

Abstract
Hemophilia A (HA) is a rare bleeding disorder caused by deficiency/dysfunction of the FVIII protein. As current therapies based on frequent FVIII infusions are not a definitive cure, long-term expression of FVIII in endothelial cells through lentiviral vector (LV)-mediated gene transfer holds the promise of a one-time treatment. Thus, here we sought to determine whether LV-corrected blood outgrowth endothelial cells (BOECs) implanted through a prevascularized medical device (Cell Pouch) would rescue the bleeding phenotype of HA mice. To this end, BOECs from HA patients and healthy donors were isolated, expanded, and transduced with an LV carrying FVIII driven by an endothelial-specific promoter employing GMP-like procedures. FVIII-corrected HA BOECs were either directly transplanted into the peritoneal cavity or injected into a Cell Pouch implanted subcutaneously in NSG-HA mice. In both cases, FVIII secretion was sufficient to improve the mouse bleeding phenotype. Indeed, FVIII-corrected HA BOECs reached a relatively short-term clinically relevant engraftment being detected up to 16 weeks after transplantation, and their genomic integration profile did not show enrichment for oncogenes, confirming the process safety. Overall, this is the first preclinical study showing the safety and feasibility of transplantation of GMP-like produced LV-corrected BOECs within an implantable device for the long-term treatment of HA.
Keywords: hemophilia A, cell and gene therapy, medical device, BOEC, lentiviral vector, FVIII, endothelial cells
Graphical abstract
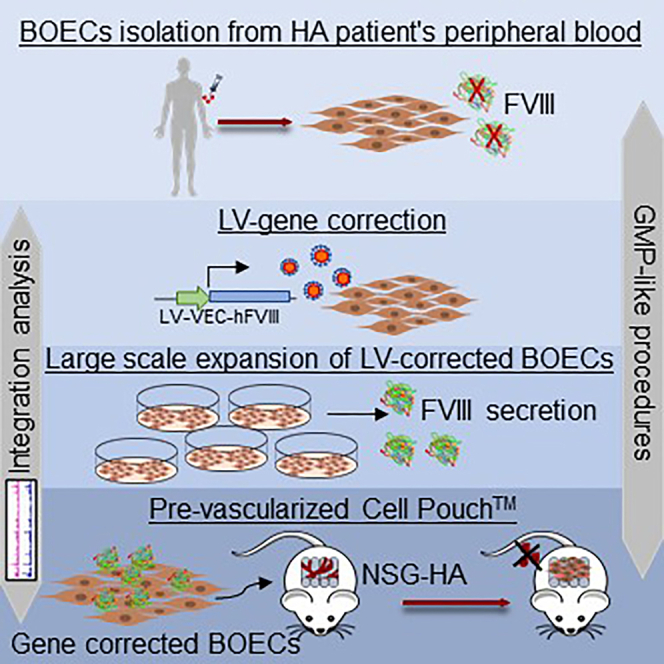
BOECs from HA patients and healthy donors were isolated, expanded, and LV-FVIII-transduced under GMP-like procedures. Amelioration of bleeding phenotype in a preclinical mouse model was reached by implantation of FVIII gene-corrected hemophilic BOECs injected in a pre-vascularized Cell Pouch reaching a long-term engraftment with a safe genomic integration profile.
Introduction
Hemophilia A (HA) is an X-linked disorder caused by mutations in the F8 gene.1,2 These mutations result in deficiency or reduced activity of the coagulation factor VIII (FVIII), leading to a life-long bleeding tendency, whose clinical severity is proportional to FVIII reduction.1 Although the current standard of care is to intravenously infuse HA patients with clotting factor concentrates, the short half-life of FVIII requires frequent and multiple infusions, with a negative impact on the patient's quality of life (QoL). A new generation of standard rFVIII was obtained by refinements of the recombinant protein through the optimization of relevant post-translational modifications, such as glycosylation, that improve the stability of the mature FVIII protein,3 or the introduction of a covalent link between the FVIII heavy and light chains, preserving FVIII from premature degradation and conferring a higher binding affinity to von Willebrand factor (vWF) with a reduction of the needed injection dose.4, 5, 6
Other new bioengineered molecules were developed with higher extended plasma half-life7 and improved pharmacokinetics by the fusion of rFVIII with the Fc portion of immunoglobulin8 or by conjugation with polyethylene glycol (PEGylation).9
However, several issues are still to be solved as the recurrent intravenous (i.v.) route of administration and the inhibitor development, common in 20%–40% of patients with the severe form,10 worsen the clinical outcome, making the treatment ineffective.11,12 Therefore, a new clinical approach emerged more recently, i.e., emicizumab, to overcome the difficulties of i.v. delivery and to improve and prolong the effectiveness of the therapy in all patients, regardless of the presence or absence of the inhibitor.13,14 However, bleeding events can still occur after trauma, requiring the use of additional hemostatic agents according to the patients' inhibitor status.15,16
This has led researchers to explore innovative cell and gene therapy strategies that may ensure continuous endogenous FVIII expression with only a one-time treatment. Another good reason for choosing gene therapy over traditional approaches is that HA is a monogenic disease, i.e., entirely ascribable to the lack of one protein, FVIII, and that a small increase in FVIII plasma levels is enough to ameliorate the bleeding phenotype of HA patients.
Given the growing number of cell and gene therapy approaches being developed, it is becoming increasingly important to identify the most suitable cell target. Even though F8 mRNA is expressed in different human and mouse organs, such as liver, spleen, lymph nodes, kidney,17, 18, 19, 20 and in hematopoietic cells,21,22 transplantation studies in hemophilic animal models have shown FVIII expression to be mainly localized in liver sinusoidal endothelial cells (LSECs),23, 24, 25 making these cells attractive targets for HA gene therapy. This is also supported by the fact that endothelial cells (ECs) secrete FVIII and can act as tolerogenic cells.24,26
Over the years, several gene therapy approaches for HA have been attempted using adeno-associated virus (AAV) vectors to induce FVIII expression in the desired cell type. Despite the encouraging preliminary results obtained in few ongoing clinical trials testing the efficacy of AAV-mediated hepatocyte-targeted FVIII expression in HA patients,27, 28, 29 some medical issues still need to be addressed, such as the use of these vectors in patients with pre-existing immunity to AAV or with FVIII inhibitors. As AAV vectors do not actively integrate into the host cell genome, they are lost upon cell division during liver growth or in case of liver disease, thus potentially limiting their use in pediatric patients and questioning their life-long maintenance. Therefore, lentiviral vectors (LVs) could represent a viable approach able to overcome some AAV limitations. Moreover, several studies have demonstrated, by the use of endothelial-specific promoters, specific expression of human FVIII in LSECs.23, 24, 25,30 Recently, we have shown that induced pluripotent stem cells (iPSCs) derived from CD34+ HA cells can be differentiated into ECs and genetically corrected by LV to express the FVIII transgene, deleted of the B domain (BDD), driven by the endothelial-specific vascular endothelial cadherin (VEC) promoter. After transplanting these cells into the liver of monocrotaline-conditioned NOD-scid IL2Rgnull HA (NSG-HA) mice, we were able to correct the bleeding phenotype of these mice and maintain a stable FVIII activity over time.31 Moreover, BDD-FVIII-transduced ECs encapsulated in microcarrier beads have been shown to survive for a prolonged time in the peritoneal cavity of NSG-HA mice secreting therapeutic levels of FVIII.31
Several studies have focused on defining different cell sources and matrices to transplant FVIII-expressing ECs.32, 33, 34 A readily available EC source is represented by patient-derived blood outgrowth endothelial cells (BOECs).35 BOECs are isolated from adult peripheral blood36 and can be fully differentiated into mature ECs. They promote neovascularization in vivo when transplanted into immunodeficient mice37 or when cultured on three-dimensional biodegradable vascular scaffolds.38, 39, 40 In addition, they can be considered a valuable source of cells to understand EC biology and model disease, and can be used in regenerative medicine due to their ability to promote neovascularization, thus representing an optimal candidate for HA cell and gene therapy. Indeed, BOECs transplanted in NSG-HA mice after gene modification for FVIII expression were able to partially rescue the hemorrhagic phenotype of these mice.35,41 Moreover, autologous transplantation of FVIII-expressing BOEC cell sheet allowed long-term phenotypic correction and survival of transplanted cells.34 Noteworthy, BOECs can promote neovascularization in vivo in combination with synthetic or natural materials.37
A combination of LV-corrected BOECs with a medical device is classified by the European Union as a combined gene therapy medicinal product (GTMP).42 The mandatory non-clinical study scheme before the first administration of a cell-based GTMP to human subjects includes the comprehensive characterization of the transduced cells and the evaluation of the medical device contribution.43 Moreover, the proof-of-concept pharmacodynamics along with the molecular mechanism of action must be identified in preclinical models in vivo and/or in vitro. These studies are deemed essential to determine the GMP cell dose to be used in clinical trials.44
Here, we show extensive characterization of LV-transduced BOECs isolated from healthy donors or HA patients for FVIII production in vivo. These cells were transplanted in a small scalable, implantable, and prevascularized medical device, namely Cell Pouch (Sernova), previously developed for diabetes treatment.45
Our findings, showing that Cell Pouch-transplanted LV-corrected HA BOECs are capable of correcting the bleeding phenotype of HA mice, open new avenues for the treatment of HA in humans.
Results
Characterization of BOECs isolated from HA patients or healthy subjects and LV-mediated FVIII gene transfer
Upon isolation and expansion in culture medium, both BOECs from healthy donors and HA patients showed the classical endothelial cobblestone-like morphology (Figure 1A). Of note, despite being all isolated from severe HA patients, HA BOECs gave rise to many colonies (Figure S1A) and there was not a significant difference in the number of isolated colonies between healthy donors and HA patients (Figure S1B). For transgene expression, isolated cells were transduced with a LV carrying the BDD form of FVIII driven by the vascular endothelial cadherin promoter (LV-VEC.hBDD-FVIII) or with an LV carrying the green fluorescent protein under the control of the same promoter (LV-VEC.GFP), both at an multiplicity of infection (MOI) of 20. FACS analysis showed 98% ± 1% GFP+ cells after transduction (Figure S1C), indicating excellent transduction efficiency. The number of integrated LV copies/cell was ∼6 and ∼3 for LV-VEC.GFP- and LV-VEC.hBDD-FVIII-transduced cells, respectively (Figure S2A). Thus, this protocol ensures a very high transduction efficiency while maintaining a safe number of integrated LV copies/cell.46,47
Figure 1.

Healthy and HA BOEC isolation, LV transduction, and in vitro FVIII detection
(A) Light microscope pictures of cultured healthy and HA BOECs at passage 3. (B) Representative RT-PCR analysis for the expression of endothelial markers. HUVECs and fibroblasts were used as positive and negative control, respectively. (C) RT-PCR for endothelial markers specific for blood endothelial cells (BECs). iPSC-derived ECs and fibroblasts were used as positive and negative control, respectively. (D) Representative histograms of healthy non-transduced (black line) and LV-VEC.hBDD-FVIII-transduced healthy BOECs (red line), showing endothelial marker expression and absence of hematopoietic markers. The filled-up histograms represent unstained BOECs. (E) Representative histograms of HA non-transduced (black line) and LV-VEC.hBDD-FVIII-transduced HA BOECs (red line) showing endothelial marker expression and absence of hematopoietic markers. The filled-up histograms represent unstained BOECs. (F) Matrigel assay confirming tubule formation of transduced BOECs. (G) RT-PCR, using primers specific for the exogenous F8 in non-transduced and LV-VEC.hBDD-FVIII BOECs. Unrelated transduced cells and fibroblast were used as positive and negative control, respectively. (H) FVIII intracytoplasmic staining on non-transduced (black line) or transduced healthy BOECs (red line). The filled-up histogram represents unstained BOECs. (I) FVIII intracytoplasmic staining on non-transduced (black line) or transduced HA BOECs (red line). The filled-up histogram represents unstained BOECs. (J) FVIII detection by immunofluorescence: blue, DAPI; red, anti-FVIII. Data are expressed as mean ± SD and are representative of four independent experiments.
We next assessed the endothelial phenotype and functionality of transduced versus non-transduced healthy or HA BOECs. As shown in Figure 1B, all cells expressed classical endothelial markers (e.g., PECAM1, KDR, TEK, CDH5, and VWF) as well as other genes specific to blood endothelial cells (BECs)48,49 (Figure 1C). The endothelial phenotype of healthy and HA BOECs was further verified at the protein level (Figures 1D and 1E, respectively), while the hematopoietic phenotype was ruled out upon CD34 and CD45 staining, which resulted negative (Figures 1D and 1E).
The endothelial functionality of the transduced cells was confirmed by their ability to form tubule networks upon Matrigel cell culture (Figure 1F). F8 mRNA expression was measured in transduced BOECs by RT-PCR (Figure 1G), while FVIII protein expression levels were detected by flow cytometry (see Figures 1H and IJ for healthy and HA BOECs, respectively) and immunofluorescence (IF) (Figure 1J). Interestingly in healthy BOECs we detected low levels of FVIII (Figures 1H and 1J). This is in accordance with previous works where FVIII in healthy BOECs was barely observed.41,50,51 When cell supernatants were subjected to activated partial thromboplastin time (aPTT) assay, we noticed a consistent shortening in LV-VEC.hBDD-FVIII-transduced BOECs (69 ± 3.5 s for transduced healthy BOECs, 66 ± 4 s for transduced HA BOECs) compared with non-transduced cells (80 ± 2.8 s for healthy BOECs, 84 ± 2.8 s for HA BOECs) (Figure S2B), in good agreement with the amount of secreted FVIII (35.9 ± 2.3 ng/mL for LV-VEC.hBDD-FVIII healthy BOECs, 4.5 ± 1.3 ng/mL for non-transduced healthy BOECs; 54 ± 7.5 ng/mL for LV-VEC.hBDD-FVIII HA BOECs, 0.15 ± 2.5 ng/mL for non-transduced HA BOECs) (Figure S2C).
To further evaluate the safety of LV transduction, healthy BOECs were transduced with different MOIs (MOI = 10, 20, 50, or 100), and HIV-1 p24 expression on cell supernatant was assessed. As shown in Figure S2C, all LV-transduced BOEC supernatants were negative for HIV-1 p24 at any of the MOIs tested 10 days after transduction, suggesting the reliability of our protocol. HIV-1 p24 on supernatant of HA BOECs transduced with an MOI of 20 showed comparable results (Figure S2D).
Secretion of the FVIII gene product by LV-transduced HA or healthy BOECs in NSG-HA mice
Since healthy and HA BOECs were both able to secrete FVIII in vitro, we evaluated their ability to survive and secrete FVIII in NSG-HA mice after intraperitoneal injection in association with Cytodex 3 microcarrier beads. Following injection, FVIII-transduced GFP+ healthy BOECs were able to partially restore FVIII activity, which reached a peak of approximately 10% at 4 weeks post injection (pi) and persisted above 5% for up to 10 weeks pi (Figure 2A). As expected, LV-VEC.GFP BOEC controls showed only basal FVIII secretion, which only lasted for 4 weeks. Importantly, mice receiving FVIII-transduced HA BOECs revealed sustained therapeutic FVIII activity (up to 10%) for up to 13 weeks pi, which persisted at a level <5% throughout the following 18 weeks (Figure 2A). Blood loss assays, run between 7 and 10 weeks pi of FVIII-transduced GFP+ HA BOECs, demonstrated partial restoration of hemostasis (Figure 2B) accompanied by detectable amounts of plasmatic FVIII antigen (12.4 ± 3.9 ng/mL) (Figure 2C). At week 13 pi, three mice in each experimental group were killed, and beads were recovered from the abdominal cavity. IF staining was performed on the recovered beads using an anti-GFP antibody that would detect the transplanted cells that were previously transduced with both LV-VEC.GFP and LV-VEC.FVIII. Therefore, the presence of GFP+ cells confirm that they are still associated with the beads and that they maintained an endothelial phenotype, as shown by the co-staining with CD31 (Figure 2D).
Figure 2.

Intraperitoneal implantation of BOECs with Cytodex microcarrier beads
(A) Kinetics of the percentage of FVIII activity measured by aPTT assay in the plasma of transplanted NSG-HA mice. BOECs used were transduced only with LV-VEC.GFP or with both LV-VEC.hBDD-FVIII and LV-VEC.GFP. Data are expressed as mean ± SD and are representative of two independent experiments using BOECs from two healthy donors (n = 7 mice), and four independent experiments using HA BOECs from four patients (n = 23 mice). (B) Blood loss evaluation on NSG-HA mice between weeks 7 and 10 (n = 4) after cell transplantation. (C) FVIII concentration in plasma of mice transplanted with transduced or non-transduced BOECs at week 16. Data are expressed as mean ± SD (∗∗∗p < 0.0001, ∗∗p < 0.001). (D) Representative immunofluorescence on beads showing cells co-expressing GFP and CD31.
Large-scale expansion of FVIII-transduced cells
With the aim to translate this approach into the clinic, we developed a protocol that would allow us to obtain a large amount of transduced HA BOECs for our in vivo experiments. LV-VEC.hBDD-FVIII-transduced HA BOECs from four patients were large-scale expanded to reach 108 cells, frozen, and sent to the partners in accordance with GMP-like procedures. Upon arrival, cells were re-cultured by simulating a centralized cell production process with long-term cryopreservation. After large-scale expansion and cryopreservation, upon thawing and reseeding, all cells showed normal cobblestone-like morphology (Figure 3A). Even though they were slightly enlarged, no significant changes in their doubling time, cell density, and length of time required for expansion were noticed (Figure 3B). In addition to maintaining expression of the classical endothelial markers (CD31, KDR, Tie-2, and VEC), expanded BOECs became CD34+, a transmembrane phosphoglycoprotein involved in cell adhesion,52 while they retained the classical CD45− phenotype (Figure 3C).
Figure 3.

Large-scale expansion of HA patient-derived BOECs
(A) Light microscope pictures of transduced HA BOECs pre- and post-expansion. (B) Cell size, cell density, culture time, and population doubling level during pre- and post-large-scale expansion. (C) Endothelial marker expression pre- and post-large-scale expansion expressed as stained cells versus cells with secondary isotype controls. (D) Tubulogenic assay to assess the functionality of transduced HA BOECs after pre- and post-large-scale expansion. (E) Kinetics of the percentage of FVIII activity measured by aPTT assay in plasma of transplanted NSG-HA mice. Data are expressed as mean ± SD and are representative of two independent experiments (n = 7).
Functionally, FVIII-transduced cells preserved their tubulogenesis activity (Figure 3D) and led to partial restoration of FVIII activity once transferred into NSG-HA mice (Figure 3E), similar to the kinetics of non-expanded BOECs. Thus, LV-VEC.hBDD-FVIII HA BOECs maintain their ability to secrete FVIII at therapeutic levels even after large-scale expansion.
Tissue matrix development and safety of LV-VEC.hBDD-FVIII-transduced BOECs within a Cell Pouch implanted in NSG-HA mice
The Cell Pouch is a medical implantation device specifically designed to enable the development of a vascularized tissue matrix environment that ensures long-term survival and function of transplanted therapeutic cells. Thus, we first evaluated the safety and survival of transduced HA BOECs within the Cell Pouch implanted in NSG-HA mice. For this purpose, 4-week implanted Cell Pouches were transplanted with one of three doses (2×, 5×, or 10 × 106) of LV-VEC.hBDD-FVIII HA BOECs isolated from two separate HA donors. The Cell Pouches transplanted with BOECs were explanted at 4, 8, or 12 weeks, and a gross pathological assessment was performed. HA BOECs were safe across doses and time points with no visible tumors observed (n = 60 total; HA1 n = 37; HA2, n = 23) (data not shown).
Overall, the tissue matrix developed within the Cell Pouch internal chamber and transplant area was viable among all groups according to time, dose, and cell lot, with no apparent signs of inflammation, hemorrhage, fibrosis, or necrosis (Figure 4A; Table S1). The center of the transplanted chamber area showed mild to moderate collagen deposition without any difference due to donor lot, time, or dose. Within the area of pre-vascularization, there was a comparative increase in established collagen, indicating that the Cell Pouch promoted, over time, the development of a natural scaffold to provide strength and structure to the environment, irrespective of the transplant. Regarding tissue vascularization, there was moderate neovascularization of the central, transplanted tissue of the Cell Pouch that was present in both donor lots, as well as the controls, along with evidence of established vessel growth, indicating that the tissue development within this area included new blood vessel formation (Figure 4B). Established vessels within the central, transplanted zone appeared to be more prominent and donor dependent at the latest time points (Figure 4A).
Figure 4.

Pathological assessment after transplantation of LV-VEC.hBDD-FVIII HA BOECs into the Cell Pouch device
(A) Sernova Cell Pouches were removed at 4, 8, or 12 weeks and stained by H&E and Masson's trichrome for blinded histopathological analysis. Histology scores and representative images at 12 weeks post-transplant with 10 × 106 LV-VEC.hBDD-FVIII BOECs (animal groups n = 2–3). (B) Quantification of H&E and Masson's trichrome for blinded histopathological analysis.
Transduced HA BOECs improve the bleeding phenotype and cell survival in mice after transplantation into the vascularized Cell Pouch
The therapeutic efficacy of LV-VEC.hBDD-FVIII HA BOECs transplanted into the Cell Pouch was evaluated by performing a tail bleeding assay on NSG-HA mice 4 months after cell transfer. Remarkably, we noticed a significantly improved presence of clotting as judged by a reduction in blood volume recovered in animals transplanted with 20 × 106 LV-VEC.hBDD-FVIII HA BOECs compared with non-transplanted mice (Figure 5A). Notably, this was not significantly different when compared with NSG mice, confirming that correction of the missing coagulation factor had been achieved in the transplanted HA mice. Relatively long-term cell survival (4 months post-transplant) was confirmed by co-staining with anti-HLA-ABC and anti-vWF antibodies as well as by the formation of blood vessels within the transplanted area (Figures 5B and 5C; Table S2).
Figure 5.

Bleeding phenotype and cell survival of LV-VEC.hBDD-FVIII HA BOECs after implantation in the Cell Pouch device
(A) Bleeding assay on mice transplanted with 10 × 106 or 20 × 106 HA and LV-VEC.hBDD-FVIII BOECs, or left untreated (n = 3–6, mean ± SEM, ∗∗p < 0.05; ns, not significant). NSG mice were used as control for bleeding assay. (B) The transplanted Cell Pouch devices were removed from the recipient NSG-HA mice, and immunofluorescence was performed to detect cell survival within the mouse tissue by human cell staining (HLA-ABC) and blood vessel formation through staining with cross-reacting human/mouse von Willebrand factor (vWF) antibody. The images shown are representative of two transplant groups (10 × 106 n = 5; 20 × 106 n = 12). (C) Quantification of HLA-ABC and blood vessel formation from blinded histopathological assessment.
Overall, these data indicate that corrected HA BOECs are able to engraft and persist for prolonged periods of time within the tissue matrix supported by the Cell Pouch and secrete enough FVIII to correct the hemophilia phenotype of the implanted NSG-HA mice.
Complex composition of BOEC clonal populations
Sonication linker-mediated PCR was performed on 53 samples of genomic DNA extracted from LV-transduced BOECs derived from 3 healthy donors (D45, D2, and D3) and 3 HA patients (pHA1, pA, and pC), collected at different expansion passages or procedure time points. By grouping the samples according to the BOEC source (i.e., healthy donors or HA patients) and the type of vector used (i.e., LV-VEC.hBDD-FVIII or LV-VEC.GFP), we obtained 4 main groups: HA.FVIII, HA.GFP, Healthy.FVIII, and Healthy.GFP. Overall, we retrieved 142,349 integration sites (ISs) (HA.FVIII, 28,069; HA.GFP, 106,554; Healthy.FVIII, 5,864; Healthy.GFP, 1,862) (Table S3). We compared the distribution of ISs of the four groups along the whole human genome and with respect to gene transcription start site. The profile of LV integrations was similar for all the groups and confirmed the marked tendency of the LV to integrate within gene bodies, without bias for promoter regions (Figures 6A and 6B), in line with previously published results.53, 54, 55, 56 Following enrichment analysis of genomic position and gene annotations, none of the ontological gene classes showed cancer or tumor suppressor gene enrichment (Table S4).
Figure 6.

Genome wide distribution of lentiviral vector ISs
(A) The pink track represents the density distribution of genes (RefSeq annotation, hg19 genome). The green tracks are the density distributions of all the ISs retrieved in the HA transduced with LV-VEC.GFP and Healthy transduced with LV-VEC.GFP groups. The blue tracks are the density distributions of all the ISs retrieved in the LV-VEC.hBDD-FVIII HA BOECs and LV-VEC.hBDD-FVIII Healthy BOECs groups. (B) Distribution of ISs of the four groups along the whole human genome and with respect to gene transcription start site (TSS).
Common insertion site (CIS) analysis showed few highly targeted genes in all datasets (e.g., NPLOC4, PACS1, and MROH1) (Table S5). The quantification of IS abundance showed only a few clones with abundance >10% in LV-VEC.hBDD-FVIII-transduced BOECs from pA and pHA1 (Figure 7). Only two ISs were retrieved from D45 cells transduced at an MOI of 30, thus resulting in both to be 50% abundant. One clone with an IS in the GNL3 gene, which may interact with p53 and may be involved in tumorigenesis, and with abundance >25%, was also observed in pHA1 BOEC, but only at a single time point (P11-UK). To address the clonality of transduced BOECs, we analyzed the diversity of the clonal population through Shannon diversity index. The highest Shannon diversity index, between 9 and 11, was observed in BOECs from pHA1 and pA, transduced with the VEC.GFP vector. All the other BOECs showed a Shannon diversity index between 4 and 8, which remained constant throughout the various cell passages. A lower diversity index directly correlated with a lower number of ISs, in particular for the HA Beads and Cell Pouch samples at different time points (Figure 8A). To better understand if, especially in the Cell Pouch samples, the clonal diversity was reduced, we compared the H index between samples grouped by type (expansion, HA beads, Cell Pouch and LV used [VEC-FVIII, VEC-GFP]) (Figure 8B). IS analysis revealed a high level polyclonality of LV-transduced BOECs, with no significant difference between the FVIII- and GFP-transduced samples. The clonal composition heterogeneity of FVIII-transduced samples remained constant over time in vitro and in vivo. Finally, Cell Pouch samples had a significant lower H index when compared with BOECs in expansion.
Figure 7.

Boxplot representation of clonal abundance
For each sample, the abundance values for each clone are represented as dots. Clones over 10% are presented as dots labeled with the closest gene symbol (RefSeq hg19).
Figure 8.

Clonal diversity comparison
(A) Shannon diversity index for each transduced cell population according to cell passage and time point. (B) H index comparison between different groups.
Discussion
Although the current therapy for HA involves the administration of plasma-derived or recombinant FVIII, there is to date no definitive cure for this inherited bleeding disorder. While several ongoing phase I–III clinical trials assessing the feasibility and safety of AAV-mediated hepatocyte-directed HA gene therapy have been able to achieve therapeutic FVIII plasma levels,57, 58, 59 further experiments are in progress to assess the long-term stability of transgene expression. In this regard, the fact that AAV vectors do not actively integrate into the host cell genome and, thus, can be lost upon cell division during liver growth or liver disease questions their life-long maintenance besides limiting their potential use in pediatric patients.
A promising alternative approach is represented by a combination of cell and gene therapy, which would, however, require the identification of a suitable cell type able to effectively secrete FVIII while meeting all the necessary conditions for successful cell transplantation. In this regard, it is widely acknowledged that the liver is the main organ producing FVIII, where LSECs appear to be the main source of FVIII60, 61, 62 and can play a tolerogenic role. In addition, because of the important role of the interaction between FVIII and vWF in the stability and activity of FVIII, LSECs may represent the most suitable target for cell- and gene therapy-based strategies aimed to correct the HA phenotype.63 Unfortunately, LSEC are not easy to obtain and maintain in vitro; therefore, in this study we explored the feasibility of using gene-corrected autologous BOECs more manageable and previously shown to be able to secrete FVIII in vivo.64 Here, we show that BOECs isolated from both healthy and HA donors can be efficiently cultured, transduced, expanded, and used to correct the bleeding phenotype of HA mice. In this regard, it is important to point out that the large number of corrected cells we obtained allowed us to reach FVIII therapeutic concentrations more quickly, thus reducing the risk of cell senescence.
The current protocols for BOEC isolation are based on the culture of mononuclear cells (MNCs) from peripheral or umbilical cord blood on collagen-coated cell culture vessels in endothelial-specific medium.65 The fact that MNCs can be isolated directly through density gradient centrifugation of blood makes these cells a safe cell source for hemophilic patients. Normally, BOEC colonies arise after 2–4 weeks of culture, and the colonies are very rare since their number in the normal peripheral blood is quite low.65 However, here we show that, under GMP-compliant conditions and using a chemically defined medium, it is possible to isolate BOECs from both healthy donors and HA patients with high efficiency and rapidly grow them to the desired amount to prevent the risk of cellular senescence once transplanted in mice.
In addition to being more easily obtainable, BOECs are fully differentiated ECs with a mature endothelial phenotype. Indeed, these cells originate from bone marrow-derived progenitors circulating in the blood or residing in the endothelium, which can be differentiated into BOECs in vitro.66 Thus, the observation that the expanded pools of BOECs from healthy donors or HA patients retained the expression of endothelial markers and were able to form vessels indicates that our GMP-compliant conditions did not alter the endothelial phenotype and function of these cells, as shown previously.66 Moreover, the healthy BOECs showed low FVIII expression, as demonstrated by IF staining and FACS analysis, in agreement with the low FVIII secretion found in the cell supernatant. This is similar to what has been shown by previous studies in which healthy BOECs isolated from both canine and human donors displayed low FVIII expression.41,50,51 Indeed, FVIII expression heterogeneity among different endothelial subpopulations has been reported, with the sinusoidal ECs shown as the main FVIII-secreting cells.67Another important aspect of this study is that we efficiently transduced BOECs with an LV carrying a functional BDD form of FVIII driven by the endothelial-specific promoter VEC. The efficiency and tissue specificity of FVIII transcription under the control of this promoter has been previously demonstrated in gene therapy approaches showing the restriction of FVIII expression in the desired cell type68 and in cell therapy by secretion of FVIII after genetic correction in target cells.31 Here, we show that LV-corrected HA BOECs transplanted in association with Cytodex 3 microcarrier beads into the peritoneum of NSG-HA mice rescues the hemophilic phenotype of these animals for up to 18 weeks, achieving 9% FVIII activity.
Importantly, we reached therapeutic levels of secreted FVIII through LV-VEC.hBDD-FVIII HA BOEC injection into a prevascularized Cell Pouch device transplanted into a preclinical murine model of severe HA. Notably, the correction of the bleeding phenotype by using LV-VEC.hBDD-FVIII HA BOECs injected into the peritoneum lasted up to 13 weeks and then slowly decreased. After 18 weeks, FVIII activity was almost absent, probably due to the death of BOECs.
Despite the encouraging results presented in this proof-of-concept study in a preclinical setting, there still remain several important issues that need to be addressed before our approach can be brought into the clinic. For instance, it will be imperative to characterize the cells within the Cell Pouch in terms of cell markers, longevity, and proliferation/senescence status. It will also be important to assess if we can increase the expression levels of FVIII using different EC-specific promoters, and if that would translate into augmented FVIII secretion and functionality ex vivo.
Overall, our findings indicate that cell transfer into a medical device is a suitable solution for cell therapy as it confers a more physiological and protected environment where cells can proliferate at an excellent rate and escape from the immune response of the transplanted organism, all the while allowing nutrient exchange and therapeutic protein secretion. Congruently, the safety and efficacy of the Cell Pouch for the transplantation of mouse pancreatic islets has been previously shown to provide insulin independence in diabetic animals in preclinical studies of type 1 diabetes mellitus.45,69 Furthermore, a phase I/II clinical trial is ongoing for the treatment of T1DM patients, the result of which may support the potential application of this device to other diseases for cell therapy approaches, such as HA.70
The Cell Pouch is a biocompatible, safe, implantable device that forms an internal vascularized tissue matrix supporting the transplanted cells. When we analyzed the Cell Pouch injected with LV-VEC.hBDD-FVIII HA BOECs after 4 weeks from cell transfer, we observed the presence of a viable vascularized tissue matrix supporting the cells, with no evidence of fibrosis-associated consequences, including inflammation and necrosis, or hemophilia-related hemorrhage episodes. Moreover, the bleeding assay demonstrated that LV-VEC.FVIII HA BOECs transplanted into the vascularized subcutaneous Cell Pouch were able to correct the clotting function of HA mice. FVIII secretion and activity measurement would support the data on hemophilic correction and strengthen our observations. These tests are planned for future studies. As previously shown in a canine model of HA, BOECs transduced with an LV carrying the canine FVIII and implanted subcutaneously allowed secretion of therapeutic levels of FVIII up to 15 weeks in Matrigel scaffolds and up to a year after omental implantation.64 Moreover, BOECs were shown to form tubule networks in vitro when plated on Matrigel71 or on the surface of synthetic vascular scaffolds72 and to promote neovascularization in vivo when transplanted into immunodeficient mice,37 suggesting that they can be directly involved in vessel formation.
In this context, our data attest the feasibility of a method to correct autologous cells based on a combined cell and gene therapy approach together with the use of a scaffold (i.e., Cell Pouch) able to guarantee long-term cell survival and, in case of need, a re-injection of new therapeutic cells. In addition to the phenotypical and functional characterization of the transduced HA BOECs, our results demonstrate the pharmacodynamics proof-of-concept in non-clinical models, which is mandatory before any GTMP can be used in human clinical trial.43,44 Thus, our next step will be to evaluate the safety and toxicity of the GTMP in vivo based on these results so as to ensure patient safety and promote product translation. Examples of required non-clinical studies are the evaluation of the potential tumorigenicity and biodistribution of the transduced BOECs with or without the medical device.73 Our molecular analysis of the integration sites in BOECs shows that no enrichment for oncogenes or expansion of clones with ISs in CISs or biases toward gene classes related to cancer genes occurred. IS analysis suggests a high level of polyclonality of LV-transduced BOECs, with no statistical difference between the FVIII- and GFP-transduced samples. The heterogeneity of the clonal composition of the FVIII-transduced samples remained constant over time (between different cell passages) also when cells were coupled to the microcarrier beads. Furthermore, Cell Pouch samples had a statistically significant lower H index when compared with BOECs in expansion.
The process of BOEC engraftment within the subcutaneous space is novel and complex and further studies will provide additional insight into the interactions between the developing tissue and the transplanted cells, elucidating the role played in the kinetics of blood vessel formation and FVIII secretion within the surrounding tissue.
In this study, we could not evaluate the immune response to the secreted factor because we used implanted cells in immunodeficient hemophilic mice. Thus, in future studies it will be interesting to evaluate antibody formation after transplantation of transduced BOECs encapsulated in the Cell Pouch into immunocompetent mice. Finally, while several gene therapy clinical trials for HA are ongoing, to our knowledge this is the first therapeutic approach that combines the GMP production of autologous human BOECs with the use of a safe ex vivo approach based on an implantable prevascularized device.
In conclusion, our findings suggest that long-term encapsulation and survival of LV-corrected BOECs by means of an implantable device may prove effective in ameliorating the QoL of HA patients. The therapeutic dose of FVIII released by these autologous genetically modified cells would in fact prevent the need of frequent infusions of FVIII and significantly reduce the morbidity and the frequency of the bleeding episodes in hemophiliacs.
Materials and methods
BOEC isolation form HA patients and healthy donors
Blood sampling from four adult severe HA patients, named pHA1, pA, pC, and pD, was performed at the hospital A.O.U. Città della Salute e della Scienza, Turin, Italy. The blood was shipped at room temperature to Università del Piemonte Orientale (UPO), Novara, Italy. Blood sampling from adult severe HA patients was approved by the Ethics Committee “Comitato Etico Interaziendale A.O.U. Maggiore della Carità” (Protocol 810/CE, study no. CE 125/17). Human BOECs were isolated as described previously,65 with the introduction of an earlier cell passaging step 7 days after initial isolation of the peripheral blood MNCs to reduce expansion time and increase the final cell yield.74 Isolated cells were cultured on CELLCOAT Collagen Type 1-coated tissue culture flasks (Greiner Bio-One) using MCDB 131 medium (Gibco, Life Technologies) containing proprietary supplements. Primary cells from adult healthy donors (named D45, D2, and D3) were isolated at Tissue Engineering and Regenerative Medicine, Würzburg, Germany, under informed consent according to ethical approval granted by the Institutional Ethics Committee of the University Hospital Würzburg (approval no. 182/10). Cell viability and count were assessed using the Countess II FL Automated Cell Counter (Thermo Fisher Scientific).
Healthy and HA BOEC transduction
Healthy and HA BOECs were plated at a 104 cells/cm2 density and after 6–8 h transduced with a lentiviral vector carrying the BDD form of FVIII under the control of the VE-cadherin promoter (LV-VEC.hBDD-FVIII) or with a lentiviral vector carrying the green fluorescent protein under the control of the same VE-cadherin promoter (LV-VEC.GFP), using an MOI of 20. After 14–16 h incubation, fresh medium was added to the cells and, 72 h later, half of the cells were harvested for subsequent analysis, while the other half was further cultured.
GMP-compliant (GMP-like) preclinical development of LV-VEC.hBDD-FVIII-transduced BOECs
BOECs were isolated and expanded using a GMP-compliant standardized approach between all partners, including a quality control strategy. The standardized expansion scheme defined within the project is based on the generation of Master Cell Banks and a Working Cell Bank, which ensures not only a controllable defined expansion for each patient's BOECs but also an in-process quality control at defined crucial steps. After isolation and expansion, cells were transduced with LV lots produced with a GMP-compliant method (TFF, see supplemental information). All freezing steps were performed using a cryopreservation solution based on compounds that are GMP-compliant free of toxic compounds (e.g., DMSO). The Cell Pouch was manufactured under GMP-compliant conditions. All steps were designed and conducted according to European GMP regulations to ensure that the product would fully comply with the quality requirements of the European authorities. The main objectives were to provide sets of design and manufacturing protocols based on current European GMP regulations and to prepare an Investigational Medicinal Product Dossier for an Investigational Medicinal Product, composed of therapeutic cells and an implantable medical device (Cell Pouch), a so-called combined Advanced Therapeutic Medicinal Product (combined ATMP).
IS analysis
ISs were retrieved from genomic DNA of LV-transduced BOEC cells by sonication linker-mediated PCR, an adaptation of a previously described method.75,76 Genomic DNA (300 ng) was sheared using a Covaris E220 Ultrasonicator (Covaris, Woburn, MA), generating fragments with a target size of 1,000 bp. The fragmented DNA was split into three parts to generate technical replicates and, by using the NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA), subjected to end repair, 3′ adenylation, and ligation to linker cassettes (Integrated DNA Technologies, Skokie, IL) containing an 8-nucleotide sequence barcode used for sample identification and a 12-random nucleotide sequence necessary for clonal abundance quantification. Ligation products were then subjected to 35 cycles of exponential PCR using primers specific for the lentiviral vector LTR and the linker cassette. The amplification product was then re-amplified with an additional 10 PCR cycles using primers specific for the linker cassette and the LTR, with the latter containing a second barcode to adopt a double barcode strategy for sample identification. The final PCR products were quantified using a KAPA Library Quantification Kit (Roche, Basil, Switzerland) and pooled in sequencing libraries with equimolar composition, avoiding repeated barcode pairs. Primers incorporate the adapter sequences required for the Illumina paired end sequencing technology (Illumina, San Diego, CA). Sequencing was performed on the Illumina MiSeq and HiSeq. Sample processing and metadata were tracked within our laboratory information management system.77,78 Sequencing reads were processed using a dedicated bioinformatics pipeline (VISPA2).78 In brief, paired sequence reads were filtered for raw read quality, then cleaned by vector genome, and the resulting cellular genomic sequence mapped on the human genome (version hg19), and the nearest RefSeq gene assigned to each unambiguously mapped ISs. Clonal abundance for each IS was estimated using the R package sonicLength,79 where the number of genomes with the same integration site is calculated by counting the number of fragments with different sizes generated by sonication belonging to each individual IS. Within each group, ISs shared between different time points of the same transduction were counted once. The relative abundance of each clone was then calculated as the percentage of genomes with a specific integration site over the total genomes. CISs were identified through the Grubbs test for outliers.80 Enrichment analysis for ontological classes among the targeted genes by vector ISs was performed using the Genomic Regions Enrichment of Annotations Tool.81
Animal procedures
Animal studies were approved by the Animal Care and Use Committee at UPO (Italian Health Ministry Authorization nos. 492/2016-PR and DBO64.5). NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (Jackson, stock no. 005557) mice with hemophilic phenotype (NSG-HA) were previously generated and maintained in our laboratory.22 Eight- to 10-week-old animals were used for cell transplantation studies. Cell Pouch implantations were conducted under additional ethical guidelines and approval from the Animal Care Committee at the University of British Columbia (Vancouver, BC, Canada) in accordance with the Canadian Council on Animal Care Guide to the Care and Use of Experimental Animals.
BOEC transplantation
For cell transplantation with beads, 5 × 106 FVIII-transduced healthy or HA BOECs were mixed with Cytodex 3 microcarrier beads (GE Healthcare Life Sciences) and intraperitoneally delivered in NSG-HA mice as described previously.23 For Cell Pouch implantation, female NSG and NSG-HA animals were anesthetized and surgically implanted with a Cell Pouch in the subcutaneous space of the lower abdomen 4 weeks before cell transplantation, allowing incorporation with vascularized tissue and forming fully developed tissue chambers suitable for cell transplantation upon removal of a space holding plug. LV-VEC.hBDD-FVIII BOECs were cultured for 3 days post-thawing and finally transplanted into the Cell Pouch. Mice received either a dose of viable BOECs (2–20 × 106) or remained untreated. All animals received a prophylactic dose (2–4 IU) of recombinant human FVIII by tail vein injection before surgical procedures.
FVIII activity
aPTT assay was performed on plasma samples of transplanted mice to assess FVIII activity. Standard curves were generated by serial dilution of recombinant human BDD-FVIII (ReFacto) in hemophilic mouse plasma. Analyses were performed using a Coatron M4 coagulometer (TECO Medical Instruments) and TEClot APTT-S kit reagents (TECO Medical Instruments).
Bleeding assay
A bleeding assay was performed on anesthetized mice. The distal portion of the tail was cut at a diameter of 2–2.5 mm. Tails were placed in a conical tube containing 14 mL of 37°C pre-warmed saline. Blood was collected for 10 min and, following centrifugation, resuspended in red blood lysis buffer (155 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA). The absorbance of the samples was measured at 575 nm. For cell transplantation experiments with Cell Pouch, the tail bleeding assay was performed using Sernova, as described previously,82,83 at the end of the experimental period, 4 months post-transplantation. In brief, mice were anesthetized, and tail tips were placed in a guide, ensuring the same diameter of 1 mm, and severed (∼a distal 10-mm segment) for each animal. The tail was immediately immersed in pre-warmed saline at 37°C. Bleeding was carried out for a maximum of 20 min, after which animals were euthanized as per approved animal use protocols. Blood loss was evaluated by determining hemoglobin concentration by lysing collected red blood cells (ACK Lysing Buffer, Gibco), and the absorbance measured at 550 nm on a Synergy Mx (BioTeck) spectrophotometer. Results were analyzed by comparing the amount of blood loss obtained from treated NSG-HA mice with control mice (untreated NSG-HA and NSG mice).
Statistical analysis
Data were expressed as means ± standard deviation (SD) or means ± standard error of the mean (SEM). Statistical significance was analyzed using Student's t test with two-tailed distribution, assuming equal SD distribution, two-way analysis of variance with Bonferroni post hoc test, or Tukey's multiple comparison post hoc tests in GraphPad Prism 6 (GraphPad software). Statistical analyses involving ISs were performed with the R software (r-project.org). Differences were considered statistically significant when p values were <0.05.
Acknowledgments
The authors thank Dr. D. Burton and Dr. A. Iftimia-Mander for their help, and V. Bruscaggin and V. Fiorio for technical assistance. We also thanks Dr. Marcello Arsura for the English revision of the manuscript. A.F., M.Z., A.S., P.M.T., and J.B. have received funding from the European Union's Horizon 2020 research and innovation program under grant agreement HemAcure no. 667421. A.F. was also supported in part by Telethon grant no. GGP19201 and by Horizon 2020, Vanguard grant no. 874700. This study was also partially funded by the Università del Piemonte Orientale (FAR 2017) to S.M., Fondazione Cariplo grant no. 2018-0253 to C.B., and by Italian Minister of Health “Ricerca Sanitaria finalizzata” Giovani Ricercatori grant no. 2018-12366399 to C.O.
Author contributions
C.O., C.B., S.M., T.B., P.B., A.B.A., N.W., K.P. and A. Cucci. performed the research and analyzed the data. A. Calabria, F.B., and E.M. performed integration analysis and analyzed the data. A.B. and B.P. collected blood samples from hemophilic patients and performed analysis. A.F., J.B., A.S., D.M.M., P.M.T., and M.Z. conceived the experiments generated funding, designed the research, and analyzed the data. C.O., C.B., S.M., and A.F. wrote the manuscript that was revised by all authors.
Declaration of interests
K.P., D.M.M., and P.M.T. are/have been employees of Sernova Corp., which holds the patent US20190240375A1.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2021.10.015.
Supplemental information
References
- 1.Bolton-Maggs P.H.B., Pasi K.J. Lancet. 2003. Haemophilias A and B; pp. 1801–1809. [DOI] [PubMed] [Google Scholar]
- 2.Roth D.A., Tawa N.E., O’Brien J.M., Treco D.A., Selden R.F. Nonviral transfer of the gene encoding coagulation factor VIII in patients with severe hemophilia A. N. Engl. J. Med. 2001;344:1735–1742. doi: 10.1056/NEJM200106073442301. [DOI] [PubMed] [Google Scholar]
- 3.Lissitchkov T., Rusen L., Georgiev P., Windyga J., Klamroth R., Gercheva L., Nemes L., Tiede A., Bichler J., Knaub S., et al. PK-guided personalized prophylaxis with Nuwiq® (human-cl rhFVIII) in adults with severe haemophilia A. Haemophilia. 2017;23:697–704. doi: 10.1111/hae.13251. [DOI] [PubMed] [Google Scholar]
- 4.Saxena K., Lalezari S., Oldenburg J., Tseneklidou-Stoeter D., Beckmann H., Yoon M., Maas Enriquez M. Efficacy and safety of BAY 81-8973, a full-length recombinant factor VIII: results from the LEOPOLD I trial. Haemophilia. 2016;22:706–712. doi: 10.1111/hae.12952. [DOI] [PubMed] [Google Scholar]
- 5.Mahlangu J., Kuliczkowski K., Karim F.A., Stasyshyn O., Kosinova M.V., Lepatan L.M., Skotnicki A., Boggio L.N., Klamroth R., Oldenburg J., et al. Efficacy and safety of rVIII-singlechain: results of a phase 1/3 multicenter clinical trial in severe hemophilia A. Blood. 2016;128:630–637. doi: 10.1182/blood-2016-01-687434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lentz S.R., Janic D., Kavakli K., Miljic P., Oldenburg J., Ozelo C.M., Santagostino E., Suzuki T., Zupancic Šalek S., et al. Long-term safety and efficacy of turoctocog alfa in prophylaxis and treatment of bleeding episodes in severe haemophilia A: final results from the guardian 2 extension trial. Haemophilia. 2018;24:e391–e394. doi: 10.1111/hae.13617. [DOI] [PubMed] [Google Scholar]
- 7.Mahlangu J., Young G., Hermans C., Blanchette V., Berntorp E., Santagostino E. Defining extended half-life rFVIII—a critical review of the evidence. Haemophilia. 2018;24:348–358. doi: 10.1111/hae.13438. [DOI] [PubMed] [Google Scholar]
- 8.Mahlangu J., Powell J.S., Ragni M.V., Chowdary P., Josephson N.C., Pabinger I., Hanabusa H., Gupta N., Kulkarni R., Fogarty P., et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood. 2014;123:317–325. doi: 10.1182/blood-2013-10-529974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Konkle B.A., Stasyshyn O., Chowdary P., Bevan D.H., Mant T., Shima M., Engl W., Dyck-Jones J., Fuerlinger M., Patrone L., et al. Pegylated, full-length, recombinant factor VIII for prophylactic and on-demand treatment of severe hemophilia A. Blood. 2015;126:1078–1085. doi: 10.1182/blood-2015-03-630897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cormier M., Batty P., Tarrant J., Lillicrap D. Advances in knowledge of inhibitor formation in severe haemophilia A. Br. J. Haematol. 2020;189:39–53. doi: 10.1111/bjh.16377. [DOI] [PubMed] [Google Scholar]
- 11.Dimichele D. Inhibitors: resolving diagnostic and therapeutic dilemmas. Haemophilia. 2002;8:280–287. doi: 10.1046/j.1365-2516.2002.00626.x. [DOI] [PubMed] [Google Scholar]
- 12.Van Den Berg H.M., Fischer K., Carcao M., Chambost H., Kenet G., Kurnik K., Königs C., Male C., Santagostino E., Ljung R. Timing of inhibitor development in more than 1000 previously untreated patients with severe hemophilia A. Blood. 2019;134:317–320. doi: 10.1182/blood.2019000658. [DOI] [PubMed] [Google Scholar]
- 13.Oldenburg J., Mahlangu J.N., Kim B., Schmitt C., Callaghan M.U., Young G., Santagostino E., Kruse-Jarres R., Negrier C., Kessler C., et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N. Engl. J. Med. 2017;377:809–818. doi: 10.1056/NEJMoa1703068. [DOI] [PubMed] [Google Scholar]
- 14.Pipe S.W., Shima M., Lehle M., Shapiro A., Chebon S., Fukutake K., Key N.S., Portron A., Schmitt C., Podolak-Dawidziak M., et al. Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given every 4 weeks in people with haemophilia A (HAVEN 4): a multicentre, open-label, non-randomised phase 3 study. Lancet Haematol. 2019;6:e295–e305. doi: 10.1016/S2352-3026(19)30054-7. [DOI] [PubMed] [Google Scholar]
- 15.Ebbert P.T., Xavier F., Seaman C.D., Ragni M.V. Emicizumab prophylaxis in patients with haemophilia A with and without inhibitors. Haemophilia. 2020;26:41–46. doi: 10.1111/hae.13877. [DOI] [PubMed] [Google Scholar]
- 16.Zimowski K.L., Batsuli G.M., Bryant P., McDaniel J., Tickle K., Meeks S.L., Sidonio R.F. Severe bleeding events in hemophilia A patients receiving emicizumab prophylaxis. Blood. 2019;134:1126. [Google Scholar]
- 17.Wion K.L., Kelly D., Summerfield J.A., Tuddenham E.G.D., Lawn R.M. Distribution of factor VIII MRNA and antigen in human liver and other tissues. Nature. 1985;317:726–729. doi: 10.1038/317726a0. [DOI] [PubMed] [Google Scholar]
- 18.Jiang H.C., Gao Y., Dai W.J., Sun B., Xu J., Qiao H.Q., Meng Q.H., Wu C.J. Ten-year experience with living related donated splenic transplantation for the treatment of hemophilia. A. Transpl. Proc. 2006;38:1483–1490. doi: 10.1016/j.transproceed.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 19.Hollestelle M.J., Thinnes T., Crain K., Stiko A., Kruijt J.K., Van Berkel T.J.C., Loskutoff D.J., Van Mourik J.A. Tissue distribution of factor VIII gene expression in vivo—a closer look. Thromb. Haemost. 2001;86:855–861. [PubMed] [Google Scholar]
- 20.Hollestelle M.J., Poyck P.P.C., Hollestelle J.M., Marsman H.A., Van Mourik J.A., Van Gulik T.M. Extra-hepatic factor VIII expression in porcine fulminant hepatic failure. J. Thromb. Haemost. 2005;3:2274–2280. doi: 10.1111/j.1538-7836.2005.01543.x. [DOI] [PubMed] [Google Scholar]
- 21.Follenzi A., Raut S., Merlin S., Sarkar R., Gupta S. Role of bone marrow transplantation for correcting hemophilia A in mice. Blood. 2012;119:5532–5542. doi: 10.1182/blood-2011-07-367680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zanolini D., Merlin S., Feola M., Ranaldo G., Amoruso A., Gaidano G., Zaffaroni M., Ferrero A., Brunelleschi S., Valente G., et al. Extrahepatic sources of factor VIII potentially contribute to the coagulation cascade correcting the bleeding phenotype of mice with hemophilia A. Haematologica. 2015;100:881–892. doi: 10.3324/haematol.2014.123117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumaran V., Benten D., Follenzi A., Joseph B., Sarkar R., Gupta S. Transplantation of endothelial cells corrects the phenotype in hemophilia A mice. J. Thromb. Haemost. 2005;3:2022–2031. doi: 10.1111/j.1538-7836.2005.01508.x. [DOI] [PubMed] [Google Scholar]
- 24.Fomin M.E., Zhou Y., Beyer A.I., Publicover J., Baron J.L., Muench M.O. Production of factor VIII by human liver sinusoidal endothelial cells transplanted in immunodeficient uPA mice. PLoS One. 2013;8:e77255. doi: 10.1371/journal.pone.0077255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Follenzi A., Benten D., Novikoff P., Faulkner L., Raut S., Gupta S. Transplanted endothelial cells repopulate the liver endothelium and correct the phenotype of hemophilia A mice. J. Clin. Invest. 2008;118:935–945. doi: 10.1172/JCI32748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Filali E. El, Hiralall J.K., Van Veen H.A., Stolz D.B., Seppen J. Human liver endothelial cells, but not macrovascular or microvascular endothelial cells, engraft in the mouse liver. Cell Transpl. 2013;22:1801–1811. doi: 10.3727/096368912X657594. [DOI] [PubMed] [Google Scholar]
- 27.Gollomp K.L., Doshi B.S., Arruda V.R. Gene therapy for hemophilia: progress to date and challenges moving forward. Transfus. Apher. Sci. 2019;58:602–612. doi: 10.1016/j.transci.2019.08.012. [DOI] [PubMed] [Google Scholar]
- 28.Peyvandi F., Garagiola I. Clinical advances in gene therapy updates on clinical trials of gene therapy in haemophilia. Haemophilia. 2019;25:738–746. doi: 10.1111/hae.13816. [DOI] [PubMed] [Google Scholar]
- 29.Samelson-Jones B.J., Arruda V.R. Translational potential of immune tolerance induction by AAV liver-directed factor VIII gene therapy for hemophilia A. Front. Immunol. 2020;11:618. doi: 10.3389/fimmu.2020.00618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merlin S., Famà R., Borroni E., Zanolini D., Bruscaggin V., Zucchelli S., Follenzi A. FVIII expression by its native promoter sustains long-term correction avoiding immune response in hemophilic mice. Blood Adv. 2019;3:825–838. doi: 10.1182/bloodadvances.2018027979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olgasi C., Talmon M., Merlin S., Cucci A., Richaud-Patin Y., Ranaldo G., Colangelo D., Di Scipio F., Berta G.N., Borsotti C., et al. Patient-specific iPSC-derived endothelial cells provide long-term phenotypic correction of hemophilia A. Stem Cell Reports. 2018;11:1391–1406. doi: 10.1016/j.stemcr.2018.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao K., Kumar P., Cortez-Toledo E., Hao D., Reynaga L., Rose M., Wang C., Farmer D., Nolta J., Zhou J., et al. Potential long-term treatment of hemophilia A by neonatal co-transplantation of cord blood-derived endothelial colony-forming cells and placental mesenchymal stromal cells. Stem Cell Res. Ther. 2019;10:3318. doi: 10.1186/s13287-019-1138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park C.-Y., Kim D.H., Son J.S., Sung J.J., Lee J., Bae S., Kim J.-H., Kim D.-W., Kim J.-S. Functional correction of large factor VIII gene chromosomal inversions in hemophilia A patient-derived iPSCs using CRISPR-cas9. Cell Stem Cell. 2015;17:213–220. doi: 10.1016/j.stem.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 34.Tatsumi K., Sugimoto M., Lillicrap D., Shima M., Ohashi K., Okano T., Matsui H. A novel cell-sheet technology that achieves durable factor VIII delivery in a mouse model of hemophilia A. PLoS One. 2013;8 doi: 10.1371/journal.pone.0083280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin Y., Weisdorf D.J., Solovey A., Hebbel R.P. Origins of circulating endothelial cells and endothelial outgrowth from blood. J. Clin. Invest. 2000;105:71–77. doi: 10.1172/JCI8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoder M.C., Mead L.E., Prater D., Krier T.R., Mroueh K.N., Li F., Krasich R., Temm C.J., Prchal J.T., Ingram D.A. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007:1801–1809. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoshi R.A., Van Lith R., Jen M.C., Allen J.B., Lapidos K.A., Ameer G. The blood and vascular cell compatibility of heparin-modified ePTFE vascular grafts. Biomaterials. 2013;34:30–41. doi: 10.1016/j.biomaterials.2012.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melero-Martin J.M., Khan Z.A., Picard A., Wu X., Paruchuri S., Bischoff J. In vivo vasculogenic potential of human blood-derived endothelial progenitor cells. Blood. 2007;109:4761–4768. doi: 10.1182/blood-2006-12-062471. [DOI] [PubMed] [Google Scholar]
- 39.Jantzen A.E., Lane W.O., Gage S.M., Jamiolkowski R.M., Haseltine J.M., Galinat L.J., Lin F.H., Lawson J.H., Truskey G.A., Achneck H.E. Use of autologous blood-derived endothelial progenitor cells at point-of-care to protect against implant thrombosis in a large animal model. Biomaterials. 2011;32:8356–8363. doi: 10.1016/j.biomaterials.2011.07.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarlon G., Zemani F., David L., Duong van Huyen J.P., Dizier B., Grelac F., Colliec-Jouault S., Galy-Fauroux I., Bruneval P., Fischer A.M., et al. Therapeutic effect of fucoidan-stimulated endothelial colony-forming cells in peripheral ischemia. J. Thromb. Haemost. 2012;10:38–48. doi: 10.1111/j.1538-7836.2011.04554.x. [DOI] [PubMed] [Google Scholar]
- 41.Lin Y., Chang L., Solovey A., Healey J.F., Lollar P., Hebbel R.P. Use of blood outgrowth endothelial cells for gene therapy for hemophilia A. Blood. 2002;99:457–462. doi: 10.1182/blood.v99.2.457. [DOI] [PubMed] [Google Scholar]
- 42.Medicines Agency, E. 2015. Reflection Paper on Classification of Advanced Therapy Medicinal Products.https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-classification-advanced-therapy-medicinal-products_en-0.pdf [Google Scholar]
- 43.Medicines Agency, E. 2020. Guideline on Quality, Non-clinical and Clinical Aspects of Medicinal Products Containing Genetically Modified Cells.https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-non-clinical-clinical-aspects-medicinal-products-containing-genetically-modified_en-0.pdf [Google Scholar]
- 44.Medicines Agency, E. 2008. Guideline on the Non-clinical Studies Required before First Clinical Use of Gene Therapy Medicinal Products.https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-non-clinical-studies-required-first-clinical-use-gene-therapy-medicinal-products_en.pdf [Google Scholar]
- 45.Pepper A.R., Pawlick R., Gala-Lopez B., MacGillivary A., Mazzuca D.M., White D.J.G., Toleikis P.M., James Shapiro A.M. Diabetes is reversed in a murine model by marginal mass syngeneic islet transplantation using a subcutaneous cell pouch device. Transplantation. 2015;99:2294–2300. doi: 10.1097/TP.0000000000000864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Charrier S., Ferrand M., Zerbato M., Précigout G., Viornery A., Bucher-Laurent S., Benkhelifa-Ziyyat S., Merten O.W., Perea J., Galy A. Quantification of lentiviral vector copy numbers in individual hematopoietic colony-forming cells shows vector dose-dependent effects on the frequency and level of transduction. Gene Ther. 2011;18:479–487. doi: 10.1038/gt.2010.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Y., Stepto H., Schneider C.K. Development of the first World Health Organization lentiviral vector standard: toward the production control and standardization of lentivirus-based gene therapy products. Hum. Gene Ther. Methods. 2017;28:205–214. doi: 10.1089/hgtb.2017.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Furuhata S., Ando K., Oki M., Aoki K., Ohnishi S., Aoyagi K., Sasaki H., Sakamoto H., Yoshida T., Ohnami S. Gene expression profiles of endothelial progenitor cells by oligonucleotide microarray analysis. Mol. Cell. Biochem. 2007;298:125–138. doi: 10.1007/s11010-006-9359-4. [DOI] [PubMed] [Google Scholar]
- 49.Nelson G.M., Padera T.P., Garkavtsev I., Shioda T., Jain R.K. Differential gene expression of primary cultured lymphatic and blood vascular endothelial cells. Neoplasia. 2007;9:1038–1045. doi: 10.1593/neo.07643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matsui H., Shibata M., Brown B., Labelle A., Hegadorn C., Andrews C., Hebbel R.P., Galipeau J., Hough C., Lillicrap D. Ex vivo gene therapy for hemophilia A that enhances safe delivery and sustained in vivo factor VIII expression from lentivirally engineered endothelial progenitors. Stem Cells. 2007;25:2660–2669. doi: 10.1634/stemcells.2006-0699. [DOI] [PubMed] [Google Scholar]
- 51.Biggelaar M.van den, Bouwens E.A.M., Kootstra N.A., Hebbel R.P., Voorberg J., Mertens K. Storage and regulated secretion of factor VIII in blood outgrowth endothelial cells. Haematologica. 2009;94:670–678. doi: 10.3324/haematol.13427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nielsen J.S., Mcnagny K.M. Novel functions of the CD34 family. J. Cell Sci. 2008;121:3683–3692. doi: 10.1242/jcs.037507. [DOI] [PubMed] [Google Scholar]
- 53.Barr S.D., Ciuffi A., Leipzig J., Shinn P., Ecker J.R., Bushman F.D. HIV integration site selection: targeting in macrophages and the effects of different routes of viral entry. Mol. Ther. 2006;14:218–225. doi: 10.1016/j.ymthe.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 54.Aiuti A., Biasco L., Scaramuzza S., Ferrua F., Cicalese M.P., Baricordi C., Dionisio F., Calabria A., Giannelli S., Castiello M.C., et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;80:341. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Biffi A., Montini E., Lorioli L., Cesani M., Fumagalli F., Plati T., Baldoli C., Martino S., Calabria A., Canale S., et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;80:341. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 56.Kvaratskhelia M., Sharma A., Larue R.C., Serrao E., Engelman A. Molecular mechanisms of retroviral integration site selection. Nucleic Acids Res. 2014;42:10209–10225. doi: 10.1093/nar/gku769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Study to Evaluate the Efficacy and Safety of PF-07055480 / Giroctocogene Fitelparvovec Gene Therapy in Moderately Severe to Severe Hemophilia A Adults (AFFINE) - ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04370054
- 58.Gene therapy for haemophilia A - full text view - ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03001830.
- 59.A gene transfer study for hemophilia A - full text view - ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03003533.
- 60.Shahani T., Covens K., Lavend’homme R., Jazouli N., Sokal E., Peerlinck K., Jacquemin M. Human liver sinusoidal endothelial cells but not hepatocytes contain factor VIII. J. Thromb. Haemost. 2014;12:36–42. doi: 10.1111/jth.12412. [DOI] [PubMed] [Google Scholar]
- 61.Everett L.A., Cleuren A.C.A., Khoriaty R.N., Ginsburg D. Murine coagulation factor VIII is synthesized in endothelial cells. Blood. 2014;123:3697–3705. doi: 10.1182/blood-2014-02-554501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fahs S.A., Hille M.T., Shi Q., Weiler H., Montgomery R.R. A conditional knockout mouse model reveals endothelial cells as the principal and possibly exclusive source of plasma factor VIII. Blood. 2014;123:3706–3713. doi: 10.1182/blood-2014-02-555151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Terraube V., O’Donnell J.S., Jenkins P.V. Factor VIII and von Willebrand factor interaction: biological, clinical and therapeutic importance. Haemophilia. 2010;16:3–13. doi: 10.1111/j.1365-2516.2009.02005.x. [DOI] [PubMed] [Google Scholar]
- 64.Ozelo M.C., Vidal B., Brown C., Notley C., Hegadorn C., Webster S., Harpell L., Ahlin J., Winterborn A., Handforth J., et al. Omental implantation of BOECs in hemophilia dogs results in circulating FVIII antigen and a complex immune response. Blood. 2014;123:4045–4053. doi: 10.1182/blood-2013-12-545780. [DOI] [PubMed] [Google Scholar]
- 65.Ormiston M.L., Toshner M.R., Kiskin F.N., Huang C.J.Z., Groves E., Morrell N.W., Rana A.A. Generation and culture of blood outgrowth endothelial cells from human peripheral blood. J. Vis. Exp. 2015;2015 doi: 10.3791/53384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hebbel R.P. Blood endothelial cells: utility from ambiguity. J. Clin. Invest. 2017;127:1613–1615. doi: 10.1172/JCI93649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hayakawa M., Sakata A., Hayakawa H., Matsumoto H., Hiramoto T., Kashiwakura Y., Baatartsogt N., Fukushima N., Sakata Y., Suzuki-Inoue K., et al. Characterization and visualization of murine coagulation factor VIII-producing cells in vivo. Sci. Rep. 2021;11:1–11. doi: 10.1038/s41598-021-94307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Merlin S., Cannizzo E.S., Borroni E., Bruscaggin V., Schinco P., Tulalamba W., Chuah M.K., Arruda V.R., VandenDriessche T., Prat M., et al. A novel platform for immune tolerance induction in hemophilia A mice. Mol. Ther. 2017;25:1815–1830. doi: 10.1016/j.ymthe.2017.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kriz J., Vilk G., Mazzuca D.M., Toleikis P.M., Foster P.J., White D.J.G. A novel technique for the transplantation of pancreatic islets within a vascularized device into the greater omentum to achieve insulin independence. Am. J. Surg. 2012;203:793–797. doi: 10.1016/j.amjsurg.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 70.A. Safety, tolerability and efficacy study of Sernova’s Cell PouchTM for clinical islet transplantation - full text view - ClinicalTrials.Gov https://clinicaltrials.gov/ct2/show/NCT03513939.
- 71.Hur J., Yoon C.H., Kim H.S., Choi J.H., Kang H.J., Hwang K.K., Oh B.H., Lee M.M., Park Y.B. Characterization of two types of endothelial progenitor cells and their different contributions to neovasculogenesis. Arterioscler. Thromb. Vasc. Biol. 2004;24:288–293. doi: 10.1161/01.ATV.0000114236.77009.06. [DOI] [PubMed] [Google Scholar]
- 72.Zhang X., Xu Y., Thomas V., Bellis S.L., Vohra Y.K. Engineering an antiplatelet adhesion layer on an electrospun scaffold using porcine endothelial progenitor cells. J. Biomed. Mater. Res. - Part A. 2011;97 A:145–151. doi: 10.1002/jbm.a.33040. [DOI] [PubMed] [Google Scholar]
- 73.Bittorf P., Bergmann T., Merlin S., Olgasi C., Pullig O., Sanzenbacher R., Zierau M., Walles H., Follenzi A., Braspenning J. Regulatory-compliant validation of a highly sensitive qPCR for biodistribution assessment of hemophilia A patient cells. Mol. Ther. - Methods Clin. Dev. 2020;18:176–188. doi: 10.1016/j.omtm.2020.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kolbe M., Dohle E., Katerla D., Kirkpatrick C.J., Fuchs S. Enrichment of outgrowth endothelial cells in high and low colony-forming cultures from peripheral blood progenitors. Tissue Eng. - Part C Methods. 2010;16:877–886. doi: 10.1089/ten.tec.2009.0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gillet N.A., Malani N., Melamed A., Gormley N., Carter R., Bentley D., Berry C., Bushman F.D., Taylor G.P., Bangham C.R.M. The host genomic environment of the provirus determines the abundance of HTLV-1-infected T-cell clones. Blood. 2011;117:3113–3122. doi: 10.1182/blood-2010-10-312926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Firouzi S., López Y., Suzuki Y., Nakai K., Sugano S., Yamochi T., Watanabe T. Development and validation of a new high-throughput method to investigate the clonality of HTLV-1-infected cells based on provirus integration sites. Genome Med. 2014;6:46. doi: 10.1186/gm568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sessa M., Lorioli L., Fumagalli F., Acquati S., Redaelli D., Baldoli C., Canale S., Lopez I.D., Morena F., Calabria A., et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet. 2016;388:476–487. doi: 10.1016/S0140-6736(16)30374-9. [DOI] [PubMed] [Google Scholar]
- 78.Spinozzi G., Calabria A., Brasca S., Beretta S., Merelli I., Milanesi L., Montini E. VISPA2: a scalable pipeline for high-throughput identification and annotation of vector integration sites. BMC Bioinform. 2017;18 doi: 10.1186/s12859-017-1937-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Berry C.C., Gillet N.A., Melamed A., Gormley N., Bangham C.R.M., Bushman F.D. Estimating abundances of retroviral insertion sites from DNA fragment length data. Bioinformatics. 2012;28:755–762. doi: 10.1093/bioinformatics/bts004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Biffi A., Bartholomae C.C., Cesana D., Cartier N., Aubourg P., Ranzani M., Cesani M., Benedicenti F., Plati T., Rubagotti E., et al. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood. 2011;117:5332–5339. doi: 10.1182/blood-2010-09-306761. [DOI] [PubMed] [Google Scholar]
- 81.McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu Y. Standardizing a simpler, more sensitive and accurate tail bleeding assay in mice. World J. Exp. Med. 2012;2:30. doi: 10.5493/wjem.v2.i2.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schuettrumpf J., Herzog R.W., Schlachterman A., Kaufhold A., Stafford D.W., Arruda V.R. Factor IX variants improve gene therapy efficacy for hemophilia B. Blood. 2005;105:2316–2323. doi: 10.1182/blood-2004-08-2990. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.