

Abstract
Water is key to protein structure and stability, yet the relationship between protein–water interactions and structure is poorly understood, in part because there are few techniques that permit the study of dehydrated protein structure at high resolution. Here, we describe liquid-observed vapor exchange (LOVE) NMR, a solution NMR-based method that provides residue-level information about the structure of dehydrated proteins. Using the model protein GB1, we show that LOVE NMR measurements reflect the fraction of the dried protein population trapped in a conformation where a given residue is protected from exchange with D2O vapor. Comparisons to solution hydrogen–deuterium exchange data affirm that the dried protein structure is strongly influenced by local solution stability and that the mechanism of dehydration protection exerted by the widely used protectant trehalose differs from its mechanism of stabilization in solution. Our results highlight the need for refined models of cosolute-mediated dehydration protection and demonstrate the ability of LOVE NMR to inform such models.
Graphical Abstract
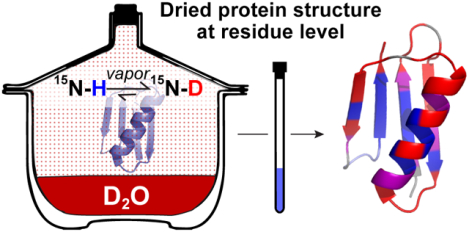
It is well-established that liquid water is necessary for proteins to realize their native structure and function,1 yet uncovering how protein–water interactions contribute to protein stability and structure remains an ongoing endeavor.2,3 Our understanding of this fundamental interaction has been limited in part by the technological inability to observe how water removal affects local protein structure.4 The same restrictions make it challenging to understand how protective molecules, collectively known as excipients,5,6 prevent dehydration-induced protein damage.
Our limited understanding of dehydrated protein structure poses a hurdle for distributing protein-based therapeutics such as vaccines, antibodies, and other biologic drugs, for which dried formulations are in high demand due to their enhanced thermostability and shelf life.7 The advent of new methods for studying dry protein structure at high resolution would inform and accelerate formulation,8 allowing more protein-based therapeutics to reach the market in freeze-dried form and reducing cold-chain costs.9
Studies performed with solid-state hydrogen–deuterium exchange mass spectrometry,10 a technique that provides peptide-level information about dried protein structure, demonstrate the predictive power afforded by high-resolution data.11,12 However, residue-level information is essential for gaining a thorough understanding of protein–water interactions and how they relate to the mechanisms of dehydration-induced unfolding and cosolute-mediated dehydration protection.
We developed liquid-observed vapor exchange (LOVE) NMR (Figure 1) to enable the study of dehydrated protein structure and protection at the residue level. Inspired by the report of Desai et al.,13 LOVE NMR uses solution NMR spectroscopy to quantify the extent of hydrogen–deuterium exchange (HDX) between D2O vapor and the unprotected amide protons of a dried protein. On the basis of the well-established principle that amide protons are less likely to exchange if involved in intra- or intermolecular H-bonds,14–16 we expect residues in structured regions of the protein, or in regions of the protein that hydrogen-bond with cosolutes, to be more protected from exchange than residues in unstructured, exposed regions.
Figure 1.
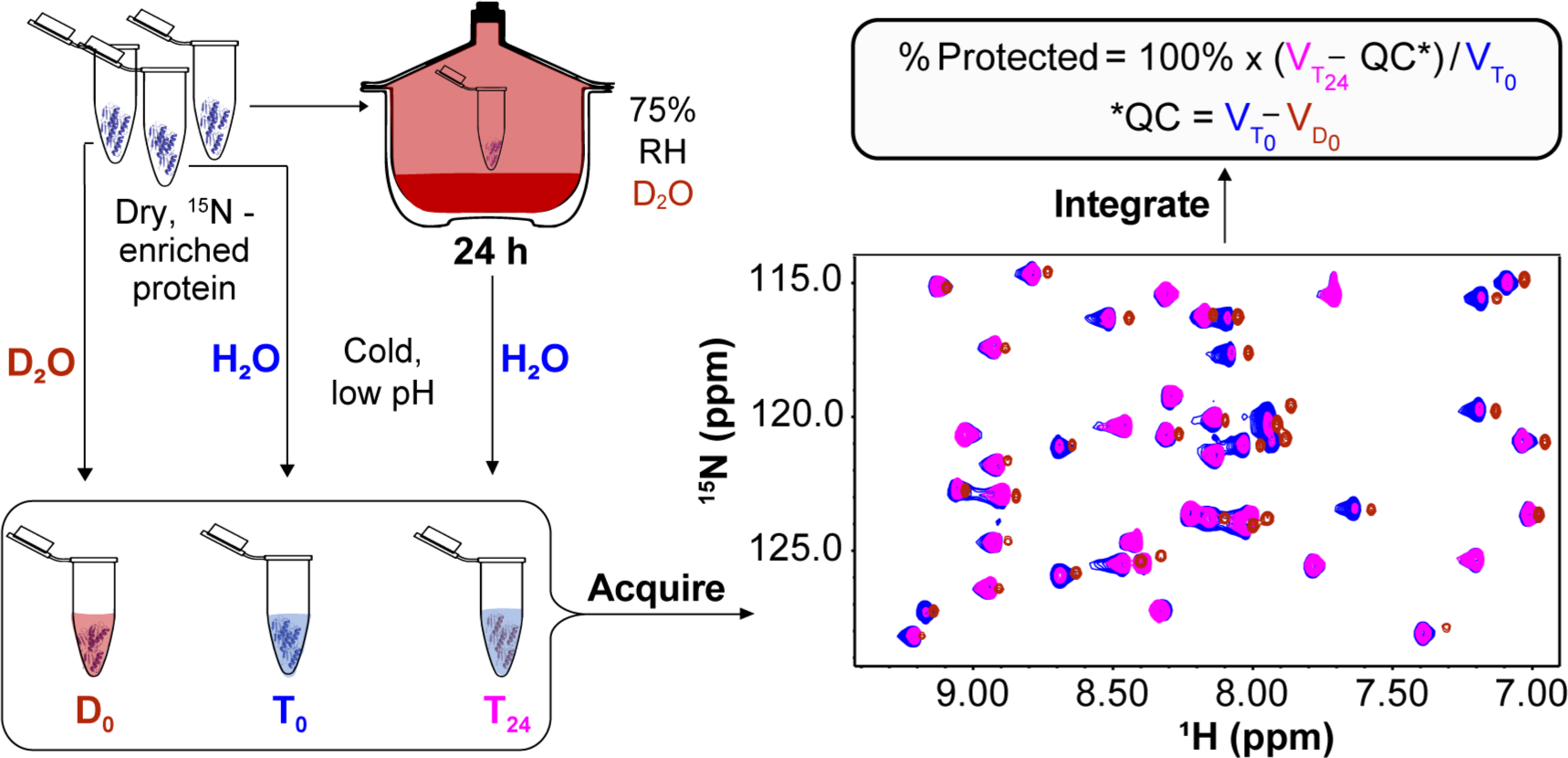
LOVE NMR. Three identical samples of 15N-enriched protein dried alone or in the presence of a cosolute are resuspended in cold, acidic buffer before (D0 and T0) or after (T24) exposure for 24 h to D2O vapor at 75% relative humidity (RH). Amide protons that are unprotected in the dry state will exchange with deuterons from the vapor, resulting in smaller cross peak volumes in the T24 15N–1H HSQC spectrum relative to the T0 spectrum (T0 and T24 cross peaks are colored blue and pink, respectively). A third sample, D0, is resuspended in D2O quench buffer (red cross peaks). The difference in volumes between corresponding peaks in the T0 and D0 spectra reflects quench labeling and is used as a quench correction [QC (see the text)], which is subtracted from the volume of the corresponding peak in the T24 spectrum . The difference is divided by and multiplied by 100% to yield % protected.
MATERIALS AND METHODS
Materials.
Ampicillin (Sigma-Aldrich), trehalose, and urea (Thermo Fisher) were used without further purification. H2O with a resistivity of >17 MΩ cm−1 was used to prepare buffers. pH values are direct readings, uncorrected for the deuterium isotope effect.17 The pET11a plasmid containing the gene encoding the T2Q variant of the B1 immunoglobulin-binding domain of streptococcal protein G was provided by L. D. Spicer’s laboratory at Duke University (Durham, NC). This variant was chosen because the mutation prevents N-terminal deamidation.18 A constant relative humidity (RH) of 75 ± 5%, as measured by a digital hygrometer (Fisherbrand Trace-ableGO Bluetooth Datalogging Hygrometer), was created by sealing a 0.5 L chamber containing 200 mL of >99% D2O (Cambridge Isotope Laboratories) saturated with Co(II)Cl2 (Agros Organics).19,20
Protein Expression and Purification.
15N-enriched GB1 was expressed in Agilent BL21 Gold (DE3) Escherichia coli grown in minimal medium supplemented with 15NH4Cl.21,22 Following expression for 2–3 h, cells from each 1 L culture were harvested via centrifugation at 4000g, the supernatant was discarded, and the pellets were stored at −20 °C. Cell pellets were thawed at room temperature, resuspended in 5 mL of 20 mM Tris (pH 7.5), and lysed via sonication for 8 min at 20% amplitude with a 33% duty cycle using a Fisher Scientific model 500 sonic dismembrator. Lysates were clarified by centrifugation at 15000g for 1 h. Clarified lysates were passed through a 0.45 μm filter (Millipore) and purified via liquid chromatography.21 The concentration of the purified protein was determined from the absorbance at 280 nm (A280) (Nanodrop One, Thermo Fisher) using an extinction coefficient of 9530 M−1 cm−1.23 Purity was confirmed by mass spectrometry (ThermoScientific, Q Exactive HF-X) in the UNC Mass Spectrometry Chemical Research and Teaching Core Laboratory (6290.32 Da expected, 6290 Da observed). Purified protein was exchanged into H2O by dialysis (ThermoScientific Snakeskin dialysis tubing, 3500 Da molecular weight cutoff) and divided into aliquots such that resuspension in 650 μL gives a protein concentration of 500 μM. Aliquots were flash-frozen, lyophilized, and stored at −20 °C.
NMR.
Experiments were performed in triplicate on Bruker Avance III HD spectrometers with cryogenic QCI probes at 1H Larmor frequencies of 600 MHz for LOVE experiments and 850 MHz for solution amide proton exchange experiments. For LOVE experiments, 15N–1H heteronuclear single-quantum coherence (HSQC) spectra were recorded in ~20 min (128 increments in the 15N dimension, eight scans per increment) with sweep widths of 3041 Hz in the 15N dimension and 8418 Hz in the 1H dimension. For residues that exchange quickly in solution, band-selective excitation short-transient (BEST) 15N–1H HSQC experiments were performed for ~3 min (128 increments in the 15N dimension, four scans per increment) with sweep widths of 3016 Hz in the 15N dimension and 13587 Hz in the 1H dimension.24 For other solution exchange experiments, traditional 15N–1H HSQC spectra were recorded for ~20 min (128 increments in the 15N dimension, eight scans per increment) with sweep widths of 4308 Hz in the 15N dimension and 11904 Hz in the 1H dimension. Spectra were processed with NMRPipe.25 Cross peaks were integrated using NMRViewJ.26
Backbone resonances were assigned [pH 4.5 and 4 °C or pH 7.5 and 22 °C (Figure S1 and Table S1)] using isotopically enriched GB1 expressed in minimal medium containing [13C]-d-glucose and 15NH4Cl (Cambridge Isotope Laboratories) as the sole sources of carbon and nitrogen, respectively, and purified as described above. HNCACB spectra were recorded with 10% sampling in the indirect dimensions using a Poisson gap scheduling scheme.27,28 Three-dimensional spectra were reconstructed using the SMILE algorithm and processed in NMRpipe.29
Solution Hydrogen–Deuterium Exchange.
Lyophilized aliquots of purified, 15N-enriched GB1 were resuspended in 650 μL of 7.5 mM HEPES (pH 6.5) with or without 100 g/L trehalose or urea, flash-frozen, and lyophilized. After 24 h, samples were removed, resuspended in 650 μL of 99% D2O, and immediately used to acquire serial NMR HSQC spectra at 22 °C. To obtain data for residues that fully exchange in <1 h (fast regime), BEST spectra were used to acquire ~10 spectra in the first 30 min with a dead time of ~3 min.24 To capture decay curves for residues that completely exchange in 2–24 h (intermediate regime), traditional 15N–1H HSQC experiments were used to acquire 10–12 spectra over ~24 h. For slowly exchanging residues (>24 h), samples were resuspended and, following acquisition of the first 15N–1H HSQC spectrum (0 h time point), placed in an incubator at 22 °C. Samples were removed every 1–3 days for spectrum acquisition and then returned to the incubator. The sample pH was measured at the end of the exchange; all samples possessed a pH of ~7.5.
The rate analysis tool in NMRViewJ was used to fit peak volumes as a function of time to the three-parameter equation V = Ae−Bt + C, where V is the peak volume in arbitrary units, t is the time in seconds, and B is the observed rate constant (kobs). For each residue, kobs was divided by the estimated intrinsic rate constant of exchange (kint) at pH 7.5 and 22 °C to approximate the opening equilibrium constant, Kop. Values of kint were obtained using the online Server Program for Hydrogen Exchange Rate Estimation.30 To promote accuracy, only cross peak volumes that decayed to ~30% or less of their initial value were analyzed. Opening free energies (ΔGop°′) were calculated as
where R is the gas constant and T is the absolute temperature.15,31
Liquid-Observed Vapor Exchange (LOVE) NMR.
For each experiment, three identical aliquots of pure, lyophilized, 15N-enriched GB1 were resuspended in 650 μL of 1.5 mM HEPES (pH 6.5) with or without 20 g/L trehalose or urea to achieve a final protein concentration of 500 μM, flash-frozen, and exposed to pressures of <0.3 mbar at ambient temperature on a LABCONCO FreeZone 1 L Benchtop Freeze-Dry System for 24 h. Following lyophilization, two samples, designated T0 and D0, were resuspended in 650 μL of cold quench buffer [100 mM citrate (pH 4.5) and 90% H2O/10% D2O for T0 or >99% D2O for D0] and immediately transferred to an NMR spectrometer for spectrum acquisition at 4 °C. The third sample, designated T24, was placed, with the cap opened, in a chamber of controlled relative D2O humidity, prepared as described above. After 24 h, the T24 sample, which remained a solid, was resuspended in 650 μL of cold quench buffer and an HSQC spectrum was acquired using the same parameters that were used for the T0 and D0 samples. The time between resuspension and initiation of spectrum acquisition was 10 min, ~8 min of which was spent at 4 °C. For the vapor exchange time course, GB1 samples dried from 650 μL of 1.5 mM HEPES (pH 6.5) were stored in the constant-humidity chamber for times stated in the caption of Figure 2.
Figure 2.
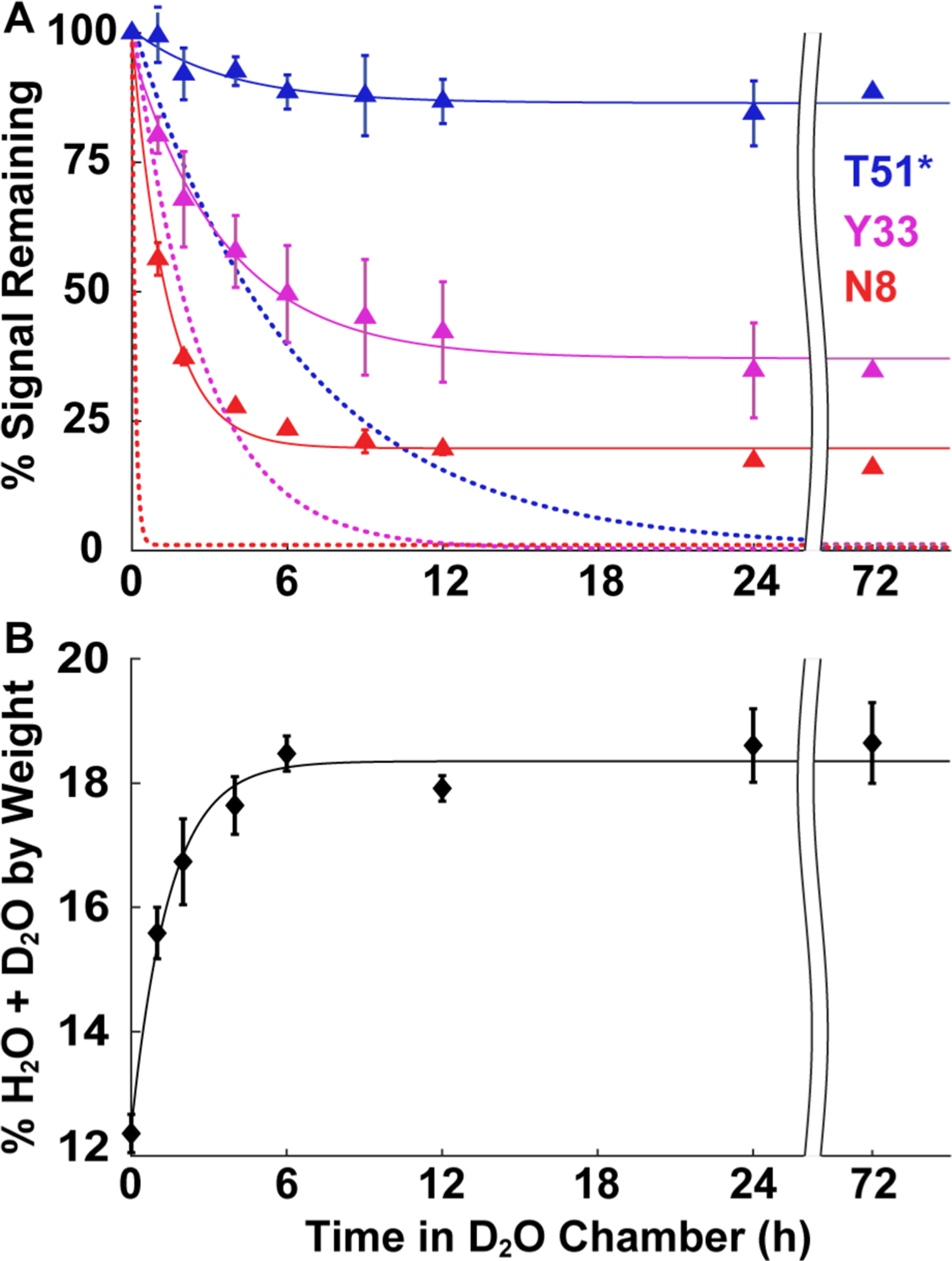
Time courses of solution HDX, vapor exchange, and vapor sorption. (A) Signal remaining as a function of time in D2O liquid (dotted lines) or vapor (75% RH, triangles) for three GB1 residues representing different solution exchange rate regimes: slow (blue, complete in >24 h), intermediate (pink, 2–24 h), and fast (red, <2 h). For T51* in solution, the abscissa is compressed 10-fold (e.g., 6 h corresponds to 60 h). Solution exchange curves are derived from the average kobs from three independent experiments in 7.5 mM HEPES (pH 7.5) (>99% D2O). For the vapor exchange time course, identical GB1 samples were freeze-dried in 1.5 mM HEPES (pH 6.5) for 24 h and resuspended in H2O-based quench buffer after vapor exchange for 0, 1, 2, 4, 6, 9, 12, 24, and 72 h at 75% RH. Spectra were acquired at 4 °C immediately upon resuspension. Vapor data are uncorrected for quench labeling. (B) Average %(H2O + D2O) content (w/w) of freeze-dried GB1 as a function of time spent at 75% RH in a D2O chamber. After 72 h at 75% RH, the accessible GB1 surface covered by H2O and D2O is ≤40% for drying from buffer alone, ≤60% for drying from buffer with 20 g/L urea, and ≤170% (approximately two layers of H2O and D2O) for drying from buffer and 20 g/L trehalose (Table S3). For vapor exchange and sorption, curves are of no theoretical significance. Error bars represent the standard deviation of the mean from three independent experiments, except for the 72 h vapor exchange data in panel A, which are from a single experiment.
To ensure that the change in cross peak volumes originates exclusively from exchange with D2O vapor, differences in GB1 concentration among the T0, D0, and T24 samples were determined after the experiment via the absorbance at 280 nm using the extinction coefficient provided above. The A280 values were used to normalize each cross peak volume across the three data sets. Using concentration-normalized cross peak volumes , , and , the percent of the dried protein population for which a given amide proton is protected from vapor exchange is calculated as
where is the quench correction. The change in % protected from drying in the presence of a cosolute is calculated as
Uncertainties from triplicate analysis and propagation of error analysis are discussed in the text and figure captions.
Thermogravimetric Analysis.
Aliquots of purified, unenriched GB1 [650 μL, 500 μM in 1.5 mM HEPES buffer (pH 6.5)] were flash-frozen and dried as described above. Samples were then placed, without caps, in a chamber with a controlled relative humidity of 75 ± 5%, created as described above. Individual tubes were removed after 0, 1, 2, 4, 6, 12, 24, 48, and 72 h and immediately analyzed using a TA Instruments model 550 thermogravimetric analyzer. The samples were loaded onto an open Pt pan and heated from 25 to 200 °C at a rate of 4 °C/min under a N2(g) sample purge of 60 mL/min and a balance purge of 40 mL/min. The well-defined mass loss ending at 140 °C was used to quantify the content of H2O and D2O.32,33
RESULTS
Quantifying Vapor Exchange with LOVE NMR.
To quantify deuterium incorporation in the solid state, two dried 15N-enriched protein samples, one exposed (T24) and one not exposed (T0) to D2O vapor for 24 h, are dissolved in cold acidic buffer, which slows solution HDX and enables the immediate acquisition of a solution HSQC spectrum (Figure S1A). Cross peak volumes (V) from the assigned resonances (Table S1) are directly proportional to the concentration of amide protons, meaning that the volumes of corresponding cross peaks in the pre- and postexchange spectra can be compared to determine the fraction of amide protons protected from exchange. That is, cross peaks from residues that exchange with D2O vapor are smaller than those from residues protected from exchange.
This approach can be applied to any protein possessing a well-dispersed 15N–1H HSQC spectrum if it folds faster than the rate of solution HDX of an unprotected amide proton. Meeting this latter condition ensures that solution HDX does not alter the difference in the 1H–15N signal between T0 and T24. Solution HDX is minimal during the 30 min between protein resuspension and completion of HSQC spectrum acquisition for the protein used here (Table S2), the T2Q variant of the 6 kDa immunoglobulin-binding domain B1 of streptococcal protein G, which we call GB1.18,34 For proteins more susceptible to solution HDX, the time in solution can be decreased by modifying acquisition parameters or by using a faster NMR experiment, such as band-selective short-transient HSQC.24
Although resuspension in aqueous buffer is necessary to acquire a well-dispersed solution NMR spectrum, it also causes a complication called quench labeling, in which labile protons or deuterons from the quench buffer immediately exchange with a fraction of surface-exposed amides upon resuspension. The quench labeling varies across the sequence, with residues in structured regions experiencing less labeling than those in unstructured regions (Figure S2). Quench labeling must be accounted for because it masks some or all the amide proton signal loss from deuterium incorporation in the dry state. To remove the signal introduced by quench labeling, a spectrum is acquired for a third, non-vapor-exchanged sample resuspended in a D2O quench buffer (D0). The isotope effect on quench labeling is small; for most residues, the signal lost due to quench labeling by is nearly identical to the signal gained by a dried, deuterium-exchanged protein due to quench labeling by H2O (Figure S2). The signal lost upon resuspension in is thus used as a quench correction (QC) that is subtracted from the volume of the corresponding peak in the T24 spectrum .
Finally, each quench-corrected peak volume is normalized by dividing by the corresponding peak volume from the non-vapor-exchanged, H2O-quenched sample and multiplied by 100% to yield the percent of the protein population for which a given amide proton is protected from vapor exchange (% protected).
Interpreting LOVE NMR Measurements.
The abilities of solution HDX and LOVE NMR to detect protein structure are derived from the same principle: H-bonding prevents exchange. However, these methods differ in how they measure protection from exchange. With solution HDX, all amide protons eventually exchange to completion (Figure 2A, dotted lines), but at different rates that, under certain conditions, can be used to estimate protection factors and opening free energies (ΔGop°′).14,31,35 In contrast, the extent of vapor exchange measured by LOVE NMR varies by residue (Figure 2A, solid lines) and the rate for all residues mirrors that of vapor sorption (Figure 2B).
Folding and HDX in the solid state are poorly understood. Nevertheless, the observation that vapor exchange plateaus at different levels for different residues suggests that, unlike solution HDX, LOVE NMR does not report on an equilibrium process. Instead, this observation suggests that the plateau values reflect the fraction of the dried protein population trapped in conformations where a given residue is protected from vapor exchange.
The similarity between the kinetics of vapor exchange and vapor sorption suggests that the D2O vapor concentration may affect LOVE NMR data. Measurements made after exposing freeze-dried GB1 to 85% RH (D2O) for 24 h confirm this idea, with all residues witnessing an ~10% reduction in signal compared with exposure to 75% RH (Figure S3). The additional signal loss at 85% RH is distributed almost evenly across the protein sequence, suggesting that a decreased level of vapor exchange in the reverse direction (N–D → N–H),36 rather than humidity-induced changes to dried protein structure, is the source of signal reduction. The preservation of trends at different humidities reinforces the idea that protection from vapor exchange is provided by dehydrated protein structure and demonstrates that LOVE NMR can be performed at different relative humidities. However, this result also suggests that experiments conducted at much lower relative humidities cannot reveal protection differences between residues in highly protected regions.
Trends in the LOVE Profile.
Plotting % protected against residue number yields the LOVE profile. Comparing the profile of GB1 to its secondary structure map shows that regions with stable secondary structure in solution tend to be more protected in the dry state (Figure 3A). The similar levels of protection experienced by regions that form tertiary contacts in the native structure (e.g., β-sheets 1 and 4)37 suggest that some GB1 molecules possess native or near-native tertiary structure in the dry state. Residues that undergo solution HDX only upon global unfolding (“global unfolders”)38 experience the most protection yet witness a 40% signal loss on average, implying that a large fraction of the GB1 population unfolds during freeze-drying.
Figure 3.
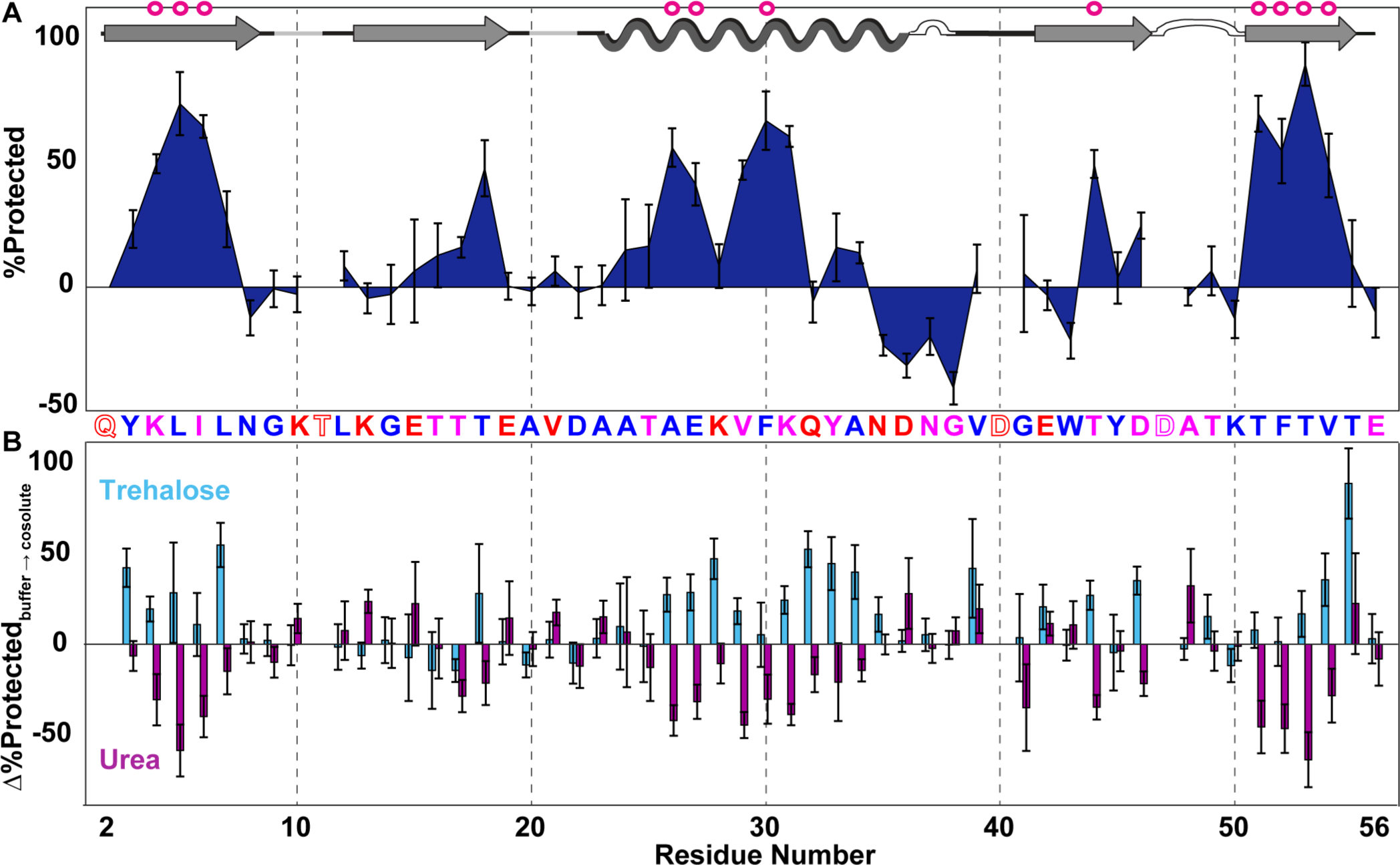
Dry-state protection of GB1 freeze-dried alone and in the presence of cosolutes. (A) LOVE profile of GB1 freeze-dried in 1.5 mM HEPES (pH 6.5). (B) Change in % protected (% protectedcosolute − % protectedbuffer) due to freeze-drying in 1.5 mM HEPES with 20 g/L trehalose or urea (pH 6.5). The primary structure of GB1 is shown between panels. Letters are colored by the total solvent-accessible surface area of the residues in the solution structure, as predicted by the online web application ProtSA for Protein Data Bank (PDB) structure 2QMT (blue, 0–50 Å2; pink, 51–100 Å2; red, >100 Å2). Empty letters indicate residues with undefined dry-state protection because they are 100% quench-labeled. GB1 secondary structures (as defined in PDB entry 2QMT; arrows, β-strands; undulations, helix; white bumps, turns; gray lines, bends) are shown at the top, with magenta circles indicating solution global unfolding residues. Error bars represent uncertainty propagated from standard deviations of the mean from triplicate analysis. For the cosolute data, D0 spectra of GB1 freeze-dried with cosolute were used to calculate QC values (Figure S4).
One region, residues 35–38, exhibits negative % protected values, indicating that the D0 sample experiences more quench labeling than the T24 sample. The quench-label profile of a sample fully exchanged into D2O before drying and then resuspended in H2O quench buffer shows similar quench labeling in this region (Figure S2), signifying that a solvent isotope effect is not responsible. Moreover, the degree of quench labeling experienced by a sample exposed to H2O vapor for 24 h followed by resuspension in D2O quench buffer is the same as that of the D0 sample (Figure S2). These data suggest that exchange with D2O vapor in the dry state is the primary source of the quench labeling artifact.
The nuances of quench labeling are not well-understood, but we suspect that interactions with D2O vapor cause this region to fold into an alternative H-bonded conformation in the dry state, thus preventing quench labeling. This explanation is consistent with the observations that these residues possess small, positive % protected values before quench correction (Figure S5) and that this region of GB1 can adopt an alternative conformation in solution.39
Cosolute Effects.
In addition to providing insight into dry protein structure, LOVE NMR can reveal residue-level effects of drying in the presence of cosolutes. We quantified the change in % protected due to freeze-drying GB1 with 20 g/L urea or trehalose by subtracting the quench-corrected LOVE profile of GB1 dried in buffer from that of GB1 dried in buffer and cosolute (Figure 3B). Drying with urea, an osmolyte that destabilizes proteins in solution via preferential interactions with the protein backbone,40,41 decreases the level of dry-state protection in general (Figure 3B and Table S4). There are, however, solvent-accessible residues near the termini of secondary structures whose level of protection increases, perhaps due to urea blocking exchange via H-bonding.
Drying with trehalose, an osmolyte that increases solution stability because it is preferentially excluded from protein backbone,42 generally increases the level of dry-state protection, with two-thirds of GB1 residues experiencing no observable vapor exchange (Table S4). Residues in unstructured areas, however, do not witness much protection from trehalose. The observation that a denaturant generally decreases the level of protection, and a stabilizer increases the level of protection, provides further confidence that LOVE NMR data reflect the presence of dried protein structure.
Comparing Dry-State Protection to Solution Stability.
To understand how dry-state protection relates to solution stability, we measured rates of solution HDX in buffer and in buffer with 100 g/L urea or trehalose and quantified the opening free energies [ΔGop°′ (Table S5)]. For GB1 dried in buffer alone, % protected correlates positively with ΔGop°′ in solution [p < 0.00001 (Figure 4A)], indicating that the native structure is a major source of dry-state protection. Intermolecular H-bonds with opening free energies of <6 kcal/mol experience little to no dry-state protection, suggesting that freeze-drying may lead to a complete loss of native structure in those regions and highlighting the importance of water to the maintenance of secondary structure.43
Figure 4.
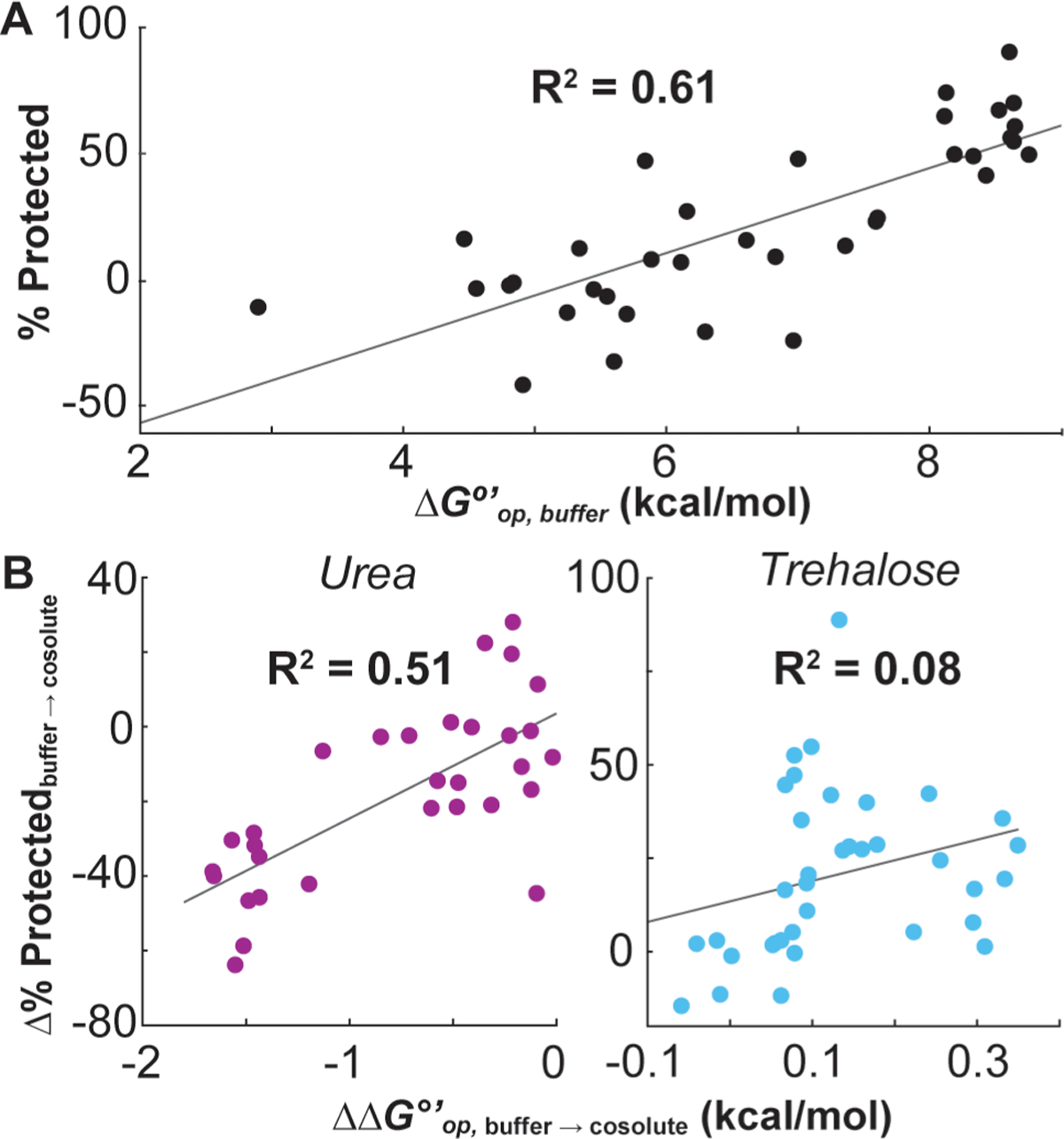
Correlations of solution- and dry-state protection. (A) % protected after freeze-drying in buffer vs average opening free energy (ΔGop°′) in buffer for the 34 GB1 residues for which solution HDX rates could be measured. (B) Average Δ% protected due to drying with 20 g/L urea or trehalose vs the change in opening free energy (ΔΔGop°′) due to the presence of the same cosolute at 100 g/L (ΔΔGop°′ = ΔGop,cosolute°′ − ΔGop,buffer°′). % protected and ΔGop°′ values are reported with their uncertainties in Tables S4 and S5, respectively.
Comparing the urea-induced change in % protected to the change in opening free energy (ΔΔGop,buffer→urea°′) in solution also reveals a positive correlation [p < 0.00001 (Figure 4B)]. This correlation corroborates the idea that the regional stability of a protein before drying influences which conformations become trapped in the dry state. By contrast, the correlation between Δ% protectedbuffer→trehalose and ΔΔGop,buffer→trehalose°′ is insignificant [p > 0.10 (Figure 4B)], suggesting that the protective mechanism during freeze-drying differs substantially from that in solution.
DISCUSSION
The observation that trehalose targets different residues in the solution and dry states (i.e., the correlation in Figure 4B is insignificant) is consistent with the observation that trehalose is one of the few osmolytes that, in addition to protecting organisms from mild osmotic stress, also protects against desiccation.44 It is therefore reasonable to suggest that the mechanism of protection exerted by trehalose under dehydrating conditions differs from its mechanism of stabilization in solution.
There are two main ideas about how trehalose and similar sugars protect proteins from dehydration-induced damage: vitrification and water replacement.45,46 The vitrification hypothesis posits that the sugar confines the protein in a glassy matrix, stabilizing the native structure by preventing large motions such as global unfolding. The water replacement hypothesis posits that the sugar maintains the native protein structure by replacing the stabilizing H-bonds usually provided by water.
If trehalose protects proteins via vitrification, global-unfolding residues should benefit most. However, even in the absence of cosolutes, the global-unfolding residues of GB1 are highly protected from vapor exchange, which means they experience an only marginal increase in protection before reaching 100%. This “saturation” of protection makes quantitative comparison of the small Δ% protectedbuffer→trehalose values of already highly protected residues unreliable, because they cannot be more protected. Nonetheless, the observation that structured regions experience more protection from trehalose than unstructured areas suggests that either trehalose does not replace water H-bonds in less structured regions or vitrification dominates.
On the contrary, if confinement-induced vitrification is the sole source of trehalose protection, we expect native H-bond donor–acceptor pairs (double arrows in Figure 5) to experience similar degrees of protection, yet there are several instances in which such pairs exhibit a large difference in Δ% protectedbuffer→trehalose [e.g., E42/T55, L7/G14 (Figure 5)], indicating that some protection arises from blocking of exchange through H-bonding outside of native contacts.
Figure 5.
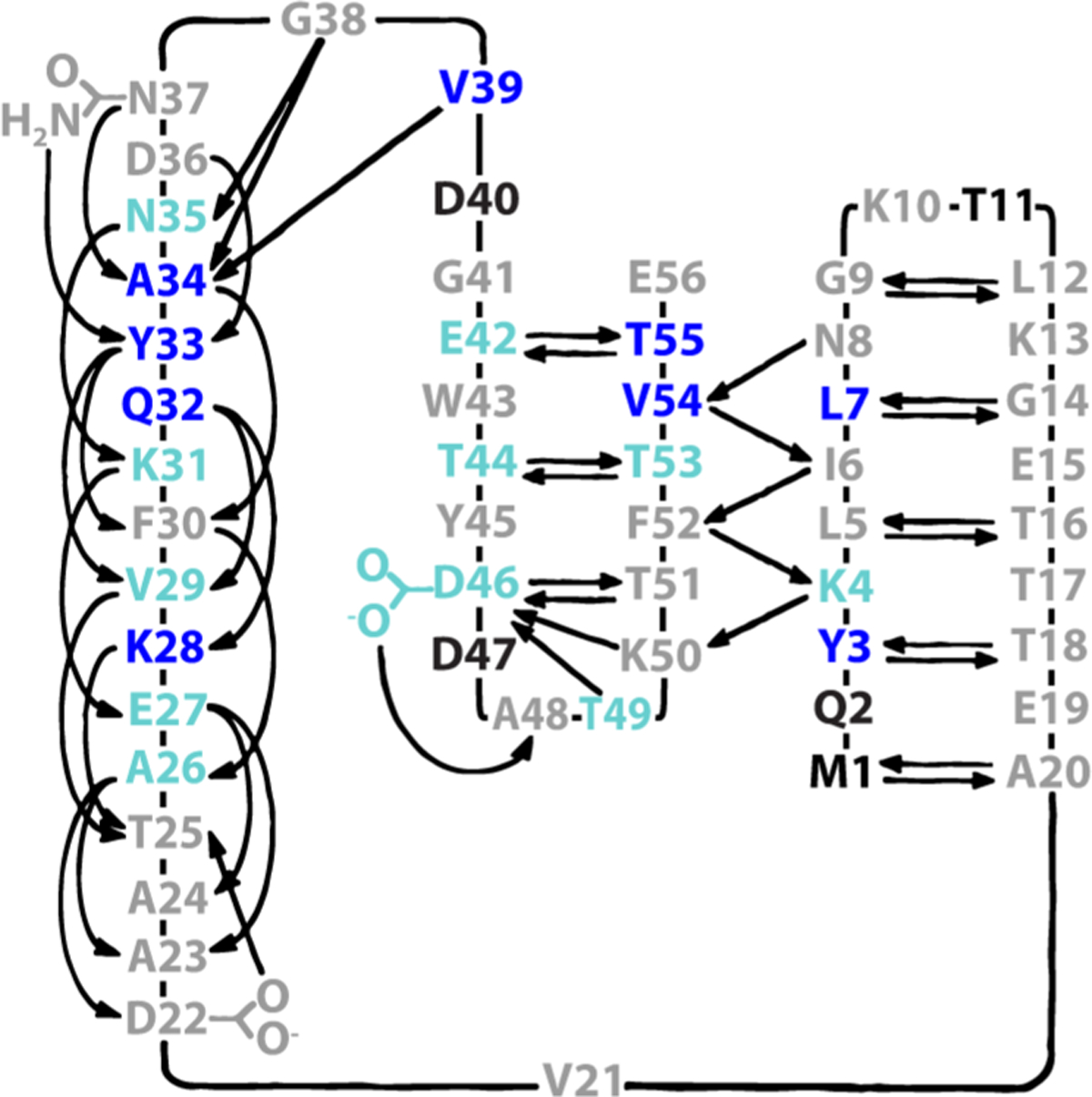
Trehalose protection mapped onto the native H-bonding pattern of GB1.37 Residues colored dark blue exhibit a Δ% protected >1 standard deviation (SD) above average, light blue values within 1 SD and not within uncertainty of zero, gray values within <1 SD or within uncertainty of zero, and black values of undefined protection (i.e., 100% quench-labeled). Adapted with permission from ref 37. Copyright 1991 American Association for the Advancement of Science.
Together, these data suggest that protection by trehalose results from a complex combination of native structure preservation and selective H-bonding between protein backbone and sugar hydroxyls. The multifaceted nature of vapor exchange protection by trehalose is confirmed by the observation that plotting Δ% protectedbuffer→trehalose values as a function of ~40 residue properties, including solvent-accessible surface area and amino acid side-chain transfer free energies, did not yield a strong linear correlation [all R2 < 0.16 (Table S6)].
CONCLUSIONS
In summary, LOVE NMR data on the model protein GB1 affirm the notion that a protein’s structure in the dry state is heavily influenced by its solution stability and demonstrate that models of cosolute-mediated dehydration protection require refinement. The application of high-resolution methods such as solid-state hydrogen–deuterium exchange mass spectrometry10 and LOVE NMR to a broad range of proteins will enable more nuanced models of dehydration protection to be proposed and tested. Such models can in turn be used to streamline the design of formulations for freeze-dried protein-based therapeutics, reducing costs and increasing accessibility to these life-saving medicines.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Stu Parnham from the UNC Biomolecular NMR Core and Brandie Ehrmann from the UNC Chemistry Mass Spectrometry Core Laboratory for equipment maintenance and advice. The authors thank Rachel D. Cohen for performing initial studies, Bo Li for use of the mass spectrometer in her laboratory, Adam Waterbury and Jack Prothero for suggestions regarding statistical analysis, the Pielak lab for helpful discussion, and Elizabeth Pielak for comments on the manuscript. The authors thank Angela Gronenborn for permission to use her hand-drawn GB1 structure in Figure 5.
Funding
This work was supported by National Institutes of Health Grant R01GM127291 to G.J.P., a National Science Foundation Graduate Research Fellowship (DGE-1650116) and a National Institutes of Health Training Grant (T32GM008570) to C.J.C., and a UNC Graduate School dissertation completion fellowship to S.P. The UNC Chemistry Mass Spec Core Laboratory is supported by the National Science Foundation (CHE 1726291).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.0c00863.
1H–15N chemical shift assignments of GB1 T2Q at pH 4.5 and 7.5, % protected and ΔGop°′ values in the presence and absence of cosolutes, water content of samples, and quench labeling controls (PDF)
Accession Codes
T2Q variant of streptococcal protein G (GB1 T2Q), UniProtKB P06654.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.biochem.0c00863
The authors declare no competing financial interest.
Contributor Information
Candice J. Crilly, Department of Chemistry, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, United States
Julia A. Brom, Department of Chemistry, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, United States
Mark E. Kowalewski, Department of Chemistry, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, United States
Samantha Piszkiewicz, Department of Chemistry, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, United States.
Gary J. Pielak, Department of Chemistry, Department of Biochemistry and Biophysics, Lineberger Cancer Center, and Integrative Program for Biological & Genome Sciences, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, United States;.
REFERENCES
- (1).Kauzmann W (1959) Some factors in the interpretation of protein denaturation. Adv. Protein Chem 14, 1–63. [DOI] [PubMed] [Google Scholar]
- (2).Ball P (2017) Water is an active matrix of life for cell and molecular biology. Proc. Natl. Acad. Sci. U. S. A 114, 13327–13335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bellissent-Funel MC, Hassanali A, Havenith M, Henchman R, Pohl P, Sterpone F, van der Spoel D, Xu Y, and Garcia AE (2016) Water determines the structure and dynamics of proteins. Chem. Rev 116, 7673–7697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Moorthy BS, Iyer LK, and Topp EM (2015) Characterizing protein structure, dynamics and conformation in lyophilized solids. Curr. Pharm. Des 21, 5845–5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ohtake S, Kita Y, and Arakawa T (2011) Interactions of formulation excipients with proteins in solution and in the dried state. Adv. Drug Delivery Rev 63, 1053–1073. [DOI] [PubMed] [Google Scholar]
- (6).Piszkiewicz S, and Pielak GJ (2019) Protecting enzymes from stress-induced inactivation. Biochemistry 58, 3825–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Crommelin DJA, Hawe A, and Jiskoot W (2019) Formulation of Biologics Including Biopharmaceutical Considerations. In Pharmaceutical Biotechnology: Fundamentals and Applications (Crommelin DJA, Sindelar RD, and Meibohm B, Eds.) 5th ed., pp 83–103, Springer International Publishing, Cham, Switzerland. [Google Scholar]
- (8).Pandya A, Howard MJ, Zloh M, and Dalby PA (2018) An evaluation of the potential of NMR spectroscopy and computational modelling methods to inform biopharmaceutical formulations. Pharmaceutics 10, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hill AB, Kilgore C, McGlynn M, and Jones CH (2016) Improving global vaccine accessibility. Curr. Opin. Biotechnol 42, 67–73. [DOI] [PubMed] [Google Scholar]
- (10).Li Y, Williams TD, Schowen RL, and Topp EM (2007) Characterizing protein structure in amorphous solids using hydrogen/deuterium exchange with mass spectrometry. Anal. Biochem 366, 18–28. [DOI] [PubMed] [Google Scholar]
- (11).Moorthy BS, Schultz SG, Kim SG, and Topp EM (2014) Predicting protein aggregation during storage in lyophilized solids using solid state amide hydrogen/deuterium exchange with mass spectrometric analysis (ssHDX-MS). Mol. Pharmaceutics 11, 1869–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Moorthy BS, Zarraga IE, Kumar L, Walters BT, Goldbach P, Topp EM, and Allmendinger A (2018) Solid-state hydrogen-deuterium exchange mass spectrometry: Correlation of deuterium uptake and long-term stability of lyophilized monoclonal antibody formulations. Mol. Pharmaceutics 15, 1–11. [DOI] [PubMed] [Google Scholar]
- (13).Desai UR, Osterhout JJ, and Klibanov AM (1994) Protein structure in the lyophilized state: A hydrogen isotope exchange/NMR study with bovine pancreatic trypsin inhibitor. J. Am. Chem. Soc 116, 9420–9422. [Google Scholar]
- (14).Hvidt A, and Nielsen SO (1966) Hydrogen exchange in proteins. Adv. Protein Chem 21, 287–386. [DOI] [PubMed] [Google Scholar]
- (15).Englander SW, and Kallenbach NR (1983) Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys 16, 521–655. [DOI] [PubMed] [Google Scholar]
- (16).Percy AJ, Rey M, Burns KM, and Schriemer DC (2012) Probing protein interactions with hydrogen/deuterium exchange and mass spectrometry-a review. Anal. Chim. Acta 721, 7–21. [DOI] [PubMed] [Google Scholar]
- (17).Glasoe PK, and Long FA (1960) Use of glass electrodes to measure acidities in deuterium oxide. J. Phys. Chem 64, 188–190. [Google Scholar]
- (18).Smith CK, Withka JM, and Regan L (1994) A thermodynamic scale for the β-sheet forming tendencies of the amino acids. Biochemistry 33, 5510–5517. [DOI] [PubMed] [Google Scholar]
- (19).Rockland LB (1960) Saturated salt solutions for static control of relative humidity between 5° and 40° C. Anal. Chem 32, 1375–1376. [Google Scholar]
- (20).Jakli G, and Van Hook WA (1981) Vapor pressure of heavy water at 283–363 K. J. Chem. Eng. Data 26, 243–245. [Google Scholar]
- (21).Monteith WB, and Pielak GJ (2014) Residue level quantification of protein stability in living cells. Proc. Natl. Acad. Sci. U. S. A 111, 11335–11340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Monteith WB, and Pielak GJ (2015) Correction for Monteith and Pielak, residue level quantification of protein stability in living cells. Proc. Natl. Acad. Sci. U. S. A 112, E7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Gill SC, and von Hippel PH (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem 182, 319–326. [DOI] [PubMed] [Google Scholar]
- (24).Schanda P, Van Melckebeke H, and Brutscher B (2006) Speeding up three-dimensional protein NMR experiments to a few minutes. J. Am. Chem. Soc 128, 9042–9043. [DOI] [PubMed] [Google Scholar]
- (25).Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Bax A (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- (26).Johnson BA, and Blevins RA (1994) NMR View: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614. [DOI] [PubMed] [Google Scholar]
- (27).Miljenović TM, Jia X, and Mobli M (2017) Nonuniform Sampling in Biomolecular NMR. In Modern Magnetic Resonance (Webb GA, Ed.) 2nd ed., pp 1–21, Springer International Publishing, Cham, Switzerland. [Google Scholar]
- (28).Hyberts SG, Takeuchi K, and Wagner G (2010) Poisson-gap sampling and forward maximum entropy reconstruction for enhancing the resolution and sensitivity of protein NMR data. J. Am. Chem. Soc 132, 2145–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ying J, Delaglio F, Torchia DA, and Bax A (2017) Sparse multidimensional iterative lineshape-enhanced (SMILE) reconstruction of both non-uniformly sampled and conventional NMR data. J. Biomol. NMR 68, 101–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhang Y-Z (1995) Protein and peptide structure and interactions studied by hydrogen exchange and NMR. Ph.D. Thesis, University of Pennsylvania, Philadelphia. [Google Scholar]
- (31).Miklos AC, Li C, and Pielak GJ (2009) Using NMR-detected backbone amide 1H exchange to assess macromolecular crowding effects on globular-protein stability. Methods Enzymol 466, 1–18. [DOI] [PubMed] [Google Scholar]
- (32).May JC, Grim E, Wheeler RM, and West J (1982) Determination of residual moisture in freeze-dried viral vaccines: Karl Fischer gravimetric and thermogravimetric methodologies. J. Biol. Stand 10, 249–259. [DOI] [PubMed] [Google Scholar]
- (33).May JC, Wheeler RM, and Grim E (1989) The gravimetric method for the determination of residual moisture in freeze-dried biological products. Cryobiology 26, 277–284. [DOI] [PubMed] [Google Scholar]
- (34).Morrone A, Giri R, Toofanny RD, Travaglini-Allocatelli C, Brunori M, Daggett V, and Gianni S (2011) GB1 is not a two-state folder: Identification and characterization of an on-pathway intermediate. Biophys. J 101, 2053–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Marmorino JL, Auld DS, Betz SF, Doyle DF, Young GB, and Pielak GJ (1993) Amide proton exchange rates of oxidized and reduced Saccharomyces cerevisiae iso-1-cytochrome c. Protein Sci 2, 1966–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kammari R, and Topp EM (2019) Solid-state hydrogen-deuterium exchange mass spectrometry (ssHDX-MS) of lyophilized poly-D,L-alanine. Mol. Pharmaceutics 16, 2935–2946. [DOI] [PubMed] [Google Scholar]
- (37).Gronenborn AM, Filpula DR, Essig NZ, Achari A, Whitlow M, Wingfield PT, and Clore GM (1991) A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science 253, 657–661. [DOI] [PubMed] [Google Scholar]
- (38).Orban J, Alexander P, Bryan P, and Khare D (1995) Assessment of stability differences in the protein G B1 and B2 domains from hydrogen-deuterium exchange: Comparison with calorimetric data. Biochemistry 34, 15291–15300. [DOI] [PubMed] [Google Scholar]
- (39).Blanco FJ, Ortiz AR, and Serrano L (1997) Role of a nonnative interaction in the folding of the protein G B1 domain as inferred from the conformational analysis of the α-helix fragment. Folding Des 2, 123–133. [DOI] [PubMed] [Google Scholar]
- (40).Yancey PH, Clark ME, Hand SC, Bowlus RD, and Somero GN (1982) Living with water stress: Evolution of osmolyte systems. Science 217, 1214–1222. [DOI] [PubMed] [Google Scholar]
- (41).Rydeen AE, Brustad EM, and Pielak GJ (2018) Osmolytes and protein-protein interactions. J. Am. Chem. Soc 140, 7441–7444. [DOI] [PubMed] [Google Scholar]
- (42).Xie G, and Timasheff SN (1997) The thermodynamic mechanism of protein stabilization by trehalose. Biophys. Chem 64, 25–43. [DOI] [PubMed] [Google Scholar]
- (43).Barlow DJ, and Poole PL (1987) The hydration of protein secondary structures. FEBS Lett 213, 423–427. [DOI] [PubMed] [Google Scholar]
- (44).Kosar F, Akram NA, Sadiq M, Al-Qurainy F, and Ashraf M (2019) Trehalose: A key organic osmolyte effectively involved in plant abiotic stress tolerance. J. Plant Growth Regul 38, 606–618. [Google Scholar]
- (45).Mensink MA, Frijlink HW, van der Voort Maarschalk K, and Hinrichs WL (2017) How sugars protect proteins in the solid state and during drying (review): Mechanisms of stabilization in relation to stress conditions. Eur. J. Pharm. Biopharm 114, 288–295. [DOI] [PubMed] [Google Scholar]
- (46).Crowe JH, Carpenter JF, and Crowe LM (1998) The role of vitrification in anhydrobiosis. Annu. Rev. Physiol 60, 73–103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.