

Abstract
Macrophages are the principal component of the innate immune system. They play very crucial and multifaceted roles in the pathogenesis of inflammatory vascular diseases. There is an increasing recognition that transcriptionally dynamic macrophages are the key players in the pathogenesis of inflammatory vascular diseases. In this context, the accumulation and aberrant activation of macrophages in the subendothelial layers govern atherosclerotic plaque development. Macrophage-mediated inflammation is an explicitly robust biological response that involves broad alterations in inflammatory gene expression. Thus, cell-intrinsic negative regulatory mechanisms must exist which can restrain inflammatory response in a spatiotemporal manner. In this study, we identified CBP/p300-interacting transactivator with glutamic acid/aspartic acid-rich carboxyl-terminal domain 2 (CITED2) as one such cell-intrinsic negative regulator of inflammation. Our in vivo studies show that myeloid-CITED2 deficient mice on the Apoe−/− background have larger atherosclerotic lesions on both control and high-fat/high-cholesterol diets. Our integrated transcriptomics and gene set enrichment analyses studies show that CITED2 deficiency elevates STAT1 and IRF1 regulated pro-inflammatory gene expression in macrophages. At the molecular level, our studies identify that CITED2 deficiency elevates IFNγ-induced STAT1 transcriptional activity and STAT1 enrichment on IRF1 promoter in macrophages. More importantly, siRNA-mediated knockdown of IRF1 completely reversed elevated pro-inflammatory target gene expression in CITED2 deficient macrophages. Collectively, our study findings demonstrate that CITED2 restrains the STAT1-IRF1 signaling axis in macrophages and limits the development of atherosclerotic plaques.
Keywords: Macrophage, Inflammation, Atherosclerosis, Gene regulation, Signal transduction
Introduction
The process of inflammation is a robust biological response that involves the recruitment of immune cells to eliminate foreign agents, helps in tissue repair, and maintains tissue homeostasis (1). The monocyte-derived macrophages are the principal innate immune cells that provide the first line of defense against infection/injury and serve as key players in orchestrating innate and adaptive immune responses (2). Inflammatory macrophages recognize foreign agents by utilizing pattern recognition receptors, including Toll-like receptors (TLRs) (3). The TLRs are the conserved single-pass membrane-spanning receptors on the sentinel cells and effectively distinguish molecular ligands of viral and microbial pathogens such as lipoproteins, bacterial flagellin, single/double-stranded RNA, and bacterial DNA (3). Similarly, macrophages recognize host-derived inflammatory signals by utilizing a battery of cell surface receptors, including interferon receptors. These receptors convey extracellular cues to the cytoplasm and nucleus to shape the appropriate immune response at cellular and molecular levels (4). Typically, these cell surface receptors recruit signaling adapters and initiate a range of intracellular signaling pathways that result in the activation of several transcriptional factors such as NFκB, HIF1α, STAT1, and IRFs (4). These transcription factors control inflammatory and metabolic gene programs that shape robust pro-inflammatory macrophage activation. However, exaggerated and uncontrolled macrophage inflammatory response could lead to excessive cytokine production, impaired tissue functions, development of chronic diseases, organ failure, and death (5). These are evident in many chronic and acute inflammatory disease conditions, including arthritis, atherosclerosis, lupus, inflammatory bowel diseases, and sepsis (5). However, macrophages contain several cell-intrinsic negative regulatory mechanisms to constrain and resolve uncontrolled pro-inflammatory gene expression in a spatiotemporal manner (6). Thus, identifying such cell-intrinsic negative regulatory mechanisms will help us to better understand pro- and anti-inflammatory signaling dynamics that are operative in disease pathogenesis. This in turn, could aid in developing new therapeutic approaches in the treatment of human ailments.
In this study, we have identified macrophage Cbp/p300 interacting transactivator with Glu/Asp rich carboxy-terminal domain 2 (CITED2) as a quintessential cell-intrinsic negative regulator of inflammatory gene expression and restrain progression of inflammatory vascular wall disease. CITED2 is exclusively localized in the nucleus and is a member of the CBP/p300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain family of transcriptional modulators (7). Studies over the years have shown that CITED2 plays a critical role in cellular development and differentiation (8). Previous studies have shown that mutations in the human CITED2 gene is associated with congenital heart defects (9). These observations were recapitulated in genetically engineered Cited2 null mice that exhibited congenital heart defects, as well as disruption in the left-right patterning of the body axis (10). Indeed, Cited2 knockout mice exhibit embryonic lethality in utero around mid to late gestation due to multiple developmental anomalies, including malformations of the adrenal gland, liver, lung, placenta, and neural tube defects (11). Studies have also shown that CITED2 plays a crucial role in maintaining the hematopoietic stem cell pool in the bone marrow and the process of hematopoiesis (12). Indeed, conditional deletion of CITED2 in the hematopoietic system resulted in substantial loss of adult hematopoietic stem cells and primitive progenitor cells that result in bone marrow failure (12). In addition, CITED2 deficient hematopoietic stem cells also exhibited impaired quiescence and reconstitution capacity (13). Our previous studies have demonstrated that CITED2 is predominantly expressed in human and murine macrophages (14). Myeloid-specific CITED2 deficient mice are highly susceptible to zymosan-induced lung inflammation (15) and LPS-induced endotoxic shock syndrome (14). At the molecular level, our previous reports have shown that CITED2 cooperates with PPARγ to elevate anti-inflammatory targets while repressing HIF1α and the NFκB functions in macrophages (14, 15). However, whether myeloid-CITED2 deficiency alters the gene programs that are involved in the development or progression of inflammatory vascular wall disease has not been previously examined. In this study, we provide evidence that CITED2 restrains the STAT1-IRF1 signaling axis in macrophages and limits the development of atherosclerotic plaques in vivo.
Materials and Methods
Experimental mouse models and atherosclerosis studies
All animal procedures were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University and conformed to the American Association for Accreditation of Laboratory Animal Care guidelines. All mice were bred and maintained under pathogen-free conditions, fed standard laboratory chow, and kept on a 12-h light/dark cycle. Myeloid CITED2-specific null mice were generated in our laboratory as described in our previous study (14). Briefly, the control (Lyz2cre/cre on C57BL/6J background) and myeloid-specific Cited2 deficient (Cited2fl/fl:Lyz2cre/cre on C57BL/6J background) were generating by breeding male and female Cited2fl/− :Lyz2cre/cre mice. The Cited2fl/fl:Lyz2cre/cre mice contained two Cited2 floxed and two Lyz2 Cre alleles (C57BL/6J background). Mice with two Cre alleles (Lyz2cre/cre) were used as the control group (C57BL/6J background). For the atherosclerosis studies, 8-week-old male and female Lyz2cre/cre:Apoe−/− (C57BL/6J) and Cited2fl/fl:Lyz2cre/cre:Apoe−/− (C57BL/6J) mice were fed a Western-style diet (TD 88137, Harlan Teklad: 21.2% fat, 0.2% cholesterol) for 20 weeks. These study mice received randomly numbered ear tags, and investigators were blinded to mice genotype information. At the end of the feeding period, total blood samples were collected for plasma cytokine and cholesterol analyses. Next, these study mice were euthanized and perfused with saline. The aortic sinuses, ascending and descending aorta were collected. The entire aorta was stained with Sudan IV, and excessive adventitial tissues were removed. For en face aortic lesion quantifications, aortas were dissected by removing all branching vessels down to the femoral bifurcation and then sliced ventrally. The images of the aortas next to a ruler were digitally captured by using Canon EOS Rebel T6 DSLR camera. Lesion areas within the entire length of the aorta were quantified using ImageJ software. Results were expressed as absolute aortic surface plaque area as well as percent lesion area relative to the total aortic surface area.
Further, aortic sinuses were cryosectioned using a Sakura Tissue-Tek Cryo 2000 cryostat at a thickness of 10 μm. The serial sections were taken starting at the aortic valve plane and covering 450 μm in intervals of 50 μm. The serial sections were placed on Superfrost Plus microscope slides, and the sections were stored at −20°C until use. These sections were stained with Oil Red O/hematoxylin staining for aortic root plaque quantification. Further, aortic sinuses cryosections were fixed with 10% of buffered formalin. These sections were subjected to antigen retrieval steps with antigen unmasking solution. Samples were treated with 0.3% of H2O2 for 30 minutes at room temperature, and the nonspecific binding was blocked with a blocking buffer. Samples were incubated with rabbit anti-F4/80 antibody at 4°C for overnight. These tissue sections were subsequently incubated with biotin-conjugated goat anti-rabbit IgG for 30 minutes at room temperature. Samples were incubated in ABC reagent, and the immunostaining was visualized using a DAB reagent. Images were acquired using a microscope, and Image J software was utilized for quantification of aortic sinus plaque and macrophage area. These quantifications included at least seven sections for each mouse. The quantification of histological data and statistical analyses were performed by two independent investigators who are blinded to sample genotypes and/or treatments.
Cell culture
The bone marrow-derived macrophages (BMDMs) were generated by ex vivo differentiation of bone marrow cells. Briefly, bone marrow cells from 8-week-old Lyz2cre/cre and Cited2fl/fl:Lyz2cre/cre mice were harvested from the femur and tibia. The isolated bone-marrow cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with recombinant mouse macrophage colony-stimulating factor (M-CSF) for seven days. The resulting BMDMs were harvested and utilized for indicated experiments. RAW264.7 cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 10 μg/ml streptomycin, and 2 mM glutamine in a humidified incubator (5% CO2 and 37°C). Mouse thioglycolate-elicited peritoneal macrophages (PMs) were obtained by inducing peritonitis with 3% thioglycolate broth in 8- to 12-week-old mice. These primary peritoneal macrophages were cultured in serum-supplemented DMEM as described above. Human monocytes were isolated from unfractionated peripheral blood mononuclear cells by CD14 negative selection using magnetic beads (Miltenyi Biotec, San Diego, CA). These cells were cultured in complete DMEM supplemented with recombinant human M-CSF (50 ng/ml) for 7 days to generate primary macrophages. These macrophages were utilized for indicated experiments. All experiments involving human samples were approved by the Case Western Reserve University Institutional Review Board. The primary macrophages from atherosclerotic aortic plaques were obtained 20 weeks after the initiation of control or Western-style diet (TD 88137, Harlan Teklad). Briefly, the entire aortic structure was collected, and periaortic adipose tissues were removed by microdissection. These aortae were washed with a copious amount of sterile, ice-cold phosphate-buffered saline to remove red blood cells. The aortic tissues were minced with scalpel blades and were incubated with 3 mg/mL collagenase-dispase mixture for 4 hours at room temperature with gentle shaking. The cellular suspension was collected by using a tissue strainer. The anti-F4/80 microbeads (130-110-443; Miltenyi Biotec, Auburn, CA) were used to purify macrophages from these cellular suspensions. The total RNA samples derived from these macrophages were utilized for RT-qPCR analyses.
RNAseq analysis
Total RNA from primary macrophages were obtained using the High Pure RNA Isolation Kit. Quality control of RNA isolated was assessed using Qubit (Invitrogen) for quantification and Agilent 2100 BioAnalyzer analysis to determine the quality using a cut-off of RIN > 7.0 to select specimens for further analysis. cDNA library for RNAseq was generated from 150 ng of total RNA using the Illumina TruSeq Stranded Total RNA kit with Ribo Zero Gold for rRNA removal according to the manufacturer’s protocol. The resulting purified mRNA was used as input for the Illumina TruSeq kit, in which libraries are tagged with unique adapter-indexes. Final libraries were validated on the Agilent 2100 BioAnalyzer, quantified via qPCR, and pooled at equimolar ratios. Pooled libraries were diluted, denatured, and loaded onto the Illumina NextSeq 550 System using a high output flowcell. STAR Aligner was used for mapping the sequencing reads to the mm10 mouse reference genome. The aligned reads were then analyzed with Cuffdiff to obtain gene-level expression data using the GENCODE gene annotation for mm10 and reported as fragments per kilobase per million reads mapped (FPKM). Differential expression analysis was also performed using the Cuffdiff package and significantly differentially expressed genes were defined using an adjusted P-value < .05 (FDR corrected). Gene expression tables for relevant pairwise comparisons were analyzed for gene set enrichment (GSEA) using GenePattern (Broad Institute) (16). We specifically utilized TFT data sets for current studies. A gene set was considered to be significantly enriched using an FWER cutoff <0.05. Heatmaps were generated using ClustVis. The sequencing data reported in this manuscript has been deposited in Gene Expression Omnibus (GSE147943).
RNA extraction, Real-time Quantitative PCR, and Western blot
Total RNA was isolated from indicated samples using the High Pure RNA Isolation Kit. The total RNA samples (one μg each) were reverse-transcribed using M-MuLV reverse transcriptase in the presence of random hexamers and oligo-dT primers. Real-time quantitative PCR was performed using Universal SYBR Green PCR Master Mix or TaqMan Universal Master Mix on Applied Biosystems (Foster City, CA) Step One Plus real-time PCR system in the presence of gene-specific primers. The list of primers utilized in this study is provided in table S1.
The primary cells and cell lines were lysed after indicated treatment in ice-cold RIPA buffer containing protease and phosphatase inhibitors. Protein concentration was measured by the bicinchoninic acid (BCA) protein assay. Equal amounts of protein lysate samples were electrophoresed using 8% or 4–15% Mini-PROTEAN TGXTM precast gels (Bio-Rad) and transferred to nitrocellulose membranes. These membranes were blocked with 5% non-fat dry milk or 5% bovine serum albumin in TBS-T for one hour at room temperature. These blots were further incubated with primary antibodies diluted in 5% non-fat dry milk in TBS-T. After overnight incubation, primary antibodies were removed by washing with TBS-T. These blots were incubated for one hour at room temperature in horseradish peroxidase-conjugated secondary antibodies. Blots were visualized using enhanced chemiluminescence Western Blotting Substrate. The densitometry analyses were performed utilizing ImageJ software. The primary antibodies were used at the following dilutions. CITED2 (1:1000), IRF1 (1:1000); STAT1, p-STAT1, (1:2500); and β-ACTIN (1:5000).
Transient transfection, luciferase assay, and Chromatin immunoprecipitation (ChIP)
Transfection of RAW264.7 cells and mice BMDMs were performed using Lipofectamine transfection reagents (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. Transfected cells were stimulated with IFNγ (10 ng/ml) or PBS and used for indicated experiments. RAW264.7 cells were transfected with STAT1 cis consensus-driven luciferase reporter plasmid or were co-transfected with pCMV-Cited2 plasmid or Cited2 specific siRNA using Lipofectamine transfection reagent. These cells were exposed to 10 ng/ml IFNγ for 18h. Luciferase reporter activity was measured and normalized according to the manufacturer’s instructions. Results are presented as relative luciferase activity over the control group.
Chromatin immunoprecipitation (ChIP) analyses were performed using the EZ-Magna ChIP G Kit according to the manufacturer’s instruction. Briefly, Lyz2cre/cre and Cited2fl/fl:Lyz2cre/cre mice BMDMs were stimulated with 10 ng/ml IFNγ. Chromatin immunoprecipitations were performed using anti-STAT1 or anti-CITED2 antibodies. Chromatin samples from these experiments were evaluated by real-time quantitative RT-PCR. DNA levels were first normalized to the internal control region in the first intron of the mouse Actb gene (forward: 5′-CGTATTAGGTCCATCTT GAGAGTAC-3′, reverse: 5′-GCCATTGAGGCGTGATCGTAGC-3′). Primer pairs flanking the STAT1 gamma interferon activation site (GAS) element were targeted to amplify the mouse Irf1 (FW, 5′-GGGAGTGGAGTGGAGCAAG-3′; REV, 5′-CCACTCGGCCTCATCATTT-3′) promoter region. ChIP performed using isotype IgG was used as a negative control. Relative enrichment was calculated by dividing the normalized levels of ChIP DNA to that of input DNA at the corresponding locus.
Quantitative and statistical analyses
All phenotype data was analyzed by groups using a combination of letter and number codes that did not specify genotypes, sex, or diet. Thus data acquisition was blinded. All data, unless indicated, are presented as the mean ± SD. All data were normally distributed passing the KS and/or D’Agostino and Pearson normality tests. Therefore, parametric statistics were used in this study. The statistical significance of differences between the two groups were analyzed by using two-way AVOVA with Bonferroni correction for multiple comparison tests. A p-value less than 0.05 were considered significant.
Supporting information
Key reagents and resources, including the source of antibodies, chemicals, reagents, and primer sequence are listed in Table S1 in the Supporting Information.
Results
CITED2 deficiency augments TLR4 mediated IRFs signaling in macrophages
TLR4 signaling is conserved across mammalian species (17). They play a central role in the recognition and effective expulsion of foreign agents. Thus, activation of TLR4 signaling activates broad inflammatory gene programs in immune cells. In this context, our previous studies have shown that CITED2 restrains LPS-induced HIF1α and NFκB signaling in macrophages (14, 15). However, whether CITED2 targets specific transcription factors to restrain broad inflammatory gene expression programs has not been examined. To evaluate this notion, we have utilized gene expression profiles derived from LPS challenged bone marrow-derived macrophages (BMDMs) from Lyz2cre/cre and Cited2fl/fl:Lyz2cre/cre mice. Western blot analyses confirmed a substantial reduction in CITED2 protein levels in Cited2fl/fl:Lyz2cre/cre mice BMDMs (Fig. 1A). To identify specific transcription factor signaling pathways that are dysregulated due to CITED2 deficiency, RNAseq data were subjected to transcription factors targets (TFT) analyses using the BROAD-Gene Set Enrichment Analysis (GSEA) program (16). Our analyses show that LPS exposure broadly enriched IRF1, IRF2, IRF_Q6, and STAT3 transcription factors target gene expression in CITED2 deficient macrophages (Fig.1B–E). As anticipated, LPS exposure significantly elevated gene expression for a large number of IRF1, IRF2, common IRFs (IRF_Q6), and canonical IRF1 (18) targets in Lyz2cre/cre mice BMDMs (Fig.1F–H). Interestingly, expression of these IRF1, IRF2, common IRFs (IRF_Q6), and canonical IRF1 targets were significantly and robustly elevated in CITED2 deficient BMDMs following LPS exposure (Fig.1 F–H). Next, we examined whether CITED2 deficiency altered the expression of key dysregulated transcription factors following LPS challenge. Our analyses show that LPS challenge robustly elevated Irf1 expression in CITED2 deficient BMDMs (Fig.2A). In addition, CITED2 deficiency also modestly elevated Irf2 and Irf9 expression following challenge (Fig.2B). However, CITED2 deficiency did not significantly alter LPS-induced Stat3 expression in macrophages (Fig.2C). Further, we probed whether CITED2 deficiency altered LPS-induced IRF1 target expression in macrophages. Accordingly, Lyz2cre/cre and Cited2fl/fl:Lyz2cre/cre mice BMDMs were stimulated with LPS, and expression of classical IRF1 target genes were evaluated by RT-qPCR analyses. As shown in figure 2D–F, LPS stimulation robustly induced IRF1 target gene expression (Batf2, Ccl8, Isg15, Ifi47, Slamf8, Irg1, Mmp13, Dnase1l3, Kynu, Gbp3, Gbp2, and Gbp5) in Lyz2cre/cre mice BMDMs. However, CITED2 deficient BMDMs exposure to LPS exhibited significant and substantial increases in these IRF1 target gene expression compared to Lyz2cre/cre mice BMDMs (Fig.2D–F). Taken together, these results demonstrate that CITED2 deficiency augments TLR4 signaling mediated IRF1 target gene expression in macrophages.
Figure 1. CITED2 deficiency augments IRF target gene expression in macrophages.

(A) Total protein extracts from Lyz2cre/cre, and Cited2fl/fl:Lyz2cre/cre mice BMDMs were evaluated for CITED2 expression by western blot. (n=3). Actin was used as housekeeping gene. (B) Lyz2cre/cre, and Cited2fl/fl:Lyz2cre/cre mice BMDMs were challenged with 100 ng/ml of LPS for four hours. The total RNA samples were subjected to RNAseq analyses. The RNAseq data were subjected to gene set enrichment analyses (GSEA) using transcription factors targets (TFT) gene sets. FWER P-value less than 0.05 was considered significant (n=3). (C-E) The enrichment plots of indicated gene set were obtained by comparing Lyz2cre/cre and Cited2fl/fl: Lyz2cre/cre mice BMDMs RNAseq data following LPS treatment in the GSEA program (n = 3). (F-H) Heatmaps of genes that are IRF1-IRF2 targets (F), IRF_Q6 targets (G), and canonical IRF1 targets (H) and are significantly upregulated in LPS-induced Cited2fl/fl:Lyz2cre/cre mice BMDMs compared to Lyz2cre/cre mice BMDMs treated with LPS. (n=3). FWER P-value *P < 0.05; **P < 0.01; ***P < 0.001.
Figure 2. CITED2 deficiency boosts IRF1 target gene expression.

Lyz2cre/cre, and Cited2fl/fl:Lyz2cre/cre mice BMDMs were stimulated with 100 ng/ml of LPS for four hours. (A-C) Total RNA samples were evaluated for expression of Irf1 (A), Irf2 and Irf9 (B), and Stat3 (C) by RT-qPCR analysis. (D-F) Lyz2cre/cre, and Cited2fl/fl:Lyz2cre/cre mice BMDMs were stimulated with 100 ng/ml of LPS for four hours. Total RNA samples were evaluated for expression of Batf2, Ccl8, Isg15, Ifi47, Slamf8, Irg1, Mmp13, Dnase1l3, Kynu, Gbp3, Gbp2, and Gbp5 by RT-qPCR. 36B4 was used as a housekeeping gene for RT-qPCR analysis. All the experiments were performed four independent times with four replicates. Values are reported as mean ± SD. Data were analyzed by ANOVA followed by Bonferroni post-testing. *P < 0.05, **P < 0.01 and ***P <0.001.
CITED2 deficiency promotes IFNγ-induced STAT1 and IRF1 targets in macrophages.
Past studies have established that IFNγ and TLR4 signaling utilize STAT1 to induce IRF1 expression that orchestrates broad pro-inflammatory gene programs (19). However, whether CITED2 deficiency affects IFNγ-induced STAT1 or IRF1 signaling has not been examined. Our analyses above (Fig.1 and 2) show that CITED2 deficiency elevates TLR4-mediated IRFs target gene expression in macrophages. Therefore, we hypothesized that CITED2 might restrain IFNγ-induced STAT1 and IRF1 target gene expression in macrophages. To test this hypothesis, Lyz2cre/cre and Cited2fl/fl:Lyz2cre/cre mice BMDMs were stimulated with IFNγ, and total RNA samples were obtained to perform gene expression profiling studies. The RNAseq data were evaluated for the enrichment of STAT1 (20) and IRF1 (18) gene targets in CITED2 deficient BMDMs utilizing the BROAD-GSEA program (16). Our analyses show that STAT1 and IRF1 inflammatory gene targets are highly enriched and elevated in IFNγ-induced CITED2 deficient BMDMs compared to Lyz2cre/cre mice BMDMs treated with IFNγ (Fig. 3A and B). As envisioned, IFNγ challenge significantly heightened a large number of STAT1 and IRF1 target gene expression in Lyz2cre/cre mice BMDMs (Fig.3C and D). Expression of these STAT1 and IRF1 target genes were significantly and robustly upregulated in CITED2 deficient BMDMs following IFNγ challenge (Fig.3C and D). Overall, these results demonstrate that CITED2 deficiency elevates IFNγ-induced STAT1 and IRF1 target gene expression in macrophages. Next, we assessed whether CITED2 deficiency influences the expression of critical transcription factors following IFNγ challenge. Our analyses show that IFNγ exposure vigorously and significantly elevated Irf1 expression in CITED2 deficient BMDMs (Fig.4A). Surprisingly, CITED2 deficiency did not significantly affect IFNγ−induced Irf2, Irf9, Stat1, and Stat3 expression in macrophages (Fig.4 B and C). Further, we evaluated whether CITED2 overexpression affects IFNγ-induced IRF1 and its target expression in macrophages. As shown in figure-4D, CITED2 overexpression significantly attenuated IFNγ-induced Irf1 expression in macrophages. Concordantly, CITED2 overexpression also substantially curtailed IFNγ-induced IRF1 target gene expression (Irg1, F3, Tmem140, and Dnase1l3) in macrophages (Fig.4E). Collectively, these results show that CITED2 plays a critical role in IFNγ-induced IRF1 and its target gene expression in macrophages.
Figure 3. CITED2 deficiency exalts STAT1 and IRF1 targets in macrophages.

Lyz2cre/cre and Cited2fl/fl:Lyz2cre/cre mice BMDMs were challenged with 10 ng/ml of IFNγ for six hours. The total RNA samples were subjected to RNAseq analyses. (A and B) The RNAseq data were subjected to gene set enrichment analyses (GSEA) using previously identified STAT1 and IRF1 gene targets. The enrichment plots of STAT1 (A) and IRF1 (B) gene sets that were obtained by comparing IFNγ-induced Lyz2cre/cre and Cited2fl/fl:Lyz2cre/cre mice BMDMs RNAseq data are shown. FWER P-value less than 0.05 was considered significant (n=3). (C and D) Heatmaps of STAT1 target genes (C) and IRF1 target genes (D) that are significantly upregulated in IFNγ-induced Cited2fl/fl:Lyz2cre/cre mice BMDMs compared to IFNγ treated Lyz2cre/cre mice BMDMs. (n=3).
Figure 4. CITED2 modulates IFNγ-induced IRF1 expression.
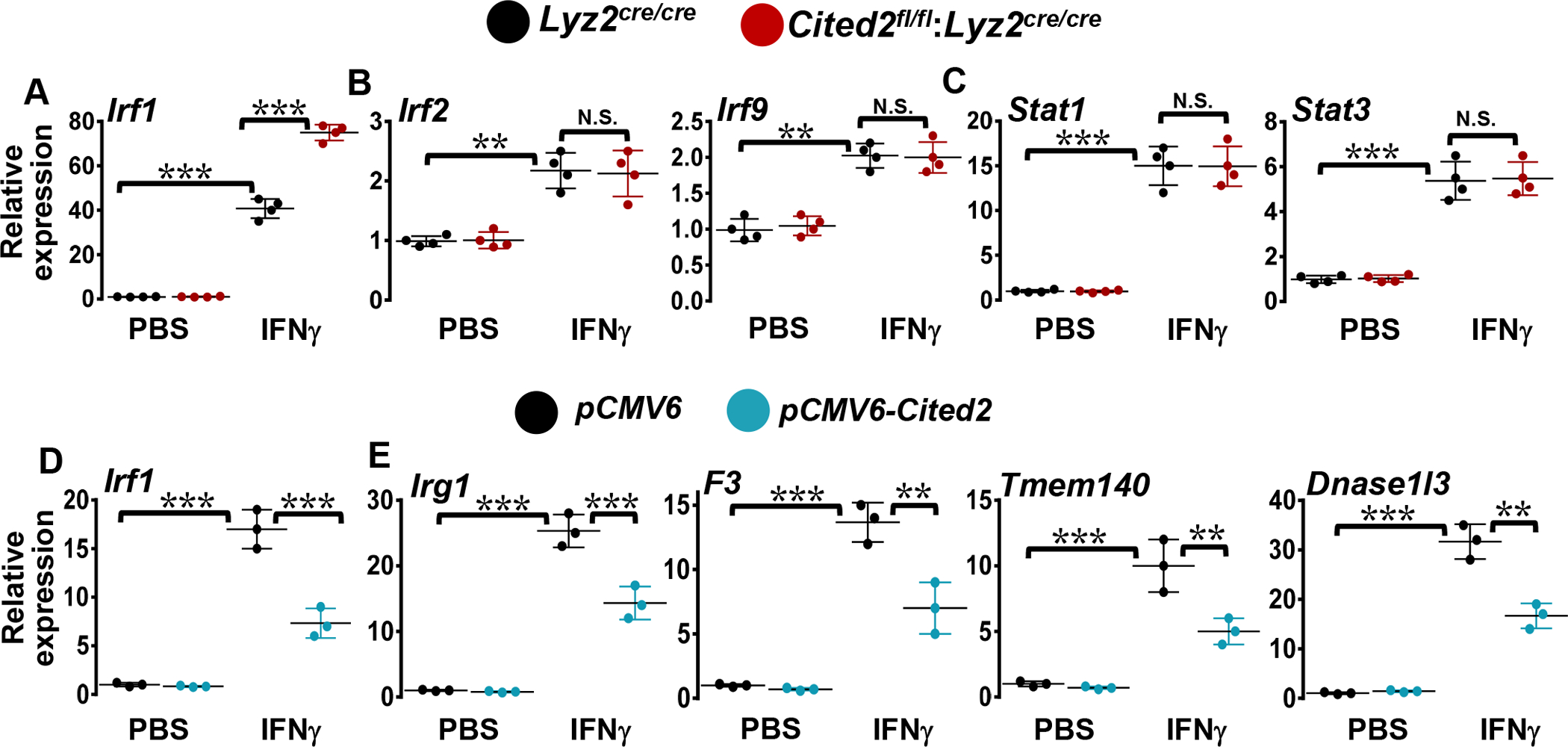
(A-C) Lyz2cre/cre and Cited2fl/fl:Lyz2cre/cre mice BMDMs were stimulated with 10 ng/ml of IFNγ for five hours. Total RNA samples were evaluated for expression of Irf1 (A), Irf2, Irf9 (B), and Stat1 and Stat3 (C) by RT-qPCR analysis (n=4). (D and E) RAW264.7 cells were transfected with control or pCMV6-Cited2 plasmid were stimulated with 10 ng/ml of IFNγ for five hours. Total RNA samples were analyzed for expression of Irf1 (D), Irg1, F3, Tmem140 and Dnase1l3 (E) by RT-qPCR (n=3). 36B4 was used as a housekeeping gene. Values are reported as mean ± SD. Data were analyzed by ANOVA followed by Bonferroni post-testing. N.S.- Not significant, **P < 0.01 and ***P <0.001.
CITED2 deficient macrophages are hyperresponsive to IFNγ
Inflammatory diseases are generally accentuated by the presence of IFNγ, which increases the production of inflammatory cytokines in macrophages. Thus, IFNγ has long been recognized as a signature pro-inflammatory cytokine that plays a central role in inflammation and disease pathogenesis (21). Our RNAseq and GSEA studies show that IFNγ substantially elevates STAT1 and IRF1 target gene expression in CITED2 deficient macrophages (Fig.3A–D). Owing to the importance of STAT1 (22) and IRF1 (23) regulated inflammatory gene expression in vascular diseases, we assessed whether CITED2 deficiency alters IFNγ-induced pro-inflammatory gene expression in macrophages. Therefore, the differentially expressed STAT1 and IRF1 target gene profiles (Fig.3A–D) were utilized as a pointer to probe the dysregulated inflammatory gene expression in CITED2 deficient macrophages. Accordingly, Lyz2cre/cre and Cited2fl/fl:Lyz2cre/cre mice BMDMs were stimulated with IFNγ, and expression of classical inflammatory gene targets were evaluated by RT-qPCR analyses. Our analyses show that IFNγ challenge significantly elevated expression of Isg15, Cmpk2, Irg1, Ccl5, Ptgs2, Dnase1l3, Ddit3, Il12rb2, Tmem140, Pfkfb3, F3, B2m, Ifit2, Ifi44, Edn1, Plau, Mmp13, Cxcl2, Il1a, and Il1b expression in Lyz2cre/cre mice BMDMs (Fig.5A–E). Interestingly, CITED2 deficient BMDMs challenged with IFNγ show a significant and substantial increase in these inflammatory target gene expression compared to Lyz2cre/cre mice BMDMs (Fig.5A–E). Collectively, these results show that CITED2 deficient macrophages are sensitive to IFNγ challenge and express substantially high levels of inflammatory mediators following the inflammatory insult.
Figure 5. CITED2 deficient macrophages are hyperresponsive to IFNγ.

(A-E) Lyz2cre/cre and Cited2fl/fl:Lyz2cre/cre mice BMDMs were stimulated with 10 ng/ml of IFNγ for five hours. Total RNA was obtained and evaluated for expression of Isg15, Cmpk2, Irg1, Ccl5, Ptgs2, Dnase1l3, Ddit3, Il12rb2, Tmem140, Pfkfb3, F3, B2m, Ifit2, Ifi44, Edn1, Plau, Mmp13, Cxcl2, Il1a, and Il1b by RT-qPCR. 36B4 was used as a housekeeping gene for RT-qPCR analysis. All the experiments were performed three independent times with three replicates. Values are reported as mean ± SD. Data were analyzed by ANOVA followed by Bonferroni post-testing. *P < 0.05, **P < 0.01 and ***P <0.001.
Myeloid-CITED2 deficiency exacerbates experimental atherogenesis
Prior studies have shown that defects in TLR4 (24), IFNγ (25), IRF1 (23), or STAT1 (22) signaling significantly curtail atherosclerotic plaque development in vivo. In this context, our analyses show that CITED2 deficiency significantly elevates TLR4, IFNγ, IRF1, and STAT1 signaling in macrophages (Fig. 1–4). However, whether myeloid-CITED2 plays any role in the development of atherosclerotic plaques has not yet been investigated. First, we examined whether high-fat diet (HFD) feeding altered Ifnγ or Cited2 expression in atherosclerotic plaque macrophages. Interestingly, the HFD challenge significantly elevated Ifnγ levels while repressing Cited2 expression in atherosclerotic plaque macrophages (Fig. 6A and B). More importantly, IFNγ exposure significantly attenuated Cited2 expression in murine as well as human primary macrophages (Fig. 6C and D). Next, we intended to examine whether myeloid-CITED2 deficiency affects the development of atherosclerotic plaques in vivo. Accordingly, at eight weeks of age, male and female myeloid-CITED2 deficient (Cited2fl/fl: Lyz2cre/cre:Apoe−/−) and control (Lyz2cre/cre:Apoe−/−) mice on Apoe−/− background were challenged with a high-fat diet (HFD) for 20 weeks. The magnitude of aortic surface atherosclerotic plaques development was assessed by en face preparation of aorta and Sudan IV staining. The quantification of aortic surface plaques area shows that myeloid-CITED2 deficient mice exhibited a significant increase in atherosclerotic lesion area in both sexes when compared to the control group (Fig.6E and F). To accommodate variations arising due to differences in total aortic surface area, the percent lesion area of the entire aortic surface area was calculated. As shown in figure-6G, myeloid-CITED2 deficient mice challenged with HFD displayed a significantly higher percentage of atherosclerotic lesion area when compared to the respective control group. Collectively, these results demonstrate that myeloid-CITED2 deficiency heightens atherosclerotic disease burden in vivo.
Figure 6. Myeloid-CITED2 deficiency aggravates atherosclerotic plaque development.

(A and B) Atherosclerotic plaque macrophages were obtained from Apoe-null mice fed on a western-style high-fat diet (HFD) or control diet for 20 weeks. Expression of Ifnγ and Cited2 were analyzed by RT-qPCR analyses. (C and D) Mouse thioglycolate-elicited peritoneal macrophages (C) and human monocyte-derived macrophages (D) were stimulated with 20ng/ml IFNγ for 8 hours. Total RNA samples were evaluated for CITED2 expression by RT-qPCR analyses. (E) Female and male Lyz2cre/cre:Apoe−/− and Cited2fl/fl:Lyz2cre/cre:Apoe−/− mice were fed on a western-style high-fat diet for 20 weeks. Aortas were harvested at the end of the feeding period and stained for lipid-filled atherosclerotic plaques by using Sudan IV stain. Representative en face preparation of entire aortas stained with Sudan IV are shown. The atherosclerotic plaques are stained in orange/red. (F and G) Quantification of atherosclerotic lesion area (F) and percent lesion area (G) of the entire isolated en face aortic area was calculated. The dot plot represents the mean with standard deviation. (n=12). Data were analyzed by ANOVA followed by Bonferroni post-testing. *** p ≤ 0.001. Scale bar=0.5 cm.
Elevated pro-inflammatory milieu in CITED2 deficient mice atherosclerotic plaques
The aortic root are highly susceptible to developing atherosclerotic lesions and exhibit hallmark changes associated with disease progression. Thus, we examined whether myeloid-CITED2 deficiency affects the architecture of atherosclerotic lesions in vivo. Accordingly, serial aortic root sections from Cited2fl/fl:Lyz2cre/cre:Apoe−/− and Lyz2cre/cre:Apoe−/− mice fed on control or HFD (20 weeks) were assessed for lipid-filled atherosclerotic plaque development with oil red O staining. Our analyses show that myeloid-CITED2 deficiency significantly elevated atherosclerotic plaque development in male and female mice fed on the control diet or HFD (Fig.7A and B). Next, we examined whether elevated cholesterol levels contribute to enhanced atherosclerotic plaque formation in myeloid-CITED2 deficient mice. Surprisingly, our analyses show that myeloid-CITED2 deficiency did not significantly impact HFD-induced total cholesterol levels (Fig.7C). Clinical and experimental studies support an important role for macrophages in the development and progression of atherosclerotic plaques (26). A hallmark event in atherosclerotic plaque development is the accumulation of inflammatory macrophages in the subendothelial layers of affected blood vessels (27). Therefore, we examined whether myeloid-CITED2 deficiency alters macrophage abundance in atherosclerotic plaque lesions. Our analyses show that myeloid-CITED2 deficiency significantly elevates macrophage abundance in aortic root atherosclerotic plaques on control as well as on HFD (Fig. 7D and E). The inflammatory macrophages are known to accelerate the progression of atherosclerotic plaque development by producing copious amounts of inflammatory cytokines, chemokines, and matrix remodeling enzymes (27). Thus, we examined whether myeloid-CITED2 deficiency alters IRF1 regulated inflammatory gene expression in atherosclerotic plaque macrophages. As shown in figure-8A, myeloid-CITED2 deficiency (Cited2fl/fl: Lyz2cre/cre:Apoe−/−) significantly and substantially elevated HFD challenged expression of Irf1 and IRF1-regulated pro-inflammatory gene targets such as Il1a, Il1b, Ccl5, Plau, F3, Mmp13, and Cxcl2 expression in atherosclerotic plaque macrophages. Next, we assessed whether this shift in the inflammatory milieu was observed at a systemic level. Accordingly, blood plasma samples derived from Cited2fl/fl:Lyz2cre/cre :Apoe−/− and Lyz2cre/cre:Apoe−/− mice fed on control or HFD (20 weeks) were evaluated for inflammatory cytokines levels. As shown in figure-8B, the blood plasma level of IL1α, IL1β, TNF, and IL6 were significantly elevated in myeloid-CITED2 deficient mice (Cited2fl/fl:Lyz2cre/cre:Apoe−/−) compared to the control mice group (Lyz2cre/cre :Apoe−/−) following HFD challenge. Together, these results show that myeloid-CITED2 deficiency significantly elevates IRF1-regulated pro-inflammatory gene expression in atherosclerotic plaque macrophages, establishes systemic pro-inflammatory milieu, and promoters macrophage abundance in lipid-filled atherosclerotic plaques.
Figure 7. Effects of Myeloid-CITED2 deficiency on aortic root atherosclerotic lesion formation.

Eight weeks old male and female Lyz2cre/cre:Apoe−/− and Cited2fl/fl:Lyz2cre/cre:Apoe−/− mice were fed on a western-style high-fat diet or control diet for 20 weeks. Aortic sinuses were harvested at the end of the feeding period and embedded in the optimal cutting temperature compound. (A and B) The aortic sinus serial sections were stained for lipid-filled atherosclerotic lesions with Oil Red O and counterstained with hematoxylin. Representative images of Oil red O stained aortic root sections from control and high-fat at the end of 20 weeks are shown (A). Scale bar represents 0.5 mm. The quantification of the aortic root lesions area were performed by using ImageJ software. The graph on the right represents quantification of the Oil Red O-positive lesion area in control and high-fat diet-fed Lyz2cre/cre:Apoe−/− and Cited2fl/fl:Lyz2cre/cre: Apoe−/− mice (B). (n=10). (C) Blood samples were collected at the end of the feeding period and total plasma cholesterol levels were quantified (n=10). The dot plot represents the mean with standard deviation. (D and E) The aortic sinus serial sections were stained for macrophages by using an anti-F4/80 antibody. Representative images of F4/80 stained aortic root sections from control and high-fat at the end of 20 weeks are shown (D). Scale bar represents 0.5 mm. The graph on the right represents quantification of the macrophage area in control and high-fat diet-fed Lyz2cre/cre:Apoe−/− and Cited2fl/fl:Lyz2cre/cre:Apoe−/− mice (E). (n=10). The box plot represents the median with first and third quartiles, and whiskers represent minimum/ maximum. Data were analyzed by two-way AVOVA. *P < 0.05, **P < 0.01 and ***P <0.001.
Figure 8. CITED2 deficiency elevates atherosclerotic plaque inflammatory milieu.

Eight weeks old Lyz2cre/cre:Apoe−/− and Cited2fl/fl:Lyz2cre/cre:Apoe−/− mice were fed on a western-style high-fat diet or control diet for 20 weeks. (A) Atherosclerotic plaque macrophages were obtained by using anti-F4/80 microbeads (n=5). Total RNA samples derived from these macrophages were evaluated for expression of Irf1, Il1a, Il1b, Ccl5, Plau, F3, Mmp13, and Cxcl2 by RT-qPCR analyses. 36B4 was used as a housekeeping gene. (B) Blood plasma samples from these mice were assessed for IL1α, IL1β, TNF, and IL6 levels by using ELISA kits (n=5). Data were analyzed by two-way AVOVA. *P < 0.05, **P < 0.01 and ***P <0.001.
CITED2 deficiency boosts STAT1 mediated IRF1 expression in macrophages
Our studies thus far have demonstrated that CITED2 deficiency elevates IRF1 target gene expression in cultured as well as atherosclerotic plaque macrophages (Fig.1–5 and 8). Therefore, we examined whether CITED2 deficiency affects IFNγ-induced IRF1 protein expression in macrophages. Accordingly, Lyz2cre/cre and Cited2fl/fl:Lyz2cre/cre mice BMDMs were stimulated with IFNγ and IRF1 protein levels were evaluated by western blot analyses. As shown in figure-9A, CITED2 deficient BMDMs exposure to IFNγ displayed a robust and substantial increase in IRF1 protein expression compared to Lyz2cre/cre mice BMDMs (Fig.9A). Prior reports have established that TLR4 and IFNγ signaling utilize STAT1 to induce IRF1 expression (19). Therefore, we hypothesized that CITED2 deficiency enhances STAT1 activation in macrophages. To test this notion, Lyz2cre/cre, and Cited2fl/fl:Lyz2cre/cre mice BMDMs were stimulated with IFNγ, and total protein extracts were evaluated for expression and phosphorylation status of STAT1 (Fig.9B). Densitometry analyses of phospho-STAT1 western blots show that CITED2 deficiency did not alter IFNγ-induced phosphorylation (Tyr-701) or expression of STAT1 in macrophages (Fig.9C). Our earlier data (Fig.1 and 3) show that CITED2 regulates STAT1 and IRF1 functions. Therefore, we examined whether altering CITED2 levels affects IFNγ-induced STAT1 transcriptional activity in macrophages. Accordingly, RAW264.7 cells were cotransfected with STAT1 cis element-driven luciferase reporter plasmid in the presence of Cited2 specific siRNA or pCMV-Cited2 plasmid. These cells were stimulated with IFNγ, and luciferase activities were recorded. Our analyses show that CITED2 knockdown augments and CITED2 overexpression attenuates IFNγ-induced STAT1 luciferase reporter activity in macrophages (Fig.9D and E). Collectively, these results indicate that CITED2 restrains STAT1 functions without modulating the phosphorylation status of STAT1. Previous studies show that transcriptional activation domains (TADs) of the STAT1 directly interact with the transcriptional adaptor zinc-binding 1 (TAZ1) domain of CBP/p300 to initiate IFNγ-induced transcription (28, 29). The CITED2 TAD domain binds to the TAZ1 domain of CBP/p300 with very high affinity (30). This high-affinity CITED2 binding could potentially displace STAT1 interaction with coactivator CBP/p300 and subsequently eject STAT1 from the transcriptional complex. Therefore, we evaluated whether CITED2 deficiency alters IFNγ-induced STAT1 enrichment on target gene promoter. Accordingly, Lyz2 cre/cre and Cited2fl/fl:Lyz2 cre/cre mice BMDMs were stimulated with IFNγ, and chromatin immunoprecipitation (ChIP) was performed using anti-STAT1 and anti-CITED2 antibodies (Fig.9F). The ChIP samples were analyzed for STAT1 and CITED2 enrichment on the IRF1 promoter (−63 to −214) region by using site-specific primers. Our analyses show that CITED2 deficiency significantly enhances IFNγ-induced STAT1 enrichment on Irf1 promoter (Fig.9F). The IFNγ challenge also elevated CITED2 recruitment on IRF1 promoter (−63 to −214) in Lyz2 cre/cre mice BMDMs, and these effects were not observed in Cited2fl/fl:Lyz2 cre/cre mice BMDMs (Fig.9F). Collectively, our analyses show that CITED2 deficiency enhances STAT1 enrichment on IRF1 promoter and elevates IRF1 mRNA/protein expression in macrophages.
Figure 9. CITED2 restrains IFNγ-induced STAT1-IRF1 signaling in macrophages.

Lyz2cre/cre and Cited2fl/fl:Lyz2cre/cre mice BMDMs were stimulated with 10 ng/ml IFNγ. (A-C) Total protein samples were analyzed for the expression of IRF1 (A), phospho-STAT1, and total STAT1 (B) by western blot. (n=3). The phospho-STAT1 levels were quantified by densitometry analyses (C). (D and E) RAW264.7 cells were transfected with a STAT1-driven luciferase reporter construct in the presence of Cited2-specific siRNA (D) or pCMV6-Cited2 plasmid (E). These cells were stimulated with 10 ng/ml of IFNγ for 18 hours, and cell lysates were analyzed for luciferase activity. (n=3). (F) Lyz2cre/cre and Cited2fl/fl:Lyz2cre/cre mice BMDMs were stimulated with 10 ng/ml of IFNγ for one hour, and ChIP analysis was performed on Irf1 promoter (−63 to −214) utilizing anti-STAT1, anti-CITED2 antibodies (n=3). ChIP analyses performed using isotype IgG were used as a negative control. (G) Lyz2cre/cre and Cited2fl/fl:Lyz2cre/cre mice BMDMs were transfected with control or Irf1-specific siRNA. These cells were stimulated with 10 ng/mL of IFNγ for 3 hours. Total RNA samples from these experiments were analyzed for expression of Tmem140, Dnase1l3, F3, and Irg1 by RT-qPCR (n=3). Actin and 36B4 were used as housekeeping genes for western blot and RT-qPCR analyses, respectively. Values are reported as mean ± SD. The box plot represents the median with first and third quartiles, and whiskers represent minimum/maximum. Data were analyzed by ANOVA followed by Bonferroni post-testing. NS not significant; *P < 0.05; **P < 0.01; ***P < 0.001
Inhibition of IRF1 abates inflammatory response in CITED2-deficient macrophages
Our analyses so far show that CITED2 deficiency elevates IFNγ-induced IRF1 mRNA/protein expression and attendant pro-inflammatory gene expression in macrophages. Therefore, we assessed whether elevated pro-inflammatory gene expression in CITED2-deficient macrophages are IRF1 dependent. We employed siRNA knockdown approaches to attenuate IRF1 expression in primary macrophages. Accordingly, Lyz2 cre/cre and Cited2fl/fl:Lyz2 cre/cre mice BMDMs were transfected with IRF1-specific siRNA. These BMDMs were stimulated with IFNγ, and classical IRF1 target gene expression was analyzed by RT-qPCR (Fig.9G). As anticipated, IFNγ challenge dramatically elevated the IRF1 target gene (Tmem140, Dnase1l3, F3, and Irg1) expression in CITED2-deficient macrophages (Fig.9G). Interestingly, loss of functional IRF1 signaling was sufficient to attenuate heightened Tmem140, Dnase1l3, F3, and Irg1 expression in CITED2 deficient macrophages (Fig.9G). Collectively, these results demonstrate that CITED2 limits pro-inflammatory gene programs by restraining STAT1-IRF1 signaling axis in macrophages (Fig.10).
Figure 10. CITED2 alleviates inflammatory disease pathogenesis.
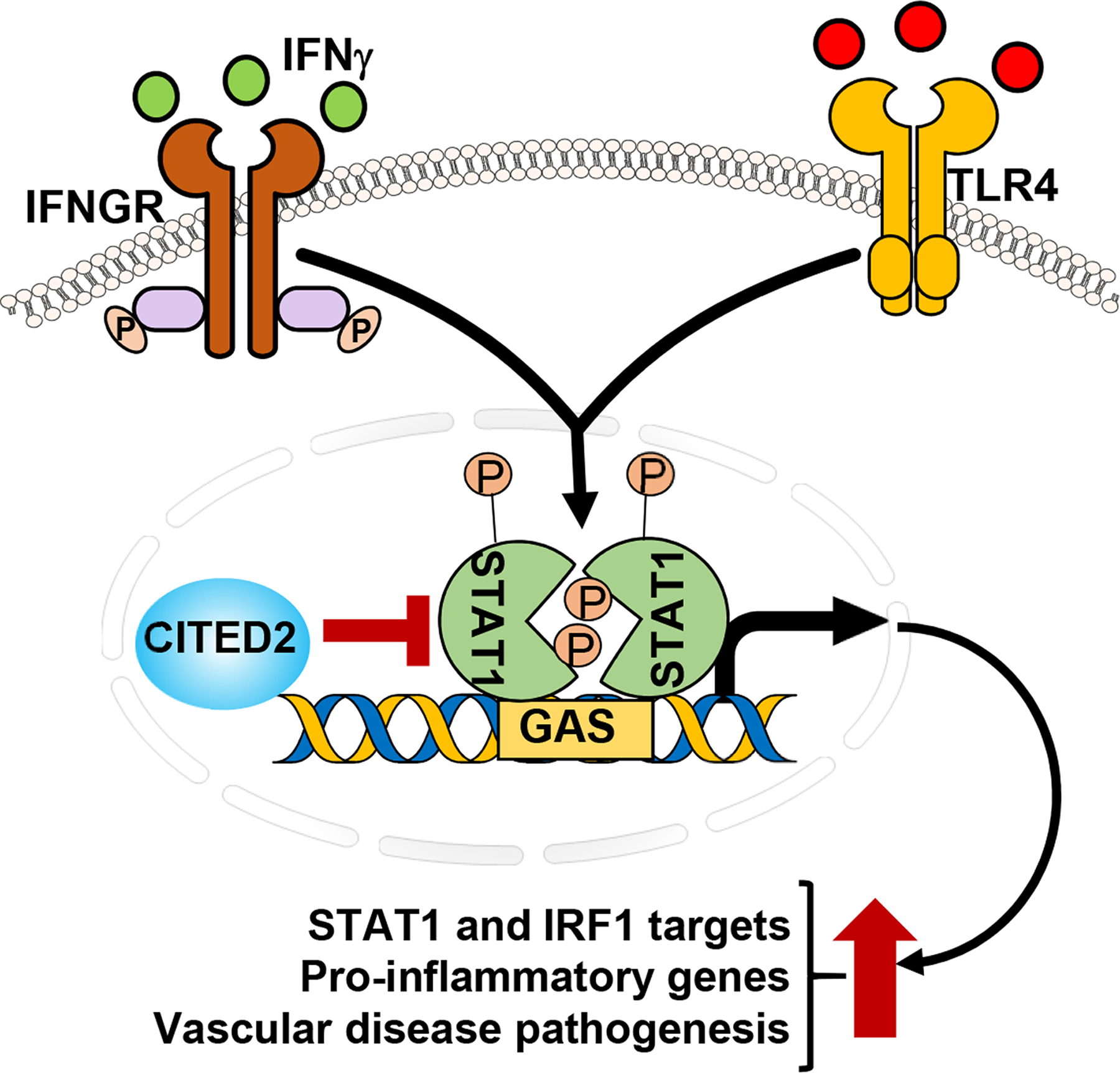
CITED2 attenuates inflammatory agent-induced STAT1-IRF1 signaling axis, pro-inflammatory gene expression, and inflammatory vascular disease pathogenesis in vivo.
Discussion
The principal findings of this study are as follows: (i) CITED2 deficiency augments TLR4 mediated IRFs target gene expression in macrophages, (ii) CITED2 deficiency boosts LPS-induced IRF1-regulated pro-inflammatory gene expression in macrophages, (iii) CITED2 deficiency heightens IFNγ-induced STAT1 and IRF1 target gene expression in macrophages, (iv) CITED2 deficient macrophages are hyperresponsive to IFNγ-challenge, (v) Myeloid-CITED2 deficiency exacerbates atherogenesis in vivo, (vi) Myeloid-CITED2 deficiency enhances macrophage abundance, and atherosclerotic lesion development in atheroprone aortic sinus regions, (vii) myeloid-CITED2 deficiency elevates IRF1-regulated pro-inflammatory gene expression in atherosclerotic plaque macrophages and establishes systemic pro-inflammatory milieu, (viii) CITED2 deficiency promotes STAT1-regulated IRF1 expression in macrophages, (ix) CITED2 deficiency elevates IFNγ-induced STAT1 enrichment on Irf1 promoter, and (x) IRF1 knockdown reverts elevated pro-inflammatory gene expression in macrophages. Overall, our study findings show that CITED2 curtails STAT1-IRF1 axis-mediated inflammatory gene expression and curb diet-induced atherogenesis in vivo (Fig.10).
Macrophages are the principal component of the innate immune system. Macrophage inflammatory response primarily depends on the tissue microenvironment that dictates their functional phenotype (31). Macrophages that are exposed to foreign agents such as inflammatory cytokines, bacterial endotoxins or DNA, robustly elevate transcriptional machinery to elicit an effective inflammatory response (31). Multiple negative regulatory mechanisms must exist in the cellular system to constrain inflammatory response in a spatiotemporal manner and curtail unwanted activation of macrophages in the resting stage to maintain the quiescence state. In this context, our previous studies have demonstrated that CITED2 serves as a cell-intrinsic negative regulator of inflammatory gene expression in macrophages and neutrophils (15). Specifically, our prior studies have shown that CITED2 deficiency elevates LPS-induced NFκB and HIF1α transcriptional activity in macrophages (14, 15). Further, we have also documented that CITED2 deficiency elevates NFκB or HIF1α dependent pro-inflammatory gene expression in innate immune cells (14, 15). In addition, our earlier studies have also shown that CITED2 interacts with PPARγ to promote IL4-induced anti-inflammatory gene expression in macrophages (14). Loss of functional CITED2 dramatically attenuated IL4 or rosiglitazone-induced PPARγ transcriptional activity and target gene expression in macrophages (14). These observations provide strong evidence that macrophages utilize CITED2 as a cell-intrinsic negative regulator inflammatory response. At the transcriptional level, it is also important to note that CITED2 interacts with a wide variety of transcription factors/modulators such as peroxisome proliferator-activated receptor alpha/gamma (PPARα/γ) (14), hepatocyte nuclear factor 4 alpha (HNF4α) (32), hypoxia-inducible factor 1 alpha (HIF1α) (33), histone deacetylase 1 (HDAC1) and estrogen receptor (ER) (34), and thereby affecting gene expression. In this context, our previous gene expression profiling studies have indicated that CITED2 deficiency elevates IFNγ as well as IFNα response in macrophages (15). These observations provided the initial notion that CITED2 could regulate IFNγ-induced inflammatory response in macrophages. It is well established that IFNγ utilizes STAT1-IRFs signaling to facilitate pro-inflammatory gene expression in macrophages. In this study, our GSEA of transcriptomics studies have shown that CITED2 deficiency significantly elevates IRF1, IRF2, and STAT3 target gene expression in macrophages. Further, our additional ex vivo analyses also show that the LPS challenge robustly elevated a large number of classical IRF1 targets in CITED2 deficient macrophages. Broadly, these results indicate that CITED2 restrains TLR4 mediated IRF1 pro-inflammatory target gene expression in macrophages.
IFNγ is a host-derived potent pro-inflammatory agent that essentially regulates all components of inflammatory disease pathogenesis (21). The binding of IFNγ to its cell surface receptors triggers tyrosine phosphorylation of STAT1, which translocates into the nucleus, and binds to the gamma interferon activation site (GAS) sequences on the target gene promoter (19). This results in the induction of a large number of inflammatory genes, including IRFs in target cells (20). The activated STAT1 could engage with IRF1 to maintain the heightened expression of inflammatory genes in macrophages. Our previous studies have implicated that CITED2 deficiency could elevate IFNγ/IFNα signaling in macrophages (15). However, whether CITED2 regulates IFNγ-induced STAT1, or IRF1 functions has never been addressed. In this context, our studies observed that CITED2 deficiency did not affect basal or IFNγ-induced STAT1 expression or STAT1 tyrosine phosphorylation in macrophages. Instead, we have identified that CITED2 deficiency elevates STAT1 mediated IRF1 mRNA/protein expression in macrophages. This resulted in an overall increase in IRF1-regulated inflammatory gene expression in CITED2 deficient macrophages. Prior biochemical studies have shown that transcriptional activation domains (TADs) of the STAT1 directly interact with the transcriptional adaptor zinc-binding 1 (TAZ1) domain of CBP/p300 to initiate the effective target gene transcription (28, 29). However, the CITED2 TAD domain also binds to the TAZ1 domain of p300/CBP with a very high affinity (30). This results in the displacement of STAT1 interaction with CPB/p300 and rapid attenuation of target gene expression. However, whether CITED2 deficiency affects STAT1 enrichment on target gene promoters in primary macrophages has not been previously reported. In this regard, our current report shows that the IFNγ challenge elevates CITED2 recruitment on IRF1 promoter and restrained STAT1 recruitment. Thus, CITED2 deficiency resulted in robust and sustained recruitment of STAT1 on the IRF1 promoter. This resulted in a substantive increase in STAT1 and IRF1 regulated inflammatory gene expression in CITED2 deficient macrophages. Thus, we postulated that CITED2 suppresses IFNγ-induced STAT1-IRF1 signaling axis in macrophages. Further, IFNγ signaling could also synergize with TLR-signaling to increase the expression and stability of mRNAs encoding for inflammatory mediators. Thus, our observations might be applicable to other inflammatory disease conditions where IFNγ and TLR signaling are operative in disease pathogenesis. Previous studies have shown that CITED2 interacts with HDAC1 following TGF-β stimulation and potentiate MYC-HDAC1 complex formation (35). However, MYC expression in macrophages are associated with anti-inflammatory gene expression (36). Pro-inflammatory cytokine exposure significantly attenuated MYC protein expression in macrophages. It is possible that anti-inflammatory cytokine exposure could induce CITED2 interaction with HDAC1 to modulate anti-inflammatory gene expression in macrophages. Further studies are needed to clarify this concept.
Earlier studies have implicated a role for IFNγ-signaling in atherogenesis (25). Immune cells within the atherosclerotic plaques produce a copious amount of IFNγ. Atherogenic effects of IFNγ have been demonstrated in murine models where exogenous administration of IFNγ enhances atherosclerotic plaque formation (25) while genetic deficiency of IFNγ reduced plaque size (37). IFNγ within the plaques affects many cell types, including macrophages, T-cells, endothelial cells, and smooth muscle cells (38). IFNγ exposure elevates cell surface adhesion molecules in leukocytes as well as endothelial cells that facilitate leukocyte rolling, binding to endothelial cells, and extravasation of leukocytes (38). IFNγ also contributes to the destabilization of mature lesions by blocking the proliferation of smooth muscle cells and collagen synthesis while elevating matrix metalloproteinase synthesis (38). In this context, our analyses show that CITED2 deficiency robustly elevates IFNγ-induced pro-inflammatory gene expression in macrophages. Further, myeloid-CITED2 deficiency significantly elevated basal and diet-induced aortic surface atherosclerotic lesions formation in vivo. Similarly, heightened macrophage abundance and atherosclerotic lesion formations were observed in aortic sinuses of myeloid-CITED2 deficient mice.
It is important to note that IFNγ and TLR4 ligands exert pro-inflammatory effects through STAT1 activation (19). Recent reports have shown that STAT1 acts as a central nexus point for the integration of TLR4 and IFNγ signaling to promote pro-atherogenic responses in human atherosclerosis (39). Prior studies have shown that STAT1 deficiency attenuates aortic root and aortic surface atherosclerotic lesions formation in vivo (22). Interestingly, STAT1 also interacts with IRF1 to facilitate the expression of pro-inflammatory gene targets. However, recent studies have also demonstrated that IRF1 deficiency is protective against diet-induced atherogenesis (23). In this regard, our analyses show that the number of IRF1 regulated pro-inflammatory target genes are significantly elevated in myeloid-CITED2 deficient mice atherosclerotic plaques. Interestingly, our analyses also show that some of the non-IRF1 inflammatory target gene expression were not significantly altered in these atherosclerotic plaque macrophages (data not shown). Therefore, our future studies will evaluate whether genetic or pharmacological inhibition of IRF1 in CITED2 deficient could reverse elevated atherosclerotic plaque formation in vivo. In addition, our studies have not identified any specific mechanisms for increased macrophage numbers that are observed in CITED2 deficient mice atherosclerotic plaques. Number of processes such as increased systemic inflammation, cellular migration, proliferation, survival, and reduced apoptosis and cellular clearance could account for these differences. CITED2 deficiency also augmented STAT1 and IRF1 target gene expression following inflammatory challenge. At the molecular level, CITED2 restrains STAT1 recruitment on the IRF1 promoter and attenuates IRF1 mRNA/protein expression in macrophages. Indeed, inhibition of IRF1 reversed elevated pro-inflammatory gene targets in CITED2 deficient macrophages. Collectively, our current studies show that CITED2 restrains STAT1-IRF1 signaling in macrophages and alleviates diet-induced atherogenesis.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant HL126626, HL141423, and Crohn’s and Colitis Foundation Senior Research Award 421904 (to G. H. M.). The National Health and Medical Research Council Fellowship ID1135886 (to S. L. D.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the National Health and Medical Research Council.
Abbreviations
- CITED2
CBP/p300-interacting transactivator with glutamic acid/aspartic acid-rich carboxyl-terminal domain 2
- BMDMs
Bone marrow-derived macrophages
- STAT1
Signal transducer and activator of transcription 1
- IRF1
Interferon regulatory factor 1
- LPS
Lipopolysaccharides
- IFNγ
Interferon-gamma
- TLRs
Toll-like receptors
- NFκB
Nuclear factor-kappa B
- GSEA
Gene set enrichment analysis
- TFT
Transcription factor targets
- HFD
High-fat diet
- TADs
Transcriptional activation domains
- TAZ1
Transcriptional adaptor zinc-binding 1
- GAS
Gamma interferon activation site
- FWER
Family-wise error rate
Footnotes
Conflict of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Koh TJ, and DiPietro LA (2011) Inflammation and wound healing: the role of the macrophage. Expert reviews in molecular medicine 13, e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laskin DL, Sunil VR, Gardner CR, and Laskin JD (2011) Macrophages and tissue injury: agents of defense or destruction? Annual review of pharmacology and toxicology 51, 267–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takeuchi O, and Akira S (2010) Pattern recognition receptors and inflammation. Cell 140, 805–820 [DOI] [PubMed] [Google Scholar]
- 4.Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, and Gordon S (2005) Macrophage receptors and immune recognition. Annual review of immunology 23, 901–944 [DOI] [PubMed] [Google Scholar]
- 5.Murray PJ, and Wynn TA (2011) Protective and pathogenic functions of macrophage subsets. Nature reviews. Immunology 11, 723–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pedraza-Alva G, Pérez-Martínez L, Valdez-Hernández L, Meza-Sosa KF, and Ando-Kuri M (2015) Negative regulation of the inflammasome: keeping inflammation under control. Immunological reviews 265, 231–257 [DOI] [PubMed] [Google Scholar]
- 7.Sun HB, Zhu YX, Yin T, Sledge G, and Yang YC (1998) MRG1, the product of a melanocyte-specific gene related gene, is a cytokine-inducible transcription factor with transformation activity. Proceedings of the National Academy of Sciences of the United States of America 95, 13555–13560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dunwoodie SL, Rodriguez TA, and Beddington RS (1998) Msg1 and Mrg1, founding members of a gene family, show distinct patterns of gene expression during mouse embryogenesis. Mechanisms of development 72, 27–40 [DOI] [PubMed] [Google Scholar]
- 9.Sperling S, Grimm CH, Dunkel I, Mebus S, Sperling HP, Ebner A, Galli R, Lehrach H, Fusch C, Berger F, and Hammer S (2005) Identification and functional analysis of CITED2 mutations in patients with congenital heart defects. Human mutation 26, 575–582 [DOI] [PubMed] [Google Scholar]
- 10.Weninger WJ, Lopes Floro K, Bennett MB, Withington SL, Preis JI, Barbera JP, Mohun TJ, and Dunwoodie SL (2005) Cited2 is required both for heart morphogenesis and establishment of the left-right axis in mouse development. Development (Cambridge, England) 132, 1337–1348 [DOI] [PubMed] [Google Scholar]
- 11.Withington SL, Scott AN, Saunders DN, Lopes Floro K, Preis JI, Michalicek J, Maclean K, Sparrow DB, Barbera JP, and Dunwoodie SL (2006) Loss of Cited2 affects trophoblast formation and vascularization of the mouse placenta. Developmental biology 294, 67–82 [DOI] [PubMed] [Google Scholar]
- 12.Kranc KR, Schepers H, Rodrigues NP, Bamforth S, Villadsen E, Ferry H, Bouriez-Jones T, Sigvardsson M, Bhattacharya S, Jacobsen SE, and Enver T (2009) Cited2 is an essential regulator of adult hematopoietic stem cells. Cell stem cell 5, 659–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du J, Chen Y, Li Q, Han X, Cheng C, Wang Z, Danielpour D, Dunwoodie SL, Bunting KD, and Yang YC (2012) HIF-1α deletion partially rescues defects of hematopoietic stem cell quiescence caused by Cited2 deficiency. Blood 119, 2789–2798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim GD, Das R, Rao X, Zhong J, Deiuliis JA, Ramirez-Bergeron DL, Rajagopalan S, and Mahabeleshwar GH (2018) CITED2 Restrains Proinflammatory Macrophage Activation and Response. Molecular and cellular biology 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pong Ng H, Kim GD, Ricky Chan E, Dunwoodie SL, and Mahabeleshwar GH (2020) CITED2 limits pathogenic inflammatory gene programs in myeloid cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 34, 12100–12113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fitzgerald KA, and Kagan JC (2020) Toll-like Receptors and the Control of Immunity. Cell 180, 1044–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langlais D, Barreiro LB, and Gros P (2016) The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. The Journal of experimental medicine 213, 585–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sikorski K, Chmielewski S, Olejnik A, Wesoly JZ, Heemann U, Baumann M, and Bluyssen H (2012) STAT1 as a central mediator of IFNγ and TLR4 signal integration in vascular dysfunction. Jak-stat 1, 241–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Semper C, Leitner NR, Lassnig C, Parrini M, Mahlakõiv T, Rammerstorfer M, Lorenz K, Rigler D, Müller S, Kolbe T, Vogl C, Rülicke T, Staeheli P, Decker T, Müller M, and Strobl B (2014) STAT1β is not dominant negative and is capable of contributing to gamma interferon-dependent innate immunity. Molecular and cellular biology 34, 2235–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boehm U, Klamp T, Groot M, and Howard JC (1997) Cellular responses to interferon-gamma. Annual review of immunology 15, 749–795 [DOI] [PubMed] [Google Scholar]
- 22.Agrawal S, Febbraio M, Podrez E, Cathcart MK, Stark GR, and Chisolm GM (2007) Signal transducer and activator of transcription 1 is required for optimal foam cell formation and atherosclerotic lesion development. Circulation 115, 2939–2947 [DOI] [PubMed] [Google Scholar]
- 23.Du M, Wang X, Mao X, Yang L, Huang K, Zhang F, Wang Y, Luo X, Wang C, Peng J, Liang M, Huang D, and Huang K (2019) Absence of Interferon Regulatory Factor 1 Protects Against Atherosclerosis in Apolipoprotein E-Deficient Mice. Theranostics 9, 4688–4703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, and Arditi M (2004) Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proceedings of the National Academy of Sciences of the United States of America 101, 10679–10684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whitman SC, Ravisankar P, Elam H, and Daugherty A (2000) Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E−/− mice. The American journal of pathology 157, 1819–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hopkins PN (2013) Molecular biology of atherosclerosis. Physiological reviews 93, 1317–1542 [DOI] [PubMed] [Google Scholar]
- 27.Moore KJ, Sheedy FJ, and Fisher EA (2013) Macrophages in atherosclerosis: a dynamic balance. Nature reviews. Immunology 13, 709–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang JJ, Vinkemeier U, Gu W, Chakravarti D, Horvath CM, and Darnell JE Jr. (1996) Two contact regions between Stat1 and CBP/p300 in interferon gamma signaling. Proceedings of the National Academy of Sciences of the United States of America 93, 15092–15096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wojciak JM, Martinez-Yamout MA, Dyson HJ, and Wright PE (2009) Structural basis for recruitment of CBP/p300 coactivators by STAT1 and STAT2 transactivation domains. The EMBO journal 28, 948–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Guzman RN, Martinez-Yamout MA, Dyson HJ, and Wright PE (2004) Interaction of the TAZ1 domain of the CREB-binding protein with the activation domain of CITED2: regulation by competition between intrinsically unstructured ligands for non-identical binding sites. The Journal of biological chemistry 279, 3042–3049 [DOI] [PubMed] [Google Scholar]
- 31.Molawi K, and Sieweke MH (2013) Transcriptional control of macrophage identity, self-renewal, and function. Advances in immunology 120, 269–300 [DOI] [PubMed] [Google Scholar]
- 32.Qu X, Lam E, Doughman YQ, Chen Y, Chou YT, Lam M, Turakhia M, Dunwoodie SL, Watanabe M, Xu B, Duncan SA, and Yang YC (2007) Cited2, a coactivator of HNF4alpha, is essential for liver development. The EMBO journal 26, 4445–4456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhattacharya S, Michels CL, Leung MK, Arany ZP, Kung AL, and Livingston DM (1999) Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes & development 13, 64–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lau WM, Doucet M, Huang D, Weber KL, and Kominsky SL (2013) CITED2 modulates estrogen receptor transcriptional activity in breast cancer cells. Biochemical and biophysical research communications 437, 261–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chou YT, Hsieh CH, Chiou SH, Hsu CF, Kao YR, Lee CC, Chung CH, Wang YH, Hsu HS, Pang ST, Shieh YS, and Wu CW (2012) CITED2 functions as a molecular switch of cytokine-induced proliferation and quiescence. Cell death and differentiation 19, 2015–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pello OM, De Pizzol M, Mirolo M, Soucek L, Zammataro L, Amabile A, Doni A, Nebuloni M, Swigart LB, Evan GI, Mantovani A, and Locati M (2012) Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood 119, 411–421 [DOI] [PubMed] [Google Scholar]
- 37.Whitman SC, Ravisankar P, and Daugherty A (2002) IFN-gamma deficiency exerts gender-specific effects on atherogenesis in apolipoprotein E−/− mice. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research 22, 661–670 [DOI] [PubMed] [Google Scholar]
- 38.Boshuizen MC, and de Winther MP (2015) Interferons as Essential Modulators of Atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology 35, 1579–1588 [DOI] [PubMed] [Google Scholar]
- 39.Chmielewski S, Olejnik A, Sikorski K, Pelisek J, Błaszczyk K, Aoqui C, Nowicka H, Zernecke A, Heemann U, Wesoly J, Baumann M, and Bluyssen HA (2014) STAT1-dependent signal integration between IFNγ and TLR4 in vascular cells reflect pro-atherogenic responses in human atherosclerosis. PloS one 9, e113318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.