

Summary
Polyamines are small, organic polycations that are ubiquitous and essential to all forms of life. Currently, how polyamines are transported across membranes is not understood. Recent studies have suggested that ATP13A2 and its close homologs, collectively known as P5B-ATPases, are polyamine transporters at endo-/lysosomes. Loss-of-function mutations of ATP13A2 in humans cause hereditary early-onset Parkinson’s disease. To understand the polyamine transport mechanism of ATP13A2, we determined high-resolution cryo-EM structures of human ATP13A2 in five distinct conformational intermediates, which together represent a near-complete transport cycle of ATP13A2. The structural basis of the polyamine specificity was revealed by an endogenous polyamine molecule bound to a narrow, elongated cavity within the transmembrane domain. The structures show an atypical transport path for a water-soluble substrate, where polyamines may exit within the cytosolic leaflet of the membrane. Our study provides important mechanistic insights into polyamine transport and a framework to understand functions and mechanisms of P5B-ATPases.
eTOC Blurb
The P-type ATPase ATP13A2 is an endo-/lysosomes polyamine transporter, defects of which are implicated in hereditary forms of Parkinson’s disease. Sim et al. report structures of human ATP13A2 in five distinct conformations, representing a near-complete catalytic cycle. This work reveals the mechanistic basis of polyamine recognition and transport by ATP13A2.
Graphical Abstract
Introduction
P-type ATPases form a large superfamily of active transporters that catalyze directional translocation of substrates across cellular membranes (Clausen et al., 2017; Dyla et al., 2019; Kuhlbrandt, 2004; López-Marqués et al., 2020; Palmgren and Nissen, 2011). To power substrate translocation, P-type ATPases undergo a series of conformational movements in their cytosolic domains mediated by ATP binding, autophosphorylation of a conserved aspartate residue, and subsequent dephosphorylation (Dyla et al., 2019; Kuhlbrandt, 2004; Palmgren and Nissen, 2011). These transitions accompany structural changes in the transmembrane domain (TMD) of the enzyme that drive substrate transport.
Based on sequence similarity, P-type ATPases can be classified into five subfamilies, P1- to P5- ATPases. Most P-type ATPases function as either ion pumps (P1- to P3- ATPases) or lipid flippases (P4-ATPases) (Bai et al., 2020; Hiraizumi et al., 2019; Morth et al., 2007; Nakanishi et al., 2020; Olesen et al., 2007; Shinoda et al., 2009; Sorensen et al., 2004; Timcenko et al., 2019; Toyoshima et al., 2000; Toyoshima et al., 2004). These transporters perform vital physiological tasks, such as generation of ionic gradients across membranes and lipid asymmetry in membrane leaflets.
Recent studies suggest that P5-ATPases, arguably the least characterized subfamily, have substrate specificities and functions that are critical for intracellular organelle homeostasis but fundamentally differ from those of P1- to P4- ATPases. Among the two subtypes—P5A and P5B (Møller et al., 2008; Sørensen et al., 2010), the P5A-ATPase (ATP13A1 in humans) seems to act as a protein quality control factor at the endoplasmic reticulum (ER), which extracts mis-targeted amino (N)- or carboxy (C)-terminally anchored transmembrane proteins from the ER membrane (Feng et al., 2020; McKenna et al., 2020; Qin et al., 2020). P5B-ATPases reside at endo-/lysosomal membranes (Sorensen et al., 2018), and humans express four isoforms, ATP13A2 to ATP13A5. Recent functional studies have suggested that ATP13A2, a prototypical P5B-ATPase, is a polyamine transporter at late endosomes and lysosomes (De La Hera et al., 2013; van Veen et al., 2020).
Polyamines are polycationic, aliphatic molecules that are essential to all species (Handa et al., 2018; Igarashi and Kashiwagi, 2010; Pegg, 2009). By binding to nucleic acids, proteins, and lipids, polyamines participate in numerous cellular processes, including modulation of chromatin structure, synthesis of proteins and nucleic acids, oxidative stress response, and ion channel functions (Handa et al., 2018; Pegg, 2009). The main naturally occurring forms are spermidine (triamine), spermine (tetraamine), and their precursor putrescine (diamine). In mammals, P5B-ATPases seem to play a key role in the uptake of extracellular polyamines by exporting polyamines from endocytic vesicles to the cytosol (De La Hera et al., 2013; Hamouda et al., 2020; Heinick et al., 2010; van Veen et al., 2020).
Defects in ATP13A2 are strongly implicated in progressive neurological disorders, such as Parkinson’s disease and hereditary spastic paraplegia (Di Fonzo et al., 2007; Estrada-Cuzcano et al., 2017; Ramirez et al., 2006; Yang and Xu, 2014). In ATP13A2 knock-out cells, lysosomes abnormally accumulate polyamines, resulting in lysosomal alkalization, dysfunction, and rupture (Dehay et al., 2012; van Veen et al., 2020). ATP13A2 deficiency also sensitizes cells to oxidative stress, suggesting a role of ATP13A2 in protecting cells against mitochondrial reactive oxygen species (Vrijsen et al., 2020). The involvement of ATP13A2 in maintaining lysosomal and mitochondrial homeostasis may explain the link between ATP13A2 defects and the pathogenesis of the diseases (Exner et al., 2012; Usenovic et al., 2012; Wallings et al., 2019). It has been suggested that small molecules that potentiate the polyamine transport activity of ATP13A2 might enhance endo-/lysosomal functions and provide therapeutic effects for neuroprotective treatments (Gitler et al., 2009; van Veen et al., 2020). However, the mechanistic basis of polyamine transport by ATP13A2 remains to be elucidated.
In the present study, we determined high-resolution cryo-electron microscopy (cryo-EM) structures of human ATP13A2 in five distinct conformations along its polyamine transport cycle. The chemical structure of polyamines is markedly different from any other characterized substrates of P-type ATPases due to their elongated structure and regularly spaced positive charges. Our study elucidated how ATP13A2 has adapted to specifically recognize these chemical signatures of polyamines and enable their membrane translocation. Together with mutagenesis analysis and molecular dynamics simulations, our work offers mechanistic insights into polyamine transport and provides a framework for further functional investigations and pharmacological targeting of P5B-ATPases.
Results
Purification and cryo-EM analysis of human ATP13A2
We expressed full-length human ATP13A2 in Spodoptera frugiperda (Sf9) cells and isolated it to apparent homogeneity in a mixture of dodecyl maltoside (DDM) and cholesteryl hemisuccinate (CHS) detergents. Purified ATP13A2 eluted largely as a single peak in size-exclusion chromatography and appeared as well-dispersed particles in cryo-EM images (Fig. S1 A–D).
To gain detailed insights into the transport mechanism of ATP13A2, we sought to determine structures of all its major catalytic intermediates (Fig. S1E). P-type ATPases are known to follow a scheme known as the Post-Albers cycle, i.e., E1-apo → E1-ATP → E1P-ADP → E1P → E2P → E2-Pi → E1-apo, where E1 and E2 represent two main conformations of the enzyme (Dyla et al., 2019; Kuhlbrandt, 2004; Palmgren and Nissen, 2011). To stabilize these intermediates, we prepared wild-type (WT) and inactive mutant ATP13A2 proteins in the presence of an adenine nucleotide (ADP, ATP, or β,γ-methyleneadenosine 5’-triphosphate [AMP-PCP]) or phosphate analog (BeF3− or AlF4−) (Bublitz et al., 2010) (Fig. S1F).
To this end, we captured structures representing six catalytic intermediates along the Post-Albers scheme indicated above (Figs. S1F and S2–4, and Tables 1 and S1). We note that the E1-ATP and E1P-ADP structures are in an essentially identical conformation, like other P-type ATPases (Bublitz et al., 2010; Nakanishi et al., 2020). Although some samples contained more than one conformational state, distinct structures could be efficiently separated by three-dimensional (3D) classifications during single-particle analysis. The cryo-EM maps were reconstructed at overall resolutions of 2.5 to 3.7 Å. Most side chains were clearly resolved in the cryo-EM maps, enabling accurate de novo structural modeling (Fig. S2A). The reaction states could be assigned based on visualized densities of bound ligands (Fig. S2 B–M), domain conformations (see below), and comparisons with other known P-type ATPase structures (Bublitz et al., 2010; Dyla et al., 2019; Hiraizumi et al., 2019; McKenna et al., 2020; Nakanishi et al., 2020).
Table 1.
Cryo-EM data collection, refinement and validation statistics
| WT E2BeF3−, E2P-like (map 3, EMD-24214; PDB 7N70) | WT E2-AlF4−, E2-Pi-like (map 6, EMD-24217; PDB 7N72) | WT E2-Pi, post-hydrolysis (map 12, EMD-24223; PDB 7N78) | WT E1P-ADP-like (map 7, EMD-24218; PDB 7N73) | D508N E1-ATP (map 8, EMD-24219; PDB 7N74) | D458N/D962N E1-apo (map 9, EMD-24220; PDB 7N75) | D458N/D962N E1-apo (map 10, EMD-24221; PDB 7N76) | D458N/D962N E1-AlF4−, E1P-like (map 11, EMD-24222; PDB 7N77) | |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Data collection and processing | ||||||||
| Magnification | 64,000x | 64,000x | 64,000x | 64,000x | 64,000x | 64,000x | 64,000x | 36,000x |
| Voltage (kV) | 300 | 300 | 300 | 300 | 300 | 300 | 300 | 200 |
| Electron exposure (e−/Å2) | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| Defocus range (μm) | −0.7 to −2.5 | −0.7 to −2.5 | −0.7 to −2.5 | −0.7 to −2.5 | −0.9 to −2.4 | −0.8 to −2.5 | −0.8 to −2.5 | −0.7 to −1.8 |
| Pixel size (Å) | 0.911 | 1.049 | 0.911 | 1.049 | 0.911 | 1.049 | 1.049 | 1.115 |
| Symmetry imposed | C1 | C1 | C1 | C1 | C1 | C1 | C1 | C1 |
| Initial particle images (no.) | 1,437,395 | 1,311,023 | n/a | 1,311,023 | 1,431,882 | 1,181,866 | 1,181,866 | 1,406,963 |
| Final particle images (no.) | 386,118 | 462,490 | 591,903 | 163,433 | 459,541 | 186,331 | 188,879 | 377,365 |
| Map resolution (Å) | 2.8 | 2.5 | 3.0 | 2.9 | 2.8 | 2.9 | 2.9 | 3.2 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 |
| Map resolution range (Å) | 2.5–12 | 2.4–8 | 2.6–8 | 2.5–11 | 2.4–8 | 2.4–8 | 2.5–8 | 2.8–8 |
| Refinement | ||||||||
| Initial model used | De novo | Model 3 | Model 6 | Model 3 | Model 7 | Model 7 | Model 7 | Model 7 |
| Model resolution (Å) | 3.0 | 2.7 | 3.1 | 3.1 | 3.0 | 3.0 | 3.0 | 3.4 |
| FSC threshold | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Map sharpening B factor (Å2) | 75 | 63 | 116 | 56 | 99 | 79 | 77 | 117 |
| Model composition | ||||||||
| Non-hydrogen atoms | 8,027 | 8,035 | 7,999 | 7,976 | 7,969 | 7,381 | 7,399 | 7,564 |
| Protein residues | 1,032 | 1,030 | 1,030 | 1,036 | 1,036 | 1,036 | 1,036 | 1,036 |
| Ligands | 4 | 11 | 11 | 6 | 4 | 0 | 0 | 4 |
| B factors (Å2) | ||||||||
| Protein | 49 | 66 | 74 | 76 | 44 | 56 | 73 | 29 |
| Ligand | 48 | 51 | 49 | 65 | 37 | - | - | 16 |
| R.m.s. deviations | ||||||||
| Bond lengths (Å) | 0.005 | 0.007 | 0.010 | 0.005 | 0.005 | 0.007 | 0.005 | 0.005 |
| Bond angles (°) | 0.649 | 0.745 | 0.783 | 0.631 | 0.735 | 0.689 | 0.647 | 0.698 |
| Validation | ||||||||
| MolProbity score | 1.43 | 1.33 | 1.55 | 1.55 | 1.46 | 1.61 | 1.53 | 1.73 |
| Clashscore | 7.93 | 5.99 | 8.15 | 7.76 | 7.46 | 7.91 | 5.80 | 9.56 |
| Poor rotamers (%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Ramachandran plot | ||||||||
| Favored (%) | 98.43 | 98.62 | 97.44 | 97.36 | 97.75 | 96.97 | 96.68 | 96.48 |
| Allowed (%) | 1.57 | 1.38 | 2.56 | 2.64 | 2.25 | 3.03 | 3.32 | 3.52 |
| Disallowed (%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Rama-Z | 0.27 | 0.87 | 1.34 | 1.31 | 0.74 | 0.75 | 0.22 | 1.56 |
| CaBLAM outliers (%) | 0.80 | 0.80 | 1.00 | 0.59 | 0.49 | 0.89 | 0.49 | 0.99 |
The 3D classifications of the catalytic intermediates in differently prepared samples suggest an interesting aspect of the reaction mechanism of ATP13A2. In our protein purification experiments, WT ATP13A2 isolated in the absence of any added ligands was found to be predominantly in a polyamine-bound E2-Pi state (Fig. S1F). This is in contrast with other P-type ATPases, in which a similar method typically results in the E1-apo state (Bai et al., 2020; Hiraizumi et al., 2019; McKenna et al., 2020; Nakanishi et al., 2020). It has previously been shown that WT ATP13A2 accumulates in cells as an autophosphorylated (E1P or E2P) form in the absence of polyamines and that addition of a polyamine to the culture greatly accelerates dephosphorylation (Holemans et al., 2015; van Veen et al., 2020). In cells, ATP13A2 probably exists in equilibrium between mainly the E1P and E2P states. During the detergent extraction step, cytosolic polyamines seem to bind to the substrate-binding pocket of the enzyme, converting the E2P into E2-Pi state. Addition of nonhydrolyzable ATP analog AMP-PCP to the enzyme after the purification did not change the state, likely because AMP-PCP could not bind to the phosphate-bound catalytic site in the E2-Pi state (Figs. S1F and S2C). However, the E2P-like structure could be obtained by incubating the membrane fraction with BeF3− before detergent extraction (Figs. S1F and S2F), probably because E2·BeF3− is more stable than the native E2P state against binding of polyamines.
Structures of four different E1 states (E1-apo, E1-ATP, E1P-ADP, and E1P) were determined by use of mutant enzymes and/or inclusion of a nucleotide and phosphate analog (Fig. S1F). A phosphorylation-defective mutant (conserved Asp508 replaced by Asn; D508N) in the presence of ATP yielded an E1-ATP structure. A substrate-binding-defective mutant (D458N/D962N; see below for the rationale of the design) produced an E1-apo structure in the absence of a ligand and an E1P-like structure in the presence of AlF4−. An E1P-ADP-like structure could be obtained from the WT enzyme prepared in the presence of ADP.
The highest-resolution (2.5 Å) structure was obtained with the WT AlF4−-bound E2-Pi-like enzyme (Figs. S1F and S3B). However, to describe the overall architecture, we will first reference the 2.8-Å-resolution BeF3−-bound E2P structure (Fig. 1 and S3A), because the cytosolic domains are better resolved in this map. The E2P structure represents a primed state of the transporter, where the substrate-binding pocket within the TMD faces the endo-/lysosomal interior prior to polyamine binding. This structure also likely represents the state that accumulates in cells at a low lumenal polyamine concentration (Holemans et al., 2015; van Veen et al., 2020). Then, we will describe the E2-Pi structure, in which a polyamine molecule is bound to a channel-like cavity, followed by the E1 structures, which represent post-translocation states in an occluded conformation.
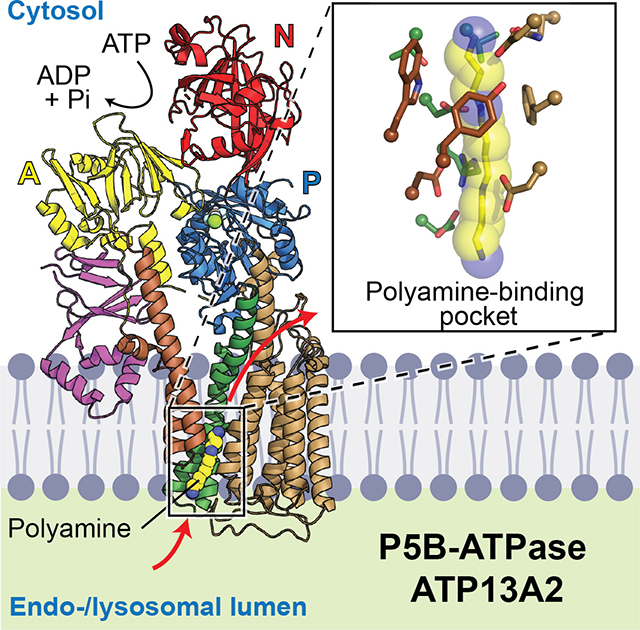
Figure 1. Cryo-EM structure of human ATP13A2 in the E2P state.

(A) Cryo-EM map of human ATP13A2 in the E2·BeF3– form (map 3). Left, a front view; right, a view from the back. Red dashed line, entrance of the substrate-binding pocket. Inset, a zoomed view into the C-terminal extension (CTE) region. Amino acid side chains at the interface between the P domain and CTE are shown as sticks. (B) Atomic model built into the map in A. (C) Domain architecture of ATP13A2. Note that the TM4 helix is unraveled in the middle (between TM4a and TM4b) by a PP(A/V)xP motif conserved among P5B-ATPases. (D) Estimated surface electrostatic potential at pH 7. Inset, electrostatics around the substrate binding pocket estimated at pH 5. (E) View of the substrate-binding pocket in the E2P structure. Dashed line indicates entrance of substrate-binding pocket. Amino acid side chains (balls and sticks) lining the cavity (dots) are shown. Pore radius is indicated by a color gradient. (F–H) Comparison between ATP13A2 and Spf1 structures. TM helices are represented by cylinders. Views are from the lumen. The substrate-binding pockets are indicated by dashed circles.
Architecture of ATP13A2
Overall, the structure of ATP13A2 follows the canonical architecture of P-type ATPases (Fig. 1 A–C). The cytosolic domains include the actuator (A), nucleotide-binding (N), and phosphorylation (P) domains, which are responsible for converting ATP energy to conformational movements required for substrate translocation. In the membrane, ATP13A2 contains a total of ten transmembrane segments (TMs 1–10), as is common for most eukaryotic P-type ATPases (Palmgren and Nissen, 2011) (Fig. 1C).
The TMD of ATP13A2 can be divided into three groups: TMs 1–2, 3–4, and 5–10 (Fig. 1C). The substrate-binding site is formed at the interface of these three groups (Fig. 1A). In the E2P structure, the pocket faces the endo-/lysosomal interior (Fig. 1 A, B, and D). Surface electrostatics calculations estimate that the pocket exhibits a negative electrostatic potential mainly due to multiple solvent-exposed aspartic and glutamic acid residues (D249, E451, and D955) (Fig. 1 D and E). While these acidic residues might be substantially neutralized in the low pH environment of the endo-/lysosomal interior, the pocket is still estimated to maintain an overall negative electrostatic potential at pH 5 (Fig. 1D, inset). These negative charges may attract positively charged polyamines into the pocket during initial polyamine binding steps. Once recruited to the surface, the polyamine molecule may further move into the sizeable (~10-Å long) cavity formed in the protein interior through a rather narrow (~1.8 Å in radius) neck (Fig. 1E).
The overall structure of ATP13A2 is similar to that of Saccharomyces cerevisiae P5A-ATPase Spf1 (a homolog of the mammalian ATP13A1), as expected from ~25% sequence identity between the two (McKenna et al., 2020). The similarity includes the arrangements of their TMs (Figs. 1 F–H and S5 A–C). However, TMs 5–10 in Spf1 are rotated away from the other TMs by ~20° along TM5 compared with ATP13A2. This widens the substrate-binding pocket of Spf1 and creates a laterally open topology to accommodate and flip a polypeptide segment attached to a lipid-embedded TM helix. By contrast, TMs 5–10 of ATP13A2 are positioned adjacent to TMs 1–2, forming a narrow cavity that is separated from the lipid phase and suited for a small, charged molecule. These structural differences provide a basis for the vast difference in substrate specificities between the two P5 subtypes. The P5A-ATPase extracts mis-inserted transmembrane proteins by flipping an ER-lumenal hydrophilic polypeptide segment (length of up to 7–10 residues; ~1,000 Da) attached to a transmembrane helix into the cytosol (McKenna et al., 2020). By contrast, polyamines are water-soluble and considerably narrower in diameter (molecular weight of spermine is ~200 Da).
ATP13A2 also shows unique features in both the N- and C- terminal regions. An ~180-residue-long N-terminal domain (NTD) contains three short hydrophobic α-helices (denoted Ha, Hb, and Hc) that are conserved in P5B-ATPases (Figs. 1 A–C and S5D). Arranged in a triangular spade shape that is attached to the A domain by a β-sandwich, these helices are embedded in the cytosolic leaflet of the lipid bilayer. By providing an additional anchor to the membrane, this feature may play a role in guiding the conformational motions between the A domain and TMs 1–2 for substrate translocation. It may also be involved in endo-/lysosomal targeting of ATP13A2, as suggested previously (Holemans et al., 2015). In the C-terminal region, a cytosolic segment after TM10 (referred to as the C-terminal extension or CTE) forms a small helix, which is bound to the P domain mainly by hydrophobic interactions (Fig. 1A, inset). Sequence comparisons suggest that this feature is also conserved in most P5B-ATPases (Fig. S5E).
Structural basis of polyamine binding
To understand the molecular basis of ATP13A2’s polyamine specificity, a polyamine-bound ATP13A2 structure needed to be determined. Serendipitously, a strong, well-resolved polyamine-like density could be seen in the substrate-binding pocket of the E2-Pi structure without supplementing exogenous polyamines (Fig. 2). Mass spectrometry analysis confirmed the presence of spermine in our WT sample but not in a substrate-binding-defective mutant sample (Fig. S6). Co-purified spermine likely originated from the insect cells used for expression. The polyamine-bound E2-Pi conformation was the predominant form of the WT enzyme in our protein preparations, as it was observed in all WT samples. When purified in the presence of AlF4− or without any added factors, WT enzyme was found to be almost exclusively in the E2-Pi conformation (Fig. S1F). More specifically, the AlF4−-bound WT enzyme is likely in the dephosphorylation transition state, whereas the other observed E2-Pi structures may represent the product (post-hydrolysis) state (Bublitz et al., 2010). However, the conformations in these two states seem indistinguishable (Fig. S2 B–E, G, and H), similar to SERCA (Bublitz et al., 2010; Dyla et al., 2019).
Figure 2. Polyamine-bound E2-Pi structure of ATP13A2.

(A and B) 2.5-Å-resolution cryo-EM map (A) and atomic model (B) of ATP13A2 in the polyamine-bound E2-Pi-like state (E2-AlF4–; map 6). The domains are colored as in Fig. 1A. Gray densities in A are lipids and detergent molecules. (C) Surface electrostatic potential calculated at pH 7. Arrow, polyamine entrance; asterisk, putative polyamine exit (see panel D). (D) Cutaway (side) view of the substrate-binding pocket in the E2-Pi structure (protein surface shown in gray). The view angle is an ~90° right-hand-rotated with respect to that of panels A–C. The observed polyamine (spermine) density is shown as a white mesh. The lipid tail and CHS molecule that sits in front of the putative polyamine exit are shown as cyan balls and sticks. Red arrowheads indicate the narrowest neck of the cavity. The asterisk indicates the position marked by the asterisk in C. Approximate membrane boundaries are also indicated. (E) Radius profile of the transport conduit. The region that spermine spans is marked by a yellow box. Red arrowheads, the narrowest (1.3 Å in radius) region corresponding to the arrowheads marked in D; asterisk, the position marked by the asterisk in C and D.
Compared with the E2P structure, the E2-Pi structure shows a deeper (~15-Å long) and more cylindrical substrate-binding cavity that is well-suited to bind an elongated polyamine molecule (Fig. 2D). This channel-like cavity forms a continuous conduit through the TMD but is constricted to ~1.3 Å in radius near the cytosolic end, which would be too narrow (< ~2Å) for the polyamine molecule to readily pass (Fig. 2D and E). Thus, the conduit is blocked on the cytosol side. However, the topology of the detected conduit suggests that the polyamine exit may be formed within the hydrophobic layer of the cytosolic leaflet of the membrane (Fig. 2 C and D). In our E2-Pi structures, this opening is further capped by a lipid acyl chain and a CHS molecule (Fig. 2D).
The bound spermine molecule is in an extended conformation and positioned along the cavity in the endo-/lysosomal lumenal leaflet. Binding of the polyamine is mediated largely by a network of ionic and cation-π interactions between three out of four amine nitrogen atoms of the spermine and the acidic and aromatic side chains that line the cavity (Fig. 3 A and B). Specifically, one middle amine nitrogen atom of spermine is coordinated by Y251, D458, and F958, and the other two outer amines are coordinated mainly by D249 and D955 on one side and D962 on the other. These amino acids are strongly conserved among P5B-ATPases, suggesting that the same or a similar mode of interaction may be used for substrate binding in other P5B-ATPases. This arrangement of acidic and aromatic amino acids and their interactions with the polyamine substrate in ATP13A2 are strikingly analogous to those observed in bacterial periplasmic polyamine-binding proteins, although their protein folds are completely different (Kashiwagi et al., 1996; Sugiyama et al., 1996) (Fig. 3 C and D). This suggests convergent evolution in polyamine recognition mechanisms.
Figure 3. Structural basis of polyamine binding in ATP13A2.

(A) Stereo view of the polyamine-binding site of ATP13A2 in the E2-Pi structure. The modelled spermine molecule is shown as yellow (carbon)/blue (nitrogen) sticks and semitransparent spheres. Side chains of ATP13A2 coordinating the spermine molecule are shown in a stick representation (brown, TMs 1–2; green TM4; tan, TM6). (B) Interactions between ATP13A2 and the polyamine (spermine) molecule. Dashed lines, ionic and cation-π interactions. (C) As in A, but with the spermidine-bound Escherichia coli PotD (PDB: 1POT). (D) As in B, but for the PotD-spermidine structure. (E and F) Spermine-induced ATPase stimulation of microsomes expressing WT ATP13A2 or mutants of polyamine-interacting aspartate (E) or aromatic (F) residues (means and s.e.m.). Lines are fitted dose-response (Michaelis-Menten) curves.
To probe the functional importance of these acidic and aromatic amino acids, we conducted mutagenesis experiments. We isolated microsomes overexpressing WT or mutant ATP13A2 from Sf9 cells (Fig. S7 A–H) and performed ATPase assays in the presence of varying concentrations of spermine (Fig. 3 E and F). As previously shown (van Veen et al., 2020), the ATPase activity of WT ATP13A2 was robustly stimulated by spermine. On the other hand, the D458N, D962N, and D458N/D962N mutants showed no spermine-dependent ATPase stimulation, suggesting their inability to bind spermine (Fig. 3E). Similarly, the W246V, Y251M, and F958V mutants also displayed little or no ATPase stimulation (Fig. 3F). These results confirm the importance of these amino acids for polyamine binding.
Conformational coupling between the cytosolic and transmembrane domains
Our E2P and E2-Pi structures suggest that in ATP13A2, binding of a polyamine molecule to its substrate-binding pocket induces a conformational change in the TMD and a transition from the phosphoenzyme (E2P) intermediate to a dephosphorylated (E2-Pi) state (Fig. 4A). This is consistent with the general mechanism of all known P-type ATPases, in which conformational changes of the TMD are tightly linked with nucleotide- and phosphorylation- state of the cytosolic domains. However, the ATP13A2 structures show interesting differences. In other P-type ATPases, the E2-Pi structures typically adopt a substrate-bound/occluded conformation (Bublitz et al., 2010; Hiraizumi et al., 2019; Nakanishi et al., 2020; Palmgren and Nissen, 2011). However, the polyamine-bound E2-Pi structure of ATP13A2 displays an outward-open conformation with part of the bound polyamine molecule exposed to the lumen. This difference might be due to the elongated structure of polyamines.
Figure 4. Movements of the cytosolic domains and TMD during the catalytic cycle of ATP13A2.

(A) Structural comparison between E2P and E2-Pi states of ATP13A2. The color scheme is the same as Figs. 1 and 2. Arrows indicate major domain movements between the compared structures. Structures were aligned based on the P domain and TMs 5–10. (B–D) As in A, but comparison between E2-Pi and E1-apo (B), between E1P-ADP and E1P (C), or between E1P and E2P states (D). For RMSDs between structures, see Fig. S8D. (E) View into the phosphorylation site (D508) in the E2P structure. A, P, and N domains are colored yellow, blue, and red, respectively. The TGES loop containing E343 is in orange with side chains shown in a stick representation. (F) As in E, but with the E2-Pi structure. Arrows, overall movements of the TGES loop and A domain from the E2P structure. (G) As in E and F, but with the E1-apo structure. Arrows, overall movements from the E2-Pi state.
To gain further insights into the mechanisms for substrate transport and coupling between the cytosolic and transmembrane domains, we determined structures of its E1 states (Fig. 4B–D, S1F, and S8). Based on the Post-Albers scheme, we reasoned that inclusion of ADP throughout protein purification might shift the equilibrium to favor an E1P-ADP state. Indeed, this method yielded an E1P-ADP-like structure from ~20% particles (Fig. S1F). In addition to this approach, a use of the phosphorylation-defective D508N mutant in the presence of ATP exclusively yielded an E1-ATP-like structure, whereas the polyamine-binding-defective D458N/D962N mutant exclusively yielded an E1-apo structure in the absence of an added ligand and an E1P-like structure in the presence of AlF4− (Fig. S1F). In all E1 structures, no polyamine-like density was observed in the substrate-binding site. A lack of an E2-Pi population in the D458N/D962N mutant, in stark contrast to WT ATP13A2, suggests that polyamine binding is necessary to form the E2-Pi state. Thus, this finding explains the dramatic stimulation of ATP13A2’s ATPase activity induced by polyamines (van Veen et al., 2020).
All E1 structures of ATP13A2 display a highly similar overall conformation, including that of the TMD (Figs. 4C and S8). One exception is the flexible N domain, which either is in a closed position when ATP or ADP is bound at the interface between the N and P domains, or swings outward in the absence of adenine nucleotide (Fig. S8C). The lack of major conformational changes in the TMD between the E1 structures and the nucleotide-dependent movement in the N domain are consistent with observations made previously with other P-type ATPases (Bai et al., 2020; Dyla et al., 2019; Hiraizumi et al., 2019; McKenna et al., 2020; Nakanishi et al., 2020).
A comparison of conformations between E2-Pi, E2P, and E1 structures of ATP13A2 further demonstrates how the enzyme couples the phosphorylation state of the P domain to the conformation of the TMD through the motion of the A domain (Fig. 4). The A domain contains the conserved Thr341-Gly-Glu-Ser344 (TGES) loop, where the side-chain carboxyl group of E343 catalyzes dephosphorylation. In both E2P and E2-Pi states, the TGES loop is in close proximity to the phosphorylated D508 of the P domain (Fig. 4 E and F). However, during the E2P to E2-Pi transition, the A domain is tilted by ~20°, because the TGES loop is displaced by ~4 Å as the side chain of E343 becomes directed toward the aspartyl phosphate for dephosphorylation (Fig. 4F). This movement seems to be coupled with the transition of the TMD from the outward-facing (E2P) to the channel-like (E2-Pi) conformation mainly through TMs 1–2, which are attached to the A-domain (Fig. 4A). In the E1 structures, the TGES loop is further displaced (~13 Å) from D508 in an orthogonal direction (Fig. 4G). This causes substantial conformational changes in the TMD that are associated with the E2-Pi to E1-apo and E1P to E2P transitions (Fig. 4 B and D; also see below).
Putative polyamine paths into the cytosol
During the E2-Pi to E1-apo transition, the polyamine molecule bound to the channel-like cavity is expected to be released into the cytosol. In fact, many P-type ATPases, including the P5A-ATPase, display an inward-open (cytosolically open) conformation in the E1 state, which is compatible with substrate release into the cytosol (Bai et al., 2020; McKenna et al., 2020; Palmgren and Nissen, 2011; Toyoshima et al., 2000; Winther et al., 2013). Contrary to this expectation, the structures of ATP13A2 in E1 states seems to adopt an “occluded” conformation (Fig. 5).
Figure 5. Structure of ATP13A2 in the E1 state.

(A and B) TMD of D458N/D962N ATP13A2 in the E1-apo state. Shown are front (A) and top (B) views. TMs are colored in the same scheme as Figs. 1 and 2. The potential polyamine paths were detected by a spherical probe and are shown in cyan and gray. Approximate membrane boundaries are indicated. The side chains of the indicated amino acids are shown in sticks and dots (panel B only). (C) Radius profiles of the paths shown in A and B. The solid and dotted lines correspond to the cyan and gray paths, respectively. Gray dashed vertical line, an average pore radius (2.1 Å) of the spermine-bound region in the E2-Pi structure (see Fig. 2E). (D) Conformational transition of TMs 1–2 (indicated by an arrow) from the E2-Pi (in gray) to E1-apo state (in color). The structures were aligned with respect to TMs 5–10. Dashed lines, TM2 axis. Gray mesh, the putative polyamine transport conduit (cyan path in panels A and B).
Compared with the E2-Pi structure, the substrate cavity in the E1 states is too narrow (with a radius of ~1 and 1.5Å) to accommodate a polyamine (Fig. 5 A–C). In fact, the cavity seems inaccessible from both sides. Thus, the E1 structures present post-translocation states prior to the enzyme becoming primed for the next round of polyamine translocation. A comparison between the E2-Pi and E1-apo structure shows that the rearrangement of the A domain (Fig. 4G) leads to mainly a translational (~5 Å downward) and rotational (~12° towards the front) movement of TMs 1–2 with respect to the TMs 3–10 (Figs. 4B and 5D). This causes a narrowing of the substrate conduit in the E2-Pi to E1 transition (Figs. 2E versus 5C).
Although the cavity in the E1 structures seems too narrow for polyamines, a continuous conduit could be detected. The conduit follows a path similar to that seen in the E2-Pi structure, and its cytosolic end is directed to the cytosolic leaflet of the membrane (Fig. 5 A and B; cyan path). Thus, the polyamine molecule bound to the E2-Pi cavity may follow this path to exit to the cytosol during the E2-Pi to E1 transition. Alternatively, the polyamine molecule may travel through another path branching out from the above-mentioned conduit around Y254 (Fig. 5 A and B; gray path). In our structures, this path is blocked by the side chains of Q239, I258, V464, and P466, but we cannot rule out the possibility that it may open during the E2-Pi to E1 transition.
Lipid interactions
The topology of the polyamine conduit seen in our E2-Pi and E1 structures suggest a possibility that the polyamine molecule may exit into the cytosolic leaflet of the membrane. Because polyamines are cations under physiological pH, their exposure to the hydrophobic phase of the membrane is expected to be energetically unfavorable. However, given the fact that polyamines can interact with negatively charged lipids (Yung and Green, 1986), the actual energetic barrier for polyamine release might be lowered by interactions with anionic lipids at the exit site.
Perhaps relevant to this issue, we noticed one prominent feature of the ATP13A2 structure—a belt of highly positively charged surface around the protein at the cytosol-membrane interface (Fig. 1D). This positively charged surface may recruit negatively charged lipids, including phosphatidic acid (PA) and phosphatidylinositol(3,5)biphosphate [PI(3,5)P2], from the cytosolic leaflet to ATP13A2. In fact, PA and PI(3,5)P2 have been shown to substantially stimulate the ATPase activity of ATP13A2 (Holemans et al., 2015; van Veen et al., 2020). The positive surface charge of ATP13A2 is mainly due to a high ratio of basic to acidic amino acids in this region (Fig. S9A). Homology modeling of other P5B-ATPases also showed similar distributions of charged amino acids, suggesting that this feature is common among P5B-ATPases (Fig. S9B). On the other hand, we did not find similar features in other P-type ATPases, such as the sarco-/endoplasmic reticulum Ca2+-ATPase (SERCA), P4-flippases, and P5A-ATPase (Fig. S9C).
To test interactions between the positively charged surface and lipids, we performed 10-μs coarse-grain molecular dynamics (MD) simulations of the E2P structure embedded in a model lysosomal membrane (Fig. S10 and Movies S1 to S4). The results show that negatively charged lipids are preferentially recruited to ATP13A2, in particular phosphoinositide (Fig. S10, “E2P/u” panels). If present, the polyanionic PI(3,5)P2 lipid strongly accumulated around ATP13A2, including the potential polyamine exit site, outcompeting less negatively charged lipids (Fig. S10, “E2P/p” panels). A similar result was also obtained with an E2-Pi structure (Fig. S10, “E2-Pi/u” and “E2-Pi/p” panels). Therefore, as hypothesized, the positive electrostatic belt causes strong lipid sorting in the simulations and enhances the local concentration of (poly)anionic lipids around ATP13A2.
Disease-associated mutations
Lastly, the structure of ATP13A2 allowed us to map known disease-associated missense mutations in order to better understand the mechanisms of these mutations (Fig. 6A and Table S2). Many mutations were mapped in buried regions of domains, suggesting that they likely cause protein folding defects. One mutation (G517V) was mapped in the connection between the P and N domains, which may impair the interdomain movements for the catalytic reaction. The A244V and R975H mutations may affect the transport function of the enzyme by altering interactions with lipids. Interestingly, two mutations were expected to affect the functions of the NTD and CTE based on our structures: the F177L mutation is located in the β-sandwich of the NTD (Fig. S5D), and the P837L mutation is at the interface between the P domain and CTE (Fig. 1A, inset). Similarly, a premature termination mutation (Q1135X) identified in a hereditary spastic paraplegia patent is expected to produce an enzyme lacking CTE (Estrada-Cuzcano et al., 2017).
Figure 6. Disease-associated mutations of ATP13A2 and functional importance of CTE.

(A) Positions of disease-associated missense mutations (colored spheres) were mapped onto the ATP13A2 E2P structure. Magenta, mutations that potentially cause protein folding defects. Orange, mutations that potentially alter the D508-phosphorylation reaction (T512I) or P-N interdomain interaction (G517V). Cyan, mutations of unknown mechanisms. Q1135X is also indicated (gray sphere). See also Table S2. (B) Spermine-induced ATPase stimulation of microsomes overexpressing WT ATP13A2 and the ΔNTD, ΔCTE, and D508N (control) mutants (means and s.e.m.). Lines are fitted dose-response curves.
The mapping of disease mutations suggested the functional importance of the NTD and CTE, both of which are unique features of P5B-ATPases compared to other P-type ATPases. To test this, we performed polyamine-dependent ATPase stimulation assay with mutants lacking the NTD (Δ2–179; ΔNTD) or CTE (Δ1149–1175; ΔCTE) (Fig. 6B). The ΔNTD mutant showed an activity comparable to WT, suggesting that the NTD itself is not required for the catalytic cycle (Fig. 6B). Instead, the F177L mutation likely causes misfolding or decreased stability of the NTD, leading to a trafficking defect at the ER and degradation (Podhajska et al., 2012). Unlike ΔNTD, the ΔCTE mutant showed essentially no ATPase activity, despite a comparable expression level (Fig. 6B and S7J). Fluorescence size-exclusion chromatography analysis indicates a moderate tendency of the ΔCTE mutant to form high molecular-weight aggregates upon detergent extraction (Fig. S7J). While the exact function is yet to be elucidated, the CTE thus seems to be important for the activity and structural stability of ATP13A2.
Discussion
Our study demonstrates that ATP13A2 is a dedicated polyamine transporter and reveals the molecular basis for its polyamine recognition and transport. Our structural analysis captured nearly all major intermediate states of the ATP13A2 transport cycle. This allows us to propose a tentative working model for polyamine export by ATP13A2 on the basis of the Post-Albers scheme (Fig. 7A). In this model, polyamine binding from the end-/lysosomal lumen and membrane translocation occurs during the transitions from the E2P to E2-Pi state and from the E2-Pi to E1 state, respectively. In the E2-Pi state, the TMD of ATP13A2 forms an outward-open elongated conduit of ~2-Å radius, which binds a polyamine molecule in an extended conformation (Fig. 2D). Because the conduit is only wide enough for one polyamine molecule, the polyamine-occupied conduit would be impermeable to other ions and molecules. During the transition from the E2-Pi to E1 state, the polyamine molecule is expected to be released to the cytosol. The conformational trajectory of this step remains unclear as our current analysis did not resolve further intermediates between the E2-Pi to E1 transition (e.g., E2). While this transition would likely involve a transient luminally-closed but cytosolically-open state, the formation of a large cytosolic opening seems unlikely, considering the closed conformation observed in the E1 states.
Figure 7. Proposed model of polyamine translocation mediated by ATP13A2.

(A) Polyamine-transport cycle of ATP13A2. All displayed structures were experimentally observed in this study. Gray arrows indicate major movements of the cytosolic domains (with respect to the previous state). (B) Putative model for the polyamine-translocation mechanism of ATP13A2. Schematics for the TMD are shown. Late endosomal and lysosomal membranes are known to have a membrane potential (ΔΨ) of ~20 to 60 mV (positive in the lumen). We note that the exact protonation states of ATP13A2 (acidic amino acids in the substrate cavity) and the exiting polyamine molecule are unknown.
The narrow, channel-like conduit and the mode of polyamine binding seen in the E2-Pi structure suggest that ATP13A2 may deviate from the canonical model of active transport. Instead of simply transitioning between outward-open, occluded, and inward-open conformations, polyamine transport by ATP13A2 likely requires the polyamine molecule to move within the narrow and elongated cavity from the lumenal entrance toward the cytosolic exit. Our structures suggest that without such movement, the lumenal gate would not be able to close, since it is blocked by the polyamine molecule near the cavity entrance. The motion of the polyamine away from the lumenal side might be driven partly by the endo-/lysosomal membrane potential, in addition to a local electrostatic gradient within the cavity and conformational changes of the cavity during the E2-Pi to E1 transition. Similar voltage-dependent passage of polyamines along pores of ion channels has been well documented (Twomey et al., 2018; Williams, 1997). A gradual upward movement would also explain how ATP13A2 binds and transports polyamines of different lengths.
The exact path through which the bound polyamine molecule travels to the cytosol remains to be elucidated. However, our structures raise a possibility that the polyamine exit may be formed within the cytosolic leaflet of the membrane. In this scenario, the polyamine molecule would move along the protein-lipid interface toward the cytosol as it exits through the opening (Fig. 7B). The membrane might locally thin around the exit site, which would facilitate the solvation of polyamine charges. The exiting polyamine molecule would also likely interact with (poly)anionic lipids (Yung and Green, 1986) recruited to the surface of ATP13A2 (Fig. S9), which may promote the release of the polyamine molecule. This mechanism could enhance efficiency and directionality of polyamine export because once completely released into the cytosol, the polyamine would be blocked from returning to the cavity by a seal of lipids. This valve-like mechanism would also help prevent ion leakage by minimizing their passage through the conduit, which could otherwise dissipate the endo-/lysosomal membrane potential and proton gradient.
A comparison of the P5A- and P5B- transporters reveals how a relatively small structural rearrangement dramatically changes their substrate specificities (Fig. 1 F–H and S5 A–C). Previously, phylogenetic analysis of P5-ATPases has shown that almost all eukaryotic species have a single P5A-ATPase (e.g., ATP13A1 in humans), whereas the P5B group had diversified into multiple isoforms in most metazoans (e.g., ATP13A2–5 in humans) (Sorensen et al., 2018). At first, this seems counterintuitive, considering that polypeptides are structurally more diverse than polyamines. However, this may be because the P5A-ATPase recognizes substrates through a combination of more general properties of its substrate polypeptides, such as hydrophobicity/hydrophilicity and overall charges (McKenna et al., 2020). On the other hand, our study shows that all P5B-ATPases likely use a pocket lined with the specifically arranged acidic and aromatic side chains to coordinate the amine groups of polyamines. Therefore, through minor structural variations of the cavity, the other P5B-ATPases may have evolved to exhibit different polyamine specificities (Hamouda et al., 2020) and/or operate at different optimal substrate concentration ranges.
In addition to P5B-ATPases, putative polyamine transporters have been identified in other membrane transporter families, such as the ATP-binding cassette (ABC) transporters and solute carriers (SLC) (Aouida et al., 2005; Fujita et al., 2012; Hiasa et al., 2014; Igarashi and Kashiwagi, 1999). To our knowledge, none of these transporters have been structurally characterized. While their transport mechanisms remain to be elucidated, the convergent evolution of polyamine-binding modes we identified here (Fig. 3 A–D) suggests that they may use similar mechanisms for substrate recognition.
Limitations of the Study
Although our study presents a near-complete catalytic cycle of ATP13A2, a structure of a potential E2-apo state is currently missing. This prevents us from precisely defining the route of polyamine release from the position in the E2-Pi structure into the cytosol. The E2-apo structure, which represents the transition state between the E2-Pi and E1 conformations, may reveal further details about the movement of the polyamine molecule along the cavity and release into the cytosol. While we propose that polyamines may exit through a gate formed laterally into the cytosolic leaflet membrane, further investigations would be necessary to corroborate this idea. This polyamine release process might be facilitated by several features of the system, including membrane potential, possible local membrane thinning, and a distribution of anionic lipids around the polyamine exit site. We also currently do not know the exact protonation state of the exiting polyamine molecule. It is possible that some of the protons initially bound to the polyamine molecule are transferred to acidic amino acids in the cavity before the polyamine is released into the cytosol. These hypotheses will need to be experimentally tested in the future.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Eunyong Park (eunyong_park@berkeley.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Cryo-EM maps for ATP13A2 (maps 1–12) have been deposited in the Electron Microscopy Data Bank (EMDB)and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Coordinates for models of ATP13A2 (map 3 and 6–12) have been deposited in the Protein Data Bank (PDB) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
The paper does not report original code.
Any additional information required to reanalyze the data reported in the paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Escherichia coli DH10Bac competent cells | Thermo | Cat#10361012 |
| Chemicals, peptides, and recombinant proteins | ||
| Cellfectin II | Thermo | Cat#10362100 |
| Polyethylenimidine (PEI MAX) | Polysciences | Cat#24765 |
| ESF 921 medium | Expression Systems | Cat#96–001 |
| n-Dodecyl-β-D-maltopyranoside (DDM) | GoldBio | Cat#DDM |
| Cholesteryl hemisuccinate | Anatrace | Cat#CH210 |
| Critical commercial assays | ||
| ADP-Glo™ Kinase Assay | Promega | Cat#V9101 |
| Deposited data | ||
| Cryo-EM structure of ATP13A2 in the E2-Pi state (map 1) | This paper | EMD-24212 |
| Cryo-EM structure of ATP13A2 in the E2-Pi state (map 2) | This paper | EMD-24213 |
| Cryo-EM structure of ATP13A2 in the E2·BeF3− state (map 3) | This paper | EMD-24214, PDB: 7N70 |
| Cryo-EM structure of ATP13A2 in the E2-Pi state (map 4) | This paper | EMD-24215 |
| Cryo-EM structure of ATP13A2 in the E2-AlF4− state (map 5) | This paper | EMD-24216 |
| Cryo-EM structure of ATP13A2 in the E2-AlF4− state (map 6) | This paper | EMD-24217, PDB: 7N72 |
| Cryo-EM structure of ATP13A2 in the E1P-ADP-like state (map 7) | This paper | EMD-24218, PDB: 7N73 |
| Cryo-EM structure of ATP13A2 D508N mutant in the E1-ATP state (map 8) | This paper | EMD-24219, PDB: 7N74 |
| Cryo-EM structure of ATP13A2 D458N/D962N mutant in the E1-apo state, Conformation 1 (map 9) | This paper | EMD-24220, PDB: 7N75 |
| Cryo-EM structure of ATP13A2 D458N/D962N mutant in the E1-apo state, Conformation 2 (map 10) | This paper | EMD-24221, PDB: 7N76 |
| Cryo-EM structure of ATP13A2 D458N/D962N mutant in the E1·AlF4− state (map 11) | This paper | EMD-24222, PDB: 7N77 |
| Cryo-EM structure of ATP13A2 in the E2-Pi state (map 12) | This paper | EMD-24223, PDB: 7N78 |
| SDS-PAGE gel image of size-exclusion chromatography fractions (Fig. S1A) | This paper | DOI:10.17632/g64mnvkgyk.1 |
| Experimental models: cell lines | ||
| Spodoptera frugiperda (Sf9) | Expression Systems | 94–001F |
| Software and algorithms | ||
| Warp | Tegunov et al., 2019 | http://www.warpem.com/warp/ |
| cryoSPARC v2 | Punjani et al., 2017 | https://cryosparc.com/ |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix | Afonine et al., 2018 | https://www.phenix-online.org/ |
| MolProbity | Chen et al., 2010 | http://molprobity.biochem.duke.edu/ |
| Adaptive Poisson-Boltzmann Solver | Baker et al., 2001; Dolinksy et al., 2004 | https://www.poissonboltzmann.org/ |
| PyMOL | Schrödinger | https://pymol.org/2/ |
| Caver 3.0 | Chovancova et al., 2012 | http://www.caver.cz/ |
| UCSF Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| UCSF ChimeraX | Goddard et al., 2018 | https://www.cgl.ucsf.edu/chimerax/ |
| R software and drc2 package | Ritz et al., 2015 | https://www.r-project.org/ |
| Martini 2.2 | de Jong et al., 2013 | http://cgmartini.nl/ |
| Gromacs 2020/3 | Abraham et al., 2015 | https://www.gromacs.org/ |
| numpy | https://www.numpy.org | |
| MDAnalysis | Gowers et al., 2016 | https://www.mdanalysis.org/ |
| matplotlib | https://www.matplotlib.org | |
| Other | ||
| Quantifoil holey carbon grid (R1.2/1.3, Au, 400 mesh) | Electron Microscopy Sciences | Cat#Q4100AR1.3 |
| Superose 6 Increase, 10/300 GL | GE Life Sciences (Cytiva) | Cat#29–0915–96 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Sf9 cells were cultured in ESF 921 medium (Expression Systems) at 27 °C.
METHOD DETAILS
DNA constructs
A coding sequence for full-length human ATP13A2 (isoform2; NCBI sequence ID NP_001135445.1; UniProt ID Q9NQ11–3) was obtained by PCR amplification of clones (Clone IDs: 5240813 and 5139467) from Mammalian Gene Collection cDNAs (Horizon Discovery) and inserted into pFastBac-1 (Thermo Fisher Scientific) by standard molecular biology techniques. The coding sequence is followed by an HRV 3C protease site and an enhanced GFP tag. Mutant ATP13A2 constructs were generated by PCR, and sequences were verified by Sanger DNA sequencing.
Baculovirus generation and Sf9 expression
Recombinant bacmids were generated from the pFastBac1 constructs using the Bac-to-Bac baculovirus expression system. DH10Bac E. coli competent cells (Thermo Fisher Scientific) were transformed with each pFastBac1-ATP13A2-GFP construct, and colonies were selected on an LB agar plate containing 50 μg/mL kanamycin, 7 μg/mL gentamycin, 10 μg/mL tetracycline, 100 μg/mL Bluo-Gal, 40 μg/mL IPTG. Bacmids were isolated and used for transfection of Sf9 cells. Sf9 cells were cultured in ESF 921 medium (Expression Systems) at 27 °C.
Baculovirus were generated by transfecting Sf9 cells using Cellfectin II reagent (Thermo Fisher Scientific), according to the standard Bac-to-Bac protocol or linear polyethylenimidine (PEI MAX; Polysciences) (Scholz and Suppmann, 2017). When necessary, baculovirus was amplified by infecting Sf9 cells at a 1:1000 volume-to-volume ratio and harvesting the culture supernatant after 4 days post-infection. For protein expression, Sf9 cells were infected at ~1.5–2.0 × 106 cells/mL. Expression of ATP13A2 was monitored by GFP fluorescence of infected cells. Cells were harvested by centrifugation (1,500g for 7 min), 2–3 days post-infection, before any substantial cell death. Cell pellets were frozen in liquid nitrogen and stored at −80 °C until use.
Preparation of microsomes
Sf9 cell pellets expressing GFP-tagged ATP13A2 constructs from 0.4 L cultures were thawed and washed twice with 15 mL of ice-cold phosphate-buffered saline (pH 7.4) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4). All subsequent steps were carried out at 4 °C. Cells were collected by centrifugation (1,500g for 7 min) and resuspended in 10 mL of hypotonic lysis buffer (10 mM Tris pH 7.5, 0.5 mM MgCl2, 2 mM DTT, 0.1 mM PMSF, 5 μ g/mL aprotinin, 5 μ g/mL leupeptin, and 1 μ g/mL pepstatin A). Cells were swollen in the lysis buffer by incubating them on ice for 10–15 min and lysed with 40 strokes of a Dounce homogenizer. The lysate was diluted using 10 mL of resuspension buffer (10 mM Tris pH 7.5, 0.5 M sucrose, 0.3 M NaCl, 2 mM DTT, and 0.5 mM PMSF) and further homogenized with 20 strokes of a Dounce homogenizer. Subcellular fractionation was subsequently performed through steps of differential centrifugation. The suspension was centrifuged at 1,000g for 10 min to remove unbroken cells and nuclear debris, and the supernatant was centrifuged at 10,000g for 20 min (Sorvall SS-34 rotor). The supernatant from this step was then subjected to ultracentrifugation at 200,000g for 35 min (Beckman Type 45 Ti rotor) to collect microsomes. The microsomal pellet was resuspended in 0.25 M sucrose containing 5 μ g/mL aprotinin, 5 μ g/mL leupeptin, 1 μ g/mL pepstatin A, and 2 mM PMSF at a concentration between 2 and 5 mg/mL. Microsome concentrations were measured based on the total protein concentration using the Bradford assay with bovine serum albumin as a standard. Aliquots of the resuspended microsomes were frozen in liquid nitrogen and stored at −80 °C until use.
Fluorescence size exclusion chromatography (FSEC)
100 μ g microsomes were solubilized in a lysis buffer containing 50 mM Tris pH 7.5, 200 mM NaCl, 1 mM EDTA, 1 mM DTT, 10% glycerol, 5 μ g/mL aprotinin, 5 μ g/mL leupeptin, 1 μ g/mL pepstatin A, and 2 mM PMSF, 1% n-dodecyl-β-D-maltopyranoside (DDM; Anatrace), and 0.2% cholesteryl hemisuccinate (CHS; Anatrace) for 2–3 h at 4 °C. Solubilized microsomes were clarified by centrifugation (17,000g for 1 h at 4 °C). The samples (equivalent to ~85 μ g microsomes) were then analyzed with an HPLC system and injected to a Superose 6 column (GE Life Sciences), equilibrated in a running buffer containing 25 mM Tris pH 7.5, 100 mM NaCl, 1 mM EDTA, 0.03% DDM, 0.006% CHS. Elution of the GFP-tagged ATP13A2 constructs was monitored by a fluorometer (λex= 475 nm; λem= 510 nm; with a fixed gain) connected to the column.
ATPase activity assay
The ATPase activity of ATP13A2 in microsomes was measured using a luminescence-based ADP detection assay (ADP Glo Max assay; Promega). Reactions were performed in a final volume of 5 μ L containing 50 mM MOPS-KOH pH 7.0, 100 mM KCl, 11 mM MgCl2, 1 mM DTT, 0.02% DDM, 1 μg microsomes, 5 mM ATP, and various concentrations of spermine (SPM). Before initiating the reaction by addition of ATP, microsomes were first allowed to equilibrate in the reaction buffer for 1.5 h on ice and for 10 min at 37 °C. The ATPase reaction was performed for 15 min at 37 °C upon addition of ATP and was terminated by heating samples for 5 min at 80 °C. The reaction was then mixed with 5 μ L of ADP-Glo Reagent for 60–80 min at 23°C, followed by addition of 10 uL of ADP-Glo Max Detection Reagent for 60 min. Luminescence was measured in a 384 multi-well plate using a luminometer (NOVOstar; BMG Labtech). Raw luminescence values were converted to ADP amounts based on a standard curve generated with 5 mM ATP/ADP mixtures at varying ratios but without microsomes. Averages and s.e.m. were calculated. Averages were corrected for the basal ATP hydrolysis activities of microsomes by subtracting the value at 0.001 mM SPM. Dose-response curves were generated using the R software with the drc (Ritz et al., 2015) and ggplot2 (https://ggplot2.tidyverse.org) packages.
Purification of human ATP13A2 for cryo-EM analysis
Sf9 cells expressing GFP-tagged ATP13A2 constructs were thawed and resuspended in a lysis buffer (Buffer L1) containing 50 mM Tris pH 7.5, 200 mM NaCl, 1 mM EDTA, 1 mM DTT, 10% glycerol, 5 μ g/mL aprotinin, 5 μ g/mL leupeptin, 1 μ g/mL pepstatin A, and 2 mM PMSF. All subsequent steps were carried out at 4 °C. Cells were mechanically lysed by douncing and unbroken cells, and large debris was removed by centrifugation at 4,000g for 10 min. Membranes were pelleted by ultracentrifugation (Beckman Type 45 Ti rotor) at 100,000g for 1.5 h and resuspended in Buffer L1. The membranes were then solubilized with 1% DDM and 0.2% CHS for 2.5 h. The lysate was clarified by ultracentrifugation (Beckman Type 45 Ti rotor) at 100,000g for 1 h, and the supernatant was mixed with Sepharose beads conjugated with anti-GFP nanobody for 2.5 h. The beads were washed with approximately 30 column volumes of a wash buffer (Buffer W1) containing 25 mM Tris pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.03% DDM, 0.006% CHS. ATP13A2 was eluted by incubating the beads with ~10 μ g/mL HRV 3C protease (in Buffer W1) for ~14 h. The eluate was concentrated using Amicon Ultra (cut-off 100k; GE Life Sciences) and injected into a Superose 6 Increase 10/300 GL column (GE Life sciences), equilibrated with Buffer W1. Peak fractions were pooled and concentrated to ~5–7 mg/mL before cryo-EM grid preparation. To obtain ATP13A2 in specific reaction intermediate states, this base procedure was modified as described below.
The WT ATP13A2 sample that yielded map 1 was purified without isolating crude membranes. Sf9 cells were directly lysed and solubilized with Buffer L1, 1% DDM, and 0.2% CHS for 3 h. All subsequent steps were the same as the base procedure.
For WT ATP13A2 prepared with AMP-PCP (sample for map 2), the crude membrane pellet was resuspended in a lysis buffer (Buffer L2) containing 50 mM Tris pH 7.5, 200 mM NaCl, 1 mM EDTA, 1 mM DTT, 10% glycerol, 10 mM MgCl2, 0.5 mM spermine (SPM; Sigma), 5 μ g/ml aprotinin, 5 μ g/ml leupeptin, 1 μ g/ml pepstatin A, and 2 mM PMSF. After detergent solubilization, binding with anti-GFP nanobody beads, and washing with Buffer W1 as described above, ATP13A2 was eluted by incubating the beads with ~10 ug/mL 3C protease in a wash buffer (Buffer W2) containing 25 mM Tris pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, 10 mM MgCl2, 0.1 mM SPM, 0.04% DDM, 0.0048% CHS, 0.0016% dipalmitoyl PA (DPPA; Echelon), and 0.001% phosphatidylinositol 3,5-bisphosphate diC16 (PI(3,5)P2-diC16; Echelon) for ~14 h. The eluate was concentrated and injected into a Superose 6 Increase column equilibrated with a running buffer (Buffer W3) containing 25 mM Tris pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, 5 mM MgCl2, 0.04% DDM, 0.0048% CHS, and 0.0016% DPPA. Purified and concentrated ATP13A2 was incubated with 1 mM AMP-PCP for 1 h at 4°C before grid preparation. We note that ATP13A2 prepared in this way was found to be in the E2-Pi state and failed to bind AMP-PCP.
For WT ATP13A2 prepared with BeF3− (sample for maps 3 and 4) and AlF4− (sample for map 5), the phosphate analogs (2 mM BeF3− or 2 mM AlF4−) and 10 mM MgCl2 were added during detergent solubilization of the crude membrane pellet and were maintained in all subsequent buffers during the purification. For the purification with AlF4−, inhibitor concentration was reduced to 1 mM AlF4− and 5 mM MgCl2 during size-exclusion chromatography.
For WT ATP13A2 prepared with ADP (sample for maps 6 and 7), 2 mM ADP and 5 mM MgCl2 was added during detergent solubilization of the crude membrane pellet and was maintained in all subsequent buffers during the purification. In addition, 2 mM AlF4− was added to the running buffer for size-exclusion chromatography.
For the D508N mutant bound to ATP (sample for map 8), ATP13A2 was purified according to the base procedure and incubated with 2 mM ATP and 5 mM MgCl2 for 1 h at 4°C before grid preparation.
The D458N/D962N mutant in its apo form (sample for maps 9 and 10) was purified according to the base procedure.
For the D458N/D962N mutant bound to AlF4− (sample for map 11), 2 mM AlF4− and 10 mM MgCl2 were added during detergent solubilization of the crude membrane pellet and maintained in all subsequent buffers during the purification, except during size-exclusion chromatography where the inhibitor concentration was reduced to 1 mM AlF4− and 5 mM MgCl2 in the running buffer.
Mass-Spectrometry
ATP13A2 samples were purified as described above in a buffer containing 25 mM Tris pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.03% DDM, 0.006% CHS and concentrated to ~1.2 mg/mL. Standards for spermidine (Sigma-Aldrich) and spermine (Sigma-Aldrich) were prepared at a concentration of 10 μ M in a buffer containing 25 mM Tris pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.1% DDM, 0.02% CHS. Samples were frozen in liquid nitrogen and stored at −80 °C until use. Each sample was diluted 1:1 into acetonitrile with 1% formic acid (volume/volume) and analyzed by nanoelectrospray ionization (nanoESI) high-resolution mass spectrometry, using an LTQ-Orbitrap-XL mass spectrometer (Thermo Fisher Scientific) at the QB3/Chemistry Mass Spectrometry Facility (University of California, Berkeley). The mass spectrometer was equipped with a nanoESI source and operated in the positive ion mode (Juraschek et al., 1999; Wilm and Mann, 1996). Mass spectra were acquired at a mass resolution setting of 100,000, as measured at mass-to-charge ratio (m/z) = 400, full width at half-maximum peak height Mass spectrometry data acquisition and processing were performed using Xcalibur software (version 2.0.7, Thermo).
Cryo-EM grid preparation and data acquisition
To prepare cryo-EM grids, 3 μL of the ATP13A2 sample were applied to a glow-discharged (PELCO easiGlow; 0.39 mBar, 25–30 mA, 40–45 s) gold holey carbon grid (Quantifoil R 1.2/1.3, 400 mesh). The grid was blotted for 3–4 s and plunge-frozen in liquid-nitrogen-cooled liquid ethane using Vitrobot Mark IV (FEI) operated at 4° C and 100% humidity. Whatman No. 1 filter paper was used to blot the samples.
The WT-BeF3−, WT-AlF4−, WT-AMP-PCP, and D508N-ATP datasets were collected on a Titan Krios G2 electron microscope (FEI), operated at an acceleration voltage of 300 kV and equipped with a Gatan Quantum Image Filter (slit width of 20 eV). The WT-ADP-AlF4− and the D458N/D962N-apo datasets were collected on a Titan Krios G3i electron microscope (FEI), operated at an acceleration voltage of 300 kV and equipped with a Gatan Quantum Image Filter (slit width of 20 eV). The D458N/D962N-AlF4− dataset was collected on a Talos Arctica electron microscope (FEI), operated at an acceleration voltage of 200 kV. Dose-fractionated images (~50 electrons per Å2 applied over 42 or 50 frames) were recorded on a K3 direct electron detector (Gatan) using the super-resolution mode. All datasets were collected using SerialEM (Mastronarde, 2005) with image-beam-shift multiple recording (typically acquiring 9 movies per stage movement with one movie per hole). Coma induced by image-beam shift was corrected by beam tilt compensation in SerialEM. The physical pixel size was 0.911 Å for the Krios G2 datasets, 1.049 Å for the Krios G3i datasets, and 1.115 Å for the Arctica dataset. Target defocus values were typically set from −0.8 to −2.0 μm.
Cryo-EM structural determination
Movies were initially preprocessed using Warp (Tegunov and Cramer, 2019), by motion-correcting and estimating contrast transfer function (CTF) and defocus parameters with 7-by-5 tiling. Micrographs were manually inspected to remove micrographs that were not suitable for image analysis, largely those containing crystalline ice. Particles were automatically picked by Warp and were extracted with a box size of 320 pixels for the WT-apo, WT-BeF3−, WT-AlF4−, WT-AMP-PCP, and D508N-ATP datasets or 300 pixels for the WT-ADP-AlF4−, D458N/D962N-apo, and D458N/D962N-AlF4− datasets. All subsequent image processing was performed in cryoSPARC v2 (Punjani et al., 2017), as described in detail below. Except for the WT-apo, D458N/D962N-apo, and D458N/D962N-AlF4− datasets where the final maps were reconstructed from particles picked in Warp, movies were reprocessed and particles were repicked in cryoSPARC. The particles selected from Warp were used to generate initial maps in cryoSPARC, which were subsequently used as a template for the second round of particle picking and heterogenous refinement in cryoSPARC.
(1). WT ATP13A2 prepared without any nucleotide or phosphate analog (map 1)
605,982 particles were automatically picked in Warp from 2,339 movies. Particles were imported into cryoSPARC for reference-free 2D classification, and classes without clear protein features (mostly empty micelles) were removed. 357,815 particles selected from the 2D classification were subjected to ab initio reconstruction, yielding three initial models (Classes 1 to 3). Clear features of ATP13A2 appeared in one of these classes (Class 1). The particles (357,815 particles) were classified by three rounds of heterogeneous refinement using the ab initio reconstructions. The final set of 164,606 particles was used for 3D reconstruction by non-uniform (NU) refinement and local and global CTF refinement to yield a map at 3.7-Å resolution.
(2). WT ATP13A2 prepared with AMP-PCP (map 2)
The initial set of 670,039 particles automatically picked in Warp from 2,374 movies was used for 2D classification. The 230,844 particles selected from the 2D classification (excluding mainly empty micelles) were subjected to ab initio reconstruction, yielding three initial models (Classes 1 to 3). Clear features of ATP13A2 appeared in one of these classes (Class 2). The particles (230,844 particles) selected from the 2D classification were classified by a round of heterogeneous refinement using the ab initio reconstructions. The resulting particles (146,852 particles) were used for 3D reconstruction by NU refinement, yielding a map at 3.7-Å resolution for the E2-Pi conformation. Raw movies (2,374 movies) were then imported into cryoSPARC for patch-based motion correction (2x pixel-binned after motion correction) and CTF estimation. 2,346 micrographs were selected, and a total of 1,001,323 particles were picked with lowpass filtered templates generated from the 3.7-Å-resolution 3D reconstruction of the E2-Pi state. Particles were extracted with a box size of 320 pixels, Fourier-cropped to 160 pixels, and subjected to a round of 2D classification. Selected particles from 2D classification (297,969 particles) were subjected to a round of heterogeneous refinement with the three initial models generated from the Warp particles. 173,886 particles were classified into Class 2 showing ATP13A2 features, and this final set of particles was subjected to NU refinement and local and global CTF refinements to yield a map at 3.2-Å resolution.
(3). WT ATP13A2 prepared with BeF3− (maps 3 and 4
A summary of the single particle-analysis is outlined in Fig. S3A. The analysis was processed similarly to WT ATP13A2 prepared with AMP-PCP. The initial set of 1,038,661 particles automatically picked in Warp from 3,431 movies was used for 2D classification. A subset (195,032 particles) of the 675,085 particles selected from the 2D classification was subjected to ab initio reconstruction, yielding three initial models (Classes 1 to 3). Clear features of ATP13A2 appeared in two structurally distinct classes (Classes 2 and 3). The selected particles (675,085 particles) were classified by a round of heterogeneous refinement using the ab initio reconstructions, where Class 1 was duplicated to aid the removal of poor-quality particles (i.e., Classes 1a, 1b, 2, and 3). Particles in Class 2 (307,364 particles) were subjected to an additional round of heterogeneous refinement. The resulting particles (281,232 particles) were used for 3D reconstruction by NU refinement and local CTF refinement, yielding a map at 3.1-Å resolution for the E2P-like conformation. Particles in Class 3 (224,085 particles) were separately subjected to an additional round of heterogeneous refinement. The resulting particles (205,001 particles) were used for 3D reconstruction by NU refinement, yielding a map at 3.7-Å resolution for the E2-Pi conformation. Raw movies (3,431 movies) were then imported into cryoSPARC for patch-based motion correction and CTF estimation. 3,395 micrographs were selected, and a total of 1,437,395 particles were picked with 2D templates generated from the 3.1-Å-resolution 3D reconstruction of the E2P-like state. Particles were extracted with a box size of 320 pixels, Fourier-cropped to 160 pixels, and subjected to2D classification. Selected particles from 2D classification (873,004 particles) were then subjected to heterogeneous refinement with the three initial models generated from the Warp particles.
Among 427,905 particles that were classified into Class 2 (E2P-like structure), a subset (200,000 particles) was subjected to a round of ab initio reconstruction to yield three maps. Two additional rounds of heterogeneous refinement (starting from all 427,905 particles) were performed to remove poor-quality particles. The final set of 386,118 particles was subjected to NU refinement and local and global CTF refinements to yield a map at 2.8-Å resolution (map 3).
311,737 particles were classified into Class 3 (E2-Pi structure) and subjected to the essentially same 3D classification procedure as described for map 3. This generated a set of 266,167 particles, which was further narrowed to 253,411 particles using class probability filtering. This final set of 253,411 particles was subjected to NU refinement and local and global CTF refinements to yield a map at 3.1-Å resolution (map 4).
(4). WT ATP13A2 prepared with AlF4− (map 5)
The analysis was processed similarly to WT ATP13A2 prepared with AMP-PCP. The initial set of 1,152,292 particles automatically picked in Warp from 3,519 movies was used for 2D classification, and 695,816 particles selected from the 2D classification were subjected to ab initio reconstruction, yielding three initial models (Classes 1 to 3). Clear features of ATP13A2 appeared in two of these classes (Classes 2 and 3), which were structurally similar. The particles (695,816 particles) were classified by two rounds of heterogeneous refinement using the ab initio reconstructions. The resulting particles (512,952 particles) were used for 3D reconstruction by NU refinement, yielding a map at 3.0-Å resolution. Raw movies (3,519 movies) were then imported into cryoSPARC for patch-based motion correction and CTF estimation. 3,420 movies were selected, and a total of 1,716,479 particles were picked with templates generated from the 3.1-Å-resolution 3D reconstruction. Particles were extracted with a box size of 320 pixels, Fourier-cropped to 160 pixels, and subjected to 2D classification. Selected particles from 2D classification (946,867 particles) were then subjected to heterogeneous refinement with the initial models generated from the Warp particles. 679,631 particles were classified into a class showing ATP13A2 features, and another round of ab initio reconstruction, heterogeneous refinement, and class probability filtering was used to further remove poor-quality particles. The final set of 596,369 particles was subjected to NU refinement and local and global CTF refinements to yield a map at 2.8-Å resolution.
(5). WT ATP13A2 prepared with ADP (maps 6 and 7)
A summary of the single particle-analysis is outlined in Fig. S3B. The analysis was processed similarly to WT ATP13A2 prepared with AMP-PCP. The initial set of 1,159,048 particles automatically picked in Warp from 3,361 movies was used for 2D classification. A subset (257,440 particles) of the 746,647 particles selected from the 2D classification were subjected to ab initio reconstruction, yielding three initial models. Clear features of ATP13A2 appeared in two of these classes (Classes 1 and 2), which were structurally distinct. These particles (746,647 particles) were classified by a round of heterogeneous refinement using the ab initio reconstructions. Particles in Class 1 (490,426 particles) were used for 3D reconstruction by NU refinement, yielding a map at 2.5-Å resolution for the E2-Pi-like conformation. Particles in Class 2 (150,444 particles) were separately used for 3D reconstruction by NU refinement, yielding a map at 3.0-Å resolution for the E1P-ADP-like conformation. Raw movies (3,361 movies) were then imported into cryoSPARC for patch-based motion correction and CTF estimation. 3,347 micrographs were selected, and a total of 1,311,023 particles were picked with templates generated from the 2.5-Å-resolution 3D reconstruction of the E2-Pi-like state. Particles were extracted with a box size of 300 pixels, Fourier-cropped to 150 pixels, and subjected to 2D classification. Selected particles from 2D classification (892,802 particles) were then subjected to heterogeneous refinement with the four initial models [Class 1 (E2-Pi-like), Class 2 (E1P-ADP-like), Class 3a (poor-quality class), Class 3b (poor-quality class)] generated from the Warp particles.
570,133 particles that were classified into Class 1 were subjected to another round of heterogeneous refinement to remove junk particles. The final set of 462,490 particles was subjected to NU refinement and local and global CTF refinements to yield a map at 2.5-Å resolution (map 6). 184,257 particles that were classified into Class 2 were subjected to another round of ab initio reconstruction and heterogeneous refinement to remove junk particles. The final set of 163,433 particles was subjected to NU refinement and local and global CTF refinements to yield a map at 2.9-Å resolution (map 7).
(6). D508N mutant prepared with ATP (map 8)
The analysis was processed similarly to WT ATP13A2 prepared with AMP-PCP. An initial set of 953,226 particles was automatically picked in Warp from 2,833 movies, and a subset of these particles (399,085 particles) was used for 2D classification 247,353 particles were selected from the 2D classification and subjected to ab initio reconstruction, yielding three initial models (Classes 1 to 3). Clear features of ATP13A2 appeared in one of these classes (Class 1). The particles (247,353 particles) were classified by a round of heterogeneous refinement using the ab initio reconstructions. The resulting particles (139,467 particles) were used for 3D reconstruction by NU refinement, yielding a map at 3.5-Å resolution for the E1-ATP-like conformation. Raw movies (2,833 movies) were then imported into cryoSPARC for patch-based motion correction and CTF estimation. 2,755 micrographs were selected, and a total of 1,431,882 particles were picked with templates generated from the 3.5-Å-resolution 3D reconstruction of the E1-ATP-like state. Particles were extracted with a box size of 320 pixels, Fourier-cropped to 160 pixels, and subjected to two rounds of 2D classification. Selected particles from 2D classification (756,595 particles) were subjected to a round of heterogeneous refinement with the three initial models [Class 1 (E1-ATP-like), Class 2 (poor-quality class), and Class 3 (poor-quality class)] generated from the Warp particles. 459,541 particles were classified into Class 1, and this final set of particles was subjected to NU refinement and local and global CTF refinements to yield a map at 2.8-Å resolution.
(7). D458N/D962N mutant in its apo form (maps 9 and 10)
The initial set of 1,181,866 particles automatically picked in Warp was used for 2D classification. 589,548 particles selected from the 2D classification were subjected to ab initio reconstruction, yielding three initial models (Classes 1 to 3). Clear features of ATP13A2 appeared in one of these classes. The particles (589,548 particles) were classified by two rounds of heterogeneous refinement using the ab initio reconstructions. The resulting particles (375,210 particles) were subjected to a final round of heterogeneous refinement to separate two different conformations of the N domain (Classes 1a and 1b). Particles in Class 1a (186,331 particles) were used for 3D reconstruction by NU refinement and local CTF refinement, yielding a map at 2.9-Å resolution for the first E1-apo conformation (map 9). Particles in Class 1b (188,879 particles) were separately used for three-dimensional (3D) reconstruction by NU refinement and local CTF refinement, yielding a map at 2.9-Å resolution for the second E1-apo conformation (map 10).
(8). D458N/D962N mutant prepared with AlF4− (map 11)
The initial set of 1,406,963 particles automatically picked in Warp from 3,214 movies was used for 2D classification. 746,533 particles selected from the 2D classification were subjected to ab initio reconstruction, yielding four initial models (Classes 1 to 4). Clear features of ATP13A2 appeared in two of these classes (Classes 1 and 2), which were structurally similar. The particles (746,533 particles) were classified by two rounds of heterogeneous refinement and an additional round of ab initio reconstruction. The final set of 377,365 particles (from Class “1+2”) was subjected to NU refinement and local and global CTF refinements to yield a map at 3.2-Å resolution (map 11).
(9). Combined post-hydrolysis E2-Pi structure of WT ATP13A2 (map 12)
To improve the quality of the post-hydrolysis E2-Pi state map, particles used to generate three maps (164,606 particles from map 1, 173,886 particles from map 2, and 253,411 particles from map 4) were combined (a total of 591,903 particles) and subjected to NU refinement to yield a map at 3.0-Å resolution.
Atomic Model Building
The initial atomic model was built de novo into the sharpened map of WT E2·BeF3 structure using Coot (Emsley et al., 2010). Models for the other structures were built after rigid-body fitting of individual domains into the corresponding maps using the model of the E2·BeF3 structure and rounds of local refinement in Coot. The following sequences were not modeled because they were poorly resolved in the density maps: N to 33, 93 to 99, 111 to 156 (a part of the NTD), 410 to 415 (connection between the A-domain and TM3), 582 to 586 (a loop in the N-domain), 595 to 613 (a part of the N-domain), and 1169 to 1175 (C-terminus). Residue side chains with poor density were truncated at the β-carbon. Model refinement was performed using Phenix (phenix.real_space_refine) (Afonine et al., 2018) with secondary structure restraints and with the refinement resolution limit set to the overall resolution of the map. Cryo-EM maps that were sharpened by the indicated sharpening factors (Table 1) were used for refinement. For E1-apo, E1P, and E2-Pi structures, the N domain was treated as a rigid body by applying a strong reference model restraint generated from the N-domain of the refined E2·BeF3− structure, because cryo-EM density features were relatively poor in this region. Structural validation was performed using MolProbity (Chen et al., 2010) in the Phenix package. Protein electrostatics were calculated using the Adaptive Poisson-Boltzmann Solver (Baker et al., 2001; Dolinsky et al., 2004) with default parameters built in PyMOL (with monovalent ion concentrations of 0.15 M each). Substrate-transport pathways were detected by Caver 3.0 (Chovancova et al., 2012). UCSF Chimera (Pettersen et al., 2004), Chimera X (Goddard et al., 2018), and PyMOL (Schrödinger) were used to prepare structural figures in the paper.
Coarse-grained simulations of ATP13A2 in a lysosomal membrane
Coarse-grained molecular dynamics simulations of ATP13A2 were performed in a model lysosomal membrane using the Martini 2.2 force field (de Jong et al., 2013). Coarse-grained Martini structures of ATP13A2 E2P and E2-Pi were prepared using the software martinize.py (de Jong et al., 2013). The protein internal structure was constrained with an elastic network with a bond force constant of 200 kJ/mol/nm2, and lower and upper bond cut-offs of 0.5 nm and 0.9 nm, respectively.
The ATP13A2 model (E2P or E2-Pi) was positioned in an asymmetric lipid bilayer with one of two lipid compositions, u (unphosphorylated) and p (phosphorylated), using the program insane.py (Wassenaar et al., 2015). The bilayer compositions mimic a lysosomal membrane with different cytosolic (upper) and lumenal (lower) leaflets as described for the plasma membrane (Lorent et al., 2020; van Meer et al., 2008). The u composition consists of 15.2% cholesterol, 19.8% phosphatidylcholine (13.2% PIPC and 6.6% POPC), 32.0% phosphatidylethanolamine (DOPE), 23.0% phosphatidic acid (DOPA), 3.0% phosphatidylserine (PAPS), and 7.0% phosphatidylinositol (PAPI) in the cytosolic leaflet, and 38.8% cholesterol, 30.8% PIPC, 15.4% POPC, and 15.0% sphingomyelin (DPSM) in the lumenal leaflet. DOPA was used as substituent for the abundant lysosomal lipid bis(monoacylglyceryl)phosphate, having the same charge and identical acyl chain length and saturation. The p composition is the same as the u composition except that half of the PAPI lipids (i.e., 3.5%) were replaced by PI(3,5)P2. We defined parameters for the non-standard lipid POPI(3,5)P2 following the procedure described previously (Lopez et al., 2013). The system was solvated with water beads using insane.py.
In the four simulation systems E2P/u, E2P/p, E2-Pi/u, and E2-Pi/p, we combined the indicated protein conformations and lipid bilayer compositions. All systems had initial box edge lengths of 25 nm in x/y-direction and 16 nm in z-direction. Gromacs 2020/3 (Abraham et al., 2015) was used to preprocess and simulate the four systems. 10% of the water beads were replaced by “anti-freeze” (WF) particles. The systems were neutralized with sodium ion beads. The energy was minimized by steepest descent up to convergence (< 1000.0 kJ/mol/nm between adjacent steps). We maintained a temperature of 300 K with the v-rescale thermostat (Bussi et al., 2007). We equilibrated the systems first in the NVT ensemble for 100 ns, then in the NPT ensemble for 100 ns using the Berendsen barostat (Berendsen et al., 1984) at 1 bar with semiisotropic pressure coupling and τc=5 ps. Production runs were carried out in the NPT ensemble for 10 μ s using the Parrinello-Rahman barostat (Parrinello and Rahman, 1981) at 1 bar with τc of 12 ps. Simulation structures were recorded every 1.5 ns.
The lipid structure around the proteins was analyzed with custom python scripts using the packages numpy (www.numpy.org) and MDAnalysis (Gowers et al., 2016). A minimum bead distance of <0.6 nm was used to define lipid-protein contacts in the upper membrane leaflet. The relative number of contacts (Crel) of a lipid species was then determined as the ratio of protein-lipid contacts of the species over the total number of lipids of that species in the leaflet. We also determined a distance-dependent enrichment (p) of each lipid species around the protein as the ratio of their respective protein-lipid distance histogram values to the sum of protein-lipid distance histograms of all species. Values of p were calculated for the entire protein as well as for the putative lipid exit site. In the latter case, we considered only lipids closer to the exit site than to any other part of the protein (with a tolerance of 0.3 nm). Plots were created with the python package matplotlib (www.matplotlib.org).
QUANTIFICATION AND STATISTICAL ANALYSIS
Refer to Method Details, Figure S3, and Figure S4 for cryo-EM processing, and Table 1 and Table S1 for model building and refinement.
Supplementary Material
Movie S1. 10-μs coarse-grain molecular dynamics simulation of ATP13A2 in the E2P state and a membrane without PI(3,5)P2. Related to Figures 1 and 5. See Fig. S10 for legend.
Movie S2. 10-μs coarse-grain molecular dynamics simulation of ATP13A2 in the E2P state and a membrane with PI(3,5)P2. Related to Figures 1 and 5. See Fig. S10 for legend.
Movie S3. 10-μs coarse-grain molecular dynamics simulation of ATP13A2 in the E2-Pi state and a membrane without PI(3,5)P2. Related to Figures 1 and 5. See Fig. S10 for legend.
Movie S4. 10-μs coarse-grain molecular dynamics simulation of ATP13A2 in the E2-Pi state and a membrane with PI(3,5)P2. Related to Figures 1 and 5. See Fig. S10 for legend.
Highlights.
High-resolution Cryo-EM structures of human ATP13A2 in five distinct conformations.
Unique features of ATP13A2 in comparison to other P-type ATPases.
Structural basis of polyamine substrate binding to the membrane domain of ATP13A2.
Conformational changes along the transport cycle and a model for polyamine transport.
Acknowledgments
We thank D. Toso and J. Remis for support for electron microscope operation, R. Zoncu and R. Shin for discussion, and J. Hurley and R. MacKinnon for critical reading of manuscript. This work was supported by the Vallee Scholars Program (E.P.) and Pew Biomedical Scholars Program (E.P.). S.I.S. was supported by an NIH training grant (5T32GM008295) and an NSF Graduate Research Fellowship (DGE 1752814). S.v.B and G.H. thank the Max Planck Society and the Michael J. Fox Foundation for Parkinson’s Research and Aligning Science Across Parkinson’s (ASAP) initiative (ASAP-000350) for support.
Footnotes
Declaration of Interests
E.P. is a consultant to EndLyz Therapeutics. The remaining authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abbas MM, Govindappa ST, Sheerin UM, Bhatia KP, and Muthane UB (2017). Exome Sequencing Identifies a Novel Homozygous Missense ATP13A2 Mutation. Mov Disord Clin Pract 4, 132–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, and Lindahl E (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. [Google Scholar]
- Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, and Adams PD (2018). Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol 74, 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouida M, Leduc A, Poulin R, and Ramotar D (2005). AGP2 encodes the major permease for high affinity polyamine import in Saccharomyces cerevisiae. The Journal of biological chemistry 280, 24267–24276. [DOI] [PubMed] [Google Scholar]
- Bai L, You Q, Jain BK, Duan HD, Kovach A, Graham TR, and Li H (2020). Transport mechanism of P4 ATPase phosphatidylcholine flippases. eLife 9, e62163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, and McCammon JA (2001). Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, and Haak JR (1984). Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics 81, 3684–3690. [Google Scholar]
- Bras J, Verloes A, Schneider SA, Mole SE, and Guerreiro RJ (2012). Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum Mol Genet 21, 2646–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bublitz M, Poulsen H, Morth JP, and Nissen P (2010). In and out of the cation pumps: P-Type ATPase structure revisited. Current Opinion in Structural Biology 20, 431–439. [DOI] [PubMed] [Google Scholar]
- Bussi G, Donadio D, and Parrinello M (2007). Canonical sampling through velocity rescaling. J Chem Phys 126, 014101. [DOI] [PubMed] [Google Scholar]
- Chen CM, Lin CH, Juan HF, Hu FJ, Hsiao YC, Chang HY, Chao CY, Chen IC, Lee LC, Wang TW, et al. (2011). ATP13A2 variability in Taiwanese Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet 156B, 720–729. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chovancova E, Pavelka A, Benes P, Strnad O, Brezovsky J, Kozlikova B, Gora A, Sustr V, Klvana M, Medek P, et al. (2012). CAVER 3.0: a tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput Biol 8, e1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen MV, Hilbers F, and Poulsen H (2017). The Structure and Function of the Na,K-ATPase Isoforms in Health and Disease. Front Physiol 8, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong DH, Singh G, Bennett WF, Arnarez C, Wassenaar TA, Schafer LV, Periole X, Tieleman DP, and Marrink SJ (2013). Improved Parameters for the Martini Coarse-Grained Protein Force Field. J Chem Theory Comput 9, 687–697. [DOI] [PubMed] [Google Scholar]
- De La Hera DP, Corradi GR, Adamo HP, and De Tezanos Pinto F (2013). Parkinson’s disease-associated human P5B-ATPase ATP13A2 increases spermidine uptake. Biochem J 450, 47–53. [DOI] [PubMed] [Google Scholar]
- Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron M-H, Doudnikoff E, Vital A, Vila M, Klein C, and Bezard E (2012). Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proceedings of the National Academy of Sciences 109, 9611–9616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G, Fabbrini G, Marconi R, Fincati E, Abbruzzese G, et al. (2007). ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 68, 1557–1562. [DOI] [PubMed] [Google Scholar]
- Djarmati A, Hagenah J, Reetz K, Winkler S, Behrens MI, Pawlack H, Lohmann K, Ramirez A, Tadic V, Bruggemann N, et al. (2009). ATP13A2 variants in early-onset Parkinson’s disease patients and controls. Mov Disord 24, 2104–2111. [DOI] [PubMed] [Google Scholar]
- Dolinsky TJ, Nielsen JE, McCammon JA, and Baker NA (2004). PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res 32, W665–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyla M, Basse Hansen S, Nissen P, and Kjaergaard M (2019). Structural dynamics of P-type ATPase ion pumps. Biochemical Society Transactions 47, 1247–1257. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estiar MA, Leveille E, Spiegelman D, Dupre N, Trempe JF, Rouleau GA, and Gan-Or Z (2020). Clinical and genetic analysis of ATP13A2 in hereditary spastic paraplegia expands the phenotype. Mol Genet Genomic Med 8, e1052. doi: 10.1002/mgg3.1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada-Cuzcano A, Martin S, Chamova T, Synofzik M, Timmann D, Holemans T, Andreeva A, Reichbauer J, De Rycke R, Chang D-I, et al. (2017). Loss-of-function mutations in the ATP13A2/ PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain 140, 287–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exner N, Lutz AK, Haass C, and Winklhofer KF (2012). Mitochondrial dysfunction in Parkinson’s disease: molecular mechanisms and pathophysiological consequences. EMBO J 31, 3038–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Zhao Y, Li T, Nie W, Yang X, Wang X, Wu J, Liao J, and Zou Y (2020). CATP-8/P5A ATPase Regulates ER Processing of the DMA-1 Receptor for Dendritic Branching. Cell Reports 32, 108101. [DOI] [PubMed] [Google Scholar]
- Fujita M, Fujita Y, Iuchi S, Yamada K, Kobayashi Y, Urano K, Kobayashi M, Yamaguchi-Shinozaki K, and Shinozaki K (2012). Natural variation in a polyamine transporter determines paraquat tolerance in Arabidopsis. Proceedings of the National Academy of Sciences of the United States of America 109, 6343–6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, Caldwell KA, Caldwell GA, Cooper AA, Rochet J-C, et al. (2009). α-Synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nature Genetics 41, 308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowers RJ, Linke J, Barnoud J, Reddy TJE, Melo MN, Seyler SL, Dotson DL, Domanski J, Buchoux S, Kenney IM, et al. (2016). MDAnalysis: A Python package for the rapid analysis of molecular dynamics simulations. Proceedings of the 15th Python in Science Conference, SciPy, 98–105. doi: 10.25080/majora-629e541a-00e. [DOI] [Google Scholar]
- Grunewald A, Arns B, Seibler P, Rakovic A, Munchau A, Ramirez A, Sue CM, and Klein C (2012). ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol Aging 33, 1843 e1841–1847. [DOI] [PubMed] [Google Scholar]
- Hamouda NN, Van den Haute C, Vanhoutte R, Sannerud R, Azfar M, Mayer R, Cortes Calabuig A, Swinnen JV, Agostinis P, Baekelandt V, et al. (2020). ATP13A3 is a major component of the enigmatic mammalian polyamine transport system. J Biol Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa AK, Fatima T, and Mattoo AK (2018). Polyamines: Bio-Molecules with Diverse Functions in Plant and Human Health and Disease. Front Chem 6, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinick A, Urban K, Roth S, Spies D, Nunes F, Phanstiel O.t., Liebau E, and Luersen K (2010). Caenorhabditis elegans P5B-type ATPase CATP-5 operates in polyamine transport and is crucial for norspermidine-mediated suppression of RNA interference. FASEB J 24, 206–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiasa M, Miyaji T, Haruna Y, Takeuchi T, Harada Y, Moriyama S, Yamamoto A, Omote H, and Moriyama Y (2014). Identification of a mammalian vesicular polyamine transporter. In Sci Rep, pp. 6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraizumi M, Yamashita K, Nishizawa T, and Nureki O (2019). Cryo-EM structures capture the transport cycle of the P4-ATPase flippase. Science 365, 1149–1155. [DOI] [PubMed] [Google Scholar]
- Holemans T, Sørensen DM, van Veen S, Martin S, Hermans D, Kemmer GC, Van den Haute C, Baekelandt V, Günther Pomorski T, Agostinis P, et al. (2015). A lipid switch unlocks Parkinson’s disease-associated ATP13A2. Proceedings of the National Academy of Sciences 112, 9040–9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi K, and Kashiwagi K (1999). Polyamine transport in bacteria and yeast. The Biochemical journal 344 Pt 3, 633–642. [PMC free article] [PubMed] [Google Scholar]
- Igarashi K, and Kashiwagi K (2010). Modulation of cellular function by polyamines. The International Journal of Biochemistry & Cell Biology 42, 39–51. [DOI] [PubMed] [Google Scholar]
- Juraschek R, Dulcks T, and Karas M (1999). Nanoelectrospray--more than just a minimized-flow electrospray ionization source. J Am Soc Mass Spectrom 10, 300–308. [DOI] [PubMed] [Google Scholar]
- Kashiwagi K, Pistocchi R, Shibuya S, Sugiyama S, Morikawa K, and Igarashi K (1996). Spermidine-preferential uptake system in Escherichia coli. Identification of amino acids involved in polyamine binding in PotD protein. J Biol Chem 271, 12205–12208. [DOI] [PubMed] [Google Scholar]
- Kuhlbrandt W (2004). Biology, structure and mechanism of P-type ATPases. Nat Rev Mol Cell Biol 5, 282–295. [DOI] [PubMed] [Google Scholar]
- López-Marqués RL, Gourdon P, Günther Pomorski T, and Palmgren M (2020). The transport mechanism of P4 ATPase lipid flippases. Biochemical Journal 477, 3769–3790. [DOI] [PubMed] [Google Scholar]
- Lopez CA, Sovova Z, van Eerden FJ, de Vries AH, and Marrink SJ (2013). Martini Force Field Parameters for Glycolipids. J Chem Theory Comput 9, 1694–1708. [DOI] [PubMed] [Google Scholar]
- Lorent JH, Levental KR, Ganesan L, Rivera-Longsworth G, Sezgin E, Doktorova M, Lyman E, and Levental I (2020). Plasma membranes are asymmetric in lipid unsaturation, packing and protein shape. Nat Chem Biol 16, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- McKenna MJ, Sim SI, Ordureau A, Wei L, Harper JW, Shao S, and Park E (2020). The endoplasmic reticulum P5A-ATPase is a transmembrane helix dislocase. Science 369, eabc5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil-Gauthier AL, Brais B, Rouleau G, Anoja N, and Ducharme S (2019). Successful treatment of psychosis in a patient with Kufor-Rakeb syndrome with low dose aripiprazole: a case report. Neurocase 25, 133–137. [DOI] [PubMed] [Google Scholar]
- Møller AB, Asp T, Holm PB, and Palmgren MG (2008). Phylogenetic analysis of P5 P-type ATPases, a eukaryotic lineage of secretory pathway pumps. Molecular Phylogenetics and Evolution 46, 619–634. [DOI] [PubMed] [Google Scholar]
- Morth JP, Pedersen BP, Toustrup-Jensen MS, Sørensen TL-M, Petersen J, Andersen JP, Vilsen B, and Nissen P (2007). Crystal structure of the sodium–potassium pump. Nature 450, 1043–1049. [DOI] [PubMed] [Google Scholar]
- Nakanishi H, Nishizawa T, Segawa K, Nureki O, Fujiyoshi Y, Nagata S, and Abe K (2020). Transport Cycle of Plasma Membrane Flippase ATP11C by Cryo-EM. Cell Reports 32, 108208. [DOI] [PubMed] [Google Scholar]
- Ning YP, Kanai K, Tomiyama H, Li Y, Funayama M, Yoshino H, Sato S, Asahina M, Kuwabara S, Takeda A, et al. (2008). PARK9-linked parkinsonism in eastern Asia: mutation detection in ATP13A2 and clinical phenotype. Neurology 70, 1491–1493. [DOI] [PubMed] [Google Scholar]
- Olesen C, Picard M, Winther AM, Gyrup C, Morth JP, Oxvig C, Moller JV, and Nissen P (2007). The structural basis of calcium transport by the calcium pump. Nature 450, 1036–1042. [DOI] [PubMed] [Google Scholar]
- Palmgren MG, and Nissen P (2011). P-Type ATPases. Annual Review of Biophysics 40, 243–266. [DOI] [PubMed] [Google Scholar]
- Park JS, Mehta P, Cooper AA, Veivers D, Heimbach A, Stiller B, Kubisch C, Fung VS, Krainc D, Mackay-Sim A, et al. (2011). Pathogenic effects of novel mutations in the P-type ATPase ATP13A2 (PARK9) causing Kufor-Rakeb syndrome, a form of early-onset parkinsonism. Hum Mutat 32, 956–964. [DOI] [PubMed] [Google Scholar]
- Parrinello M, and Rahman A (1981). Polymorphic transitions in single crystals: A new molecular dynamics method. Journal of Applied Physics 52, 7182–7190. [Google Scholar]
- Pegg AE (2009). Mammalian polyamine metabolism and function. IUBMB Life 61, 880–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Podhajska A, Musso A, Trancikova A, Stafa K, Moser R, Sonnay S, Glauser L, and Moore DJ (2012). Common pathogenic effects of missense mutations in the P-type ATPase ATP13A2 (PARK9) associated with early-onset parkinsonism. PLoS One 7, e39942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Qin Q, Zhao T, Zou W, Shen K, and Wang X (2020). An Endoplasmic Reticulum ATPase Safeguards Endoplasmic Reticulum Identity by Removing Ectopically Localized Mitochondrial Proteins. Cell Reports 33, 108363. [DOI] [PubMed] [Google Scholar]
- Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat A-L, Roeper J, et al. (2006). Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nature Genetics 38, 1184–1191. [DOI] [PubMed] [Google Scholar]
- Ritz C, Baty F, Streibig JC, and Gerhard D (2015). Dose-Response Analysis Using R. PLoS One 10, e0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro L, Breedveld GJ, Manganelli F, Iodice R, Pisciotta C, Nolano M, Punzo F, Quarantelli M, Pappata S, Di Fonzo A, et al. (2011). Novel ATP13A2 (PARK9) homozygous mutation in a family with marked phenotype variability. Neurogenetics 12, 33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz J, and Suppmann S (2017). A new single-step protocol for rapid baculovirus-driven protein production in insect cells. BMC Biotechnol 17, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda T, Ogawa H, Cornelius F, and Toyoshima C (2009). Crystal structure of the sodium-potassium pump at 2.4 A resolution. Nature 459, 446–450. [DOI] [PubMed] [Google Scholar]
- Sørensen DM, Buch-Pedersen MJ, and Palmgren MG (2010). Structural divergence between the two subgroups of P5 ATPases. Biochimica et Biophysica Acta (BBA) - Bioenergetics 1797, 846–855. [DOI] [PubMed] [Google Scholar]
- Sorensen DM, Holemans T, van Veen S, Martin S, Arslan T, Haagendahl IW, Holen HW, Hamouda NN, Eggermont J, Palmgren M, et al. (2018). Parkinson disease related ATP13A2 evolved early in animal evolution. PLoS One 13, e0193228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen TL, Moller JV, and Nissen P (2004). Phosphoryl transfer and calcium ion occlusion in the calcium pump. Science 304, 1672–1675. [DOI] [PubMed] [Google Scholar]
- Spataro R, Kousi M, Farhan SMK, Willer JR, Ross JP, Dion PA, Rouleau GA, Daly MJ, Neale BM, La Bella V, et al. (2019). Mutations in ATP13A2 (PARK9) are associated with an amyotrophic lateral sclerosis-like phenotype, implicating this locus in further phenotypic expansion. Hum Genomics 13, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama S, Matsuo Y, Vassylyev DG, Matsushima M, Morikawa K, Maenaka K, Kashiwagi K, and Igarashi K (1996). The 1.8-Å X-ray structure of the Escherichia coli PotD protein complexed with spermidine and the mechanism of polyamine binding. Protein Science 5, 1984–1990. doi: 10.1002/pro.5560051004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suleiman J, Hamwi N, and El-Hattab AW (2018). ATP13A2 novel mutations causing a rare form of juvenile-onset Parkinson disease. Brain Dev 40, 824–826. [DOI] [PubMed] [Google Scholar]
- Tegunov D, and Cramer P (2019). Real-time cryo-electron microscopy data preprocessing with Warp. Nat Methods 16, 1146–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timcenko M, Lyons JA, Januliene D, Ulstrup JJ, Dieudonné T, Montigny C, Ash M-R, Karlsen JL, Boesen T, Kühlbrandt W, et al. (2019). Structure and autoregulation of a P4-ATPase lipid flippase. Nature 571, 366–370. [DOI] [PubMed] [Google Scholar]
- Toyoshima C, Nakasako M, Nomura H, and Ogawa H (2000). Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature 405, 647–655. [DOI] [PubMed] [Google Scholar]
- Toyoshima C, Nomura H, and Tsuda T (2004). Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature 432, 361–368. [DOI] [PubMed] [Google Scholar]
- Twomey EC, Yelshanskaya MV, Vassilevski AA, and Sobolevsky AI (2018). Mechanisms of Channel Block in Calcium-Permeable AMPA Receptors. Neuron 99, 956–968.e954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usenovic M, Tresse E, Mazzulli JR, Taylor JP, and Krainc D (2012). Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J Neurosci 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Warrenburg BP, Schouten MI, de Bot ST, Vermeer S, Meijer R, Pennings M, Gilissen C, Willemsen MA, Scheffer H, and Kamsteeg EJ (2016). Clinical exome sequencing for cerebellar ataxia and spastic paraplegia uncovers novel gene-disease associations and unanticipated rare disorders. Eur J Hum Genet 24, 1460–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, and Feigenson GW (2008). Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Veen S, Martin S, Van den Haute C, Benoy V, Lyons J, Vanhoutte R, Kahler JP, Decuypere J-P, Gelders G, Lambie E, et al. (2020). ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 578, 419–424. [DOI] [PubMed] [Google Scholar]
- Vrijsen S, Besora-Casals L, van Veen S, Zielich J, Van den Haute C, Hamouda NN, Fischer C, Ghesquière B, Tournev I, Agostinis P, et al. (2020). ATP13A2-mediated endo-lysosomal polyamine export counters mitochondrial oxidative stress. Proceedings of the National Academy of Sciences, 201922342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallings RL, Humble SW, Ward ME, and Wade-Martins R (2019). Lysosomal Dysfunction at the Centre of Parkinson’s Disease and Frontotemporal Dementia/Amyotrophic Lateral Sclerosis. Trends Neurosci 42, 899–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Tan J, Chen T, Han H, Tian R, Tan Y, Wu Y, Cui J, Chen F, Li J, et al. (2019). ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome-lysosome fusion. J Cell Biol 218, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassenaar TA, Ingolfsson HI, Bockmann RA, Tieleman DP, and Marrink SJ (2015). Computational Lipidomics with insane: A Versatile Tool for Generating Custom Membranes for Molecular Simulations. J Chem Theory Comput 11, 2144–2155. [DOI] [PubMed] [Google Scholar]
- Williams K (1997). Interactions of polyamines with ion channels. Biochemical Journal 325, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm M, and Mann M (1996). Analytical properties of the nanoelectrospray ion source. Anal Chem 68, 1–8. [DOI] [PubMed] [Google Scholar]
- Winther AM, Bublitz M, Karlsen JL, Moller JV, Hansen JB, Nissen P, and Buch-Pedersen MJ (2013). The sarcolipin-bound calcium pump stabilizes calcium sites exposed to the cytoplasm. Nature 495, 265–269. [DOI] [PubMed] [Google Scholar]
- Yang X, and Xu Y (2014). Mutations in the ATP13A2 gene and Parkinsonism: a preliminary review. Biomed Res Int 2014, 371256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung MW, and Green C (1986). The binding of polyamines to phospholipid bilayers. Biochem Pharmacol 35, 4037–4041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. 10-μs coarse-grain molecular dynamics simulation of ATP13A2 in the E2P state and a membrane without PI(3,5)P2. Related to Figures 1 and 5. See Fig. S10 for legend.
Movie S2. 10-μs coarse-grain molecular dynamics simulation of ATP13A2 in the E2P state and a membrane with PI(3,5)P2. Related to Figures 1 and 5. See Fig. S10 for legend.
Movie S3. 10-μs coarse-grain molecular dynamics simulation of ATP13A2 in the E2-Pi state and a membrane without PI(3,5)P2. Related to Figures 1 and 5. See Fig. S10 for legend.
Movie S4. 10-μs coarse-grain molecular dynamics simulation of ATP13A2 in the E2-Pi state and a membrane with PI(3,5)P2. Related to Figures 1 and 5. See Fig. S10 for legend.
Data Availability Statement
Cryo-EM maps for ATP13A2 (maps 1–12) have been deposited in the Electron Microscopy Data Bank (EMDB)and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Coordinates for models of ATP13A2 (map 3 and 6–12) have been deposited in the Protein Data Bank (PDB) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
The paper does not report original code.
Any additional information required to reanalyze the data reported in the paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Escherichia coli DH10Bac competent cells | Thermo | Cat#10361012 |
| Chemicals, peptides, and recombinant proteins | ||
| Cellfectin II | Thermo | Cat#10362100 |
| Polyethylenimidine (PEI MAX) | Polysciences | Cat#24765 |
| ESF 921 medium | Expression Systems | Cat#96–001 |
| n-Dodecyl-β-D-maltopyranoside (DDM) | GoldBio | Cat#DDM |
| Cholesteryl hemisuccinate | Anatrace | Cat#CH210 |
| Critical commercial assays | ||
| ADP-Glo™ Kinase Assay | Promega | Cat#V9101 |
| Deposited data | ||
| Cryo-EM structure of ATP13A2 in the E2-Pi state (map 1) | This paper | EMD-24212 |
| Cryo-EM structure of ATP13A2 in the E2-Pi state (map 2) | This paper | EMD-24213 |
| Cryo-EM structure of ATP13A2 in the E2·BeF3− state (map 3) | This paper | EMD-24214, PDB: 7N70 |
| Cryo-EM structure of ATP13A2 in the E2-Pi state (map 4) | This paper | EMD-24215 |
| Cryo-EM structure of ATP13A2 in the E2-AlF4− state (map 5) | This paper | EMD-24216 |
| Cryo-EM structure of ATP13A2 in the E2-AlF4− state (map 6) | This paper | EMD-24217, PDB: 7N72 |
| Cryo-EM structure of ATP13A2 in the E1P-ADP-like state (map 7) | This paper | EMD-24218, PDB: 7N73 |
| Cryo-EM structure of ATP13A2 D508N mutant in the E1-ATP state (map 8) | This paper | EMD-24219, PDB: 7N74 |
| Cryo-EM structure of ATP13A2 D458N/D962N mutant in the E1-apo state, Conformation 1 (map 9) | This paper | EMD-24220, PDB: 7N75 |
| Cryo-EM structure of ATP13A2 D458N/D962N mutant in the E1-apo state, Conformation 2 (map 10) | This paper | EMD-24221, PDB: 7N76 |
| Cryo-EM structure of ATP13A2 D458N/D962N mutant in the E1·AlF4− state (map 11) | This paper | EMD-24222, PDB: 7N77 |
| Cryo-EM structure of ATP13A2 in the E2-Pi state (map 12) | This paper | EMD-24223, PDB: 7N78 |
| SDS-PAGE gel image of size-exclusion chromatography fractions (Fig. S1A) | This paper | DOI:10.17632/g64mnvkgyk.1 |
| Experimental models: cell lines | ||
| Spodoptera frugiperda (Sf9) | Expression Systems | 94–001F |
| Software and algorithms | ||
| Warp | Tegunov et al., 2019 | http://www.warpem.com/warp/ |
| cryoSPARC v2 | Punjani et al., 2017 | https://cryosparc.com/ |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix | Afonine et al., 2018 | https://www.phenix-online.org/ |
| MolProbity | Chen et al., 2010 | http://molprobity.biochem.duke.edu/ |
| Adaptive Poisson-Boltzmann Solver | Baker et al., 2001; Dolinksy et al., 2004 | https://www.poissonboltzmann.org/ |
| PyMOL | Schrödinger | https://pymol.org/2/ |
| Caver 3.0 | Chovancova et al., 2012 | http://www.caver.cz/ |
| UCSF Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| UCSF ChimeraX | Goddard et al., 2018 | https://www.cgl.ucsf.edu/chimerax/ |
| R software and drc2 package | Ritz et al., 2015 | https://www.r-project.org/ |
| Martini 2.2 | de Jong et al., 2013 | http://cgmartini.nl/ |
| Gromacs 2020/3 | Abraham et al., 2015 | https://www.gromacs.org/ |
| numpy | https://www.numpy.org | |
| MDAnalysis | Gowers et al., 2016 | https://www.mdanalysis.org/ |
| matplotlib | https://www.matplotlib.org | |
| Other | ||
| Quantifoil holey carbon grid (R1.2/1.3, Au, 400 mesh) | Electron Microscopy Sciences | Cat#Q4100AR1.3 |
| Superose 6 Increase, 10/300 GL | GE Life Sciences (Cytiva) | Cat#29–0915–96 |