

PURPOSE
Patients with myelodysplastic syndromes (MDS) have a survival that can range from months to decades. Prognostic systems that incorporate advanced analytics of clinical, pathologic, and molecular data have the potential to more accurately and dynamically predict survival in patients receiving various therapies.
METHODS
A total of 1,471 MDS patients with comprehensively annotated clinical and molecular data were included in a training cohort and analyzed using machine learning techniques. A random survival algorithm was used to build a prognostic model, which was then validated in external cohorts. The accuracy of the proposed model, compared with other established models, was assessed using a concordance (c)index.
RESULTS
The median age for the training cohort was 71 years. Commonly mutated genes included SF3B1, TET2, and ASXL1. The algorithm identified chromosomal karyotype, platelet, hemoglobin levels, bone marrow blast percentage, age, other clinical variables, seven discrete gene mutations, and mutation number as having prognostic impact on overall and leukemia-free survivals. The model was validated in an independent external cohort of 465 patients, a cohort of patients with MDS treated in a prospective clinical trial, a cohort of patients with paired samples at different time points during the disease course, and a cohort of patients who underwent hematopoietic stem-cell transplantation.
CONCLUSION
A personalized prediction model on the basis of clinical and genomic data outperformed established prognostic models in MDS. The new model was dynamic, predicting survival and leukemia transformation probabilities at different time points that are unique for a given patient, and can upstage and downstage patients into more appropriate risk categories.
INTRODUCTION
Myelodysplastic Syndromes (MDS) are clonal hematopoietic disorders that lead to bone marrow failure and a risk of progression to acute myeloid leukemia (AML).1 The outcomes of patients with MDS are heterogeneous, with some alive more than a decade following diagnosis and others dying within a few months. Accurately predicting outcome can help patients manage expectations for their disease trajectory and help physicians identify appropriate therapies.2,3
CONTEXT
Key Objective
Build and validate a personalized prediction model using artificial intelligence.
Knowledge Generated
A personalized genoclinical model outperformed established prognostic models in myelodysplastic syndromes. The new model was dynamic, predicting survival and leukemia transformation probabilities at different time points that are unique for a given patient.
Relevance
The new model can be used as a stand-alone model or in conjunction with other established models to improve the predictability of outcomes in patients with myelodysplastic syndromes. It can also be used as a risk stratification tool for clinical trials enrollment.
Established prognostic models rely primarily on clinical variables that are derived from bone marrow pathology and peripheral blood counts and divide patients into a handful of risk categories.4 The most commonly used models in clinical practice and for clinical trial eligibility are the International Prognostic Scoring System (IPSS) and the revised IPSS (IPSS-R).5,6 The recent addition of molecular data to these scoring systems has enhanced the accuracy of these models, allowing upstaging and downstaging of patients into more appropriate risk categories, although the increment in improving their accuracy is modest.7,8 Furthermore, these prognostic models, many of which were developed in untreated patients, may underestimate or overestimate the actual survival of a patient, affecting treatment recommendations and prediction of disease course.7,8
In this study, we took advantage of a machine learning algorithm that can take into account clinical, pathologic, and molecular variables, as well as their interactions with each other, and developed and validated a prediction model that can provide a personalized prognosis that is specific for a given patient.
METHODS
Patient Cohort
Peripheral blood and bone marrow samples from patients diagnosed with MDS according to 2016 WHO criteria9 (criteria were reviewed and updated for patients who were diagnosed before 2016) and evaluated at the Cleveland Clinic and Munich Leukemia laboratory between 2001 and 2018 were included in the training cohort. The training cohort was used to build the model that was subsequently validated in an independent cohort of patients with MDS treated at the Moffitt Cancer Center between 2005 and 2018. Karyotypes were classified using the International System for Cytogenetic Nomenclature Criteria. The IPSS and IPSS-R were calculated as described previously.5,6
All patients signed informed consent for their samples and data to be used in future studies, and the study was approved by each participating center's Institutional Review Board committee in accordance with Declaration of Helsinki. More details regarding the patient cohorts and samples are summarized in the Methods section of the Data Supplement (online only).
DNA Sequencing and Analysis
Targeted deep sequencing was performed on 38 genes that are commonly reported in commercial laboratories’ genomic panels and have shown to have clinical impact in MDS and other myeloid malignancies. Annotation of the mutations was blinded from the clinical variables and performed by individuals not associated with the study. Detailed sequencing information is provided in the Methods section in the Data Supplement.
Statistical Analyses
Overall survival (OS) was defined as the time from diagnosis until death from any cause or censoring at the time that the patient was last known to be alive. Patients who underwent an allogeneic stem-cell transplant were censored at the time of the transplant. Stepwise Cox proportional hazard analyses were conducted to evaluate the prognostic impact of each mutation on OS and AML transformation in univariate analyses and after controlling for age and IPSS-R score. For AML transformation, death without AML transformation was considered to be a competing risk. A Bonferroni correction was used to identify significant mutations (a P value < .002 is considered significant instead of .05). To build the new model, clinical and mutational data were entered into random survival forest algorithm.10 The algorithm randomly bootstraps the original data into two thirds, where the model is developed, and one third, where the model is internally validated. The process is repeated multiple times to assure reproducibility of the final result. Furthermore, the algorithm includes all the variables and takes into account the relationship between each variable and other variables and the desired outcome (Data Supplement). Missing variables are imputed using an internal function built into the algorithm (more details are given in the Methods section in the Data Supplement). To produce an easy-to-use and reliable model, only variables with a significant impact on the desired outcome were included using variable importance analysis (Data Supplement). The top variables that affected the desired outcome were chosen to build the final model. Given a significant overlap between the top variables that affected OS and leukemia transformation, we decided to have one combined model rather than two separate models to ease the communication and implementation of the final model. The performance of the proposed model, compared with other models, was assessed by Harrell concordance index (c-index). More details regarding the statistical analyses are included in the Statistical Analysis section in the Data Supplement.
RESULTS
Patient Characteristics
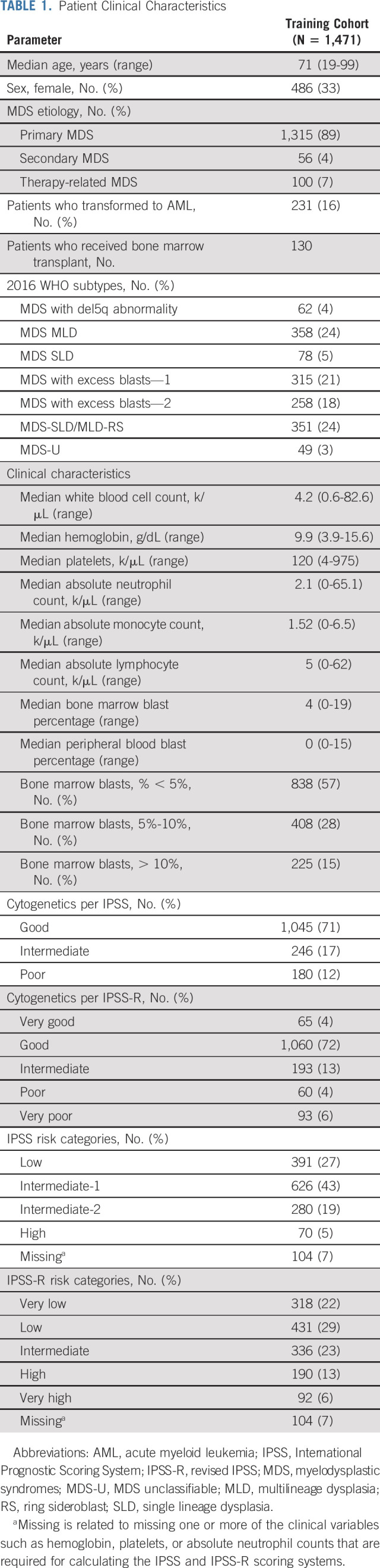
Data on 1,471 patients were used to build the new model. The clinical characteristics of the cohort are summarized in Table 1. The median age was 71 years (range, 19-99 years). Cytogenetic risk categories per IPSS-R included 65 patients (4%) with very good, 1,060 (72%) with good, 193 (13%) with intermediate, 60 (4%) with poor, and 93 (6%) with very poor risk. Risk stratification per IPSS-R included 749 (51%) with very low or low risk, 336 (23%) with intermediate, 182 (19%) with higher or very high risk, and 7% not calculated because of missing values. The clinical and mutational characteristics for the validation cohort compared with the training cohort as well the treatment modalities in each cohort are summarized in the Data Supplement. The validation cohort has more patients with higher-risk disease and differs in the presence of some mutations compared with the training cohort (Data Supplement).
TABLE 1.
Patient Clinical Characteristics
Molecular Landscape of MDS and Mutations Associations
To identify mutations associated with OS and AML transformation and to standardize the definition of mutation number or sample, we focused the analyses on 24 genes that were mutated in at least 30 patients in the study cohort (these genes are commonly included in commercial laboratory panels and have been shown to affect outcomes in our study and others11,12; Fig 1 and the Data Supplement). We identified at least one mutation of these genes in 1,149 patients (78%) with a median number of mutations per sample being 2 (range, 0-8). Gene mutation frequencies were consistent with those reported in previous studies.11,12 We then evaluated the correlation of these genes with each other. Strong correlations were observed for TP53 mutations and complex karyotype, TP53 and chromosome seven abnormalities, and STAG 2 or RUNX1 with ASXL1 mutations (Fig 1). SF3B1 mutations were mutually exclusive with TP53 mutations, complex karyotype, chromosome seven abnormalities, and ASXL1/SRSF2/U2AF1 (Fig 1).
FIG 1.
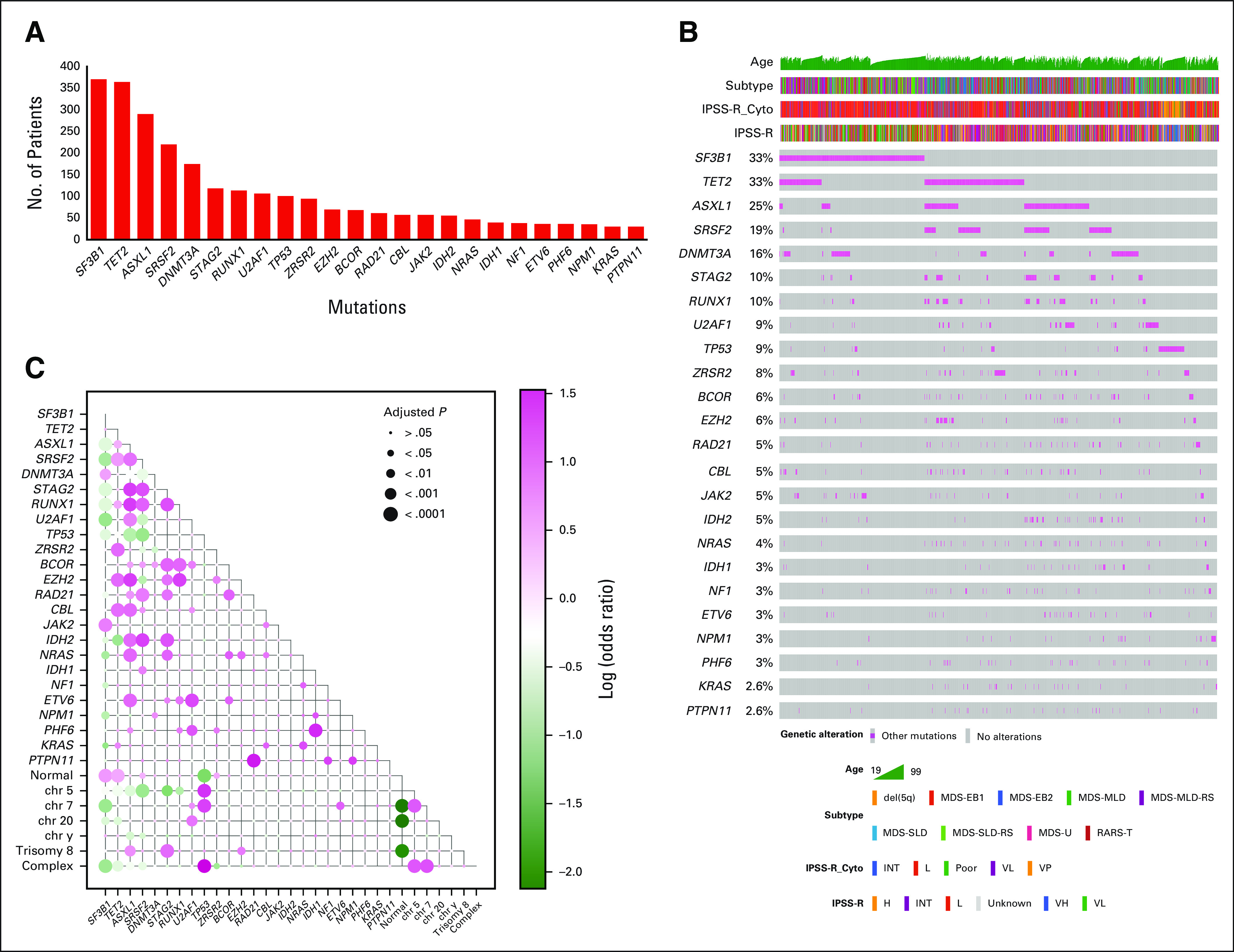
Genomic landscape of the training cohort: (A) the graph represents frequencies of recurrent gene mutations in this cohort, (B) the mosaic plot represents distributions of recurrent gene mutations in 1,471 patients, and (C) exclusivity and co-occurrence between different gene mutations. The circles were sized and colored according to their log (odds) from Fisher's exact test and person correlation (pink color represents co-occurrence, and green color represents exclusivity). H, high; INT, intermediate; IPSS-R, revised International Prognostic Scoring System; L, low; MDS, myelodysplastic syndromes; MDS-EB, MDS-excess blasts; MDS-U, MDS unclassifiable; MLD, multilineage dysplasia; OS, overall survival; RARS, refractory anemia with ring sideroblasts; RARS-T, RARS with thrombocytosis; RS, ring sideroblast; SLD, single lineage dysplasia; VH, very high; VP, very poor; VL, very low.
Impact of Mutations on OS
With a median follow-up of 43.6 months, the median OS was 32.2 months (range, 0.03-221.8 months). The impact of mutations on OS in univariate analyses and after adjustment for age and IPSS-R scores is summarized in Table 2. Using the Bonferroni correction, 10 mutations (ASXL1, EZH2, IDH2, KRAS, NRAS, RAD21, RUNX1, SRSF2, STAG2, and TP53) had a significant negative prognostic impact on OS, whereas only SF3B1 was associated with favorable outcomes (Table 2 and Fig 2). Only seven mutations (with the exception of KRAS, RUNX1, and SRSF2) remained significant after adjustment for age and IPSS-R scores (Table 2 and Fig 2). In a multivariate analysis that included age, IPSS-R risk categories, and significant mutations in univariate analysis versus all 24 gene mutations regardless of their significance, the prognostic impact of some of these mutations fluctuated depending on the variables included in the model. For example, only mutations in ASXL1, EZH2, KRAS, NRAS, RAD21, SF3B1, and TP53 were significant when only significant mutations in univariate analysis were added to the multivariate analysis, whereas more mutations (ASXl1, CBL, EZH2, NRAS, RA21, SF3B1, TET2, and TP53) were significant when adding all the 24 genes (Data Supplement).
TABLE 2.
Impact of Mutations on OS and Leukemia Transformation in Cox Regression Model

FIG 2.
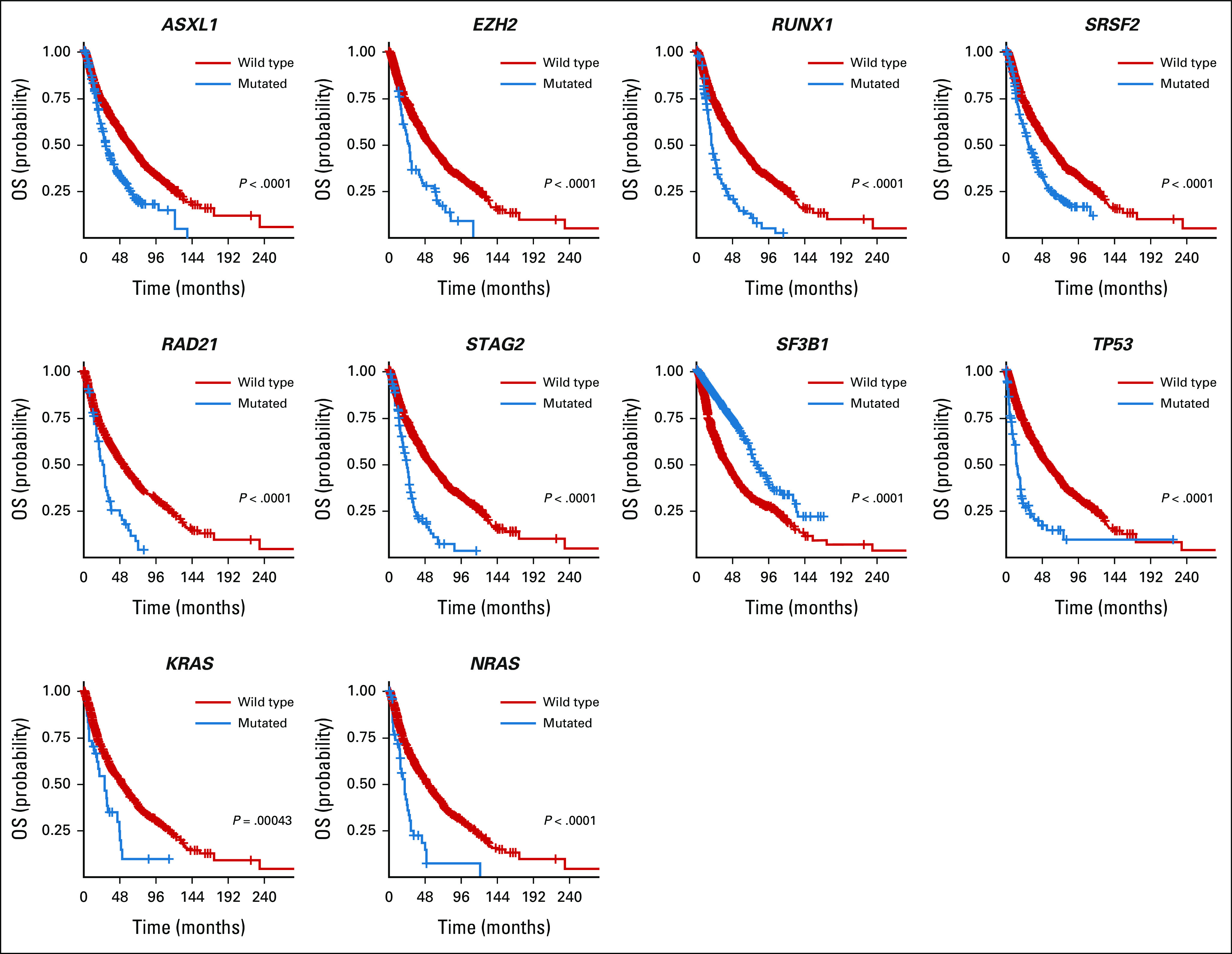
Impact of mutations on OS. Kaplan-Meier curves comparing the OS of patients with a mutated gene (blue) compared with a wild type (red). Only significant mutations after Bonferroni correction are shown. OS, overall survival.
We also investigated the impact of variant allelic frequency (VAF) of each mutation on OS. When VAF was used as a continuous variable, only EZH2 (hazard ratio [HR] 6.27; 95% CI, 2.12 to 18.50; P < .001) had a negative adjusted (for age, IPSS-R risk categories, and the Bonferroni correction) impact on OS (Data Supplement).
Impact of Mutations on AML Transformation
A total of 231 patients (16%) had AML transformation during their disease course. The leukemia transformation rate in our cohort correlates with previous reports.6,11 The impact of mutations on leukemia transformation in univariate analysis and after adjustment with age and IPSS-R is summarized in Table 2. In a multivariate analysis that included age, IPSS-R risk categories, and significant mutations in univariate analysis (after the Bonferroni correction) versus all 24 gene mutations regardless of their significance, the prognostic impact of some of these mutations on leukemia transformation also changed depending on the variables included in the model. For example, only mutations in ASXL1, IDH2, PHF6, PTPN11, RAD21, RUNX1, SF3B1, STAG2, and TP53 were significant when only significant mutations in univariate analysis were added to the multivariate analysis, whereas more mutations (ASXl1, IDH2, RAD21, RUNX1, SF3B1, SRSF2, STAG2, and TP53) were significant when adding all the 24 genes (Data Supplement).
We also investigated the impact of VAF (as a continuous variable) on AML transformation. Only mutations in ASXL1 (HR 1.08; 95% CI, 1.05 to 1.11; P < .001), NPM1 (HR 12.02; 95% CI, 1.40 to 103; P = .023), RUNX1 (HR 3.07; 95% CI, 0.26 to 35.8; P = .019), and TET2 (HR 4.67; 95% CI, 1.38 to 15.83; P = .013) led to a higher likelihood of AML transformation (Data Supplement).
Mutation Number As an Independent Prognostic Variable
Mutation number can measure the mutational load of MDS, although its number depends on the number of genes that are included in the panel. Using the 24 genes that were mutated in ≥ 30 patients in our cohort, we found that the number of mutations had a significant impact on OS and leukemia transformation (Fig 3). This impact remained significant even after adjustment for age and IPSS-R categories (OS, HR 1.13; 95% CI, 1.07 to 1.19; P < .001) and (AML transformation, HR 1.45; 95% CI, 1.33 to 1.57; P < .001). More importantly, mutation number retained its prognostic impact even after the removal of SF3B1 (OS, HR 1.29; 95% CI, 1.23 to 1.35; P < .001) (leukemia-free survival, HR 1.59; 95% CI, 1.48 to 1.71; P < .001) and TP53 mutations (OS, HR 1.20; 95% CI, 1.14 to 1.26; P < .001) (AML transformation, HR 1.50; 95% CI, 1.39 to 1.62; P < .001).
FIG 3.

OS and leukemia-free survival on the basis of mutation number: (A) Kaplan-Meier curves for OS on the basis of mutation number and (B) Kaplan-Meier curves for leukemia-free survival on the basis of mutation number. NR, no response; OS, overall survival.
Personalized Prediction Model
Variables that affected OS and leukemia transformation are shown in Figure 4 (ranked from the most to the least important). As expected, cytogenetic risk groups per IPSS-R were the most important variable for survival, whereas blast percentage was most important for AML transformation. Although there is a partial interaction between blast percentage and 2016 WHO criteria, both variables remained significant for OS and leukemia transformation and affected the final model accuracy. A total of seven mutations, along with mutation number, affected OS and AML transformation (Fig 4). The final model internal cross-validation showed the c-index score of 0.74 (95% CI, 0.73 to 0.75) for OS and 0.81 (95% CI, 0.80 to 0.82) for leukemia transformation in the training cohort and 0.71 (95% CI, 0.73 to 0.75) and 0.84 (95% CI, 0.73 to 0.75) compared with IPSS 0.66 (95% CI, 0.62 to 0.67) and IPSS-R 0.67 (95% CI, 0.62 to 0.68) in the validation cohort, respectively (Fig 4), which included patients diagnosed at an independent center, who underwent sequencing analyses that used a different set of genes and sequencing method. The new model outperformed the IPSS and IPSS-R even when mutations were added to the scoring systems as described previously7,8 (Fig 4). Furthermore, the proposed model was very well-calibrated in the training and validation cohorts for OS and leukemia transformation (Data Supplement).
FIG 4.
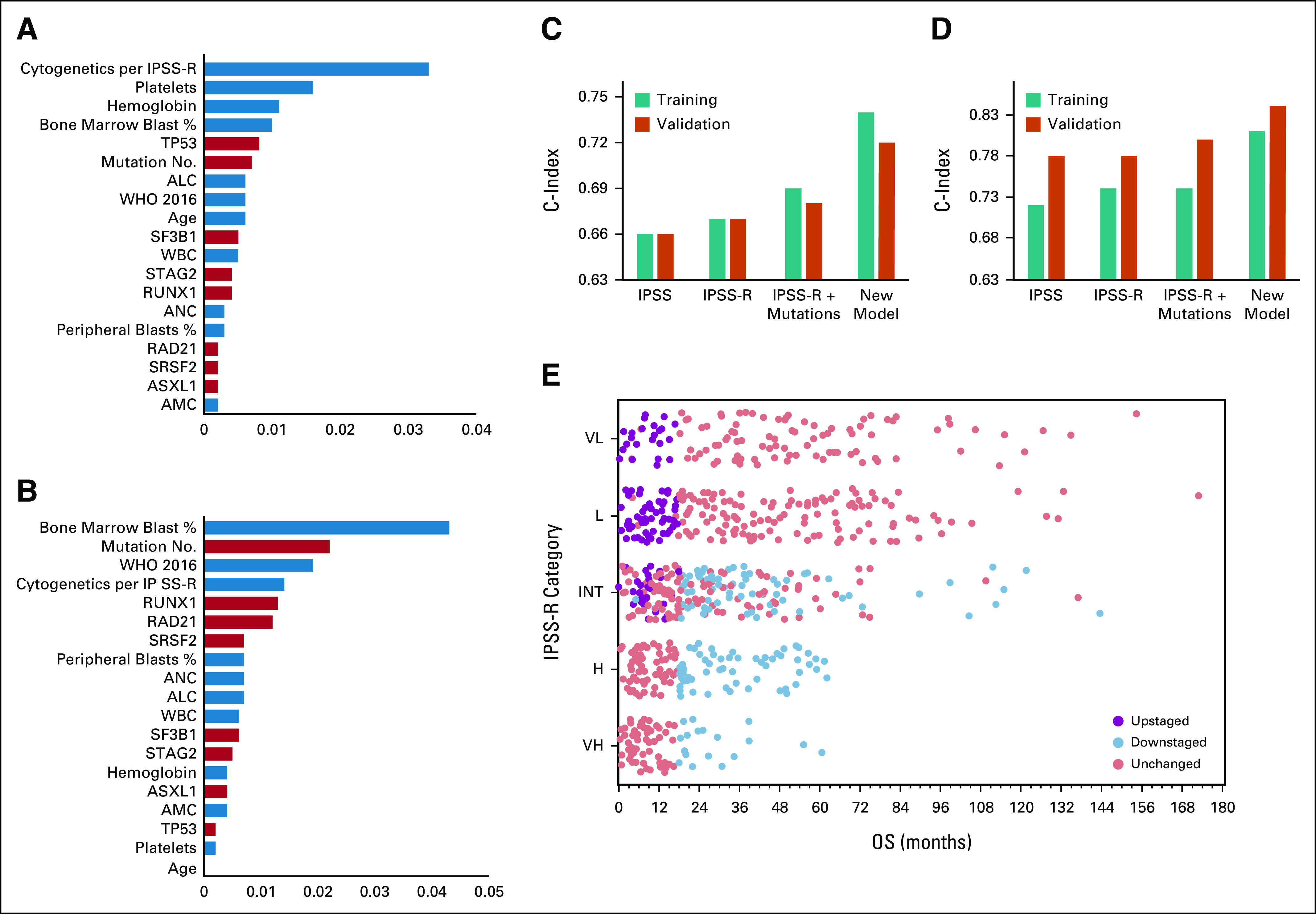
New model variables and performance matrix: (A) variable importance analysis showing the top variables from the most important to the least important that affect OS in the new model, the blue color represents clinical variables, and the orange color represents mutations; (B) variable importance analysis showing the top variables from the most important to the least important that affect leukemia transformation in the new model; the concordance index of the new model compared with that of IPSS and IPSS-R for (C) OS and (D) leukemia transformation in the training and validation cohorts; and (E) survival time from diagnosis to death in the training cohort on the basis of the IPSS-R risk categories; dots represent patients and their survival in each risk categories (only patients who died are included in this figure). Purple dots represent patients who were upstaged by the new model, and blue dots represent patients who were downstaged by the new model. ALC, absolute lymphocyte count; AMC, absolute monocyte count; ANC, absolute neutrophil count; H, high; INT, intermediate; IPSS, International Prognostic Scoring System; IPSS-R, revised IPSS; L, low; OS, overall survival; VL, very low; VH = very high.
To translate this model into a useful clinical tool, we built an online calculator13 that can generate the survival and leukemia transformation probabilities at different time points in a patient's disease course using available clinical, laboratory, and molecular data.
Clinical Implications of the Proposed Model
As shown in Figure 4D, OS survival rates varied substantially even among patients in the same IPSS-R risk group. Applying the new model, patients with a survival probability < 50% at 18 months (patients with a median OS < 18 months) were considered higher-risk. Using the new model, 306 patients (20%) in the training cohort were identified as a higher-risk group; among them, 148 (48%) were classified as lower-risk per IPSS and 107 (35%) per IPSS-R (score ≤ 3.5)14 (Fig 4D). Similarly, 73 patients (16%) in the validation cohort were classified as a higher-risk and 28 (39%) of them were classified as lower-risk per IPSS and 33 (45%) per IPSS-R (Fig 4D).
Validations of the New Model in Different Settings
To further assure the reproducibility of our model in different settings, we validated it in three different cohorts. First, we obtained clinical and mutational data from patients who participated in the prospective, phase II, North American Intergroup S1117 trial in which patients with higher-risk MDS and chronic myelomonocytic leukemia received azacitidine monotherapy or azacitidine combined with either lenalidomide or vorinostat. The study cohort and outcomes were described previously and are summarized in the Methods section in the Data Supplement. After excluding patients with chronic myelomonocytic leukemia, we identified 75 MDS patients with available clinical and genomic data. The clinical and mutational characteristics of this patient cohort are summarized in the Data Supplement. The cohort did not include absolute monocyte counts or absolute lymphocyte counts (not collected), and 37 patients (49%) had no karyotype results reported by the participating centers despite IPSS scores being reported for all patients. We compared our model with the IPSS scoring system only to assure the accuracy of the comparison.
When applying our model to this patient cohort, the c-index for our model was 0.68 compared with 0.57 for the IPSS scoring system. Although this trial targeted patients with higher-risk MDS per the IPSS scoring system, our model identified 24 patients (32%) as being higher-risk (considering patients with a survival probability of < 50% at 18 months) and 51 (68%) as lower-risk. In total, 37 patients (64%) with higher-risk disease per the IPSS (26 of 39 with intermediate-2 risk and 11 or 19 in high-risk categories) were downstaged by our model, whereas 3 of 17 patients in the Intermediate-1 risk group were upstaged to a higher-risk category. Interestingly, the median OS for the patients who were downstaged using our model was 26.1 months (95% CI, 12.5 to 45.6) compared with 16 months (95% CI, 12.3 to 35.3; P = .33) for patients without changes in their risk category. Furthermore, the median OS for patients who were identified as higher-risk by our model and by the IPSS scoring system (unchanged category) was 12.5 months for patients (n = 7) who received azacitidine treatment compared with 25.8 months for patients who received azacitidine plus lenalidomide (n = 9) and 14.9 months for patients who received azacitidine plus vorinostat (n = 8). However, the median OS for patients who were downstaged by our model was 28, 16.6, and 36.1 months, respectively. These findings suggest that patients with lower-risk disease by our model had a shorter survival when they received azacitidine plus lenalidomide, whereas truly higher-risk patients might have benefitted from the combination, although small numbers precluded significant conclusions.
We also validated our model in a cohort of patients with paired samples from different time points during the disease course and another cohort of patients who underwent allogeneic hematopoietic stem-cell transplantation (HCT). More details on the characteristics of these cohorts and the performance of our model compared with the IPSS/IPSS are summarized in the Data Supplement.
DISCUSSION
In this study, we developed a prognostic model that uses clinical and mutational data to provide estimates of the risk of death or progression to AML that are specific for a given patient. The proposed model significantly outperformed the IPSS and IPSS-R scoring systems (the most widely used systems in clinical practice and trial eligibility). To assure the reproducibility and generalizability of the model and its applicability in routine or real-world patients, our training cohort represents a diverse MDS population treated at centers in the United States and Europe and was validated in an independent cohort and a cohort of patients with MDS who were enrolled on a prospective clinical trial. In these cohorts, our model significantly outperformed the IPSS and IPSS-R scoring systems and indicated a possible different impact of treatment on the basis of the new classification by our model. We also validated our model in a patient cohort with paired samples at different time points and demonstrated the stability of our model performance over time compared with the IPSS/IPSS-R scoring systems. Although our model c-index was better compared with that of the IPSS/IPSS-R when applied to a cohort who received HCT, the drop in the performance of all models may be related to the lack of clinical and mutational variables that affect transplant-related outcomes, such as graft versus host disease. We have previously shown that transplant-related factors such as donor age, graft type, conditioning regimen, and others have a significant impact on the personalized prediction for patients with MDS before the transplant.
Recent efforts to improve the accuracy of established clinical prognostic scoring systems included the addition of molecular data or the development of gene-only prognostic models.7,8,12,15 These approaches have shown that the addition of selected gene mutations to established models such as the IPSS and IPSS-R can improve their predictive power and can upstage or downstage patients into more appropriate risk categories. Another study tried to build a gene-only model after controlling for clinical variables, but the model underperformed compared with a genoclinical model.12 The incremental improvement in the model's performance when molecular data were added was modest, though, suggesting that ad hoc inclusion of other variables, analyzed using traditional statistical methods, is not enough.
Our study has some limitations. Recent studies have shown that patient-reported outcomes and frailty scores have a significant impact on OS and that adding such variables can improve the accuracy of prognostication. Such data were not available for inclusion in the final model. Although our patient cohort is large, the significant impact of infrequent mutations on outcomes can be too small and misleading. Thus, we focused our analyses on the top 24 genes that are mutated in more than 30 patients and the seven genes that were included in our final model were among the top 10 mutated genes in our cohort and other previous reports.11,12 Although we controlled for patients who proceeded with HCT by censoring at the time of transplant, we did not control for specific treatment modalities, since not all patients had treatment data recorded in our data set. The impact of treatments (such as hypomethylating agents) on the performance of the model is minimal and only seen in a subset of higher-risk patients. Furthermore, our model was validated in four different validation cohorts at different settings including patients who received hypomethylating agents on a clinical trial. Finally, our model includes age as an important prognostic variable although younger patients with MDS generally have a greater loss of life (in years) than older patients. Thus, including age in prognostic scores may lead to less intensive treatment in younger patients since they have better OS.
In conclusion, we built and validated a personalized prediction model that can provide survival and leukemia transformation probability at MDS diagnosis and throughout a patient's disease course. The model outperformed other existing models that are used in clinical practice, for clinical trial eligibility, and the timing of transplant. This model can be used as a stand-alone model or in conjunction with the IPSS/IPSS-R scoring systems to improve their accuracy.
Aziz Nazha
Stock and Other Ownership Interests: Amazon
Honoraria: DCI Pharmaceuticals
Consulting or Advisory Role: Karyopharm Therapeutics, Tolero Pharmaceuticals
Speakers' Bureau: Novartis, Incyte
Research Funding: Jazz Pharmaceuticals
Rami Komrokji
Stock and Other Ownership Interests: AbbVie
Consulting or Advisory Role: Novartis, Bristol Myers Squibb, Jazz Pharmaceuticals, AbbVie, Geron, Acceleron Pharma
Speakers' Bureau: Jazz Pharmaceuticals, Bristol Myers Squibb, Agios
Travel, Accommodations, Expenses: Jazz Pharmaceuticals, Bristol Myers Squibb, Agios
Manja Meggendorfer
Employment: MLL Munich Leukemia Laboratory
Betty K. Hamilton
Consulting or Advisory Role: Syndax, Equilium
Sudipto Mukherjee
Honoraria: Aplastic Anemia and MDS International Foundation, Celgene/Bristol Myers Squibb, McGraw-Hill Education, Partnership for Health Analytic Research, EUSA Pharma
Consulting or Advisory Role: Novartis, Celgene
Research Funding: Novartis, BMS (formerly Celgene)
Travel, Accommodations, Expenses: BMS (formerly Celgene), Novartis, EUSA Pharma
Wencke Walter
Employment: Munich Leukemia Laboratory
Stephan Hutter
Employment: MLL Munich Leukemia Laboratory
Eric Padron
Honoraria: Stemline Therapeutics, Blueprint Medicines
Speakers' Bureau: Novartis, Taiho Pharmaceutical
Research Funding: Incyte, Bristol Myers Squibb, Kura Oncology
David Sallman
Consulting or Advisory Role: Celyad, Agios, AbbVie, Aprea AB, Bristol Myers Squibb, Gilead Sciences, Intellia Therapeutics, Kite, a Gilead Company, Magenta Therapeutics, Novartis, Syndax
Speakers' Bureau: Agios, Incyte, Bristol Myers Squibb
Research Funding: Celgene, Jazz Pharmaceuticals
Patents, Royalties, Other Intellectual Property: Intellectual Property Patent for LB-100 in MDS
David P. Steensma
Employment: Novartis Institutes for BioMedical Research
Stock and Other Ownership Interests: Arrowhead Pharmaceuticals
Research Funding: Aprea AB, Celgene/BMS, H3 Biomedicine
Amy Dezern
Consulting or Advisory Role: Celgene, Novartis, Gilead Sciences
Research Funding: Celgene, Astex Pharmaceuticals
Travel, Accommodations, Expenses: AbbVie
Gail Roboz
Consulting or Advisory Role: Janssen, Amgen, Astex Pharmaceuticals, Celgene, MedImmune, Novartis, Pfizer, AbbVie, Bayer, Celltrion, Jazz Pharmaceuticals, Genentech/Roche, Sandoz, Actinium Pharmaceuticals, Astellas Pharma, Bayer, Daiichi Sankyo, MEI Pharma, Otsuka, Takeda, Roche, Agios, GlaxoSmithKline, Bristol Myers Squibb, Helsinn Therapeutics, Mesoblast, Jasper Therapeutics
Research Funding: AbbVie, Agios, Astex Pharmaceuticals, Celgene, CTI, Karyopharm Therapeutics, MedImmune, MEI Pharma, Moffitt, Novartis, Onconova Therapeutics, Pfizer, Sunesis Pharmaceuticals, Tensha Therapeutics, Cellectis, Cellectis, Janssen, Amphivena
Travel, Accommodations, Expenses: Amphivena, Astex Pharmaceuticals, Janssen, Pfizer, Array BioPharma, Novartis, AbbVie, Jazz Pharmaceuticals, Celgene, Celltrion, Roche/Genentech, Sandoz, Bayer, Clovis Oncology, Amgen, Sunesis Pharmaceuticals, Eisai, Agios
Guillermo Garcia-Manero
Honoraria: Celgene, Astex Pharmaceuticals, Acceleron Pharma, Helssin, AbbVie
Consulting or Advisory Role: Celgene, Astex Pharmaceuticals, Acceleron Pharma, Jazz Pharmaceuticals, Bristol Myers Squibb, Helsinn Therapeutics
Research Funding: Celgene, Astex Pharmaceuticals, Amphivena, Helsinn Therapeutics, Novartis, AbbVie, Bristol Myers Squibb, Onconova Therapeutics, H3 Biomedicine, Merck
Harry Erba
Consulting or Advisory Role: Agios, Astellas Pharma, Amgen, Celgene, Daiichi Sankyo, Glycomimetics, Immunogen, Incyte, Jazz Pharmaceuticals, Macrogenics, Novartis, AbbVie/Genentech, Janssen Oncology, Pfizer, Trillium Therapeutics, Takeda, Kura Oncology
Speakers' Bureau: Agios, Celgene, Incyte, Jazz Pharmaceuticals, Novartis, AbbVie/Genentech
Research Funding: AbbVie, Agios, Amgen, Daiichi Sankyo, FORMA Therapeutics, Gilead/Forty Seven, Immunogen, Jazz Pharmaceuticals, Macrogenics, Novartis, PTC Therapeutics, AbbVie, Glycomimetics, ALX Oncology
Other Relationship: Glycomimetics, Celgene
Uncompensated Relationships: Daiichi Sankyo
Claudia Haferlach
Employment: MLL Munich Leukemia Laboratory
Leadership: MLL Munich Leukemia Laboratory
Stock and Other Ownership Interests: MLL Munich Leukemia Laboratory
Honoraria: AstraZeneca
Jaroslaw P. Maciejewski
Consulting or Advisory Role: Celgene, Novartis
Torsten Haferlach
Employment: MLL Munich Leukemia Laboratory
Leadership: MLL Munich Leukemia Laboratory
Consulting or Advisory Role: Illumina
Mikkael A. Sekeres
This author is a member of the JCO Editorial Board. Journal policy recused the author from having any role in the peer review of this manuscript.
Consulting or Advisory Role: Celgene, Millennium, Pfizer, Novartis
Research Funding: Takeda, Pfizer, Bristol Myers Squibb
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented in part as an oral abstract at the American Society of Hematology Annual Meeting, San Diego, CA, 2018.
AUTHOR CONTRIBUTIONS
Conception and design: Aziz Nazha
Administrative support: Najla Al Ali, Mikkael A. Sekeres
Provision of study materials or patients: Aziz Nazha, Manja Meggendorfer, Najla Al Ali
Collection and assembly of data: Aziz Nazha, Rami Komrokji, Manja Meggendorfer, Nathan Radakovich, Jacob Shreve, Yasunubo Nagata, Najla Al Ali, Teodora Kuzmanovic, Cassandra Kerr, Vera Adema
Data analysis and interpretation: Aziz Nazha, Xuefei Jia, Nathan Radakovich, C. Beau Hilton, Yasunubo Nagata
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Personalized Prediction Model to Risk Stratify Patients With Myelodysplastic Syndromes
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Aziz Nazha
Stock and Other Ownership Interests: Amazon
Honoraria: DCI Pharmaceuticals
Consulting or Advisory Role: Karyopharm Therapeutics, Tolero Pharmaceuticals
Speakers' Bureau: Novartis, Incyte
Research Funding: Jazz Pharmaceuticals
Rami Komrokji
Stock and Other Ownership Interests: AbbVie
Consulting or Advisory Role: Novartis, Bristol Myers Squibb, Jazz Pharmaceuticals, AbbVie, Geron, Acceleron Pharma
Speakers' Bureau: Jazz Pharmaceuticals, Bristol Myers Squibb, Agios
Travel, Accommodations, Expenses: Jazz Pharmaceuticals, Bristol Myers Squibb, Agios
Manja Meggendorfer
Employment: MLL Munich Leukemia Laboratory
Betty K. Hamilton
Consulting or Advisory Role: Syndax, Equilium
Sudipto Mukherjee
Honoraria: Aplastic Anemia and MDS International Foundation, Celgene/Bristol Myers Squibb, McGraw-Hill Education, Partnership for Health Analytic Research, EUSA Pharma
Consulting or Advisory Role: Novartis, Celgene
Research Funding: Novartis, BMS (formerly Celgene)
Travel, Accommodations, Expenses: BMS (formerly Celgene), Novartis, EUSA Pharma
Wencke Walter
Employment: Munich Leukemia Laboratory
Stephan Hutter
Employment: MLL Munich Leukemia Laboratory
Eric Padron
Honoraria: Stemline Therapeutics, Blueprint Medicines
Speakers' Bureau: Novartis, Taiho Pharmaceutical
Research Funding: Incyte, Bristol Myers Squibb, Kura Oncology
David Sallman
Consulting or Advisory Role: Celyad, Agios, AbbVie, Aprea AB, Bristol Myers Squibb, Gilead Sciences, Intellia Therapeutics, Kite, a Gilead Company, Magenta Therapeutics, Novartis, Syndax
Speakers' Bureau: Agios, Incyte, Bristol Myers Squibb
Research Funding: Celgene, Jazz Pharmaceuticals
Patents, Royalties, Other Intellectual Property: Intellectual Property Patent for LB-100 in MDS
David P. Steensma
Employment: Novartis Institutes for BioMedical Research
Stock and Other Ownership Interests: Arrowhead Pharmaceuticals
Research Funding: Aprea AB, Celgene/BMS, H3 Biomedicine
Amy Dezern
Consulting or Advisory Role: Celgene, Novartis, Gilead Sciences
Research Funding: Celgene, Astex Pharmaceuticals
Travel, Accommodations, Expenses: AbbVie
Gail Roboz
Consulting or Advisory Role: Janssen, Amgen, Astex Pharmaceuticals, Celgene, MedImmune, Novartis, Pfizer, AbbVie, Bayer, Celltrion, Jazz Pharmaceuticals, Genentech/Roche, Sandoz, Actinium Pharmaceuticals, Astellas Pharma, Bayer, Daiichi Sankyo, MEI Pharma, Otsuka, Takeda, Roche, Agios, GlaxoSmithKline, Bristol Myers Squibb, Helsinn Therapeutics, Mesoblast, Jasper Therapeutics
Research Funding: AbbVie, Agios, Astex Pharmaceuticals, Celgene, CTI, Karyopharm Therapeutics, MedImmune, MEI Pharma, Moffitt, Novartis, Onconova Therapeutics, Pfizer, Sunesis Pharmaceuticals, Tensha Therapeutics, Cellectis, Cellectis, Janssen, Amphivena
Travel, Accommodations, Expenses: Amphivena, Astex Pharmaceuticals, Janssen, Pfizer, Array BioPharma, Novartis, AbbVie, Jazz Pharmaceuticals, Celgene, Celltrion, Roche/Genentech, Sandoz, Bayer, Clovis Oncology, Amgen, Sunesis Pharmaceuticals, Eisai, Agios
Guillermo Garcia-Manero
Honoraria: Celgene, Astex Pharmaceuticals, Acceleron Pharma, Helssin, AbbVie
Consulting or Advisory Role: Celgene, Astex Pharmaceuticals, Acceleron Pharma, Jazz Pharmaceuticals, Bristol Myers Squibb, Helsinn Therapeutics
Research Funding: Celgene, Astex Pharmaceuticals, Amphivena, Helsinn Therapeutics, Novartis, AbbVie, Bristol Myers Squibb, Onconova Therapeutics, H3 Biomedicine, Merck
Harry Erba
Consulting or Advisory Role: Agios, Astellas Pharma, Amgen, Celgene, Daiichi Sankyo, Glycomimetics, Immunogen, Incyte, Jazz Pharmaceuticals, Macrogenics, Novartis, AbbVie/Genentech, Janssen Oncology, Pfizer, Trillium Therapeutics, Takeda, Kura Oncology
Speakers' Bureau: Agios, Celgene, Incyte, Jazz Pharmaceuticals, Novartis, AbbVie/Genentech
Research Funding: AbbVie, Agios, Amgen, Daiichi Sankyo, FORMA Therapeutics, Gilead/Forty Seven, Immunogen, Jazz Pharmaceuticals, Macrogenics, Novartis, PTC Therapeutics, AbbVie, Glycomimetics, ALX Oncology
Other Relationship: Glycomimetics, Celgene
Uncompensated Relationships: Daiichi Sankyo
Claudia Haferlach
Employment: MLL Munich Leukemia Laboratory
Leadership: MLL Munich Leukemia Laboratory
Stock and Other Ownership Interests: MLL Munich Leukemia Laboratory
Honoraria: AstraZeneca
Jaroslaw P. Maciejewski
Consulting or Advisory Role: Celgene, Novartis
Torsten Haferlach
Employment: MLL Munich Leukemia Laboratory
Leadership: MLL Munich Leukemia Laboratory
Consulting or Advisory Role: Illumina
Mikkael A. Sekeres
This author is a member of the JCO Editorial Board. Journal policy recused the author from having any role in the peer review of this manuscript.
Consulting or Advisory Role: Celgene, Millennium, Pfizer, Novartis
Research Funding: Takeda, Pfizer, Bristol Myers Squibb
No other potential conflicts of interest were reported.
REFERENCES
- 1.Tefferi A, Vardiman JW: Myelodysplastic syndromes. N Engl J Med 361:1872-1885, 2009 [DOI] [PubMed] [Google Scholar]
- 2.Greenberg PL, Stone RM, Al-Kali A, et al. : Myelodysplastic syndromes, version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 15:60-87, 2017 [DOI] [PubMed] [Google Scholar]
- 3.Fenaux P, Haase D, Sanz GF, et al. : Myelodysplastic syndromes: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 25:iii57-iii69, 2014. (suppl 3) [DOI] [PubMed] [Google Scholar]
- 4.Nazha A: The MDS genomics-prognosis symbiosis. Hematology Am Soc Hematol Educ Program 2018:270-276, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greenberg P, Cox C, LeBeau MM, et al. : International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 89:2079-2088, 1997 [PubMed] [Google Scholar]
- 6.Greenberg PL, Tuechler H, Schanz J, et al. : Revised international prognostic scoring system for myelodysplastic syndromes. Blood 120:2454-2465, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nazha A, Narkhede M, Radivoyevitch T, et al. : Incorporation of molecular data into the Revised International Prognostic Scoring System in treated patients with myelodysplastic syndromes. Leukemia 30:2214-2220, 2016 [DOI] [PubMed] [Google Scholar]
- 8.Nazha A, Al-Issa K, Hamilton BK, et al. : Adding molecular data to prognostic models can improve predictive power in treated patients with myelodysplastic syndromes. Leukemia 31:2848-2850, 2017 [DOI] [PubMed] [Google Scholar]
- 9.Arber DA, Orazi A, Hasserjian R, et al. : The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127:2391-2405, 2016 [DOI] [PubMed] [Google Scholar]
- 10.Ishwaran H, Kogalur UB, Chen X, et al. : Random survival forests for high-dimensional data. Stat Anal Data Mining 4:115-132, 2011 [Google Scholar]
- 11.Papaemmanuil E, Gerstung M, Malcovati L, et al. : Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122:3616-3627, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haferlach T, Nagata Y, Grossmann V, et al. : Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28:241-247, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Personalized Prediction Model for Myelodysplastic Syndromes. https://azizn38.shinyapps.io/final/
- 14.Pfeilstocker M, Tuechler H, Sanz G, et al. : Time-dependent changes in mortality and transformation risk in MDS. Blood 128:902-910, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bejar R, Stevenson K, Abdel-Wahab O, et al. : Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 364:2496-2506, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]