

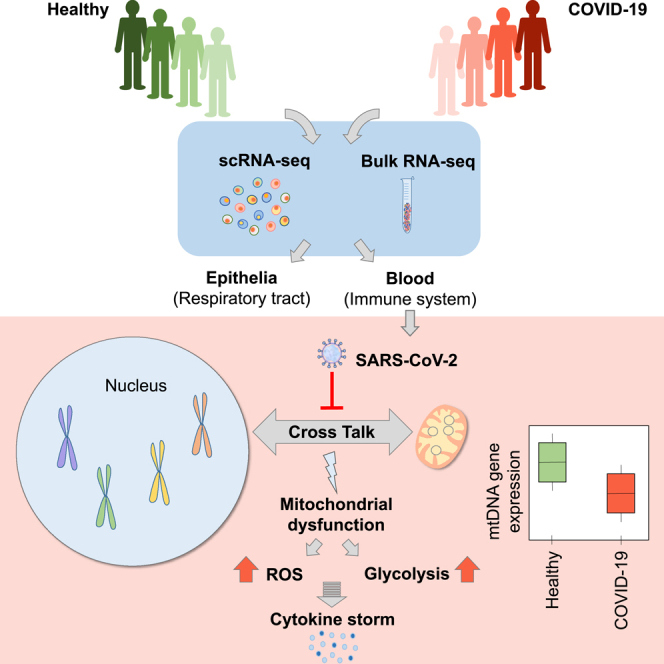
Summary
Mitochondria are pivotal for bioenergetics, as well as in cellular response to viral infections. Nevertheless, their role in COVID-19 was largely overlooked. Here, we analyzed available bulk RNA-seq datasets from COVID-19 patients and corresponding healthy controls (three blood datasets, N = 48 healthy, 119 patients; two respiratory tract datasets, N = 157 healthy, 524 patients). We found significantly reduced mtDNA gene expression in blood, but not in respiratory tract samples from patients. Next, analysis of eight single-cells RNA-seq datasets from peripheral blood mononuclear cells, nasopharyngeal samples, and Bronchoalveolar lavage fluid (N = 1,192,243 cells), revealed significantly reduced mtDNA gene expression especially in immune system cells from patients. This is associated with elevated expression of nuclear DNA-encoded OXPHOS subunits, suggesting compromised mitochondrial-nuclear co-regulation. This, together with elevated expression of ROS-response genes and glycolysis enzymes in patients, suggest rewiring toward glycolysis, thus generating beneficial conditions for SARS-CoV-2 replication. Our findings underline the centrality of mitochondrial dysfunction in COVID-19.
Subject areas: Immune system, Virology, Genomics
Graphical abstract
Highlights
-
•
mtDNA gene expression is downregulated in COVID-19 blood, but not in respiratory tract
-
•
Decreased mtDNA gene expression disrupts mito-nuclear coordination
-
•
mtDNA is downregulated and rewired toward glycolysis especially in immune system cells
-
•
Mitochondrial dysfunction is central to the etiology of COVID19
Immune system; Virology; Genomics
Introduction
A significant number of COVID-19 patients develop severe fatal consequences of the respiratory tract infection, attributed to catastrophic inflammatory events, described jointly as a 'cytokine storm'. This inflammatory state is associated with deleterious oxidative stress, and elevated production of reactive oxygen species (ROS)– both point toward altered mitochondrial activity (Singh et al., 2020). This is further supported by evidence suggesting a central role for the mitochondria in modulating the activity of the innate immune system (Bordon 2018; Zuo and Wan 2019; Ganji and Reddy 2020). Specifically, once viral genomes are recognized by the innate immune system, the retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), RIG-I, and MDA5, interact with mitochondrial antiviral-signaling protein (MAVS). MAVS, in turn, activates the intracellular signaling cascade that induces the transcription of genes encoding type I interferons (Rehwinkel and Gack 2020).
Several lines of evidence underline the mitochondria as a pivotal cellular target during SARS-CoV-2 infection: Firstly, the discovery of high sequence similarity between SARS-CoV-2-encoded proteins and mitochondrial-localized proteins encoded by SARS-CoV-1 (3/29 protein coding genes) (Shi et al., 2014; Gordon et al., 2020), which were shown to impact mitochondrial gene expression (Shao et al., 2006) and components of the innate immunity (Burtscher et al., 2020). Secondly, recent extensive computational analyses predicted preferential mitochondrial localization of SARS-CoV-2 RNAs and sgRNAs (Wu et al., 2020) and experimental evidence revealed interaction of SARS-CoV-2-encoded proteins with the mitochondria (Gordon et al., 2020). Third, recent evidence revealed alteration of mitochondrial dynamics in COVID-19 patients, which is associated with increased fusion, and inhibition of mitochondrial fission (Holder and Reddy 2021). It is thus likely that mitochondrial function is hijacked, and altered, upon SARS-CoV-2 infection (Gatti et al., 2020; Singh et al., 2020). Despite the known regulatory involvement of the mitochondria in the immune system, the regulatory response of the mitochondria to SARS-CoV-2 infection has been largely overlooked. Recent RNA-seq analysis in airway clinical samples and in lung-associated cell lines suggested little or no response of mitochondrial gene expression, either in the mitochondrial genome (mtDNA) or in the nucleus, to SARS-CoV-2 infection (Miller et al., 2021). This finding, which was reported during the course of our work, may lead to the conclusion that mitochondrial gene expression and subsequent mitochondrial activities are not significantly affected in COVID-19 patients. However, such findings do not dismiss possible tissue/cell type specific regulatory response of the mitochondria to SARS-CoV-2 infection.
Here, by analyzing both bulk and single cells RNA-seq (scRNA-seq) data in COVID-19 patients as compared to controls we discovered significant reduction in mtDNA gene expression levels in patient samples from blood, in contrast to inconsistent changes in samples from the respiratory tract. Consistent with this finding, scRNA-seq analysis also revealed that such altered mtDNA gene expression preferentially occurred in immune system cells, whereas respiratory tract epithelial cells tended to display less prominent alterations. The response of mitochondrial-related pathways in the nucleus (such as ROS generation, TCA, glycolysis) is discussed in light of the apparent dependence of SARS-CoV-2 replication on glycolysis (Codo et al., 2020), likely on the expense of the mitochondrial oxidative phosphorylation (OXPHOS). Our findings underline a tissue-type and cell-type dependent negative response of mitochondrial gene expression regulation in COVID-19 patients.
Results
Mitochondrial gene expression in COVID-19 patients is significantly reduced in peripheral blood, but not in the upper respiratory tract
To examine whether SARS-CoV-2 infection associates with altered mitochondrial regulation, we assessed mtDNA-encoded genes' expression in thirteen publicly available datasets from healthy and COVID-19 patients: five bulk RNA-seq datasets from naso-oropharyngeal (NP/OP) swab (Lieberman et al., 2020; Mick et al., 2020), peripheral blood and whole blood (Bernardes et al., 2020; Levy et al., 2021; Thair et al., 2021), as well as eight scRNA-seq datasets from peripheral blood mononuclear cells (PBMC) (Bernardes et al., 2020; Wilk et al. 2020, Wilk et al., 2021; Ren et al., 2021; Stephenson et al., 2021), Bronchoalveolar lavage fluid samples (BALF) (Liao et al., 2020), and nasopharyngeal (NP) samples (Chua et al., 2020; Ziegler et al., 2021) (Figure 1, Table 1 notice dataset numbering, used below).
Figure 1.

Workflow of this study
Several different publicly available bulk and single-cell RNA-seq datasets from healthy and COVID-19 patients were obtained for analysis (see Table 1 for resources). For scRNA-seq data quality control, cells were filtered to retain only those with read counts in mtDNA encoded-protein genes. Finally, differential expression analysis of mitochondrial genes was performed. ∗Dataset III was used both for bulk and scRNA RNA-seq analyses, and hence was considered two separate datasets.
Table 1.
Datasets sample size and resources
Bulk or Single cells RNA-seq is indicated per dataset.
In consistence with previous findings, differential expression analysis of mtDNA genes in bulk RNA-Seq from the upper airway in healthy individuals as compared to patients who were positively diagnosed for SARS-CoV-2 (Datasets I, IV) did not show any significant difference in mtDNA gene expression in the patients (Miller et al., 2021) (N = 524 SARS-CoV-2 patients, N = 157 healthy control; see Table 1) (Figure S1). In contrast, analysis of three independent bulk RNA-seq datasets from peripheral blood of healthy individuals and COVID-19 patients (N = 34 healthy individuals and N = 106 COVID-19 patients; see Table 1 in STAR Methods), revealed significant reduction in the expression of 8/13 and 11/13 mtDNA-encoded protein-coding transcripts, respectively (Figure 2 and S2, Table S1; Datasets II, V https://doi.org/10.17632/8kd3xjfrh4.1).
Figure 2.

Decreased mtDNA gene expression levels as a feature of COVID-19 in peripheral blood
(A) Box plot of bulk RNA-seq analysis in peripheral blood displays lower mtDNA gene expression in COVID-19 patients as compared to healthy controls (Dataset II).
(B) Box plot displaying mtDNA gene expression across COVID-19 pseudotimes as compared to control (Dataset III – bulk RNA-seq). X axis – mtDNA genes, Y axis – normalized read counts, which account for expression levels. Significance: ∗ - p < 0.05, ∗∗ - p < 0.005, ∗∗∗ - p < 0.0005.
See also Figures S1 and S2 and Table S1.
We next analyzed bulk RNA-seq data (Dataset III) collected from 14 healthy individuals, samples from 13 hospitalized COVID-19 patients, each having up to five samples collected during disease progression, termed pseudotime points (Bernardes et al., 2020). Since the sample size of pseudotime 7 (recovery long-term follow-up) was very small (N = 2), it was excluded from further analysis. The pseudotime points were originally defined based on inflammatory markers and ventilation requirements as follows: pseudotimes 1,2 and 3 were considered ‘severe’, pseudotimes 4 and 5 were considered early/moderate and late convalesces, respectively, and pseudotime 6 were from recovered individuals (see STAR Methods). Notably, to avoid minute sample sizes in bulk RNA-seq of Dataset III, we grouped pseudotimes 1, 2 and 3 (all originally defined as 'severe'). Pairwise comparison of gene expression between control and each of the remaining disease trajectory phases revealed significant reduction in the expression of all mtDNA protein-coding transcripts in the severe, moderate/early convalescent as well as in late convalescent individuals (Figure 2B, Table S1). Interestingly, no significant change was observed in mtDNA protein-coding transcript levels while comparing healthy controls to recovered individuals (pseudotime 6; Figure 2B). Hence, our results suggest that mtDNA gene expression is consistently reduced in patients' blood, but not in the respiratory tract, and that such altered expression is likely reversible upon recovery.
Expression of nuclear DNA-encoded OXPHOS, ROS formation, TCA, glycolysis and mitochondria-related viral response genes is generally elevated in COVID-19 patients
As positive co-expression of mitochondrial and nuclear DNA-encoded OXPHOS genes has been observed across many human tissues (Barshad et al., 2018) we asked whether the reduced mtDNA gene expression in COVID-19 patients correlate with changes in nuclear DNA-encoded OXPHOS genes and related pathways. To this end, we subjected Datasets II, III and V to assessment of differentially expressed genes involved in the following biochemical pathways: OXPHOS structural and assembly genes, mitochondrial ribosome (Wolf and Mootha 2014; Barshad et al., 2018). We also analyzed the following mitochondria-related Gene Ontology (GO) terms: tricarboxylic acid (TCA), glycolysis enzymes including L-lactate dehydrogenase, response to type I interferons, mitochondrial antiviral signaling, including genes involved in RLR-MAVS pathway (hereby annotated as MAVS pathway; Table S2) (Burtscher et al., 2020; Rehwinkel and Gack 2020), generation and response to reactive oxygen species (ROS) formation, along with ROS scavenging enzymes. Notably, since measurement of the average expression of a given pathway may mask the impact of SARS-CoV-2 infection on individual genes, we performed a gene-by-gene differential expression analysis (while stringently correcting for multiple testing) (Figure 3A). We screened for differentially expressed genes that were consistently significant in Datasets II, III, and V, and considered only genes whose responses were consistent with at least one disease condition from Dataset III. Notably, while considering Dataset III, we focused on conditions that displayed significantly reduced mtDNA genes expression analysis. The analysis revealed a tendency toward elevated expression among OXPHOS subunits (7 elevated genes), TCA (6 elevated genes) and glycolysis (19 elevated genes, 6 reduced genes) in Datasets II and V and in severe, moderate/early and late convalescent individuals from Dataset III (Figure 3A; Table S3). Specifically, while considering glycolysis, expression levels of a key glycolysis enzyme - glyceraldehyde-3-phosphate dehydrogenase (GAPDH), were elevated in Datasets II and V as well as in severe and moderate/early convalescent patients of Dataset III (Figure 3B). It is worth noting that gene expression analysis of mitochondrial ribosome subunits revealed a mixed response in patients, with only few statistically significant values (Table S3).
Figure 3.

Expression of nuclear DNA genes involved in mitochondrial function or regulation of mitochondrial gene expression was consistently altered in peripheral blood from patients
(A) Heatmaps of significant differentially expressed genes in mitochondria-related biochemical pathways. Color bar representing the log-fold-change (logFC) of significant and consistent genes' expression in COVID-19 patients. Genes with logFC higher than 0.2 or less than −0.2 are shown. Red: positive logFC, purple: negative logFC.
(B) Box plots of GAPDH, LDHA and LDHB and (C) JUND, JUN (i.e., c-Jun), POLRMT expression levels in Datasets II (left panel) and Dataset III- bulk RNA-seq (right panel) (see Figure S2 for Dataset V). X axis – gene names; Y axis – normalized read counts as in Figure 2. Significance: ∗ - p < 0.05, ∗∗ - p < 0.005, ∗∗∗ - p < 0.0005.
See also Figure S2 and Tables S2 and S3 (https://doi.org/10.17632/8kd3xjfrh4.1).
Notably, we found that the expression of two subunits of lactate dehydrogenase (LDH), a key enzyme that regulates pyruvate generation by glycolysis, responded differently in the peripheral blood samples of patients versus controls, as follows: The expression of LDHA, a subunit which is more abundant in glycolytic tissues (Read et al., 2001; Porporato et al., 2011), was elevated in patients from Datasets II and V and in severe, moderate/early and late convalescent of Dataset III (Figure 3B). In contrast, the expression of LDHB, a subunit that is more abundant in high OXPHOS tissues (Read et al., 2001; Porporato et al., 2011), was reduced in patients from Datasets II and V and in severe, moderate/early convalescent samples of Dataset III (Figure 3B). As LDH activity governs the choice between generation of pyruvate (which promotes OXPHOS function) versus lactate, these results further support the interpretation that OXPHOS function is likely compromised and rewired to glycolysis in patients versus controls.
Lastly, genes that participate in type I interferon pathway (likely a reflection of the so-called mitochondria-related cytokine storm), genes involved in MAVS pathway, ROS formation and ROS response, mostly displayed elevated expression in COVID-19 patients in Dataset II, V and in severe, moderate/early and late convalescent samples of Dataset III (Figure 3A). Specifically, there were 10 elevated genes in the IFN1 pathway, and three genes involved in the MAVS pathway. Moreover, the expression of retinoic acid-inducible gene I (RIG-I), melanoma differentiation-associated protein 5 (MDA5) (DDX58 and IFIH1, respectively) and TANK-binding kinase 1 (TBK1), was consistently elevated in COVID-19 patients (Figure 3A, Table S3), supporting the increase in type I interferon pathway. Next, most of the significant genes from the ROS formation pathway (30 out of 34) displayed elevated expression in patients; similarly, most ROS response pathway genes (49 out of 66) displayed elevated expression as well. Notably, the expression of certain ROS scavenging enzymes (e.g., Peroxiredoxin 3 (PRDX3) and superoxide dismutase 2 (SOD2) (Yoboue et al., 2018)) was also significantly elevated in COVID-19 patients in 2/3 Datasets (Table S3). Taken together, these findings suggest that along with the apparent rewiring toward glycolysis, COVID-19 patients also suffer from possible oxidative stress.
Identifying candidate regulatory factors to explain changes in mtDNA gene expression in patients
We hypothesized that the reduction of mtDNA gene expression levels in COVID-19 patients could be regulated, rather than spontaneous. To identify candidate regulatory factors that potentially explain such putative downregulation, we assessed differential expression of a set of known and candidate mtDNA regulatory factors of transcription, regulatory factors of mtDNA replication, and nuclear DNA-encoded factors with known mitochondrial RNA-binding activity (Wolf and Mootha 2014; Cohen et al., 2016), as well as RNA and DNA-binding proteins that were recently identified in human mitochondria (Ardail et al., 1993; Fernandez-Vizarra et al., 2008; She et al., 2011; Blumberg et al., 2014; Lambertini et al., 2015; Chatterjee et al., 2016) (Table S2). Our analysis revealed that in all peripheral and whole blood bulk RNA-seq datasets (Datasets II, III, and V), the expression of JUN (c-Jun) was significantly elevated while the expression levels of JunD and of mitochondrial RNA polymerase (POLRMT) were reduced especially in peripheral blood datasets (Datasets II, III) (Figures 3C and S2, Table S3). The reduction in POLRMT expression in patients, the only known mitochondrial RNA polymerase, provides an attractive explanation to the reduction in mtDNA gene expression (Figures 3C and S2). The altered expression of c-Jun and JunD is intriguing – these are known nuclear gene expression regulators that were also found to bind human mtDNA in vivo (Blumberg et al., 2014). Our results suggest that these are candidate regulators of gene expression in both the nuclear and mitochondrial genomes (see Discussion).
Single cells RNA-seq analysis further support reduction in mtDNA gene expression in COVID-19 patients, especially in immune system cells
Our above-described analysis of bulk RNA-seq data suggest that the response of mitochondrial genes' expression in patients' peripheral blood cells is more consistent than cells coming from the respiratory tract. As blood cells are enriched by immune system cell types, it is possible that cells belonging to the immune system went through a more prominent response of mtDNA gene expression to COVID-19, than other cell types. To address this possibility, we analyzed eight scRNA-seq datasets including a total of 1,192,243 high-quality and annotated cells (Tables 1 and S4). In total, we analyzed cells from 77 critical/severe COVID-19 patients, 65 COVID-19 patients with a moderate phenotype, 65 healthy individuals and scRNA-seq data from the patients studied in Dataset III (above analyzed also for bulk RNA-seq in peripheral blood) (see Table 1 in STAR Methods). To avoid technical noise due to known elevated proportions of zero read counts in scRNA-seq data (Vallejos et al., 2017), we included in our analysis only cells whose sequencing read counts were higher than zero for each of our analyzed mtDNA genes (Table S4). Notably, since scRNA-seq libraries were prepared while enriching for poly(A) mRNAs, we focused our analysis on nine mtDNA genes with known longer 3′ poly(A)(ND1-4, CO1-3, CytB, and ATP6) (Slomovic et al., 2005), and hence reduced the false discovery rates of lower mtDNA gene expression in certain genes. Finally, we considered a finding biologically meaningful if it was observed in at least two independent datasets.
Firstly, in consistence with our analysis of bulk RNA-seq from peripheral blood, significantly reduced mtDNA-encoded genes' expression was observed in several T cells sub-types (Table S1, https://doi.org/10.17632/8kd3xjfrh4.1). Specifically, reduced mtDNA gene expression was consistently observed in CD8+ T cells from patients in BALF samples (Dataset VI) and from CD8 T cells, including CD8 memory T cells, from PBMC (Datasets VII, XI) (Figure 4A, Table S1 https://doi.org/10.17632/8kd3xjfrh4.1). A smaller effect on mtDNA gene expression (measured by the fraction of mtDNA genes which displayed a significant change in patients), yet still reduction in expression, was observed in CD8 effector cells from PBMC (Datasets VII, XII) (Figure 4B, Table S1 https://doi.org/10.17632/8kd3xjfrh4.1). Similarly, significantly reduced mtDNA gene expression in patients was also observed in CD4 T cells from 2/5 PBMC datasets, CD4 memory T cells (Dataset VII) (Figure 4B) and additional sub-types of T cells that were represented only in one dataset each, such as CCR7+ T cells and Follicular helper CD4 T (Dataset VI and Dataset XII, respectively) (Table S1). Significantly reduced expression of most mtDNA-encoded genes was also observed in patients' NK cells in BALF (Figure 4A), as well as in NK cells from PBMC samples (3/4 datasets) regardless of their severity level (Figure 4B, Table S1 https://doi.org/10.17632/8kd3xjfrh4.1). It is worth noting that in both CD4 T and NK cells we observed a significant reduction in mtDNA gene expression (Dataset X) in moderate patients as compared to healthy individuals. Reduction in mtDNA gene expression was also observed in other cell types from patients such as Monocytes, IgA PB, Dendritic cells (DC), and B cells (PBMC), although with weaker effect, confined to certain datasets (Figure 4B, https://doi.org/10.17632/8kd3xjfrh4.1). Notably, certain cell types showed inconsistent response among the analyzed datasets: mtDNA expression in platelets of COVID-19 patients was significantly elevated only in one out of two datasets (Table S1 https://doi.org/10.17632/8kd3xjfrh4.1) and proliferating T cells and Macrophages displayed a mixed tendency in mtDNA gene expression in patients (Table S1 https://doi.org/10.17632/8kd3xjfrh4.1). As currently we consider significance only for results that were replicated by at least two datasets, the biological importance of the mixed response should be evaluated once additional RNA-seq datasets become available. Taken together our analysis strongly suggests a consistent reduction in mtDNA gene expression in COVID-19, especially in immune system cells.
Figure 4.

The change in mtDNA genes expression in COVID-19 patients varies among cell types, and is more prominent in immune system cells
(A) Box Plots of mtDNA gene expression from healthy and COVID-19 patients in (i) CD8 T BALF cells (Dataset VI), (ii) CD8 T memory PBMC cells (Dataset VII), (iii) NK BALF cells (Dataset VI) (iv) Ciliated NP cells (Dataset VIII). X axis - gene names; Y axis – normalized UMI (unique molecular identifier) counts.
(B) Bar plot presenting the fraction of significantly altered mtDNA genes' expression per cell type. X axis: cell types present in at least two datasets, Y axis: fraction of mtDNA genes with significantly altered expression. Orange - Epithelial cells, Green – Immune system cells. Dataset numbers are indicated in parenthesis. The dashed line represents a threshold of significance in half of the analyzed mtDNA genes.
See also Tables S1 and S4 (https://doi.org/10.17632/8kd3xjfrh4.1).
Disease severity does not clearly impact the reduction in mtDNA gene expression in patients
While considering Datasets IV and IX (NP), division into individual cell types belonging to the immune system led to cell sample sizes below our threshold for further analysis. To overcome this problem, we grouped all cell types belonging to the immune system for further comparisons of differential expression between patients and controls; this analysis revealed consistent and significant reduction in mtDNA genes' expression in patients (https://doi.org/10.17632/8kd3xjfrh4.1). Finally, we took advantage of available expression data for B-cells and monocytes from scRNA-seq in the frame of Dataset III. Such analysis revealed significantly reduced expression of most mtDNA analyzed genes in CD14 + monocytes (pseudotime points 1,5), in CD16 + monocytes (mainly in pseudotime points 1,4, and 5), and in B cells (mainly in pseudotime points 1,4) (https://doi.org/10.17632/8kd3xjfrh4.1). Notably, while comparing mtDNA gene expression of the immune system cell types between the pseudo time points (Dataset III- scRNA-seq), and between the severity levels (e.g., moderate and critical) in Datasets VI-XII (Table S1), no significance was observed between most of the pseudo time points. These results further suggest that the reduced mtDNA gene expression in COVID-19 patients occur in a variety of cell types from the immune system, with apparently inconsistent effect of disease severity.
Respiratory tract harbor immune system cells which display mtDNA gene expression reduction in COVID-19 patients
Our analysis of bulk RNA-seq from upper respiratory tract samples did not reveal any significant changes in COVID-19 patients as compared to healthy controls. Nevertheless, it is possible that either (A) mtDNA gene expression did not change in cells from the respiratory tract of the patients, or that (B) the response of mtDNA gene expression in the respiratory tract of patients varies among cell types. Our analysis of scRNA-seq imply weaker, yet significantly reduced mtDNA gene expression (i.e., evident only in ND2, ND4, and CO3 transcripts) in epithelial cells from patients' respiratory tract samples (BALF, Dataset VI). This finding contrasts with the observed stronger reduction in the expression of most mtDNA genes in T cells and NK cells isolated from the same BALF samples (Table S1). Secondly and similarly, analysis of respiratory tract samples from Dataset VIII revealed significantly reduced expression of four mtDNA genes (ND1, CO3, CYB, and ATP6) in Ciliated, Secretory, and Secretory-differentiated epithelial cells in COVID-19 patients, yet none of the genes displayed significant change in Squamous cells from this dataset. However, all epithelial cells (such as Squamous, Ciliated and Secretory) from Dataset IX, except for Goblet cells did not show any significant change in the expression of mtDNA encoded genes (Figures 4A and 4B, Table S1 https://doi.org/10.17632/8kd3xjfrh4.1). Finally, IFNG-responsive epithelial cells (IRC), displayed a significant reduction in the expression of only a single mtDNA gene (ATP6) (https://doi.org/10.17632/8kd3xjfrh4.1). These results suggest that whereas cells belonging to the immune system consistently show reduction in mtDNA gene expression, whether isolated from the peripheral blood or from the respiratory tract, such response is weaker, and notably varies among respiratory tract epithelial cells.
Analysis of scRNA-seq databases identify candidate regulators that explain reduced mtDNA gene expression in COVID-19 patients
We next asked whether reduction in mtDNA gene expression in certain cell types from patients is associated with expression changes in genes with mitochondrial function encoded by the nuclear genome. To address this question, we performed differential expression analysis of OXPHOS genes and mitochondrial-related pathways that were analyzed in the bulk RNA-seq datasets (see above). We found that in cell types which displayed reduction in the expression of most mtDNA genes, the significant expression of most nuclear DNA-encoded OXPHOS genes showed elevated expression (in all tested Datasets) (Table S3, https://doi.org/10.17632/8kd3xjfrh4.1). Similar to the analysis of bulk RNA-seq, our analysis of mitochondria-related pathways revealed that such cells displayed mostly elevated expression of genes belonging to glycolysis, response to type I interferon and MAVS pathway genes (https://doi.org/10.17632/8kd3xjfrh4.1).
To further test our hypothesis that the reduced mtDNA gene expression in certain cell types from patient samples reflects downregulation of mtDNA gene expression upon SARS-CoV-2 infection, we assessed changes in regulatory factors of mtDNA genes' expression (Table S3) in the scRNA-seq datasets from patients and controls. This analysis revealed that in consistence with the analysis of bulk RNA-seq, significantly reduced expression was observed in JunD in the following cell types: CD8 T cells, T cells, CCR7+ T cells (Dataset VI BALF), NK cells (Datasets VI BALF, XI PBMC), DC, Monocyte (Dataset XI PBMC), CD4n T cells (Dataset VII PBMC), in all immune cells from Dataset VIII NP and in CD14 + Monocytes (Dataset III) (Table S3 in all pseudotimes except for pseudotime 2). Similarly, in consistence with the analysis of bulk RNA-seq, the expression of c-Jun was elevated in B cells (XI PBMC), mDC, CCR7+ T, all T cells from Dataset VI (BALF) (Table S3) and non-classical CD16 + Monocytes (Dataset III; in all pseudotimes except for pseudotime 2). These results support the functional involvement of these two members of the activator protein 1 (AP1) transcription factors family in the regulatory response of mtDNA gene expression to SARS-CoV-2 infection.
Discussion
Our results indicate a tissue- and cell-type-dependent reduction in mtDNA-encoded gene expression in response to SARS-CoV-2 infection. Firstly, bulk RNA-seq analysis of samples coming from the peripheral and whole blood showed such reduction, in contrast to samples coming from the respiratory tract that did not. Secondly, analysis of scRNA-seq revealed reduced mtDNA gene expression levels in several tested immune system cells, as compared to little or no reduction of mtDNA gene expression in respiratory tract epithelial cells. These findings support cell-type and tissue-dependent differences in the magnitude of mitochondrial gene expression regulatory effect in COVID-19 patients. We are tempted to interpret these results, as partially explaining the apparent lack of change in mtDNA gene expression in patients' respiratory tract bulk RNA-seq samples: the weaker mtDNA gene expression differences in epithelial cells from patients (as well as in some immune system cells isolated from the respiratory tract) might have masked the difference in mtDNA gene expression in the respiratory tract, but not in blood. Because two out of the five analyzed bulk RNA-seq datasets stem from different sequencing platforms (Datasets IV, V), the differences in the impact on mtDNA gene expression could be attributed to the different tissues of origin, although variation in sample collection methods and/or sequencing platforms cannot be excluded. Another parameter that may affect gene expression differences in the mitochondria is mtDNA genetic backgrounds (haplogroups) (Cohen et al., 2016). It would therefore be of interest to study the contribution of haplogroups to gene expression differences between healthy and COVID-19 samples once larger sample sizes become available.
We hypothesized that reduction in mtDNA gene expression in samples from COVID-19 patients stem from a general alteration in OXPHOS gene co-regulation. The general tendency toward increased expression in nuclear DNA-encoded OXPHOS genes in COVID-19 patients supports our hypothesis, as by-and-large healthy human tissues display positive co-expression of mitochondrial and nuclear DNA-encoded members of the OXPHOS machinery (Barshad et al., 2018). This leads to two possible interpretations: the altered coordination of mito-nuclear gene expression in COVID-19 patients may (A) reflect a compromised mito-nuclear co-regulation, and/or (B) controlled rewiring of the OXPHOS machinery to glycolysis. Indeed, we found that COVID-19 patients exhibited a general increase in the expression of genes encoding glycolysis enzymes, reduced expression of LDHB, and increased expression of LDHA. This is consistent the observed dependence of SARS-CoV-2 replication in monocytes on active glycolysis (Codo et al., 2020), suggesting that OXPHOS malfunction in COVID-19 patients primarily enables generation of a glycolytic environment, which in turn promotes the cytokine storm. This interpretation is supported by our observed increased expression of genes involved in the IFN pathway, as well as increased expression of genes involved in RIG-I-like receptors and MAVS pathway as previously shown (Gordon et al., 2020; Rui et al., 2021). The latter might lead to activation of a signaling cascade that positively induces the genes encoding type I interferons (Rehwinkel and Gack 2020). We speculate that our observed consistent alteration in mtDNA gene expression and related pathways enabled the viral induced cytokine storm, and hence is fundamental to the disease etiology. Testing for this possibility requires a future time course controlled experiment in cells (preferably from the immune system) in which gene expression is assessed in time intervals following SARS-CoV-2 infection.
We noticed, that cells and tissues that displayed a consistently significant difference in mitochondrial gene expression between patients and controls (in at least two independent datasets), displayed reduction in mtDNA gene expression in patients (both in bulk RNA-seq and scRNA-seq), regardless of difference between sample collection sites and sequencing platforms used. In contrast, elevated mtDNA gene expression in patients was inconsistent among databases (such as the observation of Platelets). This finding strongly suggests that SARS-CoV-2 affects mitochondrial regulation in a similar manner in different cells. Nevertheless, although the direction of the effect is consistent (e.g., reduction in mtDNA gene expression in COVID-19 patients) the regulatory phenomenon is influenced by cell-types, which raises an interesting question as to the role of the affected cell types in the disease etiology.
As mentioned above, our findings indicate that bulk RNA-seq from blood, but not from the respiratory tract, showed significant reduction in mtDNA gene expression. Apart from the apparent cell type composition, and differences between these two types of samples, extracellular intact cell-free active mitochondria have been found in blood (Al Amir Dache et al., 2020). It will therefore be of interest to measure the number of extracellular mitochondria in both blood and respiratory tract samples from COVID-19 patients as compared to control, and assess their transcriptional signatures. This may add another layer to our understanding of the alteration in mitochondrial gene expression patterns in COVID-19 patients.
To identify first clues for the candidate mechanism underlying reduced mtDNA gene expression in COVID-19 patients we took a candidate gene approach and sought for association between altered mtDNA gene expression in patients with known regulators of mtDNA transcription, post transcription, and replication. Our findings indicate that in patient peripheral and whole blood samples, reduction in mtDNA gene expression is associated with reduced expression of the mitochondrial RNA-polymerase (POLRMT), along with elevated expression of c-Jun, and reduced expression of JunD. The reduction in POLRMT expression provides a simple explanation for the reduced mtDNA gene expression in the patients' peripheral blood, but this observation was not clearly identified in single cells. Interestingly, unlike POLRMT, the altered expression of c-Jun and JunD was consistently found in both the bulk RNA-seq and sc-RNA-seq datasets, e.g., the expression of c-Jun increased and JunD decreased in patients in cell types that showed significantly reduced mtDNA gene expression. This particularly attracted our attention, as c-Jun and JunD binding in human mtDNA was identified in certain cell lines (Blumberg et al., 2014), thus suggesting their possible involvement in mtDNA transcriptional regulation. Our identified association of altered c-Jun and JunD expression in patient samples suggests that they likely act as a repressor and activator, respectively, of mtDNA gene expression, and that such impact is induced in patients. As c-Jun and JunD are well-known for their regulatory targets in the nucleus, it will be of great interest to experimentally assess their mechanism of action while in the mitochondria, their possible involvement in mito-nucelar co-regulation, and whether their mitochondrial localization is altered in response to SARS-CoV-2 infection.
While further considering the mechanism by which mtDNA gene expression was down regulated in COVID-19 patients, one should consider additional factors, such as microRNAs. Indeed, recent microRNA-seq GO-term analysis of peripheral blood (Li et al., 2020) revealed altered expression of microRNAs in COVID-19 patients that target genes which associate with the mitochondrial matrix. Furthermore, microRNA-seq differential expression analysis from blood plasma (Farr et al., 2021) revealed that the expression levels of hsa-miR-542-5p, a miRNA which likely co-localize with human mitochondria (Borralho et al., 2015) and was previously associated with mitochondrial dysfunction (Garros et al., 2017), was significantly reduced in COVID-19 patients. Third, the expression of hsa-miR-483-5p which binds and represses the activity of FIS1 (Purohit and Saini 2021), a key mitochondrial fission component (James et al., 2003), was significantly elevated in COVID-19 patients who were treated by supplemental oxygen. Hence, there is room for future investigation of the role of mitochondrial-targeted microRNAs in mtDNA regulation in general, and in COVID-19 in particular.
Along with the observed reduced expression of mitochondrial genes we identified elevated expression of several genes involved in the IFN1 response pathway. This is interesting, as it was previously shown that treatment of mammalian cells with IFN leads to reduction in mtDNA gene expression, thus suggesting a regulatory impact of IFN on mtDNA transcripts (Shan et al., 1990). This offers an attractive explanation for the connection between the cellular response to SARS-CoV-2 infection and changes in mitochondrial regulation. As our results indicate that such changes occur in a cell type-dependent manner, mostly apparent in immune system cells, it is not surprising that previous analyses of (lung) epithelial cells following SARS-CoV-2 infection displayed a rather low response of type I and III interferon (Blanco-Melo et al., 2020), and inconsistent changes in mtDNA genes expression (Miller et al., 2021). Hence, it will be of great interest to carefully assess in the future the underlying mechanism by which IFN modulates mitochondrial gene expression regulation.
In summary, we observed reduced levels of mtDNA gene expression in multiple cell types, yet preferentially in cells belonging to the immune system, regardless of collection from blood or from the respiratory tract. This finding, along with apparent opposite gene expression changes in nuclear-encoded OXPHOS genes in COVID-19 patients suggest departure from co-expression regulation of the mitochondrial and nuclear genomes. This interpretation may explain the elevated expression of genes involved in ROS production, which likely reflect cellular response to mitochondrial dysfunction. This change was also accompanied by elevated expression of glycolytic enzymes, especially LDHA, and reduction of LDHB expression. Such findings suggest that upon SARS-CoV-2 infection, cells which particularly belong to the immune system, rewire to glycolysis. Analysis of mtDNA gene expression in the unique set of patients who provided samples during COVID-19 disease progression suggests that the reduction of mtDNA gene expression is reversible upon recovery. It is thus possible that recovery of mitochondrial function predicts better health conditions of COVID-19 patients, thus underlining the mitochondria as an important drug target to ameliorate patients' health conditions.
Limitations of the study
Our work is based on analysis of gene expression at the RNA level. Nevertheless, expression variability in COVID-19 patients may be also observed at the protein level, which would be interesting to assess, yet obviously cannot be observed in analysis of RNA-seq. Secondly, RNA-seq analysis, which corresponds to the steady-state RNA levels, cannot decipher whether the observed changes in mitochondrial gene expression in COVID-19 patients were due to transcriptional regulation, RNA decay, or both. Clues to such were identified by observing correlation between the changes in mitochondrial gene expression in patients and potential regulators of mitochondrial transcription (POLRMT, c-Jun and JunD). Such associations should be considered with caution until subsequent experimental validations are performed.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Dan Mishmar (dmishmar@bgu.ac.il).
Materials availability
This study did not generate new unique reagents.
Method details
Data acquisition and quality control
Bulk RNA-seq raw data (read) counts were downloaded from GEO and Github (Table 1) for Datasets I-V. To compare Healthy and COVID-19 patients in bulk RNA-seq Dataset III, samples belonging to pseudotimes 1, 2, 3 (with less than four samples each) were merged, similarly to the stages of severe diseases, as previously explained (Bernardes et al. 2020). Pseudotime phases were used as previously defined: 0- Healthy, 1 - incremental(where clinical symptoms and inflammatory markers were increasing, ICU or non-ICU), 2 –critical (ICU, mechanically ventilated with signs of ARDS), 3 – complicated (a state with severe signs of a systemic inflammatory response, ICU, high-flow oxygen, intubation readiness), 4 - moderate or early convalescent (patients with supplemental oxygen, significant signs of systemic inflammation), 5 - late convalescent (patients with intermittent supplemental oxygen, minor signs of inflammation), 6 - recovery/pre-discharge, no supplemental oxygen, absence of inflammation markers.
scRNA-seq data
scRNA-seq Seurat processed objects and sparse count matrices were obtained from each respective study (Stuart et al., 2019) (Table 1). Severity levels of cells in each dataset were determined previously. Due to small sample size and to avoid inconsistency between datasets, severity level 'mild' (WHO scores 1–3) was removed from further analyses. For Dataset VII, ventilated patients who were diagnosed with acute respiratory distress syndrome were annotated here as 'critical', and patients who were not ventilated at the time of sample collection were annotated as 'moderate'. While considering Datasets XI and XII we noticed that the samples were collected from individuals from several locations in China (Dataset XI) and several UK locations (Dataset XII). Specifically, to avoid batch effect and small sample sizes (e.g., of individuals and cells) we analysed samples collected only in Beijing in Dataset XI, and samples collected from Newcastle and Cambridge but not from London in Dataset XII. Furthermore, for Dataset VI, a filtered integrated matrix was created from the filtered expression matrices using Seurat v3 pipeline as previously described to remove batch effects across all donors (Butler et al., 2018). For all datasets Seurat objects/h5ad objects were analysed with the same cell type annotations. For the sake of consistency, Dataset XII that was downloaded as h5ad objects was converted into Seurat objects using “sceasy” R package (Cakir et al. 2020) and h5 objects (Dataset VI) were converted into Seurat objects using “Read10X_h5” Seurat function and was processed as previously described (Liao et al. 2020). Additionally, expression matrices (Dataset IX, XI) were converted into Seurat objects using high quality annotated cells as performed previously (Ren et al. 2021; Ziegler et al. 2021).
Five datasets (Datasets III, VI, VIII, XI and XII) used 10X Genomics sequencing platform while datasets VII, IX, X, utilized the seq-well sequencing platform. We used the following Gene Ontology (GO) Terms to download and perform differential expression analysis: Genes under the “TCA” (GO:0006099), glycolytic process (GO:0006096), suppression by virus of host viral-induced cytoplasmic pattern recognition receptor signalling pathway via inhibition of MAVS activity (GO:0039545), response to type I interferon (GO0034340), respond to ROS (GO:0000302), ROS formation (GO:1903409) and L-lactate dehydrogenase activity (GO:0004459).
To avoid technical noise, cells with at least one mtDNA gene with zero reads were removed from further analysis for all scRNA-seq datasets. Notably, each scRNA-seq dataset were separately analysed.
Quantification and statistical analysis
Bulk RNA-seq raw expression counts matrices were normalized using DESeq normalization method available from the DESeq2 R package (Love et al., 2014). Differential expression of genes that participate in mitochondrial activities was assessed either considering healthy and COVID-19 patients in each dataset, or while comparing healthy individuals to each of the disease pseudotime samples (Dataset III- bulk RNA-seq), using DESeq2. Correction for each specified set of genes was performed using Benjamini–Hochberg (BH) adjusted p-value value of 0.05.
For scRNA-seq datasets, high quality cells (already annotated in the original studies) were used for each respective dataset. Since high proportions of zero read counts in scRNA-seq data can skew the analysis due to technical noise (Vallejos et al. 2017), we included in our analysis only cells whose sequencing read counts were higher than zero in our analysis of mtDNA genes. For each respective Seurat object, the analysis was performed per cell type. Permutation of cells from each cell type was applied to control for variation in cell numbers collected from healthy individuals and patients. Equal number of cells was sampled (n=50 cells, for each group compared) to generate subset of Seurat object 1000 times per cell type. To avoid small sample sizes, we removed cell types represented by less than 50 cells per group of individuals (i.e., control healthy individuals, moderate and severe/critical patients or in each of the pseudotime points in Dataset III) and with at least 300 cells in total. Two comparisons were performed: in the first analysis we compared gene expression levels between cells from healthy individuals and COVID-19 patients without considering severity levels (sampling 1000 times n=50 cells for each group). The second analysis was performed using pairwise comparison of gene expression levels between cells from different conditions. In each iteration “FindAllMarkers” function of Seurat was used (performing Wilcoxon signed rank test), followed by BH (false discovery rate) correction. P-values for all iterations were computed by dividing the number of insignificant p-values (p > 0.05) by 1000. For gene-by-gene nuclear differential expression analysis “FindAllMarkers” function was applied to all cells per cell type with min.pct = 0.25 parameter. Genes from each group of genes were extracted from the resulted list of genes and were corrected with (BH) correction for multiple testing. Heatmaps were generate using the 'ComplexHeatmap' R package (Gu et al., 2016) using consistent genes with log fold change greater than 0.2 or less than −0.2. Box plot and bar plot graphs were generated using “ggplot2” (Wickham 2009) and RColorBrewer R packages. Tables were generated using “openxlsx”, “tidyr” and “reshape2” (Zhang 2016) R packages.
Acknowledgments
This study was funded by grants from the Israel Science Foundation (ISF 372/17, and 404/21) and from the US Army Life Sciences Division (LS67993) awarded to DM. This study was stimulated by early discussion with Dorit and Menashe Sonnenschein.
Author contributions
HM analyzed the data and participated in writing the manuscript; AZ Analyzed bulk RNA-seq data; DM conceived the study, wrote the paper and supervised the analysis.
Declaration of interests
The authors declare no competing interests. The manuscript is based on analyses of publicly available data. The source of each dataset is mentioned within the STAR Methods section.
Published: December 17, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103471.
Supplemental information
Yellow background - genes with log fold change higher than 0.2 or less than −0.2, Bold: genes with log fold change higher than 0.5 or less than −0.5.
Data and code availability
Links of the source data used in this paper are available in the Key Resources Table. Additional Supplemental Items are available from Mendeley Data at https://doi.org/10.17632/8kd3xjfrh4.1. DOI: doi:10.17632/8kd3xjfrh4.1.
The bulk and scRNA-seq codes used for data analysis are available from GitHub https://github.com/medinih/COVID_mishmarlab.
References
- Al Amir Dache Z., Otandault A., Tanos R., Pastor B., Meddeb R., Sanchez C., Arena G., Lasorsa L., Bennett A., Grange T., et al. Blood contains circulating cell-free respiratory competent mitochondria. FASEB. 2020;34:3616–3630. doi: 10.1096/fj.201901917RR. [DOI] [PubMed] [Google Scholar]
- Ardail D., Lerme F., Puymirat J., Morel G. Evidence for the presence of alpha and beta-related T3 receptors in rat liver mitochondria. Eur. J. Cell Biol. 1993;62:105–113. [PubMed] [Google Scholar]
- Ballestar E., Farber D.L., Glover S., Horwitz B., Meyer K., Nikolić M., Ordovas-Montanes J., Sims P., Shalek A., Vandamme N., et al. Single cell profiling of COVID-19 patients: an international data resource from multiple tissues. medrxiv. 2020 doi: 10.1101/2020.11.20.20227355. [DOI] [Google Scholar]
- Barshad G., Blumberg A., Cohen T., Mishmar D. Human primitive brain displays negative mitochondrial-nuclear expression correlation of respiratory genes. Genome Res. 2018;28:952–967. doi: 10.1101/gr.226324.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardes J.P., Mishra N., Tran F., Bahmer T., Best L., Blase J.I., Bordoni D., Franzenburg J., Geisen U., Josephs-Spaulding J., et al. Longitudinal multi-omics analyses identify responses of megakaryocytes, erythroid cells, and plasmablasts as hallmarks of severe COVID-19. Immunity. 2020;53:1296–1314 e9. doi: 10.1016/j.immuni.2020.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Melo D., Nilsson-Payant B.E., Liu W.C., Uhl S., Hoagland D., Moller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045 e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg A., Sailaja B.S., Kundaje A., Levin L., Dadon S., Shmorak S., Shaulian E., Meshorer E., Mishmar D. Transcription factors bind negatively-selected sites within human mtDNA genes. Genome Biol. Evol. 2014;6:2634–2646. doi: 10.1093/gbe/evu210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordon Y. mtDNA synthesis ignites the inflammasome. Nat. Rev. Immunol. 2018;18:539. doi: 10.1038/s41577-018-0049-8. [DOI] [PubMed] [Google Scholar]
- Borralho P.M., Rodrigues C.M., Steer C.J. microRNAs in mitochondria: an unexplored niche. Adv. Exp. Med. Biol. 2015;887:31–51. doi: 10.1007/978-3-319-22380-3_3. [DOI] [PubMed] [Google Scholar]
- Burtscher J., Cappellano G., Omori A., Koshiba T., Millet G.P. Mitochondria: in the cross fire of SARS-CoV-2 and immunity. iScience. 2020;23:101631. doi: 10.1016/j.isci.2020.101631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cakir B., Prete M., Huang N., van Dongen S., Pir P., Kiselev V.Y. Comparison of visualization tools for single-cell RNAseq data. NAR Genom. Bioinform. 2020;2 doi: 10.1093/nargab/lqaa052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A., Seyfferth J., Lucci J., Gilsbach R., Preissl S., Bottinger L., Martensson C.U., Panhale A., Stehle T., Kretz O., et al. MOF acetyl transferase regulates transcription and respiration in mitochondria. Cell. 2016;167:722–738 e3. doi: 10.1016/j.cell.2016.09.052. [DOI] [PubMed] [Google Scholar]
- Chua R.L., Lukassen S., Trump S., Hennig B.P., Wendisch D., Pott F., Debnath O., Thurmann L., Kurth F., Volker M.T., et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020;38:970–979. doi: 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- Codo A.C., Davanzo G.G., Monteiro L.B., de Souza G.F., Muraro S.P., Virgilio-da-Silva J.V., Prodonoff J.S., Carregari V.C., de Biagi Junior C.A.O., Crunfli F., et al. Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1alpha/Glycolysis-Dependent Axis. Cell Metab. 2020;32:437–446 e435. doi: 10.1016/j.cmet.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen T., Levin L., Mishmar D. Ancient out-of-Africa mitochondrial DNA variants associate with distinct mitochondrial gene expression patterns. PLoS Genet. 2016;12 doi: 10.1371/journal.pgen.1006407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr R.J., Rootes C.L., Rowntree L.C., Nguyen T.H.O., Hensen L., Kedzierski L., Cheng A.C., Kedzierska K., Au G.G., Marsh G.A., et al. Altered microRNA expression in COVID-19 patients enables identification of SARS-CoV-2 infection. PLoS Pathog. 2021;17 doi: 10.1371/journal.ppat.1009759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Vizarra E., Enriquez J.A., Perez-Martos A., Montoya J., Fernandez-Silva P. Mitochondrial gene expression is regulated at multiple levels and differentially in the heart and liver by thyroid hormones. Curr. Genet. 2008;54:13–22. doi: 10.1007/s00294-008-0194-x. [DOI] [PubMed] [Google Scholar]
- Ganji R., Reddy P.H. Impact of COVID-19 on mitochondrial-based immunity in aging and age-related diseases. Front Aging Neurosci. 2020;12:614650. doi: 10.3389/fnagi.2020.614650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garros R.F., Paul R., Connolly M., Lewis A., Garfield B.E., Natanek S.A., Bloch S., Mouly V., Griffiths M.J., Polkey M.I., et al. MicroRNA-542 promotes mitochondrial dysfunction and SMAD activity and is elevated in intensive care unit-acquired weakness. Am. J. Respir. Crit. Care Med. 2017;196:1422–1433. doi: 10.1164/rccm.201701-0101OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti P., Ilamathi H.S., Todkar K., Germain M. Mitochondria targeted viral replication and survival strategies-prospective on SARS-CoV-2. Front. Pharmacol. 2020;11:578599. doi: 10.3389/fphar.2020.578599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O'Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020 doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- Holder K., Reddy P.H. The COVID-19 effect on the immune system and mitochondrial dynamics in diabetes, obesity, and dementia. Neuroscientist. 2021;27:331–339. doi: 10.1177/1073858420960443. [DOI] [PubMed] [Google Scholar]
- James D.I., Parone P.A., Mattenberger Y., Martinou J.C. hFis1, a novel component of the mammalian mitochondrial fission machinery. J. Biol. Chem. 2003;278:36373–36379. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- Lambertini E., Penolazzi L., Morganti C., Lisignoli G., Zini N., Angelozzi M., Bonora M., Ferroni L., Pinton P., Zavan B., et al. Osteogenic differentiation of human MSCs: specific occupancy of the mitochondrial DNA by NFATc1 transcription factor. Int. J. Biochem. Cell Biol. 2015;64:212–219. doi: 10.1016/j.biocel.2015.04.011. [DOI] [PubMed] [Google Scholar]
- Levy Y., Wiedemann A., Hejblum B.P., Durand M., Lefebvre C., Surenaud M., Lacabaratz C., Perreau M., Foucat E., Dechenaud M., et al. CD177, a specific marker of neutrophil activation, is associated with coronavirus disease 2019 severity and death. iScience. 2021;24:102711. doi: 10.1016/j.isci.2021.102711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Hu X., Li L., Li J.H. Differential microRNA expression in the peripheral blood from human patients with COVID-19. J. Clin. Lab. Anal. 2020;34 doi: 10.1002/jcla.23590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F., et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- Lieberman N.A.P., Peddu V., Xie H., Shrestha L., Huang M.L., Mears M.C., Cajimat M.N., Bente D.A., Shi P.Y., Bovier F., et al. In vivo antiviral host transcriptional response to SARS-CoV-2 by viral load, sex, and age. PLoS Biol. 2020;18 doi: 10.1371/journal.pbio.3000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mick E., Kamm J., Pisco A.O., Ratnasiri K., Babik J.M., Calfee C.S., Castaneda G., DeRisi J.L., Detweiler A.M., Hao S., et al. Upper airway gene expression differentiates COVID-19 from other acute respiratory illnesses and reveals suppression of innate immune responses by SARS-CoV-2. medRxiv. 2020 doi: 10.1101/2020.05.18.20105171. [DOI] [Google Scholar]
- Miller B., Silverstein A., Flores M., Cao K., Kumagai H., Mehta H.H., Yen K., Kim S.J., Cohen P. Host mitochondrial transcriptome response to SARS-CoV-2 in multiple cell models and clinical samples. Scientific Rep. 2021;11:3. doi: 10.1038/s41598-020-79552-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porporato P.E., Dhup S., Dadhich R.K., Copetti T., Sonveaux P. Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front Pharmacol. 2011;2:49. doi: 10.3389/fphar.2011.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit P.K., Saini N. Mitochondrial microRNA (MitomiRs) in cancer and complex mitochondrial diseases: current status and future perspectives. Cell Mol. Life Sci. 2021;78:1405–1421. doi: 10.1007/s00018-020-03670-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read J.A., Winter V.J., Eszes C.M., Sessions R.B., Brady R.L. Structural basis for altered activity of M- and H-isozyme forms of human lactate dehydrogenase. Proteins. 2001;43:175–185. doi: 10.1002/1097-0134(20010501)43:2<175::aid-prot1029>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Rehwinkel J., Gack M.U. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020;20:537–551. doi: 10.1038/s41577-020-0288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X., Wen W., Fan X., Hou W., Su B., Cai P., Li J., Liu Y., Tang F., Zhang F., et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell. 2021;184:1895–1913 e1819. doi: 10.1016/j.cell.2021.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui Y., Su J., Shen S., Hu Y., Huang D., Zheng W., Lou M., Shi Y., Wang M., Chen S., et al. Unique and complementary suppression of cGAS-STING and RNA sensing- triggered innate immune responses by SARS-CoV-2 proteins. Signal Transduct. Target. Therapy. 2021;6:123. doi: 10.1038/s41392-021-00515-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan B., Vazquez E., Lewis J.A. Interferon selectively inhibits the expression of mitochondrial genes: a novel pathway for interferon-mediated responses. EMBO J. 1990;9:4307–4314. doi: 10.1002/j.1460-2075.1990.tb07879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H., Lan D., Duan Z., Liu Z., Min J., Zhang L., Huang J., Su J., Chen S., Xu A. Upregulation of mitochondrial gene expression in PBMC from convalescent SARS patients. J. Clin. Immunol. 2006;26:546–554. doi: 10.1007/s10875-006-9046-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She H., Yang Q., Shepherd K., Smith Y., Miller G., Testa C., Mao Z. Direct regulation of complex I by mitochondrial MEF2D is disrupted in a mouse model of Parkinson disease and in human patients. J. Clin. Invest. 2011;121:930–940. doi: 10.1172/JCI43871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C.S., Qi H.Y., Boularan C., Huang N.N., Abu-Asab M., Shelhamer J.H., Kehrl J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014;193:3080–3089. doi: 10.4049/jimmunol.1303196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K.K., Chaubey G., Chen J.Y., Suravajhala P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am. J. Physiol. Cell Physiol. 2020;319:C258–C267. doi: 10.1152/ajpcell.00224.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slomovic S., Laufer D., Geiger D., Schuster G. Polyadenylation and degradation of human mitochondrial RNA: the prokaryotic past leaves its mark. Mol. Cell. Biol. 2005;25:6427–6435. doi: 10.1128/MCB.25.15.6427-6435.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson E., Reynolds G., Botting R.A., Calero-Nieto F.J., Morgan M.D., Tuong Z.K., Bach K., Sungnak W., Worlock K.B., Yoshida M., et al. Single-cell multi-omics analysis of the immune response in COVID-19. Nat. Med. 2021;27:904–916. doi: 10.1038/s41591-021-01329-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902 e1. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thair S.A., He Y.D., Hasin-Brumshtein Y., Sakaram S., Pandya R., Toh J., Rawling D., Remmel M., Coyle S., Dalekos G.N., et al. Transcriptomic similarities and differences in host response between SARS-CoV-2 and other viral infections. iScience. 2021;24:101947. doi: 10.1016/j.isci.2020.101947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejos C.A., Risso D., Scialdone A., Dudoit S., Marioni J.C. Normalizing single-cell RNA sequencing data: challenges and opportunities. Nat. Methods. 2017;14:565–571. doi: 10.1038/nmeth.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. Springer; 2009. Ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- Wilk A.J., Lee M.J., Wei B., Parks B., Pi R., Martinez-Colon G.J., Ranganath T., Zhao N.Q., Taylor S., Becker W., et al. Multi-omic profiling reveals widespread dysregulation of innate immunity and hematopoiesis in COVID-19. J. Exp. Med. 2021;218:e20210582. doi: 10.1084/jem.20210582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk A.J., Rustagi A., Zhao N.Q., Roque J., Martinez-Colon G.J., McKechnie J.L., Ivison G.T., Ranganath T., Vergara R., Hollis T., et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020;26:1070–1076. doi: 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf A.R., Mootha V.K. Functional genomic analysis of human mitochondrial RNA processing. Cell Rep. 2014;7:918–931. doi: 10.1016/j.celrep.2014.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K.E., Fazal F.M., Parker K.R., Zou J., Chang H.Y. RNA-GPS predicts SARS-CoV-2 RNA residency to host mitochondria and nucleolus. Cell Syst. 2020 doi: 10.1016/j.cels.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoboue E.D., Sitia R., Simmen T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018;9:331. doi: 10.1038/s41419-017-0033-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. Reshaping and aggregating data: an introduction to reshape package. Ann. Transl. Med. 2016;4:78. doi: 10.3978/j.issn.2305-5839.2016.01.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler C.G.K., Miao V.N., Owings A.H., Navia A.W., Tang Y., Bromley J.D., Lotfy P., Sloan M., Laird H., Williams H.B., et al. Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19. Cell. 2021 doi: 10.1016/j.cell.2021.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo H., Wan Y. Metabolic reprogramming in mitochondria of myeloid cells. Cells. 2019;9 doi: 10.3390/cells9010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Yellow background - genes with log fold change higher than 0.2 or less than −0.2, Bold: genes with log fold change higher than 0.5 or less than −0.5.
Data Availability Statement
Links of the source data used in this paper are available in the Key Resources Table. Additional Supplemental Items are available from Mendeley Data at https://doi.org/10.17632/8kd3xjfrh4.1. DOI: doi:10.17632/8kd3xjfrh4.1.
The bulk and scRNA-seq codes used for data analysis are available from GitHub https://github.com/medinih/COVID_mishmarlab.