

Abstract
The genomes of virtually all organisms, including humans, contain repetitive sequences generated by the activity of transposable elements (transposons). Transposons are mobile genetic elements that can move from one genomic location to another; in this process, they amplify and increase their presence in genomes, sometimes to very high copy numbers. Transposons have been coevolving with their host genomes since the dawn of life. This relationship has been largely competitive, and transposons have earned epithets such as ‘junk DNA’ and ‘molecular parasites.’ Much of our knowledge of transposon evolution reflects their activity in the germline and is evident from genome sequence data. Recent research has revealed a wealth of information on the activity of transposons in somatic tissues during the lifetime of an individual, the underlying molecular mechanisms, and the manner in which these processes intersect with our own physiology, health and well-being. We discuss here new evidence and ideas that transposon activity influences and even promotes the process of aging and age-related diseases.
Transposons belong to two main groups: those that move using a DNA intermediate (‘DNA transposons’) in a ‘cut and paste’ mechanism, and retrotransposable elements (retrotransposons) that move using a ‘copy and paste’ mechanism that involves an RNA intermediate1. Thirty five percent of the human genome is comprised of retrotransposon DNA sequences. Retrotransposons are further divided into Long Terminal Repeat (LTR) elements, derived from exogenous retrovirus infections, and more primitive and ancient non-LTR elements with an obligate intra-cellular life cycle (Fig. 1). Non-LTR retrotransposons consist of two main groups: the Long INnterspersed Elements (LINEs), which encode their own proteins necessary for retrotransposition, and the Short INnterspersed Elements (SINEs), which are short, non-coding RNAs that hijack the LINE protein machinery2.
Fig. 1 |. L1 life cycle.
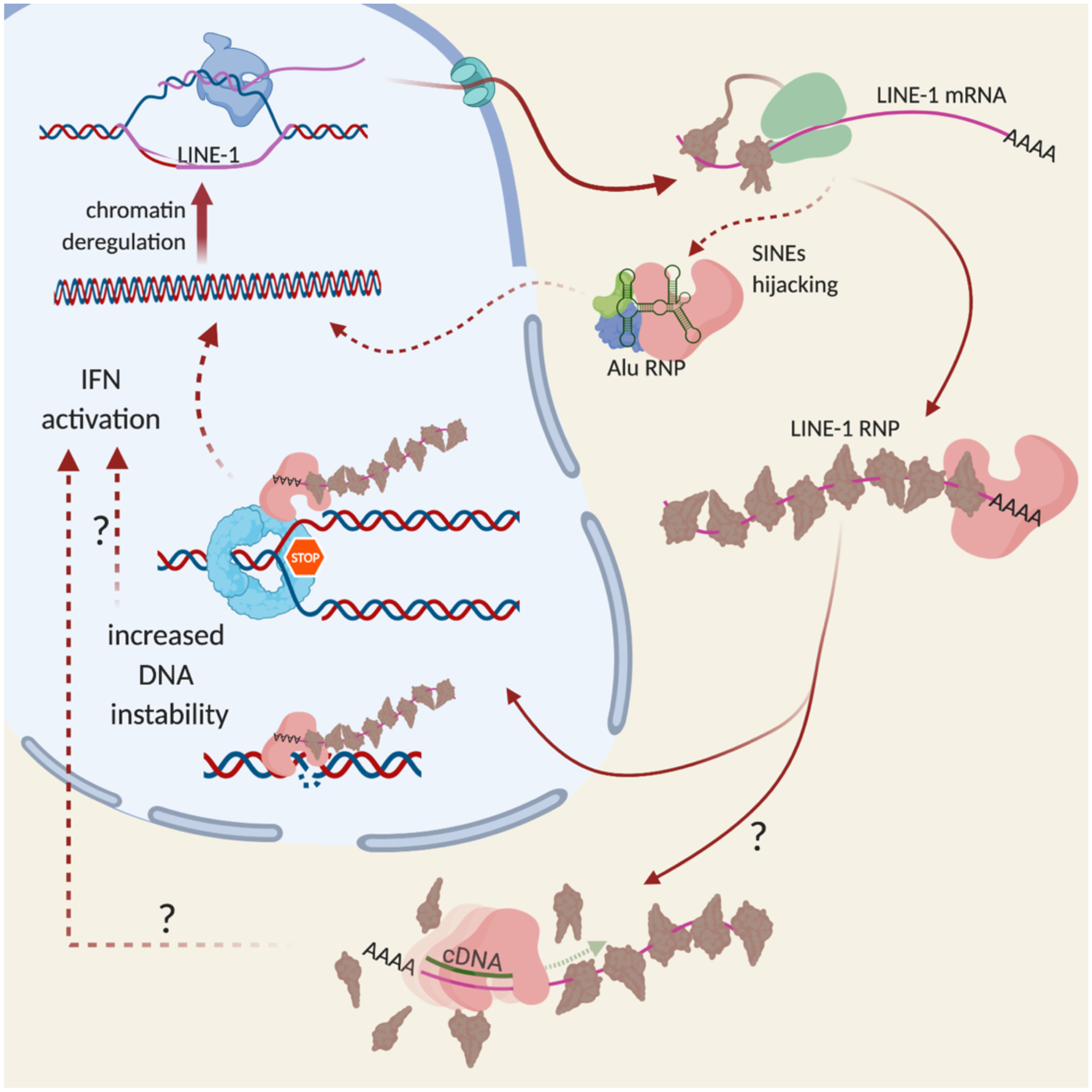
L1s are transcribed by host RNA polymerase II. The mRNAs are translated (green ribosome) into ORF1 (brown) and ORF2 (pink) proteins. Multiple trimers of ORF1 and one ORF2 bind the mRNA in cis to form RNPs. SINEs (Alu) hijack ORF2 in trans to form their own RNPs. RNPs interact with the DNA replication machinery to retrotranspose into the genome. How L1 cDNA is produced in non-replicating cells is not clear.
A retrotransposon onslaught can be profoundly deleterious and hence the germline is guarded with multiple defenses3. However, retrotransposons can also be agents of evolution4. In a process known variously as cooption, exaptation or preadaptation, over evolutionary time the hosts have ‘borrowed’ from retrotransposons and vice versa. Some examples of beneficial interactions are the establishment of chromatin states necessary for development in preimplantation mouse embryos by the transcriptional activation of distinct LTR and LINE elements4, the maintenance of telomeres in Drosophila by specific non-LTR retrotransposons5, and the use of LTR enhancers to regulate important genes in human innate immunity pathways6. The focus of this review is the unintended activation of retrotransposons in adult somatic tissues, in particular with advancing age, which appears to be deleterious7.
Host defenses are highly effective, and hence the majority of retrotransposons in our genome are passive passengers, slowly accumulating mutations, deletions or other rearrangements1. While some older elements can still affect host function through cis-acting gene regulatory or recombinational mechanisms8, 9, the deleterious effects of retrotransposons increasingly being linked with aging appear to be largely dependent on the activities of their encoded proteins, and fall into three general mechanisms: genetic and epigenetic effects associated with retrotransposition, DNA damage associated with active or abortive retrotransposition, and activation of immune pathways associated with detection of retrotransposon nucleic acids. The last topic has received considerable recent interest in the context of multiple age-related pathologies and diseases.
Retrotransposon life cycles
Different species harbor diverse retrotransposon portfolios whose activities evolve over time. The genomes of Drosophila and mice contain active LINE and LTR retrotransposons, whereas the human genome shows current evidence of only LINE-1 (and associated SINE) activity. LINE-1 (L1) comprises 17% of the human genome (~500,000 copies); however, only some 100–150 evolutionarily recent element copies are full-length and lack mutations that would be expected to affect function10–12.
L1 consists of a 5’UTR with an internal promoter, two open reading frames, ORF1 and ORF2, and a 3’UTR with a polyadenylation signal (Fig. 1). ORF1 encodes an RNA chaperone and ORF2 an enzyme with single-strand endonuclease and reverse transcriptase activities. A small ORF of unknown function, ORF0, was recently discovered in the antisense orientation in the 5’UTR13. The L1 life cycle starts with its transcription by host RNA polymerase II. After the capped and polyadenylated bicistronic L1 mRNA is translated in the cytoplasm, the two encoded proteins interact in cis with the L1 mRNA forming ribonucleoprotein particles (RNPs)14. L1 RNPs consist of many ORF1 trimers15 coating the mRNA and likely just one or two ORF2 molecules bound to the A-rich tail of the 3’UTR16.
L1 RNPs gain access to the nucleus primarily during mitosis17. The endonuclease nicks the bottom strand of genomic DNA at A/T rich sites (TTTT/AA consensus) to initiate a process known as target-primed reverse transcription (TPRT) (Fig. 2)18. The polyA tail of the L1 mRNA then anneals to the thymidines adjacent to the cut site, enabling reverse transcription from the 3’ end of the DNA flap. Subsequent steps are not well understood, but likely involve host factors to produce a double-stranded DNA (dsDNA) L1 element ligated to host DNA and flanked by target site duplications. These events occur during host DNA replication and take advantage of interactions with the replication fork11, 12, 19, 20.
Fig. 2 |. Retrotransposition mechanisms.
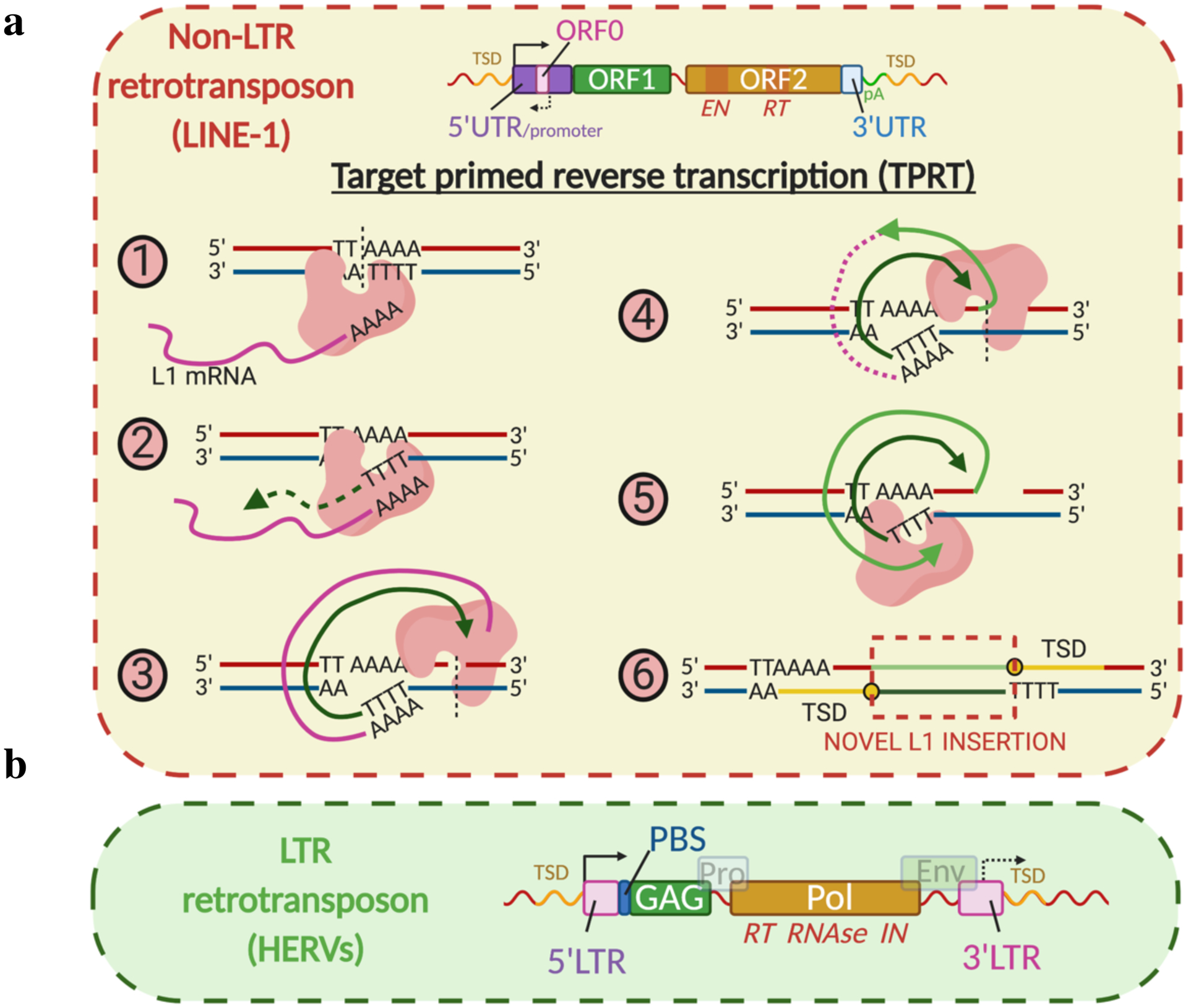
a, Non-LTR elements. L1 structure: 5’UTR with internal promoters, purple; ORF0, pink; ORF1, green; ORF2, orange; EN, endonuclease domain of ORF2; RT, reverse transcriptase domain of ORF2; 3’UTR with polyadenylation signals (pA), blue; TSD, target site duplications. The steps (1–6) of TPRT are illustrated. b, LTR elements. HERV structure: LTRs with internal promoters, pink; PBS, tRNA primer binding site; Gag (capsid protein), green; Pro (protease), grey-blue; Pol (polymerase), brown; Env (envelope protein), grey-green. Many HERVs lack Env. Pol has domains with RT, ribonuclease H (RNase) and integrase (IN) activities.
LTR retrotransposons move by a distinct process which is very similar to that used by retroviruses (Fig. 2). They encode Gag-like proteins, which assemble into capsid-like particles in the cytoplasm. Reverse transcription occurs inside these structures, typically using a host tRNA as a primer, generating a double-stranded cDNA product that, after entering the nucleus, is inserted into the genome by an integrase activity encoded by the element. Although LTR retrotransposons (including endogenized retroviruses) comprise approximately 8% of the human genome, all known sequences are mutated and no single element appears to be capable of retrotransposition. The human endogenous retrovirus K (HERV-K) appears to have become ‘extinct’ very recently, with the Gorilla genome still harboring some intact insertions21. However, several HERV elements in the human genome contain some intact ORFs, and expression of their envelope (Env) proteins in particular has been suggested to contribute to neurological diseases such as amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS)22.
Mechanisms of retrotransposon silencing
Host defense mechanisms recognize retrotransposon DNA sequences in their genomes and induce heterochromatization at these locations very early in development23, 24, 25 (Fig. 3). Heterochromatinization of retrotransposons requires the recruitment of chromatin remodeler enzymes and effector proteins to their sequences. These include histone methyltransferases (HMTs) such as SUV39H (suppressor of variegation 3–9 homolog) and SETDB1 (SET domain bifurcated 1), which are important for H3K9 (histone 3 lysine 9) methylation26, 27 and recruitment of HP1 (heterochromatin protein 1) to constitutive heterochromatin28; EZH2 (enhancer of zeste 2), which mediates trimethylation of H3K27 (histone 3 lysine 27) and maintenance of the Polycomb complex associated with facultative heterochromatin29; and several other factors including the human silencing hub (HUSH) complex19, 30 and retinoblastoma protein (RB1)29. TRIM28 (tripartite motif containing 28), also known as KAP1 (krüppel-associated box (KRAB) associated protein 1), acts as a co-repressor for multiple proteins that target retrotransposons, such as KRAB domain-containing zinc-finger proteins (KZFPs) and SIRT6 (silent mating type information regulator 2 (sirtuin) homolog 6), by recruiting SETDB1 and HP131, 32. Loss of these factors reduces heterochromatin at retrotransposons and allows their transcriptional derepression26, 29, 31, 33–35.
Fig. 3 |. Surveillance of retrotransposons.
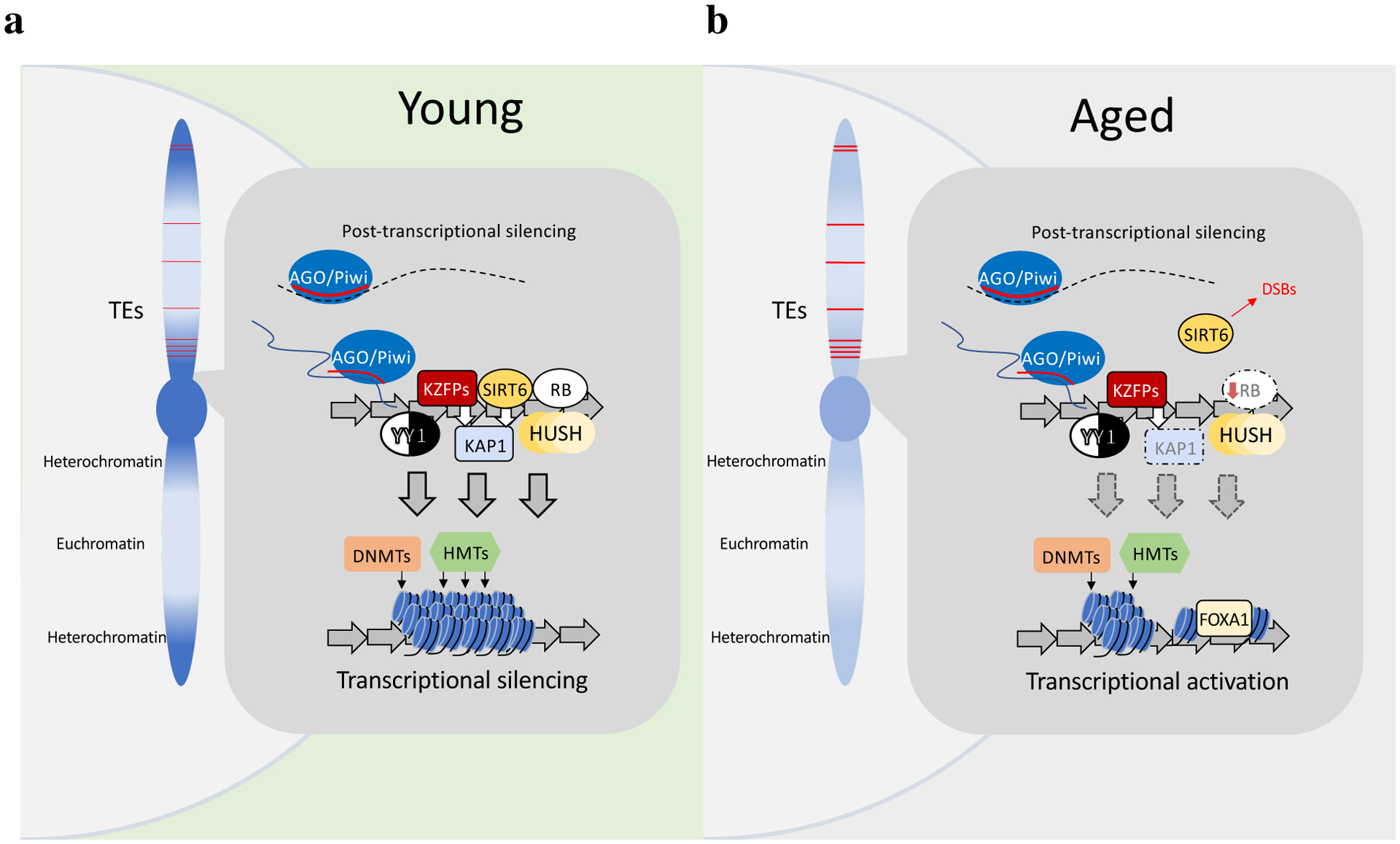
Heterochromatin, dark blue; euchromatin, light blue; retrotransposons, red lines. a, Young condition. Transcriptional regulators (KZFPs, RB, SIRT6, HUSH, YY1) target retrotransposon elements (repeating arrows) in the genome. SIRT6 and KZFPs both recruit (white arrows) the co-repressor KAP1. AGO and PIWI are targeted to nascent retrotransposon transcripts by siRNAs or piRNAs (red lines). Collectively these effectors recruit DNMTs and repressive HMTs to promote heterochromatin formation. Retrotransposon transcripts (dashed black lines) are also degraded by argonaute slicer complexes. b, Aged condition. Surveillance by regulators such as RB and SIRT6 is diminished, resulting in decreased KAP1 recruitment and reduced DNA methylation and repressive heterochromatin modification at retrotransposons. This allows the binding of transcriptional activators (FOXA1).
DNA cytosine methylation is an important mechanism of transcriptional silencing that exists in close relationship with histone modification-based mechanisms36. In mammals, retrotransposons are hypermethylated at CpG sites in adult somatic cells and in many (but not all) cases hypomethylation has been implicated in their de-repression37, 38. Deletion or inhibition of DNA methyltransferase (DNMT) enzymes leads to up-regulation of L1 elements39, 40. Conversely, SINEs are not de-repressed by Dnmt1 deletion or treatment with DNA demethylating drugs and appear to be regulated primarily by histone-methylation41. Interestingly, in Drosophila, cytosine methylation is absent and while N6-adenine methylation is found it does not appear to play a major role in retrotransposon repression42.
Histone and DNA methyltransferases are targeted to retrotransposon sequences through several pathways and effector proteins. KZFPs, of which there are over 400 in the human genome, co-evolve with retrotransposons as part of an ongoing ‘arms race’25. Many of these KZFPs bind to retrotransposon elements in the genome and promote their heterochromatinization via the recruitment of KAP143. Retrotransposons mutate to avoid this surveillance, and the host organisms evolve variant KZFPs that can again repress the new generation of retrotransposons24,44. In mouse embryonic stem cells (ESCs), some LTR retrotransposons are repressed by KAP1-mediated recruitment of SETDB1 and not DNA methylation38, 45. Evolutionarily young L1s in human ESCs are bound by the transcription factor YY1 (yin-yang-1) at a site in their 5’ UTR; in neuronal cells loss of this site is associated with hypomethylation and L1 activation46. Older L1 families are bound by KAP1, and loss of DNMTs increases expression of younger L1s but not older ones24. The KAP1- and DNMT-dependent pathways in many cases thus play complementary roles specific to individual retrotransposon families.
Short interfering RNA (siRNA) and the PIWI (P-element induced wimpy testis)-interacting RNA (piRNA) pathways recruit chromatin repressive factors to retrotransposons by a mechanism based on base pairing between the small RNAs and their target transcripts. siRNAs are derived from nuclear double-stranded RNA (dsRNA) species arising from convergent transcription or annealing of expressed retrotransposon sequences, which are processed by RNase-III-like Dicer enzymes. siRNAs are then loaded onto Argonaute (AGO) effector proteins to form silencing complexes that bind to nascent retrotransposon transcripts to recruit heterochromatin factors47. In the piRNA system, PIWI protein binds to piRNAs derived from piRNA clusters, non-coding transcriptional units that contain embedded retrotransposon sequences. Both siRNAs and piRNAs also contribute to post-transcriptional silencing by mediating transcript degradation.
Many additional factors play roles in retrotransposon surveillance48: 1) RNA editing enzymes such as the cytidine deaminase APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) family49 or ADAR (adenosine deaminase acting on RNA)50, 51; 2) the RNA helicase MOV10 (Moloney leukemia virus 10 homolog)48; 3) homologous recombination repair factors such as BRCA1 (breast cancer gene 1)20; 4) the nucleotide triphosphate hydrolase SAMHD1 (SAM domain and HD domain containing protein 1)52; and 5) the three-prime repair exonuclease 1 (TREX1)53. Some of these factors are defenses against exogenous viruses and act by diverse mechanisms such as editing of viral/retrotransposon genomes to mis-code the protein sequences (APOBEC), decreasing nucleotide triphosphate pools to limit viral/retrotransposon cDNA synthesis (SAMHD1) or degrading viral/retrotransposon nucleic acids (TREX1).
Loss of silencing with age
Aging is associated with extensive remodeling of the epigenome54. These changes are complex and generally cell-type specific, but a consistent trend is the loss of heterochromatin and an associated transcriptional de-repression at sites across the genome, which has been observed in species from yeast to humans55–57. Several studies have provided evidence that decreases of heterochromatin or heterochromatin-establishing factors contribute to elevated retrotransposon activity with age. In Drosophila, H3K9me3 and HP1 decline with age in pericentric regions and islands of heterochromatin58. Retrotransposons are enriched in these regions and later studies have confirmed their age-related activation35, 59, 60. Genetic interventions that promote heterochromatin formation and/or retrotransposon silencing attenuate age-related increases in retrotransposon expression and, remarkably, can increase life span35.
Cellular senescence is an irreversible arrest of proliferation that can be elicited by a variety of stresses, in particular DNA damage61. While senescence has beneficial functions (such as tumor suppression), senescent cells accumulate in most tissues with age, and they are important components of the overall aging process62. In particular, due to their profound proinflammatory phenotype63, senescent cells have been causally linked with many age-related pathologies and diseases64. Senescence is accompanied by complex changes and rearrangements of chromatin65. Relative decreases of heterochromatin have been mapped to retrotransposons and associated with their transcriptional derepression66. RB1 expression and binding to L1 elements were reduced in senescent cells and were associated with loss of H3K9me3 and H3K27me3 at L1 loci33. TREX1 was also downregulated, and the transcription factor FOXA1 was upregulated and bound to the L1 5’ UTR. Knockdown of RB1 and TREX1 combined with FOXA1 overexpression led to L1 derepression in human fibroblasts33.
However, senescent cells comprise only a small fraction of cells in aged tissues61 and are unlikely to be responsible for all of the retrotransposon activation that has been observed. Retrotransposon activation is found in diverse species, including nematodes67 and Drosophila35, which appear to be devoid of cell senescence. In mice, tissues with few senescent cells can show robust activation of retrotransposons with age (such as skeletal muscle)33, 68. One possible senescence-independent mechanism involves SIRT6, which promotes L1 silencing by binding to the L1 5’ UTR and mono-ADP ribosylating KAP1, which then recruits additional heterochromatin factors HP1a and MeCP231. In aged mouse tissues, SIRT6 is lost from L1 elements, likely due to increased recruitment to sites of oxidative DNA damage, and its overexpression opposes age-related increases of L1 expression in diverse tissues34. A similar mechanism has been described for SIRT1 in mice69, and the SIRT1 homolog in Drosophila is also involved in the regulation of retrotransposons35. Importantly, the extent of retrotransposon activation in chronologically aged versus senescent cell types of complex mammalian tissues is not known.
Cytosine methylation tends to decrease slowly with aging over large, gene-poor regions of the genome that are enriched for heterochromatin and repetitive sequences, whereas methylation at gene-associated CpG islands can increase or decrease more dramatically, with pronounced tissue-specific effects70, 71. Some of these changes are conserved between individuals of the same age and subsets of sites can be used to devise ‘methylation clocks’ that are highly correlated with aging72. Senescent cells show similar overall, but even more pronounced changes, which interestingly resemble changes observed in cancer cell genomes73. Given the links between DNA methylation, heterochromatin, and retrotransposon expression, and reports that interventions that slow down aging also decrease the progression of methylation changes74 and retrotransposon activation75, it is tempting to speculate that these events are causally connected. However, DNA methylation changes are early events in senescence73, whereas retrotransposon activation occurs only much later33. In cancer cells retrotransposon activation is quite sporadic between different cancer types as well as cases7. One possible explanation is that aging also increases methylome disorder76, which could lead to a stochastic and progressive activation of retrotransposons.
Some evidence suggests that alterations in small RNA pathways occur with age7, 77 and these changes may contribute to increased retrotransposon expression. Other than chromatin, however, most systems involved in retrotransposon regulation have not been well studied in the context of aging and thus provide interesting avenues for future exploration.
Consequences of retrotransposition
Over 120 L1-mediated germline insertions have been linked to cases of human genetic diseases78. Insertions of the human SINE Alu into the NF1 (neurofibromatosis type I) gene have been linked to the development of neurofibromas79. The NF1 gene appears to be a hotspot for Alu insertions, with 18 independent patients having been identified. However, retrotransposition events were estimated at ~0.4% of all NF1 mutations, and hence are a small fraction of all mutational events affecting this gene. Retrotransposition during development or adulthood would lead to somatic mosaicism, and has been estimated to occur in adult neurons by qPCR or single-cell genomic studies at varying (but generally low) frequencies (events/cell): < 0.04–0.680, 13.781, 0.58–182, 0.283 and 0.63–1.6684. While retrotransposition frequencies in other tissues have not been investigated in detail, they appear to be significantly lower, and hence the adult human brain might be a particularly permissive site for L1 activation84, 85.
Cancer is characterized by genomic instability and in many cases increased expression of L1 elements86. Subsequent efforts identified multiple de novo somatic insertions and associated rearrangements in human cancers, in some cases numbering in the hundreds for an individual tumor86–89. Some insertions into tumor suppressor genes such as APC (adenomatous polyposis coli) in colon cancer and PTEN (phosphatase and tensin homolog) in endometrial carcinoma have been found and linked to the development of the cancer87. However, the majority of de novo insertions in cancers are located in non-coding sequences and do not show hotspots near tumor suppressor or oncogene loci7, 90. Thus, there is little current evidence that new L1 insertions play a major role in tumorigenesis7.
Our knowledge is still limited on the consequences of insertions in intergenic regions, such as epigenetic effects (epimutations) on distal enhancers, long non-coding RNAs or architectural chromatin domains, especially over longer periods of time. It is plausible that such insertions could contribute to tumor evolution or development of therapy resistance. It would thus be interesting to test whether inhibition of retrotransposition might slow down relapse after therapy. However, in the absence of evidence to the contrary, we can tentatively conclude that the burden of disease attributable to L1 or Alu insertions, with the possible exception of some neurodegenerative diseases, is likely to be low.
Activation of innate immunity
Free DNA or dsRNA in the cytoplasm are generally perceived as invading pathogens. In vertebrates their presence triggers the interferon system, which then orchestrates immune responses. The signaling cascades start with a variety of sensors, such as RIG-I (retinoic acid-inducible gene I) and MDA5 (melanoma differentiation-associated protein 5) to detect cytoplasmic dsRNA, cGAS (cyclic GMP-AMP synthase) and AIM2 (absent in melanoma 2) to detect cytoplasmic DNA, and TLRs (toll-like receptors) to detect a variety of nucleic acids in the endosomal compartment91. These signals are propagated to the nucleus where they initiate the type-I interferon (IFN-I) response: a large number of factors that interfere with viral replication, expression of interferons α and β to alarm neighboring cells, a variety of cytokines and chemokines to communicate with the immune system, and in some cases, cell death pathways.
Increasing evidence indicates that retrotransposon DNA sequences accumulate in the cytoplasm under certain conditions. Pioneering work by Stetson et at.53 found that retrotransposon DNA was present in the cytoplasmic fractions of hearts from TREX1 mutant mice. Sequencing showed that L1, SINE, and LTR retrotransposon DNAs were significantly enriched compared to normal mice. Subsequent work found extra-chromosomal DNA in TREX1-deficient human neural cells, with L1 as the major source92.
Recently, cytoplasmic L1 DNA was found in normal cells in association with cellular senescence and aging33, 34. Cytoplasmic DNA was also present in multiple tissues of SIRT6 knockout mice34 that display premature aging and derepression of L1 elements31. Importantly, L1 DNA was identified in association with the DNA sensor cGAS34. Upon binding to DNA, cGAS catalyzes a reaction of GTP with ATP to form cyclic GMP-AMP (cGAMP)93. cGAMP then binds to STING (stimulator of interferon genes), which subsequently triggers the phosphorylation of the transcription factor IRF3 (interferon regulatory factor 3) by the kinase TBK1 (TRAF family member-associated NF-kappa-B activator (TANK)-binding kinase 1). IRF3 then translocates to the nucleus where it activates the IFN-I response (Box 1).
Box 1.
Diverse sources of cytoplasmic ‘self’ DNA.
cGAS-STING signaling can be activated by a variety of DNA species that leak into the cytoplasm, such as DNA fragments arising during DNA damage repair, fragments of telomeric DNA, ruptured micronuclei, and cytoplasmic chromatin fragments (CCF) formed by a nuclear autophagic process in senescent cells149, 150. Another source of cytoplasmic DNA is compromised mitochondria that can leak their DNA and prime innate antiviral responses151. Mitochondrial dysfunction was required for the progression of cellular senescence and the induction of its proinflammatory phenotype152. Another report found that mitochondrial dysfunction was required for the formation of CCF and induction of inflammatory signaling in senescent cells153. How these diverse cytoplasmic DNA species are functionally interrelated in triggering cGAS-STING signaling, in which cell types and under what conditions, is not well understood.
In all conditions discussed above – TREX1 or SIRT6 deficiency, cellular senescence, or natural organismal aging – cytoplasmic L1 DNA was correlated with activation of an IFN-I response. Importantly, treatment with nucleoside reverse transcriptase inhibitor (NRTI) drugs that inhibit L1 reverse transcriptase, or a knockdown of L1 expression with shRNAs against active L1 families, decreased cytoplasmic L1 DNAs and ameliorated cGAS and IFN-I activation33, 34, 92. This finding indicates that cytoplasmic L1 cDNAs, produced by their reverse transcriptase activity, can be potent triggers of an IFN-I response.
The mechanisms by which cytoplasmic L1 cDNAs are produced are uncertain. In the TPRT model L1 reverse transcription occurs in the nucleus. One possibility is that cytoplasmic L1 DNA species are abortive TPRT products that are somehow exported from the nucleus. However, in postmitotic senescent cells one would expect L1 RNPs to be largely excluded from the nucleus. Retrotransposition is greatly diminished in non-dividing cells94, although low levels could be detected in senescent cells33 or neurons92, and it is possible that abortive products of these processes could accumulate. Alternatively, reverse transcription could occur directly in the cytoplasm. Both ORF2 and L1 RNPs can reverse transcribe in vitro if provided with a primer14, 18. In senescent cells L1 RNPs could accumulate in the cytoplasm because of their inability to access the nucleus. Since reverse transcriptases can use both DNA and RNA primers and templates, the cytoplasmic RNPs could generate cDNA using as yet unidentified primers. Alu self-priming in the cytoplasm has recently been reported in age-related macular degeneration (AMD)95.
Evidence has also been presented that retrotransposons can be detected by RNA sensors. Retrotransposon dsRNA species are processed into siRNAs in the nucleus and normally do not reach the cytoplasm in sufficient numbers to overcome self-tolerance. In some circumstances, such as treatment of cancer cells with demethylating agents, retrotransposon upregulation overwhelms the system and is perceived by MDA596 or TLR397 sensors to trigger an IFN-I response. Major sources of dsRNA, inverted Alu repeats, are normally deaminated by ADAR to reduce their double-stranded character; decreased processing can be sensed by MDA5, resulting in IFN-I activation50, 51, 98. Overexpression of L1 can elicit an IFN-I response through either MDA5 or RIG-I99, 100, possibly mediated by some secondary structure of the L1 mRNA.
The relative contributions of DNA and RNA sensing of retrotransposons to senescence, aging, and age-related diseases remain to be thoroughly investigated. Much work to date has relied on L1 overexpression, cell lines, or situations where mechanisms that restrict retrotransposons were compromised. Interestingly, in one report a robust L1-mediated IFN-I induction in a cell line that does not express cGAS-STING was abrogated by NRTI treatment19. This might be explained by activation of cytoplasmic RNA sensors by RNA:DNA hybrids101, or the processing of L1 RNPs through lysosomes where L1 cDNA could activate TLR991. Another possibility is the existence of self-tolerance thresholds to which both DNA and RNA sensors contribute: for example, TBK1, which is upstream of IRF3, can be activated by both cGAS-STING and RIG-I or MDA593, 101.
Promotion of DNA damage
DNA lesions, in particular dsDNA breaks (DSB), can arise as ‘collateral damage’ associated with retrotransposition19, 20, 102. In Drosophila this is likely to be of particular importance since this species lacks an interferon system. More primitive mechanisms based on RNA sensing though Dicer-2 and a TLR are present, as is the NF-kB pathway, which activates the production of antimicrobial peptides; however, DNA sensing and the cGAS-STING axis are absent103. Retrotransposon activation in Drosophila has been clearly linked with DNA damage, which is alleviated by interventions such as promotion of heterochromatin35, 59, 60. DNA damage-mediated cell death104 might thus be a predominant cause of deleterious effects exerted by retrotransposons in flies. However, the basic innate immunity mechanisms of this organism remain to be examined in this context.
In mammals, retrotransposons, DNA damage, interferon and inflammation are very intricately intertwined. For example, retrotransposons can promote DNA damage, but DNA damage is also documented to activate retrotransposon expression105. Inflammation can drive DNA damage by diverse processes, but DNA damage also elicits inflammation, as in cell senescence. Retrotransposons trigger innate immune sensors, but interferon mechanisms can also regulate retrotransposons106. In many of the disease examples discussed below, inflammation and DNA damage coexist and have not been disentangled in sufficient detail. Given the current paucity of quantitative data, the frequency of DNA damage that can directly be attributed to retrotransposons is uncertain, and might be quite low, especially during natural aging.
Retrotransposons in aging
By increasing susceptibility to a wide range of diseases aging is the main cause of mortality and morbidity in the developed world. Could retrotransposons, as genomic parasites, be contributing to the process of aging (Fig. 4a)? Infestation with many parasites, such as nematodes or the plasmodium parasite that causes malaria, is clearly deleterious. The recent appreciation that retrotransposons are likely to be more active in somatic tissues than hitherto appreciated brings into focus the possibility that retrotransposon activity might have effects on the life span of a single individual.
Fig. 4 |. Retrotransposons as agents of aging and disease.
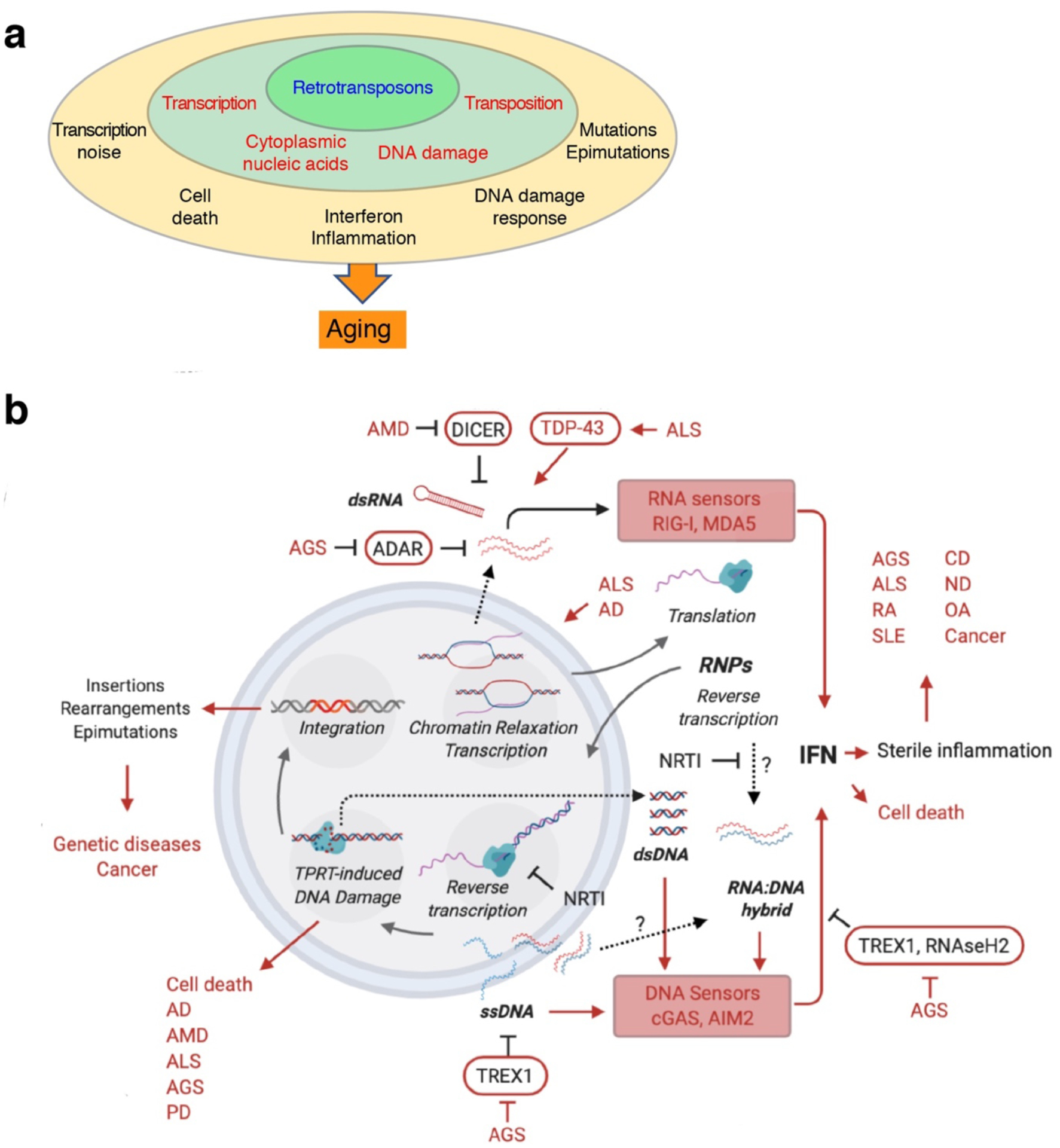
a, Overview of retrotransposon–aging interactions. Defense mechanisms are weakened with age, allowing the deprepression of retrotransposons (inner layer). The direct molecular consequences of retrotransposon activity (middle layer) drive several cellular and tissue-level processes (outer layer) which collectively promote aging. b, Specific retrotransposon–disease interactions. L1 transcription and integration are shown inside the nucleus (double circle), translation in cytoplasm, and reverse transcription in both compartments. DNA strands are shown in blue and RNA strands in red. Diseases are abbreviated in capital red letters (terms not defined in text: CD, cardiovascular diseases; ND, neurodegenerative diseases; OA, osteoarthritis; PD, Parkinson’s disease; RA, rheumatoid arthritis). Arrows: activating effects; lines ending in bars: inhibitory interactions; dashed arrows/question marks: hypothetical or uncertain processes.
Retrotransposons become reactivated in cell senescence and with age in both mammals and Drosophila58, 60, 66, 68. Remarkably, interventions that silence retrotransposons extended normal life span in flies, and treatments with NRTIs partially rescued the shortened lifespans of several fly mutations that de-repress retrotransposons35, 59. In mice, treatment with NRTIs doubled the lifespan of progeroid SIRT6-deficient mice, and improved overall health including bone density, muscle mass, intestinal function and exercise performance34. Treatment of middle-aged normal mice with NRTIs retarded the progression of aging biomarkers, in particular DNA methylation age and p16INK4A expression34. Collectively, these studies suggest that retrotransposons causally contribute to the aging process, and that interventions that oppose retrotransposon activity might improve healthy longevity.
Studies of long-lived animals have provided valuable insights into the mechanisms of aging107. Interestingly, the genome of the longest-lived rodent, the naked mole rat, contains fewer retrotransposons than other rodent genomes108; whereas the genomes of long-lived bats are replete with transposable elements109. Bats, however, have evolved dampened responses to cytoplasmic DNA110. Hence, there may not be a direct correlation between life span and transposon abundance but instead long-lived species may evolve better ways to respond to retrotransposons and associated inflammation.
Inflammation has emerged as a factor in multiple age-related pathologies, such as cancer, cardiovascular diseases or diabetes, and is a strong theme in many neurodegenerative disorders. Chronic low-level stimulation of the immune system is now viewed as a hallmark of aging111 and is often referred to as ‘sterile’ inflammation, since occurs in the absence of obvious infection with pathogens. The causes of sterile inflammation are probably many and remain inadequately defined. We suggest that mis-recognition of retrotransposons activated during aging as invading pathogens is an important driver of sterile inflammation, which in turn contributes to many aging-associated diseases. As an example, epidemiological evidence suggests that NRTIs reduce the incidence of type 2 diabetes112.
Diseases linked to retrotransposons
A pro-inflammatory role of retrotransposons has been noted in several autoimmune diseases (Fig. 4b). Elevated L1 transcripts were found in the synovial fluid of rheumatoid arthritis patients113. L1s are hypomethylated and their transcripts increased in patients with systemic lupus erythematosus (SLE) and Sjogren’s syndrome114, disorders also characterized by IFN-I induction. TREX1 mutations have been identified in SLE115 and familial chilblain lupus116, implicating TREX1 as a safeguard against autoimmune inflammation. Antibodies against DNA117 are often detected in SLE117 – could the production of such antibodies be triggered by L1 cDNA? A recent study identified antibodies to the L1 ORF1 protein in SLE patients with severe and active disease118. It remains to be determined whether L1 cDNA is an early trigger of disease or arises later during its course. It would thus be interesting to test whether suppression of L1 cytoplasmic DNA could prevent the onset of autoimmunity in genetically susceptible individuals or attenuate inflammation in established disease.
Aicardi–Goutières syndrome (AGS) is a childhood encephalopathy with similarities to SLE and strong IFN-I induction119. It is caused by recessive mutations in TREX1, SAMHD1, ADAR and RNaseH2 or dominant mutations in MDA5, all of which are involved in nucleic acid processing or sensing, DNA repair, or retrotransposon restriction. TREX1 degrades cytoplasmic L1 DNA53 and its loss initiates a cGAS/STING response. Treating TREX-deficient mice with a cocktail of NRTIs rescued their mortality120. SAMHD1 interacts with L1 in multiple ways52 and might sequester ORF1 into stress granules121. RNaseH2 digests the RNA strands of RNA:DNA hybrids122 and might reduce the levels of L1 nucleic acid species that interact with interferon sensors123. ADAR edits Alu RNA51, and gain of function mutations in MDA5 suggest that RNA-sensing mechanisms are involved in some AGS cases119. The extent to which the nucleic acids that activate interferon sensors are derived from chronic DNA damage or retrotransposons in not known; however, a clinical trial with NRTIs produced promising results124.
The role of L1 activation in cancer is not well understood. Inflammation may promote cancer by inducing cell proliferation; alternatively, the L1-triggered interferon response or DNA damage could promote cell death. L1s are activated by loss of DNA methylation, which often occurs during premalignant hyperplasia125; the resulting IFN-I response could then trigger cell death pathways. The IFN-I response could also expose the tumor cells to surveillance by the immune system96, 97. Hence, L1s, and retrotransposons more generally, could play a tumor suppressor role. Chronic IFN signaling can also promote an immune suppressive environment126.
Retrotransposons have also been implicated in several age-related neurodegenerative diseases127. AMD has been associated with the downregulation DICER1, and the consequent elevation of Alu RNA levels activates the NLRP3 (NOD, LRR and pyrin domain containing protein 3) inflammasome in retinal pigmented epithelial cells, leading to cytotoxicity and degeneration128. Alu RNA was reverse transcribed in the cytoplasm and the cDNA activated cGAS/STING signaling95. The ensuing IFN-I and inflammatory responses were mitigated by treatment with NRTIs95, 129.
ALS and frontotemporal dementia (FTD) are characterized by cytoplasmic aggregates of TDP-43 (transactivation response DNA-binding protein of 43 kDa). TDP-43 is normally localized in the nucleus, and although its function is not fully understood, its cytoplasmic aggregation has been strongly linked with disease. In post-mortem human brain, nuclear TDP-43 loss has been associated with opening of chromatin regions enriched in retrotransposons130. The permissive chromatin environment was proposed to enable L1 and HERV expression, also noted by others131, 132. Elevated interferon and inflammatory markers have been noted in ALS patients and mouse models of ALS132–134. Overexpression of human TDP-43 in the Drosophila brain induced the derepression of retrotransposons, including the retrotransposition-competent LTR element Gypsy135, 136. Retrotransposons appear to mediate the ensuing DNA damage-associated cytotoxicity, since these effects were rescued with NRTIs.
Neurofibrillary tangles, formed by insoluble aggregates of MAPT (microtubule-associated protein tau) are a hallmark of Alzheimer’s disease (AD) and other tauopathies. Postmortem AD brain samples exhibited increased markers of open chromatin137 and elevated expression of retrotransposons138, 139. Expression of a disease-associated human Tau protein in Drosophila neurons elicited loss of H3K9me2, widespread retrotransposon activation, DNA damage, and neuronal cell death that could be rescued with NRTIs139. Retrotransposon expression was positively associated with tangle burden in human cortical samples, as well as enrichment of the open chromatin mark H3K9ac at some HERV loci138. Sporadic AD was recently associated with the insertion of processed pseudogenes of APP (amyloid precursor protein), some possibly expressing disease-associated variants, into adult neuron genomes140 in a process that required reverse transcriptase activity.
Cytoplasmic retrotransposon DNAs have been detected in brain tissue of ataxia-telangiectasia (A-T) patients141, a neurodegenerative and cancer-prone syndrome caused by mutation of the ATM (A-T mutated) gene, which encodes a protein kinase that regulates cell cycle checkpoints, DNA repair and apoptosis in response to DSB. ATM kinase inhibition in glial cells of Drosophila activated an innate immune response and elicited neurodegeneration142. Loss of ATM was associated with increased L1 retrotransposition in human neural progenitor cells141. Loss of ATM in microglia led to accumulation of cytoplasmic DNA, activation of the cGAS-STING axis and production of neurotoxic cytokines143. The extent to which these responses are driven by retrotransposons or nuclear DNA fragments emanating from compromised DNA repair remains to be determined.
Therapeutic opportunities
The implication of retrotransposons in aging and age-related diseases provides an opportunity to develop novel treatments. Autoimmune disorders are typically treated with anti-inflammatory drugs, such as TNF antagonists, which target more distal aspects of the response. Small molecule inhibitors of STING have recently been reported144 and found to ameliorate STING-mediated inflammatory conditions in mouse models. cGAS inhibitors are being developed145. Interestingly, some old drugs such as aspirin146 and antimalarials quinacrine and chloroquine147 inhibit cGAS, further suggesting that downregulating this pathway might confer health benefits. The disadvantage of treatments that overly target immunity mechanisms is that they might increase susceptibility to infections.
One consistent feature in multiple models and situations is the observed efficacy of NRTIs to alleviate the effects of retrotransposon activation. This finding is intriguing given that some aspects of the retrotransposon lifecycle, such as transcription of the elements or stimulation of RNA sensors, should not be directly affected by NRTIs. One explanation might be downstream cross-talk between RNA and DNA sensing pathways and the existence of overall self-tolerance thresholds. Another possibility is the reported anti-inflammatory property of NRTIs to alleviate activation of the NLRP3 inflammasome without engaging reverse transcriptase129. Many NRTI drugs have been FDA approved for the treatment of HIV (human immunodeficiency virus) infections and are generally well-tolerated. However, NRTIs are not without side effects, as they can be recognized by cellular polymerases such as mitochondrial DNA polymerase or telomerase.
The most proximal approach to inhibiting retrotransposons would be to strengthen the epigenetic mechanisms responsible for their silencing, especially those that become compromised with age. One such epigenetic regulator is SIRT631. Male mice overexpressing SIRT6 show increased life span148, which may be due in part to more efficient silencing of L1 elements and reduced inflammation. SIRT6 is involved in multiple processes which include DNA repair, telomere maintenance and metabolism. Small molecule activators of SIRT6 are being developed and may provide an array of health benefits, including improved retrotransposon silencing.
Recent discoveries reviewed here show a convergent theme in which advancing age weakens host mechanisms that control retrotransposons, leading to their derepression and eventual activation of innate immune pathways, toxicity and degeneration. The initiating events are diverse and include genetic predisposition, environmental influences, and the natural aging process itself. Activation of interferon pathways has only recently emerged as a major downstream mechanism of retrotransposon activation and provides an exciting new framework for the relationship of retrotransposons with their hosts. Activation of innate immune signaling by retrotransposons can take place in all somatic cells, including postmitotic cells, without the need for actual retrotransposition events. The promotion of inflammatory processes resonates well with our emerging understanding of the central role inflammation plays in aging and many age-related diseases.
Outlook
Much work remains to be done on the basic biology side to understand the mechanisms and consequences of retrotransposon activation in adult somatic tissues. Heterochromatin has emerged as an important mechanism of retrotransposon repression. The loss and redistribution of heterochromatin that occur during aging thus warrant further investigation. Other pathways of retrotransposon suppression (such as siRNAs and piRNAs) need to be more fully investigated, especially in mammals. The upstream stimuli that initiate compromise of surveillance systems are beginning to come into view (for example, SIRT6 relocalization to sites of DNA damage31, or L1 chromatin opening due to TDP-43 relocalization into the cytoplasm130), but our understanding of these mechanisms is still very primitive. What is especially needed is a more holistic view of how aging mechanisms contribute to disease and vice versa.
Retrotransposon activation in senescent cells is well established, but the extent and mechanism of activation in postmitotic cells (such as neurons or myocytes) is not known. It appears that L1 has alternative lifecycles in proliferating and senescent/postmitotic cells, but the latter are not understood at all. Once retrotransposons are derepressed, is interferon activation the major aging- and disease-driving consequence, and what are the relative roles of DNA damage, responses to it, and cell death? For the pro-inflammatory effects, what are the relative roles DNA-sensing and RNA-sensing pathways? L1 has emerged as an important driver of IFN-I activation, but the genesis of the cytoplasmic L1 cDNAs that stimulate cGAS/STING signaling is not yet known. What are the relative contributions of L1s and HERVs to human diseases? It is already apparent that many species-, cell type- and disease-specific components exist, and disentangling which retrotransposons are being activated and how they are being sensed in different situations will have to be a major effort.
On the side of translation, an important consideration will be to identify patients in whom retrotransposon activation is triggering disease. For this it will be necessary to develop biomarkers that can reliably detect retrotransposon activation for patient stratification purposes. In terms of potential therapy, NRTIs have showed early promise124. Given the good safety record of recent NRTIs in HIV treatment, repurposing these drugs for a variety of indications is feasible and would generate a wealth of new information. Better-tolerated drugs will be desirable for long-term treatments of age-related pathologies, especially in geriatric populations. One strategy would be to develop new, high efficacy NRTIs specifically targeted to retrotransposon reverse transcriptases. Non-NRTI inhibitors that bind to non-catalytic sites on the HIV reverse transcriptase have been quite successful, and the same strategy can be pursued with retrotransposons. Drugs that target other functions, such as L1 endonuclease, or that destabilize RNP complexes can be contemplated. Finally, combination therapies that include drugs that target multiple pathways engaged by retrotransposons, such as DNA damage, are worth investigating.
Acknowledgments.
The following funding sources are acknowledged:
V.G. and A.S., NIH R37 AG046320, R01 AG027237, P01 AG047200, P01 AG051449; P.M. and J.D.B., NIH P01 AG051449, R21 CA235521; F.H.G., NIH R01 AG056306, R01 AG057706, American Heart Association, Paul G. Allen Frontiers Group Grant #19PABHI34610000, Grace Foundation, JPB Foundation, and Annette C. Merle-Smith; J.A.K., NIH P20 GM119943, P01 AG051449; J.R.T., NIH K99 AG057812; S.L.H., NIH R01 AG024353, P01 AG051449, R01 AG067306; J.M.S., NIH R01 AG016694, P01 AG051449.
Footnotes
Competing Financial Interests.
V.G. and A.S. are cofounders of Persimmon Bio. Inc.; V.G. is SAB member of DoNotAge Inc., Centaura Inc., and Elysium Inc.; J.D.B. is founder of Neochromosome, Inc., founder and director of CDI Labs, Inc., founder and SAB member of ReOpen Diagnostics, and SAB member of Sangamo, Inc., Modern Meadow, Inc., Sample6, Inc. and the Wyss Institute; F.H.G. is SAB member of Transposon Therapeutics, Inc.; J.M.S. is a cofounder and SAB chair of Transposon Therapeutics, Inc. and consults for Atropos Therapeutics, Inc., Gilead Sciences, Inc. and Oncolinea Inc.
References
- 1.Huang CR, Burns KH & Boeke JD Active transposition in genomes. Annu. Rev. Genet 46, 651–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bourque G et al. Ten things you should know about transposable elements. Genome Biol 19, 199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Molaro A & Malik HS Hide and seek: how chromatin-based pathways silence retroelements in the mammalian germline. Curr. Opin. Genet. Dev 37, 51–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cosby RL, Chang NC & Feschotte C Host-transposon interactions: conflict, cooperation, and cooption. Genes. Dev 33, 1098–1116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pardue ML & DeBaryshe PG Retrotransposons that maintain chromosome ends. Proc. Natl. Acad. Sci. USA 108, 20317–20324 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chuong EB, Elde NC & Feschotte C Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 351, 1083–1087 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burns KH Our conflict with transposable elements and its implications for human disease. Annu. Rev. Pathol 15, 51–70 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Jacques PE, Jeyakani J & Bourque G The majority of primate-specific regulatory sequences are derived from transposable elements. PLoS Genet 9, e1003504 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mita P & Boeke JD How retrotransposons shape genome regulation. Curr. Opin. Genet. Dev 37, 90–100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brouha B et al. Hot L1s account for the bulk of retrotransposition in the human population. Proc. Natl. Acad. Sci. USA 100, 5280–5285 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flasch DA et al. Genome-wide de novo L1 retrotransposition connects endonuclease activity with replication. Cell 177, 837–851 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sultana T et al. The landscape of L1 retrotransposons in the human genome is shaped by pre-insertion sequence biases and post-insertion selection. Mol. Cell 74, 555–570 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Denli AM et al. Primate-specific ORF0 contributes to retrotransposon-mediated diversity. Cell 163, 583–593 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Kulpa DA & Moran JV Cis-preferential LINE-1 reverse transcriptase activity in ribonucleoprotein particles. Nat. Struct. Mol. Biol 13, 655–660 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Khazina E et al. Trimeric structure and flexibility of the L1 ORF1 protein in human L1 retrotransposition. Nat. Struct. Mol. Biol 18, 1006–1014 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Doucet AJ, Wilusz JE, Miyoshi T, Liu Y & Moran JV A 3’ poly(A) tract is required for LINE-1 retrotransposition. Mol. Cell 60, 728–741 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mita P et al. LINE-1 protein localization and functional dynamics during the cell cycle. Elife 7, e30058 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cost GJ, Feng Q, Jacquier A & Boeke JD Human L1 element target-primed reverse transcription in vitro. EMBO J 21, 5899–5910 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]; Using elegant biochemical studies this paper developed the TPRT model of L1 retrotransposition.
- 19.Ardeljan D et al. Cell fitness screens reveal a conflict between LINE-1 retrotransposition and DNA replication. Nat. Struct. Mol. Biol 27, 168–178 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mita P et al. BRCA1 and S phase DNA repair pathways restrict LINE-1 retrotransposition in human cells. Nat. Struct. Mol. Biol 27, 179–191 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holloway JR, Williams ZH, Freeman MM, Bulow U & Coffin JM Gorillas have been infected with the HERV-K (HML-2) endogenous retrovirus much more recently than humans and chimpanzees. Proc. Natl. Acad. Sci. USA 116, 1337–1346 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kury P et al. Human endogenous retroviruses in neurological diseases. Trends Mol. Med 24, 379–394 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolf D & Goff SP Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature 458, 1201–1204 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castro-Diaz N et al. Evolutionally dynamic L1 regulation in embryonic stem cells. Genes Dev 28, 1397–1409 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacobs FM et al. An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature 516, 242–245 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; The above three papers show that KZFPs provide DNA sequence specificity to target retrotransposon silencing in mammalian genomes, and uncover the role of KZFPs in the co-evolutionary ‘arms race’ of primate L1s with their hosts.
- 26.Bulut-Karslioglu A et al. Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol. Cell 55, 277–290 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Kato M, Takemoto K & Shinkai Y A somatic role for the histone methyltransferase Setdb1 in endogenous retrovirus silencing. Nat. Commun 9, 1683 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Wit E, Greil F & van Steensel B Genome-wide HP1 binding in Drosophila: developmental plasticity and genomic targeting signals. Genome Res 15, 1265–1273 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishak CA et al. An RB-EZH2 complex mediates silencing of repetitive DNA sequences. Mol. Cell 64, 1074–1087 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu N et al. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature 553, 228–232 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Meter M et al. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat. Commun 5, 5011 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Helleboid PY et al. The interactome of KRAB zinc finger proteins reveals the evolutionary history of their functional diversification. EMBO J 38, e101220 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Cecco M et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that L1 upregulation triggers an IFN-I response in senescent cells and aged mice that can be therapeutically ameliorated with NRTIs.
- 34.Simon M et al. LINE1 derepression in aged wild-type and SIRT6-deficient mice drives inflammation. Cell Metab 29, 871–885 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that L1 upregulation triggers an IFN-I response in SIRT6 knockout and normal aged mice, and that pathologies and life span of SIRT6 progeroid mice can be improved with NRTIs.
- 35.Wood JG et al. Chromatin-modifying genetic interventions suppress age-associated transposable element activation and extend life span in Drosophila. Proc. Natl. Acad. Sci. USA 113, 11277–11282 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; Using chromatin-modifying genetic interventions and NRTI treatments in Drosophila this study causally links retrotransposon activation with aging.
- 36.Deniz O, Frost JM & Branco MR Regulation of transposable elements by DNA modifications. Nat. Rev. Genet 20, 417–431 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Karimi MM et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 8, 676–687 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walter M, Teissandier A, Perez-Palacios R & Bourc’his D An epigenetic switch ensures transposon repression upon dynamic loss of DNA methylation in embryonic stem cells. Elife 5, e11418 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walsh CP, Chaillet JR & Bestor TH Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet 20, 116–117 (1998). [DOI] [PubMed] [Google Scholar]
- 40.Woodcock DM, Lawler CB, Linsenmeyer ME, Doherty JP & Warren WD Asymmetric methylation in the hypermethylated CpG promoter region of the human L1 retrotransposon. J. Biol. Chem 272, 7810–7816 (1997). [DOI] [PubMed] [Google Scholar]
- 41.Varshney D et al. SINE transcription by RNA polymerase III is suppressed by histone methylation but not by DNA methylation. Nat. Commun 6, 6569 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang G et al. N6-methyladenine DNA modification in Drosophila. Cell 161, 893–906 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Imbeault M, Helleboid PY & Trono D KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 543, 550–554 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Wolf G et al. KRAB-zinc finger protein gene expansion in response to active retrotransposons in the murine lineage. Elife 9, e56337 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rowe HM et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463, 237–240 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Sanchez-Luque FJ et al. LINE-1 evasion of epigenetic repression in humans. Mol. Cell 75, 590–604 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Castel SE & Martienssen RA RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat. Rev. Genet 14, 100–112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goodier JL Restricting retrotransposons: a review. Mob. DNA 7, 16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feng Y, Goubran MH, Follack TB & Chelico L Deamination-independent restriction of LINE-1 retrotransposition by APOBEC3H. Sci. Rep 7, 10881 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chung H et al. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell 172, 811–824 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rice GI et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat. Genet 44, 1243–1248 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao K et al. Modulation of LINE-1 and Alu/SVA retrotransposition by Aicardi-Goutieres syndrome-related SAMHD1. Cell Rep 4, 1108–1115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stetson DB, Ko JS, Heidmann T & Medzhitov R Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work shows that loss of TREX1 in AGS results in accumulation of retrotransposon ssDNA that drives an IFN-I response, and implicates for the first time somatic retrotransposon activation as a causal factor in human disease.
- 54.Lopez-Otin C, Blasco MA, Partridge L, Serrano M & Kroemer G The hallmarks of aging. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Booth LN & Brunet A The aging epigenome. Mol. Cell 62, 728–744 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pal S & Tyler JK Epigenetics and aging. Sci. Adv 2, e1600584 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sen P, Shah PP, Nativio R & Berger SL Epigenetic mechanisms of longevity and aging. Cell 166, 822–839 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wood JG et al. Chromatin remodeling in the aging genome of Drosophila. Aging Cell 9, 971–978 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jones BC et al. A somatic piRNA pathway in the Drosophila fat body ensures metabolic homeostasis and normal lifespan. Nat. Commun 7, 13856 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen H, Zheng X, Xiao D & Zheng Y Age-associated de-repression of retrotransposons in the Drosophila fat body, its potential cause and consequence. Aging Cell 15, 542–552 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gorgoulis V et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Childs BG et al. Senescent cells: an emerging target for diseases of ageing. Nat. Rev. Drug Discov 16, 718–735 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Coppe JP et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6, 2853–2868 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reports the discovery of the senescence associated secretory phenotype (SASP) in senescent cells and implicates its proinflammatory effects as the major deleterious consequence of senescent cell accumulation in aging.
- 64.Pignolo RJ, Passos JF, Khosla S, Tchkonia T & Kirkland JL Reducing senescent cell burden in aging and disease. Trends Mol. Med 26, 630–638 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chandra T & Kirschner K Chromosome organisation during ageing and senescence. Curr. Opin. Cell Biol 40, 161–167 (2016). [DOI] [PubMed] [Google Scholar]
- 66.De Cecco M et al. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 12, 247–256 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.LaRocca TJ, Cavalier AN & Wahl D Repetitive elements as a transcriptomic marker of aging: Evidence in multiple datasets and models. Aging Cell 19, e13167 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Cecco M et al. Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging (Albany NY) 5, 867–883 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oberdoerffer P et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 135, 907–918 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Issa JP Aging and epigenetic drift: a vicious cycle. J. Clin. Invest 124, 24–29 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Klutstein M, Nejman D, Greenfield R & Cedar H DNA methylation in cancer and aging. Cancer Res 76, 3446–3450 (2016). [DOI] [PubMed] [Google Scholar]
- 72.Horvath S & Raj K DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet 19, 371–384 (2018). [DOI] [PubMed] [Google Scholar]
- 73.Cruickshanks HA et al. Senescent cells harbour features of the cancer epigenome. Nat. Cell Biol 15, 1495–1506 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cole JJ et al. Diverse interventions that extend mouse lifespan suppress shared age-associated epigenetic changes at critical gene regulatory regions. Genome Biol 18, 58 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wahl D, Cavalier AN, Smith M, Seals DR & LaRocca TJ Healthy aging interventions reduce repetitive element transcripts. J. Gerontol. A Biol. Sci. Med. Sci, glaa302 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang T et al. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol 18, 57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abe M et al. Impact of age-associated increase in 2’-O-methylation of miRNAs on aging and neurodegeneration in Drosophila. Genes Dev 28, 44–57 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hancks DC & Kazazian HH Jr. Roles for retrotransposon insertions in human disease. Mob. DNA 7, 9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wimmer K, Callens T, Wernstedt A & Messiaen L The NF1 gene contains hotspots for L1 endonuclease-dependent de novo insertion. PLoS Genet 7, e1002371 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Evrony GD et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 151, 483–496 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Upton KR et al. Ubiquitous L1 mosaicism in hippocampal neurons. Cell 161, 228–239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Erwin JA et al. L1-associated genomic regions are deleted in somatic cells of the healthy human brain. Nat. Neurosci 19, 1583–1591 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Evrony GD, Lee E, Park PJ & Walsh CA Resolving rates of mutation in the brain using single-neuron genomics. Elife 5, e12966 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhao B et al. Somatic LINE-1 retrotransposition in cortical neurons and non-brain tissues of Rett patients and healthy individuals. PLoS Genet 15, e1008043 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Coufal NG et al. L1 retrotransposition in human neural progenitor cells. Nature 460, 1127–1131 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports an increase in L1 DNA in adult human neurons and hippocampus and implicates the brain as a particularly permissive organ for L1 activation with age.
- 86.Lee E et al. Landscape of somatic retrotransposition in human cancers. Science 337, 967–971 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This sequencing study from the Cancer Genome Atlas Research Networks reveals for the first time the profound activation and retrotransposition of L1s in diverse human cancers.
- 87.Helman E et al. Somatic retrotransposition in human cancer revealed by whole-genome and exome sequencing. Genome Res 24, 1053–1063 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tubio JMC et al. Mobile DNA in cancer: Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science 345, 1251343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rodriguez-Martin B et al. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nat. Genet 52, 306–319 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mir AA, Philippe C & Cristofari G euL1db: the European database of L1HS retrotransposon insertions in humans. Nucleic Acids Res 43, D43–47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hartmann G Nucleic acid immunity. Adv. Immunol 133, 121–169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Thomas CA et al. Modeling of TREX1-dependent autoimmune disease using human stem cells highlights L1 accumulation as a source of neuroinflammation. Cell Stem Cell 21, 319–331 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hopfner KP & Hornung V Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell. Biol 21, 501–521 (2020). [DOI] [PubMed] [Google Scholar]
- 94.Shi X, Seluanov A & Gorbunova V Cell divisions are required for L1 retrotransposition. Mol. Cell. Biol 27, 1264–1270 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fukuda S et al. Cytoplasmic synthesis of endogenous Alu complementary DNA via reverse transcription and implications in age-related macular degeneration. Proc. Natl. Acad. Sci. USA 118, e2022751118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Roulois D et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162, 961–973 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chiappinelli KB et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162, 974–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ahmad S et al. Breaching self-tolerance to Alu duplex RNA underlies MDA5-mediated inflammation. Cell 172, 797–810 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tunbak H et al. The HUSH complex is a gatekeeper of type I interferon through epigenetic regulation of LINE-1s. Nat. Commun 11, 5387 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhao K et al. LINE1 contributes to autoimmunity through both RIG-I- and MDA5-mediated RNA sensing pathways. J. Autoimmun 90, 105–115 (2018). [DOI] [PubMed] [Google Scholar]
- 101.Brisse M & Ly H Comparative structure and function analysis of the RIG-I-like receptors: RIG-I and MDA5. Front. Immunol 10, 1586 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gasior SL, Wakeman TP, Xu B & Deininger PL The human LINE-1 retrotransposon creates DNA double-strand breaks. J. Mol. Biol 357, 1383–1393 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Martin M, Hiroyasu A, Guzman RM, Roberts SA & Goodman AG Analysis of Drosophila STING reveals an evolutionarily conserved antimicrobial function. Cell Rep 23, 3537–3550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li W et al. Activation of transposable elements during aging and neuronal decline in Drosophila. Nat. Neurosci 16, 529–531 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This works connects the age-associated activation of retrotransposons in Drosophila with cognitive decline and proposes the ‘retrotransposon storm’ theory of neurodegeneration.
- 105.Hagan CR, Sheffield RF & Rudin CM Human Alu element retrotransposition induced by genotoxic stress. Nat. Genet 35, 219–220 (2003). [DOI] [PubMed] [Google Scholar]
- 106.Yu Q et al. Type I interferon controls propagation of long interspersed element-1. J.Biol. Chem 290, 10191–10199 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Seluanov A, Gladyshev VN, Vijg J & Gorbunova V Mechanisms of cancer resistance in long-lived mammals. Nat. Rev. Cancer 18, 433–441 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim EB et al. Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature 479, 223–227 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ray DA et al. Multiple waves of recent DNA transposon activity in the bat, Myotis lucifugus. Genome Res 18, 717–728 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gorbunova V, Seluanov A & Kennedy BK The world goes bats: Living longer and tolerating viruses. Cell Metab 32, 31–43 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Franceschi C, Garagnani P, Parini P, Giuliani C & Santoro A Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol 14, 576–590 (2018). [DOI] [PubMed] [Google Scholar]
- 112.Ambati J et al. Repurposing anti-inflammasome NRTIs for improving insulin sensitivity and reducing type 2 diabetes development. Nat. Commun 11, 4737 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Neidhart M et al. Retrotransposable L1 elements expressed in rheumatoid arthritis synovial tissue: association with genomic DNA hypomethylation and influence on gene expression. Arthritis Rheum 43, 2634–2647 (2000). [DOI] [PubMed] [Google Scholar]
- 114.Mavragani CP et al. Defective regulation of L1 endogenous retroelements in primary Sjogren’s syndrome and systemic lupus erythematosus: Role of methylating enzymes. J. Autoimmun 88, 75–82 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lee-Kirsch MA et al. Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet 39, 1065–1067 (2007). [DOI] [PubMed] [Google Scholar]
- 116.Rice G et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am. J. Hum. Genet 80, 811–815 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Isenberg DA, Manson JJ, Ehrenstein MR & Rahman A Fifty years of anti-ds DNA antibodies: are we approaching journey’s end? Rheumatology (Oxford) 46, 1052–1056 (2007). [DOI] [PubMed] [Google Scholar]
- 118.Carter V et al. High prevalence and disease correlation of autoantibodies against p40 encoded by Long Interspersed Nuclear Elements in systemic lupus erythematosus. Arthritis Rheumatol 72, 89–99 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Crow YJ & Manel N Aicardi-Goutieres syndrome and the type I interferonopathies. Nat. Rev. Immunol 15, 429–440 (2015). [DOI] [PubMed] [Google Scholar]
- 120.Beck-Engeser GB, Eilat D & Wabl M An autoimmune disease prevented by anti-retroviral drugs. Retrovirology 8, 91 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hu S et al. SAMHD1 inhibits LINE-1 retrotransposition by promoting stress granule formation. PLoS Genet 11, e1005367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Uehara R et al. Two RNase H2 mutants with differential rNMP processing activity reveal a threshold of ribonucleotide tolerance for embryonic development. Cell Rep 25, 1135–1145 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bartsch K et al. RNase H2 loss in murine astrocytes results in cellular defects reminiscent of nucleic acid-mediated autoinflammation. Front. Immunol 9, 587 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rice GI et al. Reverse-transcriptase inhibitors in the Aicardi-Goutieres Syndrome. N. Engl. J. Med 379, 2275–2277 (2018). [DOI] [PubMed] [Google Scholar]; This phase I open-label clinical trial provides the first evidence that treatment with NRTIs reduces IFN-I activation in AGS patients.
- 125.Kitkumthorn N & Mutirangura A Long interspersed nuclear element-1 hypomethylation in cancer: biology and clinical applications. Clin. Epigenetics 2, 315–330 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Benci JL et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 167, 1540–1554 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Terry DM & Devine SE Aberrantly high levels of somatic LINE-1 expression and retrotransposition in human neurological disorders. Front. Genet 10, 1244 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tarallo V et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 149, 847–859 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fowler BJ et al. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 346, 1000–1003 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu EY et al. Loss of nuclear TDP-43 is associated with decondensation of LINE retrotransposons. Cell Rep 27, 1409–1421 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Prudencio M et al. Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum. Mol. Genet 26, 3421–3431 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tam OH et al. Postmortem cortex samples identify distinct molecular subtypes of ALS: Retrotransposon activation, oxidative stress, and activated glia. Cell Rep 29, 1164–1177 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hu Y et al. Increased peripheral blood inflammatory cytokine levels in amyotrophic lateral sclerosis: a meta-analysis study. Sci. Rep 7, 9094 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang R, Yang B & Zhang D Activation of interferon signaling pathways in spinal cord astrocytes from an ALS mouse model. Glia 59, 946–958 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chang YH & Dubnau J The Gypsy endogenous retrovirus drives non-cell-autonomous propagation in a Drosophila TDP-43 model of neurodegeneration. Curr. Biol 29, 3135–3152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Krug L et al. Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP-43 model of ALS. PLoS Genet 13, e1006635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Frost B, Hemberg M, Lewis J & Feany MB Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci 17, 357–366 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Guo C et al. Tau activates transposable elements in Alzheimer’s Disease. Cell Rep. 23, 2874–2880 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sun W, Samimi H, Gamez M, Zare H & Frost B Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat. Neurosci 21, 1038–1048 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lee MH et al. Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 563, 639–645 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Coufal NG et al. Ataxia telangiectasia mutated (ATM) modulates long interspersed element-1 (L1) retrotransposition in human neural stem cells. Proc. Natl. Acad. Sci. USA 108, 20382–20387 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Petersen AJ, Rimkus SA & Wassarman DA ATM kinase inhibition in glial cells activates the innate immune response and causes neurodegeneration in Drosophila. Proc. Natl. Acad. Sci. USA 109, E656–664 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Song X, Ma F & Herrup K Accumulation of cytoplasmic DNA due to ATM deficiency activates the microglial viral response system with neurotoxic consequences. J. Neurosci 39, 6378–6394 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Haag SM et al. Targeting STING with covalent small-molecule inhibitors. Nature 559, 269–273 (2018). [DOI] [PubMed] [Google Scholar]
- 145.Lama L et al. Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat. Commun 10, 2261 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dai J et al. Acetylation blocks cGAS activity and inhibits self-DNA-induced autoimmunity. Cell 176, 1447–1460 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Piscianz E et al. Reappraisal of antimalarials in interferonopathies: New perspectives for old drugs. Curr. Med. Chem 25, 2797–2810 (2018). [DOI] [PubMed] [Google Scholar]
- 148.Kanfi Y et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature 483, 218–221 (2012). [DOI] [PubMed] [Google Scholar]
- 149.Dou Z et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mackenzie KJ et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.West AP et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Correia-Melo C et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 35, 724–742 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Vizioli MG et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev 34, 428–445 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]