

Abstract
Endogenous capability of the post-mitotic human heart holds great promise to restore the injured myocardium. Recent evidence indicates that the extracellular vesicles (EVs) regulate cardiac homeostasis and regeneration. Here, we investigated the molecular mechanism of EVs for self-repair. We isolated EVs from human iPSC-derived cardiomyocytes (iCMs), which were exposed to hypoxic (hEVs) and normoxic conditions (nEVs), and examined their roles in in vitro and in vivo models of cardiac injury. hEV treatment significantly improved the viability of hypoxic iCMs in vitro and cardiac function of severely injured murine myocardium in vivo. Microarray analysis of the EVs revealed significantly enriched expression of the miR-106a–363 cluster (miR cluster) in hEVs vs. nEVs. This miR cluster preserved survival and contractility of hypoxia-injured iCMs and maintained murine left-ventricular (LV) chamber size, improved LV ejection fraction, and reduced myocardial fibrosis of the injured myocardium. RNA-Seq analysis identified Jag1-Notch3-Hes1 as a target intracellular pathway of the miR cluster. Moreover, the study found that the cell cycle activator and cytokinesis genes were significantly up-regulated in the iCMs treated with miR cluster and Notch3 siRNA. Together, these results suggested that the miR cluster in the EVs stimulated cardiomyocyte cell cycle re-entry by repressing Notch3 to induce cell proliferation and augment myocardial self-repair. The miR cluster may represent an effective therapeutic approach for ischemic cardiomyopathy.
Keywords: Myocardial Infarction, IPSCs, EVs, MiRNAs, Endogenous cardiac repair mechanism, Cell cycle re-entry
Introduction
Myocardial infarction (MI) and heart failure (HF) represent the leading cause of death and hospital admission in the US, respectively [43]. Approximately, 1 million patients suffer from MI in the US annually, resulting in cardiomyocyte injury and subsequent cardiac dysfunction. Early prevention, effective medical care, and timely intervention of MI have decreased the resultant mortality; however, the survivors of MI must endure a life-long commitment to medicine and device therapy. Furthermore, they bear an increased risk of cardiovascular complications and high mortality due to scar formation, pathological remodeling, and ventricular arrhythmia.
The intrinsic reparative capability of an adult heart is insufficient to restore the injured myocardium fully following an MI [43, 45]. Although iPSC-derived cardiomyocytes (iCMs) have the potential to address this issue [31], studies revealed poor survival, reduced plasticity, and suboptimal engraftment in the ischemic myocardium, prohibiting robust clinical translation [26, 28]. Studies have shown that the paracrine factors from the iCMs restore the injured heart [26, 28, 58]. These findings suggest that the trophic factors produced by the host cardiomyocytes may activate endogenous tissue repair and regeneration.
Extracellular vesicles (EVs) provide an effective cell-free alternative to traditional cell-based therapy by transferring bioactive molecules to the injured recipient cells directly [29]. Studies have reported that the pleiotropic effects of EVs attenuate pathological inflammation, apoptosis, necrosis, remodeling, and fibrosis [25, 37, 52, 60]. Our findings demonstrated that the beneficial effects of stem cells are recapitulated by administering stem cell-derived EVs to restore the vulnerable peri-infarct region (PIR) of the injured myocardium [16, 56, 58]. In this study, we investigated the mechanism of action of the EVs from hypoxia-injured iCMs.
microRNAs (miRNAs) carried by the EVs are intercellular messengers, which modulate the post-transcriptional regulation of gene expression [11]. Selective packaging of miRNAs in the EVs depend on the parents cells and their specific physiological environment [62]. A number of miRNAs have been identified to modulate cardiac function, providing a rationale for their therapeutic role [7, 12, 66]. Furthermore, several miRNAs were identified to regulate the survival of the native cardiomyocytes through enhanced cardiac repair and regeneration [7, 24]. We, therefore, hypothesized that the miRNAs in EVs play a critical role in restoring the injured myocardium. The exact mechanism of action of the EV derived-miRNAs, however, is poorly understood.
A miRNA cluster is defined as adjacent miRNA genes on a chromosome, transcribed as one long pre-miRNA transcript, and then cleaved into individual miRNAs. Several miRNA clusters have been found to be important in embryonic development and pathophysiology [3, 64]. Here, we identified miR-106a–363 cluster (miR cluster), consisting of 6 miRNAs (miR-106a, −18b, 19b, −20b, −92a, and −363). It is one of the paralogues of the miR-17–92 cluster and well conserved in both human and murine genome [63]. The role of miR cluster has been reported mostly in T-cell lymphoma [34], breast cancer [38], and Ewing Sarcoma [18]. To the best of our knowledge, this miR cluster has yet to be investigated in cardiovascular disease. In this study, we found that the miR cluster is consistently enriched in the EVs secreted from hypoxia-injured iCMs. RNA-sequencing revealed that the miR cluster directly repressed the Notch3 signaling pathway, a highly conserved pathway in the vertebrates, and significantly improved cardiac function following an acute MI. Our in vivo data demonstrated that the miR cluster modulated the Notch3 pathway to stimulate cell cycle re-entry and cardiomyocyte proliferation to restore the injured myocardium.
Materials and methods
A detailed description of the entire in vitro experiments (cell biology assays, molecular assays, and EVs isolation and characterization) and in vivo experiments (mice surgery, analysis of myocardial infarction models), bioinformatics, and statistical analysis are presented in the Supplemental Materials and Methods section.
Results
EVs derived from hypoxic iCMs (hEVs) improve cell survival in vitro and cardiac function in vivo after ischemic injury
We isolated, purified, and characterized the EVs generated from serum-free conditioned media of highly purified iCM culture (~ 98% of cTnT +) (Figure S1A, B). These EVs were characterized using transmission electron microscopy (TEM) (Figure S1C) and western blot analysis for EV-specific markers according to MISEV2018 recommendations. EVs were compared to the iCMs and microvesicles (MVs: > 250 nm) by measuring endosomal proteins and lipid membrane proteins. The result demonstrated that the EVs did not express the endosomal proteins including ER, Golgi, and mitochondria, while the iCMs and MVs did demonstrate them (Figure S1D). To assess whether EV production and cargo may be influenced by the physiologic factors and micro-environments, myocardial ischemia was simulated in vitro and EVs were isolated from the iCM supernatants under normal (nEVs: ~ 20% O2 for 24 h) and hypoxic (hEVs: < 1% O2 for 24 h) culture conditions. The quantity and size of EVs were measured using Nanosight (Figure S1E). hEVs were smaller when compared to nEVs (Figure S1F). The optimal hypoxic duration for iCM viability experiments and the effective EV dose were decided by time- and dose-dependent experiments (Figure S2A, B). The normal contractile iCMs were then treated with autologous nEV and hEV and exposed to hypoxic injury (< 1% O2 for 18 h). Our data showed that the hEV treatment group significantly improved cell viability after hypoxic injury when compared to the nEV- and non-EV (control) treatment groups (Fig. 1a). We then injected nEVs and hEVs (4 × 108 particles/mice; approximately 200 μg/mice) at three different sites of the PIR immediately after induction of acute MI by left anterior descending coronary artery (LAD) ligation. Two and four weeks later, we observed markedly improved myocardial viability and left-ventricular ejection fraction (LVEF), and end-diastolic (LVEDV) and end-systolic volumes (LVESV) of hEV vs. normal saline-treated control group (Fig. 1b-e). These results demonstrated that hEV had significant therapeutic effects and contributed to improve cardiac function after MI.
Fig. 1.
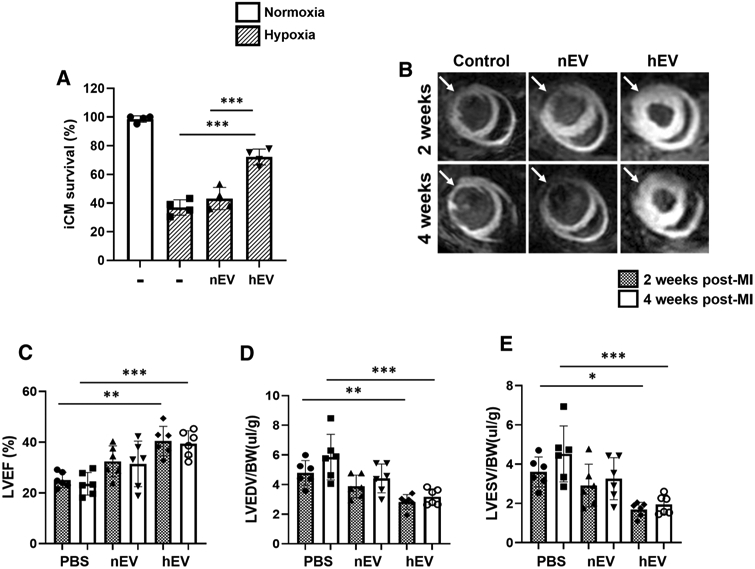
Hypoxic iCM-derived EVs enhance iCM survival and murine myocardial viability. a Survival rate of iCMs was measured by MTT analysis after hypoxic insult. EVs were isolated from the in vitro supernatant of iCMs exposed to hypoxic conditions (< 1% O2; hEVs), and normoxic condition (20% O2; nEVs). The iCMs were exposed to 50 μg of the corresponding EVs during hypoxic insult (< 1% O2, 5% CO2, 94% N2) for 18 h. The experimental groups consisted of four conditions: normoxia control, hypoxia control, hypoxia with nEV treatment, and hypoxia with hEV treatment. Statistical significance was determined by one-way ANOVA with multiple comparisons and mean data ± SD (n = 4). b Manganese-enhanced MRI of SCID mice myocardium was performed at weeks 2–4 post-MI. The treatment groups consisted of (1) normal saline control, (2) nEVs, and (3) hEVs (white arrows point to the infarcted area). c-e Left-ventricular ejection fraction (LVEF), left-ventricular end-diastolic volume (LVEDV), and left-ventricular end-systolic volume (LVESV) measurements of saline control (n = 6), nEVs (n = 6), and hEVs (n = 6) treatment groups. Statistical significance was determined by two-way ANOVA with multiple comparisons and mean data ± SD. Statistically non-significant comparisons are not shown. *p < 0.05, **p < 0.01, ***p < 0.001
EV derived-miR-106a–363 cluster improves cell survival and preserves contractile rate of iCMs after ischemic injury
We speculated that the hEVs bear a unique cargo specific to the parental iCMs exposed to hypoxic environment. Our nanosight data indicated that the number of hEVs was approximately up to 30 times more than the normoxia (Fig. 2a). However, the presence of stimuli-specific EVs and their unique molecular cargo have not been reported. To identify the putative miRNAs in hEVs, we performed microarray analysis of the miRNAs inside the EVs to compare the miRs generated from the payload of hEVs vs. nEVs. Biologically significant expressions of miRNAs (> two-fold, p < 0.05) were selected using a total of 6631 homo sapiens probe set. Among the significantly altered expression of 189 miRNAs in hEVs, 53 miRNAs were down-regulated, and 136 miRNAs were up-regulated (Fig. 2b). Among these miRNA alterations, we identified a consistent group of significantly enriched 6 miRNAs, also known as miRNA 106a–363 cluster (miR cluster) in hEVs when compared to nEVs (Fig. 2b and S3A). We also examined whether this miR cluster was up-regulated after MI not only in EVs but also in mouse heart tissues. The results showed that the miR-20b, −92a, −106a, and −363 were significantly up-regulated in the PIR at day 3 post-MI (Normal control (n =4) vs. post-MI day 3 (n = 3)) (Figure S3B).
Fig. 2.

EV-derived-miR-106a–363 cluster improves iCM viability, decreases oxidative stress, and preserves contractile rate after ischemic injury. a The number of EVs secreted from normoxic and hypoxic conditions was measured. EVs were analyzed by Nanosight (vesicles/mL). Statistical significance was determined by unpaired two-tailed t test and mean data ± SD are shown (n = 4). b Venn Diagram of microarray profiling of EV derived-miRNAs shows that 53 miRNAs were down-regulated and 136 miRNAs were up-regulated in the hEVs vs. nEVs from a total of 6631 homo sapience probe set (> twofold change). The red dots indicate 6 miRNAs of miR-106a–363 cluster, which are consistently, and significantly increased in hEV. c Transfection of the contractile iCMs by FAM-labeled miR-mimic was confirmed (immunostained with cTnT antibody). Green fluorescence from FAM in the cytoplasm was captured 48 h post-transfection (White arrows). Scale bars: 50 μm. d Survival rate of hypoxia-injured iCMs was measured by MTT assay after 18 h of hypoxia. iCMs were transfected with 50 nM of the following: (1) negative control miR-mimic (Ctrl); (2) individual miR-92a, miR-20b, and miR-363 mimics; and (3) combined miR-92a, −20b, and −363 mimics. Statistical significance was determined by one-way ANOVA with multiple comparisons and mean data ± SD (n = 5). e The number of viable and dead cells in the Core-vs. Ctrl-miR transfected group was measured by ApoLive-Glo™ Multiplex Assay after exposure to 18 h of hypoxic condition. Statistical significance was determined by unpaired two-tailed t test and mean ± SD (n = 4). f Oxidative stress was evaluated by lactate dehydrogenase (LDH) release assay after 18 h of hypoxia. iCMs were transfected with 50 nM of the following: (1) negative control miR-mimic (Ctrl); (2) individual miR-92a, miR-20b, and miR-363 mimics; and (3) combined miR-92a, −20b, and −363 mimics. Statistical significance was determined by one-way ANOVA with multiple comparisons and mean data ± SD (n = 4). g Survival rate of hypoxia-injured iCMs was measured by MTT assay after 18 h of hypoxia. iCMs were either treated with hEVs or transfected with the following: (1) negative control miR inhibitors (Ctrl); (2) Core-miR inhibitors. Statistical significance was determined by one-way ANOVA with multiple comparisons and mean data ± SD (n = 4). h Measurements of the peak divergence (magnitude of contractile movement and rate) of the control (normoxia) iCMs, hypoxia-injured iCM transfected with Ctrl-miR, and Core-miRs (n = 6) were performed. Statistical significance was determined by one-way ANOVA with multiple comparisons. i Contractile rate of the iCMs in the following treatment conditions was measured: (1) Normoxic, (2) Hypoxic, (3) Ctrl-miR transfection, and (4) Core-miR transfection. Data were normalized with the normoxic control iCMs. Statistical significance was determined by one-way ANOVA with multiple comparisons and mean ± SD (n = 6). Statistically non-significant comparisons are not shown. * p < 0.05, ** p < 0.01, *** p < 0.001, ****p < 0.0001
This miR cluster is one of the paralogues of the miR-17–92 cluster and located on chromosome X in mice and humans. This cluster encodes six miRNAs: miR-106a, miR-18b, miR-19b, miR-20b, miR-92a, and miR-363 (Figure S3C). We anticipated that several key miRs may underlie the major pleiotropic effects of this miR cluster [49] and identified 3 most highly expressed miRs in hEVs (miR-hEV) compared to the miR-nEVs from microarray analysis and designated miR-20b, miR-92a, and miR-363, Core-miR. To verify the effects of miR cluster, iCMs were successfully transfected by FAM-labeled Core-miR-mimics (Fig. 2c). The optimal dose of miR-mimic transfection was decided by dose-dependent experiments (Figure S3D) and their expression levels were measured by quantitative real-time PCR (qRT-PCR) after 72 h post-transfection (Figure S3E). The cell survival of the iCMs was measured by MTT assays after hypoxic injury. Our data showed significantly increased survival rate of the Core-miR transfected iCMs when compared to the individual miR transfected groups, untransfected, or control miR-mimic (Control) transfected iCMs (Fig. 2d). We then confirmed the viability of the Core-miR transfected iCMs by ApoLive-Glo assay. The result revealed significant improvement of the viability of the Core-miR transfected group compared to the Control-miR transfected group. Furthermore, apoptosis of the Core-miR transfected iCMs also decreased significantly (Fig. 2e). Subsequently, we then measured the mitochondrial transmembrane potential (MTP) of iCMs after hypoxic injury. MTP is an important parameter of mitochondrial function used as an indicator of cell viability. We observed significantly reduced level of apoptosis in the Core-miR transfected iCMs when compared to the Control-miR transfected group after hypoxic injury (Figure S3F). Cellular toxicity after hypoxia injury assessed by a lactate dehydrogenase (LDH) release assay indicated that the Core-miR transfected iCMs were highly resistant to the hypoxic insults when compared to individual miR transfected, untransfected, and Control-miR transfected iCM groups (Fig. 2f). To test whether Core-miR is the main mediator of the therapeutic effects of hEVs, we transfected iCMs with Core-miR inhibitors. Cell viability showed a significant decrease in the Core-miR inhibitor group after hypoxia compared to the control groups. While hEV group showed significant improvement in cell viability, Core-miR inhibitor transfected iCMs treated concurrently with the hEV group showed no statistically significant improvement in cell viability (Fig. 2g). These gain/loss-of-function data indicated that Core-miR mediated the beneficial effects of hEVs in hypoxic injury. Next, we measured the contractile rate of the Core-miR transfected iCMs and consistently found that the hypoxia-injured iCMs transfected with Core-miR have significantly preserved contractile rate compared to the non-transfected hypoxic control and Control-miR transfected groups (Fig. 2h, i). Remarkably, the contractile rate of Core-miR transfected group was restored to the level almost similar to the normal control iCMs (Supplementary movie 1-3). These data confirmed that the hEVs delivered specific bioactive molecules to protect the heart from ischemic damages. This miR cluster could be one of the key mediators of the therapeutic effects of hEVs.
EV derived-miR-106a–363 cluster enhances cardiac function and preserves chamber size in ischemia-injured heart
Next, we hypothesized that the exogenous injection of the Core-miRs may promote functional restoration of the ischemia-injured murine myocardium. To address the short half-life of synthetic miR-mimics (~ 28 h) and potential off-target effects of systemic injection [47], we encapsulated the miR-mimics in an inert, biodegradable, time-release polymer-nanoparticle (PNP) [4]. The miR-mimic PNP hydrogel complex (miR-PNP, 20ul/mice) was injected into three different sites in the PIR immediately following LAD ligation and allowed for slow, local release of miRs into the injured heart. To quantify the release of miR-mimics from the miR-PNP hydrogel, we measured the release kinetics using an in vitro assay. We observed that the miR-PNP gradually released the miRs over 19 days (Figure S4A, B). We directly injected miR-PNP to the PIR of the injured myocardium immediately following LAD ligation. The functional improvement of the injured myocardium was assessed by measuring scar size, LV volumes, and LVEF, using cardiac MRI at weeks 2–4 (Fig. 3a). We confirmed that the delivered miRs were found in the intracellular cytoplasm of various cell types in the heart at 24 h post-MI (Figure S5A). We observed that the injection of the Core-miR into the PIR of the injured mouse heart significantly improved LVEF at weeks 2–4 compared to the Control-miR injected group (Fig. 3b). Furthermore, LVEDV and LVESV of the Core-miR injected hearts were significantly decreased compared to the Control-miR injected groups at weeks 2–4, respectively (Fig. 3c, d). H and E staining showed the morphological and structural differences between the groups treated with Control-miR vs. Core-miR at week-4 post-MI (Figure S6A). Also, the number of active caspase-3 + cells was significantly attenuated in the PIR regions of the Core-miR- vs Control-miR-treated groups, indicating reduced apoptosis in the Core-miR-treated heart (Figure S6B). We then assessed the histological evidence of viable cardiomyocytes in the infarct and PIR of the injured heart by measuring the cTnT + cardiomyocytes by immunofluorescence. We observed significantly increased number of viable cardiomyocytes in both infarct and PIR areas of the Core-miR-treated group compared to the Control-miR-treated group (Fig. 3e). To test whether the Core-miR may effect angiogenesis in the injured myocardium, we evaluated endothelial cells in infarct and PIR regions at week-4 post-MI. The data showed no statistical differences in the distribution of endothelial cells between Control vs. Core-miR-treated groups (Figure S7A, B). However, Masson’s-Trichrome staining analysis of the Core-miR-treated group at week-4 post-MI showed remarkably less fibrotic scar compared to the Control-miR-treated group (Fig. 3f). Quantitative real-time PCR analysis confirmed significantly increased mRNA expression level of cardiac troponin T2 (Tnnt2) in the Core-miR-treated hearts compared to the control groups (Fig. 3g). These data suggested that this miR cluster preserved cardiac function and chamber size after ischemic injury.
Fig. 3.

EV-derived-miR-106a–363 cluster enhances LV function and preserves chamber size in ischemia-injured heart. a In vivo study schematic. b–d LVEF, LV end-diastolic volume indexed to body weight (LVEDV), and LV end-systolic volume indexed to body weight (LVESV) measurements of Core-miR (miR-92a, −20b, and −363) vs. Control-miR-treated mice (n = 6) at weeks 2 and 4 post-MI. Statistical significance was determined by unpaired two-tailed t test and mean ± SD. e Measurements of cTnT (cardiomyocytes) expression in the infarct and peri-infarct region (PIR) of the ischemia-injured hearts. cTnT column: confocal microscopic images of the Ctrl-miR vs. Core-miR-injected groups demonstrated cTnT + cardiomyocytes labeled with green fluorescent protein. DAPI column: nucleus was labeled with DAPI (blue). MERGE column: cTnT + cardiomyocytes and DAPI nucleus were co-localized. The images captured eight random areas per each group. Quantitative graphs showed (right panel) the ratio of cTnT + intensity per area of the FOV (field of view). Scale bar: 100 μm. Error bars mean ± SD (Control-miR, n = 6; Core-miR, n = 6). f Trichrome-mason stain of ischemia-injured mouse heart tissue compared the extent of fibrosis in Control-miR and Core-miR injected hearts. A quantitative graph showed the percentage of scar area in the LV. Scale bar: 1 mm. Error bars mean ± SD (Control-miR, n = 6; Core-miR, n = 6). g Tnnt2 (cardiomyocyte) mRNA expression in murine myocardial tissue was measured by qRT-PCR after 4 weeks (day 28) post-MI in the following groups: (1) WT Ctrl: non-MI normal control; (2) MI Ctrl: 4 wks post-MI control; (3) MI + Control-miR: 4 wks post-MI injected with Ctrl-miR; and 4) MI + Core-miRs: 4 wks post-MI injected with Core-miRs. The expression levels were normalized to Gapdh. Statistical significance was determined by one-way ANOVA with multiple comparisons and mean ± SD (n = 4). Statistically non-significant comparisons are not shown. *p < 0.05, **p < 0.01, *** p < 0.001
Notch3 signaling pathway is a direct target of hEV derived-miR-106a–363 cluster in the cardiomyocytes
One of the key aspects of the miR cluster is that they jointly co-target different genes of the same molecular pathway [22]. To determine the mechanism by which the miR cluster mitigates ischemic injury, we examined their putative target gene expressions by analyzing the mRNA expression in iCMs using RNA-Seq quantification by Illumina HiSeq platforms with pair-end 150 bp (PE 150) sequencing. Among the 24,081 genes, the expression levels of 59 mRNAs were significantly altered in the Core-miR transfected iCMs compared to the Control-miR transfected group following exposure to hypoxia (Fig. 4a). Pearson correlation analysis among the samples, an important indicator to test the reliability of the experiments, demonstrated correlation coefficient larger than 0.92 (Figure S8A) [23]. Gene Ontology (GO) enrichment analysis identified the most differentially expressed genes, which included Notch3, cell-surface receptor, and oxidative stress signaling. The remaining GO terms included extracellular matrix (ECM) deposition such as binding and collagen trimer (Fig. 4b). We then performed the Gene Set Enrichment Analysis (GSEA) to verify the classes of genes that are differentially expressed under two different conditions. The enrichment gene set data indicated that cell cycle G1/S transition, cellular respiration, and DNA replication genes were up-regulated, while extracellular structure and collagen fibril organization genes were down-regulated by the Core-miR (Figure S8B).
Fig. 4.

EV-derived-miR-106a–363 cluster directly targets the Notch3 signaling pathway in the injured myocardium. a Heat map of the RNA-Seq data. Hierarchical clustering analysis was carried out with the log10 (FPKM + 1) of union differential expression genes of all comparison groups under different experimental conditions. Corrected P value of 0.005 and log2 (Fold change) of 1 were set as the threshold for significant differential expression. The X-axis indicates the experimental conditions: Core-miR-mimic and Ctrl-miR-mimic transfected cells. Y-axis indicates relative expression levels. Data are representative of two independent biological replicates. All samples were collected after 18 h of hypoxia (< 1% O2) following 72 h post-transfection period of each group. b Pie Chart shows Gene Ontology (GO) enrichment in the three GO terms of down-regulated genes by the core-miRs transfected iCMs. GO analysis of differentially expressed genes was implemented by the GOseq R package in which gene length bias was corrected. GO terms with corrected P value less than 0.05 were considered significantly enriched by the differentially expressed genes. c Schematic of well-established Notch signal transduction. Briefly, once the notch extracellular domain interacts with a ligand (such as, Jagged 1,2 or Delta-like 1,3,4), the enzyme, γ-secretase, cleaves and releases the intracellular domain of the notch protein. This releases the intracellular domain of the notch protein (NICD), which moves to the nucleus to activate transcription of Notch target genes via transcription factor, CSL (CBF1, Suppressor of Hairless, Lag-1) complex. Red stars indicate the genes, which are down-regulated by Core-miRs in the pathway. d Luciferase assay of wild-type Notch3 3′UTR or mutated Notch3 3′UTR in the predicted binding site of miR-20b, and miR-92a/363 (upper panel) when transfected with Control-miR and each Core-miR. Statistical significance was determined by one-way ANOVA with multiple comparisons and mean ± SD (n = 4). e Measurement of cell survival rate of siNotch3 transfected iCMs after 18 h of hypoxia insults by MTT analysis. Statistical significance was determined by one-way ANOVA with multiple comparisons and mean ± SD (n = 5). f Immunofluorescence stain evaluated Notch3 expression in both Control-miR and Core-miR injected mouse heart tissue. A quantitative graph showed the ratio of Notch3 + cardiomyocytes (cTnT + double-positive cells) in the PIR per FOV. Scale bar: 100 μm. Statistical significance was determined by unpaired two-tailed t test and mean ± SD (n = 6). g Survival rate of hypoxia-injured iCMs was measured by MTT assay after 18 h of hypoxia. iCMs were transfected with either 50 nM of miR-mimic or 2 ug of DNA plasmid: (1) negative control miRNA mimic (Ctrl); (2) combined miR-92a, −20b, and −363 (Core-miR) mimic; and (3) Notch3 plasmid. Statistical significance was determined by one-way ANOVA with multiple comparisons and mean data ± SD (n = 4). Statistically non-significant comparisons are not shown. *p < 0.05, **p < 0.01, ***p < 0.001
This intriguing result gave rise to the possibility of increased cell cycle activity in the Core-miR-treated hearts. Our observation of significant increase in cTnT expression in vivo after Core-miR treatment (Fig. 3g) may indicate the following: (1) more cardiomyocytes survived in the PIR and/or (2) cardiomyocytes proliferated in the PIR [44]. While miR-17–92 cluster has been reported to regulate cell cycle [39, 49, 63], the role of the miR-106a–363 cluster in cell cycle regulation is poorly understood. MYC is known to be the direct transcription activator of the miR-17–92 cluster and E2F to promote the transcription factors of the cell-cycle progression and DNA replication genes [1, 10]. Furthermore, previous studies demonstrated that E2F directly bind to the promoter region of miR-17–92 and 106a–363 clusters and activate the cell cycle to promote re-entry of the quiescent cells into the G1/S phase [41]. miR-17–92, miR-106b-25, and miR-106a–363 cluster sequences have high homology, perform similar biological functions, and share target genes [30].
Our bioinformatics data (Figure S8B) indicate that the cell cycle G1/S transition and DNA replication-related gene sets were up-regulated by Core-miR treatment. We hypothesized that miR cluster regulates the cell cycle of the PIR cardiomyocytes. Thus, we examined E2f1-3 mRNA level in the mouse PIR after MI and found a significant increase of the E2f1-3 mRNA expression in the Core-miR injected hearts compared to the Control groups (Figure S8C-E). These data suggested that the E2F activators may be transcriptionally up-regulated by miR-106a–363 cluster [53].
From various differential gene analyses, we also identified significantly down-regulated three Notch-related genes (Jag1, Notch3, and Hes1), which are located sequentially in the same Notch3 signaling pathway (Fig. 4c). While other candidate signaling pathways from KEGG analysis such as ECM–receptor interaction, focal adhesion, or PI3K-Akt signaling were considered, these down-regulated genes were not located sequentially (Figure S9A-D). Co-targeting of individual mRNAs sequentially by miRNAs induces specific cellular activity and strong complex patterns of repression [3, 22, 54]. The reduced mRNA expression level of these genes was confirmed by qRT-PCR (Figure S10A), and repressed Notch3 protein expression was confirmed by western blot (Figure S10B). We also confirmed that the expression of other Notch families, including Notch1, Notch2, and Notch4, were not down-regulated by the Core-miR in vitro (Figure S10C) or in vivo (Figure S10D).
Notch signaling is crucial for cell-to-cell interaction during cardiovascular development and Notch pathway components have been well-known signaling molecules to control embryonic cardiomyocyte proliferation, vascular development, and tissue homeostasis [15, 35, 46, 67]. However, the exact role of the Notch3 signaling in the ischemia-injured myocardium has yet to be clarified. We confirmed the binding sequence of the Core-miR in Notch3 3′UTR, while we did not locate the sequence in the 3′UTR of Jag1 and Hes1. To confirm the direct regulation of Notch3 by Core-miR, we verified those regulations by conducting a luciferase reporter assay. The 3′UTR of Notch3 was subcloned into pmirGLO Dual-Luciferase miRNA Target Expression Vector. The assay demonstrated that the ectopic expression of Core-miR suppressed the luciferase activity. The luciferase activity was rescued in the mutant deletion of the binding sites for the Core-miR, indicating that the Core-miR directly suppressed Notch3 expression through 3′UTR (Fig. 4d). Thus, we sought to investigate the role of Notch3 in cell survival after hypoxic injury by siRNA knock-down experiment in vitro. siRNA-Notch3 transfection efficiency was confirmed by western blot (Figure S11A). 72 h following siRNA transfection, the iCMs were exposed to hypoxia for 18 h, < 1% O2. Our data showed that siNotch3 transfected iCMs significantly improved survival rate after hypoxia compared to the siRNA control transfected iCMs (Fig. 4e). We then evaluated the expression level of Notch3 in vivo in the murine PIR tissue by counting the cTnT + /Notch3 + double-positive cells. The expression level of Notch3 positive cardiomyocytes was significantly decreased in the Core-miR vs. Control-miR-treated hearts (Fig. 4f). Next, we examined the effects of Notch3 overexpression on iCM survival after hypoxic insult. Notch3 plasmid transfection efficiency was evaluated by Western blot (Figure S12A). Cell viability showed a significant decrease in the Notch3 overexpression group after hypoxia compared to the control groups. The Core-miR group showed significant improvement of cell viability, while concurrent transfection of Core-miR and Notch3 showed statistically no improvement of cell viability (Fig. 4g). These findings confirmed the direct interaction between miR cluster and Notch3 signaling pathway to attenuate the ischemic injury both in vitro in the hypoxic iCMs and in vivo in the ischemic myocardium.
miR106a-363 cluster reconfigures the expression of cell cycle genes in iCMs via Notch3 signaling pathway in response to hypoxic injury
It is well known that the cardiomyocytes in mammals rapidly proliferate during the embryonic stage but exit the cell cycle soon after birth [2]. The inability of adult cardiomyocytes to regenerate the injured myocardium represents a major limitation in cardiac restoration. Thus, there have been efforts to re-activate the cardiomyocytes to proliferate [24, 44, 61].
To confirm the molecular pathway, we studied whether the miR cluster could activate the cell cycle re-entry from the quiescent stage through Notch3 repression. Cell cycle arrest in cardiomyocytes is normally accompanied by down-regulation of positive regulators and up-regulation of negative regulators (cyclin-dependent kinases inhibitors, CDKIs) [2, 6]. We examined the expression of these genes in the iCMs exposed to hypoxic injury and treated with Control-miR vs. Core-miR and to normoxic iCMs. Compared to the Control-miR, Core-miR transfected iCMs had significantly increased mRNA levels of positive cell cycle regulators involved in G1/S transition (Cyclin D, CDK4) and G2/M phase (PLK-1, Cyclin A), and significantly decreased expression of CDKIs, including p21cip1 and p27kip1 (Fig. 5a). This finding correlated with the significantly increased expression of the Tnnt2 and E2f1-3 genes in the Core-miR-treated heart (Fig. 3g, Figure S8C-E). To test whether the miR cluster is able to activate cardiomyocyte proliferation, we performed EdU incorporation to the iCMs transfected with Core-miR and Control-miR-mimic. We examined other cell proliferation marker expression, including Ki-67 and Aurora B, by subsequent immunofluorescence staining. EdU detects newly synthesized DNA mostly during the active S phase [55]; Ki-67 is employed to confirm the activation of cell cycle at G1, S, G2, and M phases [8]; and Aurora B is utilized to assess the activation of M phase [14]. The results showed significantly increased EdU+ signal in the Core-miR transfected iCMs (cTnT+) at 72 h post-transfection when compared to the normoxic control and Control-miR transfected iCM groups. Similarly, we observed significantly increased Ki-67 and Aurora B signal in Core-miR transfected iCMs when compared to the control groups (normoxic and Ctrl-miR transfection, Fig. 5b), confirming that miR cluster stimulates cell cycle re-entry of the iCMs. To assess the cell cycle distribution and cell proliferation, we performed the Propidium Iodide (PI) flow cytometry analysis to analyze DNA content and cell cycle activation. The stagger offset histogram exhibited the distribution states of each cycle. Compared to the control, a significant increase in G2/M population was found in the group of iCMs transfected with the Core-miR. Mean-while, the G0/G1 population of the Core-miR transfected iCMs was proportionally decreased. Although the S phase of the Core-miR transfected iCMs was slightly increased, it was not statistically significant (Figure S13A). To verify whether the regulation of cell cycle genes is mediated by the miR cluster-Notch3 axis, we tested the expression levels of cell cycle-related genes in siRNA-Notch3 knock-down iCMs. Notably, we observed the similar changes in all cell cycle-related gene expressions, including positive cell cycle regulators and CDKIs, in the siRNA-Notch3 (siNotch3) group compared to the non-transfected and siRNA control groups (Fig. 5c). Similarly, siNotch3 iCMs demonstrated up-regulation of EdU, Ki-67, and Aurora B (Fig. 5d). We also confirmed the expression of cell-cycle regulator genes in Notch3 over-expressed iCMs. The results showed that the Notch3 over-expressed iCMs had significantly decreased mRNA levels of positive cell cycle regulators (Cyclin D, CDK4, and PLK-1, Cyclin A) and significantly increased expression of CDKIs (p21cip1, p27kip1) compared to controls (Figure S12B). Finally, PI labeling of siNotch3 iCMs revealed an analogous cell cycle distribution to the Core-miR transfected group (Figure S13B). Together, these data suggested that miR cluster stimulates iCM cell cycle activity via Notch3 signaling pathway.
Fig. 5.

EV derived-miR-106a–363 cluster reconfigures the cell cycle regulators of cardiomyocytes via Notch3 repression. mRNA expression analysis of cell cycle positive regulators (G1/S and G2/M) and negative regulators (CDKIs) in iCMs in vitro. a Control-miR and Core-miR transfected iCMs were exposed to hypoxic insult for 18 h at 72 h post-transfection. qRT-PCR analysis evaluated the expression level of cell-cycle genes. The expression levels were normalized to GAPDH. Statistical significance was determined by one-way ANOVA with multiple comparison and mean ± SD (n = 4). b Cell-cycle protein expression immunofluorescent markers were measured: (1) EdU (5-ethynyl-2′-deoxyuridine) for S Phase, (2) Ki-67 for G1/S/G2/M Phase, and (3) Aurora B for M Phase. All groups were analyzed at 72 h post-transfection of Control-miR and Core-miR. Fluorescence colors for each antibody are indicated in the figure panels: nucleus (DAPI blue), cell cycle (Edu, Ki-67, and Aurora B), and cTnT. All cell cycle markers were located in the nucleus. Other green dot signals present in cytoplasm area are FAM-labeled miRs. Only the cell cycle marker+/cTnT+ double-positive mononuclear cells were counted from eight random areas per each group (white arrows point to the double-positive cells). Scale bars: 100 μm. All statistical significance was determined by one-way ANOVA with multiple comparisons and mean ± SD (n = 3). c siNotch3 and control siRNA transfected iCMs were exposed to hypoxia insult for 18 h at 72 h post-transfection. qRT-PCR analysis evaluated the expression level of cell-cycle genes. The expression levels were normalized to GAPDH. Statistical significance was determined by one-way ANOVA with multiple comparisons and mean ± SD (n = 4). d Expression of cell cycle markers (EdU: S Phase, Ki-67: G1/S/G2/M Phase and Aurora B: M Phase) were analyzed by immunofluorescence staining. All groups were analyzed at 72 h post-transfection of sicontrol, and siNotch3 transfected iCMs. Fluorescence color for each antibody is indicated in the figure panel: nucleus (Blue), cell cycle (Edu, Ki-67, and Aurora B), and cTnT. Only the cell cycle marker+ and cTnT+ double-positive mononuclear cells were counted from eight random areas per each group (white arrows point to the double-positive cells). Scale bars: 100 μm. All statistical significance was determined by one-way ANOVA with multiple comparisons and mean ± SD (n = 3). Statistically non-significant comparisons are not shown. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
miR106a-363 cluster promoted cell cycle re-entry and proliferation of cardiomyocytes via Notch3 signaling pathway in the injured murine myocardium
We next tested whether Core-miR could stimulate cardiomyocyte cell cycle activation and proliferation in the adult murine heart. The mice underwent acute MI, treated with Control- or Core-miR, and received 50 μg/g of EdU at day 3 post-MI. Animals were sacrificed 24 h later. We evaluated EdU incorporation and Ki-67 and Aurora B expression in cTnT+ cardiomyocytes ex vivo. Compared to the Control-miR group, the mice treated with Core-miR had on average 10*-, 15*-, and 43*-fold increase in EdU+, Ki67+, and Aurora B+ cardiomyocytes (*p < 0.001), respectively (Fig. 6a-c). qRT-PCR analysis of the myocardium at days 4 and 28 post-MI revealed significant up-regulation of positive cell cycle regulators (G1/S and G2/M) in the Core-miR group, suggesting sustained cell cycle activation. CDKIs were significantly down-regulated in the Core-miR group (Fig. 7a, b) on day 4, but up-regulated on day 28. At this timepoint, the fold-change difference in CDKIs was remarkably higher than that of positive regulator expression (> 30*- vs. > eightfold increase, *p < 0.001). We speculate that the CDKIs were reactivated to arrest the cell cycle by the 4th week post-MI to maintain homeostasis of the heart by a physiologic bistability system to balance the positive and negative feedback loops, representing a synchronous biochemical switch for cell cycle regulations [50, 57]. Together, the exogenous administration of Core-miR to the adult ischemic heart stimulated cardiomyocyte cell cycle re-entry and proliferation of the pre-existing, injured cardiomyocytes in the PIR to mitigate potentially irreversible damage.
Fig. 6.

EV-derived-miR-106a–363 cluster activates cell cycle re-entry and proliferation in the injured myocardium. EdU incorporation of Control-miR and Core-miR injected mouse heart tissue was analyzed. EdU (50 μg/g) was injected via IP (intra-peritoneal) at day 3 post-MI. After 24 h, mice were sacrificed and their heart was collected. Cryo-sectioned tissue (8 μm/section) was stained by immunofluorescence specific for a EdU (green) and cTnT (red), b Ki-67 (green) and cTnT (red), and c Aurora B (green) and cTnT (red). Cells were counted from six random areas per each group (white arrows point to the double-positive cells). All quantitative graph showed the measurements of the number of cell cycle markers and cTnT double-positive cells in the peri-infarct region (PIR). Scale bar: 50 μm. Statistical significance was determined by unpaired two-tailed t-test and mean ± SD (n = 3). **P < 0.01
Fig. 7.
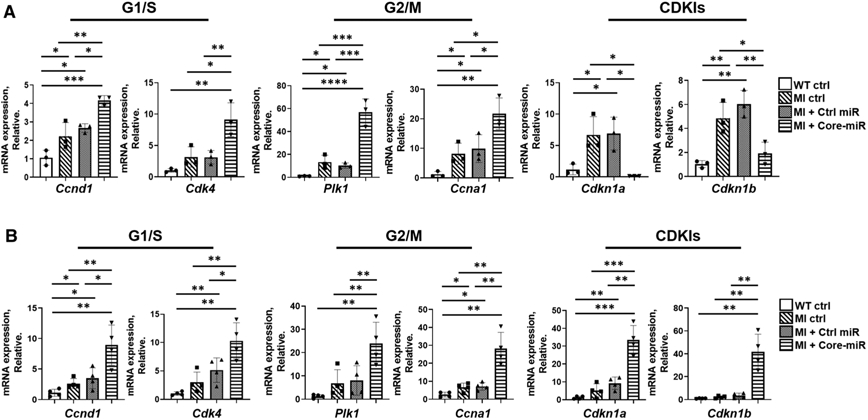
EV-derived-miR-106a–363 cluster activates cell cycle re-entry and proliferation in the injured myocardium until week-4 post-MI. a mRNA expression of cell cycle (G1/S, and G2/M) positive regulators and negative regulators (CDKIs) were analyzed in vivo. qRT-PCR analysis of in vivo mouse heart tissue was performed. Samples collected and analyzed at day 4 post-MI. Statistical significance was determined by one-way ANOVA with multiple comparisons and mean ± SD (n = 3). b qRT-PCR analysis of cell cycle marker in mouse heart tissues at week 4 (day 28) post-MI: (1) WT Ctrl: non-MI normal control; (2) MI Ctrl; (3) MI + Ctrl-miR; and (4) MI + Core-miRs. The expression levels were normalized to Gapdh. Statistical significance was determined by one-way ANOVA with multiple comparisons and mean ± SD (n = 4). Statistically non-significant comparisons are not shown. *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
Homeostatic response is triggered after an ischemic injury to the heart. This process includes structural and functional reactions such as myocardial hypertrophy, fibrosis, and remodeling to compensate for the failing heart. Proliferation of the cardiomyocytes has been investigated as a potential mechanism to restore and/or regenerate the injured myocardium [19]. Recent findings have demonstrated the ability of cardiomyocytes to trigger cell cycle re-entry, proliferation, and cardiac regeneration [24, 44]. These discoveries led to rigorous studies of novel therapeutic approaches to activate cardiomyocyte proliferation to preserve myocardial function.
EV contents vary depending on the physiologic condition of the parent cells. Our studies suggest that the iCMs secrete EVs with varying payload depending on their maturity. In our previous study by Santoso et al., we used relatively immature iCMs (< 30 (average 15) days of spontaneous contractility) for the experiments [56]. In contrast, this study employed more mature iCMs [> 30 (average 50) days of spontaneous contractility]. Furthermore, our most recent study by Ikeda et al. confirmed the differential therapeutic effects of EVs from immature vs. mature iCMs [JACC, 2021, in press]. EVs from the immature iCMs showed higher therapeutic efficacy in vitro in hypoxia-injured iCMs and in vivo in ischemia-injured murine myocardium. These studies confirmed the differential effects of EVs based on their maturity.
Moreover, our study indicated that in vivo delivery methods may result in variable effects of EVs based on their release kinetics and dynamics. We observed differential therapeutic effects of EVs when using biodegradable PNP hydrogels compared to matrigel in our previous study by Santoso et al. Encapsulation of EVs for sustained release has been studied using various formulations. These studies showed that each material has variable release kinetics and duration [9, 51], suggesting that the delivery dynamics of EVs should be considered as an important parameter for effective in vivo delivery.
In this study, we analyzed the molecular cargo of EVs and verified the putative role of a specific miRNA 106a–363 cluster to protect the ischemia-injured heart. The expression level of this cluster is increased during embryonic development while mostly silenced in the post-embryonic somatic cells. This cluster, often observed in cancer, modulates the epigenetic changes during the disease progression and regulates the downstream signaling of cell growth, proliferation, and cell-cycle re-entry [18, 34, 63]. Our study demonstrated that the EVs derived from iCMs contain a specific miR cluster, which stimulates cell cycle re-entry of cardiomyocytes by repressing the Notch3 signaling pathway. Exogenous administration of the miR cluster into the injured myocardium triggered cardiomyocyte proliferation.
Notch signaling pathway is highly conserved in all vertebrates and represents a critical molecular pathway for angiogenesis [33], cardiomyogenesis [42], and cardiac remodeling [20]. Moreover, Notch3 is closely associated with cardiac fibrosis and remodeling in chronic heart failure [48, 66] and with vascular smooth muscle cell maturation in post-embryonic state [17]. Several studies reported that Notch3 overexpression induces cell cycle arrest in breast cancer [13], trophoblast [68], and hemangioma-derived pericytes [27]. These studies implicate an essential role for Notch3 in the proliferative response to pathophysiological alteration during the post-natal stage. Although our data confirmed the important role of miR-106a–363 cluster and Notch3, some challenges still remain.
Limitation
Our data identified Core-miR as the 3 dominant miRs in the cluster. Investigation of all 6 miRs is needed to evaluate the definite role of miR cluster. Furthermore, Core-miR treatment significantly reduced CDKI expression at day 4 post-MI, which then increased at day 28. While this could explain how cell cycle-related genes are tightly regulated, this key homeostatic response requires further investigation. The temporal profile of this proliferatve response and its associated genetic modulation are needed to understand the specific physiologic effects of this novel finding [21].
Another limitation in measuring the cardiomyocyte division is the differential responses to the tightly regulated microenvironment. Studies have reported that the cardiomyocytes undergo karyokinesis without cytokinesis and become bi-, multi-nucleated after cell cycle re-entry [5, 36]. However, there is no clear explanation for the cell cycle arrest phenomenon. Various signaling pathways regulate the cell cycle genes and maintain homeostatic balance of the heart. We carefully counted and analyzed only the mononuclear iCMs and excluded the multi-nucleated iCMs since only the mononuclear cardiomyocytes divide properly during cytokinesis [5]. Although the current study showed a significant increase in the expression of proliferation markers in the cardiomyocytes of miR-treated mouse hearts, we acknowledge the inherent difficulties in reliably localizing EdU, Ki-67, and Aurora B specifically to cardiomyocyte nuclei in simple tissue sections. We will investigate the cardiomyocyte proliferation in isolated cardiomyocytes from genetically labeled adult mice such as the mosaic analysis with double marker (MADM) mouse model, which allow two daughter cells of a dividing cell to be indelibly and uniquely labeled by either GFP or RFP [69].
Finally, in this study, we used PEG precipitation for EV isolation. PEG precipitation has been long used to isolate proteins, nucleic acids, and other small particles [65]. This is a cost-effective and simple procedure, and preserves high ion concentration and EV integrity. However, the main limitation of this method is the possible contamination by other proteins, non-EV nucleoproteins, and remaining polymer, which limits the reproducibility of EV protein content measurement [32, 40]. These impurities are removed by subsequent centrifugation or filtration [59].
Conclusion
The significant improvement in cardiac function of mice hearts following an ischemic insult could be due to repression of Notch3 signaling pathway by exogenous miR cluster treatment. Our study demonstrated a potential mechanistic link between cell cycle re-entry and augmentation of cardiac restoration. This finding may provide a new therapeutic target to treat HF patients.
Supplementary Material
Acknowledgements
We thank Dr. Kyuho Han (Department of Genetics, Stanford University), and Dr. Hokyung Kay Chung (Department of Biology, Stanford University) for critical discussion in this project and sharing experimental protocols and reagents.
Funding
This work was supported by National Heart, Lung, and Blood Institute (NHLBI), National Institutes of Health (NIH) [K24 HL130553; UM1 HL113456] and the American Heart Association (AHA) [18POST34080005; 19CSLOI34700047-l].
Abbreviations
- iPSC
Induced pluripotent stem cell
- iCM
IPSC-derived iCM
- EVs
Extracellular vesicles
- nEV
EVs from normoxic iCM
- hEV
EVs from hypoxic iCM
- miR cluster
MiR-106a–363 cluster
- Core-miR
MiR-20b, miR-92a, miR-363
- MI
Myocardial infarction
- PIR
Peri-infarct region
- EdU
5-Ethynyl-2′-deoxyuridine
Footnotes
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s00395-021-00858-8.
Conflict of interest None declared.
References
- 1.Aguda BD, Kim Y, Piper-Hunter MG, Friedman A, Marsh CB (2008) MicroRNA regulation of a cancer network: consequences of the feedback loops involving miR-17-92, E2F, and Myc. Proc Natl Acad Sci USA 105:19678–19683. 10.1073/pnas.0811166106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahuja P, Sdek P, MacLellan WR (2007) Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev 87:521–544. 10.1152/physrev.00032.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altuvia Y, Landgraf P, Lithwick G, Elefant N, Pfeffer S, Aravin A, Brownstein MJ, Tuschl T, Margalit H (2005) Clustering and conservation patterns of human microRNAs. Nucleic Acids Res 33:2697–2706. 10.1093/nar/gki567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Appel EA, Tibbitt MW, Webber MJ, Mattix BA, Veiseh O, Langer R (2015) Self-assembled hydrogels utilizing polymer-nanoparticle interactions. Nat Commun 6:6295. 10.1038/ncomms7295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bersell K, Arab S, Haring B, Kühn B (2009) Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138:257–270. 10.1016/j.cell.2009.04.060 [DOI] [PubMed] [Google Scholar]
- 6.Besson A, Dowdy SF, Roberts JM (2008) CDK Inhibitors: cell cycle regulators and beyond. Dev Cell 14:159–169. 10.1016/j.devcel.2008.01.013 [DOI] [PubMed] [Google Scholar]
- 7.Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S, Kaluza D, Tréguer K, Carmona G, Bonauer A, Horrevoets AJG, Didier N, Girmatsion Z, Biliczki P, Ehrlich JR, Katus HA, Müller OJ, Potente M, Zeiher AM, Hermeking H, Dimmeler S (2013) MicroRNA-34a regulates cardiac ageing and function. Nature 495:107–110. 10.1038/nature11919 [DOI] [PubMed] [Google Scholar]
- 8.Bruno S, Darzynkiewicz Z (1992) Cell cycle dependent expression and stability of the nuclear protein detected by Ki-67 antibody in HL-60 cells. Cell Prolif 25:31–40. 10.1111/j.1365-2184.1992.tb01435.x [DOI] [PubMed] [Google Scholar]
- 9.Bunggulawa EJ, Wang W, Yin T, Wang N, Durkan C, Wang Y, Wang G (2018) Recent advancements in the use of exosomes as drug delivery systems. J Nanobiotechnol 16:81. 10.1186/s12951-018-0403-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cam H, Dynlacht BD (2003) Emerging roles for E2F: beyond the G1/S transition and DNA replication. Cancer Cell 3:311–316. 10.1016/S1535-6108(03)00080-1 [DOI] [PubMed] [Google Scholar]
- 11.Camussi G, Deregibus M-C, Bruno S, Grange C, Fonsato V, Tetta C (2011) Exosome/microvesicle-mediated epigenetic reprogramming of cells. Am J Cancer Res 1:98–110 [PMC free article] [PubMed] [Google Scholar]
- 12.Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang M-L, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MVG, Høydal M, Autore C, Russo MA, Dorn GW, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G (2007) MicroRNA-133 controls cardiac hypertrophy. Nat Med 13:613–618. 10.1038/nm1582 [DOI] [PubMed] [Google Scholar]
- 13.Chen C-F, Dou X-W, Liang Y-K, Lin H-Y, Bai J-W, Zhang X-X, Wei X-L, Li Y-C, Zhang G-J (2015) Notch3 overexpression causes arrest of cell cycle progression by inducing Cdh1 expression in human breast cancer cells. Cell Cycle 15:432–440. 10.1080/15384101.2015.1127474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, Sen S, Allis CD, Sassone-Corsi P (2002) Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol 22:874–885. 10.1128/mcb.22.3.874-885.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D’Amato G, Luxán G, del Monte-Nieto G, Martínez-Poveda B, Torroja C, Walter W, Bochter MS, Benedito R, Cole S, Martinez F, Hadjantonakis A-K, Uemura A, Jiménez-Borreguero LJ, de la Pompa JL (2016) Sequential Notch activation regulates ventricular chamber development. Nat Cell Biol 18:7–20. 10.1038/ncb3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dash R, Kim PJ, Matsuura Y, Ikeno F, Metzler S, Huang NF, Lyons JK, Nguyen PK, Ge X, Foo CWP, McConnell MV, Wu JC, Yeung AC, Harnish P, Yang PC (2015) Manganese-enhanced magnetic resonance imaging enables in vivo confirmation of peri-infarct restoration following stem cell therapy in a porcine ischemia-reperfusion model. J Am Heart Assoc, 10.1161/JAHA.115.002044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, Krebs LT, Klonjkowski B, Berrou E, Mericskay M, Li Z, Tournier-Lasserve E, Gridley T, Joutel A (2004) Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev 18:2730–2735. 10.1101/gad.308904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dylla L, Jedlicka P (2013) Growth-promoting role of the miR-106a~363 Cluster in Ewing Sarcoma. PLoS ONE 8:e63032. 10.1371/journal.pone.0063032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engel FB (2005) Cardiomyocyte proliferation: a platform for mammalian cardiac repair. Cell Cycle 4:1360–1363. 10.4161/cc.4.10.2081 [DOI] [PubMed] [Google Scholar]
- 20.Ferrari R, Rizzo P (2014) The Notch pathway: a novel target for myocardial remodelling therapy? Eur Heart J 35:2140–2145. 10.1093/eurheartj/ehu244 [DOI] [PubMed] [Google Scholar]
- 21.Gabisonia K, Prosdocimo G, Aquaro GD, Carlucci L, Zentilin L, Secco I, Ali H, Braga L, Gorgodze N, Bernini F, Burchielli S, Collesi C, Zandonà L, Sinagra G, Piacenti M, Zacchigna S, Bussani R, Recchia FA, Giacca M (2019) MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature 569:418–422. 10.1038/s41586-019-1191-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grün D, Wang Y-L, Langenberger D, Gunsalus KC, Rajewsky N (2005) microRNA target predictions across seven Drosophila species and comparison to mammalian targets. PLoS Comput Biol 1:e13. 10.1371/journal.pcbi.0010013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hansen KD, Irizarry RA, Wu Z (2012) Removing technical variability in RNA-seq data using conditional quantile normalization. Biostat Oxf Engl 13:204–216. 10.1093/biostatistics/kxr054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang W, Feng Y, Liang J, Yu H, Wang C, Wang B, Wang M, Jiang L, Meng W, Cai W, Medvedovic M, Chen J, Paul C, Davidson WS, Sadayappan S, Stambrook PJ, Yu X-Y, Wang Y (2018) Loss of microRNA-128 promotes cardiomyocyte proliferation and heart regeneration. Nat Commun 9:700. 10.1038/s41467-018-03019-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hulsmans M, Holvoet P (2013) MicroRNA-containing microvesicles regulating inflammation in association with atherosclerotic disease. Cardiovasc Res 100:7–18. 10.1093/cvr/cvt161 [DOI] [PubMed] [Google Scholar]
- 26.Ibrahim AG-E, Cheng K, Marbán E (2014) Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Rep 2:606–619. 10.1016/j.stemcr.2014.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji Y, Chen S, Xiang B, Li Y, Li L, Wang Q (2016) Jagged1/Notch3 signaling modulates hemangioma-derived pericyte proliferation and maturation. Cell Physiol Biochem 40:895–907. 10.1159/000453148 [DOI] [PubMed] [Google Scholar]
- 28.Jung J-H, Fu X, Yang PC (2017) Exosomes generated from iPSC-derivatives: new direction for stem cell therapy in human heart diseases. Circ Res 120:407–417. 10.1161/CIRCRESAHA.116.309307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khan M, Nickoloff E, Abramova T, Johnson J, Verma SK, Krishnamurthy P, Mackie AR, Vaughan E, Garikipati VNS, Benedict C, Ramirez V, Lambers E, Ito A, Gao E, Misener S, Luongo T, Elrod J, Qin G, Houser SR, Koch WJ, Kishore R (2015) Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circ Res 117:52–64. 10.1161/CIRCRESAHA.117.305990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khuu C, Utheim TP, Sehic A (2016) The three paralogous MicroRNA clusters in development and disease, miR-17-92, miR-106a–363, and miR-106b-25. Scientifica. 10.1155/2016/1379643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim PJ, Mahmoudi M, Ge X, Matsuura Y, Toma I, Metzler S, Kooreman NG, Ramunas J, Holbrook C, McConnell MV, Blau H, Harnish P, Rulifson E, Yang PC (2015) Direct evaluation of myocardial viability and stem cell engraftment demonstrates salvage of the injured myocardium. Circ Res 116:e40–50. 10.1161/CIRCRESAHA.116.304668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Konoshenko MY, Lekchnov EA, Vlassov AV, Laktionov PP (2018) Isolation of Extracellular Vesicles: General Methodologies and Latest Trends. In: BioMed Res. Int https://www.hindawi.com/journals/bmri/2018/8545347/. Accessed 9 Jan 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kume T (2012) Ligand-dependent Notch signaling in vascular formation. Adv Exp Med Biol 727:210–222. 10.1007/978-1-4614-0899-4_16 [DOI] [PubMed] [Google Scholar]
- 34.Landais S, Landry S, Legault P, Rassart E (2007) Oncogenic potential of the miR-106-363 Cluster and Its implication in human T-Cell Leukemia. Cancer Res 67:5699–5707. 10.1158/0008-5472.CAN-06-4478 [DOI] [PubMed] [Google Scholar]
- 35.Lawson ND, Weinstein BM (2002) Arteries and veins: making a difference with zebrafish. Nat Rev Genet 3:674–682. 10.1038/nrg888 [DOI] [PubMed] [Google Scholar]
- 36.Leone M, Magadum A, Engel FB (2015) Cardiomyocyte proliferation in cardiac development and regeneration: a guide to methodologies and interpretations. Am J Physiol Heart Circ Physiol 309:H1237–1250. 10.1152/ajpheart.00559.2015 [DOI] [PubMed] [Google Scholar]
- 37.Li J, Li Y, Jiao J, Wang J, Li Y, Qin D, Li P (2014) Mitofusin 1 is negatively regulated by microRNA 140 in cardiomyocyte apoptosis. Mol Cell Biol 34:1788–1799. 10.1128/MCB.00774-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li M, Zhou Y, Xia T, Zhou X, Huang Z, Zhang H, Zhu W, Ding Q, Wang S (2018) Circulating microRNAs from the miR-106a–363 cluster on chromosome X as novel diagnostic biomarkers for breast cancer. Breast Cancer Res Treat 170:257–270. 10.1007/s10549-018-4757-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu XS, Chopp M, Wang XL, Zhang L, Hozeska-Solgot A, Tang T, Kassis H, Zhang RL, Chen C, Xu J, Zhang ZG (2013) MicroRNA-17-92 Cluster mediates the proliferation and survival of neural progenitor cells after stroke. J Biol Chem 288:12478–12488. 10.1074/jbc.M112.449025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lobb RJ, Becker M, Wen SW, Wong CSF, Wiegmans AP, Leimgruber A, Möller A (2015) Optimized exosome isolation protocol for cell culture supernatant and human plasma. J Extracell Vesicles 4:27031. 10.3402/jev.v4.27031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo W, Li G, Yi Z, Nie Q, Zhang X (2016) E2F1-miR-20a-5p/20b-5p auto-regulatory feedback loop involved in myoblast proliferation and differentiation. Sci Rep 6:27904. 10.1038/srep27904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacGrogan D, Nus M, de la Pompa JL (2010) Notch signaling in cardiac development and disease. Curr Top Dev Biol 92:333–365. 10.1016/S0070-2153(10)92011-5 [DOI] [PubMed] [Google Scholar]
- 43.Mercola M, Ruiz-Lozano P, Schneider MD (2011) Cardiac muscle regeneration: lessons from development. Genes Dev 25:299–309. 10.1101/gad.2018411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mohamed TMA, Ang Y-S, Radzinsky E, Zhou P, Huang Y, Elfenbein A, Foley A, Magnitsky S, Srivastava D (2018) Regulation of cell cycle to stimulate adult cardiomyocyte proliferation and cardiac regeneration. Cell 173:104–116.e12. 10.1016/j.cell.2018.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nadal-Ginard B, Ellison GM, Torella D (2014) The cardiac stem cell compartment is indispensable for myocardial cell homeostasis, repair and regeneration in the adult. Stem Cell Res 13:615–630. 10.1016/j.scr.2014.04.008 [DOI] [PubMed] [Google Scholar]
- 46.Niessen K, Karsan A (2008) Notch signaling in cardiac development. Circ Res 102:1169–1181. 10.1161/CIRCRESAHA.108.174318 [DOI] [PubMed] [Google Scholar]
- 47.Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, Fu C, Lindow M, Stenvang J, Straarup EM, Hansen HF, Koch T, Pappin D, Hannon GJ, Kauppinen S (2011) Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet 43:371–378. 10.1038/ng.786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Øie E, Sandberg WJ, Ahmed MS, Yndestad A, Lærum OD, Attramadal H, Aukrust P, Eiken HG (2010) Activation of Notch signaling in cardiomyocytes during post-infarction remodeling. Scand Cardiovasc J SCJ 44:359–366. 10.3109/14017431.2010.511256 [DOI] [PubMed] [Google Scholar]
- 49.Olive V, Bennett MJ, Walker JC, Ma C, Jiang I, Cordon-Cardo C, Li Q-J, Lowe SW, Hannon GJ, He L (2009) miR-19 is a key oncogenic component of mir-17-92. Genes Dev 23:2839–2849. 10.1101/gad.1861409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pomerening JR, Sontag ED, Ferrell JE (2003) Building a cell cycle oscillator: hysteresis and bistability in the activation of Cdc2. Nat Cell Biol 5:346–351. 10.1038/ncb954 [DOI] [PubMed] [Google Scholar]
- 51.Riau AK, Ong HS, Yam GHF, Mehta JS (2019) Sustained delivery system for stem cell-derived exosomes. Front Pharmacol. 10.3389/fphar.2019.01368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Rooij E, Olson EN (2009) Searching for miR-acles in cardiac fibrosis. Circ Res 104:138–140. 10.1161/CIRCRESAHA.108.192492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.del Rosario RCH, Damasco JRCG, Aguda BD (2016) MicroRNA inhibition fine-tunes and provides robustness to the restriction point switch of the cell cycle. Sci Rep 6:32823. 10.1038/srep32823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ryazansky SS, Gvozdev VA, Berezikov E (2011) Evidence for post-transcriptional regulation of clustered microRNAs in Drosophila. BMC Genomics 12:371. 10.1186/1471-2164-12-371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salic A, Mitchison TJ (2008) A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci 105:2415–2420. 10.1073/pnas.0712168105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santoso MR, Ikeda G, Tada Y, Jung J-H, Vaskova E, Sierra RG, Gati C, Goldstone AB, von Bornstaedt D, Shukla P, Wu JC, Wakatsuki S, Woo YJ, Yang PC (2020) Exosomes from induced pluripotent stem cell-derived cardiomyocytes promote autophagy for myocardial repair. J Am Heart Assoc 9:e014345. 10.1161/JAHA.119.014345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Skotheim JM, Di Talia S, Siggia ED, Cross FR (2008) Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature 454:291–296. 10.1038/nature07118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tachibana A, Santoso MR, Mahmoudi M, Shukla P, Wang L, Bennett M, Goldstone AB, Wang M, Fukushi M, Ebert AD, Woo YJ, Rulifson E, Yang PC (2017) Paracrine effects of the pluripotent stem cell-derived cardiac myocytes salvage the injured myocardium. Circ Res 121:e22–e36. 10.1161/CIRCRESAHA.117.310803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taylor DD, Shah S (2015) Methods of isolating extracellular vesicles impact down-stream analyses of their cargoes. Methods San Diego Calif 87:3–10. 10.1016/j.ymeth.2015.02.019 [DOI] [PubMed] [Google Scholar]
- 60.Teng X, Chen L, Chen W, Yang J, Yang Z, Shen Z (2015) Mesenchymal stem cell-derived exosomes improve the microenvironment of infarcted myocardium contributing to angiogenesis and anti-inflammation. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol 37:2415–2424. 10.1159/000438594 [DOI] [PubMed] [Google Scholar]
- 61.Tian Y, Liu Y, Wang T, Zhou N, Kong J, Chen L, Snitow M, Morley M, Li D, Petrenko N, Zhou S, Lu M, Gao E, Koch WJ, Stewart KM, Morrisey EE (2015) A microRNA-Hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci Transl Med 7:279ra38. 10.1126/scitranslmed.3010841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9:654–659. 10.1038/ncb1596 [DOI] [PubMed] [Google Scholar]
- 63.Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, Jaenisch R, Sharp PA, Jacks T (2008) Targeted deletion reveals essential and overlapping functions of the miR-17~92 family of miRNA clusters. Cell 132:875–886. 10.1016/j.cell.2008.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu J, Bao J, Kim M, Yuan S, Tang C, Zheng H, Mastick GS, Xu C, Yan W (2014) Two miRNA clusters, miR-34b/c and miR-449, are essential for normal brain development, motile ciliogenesis, and spermatogenesis. Proc Natl Acad Sci USA 111:E2851–2857. 10.1073/pnas.1407777111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamamoto KR, Alberts BM, Benzinger R, Lawhorne L, Treiber G (1970) Rapid bacteriophage sedimentation in the presence of polyethylene glycol and its application to large-scale virus purification. Virology 40:734–744. 10.1016/0042-6822(70)90218-7 [DOI] [PubMed] [Google Scholar]
- 66.Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B, Zhang Y, Xu C, Bai Y, Wang H, Chen G, Wang Z (2007) The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med 13:486–491. 10.1038/nm1569 [DOI] [PubMed] [Google Scholar]
- 67.Zhao L, Borikova AL, Ben-Yair R, Guner-Ataman B, MacRae CA, Lee RT, Burns CG, Burns CE (2014) Notch signaling regulates cardiomyocyte proliferation during zebrafish heart regeneration. Proc Natl Acad Sci 111:1403–1408. 10.1073/pnas.1311705111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao W-X, Wu Z-M, Liu W, Lin J-H (2017) Notch2 and Notch3 suppress the proliferation and mediate invasion of trophoblast cell lines. Biol Open 6:1123–1129. 10.1242/bio.025767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zong H, Espinosa JS, Su HH, Muzumdar MD, Luo L (2005) Mosaic analysis with double markers in mice. Cell 121:479–492. 10.1016/j.cell.2005.02.012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.