

Abstract
The reduction of dinitrogen to ammonia by nitrogenase reflects a complex choreography involving two component proteins, MgATP, and reductant. At center stage of this process resides the active site cofactor, a complex metallocluster organized around a trigonal prismatic arrangement of iron sites surrounding an interstitial carbon. As a consequence of the choreography, electrons and protons are delivered to the active site for transfer to the bound N2. While the detailed mechanism for the substrate reduction remains enigmatic, recent developments highlight the role of hydrides and the privileged role for two irons of the trigonal prism in the binding of exogenous ligands. Outstanding questions concern the precise nature of the intermediates between N2 and NH3, and whether the cofactor undergoes significant rearrangement during turnover; resolution of these issues will require the convergence of biochemistry, structure, spectroscopy, computation and model chemistry.
Graphical Abstract
1. INTRODUCTION
Nitrogenase, the enzyme solely responsible for replenishing the nitrogen cycle from atmospheric dinitrogen, catalyzes the ATP-dependent reduction of N2 to ammonia. The optimal turnover frequency of nitrogenase is approximately one N2 per second, with an effective second order rate constant kcat/Km ~104 M−1 s−1.1 While this is several orders of magnitude slower than diffusion limited enzymatic reactions,2 given the stability of the N≡N triple bond and the immeasurably slow rate of the uncatalyzed reaction, this is a remarkable achievement. Since the physiological substrate of nitrogenase is readily available and since nitrogenase proceeds at a rather leisurely pace relative to faster and more complicated enzyme systems (including such massive assemblies as the ribosome or polymerases that are approximately 10–100 times faster adding monomers),3,4 one could reasonably expect that just about everything to know about nitrogenase would be known by now. Ironically, the small size of the substrate and the complexity of the active site clusters has made it challenging to define the N2 reduction mechanism in atomic detail. Recently, however, there have been exciting advances based on structural, spectroscopic and biochemical probes of the mechanism that have established important details of how substrates interact with the active site under turnover conditions. This review discusses the current state of understanding of the nitrogenase mechanism and highlights some of the outstanding issues.
Nitrogenase consists of two component metalloproteins, the iron (Fe) protein and the molybdenum-iron (MoFe) protein [or the homologous alternative vanadium (VFe) and iron-only (FeFe) nitrogenases].5 The MoFe protein is organized as an α2β2 tetramer NifD2K2, of molecular weight approximately 230 kDa (Fig. 1), where the α and β subunits NifD and NifK have similar folds, pointing towards an evolutionary relationship (see 2.2.1). Coordinated to the MoFe protein are two copies each of two extraordinary metalloclusters designated the FeMo cofactor and the P-cluster. Functionally, the FeMo cofactor represents the site of substrate reduction, while the P-cluster serves as the initial acceptor of electrons from the Fe protein. Overall, the MoFe protein tetramer may be considered to be composed of a pair of αβ subunits that coordinate one FeMo cofactor (located in a cleft between the three domains of the α subunit) and one P-cluster buried at the interface between the α and β subunits (Fig. 1). The Fe protein, NifH2, is a dimer of identical subunits (total molecular weight ~60 kDa) that symmetrically coordinate one [4Fe:4S] cluster, with the ATP binding sites at the dimer interface (Fig. 2).
Figure 1:
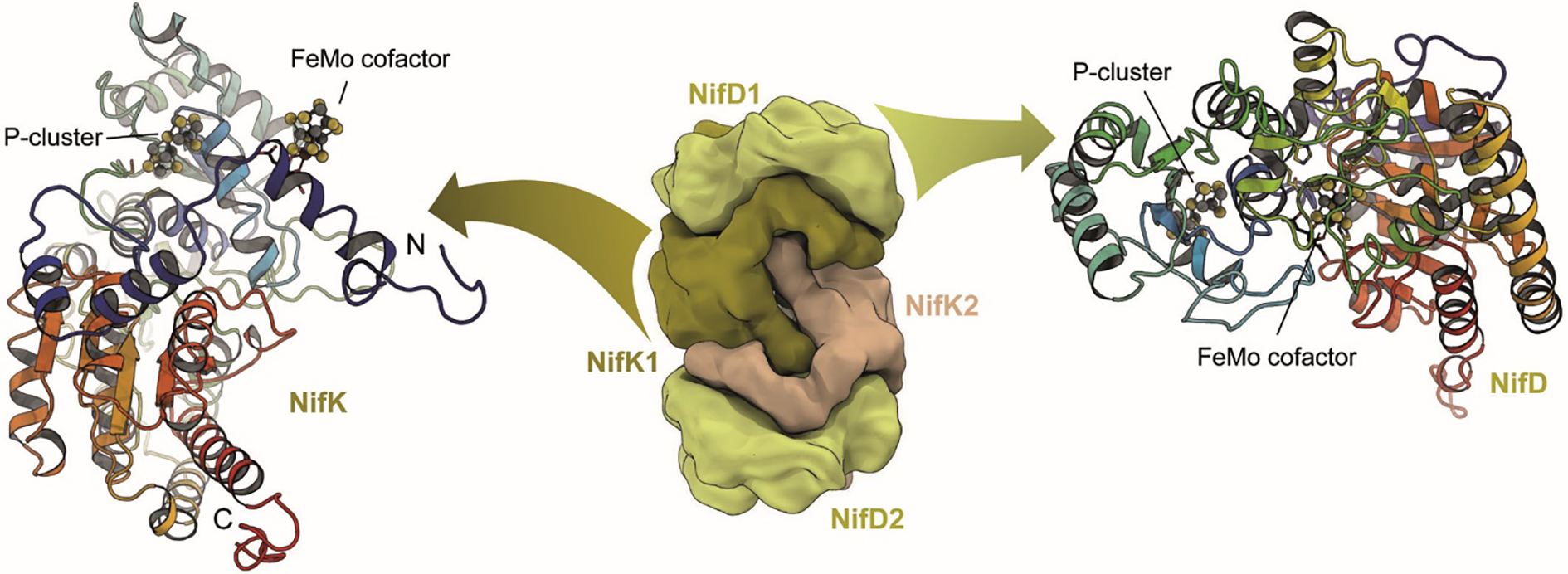
Architecture of molybdenum nitrogenase MoFe protein NifDK (PDB 3U7Q). The dinitrogenase component is a NifD2K2 heterotetramer that contains a set of two complex iron-sulfur clusters in each αβ dimer. While the electron-transferring [8Fe:7S] P-cluster is located at the interface of NifD and NifK, the active site FeMo cofactor, a unique [Mo:7Fe:9S:C]:homocitrate moiety, is buried within NifD.
Figure 2:

The Fe protein of molybdenum nitrogenase. A) The NifH protein forms a homodimer, with each 30 kDa monomer binding ATP or ADP (in stick representation). A conformational change is related to the different ligand-bound states, as Fe protein is a P-loop NTPase. It requires docking to MoFe protein to trigger ATP hydrolysis and electron transfer from its metal site to P-cluster. B) The [4Fe:4S] cluster of Fe protein is coordinated symmetrically at the dimer interface, coordinated by two cysteines from each monomer. Figure generated from PDB 6N4L.
While we are focused in this review on the molecular mechanism of nitrogenase, the cumulative effect of the fixation of N2 by microorganisms is to drive the global nitrogen cycle, and it is instructive to consider the scale of N2 fixed annually by nitrogenase. Although the overall flux is not straightforward to measure,6 an order of magnitude estimate is ~1014 g [N] fixed y−1, representing less than 10−7 of the total atmospheric N2. At the maximum velocity, nitrogenase reduces about one N2 per active site, producing four NH3 per MoFe protein per second at 30 °C. Working through the conversion factors, 3·1010 g MoFe-protein operating at maximal velocity will generate 1014 g of fixed [N] in one year. Of course, this amount of MoFe-protein represents a lower boundary, since under physiological conditions of temperature and the general metabolic state with sub-maximal growth rates, the rate of N2 fixed by nitrogenase will undoubtedly be less than 1 s−1. An analysis of the global mass of Rubisco, the enzyme responsible for CO2 fixation, suggested that the average flux through this pathway for terrestrial organisms was ~1% of the maximal rate.7 A similar effect for nitrogenase would imply that the global mass of nitrogenase is ~1012 g, which can serve as an upper limit. For reference, the global mass of Rubisco is estimated as 1015 g, with an estimated 1017 g [C] fixed yr−1.7 The volume associated with 1012 g MoFe protein would be roughly 1012 cm3 or (100 m)3. Of this, ~1% is occupied by the active site cofactor. Until the advent during the past century of the large-scale industrial Haber Bosch process, this scale of biological nitrogen fixation was required to sustain life on Earth.
Understanding how the component proteins of nitrogenase work together to reduce N2 has required a multidisciplinary effort involving biochemistry, molecular biology, spectroscopy, crystallography, inorganic chemistry and computational chemistry. Each approach has its own strengths – and limitations – for these studies. The critical advantage of a structural biology approach to the study of nitrogenase is the ability to define this system in three-dimensional, atomic detail. As described in the following sections, one of the rewarding aspects of the structural characterization of the nitrogenase has been the opportunity to establish the unprecedented structures of the associated metalloclusters and how ligands bind to the active site.
2. MOLYBDENUM NITROGENASE
2.1. Crystallization of Nitrogenase Components
The propensity of the MoFe protein to crystallize was recognized several years after the first purifications of the component proteins were reported.8,9 The crystallizability of the MoFe protein was integrated into early purification protocols 10,11 until they were replaced by improved chromatography methods.12,13 The promise of high resolution structural information on nitrogenase was fueled by the 1982 report from the Mortenson group describing crystals of the Clostridium pasteurianum MoFe protein diffracting to a resolution of 2.4 Å.14 This was subsequently followed by an analysis with Rossmann of the molecular symmetry demonstrating the homology between the α- and β-subunits.15 Crystals of the Fe protein were published in 1983.16 Despite the presence of metalloclusters that would make the structure determination from these crystals straightforward at a present-day synchrotron source, solutions of the phase problems for these two proteins took a decade to achieve. During this period, an important insight was the finding by Bolin 17 that the C. pasteurianum MoFe protein contained two copies each of the FeMo cofactor and P-cluster, arranged such that the two FeMo cofactors were separated by ~70 Å and hence could not simultaneously interact with the same substrate. This observation contrasted with the findings of the first published report of a MoFe protein crystal structure determination (at 8 Å resolution, phased by direct methods) that indicated the two FeMo cofactors were sufficiently close to be bridged by N2.18
The first crystal structures for the nitrogenase proteins were published in 1992 for the Azotobacter vinelandii MoFe protein 19,20 and Fe protein,21 followed shortly by structures for the C. pasteurianum MoFe protein.22,23 The resolutions of these structures were insufficient to unambiguously define the metallocluster architecture, and so the initial models integrated elemental analysis on the MoFe protein and isolated FeMo cofactor, stereochemical information from small molecule iron-sulfur cluster structures, and spectroscopic inferences. As the resolution of the crystal structures increased due to improvements in crystal quality, synchrotron beamlines, X-ray detectors, refinement algorithms and computational resources, errors in the original models were corrected. These changes included the existence of an interstitial ligand in the center of the FeMo cofactor (see 3.1)24 and the presence of 7, not 8, sulfurs in the P-cluster (vide infra).25 The original indication that one of the FeMo cofactor belt positions, initially identified as “Y” and now assigned as S5A, was lighter than sulfur did not survive scrutiny at high resolution. Ironically, this is the one belt sulfur position that has not subsequently been observed to be stoichiometrically substituted with lighter ligands in certain forms of either the MoFe protein or the VFe protein (see 3.3, 4.4).
2.2. MoFe Protein – The Dinitrogenase
2.2.1. Structure of MoFe Protein
In spite of the use of distinct structural genes clustered separately in the genome of A. vinelandii, all three nitrogenase systems are remarkably similar in architecture. The core of each of the three catalytic components is formed by a NifD2K2 heterotetramer of approximately 230–240 kDa (Fig. 1), arranged around a twofold symmetry axis (Fig. 3A). The two subunits are the 55 kDa NifD and the 53 kDa NifK. Interestingly, the nifD and nifK genes themselves show significant sequence homology, hinting at an early duplication of a common precursor gene. The D and K subunits are further structured into three globular domains with a canonical βαβ-fold (Rossmann fold), where a four- or five-stranded parallel β-sheet is flanked by connecting α-helices (Fig. 3B).19 In each of these domains, the C-terminal loops emanating from the β-strands interact with the two metal clusters of the protein, as is typical for this fold (Fig. 3C). The Rossmann-domains of the nitrogenase proteins are themselves phylogenetically related, suggesting that the complex nitrogen-fixing systems known today can be traced back to an original single-domain protein, possibly already functioning as ligand for an iron-sulfur cluster. This primordial ferredoxin then extended its possibilities through a gene triplication, and the dimerization of two such units further generated the possibility to bind cofactors at the dimer interface. This presumed history of nitrogenase is supported by recent studies on structurally related enzyme systems that are considered to be ancestral to the nitrogen-fixing machinery. A pair of genes that was originally described to be ‘Nif-like’ (nfl) was recently identified to play a role in the biogenesis of the tetrapyrrole cofactor F430, the unique Ni-porphyrin that forms the active site of methyl-coenzyme M reductase, a key enzyme in archaeal methanogenesis.26,27 In the Nif-like enzyme CfbDC, the CfbD proteins forms a dimeric reductase with a bridging [4Fe:4S] cluster and strong homologies to the Fe protein of nitrogenases and its relatives.26 CfbD interacts with CfbC that shows the very same three-domain architecture observed in NifD and NifK, but then forms a simple homodimer as the catalytically active component.
Figure 3:
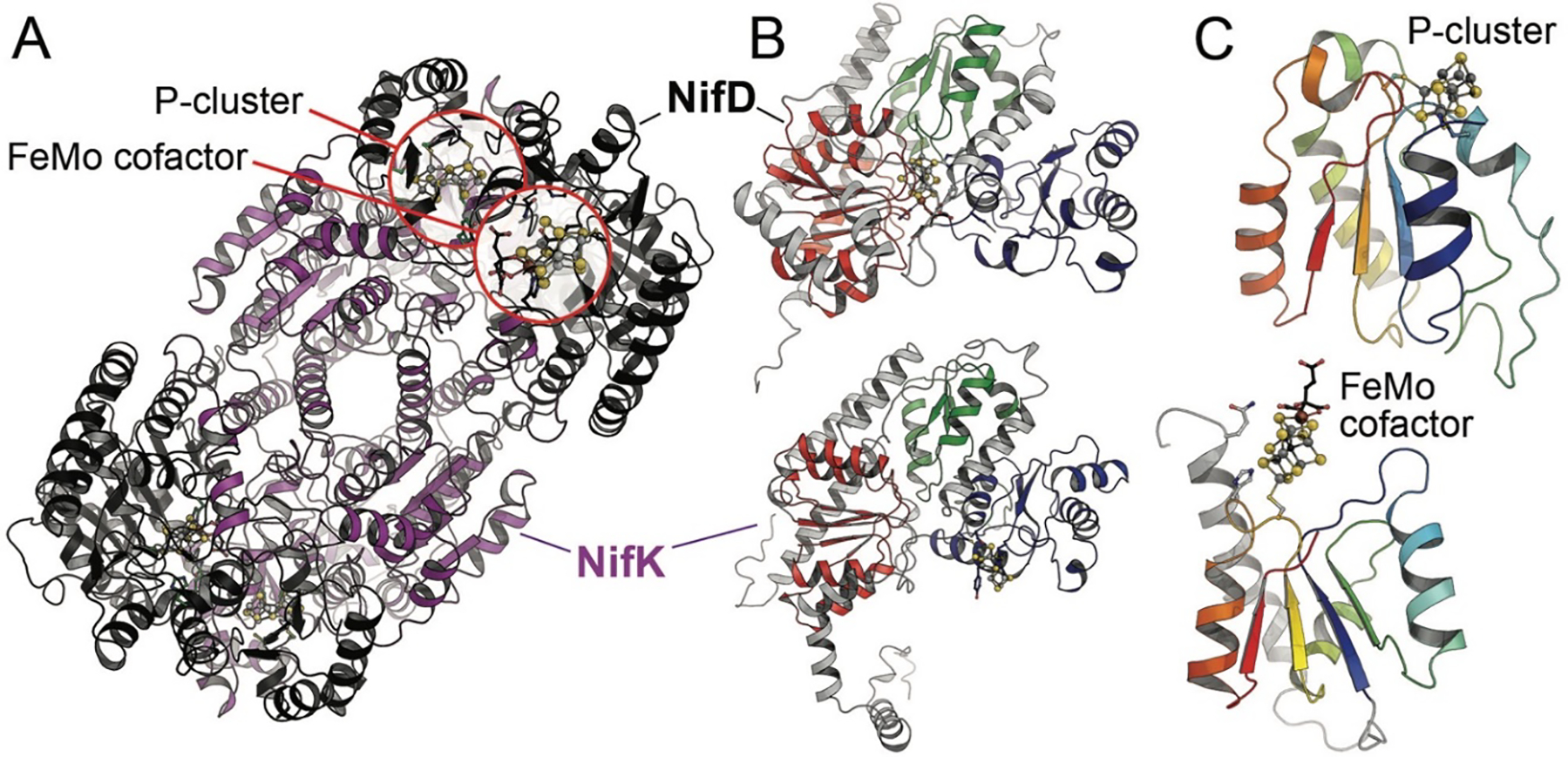
The structure of MoFe protein. A) MoFe protein forms an α2β2 heterotetramer in which two αβ units (NifDK) are connected solely via the NifK peptides. Each αβ unit holds a FeMo cofactor and a P-cluster (PDB 3U7Q). B) NifD (above) and NifK (below) are structurally and evolutionarily related and consist of three consecutive Rossmann-fold domains. The domains are highlighted in red, green and blue according to their occurrence in the chain. In NifD, all three domains cradle the FeMo cofactor in their center, while the P-cluster is symmetrically coordinated between the third domains of each chain. C) As typical for this fold, the P-cluster and FeMo cofactor are coordinated at the loop regions at the C-terminus of the parallel β-sheet of a Rossmann domain.
CfbDC thus has already evolved cofactor binding in a three-domain protein, but has not yet differentiated into two distinct subunits for the catalytic part. This differentiation is not unique to nitrogen fixation, however. Two further remarkable homologs of nitrogenases are found in the biosynthesis of bacteriochlorophyll, another tetrapyrrole cofactor that predates (and that eventually heralded) the aerobic era in the evolution of life. Here, the dark-operative protochlorophyllide reductase (DPOR, Fig. 4) and the downstream-acting chlorophyllide oxidoreductase (COR) form homologous α2β2 heterotetramers that each interact with a designated, dimeric reductase. A recent structural analysis of DPOR revealed that the enzyme contains a regular [4Fe:4S] cluster at the interface of its two subunits.28 It lacks the two unique iron-sulfur sites of nitrogenases (vide infra) and binds its substrate, a bulky tetrapyrrole, at a position close to the binding site of the catalytic cofactor in nitrogenases. The origin of nitrogenases can thus be traced back to tetrapyrrole-modifying systems from the anaerobic world (Fig. 4).29 These were then repurposed as a scaffold to accommodate a large and intricate iron-sulfur moiety, the catalytic cofactor that mediates the cleavage of the N2 triple bond.
Figure 4:
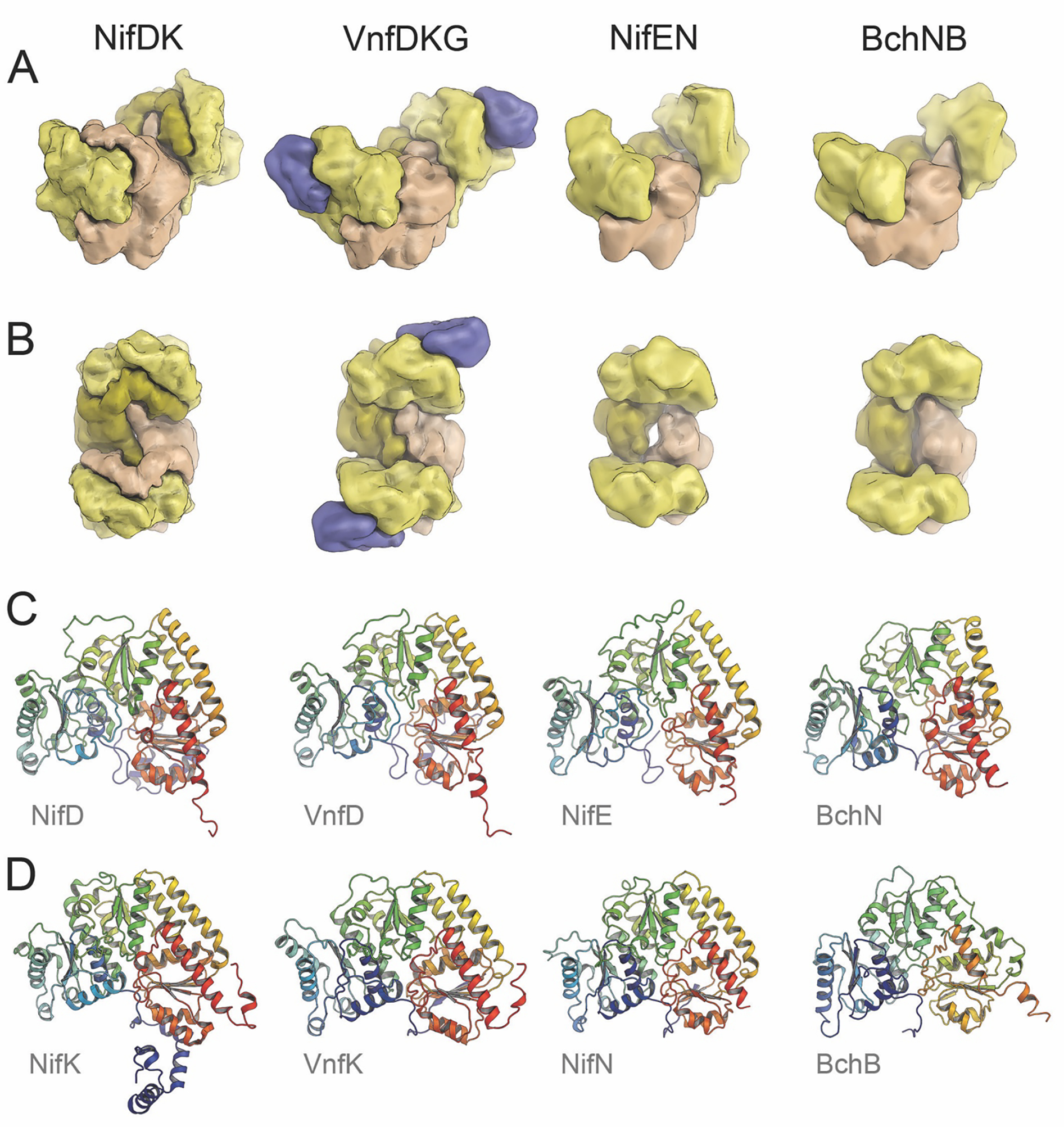
A structural comparison of Mo- and V-nitrogenases and their orthologs NifEN and the protochlorophyllide reductase BchNB (DPOR). A) Subunit arrangement of the four orthologs. Coloring scheme as in Fig.1. B) Top view of the complexes, with corresponding coloring of homologous subunits. C) The respective α-subunits of each enzyme in cartoon representation, colored from blue at the N-terminus to red at the C-terminus. Each chain contains three consecutive Rossmann-fold domains (blue, green, red). D) The respective β-subunits in the same orientation as in C). The figure was generated from the PDB entries 3U7Q (NifDK), 5N6Y (VnfDKG), 3PDI (NifEN) and 3AEK (BchNB).
Mo-nitrogenase – like the other variants – thus contains two types of large iron-sulfur clusters in each DK dimer, forming two presumably independent, functional units in the heterotetrameric assembly. The position of the [4Fe:4S] cluster in DPOR and COR is occupied by the P-cluster, a unique [8Fe:7S] moiety that is formed through the fusion of two canonical [4Fe:4S] centers during the maturation of the enzyme.30,31 It functions as an electron relay between the reductase, Fe protein, and the second of the metal clusters of nitrogenase where reductive catalysis takes place. All three known variants of nitrogenase contain this P-cluster, and at least in the two structurally characterized proteins utilizing molybdenum and vanadium, respectively, its architecture is highly conserved (see 2.2.2, Figs. 5, 13C). In contrast, the catalytic cofactor, an even larger, iron-sulfur-based metal cluster, differs among the classes of nitrogenases. Historically, Mo-nitrogenase is by far the most thoroughly characterized enzyme. It contains the FeMo cofactor at its active center, a [Mo:7Fe:9S:C] cluster of high symmetry complemented with an organic R-homocitrate ligand (see 2.2.3). As a key difference, V-nitrogenases replace the molybdenum ion with vanadium, while in Fe-nitrogenases, iron is employed exclusively. Recent years have seen substantial progress in understanding the atomic and electronic structure of FeMo cofactor, forming the basis for most current hypotheses regarding the functionality of nitrogenases.
Figure 5:
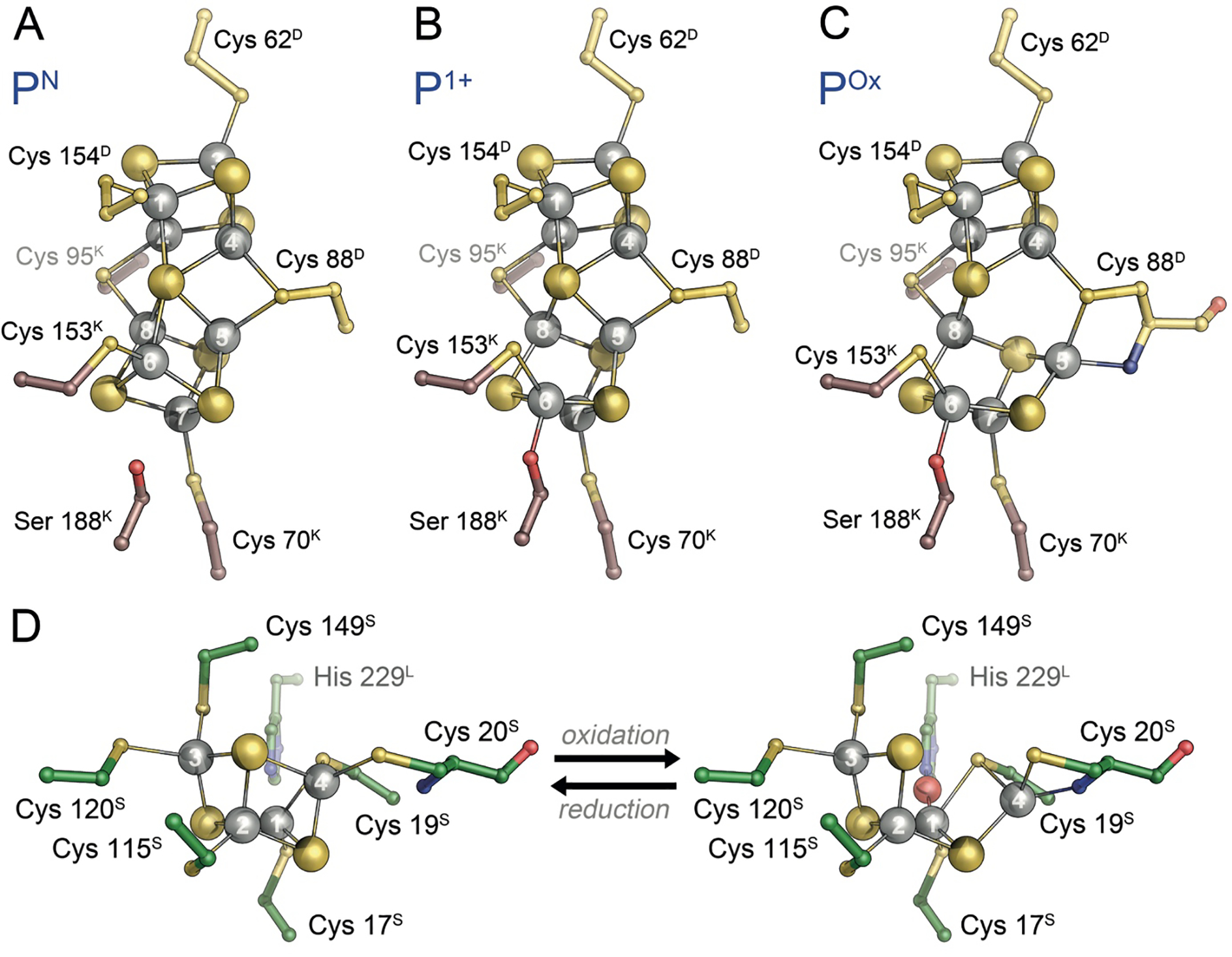
Redox-dependent conformational states of P-cluster. A) In the PN state obtained upon reduction with sodium dithionite, the [8Fe:7S]2+ cluster is all-ferrous and symmetrically arranged around a central, six-coordinate sulfide. B) The one-electron-oxidized P1+ state shows a rearrangement of Fe6 only, releasing its interaction with the central sulfide and orienting towards a nearby serine, the largely conserved Ser 188K. C) Two-electron oxidation of the PN state leads to POx, a [8Fe:7S]4+ state in which Fe5, in addition to Fe6, re-orients away from the central sulfide, this time towards a backbone amide. Notably, all observed changes are reversible. D) Redox-dependent structural changes in the [4Fe:3S] cluster of O2-tolerant hydrogenase of C. necator. The shift of Fe4 towards a backbone amide is highly reminiscent of the shift of Fe6 upon oxidation of P-cluster. Panels A)-C) generated from PDB entries 1M1N and 3U7Q, panel D) generated from PDB entry 4IUD.
Figure 13:
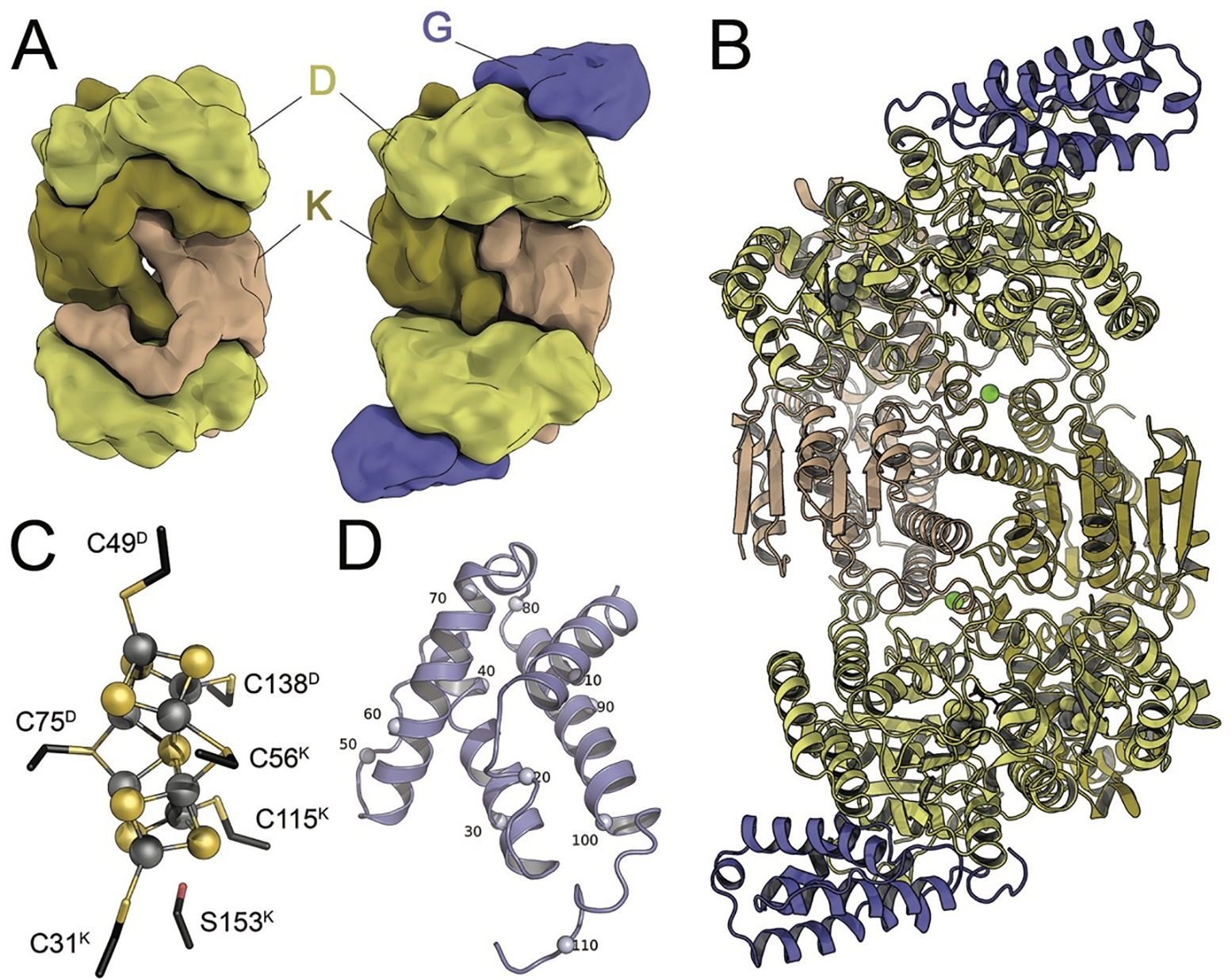
The vanadium-iron protein from A. vinelandii. A) Similar to the NifD2K2 heterotetramer (left), the core of VFe protein is a VnfD2K2 assembly with high structural homology to MoFe protein. It additionally contains a further subunit, VnfG, which is in exclusive contact with VnfD. B) Cartoon representation of A. vinelandii VFe protein (PDB 5N6Y). Green spheres denote the bridging Mg2+ ions. C) The P-cluster of VFe protein bridges the D- and K-subunits. In its reduced state it is nearly identical to its MoFe protein counterpart. D) The VnfG subunit forms a four-helix bundle and does not contain additional cofactors. Every tenth residue of VnfG is denoted by a small sphere.
2.2.2. The Role of the [8Fe:7S] P-cluster
Among the peculiarities of P-cluster, a site that according to current knowledge is entirely unique to the three classes of nitrogenase enzymes, is that it is typically isolated in an all-ferrous state, i.e. formally with 8 Fe2+, conveying a total charge of +2 (Fig. 5A). All-ferrous iron-sulfur clusters are rare, but interestingly, a second occurrence of such a highly reduced site is found in the corresponding reductase, Fe protein (see 2.3.1). Fe protein serves as the electron donor to MoFe protein, with the P-cluster acting as a relay for a single electron moving from the [4Fe:4S] site in Fe protein to the active site, FeMo cofactor. However, with P-cluster in an all-ferrous state, the addition of another electron from Fe protein is far from trivial. According to the current state of knowledge, this problem is solved through an intricate sequence of events triggered by the formation of a complex between ATP-bound Fe protein and the catalytic MoFe protein that starts with a single-electron transfer from P-cluster to the cofactor that only then is followed by the oxidation of the [4Fe:4S] cluster of Fe protein.32 This has been designated a ‘deficit-spending’ mechanism, and it implies that upon docking to MoFe protein, Fe protein has a means of lowering the reduction potential of P-cluster such that it can reduce the cofactor.33,34 Whether complex formation in itself is sufficient to achieve this or ATP hydrolysis is already required at this point remains a matter of debate. A kinetic study by Seefeldt and co-workers concluded that the hydrolysis of ATP would only occur after the oxidation of Fe protein and that the subsequent release of inorganic phosphate from hydrolyzed ATP constitutes the rate-limiting step of the process,35 although this model is challenged by the finding that the known structures of complexes of MoFe and Fe proteins do not show any structural changes within the catalytic component that were triggered by complex formation alone.
A further significant feature of the P-cluster was found through structural analysis, showing that the cluster as isolated (i.e. reduced with sodium dithionite that is used throughout the purification procedure, designated PN) shows a different conformation than a POx-form that is 2-electron oxidized, either by chemical oxidants such as phenosafranine, indigo carmine or ascorbate or by short air exposure. The PN state of P-cluster is highly symmetric, with the central sulfide S1 placed almost precisely on the pseudo-twofold symmetry axis relating the D and K subunits of the MoFe protein, reflecting their common origin. Upon oxidation to the POx state, however, this symmetric arrangement is broken by two of the Fe ions, Fe5 and Fe6, which shift position to release their coordination by S1 and approach new ligands. In the case of Fe6 this is a largely conserved serine residue, Ser 188K in the β-subunit (NifK) of the A. vinelandii MoFe protein, while in another group of nitrogenases this oxygen-based ligand is instead provided by a tyrosine, as in the case of Tyr 98K of the enzyme from Gluconacetobacter diazotrophics, where this leads to a repositioning of Fe8 rather than Fe6.36 In addition, Fe5 in both groups moves to interact with the backbone amide of residue Cys 88D in the α-subunit (NifD, A. vinelandii numbering), notably requiring deprotonation of the amide nitrogen atom (Fig. 5C), while a recent computational study has found that in A. vinelandii MoFe protein, Ser 188K is also deprotonated in the POx state.37 Various structural analyses have shown that this conformational change is reversible and that it is found in V-nitrogenase as well as in the Mo-dependent variant.38,39 The transition from PN to POx represents a 2-electron oxidation, while the mechanism of N2 reduction by the enzyme is commonly discussed in single-electron steps. A one-electron oxidized P1+ state of P-cluster has been identified spectroscopically, but has long remained elusive, until a recent structural analysis of redox-poised crystals of MoFe protein by Peters and co-workers revealed that in this state only Fe6 shows the previously defined ligand exchange for Ser 188K, while Fe5 retains the position observed in the PN state (Fig. 5B). This finding was corroborated by the first crystal structure of a vanadium nitrogenase, where an otherwise largely identical P-cluster was isolated in a mixed state between PN and a P1+ with a shift only of Fe6.38 The unusual conformational rearrangement of P-cluster is further reminiscent of an observation made in the oxygen-tolerant hydrogenase form Cupriavidus necator (previously Ralstonia eutropha), where a single Fe site of a unique [4Fe:3S] cluster undergoes a movement that is highly analogous to what is found for P-cluster (Fig. 5D).40,41 Such structural flexibility in a metal site is likely to be of functional significance, and in nitrogenase it seems safe to assume that this feature is mechanistically linked to the ‘deficit-spending’ electron release from the PN state that only occurs after the docking of a reduced Fe protein dimer. A conformational change in P-cluster not only alters its midpoint potential for re-reduction drastically, but also relays information about its oxidation state to affect the affinity of both protein components and trigger ATP hydrolysis.
The fact that both the [4Fe:4S] cluster of Fe protein and P-cluster were found to accept and release one or two electrons has led to considering the possibility that such 2-electron transfer events might occur under physiological conditions.25,42–44 This would be of major mechanistic significance, as the interaction of Fe protein with the dinitrogenase is linked to the hydrolysis of two ATP molecules, so that a concerted transfer of two electrons would effectively reduce the ATP requirement of the enzyme by 50 percent. While no instance of physiological two-electron transfer has yet been found in any functional assay or in vivo, the known flexibility of the nitrogenase system does not allow to generally rule out this type of mechanism.
Surprisingly, mutagenesis studies of residues coordinating the P-cluster have established that at least certain substitutions, or even deletions, can be tolerated, while maintaining the ability to fix nitrogen at high enough levels so that the cells can grow diazotropically.36,45–49 Structural studies from the Tezcan group have further demonstrated the compositional lability of the P-cluster upon mutagenesis of surrounding residues, where despite the loss of one or two Fe, nitrogen reduction activity is retained.36,49 One of the challenges in understanding the role of the P-cluster in nitrogen fixation is how to reconcile the evident sensitivity of this metallocluster to structural perturbations, yet at the same time, the modified nitrogenases can still fix dinitrogen at levels sufficient to support cell growth.
2.2.3. The Catalytic Cofactor of Nitrogenase, FeMoco
The second cluster of the nitrogenases constitutes the active site for the reduction of a variety of small-molecule substrates. Like the P-cluster it is a variation on the theme of iron-sulfur clusters, but differs starkly from all other metal sites within this family. Its structure has been of long-standing interest (see 3), and is now recognized to have the composition [Mo:7Fe:9S:C]:homocitrate in the case of Mo-nitrogenase, where it is designated ‘FeMo cofactor’ (Fig. 6).50 This cluster complements 7 iron ions with a single molybdenum, digresses from regular iron-sulfur clusters by containing one more acid-labile sulfide than it has metal ions and features a μ6-carbide that is unprecedented in biology. It also incorporates an organic homocitrate molecule that serves as a ligand to molybdenum and is synthesized by a homocitrate synthase, the NifV protein, from acetyl-coenzyme A and 2-oxoglutarate.51,52 In the FeMo cofactor, the carbide holds a central position, fusing two cubane-like half clusters that are additionally connected by three μ2-bridging sulfides. The entire cluster is D3-pseudosymmetric, with only the apical Mo ion breaking the point group symmetry.
Figure 6:
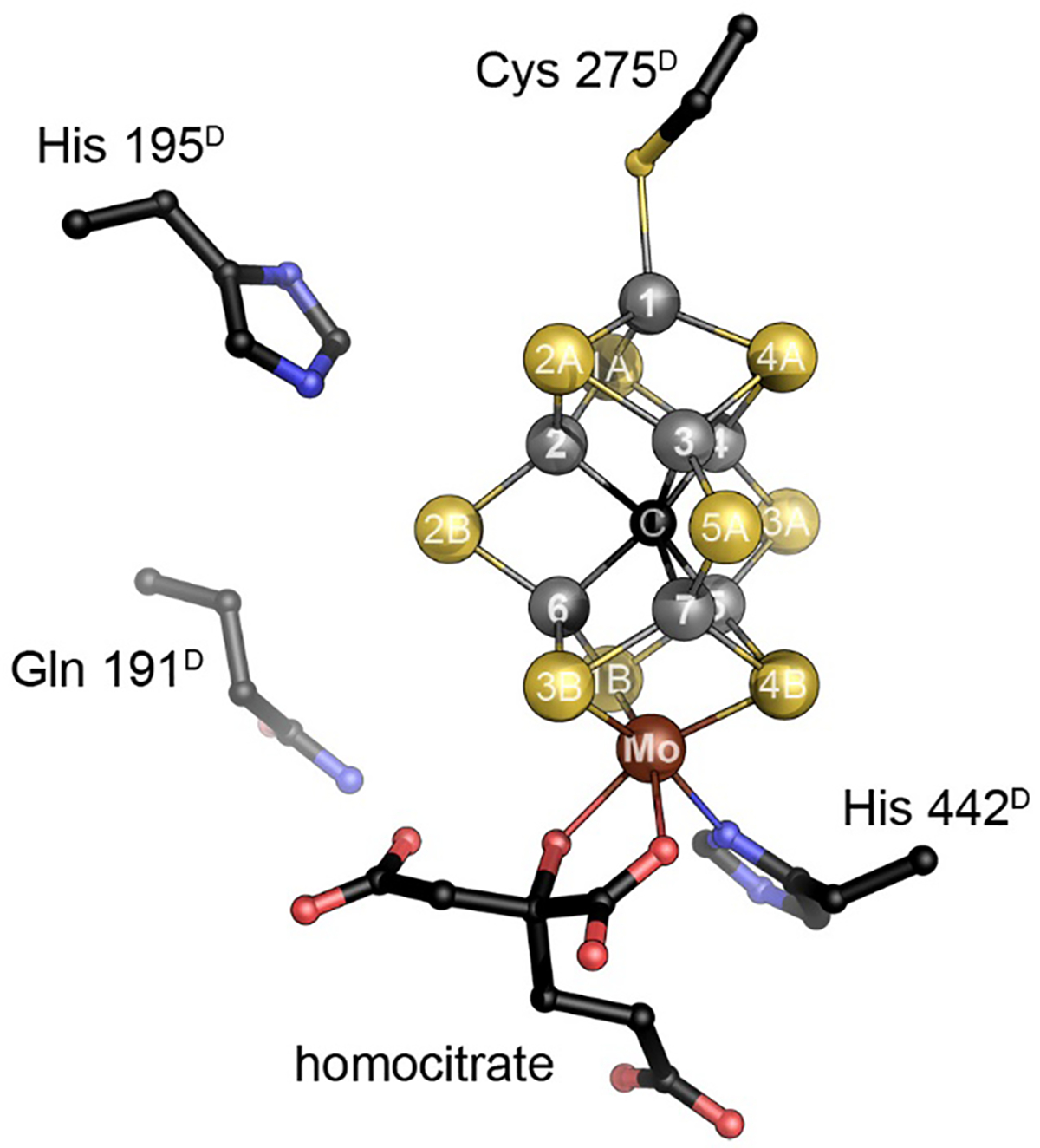
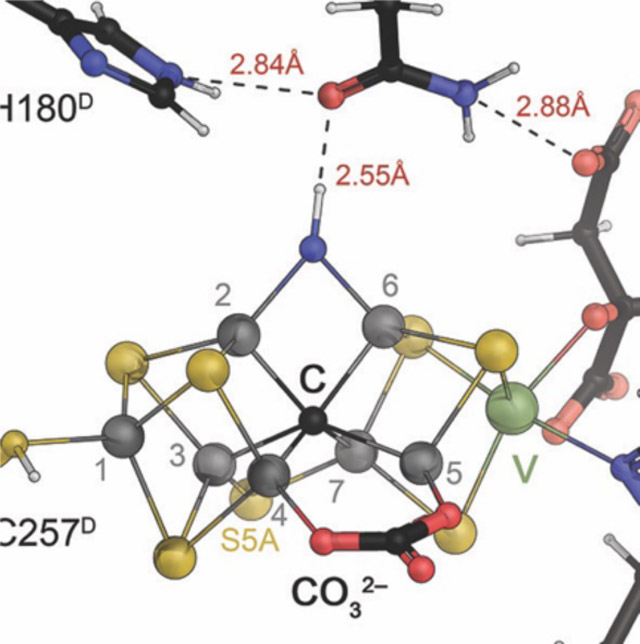
The catalytic FeMo cofactor of Mo nitrogenase. This complex iron-sulfur cluster prominently contains molybdenum at an apical position as a heterometal, bidentally coordinated by a homocitrate molecule. It obtains its highly symmetric structure through the insertion of a central carbide (formally C4−) that originates from S-adenosyl methionine. The [Mo:7Fe:9S:C]:homocitrate cluster is coordinated only by two residues of the NifD subunit, Cys 275 and His 442, and all eight metal ions are coordinatively saturated. Figure made from PDB entry 3U7Q.
More importantly, with a coordination to the protein via a cysteine residue to Fe1 and a histidine to molybdenum, all seven iron ions are tetrahedrally four-coordinate, while molybdenum is octahedrally coordinated through three sulfide ligands from the cluster, homocitrate and the aforementioned histidine. At first glance this implies that the entire cluster does not contain any free coordination site for a ligand, indicating that some kind of structural change or rearrangement should occur during catalysis. Since the first structural models for FeMo cofactor became available in 1992,20 a multitude of proposals based on spectroscopy, theory or model chemistry were put forward to address the questions regarding substrate binding and mechanism. The present review focuses on the implications of these models and on more recent findings that have led to substantial progress in understanding the chemical properties and functional features of this cofactor.
2.3. Fe Protein / Nitrogenase Reductase is a P-loop NTPase
2.3.1. Structure of Fe protein and Functional Implications
The Fe protein consists of a dimer of two highly conserved subunits that symmetrically coordinate a [4Fe:4S] cluster (Fig. 2A). As sequence information became available, regions and residues could be identified that were important for the function of the Fe protein in coupling ATP hydrolysis to electron transfer to the MoFe protein. Anticipating that the cluster ligands were plausibly cysteines, the coordinating residues were identified as Cys 97 and Cys 132 (residue numbering of the A. vinelandii protein sequence) through sequence conservation and protein chemistry,53 and subsequently confirmed by site directed mutagenesis.54 It was further evident that regions of the Fe protein exhibited significant sequence similarities to other proteins known to interact with nucleotides.55 Of particular note was the region near the N-terminus now recognized as the P-loop or Walker motif A with a characteristic sequence motif GKGGXGKS that interacts with the terminal phosphates of ATP of the non-hydrolyzable analog AMPPCP (Fig. 7A) and ADP (Fig. 7B). The Fe protein was subsequently found to be a member of a distinct branch of dimeric P-loop containing ATPases and GTPases including proteins mediating protein targeting, DNA replication and transport that are characterized by a Walker A motif with the two conserved lysines.56–58 A related family of P-loop-containing GTPases includes Ras and other G-proteins; both families are characterized by the P-loop and switch I and II regions that couple the protein conformation and the nucleotide state.
Figure 7:
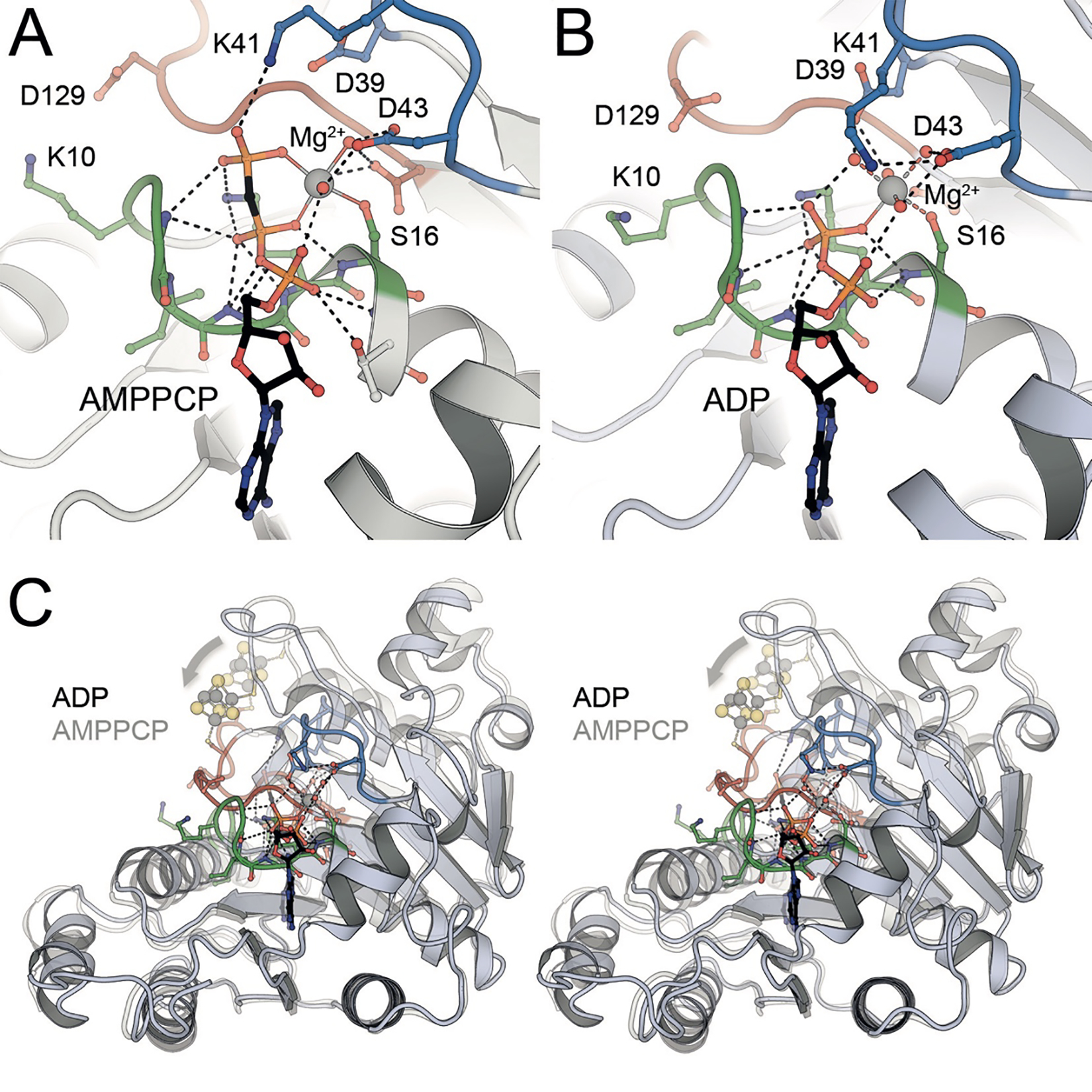
Nucleotide binding and conformational changes in the Fe protein NifH from A. vinelandii. A) The three phosphate groups of the ATP analog AMPPCP are cradled in the P-loop (green) of the protein, with a bound Mg2+ cation liganded by the β- and γ-phosphate and residues from the switch I (blue) and switch II (red) loops (PDB 4WZB). B) In the ADP-bound state after dissociation of the γ-phosphate, residue K41 from the switch I region (blue) becomes a ligand to Mg2+, leading to a major conformational change (PDB 6N4L). C) In an overlay of the NifH monomers in the AMPPCP- and ADP-bound states, the effect of the conformational change of the switch I (blue) and switch II (red) regions is seen as a displacement of the [4Fe:4S] cluster at the dimer interface (arrow).
The crystal structure of the A. vinelandii Fe protein confirmed these predictions and revealed the detailed molecular architecture.21 The Fe protein dimer exhibits approximately two-fold molecular symmetry, with the rotation axis passing through the [4Fe:4S] cluster at one end of the dimer surface. The two chemically identical subunits fold as a single α/β domain of a type frequently observed in nucleotide binding proteins with a predominantly parallel β-sheet flanked by α-helices. Two residues (Cys 97 and 132) from each subunit coordinate the [4Fe:4S] cluster (Fig. 2B). Significantly, conserved nucleotide binding motifs from each subunit are present at the dimer interface, immediately suggesting that the Fe protein conformation would be intimately coupled to the nucleotide state (Fig. 7C). These features are conserved in the structurally characterized NifH Fe proteins from C. pasteurianum 59 and Methanosarcina acetivorans,60 and the VnfH protein from A. vinelandii (see 4.3).61
The [4Fe:4S] cluster is solvent exposed at the surface that is coordinated by Cys 97. Remarkably for protein bound [4Fe:4S] clusters, the Fe protein can exist in three distinct oxidation states, that include the oxidized [4Fe:4S]2+ and dithionite-reduced [4Fe:4S]1+ forms identified in early work on nitrogenase, as well as the all-ferrous [4Fe :4S]0 form that can be obtained after incubation with Ti(III),62,63 but also with the physiological electron donor, the flavodoxin NifF.64 The electron transfer reaction between the Fe protein and MoFe protein is generally envisioned as involving the 2+ to 1+ couple, although a role for the all-ferrous form has been proposed, implying two-electron transfer and thus an ATP/e− ratio of 1:1 under certain conditions.62,64 The Fe in [4Fe:4S] clusters may be assigned as either valence-localized sites (Fe2+ or Fe3+) or as valence delocalized Fe2.5+:Fe2.5+ pairs from the isomer shifts observed in Mössbauer spectra.65,66 The Fe protein in the S=1/2 spin state of the [4Fe:4S]1+ form is composed of a delocalized Fe2.5+ pair and a pair of Fe2+. A recent analysis of the site-specific oxidation state assignments in the Fe protein using spatially refined anomalous dispersion (SpReAD, see 3.2.2) identified the delocalized Fe2.5+ pair at the solvent exposed face of the cluster, coordinated by Cys97, while the buried Fe coordinated to Cys132 are ferrous. As the form of the Fe protein competent to transfer electrons to the MoFe protein might be expected to have the reduced Fe nearer to the surface, the role of MgATP could be to promote an internal redox rearrangement such that the positions of the Fe2+ and Fe2.5+ pairs are switched. This may be promoted by the dipole moment of a long α-helix pointed directly at the cluster in both monomers (Fig. 2A).67
2.4. Nitrogenase Complexes
Complex formation between the MoFe protein and Fe protein plays a critical role in the substrate reduction mechanism as this is the species in which intermolecular electron transfer is coupled to ATP hydrolysis. Complexes between the two component nitrogenase proteins from A. vinelandii have been characterized crystallographically in the presence and absence of various nucleotides. The initial structure determination was of the ADP·AlF4−-stabilized complex with two Fe protein dimers bound to one MoFe protein tetramer (Fig. 8A).68,69 The molecular twofold axis of the Fe protein coincided with the pseudo-two fold axis relating the α- and β-subunits of the MoFe protein, such that the [4Fe:4S] cluster was positioned near and aligned with the P-cluster. Consequently, the relative positions of the metalloclusters indicated that electron transfer from the Fe protein to the FeMo cofactor proceeds through the P-cluster. Two ADP·AlF4− complexes per Fe protein dimer are bound at the interface between subunits. The nucleotide binding interactions are similar to those observed in other nucleotide switch proteins, with the phosphates bound to the P-loops and the Mg2+ coordinated by residues in the switch I and switch II regions. While the MoFe protein conformation is essentially unchanged from the uncomplexed structure, significant structural changes occur in the Fe protein that may be described as a ~13˚ rotation of each subunit towards the dimer interface in the complex. The [4Fe:4S] cluster is not a hinge point of this motion, but rather the cluster shifts ~4Å towards the MoFe protein as the switch II region undergoes extensive rearrangements between the free and complexed forms of the Fe protein.
Figure 8:

Nitrogenase complexes. Electron transfer from Fe protein to the catalytic MoFe protein requires the formation of a stoichiometric complex of both components (PDB 1N2C). A) With bound Mg-ADP·AlF4−, the [4Fe:4S] of Fe protein is positioned at a distance from P-cluster that is suitable for electron transfer. B) Different nucleotide-bound or -free states of Fe protein bind in different orientations on MoFe protein, but without nucleotide (PDB 2AFH) or with ADP (PDB 2AFI), the distance between the [4Fe:4S] cluster and P-cluster is increased with respect to the form with bound ATP analogs AMPPCP (PDB 4WZB) or ADP·AlF4−.
The transition between different conformational states of nucleotide switch proteins can be controlled by the rate of nucleotide hydrolysis serving as a timing mechanism. Ultimately, the rate of nucleotide hydrolysis reflects the ability of the protein to create the appropriate catalytic site.70 Efficient hydrolysis of ATP requires the appropriate positioning of two catalytic residues, a general base in the switch II region near the γ-phosphate and a positively charged residue near the β-phosphate. One of the fascinating aspects of these systems is that the rate of nucleotide hydrolysis can be controlled by the interaction of various external factors; for G-proteins, these include GTPase Activating Proteins (GAPs) that provide the positively charged arginine finger, and Regulators of G-protein Signaling, or RGS proteins that stabilize the conformation that is catalytically competent for nucleotide hydrolysis.71,72 In the ADP·AlF4−-stabilized nitrogenase complex, the catalytically critical residues are Asp 129 and Lys 10 of NifH, which unexpectedly interact with the nucleotide predominantly bound to the other subunit. Hence, each Fe protein subunit serves as the GAP (or more appropriately AAP) for the other subunit, while the MoFe protein stabilizes the catalytically competent form and hence is functionally equivalent to the RGS protein. The lack of significant ATP hydrolysis by the isolated Fe protein likely reflects the requirement of binding to MoFe protein to stabilize the catalytically competent conformation.
By cocrystallization using near-physiological MoFe protein and Fe protein concentrations and ionic strengths, nitrogenase complexes were prepared and structurally characterized in the nucleotide-free state, in the presence of the non-hydrolyzable ATP analog MgAMPPCP, and in the presence of the MgADP product (Fig. 8B).73 These structures showed the Fe protein occupying distinct, but mutually exclusive and overlapping docking modes showing a variation in distance between the [4Fe:4S] cluster and P-cluster. By co-crystallization in the presence of both MgADP and MgAMPPCP, a structure with two nucleotides asymmetrically bound to the Fe protein was prepared, suggesting that ATP hydrolysis and phosphate release may proceed by a stepwise mechanism.74 The conformational richness of the Fe protein underlies the coupling of protein structure and nucleotide state central to the orchestration of the sequence of electron transfer reaction. The ability in turn largely reflects the quaternary architecture of the Fe protein with the two subunits linked through the [4Fe:4S] cluster and the nucleotide binding sites at the subunit-subunit interface, so that changes in nucleotide state can be linked to intermolecular electron transfer processes.
2.5. Mo-Nitrogenases from Other Species
While the majority of the structural studies of the MoFe-protein have utilized the protein isolated from A. vinelandii, structural studies have also been reported for the orthologs from C. pasteurianum,23,75 Klebsiella pneumoniae 76,77 and G. diazotrophicus.36 Highlights of these studies include how the C. pasteurianum MoFe-protein accommodates a ~50-residue insertion in the a subunit positioned over the FeMo-cofactor, along with an ~50-residue deletion in the b subunit. This reflects the reported subclassification of nitrogenase genes into four different groups (excluding the Vnf and Anf systems), as defined in 2013, where A. vinelandii, K. pneumoniae and G. diazotrophicus belong to group I, while the 50-residue insertion is a hallmark of group II enzymes such as the one from C. pasteurianum.78 Different changes at low pH in the active sites of the A. vinelandii and C. pasteurianum MoFe-proteins have been reported 79 that presumably reflect their variations in the FeMo-cofactor environment. The structure of the NifV− variant of the K. pneumoniae MoFe protein directly established that citrate coordinates the Mo in place of homocitrate and also served as the first nitrogenase mutant to be structurally characterized. An unusual feature of the G. diazotrophicus MoFe-protein is that the residue corresponding to Ser188K in the A. vinelandii MoFe-protein is an alanine; as a compensating change in the POx state, the side chain of a non-equivalent Tyr residue, Tyr 99K, coordinates Fe8. These two changes covaried in about 20 sequences within the 2013 study and are consequently likely to represent a widespread variation.78
3. UNDERSTANDING FEMOCO
When isolated MoFe protein became available and various spectroscopic techniques were applied to generate an increasing amount of data,80 the uniqueness of the metal cofactors nitrogenase became apparent early on. In the absence of a three-dimensional structure, synthetic chemistry made many attempts to obtain small-molecule models that reproduce all or some of the observable features of the enzyme. Based on elemental analysis, a molybdenum site within (or directly coupled to) an iron-sulfur-based scaffold was generally assumed.81–83 For the P-cluster, such analyses postulated a pair of [4Fe:4S] clusters, coming very close to the actual structure.84 However, the highly unusual FeMo cofactor could not be convincingly modelled, so that understanding its structural features required the determination of a crystal structure at high resolution.
3.1. Completing the Atomic Structure of FeMoco
When the first crystal structure of Mo-nitrogenase was reported in 1992, the structure of FeMo cofactor defied expectations in many aspects. The initial structural model of A. vinelandii NifDK at a resolution of 2.7 Å consisted of two incomplete cubane-type subclusters, a [4Fe:3S] and a [Mo:3Fe:3S] unit, μ2-bridged by three non-protein ligands.19,20 Two of these were modelled as sulfides, while the third was designated ‘Y’.19 The structure also revealed that the entire cluster was only liganded by the protein through its apical atoms, coordinated to two amino acid side chains, Cys 275D to Fe1 and His 442D to Mo. The previously identified homocitrate molecule exclusively coordinated the molybdenum ion.20 In its center, the novel metal site contained a surprisingly large, internal cavity with a diameter of approximately 4 Å, surrounded by six central and coordinatively undersaturated Fe ions. While possibly quite small for accommodating N2, this unusual position presented itself as a prime candidate for a substrate binding site,85 but at the same time it raised the question how this cluster was able to retain its structural integrity. FeMo cofactor can be extracted from the protein into N-methylformamide, following denaturation under acidic conditions and used to reconstitute active nitrogenase by adding it to separately purified apoprotein.86–93 Also, the resting state that was represented in the crystal structure is unable to bind N2 prior to activation by a 3- or 4-electron reduction (see 5.4).94 At first glance, the structure could not readily explain the stability and relative chemical inertness of the cofactor. Regarding structure, subsequent computational studies tended to include metal-metal bonding interactions that helped to reproduce the atomic structure observed in the crystal.95 The central cavity of the cofactor thus did not seem to be a high-affinity substrate binding site, and the function of the cluster – for the time being – remained largely unknown.
3.1.1. Identification of a Central Ligand in FeMoco
In the following years, A. vinelandii Mo-nitrogenase continued to be the most highly resolved structure, but underwent successive improvements from the initial analysis at 2.7 Å,19,20 a further refinement to 2.2 Å,85 and eventually a model at 2.0 Å.25 For the understanding of the accessory P-cluster this progress was crucial, allowing to correct the original assignment of a [8Fe:8S] moiety to [8Fe:7S] and revealing the structural changes it undergoes upon change of redox state (see 2.2.2, Fig. 5). For FeMo cofactor, however, the structural model remained unchanged with the exception of identifying the bridging ‘Y’ as a third μ2-sulfide, S5A. A more highly resolved 1.6 Å structure for the enzyme from K. pneumoniae showed an identical FeMo cofactor.77 In the 2Fo–Fc difference electron density maps of all these analyses, the central cavity of the cluster was well-defined and quite obviously empty, both in the presence and absence of substrates.
In 2002, the change of crystallization protocols from batch to vapor diffusion then produced single crystals of unprecedented quality, allowing for a re-determination of the structure of A. vinelandii MoFe protein at a truly atomic resolution of 1.16 Å that radically changed the picture of the cofactor.24 At this resolution, the central cavity of the cofactor was found to contain a previously unseen, well-defined electron density maximum. It was modeled as a μ6-coordinated light atom that according to its intensity could represent either a carbide (C4−), nitride (N3−) or oxide (O2−). The sudden appearance of this atom raised the question whether the same species had been present, but overlooked, in earlier analyses, considering that the best available structures of MoFe proteins from A. vinelandii (2.0 Å) and K. pneumoniae (1.6 Å) were highly resolved already and depicted the very same, dithionite-reduced resting state. It was then recognized that the unusual structure of the cofactor itself led to the obfuscation of the central atom in a unique, resolution-dependent manner. The theoretical basis for this, in brief, is as follows: Each atom in FeMo cofactor individually scatters the incident X-rays in a diffraction experiment, and the diffracted photons interfere to generate an observable diffraction pattern that can be converted into a real-space electron distribution function by means of a Fourier transform. The diffraction data provides structure factors, i.e. the Fourier coefficients, and in theory, the synthesis of an infinite number of structure factors will yield a perfect representation of the electron distribution in real space. In practice, of course, any Fourier synthesis is carried out with a finite number of coefficients, leading to a discrepancy between the experimental map and the real structure. More precisely, a limited number of structure factors leads to Fourier series termination artifacts that manifest as periodic noise (known as ‘ripples’). In a typical electron density map of a protein molecule that overwhelmingly consists of light atoms from H to C, N or O, such ripples are small and tend to cancel each other out. The noticeable exception are individual heavy atoms in a structure, such as metal ions or – more severely – large metal clusters.
FeMo cofactor is the largest biological metal cluster known to date, and in addition it features an unmatched structural symmetry that is focused exactly on the central position. It is surrounded by the six iron ions Fe2-Fe7, arranged as a trigonal prism, and in addition it is equidistant to all nine sulfide ions present in the cluster. As a consequence, the minor, individual ripple effects of six iron and nine sulfides add up to a severe and resolution-dependent distortion of the electron density only at this very position. This leads to a defined profile of electron density vs. resolution that actually represents an artifact attributable to the unique cofactor geometry. Unexpectedly, the result of this additive ripple effect at the center of the cofactor was found to be a negative electron density feature. It occurs in a resolution range between 2.2 Å and 1.55 Å and is of a magnitude that is sufficient to mask the presence of a light atom at this point (Fig. 9A). The calculated limit of 1.55 Å was precisely reflected in the fact that this atom was not yet visible in the structure of K. pneumoniae MoFe protein determined to 1.6 Å resolution, but was clearly defined at 1.16 Å in A. vinelandii MoFe protein (Fig. 9D).24 It has since been established that the central atom is an integral structural component of nitrogenase cofactors, although for a long time after the discovery of its presence, its chemical nature and function remained under debate.50
Figure 9:
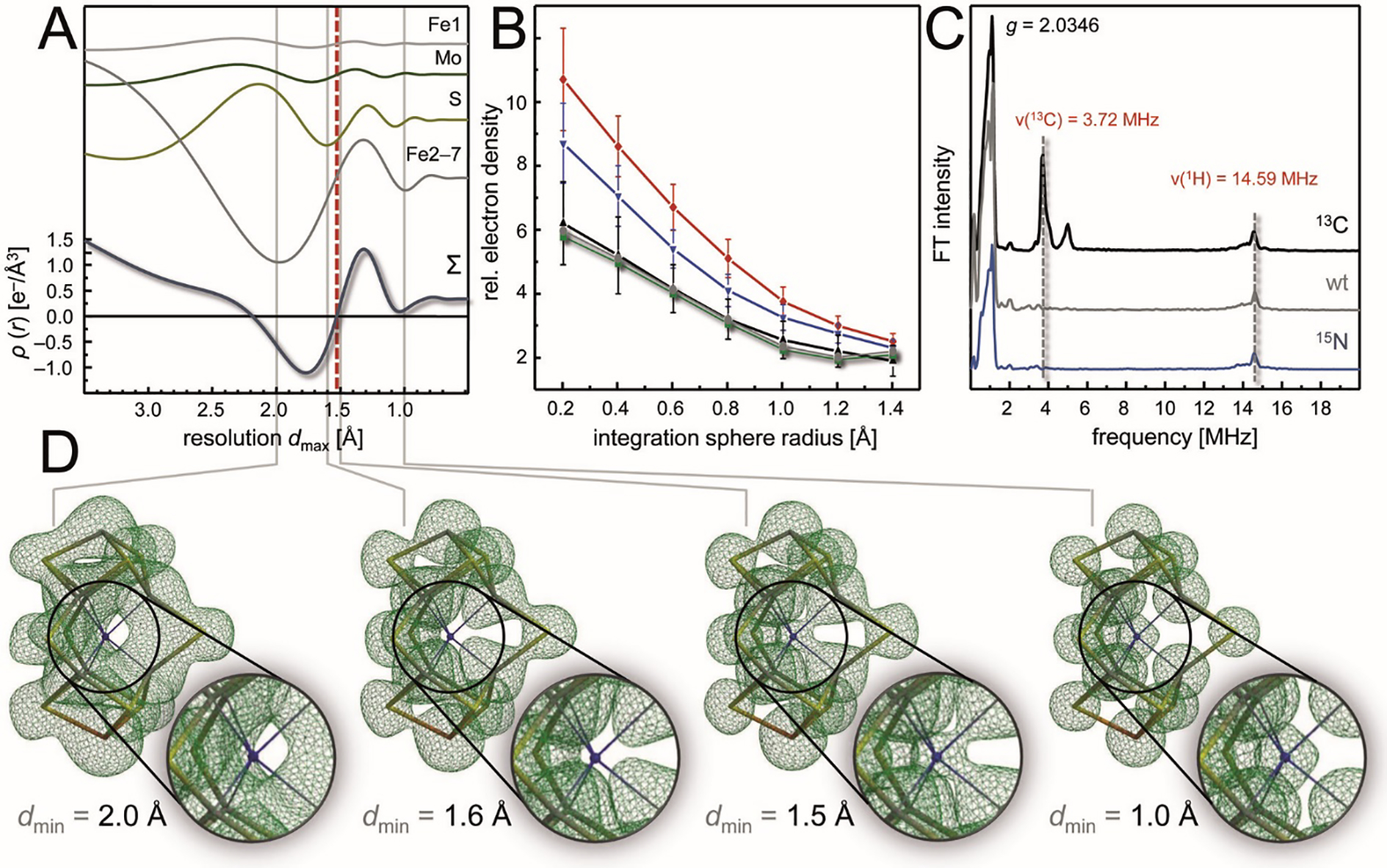
Discovery and Identification of the central carbide in FeMo cofactor. A) Calculated, resolution-dependent electron density profiles of the central position of FeMo cofactor, highlighting that the different surrounding atoms (Fe1, Mo, all nine S and the six remaining Fe) have varying effects that sum up to a profile (below) that highlights a negative electron density artifact in the resolution range between 2.2 and 1.55 Å. B) At 1.0 Å resolution, a statistical evaluation of the diffraction behavior of all carbon (black), nitrogen (blue) and oxygen (red) atoms of the structure (PDB 3U7Q), the central light atoms of the two cofactors of the MoFe protein clearly lined up with carbon. C) The result from (B) was corroborated by ESEEM spectroscopy, where only 13C, but not 15N-labelling generated a new signal at the correct Larmor frequency ν. D) Fcalc electron density maps calculated to the stated limiting resolutions underline that the maximum indicating the central carbide indeed disappears at resolutions lower than 1.55 Å, as a direct consequence of the distorting ripple effects of the surrounding atoms as dissected in (A).
After its discovery, the central ligand of FeMo cofactor was designated as a ‘light atom X’ (with X being either C, N, or O).24 In the initial characterization at 1.16 Å resolution, a careful quantification of the observed electron density feature was interpreted as N being the most likely candidate for the μ6-ligated interstitial atom, but even at this resolution, a definitive answer could not be given. Considering the physiological reactivity of the enzyme, it was obviously tempting to speculate whether an interstitial nitrogen atom might originate from the cleavage of N2, in line with the earlier concept of the central position representing a substrate binding site. However, during the following years, spectroscopic studies using ENDOR and ESEEM by Hoffman and co-workers first could not detect evidence for an exchangeable N atom,96 and subsequently pointed out that in any case the presence of nitrogen should have been recorded in this approach, so that the central atom should be of a different nature.97 In parallel, density functional calculations of the cofactor had been established through the work of Noodleman and co-workers 98,99 that subsequently investigated the possible nature of a central ligand, concluding that neither O2− or C4− were likely candidates for the central atom,100 in line with the Hoffman group that also could not conclusively assign the central ligand X.101 In the following years, a series of attempts were undertaken to clarify this questions, and nearly a decade later it was the combination of a broad range of methodologies that laid the issue to rest. DeBeer and co-workers presented a study using Fe Kβ X-ray emission spectroscopy, where they showed that an experimental valence-to-core X-ray emission difference spectrum of intact MoFe protein and a cofactor-free variant (ΔnifB MoFe) was in far better agreement with the calculated signature of an interstitial C than with those of an N or an O.102 Similarly, the same authors also found that this atom is already present in the NifEN-bound precursor of the cofactor.103 This work was complemented with a new crystallographic analysis of MoFe protein at 1.0 Å resolution (Fig. 9B) and with ESEEM studies of MoFe protein labelled with either 15N or 13C (Fig. 9C). All these techniques provided consistent evidence for an interstitial C4−.104 Important information regarding the origin of the central carbide then came through work of Ribbe and Hu, who confirmed S-adenosylmethionine as the carbon donor during cofactor biogenesis, mediated by the radical/SAM enzyme NifB.105 However, even with the complete atomic structure of the cofactor finally established, little was revealed about the mechanism and location of substrate binding and activation. As a carbon, the central atom clearly was not an intermediate of N2 reduction, and a series of theoretical studies by several groups that ensued from the new data also came to widely diverging conclusions regarding the its role for the reactivity of the cluster.106–109
3.1.2. Implications for Structure and Function
The discovery of the central carbide in nitrogenase FeMo cofactor provided an explanation for the relative stability and chemical inertness of the cluster in its resting (as isolated) state. Without the requirement to explain the presence of a large, open central cavity, it was easier to conceive that the entire cluster could be extracted from the protein after acid denaturation (pH 2) into N-methylformamide and retain sufficient structural integrity to be reconstituted into apo-nitrogenase, yielding active protein.110 At the same time, the completed structure now left every single metal ion in the cluster in a coordinatively saturated state, indicating that a conformational rearrangement or even a ligand exchange was required prior to the binding of N2. Multiple suggestions were put forward in the following years, covering all bases from a substantial enhancement of cluster stability by the central ligand to a flexibilization that would allow for major rearrangements and an opening of the core to bind substrate.111–115 Unfortunately, the different theoretical approaches towards FeMo cofactor did not converge to a generally accepted model, and if anything, the confusion in the field increased. Not in the least this was due to the problem that the theoretical description, in particular of the electronic structure of the cofactor, was itself far from settled. In the absence of clarifying experimental data, several laboratories therefore set out to accumulate more information on the enzyme and its metal clusters, although for the time being only the resting state was accessible.
3.2. Electronic Structure of FeMo Cofactor
Early on, spectroscopic studies indicated that FeMo cofactor is a highly reduced site. EPR spectroscopy showed that in its resting state, E0, the cluster is an S = 3/2 system. Iron ions most likely are ferric (Fe3+) or ferrous (Fe2+), containing five or six d-electrons, respectively. As is the case for other iron-sulfur clusters, in a tetrahedral environment with weak-field sulfide ligands all seven Fe ions will be in high-spin configuration, but may couple ferro- or antiferromagnetically. This leads to a large number of combinatorial possibilities for the electron arrangement in the cluster, as well as for the distribution of the additional electrons for ferrous sites. Finally, the oxidation state of molybdenum in biological systems in most cases is Mo4+ or Mo6+, adding up to a total cluster charge that is compensated by the 9 formal sulfides (S2−, total charge contribution −18) and the carbide (C4−). Independent of the actual charge distribution in detail, all electrons in question have to add up to the observed S = 3/2 system, while leaving the cofactor with a total charge that is chemically reasonable.
3.2.1. Broken-Symmetry Approaches toward Coupling Interactions in FeMo Cofactor
Much spectroscopic data probing the electronic properties of FeMo cofactor were compiled over decades, but a comprehensive description of the electronic structure of the site was crucially dependent on the development of appropriate theoretical methods. Noodleman and co-workers made a highly significant contribution by introducing spin-polarized broken-symmetry density functional theory (BS-DFT) to interpret the possible combinations of relatively localized electronic configurations. Among a series of chemically reasonable solutions, they favored a coupling scheme termed BS7 that has been widely used since and is currently the basis for most proposed electronic models.99 In the BS7 scheme, four Fe sites in spin-up configuration couple to three Fe sites in spin-down, maximizing the amount of antiferromagnetic coupling of each site with its neighbors. The molybdenum ion was investigated by 95Mo ENDOR. It was found to be highly reduced and was consequently assigned as a Mo4+, in line with other Mo sites in biological systems.116 Within these boundaries, the question how many ferrous sites then are required to result in a total spin of 3/2 is a matter of combinatorics: Based on 57Fe Mössbauer spectroscopy – but prior to the discovery of the central carbide – Burgess and Münck proposed a configuration of 4Fe2+ and 3Fe3+,117 but combinations of 2Fe2+:5Fe3+:Mo4+ and 6Fe2+:1Fe3+:Mo4+ would yield the correct spin state as well. The resulting ambiguity regarding the charge assignments within the cofactor prevented any model from gaining general acceptance, and furthermore contributed to the divergence in the results of theoretical studies due to the lack of a guiding electronic description.
3.2.2. SpReAD Assignment of Individual Oxidation States to the Cluster Metals
The element-specific absorption of X-rays by core electrons of any given element provides an elegant approach for the assignment of charges in metal clusters. In addition, the element iron is also amenable to Mössbauer spectroscopy, where individual iron sites manifest characteristic quadrupole splitting and isomer shifts that reveal details about their oxidation state and chemical environment. Of the two types of metal ions present in FeMo cofactor, molybdenum was the more straightforward to address, as it is present in only a single copy. In biology, the element molybdenum is most commonly used to support two-electron transfer reactions or oxygen atom transfers, as in the reactions of formate dehydrogenase or nitrate reductase.118,119 The element is found in two stable redox states, Mo4+ (4d2) and Mo6+ (4d0), both of which are diamagnetic. The original assignment of the Mo site in FeMo cofactor as Mo4+ thus was canonical and in line with expectations. It would also determine the arithmetic for the charge distribution of the iron sites, as with two unpaired electrons on Mo, the observed S = 3/2 system of the resting state of the enzyme then implied either 2, 4 or 6 Fe2+ sites (3d6). The complication in charge assignment to the Fe sites is in their number: Nitrogenase MoFe protein contains 7 iron ions in the cofactor, and another 8 in P-cluster, and both X-ray absorption and Mössbauer spectroscopies invariably only detect the cumulated spectroscopic signature of all these sites. Münck and co-workers had published an extensive Mössbauer analysis of nitrogenase, but were left with an ambiguity in their assignment,117 and X-ray absorption spectra are not sufficiently feature-rich to deconvolute 15 individual sites. For molybdenum, however, the technique was ideally suited. DeBeer and co-workers investigated both the enzyme and several model compounds of known electronic state by high energy resolution fluorescence-detected (HERFD) X-ray absorption spectroscopy (XAS) at the K-edge of Mo, and found that the pre-edge and also the rising edge features of the enzyme were not consistent with an Mo4+ assignment. An analogous finding was made for an important model, a (Et4N)[(Tp)MoFe3S4Cl3] complex originally synthesized by Holm.120 In both cases the lower energy of the edge position indicated a more reduced species, and in the case of the smaller model compound, DFT calculations supported an assignment as Mo3+ (4d3). Interestingly, the coupling of the spin system of Mo with the partially delocalized Fe sites in the complex led to a highly unusual, non-Hund ααβ ground state.121
A Mo3+ site in a biological system is unprecedented, which further highlighted the requirement to understand the charge distribution among the Fe sites within the cluster. The solution here was to exploit the effect that X-rays are diffracted by the electron shell of an atom. When in close proximity to an absorption edge, X-rays are absorbed by the core electrons of an atom (the basis for XAS), which will have a profound effect on the diffraction properties of this particular element. The interaction of an ejected core electron with the remaining particles in the shell of the atom breaks the internal centrosymmetry of the diffraction process that is described by Friedel’s law.122 It states that the intensity of a reciprocal lattice point at a position (h, k, l) is identical to one at position (–h, –k, –l), albeit with opposite phase angles. In the proximity of an absorption edge this is no longer true, giving rise to an anomalous difference Dano in intensity. This is directly proportional to the absorption coefficient for X-rays, so that it provides an information equivalent to an X-ray absorption spectrum. The advantage of reading this information out of a diffraction process, however, is that a three-dimensional reciprocal lattice is recorded that preserves spatial resolution in addition to the absorbance measurement.123 The contribution of each individual absorbing site to the magnitude of Dano can be determined during the structure refinement. If a series of data sets is collected along the absorption edge, the refined values of Dano thus approximate the individual absorption edges of single atoms in space. This spatially refined anomalous dispersion (SpReAD) was first applied to the [2Fe:2S] ferredoxin Fd4 from A. aeolicus that contains localized, antiferromagnetically coupled Fe(II) and Fe(III) sites.123 In the SpReAD analysis, the individual edge positions for Fe(III) were higher in energy by approximately 2 eV with respect to those for Fe(II), in line with expectations from XAS and theory.
This simple test case contained a dimer of the single-cluster protein Fd4 in the asymmetric unit, with a total of four individual Fe sites to be refined. Applying the SpReAD method to nitrogenase involved at least a complete NifD2K2 heterotetramer with two copies each of the P-cluster and FeMo cofactor, amounting to 30 Fe sites. The SpReAD analysis of the resting state of FeMo cofactor showed that Fe1, Fe3 and Fe7 were more reduced (lower energy edges), while Fe2, Fe4, Fe5 and Fe6 were more oxidized (Fig. 10A). In addition, the presence of P-cluster in the all-ferrous PN state provided a precise internal reference for Fe2+, aligning fully with the profiles of Fe1, Fe3 and Fe7. If, accordingly, the remaining four Fe ions are assumed to be ferric the system indeed added up to the observed total spin of S = 3/2,124 as FeMo cofactor is a S = 3/2 state as isolated, and the BS7 coupling scheme established by Noodleman and co-workers 98 in conjunction with a spin-coupled Mo(III) site in a non-Hund ground state 121 implies a formal distribution of 4 Fe(III) and 3 Fe(II). The data was in agreement with the valence electrons of the system being largely localized. This could be rationalized through the maximized antiferromagnetic coupling implied in the BS7 scheme, although model complexes show that in canonical iron-sulfur clusters the delocalization of spins across ferromagnetically coupled metal centers should be the rule.121 Based on the re-assignment of Mo as Mo3+ and the SpReAD results with respect to the Fe ion, Björnsson and DeBeer re-evaluated Münck’s Mössbauer and largely confirmed the new interpretation. In the accompanying DFT calculations, although the respective ferromagnetic coupling of Fe3/Fe4 and Fe5/Fe7 resulted in spin delocalization that was not reproduced in the SpReAD data.125 Importantly, in both approaches Fe2 and Fe6 were identified as the most oxidized sites in the E0 state of FeMo cofactor (Fig. 10B).126
Figure 10:

Analysis of the E0 state of FeMo cofactor by spatially resolved anomalous dispersion (SpReAD). A) In an experiment involving 17 data sets along the K-edge of iron, SpReAD refinement of the 7 individual iron sites of FeMo cofactor revealed the existence of a set of more oxidized Fe sites (Fe2, 4, 5, 6, red average) and a more reduced set (Fe1, 3, 7, green average). The edge position for the latter was very well in line with the all-ferrous P-cluster (blue average). As complementary information, the cofactor structures below show anomalous difference electron density maps for the data sets recorded at the indicated positions along the edge. B) Structure of FeMo cofactor with the more reduced Fe sites in green and the more oxidized ones in red. The axes indicate the principal components of the oriented magnetic g tensor (see Fig. 11A).
3.2.3. Spatial Analysis of the Magnetic Properties of FeMo cofactor
In its as isolated state, Mo nitrogenase shows a surprisingly simple spectral signature in continuous-wave electron paramagnetic resonance spectroscopy (EPR). With P-cluster in the PN state, FeMo cofactor exhibits an S = 3/2 signal with a rhombic g tensor with apparent principal components gx= 2.01, gy= 3.65 and gz = 4.31. This feature is not straightforward to reconcile with the structure of the cofactor and its D3 pseudosymmetry. An alignment of the magnetic tensor and the crystal structure was made by X-band EPR spectroscopy of single crystals, where only the respective cross-section of the g tensor ellipsoid with the magnetic field of the spectrometer absorbs microwaves, so that the rotation of the crystal in the magnet allows for scanning the tensor orientation that can subsequently be correlated to the crystal orientation that is obtained by X-ray diffraction on the same crystal.127 As expected, the longest apparent principal component, gz, oriented along the threefold cluster axis. Of the other principal axes, gx = 2.01 was in the plane formed by Fe1, Fe2, Fe6 and Mo in FeMo cofactor, providing another indication that the three-fold symmetry of the cluster is not reflected in its electronic properties and that Fe2 and Fe6, together with the apical metals of this moiety, play a prominent role (Fig. 10B, 11A).
Figure 11:

The electronic structure of the resting state E0 of FeMo cofactor. A) Orientation of the S = 3/2 g tensor of resting-state MoFe protein with respect to the cluster structure from single-crystal EPR analysis. The longest principal component, gz = 4.31, orients along the pseudo-threefold axis of the cofactor. Notably, the gx = 2.01 component is directed towards the Fe2-Fe6 edge that later also proved to be the site of ligand binding. B) Spin-localized model for the S = 3/2 E0 state of MoFe protein. The maximized antiferromagnetic coupling of this model leads to largely localized charges, leaving Fe2 and Fe6 as the most oxidized metal sites in the cluster.
3.2.4. An Electronic Structure for the Resting State
Through the combination of several data sources regarding the structural, spectroscopic and thus electronic features of FeMo cofactor as discussed in the previous sections, a comprehensive model for the resting state E0 of the enzyme has emerged and gained increasing acceptance. Interestingly, the three-fold symmetry that gives the cofactor its unique appearance is not reflected in its internal electronic properties. Instead, two adjacent Fe positions, Fe2 and Fe6, are distinguished as the most highly oxidized sites in the resting state. A possible reason for finding these sites so clearly localized is in the complex electrostatic environment provided by the protein matrix. It embeds the cofactor in its entirety, limiting access of solvent molecules to defined pathways and providing positively charged amino acid side chains that stabilize more reduced Fe sites opposing the Fe2-Fe6 edge. This distribution furthermore depends on the complex antiferromagnetic coupling scheme revealed by the broken-symmetry model BS7.98 In order to maintain the asymmetric electron distribution in the cofactor, this mechanism effectively counteracts charge delocalization within the cluster. While the analysis of FeMo cofactor thus had not revealed an obvious binding site for substrate molecules, it provided a convincing rationale for an internal asymmetry of the cluster that already hinted at very specific binding events rather than a catalysis based on the extensive metal-sulfur surface of FeMo cofactor. However, these considerations did not yet address the pressing issue that in a cofactor structure with a central carbide all individual metal seems seem to be entirely coordinatively saturated, so that a binding position for any ligand – inhibitor or substrate – is not readily apparent.
3.3. Ligand Binding to FeMoco
Even before a three-dimensional structure of the enzyme had become available in 1992, inorganic model complexes had provided multiple suggestions regarding the nature of the metal clusters in nitrogenase. Holm and co-workers produced large iron-sulfur systems with obvious similarities to the metal sites found in the first crystal structures,19,20,128, and Tatsumi and co-workers assembled a topological equivalent of P-cluster with apical, symmetric Mo sites in a single step from [2Fe:2S] precursors.129 Early on, the six central iron atoms of the cluster were considered as the most probable binding sites for a ligand.130 These positions show a complete tetrahedral ligand environment, but the Fe ion is closer to the plane formed by its three sulfide ligands than to the geometric center of the tetrahedron, so that an external ligand could be envisioned to distort the site towards a pentacoordinate trigonal bipyramid. This idea was the basis for the successful series of model complexes by Peters and co-workers that eventually yielded compounds competent in the catalytic reduction of N2 at a single, reduced iron ion. The Peters compounds used three coplanar phosphine ligands that in later studies were replaced with thiolates and featured axial Si, B or C ligands, resulting in some of the most efficient small-molecule catalysts for activation of N2 under mild conditions.131 They require a highly reduced Fe site as well as low-potential electrons, and while such conditions are not readily compatible with an aqueous environment they emphasize the primary boundary conditions for N2 activation that any catalytic system must address (vide infra). Interestingly, the discovery of a Mo3+ site in FeMo cofactor raised new considerations regarding a catalytic role of the heterometal that had been proposed early on to be the actual catalytic site of the enzyme by Coucouvanis and others.132 Indeed, molybdenum was the metal used in the first actual small-molecule catalysts for dinitrogen reduction by Schrock133 and Nishibayashi.134 These compounds exploited the wide range of oxidation states that the heterometal is able to attain. A reduced Mo3+ species is the site for N2 binding, while the stabilization of a Mo6+ nitride is required after N-N bond cleavage.135,136 At first glance, both types of complexes feature a free coordination site on the metal cation that is obviously not present in the octahedrally coordinated Mo3+ ion of the FeMo cofactor. However, considering that two of the six coordination positions are occupied by the organic homocitrate ligand that is located in a solvent-filled cavity within the protein matrix, it cannot be excluded that a structural rearrangement of the ligand exposes a coordination site on Mo. From a chemist’s perspective, both Mo and Fe thus are suitable metals for the task, but the coordination of a ligand invariably requires an accessible coordination site, so that a distortion of the cluster from its resting state conformation, or even the release or exchange of a ligand are necessary before N2 can bind. For long, none of the available crystal structures of nitrogenases provided any indication as to what kind of change might occur and what actual sites and modes of binding ligands are possible for this cofactor.
3.3.1. Ligand Binding: CO and Selenide
In enzymology, a frequently used strategy to evaluate ligand binding modes in enzymes that rapidly turn over their actual substrates is to generate stable complexes with inhibitors instead and investigate the possible mechanistic relevance of their observable binding mode. As nitrogenase is a reductase of extraordinary strength, only few molecules are known to inhibit the enzyme, the most relevant being carbon monoxide (CO) that blocks the reduction of any other known substrate, with the notable exception of protons.137 CO binds to a range of metal cations as a terminal or μ-bridging strong-field ligand, and in nitrogenase the inhibition of N2 reduction by CO was found to be non-competitive.138 Mechanistically, this type of inhibition implies that the inhibitor binds to a site where substrate cannot bind. This may be a physically different site, making the inhibition allosteric, or it might be a different state, as is the case here. As a critical part of the catalytic mechanism, the binding of N2 to nitrogenase requires reduction of the enzyme by four electrons, while CO binds after a two-electron reduction of the cofactor.139 Substrate and inhibitor therefore do not compete for binding, even if their binding sites were identical. In addition, EPR and infrared spectroscopy have differentiated two separate CO binding events that are commonly designated the low-CO and high-CO states and are assumed to have one or two molecules of CO bound to the cofactor, respectively.138,140–146 Part of the challenge in interpreting the inhibitory mechanism of CO is that while the Ki is ~1–10·10−4 atm,137 the concentrations used in biophysical studies are typically 1000-fold higher (0.1–1 atm). Furthermore, the time scale for development of the characteristic EPR signals following CO addition is several orders of magnitude longer than for CO inhibition of dinitrogen reduction.141 Obtaining such structures is severely complicated by the inability of Mo-nitrogenase to bind CO in its resting state. The relevant two-electron-reduced state E2 is only reached under turnover conditions, and the dynamic formation of the electron transfer complex with Fe protein makes this virtually impossible to achieve in a crystal.
The solution to this problem was found by inhibiting the enzyme under turnover and subsequently isolating MoFe protein, growing crystals and measuring diffraction data, leading to a 1.5 Å resolution crystal structure of the MoFe protein of nitrogenase inhibited by a single molecule of CO, i.e. the low-CO state of the enzyme.147 In this structure, the CO ligand could be unambiguously assigned, but its binding position was entirely unexpected. Replacing the μ2-bridging sulfide S2B at Fe2 and Fe6 of the cofactor, the CO ligand caused a structural change of the cluster and formed a bridging carbonyl that is chemically reasonable, but would have been impossible to predict correctly in the absence of three-dimensional structural information (Fig. 12A). When CO was removed under continued turnover, a resulting crystal structure showed that sulfide S2B had returned to its position, with the resulting structure being indistinguishable from the known resting state.147 In spite of an anomalous electron density maximum detected approximately 20 Å away from the cluster, the fate of S2B in the CO complex remained unknown, but the binding position of CO at the cluster inspired further hypotheses on mechanistic relevance. Importantly, Holland and co-workers reported a small-molecule Fe:S-based compound that bound N2 under dissociation of an Fe-S bond, emphasizing that a connection between Fe-S bond cleavage and substrate binding may well be chemically reasonable.148
Figure 12:

Ligand complexes of FeMo cofactor. A) CO complex of A. vinelandii MoFe protein obtained under turnover conditions (PDB 4TKV). The ligand replaced bridging sulfide S2B, but induced no other discernible changes at the active site. B) Under turnover with SeCN−, selenide also replaced sulfide S2B (PDB 5BVG). Under continuous turnover, replacement of the other bridging sulfides was also observed, albeit to a lesser degree.
The observed replacement of a μ2-bridging sulfide with CO and the reinstatement of the original ligand upon removal of the inhibitor then prompted experiments aimed at replacing atom S2B with a spectroscopically and structurally more easily detectable selenide anion, Se2−. While eventually successful, this could not be achieved by simple addition of selenide under turnover conditions, but rather required the alternative substrate SeCN−, whose reduction yields the products methane and ammonium, leaving the enzyme in a formal resting state, but with the exchangeable ligand as a Se2B bridging Fe2 and Fe6 (Fig. 12B).149 Under turnover in the presence of acetylene, the selenide initially bound at the 2B position was additionally found to migrate through the remaining μ2-sulfides S3A and S5A. Remarkably, although the belt sulfur positions are separated by 5.7 Å in the resting state of the cofactor, these sites can all interchange during acetylene reduction. After several thousand catalytic turnovers, the total Se was lost and apparently replaced by S, leading to a recovery of the original FeMo cofactor. Neither the source of this S, nor the pathway(s) by which Se exits the MoFe protein, were identified. The observation that Se2B can migrate to S5A and S3A and is ultimately chased from the cofactor provided clear evidence that structural flexibility is an intrinsic feature of the nitrogenase cofactor. The coordination of the cofactor through only two protein ligands may facilitate the underlying metallocluster rearrangements.
3.3.2. CO as a Substrate for Nitrogenases
The strong-field ligand CO forms stable complexes with a range of synthetic compounds and metalloenzyme active sites. Its binding to FeMo cofactor highlights the flexibility of the complex cluster, but at second glance the fact that an enzyme able to activate and reduce N2 is inhibited by CO is not immediately obvious. Accordingly, Ribbe, Hu and co-workers found that the alternative vanadium nitrogenase from A. vinelandii did indeed reduce CO, albeit at lower rates.150 The product was not primarily methane, but instead the reduction overwhelmingly involved a C-C bond formation leading to 93% of the carbon to be found as ethylene, C2H4.151 In order to achieve this, the enzyme most likely has to bind two molecules of CO, conveying potential mechanistic significance to the high-CO state mentioned previously.152 Methane was found at yields below 1% in these reactions, even less than the further products ethane (3%) and propane (2.5%). The formation of hydrocarbons from CO is obviously reminiscent of the well-established Fischer-Tropsch process, but there the main product is the comparatively low-value methane that requires further, energy-intensive conversion to more chemically versatile products, while the enzyme’s standard mode of operation already includes the formation of carbon chains. It is not surprising that this ability of vanadium nitrogenase has raised immediate interest regarding the potential for biofuel production from CO, and subsequent studies showed that this reactivity can be driven in an ATP-independent fashion, in contrast to N2 fixation. Using isolated cofactor and chemical reductants, the product chain length could be extended up to C7, although ethylene mostly remained the primary product.153 Using highly sensitive analytical methods, even Mo-nitrogenase was shown to reduce CO, but the rates observed here were more than 700-fold lower than for the V-dependent enzyme.154 CO is therefore correctly described as an inhibitor of substrate reduction by nitrogenase (with the important exception of proton reduction to form H2). The fact that MoFe and VFe proteins exhibit such striking differences in their ability to reduce this alternative substrate is an important reminder that the two variants of nitrogenase differ in key aspects of their functionality.
4. VANADIUM NITROGENASE
4.1. Isolation of native VFe Protein
The observation that A. vinelandii retains the ability to fix N2 in the absence of available molybdate goes back to 1971,155 but was initially suspected to be based on traces of molybdenum in the growth media or the used vanadium salts.156 Bishop and co-workers then presented evidence for a second enzymatic system for nitrogen fixation that was based on vanadium in 1980,157 and its presence was subsequently confirmed through the construction of a ΔnifDK strain that was still able to fix N2 in the presence of vanadium, although it lacked the structural genes for molybdenum nitrogenase.158,159 Such deletion strains allowed for the isolation of the component proteins of vanadium nitrogenase from A. vinelandii and the closely related Azotobacter chroococcum.160,161 More recently, Hu and Ribbe reported the A. vinelandii variant YM68A that contained a hexahistidine-tagged VnfK in a ΔnifDK background, allowing for straightforward isolation of the protein,162 but still retaining the non-natural situation of a nif-deletion strain. The early reports on vanadium nitrogenase indicated that the removal of molybdenum was non-trivial to start with, as A. vinelandii contains a Mo-storage protein to counteract phase of metal depletion. In order to counteract this effect in the A. vinelandii type strain,163 molybdate was omitted from all growth and selection media for at least five complete cycles of cell growth and individualization on agar plates. Eventually, diazotrophically growing were obtained cells that almost exclusively produced the vanadium-based nitrogen fixation system.164 Both the catalytic VFe protein and its cognate Fe protein VnfH were isolated by anion exchange and size exclusion chromatography, forming the basis for the determination of the three-dimensional structures described in the following sections.
4.2. Structure of VFe Protein
Based on the optimized isolation protocols for unmodified VFe protein from its native A. vinelandii, well-diffracting single crystals of the enzyme were obtained that allowed for the determination of a three-dimensional structure to a resolution of 1.35 Å. As expected, the protein was based on a heterotetrameric D2K2 assembly that underlined its close relationship with MoFe protein (Fig. 13A, B).38 As in the latter, vanadium dinitrogenase contained an [8Fe:7S] P-cluster at each DK-interface, and contrary to reports suggesting a different architecture for this cluster, the observed dithionite-reduced state had the exact same arrangement of cubane-type subclusters sharing a μ6-sulfide as found in MoFe protein (Fig. 13C). The active site FeV cofactor was also cradled in the three Rossmann-subdomains of VnfD, as is the case with FeMo cofactor in MoFe protein. With this high overall similarity, the major structural difference between the two nitrogenases was an N-terminal extension of NifK that wraps around the surface of the adjacent NifD in three extended α-helices (Fig. 13A). In VnfK this extension is missing, and is replaced by a β-strand that adds to the central β-sheet of the third Rossmann-domain of VnfD. This visible difference to the outward appearance of both proteins actually obfuscates the high degree of conservation of the relevant functional features of both enzymes. The K-subunits of the VFe protein are bridged by two symmetrically bridging metal cations. While in the MoFe protein the analogous sites were modelled as Ca2+,19,24 but later shown to be at least partly replaced by a 16th Fe,165 the 1.35 Å resolution structure of the VFe protein strongly indicated that in this case the ion is an octahedral Mg2+.38 In both enzymes these sites are presumed to play a structural role in stabilizing the heterotetramer, but beyond this, a function related to the observed half-side reactivity of the enzyme cannot be ruled out, 166,167 implying some degree of conformational communication between the two DK-heterodimers.
Beyond these strong similarities, VFe protein differs from its Mo-containing counterpart through the presence of an additional subunit, encoded by the vnfG gene that is located between the structural genes vnfD and vnfK in the genome of A. vinelandii.168 Before the availability of a crystal structure, only little was known about structure, role, and even stoichiometry of this additional subunit.38 The 113 aa VnfG forms a globular four α-helix-bundle (Fig. 13D) that occurs in two copies exclusively contacting with VnfD on opposing sides of the resulting 240 kDa VnfD2K2G2 heterohexamer (Fig. 13B). VnfG contains no cofactor and is located sufficiently remote from the putative docking site for the Fe protein VnfH to not interfere in the formation of an electron transfer complex during substrate reduction at VFe protein.38
4.2.1. Properties of FeV cofactor
As the cofactors of all three known variants of the nitrogenase enzyme derive from a common precursor, the ‘NifB cofactor’ or ‘L-cluster’ produced by the radical/SAM enzyme NifB, the three-dimensional structure of VFe protein fulfilled expectations in that FeV cofactor indeed retained the overall bridged double-cubane structure of its Mo-containing analog, of course with the apical heterometal molybdenum replaced by vanadium (Fig. 14). The biogenesis pathways of the cofactors branch after the action of NifB, and the insertion of the heterometal then occurs on the EN complex, a scaffold protein that is a structural homolog of the enzymatic component itself,169 and that in the nif cluster comprises a NifEN machinery, while in the vnf cluster a separate VnfEN system is used.168 Consequently, FeV cofactor is a heterometal cluster with a central, interstitial carbide, as predicted by XAS,170 that contains a single vanadium ion taking the position of Mo in the FeMo cofactor. V is coordinated by a single histidine residue from the VnfD subunit and the organic ligand R-homocitrate, the product of the enzyme NifV. In spite of their different sizes, the Mo ion in FeMo cofactor and the vanadium ion in the cluster of VFe protein show almost identical bond distances to their respective ligands, and both clusters are structurally very similar.38 One difference between the two moieties was, however, entirely unexpected. Rather than sharing the three μ2-sulfides of FeMo cofactor, S3A, S5A and S2B that form the characteristic edges of the cluster,24 FeV cofactor retained only S2B and S5A, while the S3A sulfide at Fe4 and Fe5 was replaced by a 1,3-bridging tetraatomic ligand (Fig. 14A).38 According to the 1.35 Å resolution electron density map this ligand was assigned as carbonate, CO32−, with identical bond distances and a hydrogen-bonding pattern that excluded protonation of its oxygen atoms. At this stage the presence of a nitrate anion, NO3− in place of CO32− could not be fully excluded based on the crystallographic analysis alone, but strong points speaking for carbonate were its availability as the hydrated product of aerobic glucose oxidation that drives diazotrophy in A. vinelandii, as well as the known immediate ‘switch-off’ of nitrogenase activity in the presence of nitrate in the cell, a signal that sufficient bioavailable nitrogen is present. More recently, computational studies have also supported the assignment as carbonate.171
Figure 14:
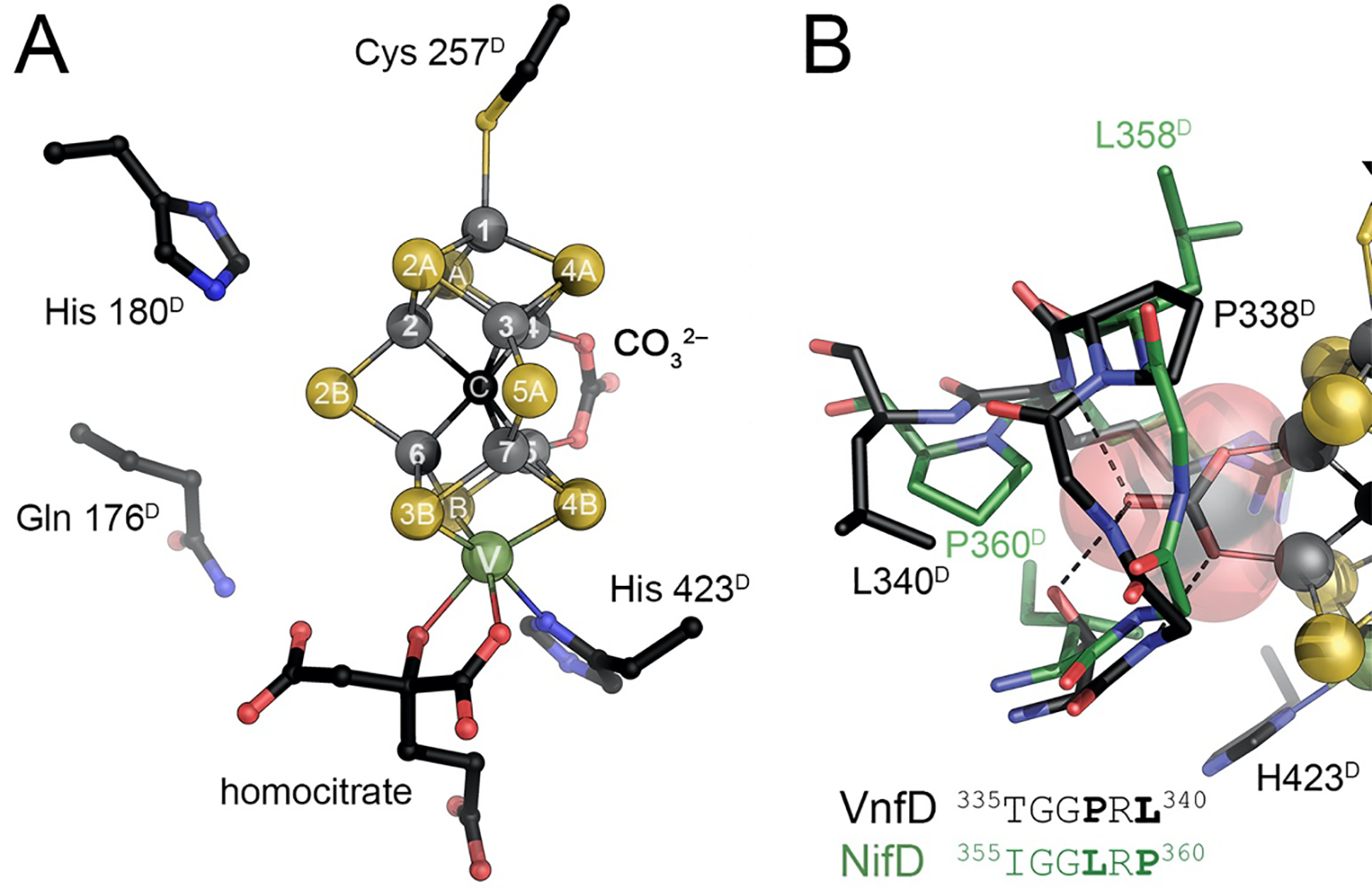
The active site FeV cofactor of vanadium nitrogenase. A) FeV cofactor is a [V:7Fe:8S:C:CO32−]:homocitrate cluster with high overall similarity to FeMo cofactor (Fig. 5). A V3+ cation replaces Mo3+, but retains its binding geometry, as well as the organic R-homocitrate ligand (PDB 5N6Y). In addition, FeV cofactor has one μ2-bridging sulfide, S3A, replaced with a μ−1,3-bridging carbonate. B) The carbonate ligand is tightly bound within a loop region of VnfD. In MoFe protein (PDB 3U7Q), the corresponding loop has a single leucine-proline swap due to which the carbonate ligand cannot be accommodated.
4.2.2. The Role of a Carbonate Ligand
The larger ligand and its binding mode increase the Fe4 - Fe5 distance to 2.76 Å,38 compared to 2.61 Å in FeMo cofactor,104 leading to the most significant distortion in metal-metal distances in the two clusters, but the functional role of this ligand change remains unclear. Carbonate is firmly embedded in a loop of the VnfD subunit (residues 335–340 in A. vinelandii) that varies from the corresponding loop in NifD by having the positions of a proline and a leucine residue swapped (Fig. 14B). This generates a cavity in VnfD that accommodates carbonate, but is blocked by the proline in NifD. As the active site cofactor of nitrogenase is formed ex situ and inserted into apo-nitrogenase only as the final step of its biosynthesis, the S3A ligand that is present in the L-cluster precursor of all cofactors must be exchanged for carbonate prior to this insertion. The protein will then bind the cluster tightly, precluding any further dissociation of the ligand. The carbonate ligand of the FeV cofactor does not seem to be directly involved in catalysis in VFe protein, including in particular the reduction of CO that occurs here.39,172 It is not currently known what factors are involved in the removal of sulfide and its replacement with carbonate. The process likely takes place on VnfEN, but may well require further maturation factors, and the effect of carbonate on the catalytic properties of FeV cofactor remains to be elucidated.
4.2.3. Biochemical and Biophysical Distinction from MoFe Protein
In the physiologically relevant reduction of dinitrogen to ammonia, V-nitrogenase only shows approximately one third of the catalytic activity of Mo-nitrogenase. One may therefore speculate that the insertion of carbonate is a posterior optimization, without which the alternative system might be performing even more poorly. On the other hand, the insertion of a carbonate might even be required to affect the electronic structure of FeV cofactor such that it becomes functional in N2 reduction in the first place. A recent XAS study revealed vanadium in FeV cofactor to reside in the 3+ oxidation state in its resting state, as is the case for Mo in FeMo cofactor.171 However, V3+ has a 3d2 configuration, while Mo in FeMo cofactor is 4d3, in an unusual, non-Hund ααβ configuration.121 Both clusters are commonly considered to have a S = 3/2 multiplet ground state, which then implies that one Fe site in FeV cofactor must be more highly reduced than in FeMo cofactor. A carbonate ligand may fundamentally influence the spin distribution within the system and thus its reactivity towards substrates, but further studies will be required to assess this effect.
4.3. VnfH, the Fe Protein of Vanadium Nitrogenase
The three nitrogenase variants of A. vinelandii use active site cofactors that differ in their namesake heterometal, but are otherwise very similar in their core structure as they are derived from a common precursor, the NifB cofactor or ‘L-cluster’.173 In contrast, the P-clusters present in all three systems seem to be very similar in all aspects. The same is true for the basic functionality of receiving electrons from the ATP-hydrolyzing Fe protein (see 2.3). Nevertheless, the distinct gene loci of the three nitrogenase variants all contain a distinct ortholog of the NifH protein of Mo-nitrogenase that is designated VnfH in the V-dependent system and AnfH in iron-only nitrogenases. The expression of these H-genes is coregulated with that of the structural genes for the cognate dinitrogenase, and yet the reductase components share the same, conserved features of a homodimeric P-loop NTPase with a bridging [4Fe:4S] cluster that obtains two cysteine ligands from each monomer. In A. vinelandii, NifH and VnfH share 91 % sequence identity, a degree of similarity that is also reflected in the recently determined structure of the ADP-bound form of VnfH (Fig. 15A).61 The two proteins are exchangeable and work with either dinitrogenase without any discernible functional difference. Moreover, the ADP-bound states of both reductases are near identical in their quaternary structure, so that this conformation – ready for nucleotide exchange – is also a defined state rather than a flexible one (Fig. 15B). From this state, Fe protein obtains an electron from its redox partner and exchanges ADP for ATP to be ready for the next round of interaction with the dinitrogenase.
Figure 15:
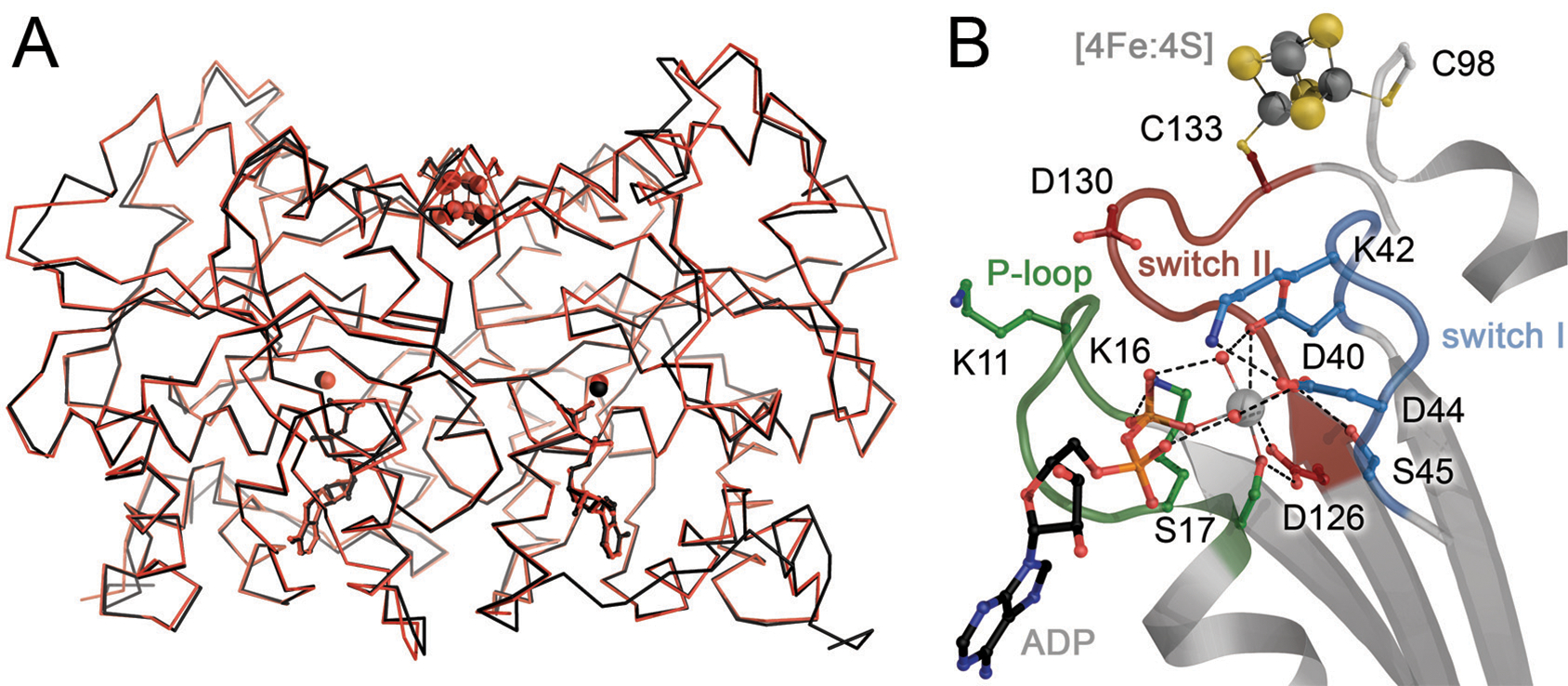
The Fe protein of V-nitrogenase, VnfH. A) Superposition of NifH (black) and VnfH (red) form A. vinelandii. The two reductases are 91% identical in sequence and can fully complement each other in function. B) The ADP-bound state of VnfH with an octahedrally coordinated Mg2+ cation shows the close spatial proximity of the nucleotide-binding P-loop, the conformationally flexible switch I and II regions and the bridging [4Fe:4S] cluster. Figure made from PDB entry 6Q93.
4.4. Ligand Binding to FeVco
Although the exchange of molybdenum for vanadium and the insertion of an additional carbonate ligand seem to infer substantial differences between FeMo and FeV cofactors, most structural features are conserved, and the clusters follow the same mechanistic principles for the reduction of N2.174 This extends to the organic homocitrate ligand, as well as to the attachment of the clusters via an apical cysteine to Fe1 and a histidine to the heterometal. The carbonate ligand that is present in FeV cofactor likely only finetunes the electronic properties of the cluster, but not to a degree that would affect its basic functionality. This is also reflected in the fact that the immediate surrounding of both cofactors remains nearly identical. The bridging sulfide S2B that was already highlighted above is in close proximity to two conserved residues, His180 and Gln176 in VFe protein and His195 and Gln191 in MoFe protein, respectively. The exchange of either residue in MoFe protein had a substantial effect on N2 reduction activities,175,176 and the histidine forms a direct hydrogen bond to S2B, connecting it to a conserved hydrogen-bonding network within the protein that extends to the nearby protein surface, suggesting a role in proton transfer to the cluster during substrate reduction.39 Note that based on theoretical approaches other pathways have also been proposed.177 In contrast, the side chain of glutamine, the other conserved residue, points away from the cofactor in all known structures, making its role less clear. Sulfide S2B is the ligand that is reversibly exchanged for CO and also for selenide in MoFe protein,147,149 and in the first structure available for VFe protein the ligand was fully in place.38 This resting state structure of the enzyme also showed the typical EPR features reported previously by others and the enzymatic activity of the protein was well within the expected range, amounting to approximately 1/3 of the activity of MoFe protein.164
When isolation protocols were refined further, however, a second, distinct species became apparent, whose EPR signature showed the same apparent g values as the resting state, but was characterized by distinct changes in the relative intensities of the signals.164 The EPR spectrum of VFe protein indeed differs significantly from the straightforward, rhombic S = 3/2 signal of the resting state (E0) of MoFe protein with its apparent g values of 2.01, 3.65, and 4.31 (see 3.2.3, Fig. 16A).127 The alignment of this g tensor with the cluster structure is compatible with the evaluation of its electronic structure by XAS and SpReAD (see 3.2.2).124 At the same time, the FeMo and FeV cofactors are very similar in structure and are suggested to follow the same principles for substrate reduction. The EPR signal of resting state VFe protein is similarly broad as for MoFe protein, nearly 3000 G, and shows signals at apparent g values similar to the ones for FeMo cofactor. In addition, however, an axial signal at 3470 G as well as a band at 1300 G are visible, indicating a mixture of several spin states in the resting state preparations (Fig. 16B).164 Münck and co-workers had previously suggested an explanation of the spectral differences based on Mössbauer and EPR studies. They described the S = 3/2 system of FeMo cofactor with a slightly axial effective g value of gx = gy = 2.003 and gz = 2.03, and used this model to rationalize the observed differences between FeMo and FeV cofactor by variations of the systems’ rhombicity parameter E/D representing the lower Kramer’s doublet of the S = 3/2 system (Fig. 16C).178 Here, an E/D of 0.05 would model the apparent g values of FeMo cofactor, while the resting state spectrum of FeV cofactor would result from an E/D of 0.30 (Fig. 16D). Alternatively, the EPR spectrum of resting state VFe protein was frequently described as a mixture of an S = 1/2 signal at a field of 3470 Gauss, the S = 3/2 signal known from FeMo cofactor and a further contribution at 1300 Gauss that was attributed to an S = 5/2 signal. The changes observed during the optimization of the isolation procedure can be explained as a change in the population of these signals, but not their positions.39
Figure 16:

Continuous-wave EPR spectra of nitrogenases. A) X-band spectrum of A. vinelandii MoFe protein in the resting state E0. B) X-band spectrum of the resting state of A. vinelandii VFe protein. C) In the S =3/2 system of the cofactor, the lower Kramer’s doublet dominates the EPR signal. D) In a rhombogram for the lower doublet, both nitrogenases can be described by an axial g-tensor, but differing in rhombicity, with E/D = 0.05 for MoFe and 0.30 for VFe protein.
The structural analysis of this state at 1.2 Å resolution then revealed an unexpected change at the FeV cofactor, in that sulfide S2B was removed from the metal cluster, as previously observed in the CO and Se adducts of the analogous FeMo cofactor (Fig. 17). The site was instead occupied with a protonated light atom, NH or OH, which in turn allowed for further rearrangements to occur in the vicinity of the Fe2-Fe6 edge of the cluster. Bound to either Fe ion at a bond distance of only 2.0 Å compared to the 2.3 Å of a sulfide or selenide, the ligand created the space for the side chain of the adjacent Q176 to rotate by 130° towards the cluster and form a short (2.8 Å) hydrogen bond with the imidazole side chain of H180. In turn, this rearrangement then opened a binding pocket that was previously occupied by the amide moiety of Q176 and that now provided a binding site for sulfide S2B, at a distance of only 7 Å from its former position in the cluster. At the same time, the carbonate ligand and the remaining μ2-sulfide S5A were retained. The presence of S2B in this binding pocket was confirmed by analysis of the anomalous scattering signal of sulfide, and the charge distribution in the surrounding protein matrix indicated that the ion was present as a hydrosulfide anion, HS−.39 The positive electrostatic potential contribution required to accommodate the anion in this pocket was provided by backbone amides of the protein chain of the D subunit, reminiscent of the arrangement of an oxyanion hole in serine proteinases that is essential for stabilizing the oxyanion in the tetrahedral transitions state of peptide bond hydrolysis in these enzymes.179
Figure 17:
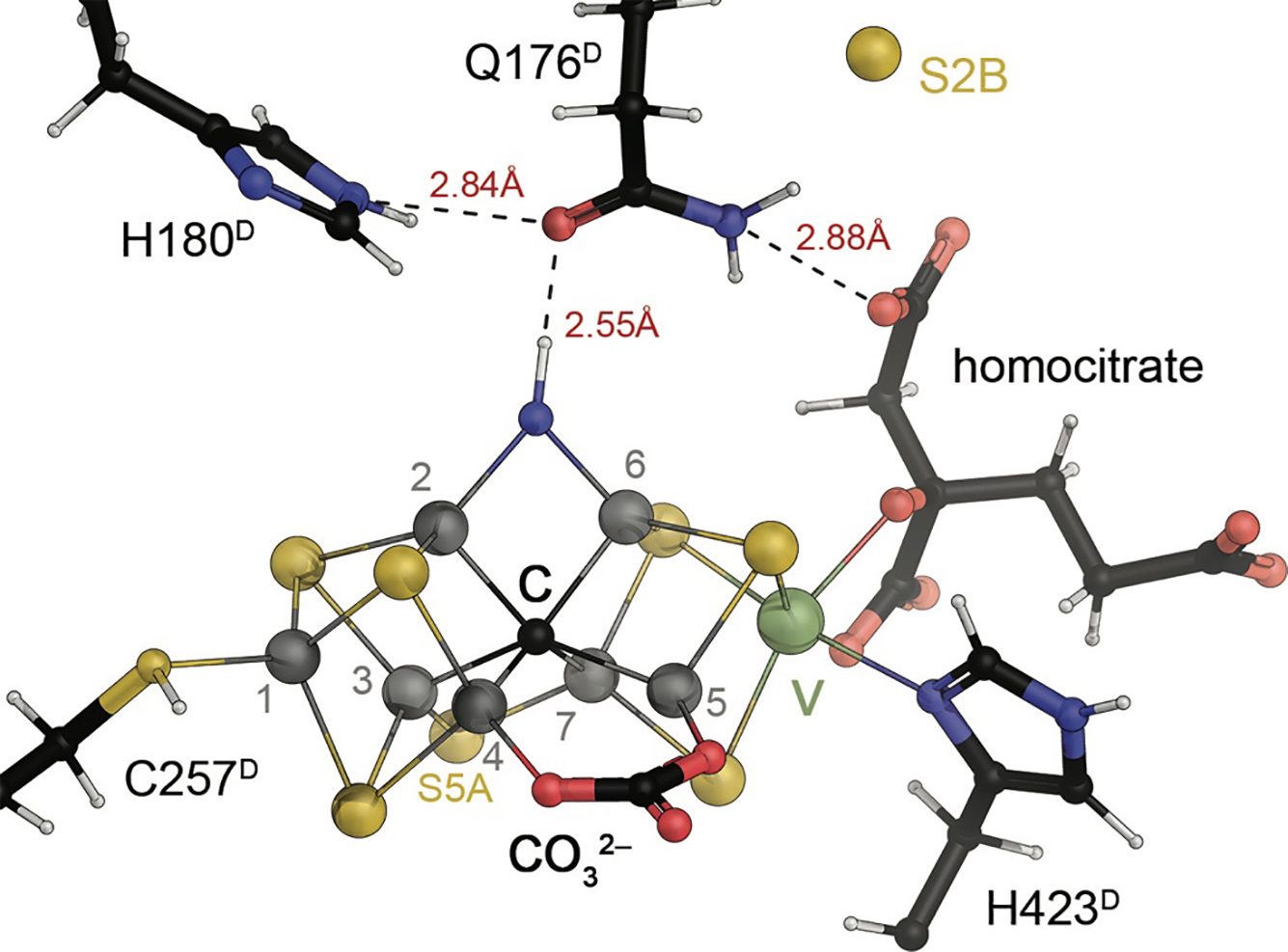
A turnover state structure for A. vinelandii VFe protein. Sulfide S2B that in the resting state E0 bridges Fe ions 2 and 6 relocated by 7 Å to a binding pocket that was created by an inward rearrangement of the sidechain of residue Q176D. It was replaced by a light atom, N or O that must be protonated according to the interatomic distances observed in the 1.2 Å resolution crystal structure. Figure generated from PDB entry 6FEA.
Importantly, a rearrangement of the residue corresponding to Q176 was not observed in the CO-bound structure of MoFe protein.147 Compared to the structure of the turnover state of VFe protein, it is the larger size of the CO ligand, whose oxygen atoms forms a stable 2.8 Å hydrogen bond with H195 (corresponding to H180 in VFe protein) that would block the rotation of the glutamine (Fig. 12A). The ligand bound to FeV cofactor in its turnover state, however, is at 2.55 Å distance from the amide oxygen of Q176 in its inward-facing position. This implies protonation of the ligand, as is also supported by an analysis of the experimental electron density maps,39 yielding a very short hydrogen bond. The nature of the ligand atom itself remains under debate, as both nitrogen and oxygen would satisfy the observed electron density maximum. As μ-bridging ligands, however, both a hydroxide, HO−, and an imide, HN2−, would represent the fully reduced forms of the respective heteroatom, and the release as either H2O or NH3/NH4+ should merely require protonation, but no further electron transfer. Given the timescale of crystallization and structure determination, the observation of this adduct in vanadium nitrogenase is quite remarkable in itself. A hydroxo ligand would imply the binding of water to the enzyme, suggesting that water would indeed compete for this binding site on the cofactor with the substrates of reduction by nitrogenase. With several lines of evidence pointing at this binding side to be relevant for substrate turnover, it seems unproductive that VFe protein should suffer from water as a competitive inhibitor, while MoFe protein does not. Moreover, binding to Fe2 and Fe6 requires dissociation of sulfide S2B, which in turn only occurs in more highly reduced states than the resting state E0, likely from E2 onwards (see 5.3). Water and ammonia are already fully reduced modifications of O and N, respectively, and would require deprotonation immediately upon binding. These protons would then have to be removed and the ligand would have to be protected from reprotonation to allow for its experimental observation. Also, a strong binding of NH3/NH4+ in vanadium nitrogenase should manifest as substantial product inhibition in the kinetic profile of the reaction, which has not been reported (see 5.3).
A possible rationalization for observing either adduct was brought forth, arguing that while the observed ligand may represent a reduced HO− or HN−, its stable association was due to the cofactor itself being in a more oxidized state than the resting state E0.39 In this model, the cluster would donate two electrons to the ligand, allowing for a substantial stabilization through backbonding interactions with the cluster. This would place an O or N ligand formally at the oxido (–II) or nitrido (–III) level, but would have the cluster in a two-electron oxidized state, corresponding to E6 in the catalytic cycle of the enzyme rather than E0. Interestingly, this proposal would apply to either O or N as a ligand with analogous mechanistic implications, but while a protonated nitrogen species could originate from reduction of N2, the presence of O2 can be safely excluded, as the high sensitivity of VFe protein and its reductase VnfH would lead to immediate inactivation of the enzyme at the stoichiometric amounts of O2 required to generate a hydroxo adduct. For these reasons, HN− was suggested the most suitable candidate for the observed ligand, but a final confirmation through corroborating analytical methods will be required to provide a definitive answer. In either case, the key finding from this work is the establishment of a dinuclear substrate binding site at Fe2 and Fe6 that was already suggested by earlier observations of CO and Se2− binding. With respect to understanding nitrogenase mechanism, it provides a structural framework essential for integrating existing data, in particular from extensive kinetic studies, spanning the work of multiple laboratories over several decades.
5. KINETIC ANALYSIS OF THE NITROGENASE REACTION
5.1. Reaction Components
The first step in establishing a reaction mechanism is to identify all the necessary components (reactants, products and catalytic species). For biological nitrogen fixation, the reactants are unquestionably N2, a source of reducing equivalents, and protons. Although the products of nitrogen fixation could be either more oxidized or more reduced than elemental N2, in pioneering studies, Wilson and Burris established that the key intermediate is ammonia.180 It is likely that there is also 1 H2 is produced per N2 reduced.181 There does not appear to be a unique electron donor to nitrogenase in vivo, and various ferredoxins 182 or flavodoxins can serve this role,183 while in vitro studies almost exclusively employ sodium dithionite (Na2S2O4).184 Finally, nitrogenase requires MgATP,182 while MgADP is a potent inhibitor.185 Nitrogenase is not uniquely specific for N2 and other substrates can be reduced, including protons to H2 (so nitrogenase is also a hydrogenase) and acetylene to ethylene (the vanadium nitrogenase 5 as well as some variants of the MoFe protein 186 can also produce ethane). Because of the relative ease of monitoring hydrogen and ethylene by gas chromatography, these two reactions are commonly used to assay enzyme activity rather than the physiologically relevant production of NH3. Other small substrates with unsaturated groups may be reduced, including acetylene, HCN, and N3−. Nitrogenase can also catalyze C-C bound formation, which was first observed during the reaction with CH3NC 187,188 and more recently with CO.154
The working model for the overall stoichiometry of N2 reduction by nitrogenase may be summarized as
| eq. 1 |
It should be emphasized that under most conditions, however, the indicated ratios of H2/N2 = 1 and MgATP / 2 e− = 4 are not observed. The reasons for this range from relatively trivial (reaction conditions have not been “optimized”, with uncoupled ATPase activity) to having profound implications for our mechanistic understanding of nitrogenase, e.g. how do we know that one H2 is produced per N2 reduced? The ubiquitous presence of protons means that nitrogenase cannot be studied in the absence of reducible substrates. A further implication is that H2/N2 ratios > 1 are nearly always observed and there is no easy way to tell if H2 production is an obligatory consequence of N2 reduction or the consequence of a parallel process of H+ reduction. MgATP / 2 e− ~5 are typically observed under optimal reaction conditions with dithionite as the electron donor, which likely reflects the contribution of some uncoupled ATPase activity to the overall stoichiometry. With other reductants such as Ti(III)citrate and flavodoxin,43,64 MgATP / 2 e− stoichiometries ~2 have been reported, although a recent evaluation of flavodoxin as the electron donor found MgATP / 2 e− ~4.35 There is also an intriguing report from Mortenson that MgATP / 2 e− ~2 is observed for (ADP)/(ATP) ratios characteristic of nitrogen-fixing cells.189
5.2. Challenges to Studying Nitrogenase Kinetics
The maximal specific activity reported for N2 reduction by the MoFe protein is ~ 600 nmol N2 min−1 mg−1 MoFe protein,190 which is equivalent to ~1 dinitrogen reduced per second per active site. To put this rate in perspective, it would take one active site over 20 min to reduce sufficient N2 to produce the amount of NH3 equivalent to that present in the constituent amino acids of one MoFe protein tetramer. Clearly, femtosecond spectroscopy is not essential to follow this reaction – so what is the problem in studying nitrogenase kinetics? There is no one dominant issue, but rather a combination of factors that contribute to these challenges. Among these are:
Oxygen sensitivity:
The nitrogenase proteins exhibit significant oxygen sensitivity and must therefore be manipulated using anaerobic techniques.191 The mechanism of inactivation is not well understood, but presumably is a consequence of oxidative damage or the formation of reactive oxygen species that can degrade the metalloclusters.
Sample heterogeneity:
The biosynthesis of nitrogenase is a complex process involving over a dozen proteins to generate the mature metalloclusters.192 As this is an ongoing process in growing cells fixing nitrogen, it is possible that partially assembled clusters are present, generating a heterogeneous mixture of proteins in the population. Oxidatively damaged clusters constitute another potential source of heterogeneity. For example, the K. pneumoniae nitrogenase samples used in the development of the Thorneley-Lowe kinetic model were reported 193 to consist of 70 % active MoFe protein (on the basis of Mo content) and 45% active Fe protein (on the basis of specific activity considerations (see below)).
Multiple intermediates during substrate reduction:
The nitrogenase mechanism involves multiple electrons (up to 8 for nitrogen reduction with obligatory hydrogen evolution) transferred from the external reductant to the active site by multiple cycles of ATP-dependent interaction between the two component proteins. Mechanistic studies must take into account the dynamic nature of the nitrogenase system, requiring multiple association and dissociation events between the two component proteins, as well as the ubiquitous presence of protons that are reduced to dihydrogen even in competition with other substrates. The resulting distribution of intermediates under turnover conditions significantly complicates the structural and spectroscopic investigation of substrate interactions. The contrast to photosystem II is striking, where electron transfer processes are triggered photochemically rather than by diffusion, and it is possible to prepare well-defined intermediate states of the system.194 Photoinitiated electron transfer systems for delivering electrons to the nitrogenase have been reported; the system developed by Syrtsova and coworkers used eosin to reduce the Fe-protein,195 while more recently, Tezcan’s group engineered covalently linked a Ru-photochemical donor to the MoFe-protein and demonstrated that substrate reduction could occur in the absence of Fe-protein and ATP.196,197 Although the reported quantum yield was low, this approach has great potential for dissecting the nitrogenase mechanism at the level of individual electron transfer steps.
Spectroscopic challenges:
Iron sulfur clusters generally have similar optical absorption spectra with broad features at ~400 nm and comparable extinction coefficients on a per Fe basis for a given average oxidation state.198 For example, the extinction coefficients at 410 nm for the as-isolated forms of the Fe protein and MoFe protein are ~10 mM−1 cm−1 and 76 mM−1 cm−1 with 4 and 30 Fe, respectively. Cluster oxidation is typically accompanied by an increased absorption; upon oxidation of the Fe protein [4Fe:4S] cluster from the +1 to +2 state, the absorption at 430 nm increases by approximately +7 mM−1 cm−1.199 More importantly, it is not possible to easily use optical spectroscopy to monitor redox changes at specific clusters since they all have similar optical properties. Iron sulfur clusters exist in multiple states that are often paramagnetic, making them accessible to study by EPR and related spectroscopies.200 While these techniques are exquisitely sensitive to changes (in appropriately paramagnetic states) and have been effectively used to monitor populations of various nitrogenase intermediates,201 the underlying chemical interpretations (structures of clusters and intermediates) can be challenging to decipher.
Determining the time course of product formation:
Standard assays for nitrogenase activity (reduction of C2H2, H+ and N2) are typically not monitored in real time, but rather by collecting samples at discrete time points for product quantitation by gas chromatography (C2H4 and H2) or colorimetrically (NH3). As a result, continuous time courses for product formation are not measured, and even if they were measured in the gas phase, the contribution of physical diffusion from solution to gas would need to be taken into account in interpreting the results. A notable exception is Burris’ use of a hydrogen electrode to monitor formation of H2 in solution.202 There are some studies, such as the use of stopped-flow IR spectroscopy to monitor the binding of certain substrates and inhibitors 203 or monitoring the consumption of dithionite 204 that can be done in real time; advances in this area would be of great significance. Insights into intermediates has been achieved using pulsed EPR methods, principally by Hoffman and collaborators,205 but due to the relaxation properties, samples must be trapped at low temperature and the assignments require heroic efforts to establish the structures of intermediates.
Dithionite:
Since the initial report by Bulen,206 dithionite has been widely used as the electron donor for nitrogenase assays, as well as a general additive to maintain oxygen-free solutions by reducing residual oxygen. It does have some potentially significant drawbacks; with nitrogenase, dithionite works as a one-electron reductant through dissociation to the radical SO2− species 193,204 and does not generate the all-ferrous form of the Fe protein;44 accumulation of the oxidation products sulfite (SO32−) and bisulfite (HSO3−) can significantly influence the reduction potentials of dithionite solutions;207 dithionite decomposes during storage and stock supplies can contain significant levels of impurities, particularly sulfite;208 and dithionite provides a source of adventitious sulfur that can complicate analyses of this crucial element of metallocluster function. It is worth keeping in mind that the apparent first order rate constant for the reduction of Fe protein by 10 mM dithionite is ~10 s−1,193,209 which is comparable to the turnover rate; given the significant problem of dithionite decomposition, this rate may be quite sensitive to the condition of the dithionite used in a particular experiment which constitutes a source of potentially uncontrolled variability.208 It is also worth noting that sulfite and potentially other dithionite-related compounds can serve as sulfur donors in cluster biosynthetic reactions.210
5.3. General Features of Nitrogenase Kinetics
A key component of the kinetic analysis of a reaction mechanism is determining the dependence of the rate of product formation on the concentrations of the various reactants. Given all the components, a comprehensive analysis of the complete time and concentration dependence of the nitrogenase reaction for multiple substrates is not feasible. Here we highlight key features of the kinetics of nitrogenase that have been established over the past half century of research.
5.3.1. The Nitrogenase Proteins Form a Dissociable Complex:
The existence of a dissociable complex formed between the component proteins was established from the concentration dependence of nitrogenase activity. For a fixed component ratio (CR; given by the molar ratio of [Fe protein] to [MoFe protein] active site), the specific activity of the individual proteins decreases non-linearly as the total concentration of proteins decreases.211 This so-called “dilution effect” was interpreted by Silverstein and Bulen 211 as indicating that nitrogenase can be considered a complex of “dissociable dissimilar subunits, neither being catalytic in the absence of the other”. Typical values of the dissociation constant estimated from activity measurements are ~ 10−7 M to 10−6 M.94,212–214
5.3.2. The Nitrogenase Proteins Dissociate After Each Cycle of Electron Transfer
Burris and Hageman 213 made the key observation that component protein dissociation after each cycle of electron transfer is an obligatory feature of the nitrogenase mechanism. By using an H2 electrode to monitor proton reduction, they observed a lag period for H2 production (but not ATP hydrolysis) under conditions of very slow electron transfer by having the MoFe protein in large excess over the Fe protein. Significantly, the lag time was proportional to the time required produce H2 at the MoFe protein active site (i.e. – the turnover time of the MoFe protein) for a given set of conditions, but not the turnover time of the Fe protein in electron transfer. The latter relationship would be expected if the catalytically competent Fe protein:MoFe protein complex remained intact during turnover. These observations are consistent with a ping-pong mechanism of the type
| eq. 2 |
where E0 and E1 represent consecutively reduced states of the MoFe protein, and the rate constants kn denote complex formation with, and electron transfer from, the Fe protein. That each step is associated with electron transfer to the MoFe protein was demonstrated by following the EPR signal from the FeMo cofactor during the lag period.201 Since electron transfer and ATP hydrolysis (but not H2 evolution) occur during the lag phase, this indicates that ATP hydrolysis is coupled to inter-protein electron transfer, but not directly to substrate reduction.
As with any kinetic study, alternative interpretations are not precluded; the requirement for cycles of dissociation/association may reflect the protein concentrations (0.01 to 3×10−6 M, relative to a dissociation constant of ~10−7 M) used in this study, so that the complex is likely to dissociate and the proteins must then re-associate for electron transfer. Whether obligatory cycles of association and dissociation are required at the concentrations found in nitrogen fixing cells (~25 to 45×10−6 M)215 is not clear. It is also possible that the initial electron transfers are required to activate an inactive (or dormant) form of the nitrogenase proteins 216,217 and that the lag phase reflects this activation event rather than turnover. An example of this type of behavior is provided by cytochrome c oxidase, where the fully oxidized form in the as-isolated resting state is distinct from the fully oxidized form produced under turnover conditions.218,219
5.3.3. Flux Through Nitrogenase Is Independent of Substrates Being Reduced
A striking feature of the reaction catalyzed by nitrogenase is that the electron flux through the system is largely independent of the substrate that is being reduced.211,220,221 Under a given set of conditions, the number of electrons transferred to substrate per active site per unit time is (nearly) the same for the reduction of dinitrogen to ammonia (the physiological reaction), the reduction of acetylene to ethylene (commonly used to assay nitrogenase activity), or the reduction of protons to dihydrogen, which occurs in the absence (or sufficiently low concentrations) of other reducible substrates. This implies that all nitrogenase substrates effectively compete for the same pool of electrons that are pumped into the MoFe protein by the Fe protein at a constant rate and with a constant rate of ATP consumption, independent of the substrate being reduced.
5.3.4. Component Protein Dependence
An important type of mechanistic analysis is provided by titration curves, where the reaction velocity is measured for a fixed concentration of one component while the second component is varied. This is typically expressed in terms of the component ratio (CR), defined as the ratio of Fe protein to MoFe protein; an important point is whether this is defined in terms of molar ratio of proteins or molar ratio of active sites. Experimentally, when the MoFe protein is titrated with Fe protein (see 222), the velocity increases with increasing CR until saturation is achieved at a MoFe protein specific activity of ~2500 nmoles C2H4 min−1 mg−1. This titration curve typically can be fit by a hyperbolic (Michaelis-Menten) type curve, although sigmoidal kinetics have been reported.202,223–225 The reverse titration of a fixed amount of Fe protein with increasing amounts of MoFe protein also shows apparent hyperbolic behavior with activity increases with increasing MoFe protein up to a maximal value of ~1500–2000 nmol H2 min−1 mg−1 (although there appears to be considerable variability in the reported Fe protein specific activities). Interestingly, as [MoFe protein] increases beyond these levels promoting maximal activity, it has been observed that the Fe protein specific activity decreases; i.e. the MoFe protein is inhibitory towards Fe protein at high concentrations.
The results of the previous two sections effectively imply that nitrogenase cycles through a series of increasingly reduced states driven by association and dissociation of nitrogenase proteins at a rate independent of the substrate. After sufficient electrons have been transferred, the product is released and nitrogenase returns to the resting state. For hydrogen evolution, the simplest form of this mechanism may be described
| eq. 3 |
where E and F designate MoFe protein and Fe protein, respectively, which cycle through different redox states. This mechanism makes several simplifying assumptions, such as neglecting the binding of Fox to MoFe protein and that the exchange of ATP for ADP on the Fe protein is fast. When kR, the rate of Fox reduction (and exchange of ATP for ADP) is fast relative to k, then the titration curve equations will be symmetrical with respect to interchange of the MoFe protein and the Fe protein concentrations. For the case where either component protein is in sufficient excess over the other, then the steady-state rate equation is hyperbolic in that component. When the concentrations of the components are comparable, the effect of complex formation on the concentration of the free component proteins may be significant, although the shapes of the titration curves remain approximately hyperbolic (i.e., they are not sigmoidal).
It is clear that a mechanism of the type described by eq 3 does not qualitatively fit the titration curve data, especially for the Fe protein titration. An important indication of this comes from comparison of the maximal specific activities of the MoFe protein and Fe protein. It may be shown that the specific activity for either component protein for this mechanism is proportional to k/2, which based on the MoFe protein specific activity = 2500 nmol H2 min−1 mg−1 yields k ~ 10 s−1. This value for k then predicts that the Fe protein specific activity should be ~4700 nmol H2 min−1 mg−1, which is over twice that typically observed. Various reasons for this significant discrepancy have been proposed, including that some other step becomes rate limiting under conditions of high Fe protein turnover (such as maintaining the Fe protein in the reduced, ATP-bound form (i.e., kR is not >> k); the Fe protein is only ~45 % active (see 5.4); that there are problems with the experimental design, such as non-constant ionic strength, that compromise the activity measurement; or that eq 3 (and the underlying assumptions) is fundamentally flawed.
An example of the last possibility is provided by the observation of sigmoidal kinetics, which have been interpreted from some of the earliest kinetic studies of nitrogenase,223 and by subsequent studies 212,226 to indicate that a complex with 2 Fe protein and 1 MoFe protein (i.e. a 2:1 complex), but not a 1:1 complex, is the active species for substrate reduction. In a detailed kinetic analysis of this point, Hageman and Burris concluded that while both 2:1 and 1:1 complexes are possible, the 1:1 complex was most consistent with their observations.202 Crystal structures of nitrogenase complexes have identified multiple discrete, but overlapping Fe protein binding sites on the MoFe protein (see 2.4, Fig. 8B),73 and, at least for the sites that have been characterized, it would not be possible for two Fe proteins to simultaneously bind adjacent to one MoFe protein active site. The observation of sigmoidal kinetics does not, of course, require that there be a 2:1 complex involved in the mechanism, but it could have distinct mechanistic origins. While the two active sites of the MoFe protein tetramer are generally assumed to be independent, sigmoidal kinetics could indicate cooperativity in Fe protein binding between the two MoFe protein active sites in a tetramer.167,227 The simplest kinetic models assume that the rate of electron transfer from the Fe protein to the MoFe protein is independent of how many electrons have already been transferred, but if that is not the case, sigmoidal kinetics could also be observed.226
5.3.5. Substrates and Inhibitors Bind to Different En States
The competition between substrates for reducing equivalents is not simply based on the relative concentrations of the different forms and their apparent affinities (Kms), but also depends on the details of the experiment, including the flux of electrons through the system, which is set by the concentration of component proteins. If the reduction of substrate S1 is preferred at low electron flux, while reduction of substrate S2 becomes favored as the electron flux increases, this is indicative that the two substrates preferentially bind to different En states, with S1 favoring less reduced and S2 more highly reduced En states. Studies of this type indicate that N2 binds to the E3 and E4 states,193,228 while C2H2 binds to E1 and E2 states,229 and CN−, CH3CN and HN3 may bind to an even more oxidized state, likely E0.230 The inhibitor/poor substrate CO binds after a two-electron reduction of the cofactor.139 An interesting consequences of this type of behavior is that the apparent Km values of substrates will depend on the electron flux through the system. An important consequence of the binding of N2 to higher Ei states is that ammonia production is favored over proton reduction (in the absence of any other reducible substrates) under conditions of high electron flux. This constraint coupled to the relatively low turnover rate of nitrogenase requires high concentrations of the nitrogenase proteins, but not too high if substrate binding and product dissociation require free MoFe protein (vide infra). The cellular concentrations of the A. vinelandii MoFe protein and Fe protein have been estimated by EPR measurements to be ~28×10−6 M (6.0 mg ml−1) and 45×10−6 M (2.8 mg ml−1), respectively, for CR = 45/(2×28) = 0.8 on the basis of MoFe protein active sites.215 This corresponds to ~ 6 % of the total cellular protein in Azotobacter, and as discussed in that reference, even higher levels (~ 10 %) have been reported.
Enzyme inhibitors can provide important insights into the nature and number of ligand binding sites on an enzyme. In principle, this information can be used to establish whether substrates bind at the same site (competitive) or distinct sites (non-competitive). However, for complex enzymes such as nitrogenase with multiple oxidation states and potential substrate binding modes, this distinction is not required.141 One of the most important inhibitors is carbon monoxide. CO is isoelectronic to the physiological substrate, only binds to partially reduced MoFe protein generated under turnover conditions, and has been found to be a non-competitive inhibitor for all substrates except protons.137,138 Intriguingly, CO can partially block proton reduction in certain MoFe protein mutants,231 as well as in the NifV− nitrogenase system,232 where the homocitrate is replaced by citrate. These cases are among the few examples of an inhibitor that reduces electron flux through nitrogenase. For the inhibition of C2H2 reduction by CO, evidence has been presented that both molecules can bind simultaneously,229 although multiple binding modes have been identified for both species, so perhaps it is to be expected that a subset of these be occupied together.
The effect of one substrate on the reduction of another substrate can be analyzed for inhibitory modes, which can illustrate the complexities of nitrogenase kinetics. As an example, C2H2 is a non-competitive inhibitor of N2 reduction, but N2 is a competitive inhibitor of C2H2 reduction.233 This is a consequence of the observation noted above that N2 binds to a more reduced form of nitrogenase than C2H2. A substrate binding to a more reduced state appears as a competitive inhibitor towards one binding to a less reduced (i.e. more oxidized) state; conversely, the second substrate appears as a mixed-type/non-competitive inhibitor of the first. On the basis of these patterns of inhibitions, attempts have been made to assign substrates and inhibitors to different binding sites (ranging from two to five different sites).137,234 Not surprisingly, a self-consistent pattern has not identified, undoubtedly due to the challenges arising from different binding modes, binding to different En states and the possibility of multiple types of binding modes for a given substrate.
Mechanism-based inhibition can be effectively used to investigate the catalytic mechanism and would provide a potent new direction for probing substrate reduction, particularly for the more highly reduced states of the MoFe protein that bind substrates. One solution would be to work at higher pH to minimize the rate of proton reduction, thereby increasing the concentrations of more highly reduced states. While it has been reported that the MoFe protein is denatured by incubation above pH 8.65,235 a subsequent analysis demonstrated instead that the MoFe protein undergoes a turnover-dependent inactivation reaction at pH 9.5, implying this is a mechanism-based inactivation process that likely involves higher reduction states.236 While the inactivated MoFe protein has distinct properties from wild type MoFe protein (including an expanded hydrodynamic radius), it retains a full complement of metals and can still interact with Fe protein and supports ATP hydrolysis, but not substrate reduction. The underlying inactivation mechanism is unknown, but perhaps reflects a rearrangement of a more reduced, deprotonated form of the FeMo cofactor.
5.3.6. N2 Reduction, H2 Evolution and HD Formation
There is clearly a special relationship between H2 and N2, although the exact relationship is complex since H+ is a ubiquitous substrate. Key observations include that N2 reduction cannot (apparently) eliminate H2 production,237 with extrapolation of limiting H2 yields under high N2 pressures 181,238 suggesting that a residual 1 H2 is produced per N2 reduced for the enzyme from A. vinelandii. H2 is a competitive inhibitor of the reduction of N2, but remarkably does not affect the reduction of any other substrate, including H+ reduction. In one of the most characteristic, but enigmatic processes, N2 is required for the nitrogenase-catalyzed exchange reaction between D2 and H+ to yield HD 239
| eq. 4 |
This reaction was interpreted by Chatt 240 as reflecting the displacement of hydride ligands as H2 upon binding of N2 as observed for small molecule complexes. In the case of nitrogenase, N2-dependent HD exchange may reflect D2 displacement of bound N2 to form deuterides at the catalytic center that are subsequently protonated to form 2HD;241,242 if N2 displaces H2 upon initial binding this overall process requires 2 electrons or 1 electron per HD, as observed.239 More detailed studies suggest that HD exchange requires not just bound N2, but a reduced form of dinitrogen, perhaps at the diimide (N2H2) level.176,239
5.4. A Kinetic Mechanism for Nitrogenase: the Thorneley-Lowe Model
The conceptual framework at the foundation for most discussions of the nitrogenase mechanism derives from two key insights of Joseph Chatt:240,243
Binding of N2 to a metal center can proceed through displacement of H2 generated by the 2-electron reduction of protons.
Metal bound N2 was proposed to be reduced to NH3 through a 6-electron process involving an alternating sequence of proton and electron transfers.
According to this framework, the reduction of N2 to NH3 by nitrogenase is an 8-electron process with obligatory H2 evolution, represented in the overall stoichiometry of Eq. 1.
The Thorneley-Lowe kinetic scheme for nitrogenase 94,244 reflects this view and builds on the kinetic foundations discussed above that the nitrogenase component proteins undergo obligatory cycles of ATP-dependent complex formation as the MoFe protein cycles through states E0 to E7. The Thorneley-Lowe model is organized around two interconnected cycles (the Fe protein and MoFe protein cycles) rooted in observations of Hageman and Burris that the nitrogenase proteins dissociate after each electron transfer:202,245,246
Fe protein cycle, where electrons are transferred from the Fe protein to the MoFe protein in an ATP dependent process. For each electron transferred, the Fe protein must undergo a cycle of nucleotide exchange and reduction by either flavodoxin or ferredoxin; this likely takes place following dissociation of the Fe protein – MoFe protein complex (Fig. 18A).
Figure 18:
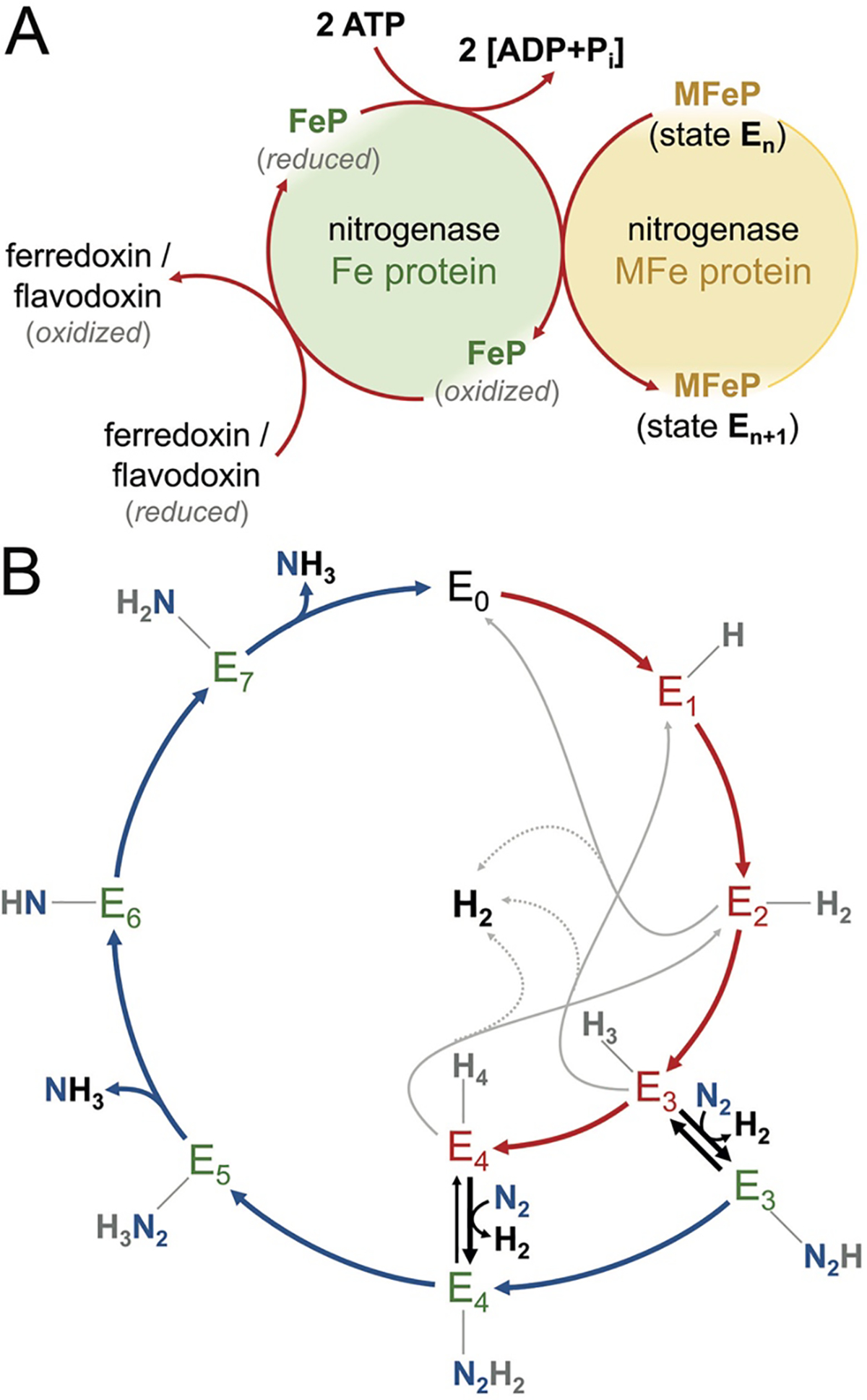
The Thorneley-Lowe model for nitrogenase. A) In a Fe protein cycle, the transfer of a single electron obtained from central metabolism via a ferredoxin or flavodoxin requires transient complex formation of the reduced Fe protein with the catalytic component (MFe protein), concomitant with the hydrolysis of 2 ATP/e−. As a result, the enzyme is advanced by one E-state in the catalytic cycle. B) The cycle of the MFe protein describes an 8-electron process and consequently cycles through 8 distinct 1-electron steps (E0–E7). An alternating transfer of electrons and protons (or vice versa) is assumed, and notably the binding of the substrate N2 requires the enzyme to be at least in state E3 or E4. From states E1–E4, unproductive H2 evolution is observed, while the exchange of N2 for H2 upon substrate binding is presumed to be a mechanistic requirement.
MoFe protein cycle, where the MoFe protein goes through a sequence of increasingly reduced states, starting with the resting (“as-isolated”) state (E0), with the subsequent states designated Ei to reflect the cumulative delivery of i electrons from the Fe protein (Fig. 18B).
Because the reduction of all known substrates by nitrogenase entails two or more electrons, the complex of the two nitrogenase proteins must turnover multiple times. Even for a simple substrate such as H+, it can be appreciated that there are a significant number of distinct states that need to be incorporated in a realistic kinetic model. Key features of the nitrogenase mechanism integrated into the Thorneley-Lowe model to model substrate interactions and the transitions between these states include:
Substrates do not bind to the resting E0 state, but rather the MoFe protein must be reduced by ~2–4 electrons to the E2 to E4 states before they can bind, with the possible exception of acetylene binding to E1.
The flux of electrons through the MoFe protein is independent of the substrate being reduced, or the number of electrons already transferred. In the absence of other reducible substrates (typically C2H2 or N2), the electron flux through nitrogenase continues at the same rate to reduce protons to H2.
Only free MoFe protein (i.e. MoFe protein not in complex with the Fe protein) can bind substrates or release products.
The MoFe protein active sites are independent.
The full form of the kinetic scheme involves 15 rate constants whose values were determined using a series of pre-steady state stop flow and steady-state kinetic experiments on the K. pneumoniae molybdenum nitrogenase at 23 °C, pH 7.4. The rate-determining step under optimized conditions in these studies was described as dissociation of the Fe protein – MoFe protein complex, with k ~6.4 s−1 at 23 °C, corresponding to maximal specific activities of MoFe and Fe protein of 1650 and 3000 nmol·min−1·mg−1, respectively.193
While the Thorneley-Lowe model is widely cited in connection with the nitrogenase mechanism, it is rarely used to simulate time courses for intermediate and product formation. Undoubtedly one important factor is that the kinetic model was explicitly evaluated for K. pneumoniae nitrogenase at 23 °C, while most contemporary nitrogenase studies use the A. vinelandii nitrogenase at 30 °C. There may be deeper reasons for this lack of quantitative application of the Thorneley-Lowe models to nitrogenase kinetics and it is worth noting two major reservations:
Independent efforts to reproduce kinetic fits using the published data were unsuccessful 247 and the rate constants needed to be reparametrized to obtain responsible fits. It is unclear why the numerical simulations could not be reproduced, but given the number of rate constants and the extent of the data, computational instabilities in correlated parameters would seem one possibility. As a related issue, rapid freeze-quench EPR studies of an intermediate described as E3 based on the kinetics of appearance 248 was reassigned to E2 based on the EPR properties; the discrepancy was ascribed to “uncertainties in the rate constants used in the kinetics analysis”.205,249 This reassignment suggests that the kinetic description is already unreliable early in the MoFe protein cycle.
The Thorneley-Lowe model requires the Fe protein to be only ~ 45 % active, based on the observed specific activity measurements. This inactive protein is assumed to be able to form complexes with the MoFe protein with the same kinetic parameters as native Fe protein. In contrast, the 30 % inactive MoFe protein is assumed to be unable to participate in complex formation with the Fe protein. It is hard to understand the molecular basis of these properties given that inactive Fe protein has never been directly characterized, but only postulated to make the specific activity measurements fit an assumed kinetic model.
One resolution to this conundrum would be the presence of Fe protein in non-reactive redox states. As a perhaps hypothetical illustration of this behavior, Watt has measured the equilibrium constant = 7.4 for the disproportionation of ATP-bound Fe protein between the oxidized (2+), reduced (1+) and all-ferrous (0) oxidation states:44
| eq 5 |
A Fe protein solution originally in the +1 state will equilibrate to a distribution of 0.42, 0.16, 0.42 between the 2+, 1+ and 0 states; assuming for the sake of this discussion that the all-ferrous form was the only form of the Fe protein active for electron transfer to the MoFe protein, then 42 % of the Fe protein would be in this form at equilibrium. The role, if any, of the all-ferrous form remains controversial, however.35
6. MECHANISTIC PRINCIPLES OF BIOLOGICAL NITROGEN FIXATION
6.1. General Mechanistic Framework for N2 Reduction
While we know now that nitrogenase produces ammonia, as a general chemical problem, the transformation of atmospheric N2 to a “fixed” form could yield a variety of different products, ranging from fully reduced ammonia (NH3) to fully oxidized nitrate (NO3−), as well as nitrite (NO2−), hydroxylamine (NH2OH), cyanic acid (HCN), urea (H2N-CO-NH2) and undoubtedly other species. A perceptive analysis in 1927 by Burk concluded that the overall free energy change for all these reactions would be generally favorable.250 “Fixation of nitrogen even with liberation of energy or free energy, will take place if either oxygen gas or hydrogen gas, or other substances, especially gases, whose standard free energies are close to zero, are involved to form either nitrates, ammonia, or cyanide, not to speak of still other compounds.” Hence, thermodynamics does not give us any direct guidance in terms of identifying a specific product of nitrogen fixation that would be uniquely thermodynamically favorable under ambient conditions. Although ammonia was early on implicated as the product of biological nitrogen fixation, it was not until the introduction of isotopically labeled 15N that Wilson and Burris firmly established that the key intermediate is indeed ammonia.180
The mechanistic challenge in the reduction of N2 to NH3 is that while the overall reaction is thermodynamically favored (when H2 or a low potential ferredoxin or flavodoxin are used as the reductant), the kinetics of the uncatalyzed reaction are highly unfavorable under ambient conditions. This behavior is due to a high activation energy for the initial reduction of the N≡N triple bond to form diimide (N2H2), which is also reflected in the highly unfavorable thermodynamics for this reaction. In the gas phase, the free energy change for the reduction of N2 to N2H2 is estimated as +210 kJ·mol−1, significantly higher than the corresponding reductions of the triple bonded compounds C2H2 to yield C2H4, or CO to H2CO (−140 kJ·mol−1 and +25 kJ·mol−1, respectively). The free energy of formation of N2H2 was estimated in an analysis by Stiefel,251 and it is likely the value he used for the enthalpy of formation of N2H2 was underestimated by ~50 kJ·mol−1 relative to contemporary values,252 making this reaction even more unfavorable. It is worth noting that this situation does not mirror the relative bond strengths of the triple bonds in these compounds, since the N2 triple bond energy (941 kJ·mol−1) is comparable to the triple bond energies in C2H2 and CO (962 and 1070 kJ·mol−1, respectively).253 An important distinction between N2 relative to C2H2 or CO is the significant additional stabilization in the N2 triple bond compared to the single and double bond forms, so that the initial reduction of N2 is significantly more unfavorable than for these other compounds.
Two general mechanistic frameworks have been considered for the reduction of N2 to NH3: 253–255
a sequence of proton-coupled electron transfers to N2, resulting in the formation of intermediates successively reduced by 1 [H] (N2H, N2H2, …, 2NH3). Protonation of N2 lowers the energetic barrier to reduction; as an example, the enthalpy change associated with N2H+ reduction in the gas phase is nearly 1000 kJ·mol−1 lower than for the reduction of N2.252 (Equivalently, it is easier to protonate N2− than N2 by the same amount). The binding of N2 to transition metals significantly lowers the barrier to reduction through back-bonding effects, and an approach to the catalytic conversion of transition metal coordinated N2 to ammonia through a sequence of proton-coupled electron transfers was pioneered by Chatt.240 The experimental realization of this general scheme at a single transition metal was subsequently achieved by Schrock with Mo,256 and by Peters with Fe.257
a multi-electron transfer dissociative process where the N≡N triple bond is split in the initial step. In the extreme case, this would involve initial reduction to form two nitrides (N3−), but it is also possible that this could involve reduction to the hydrazido level (N24−). Following transfer of the requisite number of protons and electrons, NH3 would be produced. To facilitate the multi-electron natures of these scenarios, catalysts are typically multi-metallic, such as the homogenous di-molybdenum system of Nishibayshi and coworkers,258 or the heterogenous system of Shilov.259 In essence, the catalyst serves as a capacitor that is charged up during an activation phase of the reaction before subsequently discharging multiple electrons to the substrate to form more highly reduced nitrogen species. The Haber-Bosch process 260,261 provides an important example where N2 dissociates into N atoms on the surface of a Fe catalyst, subsequent to reacting with dissociated H atoms to form NH3.
It should be noted that other mechanistic approaches to nitrogen fixation are possible. For example, the Frank-Caro cyanamide process was an early large-scale method for fixing nitrogen that involved the reaction of calcium carbide with N2 at high temperature to form the calcium salt of the cyanamide anion (NCN2−) and graphite.262 As with the Haber-Bosch process, the reactants and reaction conditions are irrelevant to nitrogenase, but the fact that there is an interstitial carbide at the heart of the FeMo cofactor suggests that an intermediate involving the reaction of the carbide with N2 to form perhaps a diazomethane or diazirine that is subsequently reduced to NH3 should not be dismissed immediately.
6.2. The Mechanism of N2 Reduction by Nitrogenase
The chemical reduction mechanism of N2 by nitrogenase is generally viewed through the mechanistic framework introduced by Chatt, integrated with the Thorneley-Lowe kinetic model. In this model, dinitrogen reduction proceeds through a sequence of single proton-coupled electron transfer steps leading from N2 to N2H to N2H2 … to 2NH3. Indirect experimental evidence supporting this model includes:
hydrazine can be detected upon acid or alkali quenching of actively fixing nitrogenase.263 This behavior strictly demonstrates that an intermediate containing N2 and hydrogens at the proper reduction level for hydrazine appears during quenching,243 but not necessarily that hydrazine is an obligatory intermediate during N2 reduction. Indeed, hydrazine can be generated during acid quenching of well-characterized N2 complexes.264
hydrazine can be detected as an intermediate in the reduction of N2 by the VFe protein.265 This observation suggests that NH3 is not formed directly from N2 (without intermediates), but it has not been demonstrated that hydrazine is an on-path intermediate.
nitrogenase can reduce N2 to NH3 under conditions where the Fe protein transfers single electrons to the MoFe protein using dithionite as a reductant, supporting the concept of a mechanistic scheme composed of single-electron transfer events. There are, however, reports that nitrogenase functions more efficiently with the all-ferrous Fe protein 266 or using reduced flavodoxin as the electron donor 202 that could serve as two electron donors. To be clear, it is not understood how the electron transfer properties of the Fe protein are related to the N2 reduction process.
Mössbauer spectroscopy studies of nitrogenase indicate that the average isomer shift of irons in the FeMo cofactor and P-cluster change little under turnover conditions in the presence of N2.178 From the correlation between average isomer shift and iron oxidation state in FeS clusters, the change is estimated to correspond to a net reduction of less than one iron per FeMo cofactor.117 A dissociative type of process with a transfer of six electrons from the metalloclusters to N2 could be expected to result in a significant net oxidation of irons in the metalloclusters during turnover conditions. That this is not observed could indicate that nitrogenase does not utilize a dissociative mechanism with N2 reduced to the nitride level in one step; it could also mean that under the experimental conditions, the enzyme was not efficiently reducing N2. NH3 formation was not reported in the experimental protocols 117 and at the low component ratio utilized in this study (~1), significant H+ reduction was likely, so the system may more reflect low En states rather than the complete nitrogen fixing cycle.
The most detailed version of this model has been developed by Hoffman, Seefeldt and Dean, who have used sophisticated pulsed EPR methodologies with various substrates and nitrogenase mutants to trap and characterize intermediates.205 These intermediates have been assigned to specific states in a modified Thorneley-Lowe mechanism (Fig. 18B) that has N2 binding to E4 (the binding of N2 to E3 is “deemphasized” in the Hoffman et al. model)267 and includes an E8 state. To date, 3 intermediates have been characterized with bound nitrogen(s) – a species with two nitrogens assigned to E4 and two species containing single nitrogens assigned to E7 and E8 in the Hoffman mechanism.205 The E4 intermediate has been identified as existing in two forms,268 with either 2 bridging hydrides or with bound N2 following displacement of bound H2 generated by hydride protonation. The replacement of two hydride ligands as H2 by N2 is well precedented in transition metal complexes.243,269 This reductive elimination reaction on nitrogenase has been shown to be reversible 270 and has been observed in all three types of nitrogenase.174 Whether or not this process is a true reductive elimination, resulting in significant Fe reduction upon H2 dissociation has been challenged.271 E4 has been termed the “Janus” intermediate, since it faces both forward and backward, either advancing to NH3 formation upon binding of N2 or returning to the resting state with loss of H2.
Despite this significant progress in identifying intermediates in the nitrogenase N2 reduction pathway, as of yet, there is still no definitive characterization of an intermediate with a reduced N≡N triple bond. Only one intermediate containing two nitrogens has been observed, assigned to the E4 state. As noted in the initial characterization of this species,268 this is “a state in which FeMo-co binds the components of diazene (an N–N moiety, perhaps N2 and two [e−/H+] or diazene itself)”, i.e. it was not possible to distinguish whether this species has a reduced N-N bond or not. In addition to E4, this mechanism predicts that two additional intermediates (E5, E6) with a reduced N-N bond should be present that have not yet been observed. Since the intermediates that have been observed to date are either not diagnostic for any specific type of reduction pathway (the E7 and E8 intermediates with a single N) or the identity of the N-N species has not been conclusively demonstrated (E4), key aspects of the chemical mechanism of N2 reduction by nitrogenase remain open.
7. FROM STRUCTURAL TO MECHANISTIC UNDERSTANDING
Structures of the resting state of the MoFe and VFe proteins highlight conserved features of the active site cofactor relevant to the substrate reduction mechanism (Figs. 6, 14).
the cofactors are coordinated to the protein through only two sidechains, a cysteine bound to the apical Fe (Fe1) and a histidine coordinated to the heterometal.
the coordination sphere of the heterometal is completed through bidentate ligation by R-homocitrate.
the six Fe sites without a protein ligand (Fe2-Fe7) form a trigonal pyramid encapsulating a biologically unprecedented interstitial carbide ligand. The two triangular faces are bridged by three non-protein ligands – either three μ2 “belt sulfurs” in the FeMo cofactor or two μ2 belt sulfurs and a planar species, likely carbonate, in the FeV cofactor.
With the orchestrated addition of electrons, protons and substrate, the cofactor becomes activated to the state catalytically competent for substrate reduction and product formation. Key features of the integration of structure and mechanism have been detailed in Rohde et al.,172 and the steps in this process may be summarized:
electron transfer:
ATP-coupled electron transfer from the Fe protein results in sequential reduction of the MoFe protein (or the alternative nitrogenases), likely by single-electron transfer events, but a role for two-electron transfer processes cannot be ruled out. Although the Fe protein reduction potential is comparable to that of ferredoxins and flavodoxins (the natural reductants of Fe protein), these low potential electron transfer proteins cannot replace the Fe protein as the electron donor to the MoFe protein. Likewise, low-potential, small molecule reductants have not been identified that can replace Fe protein for the efficient reduction of N2. This suggests that the ATP requirement for substrate reduction reflects the importance of the proper timing of the electron transfer events more than the thermodynamic driving force. Indeed, the flux of electrons through the system and ATP/e− ratio is independent of the substrate reduced, indicative of an intermolecular electron transfer process where the electrons are delivered at constant potential from the Fe protein.
Mössbauer studies detailed above indicate that there is little change during turnover in the average oxidation state of MoFe protein irons, relative to the resting state. This indicates that there is neither a significant accumulation of electrons on the nitrogenase metalloclusters during turnover, nor is the substrate reduced by a significant number of electrons in one step. The resolution of this paradox of the transfer of electrons without the resulting accumulation of electrons on the metalloclusters was the key finding by Hoffman, Seefeldt and Dean that the electrons are used to form metal hydrides on the clusters.249,268,272–274 In this fashion, reducing equivalents could be accumulated without a build of charge that would thermodynamically disfavor successive electron transfers from the Fe protein.
proton transfer:
Including obligatory H2 production and protonation to form NH4+, the reduction of dinitrogen by nitrogenase is associated with the transfer of 10 protons to the buried active site. In addition to the R-homocitrate, the other potentially ionizable groups on the cofactor include the cluster sulfurs (based on the pH titration properties of synthetic and protein-based clusters)275–278 and the carbonate ligand in the VFe protein. Structural changes observed in the crystallographic analysis of MoFe proteins at low pH suggest that the belt sulfurs S3A and S5A are potential protonation sites.79 In the immediate protein environment, the sole group with an expected pKa near neutrality is His 195. Other hydrogen bonding interactions between the protein and cofactor belt sulfurs involve main chain NH groups and the guanidinium groups of nearby arginine residues; while these groups are not appreciably ionized at neutral pH, they could participate in proton transfer processes. No water molecules are within hydrogen bonding distance of the cluster sulfurs, although the long arm of homocitrate (i.e. the carboxylate arm in homocitrate with the additional methylene group relative to citrate) is surrounded by an extensive network of waters. Proton transfer pathways involving water channels have been identified.177,279,280 The solvation of homocitrate by this buried water pool is suggestive of a role for homocitrate in proton transfer. Still unclear is why homocitrate is essential for N2 fixation relative to citrate, as the “long” arm of the homocitrate points away from the cofactor, precluding any direct interaction between this feature and the cofactor in the absence of any rearrangements.
substrate binding:
Multiple potential access pathways have been identified for substrates to reach the active site, making it unlikely there is a unique path for this process.177,279–284 Since nitrogenase has a relatively leisurely turnover rate of about 1 N2 s−1 per active site, migration through the protein scaffold in the absence of permanent pathways should not be rate limiting, given the nanosecond-scale structural fluctuations that facilitate gas diffusion through proteins.285 Substrate access to the active site is insufficient for binding, however, as the cofactor needs to be reduced to the appropriate level. A routine demonstration of this requirement is that despite immersing crystals of the resting state forms of the MoFe protein or VFe protein in liquid substrate (liquid nitrogen) prior to data collection, binding of N2 to the active site (or indeed anywhere in the protein) has not been observed. Also, the observed removal of sulfide S2B in all ligand-bound structures obtained to date not only describes a possible binding site for N2, but the observed geometry is also very well suited to accommodate a bridging hydride at the very same position when modeled according to the bond distances and geometries observed in other metal-bridging hydride structures (Fig. 19). Importantly, in this conformation the inward-facing side chain of residue Gln 176 in the VFe protein structure from A. vinelandii forms a short hydrogen bond with the Nε nitrogen of His 180, stabilizing the protonated form of the imidazole moiety and thus preventing proton transfer from this like entry point for H+ during substrate reduction. In addition to the direct, stabilizing effect of the amide oxygen interaction with the positively polarized side of the hydride, this protective effect would extend the lifetime of the E2 state – at least under conditions of high electron flow – to allow for the next reduction steps to occur before H2 is lost due to unwanted protonation.
Figure 19:
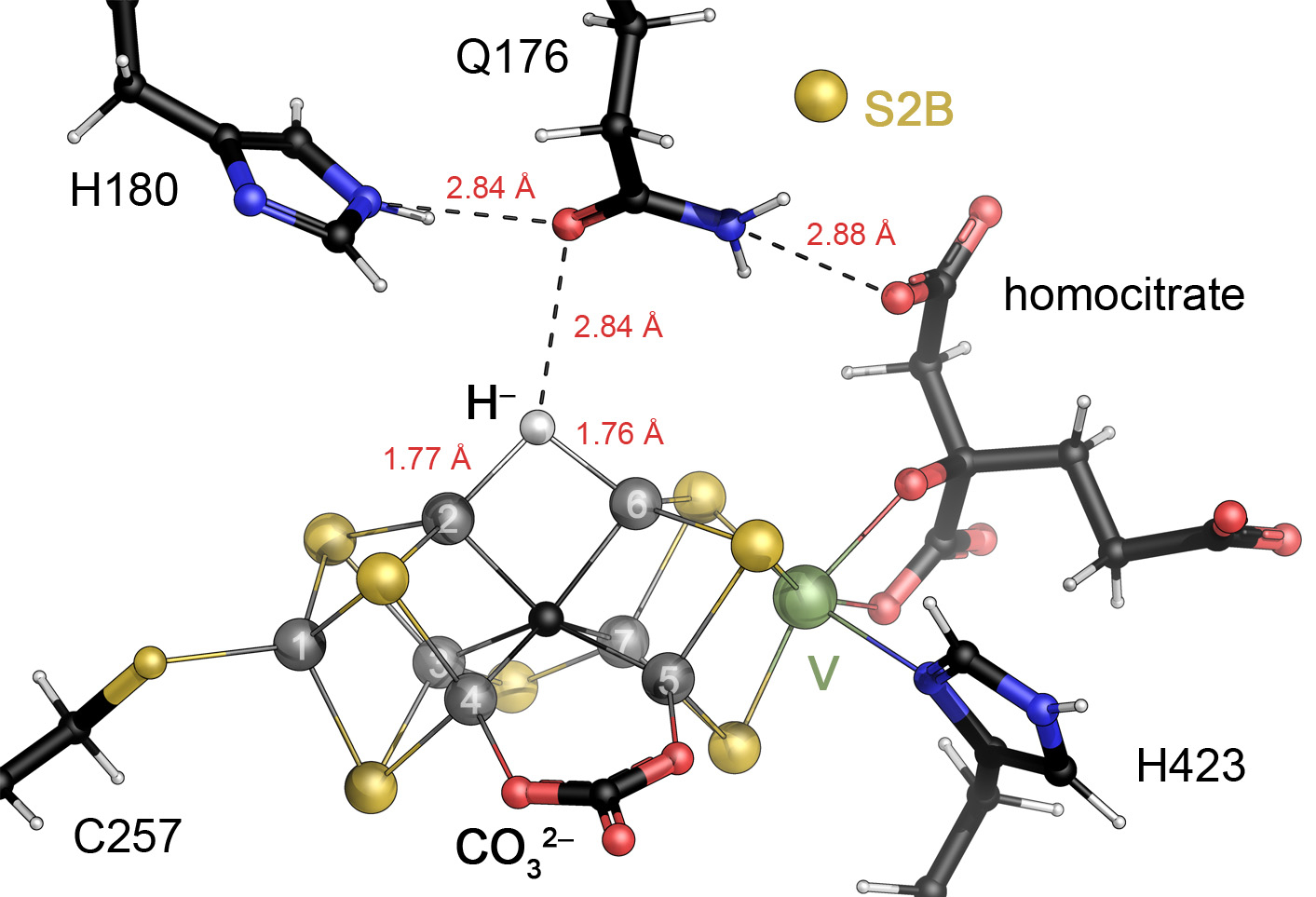
A model for hydride binding to Fe2 and Fe6 in FeV cofactor. Based on the turnover state structure of VFe protein from A. vinelandii, a bridging hydride can be modeled in place of the NH (or OH) ligand observed in the electron density map. Here, the positioning of residue Gln 176 between the likely proton source His 180 and the substrate binding site may be instrumental for the stabilization of the bridging hydride.
Furthermore, substrate binding to Fe2 and/or Fe6 is likely associated with the reduction of these sites, plausibly either by direct reduction following electron transfer from the Fe protein, or by the reductive elimination process described above. Protonation of the S2B sulfide will facilitate dissociation from the reduced Fe, thereby opening up these sites for substrate binding. The lack of reactivity of the FeMo cofactor in the resting state is likely due to the incorrect redox state of the cofactor, together with the belt sulfurs effectively serving as protecting groups that reversibly block substrate-binding sites. Given the rearrangements observed in the selenated FeMo cofactor under turnover conditions (Fig. 12B),149 it is possible that additional rearrangements may occur such that other iron sites participate in ligand binding,217 but there is as yet no direct experimental evidence for this.
The sequence of events following N2 binding have yet to be uniquely defined in terms of the specifics of the electron and proton transfers, including whether the N-N bond is reduced in 1-electron steps or possibly 2- or more electron steps, and identification of the specific sites of protonation of bound N2 that distinguish different reduction mechanisms, such as the distal vs alternating vs hybrid reduction mechanisms.253–255 Related questions are whether the same (or at least similar) mechanisms are utilized by nitrogenase during the reduction of N2 and such alternative substrates as C2H2 or substrates that undergo CC coupling reactions such as CO and CH3NC.
Experimental elucidation the atomic mechanism of N2 reduction will be ultimately limited by the transient nature of catalytic intermediates and the challenges in unambiguously establishing the temporal sequence of intermediate structures in a reaction mechanism. Computation is essential for the integration of structure, energetics and dynamics and studies to date have been valuable for exploring possible reaction pathways. At some future point, the nitrogenase mechanism will be amenable to ab initio studies, but in the meantime the integration of structure and experiment is essential for moving the problem forward.
8. CONCLUSIONS AND OUTLOOK
The past 5 years have seen major advances in our understanding of the mechanism of substrate reduction by nitrogenase, particularly in the identification of hydride and nitrogen containing intermediates by pulsed EPR methods 205 and in the crystallographic characterization of forms of nitrogenase establishing reversible displacement of a belt sulfur in the active site cofactor by exogenous ligands.39,147,149 Keeping in mind Halpern’s tenet “if you can identify a compound from a catalytic system, it is probably not the catalyst”,286 the challenge is to relate these observations to the nitrogenase catalytic mechanism. A related view on the mechanistic analysis of nitrogen fixing reactions was summarized by Chatt: “Unfortunately, it is not in general possible to isolate intermediates from these systems, and the proposed mechanisms are based on kinetic measurements, overall stoichiometry, and, not least, chemical intuition”.240 The latter quality has been the most elusive, but fortunately, advances in small molecule catalysts and computational chemistry are complementing multidisciplinary approaches on the enzymatic system to provide an increasingly detailed view inside the nitrogenase black box.
Acknowledgment
Research in the Einsle group was supported by Deutsche Forschungsgemeinschaft, RTG 1976 (project ID 235777276) and PP 1927 (project ID 273919336) and the European Research Council (grant no. 310656). Research in the Rees group was supported by US National Institutes of Health grant GM045162 and the Howard Hughes Medical Institute. Stimulating discussions with James B. Howard and members of the Einsle and Rees research groups are gratefully acknowledged.
Biography
Oliver Einsle obtained a diploma in Biology from Konstanz University, Germany, and a doctorate of natural sciences under the supervision of Peter Kroneck and Robert Huber at the Max-Planck-Institute for Biochemistry, Martinsried, Germany. He joined the group of Doug Rees at Caltech as a postdoctoral fellow in 2001 and was appointed junior professor for protein crystallography at the University of Göttingen, Germany, in 2003. Since 2008 he is the chair of Biochemistry at the Faculty for Chemistry and Pharmacy at the University of Freiburg, Germany, where he studies the structural and functional properties of metalloproteins and integral membrane proteins. He is currently the director of the Institute for Biochemistry and Dean of the Faculty of Chemistry and Pharmacy.
Douglas C. Rees received his BS in Molecular Biophysics and Biochemistry from Yale College and his PhD in Biophysics working with William Lipscomb at Harvard University. As a graduate student, Rees was introduced to nitrogenase through James B. Howard, at that time a sabbatical visitor with Lipscomb. Following postdoctoral work with Howard at the University of Minnesota, Rees joined the UCLA faculty in the Department of Chemistry and Biochemistry in 1982. He moved to the California Institute of Technology in 1989, where his group studies the structures and functions of metalloproteins and integral membrane proteins. Rees is an investigator of the Howard Hughes Medical Institute and is currently Dean of Graduate Studies at Caltech.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Guth JH; Burris RH Inhibition of Nitrogenase-Catalyzed NH3 Formation by H2. Biochemistry 1983, 22 (22), 5111. [DOI] [PubMed] [Google Scholar]
- (2).Snider MG; Temple BS; Wolfenden R The path to the transition state in enzyme reactions: a survey of catalytic efficiencies. J. Phys. Org. Chem. 2004, 17 (6–7), 586. [Google Scholar]
- (3).Ingolia NT; Lareau LF; Weissman JS Ribosome Profiling of Mouse Embryonic Stem Cells Reveals the Complexity and Dynamics of Mammalian Proteomes. Cell 2011, 147 (4), 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Garcia HG; Tikhonov M; Lin A; Gregor T Quantitative Imaging of Transcription in Living Drosophila Embryos Links Polymerase Activity to Patterning. Curr. Biol. 2013, 23 (21), 2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Eady RR Structure-function relationships of alternative nitrogenases. Chem. Rev. 1996, 96 (7), 3013. [DOI] [PubMed] [Google Scholar]
- (6).Vitousek PM; Menge DNL; Reed SC; Cleveland CC In Philos T Roy Soc B, 2013; Vol. 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bar-On YM; Milo R The global mass and average rate of rubisco. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bulen WA; LeComte JR The nitrogenase system from Azotobacter: two enzyme requirements for N2 reduction, ATP dependent H2 evolution and ATP hydrolysis. Proc. Natl. Acad. Sci. USA 1966, 56, 979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mortenson LE; Morris JA; Jeng DY Purification, metal composition and properties of molybdo-ferredoxin and azoferredoxin, two of the ocmponents of the nitrogen-fixing system of Clostridium pasteurianum. Biochim. Biophys. Acta 1967, 141, 516. [DOI] [PubMed] [Google Scholar]
- (10).Burns RC; Holsten RD; Hardy RWF Isolation by crystallization of the Mo-Fe protein of Azotobacter nitrogenase Biochem. Biophys. Res. Commun. 1970, 39 (1), 90. [DOI] [PubMed] [Google Scholar]
- (11).Shah VK; Brill WJ Nitrogenase. IV. Simple method of purification to homogeneity of nitrogenase components from Azotobacter vinelandii. Biochim. Biophys. Acta 1973, 305, 445. [DOI] [PubMed] [Google Scholar]
- (12).Burgess BK; Jacobs DB; Steifel EI Large scale purification of high activity Azotobacter vinelandii nitrogenase. Biochim. Biophys. Acta 1980, 614, 196. [DOI] [PubMed] [Google Scholar]
- (13).Wolle D; Kim C; Dean D; Howard JB Ionic interactions in the nitrogenase complex. Properties of Fe-protein containing substitutions for Arg-100. J. Biol. Chem. 1992, 267, 3667. [PubMed] [Google Scholar]
- (14).Weininger MS; Mortenson LE Crystallographic Properties of the MoFe Proteins of Nitrogenase from Clostridium pasteurianum and Azotobacter vinelandii. Proc. Natl. Acad. Sci. U. S. A. 1982, 79 (2), 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yamane T; Weininger MS; Mortenson LE; Rossmann MG Molecular symmetry of the MoFe protein of nitrogenase - structural homology/nitrogen fixation/ X-ray crystallography. J. Biol. Chem. 1982, 257 (3), 1221. [PubMed] [Google Scholar]
- (16).Rees DC; Howard JB Crystallization of the Azotobacter vinelandii nitrogenase iron protein. J. Biol. Chem. 1983, 258, 12733. [PubMed] [Google Scholar]
- (17).Bolin JT; Ronco AE; Morgan TV; Mortenson LE; Xuong NH The unusual metal clusters of nitrogenase: Structural features revealed by x-ray anomalous diffraction studies of the MoFe protein from Clostridium pasteurianum. Proc. Natl. Acad. Sci. U. S. A. 1993, 90 (3), 1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sosfenov NI; Andrianov VI; Vagin AA; Strokopytov BV; Vainshtein BK; Shilov AE; Gvozdev RI; Likhtenstein GI; Mitsova IZ; Blazhchuk IS X-ray diffraction study of the MoFe protein nitrogenase from Azotobacter vinelandii. Soviet Physics-Doklady; 1986, 31, 933. [Google Scholar]
- (19).Kim JS; Rees DC Crystallographic Structure and Functional Implications of the Nitrogenase Molybdenum Iron Protein from Azotobacter vinelandii. Nature 1992, 360 (6404), 553. [DOI] [PubMed] [Google Scholar]
- (20).Kim JS; Rees DC Structural Models for the Metal Centers in the Nitrogenase Molybdenum-Iron Protein. Science 1992, 257 (5077), 1677. [DOI] [PubMed] [Google Scholar]
- (21).Georgiadis MM; Komiya H; Chakrabarti P; Woo D; Kornuc JJ; Rees DC Crystallographic Structure of the Nitrogenase Iron Protein from Azotobacter vinelandii. Science 1992, 257 (5077), 1653. [DOI] [PubMed] [Google Scholar]
- (22).Kim J; Woo D; Rees DC X-Ray Crystal-Structure of the Nitrogenase Molybdenum Iron Protein from Clostridium pasteurianum at 3.0 Angstrom Resolution. Biochemistry 1993, 32 (28), 7104. [DOI] [PubMed] [Google Scholar]
- (23).Bolin JT; Campobasso N; Muchmore SW; Morgan TV; Mortenson LE In Molybdenum Enzymes, Cofactors and Model Systems. ACS Symposium Series No. 535; Stiefel EI;Coucouvanis D;Newton WE, Eds.; American Chemical Society: Washington, DC, 1993; Vol. 535. [Google Scholar]
- (24).Einsle O; Tezcan FA; Andrade SLA; Schmid B; Yoshida M; Howard JB; Rees DC Nitrogenase MoFe-protein at 1.16 Å resolution: A central ligand in the FeMo-cofactor. Science 2002, 297 (5587), 1696. [DOI] [PubMed] [Google Scholar]
- (25).Peters JW; Stowell MHB; Soltis SM; Finnegan MG; Johnson MK; Rees DC Redox-dependent structural changes in the nitrogenase P-cluster. Biochemistry 1997, 36 (6), 1181. [DOI] [PubMed] [Google Scholar]
- (26).Moore SJ; Sowa ST; Schuchardt C; Deery E; Lawrence AD; Ramos JV; Billig S; Birkemeyer C; Chivers PT; Howard MJ et al. Elucidation of the biosynthesis of the methane catalyst coenzyme F430. Nature 2017, 543 (7643), 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zheng KY; Ngo PD; Owens VL; Yang XP; Mansoorabadi SO The biosynthetic pathway of coenzyme F430 in methanogenic and methanotrophic archaea. Science 2016, 354 (6310), 339. [DOI] [PubMed] [Google Scholar]
- (28).Muraki N; Nomata J; Ebata K; Mizoguchi T; Shiba T; Tamiaki H; Kurisu G; Fujita Y X-ray crystal structure of the light-independent protochlorophyllide reductase. Nature 2010, 465 (7294), 110. [DOI] [PubMed] [Google Scholar]
- (29).Boyd ES; Peters JW New insights into the evolutionary history of biological nitrogen fixation. Front Microbiol 2013, 4, article 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Rupnik K; Lee CC; Hu YL; Ribbe MW; Hales BJ A VTVH MCD and EPR Spectroscopic Study of the Maturation of the “Second” Nitrogenase P-Cluster. Inorg. Chem. 2018, 57 (8), 4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Jimenez-Vicente E; Yang ZY; Ray WK; Echavarri-Erasun C; Cash VL; Rubio LM; Seefeldt LC; Dean DR Sequential and differential interaction of assembly factors during nitrogenase MoFe protein maturation. J. Biol. Chem. 2018, 293 (25), 9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Howard JB; Rees DC Nitrogenase - a Nucleotide-Dependent Molecular Switch. Annu. Rev. Biochem. 1994, 63, 235. [DOI] [PubMed] [Google Scholar]
- (33).Danyal K; Dean DR; Hoffman BM; Seefeldt LC Electron Transfer within Nitrogenase: Evidence for a Deficit-Spending Mechanism. Biochemistry 2011, 50 (43), 9255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lanzilotta WN; Seefeldt LC Changes in the midpoint potentials of the nitrogenase metal centers as a result of iron protein molybdenum-iron protein complex formation. Biochemistry 1997, 36 (42), 12976. [DOI] [PubMed] [Google Scholar]
- (35).Yang ZY; Ledbetter R; Shaw S; Pence N; Tokrnina-Lukaszewska M; Filers B; Guo QJ; Pokhrel N; Cash VL; Dean DR et al. Evidence That the Pi Release Event Is the Rate-Limiting Step in the Nitrogenase Catalytic Cycle. Biochemistry 2016, 55 (26), 3625. [DOI] [PubMed] [Google Scholar]
- (36).Owens CP; Katz FEH; Carter CH; Oswald VF; Tezcan FA Tyrosine-Coordinated P-Cluster in G. diazotrophicus Nitrogenase: Evidence for the Importance of O-Based Ligands in Conformationally Gated Electron Transfer. J. Am. Chem. Soc. 2016, 138 (32), 10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Cao LL; Borner MC; Bergmann J; Caldararu O; Ryde U Geometry and Electronic Structure of the P-Cluster in Nitrogenase Studied by Combined Quantum Mechanical and Molecular Mechanical Calculations and Quantum Refinement. Inorg. Chem. 2019, 58 (15), 9672. [DOI] [PubMed] [Google Scholar]
- (38).Sippel D; Einsle O The structure of vanadium nitrogenase reveals an unusual bridging ligand. Nat Chem Biol 2017, 13 (9), 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sippel D; Rohde M; Netzer J; Trncik C; Gies J; Grunau K; Djurdjevic I; Decamps L; Andrade SLA; Einsle O A bound reaction intermediate sheds light on the mechanism of nitrogenase. Science 2018, 359 (6383), 1484. [DOI] [PubMed] [Google Scholar]
- (40).Fritsch J; Scheerer P; Frielingsdorf S; Kroschinsky S; Friedrich B; Lenz O; Spahn CMT The crystal structure of an oxygen-tolerant hydrogenase uncovers a novel iron-sulphur centre. Nature 2011, 479 (7372), 249. [DOI] [PubMed] [Google Scholar]
- (41).Shomura Y; Yoon KS; Nishihara H; Higuchi Y Structural basis for a [4Fe-3S] cluster in the oxygen-tolerant membrane-bound [NiFe]-hydrogenase. Nature 2011, 479 (7372), 253. [DOI] [PubMed] [Google Scholar]
- (42).Keable SM; Zadvornyy OA; Johnson LE; Ginovska B; Rasmussen AJ; Danyal K; Eilers BJ; Prussia GA; LeVan AX; Raugei S et al. Structural characterization of the P1+ intermediate state of the P-cluster of nitrogenase. J. Biol. Chem. 2018, 293 (25), 9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Nyborg AC; Johnson JL; Gunn A; Watt GD Evidence for a two-electron transfer using the all-ferrous Fe protein during nitrogenase catalysis. J. Biol. Chem. 2000, 275 (50), 39307. [DOI] [PubMed] [Google Scholar]
- (44).Jacobs D; Watt GD Nucleotide-Assisted [Fe4:S4] Redox State Interconversions of the Azotobacter vinelandii Fe Protein and Their Relevance to Nitrogenase Catalysis. Biochemistry 2013, 52 (28), 4791. [DOI] [PubMed] [Google Scholar]
- (45).Kent HM; Ioannidis I; Gormal C; Smith BE; Buck M Site-Directed Mutagenesis of the Klebsiella pneumoniae Nitrogenase - Effects of Modifying Conserved Cysteine Residues in the alpha- and beta-Subunits. Biochem. J. 1989, 264 (1), 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Kent HM; Baines M; Gormal C; Smith BE; Buck M Analysis of Site-Directed Mutations in the alpha- and beta-Subunits of Klebsiella pneumoniae Nitrogenase. Mol. Microbiol. 1990, 4 (9), 1497. [PubMed] [Google Scholar]
- (47).Dean DR; Setterquist RA; Brigle KE; Scott DJ; Laird NF; Newton WE Evidence That Conserved Residues Cys-62 and Cys-154 within the Azotobacter vinelandii Nitrogenase MoFe Protein alpha-Subunit Are Essential for Nitrogenase Activity but Conserved Residues His-83 and Cys-88 Are Not. Mol. Microbiol. 1990, 4 (9), 1505. [PubMed] [Google Scholar]
- (48).May HD; Dean DR; Newton WE Altered Nitrogenase Mofe Proteins from Azotobacter vinelandii - Analysis of MoFfe Proteins Having Amino-Acid Substitutions for the Conserved Cysteine Residues within the Beta-Subunit. Biochem. J. 1991, 277, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Rutledge HL; Rittle J; Williamson LM; Xu WQA; Gagnon DM; Tezcan FA Redox-Dependent Metastability of the Nitrogenase P-Cluster. J. Am. Chem. Soc. 2019, 141 (25), 10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Einsle O Nitrogenase FeMo cofactor: an atomic structure in three simple steps. J. Biol. Inorg. Chem. 2014, 19 (6), 737. [DOI] [PubMed] [Google Scholar]
- (51).Gronberg KLC; Gormal CA; Durrant MC; Smith BE; Henderson RA Why R-Homocitrate is essential to the reactivity of FeMo-cofactor of nitrogenase: Studies on NifV(−)-extracted FeMo-cofactor. J. Am. Chem. Soc. 1998, 120 (41), 10613. [Google Scholar]
- (52).Kennedy C; Dean D The NifU, NifS and NifV Gene-Products Are Required for Activity of All 3 Nitrogenases of Azotobacter vinelandii. Mol. Gen. Genet. 1992, 231 (3), 494. [DOI] [PubMed] [Google Scholar]
- (53).Hausinger RP; Howard JB Thiol Reactivity of the Nitrogenase Fe-Protein from Azotobacter vinelandii. J. Biol. Chem. 1983, 258 (22), 3486. [PubMed] [Google Scholar]
- (54).Howard JB; Davis R; Moldenhauer B; Cash VL; Dean D Fe-S Cluster Ligands Are the Only Cysteines Required for Nitrogenase Fe-Protein Activities. J. Biol. Chem. 1989, 264 (19), 11270. [PubMed] [Google Scholar]
- (55).Robson RL Identification of Possible Adenine Nucleotide-Binding Sites in Nitrogenase Fe-Proteins and MoFe-Proteins by Amino-Acid-Sequence Comparison. FEBS Lett. 1984, 173 (2), 394. [DOI] [PubMed] [Google Scholar]
- (56).Koonin EV A Superfamily of ATPases with Diverse Functions Containing Either Classical or Deviant ATP-Binding Motif. J. Mol. Biol. 1993, 229 (4), 1165. [DOI] [PubMed] [Google Scholar]
- (57).Leipe DD; Wolf YI; Koonin EV; Aravind L Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol. 2002, 317 (1), 41. [DOI] [PubMed] [Google Scholar]
- (58).Bange G; Sinning I SIMIBI twins in protein targeting and localization. Nat Struct Mol Biol 2013, 20 (7), 776. [DOI] [PubMed] [Google Scholar]
- (59).Schlessman JL; Woo D; Joshua-Tor L; Howard JB; Rees DC Conformational variability in structures of the nitrogenase iron proteins from Azotobacter vinelandii and Clostridium pasteurianum. J. Mol. Biol. 1998, 280 (4), 669. [DOI] [PubMed] [Google Scholar]
- (60).Rettberg LA; Kang W; Stiebritz MT; Hiller CJ; Lee CC; Liedtke J; Ribbe MW; Hu YL Structural Analysis of a Nitrogenase Iron Protein from Methanosarcina acetivorans: Implications for CO2 Capture by a Surface-Exposed [Fe4S4] Cluster. Mbio 2019, 10 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Rohde M; Trncik C; Sippel D; Gerhardt S; Einsle O Crystal structure of VnfH, the iron protein component of vanadium nitrogenase. J. Biol. Inorg. Chem. 2018, 23, 1049. [DOI] [PubMed] [Google Scholar]
- (62).Watt GD; Reddy KRN Formation of an All-Ferrous Fe4S4 Cluster in the Iron Protein Component of Azotobacter vinelandii Nitrogenase. J. Inorg. Biochem. 1994, 53 (4), 281. [Google Scholar]
- (63).Angove HC; Yoo SJ; Munck E; Burgess BK An all-ferrous state of the Fe protein of nitrogenase - Interaction with nucleotides and electron transfer to the MoFe protein. J. Biol. Chem. 1998, 273 (41), 26330. [DOI] [PubMed] [Google Scholar]
- (64).Lowery TJ; Wilson PE; Zhang B; Bunker J; Harrison RG; Nyborg AC; Thiriot D; Watt GD Flavodoxin hydroquinone reduces Azotobacter vinelandii Fe protein to the all-ferrous redox state with a S=0 spin state. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (46), 17131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Beinert H; Holm RH; Münck E Iron-sulfur clusters: Nature’s modular, multipurpose structures. Science 1997, 277 (5326), 653. [DOI] [PubMed] [Google Scholar]
- (66).Noodleman L; Peng CY; Case DA; Mouesca JM Orbital Interactions, Electron Delocalization and Spin Coupling in Iron-Sulfur Clusters. Coord. Chem. Rev. 1995, 144, 199. [Google Scholar]
- (67).Wenke BB; Spatzal T; Rees DC Site-Specific Oxidation State Assignments of the Iron Atoms in the [4Fe:4S](2+/1+/0) States of the Nitrogenase Fe-Protein. Angew Chem Int Edit 2019, 58 (12), 3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Schindelin H; Kisker C; Sehlessman JL; Howard JB; Rees DC Structure of ADP • AIF4−-stabilized nitrogenase complex and its implications for signal transduction. Nature 1997, 387 (6631), 370. [DOI] [PubMed] [Google Scholar]
- (69).Schmid B; Einsle O; Chiu HJ; Willing A; Yoshida M; Howard JB; Rees DC Biochemical and structural characterization of the cross-linked complex of nitrogenase: Comparison to the ADP-AIF4−-stabilized structure. Biochemistry 2002, 41 (52), 15557. [DOI] [PubMed] [Google Scholar]
- (70).Wittinghofer A Signaling mechanistics: Aluminum fluoride for molecule of the year. Curr. Biol. 1997, 7 (11), R682. [DOI] [PubMed] [Google Scholar]
- (71).Stanley RJ; Thomas GMH Activation of G Proteins by Guanine Nucleotide Exchange Factors Relies on GTPase Activity. Plos One 2016, 11 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Goody RS How not to do kinetics: examples involving GTPases and guanine nucleotide exchange factors. Febs J 2014, 281 (2), 593. [DOI] [PubMed] [Google Scholar]
- (73).Tezcan FA; Kaiser JT; Mustafi D; Walton MY; Howard JB; Rees DC Nitrogenase complexes: Multiple docking sites for a nucleotide switch protein. Science 2005, 309 (5739), 1377. [DOI] [PubMed] [Google Scholar]
- (74).Tezcan FA; Kaiser JT; Howard JB; Rees DC Structural Evidence for Asymmetrical Nucleotide Interactions in Nitrogenase. J. Am. Chem. Soc. 2015, 137 (1), 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Kim J; Woo D; Rees DC X-ray crystal structure of the nitrogenase molybdenum-iron protein from Clostridium pasteurianum at 3.0 Å resolution. Biochemistry 1993, 32, 7104. [DOI] [PubMed] [Google Scholar]
- (76).Mayer SM; Gormal CA; Smith BE; Lawson DM Crystallographic analysis of the MoFe protein of nitrogenase from a nifV mutant of Klebsiella pneumoniae identifies citrate as a ligand to the molybdenum of iron molybdenum cofactor (FeMoco). J. Biol. Chem. 2002, 277 (38), 35263. [DOI] [PubMed] [Google Scholar]
- (77).Mayer SM; Lawson DM; Gormal CA; Roe SM; Smith BE New insights into structure-function relationships in nitrogenase: A 1.6 Å resolution X-ray crystallographic study of Klebsiella pneumoniae MoFe-protein. J. Mol. Biol. 1999, 292 (4), 871. [DOI] [PubMed] [Google Scholar]
- (78).Howard JB; Kechris KJ; Rees DC; Glazer AN Multiple Amino Acid Sequence Alignment Nitrogenase Component 1: Insights into Phylogenetics and Structure-Function Relationships. Plos One 2013, 8 (9), e72751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Morrison CN; Spatzal T; Rees DC Reversible protonated resting state of the nitrogenase active site. J. Am. Chem. Soc. 2017, 139 (31), 10856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Shah VK; Brill WJ Isolation of an Iron-Molybdenum Cofactor from Nitrogenase. Proc. Natl. Acad. Sci. U. S. A. 1977, 74 (8), 3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Holm RH Metal-Clusters in Biology - Quest for a Synthetic Representation of the Catalytic Site of Nitrogenase. Chem. Soc. Rev. 1981, 10 (4), 455. [Google Scholar]
- (82).Coucouvanis D Fe-M-S Complexes Derived from Ms42− Anions (M=Mo,W) and Their Possible Relevance as Analogs for Structural Features in the Mo Site of Nitrogenase. Acc. Chem. Res. 1981, 14 (7), 201. [Google Scholar]
- (83).Smith BE; Eady RR Metalloclusters of the Nitrogenases. Eur. J. Biochem. 1992, 205 (1), 1. [DOI] [PubMed] [Google Scholar]
- (84).Kurtz DM; Mcmillan RS; Burgess BK; Mortenson LE; Holm RH Identification of Iron-Sulfur Centers in the Iron-Molybdenum Proteins of Nitrogenase. Proc. Natl. Acad. Sci. U. S. A. 1979, 76 (10), 4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Chan MK; Kim JS; Rees DC The Nitrogenase Femo-Cofactor and P-Cluster Pair - 2.2 Å Resolution Structures. Science 1993, 260 (5109), 792. [DOI] [PubMed] [Google Scholar]
- (86).Yang SS; Pan WH; Friesen GD; Burgess BK; Corbin JL; Stiefel EI; Newton WE Iron-Molybdenum Cofactor from Nitrogenase - Modified Extraction Methods as Probes for Composition. J. Biol. Chem. 1982, 257 (14), 8042. [PubMed] [Google Scholar]
- (87).Mclean PA; Wink DA; Chapman SK; Hickman AB; Mckillop DM; Orme-Johnson WH A New Method for Extraction of Iron Molybdenum Cofactor (FeMoco) from Nitrogenase Adsorbed to DEAE-Cellulose .1. Effects of Anions, Cations, and Preextraction Treatments. Biochemistry 1989, 28 (24), 9402. [DOI] [PubMed] [Google Scholar]
- (88).Wink DA; Mclean PA; Hickman AB; Orme-Johnson WH A New Method for Extraction of Iron Molybdenum Cofactor (FeMoco) from Nitrogenase Adsorbed to DEAE-Cellulose .2. Solubilization of FeMoco in a Wide Range of Organic Solvents. Biochemistry 1989, 28 (24), 9407. [DOI] [PubMed] [Google Scholar]
- (89).Robinson AC; Burgess BK; Dean DR Activity, Reconstitution, and Accumulation of Nitrogenase Components in Azotobacter vinelandii Mutant Strains Containing Defined Deletions within the Nitrogenase Structural Gene-Cluster. J. Bacteriol. 1986, 166 (1), 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Robinson AC; Chun TW; Li JG; Burgess BK Iron-Molybdenum Cofactor Insertion into the Apo-MoFe Protein of Nitrogenase Involves the Iron Protein-MgATP Complex. J. Biol. Chem. 1989, 264 (17), 10088. [PubMed] [Google Scholar]
- (91).Fay AW; Hu Y; Schmid B; Ribbe MW Molecular insights into nitrogenase FeMoco insertion - The role of His 274 and His 451 of MoFe protein alpha subunit. J. Inorg. Biochem. 2007, 101 (11–12), 1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Fay AW; Blank MA; Lee CC; Hu YL; Hodgson KO; Hedman B; Ribbe MW Characterization of Isolated Nitrogenase FeVco. J. Am. Chem. Soc. 2010, 132 (36), 12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Fay AW; Lee CC; Wiig JA; Hu YL; Ribbe MW Protocols for Cofactor Isolation of Nitrogenase. Nitrogen Fixation: Methods and Protocols 2011, 766, 239. [DOI] [PubMed] [Google Scholar]
- (94).Thorneley RNF; Lowe DJ In Molybdenum Enzymes; Spiro TG, Ed.; Wiley-Interscience: New York, 1985; Vol. 1. [Google Scholar]
- (95).Deng HB; Hoffmann R How N2 Might Be Activated by the FeMo-Cofactor in Nitrogenase. Angew Chem Int Edit 1993, 32 (7), 1062. [Google Scholar]
- (96).Lee HI; Benton PMC; Laryukhin M; Igarashi RY; Dean DR; Seefeldt LC; Hoffman BM The interstitial atom of the nitrogenase FeMo-cofactor: ENDOR and ESEEM show it is not an exchangeable nitrogen. J. Am. Chem. Soc. 2003, 125 (19), 5604. [DOI] [PubMed] [Google Scholar]
- (97).Yang TC; Maeser NK; Laryukhin M; Lee HI; Dean DR; Seefeldt LC; Hoffman BM The interstitial atom of the nitrogenase FeMo-Cofactor: ENDOR and ESEEM evidence that it is not a nitrogen. J. Am. Chem. Soc. 2005, 127 (37), 12804. [DOI] [PubMed] [Google Scholar]
- (98).Lovell T; Li J; Case DA; Noodleman L FeMo cofactor of nitrogenase: energetics and local interactions in the protein environment. J. Biol. Inorg. Chem. 2002, 7 (7–8), 735. [DOI] [PubMed] [Google Scholar]
- (99).Lovell T; Li J; Liu TQ; Case DA; Noodleman L FeMo cofactor of nitrogenase: A density functional study of states MN, MOX, MR, and MI. J. Am. Chem. Soc. 2001, 123 (49), 12392. [DOI] [PubMed] [Google Scholar]
- (100).Lovell T; Liu TQ; Case DA; Noodleman L Structural, spectroscopic, and redox consequences of central ligand in the FeMoco of nitrogenase: A density functional theoretical study. J. Am. Chem. Soc. 2003, 125 (27), 8377. [DOI] [PubMed] [Google Scholar]
- (101).Lukoyanov D; Pelmenschikov V; Maeser N; Laryukhin M; Yang TC; Noodleman L; Dean DR; Case DA; Seefeldt LC; Hoffman BM Testing if the interstitial atom, X, of the nitrogenase molybdenum-iron cofactor is N or C: ENDOR, ESEEM, and DFT studies of the S=3/2 resting state in multiple environments. Inorg. Chem. 2007, 46 (26), 11437. [DOI] [PubMed] [Google Scholar]
- (102).Lancaster KM; Roemelt M; Ettenhuber P; Hu YL; Ribbe MW; Neese F; Bergmann U; DeBeer S X-ray Emission Spectroscopy Evidences a Central Carbon in the Nitrogenase Iron-Molybdenum Cofactor. Science 2011, 334 (6058), 974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Lancaster KM; Hu YL; Bergmann U; Ribbe MW; DeBeer S X-ray Spectroscopic Observation of an Interstitial Carbide in NifEN-Bound FeMoco Precursor. J. Am. Chem. Soc. 2013, 135 (2), 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Spatzal T; Aksoyoğlu M; Zhang LM; Andrade SLA; Schleicher E; Weber S; Rees DC; Einsle O Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor. Science 2011, 334 (6058), 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Wiig JA; Hu YL; Lee CC; Ribbe MW Radical SAM-Dependent Carbon Insertion into the Nitrogenase M-Cluster. Science 2012, 337 (6102), 1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Hinnemann B; Nørskov JK Modeling a central ligand in the nitrogenase FeMo cofactor. J. Am. Chem. Soc. 2003, 125 (6), 1466. [DOI] [PubMed] [Google Scholar]
- (107).Varley JB; Wang Y; Chan K; Studt F; Norskov JK Mechanistic insights into nitrogen fixation by nitrogenase enzymes. Phys Chem Chem Phys 2015, 17 (44), 29541. [DOI] [PubMed] [Google Scholar]
- (108).Kästner J; Blöchl PE Towards an understanding of the workings of nitrogenase from DFT calculations. Chemphyschem 2005, 6 (9), 1724. [DOI] [PubMed] [Google Scholar]
- (109).Siegbahn PEM Model Calculations Suggest that the Central Carbon in the FeMo-Cofactor of Nitrogenase Becomes Protonated in the Process of Nitrogen Fixation. J. Am. Chem. Soc. 2016, 138 (33), 10485. [DOI] [PubMed] [Google Scholar]
- (110).Hawkes TR; Smith BE The Inactive Mofe Protein (NifB−Kp1) of the Nitrogenase from nif B Mutants of Klebsiella pneumoniae - Its Interaction with Femo-Cofactor and the Properties of the Active Mofe Protein Formed. Biochem. J. 1984, 223 (3), 783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Rod TH; Nørskov JK Modeling the nitrogenase FeMo cofactor. J. Am. Chem. Soc. 2000, 122 (51), 12751. [Google Scholar]
- (112).Hinnemann B; Norskov JK Chemical activity of the nitrogenase FeMo cofactor with a central nitrogen ligand: Density functional study. J. Am. Chem. Soc. 2004, 126 (12), 3920. [DOI] [PubMed] [Google Scholar]
- (113).Schimpl J; Petrilli HM; Blöchl PE Nitrogen binding to the FeMo-cofactor of nitrogenase. J. Am. Chem. Soc. 2003, 125 (51), 15772. [DOI] [PubMed] [Google Scholar]
- (114).Kästner J; Hemmen S; Blöchl PE Activation and protonation of dinitrogen at the FeMo cofactor of nitrogenase. J. Chem. Phys. 2005, 123 (7). [DOI] [PubMed] [Google Scholar]
- (115).Siegbahn PEM; Westerberg J; Svensson M; Crabtree RH Nitrogen fixation by nitrogenases: A quantum chemical study. J Phys Chem B 1998, 102 (9), 1615. [Google Scholar]
- (116).Lee HI; Hales BJ; Hoffman BM Metal-ion valencies of the FeMo cofactor in CO-inhibited and resting state nitrogenase by 57Fe Q-band ENDOR. J. Am. Chem. Soc. 1997, 119 (47), 11395. [Google Scholar]
- (117).Yoo SJ; Angove HC; Papaefthymiou V; Burgess BK; Münck E Mössbauer study of the MoFe protein of nitrogenase from Azotobacter vinelandii using selective 57Fe enrichment of the M-centers. J. Am. Chem. Soc. 2000, 122, 4926. [Google Scholar]
- (118).Mendel RR; Bittner F Cell biology of molybdenum. Bba-Mol Cell Res 2006, 1763 (7), 621. [DOI] [PubMed] [Google Scholar]
- (119).Mendel RR The Molybdenum Cofactor. J. Biol. Chem. 2013, 288 (19), 13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Fomitchev DV; McLauchlan CC; Holm RH Heterometal cubane-type MFe3S4 clusters (M = Mo, V) trigonally symmetrized with hydrotris(pyrazolyl)borate(1−) and tris(pyrazolyl)methanesulfonate(1−) capping ligands. Inorg. Chem. 2002, 41 (4), 958. [DOI] [PubMed] [Google Scholar]
- (121).Björnsson R; Lima FA; Spatzal T; Weyhermüller T; Glatzel P; Bill E; Einsle O; Neese F; DeBeer S Identification of a spin-coupled Mo(III) in the nitrogenase iron-molybdenum cofactor. Chem Sci 2014, 5 (8), 3096. [Google Scholar]
- (122).Friedel G Sur les symétries cristallines que peut révéler la diffraction des rayons Röntgen. Comptes Rendus de l’Academie des Sciences. 1913, 157, 1533. [Google Scholar]
- (123).Einsle O; Andrade SL; Dobbek H; Meyer J; Rees DC Assignment of individual metal redox states in a metalloprotein by crystallographic refinement at multiple X-ray wavelengths. J. Am. Chem. Soc. 2007, 129 (8), 2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Spatzal T; Schlesier J; Burger EM; Sippel D; Zhang LM; Andrade SLA; Rees DC; Einsle O Nitrogenase FeMoco investigated by spatially resolved anomalous dispersion refinement. Nat. Commun. 2016, 7, 10902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Björnsson R; Neese F; DeBeer S Revisiting the Mössbauer Isomer Shifts of the FeMoco Cluster of Nitrogenase and the Cofactor Charge. Inorg. Chem. 2017, 56, 1470. [DOI] [PubMed] [Google Scholar]
- (126).Björnsson R; Neese F; Schrock RR; Einsle O; DeBeer S The discovery of Mo(III) in FeMoco: reuniting enzyme and model chemistry. J. Biol. Inorg. Chem. 2015, 20 (2), 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Spatzal T; Einsle O; Andrade SL Analysis of the Magnetic Properties of Nitrogenase FeMo Cofactor by Single-Crystal EPR Spectroscopy. Angew. Chem. 2013, 52, 10116. [DOI] [PubMed] [Google Scholar]
- (128).Zhang YG; Zuo JL; Zhou HC; Holm RH Rearrangement of symmetrical dicubane clusters into topological analogues of the P-cluster of nitrogenase: Nature’s choice? J. Am. Chem. Soc. 2002, 124 (48), 14292. [DOI] [PubMed] [Google Scholar]
- (129).Ohki Y; Ikagawa Y; Tatsumi K Synthesis of new [8Fe-7S] clusters: A topological link between the core structures of P-cluster, FeMo-co, and FeFe-co of nitrogenases. J. Am. Chem. Soc. 2007, 129 (34), 10457. [DOI] [PubMed] [Google Scholar]
- (130).Rittle J; Peters JC Fe-N2/CO complexes that model a possible role for the interstitial C atom of FeMo-cofactor (FeMoco). Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (40), 15898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Takaoka A; Mankad NP; Peters JC Dinitrogen Complexes of Sulfur-Ligated Iron. J. Am. Chem. Soc. 2011, 133 (22), 8440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Demadis KD; Malinak SM; Coucouvanis D Catalytic reduction of hydrazine to ammonia with MoFe3S4-polycarboxylate clusters. Possible relevance regarding the function of the molybdenum-coordinated homocitrate in nitrogenase. Inorg. Chem. 1996, 35 (13), 4038. [DOI] [PubMed] [Google Scholar]
- (133).Yandulov DV; Schrock RR Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Science 2003, 301 (5629), 76. [DOI] [PubMed] [Google Scholar]
- (134).Arashiba K; Miyake Y; Nishibayashi Y A molybdenum complex bearing PNP-type pincer ligands leads to the catalytic reduction of dinitrogen into ammonia. Nat Chem 2011, 3 (2), 120. [DOI] [PubMed] [Google Scholar]
- (135).Weare WW; Dai X; Byrnes MJ; Chin JM; Schrock RR; Muller P Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (46), 17099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Schrock RR Catalytic reduction of dinitrogen to ammonia at well-defined single metal sites. Philos. Trans. R. Soc. London, A 2005, 363 (1829), 959. [DOI] [PubMed] [Google Scholar]
- (137).Hwang JC; Chen CH; Burris RH Inhibition of Nitrogenase-Catalyzed Reductions. Biochim. Biophys. Acta 1973, 292 (1), 256. [DOI] [PubMed] [Google Scholar]
- (138).Cameron LM; Hales BJ Investigation and CO binding and release from Mo-nitrogenase during catalytic turnover. Biochemistry 1998, 37, 9449. [DOI] [PubMed] [Google Scholar]
- (139).Lee HI; Sorlie M; Christiansen J; Yang TC; Shao JL; Dean DR; Hales BJ; Hoffman BM Electron inventory, kinetic assignment (En), structure, end bonding of nitrogenase turnover intermediates with C2H2 and CO. J. Am. Chem. Soc. 2005, 127 (45), 15880. [DOI] [PubMed] [Google Scholar]
- (140).Lowe DJ; Eady RR; Thorneley RNF Electron-paramagnetic-resonance studies on nitrogenase of Klebsiella pneumonia. Biochem. J. 1978, 173, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Davis LC; Henzl MT; Burris RH; Orme-Johnson WH Iron-Sulfur Clusters in the Molybdenum-Iron Protein-Component of Nitrogenase - Electron-Paramagnetic Resonance of the Carbon-Monoxide Inhibited State. Biochemistry 1979, 18 (22), 4860. [DOI] [PubMed] [Google Scholar]
- (142).Lee H-I; Cameron LM; Hales BJ; Hoffman BJ CO binding to the FeMo cofactor of CO-inhibited nitrogenase: 13CO and 1H Q-band ENDOR investigation. J. Am. Chem. Soc. 1997, 119, 10121. [Google Scholar]
- (143).Pollock RC; Lee HI; Camereon LM; DeRose VJ; Hales BJ; Orme-Johnson WH; Hoffman BM Investigation of CO bound to inhibited forms of nitrogenase MoFe protein by 13C ENDOR. J. Am. Chem. Soc. 1995, 117, 8686. [Google Scholar]
- (144).George SJ; Ashby GA; Wharton CW; Thorneley RNF Time-resolved binding of carbon monoxide to nitrogenase monitored by stopped-flow infrared spectroscopy. J. Am. Chem. Soc. 1997, 119, 6450. [Google Scholar]
- (145).Maskos Z; Hales BJ Photo-lability of CO bound to Mo-nitrogenase from Azotobacter vinelandii. J. Inorg. Biochem. 2003, 93 (1–2), 11. [DOI] [PubMed] [Google Scholar]
- (146).Yan LF; Pelmenschikov V; Dapper CH; Scott AD; Newton WE; Cramer SP IR-monitored photolysis of CO-inhibited nitrogenase: A major EPR-silent species with coupled terminal CO ligands. Chemistry-a Eur. J. 2012, 18 (51), 16349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Spatzal T; Perez KA; Einsle O; Howard JB; Rees DC Ligand binding to the FeMo-cofactor: structures of CO-bound and reactivated nitrogenase. Science 2014, 345 (6204), 1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Coric I; Mercado BQ; Bill E; Vinyard DJ; Holland PL Binding of dinitrogen to an iron-sulfur-carbon site. Nature 2015, 526 (7571), 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Spatzal T; Perez KA; Howard JB; Rees DC Catalysis-dependent selenium incorporation and migration in the nitrogenase active site iron-molybdenum cofactor. Elife 2015, 4, e11620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Lee CC; Hu YL; Ribbe MW Vanadium Nitrogenase Reduces CO. Science 2010, 329 (5992), 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Lee CC; Tanifuji K; Newcomb M; Liedtke J; Hu YL; Ribbe MW A Comparative Analysis of the CO-Reducing Activities of MoFe Proteins Containing Mo- and V-Nitrogenase Cofactors. Chembiochem 2018, 19 (7), 649. [DOI] [PubMed] [Google Scholar]
- (152).Lee CC; Wilcoxen J; Hiller CJ; Britt RD; Hu YL Evaluation of the Catalytic Relevance of the CO-Bound States of V-Nitrogenase. Angew Chem Int Edit 2018, 57 (13), 3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Lee CC; Hu YL; Ribbe MW ATP-Independent Formation of Hydrocarbons Catalyzed by Isolated Nitrogenase Cofactors. Angew Chem Int Edit 2012, 51 (8), 1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Hu YL; Lee CC; Ribbe MW Extending the Carbon Chain: Hydrocarbon Formation Catalyzed by Vanadium/Molybdenum Nitrogenases. Science 2011, 333 (6043), 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Burns RC; Fuchsman WH; Hardy RWF Nitrogenase from Vanadium-Grown Azotobacter - Isolation, Characteristics, and Mechanistic Implications. Biochemical and Biophysical Research Communications 1971, 42 (3), 353. [DOI] [PubMed] [Google Scholar]
- (156).Benemann JR; Kamen MD; Traylor TG; Lie RF; McKenna CE Vanadium Effect in Nitrogen Fixation by Azotobacter. Biochim. Biophys. Acta 1972, 264 (1), 25. [DOI] [PubMed] [Google Scholar]
- (157).Bishop PE; Jarlenski DML; Hetherington DR Evidence for an Alternative Nitrogen Fixation System in Azotobacter vinelandii. Proc. Natl. Acad. Sci. U. S. A. 1980, 77 (12), 7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Bishop PE; Hawkins ME; Eady RR Nitrogen fixation in molybdenum-deficient continuous culture by a strain of Azotobacter vinelandii carrying a deletion of the structural genes for nitrogenase (nifHDK). Biochem. J. 1986, 238 (2), 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Bishop PE; Premakumar R; Dean DR; Jacobson MR; Chisnell JR; Rizzo TM; Kopczynski J Nitrogen Fixation by Azotobacter vinelandii Strains Having Deletions in Structural Genes for Nitrogenase. Science 1986, 232 (4746), 92. [DOI] [PubMed] [Google Scholar]
- (160).Hales BJ; Case EE; Morningstar JE; Dzeda MF; Mauterer LA Isolation of a New Vanadium-Containing Nitrogenase from Azotobacter vinelandii. Biochemistry 1986, 25 (23), 7253. [DOI] [PubMed] [Google Scholar]
- (161).Eady RR; Robson RL; Richardson TH; Miller RW; Hawkins M The Vanadium Nitrogenase of Azotobacter chroococcum - Purification and Properties of the VFe Protein. Biochem. J. 1987, 244 (1), 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Lee CC; Hu YL; Ribbe MW Unique features of the nitrogenase VFe protein from Azotobacter vinelandii. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (23), 9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Lipman JG Experiments on the transformation and fixation of nitrogen by bacteria. Rep.New Jersey Agric.Exper.Stat 1903, 24, 217. [Google Scholar]
- (164).Sippel D; Schlesier J; Rohde M; Trncik C; Decamps L; Djurdjevic I; Spatzal T; Andrade SLA; Einsle O Production and isolation of vanadium nitrogenase from Azotobacter vinelandii by molybdenum depletion. J. Biol. Inorg. Chem. 2017, 22 (1), 161. [DOI] [PubMed] [Google Scholar]
- (165).Zhang LM; Kaiser JT; Meloni G; Yang KY; Spatzal T; Andrade SLA; Einsle O; Howard JB; Rees DC The Sixteenth Iron in the Nitrogenase MoFe Protein. Angew Chem Int Edit 2013, 52 (40), 10529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Clarke TA; Fairhurst S; Lowe DJ; Watmough NJ; Eady RR Electron transfer and half-reactivity in nitrogenase. Biochem. Soc. Trans. 2011, 39, 201. [DOI] [PubMed] [Google Scholar]
- (167).Danyal K; Shaw S; Page TR; Duval S; Horitani M; Marts AR; Lukoyanov D; Dean DR; Raugei S; Hoffman BM et al. Negative cooperativity in the nitrogenase Fe protein electron delivery cycle. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (40), E5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Setubal JC; dos Santos P; Goldman BS; Ertesvag H; Espin G; Rubio LM; Valla S; Almeida NF; Balasubramanian D; Cromes L et al. Genome Sequence of Azotobacter vinelandii, an Obligate Aerobe Specialized To Support Diverse Anaerobic Metabolic Processes. J. Bacteriol. 2009, 191 (14), 4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Kaiser JT; Hu YL; Wiig JA; Rees DC; Ribbe MW Structure of Precursor-Bound NifEN: A Nitrogenase FeMo Cofactor Maturase/Insertase. Science 2011, 331 (6013), 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Rees JA; Björnsson R; Schlesier J; Sippel D; Einsle O; DeBeer S The Fe-V Cofactor of Vanadium Nitrogenase Contains an Interstitial Carbon Atom. Angew Chem Int Edit 2015, 54 (45), 13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Rees JA; Björnsson R; Kowalska JK; Lima FA; Schlesier J; Sippel D; Weyhermüller T; Einsle O; Kovacs JA; DeBeer S Comparative electronic structures of nitrogenase FeMoco and FeVco. Dalton T 2017, 46 (8), 2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Rohde M; Sippel D; Trncik C; Andrade SLA; Einsle O The Critical E4 State of Nitrogenase Catalysis. Biochemistry 2018, 57 (38), 5497. [DOI] [PubMed] [Google Scholar]
- (173).Ribbe MW; Hu YL; Hodgson KO; Hedman B Biosynthesis of Nitrogenase Metalloclusters. Chem. Rev. 2014, 114 (8), 4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Harris DF; Lukoyanov DA; Kallas H; Trncik C; Yang ZY; Compton P; Kelleher N; Einsle O; Dean DR; Hoffman BM et al. Mo-, V-, and Fe-Nitrogenases Use a Universal Eight-Electron Reductive-Elimination Mechanism To Achieve N2 Reduction. Biochemistry 2019, 58 (30), 3293. [DOI] [PubMed] [Google Scholar]
- (175).Fisher K; Dilworth MJ; Kim CH; Newton WE Azotobacter vinelandii nitrogenases containing altered MoFe proteins with substitutions in the FeMo-cofactor environment: Effects on the catalyzed reduction of acetylene and ethylene. Biochemistry 2000, 39 (11), 2970. [DOI] [PubMed] [Google Scholar]
- (176).Fisher K; Dilworth MJ; Newton WE Differential effects on N2 binding and reduction, HD formation, and azide reduction with alpha-195(His)- and alpha-191(Gln)-substituted MoFe proteins of Azotobacter vinelandii nitrogenase. Biochemistry 2000, 39 (50), 15570. [DOI] [PubMed] [Google Scholar]
- (177).Dance I The controlled relay of multiple protons required at the active site of nitrogenase. Dalton T 2012, 41 (25), 7647. [DOI] [PubMed] [Google Scholar]
- (178).Münck E; Rhodes H; Orme-Johnson WH; Davis LC; Brill WJ; Shah VK Nitrogenase VIII. Mössbauer and EPR Spectroscopy - MoFe Protein Component from Azotobacter vinelandii. Biochim. Biophys. Acta 1975, 400 (1), 32. [DOI] [PubMed] [Google Scholar]
- (179).Menard R; Storer AC Oxyanion Hole Interactions in Serine and Cysteine Proteases. Biological Chemistry Hoppe-Seyler 1992, 373 (7), 393. [DOI] [PubMed] [Google Scholar]
- (180).Wilson PW; Burris RH The mechanism of biological nitrogen fixation. Bact. Rev. 1947, 11 (1), 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).Simpson FB; Burris RH A nitrogen pressure of 50 atmospheres does not prevent evolution of hydrogen by nitrogenase. Science 1984, 224, 1095. [DOI] [PubMed] [Google Scholar]
- (182).Mortenson LE Ferredoxin and ATP Requirements for Nitrogen Fixation in Cell-Free Extracts of Clostridium pasteurianum. Proc. Natl. Acad. Sci. U. S. A. 1964, 52 (2), 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Martin AE; Burgess BK; Iismaa SE; Smartt CT; Jacobson MR; Dean DR Construction and characterization of an Azotobacter vinelandii strain with mutations in the genes encoding flavodoxin and ferredoxin I. J. Bacteriol. 1989, 171 (6), 3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Bulen WA Biological Nitrogen Fixation. Science 1965, 147 (3655), 310. [DOI] [PubMed] [Google Scholar]
- (185).Moustafa E; Mortenson LE Acetylene Reduction by Nitrogen Fixing Extracts of Clostridium pasteurianum: ATP Requirement and Inhibition by ADP. Nature 1967, 216 (5121), 1241. [DOI] [PubMed] [Google Scholar]
- (186).Scott DJ; Dean DR; Newton WE Nitrogenase-Catalyzed Ethane Production and Co-Sensitive Hydrogen Evolution from Mofe Proteins Having Amino-Acid Substitutions in an Alpha-Subunit Femo Cofactor-Binding Domain. J. Biol. Chem. 1992, 267 (28), 20002. [PubMed] [Google Scholar]
- (187).Kelly M; Postgate JR; Richards RL Reduction of Cyanide and Isocyanide by Nitrogenase of Azotobacter chroococcum. Biochem. J. 1967, 102 (1), C1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Rubinson JF; Corbin JL; Burgess BK Nitrogenase Reactivity - Methyl Isocyanide as Substrate and Inhibitor. Biochemistry 1983, 22 (26), 6260. [DOI] [PubMed] [Google Scholar]
- (189).Mortenson LE; Upchurch RG In Current Perspectives in NItrogenase Fixation; Gibson AH;Newton WE, Eds.; Elsevier: Amsterdam, 1981. [Google Scholar]
- (190).Winter HC; Burris RH Nitrogenase. Annu. Rev. Biochem. 1976, 45, 409. [DOI] [PubMed] [Google Scholar]
- (191).Carnahan JE; Mortenson LE; Mower HF; Castle JE Nitrogen Fixation in Cell-Free Extracts of Clostridium-Pasteurianum. Biochim. Biophys. Acta 1960, 44 (3), 520. [DOI] [PubMed] [Google Scholar]
- (192).Hu Y; Ribbe MW Biosynthesis of the Metalloclusters of Nitrogenases. Annual Reviews of Biochemistry 2016, 85, 455. [DOI] [PubMed] [Google Scholar]
- (193).Thorneley RNF; Lowe DJ The Mechanism of Klebsiella pneumoniae Nitrogenase Action - Pre-Steady-State Kinetics of an Enzyme-Bound Intermediate in N2 Reduction and of NH3 Formation. Biochem. J. 1984, 224 (3), 887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Kok B; Forbush B; McGloin M Cooperation of Charges in Photosynthetic O2 Evolution - 1. A Linear Four Step Mechanism. Photochemistry and Photobiology 1970, 11 (6), 457. [DOI] [PubMed] [Google Scholar]
- (195).Syrtsova LA; Nadtochenko VA; Denisov NN; Timofeeva EA; Shkondina NI; Gak VY Kinetics of elementary steps of electron transfer in nitrogenase in the presence of a photodonor. Biochemistry-Moscow 2000, 65 (10), 1145. [PubMed] [Google Scholar]
- (196).Roth LE; Nguyen JC; Tezcan FA ATP- and Iron-Protein-Independent Activation of Nitrogenase Catalysis by Light. J. Am. Chem. Soc. 2010, 132 (39), 13672. [DOI] [PubMed] [Google Scholar]
- (197).Roth LE; Tezcan FA ATP-Uncoupled, Six-Electron Photoreduction of Hydrogen Cyanide to Methane by the Molybdenum-Iron Protein. J. Am. Chem. Soc. 2012, 134 (20), 8416. [DOI] [PubMed] [Google Scholar]
- (198).Orme-Johnson WH Molecular Basis of Biological Nitrogen Fixation. Annu. Rev. Biophys. Biophys. Chem. 1985, 14, 419. [DOI] [PubMed] [Google Scholar]
- (199).Anderson GL; Howard JB Reactions with the Oxidized Iron Protein of Azotobacter vinelandii Nitrogenase - Formation of a 2Fe Center. Biochemistry 1984, 23 (10), 2118. [DOI] [PubMed] [Google Scholar]
- (200).Iron-Sulfur Proteins; Lovenberg W, Ed.; Academic Press: New York, 1973. [Google Scholar]
- (201).Hageman RV; Burris RH Changes in the EPR Signal of Dinitrogenase from Azotobacter vinelandii during the Lag Period before Hydrogen Evolution Begins. J. Biol. Chem. 1979, 254 (22), 1189. [PubMed] [Google Scholar]
- (202).Hageman RV; Burris RH Kinetic studies on electron transfer and interaction between nitrogenase components from Azotobacter vinelandii. Biochemistry 1978, 17, 4117. [DOI] [PubMed] [Google Scholar]
- (203).George SJ; Ashby GA; Wharton CW; Thorneley RNF Time-resolved binding of carbon monoxide to nitrogenase monitored by stopped-flow infrared spectroscopy. J. Am. Chem. Soc. 1997, 119 (27), 6450. [Google Scholar]
- (204).Watt GD; Burns A Kinetics of Dithionite Ion Utilization and ATP Hydrolysis for Reactions Catalyzed by Nitrogenase Complex from Azotobacter vinelandii. Biochemistry 1977, 16 (2), 264. [DOI] [PubMed] [Google Scholar]
- (205).Hoffman BM; Lukoyanov D; Yang ZY; Dean DR; Seefeldt LC Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem. Rev. 2014, 114 (8), 4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Bulen WA; Burns RC; Lecomte JR Nitrogen Fixation - Hydrosulfite as Electron Donor with Cell-Free Preparations of Azotobacter vinelandii and Rhodospirillum rubrum. Proc. Natl. Acad. Sci. U. S. A. 1965, 53 (3), 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Mayhew SG Redox Potential of Dithionite and SO2− from Equilibrium Reactions with Flavodoxins, Methyl Viologen and Hydrogen Plus Hydrogenase. Eur. J. Biochem. 1978, 85 (2), 535. [DOI] [PubMed] [Google Scholar]
- (208).Mckenna CE; Gutheil WG; Wei S A Method for Preparing Analytically Pure Sodium Dithionite - Dithionite Quality and Observed Nitrogenase-Specific Activities. Biochim. Biophys. Acta 1991, 1075 (1), 109. [DOI] [PubMed] [Google Scholar]
- (209).Wilson R; Bunker J; Lowery TJ; Watt GD Reduction of nitrogenase Fe protein from Azotobacter vinelandii by dithionite: quantitative and qualitative effects of nucleotides, temperature, pH and reaction buffer. Biophys. Chem. 2004, 109 (2), 305. [DOI] [PubMed] [Google Scholar]
- (210).Tanifuji K; Lee CC; Sickerman NS; Tatsumi K; Ohki Y; Hu YL; Ribbe MW Tracing the ‘ninth sulfur’ of the nitrogenase cofactor via a semi-synthetic approach. Nat Chem 2018, 10 (5), 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Silverstein R; Bulen WA Kinetic Studies of Nitrogenase-Catalyzed Hydrogen Evolution and Nitrogen Reduction Reactions. Biochemistry 1970, 9 (19), 3809. [DOI] [PubMed] [Google Scholar]
- (212).Orme-Johnson WH; Henzl MT; Averill BA; Wyeth P; Munck E Electron-Transfer Centers in Nitrogenase Component Proteins. Fed. Proc. 1977, 36 (3), 880. [Google Scholar]
- (213).Hageman RV; Burris RH Nitrogenase and Nitrogenase Reductase Associate and Dissociate with Each Catalytic Cycle. Proc. Natl. Acad. Sci. U. S. A. 1978, 75 (6), 2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (214).Deits TL; Howard JB Effect of Salts on Azotobacter vinelandii Nitrogenase Activities - Inhibition of Iron Chelation and Substrate Reduction. J. Biol. Chem. 1990, 265 (7), 3859. [PubMed] [Google Scholar]
- (215).Jacobs D; Mitchell D; Watt GD The concentration of cellular nitrogenase proteins in Azotobacter vinelandii whole cells as determined by activity measurements and electron paramagnetic resonance spectroscopy. Archives of Biochemistry and Biophysics 1995, 324 (2), 317. [DOI] [PubMed] [Google Scholar]
- (216).Liang J; Burris RH Hydrogen burst associated with nitrogenase-catalyzed reactions. Proc. Natl. Acad. Sci. USA 1988, 85, 9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Buscagan TM; Rees DC Rethinking the Nitrogenase Mechanism: Activating the Active Site. Joule 2019, 3 (11), 2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Kaila VRI; Verkhovsky MI; Wikstrom M Proton-Coupled Electron Transfer in Cytochrome Oxidase. Chem. Rev. 2010, 110 (12), 7062. [DOI] [PubMed] [Google Scholar]
- (219).Yoshikawa S; Muramoto K; Shinzawa-Itoh K Proton-pumping mechanism of cytochrome c oxidase. Annu. Rev. Biophys. 2011, 40, 205. [DOI] [PubMed] [Google Scholar]
- (220).Hageman R; Burris RH Effect of Varying Electron Flux on Electron Allocation to Alternative Substrates by Nitrogenase. Fed. Proc. 1978, 37 (6), 1420. [Google Scholar]
- (221).Wherland S; Burgess BK; Stiefel EI; Newton WE Nitrogenase Reactivity - Effects of Component Ratio on Electron Flow and Distribution during Nitrogen-Fixation. Biochemistry 1981, 20 (18), 5132. [DOI] [PubMed] [Google Scholar]
- (222).Johnson JL; Nyborg AC; Wilson PE; Tolley AM; Nordmeyer FR; Watt GD Mechanistic interpretation of the dilution effect for Azotobacter vinelandii and Clostridium pasteurianum nitrogenase catalysis. Biochim. Biophys. Acta 2000, 1543 (1), 36. [DOI] [PubMed] [Google Scholar]
- (223).Bergersen FJ; Turner GL Kinetic studies of nitrogenase from soya-bean root-nodule bacteroids. Biochem. J. 1973, 131, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Orme-Johnson WH; Davis LC; Henzl MT; Averill BA; Orme-Johnson NR; Munck E; Zimmerman R In Recent Developments in Nitrogen Fixation; Newton WE;Postgate JR;Rodriguez-Barrueco C, Eds.; Academic Press: London, 1977. [Google Scholar]
- (225).Johnson JL; Nyborg AC; Wilson PE; Tolley AM; Nordmeyer FR; Watt GD Analysis of steady state Fe and MoFe protein interactions during nitrogenase catalysis. Biochim. Biophys. Acta 2000, 1543 (1), 24. [DOI] [PubMed] [Google Scholar]
- (226).Johnson JL; Nyborg AC; Wilson PE; Tolley AM; Nordmeyer FR; Watt GD Analysis of steady state Fe and MoFe protein interactions during nitrogenase catalysis. Biochimica Et Biophysica Acta-Protein Structure and Molecular Enzymology 2000, 1543 (1), 24. [DOI] [PubMed] [Google Scholar]
- (227).Clarke TA; Maritano S; Eady RR Formation of a tight 1 : 1 complex of Clostridium pasteurianum Fe protein-Azotobacter vinelandii MoFe protein: Evidence for long-range interactions between the Fe protein binding sites during catalytic hydrogen evolution. Biochemistry 2000, 39 (37), 11434. [DOI] [PubMed] [Google Scholar]
- (228).Lowe DJ; Thorneley RNF The Mechanism of Klebsiella pneumoniae Nitrogenase Action - Pre-Steady-State Kinetics of H2 Formation. Biochem. J. 1984, 224 (3), 877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Lowe DJ; Fisher K; Thorneley RNF Klebsiella pneumoniae Nitrogenase - Mechanism of Acetylene Reduction and Its Inhibition by Carbon Monoxide. Biochem. J. 1990, 272 (3), 621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Lowe DJ; Fisher K; Thorneley RNF; Vaughn SA; Burgess BK Kinetics and Mechanism of the Reaction of Cyanide with Molybdenum Nitrogenase from Azotobacter vinelandii. Biochemistry 1989, 28 (21), 8460. [DOI] [PubMed] [Google Scholar]
- (231).Maskos Z; Fisher K; Sorlie M; Newton WE; Hales BJ Variant MoFe proteins of Azotobacter vinelandii: effects of carbon monoxide on electron paramagnetic resonance spectra generated during enzyme turnover. J. Biol. Inorg. Chem. 2005, 10 (4), 394. [DOI] [PubMed] [Google Scholar]
- (232).McLean PA; Smith BE; Dixon RA Nitrogenase of Klebsiella pneumoniae nif V Mutants - Investigation of the Novel Carbon Monoxide-Sensitivity of Hydrogen Evolution by the Mutant Enzyme. Biochem. J. 1983, 211 (3), 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Davis LC; Wang YL In Vivo and In Vitro Kinetics of Nitrogenase. J. Bacteriol. 1980, 141 (3), 1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Burgess BK In Molybdenum Enzymes; Spiro TG, Ed.; Wiley-Interscience: New York, 1985. [Google Scholar]
- (235).Pham DN; Burgess BK Nitrogenase Reactivity - Effects of pH on Substrate Reduction and CO Inhibition. Biochemistry 1993, 32 (49), 13725. [DOI] [PubMed] [Google Scholar]
- (236).Yang KY; Haynes CA; Spatzal T; Rees DC; Howard JB Turnover-Dependent Inactivation of the Nitrogenase MoFe-Protein at High pH. Biochemistry 2014, 53 (2), 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Hadfield KL; Bulen WA Adenosine Triphosphate Requirement of Nitrogenase from Azotobacter vinelandii. Biochemistry 1969, 8 (12), 5103. [DOI] [PubMed] [Google Scholar]
- (238).Rivera-Ortiz JH; Burris RH Interactions among substrates and inhibitors of nitrogenase. J. Bact. 1975, 123, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Burgess BK; Wherland S; Newton WE; Stiefel EI Nitrogenase reactivity: insight into the nitrogen-fixing process through hydrogen-inhibition and HD-forming reactions. Biochemistry 1981, 20, 5140. [DOI] [PubMed] [Google Scholar]
- (240).Chatt J; Dilworth JR; Richards RL Recent Advances in Chemistry of Nitrogen Fixation. Chem. Rev. 1978, 78 (6), 589. [Google Scholar]
- (241).Guth JH; Burris RH Inhibition of nitrogenase-catalyzed NH3 formation by H2. Biochemistry 1983, 22, 5111. [DOI] [PubMed] [Google Scholar]
- (242).Jensen BB; Burris RH Effects of high pN2 and high pD2 on NH3 production, H2 evolution, and HD formation by nitrogenases. Biochemistry 1985, 24, 1141. [DOI] [PubMed] [Google Scholar]
- (243).Chatt J In Nitrogen Fixation: Proceedings of the Phytochemical Society of Europe Symposium, Sussex, September, 1979; Stewart WDP;Gallon JR, Eds.; Academic Press: London, 1980. [Google Scholar]
- (244).Burgess BK; Lowe DJ Mechanism of molybdenum nitrogenase. Chem. Rev. 1996, 96 (7), 2983. [DOI] [PubMed] [Google Scholar]
- (245).Hageman RV; Burris RH Changes in the EPR signal of dinitrogenase from Azotobacter vinelandii during the lag period before hydrogen evolution begins. J. Biol. Chem. 1979, 254, 1189. [PubMed] [Google Scholar]
- (246).Hageman RV; Burris RH Nitrogenase and nitrogenase reductase associate and dissociate with each catalytic cycle. Proc. Natl. Acad. Sci. USA 1978, 75, 2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Wilson PE; Nyborg AC; Watt GD Duplication and extension of the Thorneley and Lowe kinetic model for Klebsiella pneumoniae nitrogenase catalysis using a MATHEMATICA software platform. Biophys. Chem. 2001, 91 (3), 281. [DOI] [PubMed] [Google Scholar]
- (248).Fisher K; Newton WE; Lowe DJ Electron paramagnetic resonance analysis of different Azotobacter vinelandii nitrogenase MoFe-protein conformations generated during enzyme turnover: Evidence for S = 3/2 spin states from reduced MoFe-protein intermediates. Biochemistry 2001, 40 (11), 3333. [DOI] [PubMed] [Google Scholar]
- (249).Lukoyanov D; Barney BM; Dean DR; Seefeldt LC; Hoffman BM Connecting nitrogenase intermediates with the kinetic scheme for N2 reduction by a relaxation protocol and identification of the N2 binding state. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (5), 1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Burk D The free energy of nitrogen fixation by living forms. J. Gen. Physiol. 1927, 10, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Stiefel EI In Recent Developments in Nitrogen Fixation; Newton W;Postgate JR;Rodriguez-Barrueco C, Eds.; Academic Press: London, New York, San Francisco, 1977. [Google Scholar]
- (252).Ruscic B; Pinzon RE; Laszewski G. v.; Kodeboyina D; Burcat A; Leahy D; Montoy D; Wagner AF Active Thermochemical Tables: thermochemistry for the 21st century. Journal of Physics: Conference Series 2005, 16, 561. [Google Scholar]
- (253).Shilov AE Catalytic reduction of molecular nitrogen in solutions. Russian Chemical Bulletin 2003, 52 (12), 2555. [Google Scholar]
- (254).Jia HP; Quadrelli EA Mechanistic aspects of dinitrogen cleavage and hydrogenation to produce ammonia in catalysis and organometallic chemistry: relevance of metal hydride bonds and dihydrogen. Chem. Soc. Rev. 2014, 43 (2), 547. [DOI] [PubMed] [Google Scholar]
- (255).van der Ham CJ; Koper MT; Hetterscheid DG Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 2014, 43 (15), 5183. [DOI] [PubMed] [Google Scholar]
- (256).Yandulov DV; Schrock RR Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Science 2003, 301, 76. [DOI] [PubMed] [Google Scholar]
- (257).Anderson JS; Rittle J; Peters JC Catalytic conversion of nitrogen to ammonia by an iron model complex. Nature 2013, 501 (7465), 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (258).Ashida Y; Arashiba K; Nakajima K; Nishibayashi Y Molybdenum-catalysed ammonia production with samarium diiodide and alcohols or water. Nature 2019, 568 (7753), 536. [DOI] [PubMed] [Google Scholar]
- (259).Shilov AE Metal Complexes in Biomimetic Chemical Reactions; CRC Press: Boca Raton, 1996. [Google Scholar]
- (260).Ertl G Primary steps in catalytic synthesis of ammonia. J. Vac. Sci. Technol. A 1983, 1, 1247. [Google Scholar]
- (261).Catalytic Ammonia Synthesis: Fundamentals and Practice; Jennings JR, Ed.; Plenum Press: New York, 1991. [Google Scholar]
- (262).Frank A; Caro FN Germany, 1895; Vol. Deutsches Reichspatent DRP 88363. [Google Scholar]
- (263).Thorneley RNF; Eady RR; Lowe DJ Biological nitrogen fixation by way of an enzyme-bound dinitrogen-hydride intermediate. Nature 1978, 272, 557. [Google Scholar]
- (264).Manriquez JM; Sanner RD; Marsh RE; Bercaw JE Reduction of molecular nitrogen to hydrazine. Structure of a dinitrogen complex of bis(pentamethylcyclopentadienyl)zirconium(II) and an 15N labeling study of its reaction with hydrogen chloride. J. Am. Chem. Soc. 1976, 98 (10), 3042. [Google Scholar]
- (265).Dilworth MJ; Eady RR Hydrazine is a product of dinitrogen reduction by the vanadium-nitrogenase from Azotobacter chroococcum. Biochem. J. 1991, 277, 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (266).Erickson JA; Nyborg AC; Johnson JL; Truscott SM; Gunn A; Nordmeyer FR; Watt GD Enhanced efficiency of ATP hydrolysis during nitrogenase catalysis utilizing reductants that form the all-ferrous redox state of the Fe protein. Biochemistry 1999, 38, 14279. [DOI] [PubMed] [Google Scholar]
- (267).Hoffman BM; Lukoyanov D; Dean DR; Seefeldt LC Nitrogenase: A draft mechanism. Acc. Chem. Res. 2013, 46 (2), 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Lukoyanov D; Yang ZY; Khadka N; Dean DR; Seefeldt LC; Hoffman BM Identification of a Key Catalytic Intermediate Demonstrates That Nitrogenase Is Activated by the Reversible Exchange of N2 for H2. J. Am. Chem. Soc. 2015, 137 (10), 3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Sacco A; Rossi M Hydride and nitrogen complexes of cobalt. Chem Commun 1967, DOI: 10.1039/c19670000316(7), 316. [DOI] [Google Scholar]
- (270).Lukoyanov D; Khadka N; Yang ZY; Dean DR; Seefeldt LC; Hoffman BM Reversible Photoinduced Reductive Elimination of H2 from the Nitrogenase Dihydride State, the E4(4H) Janus Intermediate. J. Am. Chem. Soc. 2016, 138 (4), 1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Dance I Misconception of reductive elimination of H2, in the context of the mechanism of nitrogenase. Dalton T 2015, 44 (19), 9027. [DOI] [PubMed] [Google Scholar]
- (272).Lukoyanov D; Khadka N; Yang ZY; Dean DR; Seefeldt LC; Hoffman BM Reductive Elimination of H2 Activates Nitrogenase to Reduce the N-N Triple Bond: Characterization of the E4(4H) Janus Intermediate in Wild-Type Enzyme. J. Am. Chem. Soc. 2016, 138 (33), 10674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Lukoyanov D; Yang ZY; Barney BM; Dean DR; Seefeldt LC; Hoffman BM Unification of reaction pathway and kinetic scheme for N2 reduction catalyzed by nitrogenase. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (15), 5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Yang ZY; Khadka N; Lukoyanov D; Hoffman BM; Dean DR; Seefeldt LC On reversible H2 loss upon N2 binding to FeMo-cofactor of nitrogenase. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (41), 16327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Bruice TC; Maskiewicz R; Job R The acid-base properties, hydrolytic mechanism, and susceptibility to O2 oxidation of Fe4S4(SR)4−2 clusters. Proc. Natl. Acad. Sci. USA 1975, 72, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (276).Chen K; Hirst J; Camba R; Bonagura CA; Stout CD; Burgess BK; Armstrong FA Atomically defined mechanism for proton transfer to a buried redox centre in a protein. Nature 2000, 405, 814. [DOI] [PubMed] [Google Scholar]
- (277).Bates K; Garrett B; Henderson RA Rates of proton transfer to Fe-S-based clusters: Comparison of clusters containing {MFe(μ2-S)2}n+ and {MFe3(μ3-S)4}n+ (M = Fe, Mo, or W) cores. Inorg. Chem. 2007, 46 (26), 11145. [DOI] [PubMed] [Google Scholar]
- (278).Dance I; Henderson RA Large structural changes upon protonation of Fe4S4 clusters: the consequences for reactivity. Dalton T 2014, 43 (43), 16213. [DOI] [PubMed] [Google Scholar]
- (279).Durrant MC Controlled protonation of iron-molybdenum cofactor by nitrogenase: a structural and theoretical analysis. Biochem. J. 2001, 355, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Dance I The pathway for serial proton supply to the active site of nitrogenase: enhanced density functional modeling of the Grotthuss mechanism. Dalton T 2015, 44 (41), 18167. [DOI] [PubMed] [Google Scholar]
- (281).Igarashi RY; Seefeldt LC Nitrogen fixation: The mechanism of the Mo-dependent nitrogenase. Critical Reviews in Biochemistry and Molecular Biology 2003, 38 (4), 351. [DOI] [PubMed] [Google Scholar]
- (282).Barney BM; Yurth MG; Dos Santos PC; Dean DR; Seefeldt LC A substrate channel in the nitrogenase MoFe protein. J. Biol. Inorg. Chem. 2009, 14 (7), 1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).Dance I A molecular pathway for the egress of ammonia produced by nitrogenase. Sci Rep-Uk 2013, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (284).Morrison CN; Hoy JA; Zhang LM; Einsle O; Rees DC Substrate Pathways in the Nitrogenase MoFe Protein by Experimental Identification of Small Molecule Binding Sites. Biochemistry 2015, 54 (11), 2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (285).Lakowicz JR; Weber G Quenching of protein fluorescence by oxygen. Detection of structural fluctuations in proteins on the nanosecond time scale. Biochemistry 1973, 12 (21), 4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Bercaw JE Jack Halpern (1925–2018): Pioneer of homogeneous catalysis. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 5049. [DOI] [PMC free article] [PubMed] [Google Scholar]