

Highlights
-
•
LC-MS methods are used to identify intact Klebsiella pneumoniae carbapenemase (KPC).
-
•
Mass spectrometry validates the mature form of KPC based on molecular weight.
-
•
Combining top-down and bottom-up proteomics confirms mature protein sequence of KPC.
-
•
This method can be applied to multiple variants, including KPC-2 through KPC-5.
-
•
The method can be easily adapted for KPC determination from any bacterial pathogen.
Abbreviations: CPO, carbapenemase-producing organisms; KPC, Klebsiella pneumoniae carbapenemase; CDC, Centers for Disease Control and Prevention; ATCC, American type culture collection; MS, mass spectrometry; MS/MS, tandem mass spectrometry; LC, liquid chromatography; MALDI, matrix-assisted laser desorption ionization; ESI, electrospray ionization; TOF, time-of-flight; MW, molecular weight; CSD, charge state distribution; PCR, polymerase chain reaction; BLAST, basic local alignment search tool; m/z, mass-to-charge ratio
Keywords: Carbapenem-resistant Enterobacteriaceae, Carbapenemase-producing organisms, Klebsiella pneumoniae carbapenemase, Mass Spectrometry, Tandem mass spectrometry
Abstract
Introduction
Carbapenemase-producing organisms (CPOs) are a growing threat to human health. Among the enzymes conferring antibiotic resistance produced by these organisms, Klebsiella pneumoniae carbapenemase (KPC) is considered to be a growing global health threat. Reliable and specific detection of this antibiotic resistance-causing enzyme is critical both for effective therapy and to mitigate further spread.
Objectives
The objective of this study is to develop an intact protein mass spectrometry-based method for detection and differentiation of clinically-relevant KPC variants directly from bacterial cell lysates. The method should be specific for any variant expressed in multiple bacterial species, limit false positive results and be rapid in nature to directly influence clinical outcomes.
Methods
Lysates obtained directly from bacterial colonies were used for intact protein detection using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). Bottom-up and top-down proteomic methods were used to characterize the KPC protein targets of interest. Comparisons between KPC-producing and KPC-non-producing isolates from a wide variety of species were also performed.
Results
Characterization of the mature KPC protein revealed an unexpected signal peptide cleavage site preceding an AXA signal peptide motif, modifying the molecular weight (MW) of the mature protein. Taking the additional AXA residues into account allowed for direct detection of the intact protein using top-down proteomic methods. Further validation was performed by transforming a KPC-harboring plasmid into a negative control strain, followed by MS detection of the KPC variant from the transformed cell line. Application of this approach to clearly identify clinically-relevant variants among several species is presented for KPC-2, KPC-3, KPC-4 and KPC-5.
Conclusion
Direct detection of these enzymes contributes to the understanding of occurrence and spread of these antibiotic-resistant organisms. The ability to detect intact KPC variants via a simple LC-MS/MS approach could have a direct and positive impact on clinical therapy, by providing both direction for epidemiological tracking and appropriate therapy.
1. Introduction
Bacterial resistance to antimicrobials has grown to be one of the most significant threats to global health in the modern era. This has become such an important issue that multiple organizations have considered it to be a crisis [1], [2], [3], [4], [5]. Since 2013, the Centers for Disease Control and Prevention (CDC) have categorized carbapenem-resistant Enterobacteriaceae (CRE) as an urgent threat [4], and in 2019 added carbapenem-resistant Acinetobacter baumannii [5]. This is due to a variety of methods by which carbapenemase-producing organisms (CPOs) are able to spread carbapenemase genes, such as through clonal propagation, horizontal gene transfer, or mutations [6], [7], [8]. Among these, Klebsiella pneumoniae carbapenemase (KPC) is considered endemic in the United States [9] and is wide-spread globally [10].
The detection of KPC-producing bacteria is often challenging due to variable low level expression, complications arising from impermeability, AmpC activity, and the presence of extended spectrum β-lactamases [11]. The Hodge test and acidimetric-based approaches employing carbapenem substrates have been used in confirming carbapenemase activity. However, in addition to requiring overnight incubation, these approaches suffer from high false-positive rates, even when modified procedures are utilized. More rapid, cost-effective, and easy-to-interpret results are critical in determining patient treatment therapy. In hospital settings, infections can occur approximately two days after admission with isolates identified in a variety of specimen types [11], [12]. Complications can also arise with treatment regimens depending upon the type of KPC variant associated with the infection. In the case of KPC-2, it has effective enzyme activity against carbapenems, penicillins, and cephalosporins with limited effectiveness against cephamycins and ceftazidime. However, of the known 30-plus KPC variants that have been characterized to date, some such as KPC-4, which is a double mutation of KPC-2, have an 80-fold increase in activity against ceftazidime-based antibiotics while maintaining the ability to hydrolyze the β-lactam ring of carbapenems and its associated structural analogs. For the single point mutations of KPC-3 and KPC-5, both show an increase in resistance to ceftazidime by four- and five-fold, respectively [13]. Therefore, the ability to determine not only if KPC is present, but to also identify the actual variant type, has a significant impact on patient treatment options.
Mass spectrometry (MS) has been applied to the detection of antibiotic resistance using matrix-assisted laser desorption ionization coupled to a time-of-flight (MALDI-TOF) MS. It was shown that the hydrolysis of β-lactam antibiotics could be used to infer the presence of enzymatic activity [14]. Carbapenemase activity was similarly identified by introducing a carbapenem to bacterial solutions, followed by monitoring the presence and abundance of both the intact antibiotic as well as hydrolysis products in Enterobacteriaceae [15] and Pseudomonas aeruginosa [16], [17], [18], with subsequent studies showing the utility of this approach with Acinetobacter baumannii [18], [19]. Commentary on the benefits and disadvantages of these approaches are discussed by Mirande [20], with reviews on the topic offered by Pulido [21] and, more recently, Maugeri [22].
Camara and Hays were among the earliest researchers using MALDI-TOF MS to directly detect proteins conferring antibiotic resistance in Gram negative bacteria [23]. Since that time, several approaches have been developed towards direct detection of resistance proteins produced in Gram negative bacteria using mass spectrometry (MS). In one such approach, bottom-up proteomics using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) was employed to determine the presence or absence of KPC based on observed peptides [24]. The use of MALDI-TOF MS was proposed to predict whether or not isolates produced KPC by observation of a protein peak at approximately mass-to-charge ratio (m/z) 11,109 [25], [26]; however, this was found to be a poor predictor for the presence of this carbapenemase [27]. Recently, a report on the direct detection of KPC-2 using a MALDI-TOF-based analysis has shown a high degree of reproducibility [27], although the authors noted the measured mass of the protein, around 28,544 Da, was found to differ from a predicted mass of 28,477 Da. The predicted mass described in that report corresponds to the average mass of the expected mature KPC-2 protein, which can be found in Uniprot (uniprot.org) entry Q9F663.
Mass spectrometric techniques applied to intact protein analysis extend beyond MALDI-TOF methods. Electrospray ionization (ESI) allows proteins in solution to be gently transferred into the gas phase and easily coupled to liquid chromatography (LC) for online separation and subsequent ionization and introduction into a mass spectrometer (LC-MS). Mass analysis can then be performed for either the intact mass (MS) or fragment ions via tandem mass spectrometry (MS/MS). This approach is often referred to as top-down proteomics [28], with a useful introduction by Kelleher [29] and a recent review on the contributions and future trajectory of this approach given by Toby [30]. The use of MS/MS allows the intact protein to be dissociated into multiple fragments, all of which contain information on mass and charge. These fragments can then be used to provide characteristic information on the intact protein, similar to combining pieces of a puzzle to form an overall picture.
Here, we demonstrate the use of combining bottom-up and top-down proteomics techniques, both providing complimentary information to each other for the development of a top-down method used for direct detection of clinically-relevant KPC variants. The combination of LC and high resolution MS methods, which offers the ability to rapidly alternate between detection of intact protein ions and fragments produced from them, provides confidence for specific identification of KPC variants in a rapid and easy to interpret fashion. Performing MS/MS on the target affords confidence in the identification of the protein to the variant level, offering both the opportunity for accurate clinical analysis as well as epidemiological surveillance of KPC variant propagation. In this report, we show the sequence of the mature KPC proteins include three additional residues at the N-terminus from the predicted sequences. Using top-down proteomics, multiple KPC variants can be detected from several species on a short LC timeframe directly from cell lysates. The method demonstrated herein is effective with samples harvested either from broth or agar, and analysis time can be optimized based on separation conditions.
2. Methods
Isolates used in this study were collected from American Type Culture Collection (ATCC) and from the Centers for Disease Control and Prevention and Food and Drug Administration Antibiotic Resistance Isolate Bank (CDC and FDA AR Isolate Bank). Gram Negative Carbapenemase Detection and Enterobacteriaceae Carbapenemase Diversity Panels were used from the CDC and FDA AR Isolate Bank.
2.1. Sample preparation for Bottom-Up (digest) and Top-Down (intact protein) analysis
Cells were grown either on tryptone soya agar or in tryptone soya broth (Oxoid, Basingstoke, UK) for a period of 16–20 h at 37 °C under aerobic conditions. For analyses from broth, cells in solution were centrifuged for 30 min at 4,000 g while held at 10 °C. Supernatants were discarded and the pellets were reconstituted with 3 mL of ice cold 0.9% NaCl. The solutions were then centrifuged again at 4,000 g for 30 min at 10 °C. Supernatants were discarded, and depending on the analysis methods, the pellets were reconstituted in either 8 M urea, 50 mM Tris, pH 8 for bottom-up, or 6 M guanidinium chloride, 50 mM Tris, pH 8 for top-down experiments. For top-down analyses, cells grown on agar were harvested with a 10 µL loop tool (approximately 10 colonies) and transferred to centrifuge tubes containing 1 mL of 6 M guanidinium chloride, 50 mM Tris, pH 7. For preparation steps, 1 mL aliquots were transferred to 1 mL lysing matrix B bead-beating tubes (MP biomedicals) and were mechanically lysed using a Fisherbrand Bead Mill 4 Homogenizer (Fisher Scientific) for 3 iterations of 5 m/s for 60 s. The tubes were then centrifuged for 5 min at 12,000 g. Supernatants for bottom-up were extracted and quantified using a Coomassie Plus (Bradford) assay. For bottom-up analyses, lysate solutions were portioned out to 100 µg/sample, reduced, alkylated, and digested with trypsin (Pierce trypsin protease, TPCK-treated, MS grade) in a 25:1 ratio of protein to enzyme. Prior to LC-MS/MS analyses, bottom-up solutions were cleaned with Pierce C18 spin columns (Thermo Fisher Scientific), following recommended protocol. In top-down experiments, supernatants were transferred directly to vials for LC-MS/MS analysis. For short elution times of KPC (e.g. < 10 min), the solutions were buffer-exchanged using Amicon 3K MWCO spin columns (Millipore Sigma), buffer-exchanged into 2% acetonitrile, 0.2% formic acid in water.
2.2. Plasmid transformation
An Escherichia coli strain containing a plasmid harboring blaKPC-4 (E. coli, CDC and FDA AR Bank #104, Gram Negative Carbapenemase Detection Panel) was cultured on sheep’s blood agar with an imipenem Kirby-Bauer disc to ensure plasmid retention. A single colony was enriched in MacConkey broth containing imipenem. The plasmid was purified using a DNA miniprep kit (Qiagen, Frederick, MD) and transformed using electroporation. Purified plasmid containing blaKPC-4 was electroporated into competent DH5α cells using a standard protocol. Cells were then plated to LB agar containing ampicillin for plasmid uptake selection. Transformed cells were confirmed for carbapenem production by performing susceptibility testing using E-tests (bioMérieux, Marcy-l’Étoile, France), carbapenem disc diffusion, and a BD Phoenix (Franklin Lakes, NJ) following recommended operating procedures. In addition, genotypic confirmation was performed using an in-house PCR (ASR primers, Cepheid Sunnyvale CA).Test results were compared to E. coli DH5α containing no plasmid and the original CDC and FDA AR bank #0104 isolate.
2.3. Liquid chromatography
Solutions of tryptic peptides (bottom-up) were separated using an EASY-nLC with a PepMap RSLC C18 75 µm × 25 cm LC column connected to an EASY-Spray ion source (Thermo Fisher Scientific). A binary gradient of Solvent A (0.2% formic acid in water) and solvent B (10% water, 10% isopropanol, 0.2% formic acid in acetonitrile) was used, increasing from 7% to 35% Solvent B over 3 h at a flow rate of 300 nL/min
LC separations of intact proteins (top-down) were performed from both an Accela and a Dionex Ultimate 3000, both using binary gradients of Solvent A (0.2% formic acid in water) and Solvent B (0.2% formic acid in acetonitrile) with flow rates of 200 µL/min using a ProSwitft (Thermo Fisher Scientific) 1 mm × 250 mm RP4H column. While the gradients can be adjusted with an increase or decrease in the slope, resulting in earlier or later elution times, the elution gradient for intact proteins can range from 20% to 45% Solvent B over 10 to 65 min. Additional examples of gradient modifications with corresponding elution times are given in the supplemental information Fig. S1. For solutions containing GHCL, the first 10 min of solvent flow through the column were diverted away from the ionization source; however, solutions that have been buffer-exchanged did not require diverting to waste.
2.4. Mass spectrometry
Bottom-up MS data were collected using a Q-Exactive (Thermo Fisher Scientific) with MS/MS data-dependent acquisition mode over the course of the peptide elution, with a m/z range of 400–2000, a resolution of 35,000 5 µscans in MS mode, and a resolution of 17,500, 5 µscans, an isolation width of 4.0 m/z, and normalized collision energy of 25 eV in MS/MS mode. Spectra were analyzed using Proteome Discoverer 2.2 (Thermo Fisher Scientific) for peptide searches against a bacterial database of K. pneumoniae protein sequences uploaded from UniProt (www.uniprot.org, accessed on 08/29/2016), as well as against a single KPC-2 protein sequence (Q9F663). The database is comprised of nearly 175,000 protein sequences having more than 75 million amino acid residues from proteins submitted and/or categorized as belonging to K. pneumoniae without redundancies removed. Sequest HT was used with search parameters set to semi-tryptic proteolysis, up to 2 missed cleavages, minimum length of 6 residues, and within 10 ppm mass tolerance. Default processing and consensus workflows for Q-Exactive mass spectrometers were used for analysis.
For top-down analyses, MS spectra were collected using a Q-Exactive HF (Thermo Fisher Scientific) with 120,000 resolution and a m/z range of 1100–2000. ESI was performed using a heated ESI (HESI) source (Thermo Fisher Scientific) set to 3.8 kV, heated metal capillary set to 325 °C. MS/MS data were collected by isolating the z = +19 charge state of intact KPC with an isolation width of m/z 2.5 centered around m/z 1513.15 for KPC-2, −3 and −4; for KPC-5 the isolation window was centered around m/z 1515.60. Fragmentation spectra were collected to include m/z 400–2000. In-source activation, set to 40 eV, was used to increase the detection sensitivity. Mass spectra were interpreted manually, based on fragment masses calculated from MS-Product utility from http://prospector.ucsf.edu/prospector/mshome.htm and with Prosight Lite [31]. Spectra were deconvoluted from m/z to molecular weight (MW) using Xtract software and based on the “Averagine” model [32], set for low sulfur content. Briefly, spectral deconvolution translates mass spectra from the mass-to-charge (m/z) domain to the neutral mass domain (in Da) by taking into account m/z, charge state (z), and isotopic distribution models. These models were based on an “Averagine” model, where an “average” amino acid was used to estimate the elemental composition of the unknown mass. The best fit isotopic model to the data allows the direct calculation of the monoisotopic mass.
2.5. Data availability
Data produced from LC-MS and LC-MS/MS evaluations are available online in a data repository stored on figshare.com. Data include spectra for all figures produced herein and for supplementary information. Software provided and maintained by Thermo Fisher Scientific was used to generate all data and files, each stored as a “.raw” file. These are readable using Xcalibur or Freestyle software programs (Thermo Fisher Scientific). Protein sequences of KPC variants can be found at Uniprot.org with the following identifiers: Q9F663 for KPC-2, Q93DC4 for KPC-3, B1PL86 for KPC-4, and B0ZSP4 for KPC-5. It should be noted there may be multiple identifiers with identical sequence information for a given variant. Sequence modifications described in this report are based on the mature protein sequence.
3. Results
3.1. Discovery of Klebsiella pneumoniae carbapenemase (KPC)
Bottom up proteomics techniques employing trypsin as a digesting agent were first used to detect the presence of KPC, using KPC-producing strains of K. pneumoniae ATCC® BAA-1903, and ATCC® BAA-1905. The KPC-non-producing strains of E. coli ATCC® 11775, and K. pneumoniae ATCC® 13883 were employed as negative controls. Interestingly, a thorough evaluation of data produced from this technique suggested the presence of additional and unique cleavage sites between the signal peptide and mature protein; however, there appears to be only a single pathway expressed in any significant abundance. Although data for the additional cleavage sites are presented as supplemental information (Figs. S2–S5 and Table S1), the remainder of this report will discuss the main pathway observed. Table 1 shows the peptides formed via tryptic proteolytic digestion that were observed from this process. The presence of the N-terminal peptide suggested that the sequence of the mature protein was actually three residues longer than predicted, based on UniProt (uniprot.org) entry Q9F663.
Table 1.
Tryptic peptides observed corresponding to KPC-2 from cell lysate of Klebsiella pneumoniae ATCC® BAA-1905. Peptide identifications were based on 10 ppm m/z accuracy for all ions.
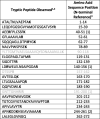 |
1Bold letters indicate C-terminal cleavage for tryptic peptide.
2Gray text indicates tryptic peptide not found.
3Values in parenthesis () indicate number of missed cleavages for trypsin.
Fig. 1 provides the fragmentation spectrum of this peptide with the additional ATA residues on the N-terminus, with Table 2 describing the fragment ions produced. The MS/MS process facilitates cleavage of peptides and proteins at the amide bond, producing fragments that correspond directly to the amino acid sequence of any peptide or protein, with b-type ions starting from the N-terminus and y-type ions starting from the C-terminus. The presence of the b2 fragment ion, combined with the known sequence of the corresponding gene, indicates that the N-terminus begins with the sequence AT, while examining the complementary y12 ion includes an alanine in position three. The MW of the tryptic peptide, in combination with the fragmentation information, accounts for the presence of the ATA amino acid residues. Using LC-MS/MS, and incorporating the ATA residues into the MW of the mature protein, a targeted search could then be performed to identify intact KPC-2.
Fig. 1.

Fragmentation spectrum of the N-terminal tryptic peptide of KPC-2. Sequence ladder produced using Prosight lite [31].
Table 2.
Sequence-specific amino acid fragments for the N-terminal tryptic peptide of KPC-2 via MS/MS.
| Δb ppm | b (theo) | b (obs) | Nterm | Sequence | Cterm | y (obs) | y (theo) | Δy ppm |
|---|---|---|---|---|---|---|---|---|
| --- | 72.0403 | --- | 1 | A | 14 | --- | --- | --- |
| 0.00 | 173.0921 | 173.0921 | 2 | T | 13 | --- | 1374.7627 | --- |
| 0.4 | 244.1292 | 244.1293 | 3 | A | 12 | 1273.7150 | 1273.7151 | −0.1 |
| 1.1 | 357.2132 | 357.2136 | 4 | L | 11 | 1202.6800 | 1202.6780 | 1.7 |
| 3.5 | 458.2609 | 458.2625 | 5 | T | 10 | 1089.5940 | 1089.5939 | 0.1 |
| −2.6 | 572.3039 | 572.3024 | 6 | N | 9 | 988.5477 | 988.5462 | 1.5 |
| 3.2 | 685.3879 | 685.3901 | 7 | L | 8 | 874.5042 | 874.5033 | 1.0 |
| 4.1 | 784.4563 | 784.4595 | 8 | V | 7 | 761.4197 | 761.4192 | 0.7 |
| – | 855.4934 | – | 9 | A | 6 | 662.3514 | 662.3508 | 0.9 |
| – | 984.5360 | – | 10 | E | 5 | 591.3149 | 591.3137 | 2.0 |
| – | 1081.5888 | – | 11 | P | 4 | 462.2713 | 462.2711 | 0.4 |
| – | 1228.6572 | – | 12 | F | 3 | 365.2180 | 365.2183 | −0.8 |
| – | 1299.6943 | – | 13 | A | 2 | 218.1503 | 218.1499 | 1.8 |
| – | – | – | 14 | K | 1 | 147.1127 | 147.1128 | −0.7 |
Bacterial cell lysates contain a high concentration of multiple protein species, such that detection of a specific protein often requires some form of separation. Fig. 2a shows the chromatogram from K. pneumoniae ATCC® BAA-1905 produced from the total ion current for intact proteins, offering a view as to the elution behavior and sample complexity. Fig. 2b shows the extracted ion chromatogram of the peak that corresponds to intact KPC-2 with the ATA residues at the N-terminus. The mass spectrum of intact KPC-2, as shown in Fig. 2c, illustrates the charge state distribution (CSD), spanning from z = +15 to +26 within the m/z range of 1100–2000. Fig. 2d, an expanded view of m/z 1512 – 1513 from Fig. 2c, illustrates the isotopic distribution that can be observed within a single charge state, with the spacing corresponding to the mass of a neutron divided by the charge. The theoretical m/z for the most abundant isotope of KPC-2, 1512.4673, is highlighted with a dashed vertical line. It should be noted that the m/z values given are of a central and single isotope out of a series from each charge state. The MW of KPC-2 can be determined from the isotopes, m/z values, and CSDs of the MS data. Fig. 2e represents the deconvoluted neutral mass of KPC-2, as derived from data in Fig. 2c, and indicates the intact MW of the protein.
Fig. 2.

Chromatographic separation of intact proteins from cell lysates show (a) the total ion current from all ionized species, and (b) the extracted ion chromatogram corresponding to KPC. Mass spectra of intact KPC-2 showing (c) the CSD of the protein; (d) expanded view of the isotopic distribution within a single charge state, average isotopic spacing of 0.0528 m/z. The theoretical m/z for the + 19 charge state, 1512.4673, is depicted with a dashed vertical line; (e) deconvoluted mass of KPC-2 from (c).
3.2. Fragmentation of intact KPC-2
Fig. 3a represents the fragmentation spectrum of KPC-2 following isolation of the z = +19 charge state (1512.5 m/z) and is the result of the MS/MS process. The sequence-specific information obtained through the MS/MS process includes fragments which contain either an intact N- or C-terminus. These fragments associated with the N- and C-termini are designated as b- or y-type ions, respectively, with their position in the amino acid sequence designated by a subscript and the charge state indicated by a superscript. Fig. 3b represents the expanded view of the higher m/z scale between the m/z range of 1450–1750. The MS/MS fragment information produced from Fig. 3 is further detailed in Table 3.
Fig. 3.

Fragmentation of KPC-2 showing (a) the range from m/z 400 to 1800, sequence coverage of KPC-2 showing cleavage sites, with a 4x magnification from m/z 400 to 1000; (b) expanded view of m/z 1450–1750 range of the fragmentation spectrum.
Table 3.
Sequence-specific amino acid fragments from intact KPC-2 obtained via MS/MS.
| Δb ppm | b (theo)1 | b (obs)1 | Nterm | Sequence | Cterm | y (obs)1 | y (theo)1 | Δy ppm |
|---|---|---|---|---|---|---|---|---|
| 1.8 | 685.38791+ | 685.38911+ | 7 | A | 265 | – | – | – |
| 2.0 | 784.45631+ | 784.45791+ | 8 | T | 264 | – | – | – |
| 1.4 | 855.49341+ | 855.49461+ | 9 | A | 263 | – | – | – |
| 1.2 | 984.53601+ | 984.53721+ | 10 | L | 262 | – | – | – |
| 1.2 | 835.46162+ | 835.46262+ | 16 | N | 256 | – | – | – |
| −1.5 | 1537.016418+ | 1537.014118+ | 261 | R | 11 | – | – | – |
| −2.9 | 1543.298818+ | 1543.294318+ | 262 | L | 10 | 957.50451+ | 957.50001+ | 4.7 |
| −0.8 | 1634.021917+ | 1634.020617+ | ||||||
| −1.1 | 1547.245318+ | 1547.243718+ | 263 | A | 9 | 886.46411+ | 886.46291+ | 1.4 |
| −2.3 | 1638.200517+ | 1638.196717+ | ||||||
| −2.0 | 1740.525116+ | 1740.521716+ | ||||||
| −2.2 | 1553.527818+ | 1553.524418+ | 264 | L | 8 | 773.38031+ | 773.37881+ | 1.9 |
| −2.9 | 1644.852517+ | 1644.847717+ | ||||||
| −1.9 | 1560.696818+ | 1560.693918+ | 265 | E | 7 | 644.33751+ | 644.33621+ | 2.0 |
| −3.1 | 1652.443317+ | 1652.438217+ | ||||||
| −2.0 | 1563.864718+ | 1563.861518+ | 266 | G | 6 | 587.31631+ | 587.31481+ | 2.6 |
| −2.3 | 1655.797517+ | 1655.793617+ | ||||||
| −2.2 | 1570.147118+ | 1570.143718+ | 267 | L | 5 | 474.23171+ | 474.23071+ | 2.1 |
| −2.9 | 1490.562019+ | 1490.557719+ | 268 | G | 4 | 417.20961+ | 417.20921+ | 0.9 |
| −2.4 | 1573.315018+ | 1573.311218+ | ||||||
| −2.7 | 1665.803717+ | 1665.799217+ | ||||||
| −2.0 | 1495.776119+ | 1495.773119+ | 269 | V | 3 | – | – | – |
| −2.6 | 1578.818818+ | 1578.814718+ | ||||||
| −3.1 | 1585.154518+ | 1585.149618+ | 270 | N | 2 | – | – | – |
| −2.9 | 1588.322418+ | 1588.317818+ | 271 | Q | 1 | – | – | – |
Fragment ion charge states for both theoretical and experimental m/z values are indicated by the numbers in the upper right corner for the b and y m/z values. Observed isotopic values fall within a range of 3 central isotopes of the theoretical distribution.
3.3. Fragmentation of intact KPC-4
Following plasmid extraction from the KPC-producing isolate and transformation into the carbapenem-susceptible strain, LC-MS/MS data were collected for all three strains, as presented in Fig. 4. Phenotypic assays involving imipenem and ertapenem disks demonstrated that the electroporated cells, along with the strain from the AR isolate bank were resistant to both, while the unmodified DH5α cells were susceptible to both (data not shown). Comparisons of both extracted ion chromatograms and tandem mass spectra between the DH5α negative control, the DH5α KPC-producing strain, and the KPC-producing strain from the CDC and FDA AR Isolate bank reveal no detection of KPC from the EIC (Fig. 4a) or the MS/MS experiment (Fig. 4b). KPC is identified in both the EIC of the DH5α KPC-producing strain (Fig. 4c) as well as from the MS/MS data (Fig. 4d). Similarly, the strain from the CDC and FDA AR Isolate Bank shows the presence of KPC in the EIC (Fig. 4e) and the MS/MS data as well (Fig. 4f).
Fig. 4.

Comparison of strains with and without KPC-4. Extracted ion chromatogram (EIC) centered on the y7+ fragment of KPC-4 from (a) DH5α carbapenem-susceptible E. coli lysate, (c) lysate from DH5α E. coli cells following transformation of a KPC + plasmid, and (e) cell lysate from KPC-4-producing K. pneumoniae, #104 from the CDC and FDA AR isolate Bank, from which the KPC + plasmid was isolated. The corresponding MS/MS spectra presenting (b) no evidence for KPC from the DH5α lysate, and fragment ions of KPC-4 from lysates of (d) DH5α KPC + and (f) the original KPC-producing isolate. MS spectra of the intact KPC-4 proteins from both (d) and (f) were also identified and are provided as supplemental information (Fig. S6).
3.4. Comparisons of KPC variants
Although there are many variants of KPC, we focus here on KPC-2 (Fig. 5a), KPC-3 (Fig. 5b), KPC-4 (Fig. 5c) and KPC-5 (Fig. 5d) as a starting point for evaluation of this method. MS/MS spectra from intact proteins, with differences described in Table 4, were collected with a single m/z precursor of 1513.15, isolation width of 2.5 m/z, except for KPC-5, which was collected with a precursor m/z of 1515.60; corresponding to the z = +19 charge state, and an isolation width of 1.5 m/z. The major fragment in all spectra, b269, a fragment formed from loss of the three C-terminal residues, provides sufficient information in most cases to identify the intact variant. A list of isolates evaluated for the presence or absence of KPC using this method is provided in Table 5.
Fig. 5.

Comparison of MS/MS spectra between KPC variants. Fragmentation spectra of (a) KPC-2, C. freundii, CDC and FDA AR Bank #116, (b) KPC-3, K. ozaenae CDC and FDA AR Bank #96, and (c) KPC-4, E. coli CDC and FDA AR Bank #104. (d) KPC-5, P. aeruginosa CDC and FDA AR Bank #90.
Table 4.
Sequence differences between selected KPC variants.
| Variant | Mutation relative to KPC-2 | Accession ID Used | Source | Δ MW |
|---|---|---|---|---|
| KPC-2 | – | Q9F663 | UniProt | 0.0000 |
| KPC-3 | H251Y | Q93DC4 | UniProt | 26.0044 |
| KPC-4 | P82R, V218G | B1PL86 | UniProt | 17.0014 |
| KPC-5 | P82R | B0ZSP4 | UniProt | 59.0483 |
Amino acid substitution positions are calculated for the mature form of KPC beginning with the sequence ATA at the N-terminus. Differences in MW correspond to the mass differences of the amino acid substitutions.
Table 5.
Isolates evaluated via MS for the presence or absence of KPC. Mass accuracy values are based on the measured b269 fragment deconvoluted MW relative to the theoretical. Theoretical values for b269 are 28383.5611 Da for KPC-2, 28409.5655 Da for KPC-3, 28400.5625 Da for KPC-4, and 28442.6094 Da for KPC-5.
| Identifier | Species | Source | KPC Marker | MS Result | ppm b269 |
|---|---|---|---|---|---|
| 32 | Enterobacter cloacae | CDC FDA AR Bank | KPC-3 | KPC-3 | 1.8154 |
| 50 | Enterobacter cloacae | CDC FDA AR Bank | KPC-4 | KPC-4 | −1.1734 |
| 61 | Escherichia coli | CDC FDA AR Bank | KPC-3 | KPC-3 | 0.8087 |
| 90 | Pseudomonas aeruginosa | CDC FDA AR Bank | KPC-5 | KPC-5 | −0.7110 |
| 96 | Klebsiella ozanenae | CDC FDA AR Bank | KPC-3 | KPC-3 | −3.2181 |
| 104 | Escherichia coli | CDC FDA AR Bank | KPC-4 | KPC-4 | 4.8230 |
| 116 | Citrobacter freundii | CDC FDA AR Bank | KPC-2 | KPC-2 | −0.2052 |
| 136 | Enterobacter cloacae | CDC FDA AR Bank | KPC-3 | KPC-3 | 0.7806 |
| 144 | Kluyvera ascorbate | CDC FDA AR Bank | KPC-3 | KPC-3 | 2.2026 |
| 147 | Klebsiella oxytoca | CDC FDA AR Bank | KPC-3 | KPC-3 | −1.4335 |
| 163 | Enterobacter cloacae | CDC FDA AR Bank | KPC-2 | KPC-2 | 0.0273 |
| BAA-1898 | Klebsiella pneumoniae | ATCC | KPC-2 | KPC-2 | 2.4055 |
| BAA-1899 | Klebsiella pneumoniae | ATCC | KPC-2 | KPC-2 | 3.2792 |
| BAA-1903 | Klebsiella pneumoniae | ATCC | KPC-2 | KPC-2 | 0.4924 |
| BAA-1905 | Klebsiella pneumoniae | ATCC | KPC-2 | KPC-2 | 1.6515 |
| 42 | Klebsiella pneumoniae | CDC FDA AR Bank | none | none | – |
| 43 | Klebsiella pneumoniae | CDC FDA AR Bank | none | none | – |
| 44 | Klebsiella pneumoniae | CDC FDA AR Bank | none | none | – |
| 47 | Klebsiella pneumoniae | CDC FDA AR Bank | none | none | – |
| 58 | Escherichia coli | CDC FDA AR Bank | none | none | – |
| 60 | Enterobacter cloacae | CDC FDA AR Bank | none | none | – |
| 64 | Pseudomonas aeruginosa | CDC FDA AR Bank | none | none | – |
| 72 | Enterobacter cloacae | CDC FDA AR Bank | none | none | – |
| 73 | Enterobacter cloacae | CDC FDA AR Bank | none | none | – |
| 77 | Escherichia coli | CDC FDA AR Bank | none | none | – |
| 11775 | Escherichia coli | ATCC | none | none | – |
| 13883 | Klebsiella pneumoniae | ATCC | none | none | – |
| 700721 | Klebsiella pneumoniae | ATCC | none | none | – |
4. Discussion
4.1. Discovery and Characterization of KPC
The search for the intact KPC was performed by making comparisons between strains that had been previously characterized as either KPC-producing or KPC-non-producing. The nature of KPC to interact with and hydrolyze β-lactams acting on the peptidoglycan layer leads this enzyme to localize in the periplasmic portion of the cell. Following translation in the cytoplasm, these proteins are likely cleaved via SPaseI as part of the secYEG protein conducting channel located in the plasma membrane [33]. Typically, the signal peptides have high alanine content for α-helix formation, as found in many Enterobacteriaceae, and cleave at the canonical AXA motif [34]. However, for KPC-2 there is a significant reduction in alanine residues in the hydrophobic region of the signal peptide. Instead, cleavage occurs at GXS (where X = F), which is directly adjacent to the AXA (where X = T) motif and consistent with the report that in prokaryotes a significant reduction in alanine content leads to the observance of novel motifs for SPaseI cleavage [34]. Based on UniProt accession number Q9F663, (www.uniprot.org) the predicted cleavage site for KPC-2 follows an AXA sequence; however, bottom-up analysis of KPC-producing strains provided evidence of an N-terminal peptide that corresponded to the inclusion of these three residues, thus identifying GXS as the major experimentally determined cleavage site. As shown in Table 1, this sequence, ATALTNLVAEPFAK, provided evidence that the major form of the mature protein of KPC-2 was three residues longer than predicted.
Adjusting for the mass addition of these residues, it became possible to search for the intact mass of KPC-2. Fig. 2c illustrates an example of the CSD of KPC-2, which is relatively low for a protein of roughly 29 kDa. This can be explained by the presence of a disulfide bond between cysteine residues at positions 41 and 216 of the mature protein, effectively restricting the number of charges due to limited protein unfolding during the ionization process. The theoretical monoisotopic mass of this protein, including a disulfide bond, is 28700.6941 Da, with an average mass of 28718.6467 Da. The deconvoluted measured mass was found to be 28700.7973 Da, indicating a 0.1032 Da difference, or a 3.60 ppm error in measurement. A further evaluation of this protein was performed by first reducing and alkylating the entire cell lysate, followed by LC-MS and LC-MS/MS (Fig. S7). The protein corresponding to two alkylations and no disulfide bonds, with a higher CSD and with an expected mass of 28816.7532 Da, was observed. The deconvoluted mass of this protein was measured to be 28816.8583 Da, corresponding to a 0.1051 Da difference, or a 3.65 ppm error in measurement. Additional strain comparisons are included in Table S2. MS/MS spectra also confirmed the identity, providing a greater extent of fragmentation than the disulfide-bonded protein (Fig. S8). While several factors play into the increased fragmentation of the reduced and alkylated protein, the reduction of the disulfide bond effectively removed a cyclic structure within the protein strand, such that a single cleavage between the cysteine residues could be observed. The increased CSD is explained by removing the structural restriction imposed by the native disulfide bonds, thereby allowing more charges to be accumulated along the unfolded protein.
Top-down proteomics approaches allow the intact protein to be further probed beyond intact mass by using the fragment ions that are produced to accurately verify the identity of the amino acid sequence with sufficient confidence. Two significant advantages of mass spectrometric techniques are that the intact mass of the protein can be easily measured with any of its corresponding charge states isolated, and fragment ions from the intact mass can be easily matched to the specific corresponding protein.
The extent of dissociation of KPC-2 is relatively restricted. This is due to the disulfide bond cyclizing the protein, thus requiring two simultaneous backbone amide bond cleavages to occur in order to obtain sequence-specific information. The MS/MS approach applied to the intact KPC variants in this study is restricted to only a single amide bond cleavage. Fig. 3 shows multiple consecutive cleavages of peptide bonds along the protein backbone, thereby generating a sequence tag, LALEGLGVNGQ. These fragments provide sufficient information to perform an alignment search using the Basic Local Alignment Search Tool (BLAST) against K. pneumoniae organisms. Results of this protein alignment search using the sequence tag above correspond to entries matching 100% to known KPC variants (using www.blast.ncbi.nlm.nih.gov/Blast.cgi, data not shown).
The main determinants for the identification of KPC are (1) the MW corresponds to that of the adjusted sequence, including ATA residues at the N-terminus; (2) MS/MS data produce KPC-specific fragment ions; (3) the CSD is indicative of the presence of a disulfide bond; reduction and alkylation data were consistent with this; and (4) this protein only appears in KPC-producing isolates.
4.2. Evaluation of multiple KPC variants
MS techniques performed up to this point were from isolates already characterized as producing a KPC variant. To further validate the ability to detect and identify KPC variants, a control study was performed using carbapenem-susceptible DH5α cells. Within the original cell line, KPC was searched for, with no observation of the protein present. Following the introduction of a plasmid harboring blaKPC-4, the modified cell line exhibited carbapenem resistance, and MS data clearly identified the presence of the corresponding protein, KPC-4, as shown in Fig. 4.
Many different variants have arisen from the initial discovery of KPC-2, with Table 4 representing a small selection. The significant advantage of an MS detection approach for these variants is that, at most, the change in detection from one variant to another is a digital change of the precursor m/z. This is a direct effect of the differing masses resulting from minor sequence variations. KPC-3 differs from KPC-2 by a single amino acid, resulting in the substitution of a tyrosine for a histidine (H251Y). This results in a mass difference of 26.0044 Da, and an m/z difference of 1.3687 at z = +19. KPC-4 differs from KPC-2 by two substitutions, resulting in P82R and V218G. This results in a mass difference of 17.0014 Da, and an m/z difference of 0.0895 at z = +19. KPC-5 differs the most from KPC-2, with a mass difference of 59.0484 Da from an amino acid substitution of P82R, resulting in an m/z difference of 3.1078 at z = +19. The m/z of additional variants can be easily determined, and in most cases, will be very similar to that of KPC-2. Differences between KPC-2 through KPC-5, as well as UniProt entries, are shown in Table 4, while Fig. 5 illustrates the similarities between these variants by presenting similar fragmentation behavior for the z = +19 ion. It should also be noted that similar charge states will result in similar fragmentation spectra among variants, and that the KPC-2 fragmentation spectrum of Fig. 3, expressed by K. pneumoniae, produces identical fragments to the KPC-2 fragmentation spectrum of Fig. 5a, expressed by C. freundii, despite expression in different species. Further evaluations of these variants expressed in additional species are presented in Table 5.
The method discussed herein takes advantage of the unique capabilities offered by mass spectrometry, with particular focus placed on the combination of appropriate ionization sources, mass selection for filtering ions, and high resolution, accurate mass analysis. ESI couples well with LC to introduce a dynamic sample into the mass spectrometer, while the use of a high resolution mass analyzer maintains speed and sensitivity. In this report, we use this application to sort through a complex cell lysate to interrogate the thousands of components, looking for a single protein in the sample, and to characterize it well enough to identify it.
Mass spectrometry is a versatile analytical method that can be applied for many different purposes, and offers the ability to maintain a balance between speed of data acquisition, resolution and sensitivity. Four KPC variants were unambiguously identified and distinguished from each other using similar m/z precursors; however, expanding the search to additional markers requires no additional consumables, but only a digital change in precursor m/z. Sensitivity in mass spectrometry can be enhanced by using MS/MS over MS alone. Each of these fragment ions will have very specific characteristics, such as m/z within a specific ppm error, and with specific charge states. Furthermore, because each protein produces a CSD, more than one charge state could be selected for MS/MS to verify the protein identity. Taking these factors into account, the likelihood for a false positive is essentially eliminated using MS/MS, based on the characteristics of fragment ions and their inherent connection to an intact protein. This positions LC-MS/MS as an extremely powerful tool for the direct detection of intact KPC from bacterial lysates, with minimal sample preparation and relatively short chromatographic timescales, and could likely be applied to detection of additional resistance proteins using similar approaches.
Declaration of Competing Interest
W. M. McGee, J. R. Neil, S. R. Kronewitter, and J. L. Stephenson, Jr. are employees of Thermo Fisher Scientific. Also, W.M. McGee, J.R. Neil, and J.L. Stephenson Jr. own stock in Thermo Fisher Scientific. There are two patents that have been filed on resistance marker identification by the authors from Thermo Fisher Scientific. M. L. Faron, B. W. Buchan and N. A. Ledeboer have received research funding from Bruker Daltonics and bioMerieux, and N. A. Ledeboer is a consultant for Thermo Fisher Scientific.
Acknowledgements
The authors would like to acknowledge Dr. Christopher Mullen, Dr. John Syka, and Dr. Romain Huguet of Thermo Fisher Scientific, San Jose, CA for helpful conversations.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.clinms.2020.07.001.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Michael C.A., Dominey-Howes D., Labbate M. The antimicrobial resistance crisis: causes, consequences, and management. Front. Public Health. 2014;2:145. doi: 10.3389/fpubh.2014.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ventola CL. The Antibiotic Resistance Crisis. P&T 2015; 40(4): 7 [PMC free article] [PubMed]
- 3.Roca I., Akova M., Baquero F., Carlet J., Cavaleri M., Coenen S., Cohen J., Findlay D., Gyssens I., Heuer O.E., Kahlmeter G., Kruse H., Laxminarayan R., Liebana E., Lopez-Cerero L., MacGowan A., Martins M., Rodriguez-Bano J., Rolain J.M., Segovia C., Sigauque B., Tacconelli E., Wellington E., Vila J. The global threat of antimicrobial resistance: science for intervention. New Microbes New Infections. 2015;6:22–29. doi: 10.1016/j.nmni.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antibiotic C.D.C. Services, CDC; 2013. Resistance Threats in the United States. Atlanta, GA: U.S. Department of Health and Human. [Google Scholar]
- 5.CDC. Antibiotic Resistance threats in the United States. Atlanta, GA: U. S. Department of Health and Human Services, CDC, 2019.
- 6.Ho J., Tambyah P.A., Paterson D.L. Multiresistant Gram-negative infections: a global perspective. Curr. Opin.. Infect Dis. 2010;23(6):546–553. doi: 10.1097/QCO.0b013e32833f0d3e. [DOI] [PubMed] [Google Scholar]
- 7.Viswanathan V.K. Off-label abuse of antibiotics by bacteria. Gut. Microbes. 2014;5(1):3–4. doi: 10.4161/gmic.28027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Read A.F., Woods R.J. Antibiotic resistance management. Evol. Med. Public Health. 2014;2014(1):147. doi: 10.1093/emph/eou024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonomo R.A., Burd E.M., Conly J., Limbago B.M., Poirel L., Segre J.A., Westblade L.F. Carbapenemase-producing organisms: a global scourge. Clin. Infect. Dis. 2018;66(8):1290–1297. doi: 10.1093/cid/cix893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Logan L.K., Weinstein R.A. The epidemiology of carbapenem-resistant enterobacteriaceae: the impact and evolution of a global menace. J. Infect. Dis. 2017;215(suppl_1):S28–S36. doi: 10.1093/infdis/jiw282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munoz-Price L.S., Poirel L., Bonomo R.A., Schwaber M.J., Daikos G.L., Cormican M., Cornaglia G., Garau J., Gniadkowski M., Hayden M.K., Kumarasamy K., Livermore D.M., Maya J.J., Nordmann P., Patel J.B., Paterson D.L., Pitout J., Villegas M.V., Wang H., Woodford N., Quinn J.P. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect. Dis. 2013;13(9):785–796. doi: 10.1016/S1473-3099(13)70190-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gasink L.B., Edelstein P.H., Lautenbach E., Synnestvedt M., Fishman N.O. Risk factors and clinical impact of Klebsiella pneumoniae carbapenemase-producing K. pneumoniae. Infect. Control Hosp. Epidemiol. 2009;30(12):1180–1185. doi: 10.1086/648451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mehta S.C., Rice K., Palzkill T. Natural Variants of the KPC-2 Carbapenemase have Evolved Increased Catalytic Efficiency for Ceftazidime Hydrolysis at the Cost of Enzyme Stability. PLoS Pathog. 2015;11(6) doi: 10.1371/journal.ppat.1004949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hooff G.P., van Kampen J.J., Meesters R.J., van Belkum A., Goessens W.H., Luider T.M. Characterization of beta-lactamase enzyme activity in bacterial lysates using MALDI-mass spectrometry. J. Proteome Res. 2012;11(1):79–84. doi: 10.1021/pr200858r. [DOI] [PubMed] [Google Scholar]
- 15.Sparbier K., Schubert S., Weller U., Boogen C., Kostrzewa M. Matrix-assisted laser desorption ionization-time of flight mass spectrometry-based functional assay for rapid detection of resistance against beta-lactam antibiotics. J Clin. Microbiol. 2012;50(3):927–937. doi: 10.1128/JCM.05737-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hrabak J., Walkova R., Studentova V., Chudackova E., Bergerova T. Carbapenemase activity detection by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J. Clin. Microbiol. 2011;49(9):3222–3227. doi: 10.1128/JCM.00984-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burckhardt I., Zimmermann S. Using matrix-assisted laser desorption ionization-time of flight mass spectrometry to detect carbapenem resistance within 1 to 2.5 hours. J. Clin. Microbiol. 2011;49(9):3321–3324. doi: 10.1128/JCM.00287-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carvalhaes C.G., Cayo R., Assis D.M., Martins E.R., Juliano L., Juliano M.A., Gales A.C. Detection of SPM-1-producing Pseudomonas aeruginosa and class D beta-lactamase-producing Acinetobacter baumannii isolates by use of liquid chromatography-mass spectrometry and matrix-assisted laser desorption ionization-time of flight mass spectrometry. J. Clin. Microbiol. 2013;51(1):287–290. doi: 10.1128/JCM.02365-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kempf M., Bakour S., Flaudrops C., Berrazeg M., Brunel J.M., Drissi M., Mesli E., Touati A., Rolain J.M. Rapid detection of carbapenem resistance in Acinetobacter baumannii using matrix-assisted laser desorption ionization-time of flight mass spectrometry. PLoS One. 2012;7(2) doi: 10.1371/journal.pone.0031676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mirande C., Canard I., Buffet Croix Blanche S., Charrier J.P., van Belkum A., Welker M., Chatellier S. Rapid detection of carbapenemase activity: benefits and weaknesses of MALDI-TOF MS. Eur. J. Clin. Microbiol. Infect. Dis. 2015;34(11):2225–2234. doi: 10.1007/s10096-015-2473-z. [DOI] [PubMed] [Google Scholar]
- 21.Pulido M.R., Garcia-Quintanilla M., Martin-Pena R., Cisneros J.M., McConnell M.J. Progress on the development of rapid methods for antimicrobial susceptibility testing. J. Antimicrob. Chemother. 2013;68(12):2710–2717. doi: 10.1093/jac/dkt253. [DOI] [PubMed] [Google Scholar]
- 22.Maugeri G., Lychko I., Sobral R., Roque A.C.A. Identification and antibiotic-susceptibility profiling of infectious bacterial agents: a review of current and future trends. Biotechnol. J. 2019;14(1) doi: 10.1002/biot.201700750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Camara J.E., Hays F.A. Discrimination between wild-type and ampicillin-resistant Escherichia coli by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal. Bioanal. Chem. 2007;389(5):1633–1638. doi: 10.1007/s00216-007-1558-7. [DOI] [PubMed] [Google Scholar]
- 24.Wang H., Drake S.K., Youn J.H., Rosenberg A.Z., Chen Y., Gucek M., Suffredini A.F., Dekker J.P. Peptide Markers for Rapid Detection of KPC Carbapenemase by LC-MS/MS. Sci. Rep. 2017;7(1):2531. doi: 10.1038/s41598-017-02749-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lau A.F., Wang H., Weingarten R.A., Drake S.K., Suffredini A.F., Garfield M.K., Chen Y., Gucek M., Youn J.H., Stock F., Tso H., DeLeo J., Cimino J.J., Frank K.M., Dekker J.P. A rapid matrix-assisted laser desorption ionization-time of flight mass spectrometry-based method for single-plasmid tracking in an outbreak of carbapenem-resistant Enterobacteriaceae. J. Clin. Microbiol. 2014;52(8):2804–2812. doi: 10.1128/JCM.00694-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cordovana M., Kostrzewa M., Glandorf J., Bienia M., Ambretti S., Pranada A.B. A Full MALDI-Based Approach to Detect Plasmid-Encoded KPC-Producing Klebsiella pneumoniae. Front. Microbiol. 2018;9:2854. doi: 10.3389/fmicb.2018.02854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Figueroa-Espinosa R., Costa A., Cejas D., Barrios R., Vay C., Radice M., Gutkind G., Di Conza J. MALDI-TOF MS based procedure to detect KPC-2 directly from positive blood culture bottles and colonies. J Microbiol Methods. 2019;159:120–127. doi: 10.1016/j.mimet.2019.02.020. [DOI] [PubMed] [Google Scholar]
- 28.Kelleher N.L., Lin H.Y., Valaskovic G.A., Aaserud D.J., Fridriksson E.K., McLafferty F.W. Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry. J Am Chem Soc. 1999;121:806–812. [Google Scholar]
- 29.Kelleher N.L. Top-down proteomics. Anal. Chem. 2004;76(11):196A–203A. [PubMed] [Google Scholar]
- 30.Toby T.K., Fornelli L., Kelleher N.L. Progress in top-down proteomics and the analysis of proteoforms. Annu. Rev. Anal. Chem. (Palo Alto Calif) 2016;9(1):499–519. doi: 10.1146/annurev-anchem-071015-041550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fellers R.T., Greer J.B., Early B.P., Yu X., LeDuc R.D., Kelleher N.L., Thomas P.M. ProSight Lite: Graphical software to analyze top-down mass spectrometry data. Proteomics. 2015;15(7):1235–1238. doi: 10.1002/pmic.201570050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Senko M.W., Beu S.C., McLafferty F.W. Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. J. Am. Soc. Mass.. Spectrom. 1995;6:229–233. doi: 10.1016/1044-0305(95)00017-8. [DOI] [PubMed] [Google Scholar]
- 33.Owji H., Nezafat N., Negahdaripour M., Hajiebrahimi A., Ghasemi Y. A comprehensive review of signal peptides: Structure, roles, and applications. Eur. J. Cell. Biol. 2018;97(6):422–441. doi: 10.1016/j.ejcb.2018.06.003. [DOI] [PubMed] [Google Scholar]
- 34.Payne S.H., Bonissone S., Wu S., Brown R.N., Ivankov D.N., Frishman D., Pasa-Tolic L., Smith R.D., Pevzner P.A. Unexpected diversity of signal peptides in prokaryotes. mBio. 2012;3(6) doi: 10.1128/mBio.00339-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data produced from LC-MS and LC-MS/MS evaluations are available online in a data repository stored on figshare.com. Data include spectra for all figures produced herein and for supplementary information. Software provided and maintained by Thermo Fisher Scientific was used to generate all data and files, each stored as a “.raw” file. These are readable using Xcalibur or Freestyle software programs (Thermo Fisher Scientific). Protein sequences of KPC variants can be found at Uniprot.org with the following identifiers: Q9F663 for KPC-2, Q93DC4 for KPC-3, B1PL86 for KPC-4, and B0ZSP4 for KPC-5. It should be noted there may be multiple identifiers with identical sequence information for a given variant. Sequence modifications described in this report are based on the mature protein sequence.