

Abstract
Proline dehydrogenase (PRODH) is a flavoenzyme that catalyzes the first step of proline catabolism, the oxidation of L-proline to Δ1-pyrroline-5-carboxylate. PRODH has emerged as a cancer therapy target because of its involvement in the metabolic reprogramming of cancer cells. Here we report the discovery of a new class of PRODH inactivator, which covalently and irreversibly modifies the FAD in a light-dependent manner. Two examples, 1,3-dithiolane-2-carboxylate and tetrahydrothiophene-2-carboxylate, have been characterized using X-ray crystallography (1.52 – 1.85 Å resolution), absorbance spectroscopy, and enzyme kinetics. The structures reveal that in the dark, these compounds function as classical reversible, proline analog inhibitors. However, exposure of enzyme-inhibitor co-crystals to bright white light induces decarboxylation of the inhibitor and covalent attachment of the residual S-heterocycle to the FAD N5 atom, locking the cofactor into a reduced, inactive state. Spectroscopic measurements of the inactivation process in solution confirm the requirement for light and show that blue light is preferred. Enzyme activity assays show that the rate of inactivation is enhanced by light and that the inactivation is irreversible. We also demonstrate the photosensitivity of cancer cells to one of these compounds. A possible mechanism is proposed involving photoexcitation of the FAD while the inhibitor is noncovalently bound in the active site, followed by electron transfer, decarboxylation, and radical combination steps. Our results could lead to the development of photopharmacological drugs targeting PRODH.
Graphical Abstract
Proline catabolism involves the 4-electron oxidation of L-proline to L-glutamate (Figure 1A). The pathway consists of two enzymes, which are highly conserved by eukaryotes and bacteria: proline dehydrogenase (PRODH) and L-glutamate-γ-semialdehyde (GSAL) dehydrogenase (GSALDH). PRODH catalyzes the FAD-dependent oxidation of proline to Δ1-pyrroline-5-carboxylate (P5C). The non-enzymatic hydrolysis of P5C produces GSAL, which is then oxidized to glutamate by the NAD+-dependent enzyme GSALDH (a.k.a. ALDH4A1).
Figure 1.
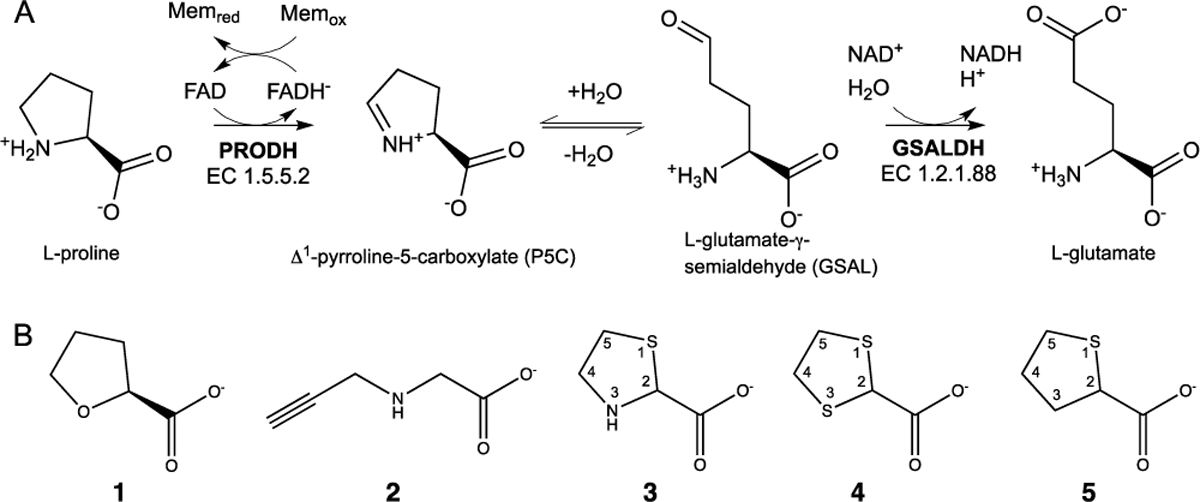
Proline catabolism and PRODH inhibitors. (A) The enzymes and reactions of proline catabolism. (B) Structures of a reversible PRODH inhibitor (1) and four covalent inactivators, which modify the N5 of PRODH (2 - 5).
Proline metabolism is important in many aspects of human health and disease, especially cancer. Alterations in proline metabolism have been observed in many cancers. This reflects the role of proline metabolism in redox stress, energy production and apoptosis.1, 2 Enzymes involved in proline metabolism are differentially expressed in cancer cells compared to normal cells.3, 4 Many studies have shown that PRODH has a special role in cancer cells, particularly in the generation of ATP and redox cofactors to help cancer cells proliferate.5, 6 The proline catabolic pathway has been proposed as a target for cancer therapeutics, with a particular interest of finding inhibitors of PRODH.1, 7 The inhibition of PRODH activity in cancer cells is expected to impair the production of ATP needed for proliferation and to disrupt cellular redox balance, leading to cell death. Indeed, these effects have been observed in breast,4, 8 lung,9 skin10 and prostate10 cancer models. For example, Elia et al showed that inhibition of PRODH with the proline analog S-(–)-tetrahydro-2-furoic acid (1 in Figure 1B) impaired the formation of lung metastases in the orthotopic 4T1 and EMT6.5 mouse models, without harming non-cancerous tissues. Liu et al showed that PRODH functions as an oncogene in non-small cell lung cancer, and treatment with 1 significantly inhibited cell proliferation and the epithelial-to-mesenchymal transition.9 Scott et al showed that the covalent inactivation of PRODH with the mechanism-based inactivator N-propargylglycine (2) induced PRODH degradation and the mitochondrial unfolded protein response, resulting in cell apoptosis in breast tumor (MCF7)-xenografted mice.8 Taken together the literature suggests that PRODH is a promising cancer drug target.11
Only a few inhibitors of PRODH are available, and improvements in potency, duration of inhibition, and selectivity are needed. The best characterized reversible PRODH inhibitor, 1, has a modest inhibition constant in the range of 0.1 – 1.6 mM.12–18 Covalent inactivation of PRODH has also been studied. The best characterized covalent inactivator is 2, a mechanism-based inactivator that locks the enzyme into the reduced state by covalently linking the FAD N5 atom to the ε-nitrogen of an active site lysine residue.19, 20 However, the apparent second-order rate constant for PRODH inactivation of 1 – 10 M−1s−1 is below the range expected for drugs.21, 22 Thus, there is a need for PRODH inhibitors with better potency and new mechanisms of inhibition for use as chemical probes to study the roles of proline metabolism in cancer and potentially as starting points for the design of new therapeutics.
The present study was inspired by our recent discovery that thiazolidine-2-carboxylate (3) is a mechanism-based covalent irreversible inactivator of PRODH.23 A high resolution crystal structure of the inactivated enzyme showed that the FAD is reduced and 3 is covalently bonded to the FAD N5 atom via its C5 atom. Although the rate of enzyme inactivation is very slow (timescale measured in days), and the mechanism of inactivation is uncertain, this discovery nevertheless motivated the investigation of other sulfur-containing proline analogs as possible covalent inhibitors of PRODH. Here we report the discovery of two new covalent inactivators of PRODH, 1,3-dithiolane-2-carboxylate (4) and tetrahydrothiophene-2-carboxylate (5), which irreversibly inactivate the enzyme through a light-mediated mechanism.
RESULTS AND DISCUSSION
Visual Evidence for Photoinduced Reduction of PRODH by S-containing Proline Analogs.
For this study, we used the bifunctional PRODH-GSALDH enzyme proline utilization A from Sinorhizobium meliloti (SmPutA), which we used previously to investigate the covalent inactivation of PRODH by 3 and the reversible inhibition by 1.23, 24 SmPutA is a good model system for inhibitor testing because of its exceptional crystallizability (routinely 1.4 – 1.9 Å resolution), the fact that the PRODH and GSALDH active sites are distinct (separated by 40 Å) and can be interrogated independently, and the high sequence conservation of PRODH active sites across bacteria and eukaryotes.7 We note that human PRODH is an inner mitochondrial membrane protein and is challenging to isolate for structural and kinetic study.
Compounds 4 and 5 were tested as inhibitors of PRODH due to their structural similarity to 3, a previously characterized covalent inactivator of PRODH that forms an irreversible adduct with the N5 atom of the FAD.23 Further, because the new compounds are not pyrrolidines, they cannot be oxidized by PRODH to an imine analogous to P5C (Figure 1A), and based on the proposed mechanism of inactivation of 3, we thought that elimination of the imine pathway would improve the kinetics of inactivation. Surprisingly, we discovered that the new compounds have a completely different mechanism of inactivation than 3.
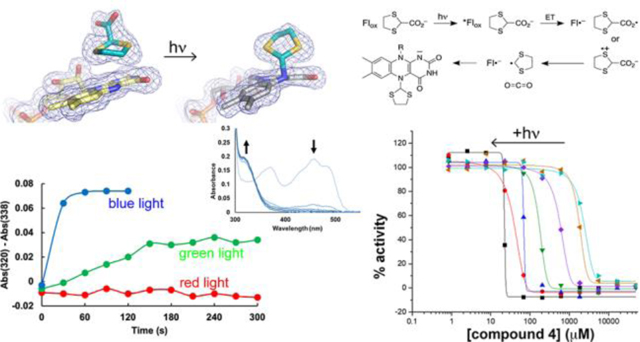
Whereas 3 immediately bleaches the yellow color from the enzyme upon mixing, the enzyme mixed with 4 or 5 remained yellow, and the crystals that grew were also yellow. We then serendipitously discovered that the yellow co-crystals grown with 4 were bleached within 10 minutes during inspection under a light microscope, suggesting that the FAD had been reduced (Supplementary Figure S1). The color change also occurred with the co-crystals of 5, but over a longer time scale of about an hour and required an additional light source (700 lumen LED bulb) to accelerate the color change. We note that bleaching did not occur with ligand-free SmPutA crystals or SmPutA-1 crystals, suggesting that 4 and 5 were playing a direct role in the photoinduced reduction of the FAD in PRODH. These observations motivated an investigation of the role of light in the interaction of 4 and 5 with PRODH.
Crystal Structures of Noncovalently-inhibited and Covalently-inactivated PRODH.
Crystal structures (1.52 – 1.85 Å resolution) were obtained for enzyme complexes with 4 and 5 from crystals harvested under low light conditions (yellow crystals) and under bright light conditions (colorless crystals) (Supplementary Table S1). The crystal structures determined from yellow crystals showed 4 and 5 bound noncovalently in the proline site of PRODH (Figure 2A–2B, left), whereas those determined from colorless crystals revealed a novel covalent modification of the FAD (Figure 2A–2B, right).
Figure 2.
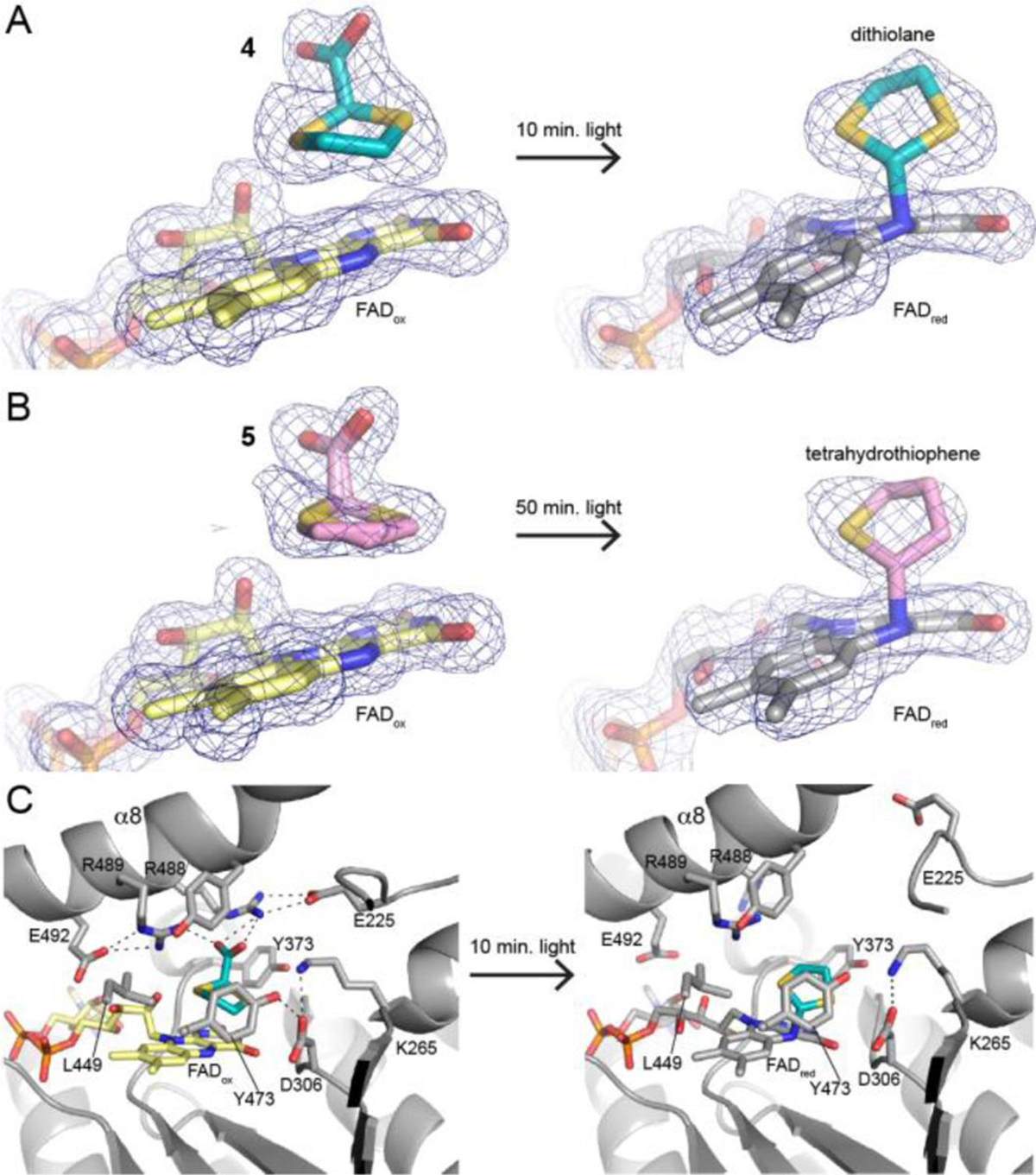
Structural basis of photoinduced covalent inactivation of PRODH. (A) Electron density for the active site inhibited by compound 4 before and after illumination with white light (polder omit, 4σ). (B) Electron density for the active site inhibited by compound 5 before and after illumination with white light (polder omit, 4σ). (C) Interactions with 4 before illumination (left) and the expanded active site after covalent modification.
The binding poses and interactions of 4 and 5 in the noncovalent complexes (dark state) resemble that of the noncovalent inhibitor 1. Both 4 and 5 sit against the middle ring of the si face of the FAD isoalloxazine. We note the density for noncovalent 5 was consistent with dual occupancy of both stereoisomers (Figure 2B, left). The carboxylate group of the noncovalent inhibitor forms ion pairs with conserved Arg488 and Arg489, as shown for 4 in Figure 2c. These ion pairs are also observed in the complex with 1 (PDB ID 5KF6).24 Thus, in the absence of light, 4 and 5 act as classical proline analog inhibitors of PRODH.
Exposure of the crystals to light profoundly affected the active site structure. The electron density maps clearly showed a strong tube of density connecting the FAD N5 to a species in the proline site, suggesting that light induced a covalent modification of the FAD (Figure 2A–2B, right). The electron density further suggested that the species bonded to the FAD is smaller than the parent compound. The density could be satisfactorily modeled as 1,3-dithiolane in the 4-inactivated enzyme and tetrahydrothiophene in the 5-inactivated enzyme. Thus, it appears that the photoinduced covalent modification of the FAD involves decarboxylation of the inhibitor.
The electron density map of the 4-inactivated enzyme was particularly useful for identifying the atom bonded to the FAD N5. The map clearly indicated the locations of the electron-rich S atoms of the bound 1,3-dithiolane, suggesting that the FAD N5 was bonded to the C2 atom of the inhibitor (Supplementary Figure S2). This assignment of the regiochemistry of inactivation was also consistent with the electron density of the 5-inactivated enzyme (Figure 2B, right).
The active site expands upon covalent inactivation by 4 and 5. The active sites of the noncovalently-inhibited enzymes are closed and crowded. The ion pair gate (Arg488-Glu225), which is conserved in PRODHs, is closed and several residues cluster around the proline analog to create a congested environment (Figure 2C, left). For example, Arg488 and Arg489 form ion pairs with the inhibitor carboxylate, while the side chains of Leu449, Lys265 and three tyrosine residues form a tight cage around inhibitor. Presumably, the crowded active site environment constrains the motion of the noncovalently bound inhibitor. Upon photoinduced covalent modification of the FAD, the ion pair gate ruptures, and the basic residues that interacted with the carboxylate of the inhibitor in the noncovalent complexes recoil from the proline site (Figure 2C, right). The electron density for the α8 helix diminishes, suggesting that inactivation increases the conformational mobility of this important active site element. The net effect is that the active site expands upon inactivation. The expansion of the active site is a natural consequence of decarboxylation of the inhibitor, since removal of the negatively-charged carboxylate likely results in electrostatic repulsion of conserved Arg488, Arg489, and Lys265.
The FAD appears to be oxidized in the noncovalent complexes and reduced in the covalently-modified structures, which is consistent with the color changes observed in the crystals. The conformation of the isoalloxazine is a reliable structural proxy for the flavin redox state in PRODH, where the oxidized isoalloxazine is planar and the 2-electron-reduced isoalloxazine has butterfly bending angles of 9–35° (si face convex).19, 20, 23, 25–27 In the two noncovalent complexes, the isoalloxazine is planar, consistent with the oxidized FAD (Figures 2A–2B, left; Supplementary Figure S3, left). In contrast, the covalently-modified FADs have convex si faces with butterfly bend angles of 27° (4) and 17° (5) (Supplementary Figure S3, right). These large butterfly angles are consistent with flavin reduction.
Spectroscopic Analysis of Photoinduced Covalent Inactivation of PRODH.
The crystal structures suggested that light induces the covalent inactivation of PRODH by 4 and 5. This phenomenon was studied further in solution using UV-visible absorbance spectroscopy.
The absorbance spectrum of SmPutA measured in the absence of PRODH inhibitors shows the typical features of an oxidized flavoenzyme, such as maxima near 379 nm and 450 nm, and the spectrum is independent of whether the sample was kept in the dark (Figure 3A) or exposed to a 700 lumen LED bulb between spectral acquisitions (Figure 3B). In contrast, the spectra of SmPutA complexed with 4 or 5 depended on the amount of light exposure. When the samples were kept in the dark, the spectra were nearly identical to that of the oxidized inhibitor-free enzyme (Figures 3C and 3E). This result agrees with the crystal structures of the noncovalent complexes, which show that the FAD is oxidized. However, when the SmPutA-inhibitor complexes were exposed to a 700 lumen LED bulb between spectral acquisitions, the features near 379 nm and 450 nm continuously decreased with increasing light exposure, while a new feature appeared near 320 nm (Figures 3D and 3F). The peak at 320 nm is more pronounced with 4 (Figure 3D). The decreases at 379 nm and 450 nm are consistent with reduction of the FAD, and the increasing feature near 320 nm is interpreted as reporting on covalent modification of the FAD N5.28–30
Figure 3.
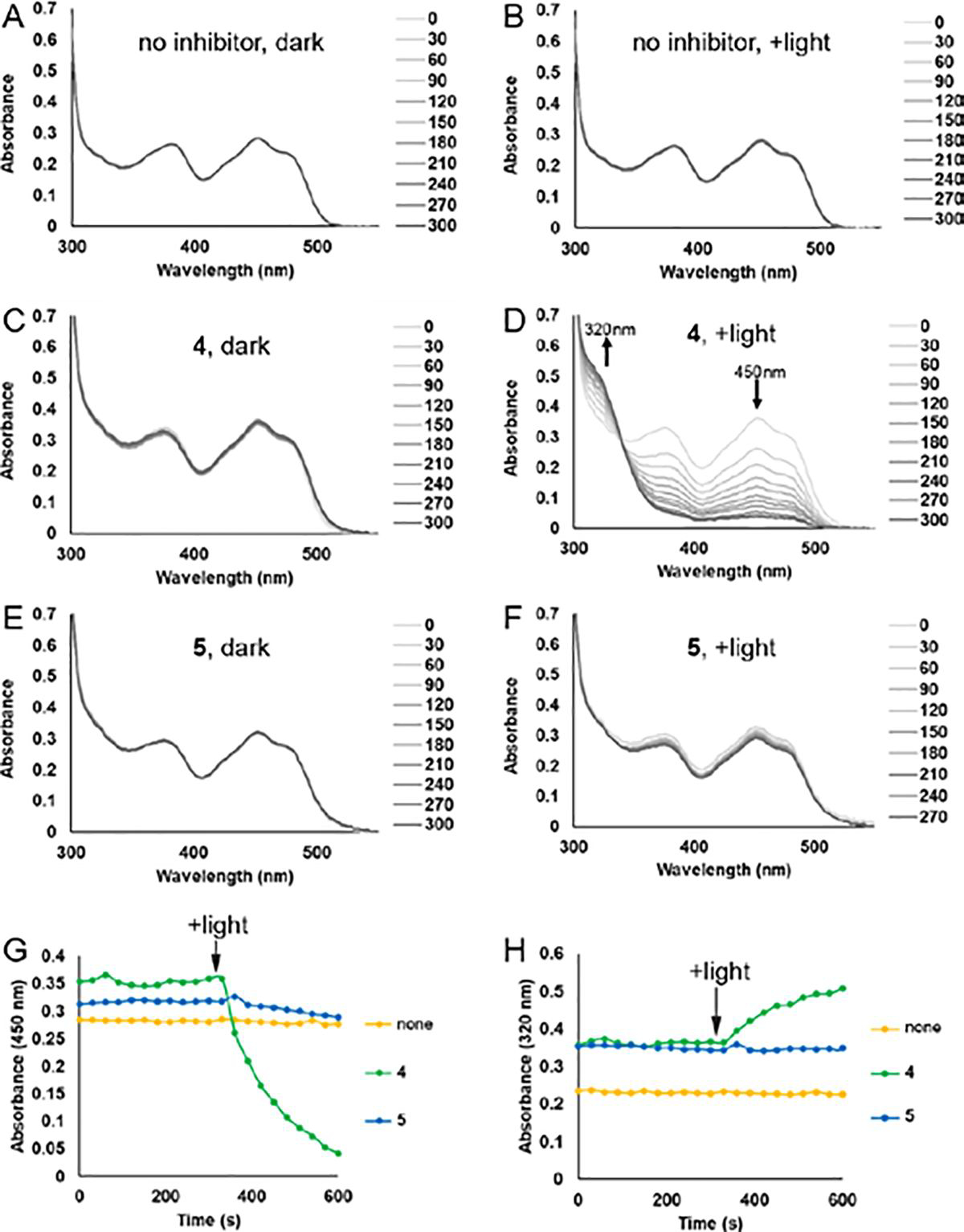
Spectroscopic evidence for photoinduced covalent inactivation of PRODH by 4 and 5. (A-F) Flavin spectral changes measured from SmPutA samples treated with various combinations of inhibitors (25 mM) and exposure to white light. Spectra were acquired at 30s intervals for a total of 300s. For panels A, C and E labeled “dark”, the sample was protected from external light during the experiment. For panels B, D and F labeled “+light”, the sample was illuminated with a 700 lumen LED bulb from a distance of 1 cm for 15 seconds between each spectral acquisition. (G) Effect of white light on the absorbance at 450 nm of SmPutA in the presence of 25 mM 4 or 5. Beginning at the indicated point, the sample was illuminated with a 700 lumen LED bulb from a distance of 1 cm for 15 seconds between each spectral acquisition. (H) Effect of white light on the absorbance at 320 nm of SmPutA in the presence of 25 mM 4 or 5. Beginning at the indicated point, the sample was illuminated with a 700 lumen LED bulb from a distance of 1 cm for 15 seconds between each spectral acquisition.
UV-Vis spectroscopy also provided information about the relative rates of inactivation by 4 and 5. Photoinduced covalent modification of the FAD occurred rapidly with 4, with almost complete inactivation of the sample achieved within 5 minutes of beginning periodic exposure to white light between spectral acquisitions (Figures 3G and 3H). Inactivation proceeded much more slowly with 5 (Figures 3F, 3G, and 3H) resulting in partial reduction of the FAD, which is consistent with the slow color changes observed in the crystals.
To demonstrate the inactivation by 5 in solution, we repeated the experiment for a longer time of 2 hours, with spectral acquisitions every 10 minutes and illumination of the sample for 9 minutes between acquisitions. Spectral changes indicative of covalent modification were clearly observed, including decreasing absorbance near 450 nm and increasing absorbance near 320 nm (Supplementary Figure S4). The spectra imply that 5 inactivates PRODH in solution, consistent with the in crystallo results (Figure 2B).
Wavelength Dependence of Photoinduced Covalent Inactivation.
To investigate the wavelength dependence of photoinduced covalent inactivation, SmPutA was incubated with 4 and illuminated with 5mW hand-held lasers emitting blue (~410 nm), green (~530 nm), or red (~650 nm) light (Figure 4). The flavin spectrum changed most rapidly with blue light (Figure 4A), followed by green light (Figure 4B). Red light produced little change in the spectrum (Figure 4C). Note that treatment with blue light resulted in complete inactivation of the sample within 60 s, as shown by A450 and A320 (Figures 4D and 4E). In contrast, just about one-half of the sample was inactivated under green light after 300 s. These results show that blue light is preferred for photoinduced covalent inactivation of SmPutA by 4.
Figure 4.
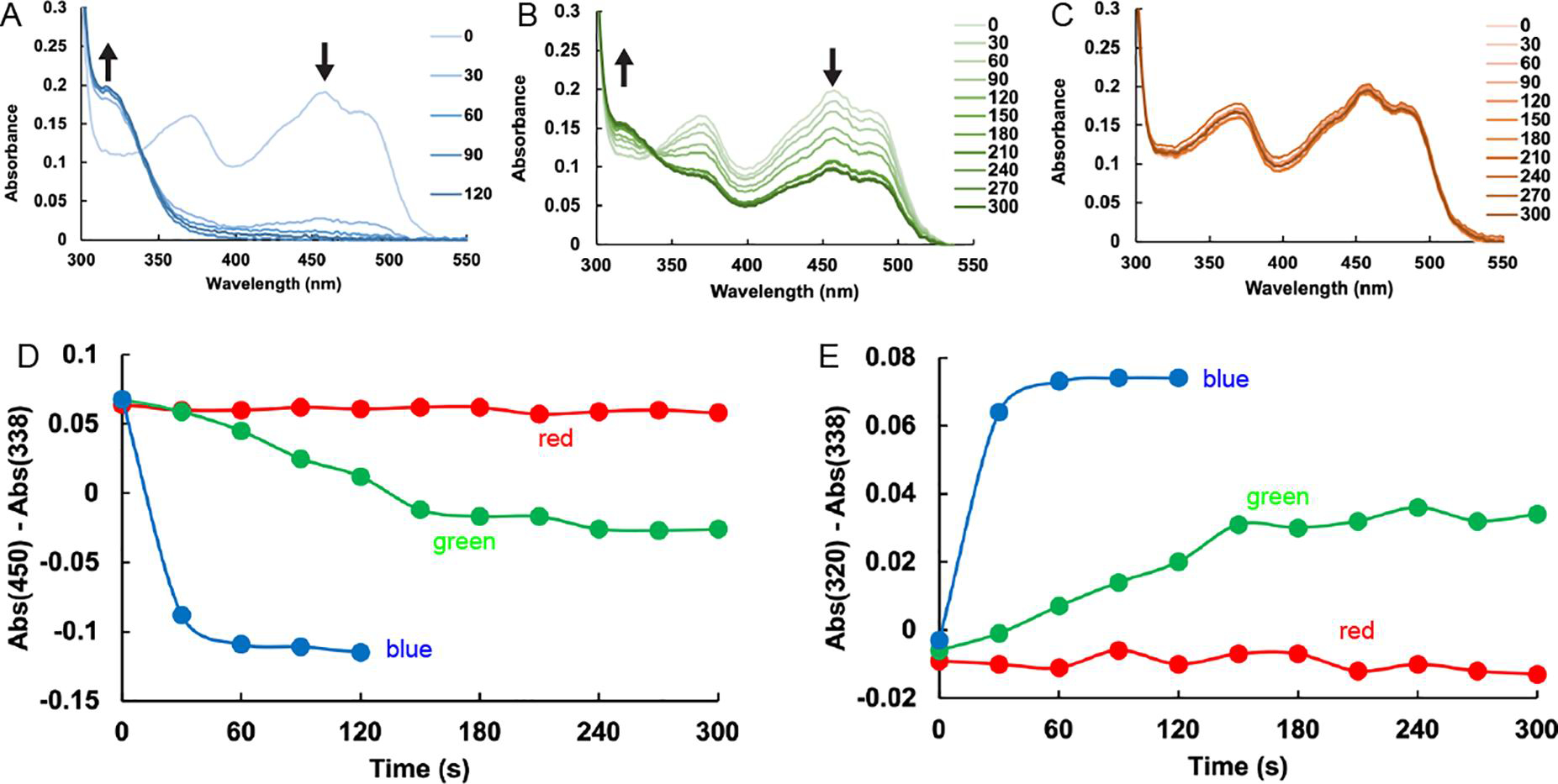
Wavelength dependence of the inactivation of SmPutA by compound 4. (A-C) Flavin spectral changes measured from SmPutA samples containing 25 mM 4 and exposed to 5 mW lasers emitting (A) blue light (410 nm ±10), (B) green light (530 nm ±10), or (C) red light (650 nm ±10). Spectra were acquired every 30s for a total of 300s. The sample was illuminated with the laser from a distance of 1 cm for 15 seconds between each spectral acquisition. (D) Time dependence of the absorbance at 450 nm for the three light sources, relative to the absorbance at the isosbestic point. The colors of the curves correspond to the color of light used. (E) Time dependence of the absorbance at 320 nm for the three light sources, relative to the absorbance at the isosbestic point. The colors of the curves correspond to the color of light used.
The observed photoinactivation was further examined by separately exposing 4 and the enzyme to a blue laser or white light source prior to mixing them. In either case, FAD reduction was not observed (data not shown). Thus, only when the enzyme and 4 are pre-mixed, and then exposed to light, does photoinactivation occur. This suggests that 4 needs to be in the PRODH active site at the time of light exposure to inactivate the enzyme. We also observed that 4 was unable to reduce the FAD of two other unrelated flavoenzymes available in our lab, the ornithine monooxygenase SidA31 and UDP-galactopyranose mutase32 either in the dark or the light (data not shown), suggesting that 4 has some specificity for PRODH.
Photoinduced Covalent Inactivation of PRODH Depends on Both Inhibitor Concentration and Illumination Time.
Enzyme activity assays were performed as functions of both the concentration of 4 and the illumination time with white light. A coupled PRODH-GSALDH assay was used in which proline is the substrate for the PRODH active site, and the production of NADH from the GSALDH active site is monitored by absorbance at 340 nm. The apparent IC50 for the inhibition of SmPutA by 4 in the absence of light is 2.3 mM (Figure 5A). This value represents the IC50 for noncovalent inhibition and shows that 4 is a weak noncovalent inhibitor. However, when the enzyme-4 mixture was exposed to white light prior to assaying the activity, the apparent IC50 steadily decreased with increasing illumination time, reaching a value of 22.4 μM at 45 minutes of illumination (Figure 5A). The apparent increase in potency with light presumably results from the covalent modification of the FAD, as seen in the crystal structure. The IC50 as a function of illumination time could be modeled satisfactorily with an exponential decay function, which yielded a time constant of 3.53 ± 0.06 min and a theoretical minimum IC50 corresponding to infinite duration of light exposure of 32 ± 8 μM (Figure 5B).
Figure 5.
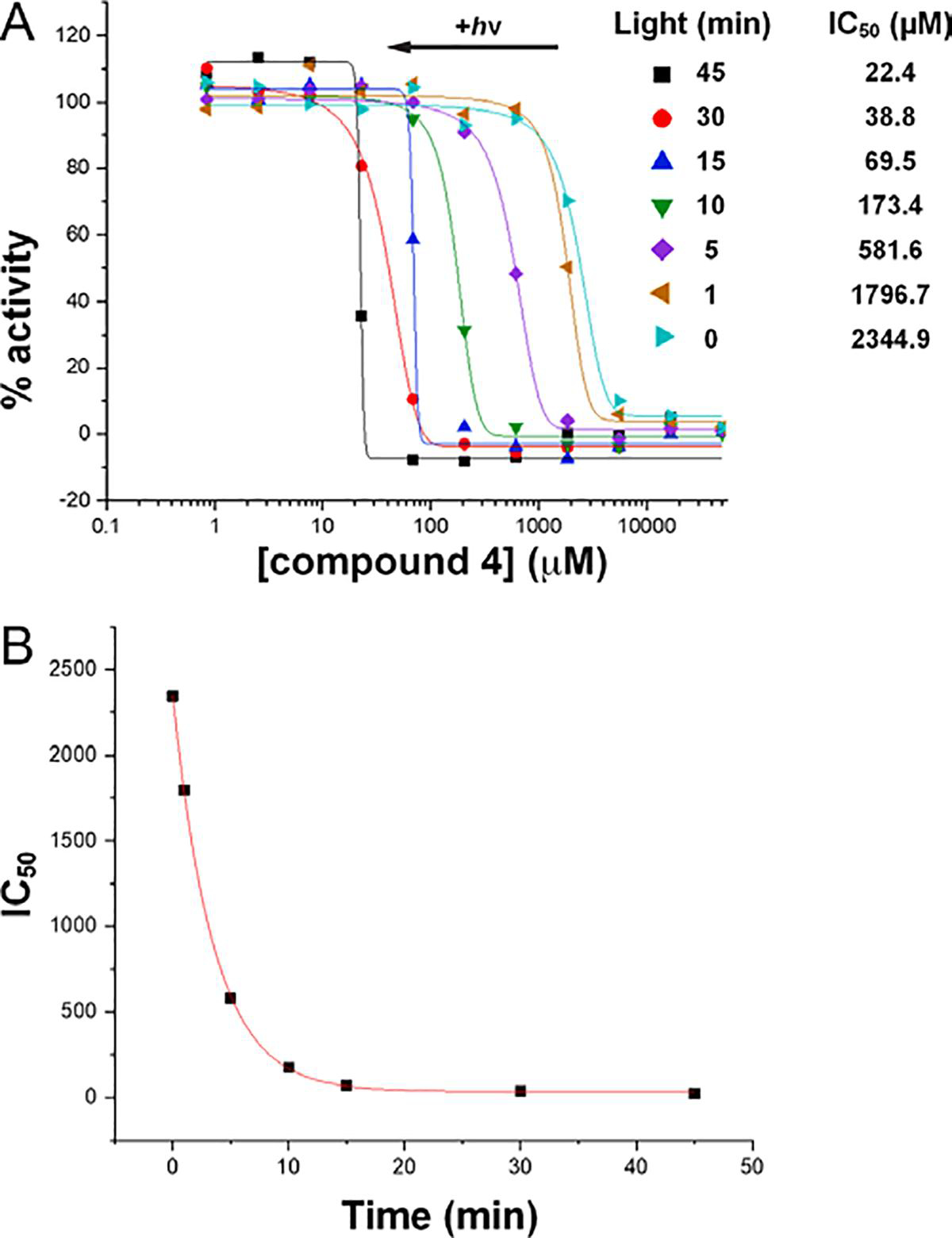
Kinetics of photoinduced covalent inactivation of PRODH by compound 4. (A) PRODH activity remaining after incubating the enzyme with 4 at various concentrations and exposing the sample to white light for various times. The curves represent fits to a dose-response function calculated with Origin software. (B) Dependence of the apparent IC50 on illumination time. The curve represents the fit to an exponential decay function (y = Aexp(−x/τ) + y0) calculated with Origin software. The decay time constant and offset from fitting are τ=3.53 ± 0.06 min and y0 = 32 ± 8 μM.
Photoinduced Covalent Inactivation of PRODH is Irreversible.
The stability of the covalent modification of the FAD was assessed by measuring the recovery of activity following size exclusion chromatography (SEC). Samples of SmPutA containing 25 mM 4 were either kept in the dark to allow only noncovalent inhibition or exposed to white light to promote covalent inactivation. Control samples lacking 4 were treated similarly. Following SEC, the enzyme-4 sample that had been kept in the dark exhibited activity similar to the controls, consistent with the noncovalently bound inhibitor dissociating during SEC (Figure 6A). We note the slightly lower activity for this sample may reflect a small amount of covalent inactivation that occurred during sample manipulation under low-light conditions. In contrast, the enzyme-4 sample that was exposed to bright light exhibited negligible activity throughout the entire range of substrate concentration (Figure 6A). This result suggests that the photoinduced covalent modification of the FAD is irreversible.
Figure 6.
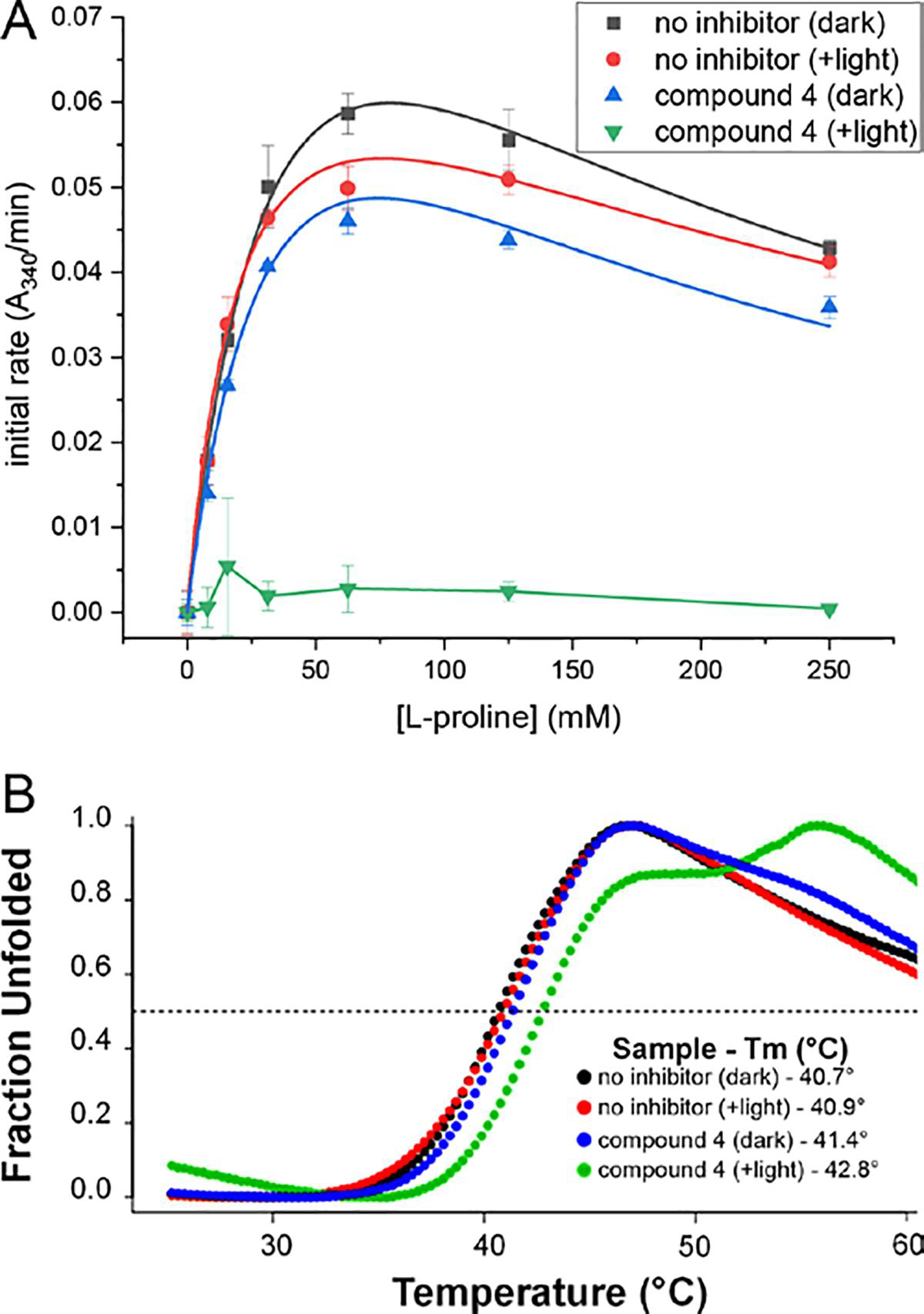
Irreversibility of inactivation of PRODH by 4. (A) Enzyme activity remaining after SEC. The symbols correspond to the following treatments prior to SEC: black squares, no inhibitor and sample was kept in the dark; red circles, no inhibitor and sample was exposed to a 700 lumen LED bulb (5 min); blue triangles, 25 mM 4 and sample was kept in the dark; green triangles, 25 mM 4 and the sample was exposed to a 700 lumen LED bulb (5 min, enzyme was colorless in this timeframe). The smooth curves for the three active samples represent fits to a substrate inhibition model. The line segments for compound 4 (+light) simply connect the points to guide the eye. (B) Melting temperature from fluorescence thermal shift assays performed after SEC. The symbols correspond to the following treatments prior to SEC: black circles, no inhibitor and sample was kept in the dark; red circles, no inhibitor and sample was exposed to a 700 lumen LED bulb (5 min); blue circles, 25 mM 4 and sample was kept in the dark; green circles, 25 mM 4 and the sample was exposed to a 700 lumen LED bulb until the enzyme had turned colorless (5 min, enzyme was colorless in this timeframe).
Irreversibility of the covalent modification was confirmed with fluorescence thermal shift assays performed after SEC. The control samples that had not been incubated with 4 exhibited a single-phase melting curves with midpoints (Tm) of 40.7 °C in the dark and 40.9 °C in the light (Figure 6B). The sample that included 25 mM 4 but had been kept in the dark had a similar single-phase melting curve and Tm of 41.4 °C. In contrast, the sample that was incubated with 4 and exposed to light exhibited a biphasic melting curve (Figure 6B). The midpoint of the first phase is 42.8 °C, which is a shift of 2.1 °C from the untreated sample that was kept in the dark. We interpret the fundamental change in the shape of the melting curve and the Tm shift as evidence of a permanent change to the enzyme structure, which was retained during SEC. Thus, we conclude that photoinduced covalent inactivation by 4 is irreversible.
The stability of the covalent modification in the dark was investigated using absorbance spectroscopy. SmPutA was treated with 4 (25 mM) and white light to generate the inactivated state. The sample was then stored in the dark. After 26 hours in the dark, the sample was quickly transferred to the spectrophotometer under low-light conditions and the spectrum was measured. The resulting spectrum was essentially identical to that of the freshly inactivated enzyme (Supplementary Figure S5). These results show that dark recovery from the N5-alkylated species does not occur on a ~24-hour timescale.
In-cellulo Activity of 4.
Metastatic prostate cancer cells (PC3M) were used to analyze the effects of different PRODH active site-directed ligands on cell growth. In these experiments, cells were grown in 2D culture, adhered to the surface of 96-well plates. The cells were incubated for three days with various concentrations of four PRODH ligands: L-proline (substrate), 1 (non-covalent inhibitor), 2 (mechanism-based covalent inactivator) and 4 (photoinduced covalent inactivator). The 96-well plates were removed from the incubator twice a day and either exposed to a bright light source (Figure 7, +light) or kept under a non-translucent cover (Figure 7, dark) for 45 minutes. On the third day, an MTT assay was performed to assess the relative number of viable cells.
Figure 7.
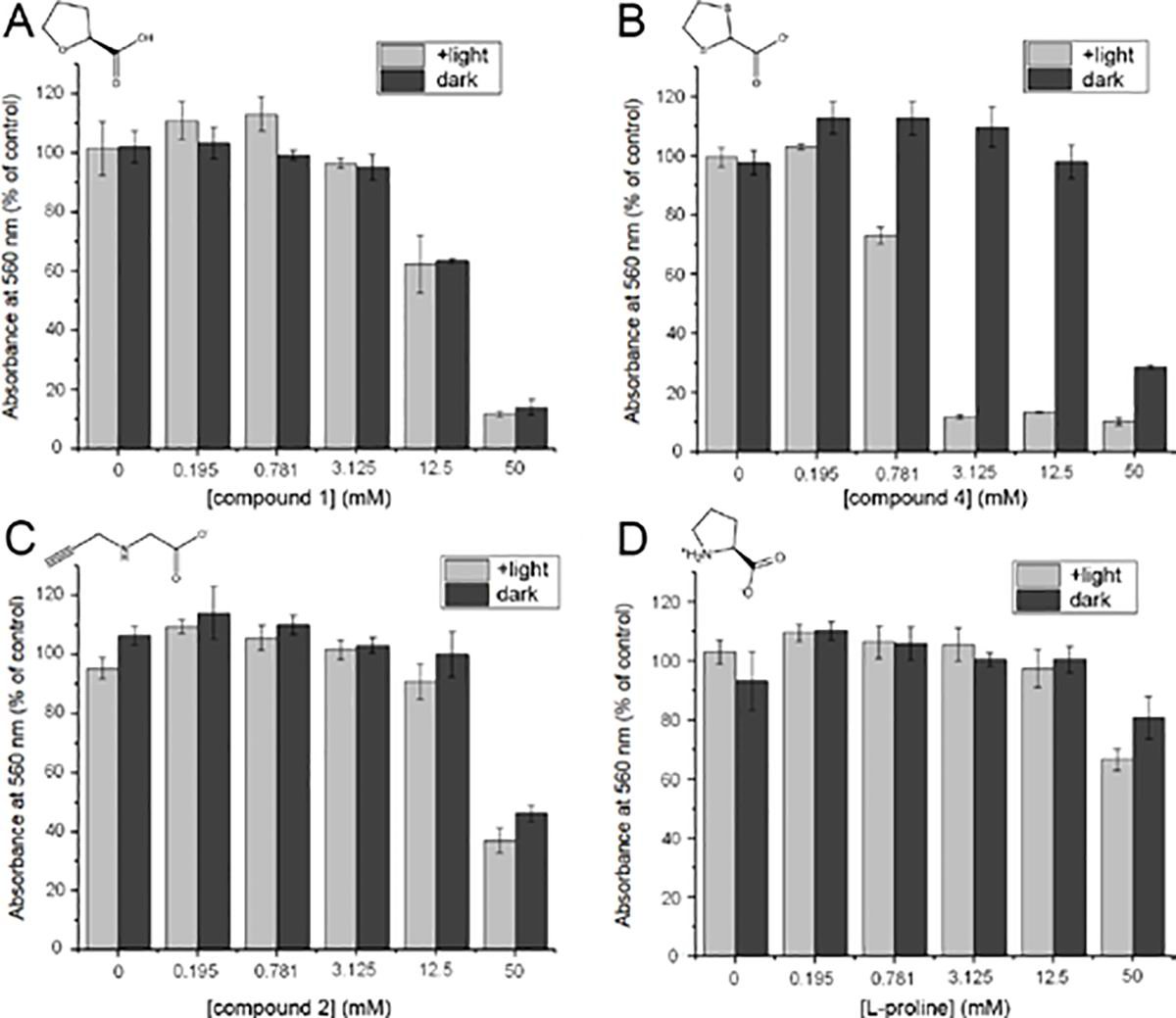
Effects of PRODH ligands on the viability of PC3M cells: (A) compound 1; (B)compound 4, (C) compound 2, (D) L-proline. The compounds were added to PC3M cells at 0, 0.195, 0.781, 3.125, 12.5 or 50 mM and exposed to two 45 min treatments (light bars) or kept in the dark (dark bars) per day for 3 days. The number of viable cells was determined using the MTT assay by absorbance at 560 nm and the data were normalized to a control sample with no added compound.
We observed a significant decrease in cell viability for cultures incubated with 4 and exposed to light treatments (Figure 7B). At moderate concentrations of 4 (0.78, 3.125, 12.5 mM) the difference between light and dark treated samples was significant. For instance, at 3.125 mM of 4, exposure of the cells to light reduced the apparent viability by a factor of ten. This demonstrates that 4 is much more cytotoxic in the presence of light than it is in the dark. Note that light alone does not affect cell viability, as the cells not treated with 4 (0 mM) showed no difference between light and dark samples. We also observed decreases in cell viability for cultures treated with high concentrations of 1, 2, or L-proline (Figures 7A, 7C, 7D). 1 was more cytotoxic than 2, and 2 was more cytotoxic than L-proline, which only had a mild cytotoxic effect at the highest concentration tested (50 mM). In contrast to 4, cell viability in the presence of 1, 2, or L-proline did not change with light treatment. This shows that the light treatment alone is not killing the cells, and that not all PRODH ligands become more cytotoxic in the presence of light. 4 was unique as it had enhanced cytotoxic effects when it was combined with light treatments. This effect mirrors what we observed with the purified enzyme.
Next, we investigated whether light could enhance the cytotoxic effects of 4 in other cell lines. Similar experiments to those in PC3M cells were performed in prostate cancer (PC3), colon cancer (HT29), melanoma (M21), and squamous carcinoma (A431) cell lines. All cell types tested had a dose-dependent response to 4, and the sensitivity to 4 was dramatically increased by treatment with light, compared to cells kept in the dark (Supplementary Figure S6).
Proposed Mechanism of Inactivation.
Here we showed that 4 and 5 are photoinducible covalent inactivators of PRODH that modify the N5 of the FAD and lock it into the reduced state. The requirement of light, particularly blue light, implicates an excited state of the FAD (FAD*) in the inactivation mechanism. Ground state optical absorption by flavins in the visible region of 400 to 500 nm (blue light) generates a short-lived singlet excited state that converts via intersystem crossing to the triplet state.33, 34 Excited flavin states are involved in the mechanisms of several flavoproteins, including flavoprotein light sensors,35 the DNA repair enzyme photolyase,36 and fatty acid photodecarboxylase (FAP).37 Excited flavins are powerful oxidants,34 a property likely leveraged in the inactivation of PRODH by 4 and 5.
The overall reaction described here – photoinduced covalent modification of the flavin N5 accompanied by decarboxylation – has been observed previously with a few other flavoenzymes. Almost 50 years ago, Ghisla and Massey reported the photoinduced covalent inactivation of lactate oxidase by oxalate and oxamate.29 With oxalate, the inhibition was reversible in the dark, analogous to what we observed with PRODH and 4/5. Upon exposure to light, the enzyme-oxalate complex was converted into a reduced flavoenzyme with CO2 covalently bound to the FMN N5, implying decarboxylation of oxalate in the process. Similarly, inactivation with oxamate produced reduced FMN with carbamate bonded to the N5. In 1979, Ghisla et al further characterized analogous reactions of lactate oxidase with several mono- and dicarboxylic acids.28 The first in crystallo characterization of this type of reaction was done with D-amino acid oxidase and the inhibitor 3-methyl-2-oxobutyric acid; the structure showed the N5 of the reduced FAD was acylated, consistent with decarboxylation of the inhibitor (PDB ID 1DAO).30 A recent example of a structurally characterized flavoenzyme which had the N5 acylated by a substrate-like compound, triggered by light, and coupled to an oxidative decarboxylation step is D-arginine acid dehydrogenase.38 The enzyme was unexpectedly found to have a covalently modified flavin when co-crystalized with a potential substrate D-leucine (PDB ID: 3SM8). The structure of the N5-adduct was subsequently confirmed in solution using photo-reduction in the presence of ketoleucine.38
A plausible mechanism for the inactivation of PRODH by 4/5 is suggested by the literature on the photoinduced modification of free34 and enzyme-bound flavins,28, 29 as well as the catalytic mechanism of FAP.39, 40 The proposed mechanism begins with the binding of the inactivator in the proline site, prior to exposure to light (Figure 8). The structures determined from crystals prepared under low-light conditions represent this state. Exposure of the complex to light generates an excited flavin state, FAD*. Single electron transfer from 4/5 to FAD* generates the reduced FAD semiquinone and either a carboxyl- or sulfur-centered radical. Precedent for the latter is suggested by studies of the photooxidation of cyclic sulfides by free flavin.41, 42 Decarboxylation generates a carbon-centered radical, which combines with the FAD semiquinone to form the covalently inactivated enzyme. We note the spectra obtained during photoinduced covalent inactivation lack features indicative of the FAD semiquinone radical. This may mean that radical recombination is very fast and/or the radical pair is never highly populated because of relative inefficiency of the first light dependent step.
Figure 8.
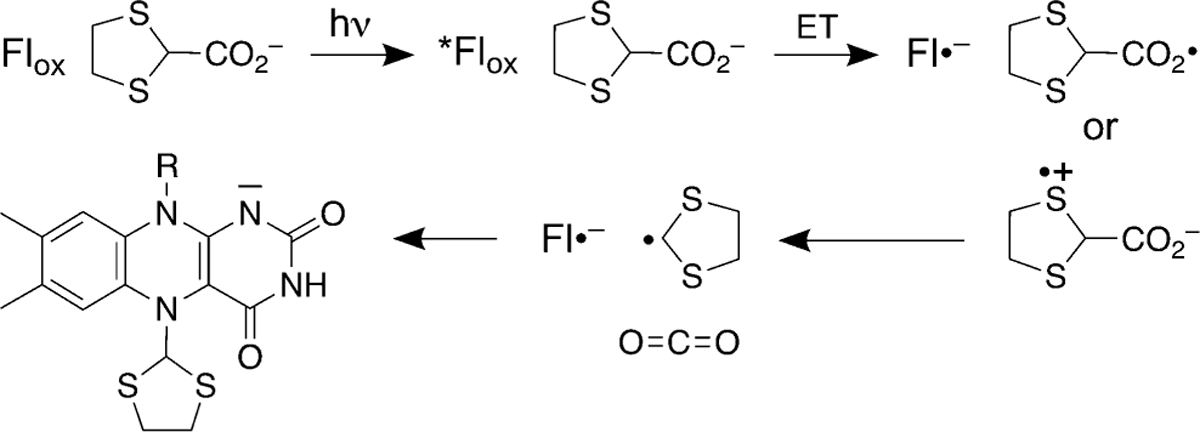
Proposed mechanism of photoinduced inactivation of PRODH by 4.
We found that 4 is much faster than 5 in the photoinduced inactivation of PRODH. It is possible that the S atoms adjacent to the site of decarboxylation stabilize the carbon-centered radical in the proposed mechanism. The stabilization of radicals by adjacent S atoms has been documented.43–45 The presence of a second sulfur in 4 provides additional resonance stabilization leading to faster inactivation kinetics. This explanation is consistent with the fact that 1 does not participate in light-dependent inactivation of PRODH.
Compound 4 and its derivatives have potential as in vivo photopharmacological agents.46 These are drugs applied as an inactive precursor, and after allowing time for them to bind their biological targets, light is applied to the tissues where a cytotoxic response is favorable (e.g., in cancerous tissues). For example, photodynamic cancer therapy agents can be applied orally, intravenously, or topically, before light is used to induce a cytotoxic oxidative response.47 This is beneficial, as appropriately delivered light is used to target cancerous tissues, and avoid harmful effects to healthy tissues. Although the attractive features of photopharmacological drugs are increasingly appreciated by the medical and scientific community, currently only a limited number of enzymes can be modulated in this way.48 The compounds discussed here could be useful as light-activated chemical probes in cancer biology research and lead compounds for the development of a new class of photopharmacological cancer drugs.
METHODS
Inhibitors, Protein Production, and Crystallization.
1,3-dithiolane-2-carboxylic acid (4, product number ENA372572762) and tetrahydrothiophene-2-carboxylic acid (5, product number SY3H3D6823B8) were purchased from Sigma-Aldrich. The bifunctional PRODH-GSALDH enzyme proline utilization A from Sinorhizobium meliloti (SmPutA) was used for crystallization and kinetics experiments. SmPutA was expressed and purified as described.23
SmPutA was co-crystallized with 4 and 5 at 13 °C using the sitting drop vapor diffusion method. Prior to crystallization, SmPutA in a buffer containing 50 mM Tris (pH 8.0), 50 mM NaCl, 5% (w/v) glycerol, and 0.5 mM Tris(2-caboxyethyl)phosphine was combined with either 4 or 5 to achieve a final ligand concentration of 50 mM. The final enzyme concentration was 6 mg/mL. Crystals were grown using a reservoir solution containing 19 % PEG-3350, 0.2 M ammonium sulfate, 0.1 M magnesium chloride, 0.1 M HEPES (pH 8.0), and 0.1 M sodium formate, and the drops were formed by combining 2 μL of the reservoir and 2 μL of the protein-ligand solution. For the noncovalent complex structures, the protein-ligand solutions and crystallization experiments were kept in the dark as much as possible, and the crystals were harvested under dim light, within ~5 minutes of bringing the crystallization tray out of the incubator. To induce covalent modification of the FAD, the crystals of the noncovalent complexes were exposed to bright light (microscope stage and 700 lumen LED bulb), until the yellow color of the crystal was bleached. Once colorless (~10 mins on the microscope stage for 4, ~50 mins using light simultaneously from both the microscope stage and a 700 lumen LED bulb for 5) the crystals were cryoprotected in reservoir buffer with 15 % additional PEG-200, and flash cooled.
X-ray Diffraction Data Collection and Refinement.
X-ray diffraction data were collected in shutterless mode at beamline 24-ID-C of the Advanced Photon Source (Pilatus 6M detector) and beamline 4.2.2 of the Advanced Light Source (Taurus-1 CMOS detector). The data were integrated and scaled using XDS.49 Intensities were converted to amplitudes using Aimless.50 All the data sets are in space group P21 and have similar unit cell dimensions of a = 101 – 102 Å, b = 102 – 103 Å, c = 126 – 127 Å, β =106°. The asymmetric unit contains a dimer of SmPutA. We note this is the same crystal form used for previous crystallographic studies of SmPutA.23, 24 Data processing statistics are listed in Supplementary Table S1.
PHENIX51 was used for refinement and Coot52 was used for model building. The starting model for refinement was derived from the structure of SmPutA complexed with 1 (PDB ID: 5KF624). SMILES strings for 4, 5, 1,3-dithiolane and tetrahydrothiophene were used as the input to ELBOW53 to generate the ligand coordinates and restraint files used during refinement. Noncovalently-bound 4 was modeled with occupancy of 0.73 in chain A and 0.81 in chain B. Noncovalently-bound 5 was modeled as dual occupancy of both stereoisomers (total occupancy of 0.74 in chain A and 0.81 in chain B). For covalently modified FAD structures the ligands were fit into the density and a covalent bond with the FAD N5 was explicitly defined in PHENIX using a target bond length of 1.80 ± 0.02 Å. Structure validation was performed using MolProbity and the wwPDB validation service.54, 55 Refinement statistics can be found in Supplementary Table S1.
Absorbance Spectroscopy.
Spectra of SmPutA (4 mg/mL) in the presence of 25 mM 4 or 5 were measured in a quartz cuvette using a Nanodrop2000c. Spectra were acquired at 30 second intervals for a total of 5 minutes. To obtain spectra of the noncovalent inhibitor-enzyme complexes, the sample was protected from external light by keeping the cuvette in its holder throughout the experiment. To obtain the spectra of the covalently inactivated enzyme, the cuvette was removed from the holder between spectral acquisitions and exposed to a 700 lumen LED bulb from a distance of 1 cm for 15 seconds (cumulative light exposure time of 2.5 minutes). An additional absorbance spectroscopy experiment with a longer observation time of 120 minutes was performed with compound 5 (25 mM). In this case, spectra were recorded every 10 minutes, and the sample was illuminated with the 700 lumen LED bulb from a distance of 1 cm for 9 minutes between spectral acquisitions.
Absorbance spectroscopy was also used to investigate the possibility of dark recovery from the N5-alkylated state. A sample of SmPutA (4 mg/mL) was treated with 4 (25 mM) and light from a 700 lumen LED bulb until the yellow color of the sample was completely bleached. The spectrum was obtained to confirm that the enzyme was inactivated. Then the sample was then stored in the dark at 4°C for 26 hours. The next day, the sample was transferred to the spectrophotometer under low light conditions and the spectrum was measured.
To study the wavelength dependence of photoinduced covalent inactivation, SmPutA was incubated with 25 mM 4 and hand-held lasers (~5 mW) with emission wavelengths in the red (650 nm ±10), green (530 nm ±10) or blue (410 nm ±10) part of the spectrum were used for illumination between spectral acquisitions. Readings were taken every 30 seconds, for a maximum of five minutes, or until the peak at 450 nm had completely diminished.
Kinetics of Enzyme Inactivation.
Enzyme activity was measured using a coupled PRODH-GSALDH assay containing 50 mM L-proline, 0.1 mM NAD+, and 15 μM menadione, the latter required for reoxidation of the FAD to allow catalytic cycling. Reaction progress was monitored by measuring NADH production from the GSALDH active site by absorbance at 340 nm. Samples of SmPutA (0.2 mg/mL) were incubated with 4 at concentrations of 0, 0.85, 2.5, 7.6, 22.9, 68.6, 205.8, 617.3, 1851.8, 5555.6, 16666.7, and 50000 μM in a 96-well flat bottom clear, black polystyrene plate (Sigma, product number CLS3603) kept in the dark and equipped with a sliding opaque lid that allowed certain sections of the plate to be uncovered and illuminated for various durations by a 700 lumen LED bulb held at a distance of 20 cm. Illumination times were of 45, 30, 15, 10, 5, 1 and 0 min. Following illumination, the enzyme-inhibitor sample was diluted 2-fold into the reaction assay buffer and the initial rate was measured. The rates were expressed as percent activity remaining by normalizing to the rate of SmPutA that had not been treated with 4. IC50 plots were analyzed using Origin software. The apparent IC50 values were plotted against the duration of light exposure and fitted to an exponential decay function.
Stability of the Covalent Modification.
The stability of the covalent modification was assessed using enzyme activity assays performed after SEC. SmPutA (6 mg/mL) was incubated with 25 mM 4 and either kept in the dark or exposed to a 700 lumen LED bulb until the solution turned colorless. The samples were run over an SEC column to separate excess and noncovalently bound 4 from the enzyme. The enzymatic activity was measured using the PRODH-GSALDH assay described above with L-proline as the variable substrate (0 – 250 mM) at fixed concentrations of 0.1 mM NAD+ and 15 μM menadione. The initial rate data as a function of L-proline concentration was fit with a substrate inhibition model. Substrate inhibition is common for PutAs and is due to L-proline binding in the GSALDH active site.56
The stability of the covalent modification was also assessed using fluorescence thermal shift assays after SEC. The samples were treated the same as for the activity assays above. Following SEC, the samples were diluted to 1 mg/mL in a buffer containing 50 mM Tris (pH 8.0), 50 mM NaCl, 5% (w/v) glycerol, and 0.5 mM Tris(2-caboxyethyl)phosphine. The enzyme solution was then mixed 1:1 with a 2 × SYPRO orange solution (Thermo Fisher Scientific) and incubated for 45 minutes in the dark at 4 °C. The total volume of each sample was 20 μL. The melting temperatures of the enzyme samples were measured in a QuantStudio version 3 real-time PCR system (Thermo Fisher Scientific) in a MicroAmp 96-well optical plate (Thermo Fisher Scientific). The heating protocol raised the temperature from 4 to 95 °C in 0.5 °C steps with a 20-s hold at each step while the fluorescence was measured. Melting curves were processed using the TSA analysis software at https://beamerlab.shinyapps.io/software/ and Tm was determined as previously described.57
In-cellulo Experiments.
The activities of 1, 2, 4 and L-proline were studied in five cancer cell lines. The human melanoma M21 cell line was kindly provided by Dr. David Cheresh (University of California, San Diego, CA). M21, human prostate cancer cell line PC3M, human prostate cancer cell line PC3 (ATCC® CRL-1435™), human colon cancer cell line HT-29 (ATCC® HTB-38™), and human epidermoid carcinoma cell line A431 (ATCC® CRL-1555™) were cultured in RPMI-1640 custom media supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 48 μg/mL gentamicin at 37°C in 5% CO2. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to assess cell viability after exposure to the compounds and light as follows. Cells were plated on a 96-well plate at 2000 cells/well and grown for 2 days. The media were removed and replaced with media containing the compound at 0, 0.012, 0.049, 0.195, 0.781, 3.125, 12.5, or 50 mM. The cells were exposed to light from a 700 lumen LED bulb, at a distance of 20 cm twice a day, each for 45 minutes. A control plate was treated identically, but was kept covered, in the dark, during the light treatments. After 3 days, the media were removed from the plate, replaced with media containing 0.5 mg/mL MTT, and incubated for one hour. The metabolic activity of viable cells reduces MTT (yellow) to formazan (purple). The amount of formazan was measured by absorbance at 560 nm.
Supplementary Material
ACKNOWLEDGMENT
We thank J. Nix and J. Schuermann for help with remote X-ray diffraction data collection at beamlines 4.2.2 and 24-ID-C, respectively. Beamline 4.2.2 of the Advanced Light Source, a DOE Office of Science User Facility under Contract No. DE-AC02-05CH11231, is supported in part by the ALS-ENABLE program funded by the National Institutes of Health, National Institute of General Medical Sciences, grant P30 GM124169-01. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Funding Sources
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award numbers R01GM065546 and R01GM132640. A.N.B. was supported by a Wayne L. Ryan Fellowship through the Ryan Foundation.
ABBREVIATIONS
- FAP
fatty acid photodecarboxylase
- GSAL
L-glutamate-γ-semialdehyde
- GSALDH
L-glutamate-γ-semialdehyde dehydrogenase
- PRODH
proline dehydrogenase
- P5C
Δ1-pyrroline-5-carboxylate
- SEC
size exclusion chromatography
- SmPutA
proline utilization A from Sinorhizobium meliloti
Footnotes
Competing interests
A patent application has been submitted by the Curators of the University of Missouri and University of Nebraska based on these results.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Table S1 and Figures S1–S6 (PDF).
Accession Codes
Coordinates and structure factor amplitudes have been deposited in the Protein Data Bank under accession codes 7MY9 (4-dark), 7MYA (4-light), 7MYB (5-dark), 7MYC (5-light).
REFERENCES
- 1.Phang JM, Proline Metabolism in Cell Regulation and Cancer Biology: Recent Advances and Hypotheses. Antioxid Redox Signal 2019, 30 (4), 635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Donald SP; Sun XY; Hu CA; Yu J; Mei JM; Valle D; Phang JM, Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res 2001, 61 (5), 1810–5. [PubMed] [Google Scholar]
- 3.Polyak K; Xia Y; Zweier JL; Kinzler KW; Vogelstein B, A model for p53-induced apoptosis. Nature 1997, 389 (6648), 300–5. [DOI] [PubMed] [Google Scholar]
- 4.Elia I; Broekaert D; Christen S; Boon R; Radaelli E; Orth MF; Verfaillie C; Grunewald TGP; Fendt SM, Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nature communications 2017, 8, 15267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phang JM; Liu W, Proline metabolism and cancer. Front Biosci (Landmark Ed) 2012, 17, 1835–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Y; Borchert GL; Surazynski A; Hu CA; Phang JM, Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: the role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene 2006, 25 (41), 5640–7. [DOI] [PubMed] [Google Scholar]
- 7.Tanner JJ; Fendt SM; Becker DF, The Proline Cycle As a Potential Cancer Therapy Target. Biochemistry 2018, 57 (25), 3433–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scott GK; Yau C; Becker BC; Khateeb S; Mahoney S; Jensen MB; Hann B; Cowen BJ; Pegan SD; Benz CC, Targeting Mitochondrial Proline Dehydrogenase with a Suicide Inhibitor to Exploit Synthetic Lethal Interactions with p53 Upregulation and Glutaminase Inhibition. Mol Cancer Ther 2019, 18 (8), 1374–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y; Mao C; Wang M; Liu N; Ouyang L; Liu S; Tang H; Cao Y; Liu S; Wang X; Xiao D; Chen C; Shi Y; Yan Q; Tao Y, Cancer progression is mediated by proline catabolism in non-small cell lung cancer. Oncogene 2020, 39 (11), 2358–2376. [DOI] [PubMed] [Google Scholar]
- 10.Natarajan SK; Zhu W; Liang X; Zhang L; Demers AJ; Zimmerman MC; Simpson MA; Becker DF, Proline dehydrogenase is essential for proline protection against hydrogen peroxide-induced cell death. Free Radic Biol Med 2012, 53 (5), 1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kononczuk J; Czyzewska U; Moczydlowska J; Surażyński A; Palka J; Miltyk W, Proline Oxidase (POX) as A Target for Cancer Therapy. Curr Drug Targets 2015, 16 (13), 1464–9. [DOI] [PubMed] [Google Scholar]
- 12.Zhu W; Gincherman Y; Docherty P; Spilling CD; Becker DF, Effects of proline analog binding on the spectroscopic and redox properties of PutA. Arch. Biochem. Biophys 2002, 408 (1), 131–6. [DOI] [PubMed] [Google Scholar]
- 13.Zhang M; White TA; Schuermann JP; Baban BA; Becker DF; Tanner JJ, Structures of the Escherichia coli PutA proline dehydrogenase domain in complex with competitive inhibitors. Biochemistry 2004, 43 (39), 12539–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moxley MA; Tanner JJ; Becker DF, Steady-state kinetic mechanism of the proline:ubiquinone oxidoreductase activity of proline utilization A (PutA) from Escherichia coli. Arch. Biochem. Biophys 2011, 516 (2), 113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White TA; Krishnan N; Becker DF; Tanner JJ, Structure and kinetics of monofunctional proline dehydrogenase from Thermus thermophilus. J. Biol. Chem 2007, 282 (19), 14316–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krishnan N; Becker DF, Oxygen Reactivity of PutA from Helicobacter Species and Proline-Linked Oxidative Stress. J. Bacteriol 2006, 188 (4), 1227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krishnan N; Becker DF, Characterization of a bifunctional PutA homologue from Bradyrhizobium japonicum and identification of an active site residue that modulates proline reduction of the flavin adenine dinucleotide cofactor. Biochemistry 2005, 44 (25), 9130–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tallarita E; Pollegioni L; Servi S; Molla G, Expression in Escherichia coli of the catalytic domain of human proline oxidase. Protein expression and purification 2012, 82 (2), 345–51. [DOI] [PubMed] [Google Scholar]
- 19.White TA; Johnson WH Jr.; Whitman CP; Tanner JJ, Structural basis for the inactivation of Thermus thermophilus proline dehydrogenase by N-propargylglycine. Biochemistry 2008, 47 (20), 5573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Srivastava D; Zhu W; Johnson WH Jr.; Whitman CP; Becker DF; Tanner JJ, The structure of the proline utilization a proline dehydrogenase domain inactivated by N-propargylglycine provides insight into conformational changes induced by substrate binding and flavin reduction. Biochemistry 2010, 49 (3), 560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strelow JM, A Perspective on the Kinetics of Covalent and Irreversible Inhibition. SLAS Discov 2017, 22 (1), 3–20. [DOI] [PubMed] [Google Scholar]
- 22.Liclican A; Serafini L; Xing W; Czerwieniec G; Steiner B; Wang T; Brendza KM; Lutz JD; Keegan KS; Ray AS; Schultz BE; Sakowicz R; Feng JY, Biochemical characterization of tirabrutinib and other irreversible inhibitors of Bruton’s tyrosine kinase reveals differences in on - and off - target inhibition. Biochim Biophys Acta Gen Subj 2020, 1864 (4), 129531. [DOI] [PubMed] [Google Scholar]
- 23.Campbell AC; Becker DF; Gates KS; Tanner JJ, Covalent Modification of the Flavin in Proline Dehydrogenase by Thiazolidine-2-Carboxylate. ACS Chem Biol 2020, 15 (4), 936–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo M; Gamage TT; Arentson BW; Schlasner KN; Becker DF; Tanner JJ, Structures of Proline Utilization A (PutA) Reveal the Fold and Functions of the Aldehyde Dehydrogenase Superfamily Domain of Unknown Function. J. Biol. Chem 2016, 291 (46), 24065–24075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh H; Arentson BW; Becker DF; Tanner JJ, Structures of the PutA peripheral membrane flavoenzyme reveal a dynamic substrate-channeling tunnel and the quinone-binding site. Proc. Nat. Acad. Sci. USA 2014, 111 (9), 3389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang W; Zhang M; Zhu W; Zhou Y; Wanduragala S; Rewinkel D; Tanner JJ; Becker DF, Redox-induced changes in flavin structure and roles of flavin N(5) and the ribityl 2’-OH group in regulating PutA-membrane binding. Biochemistry 2007, 46 (2), 483–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo M; Arentson BW; Srivastava D; Becker DF; Tanner JJ, Crystal structures and kinetics of monofunctional proline dehydrogenase provide insight into substrate recognition and conformational changes associated with flavin reduction and product release. Biochemistry 2012, 51 (50), 10099–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghisla S; Massey V; Choong YS, Covalent adducts of lactate oxidase. Photochemical formation and structure identification. J Biol Chem 1979, 254 (21), 10662–9. [PubMed] [Google Scholar]
- 29.Ghisla S; Massey V, Mechanism of inactivation of the flavoenzyme lactate oxidase by oxalate. J Biol Chem 1975, 250 (2), 577–84. [PubMed] [Google Scholar]
- 30.Todone F; Vanoni MA; Mozzarelli A; Bolognesi M; Coda A; Curti B; Mattevi A, Active site plasticity in D-amino acid oxidase: a crystallographic analysis. Biochemistry 1997, 36 (19), 5853–60. [DOI] [PubMed] [Google Scholar]
- 31.Robinson RM; Klancher CA; Rodriguez PJ; Sobrado P, Flavin oxidation in flavin-dependent N-monooxygenases. Protein Sci 2019, 28 (1), 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanner JJ; Boechi L; Andrew McCammon J; Sobrado P, Structure, mechanism, and dynamics of UDP-galactopyranose mutase. Arch. Biochem. Biophys 2014, 544 128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kowalczyk RM; Schleicher E; Bittl R; Weber S, The photoinduced triplet of flavins and its protonation states. J Am Chem Soc 2004, 126 (36), 11393–9. [DOI] [PubMed] [Google Scholar]
- 34.Heelis PF, The photophysical and photochemical properties of flavins (isoalloxazines). Chemical Society Reviews 1982, 11 (1), 15–39. [Google Scholar]
- 35.Conrad KS; Manahan CC; Crane BR, Photochemistry of flavoprotein light sensors. Nature chemical biology 2014, 10 (10), 801–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weber S, Light-driven enzymatic catalysis of DNA repair: a review of recent biophysical studies on photolyase. Biochimica et biophysica acta 2005, 1707 (1), 1–23. [DOI] [PubMed] [Google Scholar]
- 37.Sorigue D; Legeret B; Cuine S; Blangy S; Moulin S; Billon E; Richaud P; Brugiere S; Coute Y; Nurizzo D; Muller P; Brettel K; Pignol D; Arnoux P; Li-Beisson Y; Peltier G; Beisson F, An algal photoenzyme converts fatty acids to hydrocarbons. Science 2017, 357 (6354), 903–907. [DOI] [PubMed] [Google Scholar]
- 38.Fu G; Yuan H; Wang S; Gadda G; Weber IT, Atomic-resolution structure of an N5 flavin adduct in D-arginine dehydrogenase. Biochemistry 2011, 50 (29), 6292–4. [DOI] [PubMed] [Google Scholar]
- 39.Heyes DJ; Lakavath B; Hardman SJO; Sakuma M; Hedison TM; Scrutton NS, Photochemical Mechanism of Light-Driven Fatty Acid Photodecarboxylase. ACS Catal 2020, 10 (12), 6691–6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang W; Ma M; Huijbers MME; Filonenko GA; Pidko EA; van Schie M; de Boer S; Burek BO; Bloh JZ; van Berkel WJH; Smith WA; Hollmann F, Hydrocarbon Synthesis via Photoenzymatic Decarboxylation of Carboxylic Acids. J Am Chem Soc 2019, 141 (7), 3116–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knappe WR; Hemmerich P, Covalent intermediates in flavin-sensitized photodehydrogenation and photodecarboxylation. Z Naturforsch B 1972, 27 (9), 1032–5. [DOI] [PubMed] [Google Scholar]
- 42.Haas W; Hemmerich P, pH-dependence, isotope effect and products of flavin-sensitized photodecarboxylation and photodehydrogenation. Z Naturforsch B 1972, 27 (9), 1035–7. [DOI] [PubMed] [Google Scholar]
- 43.Bernasconi CF; Kittredge KW, Carbanion Stabilization by Adjacent Sulfur: Polarizability, Resonance, or Negative Hyperconjugation? Experimental Distinction Based on Intrinsic Rate Constants of Proton Transfer from (Phenylthio)nitromethane and 1-Nitro-2-phenylethane. The Journal of Organic Chemistry 1998, 63 (6), 1944–1953. [Google Scholar]
- 44.Saeva FD; Olin GR, Electron-donating properties of oxygen vs. sulfur. Redox potentials for some pyrylium and thiapyrylium salts. Journal of the American Chemical Society 1980, 102 (1), 299–303. [Google Scholar]
- 45.Byers JH; Whitehead CC; Duff ME, Addition of a sulfur-stabilized radical to electron-deficient alkenes via phenyl selenide transfer. Tetrahedron Letters 1996, 37 (16), 2743–2744. [Google Scholar]
- 46.Bacellar IO; Tsubone TM; Pavani C; Baptista MS, Photodynamic Efficiency: From Molecular Photochemistry to Cell Death. Int J Mol Sci 2015, 16 (9), 20523–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hull K; Morstein J; Trauner D, In Vivo Photopharmacology. Chem Rev 2018, 118 (21), 10710–10747. [DOI] [PubMed] [Google Scholar]
- 48.Lerch MM; Hansen MJ; van Dam GM; Szymanski W; Feringa BL, Emerging Targets in Photopharmacology. Angew Chem Int Ed Engl 2016, 55 (37), 10978–99. [DOI] [PubMed] [Google Scholar]
- 49.Kabsch W, XDS. Acta Crystallogr. D Biol. Crystallogr 2010, 66 (Pt 2), 125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evans PR; Murshudov GN, How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr 2013, 69 (Pt 7), 1204–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Afonine PV; Grosse-Kunstleve RW; Echols N; Headd JJ; Moriarty NW; Mustyakimov M; Terwilliger TC; Urzhumtsev A; Zwart PH; Adams PD, Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr 2012, 68 (Pt 4), 352–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emsley P; Lohkamp B; Scott WG; Cowtan K, Features and development of Coot. Acta Cryst. D Biol. Crystallogr 2010, 66 (Pt 4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moriarty NW; Grosse-Kunstleve RW; Adams PD, electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr 2009, 65 (Pt 10), 1074–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen VB; Arendall WB 3rd; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 2010, D66 (Pt 1), 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gore S; Sanz Garcia E; Hendrickx PMS; Gutmanas A; Westbrook JD; Yang H; Feng Z; Baskaran K; Berrisford JM; Hudson BP; Ikegawa Y; Kobayashi N; Lawson CL; Mading S; Mak L; Mukhopadhyay A; Oldfield TJ; Patwardhan A; Peisach E; Sahni G; Sekharan MR; Sen S; Shao C; Smart OS; Ulrich EL; Yamashita R; Quesada M; Young JY; Nakamura H; Markley JL; Berman HM; Burley SK; Velankar S; Kleywegt GJ, Validation of Structures in the Protein Data Bank. Structure 2017, 25 (12), 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Korasick DA; Pemberton TA; Arentson BW; Becker DF; Tanner JJ, Structural Basis for the Substrate Inhibition of Proline Utilization A by Proline. Molecules 2017, 23 (1), 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andreotti G; Monticelli M; Cubellis MV, Looking for protein stabilizing drugs with thermal shift assay. Drug Test. Anal 2015, 7 (9), 831–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.