

Abstract
Monocytes and macrophages are cellular forces that drive and resolve inflammation triggered by myocardial ischemia. One of the most important yet least understood regulatory mechanisms is how these cells integrate cues from micro-milieu with their response that eventually determines the outcome of myocardial repair. In the current study, we investigate if the mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) plays this role. We present evidence that support a robustly activated mTORC1 pathway in monocytes and macrophages in the infarcting myocardium, a surprising finding considering these cells resided in an environment that was hypoxic and lack of energy and nutrients. Our data suggests that the cardiac monocytes and macrophages gained effector functions predominantly in the ischemic niche, further supporting the crucial role of the microenvironment in deciding their functions. Specific mTORC1 inhibition transformed the landscape of cardiac monocytes and macrophages into reparative cells that promoted myocardial healing. As the result, mTORC1 inhibition diminished remodeling and reduced mortality from acute ischemia by 80%. In conclusion, our data suggest a critical role of mTORC1 in regulating the functions of cardiac monocytes and macrophages, and specific mTORC1 inhibition protects the heart from inflammatory injury in acute ischemia. As mTOR/mTORC1 is a master regulator that integrates external signals with cellular responses, the study sheds light on how the cardiac monocytes and macrophages sense the ischemia milieu and adjust their function.
Keywords: myocardial infarction, remodeling, macrophage, mTORC1, rapamycin
Graphical Abstract
INTRODUCTION
In the setting of myocardial ischemia, the peripheral monocytes are recruited to the ischemic region and differentiate towards macrophages[1]. These cells orchestrate both the inflammatory response and the reparative action that occur in the first and second half of the first week after myocardial infarction (MI), respectively. Accordingly, the monocytes and macrophages in these stages are described as inflammatory and reparative cells. Although both types of cells are critical to the repair of the myocardial wound [1–4], prolonged presence of inflammatory cells hinders this process. Reparative macrophages, on the other hand, limits inflammatory injury and promotes healing. Unlike other organs such as the skeletal muscle and skin, the cardiac parenchyma dictates the final outcome of repair in the form of scar formation. This highlights the importance of efficient repair to minimize the damage in order to reduce ventricular remodeling that eventually culminates in clinical heart failure (HF).
The microenvironment that monocytes and macrophage are recruited to and resides is one of the most important determinants of their function[5]. For example, as soon as they were recruited to the injury site generated by mechanical manipulation of the small intestine, monocytes immediately changed their transcriptomes [6]. The heterogeneous population of cardiac monocytes and macrophages in the ischemic myocardium [7–10] suggested the influence from the various environments these cells reside. However, it remains largely uncharacterized and challenging to decipher how the environmental cues are sensed and responded by the monocytes and macrophages (referred as cardiac mononuclear phagocytes (cMP) for simplicity thereafter) in the ischemic myocardium. The mechanistic target of rapamycin (mTOR) is a master regulator that integrates the environment cues and modulates cellular functions accordingly. mTOR exists in two structurally distinct complexes denoted as mTOR complex 1(mTORC1) and mTORC2. In the presence of growth factors plus abundant nutrients and energy[11], mTORC1 is activated and promotes cell growth, proliferation, and acquisition of effector functions. Activated mTORC1 promotes protein synthesis by mediating the phosphorylation of ribosomal protein S6 kinase beta-1 also known as P70S6K and eukaryotic translation initiation factor 4E (eIF4E) binding protein 1(4EBP1)[12], de novo lipid production by activating the sterol responsive element binding protein (SREBP) transcription factor, and the synthesis of nucleotides by increasing key enzymes responsible for the synthesis of purine and pyrimidine[13, 14]. As monocytes differentiate towards macrophages, the cells become bigger and gain complex subcellular structures especially lysosomes, endoplasmic reticulum, and Golgi network. These structural complexity supports their effector functions at a higher echelon of the MP system [15]. One example is that macrophages possessed higher capacity of phagocytosis and were able to produce higher amount of inflammatory cytokines [15]. These structural and functional changes may indicate a role of mTORC1 during differentiation. Indeed, interruption of mTORC1 signaling by deleting Ras homolog enriched in brain (Rheb1), which is a direct activator of mTORC1, hindered monocyte to macrophage differentiation [16]. In addition to the monocyte-to-macrophage differentiation, mTORC1 plays complex roles in the activation of macrophages and is implicated in promoting both pro- and anti-inflammatory phenotypes of these cells [17, 18]. Currently, there are limited data regarding mTORC2. However, these evidence are consistent in suggesting that mTORC2 promotes anti-inflammatory macrophages [19, 20], which have also been referred as resolving/reparative cells [19, 21].
Currently, it is not known if mTORC1 governs the differentiation process (monocytes towards macrophages) and the transition process (inflammatory to reparative cMPs) in the infarcting myocardium. mTORC1 is a tightly regulated process that requires strictly both growth factors/cytokines and nutrients for activation [22]. The infarct tissue, which is hypoxic with limited energy production and lacks nutrients due to non-functional vessels, is seemingly not the environment suitable for mTORC1 activation. Prior works regarding mTOR have focused mainly on cardiomyocytes (comprehensively reviewed in [23]). In addition, treatment with mTORC1 inhibitor rapamycin during the first week of experimental MI demonstrated no benefit [24]. In contrast, prolonged treatment (4 to 6 weeks) improved cardiac function [24, 25]. These data suggest that the benefit is limited to the chronic remodeling phase. From these evidence, it could also be inferred that mTOCR1 has no role in controlling the function of monocytes and macrophages because the most dynamic changes of these cells occur in the first week after MI and manipulation of their behavior in the same time window had impact on survival and remodeling[2, 26, 27]. Rapamycin predominantly inhibits mTORC1. However, the specificity is lost and mTORC2 becomes a target when rapamycin is used at higher doses (similar to that used in the prior MI studies [24, 25]) and for longer durations [28, 29]. As some evidence suggest that mTORC1 and mTORC2 polarized macrophage towards the inflammatory and pro-resolving/anti-inflammatory cells, which stymies or promotes the myocardial repair, respectively, it is therefore possible that duo inhibition of mTORC1 and mTORC2 achieved by high dose rapamycin [24] may have canceled the effect of inhibiting individual complex and resulted in the null effect in the acute phase of ischemia.
In the current study, we determined if mTORC1 played a key role in sensing the environment cues and regulating the function of cMPs in the infracting myocardium. We uncovered potential un-conventional source of nutrients that could be used for mTORC1 activation. In addition, we demonstrated where the cMPs gained their effector function in the setting of myocardial ischemia. Specifically, the effector function discussed here is referred to as cytokine production and phagocytosis. We also defined a strategy to achieve selective inhibition of mTORC1 without affecting the activity of mTORC2.
MATERIALS AND METHODS
Animals
The investigation conforms to the Guide for Care and Use of Laboratory animals published by the US National Institutes of Health (NIH Publication No.85–23, revised 2011). Animal experiments were approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center (approved and active protocol Numbers: 2019–102736). Male mice (body weight from 23g to 28g) were anaesthetized via intraperitoneal injection of ketamine and xylazine at 100mg/kg and 5mg/kg, respectively. During the surgery, anesthesia was maintained with 0.6 to 1% isoflurane via Precision vaporizer. Euthanasia was performed by inhalational overdose of isoflurane followed by cervical dislocation. The cardiac tissue was collected immediately after euthanasia. All procedures were approved by Institutional Animal Care and Use Committee at the University Of Texas Southwestern Medical Center. Animals were maintained in a pathogen-free environment with free access to food and water. They were maintained on a 12-hour light/dark cycle from 6 am to 6 pm.
Antibodies
Antibodies used for immunoblot are listed here based on where they are acquired. 1. From Cell Signaling Technology (Danvers, MA): anti-pS6 (4858); anti-pS6K (9204); anti-S6 (2317); anti-p4E-BP1 (2855); anti-4E-BP1(9452); anti-mTOR (2983); anti-pAkt (Ser473) (4060); anti-pAkt (Thr308) (13038); anti-Akt (2920). 2. From BioRad (Hercules, California): anti-CD68 (MCA19571); 3. From Biolegend (San Diego, California): Alexa fluoro-labeled anti-CD 206 (141709); APC anti-mouse Ly-6G Antibody (127614); PE anti-mouse F4/80 Antibody (123110); Pacific Blue™ anti-mouse/human CD11b Antibody (101224); PE/Cy7 anti-mouse CD45 (103114); Brilliant Violet 711™ anti-mouse Ly-6C Antibody (128037); PerCP/Cy5.5 anti-mouse CD206 (MMR) Antibody (141716). 4. From Santa Cruz Biotechnology (Santa Cruz, CA): mouse anti-GAPDH antibody; anti-LAMP1 (sc-20011). 5. From R&D Systems (Minneapolis, MN): goat anti-CD31 (AF3628). 6. From Cedarlane (Burlington, Ontario, Canada): anti-Mouse Mac-3 (CL8993AP).
Bone marrow-derived macrophage
Isolation of bone marrow cells and differentiation of macrophages were performed based on established protocols.20, 21 Briefly, bone marrow cells were cultured on untreated petri dishes in RPMI 1640 medium (Cellgro Mediatech, No.15–040-CV) supplemented with 10% FBS and 10 ng/mL macrophage colony stimulating factor (M-CSF, Sigma, SRP3221). On day 3 after initial plating, 50% fresh medium and M-CSF were added. After 5 to 7 days of differentiation, macrophages were disassociated with PBS plus 0.6 mM EDTA, washed in culture medium, and plated onto 12-well cell culture plates (2.5 × 105/mL/well).
Mouse myocardial infarction model and rapamycin injection
A murine model of myocardial infarction was employed involving permanent ligation of the left anterior descending coronary artery (LAD) as described previously [26]. Briefly, 12–14-week-old WT mice were anesthetized with 2.4% isoflurane and positioned supine on a heating pad (37°C). Tracheal intubation was performed with a 19G stump needle, and animals ventilated with room air using a MiniVent mouse ventilator (Hugo Sachs Elektronik; stroke volume, 250 μL; respiratory rate, 210 breaths per minute). Following left thoracotomy between the fourth and fifth ribs, the LAD was visualized under a microscope and ligated with 6–0 Prolene suture. The suture was placed at the LAD segment corresponding to 1.5 mm to 2 mm below the lower edge of the left atrium. Regional ischemia was confirmed by visual inspection of discoloration of myocardium distal to the occlusion under a dissecting microscope (Leica). Sham-operated animals underwent the same procedure without occlusion of the LAD. Area at risk (AAR) is determined by perfusing 2% Evans blue after LAD ligation. The LV is cross-sectioned into 6 slices and the total and non-stained area (AAR) are determined by Image J. The percentage of AAR/LV is calculated. After LAD ligation, hearts were collected at 3-day and 7-day. Left ventricles were separated into remote and infarct-at-risk zones and then flash frozen for further analysis. For rapamycin treatment, mice were randomly divided into DMSO or rapamycin group 24 hours after the LAD ligation and rapamycin was injected sub-cutaneous at 0.25mg/kg/d for 2 or 6 days. The control cohort was injected with the same amount of DMSO.
Murine Echocardiography
Mouse echocardiograms were performed as described previously [26]. Briefly, non-sedated mice were gently restrained and echo images were obtained using a Vevo 2100 system and an 18-MHz linear probe. A short-axis view of the LV at the level of the papillary muscles was obtained, and M-mode recordings were obtained from this view. Heart rate was closely monitored during image acquisition. Left ventricular internal diameter at end-diastole (LVID;d) and endsystole (LVID;s) were measured from M-mode recordings. Fractional shortening was calculated as (LVIDd − LVIDs)/LVIDd (%).
Isolation of immune cells and cMP enrichment
Cardiac immune cells were isolated as described previously [1, 3, 26] with some modifications. The heart was exposed by left thoracotomy followed by perfusion with 20 ml ice cold PBS. After perfusion, the hearts were separated and placed in cold PBS on ice. We examined all hearts under a dissection microscope to ensure the procedure inducing infarction was successful. The infarct region and remote area was separated also under the dissection microscope. The hearts were examined and tissue was finely minced and extensively washed in ice-cold PBS. The minced tissues were then incubated at 37°C for 45 to 50 minutes on an orbital shaker at a speed of 75 rpm in the digestion buffer that contained the following enzymes obtained from Sigma (St. Louis, MI): collagenase (C0130) at 450 U/mL, hyaluronidase type I-S (H3506) at 60U/ml, and DNase (D4513) at 60U/ml. We use 1 ml of the digestion buffer per remote or infarct tissues. After digestion, the tissues were then triturated and passed through a 40 μm strainer. The strainer was washed with 1 ml ice cold PBS. The cells were pelleted by centrifugation at 250 × g for 15 min and washed with Opti-MEM (ThermoFisher, Waltham, MA). The cell pellet was suspended in Opti-MEM and seeded onto Corning cell culture dishes. After 2-hour incubation, the unattached cells were removed by gentle pipetting followed by washing with PBS. The attached cells were collected for RNA or protein isolation. For confocal microscopy, the cell mixture was dispended on coverslips coated with SureCoat (sc-9035) obtained from Cellutron (Rosedale, MD). ,
Fluorescence activated cell sorting (FACS)
The immune cells isolated as described above were treated with RBC lysis buffer for 5 minutes followed by inactivation of the process by adding 5–8 mL of cold Opti-MEM. Centrifuge the samples at 400 x g for 5 min at 4 °C. Aspirate off supernatant and suspend the cell pellet with 1 mL of FACS buffer that contained 2% FBS and 1 mM Ethylenediaminetetraacetic acid (EDTA, pH 8.0) in PBS. Cells were labeled with the following antibodies at indicated dilutions: CD11b-PB, 1:200; CD45.2-FITC 1:200; F4/80-PE, 1:100; Ly-6G-APC 1:250; Ly-6C-BV711 1:200; CD206- PerCP/Cy5.5 : 1:200. After the 30-minute incubation, the cells were washed and suspended in FACS scan buffer. Gating strategies are described in the results section.
Isolation of monocytes from the spleen
The spleen is harvest and gently crashed into ice cold PBS. The tissue suspension is trifurcated by pipetting to further disassociate the tissue into single cell suspension. The resulting solution is filtered through 70μM filter to remove large tissue debris. Red blood cells are removed from the cell suspension by incubating with RBC lysis buffer (cat# 00–4333-57, ThermoFisher) for 5 minutes followed by spinning down at 400g for 5 minutes at 4°C. The monocytes are purified using a monocyte isolation kit from according to manufacturer’s instruction (Cat.# 19861, STEMCELL, Cambridge, MA). The monocytes prepared are subjected with RNA isolation or flow cytometry.
Isolation of myocardium-derived particle
Mice were anesthetized with isoflurane followed by cervical dislocation. 70% ETOH was sprayed onto the chest before the heart was exposed. The infarct tissue or the remote myocardium 1 day after LAD ligation were collected and weighed. The tissue was then transferred to sterile cell culture hood and extensively rinsed with ice cold PBS. Procedures from now on were performed using sterile techniques and instruments. The heart tissue was minced into 1–2 mm dimension pieces in cold PBS and rinsed again for 3x with cold sterile PBS. The tissue was then transferred to Dunce tissue homogenizer that contained cold particle isolation buffer that has 320 mM sucrose, 20 mM Tris, 1 mM EGTA, 0.2 % BSA, and pH adjusted to 7.2. The ration of tissue weight (mg) to buffer volume (ml) is 100 to 1. First, the tissue was broke by stroking pistol A (1up +1 down) for 10–15x followed by pistol B 10–15x. The resulting tissue homogenate was spun down at 1000g for 5 minutes at 4 degree. The supernatant was collected into a new pre-cooled tube. Suspend the pellet in 1ml (same ratio as the above) isolation buffer and perform 10–15 strokes with pistol B. The second homogenate was spun down at 1000g for 5 minutes at 4 degree. Combine the supernatant obtained as described above and spin down for 10 minutes at 12,000g at 4°C. Discard the supernatant and wash the pellet with 1.5 ml cold PBS. Repeat centrifugation at 12,000g for 10 minutes at 4°C. Particles prepared using this method are enriched with mitochondria with sizes ranging from 0.5 μm in diameter and up to 7 μm in length, ideal for phagocytosis assay. For the purpose of easier to describe, we name the as mitochondria-enriched myocardium-derived particles Mito-MyoDP.
Phagocytosis Assay
The advantage of using the Mito-MyoDP in our phagocytosis assay is that they are physiological relevant to the phagocytotic process in vivo, comparing to the commonly used opsonized latex beads that cannot be digested by macrophages. In addition, we developed a method of tracking these particles by staining them first with a MitoTracker dye, as the particles purified are enriched with mitochondria. The Mito-MyoDP was suspended in 200 μl Opti-MEM containing 100 nM MitoTracker Red (M7512, ThermoFisher) at 37°C for 30 minutes. After incubation, the particles were collected by spinning down at 12,000g for 5 minutes. The Mito-MyoDP was washed by resuspending the pellet in 500 μl Opti-MEM and spinning down using the same parameters. The labeled Mito-MyoDP was added to macrophages cultured on coverslip and incubated in cell culture incubator at 37°C for 10-, 20-, 30-, and 40-minutes. After the incubation, the cells were washed with PBS followed by fixation with 4% PFA for 10 minutes in room temperature. The engulfed Mito-MyoDP can be visualized under confocal microscope.
Immunoblot analysis
The protocol for immunoblot analysis was as described previously with minor modifications [26]. Briefly, tissues from the remote and infarct areas of myocardial tissue were homogenized in T-PER lysis buffer from ThermoFisher (Waltham, MA) supplemented with protease inhibitors and phospho-STOP (Roche, Indianapolis, IN) using a Dounce homogenizer. Tissue lysates were centrifuged at 12,000g for 12 minutes. The supernatant was collected, and the protein concentration was measured by Bradford assay (Bio-Rad, Hercules, CA). Fifteen micrograms of total protein was loaded onto an SDS-PAGE gel and ultimately transferred to a nitrocellulose membrane. The membrane was blocked with 5% nonfat dry milk, followed by incubation with primary antibodies (overnight, 4°C) and secondary antibodies (1h, RT). The membrane was scanned and quantified using an Odyssey scanner (LI-COR, Lincoln, NE).
Immunohistochemistry
Sham- or MI-operated mice at indicated time points were anesthetized with isoflurane. Immediately after cervical dislocation, the heart was exposed and the right atrium was nicked with a scalpel. The heart was perfused with 10 ml ice-cold PBS to flush out the residue blood in the coronary circulation. The heart was fixed by perfusing ice-cold 4% paraformaldehyde in PBS. After trimming away the atria and great vessels, the heart was embedded in OCT compound (VWR International) and frozen in isopentane chilled in liquid nitrogen. The frozen tissue sections were obtained in transverse orientation starting at the level of LAD suture and progressing toward the apex at 0.5 mm intervals. Sections were incubated in PBS containing 0.1% Triton X (5 min, RT), and non-specific antibody binding sites were pre-blocked with PBS plus 5% BSA. Sections were then incubated with primary antibodies in PBS with 5% BSA (overnight, 4°C) in a humidified chamber. After rinsing with PBS for 10 times, the sections were next incubated with appropriate fluorophore-conjugated secondary antibodies for 1 hour at room temperature. Stained sections were mounted with anti-fade mounting medium before proceeding to fluorescent microscopy.
RNA isolation and quantitative RT-PCR
Total RNA was isolated from heart tissues and cultured cells using the Aurum kit for total RNA isolation (Bio-Rad, Hercules, CA). 100 ng RNA was used for reverse transcription using Superscript (ThermoFisher, Waltham, MA). The resulted cDNA was diluted 5x, and 2 μL of the final cDNA solution was used in the subsequent quantitative polymerase chain reaction analysis (qPCR) with Sybergreen according to manufacturer’s instructions (Roche, Indianapolis, IN). Primer sequences are as follows.
Mouse 18s Forward AAACGGCTACCACATCCAAG
Mouse 18s Reverse TACAGGGCCTCGAAAGAGTC
Mouse IL-6 Forward TCCATCCAGTTGCCTTCTTG
Mouse IL-6 Reverse GGTCTGTTGGGAGTGGTATC
Mouse TNF-α Forward CCTCCCTCTCATCAGTTCTATGG
Mouse TNF-α Reverse GGCTACAGGCTTGTCACTCG
Mouse MCP1 Forward AGGTCCCTGTCATGCTTCTG
Mouse MCP1 Reverse TGGGATCATCTTGCTGGTG
Mouse CCL5 Forward ACGTCAAGGAGTATTTCTACAC
Mouse CCL5 Reverse GATGTATTCTTGAACCCACT
Mouse IL1β Forward CCAAGCAACGACAAAATACC
Mouse IL1β Reverse GTTGAAGACAAACCGTTTTTCC
Mouse CD14 Forward CTCTGTCCTTAAAGCGGCTTAC
Mouse-CD14 Reverse GTTGCGGAGGTTCAAGATGTT
Mouse Arginase1 Forward CTCCAAGCCAAAGTCCTTAGAG
Mouse Arginase1 Reverse AGGAGCTGTCATTAGGGACATC
Mouse MMP2 Forward GATAACCTGGATGCCGTCGTG
Mouse MMP2 Reverse CTTCACGCTCTTGAGACTTTGGTTC
Mouse MMP9 Forward GCCCTGGAACTCACACGACA
Mouse MMP9 Reverse TTGGAAACTCACACGCCAGAAG
Mouse CCL3 Forward TGAAACCAGCAGCCTTTGCTC
Mouse CCL3 Reverse AGGCATTCAGTTCCAGGTCAGTG
Mouse CCL4 Forward CCATGAAGCTCTGCGTGTCTG
Mouse CCL4 Reverse GGCTTGGAGCAAAGACTGCTG
Statistical analyses
Findings are expressed as mean ± SD. The data were analyzed using statistical software (GraphPad Prism, version 7.01; GraphPad Software, San Diego, CA). An unpaired Student t test was performed to analyze 2 independent groups. One-way ANOVA coupled with the Tukey post-hoc test was used for pairwise comparisons. In representative datasets, we have also employed nonparametric tests (Wilcoxon rank sum test, Wilcoxon two-sample test, Kruskal-Wallis test). A value of p < 0.05 was considered statistically significant and results are depicted throughout as follows: *p<0.05; **p<0.01, ***p<0.001.
RESULT
Robust activation of mTORC1 in the mononuclear phagocytes in the infarcting myocardium.
mTORC1 activation is gauged by phosphorylation of its downstream targets including ribosomal protein S6 kinase beta-1 (P70S6K), S6 ribosomal protein (S6), and eIF4E binding protein 1 (4EBP1). We first determined the overall mTORC1 activity in the remote and infarcting myocardium. Supplemental(S)-Figure 1A demonstrates the p-P70S6K level in sham hearts, infarct tissues, or remote myocardium 3- or 7-day after the MI procedure. The increase in p-P70S6K predominantly occurred in the infarcting myocardium, suggesting that ischemia triggered the activation of P-70S6K. The summarized data from different cohorts revealed a 13 to 17 fold increase in pP70S6K when compared to that in sham (S-Figure 1C). We observed a modest increase in p-P70S6K in the remote myocardium but it did not reach statistical significance (Figure 1C). S-Figure 1B is a representative immunoblot demonstrating the increase in p-S6 in the infarcting myocardium. The p-S6 levels from different MI cohorts was summarized in S-Figure 1D. S-Figure 1E depicts the phosphorylation pattern of 4EBP1. We detected both hypo- and hyper-phosphorylated 4EBP1 in all samples. The hypo-phosphorylated isoform dominated in sham-operated hearts (S-Figure 1E). In contrast, the hyper-phosphorylated 4EBP1 became more prominent in the infarct tissue especially at the 1 week post MI. As binding of hypo-phosphorylated 4EBP1 with eIF4E prevents its interaction with eIF4G and subsequently inhibits complex assembly leading to translation suppression, the shift from hypo- to hyper-phosphorylated isoform in the MI tissues is indicative of activation. While quantification of the phosphorylation pattern was difficult when using t-4EBP1 as loading control, we did observe a two fold increase in p-4EBP1 after normalizing it to GAPDH (S-Figure 1F).
Figure 1.


C57BL/6J mice were subjected to sham procedures or LAD ligation. Immunohistochemistry was performed on cryosections prepared from MI hearts. Immunoblot analysis was performed using total protein extracted from cMPs isolated from sham hearts or MI tissues. A. Tissue sections from hearts three days after LAD ligation were labeled with antibodies made against p-S6 and monocyte/macrophage marker CD68. Images were taken from both the infarct and remote regions. n=3 hearts. B. Tissue sections from the same MI cohorts as in A were labeled with neutrophil and p-S6 antibodies. C. Quantification of p-S6+ and CD68+ areas relative to the total area using ImageJ. The percentage of p-S6+CD68+ area to the total p-S6+ area was plotted on the right axis. n=3 hearts. D. Immunoblot analysis of p-P70S6K and p-S6 using cMPs isolated from MI tissue 3-day and 7-day after LAD ligation. E. Quantification of p-P70S6K and p-S6 from different experiments performed as in D. Each data point in sham represents 3 to 4 hearts combined, total 10 to 12 hearts used. In the infarct cohorts, some data points are from pooled infarct tissue. Total n= 6 to 9 hearts at each time point. * p<0.05 ** p<0.01.
In order to determine if the cardiac mononuclear phagocytes (cMPs) contributed to the mTORC1 activity, we co-labeled tissue sections with antibodies against p-S6 and CD68 (a cell surface marker for monocytes and macrophages). We also determined mTORC1 activation using isolated cMPs. Figure 1A demonstrates the staining patterns of p-S6 and CD68 on cryosections prepared from MI hearts 3-day after LAD ligation. The majority of the cMPs in the infarct region was strongly positive for p-S6 staining. In contrast, there was very limited p-S6 positivity in the remote myocardium (Figure 1A). Interestingly, neutrophils completely lacked p-S6 (Figure 1B), suggesting that mTORC1 was not activated in these cells. We then calculated the area ratio of CD68+p-S6+ to p-S6+ using ImageJ. As demonstrated in Figure 1C, cMPs contributed to about 60% of the overall p-S6 staining. We next determined if mTORC1 was activated in myofibroblasts by staining tissue sections with αSMA and p-S6 antibodies. As demonstrated in S-Figure 2A, we did not detect p-S6 in αSMA positive cells. Representative HE staining of the infarct region where images were acquired (as in Figure 1A, 1B, S-Figure 2A) and quantified (Figure 1C) are presented in S-Figure 2B and 2C. To further confirm our observation, we evaluated the levels of p-P70S6K and p-S6 using protein samples prepared from cells enriched with cMPs. As demonstrated in Figure 1D, p-P70S6K and p-S6 in cMPs isolated from the infarcting tissue were higher than cMPs isolated from sham heart. Figure 1E was the summarized data generated using cMPs isolated from different MI cohorts. Taken together, our results strongly support that mTORC1 is robustly and persistently activated in the cMPs in the infarcting myocardium, suggesting an important role of this pathway in regulating the functions of these cells.
Digesting tissue debris activates mTORC1.
mTORC1 activation requires both nutrients and growth factors or cytokines such as TNFα [30–32]. While TNFα is abundantly produced in the infarcting myocardium, the source of nutrient is not known and unlikely from blood stream as sparse vasculature was observed in the infarct region within the first week of MI [33]. Here, we investigated if tissue debris could be a source of nutrients in mTORC1 activation. We observed that the mTORC1 activation in cMPs had a distinct spatial distribution. As demonstrated in S-Figure 3A and 3B, cMPs surrounding the unresolved dead tissue core had higher p-S6 levels. The images were taken from either the center (3A) or the border (3B) of the infarct, which were marked on the cross sections with HE staining. S-Figure 3C confirms that most p-S6+ cells were CD68+ cMPs. In contrast, CD68+ cMPs in the region where the necrotic tissue had been resolved demonstrated minimal p-S6 positivity (S-Figure 3D).
The evidence presented suggests that the cMPs in the infarct tissue could have turned the damaged tissue into nutrients that in turn activate the mTORC1 pathway. We hypothesize that the nutrients used to activate mTORC1 are derived intracellularly through the phagolysosomal degradation of tissue debris. To test this hypothesis, we designed an in vitro model in which we treated bone marrow derived macrophages (BMDM) with particles derived from the infarct area to simulate in vivo phagocytosis. To generate myocardium-derived particles (Myo-DP) suitable for phagocytosis, we modified a protocol used to enrich mitochondria from tissues[34]. We isolated the particles from myocardium and incubated them with BMDM for 0-, 10-, 20– 30-, and 40-minute. S-Figure 3E and 3F are images from 10- and 40-min treatment group, respectively. The engulfed mitochondrial particles were in red and endogenous mitochondria were labeled with anti-Tom20 (green). S-Figure 3G demonstrates increased p-S6 after the BMDMs were treated with Myo-DP for 6 hours. Myo-DP treatment also induced hyper-phosphorylation of 4EBP1 (S-Figure 3G). Summarized data for p-S6 from different experiments demonstrated a more than 3.5 fold increase in p-S6 by Myo-DP treatment (S-Figure 3H). As expected, the increased mTORC1 activation was sensitive to rapamycin treatment, a classic mTORC1 inhibitor (S-Figure 3G, 3H). Next, we tested our hypothesis using peritoneal macrophages. S-Figure 3I is the representative immunoblot of p-S6 level of peritoneal macrophages after they were treated with Myo-DP for 15 hours. Similar to BMDM, Myo-DP activated mTORC1 in peritoneal macrophages (S-Figure 3I). Results from different experiments were summarized in S-Figure 4J. We next tested if phagocytosis of indigestible beads stimulates mTORC1 activation. We incubated the BMDM with latex beads for 6 hours (S-Figure 3K) and determined the phosphorylation of P70S6K and S6. As demonstrated in S-Figure 3L, only treatment with biodegradable Myo-DP increased p-P70S6K and p-S6.
We next investigated if the cMPs in the infarcting myocardium had sufficient intracellular nutrients such as amino acids, a requirement for mTORC1 activation. Amino acids inside the lysosomal lumen activated mTORC1 by recruiting the complex to the lysosomal membrane via Ras-related small GTPase (Rag) [35, 36]. During this process, the staining pattern of mTOR changed from diffusely localized in the cytosol to punctuated structures [37]. S-Figure 4A demonstrates mTOR as the punctate-like structure that co-localize with the CD68+ cMPs in the infarct region. To determine if the mTOR complex was located to the lysosomal membrane, we first defined the lysosomal structure on the tissue section by labeling the lysosomal membrane protein MAC3 (S-Figure 4B). S-Figure 4B Inlet shows the lysosomes in circular structure (indicated by arrows). S-Figure 4C demonstrates the co-localization of mTOR with lysosomes and the visualization of the mTOR complex on the lysosomal membrane (indicated by arrows, the lysosome lumens were marked with *). Furthermore, we labeled isolated cMP with MAC3 and mTOR. As demonstrated in S-Figure 4D, mTOR was localized to the lysosome membrane (some were indicated by arrows). In summary, our data suggest that cMPs in the infarct region could transform the “waste”/dead tissues into signals that supports mTORC1 activation.
The ischemic environment determines the effector function of cMPs.
Myocardial infarction is also a systemic inflammatory disease[38]. One example is that IL1β released from the infarct tissue promoted leukocyte production from bone marrow and spleen that in turn intensified the inflammation-mediated cardiac damage [39]. Similarly, monocytes could become activated before they were recruited to the infarcting tissue. To test our hypothesis that the microenvironmental signals drive the cMPs to develop their effector function, we tracked the transcriptome activation by comparing the cMPs with monocytes isolated from spleen from both sham and MI cohorts. We first determined the composition of cell population prepared from the infarct tissue by labeling them with mononuclear phagocyte markers CD68 and CD11b. We counter-stained the same samples with universal structural proteins Actin and Tom20 to detect all intact cells. As demonstrated in Figure 2A and 2B, the majority of cells were positive for both CD68 and CD11b. Consistent with the role of CD11b as a myeloid integrin (αMβ2, CD11b/CD18), the focal adhesion structure was easily visualized when the cells were examined under higher magnifications (Figure 2C and the inlet). Interestingly, we noticed DAPI positive structures (some were indicated by arrows) that lacked mitochondria and actin. We demonstrated in S-Figure 5A with multiple views and higher magnification that these were fragmented and condensed nuclei. The percentages of CD68+ or CD11b+ cells to total number of cells was plotted in Figure 2D. In order to further confirm the cells prepared were enriched with cMPs, we determined the protein expression of cGAS (cyclic GAMP synthase), a DNA receptor most abundantly expressed in monocytes/macrophages, using samples prepared from normal myocardium tissue (sham), cMP-enriched cells isolated from infarct tissue, mouse cardiomyocytes and cardiac fibroblasts. As demonstrated in S-Figure 5B, cGAS was easily detectable in cells enriched with cMPs prepared from MI tissues of WT but not cGAS knockout mice. In contrast, no cGAS was detected from cardiomyocytes and fibroblasts (S-Figure 5C). The resident macrophages in the normal heart likely contributed to the cGAS signal observed at tissue level (sham). In addition, we stained the cells with CD45, another myeloid marker that was used to exclude fibroblasts, and the endothelial marker CD31. S-Figure 5D demonstrates the majority of the cells prepared were CD45+ and CD31- cells. Taken together, these data suggest that cells we prepared from the infarct tissue are highly enriched with cMPs.
Figure 2.


Immune cells were isolated from the infarct tissue 3-day or 7-day after LAD ligation. Cells were seeded on coverslips for two hours before they were fixed with 4%PFA for 10 minutes. RTqPCR was performed using RNAs purified from spleen monocytes (sMo) and cMPs isolated from infarct tissue. A and B. Cells isolated from infarcting myocardium 7-day post MI were labeled with CD68 or CD11b and counter-stained with Tom20 or Actin. C. The focal adhesion structures formed by myeloid integrin CD11b under a higher magnification. D. Percentages of cMPs (CD68+Tom20+or CD11b+Actin+) to total cells (Tom20+ n=211 or Actin+ n=238). Cells were isolated from 4 MI hearts. E. Transcripts of inflammatory cytokines IL1β, TNFα, and IL6 from sMo and cMPs were determined using RT-qPCR. F. Expression of chemokine MCP-1, CCL3, and CCL4 from sMo and cMP. G. Expression of Arg1 and iNOS from the same set of samples as in F. H. The ratio of Arg1 to iNOS. I. Scavenger receptor CD14 expression in sMo and cMPs from sham and MI hearts. J. MMP2 and MMP9 expression from cMP samples as indicated. For RT-qPCR performed in E to J, n=3 to 4 spleens or 10 to 12 MI hearts (some data points were from pooled MI tissues). * p<0.05 ** p<0.01 *** p<0.001.
We next examined the expression of inflammatory cytokines, chemokines, scavenger receptors involved in phagocytosis, and matrix metalloproteinases (MMP) that are essential for matrix remodeling - aspects reflecting the effector function spectrum of cMP. We also determined the expression of arginase 1 (Arg1) and iNOS, which have been extensively used in in vitro macrophage polarization experiments[40]. Figure 2E demonstrates that the expression of IL1β, TNFα and IL6 were similar in monocytes isolated from spleens (spleen monocytes: sMo) regardless of the type of surgical procedure (sham or LAD ligation). In contrast, cytokine expression was increased in the cMPs isolated from the infarct tissue. Monocyte chemoattractant protein 1 (MCP-1, also known as CCL2) is a potent chemokine that attracts blood monocytes. The main activity of Chemokine (C-C motif) ligand 3 (CCL3) and CCL4 is to recruit and activate polymorph nuclear leukocytes [41, 42]. As demonstrated in Figure 2F, the robust increase in MCP1, CCL3, and CCL4 all occurred in cMPs. One commonly used standard to separate inflammatory from alternatively activated macrophages in vitro is the ratio of Arg1 to iNOS. While inflammatory macrophages express high levels of iNOS therefor lower ratio of Arg1/iNOS, the alternatively activated macrophages (which is anti-inflammatory) is the opposite in expression levels of these two markers[40]. We examined both markers in our ex vivo experimental setting. Figure 2G demonstrates the increase of Arg1 in cMPs that reached the highest level 3-day after MI. Interestingly, iNOS expression not only had much less dynamic changes comparing to Arg1 but it was decreased in cMPs. The ratio of Arg1/iNOS peaked at 3-day post MI and decreased at 1 week time point (Figure 2H). As Arg1 is considered a prototypical marker for alternatively activated/anti-inflammatory macrophages and cMPs in the first three days after MI procedure are considered inflammatory cells, the data suggest cMPs in the heart are drastically different than macrophages polarized in in vitro conditions. We next examined the changes of scavenger receptor CD14 that was involved in phagocytosis [43]. Interestingly, we observed a small but significant increase in CD14 in spleen monocytes isolated from MI mice (Figure 2I). Similar to the changes in cytokines and chemokines, the increase in CD14 predominantly occurred in cMPs (Figure 2I). Consistent with a prior observation that cMPs at latter half of the first week post MI had enhanced phagocytotic activity [3], we observed a higher CD14 level 7-day post MI. Matrix metallopeptidases (MMP) are important in remodeling of the provisional matrix formed after MI [44]. We observed that both MMP2 and MMP9 were increased in cMPs but with different dynamics – MMP2 had more prominent changes and peaked at 7-day while MMP9 started to decrease 3-day after the MI procedure (Figure 2J). These data overwhelmingly support that the cardiac ischemic environment drives the acquisition of effector functions critical to the inflammatory injury and the repair process.
mTORC1 inhibition by rapamycin protects the heart from ischemic injury.
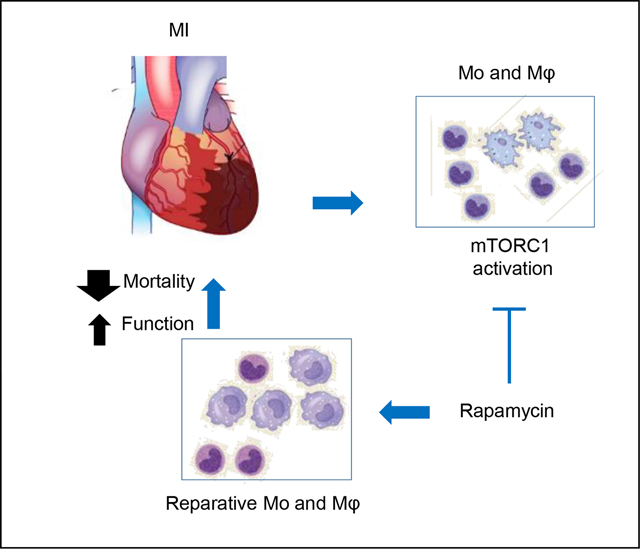
Our data presented in Figure 1 demonstrate a robust mTORC1 activation in the cMPs. We next determined the impact of mTORC1 inhibition on ischemia-induced cardiac remodeling and mortality. Twenty four hours after the LAD ligation, animal cohorts were randomly divided into groups receiving either DMSO or rapamycin at 0.25mg/kg/day for a total of 6 days. The experimental scheme was outlined in Figure 3A. Figure 3B demonstrated a much lower p-S6 level after rapamycin treatment, indicating effective mTORC1 inhibition. Cross sections stained with Trichrome C (TC) or Picrosirius red (PSR) one week after MI are presented in Figure 3C. Using ImageJ, the percentage of infarct area to LV was estimated at 35% and 26% for DMSO and rapamycin group, respectively. The parameters of gravimetric analysis, including heart weight to body weight or tibia length ratio, were also reduced in rapamycin-treated animals (Figure 3D). Acute myocardial ischemia immediately induces hemodynamic stress to the remaining functional myocardium. The severity of the ischemia-induced injury is reflected by the hemodynamic stress that increases the hypertrophy markers in the remote myocardium. Figure 3E demonstrated the profiles of ANF, BNP, αSMA, and βMHC from the remote myocardium. We observed a striking decrease in ANF level after mTORC1 was inhibited. Similarly, the BNP level was also reduced in rapamycin-treated group. We did not observe changes in αSMA and βMHC between DMSO and rapamycin treated animals. Interestingly, αSMA level in the infarct region, which is often used as an indicator for infarct size, was reduced by rapamycin treatment (Figure 3F). Consistently, the infarct size, estimated by the percentage of infarct to left ventricle (LV), was reduced by rapamycin treatment (Figure 3G). We next compared the mortality in the first week of MI, a time window when more than 95% of the mortality occurs in experimental MI model. Figure 3H demonstrates improved survival in the mouse cohort treated with rapamycin. Importantly, we compared the cardiac function of DMSO- and rapamycin-treated groups 4 weeks after MI. Even though the treatment was stopped after the first week (a total of 6 injections), fraction shortening (FS) was improved in rapamycin group 4 weeks after MI (Figure 3I). Consistently, left ventricle chamber dimensions were smaller in rapamycin group (Figure 3I). These data suggest that mTORC1 inhibition in the acute phase of MI has sustaining beneficial effect on cardiac function and remodeling in addition to the mortality reduction. In aggregate, our results strongly support a critical role of mTORC1 activation in mediating ischemia-induced remodeling and mTORC1 inhibition via rapamycin at 0.25mg/kg/day in the first week of MI provides cardiac protection.
Figure 3.

Myocardial infarction was generated by LAD ligation. Twenty four hours after the procedure, mice were randomly divided into groups that received subcutaneous injection of DMSO or rapamycin at 0.25mg/kg/day. Hearts were harvested either 3-day or 7-day post MI procedure, resulting a total of 2-day or 6-day injection respectively. A. A diagram of the experimental design. B. Protein samples from sham or infarct tissue (7-day post MI) were subjected to immunoblot analysis to detect p-S6 to assess the effectiveness of rapamycin. n=2 hearts from each group. C. Cross sections from 1-week MI hearts were stained with Trichrome C (TC) or Picrosirius red (PSR). Infarct area is estimated using ImageJ software. The percentages of IA (ischemic area) to LV (left ventricle) were calculated. D. Gravimetric data reflecting cardiac remodeling 1-week after LAD ligation. Both heart weight to body weight and heart weight to tibia length ratios were included. n=3 to 7 hearts. E. Expression of cardiac hypertrophy markers in the remote myocardium from mice treated with DMSO or rapamycin. n=3 to 4 hearts. F. αSMA protein levels in the infarct tissues from DMSO- or rapamycin-treated group. n=2 hearts from each group. G. The percentage of infarct to left ventricle was calculated using the tissue wet weight. The infarct tissues were meticulously separated under a dissection microscope. n=10 to 22 MI hearts. H. Survival curves of mouse cohorts receiving either DMSO or rapamycin treatment. n=19 to 33 mice. I. Cardiac function (FS), LV dimensions at the end of diastole (LVID;d) and systole (LVID;s), and heart rate (HR) were measured 4 weeks after MI. Mice were treated with either DMSO or rapamycin for 6 days starting at the second day after LAD ligation. n=4. * p<0.05 ** p<0.01 *** p<0.001.
The heterogeneity of cMPs in ischemic environment and the role of mTORC1 in promoting the accumulation of mature macrophages.
The effector functions are gained/amplified when monocytes differentiate towards macrophages in the ischemic microenvironment. This is an important process yet our understanding of it is quite limited. Interestingly, it was reported that the mean resident time of cMPs in the infarcting myocardium was around 20 hours [45], making differentiation a very challenging task to accomplish in such short period of time. Separating monocytes from macrophages via flow cytometry is inherently difficult because both cell types share most of the cell surface markers. On the other hand, the morphological criteria that distinguishing macrophages from monocytes are well-established and include: 1) macrophages are larger (greater than 20 μm); 2) round or oval nuclei; 3) nuclei to cytoplasm ratio less than 1; 4) sophisticated organelles especially abundant lysosomes. Here, we performed morphology sorting to characterize the subpopulations of cMPs 3-day and 7-day after MI. The single cell solution yielded from digesting the infarct tissue was seeded onto coverslips for 2 hours before fixation and immune fluorescent study.
In Figure 4A, we demonstrate the morphological spectrum of cMPs in four sub-groups, cMP1, cMP2, cMP3, and cMP4, based on the criteria discussed above. Lysosome and mitochondria, labeled by MAC3 and Tom20, respectively, were used to gauge the complexity of intracellular organelles. cMP1 resembled the circulating monocytes and represented cMP recently recruited from the circulation. The cMP2 represented a population that had differentiated but had not yet evolved into mature macrophages. cMP3 and cMP4 represented mature macrophages and cMP4 possessed more mitochondria comparing to cMP3. The vacuoles (indicated by arrows) were typical structures seen in mononuclear phagocytes and represented phagosomes. To correlate the morphology with function, we stained these cells with CD206, a bona fide marker for anti-inflammatory/reparative macrophages that promoted myocardial repair [27] and accumulated in the second half of the week post MI [2, 4, 26, 27]. As demonstrated in Figure 4B, CD206 was barely detectable in cMP1 but became the strongest in cMP4, suggesting a correlation of CD206 with the differentiation of macrophages. Figure 4C summarized the percentage of cMP1 to cMP4 in the infarct tissue. The cMPs isolated from the infarcting tissue 3-day after MI were predominantly cMP1s and cMP2s that accounted for about 80% of total cMPs. One week after MI, the percentage of cMP1 dropped from 42% (at 3-day post MI) to 27%, and the percentage of cMP2 on the other hand increased from 38% to 45%. However, the cMP1 and cMP2 subpopulations still constituted the majority of the cMPs at the 1 week time point. Inhibiting mTORC1 by rapamycin did not alter this dynamic that was the majority of cMPs were from cMP1 and cMP2 at either time points (Figure 4C). However, the total percentage of cMP1s and cMP2s was lower whereas the cMP3 population was larger in rapamycin group (Figure 4C). To connect mTORC1 activity and the morphology changes to functionality, we next co-labeled isolated cMPs with CD206 and p-S6 antibodies. We demonstrate in Figure 4D that mature macrophages (cMP3 and cMP4) had lower mTORC1 activity (p-S6) but much higher level of CD206. Consistent with the finding that mTORC1 inhibition led to a larger and the more mature cMP3 population (Figure 4C), we observed more CD206+ cMPs in rapamycin treated group (Figure 4E). Next, we examined the MerTK on tissue sections obtained from either DSMO or rapamycin treated group. MerTK is one of the markers that distinctly and universally associated with mature tissue macrophages that mediated the phagocytosis of apoptotic cells, a process known as efferocytosis [46]. MerTK was expressed at a higher level in alternatively activated macrophages [47] that were cells similar to the pro-resolving/pro-reparative macrophages. Not surprisingly, we did not observe strong MerTK signal at 3-day post MI time point (not shown). One week after MI, the MerTK signal co-localized with CD68 (Figure 4F), and mTORC1 inhibition expanded the macrophage population expressing higher level of MerTK (Figure 4F). Figure 4G demonstrates the summarized data of experiments performed as in Figure 4F. In summary, our data demonstrate that the cMP in the infarcting myocardium is a morphologically and functional heterogeneous population and the majority of these cells are not mature macrophages, suggesting limited differentiate in the first week of acute ischemia. Inhibition of mTORC1 by rapamycin expands the subpopulation of mature macrophage. In addition, our data reveal that CD206 is predominantly expressed in mature macrophage (cMP3 and cMP4) and negatively correlates with mTORC1 activation. These evidence suggest mTORC1 activation diminishes the accumulation of mature macrophages. As the primary function of mTOR/mTORC1 is pro-growth, proliferation, and survival, the result implies that mTORC1 activation could have interfered the differentiation from monocyte to macrophage. Additional studies with fate mapping and lineage tracing are needed to detailed underlying mechanism.
Figure 4.


Myocardial infarction was generated via LAD ligation. Twenty four hours after the procedure, mice are randomly divided into groups that receive subcutaneous injection of DMSO or rapamycin at 0.25mg/kg/day. Cardiac immune cells were isolated from infarct tissue 3-day or 7-day after MI and prepared for ICC. A. Morphological spectrum of cMPs. Cells were labeled with lysosome marker MAC3 and mitochondria protein Tom20. Representative cMPs from the infarct tissue were classified into cMP1, cMP2, cMP3, and cMP4 based on criteria that separate monocytes from mature macrophages. B. cMPs with morphological criteria of cMP1, cMP2, cMP3, and cMP4 were labeled with CD206. C. Morphology sorting of cMPs isolated from the infarct tissues 3-day and 7-day after LAD ligation and treated with or without DMSO or rapamycin. The percentages of the cMP1, cMP2, cMP3, and cMP4 populations and total cells examined were depicted. D and E. cMPs from DMSO (D) or rapamycin (E) treated hearts were labeled with p-S6 and CD206 antibodies. F. Cryosections of the hearts were labeled with CD68 and MerTK. G. Quantification of MerTK+ cells. n=8 MI hearts for A,B,C,D,E. n=3 MI hearts for F and G. *p<0.05
mTORC1 inhibition shifts the landscape of cMPs to reparative cells.
We performed flow cytometry to determine if mTORC1 regulated the transition from inflammatory to reparative cMPs in acute ischemia. Figure 5A is the scatter plot that demonstrates the cMPs (CD45+CD11b+Ly6Glow) population further separated by CD64 and CD206, markers for inflammatory [48, 49] and reparative cMPs, respectively. mTORC1 inhibition by rapamycin shifted the majority of cMPs to CD206high CD64low cells (72.3% in rapamycin group and 14.1% in DMSO group). We further characterized the cMPs using Ly6C and F4/80. Ly6C was a classical inflammatory marker [1, 2]. Figure 5B revealed a close positive correlation between F4/80 with CD206, suggesting F4/80 can be used as a marker for reparative cMPs. Figure 5C demonstrates that mTORC1 inhibition with rapamycin shifted the cMPs towards F4/80highLy6Clow cells (11.5% vs 52% in DMSO and rapamycin group, respectively). These data strongly support mTORC1 inhibition promoted the transition from inflammatory cells to reparative cMPs. Figure 5D depicts the comprehensive comparison of all cell surface makers used to characterize the cMPs isolated 1 week after the MI and highlighted the landscape shift of cMPs when mTORC1 was inhibited. Figure 5E depicts the heat map of the median fluorescent intensity (MFI) that quantified the decrease in the inflammatory marker (CCR2, CD64, Ly6C) and increase in reparative cell markers (CD206, F4/80) and the marker that was indicative of maturation of cMPs (MHCII) by rapamycin treatment. Next, we evaluated if mTORC1 inhibition by rapamycin affected the cytokine expression from cMPs. Intriguingly, we observed higher levels of inflammatory cytokines (TNFα, IL1β, and IL6) in cMPs isolated from rapamycin treated group (Figure 5F and 5G). These data suggests mTORC1 inhibition may promote a group of cMPs that phenotypically resembles a subtype of alternatively activated macrophage M2b [50]. Interestingly, recent studies suggested the M2b subtype macrophage protected the heart from ischemic or I/R injury [51, 52].
Figure 5.


C57BL/6J mice were subjected to sham or LAD ligation surgeries. Twenty four hours after the MI procedure, mice were randomly divided into groups that received subcutaneous injection of vehicle or rapamycin at 0.25mg/kg/day. Cardiac immune cells were isolated from pooled infarct tissues from three mice (n=3 MI hearts from each group) 7-day after MI for flow cytometry. Cardiac tissues from the same cohorts were subjected to immunoblot analysis to detect the phosphorylated and total Akt. A. The immune cells isolated from DMSO- or rapamycin-treated hearts were labeled with CD45, CD11b, CCR2, Ly6G, CD206, CD64, F4/80, Ly6C, and MHCII. After forward and side scatter selection, cMPs were gated as CD45+CD11b+Ly6Glow cells. The selected cMP population was gated further with CD206 and CD64 and is presented individually as DMSO or rapamycin group or with the two group over-layed. The percentage of CD206highCD64low or CD206lowCD64high were indicated. B. Positive correlation F4/80 with CD206. C. The CD45+CD11b+Ly6Glow population was further gated based on Ly6C and F4/80. Percentage of Ly6ClowF4/80high, Ly6CmidF4/80mid, and Ly6ChighF4/80mid were 11.5%, 33.2%, and 9.2% for DMSO-treated hearts and 52%, 12.8%, and 0.4% for rapamycin-treated group. D. The landscape changes of cMPs. E. Heat map of the median fluorescent intensity (MFI) of the inflammatory cell surface markers (CCR2, Ly6C, CD64), antiinflammatory markers (CD206, F4/80), macrophage maturation marker (MHCII) and myeloid marker (CD45) of cMPs from DMSO- or rapamycin-treated hearts. F. Expression of IL1β or TNFα from cMPs isolated from DMSO- or rapamycin-treated MI hearts at indicated time points. n=6 to 8 MI hearts (some data points are from pooled MI tissue). G. IL6 expression from cMPs isolated from DMSO- or rapamycin-treated hearts 7-day after MI. H. Protein samples extracted from hearts 7-day after MI were subjected to immunoblot analysis. Individual lanes represent different hearts. The activation of Akt was determined by the levels of p-AktSer473 and p-AktThr308. I and J. Summarized data from experiments performed as in H. n=4–6 MI hearts. * p<0.05 ** p<0.01.
Prior studies suggested mTORC2 activity was essential for the development of alternative/anti-inflammatory macrophages [19, 20]. Chronic rapamycin treatment at 2mg/kg for 14 to 28 days inhibited mTORC2 that led to glucose intolerance and insulin resistance [29]. In addition, rapamycin treatment after MI at 2mg/kg to 10mg/kg suppressed mTORC2 activation [24]. We determined next if the rapamycin used in the current study had any impact on mTORC2 activity. We demonstrates in Figure 5H that there were no differences in p-Akt473 or p-Akt308 from remote or infarct region in DMSO- or rapamycin-treated hearts, suggesting rapamycin treatment in the current study selectively inhibited mTORC1 and had no impact on mTORC2. The summarized result for pAkt473 or pAkt308 is shown in Figure 5I and Figure 5J, respectively. In summary, our data support that mTORC1 promotes inflammatory cMPs in acute ischemia. Inhibition of mTORC1 by rapamycin shifts the landscape of the cMPs into a cell population that promotes the resolution of inflammation and myocardial repair.
DISCUSSION
The critical role of the monocytes and macrophages in repairing the damaged myocardium from ischemic insult is well established by prior studies [1–3, 8, 53]. However, we still lack effective ways to reduce inflammatory injury by targeting these cells. More work is needed to define how the MPs take “cues” from the microenvironment and tune their effector functions. In the current study, we present evidence to support a critical role of mTORC1 in regulating the function of the mononuclear phagocytes in the infarcting myocardium. First, we demonstrate that mTORC1 was robustly activated in the cMPs in the infarcting myocardium – an environment seemingly not compatible for mTORC1 activation on the surface. Selective Inhibition of mTORC1 led to phenotypical transition to anti-inflammatory and reparative cMPs. As a result, there was a greater than 80% reduction in mortality and cardiac remodeling was mitigated. As the mTORC1 is a master regulator that integrates signals from the environment and serves as nexus point for cellular signals that controls cellular function, we identify that mTORC1 plays this critical role in the cMP population in the ischemic environment. To our best knowledge, this study is the first to report such observation.
Activation of mTORC1 in the setting of ischemia.
mTORC1 is a tightly regulated process that requires the presence of both growth factors/cytokines and abundant nutrients. Activation signals must be „„licensed‟‟ by nutrient availability. While TNFα is abundantly produced in the ischemic tissue and was known to activate mTOR/mTORC1[30–32], the infarcting myocardium, on the other hand, is a hypoxic environment with low energy and lack of conventional source of nutrients, all of which suppress mTORC1 activation (reviewed in[11]). Contrary to what was perceived, mTORC1 was robustly activated in the cMPs in the infarct tissue. The translocation of mTOR to the lysosome membrane was consistent with an adequate intracellular amino acid level required for mTROC1 activation [37]. Potentially, nutrients could be delivered from newly formed vessels. However, this may not be feasible because the infarct center lacked vasculature in the first week of MI [33]. Our data suggest cMPs could generate the required nutrients by digesting the tissue debris. The concept of turning dead tissue/cells into signals that regulate cellular function is supported by evidence presented in a recent study, in which fatty acid generated by efferocytosis promoted anti-inflammatory macrophage polarization in MI by fueling mitochondria respiration and activating NAD+-dependent signaling [54]. However, comprehensive analysis including metabolic studies are needed to provide detailed mechanisms that is beyond the scope of the current study. Nonetheless, our findings shed light on how the dead tissue could be used as a way to critically regulate cMP function and could guide future studies to dissect molecular events.
mTORC1 in cardiac MP differentiation and phenotype transition in the ischemic environment.
1. Monocytes to macrophage differentiation.
Evidence presented in this study demonstrate that the mononuclear phagocytes accumulated in the infarct tissue were heterogeneous cells with vastly different morphology. Morphology sorting using well established criteria for macrophages plus the complexity of intracellular organelles revealed that the majority of the cMP population did not differentiate into mature macrophages in the first week of MI. The advantage in this approach is that it does not rely on the cell surface markers that are mostly shared between monocytes and macrophages, especially in the setting of MI when peripheral monocytes are constantly recruited into the infarct tissue and are precursor cells of cMPs. By strict criteria, the majority of these cells are not macrophages but a heterogeneous population at different stages of differentiation. In fact, although there was evidence of differentiation at 1 week after the MI procedure, the time when the anti-inflammatory/reparative “macrophages” dominate, the majority of cells were still morphologically closer to monocytes. This finding is consistent with a prior study performed on other organs that monocytes recruited to acute inflammation site rarely differentiated beyond a phase similar to the cMP2 defined in the current study [55]. Our result here suggest that perhaps a different term should be used to describe the heterogeneous cMP population. In contrast to the cells that remained in the early stage of differentiation, we observed a population of cMPs that have mature macrophage morphology (cMP3 and cMP4) and were strongly positive for CD206 (CD206+++), a bona fide marker for reparative cMPs. Because CD206 cMPs were derived from recruited CCR2+ monocytes[8], our finding suggests certain cMPs, although at a small percentage, might be able to differentiate into mature macrophages within a limited time (3-day after MI). Importantly, mTORC1 inhibition expanded this population of cMPs, suggesting interventions targeting this specific group of cMPs could reduce inflammation-induced injury and promote repair. As the primary function of mTOR/mTORC1 is to promote growth, cell survival and proliferation, it is counterintuitive to consider that inhibiting mTORC1 by rapamycin could promote proliferation and survival that lead to such accumulation. However, the limitation of our current study is that these processes cannot be rule out. Additional studies that characterize the dynamics of the cMPs, lineage tracing, and fate mapping are needed to delineate what dictates the differentiation and if a specific set of monocytes is “destined” to become the CD206+++ macrophages in the ischemic environment.
2. The phenotype transition of cardiac mononuclear phagocytes in acute ischemia.
mTORC1 activation in in vivo inflammatory models such as arthritis and nephritis promoted or inhibited inflammation, respectively[17, 18], suggesting a complex and context-dependent role of mTORC1 in macrophage effector functions (reviewed in[56]). In the myocardial ischemia model, our data suggested that the cMPs were turned into reparative cells by mTORC1 inhibition. Intriguingly, cMPs isolated from rapamycin treated MI hearts had increased expression of inflammatory cytokines (IL6, TNFα and IL1β). Although higher inflammatory cytokine is often used as a barometer of inflammatory macrophages, we detected no difference in IL6, TNFα and IL1β from cMPs isolated 3-day after MI than those from cMPs purified from 7-day MI tissue, the two time points with peak inflammatory (3-day) and reparative/anti-inflammatory (7-day) cells. Furthermore, a prior study demonstrated that, although TNFα and IL1β (specified as inflammatory macrophage markers by the investigators), was down regulated after neutrophil depletion in MI, the ischemia-induced remodeling was worse [57]. Together, these evidence suggest that the inflammatory cytokines by themselves may not accurately reflect the complex phenotype of cMPs. In addition, this “paradoxical correlation” of anti-inflammatory cMPs with relatively high inflammatory cytokines is intriguing and suggests the cMPs in the 7-day infarcting tissue resemble M2b (alternatively activate macrophage (M2) type b). mTORC1 inhibition enhanced the M2b-type like phenotype. The M2b macrophages were considered immunity-regulating and were induced by IL-1 receptor agonists, LPS and immune complexes [58]. Prior evidence demonstrated mTORC1 inhibition promoted NF-κB p65-RelA-mediated inflammatory cytokine transcription [59, 60]. In addition, inhibition of mTORC1 favored the translation of abundant transcripts that included IL1β, TNFα, and IL6 through suppressing the eIF4E-initiated CAP-dependent translation [61]. It is therefore plausible that the resulting increase in IL1β in the infarct tissue by rapamycin treatment could promote the development of M2b-type cMPs. Interestingly, injecting M2b macrophages developed in vitro by LPS and immune complex stimulation attenuated I/R injury and decreased infarct size [52].
In addition to the possible mechanism that IL1β could promote cMPs transition to reparative cells and improve repair, it is also possible that effector functions other than producing inflammatory cytokines may be more detrimental to the heart after MI. One such candidate could be phagocytosis of viable cardiomyocytes especially in the border region[62]. Phagocytosis is an effector function that was upregulated as the cMPs evolved within the infarct tissue [3]. Interestingly, phagocytosis stimulated by TLR4 ligation required de novo lipid synthesis mediated by sterol regulatory element binding proteins downstream of mTORC1 activation[63], suggesting mTORC1 may also facilitate the phagocytosis of live cardiomyocytes. Future work on other cMP effector functions such as phagocytosis as mechanisms of inflammatory injury to the heart is needed.
Inhibition of mTORC1 by rapamycin in myocardial infarction.
We demonstrated that mTORC1 inhibition by rapamycin starting from the second day after MI procedure reduced mortality by more than 80% and mitigated remodeling. Our findings extended prior observations that rapamycin protected the heart in the chronic stage of MI (4 to 5 weeks) [24, 25]. Interestingly, similar treatment duration (1 week post MI procedure) did not protect the heart compared to the long term treatment groups [24]. The difference between our study and the prior report could be the different dosage of rapamycin administered, which were 0.25mg/kg/day subcutaneous injection versus 2 to 10mg/kg/d via intraperitoneal injection, respectively. Although rapamycin selectively inhibits mTORC1, pro-longed treatment and higher concentrations of rapamycin suppressed mTORC2. mTOR2 complex was vital in promoting cell survival through phosphorylating Akt473 and suppressing MST1 kinase [64, 65]. Also importantly, inhibition of mTORC2 prevented that activation of alternatively activated macrophages [19, 20], the macrophage phenotype similar to the reparative cardiac macrophages after MI. While mTORC2 activity was completely suppressed when rapamycin was given at 2 or 10mg/kg/d after MI for 1 week [24], we observed minimal changes in mTORC2 activity when the rapamycin was reduced to 0.25mg/kg/d. Rapamycin does not directly bind mTOR, instead, it binds FK506 binding protein (FKBP) and forms a complex specifically interacts with the FRB (FK506-rapamycin binding) domain of mTOR, allosterically inhibiting the kinase activity[66]. Interestingly, phosphatidic acid (PA), a type of phospholipid produced by cells naturally, bound the same region where the FK506-rapamycin interacted with mTOR. PA binding to mTOR promoted both mTORC1 and 2 activation [67, 68]. Based on these evidence, a model proposed recently suggests a tighter binding between mTORC2 and PA rendered mTORC2 less sensitive to rapamycin [28]. The differential inhibitory action of rapamycin at different concentrations on mTORC1 and mTORC2 could be from the interaction dynamics between FK506-rapamycin, phosphatidic acid, and mTOR complexes. Our study provides evidence that supports a lower, non-mTORC2 suppressing rapamycin dose is profoundly beneficial and protects the heart from acute ischemia. This could serve as an important insight to harness the benefit of mTORC1 inhibition and avoid the off target mTORC2 inhibition that is detrimental.
In summary, we presents evidence that supports a robust activation of mTORC1 in the mononuclear phagocytes in the infarcting myocardium. Inhibiting mTORC1 with rapamycin leads to a shift of cMP population towards reparative cells that contribute at least in part to the improved myocardial repair. Importantly, rapamycin treatment reduced mortality by more than 80% and improved cardiac function. This is achieved using rapamycin at a much lower dose that selectively inhibits mTORC1 but not mTORC2. Our data uncovers mTORC1 as a crucial pathway that links the microenvironment to cellular response, a complex and crucial factor that determines the cMP function but yet is the least understood. More studies are necessary to dissect the role of mTORC1 in the metabolism, survival, differentiation, and effector function regulation of cMPs that could lead to novel treatment of inflammatory injury.
Supplementary Material
Highlights.
mTORC1 is robustly activated in cardiac monocytes and macrophages in the infarcting myocardium.
Selective inhibition of mTORC1 by low dose rapamycin remarkably reduces mortality and improves cardiac function after myocardial infarction.
Cardiac protection by mTORC1 inhibition is achieved via regulating the effector functions of monocytes and macrophages in the ischemic niche.
Acknowledgments
SOURCES OF FUNDING
This work was supported by grants from the National Institutes of Health (R01 HL145298 to DC, K08 HL116801 to DC, VA Merit 5 I01 BX004562 to DC).
Footnotes
DISCLOSURES
None.
The authors have declared that no conflict of interest exists
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ, The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions, J Exp Med 204(12) (2007) 3037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer-Crosbie M, Hedrick CC, Libby P, Nahrendorf M, Weissleder R, Swirski FK, Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium, Circ Res 114(10) (2014) 1611–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mouton AJ, DeLeon-Pennell KY, Rivera Gonzalez OJ, Flynn ER, Freeman TC, Saucerman JJ, Garrett MR, Ma Y, Harmancey R, Lindsey ML, Mapping macrophage polarization over the myocardial infarction time continuum, Basic Res Cardiol 113(4) (2018) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, Yamamoto T, Takeshima A, Shinmura K, Shen W, Fukuda K, Sano M, Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction, J Mol Cell Cardiol 62 (2013) 24–35. [DOI] [PubMed] [Google Scholar]
- [5].Watanabe S, Alexander M, Misharin AV, Budinger GRS, The role of macrophages in the resolution of inflammation, J Clin Invest 129(7) (2019) 2619–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Desalegn G, Pabst O, Inflammation triggers immediate rather than progressive changes in monocyte differentiation in the small intestine, Nat Commun 10(1) (2019) 3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dal-Secco D, Wang J, Zeng Z, Kolaczkowska E, Wong CH, Petri B, Ransohoff RM, Charo IF, Jenne CN, Kubes P, A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury, J Exp Med 212(4) (2015) 447–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, Mohan J, Ivey B, Hsiao HM, Weinheimer C, Kovacs A, Epelman S, Artyomov M, Kreisel D, Lavine KJ, Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury, Circ Res 124(2) (2019) 263–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, Chen J, Kantores C, Hosseinzadeh S, Aronoff L, Wong A, Zaman R, Barbu I, Besla R, Lavine KJ, Razani B, Ginhoux F, Husain M, Cybulsky MI, Robbins CS, Epelman S, Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction, Nat Immunol 20(1) (2019) 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, Ho JW, Nordon RE, Harvey RP, Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury, Elife 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Saxton RA, Sabatini DM, mTOR Signaling in Growth, Metabolism, and Disease, Cell 169(2) (2017) 361–371. [DOI] [PubMed] [Google Scholar]
- [12].Holz MK, Ballif BA, Gygi SP, Blenis J, mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events, Cell 123(4) (2005) 569–80. [DOI] [PubMed] [Google Scholar]
- [13].Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD, mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle, Science 351(6274) (2016) 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ben-Sahra I, Howell JJ, Asara JM, Manning BD, Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1, Science 339(6125) (2013) 1323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ecker J, Liebisch G, Englmaier M, Grandl M, Robenek H, Schmitz G, Induction of fatty acid synthesis is a key requirement for phagocytic differentiation of human monocytes, Proc Natl Acad Sci U S A 107(17) (2010) 7817–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang X, Li M, Gao Y, Gao J, Yang W, Liang H, Ji Q, Li Y, Liu H, Huang J, Cheng T, Yuan W, Rheb1-mTORC1 maintains macrophage differentiation and phagocytosis in mice, Exp Cell Res 344(2) (2016) 219–28. [DOI] [PubMed] [Google Scholar]
- [17].Kirsch AH, Riegelbauer V, Tagwerker A, Rudnicki M, Rosenkranz AR, Eller K, The mTOR-inhibitor rapamycin mediates proteinuria in nephrotoxic serum nephritis by activating the innate immune response, Am J Physiol Renal Physiol 303(4) (2012) F569–75. [DOI] [PubMed] [Google Scholar]
- [18].Cejka D, Hayer S, Niederreiter B, Sieghart W, Fuereder T, Zwerina J, Schett G, Mammalian target of rapamycin signaling is crucial for joint destruction in experimental arthritis and is activated in osteoclasts from patients with rheumatoid arthritis, Arthritis Rheum 62(8) (2010) 2294–302. [DOI] [PubMed] [Google Scholar]
- [19].Hallowell RW, Collins SL, Craig JM, Zhang Y, Oh M, Illei PB, Chan-Li Y, Vigeland CL, Mitzner W, Scott AL, Powell JD, Horton MR, mTORC2 signalling regulates M2 macrophage differentiation in response to helminth infection and adaptive thermogenesis, Nat Commun 8 (2017) 14208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, Pearce EJ, Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation, Immunity 45(4) (2016) 817–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Haloul M, Oliveira ERA, Kader M, Wells JZ, Tominello TR, El Andaloussi A, Yates CC, Ismail N, mTORC1-mediated polarization of M1 macrophages and their accumulation in the liver correlate with immunopathology in fatal ehrlichiosis, Sci Rep 9(1) (2019) 14050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Saxton RA, Sabatini DM, mTOR Signaling in Growth, Metabolism, and Disease, Cell 168(6) (2017) 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sciarretta S, Forte M, Frati G, Sadoshima J, New Insights Into the Role of mTOR Signaling in the Cardiovascular System, Circ Res 122(3) (2018) 489–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Di R, Wu X, Chang Z, Zhao X, Feng Q, Lu S, Luan Q, Hemmings BA, Li X, Yang Z, S6K inhibition renders cardiac protection against myocardial infarction through PDK1 phosphorylation of Akt, Biochem J 441(1) (2012) 199–207. [DOI] [PubMed] [Google Scholar]
- [25].Buss SJ, Muenz S, Riffel JH, Malekar P, Hagenmueller M, Weiss CS, Bea F, Bekeredjian R, Schinke-Braun M, Izumo S, Katus HA, Hardt SE, Beneficial effects of Mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction, J Am Coll Cardiol 54(25) (2009) 2435–46. [DOI] [PubMed] [Google Scholar]
- [26].Cao DJ, Schiattarella GG, Villalobos E, Jiang N, May HI, Li T, Chen ZJ, Gillette TG, Hill JA, Cytosolic DNA Sensing Promotes Macrophage Transformation and Governs Myocardial Ischemic Injury, Circulation 137(24) (2018) 2613–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shiraishi M, Shintani Y, Shintani Y, Ishida H, Saba R, Yamaguchi A, Adachi H, Yashiro K, Suzuki K, Alternatively activated macrophages determine repair of the infarcted adult murine heart, J Clin Invest 126(6) (2016) 2151–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mukhopadhyay S, Frias MA, Chatterjee A, Yellen P, Foster DA, The Enigma of Rapamycin Dosage, Mol Cancer Ther 15(3) (2016) 347–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, Guertin DA, Sabatini DM, Baur JA, Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity, Science 335(6076) (2012) 1638–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Glantschnig H, Fisher JE, Wesolowski G, Rodan GA, Reszka AA, M-CSF, TNFalpha and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase, Cell Death Differ 10(10) (2003) 1165–77. [DOI] [PubMed] [Google Scholar]
- [31].Zhou JX, Fan LX, Li X, Calvet JP, Li X, TNFalpha signaling regulates cystic epithelial cell proliferation through Akt/mTOR and ERK/MAPK/Cdk2 mediated Id2 signaling, PLoS One 10(6) (2015) e0131043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liu Y, Cao GF, Xue J, Wan J, Wan Y, Jiang Q, Yao J, Tumor necrosis factor-alpha (TNF-alpha)-mediated in vitro human retinal pigment epithelial (RPE) cell migration mainly requires Akt/mTOR complex 1 (mTORC1), but not mTOR complex 2 (mTORC2) signaling, Eur J Cell Biol 91(9) (2012) 728–37. [DOI] [PubMed] [Google Scholar]
- [33].Kobayashi K, Maeda K, Takefuji M, Kikuchi R, Morishita Y, Hirashima M, Murohara T, Dynamics of angiogenesis in ischemic areas of the infarcted heart, Sci Rep 7(1) (2017) 7156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Parone PA, Da Cruz S, Cleveland DW, Mitochondrial Isolation and Purification from Mouse Spinal Cord, Bio Protoc 3(21) (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM, mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase, Science 334(6056) (2011) 678–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, Vander Heiden MG, Sabatini DM, mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient, Cell 171(3) (2017) 642–654 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM, The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1, Science 320(5882) (2008) 1496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Libby P, Nahrendorf M, Swirski FK, Leukocytes Link Local and Systemic Inflammation in Ischemic Cardiovascular Disease: An Expanded “Cardiovascular Continuum”, J Am Coll Cardiol 67(9) (2016) 1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sager HB, Heidt T, Hulsmans M, Dutta P, Courties G, Sebas M, Wojtkiewicz GR, Tricot B, Iwamoto Y, Sun Y, Weissleder R, Libby P, Swirski FK, Nahrendorf M, Targeting Interleukin-1beta Reduces Leukocyte Production After Acute Myocardial Infarction, Circulation 132(20) (2015) 1880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Rath M, Muller I, Kropf P, Closs EI, Munder M, Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages, Front Immunol 5 (2014) 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].McDonald B, Kubes P, Chemokines: sirens of neutrophil recruitment-but is it just one song?, Immunity 33(2) (2010) 148–9. [DOI] [PubMed] [Google Scholar]
- [42].Chou RC, Kim ND, Sadik CD, Seung E, Lan Y, Byrne MH, Haribabu B, Iwakura Y, Luster AD, Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis, Immunity 33(2) (2010) 266–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, Granucci F, Kagan JC, CD14 controls the LPS-induced endocytosis of Toll-like receptor 4, Cell 147(4) (2011) 868–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].DeLeon-Pennell KY, Meschiari CA, Jung M, Lindsey ML, Matrix Metalloproteinases in Myocardial Infarction and Heart Failure, Prog Mol Biol Transl Sci 147 (2017) 75–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Leuschner F, Rauch PJ, Ueno T, Gorbatov R, Marinelli B, Lee WW, Dutta P, Wei Y, Robbins C, Iwamoto Y, Sena B, Chudnovskiy A, Panizzi P, Keliher E, Higgins JM, Libby P, Moskowitz MA, Pittet MJ, Swirski FK, Weissleder R, Nahrendorf M, Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis, J Exp Med 209(1) (2012) 123–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma’ayan A, Chua WJ, Hansen TH, Turley SJ, Merad M, Randolph GJ, Immunological Genome C, Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages, Nat Immunol 13(11) (2012) 1118–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zizzo G, Hilliard BA, Monestier M, Cohen PL, Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction, J Immunol 189(7) (2012) 3508–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Akinrinmade OA, Chetty S, Daramola AK, Islam MU, Thepen T, Barth S, CD64: An Attractive Immunotherapeutic Target for M1-type Macrophage Mediated Chronic Inflammatory Diseases, Biomedicines 5(3) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hristodorov D, Mladenov R, von Felbert V, Huhn M, Fischer R, Barth S, Thepen T, Targeting CD64 mediates elimination of M1 but not M2 macrophages in vitro and in cutaneous inflammation in mice and patient biopsies, MAbs 7(5) (2015) 853–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Na YR, Stakenborg M, Seok SH, Matteoli G, Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD, Nat Rev Gastroenterol Hepatol 16(9) (2019) 531–543. [DOI] [PubMed] [Google Scholar]
- [51].Eghbalzadeh K, Georgi L, Louis T, Zhao H, Keser U, Weber C, Mollenhauer M, Conforti A, Wahlers T, Paunel-Gorgulu A, Compromised Anti-inflammatory Action of Neutrophil Extracellular Traps in PAD4-Deficient Mice Contributes to Aggravated Acute Inflammation After Myocardial Infarction, Front Immunol 10 (2019) 2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yue Y, Yang X, Feng K, Wang L, Hou J, Mei B, Qin H, Liang M, Chen G, Wu Z, M2b macrophages reduce early reperfusion injury after myocardial ischemia in mice: A predominant role of inhibiting apoptosis via A20, Int J Cardiol 245 (2017) 228–235. [DOI] [PubMed] [Google Scholar]
- [53].Ma Y, Mouton AJ, Lindsey ML, Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction, Transl Res 191 (2018) 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhang S, Weinberg S, DeBerge M, Gainullina A, Schipma M, Kinchen JM, Ben-Sahra I, Gius DR, Yvan-Charvet L, Chandel NS, Schumacker PT, Thorp EB, Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair, Cell Metab 29(2) (2019) 443–456 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Tamoutounour S, Guilliams M, Montanana Sanchis F, Liu H, Terhorst D, Malosse C, Pollet E, Ardouin L, Luche H, Sanchez C, Dalod M, Malissen B, Henri S, Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin, Immunity 39(5) (2013) 925–38. [DOI] [PubMed] [Google Scholar]
- [56].Jones RG, Pearce EJ, MenTORing Immunity: mTOR Signaling in the Development and Function of Tissue-Resident Immune Cells, Immunity 46(5) (2017) 730–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O, Steffens S, Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype, Eur Heart J 38(3) (2017) 187–197. [DOI] [PubMed] [Google Scholar]
- [58].Abdelaziz MH, Abdelwahab SF, Wan J, Cai W, Huixuan W, Jianjun C, Kumar KD, Vasudevan A, Sadek A, Su Z, Wang S, Xu H, Alternatively activated macrophages; a double-edged sword in allergic asthma, J Transl Med 18(1) (2020) 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P, Schmid MC, Pink M, Winkler DG, Rausch M, Palombella VJ, Kutok J, McGovern K, Frazer KA, Wu X, Karin M, Sasik R, Cohen EE, Varner JA, PI3Kgamma is a molecular switch that controls immune suppression, Nature 539(7629) (2016) 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Weichhart T, Costantino G, Poglitsch M, Rosner M, Zeyda M, Stuhlmeier KM, Kolbe T, Stulnig TM, Horl WH, Hengstschlager M, Muller M, Saemann MD, The TSC-mTOR signaling pathway regulates the innate inflammatory response, Immunity 29(4) (2008) 565–77. [DOI] [PubMed] [Google Scholar]
- [61].Ivanov SS, Roy CR, Pathogen signatures activate a ubiquitination pathway that modulates the function of the metabolic checkpoint kinase mTOR, Nat Immunol 14(12) (2013) 1219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Frangogiannis NG, The inflammatory response in myocardial injury, repair, and remodelling, Nat Rev Cardiol 11(5) (2014) 255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Lee JH, Phelan P, Shin M, Oh BC, Han X, Im SS, Osborne TF, SREBP-1a-stimulated lipid synthesis is required for macrophage phagocytosis downstream of TLR4-directed mTORC1, Proc Natl Acad Sci U S A 115(52) (2018) E12228–E12234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kazyken D, Magnuson B, Bodur C, Acosta-Jaquez HA, Zhang D, Tong X, Barnes TM, Steinl GK, Patterson NE, Altheim CH, Sharma N, Inoki K, Cartee GD, Bridges D, Yin L, Riddle SM, Fingar DC, AMPK directly activates mTORC2 to promote cell survival during acute energetic stress, Sci Signal 12(585) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sciarretta S, Zhai P, Maejima Y, Del Re DP, Nagarajan N, Yee D, Liu T, Magnuson MA, Volpe M, Frati G, Li H, Sadoshima J, mTORC2 regulates cardiac response to stress by inhibiting MST1, Cell Rep 11(1) (2015) 125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Marz AM, Fabian AK, Kozany C, Bracher A, Hausch F, Large FK506-binding proteins shape the pharmacology of rapamycin, Mol Cell Biol 33(7) (2013) 1357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J, Phosphatidic acid-mediated mitogenic activation of mTOR signaling, Science 294(5548) (2001) 1942–5. [DOI] [PubMed] [Google Scholar]
- [68].Foster DA, Salloum D, Menon D, Frias MA, Phospholipase D and the maintenance of phosphatidic acid levels for regulation of mammalian target of rapamycin (mTOR), J Biol Chem 289(33) (2014) 22583–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.