

Summary
Cellular morphology has the capacity to serve as a surrogate for cellular state and functionality. However, primary cardiomyocytes, the standard model in cardiovascular research, are highly heterogeneous cells and therefore impose methodological challenges to analysis. Hence, we aimed to devise a robust methodology to deconvolute cardiomyocyte morphology on a single-cell level: C-MORE (cellular morphology recognition) is a workflow from bench to data analysis tailored for heterogeneous primary cells using our R package cmoRe. We demonstrate its utility in proof-of-principle applications such as modulation of canonical hypertrophy pathways and linkage of genotype-phenotype in human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs). In our pilot study, exposure of cardiomyocytes to blood plasma prior to versus after aortic valve replacement allows identification of a disease fingerprint and reflects partial reversibility following therapeutic intervention. C-MORE is a valuable tool for cardiovascular research with possible fields of application in basic research and personalized medicine.
Keywords: cellular morphology, high-content imaging, high-throughput screening, human induced pluripotent stem cell-derived cardiomyocytes, hypertrophic cardiomyopathy, neonatal rat cardiomyocytes, primary cells, phenoprint, phenotype, pattern recognition
Graphical abstract
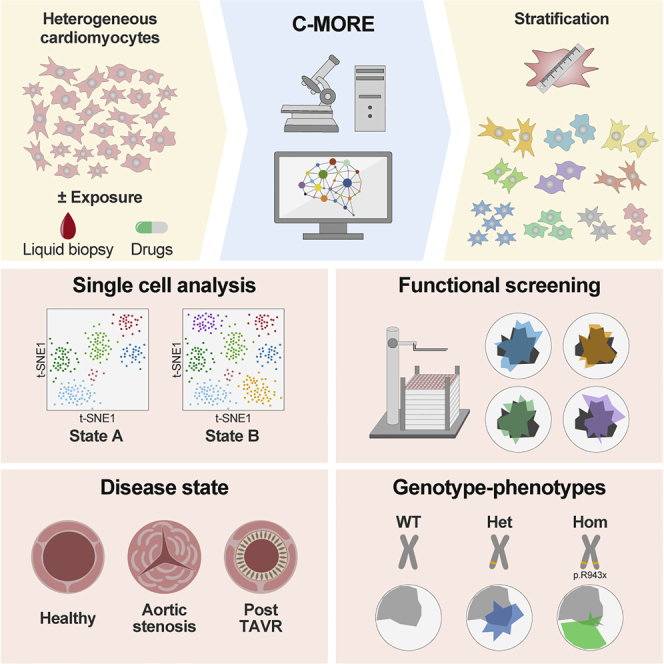
Highlights
C-MORE deconvolutes cardiomyocyte morphology on single-cell level
C-MORE could be utilized for high-throughput pharmacological screens
C-MORE detects pathological phenotypes caused by genetic alterations
C-MORE liquid biopsy setup reflects disease state in individuals with aortic stenosis
Furkel et al. present an integrative single-cell morphology-based strategy (C-MORE) to detect cardiomyocyte activation status and response to pharmacological treatment. Deconvolution of disease state in cardiomyocyte cultures exposed to liquid biopsies and in genetically engineered models by C-MORE may pave the way for development of next-generation personalized cardiovascular medicine.
Introduction
Heart failure remains the leading cause of mortality in western countries and is the mutual final stage of a wide range of cardiac pathologies. To prevent progression, early diagnosis and adapted treatment with regard to the specific underlying cause is crucial.1 However, diagnosis and estimation of prognosis remain challenging, and invasive diagnostics (e.g., biopsy) are often indispensable.2 Novel methods with potential to elucidate the modes of action of cardiac diseases as well as clinical decision-making are needed.
Image-based high-content morphology assessment and its proficiency to serve as a surrogate for cell state and functionality has become an integral readout in cell line-based screenings in drug discovery:3,4 high-throughput fluorescence microscopes allow automated acquisition of images from 96- or 384-well plates in multiple fluorescence channels, and raw images can then be analyzed to identify cells and compute single-cell morphological multi-feature profiles (e.g., shape, intensity, and texture, including Haralick features).5,6
However, in the cardiac context, heterogeneity and a higher variability of model cell types under investigation—namely, primary neonatal rat ventricular cardiomyocytes (NRCMs) or human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs)—have impeded implementation of robust morphology assessment.7,8 Morphological readout in cardiovascular research is still limited to cell size and a few selected features (e.g., perimeter, elongation, and form factor).9,10 Alternative methods focus on sarcomere organization, 3D volume/shape, proliferation, or reporter readouts.11, 12, 13, 14 However, these established methods address only specific parts of morphology, and integrated approaches are not established for use in cardiovascular research.
In the clinical setting, manual morphological assessment of tissue and cellular biopsy material is a traditional method to classify disease state and estimate prognosis. In oncological research, first advances toward integration of automated morphological assays on tumor cells in clinical personalized medicine have been taken. For example, cellular morphology has enabled prediction of responsiveness to therapeutic agents.15, 16, 17, 18 In cardiovascular research, however, no high-content morphology assay for translational applications has been established yet. Hence, we aimed to devise a robust in vitro methodology to deconvolute CM morphology for use in basic research and translational medicine.
We present C-MORE (cellular morphology recognition), a ready-to-use workflow for single-cell morphology analysis in CMs. The C-MORE workflow comprises a standardized experimental protocol including high-throughput image acquisition and morphological feature extraction with CellProfiler.5 With our R package cmoRe, we present data preprocessing and curation tailored for CM-specific biology, followed by data analysis for highly heterogeneous cell types such as NRCMs and hiPSC-CMs. We validate C-MORE against manual assessment and benchmark C-MORE performance against alternative analysis approaches. Last, we evaluate C-MORE for use in basic and clinical cardiovascular research and demonstrate the applicability of C-MORE with four examples.
Results
Experimental workflow, imaging, and feature extraction
We present an overview of our complete workflow in Figure 1A. NRCMs were isolated and seeded in 96-well imaging plates. On the following day, NRCMs can optionally be transduced with a reporter construct to add specific biological information to the assay; e.g., the subcellular location of a protein of interest. As a proof of principle, we chose a viral reporter for NFAT (nuclear factor of activated T cells), a transcription factor known to translocate to the nuclear compartment upon activation of the pro-hypertrophic Calcineurin-NFAT pathway but otherwise located in the cytoplasm.19 Cells were then treated with test substances for 48 h; e.g., hypertrophic stimuli, inhibitors, or plasma. The cytoskeleton and the nuclei of cells were stained fluorescently, and NFAT was detected by native green fluorescence. After image acquisition with the IN Cell Analyzer 2200 microscopy system, segmentation and computing of single-cell morphological features were performed using the freely available software CellProfiler.5 We adapted a standard pipeline comprising nucleus segmentation with the Otsu algorithm and cell segmentation using the propagation algorithm (Data S1 and S2; Figures S1A–S1D). For initial development purposes, we worked with the two canonical hypertrophic stimuli phenylephrine (PE) and insulin (INS).20,21 In Figure 1B, we show representative immunofluorescence images for control, PE, and INS treatment together with representative feature values in Figure 1C. PE is known to activate the Calcineurin-NFAT pathway and, therefore, served as a positive control for the reporter readout (Figure 1B, GFP/fluorescein isothiocyanate [FITC] images).
Figure 1.

C-MORE experimental and data analysis workflow with NFAT-GFP reporter readout
(A) Overview of the C-MORE workflow. QC, quality control.
(B) Representative images of CMs without stimulation (ctrl) and with phenylephrine (PE) and insulin (INS) treatment. In the lower left corner of the GFP/FITC channel, we show a representative cell magnified to demonstrate NFAT-GFP enrichment in the nucleus after PE treatment. For ctrl and INS treatment, NFAT-GFP is localized predominantly in the cytoplasm, with low GFP-intense nuclei appearing as black holes within the GFP intense cytoplasm. Scale bar, 100 μm.
(C) The image features area, perimeter, and eccentricity and the NFAT score are shown for ctrl, INS, and PE. The boxplots show z-transformed feature values of 5 independent cell preparations, the box center shows the median value, and the box limits show the 25th and 75th percentiles. The p values were calculated with linear mixed effects models; ∗∗p < 0.01, ∗∗∗p < 0.001. NS, not significant. The NFAT score was calculated via our cmoRe thresholding function on the feature Intensity_MedianIntensity_GFP of the nucleus.
Data preprocessing: In silico cell sorting and cell cycle features
CellProfiler output consists of a large data matrix with primary features for each measured cell (nucleus, cytoplasm, and custom readouts). These can be analyzed with our custom R package cmoRe. The main steps include (1) data loading and quality control, (2) cell cycle analysis and secondary feature calculation, (3) cell filtering, and (4) data analysis of aggregated or non-aggregated single cells, as outlined in Figure 2A (detailed outline in Figure S2).
Figure 2.

Preprocessing and data analysis with cmoRe
(A) Overview of cmoRe package functionality. (a) Preprocessing comprises data loading, QC, filtering steps, and addition of secondary features. Data analysis can be done at the single-cell level or bulk population level. (b) An exemplary QC plot for a 96-well plate regarding the number of cells per well. (c) A representative cell cycle plot as used for cell cycle-based filtering of apoptotic cells and to assign cell cycle status as a secondary feature to each cell. Vertical lines indicate determined cutoffs; dashed lines limit high-certainty regions of cutoffs.
(B) This boxplot shows the z-transformed cell area for NRCMs incubated with three concentrations of PE in comparison with the control (ctrl). The red line depicts a fitted linear mixed model assuming equidistance between ctrl/dose-levels, as applied in our analyses. The box center shows the median value, and the box limits show the 25th and 75th percentiles.
(C) Hierarchical cluster analysis of selected features (ward.D2) for meta-feature construction. Each cluster corresponds to a meta-feature (color coded).
(D) The PE phenotype. Radar plots show meta-feature values for PE (colored) against meta-feature values of an unstimulated control (gray) ordered in a circle. For PE, three concentrations are shown (red, low; yellow, intermediate; green, high; control, gray).
(E) Numbers of retained features after each selection step for the canonical hypertrophic stimuli: norepinephrine (NE), adrenaline (A), isoproterenol (ISO), endothelin (ET), and angiotensin (AT); Data of 5 independent cell preparations are shown.
cmoRe expects a specific folder structure for data loading (data organization). With these data, an initial quality control (QC) is performed: QC plots depict the number of cells per well on the plate layout (Figure 2Ab). To assure absence of potentially interfering edge effects or cytotoxicity, we checked whether at least 500 cells per well were present (Figure 2Aa). Further QC plots or criteria can be set generated on any feature or metric of interest.
In the next step, specific features of interest are assessed for filtering steps and deriving biologically relevant information; e.g., the cell cycle state. For cell cycle analysis, we utilized the well-established approach of quantifying DNA content via DAPI intensity (integrated DAPI intensity within the nucleus), as used commonly in flow cytometry.22,23 For filtering and cell cycle analysis, distributions of the respective feature measurements are evaluated per biological replicate, plate, and treatment to identify reasonable cutoffs under a set of constraints (prior knowledge; e.g., an expected range for a cutoff; Methods S1). Regarding the cell cycle, the most abundant fraction of cells are G1/G0 cells. Therefore, the highest peak of nuclear DAPI signal in a histogram/estimated density will correspond to G1/G0 cells. cmoRe identifies this peak as global maximum in the corresponding density and a (local) minimum right (mR) and minimum left (mL) from this value utilizing a resampling method. These identified minima are used as thresholds to separate dead cells (left from mL) and G2/M cells (right from mR) (Figure 2Ac; Methods S1). Additional filters, using the same methodology, are implemented to remove improperly attached cells characterized by a high nuclear to cellular area ratio and median DNA intensity of the nucleus to discriminate CMs from non-CMs (see also Ali et al.24). These filters were developed and validated on a manually rated dataset, as described in Figure S3 (filter steps). Cell cycle fractions are a prime example of secondary features and are calculated in cmoRe by default. Defined by the user, any feature can be processed analogously, yielding custom secondary features and, thus, extending the number of features originally provided by CellProfiler as, e.g., the NFAT score. The NFAT score was calculated via the cmoRe thresholding function on the feature Intensity_MedianIntensity_GFP of the nucleus to quantify NFAT translocation to the nucleus. For single-cell analysis, these cutoffs, as, e.g., the nuclear DNA intensity, can be used to assign single cells to specific groups (e.g., CMs or non-CMs) for cell-type-specific analysis/filtering (see below).
Finally, data of pure, vital, and properly attached CMs can be analyzed on the single-cell level or after median aggregation of features per well. Typically, aggregated data are z-transformed per experiment.
Data analysis: Feature selection
To analyze well-aggregated data for differential treatment effects, we use a number of steps to ensure robustness of identified features and implement redundancy reduction by generation of meta-features. We utilize (1) cross-validation, (2) multiple test correction, and (3) dimensionality reduction in cmoRe. (1) and (2) are performed with linear mixed models with random effects of biological replicate and plate to adjust for unwanted variability. For meta-feature calculation, hierarchical cluster analysis with adaptive selection of numbers of clusters was utilized.
Figure 2A demonstrates analysis steps, and Figures 2B–2E show representative analysis results for identification of PE-induced morphological feature alterations. After preprocessing and secondary feature generation, the data matrix consisted of 1,338 morphological features for PE and approximately 110,000 cells per plate. These were each evaluated with a linear mixed model, assuming a linear equidistant dependency between the control (ctrl) and lowest concentration as well as between dose levels (Figure 2B; Methods S1). Cross-validation ensured only retaining features with high reproducibility (Figure 2E; Cross-validated coefficient of determination (R²cv) > 0.7), and multiplicity adjustment was performed (Benjamini-Hochberg). For PE, 112 features were retained (Figure 2E; adjusted p value (p-adj) < 0.05).
To summarize highly correlated features and to ease interpretability, dimensionality reduction was performed using hierarchical clustering (Figure 2C). Numbers of clusters were iteratively incremented within a fixed range, and the intra-cluster variability was calculated (Figures S4A–S4C). This knee plot gives a first orientation for a suitable cutoff, but we used the change in intra-cluster variability for cutoff selection (Figures S4A–S4C; Methods S1). For PE, this approach identified 38 clusters/meta-features (Figure 2E). The latter can be visualized with radar plots (Figure 2D).
An alternative to the aggregated bulk population analysis constitutes single cell analysis, which might help with identification of (functional) subpopulations of cells (see below).
Validation and benchmarking
We present an overview of established morphological tools with regard to cardiology or integrated analysis in general in Table S1. Because NRCMs are cells with high morphological heterogeneity, thus hindering established cell morphology analysis approaches, we aimed to assure that cmoRe-analyzed data are reliable and, therefore, manually evaluated key morphological features (1). Further, we compare cmoRe with naive analysis approaches on Cell Profiler output data (2).
-
(1)
Cell sizes used in cmoRe analyses were compared with manually contoured cell size measurements, revealing a high overlap (Figures S1E and S4E). The number of NFAT-GFP-positive nuclei showed high concordance between manual assignment and cmoRe-based labels (Figure S1F).
CellProfiler is a versatile tool for cell segmentation and extraction of morphological features from images and an integral element in our C-MORE workflow. However, we aimed to assess the added value of combined use of CellProfiler and cmoRe.
-
(2)
We evaluated pairwise comparison results of ctrl and INS/PE with three dose levels against the full cmoRe workflow (preprocessing and filtering of cells, linear mixed model analyses to naive linear model analyses using filtered and non-filtered Cell Profiler data with per-well aggregated data) (Figure S5).
Cell size alterations induced by a strong stimulus such as PE were detected with all analysis approaches (Figures S5A and S5B). For weaker stimuli such as INS, only cmoRe was able to identify significant differences between control and treated cells for all tested dose levels. Assessment of all cellular and nuclear morphological features in PE/INS-treated cells revealed a small benefit of using filtered versus non-filtered data with naive analysis approaches in detecting differences, but a major increase in detected differential features was observed for cmoRe (Figure S5C).
As a proof of principle, after establishing C-MORE, we aimed to test its usability for basic research questions.
Morphological patterns of canonical hypertrophic stimuli
A potentially reversible preliminary step of heart failure is hypertrophic growth of the heart, the primary response to an increased workload.25 Key signaling pathways involved in cardiac hypertrophy have been identified.26 We chose these well-described canonical hypertrophy stimuli to model differing modes of actions and to generate a broad range of phenotypes to train C-MORE.9,26 Following the C-MORE workflow, we incubated NRCMs with PE, INS, norepinephrine (NE), epinephrine/adrenaline (A), isoproterenol (ISO), endothelin ET) 1, and angiotensin (AT) II in three respective concentrations (Table S2; Methods S1). Figure 2E shows differences in the numbers of selected features for each selection step with cmoRe. Initially, hundreds of altered features are identified. However, fewer of these features meet the cmoRe criteria for robustness (R²cv > 0.7). Further dimensional reduction is achieved by multiplicity adjustment and meta-feature aggregation. In Figure 3A, we show meta-features for all stimuli under three concentrations (analogous to Figure 2D). We ordered the stimuli by similarity in previously described adrenergic receptor activation (alpha, beta, and none). Direct comparison of the feature pattern between the tested stimuli revealed that each stimulus pattern comprises a set of stimulus-specific features and a set of shared features with other stimuli (Figure 3B; Figure S4D). Biologically related stimuli, (A and ISO, for example) share a great amount of features (Figure 3B). Additionally, we could validate that the NFAT reporter was functional because it showed translocation to the nucleus for stimuli known to activate the respective pathway (Figure S4F).
Figure 3.

Population-level and single-cell phenotyping of canonical hypertrophic stimuli
(A) Radar plots show meta-feature values for the canonical hypertrophic stimuli aggregated on a population level (three concentrations: red, low; yellow, intermediate; green, high; untreated control, gray).
(B) Comparative analysis of the features selected for the canonical hypertrophic stimuli. For each stimulus, the dose-dependent dynamic is color coded (red indicates an increase and blue a decrease; clusters were calculated on an unstimulated control). See also Figure S4D.
(C) Single-cell phenotyping of the canonical hypertrophic stimuli: t-distributed stochastic neighbor embedding (t-SNE) of all single cells measured in one experiment (highest substance concentrations, substances are color coded). Marginal densities for each stimulus over the t-SNE plot are shown by colored lines to the right and at the top of the t-SNE plot.
(D) Densities per stimulus on the t-SNE plots from (C). Visually, areas of notably higher density represent subpopulations of potential interest and are circled for ISO and for INS are indicated with a dotted line.
(E) Quantification of stimulus-induced differences, pairwise differences of binned and normalized data (50 × 50 grid, rate differences were calculated: x − y axis). The most extreme differences are colored in red (upper 5%) and green (lowest 5%). Quantitatively identified substance-specific subpopulations are circled for ISO and for INS are circled with a dotted line.
In (A) and (B), data are shown for 5 independent experiments. In (C)–(E), single-cell analysis of 1 experiment is shown.
Single-cell phenotyping
High inherent variability of the primary cell types and the observed non-uniform morphology following stimulation highlight the importance of single-cell analysis for detection and characterization of subpopulations or subtypes of cells. Especially less abundant cell populations might easily be missed with per-well aggregated population analyses. Therefore, we assessed low-dimensional t-distributed stochastic neighbor embedding (t-SNE) (Figures 3C–3E) and uniform manifold approximation and projection (UMAP) (Figures S6A and S6B) representations of our high-dimensional morphology measurements of NRCMs treated with canonical stimuli. Different areas in these representation plots represent specific morphological phenotypes, and pairwise comparisons allow fast identification of enriched or reduced regions (corresponding to specific subpopulations) following treatment (e.g., circles and dotted circles in Figures 3D and 3E).
Screening in primary CMs
Automated image analysis opens the possibility of high-throughput screening. As a second exemplary application, we set up a screening setting for which we treated PE-stimulated NRCMs with a selection of inhibitors of canonical hypertrophy pathways27 (Methods S1; Table S2). The resulting phenotypes are shown in Figure 4A, and with increasing inhibitor concentration, a phenotypic shift from the PE phenotype back to the control phenotype was observed. For screening purposes, we evaluated differences between meta-features in positive control (PE) versus positive control and inhibitor-treated cells (Figure 4B). Plotting a volcano plot with effect size and p value further simplifies evaluation, and the user is able to set the p value cutoff depending on the screening purpose (Figure 4B). C-MORE provides tools for hit detection in large-scaled screening approaches as commonly used for drug development.
Figure 4.

Inhibition of PE-induced hypertrophic growth by canonical pathway inhibitors
(A) Radar plots depict phenotypical changes upon concomitant application of PE and inhibitors of main cardiac hypertrophy pathways in three concentrations (low, red; intermediate, yellow; high, pink). A positive PE-stimulated control (PE stimulation without any inhibitor) is shown in turquoise, and an untreated control condition is shown in gray. Stars indicate whether significant differences between PE-stimulated and inhibitor-treated cells with PE stimulation can be observed (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, linear mixed effects model; evaluation of absolute differences in meta-features between PE stimulated ± inhibitor-treated cells).
(B) Volcano plot of effect size and p value (linear mixed effects model) of concomitantly inhibitor- and PE-treated phenotype versus PE-treated phenotype. Data are shown for 4 independent experiments.
Liquid biopsy: Aortic valve stenosis phenotype
In a third example, we aim to demonstrate the potential clinical applicability of C-MORE using blood-treated NRCMs. Aortic valve stenosis (AS) is a cardiac disease with high prevalence in elderly patients and commonly treated with transcatheter aortic valve replacement (TAVR)28. We incubated NRCMs with blood plasma from individuals with AS before and within 1 week following TAVR; blood plasma from healthy individuals served as controls (ctrl). Applied selection steps with C-MORE and numbers of selected features are presented in Figure 5A. The prominent aortic valve stenosis phenotype and the attenuated post-TAVR phenotype are shown in Figures 5C and 5D. For individuals with aortic valve stenosis in comparison with controls, 224 features were altered significantly; after TAVR treatment, 88 of these 224 features (39%) did not differ significantly from the control. In the next step, as a tool for systematic investigation of disease mechanisms, cmoRe allows cross-experimental comparison. As shown in Figure 5B, we quantified shared features of the canonical hypertrophic stimulus experiment and the aortic valve stenosis phenotype as well as TAVR-reversible features. To evaluate soundness of the selected features, we conducted reclassification using a random forest classifier that showed excellent performance in separating and predicting the conditions AS, post-TAVR, and ctrl (Figures S6C and S6D). In the setting of a liquid biopsy, C-MORE allowed us to detect morphological correlates of a cardiac disease and therapeutic effects with predictive properties.
Figure 5.

Morphological pattern of aortic valve stenosis (aortic stenosis/AS) and reversibility upon TAVR
(A) Preprocessing and data analysis steps and retained number of features in each step (∗, reversible after TAVR).
(B) Venn-like diagram of substance-specific and AS-specific features. AS-specific TAVR-reversible features are counted separately and highlighted in lime. Overlaps within substance-specific features are not shown.
(C) Heatmap of model-predicted batch-corrected differential morphological features (hierarchical clustering, ward.D2).
(D) Radar plots show differentially regulated meta-features for AS and TAVR. Data are shown for 3 independent experiments.
Phenotypic characterization of hiPSC-CMs
Generation of organ-specific tissue in vitro from specific induced progenitor stem cells from affected individuals opens up a multitude of applications.29 However, hiPSC-CMs are highly heterogeneous in their appearance and, like NRCMs, challenge morphological analysis. HiPSC-CM differentiation is a delicate process and not a specialty of our group; therefore, we re-analyzed plates from an already published research article for this fourth example. Seeger et al.30 published an article about the mutation p.R943x in myosin binding protein C3 (MYBPC3) in individuals with hypertrophic cardiomyopathy regarding calcium handling and contractility. In this study, hiPSC-CMs derived from individuals with the heterozygous (het) mutation genotype were compared with healthy wild-type (WT) genotype controls. Additionally, in this study, hiPSC-CMs were genetically modified to generate an induced het genotype (het-ind) and an artificial homozygous genotype (hom) from WT hiPSC-CMs. Comparison of cell size showed no difference in this study; however, no high-content morphology analyses were conducted. We were kindly provided with the very exact same plates and cells used for this publication and re-evaluated cell size with cmoRe (Figures 6A and 6C). We could confirm no significant change in cell size; however, applying full cmoRe analysis, we observed a prominent phenotype for hom and an attenuated form for het in comparison with the WT (Figures 6B, 6D, and 6E). cmoRe was applied successfully in a setting, where traditional morphological readouts such as cell size show no change; however, we were able to identify a prominent genotype-related phenotype using cmoRe.
Figure 6.

Morphological pattern of wild-type (WT), heterozygous (het), and homozygous (hom) MYBPC3 pR943x genotype hiPSC-CMs
(A) Preprocessing and data analysis steps with retained number of features in each step.
(B) Representative images of WT, het, induced/engineered het (het-ind), and hom genotype hiPSC-CMs regarding the MYBPC3 pR943x mutation. In the lower left corner of the Troponin T (TnT) images, the indicated image section is shown magnified. Scale bar, 30 μm.
(C) Cell area in MYBPC3 pR943x WT, het, and hom cells (n = 4 patients, linear mixed effects model, z-transformed data).The boxplots show z-transformed values of cell area, the box center shows the median value, and the box limits show the 25th and 75th percentiles.
(D) Radar plots showing meta-feature values for the pR943x phenotype (het, het-ind, and hom genotypes, colored) against the WT (gray). p values: linear mixed effects model.
(E) Heatmap of model-predicted differential morphological features for the genotypes WT, het, and hom (hierarchical clustering, ward.D2). n = 4 hiPSC-CM cell lines derived from affected individuals, n = 2 from pR943x het mutated individuals, n = 2 from pR943x WT and phenotypically healthy persons.
Discussion
Cellular morphology and its proficiency to serve as a surrogate for cell state and functionality is a powerful tool typically used for drug screening and increasingly for personalized medicine in oncology.4,15 In cardiology research, primary NRCMs are the most frequently used cell model for preclinical studies. NRCMs and hiPSC-CMs, however, exhibit a high degree of intercellular morphological variability, making identification of specific morphological phenotypes challenging.7 We therefore developed C-MORE, an experimental pipeline and analysis platform, to enable researchers to make use of the full potential of CM morphology assessment. Main advantages comprise the high number of morphological features being made available (including customizable secondary features) and the high power of the approach to detect small but nevertheless robust effects in highly variable data.
For the experimental part, we aimed to establish a lean workflow and therefore chose cytoskeletal and nucleic staining suitable for shape and texture analysis. This marker panel can be adapted to the specific purpose of C-MORE application; e.g., staining of specific intracellular proteins of interest. NRCM isolation optimally yields a highly pure population of more than 90% CMs. However, the presence of a minor fraction of non-CMs needs to be considered for accurate analysis. In CM-specific morphology readouts, this has been taken into account, as reported, e.g., by Jentzsch et al.31 However, these approaches typically require additional fibroblast-specific staining and were limited to a few morphological features.32 In contrast, cmoRe identifies CMs and non-CMs based on DAPI intensity, as reported previously by Ali et al.,24 and further curates data from dead and non-attached cells; the automated and adaptive thresholding function is able to accommodate changing cell densities and highly heterogeneous cell sizes following specific treatments. By applying the cmoRe threshold function on the integrated intensity of DAPI as a surrogate for DNA content, we integrate a classical cell cycle analysis approach into our automated analysis, circumventing the need for manual gating as in flow cytometry.22,23
Recently, the importance of cross-talk between different cell types in the heart has been investigated.7 With the ability to robustly differentiate CMs from non-CMs, cmoRe opens possibilities to analyze cross-talk between the different cell types in the heart; e.g., using co-culture experiments or even whole-heart cell suspensions.
Our data preprocessing tools ensure identification of cells of interest and appropriate handling of features with skewed or variable distributions and can acquire additional biologically valuable information.
For data analysis, we used linear mixed models to account for variability between biological replicates. Canonical hypertrophy stimulus and inhibitor experiments were performed with a number of increasing dose levels for which we assumed a linear dose dependency over ascending treatment dosages. Although this serves as a robust base analysis, alternative dependencies (foremost sigmoidal) should be tested in further work. We implemented a cross-validation step, multiplicity adjustment and dimensional aggregation to meta-features, to provide a highly sensitive and robust analysis tool. Benchmarking showed superior performance of our proposed C-MORE workflow compared with naive analysis approaches using CellProfiler output data but lacking essential characteristics of C-MORE. Application of C-MORE was especially beneficial in settings where morphological changes were very subtle (a weaker stimulus such as INS or lower concentrations of stimuli). cmoRe allows automated data curation (filtered cells) and is characterized by powerful detection of subtle changes in settings of low signal to noise. We might be able to apply C-MORE to other types of heterogeneous primary cells in the future.
Cardiac hypertrophic growth is a heterogeneous process and can involve mechanistically different pathways. However, more work is needed to better understand hypertrophic growth as a potential target for therapy.33 Because our understanding of cardiac remodeling deepens on the molecular level, measuring a few morphological parameters without application of appropriate preprocessing steps risks missing relevant morphological effects.34 We applied C-MORE to four exemplary applications for basic research and translational medicine to evaluate its performance but without focusing on biological interpretation.
To evaluate C-MORE as a diagnostic tool, NRCMs were incubated with blood plasma from individuals with aortic valve stenosis before and after TAVR and from healthy persons. In this pilot study, we observed excellent classification accuracy in a representative random forest analysis. This implicates that the morphology of cells incubated with material from affected individuals might reflect a complex disease state better than conventional blood tests and could be utilized to support non-invasive diagnostics and clinical decision-making in the future. Linking results cross-experimentally, as demonstrated for aortic valve stenosis and the canonical hypertrophy stimuli, can be a valuable tool for investigating mechanisms of actions in cardiological disease.
hiPSCs from affected individuals are an important tool for disease modeling and can facilitate implementation of precision medicine. However, differentiation of hiPSCs into CMs to study cardiovascular disease is a lengthy and highly variable process.34,35 Seeger et al.30 demonstrated that hiPSC-CMs derived from MYBPC3 mutant individuals showed no significant difference in cell size compared with healthy controls. However, the ability of cmoRe to detect a prominent phenotype for the hom genotype and an attenuated phenotype (het) emphasizes the benefits of our approach. Applying C-MORE to patient-derived hiPSC-CMs may help deepen our understanding of individual pathology and enable individualized drug screening in the frame of personalized medicine. Furthermore, culture and maturation of hiPSC-CMs is a very delicate process under intense investigation. In general, fine-tuning of the attachment as lamellipodium formation, spreading or establishing focal adhesion are biological meaningful process of cell biology, reflecting the functional status of a cell in a detectable morphological readout. C-MORE might help to further our understanding of the underlying biology and standardize these processes.
We present C-MORE as an open-source tool to identify morphological patterns in basic research and cardiovascular disease in translational approaches. C-MORE is compatible with both primary cell types used most frequently in cardiac research and comprises preprocessing steps and data analysis suited for extracting relevant information out of data with a low signal-to-noise ratio. The demonstrated versatility of C-MORE makes it ideal for quick and easy application in drug screening and in-depth characterization of single cells for investigating CM biology. Future applications of C-MORE may include investigation of disease mechanisms and drug screening in CMs, and might also be applicable to other primary cell types beyond the niche of CMs. In translational medicine, C-MORE shows applicability for material from affected individuals with potential to support clinical decision-making in the frame of personalized medicine.
Limitations of the study
With our experimental setup, we present a lean C-MORE experimental workflow, but it is possible to integrate additional custom settings, such as staining for other target proteins of interest or other reporter readouts. As a proof of principle, we integrated the NFAT reporter only. Furthermore, staining of the cell membrane might improve exact quantification of cell area or cell volume compared with staining of structural proteins such as Desmin or Troponin as used here. The applied microscope setup only allows acquisition of three fluorescence channels. Using more markers or even applying a multiplexing approach would be possible and would only be limited by the specifics of the microscope in use and computational power available.
Our density-based algorithm used for the preprocessing steps offers adaptive thresholding and can accommodate variation and skewed populations. However, in its current implementation, it is limited to detection of a maximum of three subpopulations (Methods S1). Also, in our study, the mixed linear effects models assume linear dose dependency over different treatment dosages. Although this shows superior performance over analyses lacking cmoRe characteristics, other dose-effect dependencies (e.g., sigmoidal) are currently not modeled explicitly.
The four representative use cases of C-MORE were designed as pilot studies to demonstrate C-MORE’s versatile fields of application and require further intense experimentation.
For the liquid biopsy approach, we currently use samples of healthy control individuals as a reference, but we expect to enable computing a virtual control sample in the future based on the growing database.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal Anti-Desmin antibody, rabbit | abcam | Cat# ab15200, RRID: AB_301744 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Life Technologies, Thermo Fisher | Cat# A-11012, RRID: AB_2534079 |
| Monoclonal Anti-troponin T antibody, rabbit | abcam | ab209813 |
| Bacterial and virus strains | ||
| AdNFATc3-GFP adenovirus |
Sanna et al.;36 | N/A |
| MacDonnell et al.37 | ||
| Biological samples | ||
| Aortic stenosis patient blood plasma | Department of Cardiology, University Hospital Heidelberg | Protocol S-587/2019 |
| Chemicals, peptides, and recombinant proteins | ||
| Cesium chloride | Carl Roth | 7878.2 |
| Suprarenin/Epinephrin, 1mg/ml | Sanofi | 6053210 |
| (R)-(-)-Phenylephrine hydrochloride | Sigma | A9525 |
| Endothelin 1 97% (HPLC), powder | sigma | E7764 |
| Insulin Insuman rapid 40 ie | Sanofi | 1843315 |
| (-) Isoproterenol hydrochloride | Sigma | I6504 |
| Arterenol 1mg/ml | Sanofi | 3870227 |
| Tricibine Akt V Inhibitor | Sigma | 124038 |
| ERK/MEK Inhibitor | Promega | U0126 |
| PF 573228 FAK inhibitor | Tocris | 3239 |
| BIO GSK3b inhibitor | Tocris | 3194 |
| Ly294002, PI3K Inhibitor | Millipore | 440202 |
| Laminin | Sigma | L2020-1MG |
| Paraformalaldehyd, 20% solution | Electron Microscopy | 15713-S |
| HEPES | Carl Roth | 9105.2 |
| Medium 199 | Sigma | M7528-500ML |
| FBS | Life Technologies | 10500064 |
| DAPI | Life Technologies | D1306 |
| DNase II Type V from Bovine spleen | Sigma | D8764-300KU |
| Experimental models: Cell lines | ||
| Neonatal rat cardiomyocytes from rattus norvegicus | Janvier | Rattus norvegicus, wistar |
| Software and algorithms | ||
| INCellAnalyzer 2200 Acquisition Software | GE | N/A |
| CellProfiler | The Broad Institute | N/A |
| R 4.0 | R Core Team | https://www.r-project.org |
| cmoRe | This paper | https://github.com/mknoll/cmoRe |
| Other | ||
| 96 Well Black with Clear Flat Bottom TC-Treated Imaging Plate with Lid | corning | 353219 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jennifer Furkel (Jennifer.furkel@med.uni-heidelberg.de).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
NRCM isolation and treatment
NRCMs were obtained from hearts of 1-2 days old neonatal rats using a trypsin based enzymatic digestion standard protocol as previously described.38 To enrich cardiomyocytes, a percoll gradient centrifugation was performed after digestion. Cells were then counted and plated onto imaging compatible 96-well plates (corning, 353219), which were coated with Laminin (sigma, L2020, diluted to 1mg/l PBS) for 2h beforehand. 20 000 cells per well were seeded in 200μl high serum medium (10% FBS in M199) for the first 24h.
24h after isolation, NRCMs were washed with PBS and incubated in 80μl of low serum medium (0.5% FCS in M199) with NFAT-GFP adenovirus for 12h. The following 48h cells were incubated with hypertrophy inducing stimuli (phenylephrine (PE), adrenaline (A), noradrenaline (NA), isoproterenol (ISO), insulin (INS), endothelin (ET), angiotensin II (AT), all reagents were diluted in water and PBS) and an unstimulated PBS control (CTRL). For the liquid biopsy/ aortic stenosis experiments NRCMs were treated for 48h with blood plasma of healthy controls or patients showing a high-grade stenosis of the aortic valve undergoing transcatheter aortic valve implantation (before TAVR, within one week after TAVR). For the inhibitor screening experiment cells were for 48h concomitantly incubated with PE and an inhibitor of major pathways involved in cardiac hypertrophy (inhibitors of protein kinase B (AKT), extracellular signal-regulated kinase (ERK), focal adhesion kinase (FAK), glykogensynthase kinase (GSK) or phosphoinositid-3-kinase (PI3K)). All inhibitors were diluted in Dimethylsulfoxid (DMSO), adequate DMSO solvent controls were performed. Detailed information of the reagents used and concentrations can be found in Table S2.
hiPSC-CMs
hiPSC generation and differentiation into hiPSC-CMs was performed by and as previously described by Seeger et al.30 The identical plates utilized in the study by Seeger et al. were kindly provided to us, permeabilized and stained with anti-Troponin T.
Method details
AdNFATc3-GFP adenovirus amplification and purification
The adNFATc3-GFP adenovirus was kindly provided by Dr. Mark Sussmann. The AdNFATc3-GFP adenovirus, as described previously was propagated on HEK293 cells.36,37,39 In brief, cells were harvested and the pellet was washed with PBS (10 minutes, 300 g, 4°C). We performed four freeze-thaw cycles to lyse the cells (thawing at room temperature, freezing on dry ice), incubated the solution with DNase I (10μg/mL, 30min, 37°C). Debris was pelleted at 750 g for 10 minutes at 4°C. The supernatant was layered onto a Cesium Chloride (CsCl) gradient (CsCl cushions of 1.4 g/ml and 1.2 g/ml). We performed one centrifugation for 3h, and two subsequent centrifugations of 18h (Beckman Coulter SW41 rotor, 22500 rpm, 4°C temperature). The purified virus was dialyzed against dialysis buffer (3% sucrose, 10mM Tris (pH 7.8 at room temperature), 150mM NaCl, 10mM MgCl2), as described previously.40 Storage temperature of the purified virus was −80°C.
Immunocytochemistry
In all NRCM experiments cells were fixed with 4% paraformaldehyde (PFA), permeabilized with 0.2% Triton X-100, then incubated in blocking solution (10% FBS in PBS) for one hour at room temperature. In all NRCM experiments cytoskeletal staining using a desmin antibody (1:800) was applied on the plates overnight at 4°C. After washing with PBS the secondary antibody (1:600) was applied for 1h. The hiPSC-CMs were stained with troponin T antibody and secondary antibody (1:400 and 1:600). Nuclear DNA was stained in all experiments using 4′,6-Diamidin-2-phenylindol (DAPI).
Image acquisition (IN Cell Analyzer 2200)
Images were acquired in an automated fashion using the IN Cell Analyzer 2200. For all NRCM experiments a centered square arrangement of a 4x4 grid of imaging fields per well was chosen and three channels were imaged (DNA - DAPI, desmin – TexasRed, NFAT- GFP native fluorescence /FITC), as outlined in Figures 1A and 1B. For hiPSC-CMs, a circular arrangement of 96 imaging fields for maximum coverage of the well was chosen and two fluorescent channels for each field were imaged (DNA - DAPI, troponin T – TexasRed). Images had a resolution of 2048x2048 pixel.
Quantification and statistical analysis
Image processing, cell segmentation and feature extraction
Image features were computed from raw images using CellProfiler v 3.1.8.5 Upon loading images into the software, images of the three fluorescent channels blue (DAPI), TexasRed (Desmin/TroponinT), GFP/FITC (NFAT) are matched by file name and image metadata such as well number and image number are extracted from file names. Then nuclei are segmented based on an intensity-based algorithm on the DAPI image (single steps: Figure S1A). Using these nuclei as seeding points, cells are segmented by the propagation algorithm, which identifies cell borders based on intensity changes (Figures S1B and S1D). The occurrence of double nucleated cells is taken into account by assigning nuclei located within 10 pixels from each other to one cell (Figure S1C). This cutoff was validated by comparing unstimulated and PE treated cells to be optimal for discrimination of binucleated cells from single nucleated cells in close proximity. Cells touching the image borders are excluded from further analysis. Nuclei and cell segmentations are used to measure multiple primary morphological features such as shape, intensity, and texture in all acquired images. Single cell measurements are stored in .txt files (one row per cell). CellProfiler pipelines are provided as Data S1 and S2.
Preprocessing
Cell and feature processing was performed as described in the main text using functions of the custom written R package cmoRe, publicly available on github (https://github.com/mknoll/cmoRe). Its general functionality is outlined in Figure 2, a detailed outline with function names is provided in Figure S2, a full handbook of all package functions and an example are provided in the Methods S1 and with the package vignette. The NFAT score was calculated by automated thresholding on the median GFP intensity of the nucleus (Figure S4F). It reflects translocation of NFAT-GFP from the cytoplasm compartment into the nuclear compartment upon activation of the Calcineurin-NFAT pathway. For cell cycle analysis we used the integrated DAPI intensity of the nucleus, for identification of non-attached cells we used the cellular/nuclear area ratio and for identification of non-cardiomyocytes we used the median DAPI intensity of the nucleus (Figure S3).
Single cell phenotyping
Filtered, non-aggregated data of highest concentrations of tested substances were analyzed with t-SNE using the FI-tSNE implementation41 and UMAP,42 similarity between substance induced phenotypes were assessed by binning the two-dimensional t-SNE data (n = 50 bins), normalizing data (rates) and calculating pairwise differences. 5% and 95% quantiles of combined unique pairwise combination differences were used to indicate significance (Figure 3E).
Population-level phenotyping
All analyses were performed in R, version 3.4.1. Mixed effect models were computed using lme443 or nlme.44 Significance level was set to alpha = 0.05 (two-sided).
Acknowledgments
We thank Dr. Katrin Rein for excellent support in creating the graphical abstract. This work was supported by the Deutsche Forschungsgemeinschaft (DFG; Ko3900/2-1 to M.H.K., Unite: SFB-1389, Project 404521405), the German Centre for Cardiovascular Research (DZHK), the Klaus-Tschira-Stiftung, and intramural funds of the National Center for Tumor Disease (NCT) and the German Cancer Consortium (DKTK). M.K. and J.F. received stipends in the frame of the MD/PhD program of the Heidelberg Faculty of Medicine (MFHD), Heidelberg University. Studies of human samples were performed under a protocol approved by the institutional review board in accordance with the declaration of Helsinki. Experimentation with primary rat cardiomyocytes was done under an approved animal protocol.
Author contributions
Conceptualization, J.F., M.K., and M.H.K.; methodology, J.F., M.K., and M.H.K.; investigation, J.F., M.K., S.D., N.V.B., T.S., and M.H.K.; writing – original draft, J.F., M.K., and M.H.K.; writing – review & editing, J.F., M.K., S.D., N.F., A.A., H.A.K., and M.H.K.; funding acquisition, M.H.K. and H.A.K.; resources, M.H.K. and H.A.K.
Declaration of interests
A.A. reports research grants (Merck and EMD, Fibrogen, and Bayer) and SAB (Merck and EMD, Fibrogen, BMS, Roche, and Bayer) and consulting roles (Bayer and BioMedX).
Inclusion and diversity
We worked to ensure sex balance in the selection of non-human subjects. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Published: November 3, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100436.
Contributor Information
Jennifer Furkel, Email: jennifer.furkel@med.uni-heidelberg.de.
Mathias H. Konstandin, Email: mathias.konstandin@med.uni-heidelberg.de.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
All original code has been deposited at github and is publicly available as of the date of publication (https://github.com/mknoll/cmoRe). CellProfiler pipelines are available as online supplemental files (Data S1 and S2).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Cleland J.G.F., van Veldhuisen D.J., Ponikowski P. The year in cardiology 2018: heart failure. Eur. Heart J. 2019;40:651–661. doi: 10.1093/eurheartj/ehz010. [DOI] [PubMed] [Google Scholar]
- 2.Chimenti C., Frustaci A. Contribution and risks of left ventricular endomyocardial biopsy in patients with cardiomyopathies: a retrospective study over a 28-year period. Circulation. 2013;128:1531–1541. doi: 10.1161/CIRCULATIONAHA.13.001414. [DOI] [PubMed] [Google Scholar]
- 3.Bougen-Zhukov N., Loh S.Y., Lee H.K., Loo L.H. Large-scale image-based screening and profiling of cellular phenotypes. Cytometry A. 2017;91:115–125. doi: 10.1002/cyto.a.22909. [DOI] [PubMed] [Google Scholar]
- 4.Boutros M., Heigwer F., Laufer C. Microscopy-Based High-Content Screening. Cell. 2015;163:1314–1325. doi: 10.1016/j.cell.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Carpenter A.E., Jones T.R., Lamprecht M.R., Clarke C., Kang I.H., Friman O., Guertin D.A., Chang J.H., Lindquist R.A., Moffat J., et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haralick R.M., Shanmugam K., Dinstein I.H. Textural Features for Image Classification. IEEE Trans. Syst. Man Cybern. 1973;SMC-3:610–621. [Google Scholar]
- 7.Litviňuková M., Talavera-López C., Maatz H., Reichart D., Worth C.L., Lindberg E.L., Kanda M., Polanski K., Heinig M., Lee M., et al. Cells of the adult human heart. Nature. 2020;588:466–472. doi: 10.1038/s41586-020-2797-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biendarra-Tiegs S.M., Secreto F.J., Nelson T.J. Addressing Variability and Heterogeneity of Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Adv. Exp. Med. Biol. 2020;1212:1–29. doi: 10.1007/5584_2019_350. [DOI] [PubMed] [Google Scholar]
- 9.Ryall K.A., Bezzerides V.J., Rosenzweig A., Saucerman J.J. Phenotypic screen quantifying differential regulation of cardiac myocyte hypertrophy identifies CITED4 regulation of myocyte elongation. J. Mol. Cell. Cardiol. 2014;72:74–84. doi: 10.1016/j.yjmcc.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hein S., Furkel J., Knoll M., Aus dem Siepen F., Schönland S., Hegenbart U., Katus H.A., Kristen A.V., Konstandin M.H. Impaired in vitro growth response of plasma-treated cardiomyocytes predicts poor outcome in patients with transthyretin amyloidosis. Clin. Res. Cardiol. 2021;110:579–590. doi: 10.1007/s00392-020-01801-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toepfer C.N., Sharma A., Cicconet M., Garfinkel A.C., Mücke M., Neyazi M., Willcox J.A.L., Agarwal R., Schmid M., Rao J., et al. SarcTrack. Circ. Res. 2019;124:1172–1183. doi: 10.1161/CIRCRESAHA.118.314505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rupert C.E., Chang H.H., Coulombe K.L. Hypertrophy changes 3D shape of hiPSC-cardiomyocytes: Implications for cellular maturation in regenerative medicine. Cell. Mol. Bioeng. 2017;10:54–62. doi: 10.1007/s12195-016-0462-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woo L.A., Tkachenko S., Ding M., Plowright A.T., Engkvist O., Andersson H., Drowley L., Barrett I., Firth M., Akerblad P., et al. High-content phenotypic assay for proliferation of human iPSC-derived cardiomyocytes identifies L-type calcium channels as targets. J. Mol. Cell. Cardiol. 2019;127:204–214. doi: 10.1016/j.yjmcc.2018.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Rooij E., Doevendans P.A., de Theije C.C., Babiker F.A., Molkentin J.D., de Windt L.J. Requirement of nuclear factor of activated T-cells in calcineurin-mediated cardiomyocyte hypertrophy. J. Biol. Chem. 2002;277:48617–48626. doi: 10.1074/jbc.M206532200. [DOI] [PubMed] [Google Scholar]
- 15.Snijder B., Vladimer G.I., Krall N., Miura K., Schmolke A.S., Kornauth C., Lopez de la Fuente O., Choi H.S., van der Kouwe E., Gültekin S., et al. Image-based ex-vivo drug screening for patients with aggressive haematological malignancies: interim results from a single-arm, open-label, pilot study. Lancet Haematol. 2017;4:e595–e606. doi: 10.1016/S2352-3026(17)30208-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gorshkov K., Chen C.Z., Marshall R.E., Mihatov N., Choi Y., Nguyen D.-T., Southall N., Chen K.G., Park J.K., Zheng W. Advancing precision medicine with personalized drug screening. Drug Discov. Today. 2019;24:272–278. doi: 10.1016/j.drudis.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breinig M., Klein F.A., Huber W., Boutros M. A chemical-genetic interaction map of small molecules using high-throughput imaging in cancer cells. Mol. Syst. Biol. 2015;11:846. doi: 10.15252/msb.20156400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simm J., Klambauer G., Arany A., Steijaert M., Wegner J.K., Gustin E., Chupakhin V., Chong Y.T., Vialard J., Buijnsters P., et al. Repurposing High-Throughput Image Assays Enables Biological Activity Prediction for Drug Discovery. Cell Chem. Biol. 2018;25:611–618.e3. doi: 10.1016/j.chembiol.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilkins B.J., Molkentin J.D. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem. Biophys. Res. Commun. 2004;322:1178–1191. doi: 10.1016/j.bbrc.2004.07.121. [DOI] [PubMed] [Google Scholar]
- 20.Shimizu I., Minamino T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016;97:245–262. doi: 10.1016/j.yjmcc.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura M., Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018;15:387–407. doi: 10.1038/s41569-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 22.Darzynkiewicz Z., Juan G. DNA content measurement for DNA ploidy and cell cycle analysis. Curr Protoc Cytom. 2001;Chapter 7:Unit 7.5. doi: 10.1002/0471142956.cy0705s00. [DOI] [PubMed] [Google Scholar]
- 23.Ferro A., Mestre T., Carneiro P., Sahumbaiev I., Seruca R., Sanches J.M. Blue intensity matters for cell cycle profiling in fluorescence DAPI-stained images. Lab. Invest. 2017;97:615–625. doi: 10.1038/labinvest.2017.13. [DOI] [PubMed] [Google Scholar]
- 24.Ali S.R., Nguyen D., Wang B., Jiang S., Sadek H.A. Deep Learning Identifies Cardiomyocyte Nuclei With High Precision. Circ. Res. 2020;127:696–698. doi: 10.1161/CIRCRESAHA.120.316672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hein S., Arnon E., Kostin S., Schönburg M., Elsässer A., Polyakova V., Bauer E.P., Klövekorn W.P., Schaper J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–991. doi: 10.1161/01.cir.0000051865.66123.b7. [DOI] [PubMed] [Google Scholar]
- 26.Mishra S., Kass D.A. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2021;18:400–423. doi: 10.1038/s41569-020-00480-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heineke J., Molkentin J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 28.Coffey S., Cairns B.J., Iung B. The modern epidemiology of heart valve disease. Heart. 2016;102:75–85. doi: 10.1136/heartjnl-2014-307020. [DOI] [PubMed] [Google Scholar]
- 29.Mummery C.L. Perspectives on the Use of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes in Biomedical Research. Stem Cell Reports. 2018;11:1306–1311. doi: 10.1016/j.stemcr.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seeger T., Shrestha R., Lam C.K., Chen C., McKeithan W.L., Lau E., Wnorowski A., McMullen G., Greenhaw M., Lee J., et al. A Premature Termination Codon Mutation in MYBPC3 Causes Hypertrophic Cardiomyopathy via Chronic Activation of Nonsense-Mediated Decay. Circulation. 2019;139:799–811. doi: 10.1161/CIRCULATIONAHA.118.034624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jentzsch C., Leierseder S., Loyer X., Flohrschütz I., Sassi Y., Hartmann D., Thum T., Laggerbauer B., Engelhardt S. A phenotypic screen to identify hypertrophy-modulating microRNAs in primary cardiomyocytes. J. Mol. Cell. Cardiol. 2012;52:13–20. doi: 10.1016/j.yjmcc.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 32.Zhou P., Pu W.T. Recounting Cardiac Cellular Composition. Circ. Res. 2016;118:368–370. doi: 10.1161/CIRCRESAHA.116.308139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schiattarella G.G., Hill J.A. Inhibition of hypertrophy is a good therapeutic strategy in ventricular pressure overload. Circulation. 2015;131:1435–1447. doi: 10.1161/CIRCULATIONAHA.115.013894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sutcliffe M.D., Tan P.M., Fernandez-Perez A., Nam Y.J., Munshi N.V., Saucerman J.J. High content analysis identifies unique morphological features of reprogrammed cardiomyocytes. Sci. Rep. 2018;8:1258. doi: 10.1038/s41598-018-19539-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peter A.K., Bjerke M.A., Leinwand L.A. Biology of the cardiac myocyte in heart disease. Mol. Biol. Cell. 2016;27:2149–2160. doi: 10.1091/mbc.E16-01-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanna B., Bueno O.F., Dai Y.S., Wilkins B.J., Molkentin J.D. Direct and indirect interactions between calcineurin-NFAT and MEK1-extracellular signal-regulated kinase 1/2 signaling pathways regulate cardiac gene expression and cellular growth. Mol. Cell. Biol. 2005;25:865–878. doi: 10.1128/MCB.25.3.865-878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacDonnell S.M., Weisser-Thomas J., Kubo H., Hanscome M., Liu Q., Jaleel N., Berretta R., Chen X., Brown J.H., Sabri A.K., et al. CaMKII negatively regulates calcineurin-NFAT signaling in cardiac myocytes. Circ. Res. 2009;105:316–325. doi: 10.1161/CIRCRESAHA.109.194035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Konstandin M.H., Völkers M., Collins B., Quijada P., Quintana M., De La Torre A., Ormachea L., Din S., Gude N., Toko H., Sussman M.A. Fibronectin contributes to pathological cardiac hypertrophy but not physiological growth. Basic Res. Cardiol. 2013;108:375. doi: 10.1007/s00395-013-0375-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chin E.R., Olson E.N., Richardson J.A., Yang Q., Humphries C., Shelton J.M., Wu H., Zhu W., Bassel-Duby R., Williams R.S. A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev. 1998;12:2499–2509. doi: 10.1101/gad.12.16.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bruder J.T., Kovesdi I. Adenovirus infection stimulates the Raf/MAPK signaling pathway and induces interleukin-8 expression. J. Virol. 1997;71:398–404. doi: 10.1128/jvi.71.1.398-404.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Linderman G.C., Rachh M., Hoskins J.G., Steinerberger S., Kluger Y. Fast interpolation-based t-SNE for improved visualization of single-cell RNA-seq data. Nat. Methods. 2019;16:243–245. doi: 10.1038/s41592-018-0308-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McInnes L., Healy J., Melville J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. ArXiv. 2018 https://arxiv.org/abs/1802.03426 arXiv:1802.03426. [Google Scholar]
- 43.Bates D., Machler M., Bolker B.M., Walker S.C. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015;67:1–48. [Google Scholar]
- 44.Pinheiro J., Bates D., DebRoy S., Sarkar D., R Core Team . 2017. nlme: Linear and Nonlinear Mixed Effects Models. R package version 3.1-131.https://cran.r-project.org/web/packages/nlme/index.html [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
All original code has been deposited at github and is publicly available as of the date of publication (https://github.com/mknoll/cmoRe). CellProfiler pipelines are available as online supplemental files (Data S1 and S2).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.