

Summary
During normal DNA replication, all cells encounter damage to their genetic material. As a result, organisms have developed response pathways that provide time for the cell to complete DNA repair before cell division occurs. In Bacillus subtilis, it is well established that the SOS-induced cell division inhibitor YneA blocks cell division after genotoxic stress; however, it remains unclear how YneA enforces the checkpoint. Here, we identify mutations that disrupt YneA activity and mutations that are refractory to the YneA-induced checkpoint. We found that YneA C-terminal truncation mutants and point mutants in or near the LysM peptidoglycan binding domain rendered YneA incapable of checkpoint enforcement. In addition, we developed a genetic method which isolated mutations in the ftsW gene that completely bypassed checkpoint enforcement while also finding that YneA interacts with late divisome components FtsL, Pbp2b and Pbp1. Characterization of an FtsW variant resulted in considerably shorter cells during the DNA damage response indicative of hyperactive initiation of cell division and bypass of the YneA enforced DNA damage checkpoint. With our results, we present a model where YneA inhibits septal cell wall synthesis by binding peptidoglycan and interfering with interaction between late arriving divisome components causing DNA damage checkpoint activation.
Keywords: YneA, DNA damage checkpoint, FtsW, cell division
Graphical Abstract
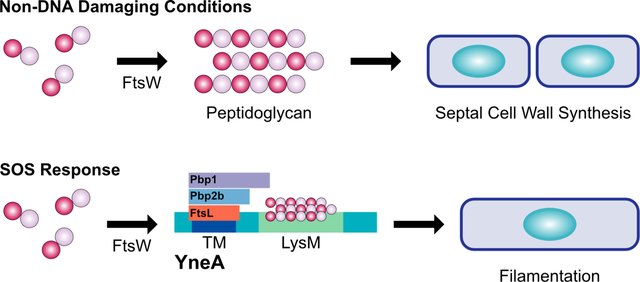
During normal cell division and non-DNA damaging conditions, the peptidoglycan polymerase FtsW assembles Lipid II into peptidoglycan during septal cell wall synthesis. In this work, we propose a new model for how YneA halts cell division in Bacillus subtilis. Following DNA damage, the SOS-induced cell division inhibitor YneA interacts with the late divisome proteins FtsL, Pbp2b and Pbp1 as well as binds peptidoglycan through its LysM domain (LysM), inhibiting septal cell wall synthesis and preventing cell division.
Introduction
When cells undergo DNA replication, they encounter a variety of spontaneous and environmental factors that damage their DNA (Friedberg, Walker et al. 2006). As a result, organisms from bacteria to humans have developed response pathways that halt cell cycle progression allowing time for accurate DNA repair to take place before cell division occurs (Bridges 1995, Sutton, Smith et al. 2000, Simmons, Grossman et al. 2007, Simmons, Foti et al. 2008, Ciccia and Elledge 2010). It is well established that eukaryotic cells induce expression of cell cycle checkpoints to delay cell cycle progression in response to DNA damage [for review (Ciccia and Elledge 2010)]. However, the process by which different bacterial species respond to genotoxic stress and pause cell cycle progression remains incompletely understood.
In bacteria, cells respond to DNA damage by activating the SOS response (Sutton, Smith et al. 2000, Walker, Smith et al. 2000, Simmons, Foti et al. 2008). This response pathway results in the upregulation of a variety of genes that relieve cellular stress and promote cell survival (Sutton, Smith et al. 2000, Walker, Smith et al. 2000, Simmons, Foti et al. 2008). Following DNA damage in Escherichia coli, RecA binds to single-stranded DNA, which promotes autocleavage of the LexA transcriptional repressor, and subsequent activation of the SOS regulon (Little 1983, Sutton, Smith et al. 2000, Lenhart, Schroeder et al. 2012). The SOS regulated cytoplasmic cell division inhibitor SulA delays cell cycle progression by directly interacting with and preventing the polymerization of FtsZ (Cole 1983, Mizusawa, Court et al. 1983, Mukherjee, Cao et al. 1998). After the DNA has been repaired, SulA is degraded by Lon protease and cell proliferation resumes (Mizusawa and Gottesman 1983, Mukherjee, Cao et al. 1998). While this process is well understood in E. coli, the mechanism used by many other bacteria remains partially understood particularly the mechanisms used to prevent cell division or release checkpoint enforcement.
Recently, it has become clear that the SulA-type DNA damage checkpoint enforcement mechanism is of limited conservation among bacteria [for review (Bojer, Frees et al. 2020, Burby and Simmons 2020). In other bacterial species the DNA damage-induced cell division inhibitor is a small membrane binding protein (Modell, Hopkins et al. 2011, Bojer, Frees et al. 2020, Burby and Simmons 2020). The best understood example of this conserved bacterial DNA damage checkpoint has been described in Caulobacter crescentus. Caulobacter contains SidA and DidA two DNA damage-inducible cell division inhibitors (Modell, Hopkins et al. 2011, Modell, Kambara et al. 2014). Both SidA and DidA are small membrane binding proteins that halt cell division following exposure to DNA damage (Modell, Hopkins et al. 2011, Modell, Kambara et al. 2014). SidA and DidA delay cell proliferation by preventing FtsW, FtsI and FtsN from forming a subcomplex that is essential for peptidoglycan synthesis at mid-cell (Modell, Hopkins et al. 2011, Modell, Kambara et al. 2014).
In Bacillus subtilis, the SOS-dependent cell division inhibitor YneA blocks cell proliferation after exposure to genotoxic stress (Kawai, Moriya et al. 2003). In the absence of damage, YneA accumulation is tightly controlled by the checkpoint recovery proteases, DdcP and CtpA, and the DNA damage checkpoint antagonist, DdcA (Burby, Simmons et al. 2018, Burby, Simmons et al. 2019). When cells are exposed to agents that halt DNA replication, YneA must reach a critical threshold to overcome these three negative regulators to activate the DNA damage checkpoint (Burby, Simmons et al. 2018, Burby, Simmons et al. 2019). After the damage is repaired, YneA is then degraded by membrane-bound proteases CtpA and DdcP allowing for cell division to resume (Burby, Simmons et al. 2018). The process controlling YneA expression and degradation is well understood; however, the mechanism underlying how YneA inhibits cell division or how cells can circumvent YneA function remains unknown.
To understand how YneA delays cell division, we used several genetic approaches to identify mutations that disrupt YneA function and extragenic mutations that bypass YneA activity. We identify mutations in yneA that prevent function including point mutations in the LysM peptidoglycan binding domain. We also isolated extragenic mutations in the ftsW gene encoding a peptidoglycan polymerase that are refractory to YneA activity. Characterization of one FtsW variant shows cell division initiates hyperactively under conditions of DNA damage bypassing the YneA-enforced checkpoint. Further, we show that ectopic or DNA damage induced expression of YneA strongly sensitizes cells to the cell wall antibiotic cephalexin and we show that YneA interacts with late divisome components FtsL, Pbp2b and Pbp1. With these results, we present a new model for YneA function where it induces checkpoint enforcement by binding peptidoglycan through its LysM domain while also interacting with and inhibiting late arriving cell division and septal cell wall synthesis proteins FtsL, Pbp2b and Pbp1.
Results
yneA C-terminal truncations impair checkpoint activation
Previous work identified a point mutation within the C-terminal tail (D95A) of yneA that caused an increase in YneA activity (Mo and Burkholder 2010). Further, it was shown that C-terminal truncations of S. aureus SosA resulted in increased growth interference (Bojer, Wacnik et al. 2019). A distinct difference between SosA and YneA is that YneA contains a LysM domain and SosA does not (Bojer, Wacnik et al. 2019). Therefore, we asked if the 15 amino acid C-terminal tail present after the LysM domain is required for YneA to block cell division in B. subtilis. We generated C-terminal truncations of YneA that lack the last five (yneAΔ5), ten (yneAΔ10) or fifteen (yneAΔ15) amino acid residues with the Δ15 truncation located near the predicted LysM domain boundary (Fig. 1A) (Mo and Burkholder 2010). We placed these truncations under the control of a highly induced IPTG regulated promoter (Phy) and integrated each allele at the ectopic amyE locus. We used an IPTG regulated promoter to uncouple yneA expression from the SOS response so that we could induce yneA without adding DNA damage and inducing expression of the other ~64 genes in the SOS regulon (Au, Kuester-Schoeck et al. 2005). We ectopically expressed WT yneA and each yneA truncation mutant in a strain that lacks endogenous yneA (ΔyneA::loxP) (Fig. 1B). We show that cells are highly sensitive to yneA overexpression in the absence of endogenous yneA (Fig. 1B). However, when we induced expression of yneAΔ5 or yneAΔ10, cell proliferation was partially impaired, showing more growth than WT yneA, but less growth compared to cells grown in the absence of induced yneA expression (Fig. 1B). Interestingly, in cells expressing yneAΔ15 growth was the same as that observed in cells lacking the IPTG induced yneA gene or cells with Phy-yneA grown in the absence of IPTG (Fig. 1B). These results show that the C-terminal 15 amino acids are required for checkpoint enforcement.
Figure 1. yneA C-terminal truncations impair checkpoint activation.
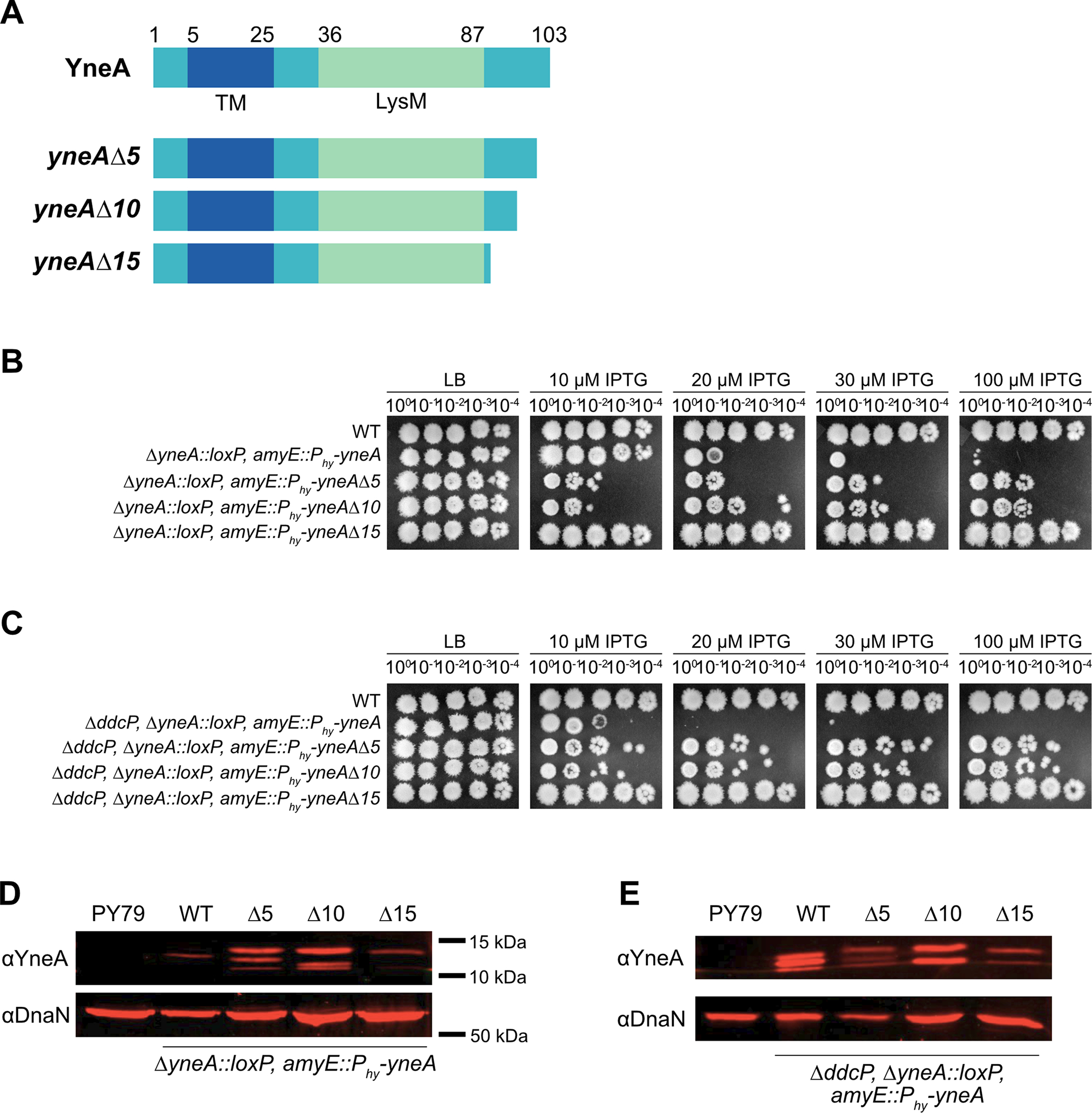
(A) Schematic of the full-length YneA protein and the C-terminal truncation mutants lacking the last five (yneAΔ5), ten (yneAΔ10) and fifteen (yneAΔ15) amino acid residues. YneA is predicted to have a transmembrane domain (TM) and a LysM binding domain (LysM). (B) Spot titer assay using B. subtilis strains WT (PY79), ΔyneA::loxP amyE::Phy-yneA (EAM46), ΔyneA::loxP amyE::Phy-yneAΔ5 (EAM53), ΔyneA::loxP amyE::Phy-yneAΔ10 (EAM54) and ΔyneA::loxP amyE::Phy-yneAΔ15 (EAM55) spotted on the indicated media. (C) Spot titer assay using B. subtilis strains WT (PY79), ΔddcP ΔyneA::loxP amyE::Phy-yneA (EAM48), ΔddcP ΔyneA::loxP amyE::Phy-yneAΔ5 (EAM83), ΔddcP ΔyneA::loxP amyE::Phy-yneAΔ10 (EAM84) and ΔddcP ΔyneA::loxP amyE::Phy-yneAΔ15 (EAM85) spotted on the indicated media. (D) Western blot using antisera against YneA (upper panel) or DnaN (lower panel) using B. subtilis strains WT (PY79), ΔyneA::loxP amyE::Phy-yneA (EAM46), ΔyneA::loxP amyE::Phy-yneAΔ5 (EAM53), ΔyneA::loxP amyE::Phy-yneAΔ10 (EAM54) and ΔyneA::loxP amyE::Phy-yneAΔ15 (EAM55) after growing in the presence of IPTG until an OD600 = 1. (E) Western blot using antisera against YneA (upper panel) or DnaN (lower panel) using B. subtilis strains WT (PY79), ΔddcP ΔyneA::loxP amyE::Phy-yneA (EAM48), ΔddcP ΔyneA::loxP amyE::Phy-yneAΔ5 (EAM83), ΔddcP ΔyneA::loxP amyE::Phy-yneAΔ10 (EAM84) and ΔddcP ΔyneA::loxP amyE::Phy-yneAΔ15 (EAM85) after growing in the presence of IPTG until an OD600 = 1.
Previous work showed that cells are more sensitive to yneA overexpression when they lack a single checkpoint recovery protease (Burby, Simmons et al. 2018). As a result, we asked if overexpression of the yneA C-terminal truncation mutants caused sensitivity when expressed in the ΔddcP single protease mutant background. We show that cells are more sensitive to yneA overexpression in the ΔddcP protease mutant background compared to expression in the yneA null strain (Fig. 1B, C). In addition, the C-terminal truncations also cause a more sensitive overexpression phenotype in the protease mutant background compared to expression in the yneA null strain (Fig. 1B, C). Nevertheless, overexpression of the yneAΔ5 or yneAΔ10 truncation caused moderate growth interference while yneAΔ15 truncation mutant was completely benign (Fig. 1C). Importantly, these results show that the C-terminal amino acids of YneA are important for activity and that these alleles do not confer a more toxic expression phenotype as observed for the S. aureus SosA C-terminal truncation mutants lacking the last 10 or 20 amino acid residues (Bojer, Wacnik et al. 2019). Further, our results show that loss of the C-terminal 15 amino acids of YneA completely blocks checkpoint enforcement indicating that complete YneA clearance is not required for cell division to resume.
Given that the C-terminal truncations attenuated the growth interference of yneA expression in both the ΔyneA::loxP and ΔyneA::loxP, ΔddcP backgrounds, we asked if YneA protein levels changed when the truncation mutants were overexpressed. When YneA is examined by Western blot multiple bands are observed because the protein is cleaved by proteases generating different sizes (Burby, Simmons et al. 2018). In the ΔyneA::loxP background, we observed an increase in YneA expression when yneAΔ5 and yneAΔ10 truncation mutants were induced relative to WT. This result indicates that although the YneAΔ5 and YneAΔ10 variants accumulate to levels higher than WT they are less toxic than WT YneA when IPTG concentrations are increased. The stabilization of YneAΔ5 and YneAΔ10 further suggests that both variants are less susceptible to proteolytic digestion by DdcP and CtpA although the deletion of these C-terminal residues impairs YneA checkpoint enforcement. To gain more insight into the susceptibility of the YneA C-terminal truncations to proteolytic cleavage we completed Western blots of YneA, YneAΔ5, YneAΔ10 and YneAΔ15 in lysates prepared from cells lacking DdcP (ΔddcP) (Fig. 1E). We show that induction of YneAΔ15 caused a reduction in YneA expression in the ΔddcP background as well. We find a lower abundance of YneAΔ15 as compared with WT or YneAΔ10 (Fig. 1E). With this data we suggest that YneA is less stable without the last fifteen amino acids, and this truncation is completely ineffective at inducing the DNA damage checkpoint. In conclusion, we show that once YneA has lost its C-terminal 15 amino acids it is inactivated demonstrating that protease cleavage of the C-terminal residues is sufficient to inactivate YneA and allow for cell division to resume without requiring complete clearance of the protein. This observation provides a mechanism for efficient inactivation of the DNA damage checkpoint after YneA expression is repressed and the C-terminus is cleaved by DdcP or CtpA (Burby, Simmons et al. 2018).
Isolation of mutations in YneA that prevent checkpoint activation
Given our results above showing the importance of the C-terminal residues to YneA activity we chose to identify single residues critical for checkpoint enforcement using a genetic selection. We show in Figure 1 that B. subtilis cells are strongly growth impaired when ectopic expression of yneA occurs from an IPTG regulated promoter. This provides an assay to select for mutations in yneA that fail to enforce the DNA damage checkpoint with the potential to identify the most important characteristics of YneA that are required for checkpoint activation (Fig. 2A). Therefore, we selected for colonies that were able to grow on LB plates containing IPTG to induce expression of WT yneA (Fig. 2A). We identified three mutations in the yneA gene located in two functional domains (Fig. 2B) (Mo and Burkholder 2010). One mutation is located in the transmembrane domain and two mutations are located in the LysM peptidoglycan binding domain. Mutations in the transmembrane domain have been extensively studied in prior work (Mo and Burkholder 2010), while mutations in the LysM domain have not been studied and only truncation of the entire LysM domain has been reported to inactivate YneA (Mo and Burkholder 2010). The important point from this selection is that our unbiased approach has identified three-point mutations within two regions, which appear to render YneA incapable of checkpoint enforcement.
Figure 2. Isolation of mutations in YneA that prevent checkpoint activation.
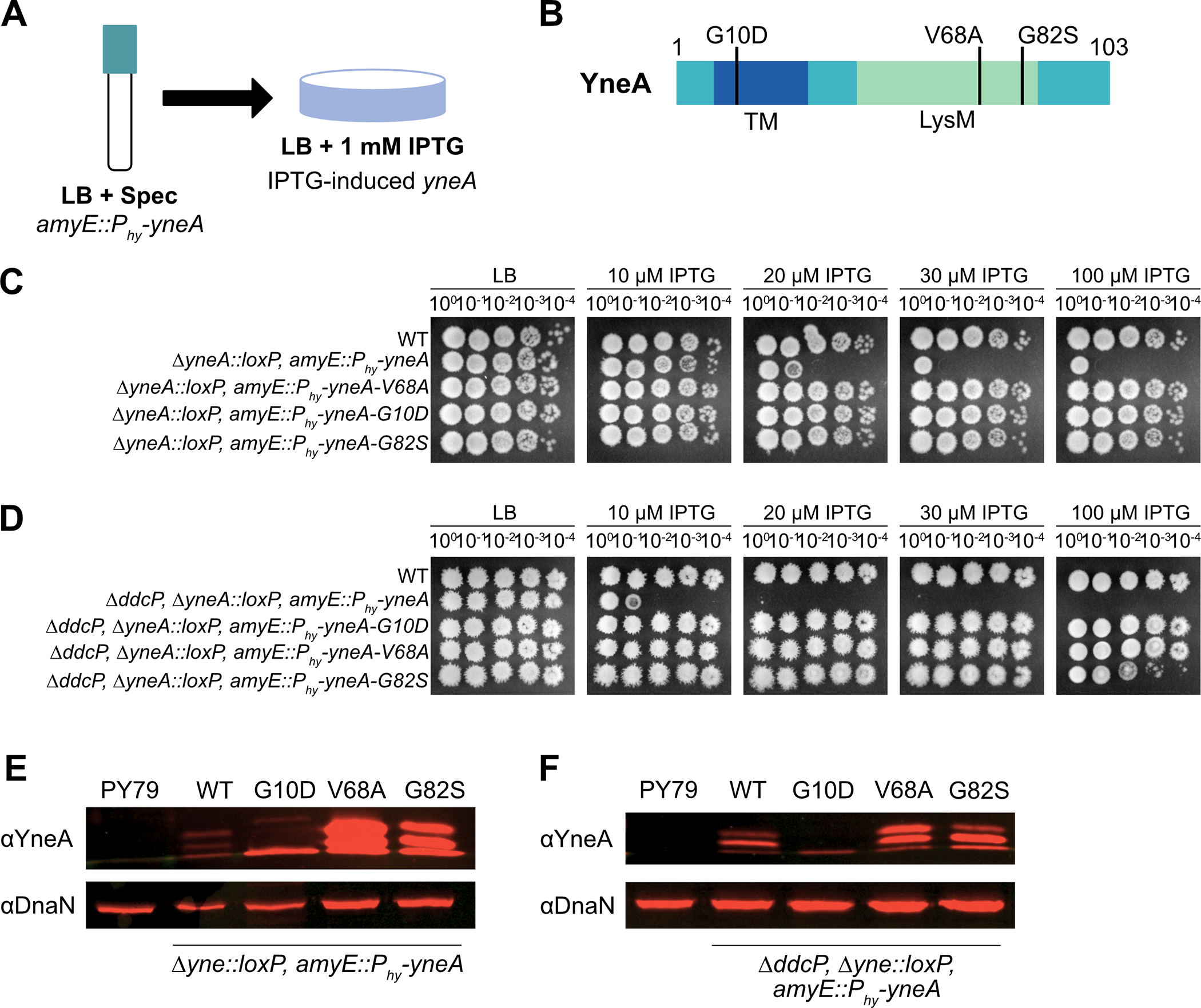
(A) Experimental design for the primary selection. Cultures were plated on LB agar containing 1 mM IPTG to induce expression of amyE::Phy-yneA. (B) Schematic of the YneA protein and the location of the suppressor mutations identified in the screen. Transmembrane domain (TM) and a LysM binding domain (LysM). (C) Spot titer assay using B. subtilis strains WT (PY79), ΔyneA::loxP amyE::Phy-yneA (EAM46), ΔyneA::loxP amyE::Phy-yneA-V68A (EAM49), ΔyneA::loxP amyE::Phy-yneA-G10D (EAM50) and ΔyneA::loxP amyE::Phy-yneA-G82S (EAM52) spotted on the indicated media. (D) Spot titer assay using B. subtilis strains WT (PY79), ΔddcP ΔyneA::loxP amyE::Phy-yneA (EAM48), ΔddcP ΔyneA::loxP amyE::Phy-yneA-G10D (EAM63), ΔddcP ΔyneA::loxP amyE::Phy-yneA-V68A (EAM78) and ΔddcP ΔyneA::loxP amyE::Phy-yneA-G82S (EAM79) spotted on the indicated media. (E) Western blot using antisera against YneA (upper panel) or DnaN (lower panel) using B. subtilis strains WT (PY79), ΔyneA::loxP amyE::Phy-yneA (EAM46), ΔyneA::loxP amyE::Phy-yneA-V68A (EAM49), ΔyneA::loxP amyE::Phy-yneA-G10D (EAM50) and ΔyneA::loxP amyE::Phy-yneA-G82S (EAM52) after growing in the presence of IPTG until an OD600 = 1. (F) Western blot using antisera against YneA (upper panel) or DnaN (lower panel) using B. subtilis strains WT (PY79), ΔddcP ΔyneA::loxP amyE::Phy-yneA (EAM48), ΔddcP ΔyneA::loxP amyE::Phy-yneA-G10D (EAM63), ΔddcP ΔyneA::loxP amyE::Phy-yneA-V68A (EAM78) and ΔddcP ΔyneA::loxP amyE::Phy-yneA-G82S (EAM79) after growing in the presence of IPTG until an OD600 = 1.
To functionally assess the novel yneA alleles, we cloned each and placed the alleles in B. subtilis at an ectopic locus in a clean genetic background and performed spot titer assays to determine if these mutations render YneA incapable of blocking cell division (Fig. 2C, D). We ectopically induced expression of each allele with increasing concentrations of IPTG in the absence of native yneA (ΔyneA::loxP). We found that each yneA allele was impaired or completely broken for checkpoint enforcement even in conjunction with deletion of the checkpoint recovery protease ddcP (ΔyneA::loxP, ΔddcP) (Fig. 2C, D). These results establish that induced expression of all three mutants renders yneA incapable of causing a block to cell division (Fig. 2C, D).
We directly assessed YneA protein levels using Western blotting, to determine if the integrity of the protein variants were compromised (Fig. 2E, F). We observed higher expression of WT in the absence of endogenous ddcP, as previously established (Burby, Simmons et al. 2018). We detected a single band when yneA-G10D was induced in the yneA null background and a complete loss of expression in the absence of endogenous ddcP (Fig. 2F). Although the reason for poor accumulation of YneAG10D is unclear, it suggests that YneAG10D is either intrinsically unstable or hypersensitive to proteolysis by other proteases in the absence of ddcP. We observe an increase in expression of LysM domain YneA variants V68A and G82S relative to WT.
We asked if YneA LysM domain mutants V68A and G82S are dominant or recessive to SOS induced yneA. We found that expression of yneA V68A and G82S are recessive to SOS induced yneA on increasing concentrations of DNA damage (Fig. S1). In addition, we created a LysM domain swap where we replaced the YneA LysM domain with the LysM from the B. subtilis sporulation protein SafA (Pereira, Nunes et al. 2019) followed by the YneA C-terminal tail. We found that expression of the yneA-safA-lysM chimera failed to interfere with growth demonstrating that the YneA LysM domain is specific for checkpoint enforcement (Fig. S2). These results establish the importance of the LysM domain for YneA activity.
Cells are more sensitive to yneA induction in the absence of the negative regulators ddcP, ctpA and ddcA
Previous work established that the checkpoint recovery proteases, DdcP and CtpA, as well as the DNA damage checkpoint antagonist, DdcA, ensure YneA activity is suppressed in the absence of DNA damage (Burby, Simmons et al. 2018, Burby, Simmons et al. 2019). Moreover, YneA expression must reach a certain threshold to overcome these negative regulators to activate the checkpoint and inhibit cell division (Burby, Simmons et al. 2018, Burby, Simmons et al. 2019). This work also showed that cell proliferation was inhibited when yneA was expressed ectopically using xylose induction in the absence of ddcA, ddcP and ctpA and in the presence of native yneA (Burby, Simmons et al. 2019). We built from these prior studies to clearly establish a system where we could drive yneA expression and cause toxicity using either xylose or an IPTG induced promoter (Fig. 1B, C). We ectopically expressed yneA using an IPTG regulated promoter in cells lacking ddcA, ddcP, ctpA, and native yneA genes (Fig. 3A). As a control we show that growth inhibition does not occur in the absence of ddcP, ctpA, ddcA and native yneA, supporting prior results that growth inhibition is dependent on induced yneA expression (Fig. 3A) (Burby, Simmons et al. 2019). Therefore, the cell proliferation defect observed with ΔddcP, ΔctpA, ΔddcA and ΔyneA::loxP is caused by induced expression of IPTG regulated yneA (Fig. 3A) (Burby, Simmons et al. 2018, Burby, Simmons et al. 2019).
Figure 3. Cells are more sensitive to yneA induction in the absence of the negative regulators ddcP, ctpA and ddcA.

(A) Spot titer assay using B. subtilis strains WT (PY79), ΔddcP ΔctpA ΔddcA (PEB639), ΔyneA::loxP (PEB439), ΔddcP ΔctpA ΔddcA ΔyneA::loxP (PEB643), ΔddcP ΔctpA ΔddcA amyE::Phy-yneA (PEB844), ΔyneA::loxP amyE::Phy-yneA (EAM46) and ΔddcP ΔctpA ΔddcA ΔyneA::loxP amyE::Phy-yneA (EAM56) spotted on the indicated media. (B) Spot titer assay using B. subtilis strains WT (PY79), ΔddcP ΔctpA ΔddcA (PEB639), ΔyneA::loxP (PEB439), ΔddcP ΔctpA ΔddcA ΔyneA::loxP (PEB643), ΔddcP ΔctpA ΔddcA amyE::Phy-yneA (PEB844), ΔyneA::loxP amyE::Phy-yneA (EAM46) and ΔddcP ΔctpA ΔddcA ΔyneA::loxP amyE::Phy-yneA (EAM56) spotted on the indicated media.
We further investigated the effect of yneA expression on cell proliferation by treating cells with increasing concentrations of the DNA damaging agent mitomycin C (MMC) (Iyer and Szybalski 1963, Noll, Mason et al. 2006) to induce native yneA in the presence or absence of the IPTG regulated yneA allele (Burby, Simmons et al. 2019) (Fig. 3B). It was previously shown that YneA inhibits cell division in B. subtilis following DNA damage (Kawai, Moriya et al. 2003, Burby, Simmons et al. 2018, Burby, Simmons et al. 2019). As a result, we expect a cell proliferation defect following MMC treatment because endogenous yneA should be activated. When we treat cells with increasing concentrations of MMC, we find that cells are sensitive to MMC and this phenotype is more severe in the absence of the ddcP, ctpA and ddcA negative regulators of YneA (Fig. 3B) as described (Burby, Simmons et al. 2019). If this phenotype is due to induction of endogenous yneA, then we would expect that loss of the native yneA gene should rescue the phenotype. Indeed, we show that cells are able to continue proliferating when treated with MMC in the absence of endogenous yneA and in the presence of IPTG regulated yneA (uninduced) (Fig. 3B). As a result, the inability to continue proliferating after MMC treatment is the result of SOS regulated expression of the native yneA gene supporting prior observations (Burby, Simmons et al. 2018, Burby, Simmons et al. 2019).
These results and those of Burby et al. establish that we can inhibit cell proliferation by inducing expression of yneA from two different locations in the genome (Burby, Simmons et al. 2019). Our ability to induce ectopic yneA or native yneA in the absence of the yneA negative regulators establishes a strong selective pressure to isolate mutations outside of the yneA gene that are refractory to checkpoint enforcement (see below).
ftsW-L148P suppresses YneA activity in the presence of DNA damage
In order to better understand how YneA inhibits cell division, we sought to identify additional factors that are involved in this process. The intent is to isolate extragenic mutations that are refractory to checkpoint enforcement. To achieve this end, we devised a method that takes advantage of yneA at two different chromosomal locations under different transcriptional regulatory control as described in Figure 3 and in (Burby, Simmons et al. 2019). We first employed a strain with yneA at its native locus under SOS control and yneA integrated at the amyE locus with expression under xylose control for tight repression to decrease the likelihood of leak expression causing growth interference. Therefore, this method favors mutations outside of the yneA gene that will overcome YneA function because to inactivate the yneA gene directly would require mutations in two separate copies of yneA located at distant chromosomal locations.
Our method identified 25 independent mutations that occurred in the ftsW gene. A striking feature of this result is that of the 25 mutations identified in the ftsW gene only three amino acids were affected and 19 out of 25 changes altered one amino acid residue (Fig. 4B). A few other spurious mutations were identified, these mutations only occurred once and are reported in Supporting Information Table S1. We interpret this result to mean that there are a select few mutations outside of yneA that can overcome checkpoint enforcement contributing to the reported difficulties in isolating such mutants (Mo and Burkholder 2010). As a follow up test, we introduced the ftsW-A99V, ftsW-L148P, ftsW-P158L and ftsW-P158S under the control of an IPTG-inducible promoter at an ectopic locus to allow for increased expression in WT and ΔddcP, ΔctpA, ΔddcA triple mutant backgrounds with native ftsW intact. This was done to determine if any of the ftsW mutants we isolated are dominant negative to WT ftsW as a stringent genetic test for integrity of the variant protein in vivo.
Figure 4. ftsW-L148P suppresses YneA activity in the presence of DNA damage.
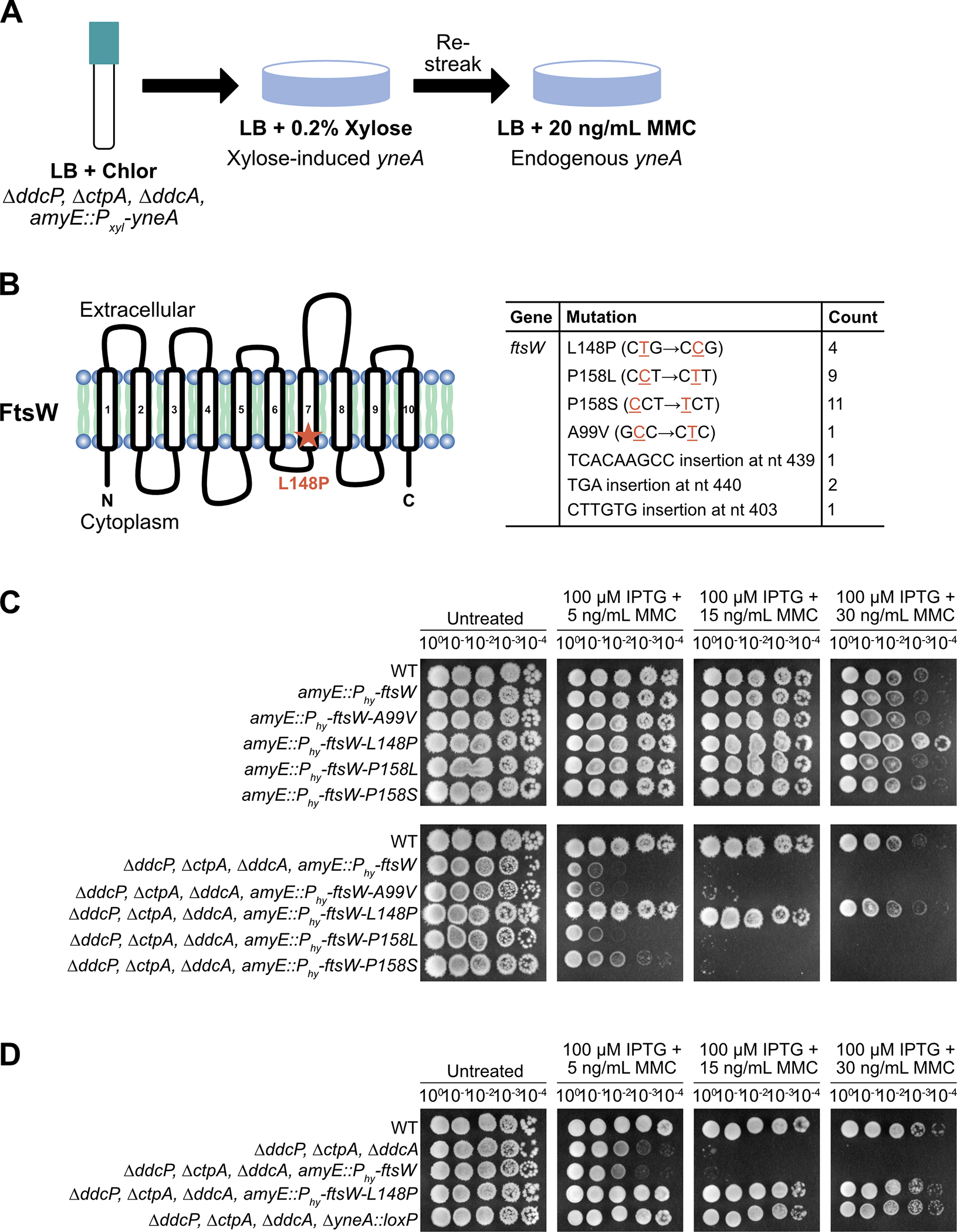
(A) Experimental design for the selection followed by secondary screen. Cultures were plated on LB agar containing 0.2% xylose to induce expression of amyE::Pxyl-yneA. Colonies were re-streaked on LB agar containing 20 ng/mL MMC to induce expression of endogenous yneA. (B) Schematic of the FtsW protein and the location of the suppressor mutation identified in the screen. FtsW is a membrane-spanning protein that is predicted to have ten transmembrane segments. Table of the ftsW point mutations and insertions identified in the screen. (C) Spot titer assay using B. subtilis strains WT (PY79), amyE::Phy-ftsW (EAM72), amyE::Phy-ftsW-A99V (EAM68), amyE::Phy-ftsW-L148P (EAM69), amyE::Phy-ftsW-P158L (EAM70), amyE::Phy-ftsW-P158S (EAM71), ΔddcP ΔctpA ΔddcA amyE::Phy-ftsW (EAM73), ΔddcP ΔctpA ΔddcA amyE::Phy-ftsW-A99V (EAM64), ΔddcP ΔctpA ΔddcA amyE::Phy-ftsW-L148P (EAM65), ΔddcP ΔctpA ΔddcA amyE::Phy-ftsW-P158L (EAM66) and ΔddcP ΔctpA ΔddcA amyE::Phy-ftsW-P158S (EAM67) spotted on the indicated media. (D) Spot titer assay using B. subtilis strains WT (PY79), ΔddcP ΔctpA ΔddcA (PEB639), ΔddcP ΔctpA ΔddcA amyE::Phy-ftsW (EAM73), ΔddcP ΔctpA ΔddcA amyE::Phy-ftsW-L148P (EAM65) and ΔddcP ΔctpA ΔddcA ΔyneA::loxP (PEB643) spotted on the indicated media.
If the FtsW variant bypasses YneA activity and is dominant to WT FtsW this would provide the best candidate for further characterization. To this end, we asked if each ftsW allele was able to suppress the yneA-dependent DNA damage checkpoint in the presence of MMC and WT ftsW. We found that IPTG-induced ftsW-L148P was refractory to YneA in an otherwise WT background or in cells lacking all three YneA negative regulators (ΔddcP, ΔctpA, ΔddcA) (Fig. 4C). The triple mutant strain alone is highly sensitive to MMC treatment; however, induction of ftsW-L148P, but not ftsW, in this background rescues growth (Fig. 4D). If this mutation is bypassing YneA activity, then we would expect that the strain expressing ftsW-L148P to phenocopy the strain without endogenous yneA. Indeed, challenged with MMC, in the absence of endogenous yneA, cells are able to continue growth (Fig. 4D). We observed that IPTG-induced ftsW-L148P rescued the sensitivity to MMC and rescued the cell proliferation defect observed in the triple mutant alone. These results establish ftsW-L148P as either encoding a form of FtsW that induces hyperactive cell division to bypass YneA inhibition or an FtsW variant that is refractory to negative regulation by impairing direct interaction between YneA and FtsW.
ftsW-L148P bypasses yneA expression
Based on the observation that induced expression of ftsW-L148P suppressed YneA activity, we hypothesized that ftsW-L148P either prevents interaction with YneA or induces hyperactive cell division bypassing the YneA-induced checkpoint due to a change in conformation of the late divisome. Because FtsW is an essential protein with ten transmembrane domains we chose to measure cell length as a proxy to initiate division hyperactively in vivo. If FtsW-L148P is a variant that causes hyperactive cell division than cells expressing this variant should be shorter in the presence of DNA damage induced YneA. Such a result would suggest that FtsW-L148P overcomes the YneA-induced checkpoint through a change in interaction with YneA, a change in peptidoglycan synthesis activity or a change in the divisome that initiates cell division hyperactively.
To test these ideas, we grew cells expressing ftsW-L148P or ftsW in a WT or ΔddcP, ΔctpA, ΔddcA triple mutant background during normal growth or in the presence of DNA damage and measured cell length. Under conditions of normal growth, we did not observe a difference in cell length compared to WT when we induced ftsW and ftsW-L148P (Fig. 5A, Supporting Fig. S3, Table S2). When we caused DNA damage with MMC, and therefore expression of native SOS-controlled yneA we found that cells expressing both ftsW-L148P and yneA were shorter in length. We found that cells expressing ftsW-L148P (7.27 ± 1.64) are nearly 30% shorter than cells expressing ftsW (10.15 ± 2.97) and this difference was significant (p=4.71E−300) (Fig. 5B, Table S3). Given that ftsW does not bypass YneA activity, we would not expect the cell length of ftsW expressing cells in the triple mutant background to be much different than the triple mutant background alone. Indeed, we did not observe a reduction in the cell length of ftsW expressing cells in the triple mutant background (15.86 ± 4.89) compared to the triple mutant alone (16.5 ± 5.21) (Fig. 5B, Supporting Fig. S4 Table S3). However, cell length is dramatically reduced in cells expressing ftsW-L148P in the triple mutant background (8.81 ± 2.43), a near 50% reduction in cell length, which is significantly different (p=2.6E−36) (Fig. 5B, Supporting Fig. S4 Table S3). With these results we suggest the FtsW is unlikely to be a direct target of YneA. Instead, we suggest that ftsW-L148P generates a form of FtsW, which causes cell division to initiate hyperactively bypassing the inhibitory effect of YneA and preventing activation of the DNA damage checkpoint. Our result showing that ftsW-L148P does not result in shorter cells in the absence of DNA damage suggests the hyperactive cell division by FtsW-L148P either requires DNA damage to observe the effect or is more pronounced during conditions of damage when cell filamentation is extreme (Fig. 5A, Table S2).
Figure 5. ftsW-L148P bypasses yneA expression.
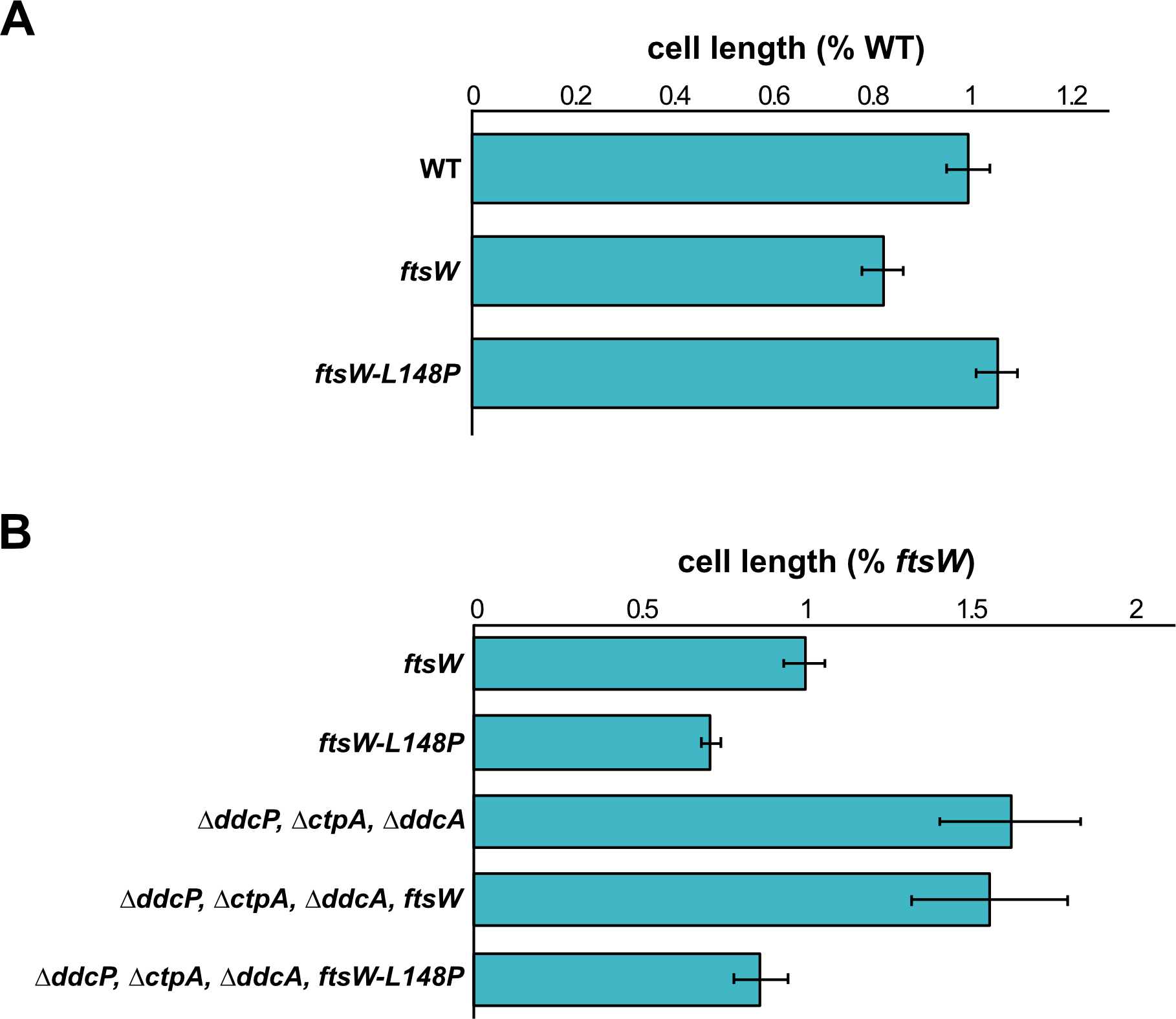
(A) Cell lengths of each strain relative to WT plotted as a bar graph. Error bars represent standard error of the mean (SEM). The significance test are as follows for a two-tailed t-test: PY79 and amyE::Phy-ftsW (p=0.988); PY79 and amyE::Phy-ftsW-L148P (p=0.959). (B) Cell lengths of each strain relative to ftsW plotted as a bar graph. Error bars represent standard error of the mean (SEM). The significance test are as follows for a two-tailed t-test. For amyE::Phy-ftsW and amyE::Phy-ftsW-L148P (p=4.71E−300); amyE::Phy-ftsW and ΔddcP, ΔctpA, ΔddcA (p=5.14E−119); amyE::Phy-ftsW and ΔddcP, ΔctpA, ΔddcA, amyE::Phy-ftsW-L148P (p=2.6E−36). The cell length measurements graphed here are also presented in supporting Tables S2 and S3.
YneA interacts with FtsL, Pbp2b and Pbp1, but not FtsW
Given that cells expressing ftsW-L148P suppress YneA activity, we hypothesized that YneA either directly interacts with FtsW to inhibit cell division as observed for the Caulobacter proteins or FtsW-L148P encodes a hyperactive form of the protein as suggested above (Modell, Hopkins et al. 2011). We chose to assess interaction using the bacterial two-hybrid system (Karimova, Pidoux et al. 1998, Karimova, Gauliard et al. 2017) as done previously to measure interaction between Caulobacter SidA and FtsW (Modell, Hopkins et al. 2011). As a positive control it was previously shown that YneA and a catalytically inactive CtpA-S297A protease variant interact by bacterial two-hybrid analysis (Burby, Simmons et al. 2018). We did not observe an interaction between YneA and FtsW or YneA and FtsW-L148P (Fig. 6A, B). Since we did not detect a direct interaction between YneA and FtsW, we asked if YneA targets FtsW indirectly by interacting with other proteins involved in the late arriving divisome affecting cell division or peptidoglycan synthesis (Kawai and Ogasawara 2006, Król, van Kessel et al. 2012, Halbedel and Lewis 2019, Morales Angeles, Macia-Valero et al. 2020). First, we failed to observe an interaction between YneA and nine other proteins known to be involved in these processes (Fig. 6A). We did however, find a weak signal between YneA and FtsL suggesting an interaction may occur. When we switched the T25 and T18 fusions, we identified an interaction between YneA and FtsL, Pbp2b and Pbp1 (Fig. 6C). FtsL is a an unstable late divisome component (Daniel and Errington 2000). Pbp2b is a transpeptidase and Pbp1 is a bifunctional transpeptidase/transglycosylase (Yanouri, Daniel et al. 1993, Popham and Setlow 1995). FtsL, Pbp2b and Pbp1 all localize to the septum late during division contributing to the divisome or septal peptidoglycan synthesis (Scheffers and Errington 2004, Bhambhani, Iadicicco et al. 2020). These results suggest that YneA could indirectly effect FtsW through a direct interaction with one of these proteins contributing to DNA damage checkpoint enforcement.
Figure 6. YneA interacts with FtsL, Pbp2b and Pbp1.
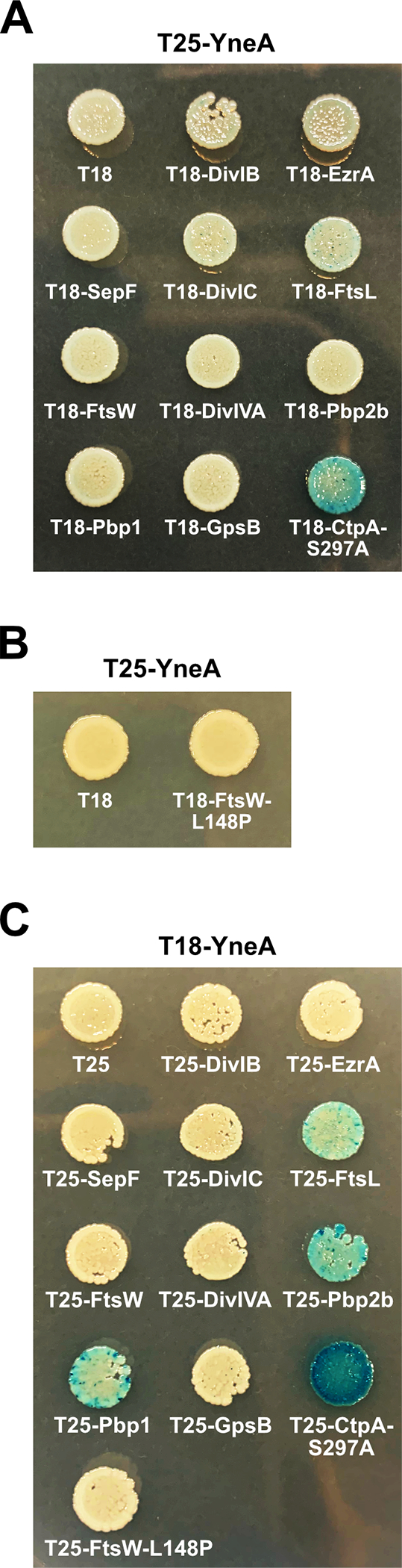
Bacterial two-hybrid assay using (A) empty vector (T18), T18 fusions and T25-YneA fusion; (B) empty vector (T18), T18-FtsW-L148P fusion and T25-YneA fusion; (C) empty vector (T25), T25 fusions and T18-YneA co-transformed into E. coli.
Mutations that prevent checkpoint activation and bypass yneA expression are less sensitive to an inhibitor of cell wall synthesis
Given that our results shown above suggest that YneA inhibits cell division by binding peptidoglycan through its LysM domain and through interaction with FtsL, Pbp2b and Pbp1, we asked if cells expressing YneA are more sensitive to a cell wall antibiotic (Fig. 7). If YneA prevents cell division by binding peptidoglycan then we would expect cells to be more sensitive to a cell wall inhibitor when YneA is expressed. Therefore, we treated cells with the inhibitor cephalexin, which restricts septal cell wall synthesis by preventing FtsI from crosslinking the glycan strands, but it does not directly damage the DNA (Modell, Kambara et al. 2014). As a result, cephalexin will impede cell division, but independent of YneA. First, we treated cells with a low concentration of cephalexin in conjunction with increasing concentrations of IPTG and performed spot titer assays to assess if yneA expression affected growth in the presence of cephalexin (Fig. 7A). We found that WT cells are unaffected by a low concentration of cephalexin; however, induction of yneA with increased concentrations of IPTG caused strong growth interference (Fig. 7A, B). When we activate native yneA following treatment with MMC, cells are more sensitive to cephalexin (compare Fig. 7B with Fig. 4D) and this proliferation defect is suppressed by ΔyneA (Fig. 7B). To assess how the novel yneA alleles respond to cephalexin, we ectopically induced expression of each allele with increasing concentrations of IPTG in the absence of native yneA. At lower concentrations of IPTG, we found that each yneA mutant phenocopied the yneA null strain and suppressed the growth interference on cephalexin treatment (Fig 7B).
Figure 7. Mutations that prevent checkpoint activation and bypass yneA expression are less sensitive to an inhibitor of cell wall synthesis.
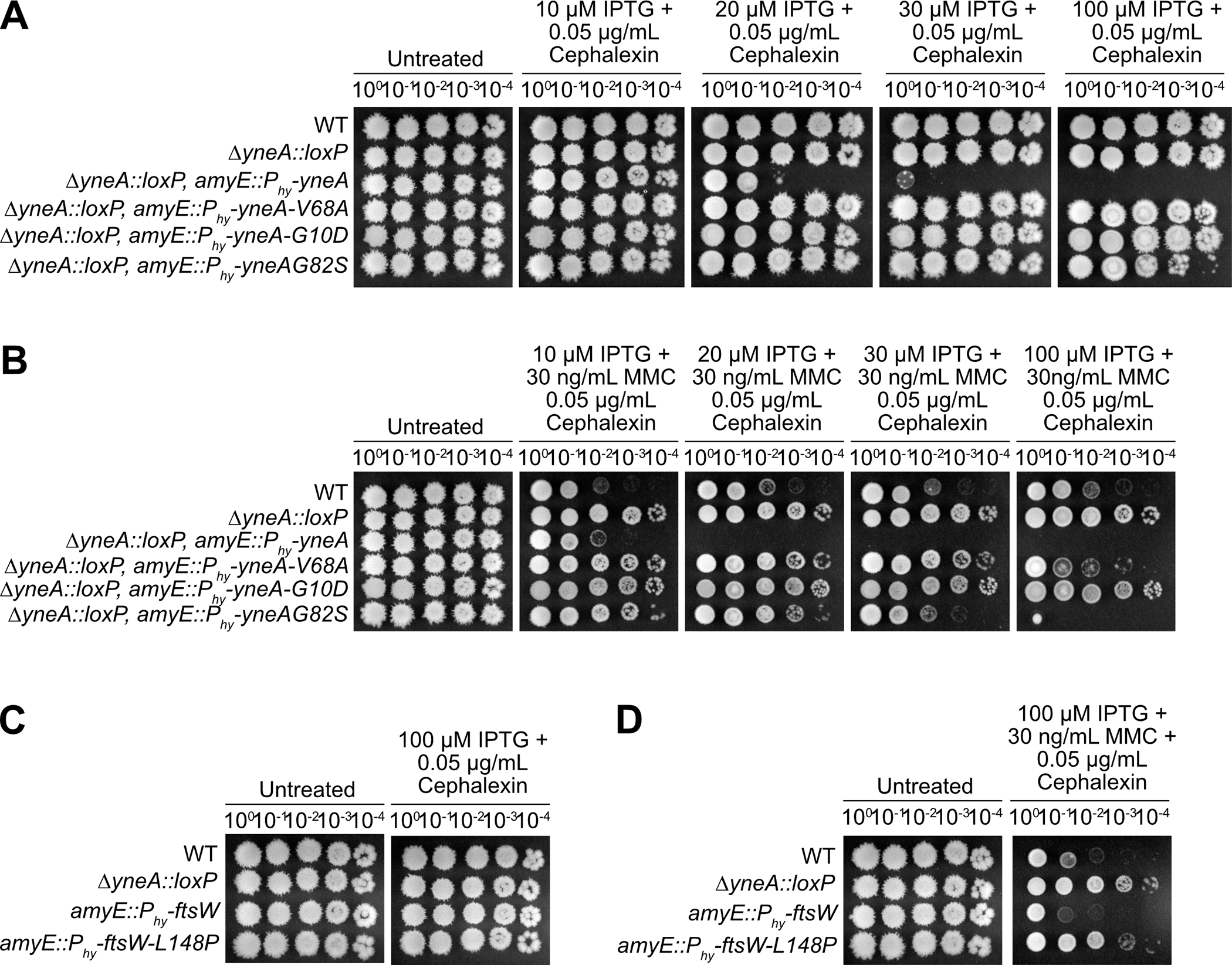
Spot titer assay using B. subtilis strains (A) and (B) WT (PY79), ΔyneA::loxP (PEB439), ΔyneA::loxP amyE::Phy-yneA (EAM46), ΔyneA::loxP amyE::Phy-yneA-V68A (EAM49), ΔyneA::loxP amyE::Phy-yneA-G10D (EAM50) and ΔyneA::loxP amyE::Phy-yneA-G82S (EAM52); (C) and (D) WT (PY79), ΔyneA::loxP (PEB439), amyE::Phy-ftsW (EAM72) and amyE::Phy-ftsW-L148P (EAM69) spotted on the indicated media.
To assess how ftsW-L148P responds to cephalexin, we ectopically induced WT ftsW and ftsW-L148P with IPTG in the presence of cephalexin. Similar to previous results, a low concentration of cephalexin does not hinder cell proliferation (Fig. 7C). With the addition of MMC, WT cells are sensitive to cephalexin and are unable to continue proliferating (compare Fig. 7D with Fig. 4D). However, induced expression of ftsW-L148P suppresses the effect of cephalexin and phenocopied the yneA null strain (Fig. 7D). These results support the conclusion that ftsW-L148P generates a hyperactive form of FtsW that is able to bypass the inhibitory effect of YneA and prevent activation of the DNA damage checkpoint. Further, our results showing that expression of YneA causes hypersensitivity to cephalexin supports the model that part of the inhibitory effect of YneA on cell division is exerted through peptidoglycan binding by the YneA LysM domain.
Discussion
DNA damage checkpoints are ubiquitous across biology. In all organisms the overarching process is to slow or arrest the cell cycle when DNA damage is detected enabling enough time for repair before chromosomes are segregated and cell division is complete. In bacteria, an SOS-induced protein enforces the DNA damage checkpoint by preventing cell division [for review (Burby and Simmons 2020)]. In E. coli, SOS induced SulA blocks cell division by preventing FtsZ polymerization (Mukherjee, Cao et al. 1998); however, in C. crescentus, SidA and DidA do not affect FtsZ assembly, but instead delay cell division through a direct interaction with FtsW and FtsN, respectively (Modell, Hopkins et al. 2011, Modell, Kambara et al. 2014). In S. aureus, SOS-induced SosA does not block the initial steps of septum formation, but prevents the final steps of cell division (Bojer, Wacnik et al. 2019). In addition, previous two-hybrid analysis indicated possible interactions between SosA and factors required for cell division suggesting that like SidA and DidA protein-protein interactions are required for SosA-dependent checkpoint enforcement (Modell, Hopkins et al. 2011, Bojer, Wacnik et al. 2019). Therefore, the most prominent mechanism of checkpoint enforcement in bacteria invokes an interaction between an SOS-induced cell division inhibitor and a component of the divisome FtsZ, FtsW or FtsN. In Caulobacter, some of the mutant forms for FtsW, I and N that overcome SidA and DidA result in a mild decrease in cell length (4–14%) suggesting that cell division can initiate hyperactively to some degree in these mutants as well (Modell, Hopkins et al. 2011). Based on our results we suggest that YneA blocks cell division by targeting peptidoglycan through its LysM domain and by contact with FtsL, Pbp2b and Pbp1 interfering with the ability of these proteins to properly function in cell division (Fig. 8).
Figure 8. Model for YneA-induced cell division inhibition.
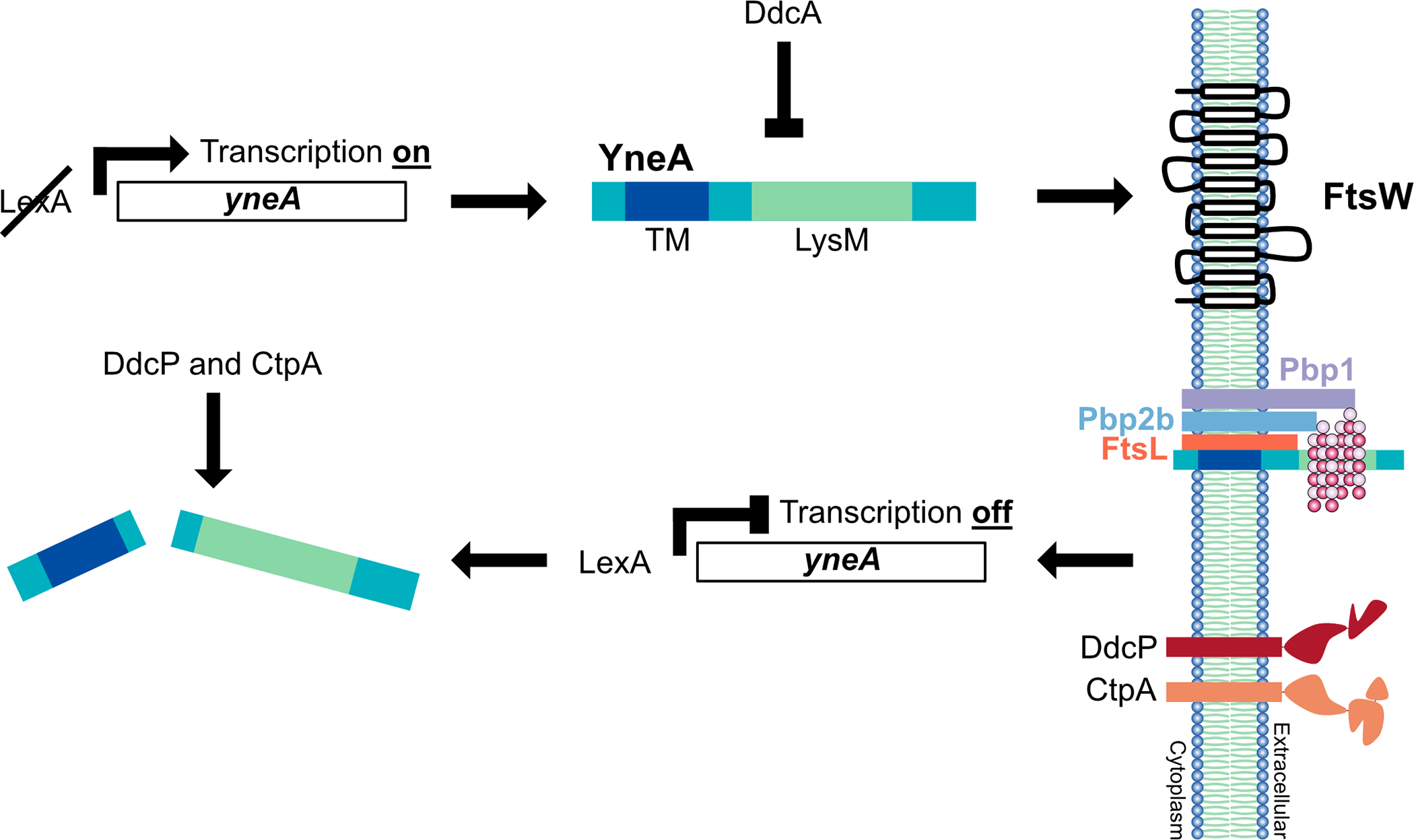
In the presence of DNA damage, cleavage of LexA allows for expression of the SOS-induced cell division inhibitor YneA. YneA expression must reach a critical threshold to bypass the negative regulators DdcA, DdcP and CtpA to activate the checkpoint. YneA localizes to the membrane, mediated by the transmembrane domain (TM), where it interacts with the late divisome proteins FtsL, Pbp2b and Pbp1 as well as binds peptidoglycan interfering with cell wall remodeling at the septum. After the DNA is repaired and YneA expression is repressed by LexA, YneA is cleared by the proteases DdcP and CtpA allowing septal cell wall synthesis to commence and cell division to resume.
Another important feature of this work is our finding that loss of amino acid residues from the C-terminal tail up to the LysM domain decrease or prevent cells from responding to checkpoint enforcement (Fig. 1). Previous work identified a point mutation in the extreme C-terminus that increased YneA stability and activity; however, removal of the entire C-terminal region of the protein, including the LysM domain, abolished YneA function (Mo and Burkholder 2010). In line with the findings that full-length YneA is important to block cell division (Mo and Burkholder 2010), we show that the Δ5 and Δ10 C-terminal truncations are stable suggesting that loss of portions of the C-terminal tail impairs YneA function. Because the C-terminal tail directly follows the LysM domain, we speculate that the truncations may impair or alter LysM domain function. Another important finding is that B. subtilis cells show rather quick recovery from the DNA damage checkpoint (Burby, Simmons et al. 2018). Our results demonstrating that loss of the C-terminal 15 amino acids ablates YneA function even though the protein can still be detected in cell extracts suggests that the quick recovery is in part mediated by protease-dependent truncation of the C-terminal tail. Therefore, we suggest that DdcP and CtpA-mediated truncation of the C-terminal tail inactivates YneA quickly allowing cells to re-enter the cell cycle without requiring complete clearance of YneA from the septum.
We present a genetic selection for yneA mutants that fail to enforce the checkpoint identifying missense mutations with the only stable variants occurring in the LysM domain (Fig. 2). Previous work identified several point mutation in the transmembrane domain that caused a reduction in YneA activity; furthermore, complete loss of the C-terminus, including the LysM domain, ablated YneA function (Mo and Burkholder 2010). The LysM domain binds peptidoglycan and proteins that have LysM domains are often involved in remodeling the peptidoglycan cell wall (Buist, Steen et al. 2008). The analogous cell division inhibitor in M. tuberculosis, Rv2719c (ChiZ), contains a LysM domain that was previously suggested to have peptidoglycan hydrolytic activity, although a recent report shows it does not (Chauhan, Lofton et al. 2006, Escobar and Cross 2018). The precise role of the LysM domain in YneA remains undetermined; however, our results show that single amino acid substitutions in the YneA LysM, or a LysM domain swap abolish checkpoint enforcement even though the protein accumulates higher than WT levels in vivo. Our results considered with those of Mo and Burkholder demonstrate that integrity of the LysM domain is required for enforcement of the DNA damage checkpoint. We wish to note that most DNA damage induced cell division inhibitors lack a LysM domain (Bojer, Frees et al. 2020, Burby and Simmons 2020). SosA, DidA and SidA lack LysM domains and these proteins have been shown to interact with the cell wall synthesis machinery through a two-hybrid analysis indicating the mechanism of checkpoint enforcement is through protein-protein interactions (Modell, Hopkins et al. 2011, Modell, Kambara et al. 2014, Bojer, Wacnik et al. 2019). Further, we show that expression of yneA strongly sensitizes cells to the cell wall antibiotic cephalexin. This result combined with our data showing that YneA LysM domain point mutants confer less sensitivity to cephalexin and ftsW-L148P phenocopies the yneA null strain on cephalexin suggests that an important part of checkpoint enforcement is interference of YneA with septal peptidoglycan synthesis.
In M. tuberculosis, it was previously shown that overexpression of ChiZ did not alter the amount of FtsZ or its activity; however, ChiZ did affect the organization of FtsZ at the septum. As a result, the authors speculated that the loss of peptidoglycan, mediated by ChiZ, at the site of cell wall synthesis could disrupt FtsZ localization and subsequent Z-ring formation (Chauhan, Lofton et al. 2006). Interestingly, like ChiZ, overexpression of YneA did not change the level of FtsZ; however, YneA expression was reported to delay the assembly of FtsZ rings and/or the number of Z rings formed (Kawai, Moriya et al. 2003, Mo and Burkholder 2010). Bacterial two-hybrid analysis showed that ChiZ directly interacted with the cell division proteins FtsI and FtsQ, but not FtsZ (Vadrevu, Lofton et al. 2011). Furthermore, a previous study was unable to detect a direct interaction between YneA and FtsZ by a two-hybrid assay; however, we detected an interaction between YneA and FtsL, Pbp2b and Pbp1 (Fig. 6B and (Kawai, Moriya et al. 2003). We suggest that an important part of the YneA checkpoint enforcement mechanism is interfering with the ability of late divisome components FtsL, Pbp2B and Pbp1 to interact. Our results suggest that part of the YneA inhibitory mechanism overlaps with that of Gp56 protein from bacteriophage SPO1 (Bhambhani, Iadicicco et al. 2020).
We developed a genetic method using a selection followed by a secondary screen that yielded mutations in the ftsW gene, with most mutations impacting just two amino acid residues. Our characterization of ftsW-L148P shows that this variant completely bypassed the YneA-enforced DNA damage checkpoint including the cephalexin sensitivity (Fig. 4 and Fig. 7). FtsW is an essential protein that is required for the polymerization of Lipid II into peptidoglycan during septal cell wall synthesis (Pastoret, Fraipont et al. 2004, Taguchi, Welsh et al. 2019). Our results combined with the interactions we identified between YneA and other late divisome proteins suggest that ftsW-L148P encodes a hyperactive form of FtsW that is able to initiate division prematurely by stabilizing the late divisome components. In support of this model, in E. coli it has been shown that mutations in FtsL and FtsA are also able to initiate cell division hyperactively (Geissler, Shiomi et al. 2007, Tsang and Bernhardt 2015). In the case of the ftsL mutant the shorter cell length is a result of stabilization of late division components involved in septal peptidoglycan synthesis (Tsang and Bernhardt 2015). For FtsA* hyperactive initiation is through an increase in interaction between FtsA* and FtsZ(Geissler, Shiomi et al. 2007). In our work, induced expression of ftsW-L148P caused a substantial reduction in cell length compared to WT during the DNA damage response (Fig. 5B). Given that we did not detect an interaction between YneA and FtsW or FtsW-L148P, but we did detect an interaction between YneA and other components of the divisome contributing to division and septal cell peptidoglycan synthesis (Fig. 6A–C), we propose a model where YneA blocks cell division through both a direct protein-protein interaction with FtsL, Pbp2b, and Pbp1 and through binding to peptidoglycan using its LysM domain (Bhambhani, Iadicicco et al. 2020). We further suggest that ftsW-L148P overcomes YneA by stabilizing assembly of the late divisome complex. Our results provide new insight into how bacterial DNA damage checkpoints function. Since YneA is present in other organisms including the clinically relevant Listeria monocytogenes these results may be applicable to understanding how certain bacteria are able to regulate cell proliferation under stress conditions.
Experimental Procedures
Bacteriological methods and chemicals
Bacterial plasmids, oligonucleotides and strains used in this study are listed in Supporting Information Tables S2, S3 and S4. Construction of individual strains is detailed in the supporting methods using double cross-over recombination as previously described (Burby and Simmons 2017, Burby, Simmons et al. 2018). All Bacillus subtilis strains are isogenic derivatives of PY79 (Youngman, Perkins et al. 1984). Bacillus subtilis strains were grown in LB (10 g/L NaCl, 10 g/L tryptone and 5 g/L yeast extract) at 30°C with shaking (200 rpm). Individual plasmids were constructed using Gibson assembly as described previously (Gibson 2011). The details of plasmid construction are described in the supporting information. Oligonucleotides used in this study were obtained from Integrated DNA technologies (IDT). Mitomycin C (Fisher bioreagents) was used at the concentrations indicated in the figures. Cephalexin (Millipore Sigma) was used at the concentration indicated in the figures. Spectinomycin (100 μg/mL) was used for selection in B. subtilis as indicated in the method details. Selection of Escherichia coli (MC1061 cells) transformants was performed using ampicillin (100 μg/mL).
Transformation of PY79
PY79 was struck out on LB agar and incubated at 37°C overnight. A single colony was used to inoculate a 2 mL LM culture (LB + 3 mM MgSO4) in a 14 mL round bottom culture tube. The culture was incubated at 37°C on a rolling rack until OD600 of approximately 1. Then, 150 μL of the LM culture was transferred to 3 mL pre-warmed MD media (1X PC buffer (107 g/L K2HPO4, 60 g/L KH2PO4, 11.8 g/L trisodium citrate dihydrate), 2% glucose, 50 μg/mL phenylalanine, 50 μg/mL tryptophan, 11 μg/mL ferric ammonium citrate, 2.5 mg/mL sodium aspartate, 3 mM MgSO4) and incubated on a rolling rack at 37°C for 4 hours. To each 3 mL competent cell culture, 2 μL of the designated plasmid or genomic DNA was added and the cultures were incubated on a rolling rack at 37°C for an additional 30 minutes. Transformations were plated on LB agar + 100 μg/mL spectinomycin (200 μL per 100 cm plate) and incubated at 30°C overnight.
Strain construction
General strain construction methods
All B. subtilis strains are isogenic derivatives of PY79 (Youngman, Perkins et al. 1984) and were generated by transforming cells with a PCR product, plasmid DNA or genomic DNA via natural competence (Harwood and Cutting 1990).
Integration of inducible constructs at the amyE locus was achieved via double cross-over recombination. For constructs containing an IPTG-inducible promoter (Phy), strains were transformed with plasmids or with genomic DNA of a strain already generated (see detailed strain construction) and transformants were selected using LB agar + 100 μg/mL spectinomycin. Isolates were colony purified by re-streaking on LB agar + 100 μg/mL spectinomycin. Incorporation via double cross-over at amyE was determined by PCR colony screen.
Individual strain construction
Plasmid construction
General cloning techniques
Plasmids were assembled using Gibson assembly (Gibson 2011). Gibson assembly reactions were 20 μL consisting of 1X Gibson assembly master mix (0.1 M Tris pH 8.0, 5% PEG-8000, 10 mM MgCl2, 10 mM DTT, 0.2 mM dNTPs, 1 mM NAD+, 4 units/mL T5 exonuclease, 25 units/mL Phusion DNA polymerase, 4,000 units/mL Taq DNA ligase) and 40–100ng of each PCR product and incubated at 50°C for 60 minutes. All PCR products were isolated via gel extraction from an agarose gel. Gibson assembly reactions were used to transform MC1061 E. coli.
Individual plasmid construction
Spot titer assays
B. subtilis strains were struck out on LB agar and incubated at 30°C overnight. The next day, a single colony was used to inoculate a 2 mL LB culture in a 14 mL round bottom culture tube, which was incubated at 37°C on a rolling rack until OD600 was 0.5–1. Cultures were normalized to OD600 = 0.5 and serial diluted. The serial dilutions were spotted (5 μL) on the agar media indicated in the figures and the plates were incubated at 30°C overnight (16–20 hours). All spot titer assays were performed at least three times.
Western blotting
For overexpression of YneA, cultures of LB were inoculated at an OD600 = 0.2 and 1 mM IPTG final concentration was added. Cultures were incubated at 30°C. Samples of an OD600 = 1 were harvested and washed one time with 1X PBS pH 7.4. Samples were re-suspended in 100 μL 1X SMM buffer (0.5 M sucrose, 0.02 M maleic acid, 0.02M MgCl2, adjusted o PH 6.5) containing 100 units/mL mutanolysin and 2X Roche protease inhibitors. Samples were incubated at 37°C for 2 hours. SDS sample buffer was added to 1X and incubated at 100°C for 7 minutes. Samples (12 μL) were separated via 10% SDS-PAGE and transferred to nitrocellulose using a Trans-Blot Turbo (BioRad) according to the manufacturer’s directions. Membranes were blocked in 5% milk in TBST (25 mM Tris, pH 7.5, 150 mM NaCl and 0.1% Tween 20) at room temperature for 1 hour. Blocking buffer was removed and primary antibodies were added in 2% milk in TBST (αYneA, 1:3000; αDnaN, 1:4000). Primary antibody incubation was performed at room temperature for 1 hour. Primary antibodies were removed and membranes were washed three times with TBST for 5 minutes at room temperature. Secondary antibodies (Licor, 1:15000) were added in 2% milk in TBST and incubated at room temperature for 1 hour. Membranes were washed three times as above and imaged using Li-COR Odyssey imaging system. All Western blot experiments were performed at least three times with independent samples. Molecular weight markers were used in the YneA and DnaN blots.
Microscopy
Strains were grown on LB agar plates at 30° overnight. Plates were washed with LB media and cultures of LB were inoculated at an OD600 = 0.1 and incubated at 30° until OD600 of about 0.2. Mitomycin C (MMC) was added at 100 ng/mL final concentration and cultures were induced with 1 mM IPTG final concentration and incubated at 30° until OD600 of 0.6 – 0.8. Samples were taken and incubated with 4 μg/mL FM4–64 for 5 minutes and transferred to pads of 1X Spizizen salts and 1% agarose. Images were captured with an Olympus BX61 microscope (Burby, Simmons et al. 2018, Burby, Simmons et al. 2019).
FtsW suppressor identification.
The parent strain for the yneA overexpression suppressor screen was PEB852, a derivative of PY79 with ddcP, ctpA, and ddcA genes deleted and ectopic expression of yneA under the control of a xylose inducible promoter inserted into the amyE locus. PEB852 was grown on an LB plate with 5 μg/ml chloramphenicol overnight. The next day, the plate were washed with LB and used to inoculate a 10 mL LB culture to an OD600 of 0.05. The culture was grown to OD ~1 and used to inoculate multiple 10 mL LB cultures to OD 0.05. These source flasks were grown to an OD600 of 2 at 30oC. At this point, cells were pelleted and stored for DNA extraction and glycerol stocks were prepared. Later 100 μL of 10−1 diluted cells were used to inoculate three 0.2% xylose LB plates per source flask. Colonies capable of growing on the initial xylose LB plates were restruck onto new 0.2% xylose LB plates and allowed to grow overnight. Those that grew on the secondary xylose plates were then struck on plates with 20 ng/mL MMC. Colonies that grew on MMC were saved as glycerol stocks and eventually sequenced. This screen was preformed twice. The first utilized five source flasks which were named WDH1–5 and yielded three mutants capable of growth in both conditions; these isolates were named WDH6–8. The second iteration of the screen was scaled up to 10 source flasks named WDH9–18 and yielded 27 mutants which were named WDH19–45.
All xylose/MMC growing mutants and the mutant-yielding source flasks were sent for NovaSeq H4K paired end 75 cycle sequencing at the University of Michigan core. The sequencing results were analyzed by breseq (Deatherage and Barrick 2014) on the Flux computing cluster. This analysis revealed that all but one of the mutants had a mutation in ftsW. The one mutant that did not, had no detectable mutations beyond baseline. Other mutations were detected in some isolates but never without an accompanying ftsW mutation and no other gene or intergenic locus was found to be mutated in more than one isolate. Of the 29 detected ftsW mutations, 20 occurred at amino acid 158 and seven occurred at or near amino acid 148 indicating two potentially important residues. All raw sequencing files are publicly available BioProject # PRJNA707525 (www.ncbi.nlm.nih.gov/bioproject)([Dataset], Hawkins et al. 2021).
Bacterial two-hybrid
Plasmids used for bacterial two-hybrid assay are listed in Supporting Information Table S4. Bacterial two-hybrid assays were performed as previously described (Burby, Simmons et al. 2018, Matthews and Simmons 2019) T18 and T25 fusion plasmids (details of the specific plasmids used can be viewed in Supporting Information Table S4) were used to co-transform BTH101 cells and co-transformants were selected on LB agar + 100 μg/mL ampicillin + 25 μg/mL kanamycin at 37°C overnight. Co-transformants were grown in 3 mL of LB media (supplemented with 100 μg/mL ampicillin and 25 μg/mL kanamycin) at 37°C until an OD600 of between 0.5 and 1.0 was reached. The cultures were adjusted to an OD600 of 0.5, diluted 1/1000 in LB and spotted (5 μL per spot) onto LB agar plates containing 40 μg/mL of X-Gal (5-bromo-4-chloro-3-indoxy-β-D-galactopyranoside), 0.5 mM IPTG, 100 μg/mL ampicillin and 25 μg/mL kanamycin. The plates were incubated for two days at 30°C followed by an additional 24 hours at room temperature while being protected from light. All two-hybrid experiments were performed a minimum of three times working from fresh co-transformation.
Data Availability
All data relevant to this study is presented in the main text or supporting information. The whole genome sequence data used to identify the ftsW mutations have been deposited at SRA and are available through BioProject # PRJNA707525. The BioProject covers accession numbers SAMN18208222 through SAMN18208263 and can be found at the following site (www.ncbi.nlm.nih.gov/bioproject)([Dataset], Hawkins et al. 2021). Any strain, sequence or other material or information used in this study is available upon request.
Supplementary Material
S1 Figure. YneA variants are recessive to native yneA with increasing concentrations of DNA damage. Spot titer assay using B. subtilis strains WT (PY79), ΔyneA::loxP ΔddcP amyE::Phy-yneA (EAM48), ΔddcP amyE::Phy-yneA (PEB681), ΔddcP amyE::Phy-yneA-V68A (EAM57) and ΔddcP amyE::Phy-yneA-G82S (EAM58) spotted on the indicated media.
S2 Figure. YneA LysM domain swap fails in checkpoint activation. Spot titer assay using B. subtilis strains WT (PY79), (A) ΔyneA::loxP amyE::Phy-yneA (EAM46) and ΔyneA::loxP amyE::Phy-yneA-safA-LysM (EAM80) (B) amyE::Phy-yneA (PEB677) and amyE::Phy-yneA-safA-LysM (EAM82) spotted on the indicated media.
S3 Figure. Representative images of WT, cells expressing FtsW or FtsW-L148P. The white bar corresponds to 10 μm.
S4 Figure. Representative images of cells expressing FtsW or FtsW-L148P under the condition indicated. The white bar corresponds to 10 μm.
S1 Table. Other mutations identified in the sequenced strains that occurred outside of ftsW.
S2 Table. Cell lengths of the strains indicated in Fig. 5A.
S3 Table. Cell lengths of the strains indicated in Fig. 5B.
S4 Table. Plasmids used in this study.
S5 Table. Oligonucleotides used in this study.
S6 Table. Strains used in this study.
S7 Table. Plasmid list for bacterial two-hybrid assay.
Acknowledgments
We would like to thank members of the Simmons lab for their helpful discussions. PEB was supported by a predoctoral fellowship #DGE1256260 from the National Science Foundation. PEB and JSL were also supported by Rackham Predoctoral Fellowships and BRG was supported in part by an MCDB Summer Undergraduate Fellowship. The National Institutes of Health grant R35 GM131772 to LAS supported this work. The authors have no conflict of interest to declare.
References
- [Dataset], Hawkins WD, Burby PE and Simmons LA (2021). “Whole-genome sequencing coverage for identification of ftsW mutations.” SRA BioProject. [Google Scholar]
- Au N, Kuester-Schoeck E, Mandava V, Bothwell LE, Canny SP, Chachu K, Colavito SA, Fuller SN, Groban ES, Hensley LA, O’Brien TC, Shah A, Tierney JT, Tomm LL, O’Gara TM, Goranov AI, Grossman AD and Lovett CM (2005). “Genetic composition of the Bacillus subtilis SOS system.” J Bacteriol 187(22): 7655–7666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhambhani A, Iadicicco I, Lee J, Ahmed S, Belfatto M, Held D, Marconi A, Parks A, Stewart CR, Margolin W, Levin PA and Haeusser DP (2020). “Bacteriophage SP01 Gene Product 56 Inhibits Bacillus subtilis Cell Division by Interacting with FtsL and Disrupting Pbp2B and FtsW Recruitment.” J Bacteriol 203(2):e00463–20.doi: 10.1128/JB.00463-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojer MS, Frees D and Ingmer H (2020). “SosA in Staphylococci: an addition to the paradigm of membrane-localized, SOS-induced cell division inhibition in bacteria.” Curr Genet 66(3): 495–499. [DOI] [PubMed] [Google Scholar]
- Bojer MS, Wacnik K, Kjelgaard P, Gallay C, Bottomley AL, Cohn MT, Lindahl G, Frees D, Veening JW, Foster SJ and Ingmer H (2019). “SosA inhibits cell division in Staphylococcus aureus in response to DNA damage.” Mol Microbiol 112(4): 1116–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges BA (1995). “Are there DNA damage checkpoints in E. coli?” Bioessays 17(1): 63–70. [DOI] [PubMed] [Google Scholar]
- Buist G, Steen A, Kok J and Kuipers OP (2008). “LysM, a widely distributed protein motif for binding to (peptido)glycans.” Mol Microbiol 68(4): 838–847. [DOI] [PubMed] [Google Scholar]
- Burby PE and Simmons LA (2017). “CRISPR/Cas9 Editing of the.” Bio Protoc 7(8):e2272. doi: 10.21769/BioProtoc.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burby PE and Simmons LA (2020). “Regulation of Cell Division in Bacteria by Monitoring Genome Integrity and DNA Replication Status.” J Bacteriol 202(2):e00408–19.doi: 10.1128/JB.00408-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burby PE, Simmons ZW, Schroeder JW and Simmons LA (2018). “Discovery of a dual protease mechanism that promotes DNA damage checkpoint recovery.” PLoS Genet 14(7): e1007512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burby PE, Simmons ZW and Simmons LA (2019). “DdcA antagonizes a bacterial DNA damage checkpoint.” Mol Microbiol 111(1): 237–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan A, Lofton H, Maloney E, Moore J, Fol M, Madiraju MV and Rajagopalan M (2006). “Interference of Mycobacterium tuberculosis cell division by Rv2719c, a cell wall hydrolase.” Mol Microbiol 62(1): 132–147. [DOI] [PubMed] [Google Scholar]
- Ciccia A and Elledge SJ (2010). “The DNA damage response: making it safe to play with knives.” Mol Cell 40(2): 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole ST (1983). “Characterization of the promoter for the LexA regulated sulA gene of Escherichia coli.” Mol. Gen. Genet. 189: 400–404. [DOI] [PubMed] [Google Scholar]
- Daniel RA and Errington J (2000). “Intrinsic instability of the essential cell division protein FtsL of Bacillus subtilis and a role for DivIB protein in FtsL turnover.” Mol Microbiol 36(2): 278–289. [DOI] [PubMed] [Google Scholar]
- Deatherage DE and Barrick JE (2014). “Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq.” Methods Mol Biol 1151: 165–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar CA and Cross TA (2018). “False positives in using the zymogram assay for identification of peptidoglycan hydrolases.” Anal Biochem 543: 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA and Ellenberger T (2006). DNA Repair and Mutagenesis: Second Edition. Washington, DC, American Society for Microbiology. [Google Scholar]
- Geissler B, Shiomi D and Margolin W (2007). “The ftsA* gain-of-function allele of Escherichia coli and its effects on the stability and dynamics of the Z ring.” Microbiology (Reading) 153(Pt 3): 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG (2011). “Enzymatic assembly of overlapping DNA fragments.” Methods Enzymol 498: 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbedel S and Lewis RJ (2019). “Structural basis for interaction of DivIVA/GpsB proteins with their ligands.” Mol Microbiol 111(6): 1404–1415. [DOI] [PubMed] [Google Scholar]
- Harwood CR and Cutting SM (1990). Molecular biological methods for Bacillus, Wiley. [Google Scholar]
- Iyer VN and Szybalski W (1963). “A molecular mechanism of mitomycin action: linking of complementary DNA strands.” Proc Natl Acad Sci U S A 50: 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimova G, Gauliard E, Davi M, Ouellette SP and Ladant D (2017). “Protein-Protein Interaction: Bacterial Two-Hybrid.” Methods Mol Biol 1615: 159–176. [DOI] [PubMed] [Google Scholar]
- Karimova G, Pidoux J, Ullmann A and Ladant D (1998). “A bacterial two-hybrid system based on a reconstituted signal transduction pathway.” Proc Natl Acad Sci U S A 95(10): 5752–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai Y, Moriya S and Ogasawara N (2003). “Identification of a protein, YneA, responsible for cell division suppression during the SOS response in Bacillus subtilis.” Mol Microbiol 47(4): 1113–1122. [DOI] [PubMed] [Google Scholar]
- Kawai Y and Ogasawara N (2006). “Bacillus subtilis EzrA and FtsL synergistically regulate FtsZ ring dynamics during cell division.” Microbiology (Reading) 152(Pt 4): 1129–1141. [DOI] [PubMed] [Google Scholar]
- Król E, van Kessel SP, van Bezouwen LS, Kumar N, Boekema EJ and Scheffers DJ (2012). “Bacillus subtilis SepF binds to the C-terminus of FtsZ.” PLoS One 7(8): e43293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenhart JS, Schroeder JW, Walsh BW and Simmons LA (2012). “DNA repair and genome maintenance in Bacillus subtilis.” Microbiol Mol Biol Rev 76(3): 530–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little JW (1983). “The SOS regulatory system: control of its state by the level of RecA protease.” J Mol Biol 167(4): 791–808. [DOI] [PubMed] [Google Scholar]
- Matthews LA and Simmons LA (2019). “Cryptic protein interactions regulate DNA replication initiation.” Mol Microbiol 111(1): 118–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizusawa S, Court D and Gottesman S (1983). “Transcription of the sulA gene and repression by LexA.” J. Mol. Biol. 171: 337–343. [DOI] [PubMed] [Google Scholar]
- Mizusawa S and Gottesman S (1983). “Protein degradation in Escherichia coli: the lon gene controls the stability of sulA protein.” Proc Natl Acad Sci U S A 80(2): 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo AH and Burkholder WF (2010). “YneA, an SOS-induced inhibitor of cell division in Bacillus subtilis, is regulated posttranslationally and requires the transmembrane region for activity.” J Bacteriol 192(12): 3159–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell JW, Hopkins AC and Laub MT (2011). “A DNA damage checkpoint in Caulobacter crescentus inhibits cell division through a direct interaction with FtsW.” Genes Dev 25(12): 1328–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell JW, Kambara TK, Perchuk BS and Laub MT (2014). “A DNA damage-induced, SOS-independent checkpoint regulates cell division in Caulobacter crescentus.” PLoS Biol 12(10): e1001977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales Angeles D, Macia-Valero A, Bohorquez LC and Scheffers DJ (2020). “The PASTA domains of.” Microbiology (Reading) 166(9): 826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A, Cao C and Lutkenhaus J (1998). “Inhibition of FtsZ polymerization by SulA, an inhibitor of septation in Escherichia coli.” Proc Natl Acad Sci U S A 95(6): 2885–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll DM, Mason TM and Miller PS (2006). “Formation and repair of interstrand cross-links in DNA.” Chem Rev 106(2): 277–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastoret S, Fraipont C, den Blaauwen T, Wolf B, Aarsman ME, Piette A, Thomas A, Brasseur R and Nguyen-Distèche M (2004). “Functional analysis of the cell division protein FtsW of Escherichia coli.” J Bacteriol 186(24): 8370–8379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira FC, Nunes F, Cruz F, Fernandes C, Isidro AL, Lousa D, Soares CM, Moran CP Jr., Henriques AO and Serrano M (2019). “A LysM Domain Intervenes in Sequential Protein-Protein and Protein-Peptidoglycan Interactions Important for Spore Coat Assembly in Bacillus subtilis.” J Bacteriol 201(4) e00642–18. doi: 10.1128/JB.00642-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popham DL and Setlow P (1995). “Cloning, nucleotide sequence, and mutagenesis of the Bacillus subtilis ponA operon, which codes for penicillin-binding protein (PBP) 1 and a PBP-related factor.” J Bacteriol 177(2): 326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffers DJ and Errington J (2004). “PBP1 is a component of the Bacillus subtilis cell division machinery.” J Bacteriol 186(15): 5153–5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons LA, Foti JJ, Cohen SE and Walker GC (2008). “The SOS Regulatory Network.” EcoSal Plus 3(1). [DOI] [PubMed] [Google Scholar]
- Simmons LA, Grossman AD and Walker GC (2007). “Replication is required for the RecA localization response to DNA damage in Bacillus subtilis.” Proc Natl Acad Sci U S A 104(4): 1360–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MD, Smith BT, Godoy VG and Walker GC (2000). “The SOS response: recent insights into umuDC-dependent mutagenesis and DNA damage tolerance.” Annu Rev Genet 34: 479–497. [DOI] [PubMed] [Google Scholar]
- Taguchi A, Welsh MA, Marmont LS, Lee W, Sjodt M, Kruse AC, Kahne D, Bernhardt TG and Walker S (2019). “FtsW is a peptidoglycan polymerase that is functional only in complex with its cognate penicillin-binding protein.” Nat Microbiol 4(4): 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang MJ and Bernhardt TG (2015). “A role for the FtsQLB complex in cytokinetic ring activation revealed by an ftsL allele that accelerates division.” Mol Microbiol 95(6): 925–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadrevu IS, Lofton H, Sarva K, Blasczyk E, Plocinska R, Chinnaswamy J, Madiraju M and Rajagopalan M (2011). “ChiZ levels modulate cell division process in mycobacteria.” Tuberculosis (Edinb) 91 Suppl 1: S128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker GC, Smith BT and Sutton MD (2000). The SOS response to DNA damage. Bacterial Stress Responses. Storz G and Hengge-Aronis R. Washington DC, The American Society for Microbiology: 131–144. [Google Scholar]
- Yanouri A, Daniel RA, Errington J and Buchanan CE (1993). “Cloning and sequencing of the cell division gene pbpB, which encodes penicillin-binding protein 2B in Bacillus subtilis.” J Bacteriol 175(23): 7604–7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman P, Perkins JB and Losick R (1984). “Construction of a cloning site near one end of Tn917 into which foreign DNA may be inserted without affecting transposition in Bacillus subtilis or expression of the transposon-borne erm gene.” Plasmid 12(1): 1–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1 Figure. YneA variants are recessive to native yneA with increasing concentrations of DNA damage. Spot titer assay using B. subtilis strains WT (PY79), ΔyneA::loxP ΔddcP amyE::Phy-yneA (EAM48), ΔddcP amyE::Phy-yneA (PEB681), ΔddcP amyE::Phy-yneA-V68A (EAM57) and ΔddcP amyE::Phy-yneA-G82S (EAM58) spotted on the indicated media.
S2 Figure. YneA LysM domain swap fails in checkpoint activation. Spot titer assay using B. subtilis strains WT (PY79), (A) ΔyneA::loxP amyE::Phy-yneA (EAM46) and ΔyneA::loxP amyE::Phy-yneA-safA-LysM (EAM80) (B) amyE::Phy-yneA (PEB677) and amyE::Phy-yneA-safA-LysM (EAM82) spotted on the indicated media.
S3 Figure. Representative images of WT, cells expressing FtsW or FtsW-L148P. The white bar corresponds to 10 μm.
S4 Figure. Representative images of cells expressing FtsW or FtsW-L148P under the condition indicated. The white bar corresponds to 10 μm.
S1 Table. Other mutations identified in the sequenced strains that occurred outside of ftsW.
S2 Table. Cell lengths of the strains indicated in Fig. 5A.
S3 Table. Cell lengths of the strains indicated in Fig. 5B.
S4 Table. Plasmids used in this study.
S5 Table. Oligonucleotides used in this study.
S6 Table. Strains used in this study.
S7 Table. Plasmid list for bacterial two-hybrid assay.
Data Availability Statement
All data relevant to this study is presented in the main text or supporting information. The whole genome sequence data used to identify the ftsW mutations have been deposited at SRA and are available through BioProject # PRJNA707525. The BioProject covers accession numbers SAMN18208222 through SAMN18208263 and can be found at the following site (www.ncbi.nlm.nih.gov/bioproject)([Dataset], Hawkins et al. 2021). Any strain, sequence or other material or information used in this study is available upon request.