

Abstract
Telomeres are highly repetitive DNA sequences found at the ends of chromosomes that protect the chromosomes from deterioration duringcell division. Here, using whole-genome re-sequencing and terminal restriction fragment assays, we found substantial natural intraspecific variation in telomere length in Arabidopsis thaliana, rice (Oryza sativa), and maize (Zea mays). Genome-wide association study (GWAS) mapping in A. thaliana identified 13 regions with GWAS-significant associations underlying telomere length variation, including a region that harbors the telomerase reverse transcriptase (TERT) gene. Population genomic analysis provided evidence for a selective sweep at the TERT region associated with longer telomeres. We found that telomere length is negatively correlated with flowering time variation not only in A. thaliana, but also in maize and rice, indicating a link between life-history traits and chromosome integrity. Our results point to several possible reasons for this correlation, including the possibility that longer telomeres may be more adaptive in plants that have faster developmental rates (and therefore flower earlier). Our work suggests that chromosomal structure itself might be an adaptive trait associated with plant life-history strategies.
Telomere length is associated with flowering time variation in Arabidopsis, rice, and maize.

Introduction
Telomeres are regions of repetitive sequences that cap the ends of eukaryotic chromosomes to protect them from deterioration and from eliciting a DNA damage response (Shay and Wright, 2019). During DNA replication, failure to fill in terminal base pairs at the lagging strand leads to the end-replication problem (Olovnikov, 1971, 1973; Watson, 1972), resulting in the shortening of chromosome ends during each cell division and the eventual loss of replicative capacity (Hayflick and Moorhead, 1961; van Deursen, 2014). To prevent this loss of chromosomal DNA at the termini, the ribonucleoprotein enzyme complex telomerase, whose core components consist of a telomerase reverse transcriptase (TERT) and RNA template (TR; Osterhage and Friedman, 2009; Song et al., 2019), binds to single-stranded telomeric DNA at the 3′-end and processively extends the telomere sequence (Wu et al., 2017). Other specialized telomere binding proteins are also recruited to prevent the telomere from being detected as damaged DNA (Fulcher et al., 2014).
Eukaryotic telomeres consist of a tandem repeat of TG-rich microsatellite sequences (Podlevsky and Chen, 2016). The core telomeric repeat sequence is conserved among species. For instance, vertebrates have the telomeric repeat TTAGGG (Meyne et al., 1989), while in most plants, the sequence is TTTAGGG (Fajkus et al., 2005). The most noticeable difference in telomeres between organisms is differences in telomere length, which can be as short as 300 bps in yeast (Saccharomyces cerevisiae; Gatbonton et al., 2006) and up to 150 kb in tobacco (Nicotiana tabacum; Fajkus et al., 1995). Within species, telomere sequences also display substantial heritable length variation. Several examples of telomere length polymorphisms and the underlying genes responsible for this variation have been identified in humans (Homo sapiens), yeast, and the roundworm Caenorhabditis elegans (Liti et al., 2009; Levy et al., 2010; Jones et al., 2012; Codd et al., 2013; Cook et al., 2016). In plants, variation in telomere length has also been observed between individuals (Burr et al., 1992; Shakirov and Shippen, 2004; Maillet et al., 2006; Fulcher et al., 2015), between organs (Kilian et al., 1995), and between cell types (González-García et al., 2015). Quantitative trait locus (QTL) studies in Arabidopsis thaliana and maize (Zea mays) have indicated that natural variation in telomere length is a heritable complex trait (Burr et al., 1992; Brown et al., 2011; Fulcher et al., 2015). In Arabidopsis, a recent QTL analysis using MAGIC lines suggested that genes involved in ribosome biogenesis and cell proliferation, such as NOP2A, RPL5A, and RPL5B, may be involved in setting telomere length set points (Abdulkina et al., 2019).
A more puzzling question is this: What is the significance of natural variation in telomere length for organisms? Telomere length variation could be neutral and result from random genetic drift or random stochasticity in the activity of the telomerase. Alternatively, telomere length differences could have fitness effects that are subject to natural selection, possibly due to their association with cellular senescence, which has been implicated in controlling lifespan in yeast and animals (Aubert and Lansdorp, 2008; Kupiec, 2014). In mammals, for example, telomere shortening correlates with between-species differences in lifespan (Whittemore et al., 2019), suggesting that telomeres are involved in the aging process (Aubert and Lansdorp, 2008). Indeed, it has been suggested that the aging trajectory of telomere lengths could be a product of optimization of a life-history tradeoff (Young, 2018). This is by no means universal, as in C. elegans, no fitness differences or clear phenotypic consequences were associated with natural variation in telomere lengths (Cook et al., 2016). A recent hypothesis proposes that shorter telomeres are associated with a faster “pace of life” in some organisms, in which energy is conserved for reproduction and away from somatic maintenance (Giraudeau et al., 2019).
While there is interest in the links between telomeres and life-history traits (e.g. aging) in animals, relatively little is known about how telomere length evolution affects plant life-history strategies. Aging in plants differs fundamentally from that in animals (Watson and Riha, 2011), and it is unclear whether the telomere-aging and “pace-of-life” models are also applicable to plants. Indeed, no specific hypotheses have been put forth to explain natural telomere length variation in plants; whether telomeres have an effect on plant life-history traits and are a target of natural selection remains an open question.
Here we describe an association between a life-history trait—flowering time—and natural telomere length variation in plants. Using whole-genome sequence data as well as experimental assays, we determine the extent of telomere length variation in three plant species: A. thaliana, rice (Oryza sativa), and maize (Z. mays). We use a genome-wide association (GWAS) mapping to identify genomic regions associated with natural telomere length variation in A. thaliana, including one that spans the telomerase gene TERT. We show that longer telomeres are found in plants that flower earlier in these three annual plant species.
Results
Genome-wide variation in A. thaliana tandem repeats
Satellite DNA sequences are repetitive sequences structured as arrays of DNA that are tandemly repeated in the genome, sometimes up to 106 copies. We examined genome-wide variation in satellite DNA repeat copy number in A. thaliana using the program k-Seek (Wei et al., 2014, 2018). k-Seek is an assembly-free method of identifying and quantifying k-mer repeats in unmapped short read sequence data, and k-mer counts are highly correlated with direct measurements of satellite repeat abundance (Wei et al., 2014).
We used whole-genome re-sequencing data from the 1001 A. thaliana Genome Consortium Project (Alonso-Blanco et al., 2016). We quantified genome-wide A. thaliana tandem repeat copy numbers by focusing on 483 individuals that were sequenced from leaves using identical protocols (designated as AraThaKmer; see Methods section for details). The quantity of each k-mer sequence is presented as copies per 1× read depth after GC normalization (Flynn et al., 2017). We set 50 bps as the minimum length of tandem repeat sequences that can be called.
Adding up k-mer copy numbers, the median total length of tandem repeats per individual is estimated at 341 kb. Across the population, individuals displayed over 25-fold differences in total tandem repeat lengths. The most abundant k-mer was the poly-A repeat, followed by the 7-mer AAACCCT (Figure 1A). Some k-mers, such as the AC repeat, had a wide range of variation between individuals, with a range of zero to thousands of copies. Our computationally based estimates were qualitatively concordant with direct estimates of repeat copy number that used DNA gel blot analysis to characterize 1- to 4-mer variation in a single A. thaliana ecotype (Depeiges et al., 1995). For instance, DNA gel blot analysis showed that A and AG repeats were the most abundant 1-mer and 2-mer, respectively, while in the 3-mer class, AAG and ATC repeats were highly abundant. All of this was seen in our estimates as well. Furthermore, DNA gel blot analysis indicated that AAG was more abundant than AAC repeat, which we also observed as well, indicating that k-Seek is highly specific in quantifying the type of k-mer repeat.
Figure 1.

Arabidopsis thaliana tandem repeat profile. (A) Top 25 most abundant k-mers from the AraThaKmer set. K-mers are ordered alphabetically, then by size. Whiskers in boxplot represent ± 1.5* interquartile range. (B) Distribution of estimates of telomere repeat copy numbers from the AraThaKmer set (above) and terminal fragment lengths from the AraThaTRF set (below). (C) Scatter plot of telomere lengths from the 112 accessions that were in common between the AraThaKmer and AraThaTRF sets. Spearman ρ and P-values are shown in the upper-right side.
Arabidopsis thaliana telomere length variation
The tandem repeat with the second highest abundance in the A. thaliana genome is the k-mer AAACCCT, which corresponds to the canonical telomere repeat sequence in plants [a reverse complement of the Arabidopsis TTTAGGG telomere repeat, followed by tandem repetition] (Fajkus et al., 2005; Watson and Riha, 2010). There is a wide range in total copy numbers for the AAACCCT repeat, from 1,257 copies in Arabidopsis ecotype Ler-1 to 38,850 copies in ecotype IP-Fel-2 (Supplemental Data Set 1), with a median of 6,411 and a mean of 7,113.6 ± 161.1 (±standard error) copies (Fig. 1B).
We compared telomere repeat copy numbers inferred from k-Seek to directly measure telomere lengths in various A. thaliana accessions using the terminal restriction fragment (TRF) method (Fitzgerald et al., 1999). We experimentally measured the telomere length by TRF in 424 A. thaliana accessions (Supplemental Figure 1) and combined this dataset with data for 229 previously analyzed accessions (Fulcher et al., 2015). In our total dataset of 653 A. thaliana accessions, designated as AraThaTRF, the mean telomere lengths ranged from 1,065.2 bp in Hov1-10 to 11,787.2 bp in IP-Pro-0 (Supplemental Data Set 2), with a median length of 3,533 bp and a mean length of 3,767.4 ± 50.1 bp (±standard error; Figure 1B).
A total of 112 accessions overlapped between the AraThaKmer and AraThaTRF sample sets, and we found a significant positive correlation in log10 telomere lengths from the two methods (Figure 1C; Pearson’s r = 0.58 and P = 2.3 × 10e−11, Spearman’s ρ = 0.55 and P = 1.9 × 10−10, Kendall’s τ = 0.39 and P = 8.8 × 10−10). We also looked at genomic data from a second set of 201 accessions that were sequenced using a different protocol from the AraThaKmer set (Supplemental Data Set 1). This second genome dataset had 140 accessions in common with AraThaTRF, and we found a significant positive correlation in estimated telomere lengths from the two methods as well (Pearson’s r = 0.33 and P = 8.5 × 10−5; Spearman’s ρ = 0.34 and P = 4.1 × 10−5; Kendall’s τ = 0.24 and P = 2.7 × 10−5). It should be noted that the estimates of total telomere lengths are generally higher using k-Seek compared to the direct TRF method, which may be attributed to interstitial telomere repeats that can be detected with the former method. Nevertheless, our analysis revealed significant correlations in estimated telomere lengths from the k-Seek versus the TRF methods, and the use of either estimate gave similar results in our downstream analyses (see below).
GWAS mapping of telomere length variation implicates telomerase
We investigated whether the natural variation in A. thaliana telomere length has a genetic basis. We analyzed the AraThaKmer and AraThaTRF sets separately by conducting genome-wide association mapping of telomere length variation. We used the FarmCPU method for GWAS analysis, which works well for identifying loci of complex traits that may be confounded by population structure (Liu et al., 2016). GWAS analysis revealed seven genomic regions with single-nucleotide polymorphisms (SNPs) significantly associated (after Bonferroni correction) with telomere length variation in the AraThaKmer set, and seven significant GWAS hits in a separate analysis of the AraThaTRF data (Figure 2 and see Supplemental Table 1 for genome coordinates).
Figure 2.

GWAS of A. thaliana telomere length variation. Manhattan plot of the genome-wide P-values testing the association of telomere copy number of the AraThaKmer set (top) and terminal fragment length of the AraThaTRF set (middle) using the FarmCPU approach. Red dotted line indicates the Bonferroni-corrected significance threshold (α = 0.05). The GWAS-significant region common in both the AraThaKmer and AraThaTRF sets is highlighted in blue. Closeup of the region with the most significant GWAS SNP is shown at the bottom. Dark blue points are GWAS P-values from the AraThaKmer set, and light blue points are GWAS p-values from the AraThaTRF set.
Of these, one SNP was detected in both analyses (P < 1 × 10−10) and is located on chromosome 5 at position 5,538,242. This SNP is found at the 3′–UTR (untranslated region) of locus AT5G16850 (Figure 2), which corresponds to the TERT gene. The TERT gene is crucial in maintaining telomere length in A. thaliana (Fitzgerald et al., 1999) and other eukaryotes (Autexier and Lue, 2006). Besides the overlapping SNP at the TERT gene, the only other potentially overlapping SNPs were two GWAS-significant SNPs on chromosome 3 position 5,136,799 from the AraThaKmer set and position 5,295,758 from AraThaTRF set (∼159 kbp apart). However, none of the nonoverlapping 12 significant SNPs from both GWAS studies were in proximity (within 200 kbp) to known telomere-regulating genes. Nonetheless, two significant SNPs (chromosome 2 position 12,466,562 from AraThaTRF dataset and chromosome 2 position 13,008,964 AraThaKmer dataset) from our GWAS analyses were also located in a QTL region on chromosome 2 identified in a recombinant inbred line mapping study (Fulcher et al., 2015; Supplemental Table 1).
Arabidopsis thaliana telomere length is associated with flowering time variation
We examined if the telomere length distribution among ecotypes had a geographical basis. We compared the telomere lengths from the AraThaKmer and AraThaTRF sets to each ecotype’s natural geolocation. We detected a significant negative correlation between telomere length and latitude for both the AraThaKmer (Spearman’s ρ = −0.182 and P = 8.8 × 10−5) and AraThaTRF (Spearman’s ρ = −0.115 and P = 0.022) datasets (Figure 3A), but for longitude, only the AraThaKmer set had a significant negative correlation [Spearman’s ρ = −0.165 and P = 3.8 × 10−4] (Figure 3B).
Figure 3.

Geographic distribution of telomere lengths in A. thaliana. Correlation between telomere length and latitude/longitude in the AraThaKmer set (A) and AraThaTRF set (B). Spearman’s ρ and P-values are shown in the upper-right side in each plot.
In A. thaliana, life-history traits are often associated with geographic adaptation (Stinchcombe et al., 2004; Montesinos-Navarro et al., 2012). We hypothesized that telomere length polymorphisms occurred as a response to adaptation to specific life-history strategies. We tested whether specific developmental traits associated with life history were correlated with a variation in telomere length by examining the developmental trait or telomere length phenotype for each accession. We compared telomere lengths from the AraThaKmer and AraThaTRF sets to seven different developmental traits (see Figure 4A and Supplemental Figure 2). The AraThaKmer set had significant negative correlations with four traits: days to flowering at 10°C (Spearman’s ρ = −0.119, P < 0.009), days to flowering at 16°C (Spearman’s ρ = −0.173, P < 1.6 × 10−4), cauline leaf number (Spearman’s ρ = −0.125, P < 0.007), and rosette leaf number (Spearman’s ρ = −0.152, P < 0.001), and positive correlation with rosette branch number [Spearman’s ρ = 0.111, P < 0.03] (Figure 4A). In the AraThaTRF set, the same four traits had significant negative correlations with telomere length: days to flowering at 10°C (Spearman’s ρ = −0.223, P < 2 × 10−6), days to flowering at 16°C (Spearman’s ρ = −0.210, P < 1 × 10−5), cauline leaf number (ρ = −0.178, P < 8 × 10−4), and rosette leaf number [Spearman’s ρ = −0.211, P < 0.0001] (Figure 4B;Supplemental Figure 2). Telomere length explains between 0.3% and 0.8% of variation in flowering time at 10°C and 2.36 and 2.46% at 16°C. It should be noted that in A. thaliana, leaf number is developmentally correlated with flowering time.
Figure 4.

Association between telomere length and developmental traits in A. thaliana. Correlation between telomere copy number or length and various A. thaliana life-history traits for the AraThaKmer set (A) and AraThaTRF set (B). Spearman’s ρ and P-values are shown in the upper-right side in each plot.
To test whether these correlations were simply due to population structure, we fit a multiple linear regression model that included the first four axes of a principal component analysis of the SNP variation as additive variables. The results showed that in the AraThaKmer set, telomere copy number was a significant negative predictor for the traits days to flowering at 16°C (P < 0.024), cauline leaf number (P < 0.034), and rosette leaf number (P < 0.003), even when accounting for population structure. Also, in the AraThaTRF set, telomere length was a significant negative predictor for days to flowering at 10°C (P < 5 × 10−4), days to flowering at 16°C (P < 0.0015), and rosette leaf number (P < 0.0076) after accounting for population structure. Telomere length in this set accounts for ∼0.5% of flowering time variation at 10°C, but this increases to ∼2.4% at 16°C. Together, these results suggest that telomere length is negatively associated with flowering time in this annual species, such that plants with longer telomeres flower earlier.
Influence of A. thaliana genome size and telomere or k-mer repeats on flowering time
A recent study showed that chromosomal features other than telomeres, such as genome size, can affect flowering time in plants (Bilinski et al., 2018). We investigated the relationship between telomere length and genome size in A. thaliana using data obtained through direct TRF measurements. A previous flow cytometry study provided genome size estimates (Long et al., 2013) for 139 accessions in our AraThaTRF set, and these samples showed no significant correlation between telomere length and genome size (Supplemental Figure 3). We also compared the copy number of ribosomal DNA, which is responsible for the majority of the A. thaliana genome size differences (Long et al., 2013), and also found no significant correlations with telomere length (Supplemental Figure 3). We did find a significant positive correlation between flowering time and genome size in this set (Spearman’s ρ = +0.228, P < 0.0042), as has been observed in maize (Bilinski et al. 2018); interestingly, this relationship is opposite that of the correlation with telomere length.
We then examined other k-mer repeats and their relationship with the telomere repeat and flowering time variation in A. thaliana. Examining the computational predictions from the AraThaKmer set, the top 10 most abundant k-mers (Figure 1A) all had significant positive correlations with the telomere repeat copy number (Supplemental Table 2). Examining each k-mer and its association with flowering time; however, there was no correlation with flowering time at 10°C, while there was a positive correlation with flowering time at 16°C for the k-mers A (ρ = 0.155, P < 6.8 × 10−4), AG (ρ = 0.134, P < 3.42 × 10−3), and AT (ρ = 0.178, P < 9.77 × 10−5). Note that the correlations for those three k-mers, like for genome size and ribosomal repeat lengths, were opposite that between telomere repeat length and flowering time. We then conducted a multiple linear regression of flowering time at 16°C with the k-mers A, AG, and AT (which were computationally estimated for abundance in this study) and the telomere repeat abundance in the model. The results showed that the telomere repeat was a significant negative predictor of flowering time at 16°C (P < 5 × 10−7), even after accounting for the abundance of the other three k-mers.
Evidence of selection within the genomic region associated with telomere length
The nonrandom geographical distribution of telomere lengths and the association with flowering time suggest that the length variation may be the result of natural selection. We conducted a selective sweep analysis focusing on the SNP region (chromosome 5 and position 5,538,242) that was associated with telomere length variation in GWAS analyses of both the AraThaKmer and AraThaTRF sets (Figure 2).
Using the entire 1,001 A. thaliana genome dataset, we calculated the integrated haplotype homozygosity score (iHS; Voight et al., 2006) and ω (Kim and Nielsen, 2004; Alachiotis et al., 2012) statistics, and found no evidence of a selective sweep across the entire population in this GWAS-significant SNP region. We then divided the sample population based on the allele status of the GWAS-significant SNP to examine group-specific evidence of selective sweeps. Our GWAS analysis indicated that individuals carrying the minor allele (frequency = 17% in the 1001 A. thaliana genome dataset) had longer telomeres than the others. We examined the site-specific extended haplotype homozygosity statistics (EHHS) between individuals carrying the major and minor alleles at the GWAS-significant SNP region. The ratio of EHHS between the populations (Rsb; Tang et al., 2007) statistic was elevated around the region encompassing the GWAS-significant SNP region (Figure 5A), and the increased EHHS occurred around the SNP for individuals carrying the minor allele (Figure 5B). We then calculated the ω statistic, which detects selective sweeps based on patterns of linkage disequilibrium (LD), and found that the GWAS-significant SNP region was a significant outlier within individuals carrying the minor SNP allele associated with longer telomeres, but not within the individuals carrying the major allele associated with shorter telomeres (Figure 5C). We also conducted a bootstrap procedure to determine the significance of this sweep signature. None of the bootstrap replicates had a ω statistic as large as what we observed for the group of individuals carrying the minor SNP allele (Supplemental Figure 4).
Figure 5.

Selection on the GWAS-significant region found in both the AraThaKmer and AraThaTRF sets. (A) Rsb statistics (log10P-value) comparing the EHHS of derived versus ancestral alleles on chromosome 5 near the GWAS-significant SNP (dotted line) region. (B) EHHS decay plot centered on site 5,538,242 of chromosome 5. Red line is for the derived allele (corresponding to the minor allele) and blue line is for the ancestral allele (corresponding to the major allele). (C) OmegaPlus-based selective sweep test statistics in 10-kbp windows for individuals carrying derived or ancestral allele. Red lines indicate the top 1% OmegaPlus statistic threshold. Red point indicates the OmegaPlus statistics for the window containing the TERT gene.
Flowering time is also negatively correlated with telomere copy number in rice and maize
The association between telomere length and flowering time was unexpected, but it suggested that individuals with different telomere lengths had contrasting life-history strategies. We investigated if this correlation is found outside A. thaliana by examining the relationship between telomere length and flowering time in O. sativa and Z. mays. For each species, we analyzed whole-genome re-sequencing data from previous studies that also reported flowering time data (Flint-Garcia et al., 2005; Wang et al., 2018).
In rice (O. sativa) and maize (Z. mays), there was a wide variation in telomere copy number, and like A. thaliana, many of the differences appear to show population stratification. In maize, data are available that incorporate both whole-genome re-sequencing and flowering time (Flint-Garcia et al., 2005), and we were able to computationally estimate telomere copy numbers in this set of 277 maize genotypes using k-Seek. An earlier study directly measured telomere lengths in maize inbred lines (Burr et. al. 1992), and 11 samples were common to both of these studies. Although the sample overlap was low, we did find that in these 11 common genotypes, the correlation between the computational versus direct measurement of telomere length was very high (Spearman’s ρ 0.95, P < 1 × 10−16), suggesting that our computational estimates also provide good estimates of telomere length in this species.
Most maize varieties are genetically classified as either from nonstiff-stalk (NSS) and stiff-stalk (SS) populations from temperate regions (Liu et al., 2003) or from the tropical/subtropical (TS) population. Our analysis of the 277 maize cultivars showed that NSS varieties had significantly higher telomere copy number than both SS and TS maize cultivars [Mann–Whitney U (MWU) test, P = 0.0304 and 0.0065, respectively] (see Figure 60.
We also analyzed data for 2,952 rice varieties (Wang et al., 2018). This species displayed the most significant differences in telomere copy numbers between subpopulations, likely due to deep population structure in rice (Huang et al., 2012; Wang et al., 2018). Most rice varieties can be divided into japonica or indica subspecies (Wang et al., 2018), which possess a significant genetic and physiological differentiation with each other (Zhao et al., 2011), so we analyzed each subpopulation separately (Figure 6). In japonica, the temperate japonica (GJtmp) group had significantly higher telomere repeat copy numbers than both subtropical (GJsubtrp) and tropical japonica (GJtrp; MWU test, P = 0.0051 and 1.34 × 10−10, respectively). In indica rice, the subpopulation XI-1A (from East Asia) had significantly higher telomere copy numbers compared to subpopulation XI-1B (modern varieties of diverse origin), XI-2 (from South Asia), and XI-3 (from Southeast Asia; MWU test, P = 0.0046, 2.38 × 10−20, and 1.83 × 10−18, respectively; see Figure 6).
Figure 6.

Association between telomere length and flowering time in rice (O. sativa) and maize (Z. mays). Telomere copy numbers per subpopulation are shown on the left. *P < 0.05, **P < 0.01, and ***P < 0.001. Correlation between telomere copy number and flowering time is shown on the right. For maize, the days to silk measured in Aurora, NY in 2007 is shown. Spearman’s ρ and P-values are shown in the lower-left side of each plot. Whiskers represent ±1.5* interquartile range.
Notably, in both rice and maize, the subpopulations with the highest telomere copy numbers (temperate japonica and NSS maize) were from temperate regions. It should be noted that in maize, the SS population is also temperate but has shorter telomeres, which may reflect the distinct recent breeding history of SS inbred maize.
Like in A. thaliana, we observed a significant negative correlation between telomere copy number and flowering time in rice (ρ = −0.084, P < 9.3 × 10−5; see Figure 6). This correlation is even more pronounced within each rice subspecies (ssp. japonica, ρ = −0.255, P < 5.3 × 10−9 and ssp. indica, ρ = −0.259, P < 2.9 × 10−15). In maize, we obtained previously measured flowering time data that were collected in five field locations in the USA and over a 3-year period in some locations; there were data for a total of nine fields/seasons (Zhao et al., 2006). In seven cases, there were significant negative correlations between telomere repeat copy number and flowering time (ρ = −0.123 to −0.169, P < 0.008 to 0.045; see Figure 6 for one example), one was marginally nonsignificant (ρ = −0.130, P < 0.057), and one still appeared negative but was nonsignificant (ρ = −0.057, P < 0.43; see Supplemental Table 3).
As in A. thaliana, telomere length explains a relatively small fraction of flowering time variation in these plants: a mean of 2.64% of the variation in maize (based on data from seven fields/seasons; see Supplemental Table 3), 5.39% of the variation for japonica, and 3.80% of the variation for indica rice. Despite the relatively low levels of flowering time variation explained, these correlations with telomere length are significant. To test whether these correlations were due simply to population structure, we fit a multiple linear regression model that included the first four axes of a principal component analysis of the SNP variation as additive variables. Similar to the case in A. thaliana, for both rice and maize, telomere length had a significantly negative effect on flowering time, even after accounting for population stratification (P < 0.02 for rice and P < 0.033 for maize).
GWAS of telomere repeat copy number variation in rice and maize using FarmCPU showed significant SNP markers in the japonica rice, indica rice, and maize populations (Supplemental Figure 5). There were 16, 11, and 9 SNPs in indica rice, japonica rice, and maize, respectively, that were significant after Bonferroni correction (Supplemental Table 4). We identified 19 rice and maize orthologs of known telomere-regulating genes (see Supplemental Table 5 for ortholog list) and compared their genomic positions to the GWAS-significant SNP markers; none of the significant SNPs were in close proximity to these telomere-regulating genes (in japonica rice, the closest GWAS-significant SNP was on chromosome 12 position 11,760,516 and near the gene TERT [< 395 kbp]; in maize, the closest GWAS-significant SNP was on chromosome 8 position 166,932,200 near the gene paralogs RPL5A/RPL5B [<3.2 Mbp]).
No overlap of GWAS peaks for telomere length and flowering time
We examined whether telomere-regulating genes were in fact previously unrecognized flowering time QTLs, and vice versa. Using both AraThaKmer and AraThaTRF individuals, we conducted GWAS of flowering time and compared the results to our GWAS results for telomere copy number (see Supplemental Tables 1 and 6 for significant SNPs from each GWAS analysis). We demarcated a 100-kb window centered on each significant telomere length SNP and examined whether any significant flowering time SNP was found within this window. No GWAS-significant SNPs were directly overlapping between the two traits (Figure 7 and see Supplemental Figure 6 for AraThaKmer set results). There was, however, a GWAS-significant SNP for telomere copy number on chromosome 5 position 15,389,625 in the AraThaKmer set and a GWAS-significant SNP for flowering time on chromosome 5 position 15,322,950 in the AraThaTRF set, which are ∼67 kb apart.
Figure 7.
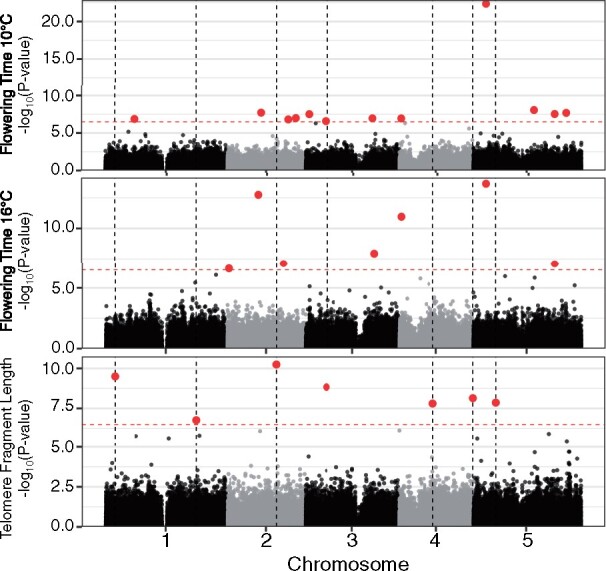
Genetic architecture underlying flowering time and telomere variation. GWAS results using the AraThaTRF set for flowering time at 10°C (top), flowering time at 16°C (middle), and terminal fragment length (bottom). Red dotted line indicates the Bonferroni-corrected significance threshold (α = 0.05). Dotted vertical lines are positions of the GWAS-significant SNPs for telomere fragment length. Red points indicate GWAS-significant SNPs.
We also examined the genetic architecture underlying flowering time variation in rice and maize. Like in A. thaliana, we did not find any directly overlapping GWAS-significant SNP positions for telomere length and flowering time variation (see Supplemental Tables 4 and 7). The distances between the closest GWAS-significant SNP for telomere length and flowering time were ∼434.2 kb for rice and ∼26 Mb for maize.
Discussion
The links between telomere length and organismal life-history traits are tantalizing, especially since telomeres are linked to cellular senescence, aging, and disease in humans. Despite its central role in chromosomal stability, the drivers of telomere length variation and their phenotypic consequences remain unclear. This is particularly relevant for plants, where telomere length variation is not easily connected to aging and senescence, as observed in animals (Watson and Riha, 2011). In our analysis, we found that natural telomere length variation in three annual plant species is related to flowering time, one of the most crucial life-history traits of plants. We found that in Arabidopsis, rice, and maize, individuals that had longer telomeres flowered earlier; finding this correlation in three distinct species from both dicots and monocots suggests that this relationship may be widespread. Indeed, we also observed a negative correlation between flowering time and telomere length in the polyploid plant rapeseed (Brassica napus), although this relationship was not significant in this species.
It should be noted that only a small amount of variation in flowering time is explained by telomere length in the three plant species. Telomere length accounts for approximately 0.5%–2.4% of flowering time variation in Arabidopsis and 3%–6% in maize and rice. The low fraction of variation in life-history traits explained by telomere length is perhaps not surprising: flowering time, for example, is a complex quantitative trait whose variation is largely explained by numerous major genes in the flowering time pathway (Salomé et al., 2011). What remains intriguing is that, despite the complex genetic architecture underlying flowering time variation, the effects of telomere length on this life-history trait were still observed, even after correcting for population structure accounting for local genetic differences. Moreover, this level of correlation seems to still be sufficiently large that it can be acted upon by natural selection, providing a key evolutionary link between telomeres and plant ecology and life history.
Moreover, while the levels of flowering time variation explained by telomere lengths may be low, similar levels are also observed for correlations of telomere length with lifespan traits in animals. For example, the variation explained between telomere length and mortality traits in dogs ranges from 0.62% to 8.8% (Fick et al., 2012). In humans, three studies showed that the percentage of telomere length variation that can be explained by age ranges from 6.8% to 16% (Njajou et al., 2007; Fitzpatrick et al., 2007; Weischer et al., 2014), although one early study had it as high as 50% (Slagboom et al., 1994); the latter appears to be an outlier. These results indicate that in both plants and animals, the amounts of variation in life-history traits explained by telomere length (or vice versa) may be low but are nevertheless significant.
The precise molecular mechanisms behind the correlation between telomere length and flowering time remain unknown, but we can suggest several possibilities. First, genes that control telomere length may have pleiotropic effects and also affect flowering time (or vice versa). Our analysis did not find any overlap between significant GWAS peaks for these two traits, but this does not rule out the possibility of multiple pleiotropic genes with small effects. Second, early flowering and rapid reproduction may somehow affect chromosome stability (precisely how is unclear), and so longer telomeres are adaptive in early flowering lines. A third possibility is that longer telomeres may be adaptive due to some other selection pressure, but given the energy expenditure for telomere maintenance, this may result in a trade-off that leads to early flowering. Finally, perhaps greater telomerase activity is adaptive in plants with rapid developmental rates and early flowering, and the longer telomeres in early flowering lines are merely an indication of higher levels of telomerase activity. In this latter case, it should be noted that in maize, for example, faster rates of cell differentiation in the shoot apical meristem are observed in plants with earlier flowering times (Bilinski et al., 2018; Leiboff et al., 2015), and telomerase is most active in differentiating tissues such as the meristem (Fitzgerald et al., 1996; Riha et al., 1998). All of these theories will need to be experimentally tested in the future.
In support of the hypothesis that the negative correlation between telomere length and flowering time may indeed be driven by adaptive evolution, we should note that there is evidence for a selective sweep at the A. thaliana TERT genomic region. Adaptation may also explain the significant latitudinal cline of telomere length variation in Arabidopsis, and we also found that longer telomeres are associated with other aspects of the spring cycling life-history strategy of this ruderal species, such as germination in response to cold. Moreover, longer telomeres are found in temperate-adapted varieties of rice (temperate japonica) and maize (non-stiff-stalk and stiff-stalk maize), which also flower significantly earlier in their growing seasons compared to TS varieties.
While we can advance these different hypotheses for the observed correlations, other explanations are possible. More work is required to understand why such correlations exist in plants. What is apparent is that the relationship of telomere length to life history is different in plants compared to other organisms. In mammals and other eukaryotic systems, shorter telomeres are generally associated with aging, in part because of the association of telomere shortening with cellular senescence (Urquidi et al., 2000). That clearly is not the case with the annual plant species we have studied. It has also been suggested that shorter telomeres are found in organisms with a faster “pace of life,” since the investment in somatic maintenance in such organisms is thought to be reduced to save energy for reproduction (Giraudeau et al., 2019). Again, our results suggest that this does not appear to be the case for these annual plants, as early flowering presumably due to faster developmental rates is associated with longer (and not shorter) telomeres.
Interestingly, the genetic architecture of telomere length variation is distinct in the three species we analyzed. In maize, an early QTL study examined telomere length variation and identified three loci associated with this trait (Burr et al., 1992), only one of which (a QTL on chromosome 4) may overlap with one of our nine GWAS hits. Natural telomere length variation has been more extensively studied in the model plant A. thaliana, and our GWAS found that three of our significant SNPs overlap with two previously identified telomere length QTL regions (Fulcher et al., 2015). Interestingly, genes previously associated with telomere set point regulation in Arabidopsis, including NOP2A, RPL5A, and RPL5B (Abdulkina et al., 2019), did not appear in our GWAS, perhaps due to differences in the mapping populations employed or their allele frequency in the population.
Our GWAS mapping, however, does span a key telomere-regulating gene in A. thaliana: the telomerase gene TERT. The A. thaliana TERT gene is involved in telomere elongation (Riha et al., 2001), and this locus also overlapped with a large QTL region for telomere length identified from a previous recombinant inbred line mapping study (Fulcher et al., 2015). TERT has also been identified in a human GWAS mapping study showing an association with leukocyte telomere length variation (Codd et al., 2013), pointing to cross-kingdom functional conservation. Determining whether it is indeed genetic variation at TERT that controls natural differences in telomere length variation in A. thaliana awaits future fine-mapping and molecular genetic investigations.
The lack of overlap between GWAS-significant SNPs across the three plant species suggests a divergence in the genetic architecture underlying telomere length regulation. However, there are caveats to our results. FarmCPU is computationally intensive. To reduce the computational burden in analyzing a large number of SNPs, we used LD pruned dataset for the GWAS. While this was suitable for A. thaliana and its small genome, for rice and maize, the LD pruning may have resulted in a coarse genome-wide landscape of the variation. However, it is worth noting that in maize, even with a reduced representation SNP set, the FarmCPU algorithm was able to detect SNPs in known flowering time-regulating genes, albeit by using a sample size that is 10-fold greater than our study (Liu et al., 2016). Hence, perhaps at least for our maize GWAS results, the analysis was underpowered due to the low sample size.
Our study complements recent work in identifying the effects of genome size and structure on life-history traits such as flowering time (Meagher and Vassiliadis, 2005). In maize, for example, genome size is positively correlated with flowering time (Jian et al., 2017), and changes in repetitive DNA sequences are associated with altitudinal adaptation (Bilinski et al., 2018). The negative correlation between genome size, repetitive DNA content, and cellular growth rate has been advanced as a plausible explanation for this phenomenon (Tenaillon et al., 2016; Bilinski et al., 2018). These studies, as well as our results on telomere variation, suggest that a variation in life-history strategy can indirectly influence chromosome and genome structure via selection. This opens up future areas of inquiry, including determining how widespread this phenomenon is, the relationship between telomere length and cell differentiation rate in plants, details of any selective advantage of telomere length in plant species with different life histories, and the precise molecular genetic mechanisms underlying telomere length polymorphisms in plant species.
Materials and methods
Plant materials and growth conditions
Seeds for the set of A. thaliana genotypes from the 1,001 Genome Project were purchased from ABRC (CS78942). Additional lines were obtained from the laboratory collection of TEJ. Seeds were sown into a mixture of three parts Promix BX mycorrhizae soil, one part Profile Field and Fairway calcined clay, and one part Turface medium stabilizer. Plants were grown in a greenhouse at UT Austin under 16/8-h light/dark (150 µmol light levels using Phantom Pro Double-ended 1,000-watt high-pressure sodium bulbs.), 21/18°C (day/night) conditions. Plant tissue for analysis was collected at the 5-week stage.
TRF analysis of telomere length analysis
Genomic DNA was extracted from individual whole plants (n = 1–3) and digested with the restriction enzyme Tru1I (Fermentas, Hanover, MD, USA) as previously described (Fitzgerald et al., 1999). [32P] 5′-end-labeled or 5′-DIG-(T3AG3)4 oligonucleotides were used as probes (Nigmatullina et al., 2016; Abdulkina et al., 2019). Radioactive signals were scanned with a Pharos FX Plus Molecular Imager (Bio-Rad), and nonradioactive signals were scanned with a GBox-F3 Imager (Syngene). Images were visualized with Quantity One v.4.6.5 software (Bio-Rad), and mean telomere length values (mean TRF used for GWAS) were calculated using the TeloTool program (Göhring et al., 2014), as described previously (Abdulkina et al., 2019). Overall, we measured telomere length in 424 A. thaliana accessions. Since our TRF method is identical to the one used in Fulcher et al. (2015), we added data for 229 accessions analyzed in their study to obtain our final dataset of 653 genotypes.
Analyzed genome sequences
We obtained the whole-genome re-sequencing data for A. thaliana, O. sativa, and Z. mays from previous published studies.
Arabidopsis thaliana: Genome sequences were obtained from the 1001 A. thaliana Genome Consortium (Alonso-Blanco et al., 2016) and are available at the NCBI SRA SRP056687. We grouped the 1,135 samples with the same genome sequencing protocols and analyzed the two most highly represented groups. This included the first group (designated as AraThaKmer) with 483 individuals prepared using leaf tissue and sequenced with 2 × 100-bp read length on the Illumina HiSeq 2000 platform. The second group consists of 201 individuals prepared using leaf tissue and sequenced as 2 × 101-bp read length on the Illumina HiSeq 2000 platform.
Oryza sativa: Genome sequencing data from Wang et al. (2018) were obtained at NCBI SRA PRJEB6180. All 3,000 samples were prepared from leaf tissue and sequenced as 2 × 83 bps using Illumina HiSeq 2000. Only samples with greater than 5× genome coverage were used.
Zea mays: We analyzed the “Buckler-Goodman 282” panel of Flint-Garcia et al. (2005), which captures the genetic diversity of maize. We analyzed the most recent sequencing batch that re-sequenced the panel to a higher depth using 2 × 150 bp on the Illumina HiSeq 10X platform (Bukowski et al., 2018). The data were obtained from the NCBI SRA PRJNA389800.
Identifying and quantifying tandem repeats
Sequencing reads were subjected to quality control using BBTools (https://jgi.doe.gov/data-and-tools/bbtools/). We used the bbduk.sh script version 37.66 using parameters minlen = 25 qtrim=rl trimq = 10 ktrim=r k = 25 mink = 11 hdist = 1 tpe tbo to trim sequencing adapters and low-quality sequences.
We used k-Seek to quantify k-mers and to identify the total copy number for a tandemly repeating k-mer in a sample of an unmapped genome sequencing library (Wei et al., 2014, 2018). k-Seek requires the minimum repeating length to be 50 bp to avoid counting small microsatellites that are scattered across the genome. We note that because k-Seek is optimized to analyze “short” Illumina sequencing reads (<150 bp), we will inevitably miss the characterization of large k-mer sequence-based satellite repeats (i.e. centromeric or ribosomal DNA repeats).
PCR-based library preparation is known to have a bias in underrepresenting high and low GC regions of the genome (Benjamini and Speed, 2012). To account for this bias, we implemented the method of Flynn et al., (2017) to correct for differences in GC content. Reads were mapped against the reference genome to first calculate the mean insert size using bamPEFragmentSize from deeptools version 3.3.0 package (Ramírez et al., 2016). The insert size was used to calculate the GC content of a given position in the genome, which was defined as the proportion of G or C bases of a given position plus the downstream fragment length of the library (Benjamini and Speed, 2012). The alignment was then used to calculate the average coverage of each GC content. We used bwa-mem version 0.7.16a-r1181 (Li, 2013) with default parameters to align paired end reads to the reference genome. The average coverage per GC content was then used to calculate the correction factor of Benjamini and Speed (2012) and applied to k-mer counts. We used scripts from Flynn et al., (2017;https://github.com/jmf422/Daphnia-MA-lines/tree/master/GC_correction) that implement the entire process.
For A. thaliana re-sequencing data, we used the reference genome TAIR10 from The Arabidopsis Information Resource. The genome sequences for O. sativa and Z. mays, however, were not ideal for implementing the GC content correction method. For O. sativa, samples were sequenced across multiple runs, suggesting that any differences in the sequencing run should also be implemented in the correction. For Z. mays, the genome coverage was relatively low (on average ∼5×), indicating that a coverage-based method of correction would not be ideal. Hence, for these two species, we only analyzed the telomere repeat, and for each sample, its telomere count was divided by average genome-wide coverage to account for differences in sequencing coverage between samples. The per-sample average coverage was obtained from Supplementary Data 2 of Wang et al., (2018) for O. sativa and was calculated using bedtools version 2.25.0 (Quinlan and Hall, 2010) genomecov program for Z. mays.
Genome-wide association study
For A. thaliana, the population VCF file was downloaded from the 1001 Genomes Project website (https://1001genomes.org/). For O. sativa, the population VCF was downloaded from the 3,000 Rice Genome Projects’ snp-seek website (https://snp-seek.irri.org/; Mansueto et al., 2017). For Z. mays, the population VCF was downloaded from the Gigascience database (http://dx.doi.org/10.5524/100339; Bukowski et al., 2018).
The SNP files were initially filtered to exclude polymorphic sites that had >10% of the individuals with a missing genotype and sites with <5% minor allele frequency. The VCF files were converted to PLINK format using vcftools version 0.1.15 (Danecek et al., 2011), and the program plink ver. 1.9 (Chang et al., 2015) was used with the parameter –indep-pairwise 100 5 0.5, which scans the file in 100 variant count windows while shifting the window in 5 variants and LD pruning pairs of variants that have r2 >0.5. In the end, we used 173,688 SNPs for AraThaKmer set, 153,845 SNPs AraThaTRF set, 585,026 SNPs for indica rice, 370,052 SNPs for japonica rice, and 3,048,120 SNPs in maize.
The LD-pruned PLINK file was converted to HAPMAP format for the GWAS using GAPIT version 2 (Tang et al., 2016). We took the log10 of the telomere lengths (corresponding to telomere repeat copy number from k-Seek-based estimate or the telomere fragment length from the TRF-based estimate) to normalize the distribution. For GWAS mapping, we used FarmCPU (Liu et al., 2016), which is a mixed linear model (MLM) incorporating population structure and kinship but is robust to false-positive and false-negative associations compared to other MLM GWAS algorithms. We used four principal components to model the underlying population structure.
Orthologs of A. thaliana telomere-regulating genes were found in the rice and maize gene annotation using Orthofinder ver 2.3.12 (Emms and Kelly, 2019, 2015).
Selective sweep analysis of A. thaliana
Evidence of selective sweep was examined using the iHS (Voight et al., 2006), Rsb (Tang et al., 2007) and OmegaPlus methods (Alachiotis et al., 2012) with SNPs extracted from chromosome 5. The SNP files that were filtered to exclude polymorphic sites that had >10% of the individuals with a missing genotype, and sites with <5% minor allele frequency were used. Missing genotype imputation and SNP phasing were conducted with Beagle version 4.1 (Browning and Browning, 2016).
To calculate the iHS and Rsb statistics, we used the filtered and phased VCF file with the R program (R Core Team, 2016) rehh package (Gautier and Vitalis, 2012). OmegaPlus statistics were calculated with OmegaPlus version 3.0.3 with -grid 2697 so that each grid would correspond to roughly 10,000 bp, and additional parameters -minwin 5000 -maxwin 3,000,000 -no-singletons. A bootstrap procedure was conducted to determine the significance of the OmegaPlus statistics. We randomly sampled individuals matching the sample size of the individuals carrying the minor allele at the GWAS-significant region on chromosome 5. OmegaPlus statistics were then calculated for this randomized group, and this procedure was repeated 200 times to generate a bootstrap distribution.
Plant phenotype analysis
For A. thaliana, various developmental traits were obtained from Arapheno (https://arapheno.1001genomes.org/; Seren et al., 2017) with phenotype names FT10 (days to flowering at 10°C), FT16 (days to flowering at 16°C), CL (cauline axillary branch number), RL (leaf number), length (stem length), RBN (primary branch number), and diameter (flower diameter). Genome size and ribosomal DNA size estimates were taken from the github repository (https://github.com/Gregor-Mendel-Institute/swedish-genomes/tree/master/files) for the original study (Long et al., 2013).
For rice, we obtained flowering time data measured as part of the 3000 Rice Genome Project (Sanciangco et al., 2018; Wang et al., 2018), which was measured as number of days at which 80% of the plants were fully headed (code HDG_80HEAD). The data are available from https://doi.org/10.7910/DVN/HGRSJG. For maize, we obtained phenotype data from the Buckler-Goodman association panel. The phenotype file (traitMatrix_maize282NAM_v15-130212.txt) was downloaded from Panzea (https://www.panzea.org/phenotypes), and we only analyzed the days to silk trait (code GDDDaystoSilk).
For each plant genotype, the phenotypes we obtained were a single representative value that was a product of summarizing the phenotype values from multiple replicates. The replicate information, however, was not readily available; hence, we considered each phenotype value to be an overall representation of the genotype.
Association between the telomere length and plant phenotypes were conducted in R. Telomere lengths were log10-converted before the linear regression modeling. The multiple linear regression analysis was conducted using the lm function, and population structure information was obtained from the four principal components that were used in the GWAS analysis.
Accession numbers
Sequence data from this article can be found in the GenBank/EMBL libraries under the following accession numbers: PRJNA273563 for A. thaliana, PRJNA389800 for Z. mays, and PRJEB6180 for O. sativa. The genome-wide K-mer abundance estimation and SNP file used for GWAS for A. thaliana, rice, and maize are available from Zenodo data repository (https://doi.org/10.5281/zenodo.4295944).
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure 1. Arabidopsis thaliana accessions display different telomere length set points.
Supplemental Figure 2. Scatter plots of A. thaliana phenotypes including cauline leaf number, diameter of rosette, rosette branch number, and length of flowering stem versus telomere copy number or terminal fragment length.
Supplemental Figure 3. Scatter plots of A. thaliana genome size and ribosomal DNA copy number versus terminal fragment length.
Supplemental Figure 4. Genome-wide ω statistic from 200 bootstrap groups.
Supplemental Figure 5. GWAS results for telomere repeat copy number in rice and maize.
Supplemental Figure 6. GWAS results for telomere copy number and days to flowering in A. thaliana.
Supplemental Table 1. GWAS-significant SNPs for telomere repeat copy number in the AraThaKmer set or telomere length in the AraThaTRF set.
Supplemental Table 2. Top 10 most abundant k-mer repeats and association with the telomere repeat or flowering time in A. thaliana.
Supplemental Table 3. Correlation between maize telomere repeat copy number and days to silk measured in 9 different environments.
Supplemental Table 4. GWAS-significant SNPs for telomere repeat copy number in rice and maize.
Supplemental Table 5. Orthologs of A. thaliana telomere-regulating genes in rice and maize.
Supplemental Table 6. GWAS-significant SNPs for days to flowering in the AraThaKmer set and the AraThaTRF set in A. thaliana.
Supplemental Table 7. GWAS of significant SNPs for days to flowering in rice and maize.
Supplemental Data Set 1. Telomere repeat copy numbers for the AraThaKmer set and the sequencing set with 101-bp read lengths.
Supplemental Data Set 2. Terminal fragment length for the AraThaTRF set.
Supplementary Material
Acknowledgments
We thank Kevin Wei with assistance in using the k-Seek method and discussions on satellite sequence evolution, and Jullien Flynn and Ian Caldas for assistance in implementing the GC-bias correction method. We also thank members of the Purugganan laboratory, especially Simon (Niels) Groen for helpful discussions.
Funding
This work was supported in part by grants from the National Science Foundation Plant Genome Research Program (IOS-1546218) and the Zegar Family Foundation (A16-0051) to M.D.P, National Institutes of Health (R01 GM127402 to E.V.S. and R01 GM065383 to D.E.S), Russian Foundation for Basic Research (18-34-00629 to L.R.A.), and funds from the Program of Competitive Growth of Kazan Federal University.
J.Y.C, T.E.J., E.V.S., and M.D.P. designed the research. L.R.A, J.Y., I.B.C., I.A.A., P.G.Y., and E.V.S. generated the telomere length data. J.Y.C, J.T.L., S.R., D.E.S., T.E.J., E.V.S., and M.D.P. analyzed the results. J.Y.C created the figures. J.Y.C, E.V.S., and M.D.P. wrote the article with contributions from all other authors.
The authors responsible for the distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plcell) are Jae Young Choi (jyc387@nyu.edu), Eugene V. Shakirov (shakirov@marshall.edu), and Michael D. Purugganan (mp132@nyu.edu).
References
- Abdulkina LR, Kobayashi C, Lovell JT, Chastukhina IB, Aklilu BB, Agabekian IA, Suescún AV, Valeeva LR, Nyamsuren C, Aglyamova GV, et al. (2019) Components of the ribosome biogenesis pathway underlie establishment of telomere length set point in Arabidopsis. Nat Commun 10: 5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alachiotis N, Stamatakis A, Pavlidis P (2012) OmegaPlus: a scalable tool for rapid detection of selective sweeps in whole-genome datasets. Bioinformatics 28: 2274–2275 [DOI] [PubMed] [Google Scholar]
- Alonso-Blanco C, Andrade J, Becker C, Bemm F, Bergelson J, Borgwardt KM, Cao J, Chae E, Dezwaan TM, Ding W, Ecker JR (2016) 1,135 genomes reveal the global pattern of polymorphism in Arabidopsis thaliana. Cell 166: 481–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert G, Lansdorp PM (2008) Telomeres and aging. Physiol Rev 88: 557–579 [DOI] [PubMed] [Google Scholar]
- Autexier C, Lue NF (2006) The structure and function of telomerase reverse transcriptase. Annu Rev Biochem 75: 493–517 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Speed TP (2012) Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res 40: e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilinski P, Albert PS, Berg JJ, Birchler JA, Grote MN, Lorant A, Quezada J, Swarts K, Yang J, Ross-Ibarra J (2018) Parallel altitudinal clines reveal trends in adaptive evolution of genome size in Zea mays. PLOS Genet 14: e1007162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AN, Lauter N, Vera DL, McLaughlin-Large KA, Steele TM, Fredette NC, Bass HW (2011). QTL mapping and candidate gene analysis of telomere length control factors in maize ( Zea mays L.). G3 (Bethesda) 1: 437–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning BL, Browning SR (2016) Genotype imputation with millions of reference samples. Am J of Hum Genet 98: 116–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukowski R, Guo X, Lu Y, Zou C, He B, Rong Z, Wang B, Xu D, Yang B, Xie C, et al. (2018) Construction of the third-generation Zea mays haplotype map. Gigascience 7: gix134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burr B, Burr FA, Matz EC, Romero-Severson J (1992) Pinning down loose ends: mapping telomeres and factors affecting their length. Plant Cell 4: 953–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codd V, Nelson CP, Albrecht E, Mangino M, Deelen J, Buxton JL, Hottenga JJ, Fischer K, Esko T, Surakka I, et al. (2013) Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet 45: 422–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DE, Zdraljevic S, Tanny RE, Seo B, Riccardi DD, Noble LM, Rockman MV, Alkema MJ, Braendle C, Kammenga JE, et al. (2016) The genetic basis of natural variation in Caenorhabditis elegans telomere length. Genetics 204: 371–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. (2011). The variant call format and VCFtools. Bioinformatics 27: 2156–2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depeiges A, Goubely C, Lenoir A, Cocherel S, Picard G, Raynal M, Grellet F, Delseny M (1995) Identification of the most represented repeated motifs in Arabidopsis thaliana microsatellite loci. Theoret Appl Genetics 91: 160–168 [DOI] [PubMed] [Google Scholar]
- van Deursen JM (2014) The role of senescent cells in ageing. Nature 509: 439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms DM, Kelly S (2019) OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms DM, Kelly S (2015) OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Bio 16: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajkus J, Kovarík A, Královics R, Bezdĕk M (1995) Organization of telomeric and subtelomeric chromatin in the higher plant Nicotiana tabacum. Mol Gen Genet 247: 633–638 [DOI] [PubMed] [Google Scholar]
- Fajkus J, Sýkorová E, Leitch AR (2005) Telomeres in evolution and evolution of telomeres. Chromosome Res 13: 469–479 [DOI] [PubMed] [Google Scholar]
- Fick LJ, Fick GH, Li Z, Cao E, Bao B, Heffelfinger D, Parker HG, Ostrander EA, Riabowol K (2012) Telomere length correlates with life span of dog breeds. Cell Rep 2: 1530–1536 [DOI] [PubMed] [Google Scholar]
- Fitzgerald MS, McKnight TD, Shippen DE (1996) Characterization and developmental patterns of telomerase expression in plants. Proc Natl Acad Sci U S A 93: 14422–14427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald MS, Riha K, Gao F, Ren S, McKnight TD, Shippen DE (1999) Disruption of the telomerase catalytic subunit gene from Arabidopsis inactivates telomerase and leads to a slow loss of telomeric DNA. PNAS 96: 14813–14818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick AL, Kronmal RA, Gardner JP, Psaty BM, Jenny NS, Tracy RP, Walston J, Kimura M, Aviv A (2007) Leukocyte telomere length and cardiovascular disease in the cardiovascular health study. Am J Epidemiol 165: 14–21 [DOI] [PubMed] [Google Scholar]
- Flint-Garcia SA, Thuillet A-C, Yu J, Pressoir G, Romero SM, Mitchell SE, Doebley J, Kresovich S, Goodman MM, Buckler ES (2005) Maize association population: a high-resolution platform for quantitative trait locus dissection. Plant J 44: 1054–1064 [DOI] [PubMed] [Google Scholar]
- Flynn JM, Caldas I, Cristescu ME, Clark AG (2017) Selection constrains high rates of tandem repetitive DNA mutation in Daphnia pulex. Genetics 207: genetics.300146.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulcher N, Derboven E, Valuchova S, Riha K (2014) If the cap fits, wear it: an overview of telomeric structures over evolution. Cell Mol Life Sci 71: 847–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulcher N, Teubenbacher A, Kerdaffrec E, Farlow A, Nordborg M, Riha K (2015) Genetic architecture of natural variation of telomere length in Arabidopsis thaliana. Genetics 199: 625–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatbonton T, Imbesi M, Nelson M, Akey JM, Ruderfer DM, Kruglyak L, Simon JA, Bedalov A (2006) Telomere length as a quantitative trait: genome-wide survey and genetic mapping of telomere length-control genes in yeast. PLoS Genet 2: e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier M, Vitalis R (2012) rehh: an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 28: 1176–1177 [DOI] [PubMed] [Google Scholar]
- Giraudeau M, Heidinger B, Bonneaud C, Sepp T (2019) Telomere shortening as a mechanism of long-term cost of infectious diseases in natural animal populations. Biol Lett 15: 20190190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göhring J, Fulcher N, Jacak J, Riha K (2014) TeloTool: a new tool for telomere length measurement from terminal restriction fragment analysis with improved probe intensity correction. Nucleic Acids Res 42: e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-García M-P, Pavelescu I, Canela A, Sevillano X, Leehy KA, Nelson ADL, Ibañes M, Shippen DE, Blasco MA, Caño-Delgado AI (2015) Single-cell telomere-length quantification couples telomere length to meristem activity and stem cell development in Arabidopsis. Cell Rep 11: 977–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L, Moorhead PS (1961) The serial cultivation of human diploid cell strains. Exp Cell Res 25: 585–621 [DOI] [PubMed] [Google Scholar]
- Huang X, Kurata N, Wang ZX, Wang A, Zhao Q, Zhao Y, Liu K, Lu H, Li W, Guo Y, et al. (2012) A map of rice genome variation reveals the origin of cultivated rice. Nature 490: 497–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian Y, Xu C, Guo Z, Wang S, Xu Y, Zou C (2017) Maize (Zea mays L.) genome size indicated by 180-bp knob abundance is associated with flowering time. Sci Rep 7: 5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AM, Beggs AM, Carvajal-Carmona L, Farrington S, Tenesa A, Walker M, Howarth K, Ballereau S, Hodgson SV, Zauber A, et al. (2012) TERC polymorphisms are associated both with susceptibility to colorectal cancer and with longer telomeres. Gut 61: 248–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian A, Stiff C, Kleinhofs A (1995) Barley telomeres shorten during differentiation but grow in callus culture. Proc Natl Acad Sci U S A 92: 9555–9559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Nielsen R (2004) Linkage disequilibrium as a signature of selective sweeps. Genetics 167: 1513–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupiec M (2014) Biology of telomeres: lessons from budding yeast. FEMS Microbiol Rev 38: 144–171 [DOI] [PubMed] [Google Scholar]
- Leiboff S, Li X, Hu H-C, Todt N, Yang J, Li X, Yu X, Muehlbauer GJ, Timmermans MCP, Yu J,. et al. (2015) Genetic control of morphometric diversity in the maize shoot apical meristem. Nat Commun 6: 8974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Neuhausen SL, Hunt SC, Kimura M, Hwang SJ, Chen W, Bis JC, Fitzpatrick AL, Smith E, Johnson AD, et al. (2010) Genome-wide association identifies OBFC1 as a locus involved in human leukocyte telomere biology. Proc Natl Acad Sci U S A 107: 9293–9298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv: 1303.3997v2
- Liti G, Haricharan S, Cubillos FA, Tierney AL, Sharp S, Bertuch AA, Parts L, Bailes E, Louis EJ (2009) Segregating YKU80 and TLC1 alleles underlying natural variation in telomere properties in wild yeast. PLoS Genet 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Goodman M, Muse S, Smith JS, Buckler E, Doebley J (2003) Genetic structure and diversity among maize inbred lines as inferred from DNA microsatellites. Genetics 165: 2117–2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Huang M, Fan B, Buckler ES, Zhang Z (2016) Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet 12: e1005767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Q, Rabanal FA, Meng D, Huber CD, Farlow A, Platzer A, Zhang Q, Vilhjálmsson BJ, Korte A, Nizhynska V, et al. (2013) Massive genomic variation and strong selection in Arabidopsis thaliana lines from Sweden. Nat Genet 45: 884–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillet G, White CI, Gallego ME (2006). Telomere-length regulation in inter-ecotype crosses of Arabidopsis. Plant Mol Biol 62: 859–866 [DOI] [PubMed] [Google Scholar]
- Mansueto L, Fuentes RR, Borja FN, Detras J, Abriol-Santos JM, Chebotarov D, Sanciangco M, Palis K, Copetti D, Poliakov A, et al. (2017) Rice SNP-seek database update: new SNPs, indels, and queries. Nucleic Acids Res 45: D1075–D1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meagher TR, Vassiliadis C (2005) Phenotypic impacts of repetitive DNA in flowering plants. New Phytol 168: 71–80 [DOI] [PubMed] [Google Scholar]
- Meyne J, Ratliff RL, Moyzis RK (1989). Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc Natl Acad Sci U S A 86: 7049–7053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesinos-Navarro A, Picó FX, Tonsor SJ (2012) Clinal variation in seed traits influencing life cycle timing in Arabidopsis thaliana. Evolution 66: 3417–3431 [DOI] [PubMed] [Google Scholar]
- Nigmatullina LR, Sharipova MR, Shakirov EV (2016) Non-radioactive TRF assay modifications to improve telomeric DNA detection efficiency in plants. BioNanoSci 6: 325–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njajou OT, Cawthon RM, Damcott CM, Wu S-H, Ott S, Garant MJ, Blackburn EH, Mitchell BD, Shuldiner AR, Hsueh W-C (2007). Telomere length is paternally inherited and is associated with parental lifespan. Proc Natl Acad Sci U S A 104: 12135–12139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olovnikov AM (1973) A theory of marginotomy: the incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol 41: 181–190 [DOI] [PubMed] [Google Scholar]
- Olovnikov AM (1971) Principle of marginotomy in template synthesis of polynucleotides. Dokl Akad Nauk SSSR 201: 1496–1499 [PubMed] [Google Scholar]
- Osterhage JL, Friedman KL (2009) Chromosome end maintenance by telomerase. J Biol Chem 284: 16061–16065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlevsky JD, Chen JJ-L (2016) Evolutionary perspectives of telomerase RNA structure and function. RNA Biol 13: 720–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2016). R: A Language and Environment for Statistical Computing. Vienna, Austria.
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T (2016) deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44: W160–W165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riha K, Fajkus J, Siroky J, Vyskot B (1998) Developmental control of telomere lengths and telomerase activity in plants. The Plant Cell 10: 1691–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riha K, McKnight TD, Griffing LR, Shippen DE (2001) Living with genome instability: plant responses to telomere dysfunction. Science 291: 1797–1800 [DOI] [PubMed] [Google Scholar]
- Salomé PA, Bomblies K, Laitinen RAE, Yant L, Mott R, Weigel D (2011) Genetic architecture of flowering-time variation in Arabidopsis thaliana. Genetics 188: 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanciangco MD, Alexandrov NN, Chebotarov D, King RD, Naredo MaEB, Leung H, Mansueto L, Mauleon RP, Orhobor OI, McNally KL (2018). Discovery of genomic variants associated with genebank historical traits for rice improvement: SNP and indel data, phenotypic data, and GWAS results. 10.7910/DVN/HGRSJG. [DOI]
- Seren Ü, Grimm D, Fitz J, Weigel D, Nordborg M, Borgwardt K, Korte A (2017) AraPheno: a public database for Arabidopsis thaliana phenotypes. Nucleic Acids Res 45: D1054–D1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakirov EV, Shippen DE (2004) Length regulation and dynamics of individual telomere tracts in wild-type arabidopsis. The Plant Cell 16: 1959–1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay JW, Wright WE (2019) Telomeres and telomerase: three decades of progress. Nat Rev Genet 20: 299–309 [DOI] [PubMed] [Google Scholar]
- Slagboom PE, Droog S, Boomsma DI (1994) Genetic determination of telomere size in humans: a twin study of three age groups. Am J Hum Genet 55: 876–882 [PMC free article] [PubMed] [Google Scholar]
- Song J, Logeswaran D, Castillo-González C, Li Y, Bose S, Aklilu BB, Ma Z, Polkhovskiy A, Chen JJ-L, Shippen DE (2019) The conserved structure of plant telomerase RNA provides the missing link for an evolutionary pathway from ciliates to humans. PNAS 116: 24542–24550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinchcombe JR, Weinig C, Ungerer M, Olsen KM, Mays C, Halldorsdottir SS, Purugganan MD, Schmitt J (2004) A latitudinal cline in flowering time in Arabidopsis thaliana modulated by the flowering time gene FRIGIDA. Proc Natl Acad Sci U S A 101: 4712–4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K, Thornton KR, Stoneking M (2007) A new approach for using genome scans to detect recent positive selection in the human genome. PLoS Biology 5: e171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Liu X, Wang J, Li M, Wang Q, Tian F, Su Z, Pan Y, Liu D, Lipka AE, Buckler ES, Zhang Z (2016) GAPIT version 2: an enhanced integrated tool for genomic association and prediction. Plant Genome 9. doi: 10.3835/plantgenome2015.11.0120 [DOI] [PubMed] [Google Scholar]
- Tenaillon MI, Manicacci D, Nicolas SD, Tardieu F, Welcker C (2016) Testing the link between genome size and growth rate in maize. PeerJ 4: e2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urquidi V, Tarin D, Goodison S (2000) Role of telomerase in cell senescence and oncogenesis. Annu Rev Med 51: 65–79 [DOI] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK (2006) A map of recent positive selection in the human genome. PLoS Biol 4: e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Mauleon R, Hu Z, Chebotarov D, Tai S, Wu Z, Li M, Zheng T, Fuentes RR, Zhang F, et al. (2018) Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 557: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JD (1972). Origin of concatemeric T7DNA. Nature New Biol 239: 197–201 [DOI] [PubMed] [Google Scholar]
- Watson JM, Riha K (2010) Comparative biology of telomeres: where plants stand. FEBS Lett 584: 3752–3759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JM, Riha K (2011) Telomeres, aging, and plants: from weeds to methuselah - a mini-review. Gerontology 57: 129–136 [DOI] [PubMed] [Google Scholar]
- Wei KH-C, Grenier JK, Barbash DA, Clark AG (2014) Correlated variation and population differentiation in satellite DNA abundance among lines of Drosophila melanogaster. Proc Natl Acad Sci U S A 111: 18793–18798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei KH-C, Lower SE, Caldas IV, Sless TJS, Barbash DA, Clark AG (2018) Variable rates of simple satellite gains across the drosophila phylogeny. Mol Biol Evol 35: 925–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischer M, Bojesen SE, Nordestgaard BG (2014) Telomere shortening unrelated to smoking, body weight, physical activity, and alcohol intake: 4,576 general population individuals with repeat measurements 10 years apart. PLoS Genet 10: e1004191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittemore K, Vera E, Martínez-Nevado E, Sanpera C, Blasco MA (2019) Telomere shortening rate predicts species life span. Proc Natl Acad Sci U S A 116: 15122–15127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RA, Upton HE, Vogan JM, Collins K (2017) Telomerase mechanism of telomere synthesis. Annu Rev Biochem 86: 439–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AJ (2018) The role of telomeres in the mechanisms and evolution of life-history trade-offs and ageing. Philos Trans R Soc Lond B Biol Sci 373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K, Tung CW, Eizenga GC, Wright MH, Ali ML, Price AH, Norton GJ, Islam MR, Reynolds A, Mezey J, et al. (2011) Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat Commun 2: 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Canaran P, Jurkuta R, Fulton T, Glaubitz J, Buckler E, Doebley J, Gaut B, Goodman M, Holland J, et al. (2006) Panzea: a database and resource for molecular and functional diversity in the maize genome. Nucleic Acids Res 34: D752–D757 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.