gnomAD total alleles | gnomAD Ethnically Matched alleles (South Asian) | Polyphen 2 (Score) |
|---|---|---|---|---|---|---|---|---|
| PCG | 3:145789071 | 17 | c.1925A>G | p.Tyr642Cys | Novel | - | - | Probably damaging (0.998) |
| JOAG | 3:145789116 | 17 | c.1880G>A | p.Arg627Gln | 17 | 250598 | 30600 | Probably damaging (0.598) |
Figure 3.

(a) The Pedigree of JOAG patient with PLOD2 (p.Arg627Gln) mutation. Affected status is shaded in black. ↑ indicates proband. - /- represents homozygous wild genotype. - /+ indicates p. Arg627Gln heterozygous mutation. Siblings (*) were not available for the genetic study. Y indicates age in years at the time of study. (b) DNA Sanger sequencing traces revealed wild allele “C” at chr3:145789116 of PLOD2 in father (II.1) and mother (II.2) and confirmed a heterozygous T allele [chr3: 145789116 C>T(p.Arg627Gln)] in the proband (III.1)
Figure 4.

(a) Schematic exon organization of PLOD2 and the exon location of R627Q and Y642C are indicated. (b) PLOD2 protein sequence alignment across multiple species showing conservation of arginine (R627) and tyrosine (Y642) residues is seen
Table 2.
Variants segregating and nonsegregating with the disease in the monozygotic twins and the JOAG patient
| Monozygotic twins | JOAG patient | |
|---|---|---|
| Segregating with disease | ||
| Total variants | n=548 | n=695 |
| Glaucoma associated | n=1 (NF1 gene)* | n=1 (PAX6 gene)** |
| Nonsegregating with disease | ||
| Total variants | n=2174 | n=1195 |
| Glaucoma associated | n=0 | n=1 (OPTN gene)*** |
*NF1 (neurofibromin 1) gene, rs191111884 an intronic variant (ClinVar: Benign). **PAX6 (paired box 6) gene, rs759391101 an inframe deletion (ClinVar: Benign). ***OPTN (optineurin) gene, rs71492279 an intronic variant (ClinVar: Benign)
We also explored the expression dynamics of the plod2 gene in the vertebrate model, zebrafish. The plod2 gene expression analysis was performed during zebrafish embryonic development using reverse transcription-polymerase chain reaction (RT-PCR) [Fig. 5a]. The presence of plod2 gene much before zygotic transcription started, indicates the essential requirement of plod2 during early embryonic development. The plod2 gene was cloned before making a DIG-labeled antisense mRNA probe for using in situ hybridization. Embryos were harvested at 24 and 48 hours of development for mRNA in situ hybridization [Fig. 5b-d]. The expression analysis revealed that the plod2 gene is induced in the regions such as the head and anterior segment of the eye during embryonic development suggesting the possibility of involvement of plod2 in the structural and functional integrity of zebrafish eye.
Figure 5.
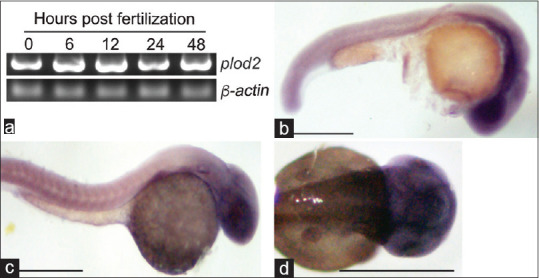
Expression of plod2 gene during zebrafish embryonic development (a) RT-PCR analysis of the plod2 at various time points post-fertilization. (b and c) Bright-field microscopy lateral view images of zebrafish embryos at 24 hpf (b) and 48 hpf. (c) embryos expressing plod2 gene as revealed by mRNA in situ hybridization. (d) Bright-field microscopy dorsal view image of the 48 hpf zebrafish head region showing plod2 gene expression. Scale bar indicates 1 mm
Discussion
This study reports the novel association of PLOD2 with glaucoma. The rare presentation of PCG in this pair of monozygotic twins gave us an opportunity to look for genetic mutations yet unknown in PCG. While rare cases of bilateral PCG in monozygotic twins are known, there is no genetic study reported for them.[32,33,34] Although discordance of glaucoma has been reported among monozygotic twins,[34] the twins in our study showed no discordance and presented with similar severity and onset of the disease. We relied on the facial similarity and blood group typing along with a 96% concordance in the Exome to establish monozygosity.
In the absence of the known Mendelian genes for POAG and PCG in our cases, as well as those that increase the risk susceptibility to glaucoma, we believe PLOD2 could be an important gene associated with de-novo or sporadic cases of PCG and JOAG. The pathogenicity of the two variants is supported by their extremely low frequency in the general population, their absence in controls along with their evolutionary conservation, and their effect on protein function. The PLOD2 gene consists of 19 exons, codes for lysyl hydroxylase2 (LH2) and the LH2 enzyme is responsible for the hydroxylation of collagen telopeptide which directs cross-linking of collagen fibrils in the extracellular matrix, necessary to provide tensile integrity and stability.[35,36] PLOD2 mutations are known to occur among patients with Brucks syndrome,[37] a rare form of osteogenesis imperfect characterized by bone fragility and congenital joint contractures. None of our patients had any orthopedic problems.
Though PLOD2 mutations are not known to have an association with causing raised IOP or glaucoma, the PLOD2 enzyme is known to have an association with the PITX2 gene, the mutations of which cause Axenfeld Reiger (AR) syndrome.[38] The PLOD2 gene is a potential downstream target for the PITX2 gene.[39,40] Although none of the three cases reported here with PLOD2 mutations had features of AR syndrome, we believe the pathways of trabecular meshwork (TM) development may have been altered due to the mutations, leading to the immaturity of the TM. The PLOD enzymes have been shown to be important in collagen cross-linking and hence may be postulated to have a role in TM development pathways.[35] The G protein-coupled receptor 48 (Gpr48) has been shown to be associated with anterior segment dysgenesis during development and deletion of the Gpr48 in mice has been shown to lead to decreased levels of both PLOD1 and 2 enzymes.[41] We also found PLOD2 expression in the ocular tissues during embryonic development of the eye as shown in our in vivo study using the zebrafish model.
The LH2 enzyme in mice has been found to be associated with embryogenesis[42] and its deficiency is related to endoplasmic reticulum stress and apoptosis.[43] In the evolutionary ladder of complex multicellular organisms, lysyl hydroxylation plays an important role in the initial steps of development as embryonic lethality was associated with LH2-null mice.[44] However, plod2 zebrafish mutants survive into adulthood despite complete loss of LH2 but with disturbed fibrillar organization.[45] It is possible that different disorders might develop, depending on the species and extent of the variation in the PLOD2 gene. PLOD2 mutations identified in Brucks syndrome2 cause a marked reduction in LH2 activity, whereas an elevated expression of LH2 has been recognized as a general phenomenon that promotes fibrosis and cancer progression.[43,46] Most PLOD2 mutations associated with Brucks syndrome involve exons 17–19 encoding the C-terminal domain with LH activity[47], and the amino acid sequence in this region has shown high homology among LH1, LH2, and LH3 across species, suggesting the region being important for lysyl hydroxylase function. In our study, both PLOD2 mutations were in exons 17 encoding the C-terminal domain for LH activity.
The PLOD2 gene also lies in the 3q24 locus, which was originally described by Wirtz et al.[48] for its association with adult-onset POAG. Because PCG, JOAG, and adult-onset POAG form a continuum, especially with regards to the overlapping genetic variations described in these three phenotypes that are distinguished mainly by their age of onset, one could postulate that the PLOD2 mutations might also play a role in adult-onset POAG. At this juncture, we can only hypothesize that the effects of PLOD2 mutations could be related to angle maldevelopment in humans.
The earlier age of onset with c.1925A>G change leading to congenital glaucoma points to the greater pathogenicity of this mutant in comparison to the c.1880G>A, which resulted in delayed onset glaucoma in the JOAG patient. While the frequency of c.1880G>A change among South Asians is 0.0005556, the c.1925A>G change was absent in South Asians.
Conclusion
In conclusion, novel missense mutations have been identified in a new gene locus, namely PLOD2 by WES. A larger sample of congenital and juvenile glaucoma patients would be needed to replicate our results. There is also a need for further research as regards the cause and effect relationship of this gene with glaucoma.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
Funding for this study was received from the Indian Council of Medical Research, New Delhi (Grant no. ISRM/12(58)/2019 ID no. 2018_2666).
References
- 1.Sarfarazi M, Akarsu AN, Hossain A, Turacli ME, Aktan SG, Barsoum-Homsy M, et al. Assignment of a locus (GLC3A) for primary congenital glaucoma (Buphthalmos) to 2p21 and evidence for genetic heterogeneity. Genomics. 1995;30:171–7. doi: 10.1006/geno.1995.9888. [DOI] [PubMed] [Google Scholar]
- 2.Bejjani BA, Lewis RA, Tomey KF, Anderson KL, Dueker DK, Jabak M, et al. Mutations in CYP1B1, the gene for cytochrome P4501B1, are the predominant cause of primary congenital glaucoma in Saudi Arabia. Am J Hum Genet. 1998;62:325–33. doi: 10.1086/301725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abu-Amero KK, Morales J, Aljasim LA, Edward DP. CYP1B1 mutations are a major contributor to juvenile-onset open angle glaucoma in Saudi Arabia. Ophthalmic Genet. 2015;36:184–7. doi: 10.3109/13816810.2013.841961. [DOI] [PubMed] [Google Scholar]
- 4.Bayat B, Yazdani S, Alavi A, Chiani M, Chitsazian F, Tusi BK, et al. Contributions of MYOC and CYP1B1 mutations to JOAG. Mol Vis. 2008;14:508–17. [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta V, Somarajan BI, Walia GK, Kaur J, Kumar S, Gupta S, et al. Role of CYP1B1, p.E229K and p.R368H mutations among 120 families with sporadic juvenile onset open-angle glaucoma. Graefes Arch Clin Exp Ophthalmol. 2018;256:355–62. doi: 10.1007/s00417-017-3853-0. [DOI] [PubMed] [Google Scholar]
- 6.Akarsu AN, Turacli ME, Aktan SG, Barsoum-Homsy M, Chevrette L, Sayli BS, et al. A second locus (GLC3B) for primary congenital glaucoma (Buphthalmos) maps to the 1p36 region. Hum Mol Genet. 1996;5:1199–203. doi: 10.1093/hmg/5.8.1199. [DOI] [PubMed] [Google Scholar]
- 7.Ali M, McKibbin M, Booth A, Parry DA, Jain P, Riazuddin SA, et al. Null mutations in LTBP2 cause primary congenital glaucoma. Am J Hum Genet. 2009;84:664–71. doi: 10.1016/j.ajhg.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Firasat S, Riazuddin SA, Hejtmancik JF, Riazuddin S. Primary congenital glaucoma localizes to chromosome 14q24.2-24.3 in two consanguineous Pakistani families. Mol Vis. 2008;14:1659–65. [PMC free article] [PubMed] [Google Scholar]
- 9.Stoilov IR, Costa VP, Vasconcellos JP, Melo MB, Betinjane AJ, Carani JC, et al. Molecular genetics of primary congenital glaucoma in Brazil. Invest Ophthalmol Vis Sci. 2002;43:1820–7. [PubMed] [Google Scholar]
- 10.Alward WL, Fingert JH, Coote MA, Johnson AT, Lerner SF, Junqua D, et al. Clinical features associated with mutations in the chromosome 1 open-angle glaucoma gene (GLC1A) N Engl J Med. 1998;338:1022–7. doi: 10.1056/NEJM199804093381503. [DOI] [PubMed] [Google Scholar]
- 11.Huang C, Xie L, Wu Z, Cao Y, Zheng Y, Pang CP, et al. Detection of mutations in MYOC, OPTN, NTF4, WDR36 and CYP1B1 in Chinese juvenile onset open-angle glaucoma using exome sequencing. Sci Rep. 2018;8:4498. doi: 10.1038/s41598-018-22337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Svidnicki PV, Braghini CA, Costa VP, Schimiti RB, de Vasconcellos JPC, de Melo MB. Occurrence of MYOC and CYP1B1 variants in juvenile open angle glaucoma Brazilian patients. Ophthalmic Genet. 2018;39:717–24. doi: 10.1080/13816810.2018.1546405. [DOI] [PubMed] [Google Scholar]
- 13.Yen YC, Yang JJ, Chou MC, Li SY. Identification of mutations in the myocilin (MYOC) gene in Taiwanese patients with juvenile-onset open-angle glaucoma. Mol Vis. 2007;13:1627–34. [PubMed] [Google Scholar]
- 14.Chakrabarti S, Kaur K, Komatireddy S, Acharya M, Devi KR, Mukhopadhyay A, et al. Gln48His is the prevalent myocilin mutation in primary open angle and primary congenital glaucoma phenotypes in India. Mol Vis. 2005;11:111–3. [PubMed] [Google Scholar]
- 15.Chen Y, Jiang D, Yu L, Katz B, Zhang K, Wan B, et al. CYP1B1 and MYOC mutations in 116 Chinese patients with primary congenital glaucoma. Arch Ophthalmol. 2008;126:1443–7. doi: 10.1001/archopht.126.10.1443. [DOI] [PubMed] [Google Scholar]
- 16.Kaur K, Reddy AB, Mukhopadhyay A, Mandal AK, Hasnain SE, Ray K, et al. Myocilin gene implicated in primary congenital glaucoma. Clin Genet. 2005;67:335–40. doi: 10.1111/j.1399-0004.2005.00411.x. [DOI] [PubMed] [Google Scholar]
- 17.Acharya M, Mookherjee S, Bhattacharjee A, Bandyopadhyay AK, Daulat Thakur SK, Bhaduri G, et al. Primary role of CYP1B1 in Indian juvenile-onset POAG patients. Mol Vis. 2006;12:399–404. [PubMed] [Google Scholar]
- 18.Saeedi O, Yousaf S, Tsai J, Palmer K, Riazuddin S, Ahmed ZM. Delineation of novel compound heterozygous variants in LTBP2 associated with juvenile open angle glaucoma. Genes (Basel) 2018;9:527. doi: 10.3390/genes9110527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Somarajan BI, Gupta S, Mahalingam K, Azmir K, Gupta V. Digenic Inheritance of MYOC and LTBP2 genes in Juvenile onset open angle glaucoma. J Pediatr Genet. 2021 doi: 10.1055/s-0040-1722213. doi:10.1055/s-0040-1722213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siggs OM, Souzeau E, Taranath DA, Dubowsky A, Chappell A, Zhou T, et al. Biallelic CPAMD8 variants are a frequent cause of childhood and juvenile open-angle glaucoma. Ophthalmology. 2020;127:758–66. doi: 10.1016/j.ophtha.2019.12.024. [DOI] [PubMed] [Google Scholar]
- 21.Ferre-Fernandez JJ, Aroca-Aguilar JD, Medina-Trillo C, Bonet-Fernandez JM, Mendez-Hernandez CD, Morales-Fernandez L, et al. Whole-exome sequencing of congenital glaucoma patients reveals hypermorphic variants in GPATCH3, a new gene involved in ocular and craniofacial development. Sci Rep. 2017;7:46175. doi: 10.1038/srep46175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labelle-Dumais C, Pyatla G, Paylakhi S, Tolman NG, Hameed S, Seymens Y, et al. Loss of PRSS56 function leads to ocular angle defects and increased susceptibility to high intraocular pressure. Dis Model Mech. 2020;13:dmm042853. doi: 10.1242/dmm.042853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Souma T, Tompson SW, Thomson BR, Siggs OM, Kizhatil K, Yamaguchi S, et al. Angiopoietin receptor TEK mutations underlie primary congenital glaucoma with variable expressivity. J Clin Invest. 2016;126:2575–87. doi: 10.1172/JCI85830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomson BR, Souma T, Tompson SW, Onay T, Kizhatil K, Siggs OM, et al. Angiopoietin-1 is required for Schlemm's canal development in mice and humans. J Clin Invest. 2017;127:4421–36. doi: 10.1172/JCI95545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kakiuchi-Matsumoto T, Isashiki Y, Ohba N, Kimura K, Sonoda S, Unoki K. Cytochrome P450 1B1 gene mutations in Japanese patients with primary congenital glaucoma (1) Am J Ophthalmol. 2001;131:345–50. doi: 10.1016/s0002-9394(00)00808-4. [DOI] [PubMed] [Google Scholar]
- 26.Micheal S, Siddiqui SN, Zafar SN, Iqbal A, Khan MI, den Hollander AI. Identification of novel variants in LTBP2 and PXDN using whole-exome sequencing in developmental and congenital glaucoma. PLoS One. 2016;11:e0159259. doi: 10.1371/journal.pone.0159259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Demenais F, Elston RC, Bonaiti C, Briard ML, Kaplan EB, Namboodiri KK. Segregation analysis of congenital glaucoma:Approach by two differential models. Am J Hum Genet. 1981;33:300–6. [PMC free article] [PubMed] [Google Scholar]
- 28.Morton NE. Heterogeneity in nonsyndromal congenital glaucoma. Am J Med Genet. 1982;12:97–102. doi: 10.1002/ajmg.1320120113. [DOI] [PubMed] [Google Scholar]
- 29.Singleton AB. Exome sequencing:A transformative technology. Lancet Neurol. 2011;10:942–6. doi: 10.1016/S1474-4422(11)70196-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit:A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez-Perez A, Lopez-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88:440–9. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ben-Zion I, Bogale A, Moore DB, Helveston EM. Bilateral primary congenital glaucoma in monozygotic twins. J Pediatr Ophthalmol Strabismus. 2010;47:124–6. doi: 10.3928/01913913-20100308-15. [DOI] [PubMed] [Google Scholar]
- 33.Rasmussen DH, Ellis PP. Congenital glaucoma in identical twins. Arch Ophthalmol. 1970;84:827–30. doi: 10.1001/archopht.1970.00990040829024. [DOI] [PubMed] [Google Scholar]
- 34.Zhang K, Jampel HD. Discordance of primary infantile glaucoma in monozygotic twins. Am J Ophthalmol. 1999;128:97–8. doi: 10.1016/s0002-9394(99)00025-2. [DOI] [PubMed] [Google Scholar]
- 35.Kivirikko KI, Myllyla R. Post-translational processing of procollagens. Ann N Y Acad Sci. 1985;460:187–201. doi: 10.1111/j.1749-6632.1985.tb51167.x. [DOI] [PubMed] [Google Scholar]
- 36.Kivirikko KI, Pihlajaniemi T. Collagen hydroxylases and the protein disulfide isomerase subunit of prolyl 4-hydroxylases. Adv Enzymol Relat Areas Mol Biol. 1998;72:325–98. doi: 10.1002/9780470123188.ch9. [DOI] [PubMed] [Google Scholar]
- 37.Lv F, Xu X, Song Y, Li L, Asan, Wang J, et al. Novel mutations in PLOD2 cause rare Bruck syndrome. Calcif Tissue Int. 2018;102:296–309. doi: 10.1007/s00223-017-0360-6. [DOI] [PubMed] [Google Scholar]
- 38.Hjalt TA, Amendt BA, Murray JC. PITX2 regulates procollagen lysyl hydroxylase (PLOD) gene expression:Implications for the pathology of Rieger syndrome. J Cell Biol. 2001;152:545–52. doi: 10.1083/jcb.152.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gould DB, Smith RS, John SW. Anterior segment development relevant to glaucoma. Int J Dev Biol. 2004;48:1015–29. doi: 10.1387/ijdb.041865dg. [DOI] [PubMed] [Google Scholar]
- 40.Holmberg J, Liu CY, Hjalt TA. PITX2 gain-of-function in Rieger syndrome eye model. Am J Pathol. 2004;165:1633–41. doi: 10.1016/S0002-9440(10)63420-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weng J, Luo J, Cheng X, Jin C, Zhou X, Qu J, et al. Deletion of G protein-coupled receptor 48 leads to ocular anterior segment dysgenesis (ASD) through down-regulation of Pitx2. Proc Natl Acad Sci U S A. 2008;105:6081–6. doi: 10.1073/pnas.0708257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salo AM, Sipila L, Sormunen R, Ruotsalainen H, Vainio S, Myllyla R. The lysyl hydroxylase isoforms are widely expressed during mouse embryogenesis, but obtain tissue- and cell-specific patterns in the adult. Matrix Biol. 2006;25:475–83. doi: 10.1016/j.matbio.2006.08.260. [DOI] [PubMed] [Google Scholar]
- 43.Kasamatsu A, Uzawa K, Hayashi F, Kita A, Okubo Y, Saito T, et al. Deficiency of lysyl hydroxylase 2 in mice causes systemic endoplasmic reticulum stress leading to early embryonic lethality. Biochem Biophys Res Commun. 2019;512:486–91. doi: 10.1016/j.bbrc.2019.03.091. [DOI] [PubMed] [Google Scholar]
- 44.Hyry M, Lantto J, Myllyharju J. Missense mutations that cause Bruck syndrome affect enzymatic activity, folding, and oligomerization of lysyl hydroxylase 2. J Biol Chem. 2009;284:30917–24. doi: 10.1074/jbc.M109.021238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gistelinck C, Witten PE, Huysseune A, Symoens S, Malfait F, Larionova D, et al. Loss of type i collagen telopeptide lysyl hydroxylation causes musculoskeletal abnormalities in a Zebrafish model of Bruck syndrome. J Bone Miner Res. 2016;31:1930–42. doi: 10.1002/jbmr.2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van der Slot AJ, Zuurmond AM, Bardoel AF, Wijmenga C, Pruijs HE, Sillence DO, et al. Identification of PLOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis. J Biol Chem. 2003;278:40967–72. doi: 10.1074/jbc.M307380200. [DOI] [PubMed] [Google Scholar]
- 47.Mumm S, Gottesman GS, Wenkert D, Campeau PM, Nenninger A, Huskey M, et al. Bruck syndrome 2 variant lacking congenital contractures and involving a novel compound heterozygous PLOD2 mutation. Bone. 2020;130:115047. doi: 10.1016/j.bone.2019.115047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wirtz MK, Samples JR, Kramer PL, Rust K, Topinka JR, Yount J, et al. Mapping a gene for adult-onset primary open-angle glaucoma to chromosome 3q. Am J Hum Genet. 1997;60:296–304. [PMC free article] [PubMed] [Google Scholar]