

Abstract
Chronic lung allograft dysfunction (CLAD) remains the major complication limiting long-term survival among lung transplant recipients (LTRs). Limited understanding of CLAD immunopathogenesis and a paucity of biomarkers remain substantial barriers for earlier detection and therapeutic interventions for CLAD. We hypothesized the airway transcriptome would reflect key immunologic changes in disease. We compared airway brush-derived transcriptomic signatures in CLAD (n = 24) versus non-CLAD (n = 21) LTRs. A targeted assessment of the proteome using concomitant bronchoalveolar lavage (BAL) fluid for 24 cytokines/chemokines and alloimmune T cell responses was performed to validate the airway transcriptome. We observed an airway transcriptomic signature of differential genes expressed (DGEs) in CLAD marked by Type-1 immunity and striking upregulation of two endogenous immune regulators: indoleamine 2, 3 dioxygenase 1 (IDO-1) and tumor necrosis factor receptor superfamily 6B (TNFRSF6B). Advanced CLAD staging was associated with a more intense airway transcriptome signature. In a validation cohort using the identified signature, we found an area under the curve (AUC) of 0.77 for CLAD LTRs. Targeted proteomic analyses revealed a predominant Type-1 profile with detection of IFN-γ, TNF-α, and IL-1β as dominant CLAD cytokines, correlating with the airway transcriptome. The airway transcriptome provides novel insights into CLAD immunopathogenesis and biomarkers that may impact diagnosis of CLAD.
Keywords: biomarker, bronchiolitis obliterans (BOS), clinical research, practice, immune regulation, immunobiology, lung (allograft) function, dysfunction, lung transplantation, pulmonology, molecular biology: mRNA, mRNA expression
1 |. INTRODUCTION
Lung transplantation is the final therapeutic option for select patients with end-stage lung disease. However, chronic lung allograft dysfunction (CLAD) is the major complication limiting long-term survival in lung transplant recipients (LTRs), resulting in a median 5-year survival of only 55% which substantially lags other solid organ transplants.1,2 Recent studies have identified two major clinical endotypes of CLAD: the bronchiolitis obliterans syndrome (BOS; obstructive CLAD) and restrictive allograft syndrome (RAS; rCLAD), with the latter portending a worse prognosis.3,4 CLAD is less commonly diagnosed by biopsy, rather using spirometry and the exclusion of other pathologies and generally responds poorly to augmentation of conventional immunosuppressive therapy.5 Moreover, the immunopathogenesis of CLAD and its clinical endotypes remain poorly understood, hence there is a major need in the lung transplant field to identify novel early molecular pathways and potential therapeutic targets to diagnose and potentially treat early CLAD.
Previous studies have explored molecular signatures of acute cellular rejection (ACR) in LTRs. Gregson et al. examined exosomal RNA profiles in BAL from LTRs in the setting of ACR.6 They predicted activation of IFN-γ and HLA-I based on the ACR profile observed. Weigt et al. conducted a proof of concept study investigating the utility of BAL gene expression in diagnosing ACR.7 They identified four genes (TOX,SAMD3,IL32, and KLRK1) related to T cell activation that classified ACR. Halloran et al. also evaluated ACR in LTRs, using lung biopsies to evaluate gene expression.8 They identified a group of genes related to macrophage activation including ADAMDEC1 that were associated with T cell–mediated rejection. Sacreas et al used the common rejection model to evaluate CLAD in lung transplant patients in a targeted fashion.9 Airway cytology brushes have previously been used in asthma to classify disease by gene expression,10 and in lung transplant to evaluate the effects of azithromycin.11 No prior studies have used airway cytology brushes to evaluate CLAD in lung transplant. The objective of this study was to determine differences in the airway transcriptome in LTRs with and without CLAD using airway brush samples. We hypothesized the airway transcriptome would reflect key immunologic changes in disease.
2 |. MATERIALS AND METHODS
2.1 |. Study cohort
This was a single center, cross-sectional study of airway brush samples to investigate the airway transcriptome in CLAD using RNAseq analyses. The study was approved by the IRB of the University of Pittsburgh (PRO14110014). Patients were selected from lung transplant registry and biorepository, with enrolled LTRs since 2016, and with transbronchial brushes since 2017. All LTRs in the registry and bioreposity were eligible for inclusion, but patients were required to provide separate consent for brushing. Patients scheduled to undergo bronchoscopy who were clinically suspected of having CLAD were invited to participate, as well as stable non-CLAD control patients scheduled for bronchoscopy. A bronchial cytologic brush sample (ConMed) was collected for each LTR at the time of routine surveillance bronchoscopy (assessed every 3–4 months for first 2 years posttransplant) or subsequent for-cause biopsy. A single brush sample was collected for sequencing per patient. BAL supernatant and cell pellets were collected and stored when available.
LTRs were classified as CLAD or non-CLAD based on the 2019 International Society of Heart and Lung Transplantation definitions.12 Patients with CLAD phenotypes consistent with the BOS phenotype were included. CLAD staging based off fall in FEV1 was completed using this definition. A validation cohort of LTRs from a separate center (University of California, San Francisco) undergoing evaluation of airway brushes for the airway transcriptome in CLAD versus controls were assessed using the top 25 genes upregulated from the Pittsburgh cohort to generate a receiver operator characteristic (ROC) curve.
2.2 |. Airway brush processing and RNAseq analysis
Bronchial brushings were obtained from the fourth- to sixth generation airways, as previously described.13 Bronchial brushings were placed directly into RLT-plus (Qiagen, Valencia, CA) during the bronchoscopy to ensure minimal RNA degradation, after which samples were stored at −80°C until RNA isolation, using Qiagen RNeasy isolation kit. RNA was quantitated using nanodrop, and integrity was determined with a total RNA nano chip (Agilent Technologies).
Total RNA was isolated and stranded total RNA-seq libraries were prepared following ribosomal RNA depletion with Illumina reagents, and libraries were sequenced with an Illumina Nextseq500 sequencer with a depth of paired-end 40 million read pairs per sample. Fastq files with high quality reads (phred score >30) were uploaded to the CLC Genomics Workbench (Qiagen). Reads were aligned to hg38 human genome. Transcript counts and differential expression analysis were carried out using the CLC Genomics Workbench.
2.3 |. Flow cytometry
Flow cytometry was completed to characterize cellular composition of the airway brush samples. Cellular composition of airway brushes (n = 12 LTRs airway brushes) was assessed using anti-HLA-DR-FITC, CD3-AlexaFluor700, CD19-BV421, CD56-PE, CD4-APCCy7, CD8-V500, CD14-BV605, E-cadherin-APC (BioLegend, eBioscience). To assess in vitro short-term stimulation and ICS for alloreactive T cell responses, total BAL cells were cultured in the presence or absence of donor irradiated (3000 Rad, Gamma irradiator) PBMC or cell lysate. CFSE-labeled (2 μM) recipient BAL cells (responders) were incubated in medium alone, or in the presence of PKH-26-labeled donor cells (1:1 ratio) or lysate donor cells for 6 h. Cells were then surface stained with the fluorochrome-labeled antibodies anti-CD3-Alexa-Fluor700, anti-CD8-V500, anti-CD4-APC-Cy7 (BD Biosciences), and Live/Dead Fixable Blue Dead-Cell Stain (Invitrogen).14 ICS was performed using anti-IFN-γ-BV605, anti-TNF-α-PE-Cy7, anti-IL-2-BV650, anti-CD107a-Pacific Blue, anti-IL-13-BV421, and anti-IL-17a-APC (BD-Biosciences). All re-stimulations for ICS were performed using 106 cells for 6 h at 37°C with brefeldin-A (10 μg/mL) and monensin (5 μg/ml) with anti-CD107a-Pac-Blue added at the beginning of culture. All cells were collected for flow cytometric analysis using an LSR Fortessa-cytometer (BD Biosciences).15 Data analysis and graphic representations were done with FlowJo v.10 (TreeStar).
2.4 |. Cytokines and chemokines analysis by MSD assay
Cytokine and chemokine analysis were completed to validate RNAseq findings. A total of n = 36 LTRs (n = 19 CLAD and n = 17 non-CLAD) BAL supernatant samples were used to detect cytokines and chemokines using Meso Scale Discovery (MSD) U-PLEX multiplex assay platforms (Meso Scale Discovery, Rockville, MD).16,17 These BAL samples were obtained from the same bronchoscopy as the brush samples. We analyzed the cytokines: (Type 1) IFNγ, TNFα, IL-2, IL-12/IL-23p40, IL-15, IL-18, IL-27, IL-1β and (Type 2): IL-4, IL-5. IL-6. IL-10, IL-13, GM-CSF and IL-17A, IL-21, IL-22, and G-CSF, IL1α, IFN-α2a, IFNβ. We analyzed the following chemokines: CXCR3 family (MIG, IP-10, I-TAC) and CCR5 family (MIP-1α, MIP–1β, RANTES) and IL-8 (CXCL8). Limit of detection measured by MSD assay for the cytokines was between 0.08 and 9.6 pg/ml and for the chemokines was between 0.15 and 7.7 pg/ml.
2.5 |. Real-Time PCR
Real-Time PCR reactions were performed on the Bio-Rad-CFX96 system using TaqMan Fast Advanced Master Mix.18 The primers were human IDO1 (Hs00984148_m1), TNFRSF6B (Hs00187070_m1), and GAPDH (Hs03929097_g1) (Thermo-Fisher-Scientific).
2.6 |. Statistical analysis
For the airway brush RNAseq analysis, differential gene expression analysis between CLAD and non-CLAD subjects was completed using CLC Genomics Workbench version 20. Additional analyses of these results were completed using Bioconductor version 3.10 for R version 3.6.3 (R CoreTeam 2019, Vienna, Austria). Genes with a false discovery rate (FDR) P ≤ .05 and absolute log2-fold change >1.5 were included in upstream regulator and pathway analyses completed with Ingenuity Pathway Analysis (Illumina).
Top differentially expressed genes identified in the primary cohort were validated in a validation cohort from a separate institution using receiver operating characteristic (ROC) curve and area under the ROC curve (AUC).
For MSD, flow cytometry, and PCR assessments, statistical analysis was performed using Graph Pad Prism and SPSS version 22. The non-parametric tests of Wilcoxon signed-rank and Mann-Whitney U were used. A two-tailed P < .05 was considered statistically significant.
3 |. RESULTS
3.1 |. Airway brush study cohort in lung transplant recipients
Airway brushes from 52 LTRs were screened for inclusion. Of these, 45 (86.5%) had sufficient quantity and quality of RNA for sequencing (Figure 1A). Our cohort consisted 24 CLAD LTRs and 21 non-CLAD control LTRs, defined using ISHLT definitions. Demographics for both groups are shown in Table 1. The most common indication for transplant was interstitial lung disease, and the overall median age was 53. The groups were similar in age, sex, transplant indication, and acute rejection at the time of sampling. The CLAD group had a greater proportion of positive cultures at the time of sampling (52.4% in CLAD group vs. 0% in control group). Most positive cultures identified bacteria (10/13) with one viral and two fungal cultures also testing positive. The median time to brush sample was 1896 days in the CLAD group versus 536 days in the control group (P < .01). The median time to CLAD was 1645 days posttransplant. Most CLAD patients had CLAD Stage 1 (15/24, 62.5%), while nine had more advanced disease (37.5%). We analyzed 12 brushes from LTRs who were not included in this study to evaluate the cellular populations in brush samples and found that epithelial cells were the predominant population (59%), followed by monocyte/macrophages (30%), with lymphocytes and NK cells comprising the minority balance of cells (Figure 1B,C; Figure S1A).
FIGURE 1.
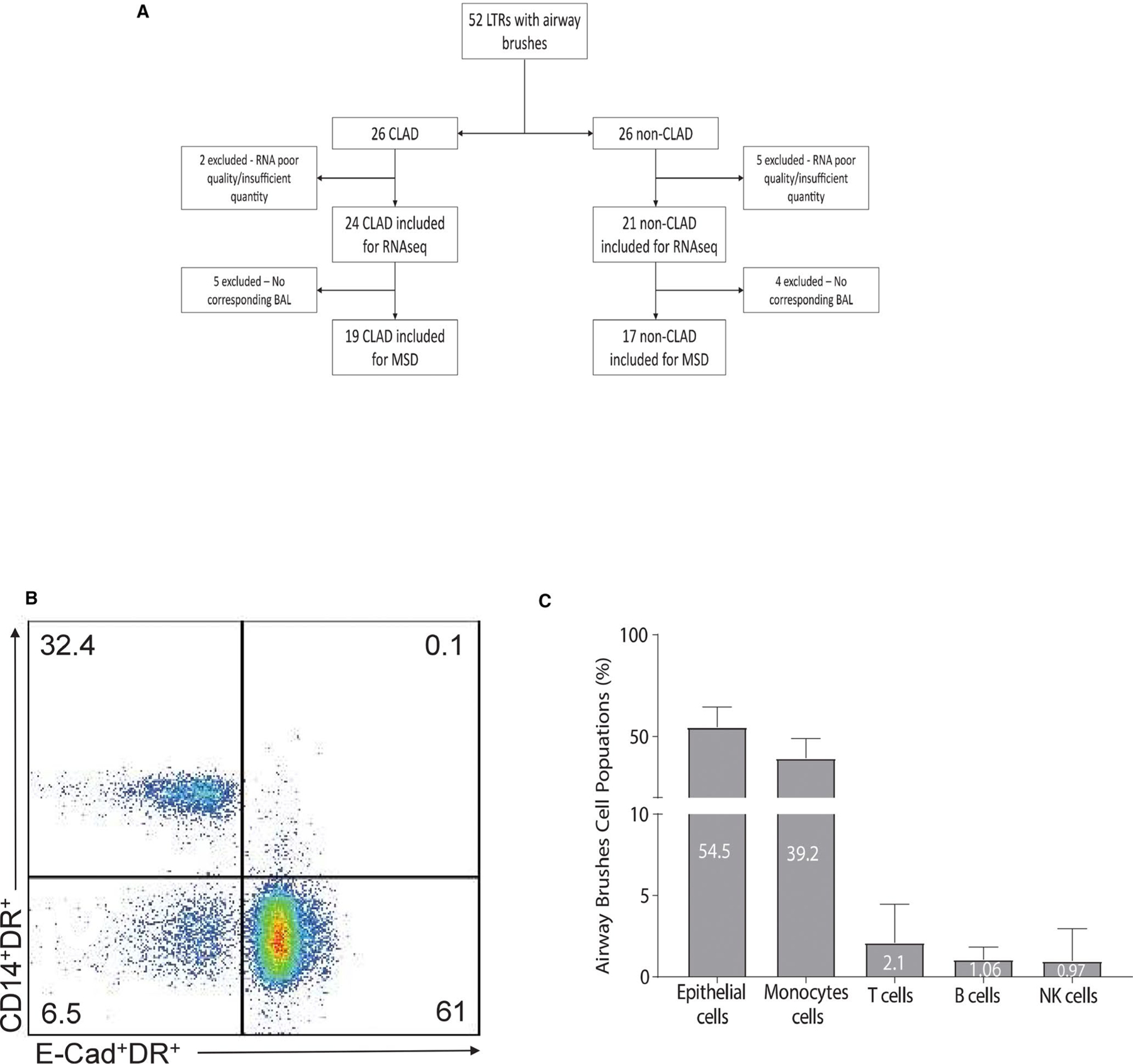
(A) Consort diagram showing included and excluded samples used in the study. The transcriptome analysis included 45 samples (24 CLAD, 21 non-CLAD) while the proteome analysis included 36 (19 CLAD, 17 non-CLAD) with corresponding bronchoalveolar lavage samples. (B) Representative flow cytometry plot showing the phenotype of airway brushes cells populations of the majority cells components such as the epithelial cells (Ecadherin+HLA-DR+) and monocytes (CD14+HLA-DR+). (C) Cumulative data from n = 12 LTRs of airway brushes cells populations phenotype. Values represent frequencies mean (%) of cells type such epithelial, monocytes, T, B, and NK cells. Statistical analysis was performed using Mann-Whitney U-test with a two-sided P-value of <.05 considered statistically significant
TABLE 1.
Patient characteristics
| CLAD (n = 24) | Control (n = 21) | P-value | |
|---|---|---|---|
| Age at transplant, median (IQR) | 52.5 (34.75–62.75) | 54 (37.0–63.5) | .59 |
| Female, n (%) | 10 (41.7) | 10 (47.6) | .16 |
| Transplant diagnosis, n (%) | .91 | ||
| Interstitial lung disease | 9 (37.5) | 7 (33.3) | |
| Cystic fibrosis | 7 (29.2) | 7 (33.3) | |
| Chronic obstructive pulmonary disease | 7 (29.2) | 6 (28.6) | |
| Retransplant | 1 (4.2) | 1 (4.8) | |
| Congenital | 1 (4.2) | 0 | |
| Cytomegalovirus serostatus | .44 | ||
| D+/R+ | 14 (36.4) | 8 (53.8) | |
| D−/R+ | 5 (22.7) | 5 (19.2) | |
| D+/R− | 5 (22.7) | 2 (7.7) | |
| D−/R− | 4 (18.2) | 5 (19.2) | |
| Anti-HLA donor-specific antibody | 6 (27.3) | 3 (11.5) | .27 |
| Acute rejection, n (%) | 9 (37.5) | 3 (14.3) | .11 |
| Acute infection, n (%) | 13 (52.4) | 0 | <.01 |
| Bacterial | 10 | — | |
| Viral | 1 | — | |
| Fungal | 2 | ||
| Days to CLAD, median (IQR) | 1645 (1133–1981) | — | |
| CLAD stage, n (%) | |||
| CLAD 1 | 15 (62.5) | — | |
| CLAD 2 | 3 (12.5) | — | |
| CLAD 3 | 6 (25.0) | — | |
| CLAD 4 | 0 |
3.2 |. Airway brush RNAseq analysis shows significant differential gene expression in the airway transcriptome between CLAD and non-CLAD LTRs
We constructed a pipeline for RNAseq analyses of airway brushes for our cohort (Figure S1B). We performed RNAseq analysis of endobronchial brush samples and found significant differential gene expression (DGE) between CLAD and controls, with a total of 2340 DGEs identified after adjustment for a false discovery rate (FDR <0.05). Of these, 513 had an absolute log2-fold change of >1.5, shown visually in the volcano plot (Figure 2A; full gene list Figure S2). The majority of DGEs were upregulated in the CLAD group. Figure 2B shows a heat map depicting differences in the visual comparison of the top 25 upregulated DGEs and the top 25 downregulated DGEs between the CLAD and non-CLAD groups. The top 25 upregulated genes are displayed in Table 2. We then assessed whether the airway transcriptome changed with CLAD progression. We found a significant intensification of the airway transcriptome signature for CLAD stage 2–3 compared to CLAD stage 1 (Figure 2C).
FIGURE 2.
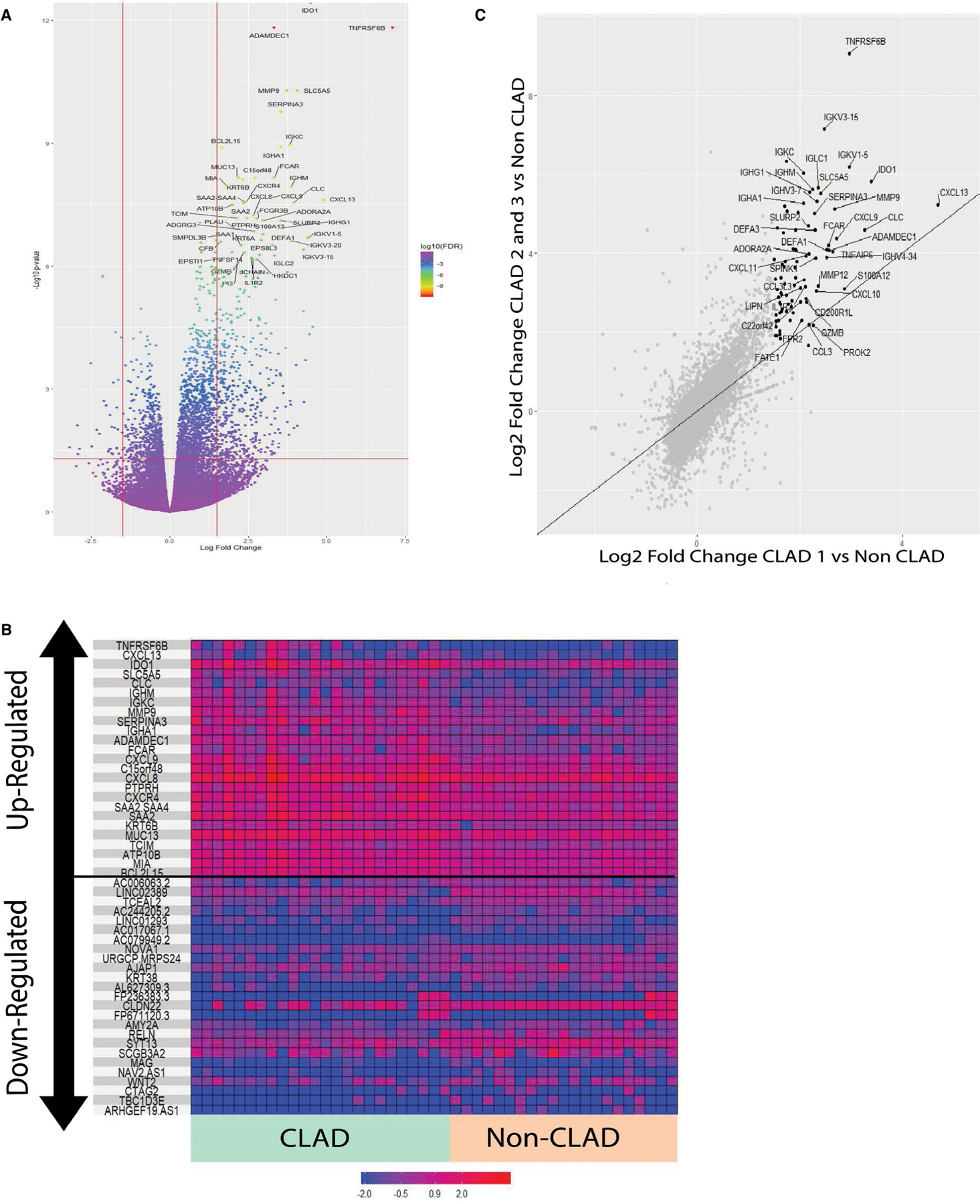
(A) Volcano plot of DGE analysis for CLAD versus non-CLAD LTRs. Genes meeting the threshold for inclusion are indicated in the top left and top right sections divided by the red lines. Top upregulated genes are labeled. Top upregulated genes are shown and labeled in black. (B) Heat map showing the top 25 upregulated and top 25 downregulated genes in the CLAD versus non-CLAD DGE analysis. (C) Scatter plot showing the Log2 fold change for gene expression in the CLAD stage 1 versus Non-CLAD comparison (x-axis) versus that of the CLAD stage 2 and 3 versus Non-CLAD comparison
TABLE 2.
Top 25 upregulated genes in CLAD versus non-CLAD
| CLAD versus non-CLAD | ||
|---|---|---|
| Gene | Fold change | FDR P-value |
| IDO1 | 22.32 | <1.00E−13 |
| ADAMDEC1 | 9.99 | 1.49E−12 |
| TNFRSF6B | 137.19 | 1.49E−12 |
| SLC5A5 | 16.68 | 5.10E−11 |
| MMP9 | 13.18 | 5.10E−11 |
| SERPINA3 | 11.63 | 1.69E−10 |
| IGKC | 14.03 | 1.10–09 |
| IGHA1 | 11.55 | 1.21E−09 |
| BCL2L15 | 3.18 | 1.28E−09 |
| MUC13 | 4.56 | 6.84E−09 |
| C15orf48 | 6.59 | 6.84E−09 |
| FCAR | 9.92 | 6.84E−09 |
| KRT6B | 5.03 | 7.54E−09 |
| MIA | 3.51 | 1.05E−08 |
| IGHM | 14.52 | 1.16E−08 |
| CXCL9 | 9.65 | 2.11E−08 |
| CXCL13 | 29.65 | 2.47E−08 |
| CXCR4 | 5.35 | 2.66E−08 |
| SAA2-SAA4 | 5.21 | 2.66E−08 |
| CLC | 15.35 | 2.88E−08 |
| SAA2 | 5.10 | 2.94E−08 |
| ATP10B | 4.00 | 3.18E−08 |
| CXCL8 | 6.41 | 5.68E−08 |
| TCIM | 4.41 | 6.73E−08 |
| PTPRH | 5.46 | 6.73E−08 |
Because of baseline differences in the rate of infection between CLAD and non-CLAD controls, infection was evaluated separately to characterize its influence on the CLAD group. Table 3 shows the DGE analyses for: (1) CLAD non-infected versus non-CLAD controls and (2) CLAD infected versus CLAD non-infected for the top 25 upregulated genes in the CLAD versus non-CLAD control comparison. DGE results for the CLAD non-infected versus non-CLAD comparison are similar to the overall results comparing CLAD versus non-CLAD. Unexpectedly, comparison of CLAD infected to CLAD non-infected showed no differences in the top 25 upregulated genes. Complete DGE analysis between CLAD non-infected and CLAD infected revealed no genes that overlapped with the DGE analysis for CLAD versus non-CLAD control at the same threshold of FDR P-value <.05 and absolute LFC >1.5. None of the 513 DGEs observed in the overall CLAD versus non-CLAD comparison were differentially expressed in the CLAD infected versus CLAD non-infected comparison. A similar approach was used to assess the effect of rejection (Figure S3). All top 25 genes remained significantly upregulated when removing ACR subjects from the analysis and comparing CLAD without ACR to non-CLAD without ACR. ADAMDEC1,MMP9,CXCL9,CXCR4,CLC,C15orf48, and CXCL8 were upregulated in CLAD with ACR versus CLAD without ACR. MMP9 and CXCL9 have been previously associated with allograft rejection.9,19,20
TABLE 3.
Differential gene analysis of top genes in those with or without infection
| CLAD no infection versus
non-CLAD |
CLAD infection versus CLAD no
infection |
|||
|---|---|---|---|---|
| Fold change | FDR P-value | Log fold change | FDR P-value | |
| IDO1 | 30.21 | <1.00E−13 | 1.51 | 1.00 |
| ADAMDEC1 | 6.02 | 1.79E−6 | 1.97 | 1.00 |
| TNFRSF6B | 243.46 | <1.00E−13 | 1.31 | 1.00 |
| SLC5A5 | 15.22 | 6.50E−8 | 2.74 | 1.00 |
| MMP9 | 6.35 | 4.16E−5 | 1.73 | 1.00 |
| SERPINA3 | 8.88 | 1.05E−7 | 2.59 | 1.00 |
| IGKC | 19.41 | 4.23E−9 | −1.64 | 1.00 |
| IGHA1 | 16.70 | 1.48E−9 | −1.44 | 1.00 |
| BCL2L15 | 3.59 | 6.50E−8 | 1.03 | 1.00 |
| MUC13 | 5.15 | 2.24E−7 | 1.06 | 1.00 |
| C15orf48 | 5.41 | 9.00E−8 | 2.22 | 1.00 |
| FCAR | 4.13 | 6.32E−3 | 4.06 | 1.00 |
| KRT6B | 4.89 | 2.44E−7 | 1.00 | 1.00 |
| MIA | 3.51 | 7.69E−7 | 1.53 | 1.00 |
| IGHM | 17.40 | 1.00E−7 | −1.44 | 1.00 |
| CXCL9 | 17.99 | 1.74E−10 | −1.38 | 1.00 |
| CXCL13 | 26.05 | 5.27E−7 | 1.08 | 1.00 |
| CXCR4 | 2.74 | 5.67E−3 | 2.61 | 1.00 |
| SAA2-SAA4 | 6.92 | 1.00E−7 | 1.89 | 1.00 |
| CLC | 6.01 | 5.44E−3 | 2.28 | 1.00 |
| SAA2 | 5.97 | 1.42E−7 | 1.54 | 1.00 |
| ATP10B | 3.79 | 8.91E−6 | 1.42 | 1.00 |
| CXCL8 | 5.78 | 3.63E−5 | 1.03 | 1.00 |
| TCIM | 4.78 | 2.16E−6 | 1.15 | 1.00 |
| PTPRH | 6.60 | 2.82E−6 | 1.02 | 1.00 |
3.3 |. The endogenous immune regulators, IDO1 and TNFRSF6, are both significantly upregulated in the airway transcriptome during CLAD
Both IDO1 and TNFRSF6B, two endogenous immune regulators, were the top upregulated genes based on fold change (22-fold and 137-fold, respectively). To further validate these, we performed quantitative PCR (qPCR) analyses on remaining brush specimens in our cohort. We detected increased qPCR levels of IDO1 and TNFRSF6B in airway brushes from CLAD LTRs versus controls with a relative enrichment for CLAD stage 2–3 in patients with higher levels (Figure 3A–B) and consistent with our data showing intensification of the CLAD transcriptomic signature in higher grade CLAD (Figure 2C). We then performed a Kendall correlation for IDO1 and TNFRSF6B within the CLAD cohort and found significant correlation between IDO1 and TNFRSF6B levels (Figure 3C). Subsequent sensitivity analyses found that infection had no effect on this relationship. Notably, other TNFRSF members were also significantly upregulated in the airway (Table 4), including two ligands for TNFRSF6B, Fas-ligand (FasL;CD95L/TNFSF6) and “homologous to lymphotoxin, exhibits inducible expression and competes with HSV glycoprotein D for binding to herpesvirus entry mediator, a receptor expressed on T lymphocytes” (LIGHT;CD258/TNFSF14) Together, these data further support upregulation of the immune regulators, IDO1 and TNFRSF6B in our CLAD cohort and suggest these factors as potential biomarkers in the airway for CLAD.
FIGURE 3.
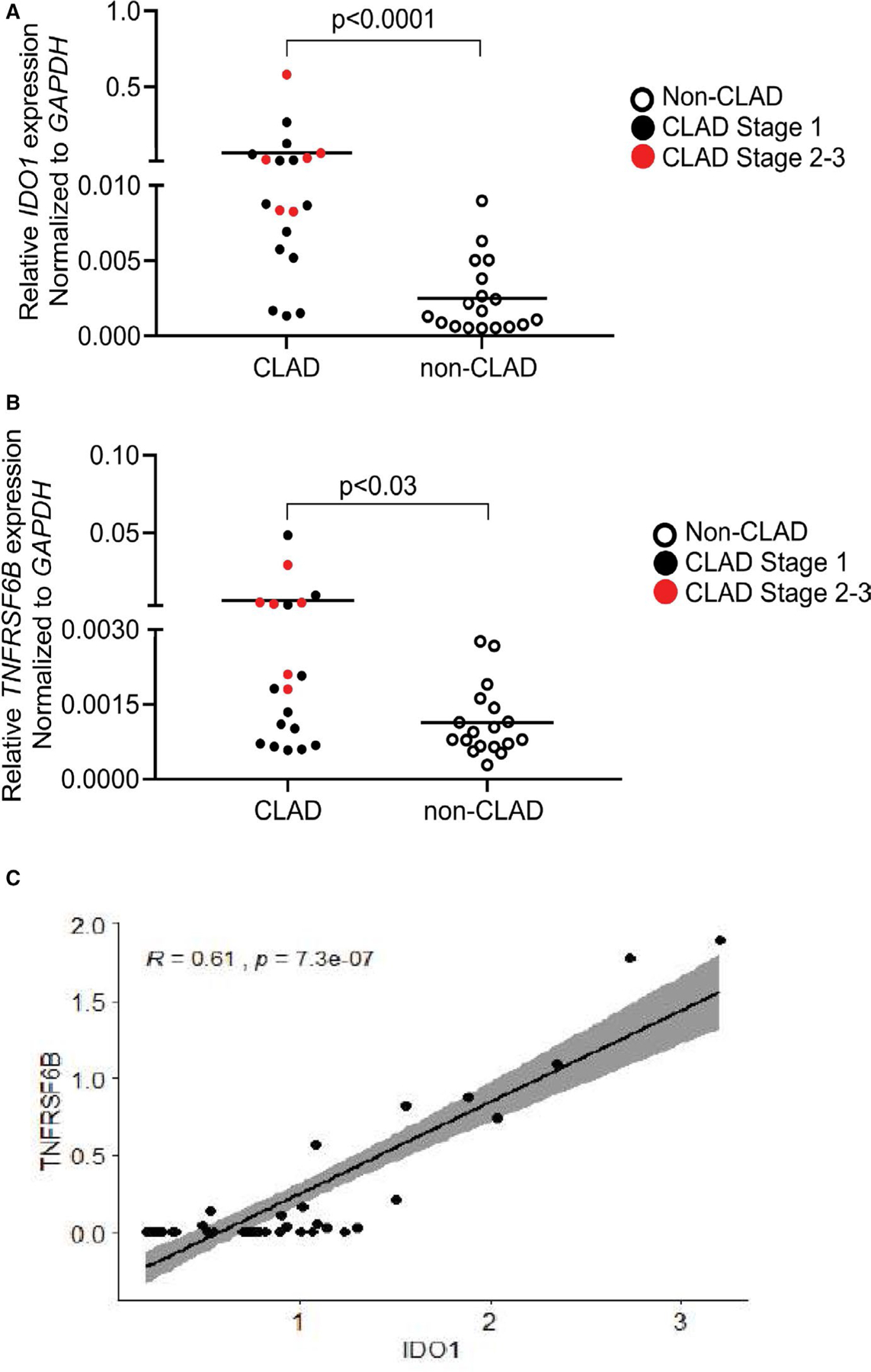
(A) Relative IDO1 expression for LTRs in cohort by qPCR. A statistically significant difference in expression was observed between CLAD and non-CLAD (Mann-Whitney U P < .001). (B) Relative TNFRSF6B expression for LTRs by qPCR. A statistically significant difference in expression was observed between CLAD and non-CLAD (Mann-Whitney U P < .03). (C) Correlation plot for TNFRSF6B versus IDO1 in LTRs. A significant correlation was observed with a Kendall correlation coefficient of 0.61
TABLE 4.
Upregulated TNFSF and TNFRSF family members in the airway transcriptome
| Name | Fold change | FDR P-value |
|---|---|---|
| TNFRSF6B (DcR3) | 137.19 | 1.49E−12 |
| TNFSF14 (LIGHT) | 3.93 | 4.46E−07 |
| TNFRSF9 (4–1BB) | 4.74 | 3.61E−06 |
| TNFRSF1B (TNFR2) | 2.77 | 6.01E−05 |
| TNFRSF8 (CD30) | 3.89 | 2.80E−04 |
| TNFAIP2 | 1.95 | 1.91E−03 |
| TNFRSF10C (DcR1) | 2.29 | 6.21E−03 |
| TNFSF10 (TRAIL) | 1.43 | 9.52E−03 |
| TNFSF13B (BAFFR3) | 2.06 | 9.73E−03 |
| TNFRSF18 (GITR) | 1.80 | 3.01E−02 |
| TNFSF6 (FASL) | 1.84 | 4.00E−02 |
| TNFSF8 (CD30L) | 1.90 | 4.31E−02 |
| TNFRSF1A (TNFR1) | 1.21 | 4.67E−02 |
| TNFRSF12A (TWEAKR) | 1.48 | 5.00E−02 |
| TNFRSF4 (OX40R) | 2.37 | 7.18E−02 |
| TNFRSF11A (TRANCER) | 1.28 | 8.03E−02 |
| TNFRSF10A (TRAILR1) | 1.27 | 8.88E−02 |
3.4 |. Upstream regulators and pathway analysis suggest an important role for IFN-γ, TNF-α, and IL-1β in the airway transcriptome during CLAD
Five hundred and thirteen differentially expressed genes were included in gene enrichment and pathway analyses. Upstream regulator analyses are shown in Table 5. Using Ingenuity Pathway Analysis (IPA), the top 3 upstream regulators predicted to be activated by z-score were TNF-α (p = 1.71E-21), IL1-β (p = 7.06E-30), and IFN-γ (p = 7.75E-20). Representative IPA pathway containing these regulators is shown in Figure 4A, with several top 25 upregulated genes including IDO1, TNFRSF6B, SERPINA3, and MMP9 as downstream to these key Type-1 cytokines. Using IPA, we determined that our 513 DGEs were enriched for multiple cellular and humoral immune genes related to cellular trafficking, cell cycle, cell movement, function, signaling, and immune-mediated diseases, as shown in Figure 4B. These numerous immune pathways were predicted to be significantly upregulated in CLAD, notably the inflammatory response (p = 1.45E-53) and humoral immune response (p = 1.47E-53), among others. Collectively these data support significant immune activation in the airway transcriptome during CLAD, with Type-1 effector cytokines playing a hierarchical predominant role.
TABLE 5.
Upstream regulators
| Upstream regulator | Predicted activation state | Activation z-score | P-value of overlap |
|---|---|---|---|
| TNF-α | Activated | 7.017 | 2E−21 |
| IL1-β | Activated | 6.412 | 7E−30 |
| IFN-γ | Activated | 6.013 | 8E−20 |
| IL−6 | Activated | 5.941 | 3E−23 |
| NFκβ (complex) | Activated | 5.921 | 4E−20 |
| IL1-α | Activated | 5.308 | 5E−22 |
| MYD88 | Activated | 4.957 | 8E−15 |
| CSF2 | Activated | 4.793 | 2E−17 |
| IL−2 | Activated | 4.631 | 8E−16 |
| TLR3 | Activated | 4.565 | 2E−12 |
FIGURE 4.
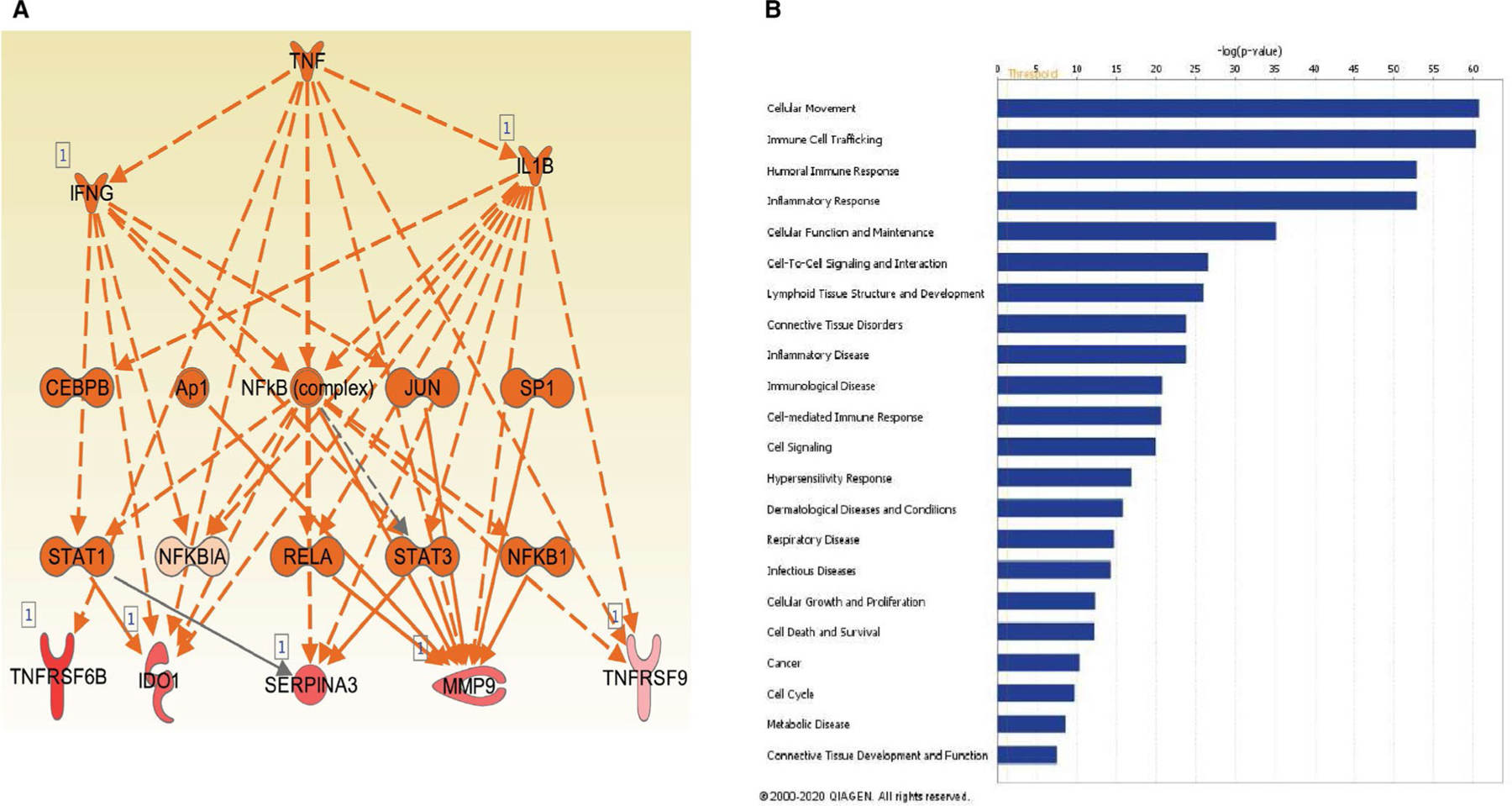
(A) Sample network showing predicted activation using Ingenuity Pathway Analysis. The network shows the relationships between the three most activated upstream pathways: IFN-γ, TNF-α, and IL-1β and IDO1 and TNFRSF6B as downstream. (B) Gene enrichment analysis of selected disease and function groups of the upregulated genes. Multiple inflammatory and immune activation function groups were among the top predicted areas of activation
3.5 |. Corroboration of the CLAD airway transcriptome signature in a separate validation cohort
An external cohort of LTRs from another institution was used to validate the signature identified in our study cohort. Characteristics of the 22 CLAD and 17 non-CLAD patients in the validation cohort are described in Table 6. The validation cohort had a numerically larger proportion of ILD patients, but characteristics were otherwise similar to the study cohort. The top 25 genes from the study cohort were used to classify CLAD in the validation cohort. As a group, these genes were significantly upregulated in the CLAD patients in the validation cohort (Figure 5A). A ROC curve was generated and showed good ability to distinguish CLAD from non-CLAD with a ROC-AUC =0.77 (Figure 5B).
TABLE 6.
Validation cohort characteristics
| CLAD (n = 22) | Control (n = 17) | P-value | |
|---|---|---|---|
| Age at transplant, median (IQR) | 54 (44.7–57.6) | 60.6 (28.8–66.5) | .94 |
| Female, n (%) | 12 (54.5) | 7 (41.1) | .61 |
| Transplant diagnosis | .55 | ||
| Cystic fibrosis, n (%) | 3 (13.6) | 5 (29.4) | |
| Interstitial lung disease, n (%) | 15 (68.2) | 10 (58.8) | |
| Chronic obstructive pulmonary disease, n (%) | 3 (13.6) | 2 (11.8) | |
| Pulmonary hypertension, n (%) | 1 (4.5) | 0 (0) | |
| Acute rejection, n (%) | 7 (31.8) | 1 (5.8) | .13 |
| Acute infection, n (%) | 11 (50.0) | 0 | <.01 |
| Bacterial | 8 | — | |
| Viral | 1 | — | |
| Fungal | 5 | ||
| Days from Tx to CLAD, median (IQR) | 955 (641–1478) | ||
| Days from CLAD onset to brush, median (IQR) | 6 (1–40) | ||
| BOS stage at brush, n (%) | |||
| BOS 0p | 3 (14) | ||
| BOS 1 | 7 (32) | — | |
| BOS 2 | 8 (36) | — | |
| BOS 3 | 4 (18) | — |
FIGURE 5.
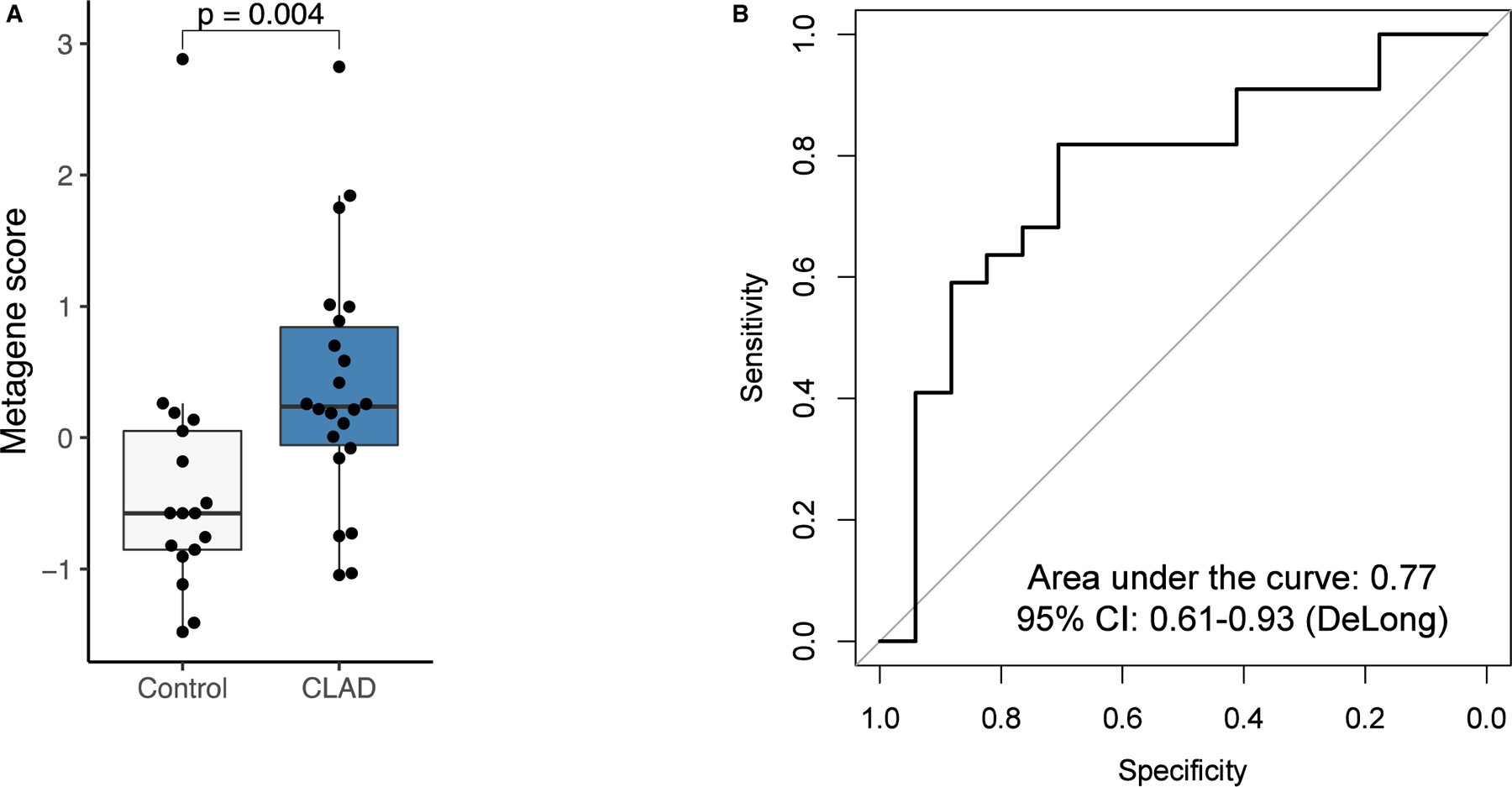
(A) Comparison of top 25 activated genes identified in the experimental cohort in the CLAD and non-CLAD control patients in the external validation set. The top 25 genes were significantly upregulated in the CLAD patients of the validation set (P = .004). (B) ROC curve applying the top 25 activated genes identified in the experimental cohort to the validation cohort. The AUC =0.77 indicating a good ability to distinguish CLAD from non-CLAD
3.6 |. Targeted proteomic assessment of the lung allograft validates enhanced Type-1 immunity in the airway transcriptome
Next, we sought to further validate our airway transcriptome findings using a targeted assessment of the lung allograft proteome. For this, we first used the multi-analyte Meso Scale Discovery system to probe cell-free BAL supernatant collected on the same day as airway brush samples and assess a targeted 28 cytokine/chemokine panel (Figure 6A–B). Here we detected increased levels of IFN-γ, TNF-α, and IL-1β protein in CLAD versus controls. In addition, the Type-2 cytokines IL-5 and granulocyte-macrophage colony-stimulating factor (GM-CSF), were both upregulated in BAL from CLAD patients compared to controls. We also tested a panel of chemokines and found that Regulated upon Activation, Normal T cell Expressed and Secreted (RANTES; CCL5) and IL-8 were both significantly increased in CLAD patients. As shown in Table 7, multiple cytokines and chemokines were upregulated in CLAD at both the transcriptomic and protein levels. Together, these data demonstrate overlapping and predominant Type-1 immune signatures between the airway transcriptome and a targeted proteome assessment in the lung allograft during CLAD.
FIGURE 6.
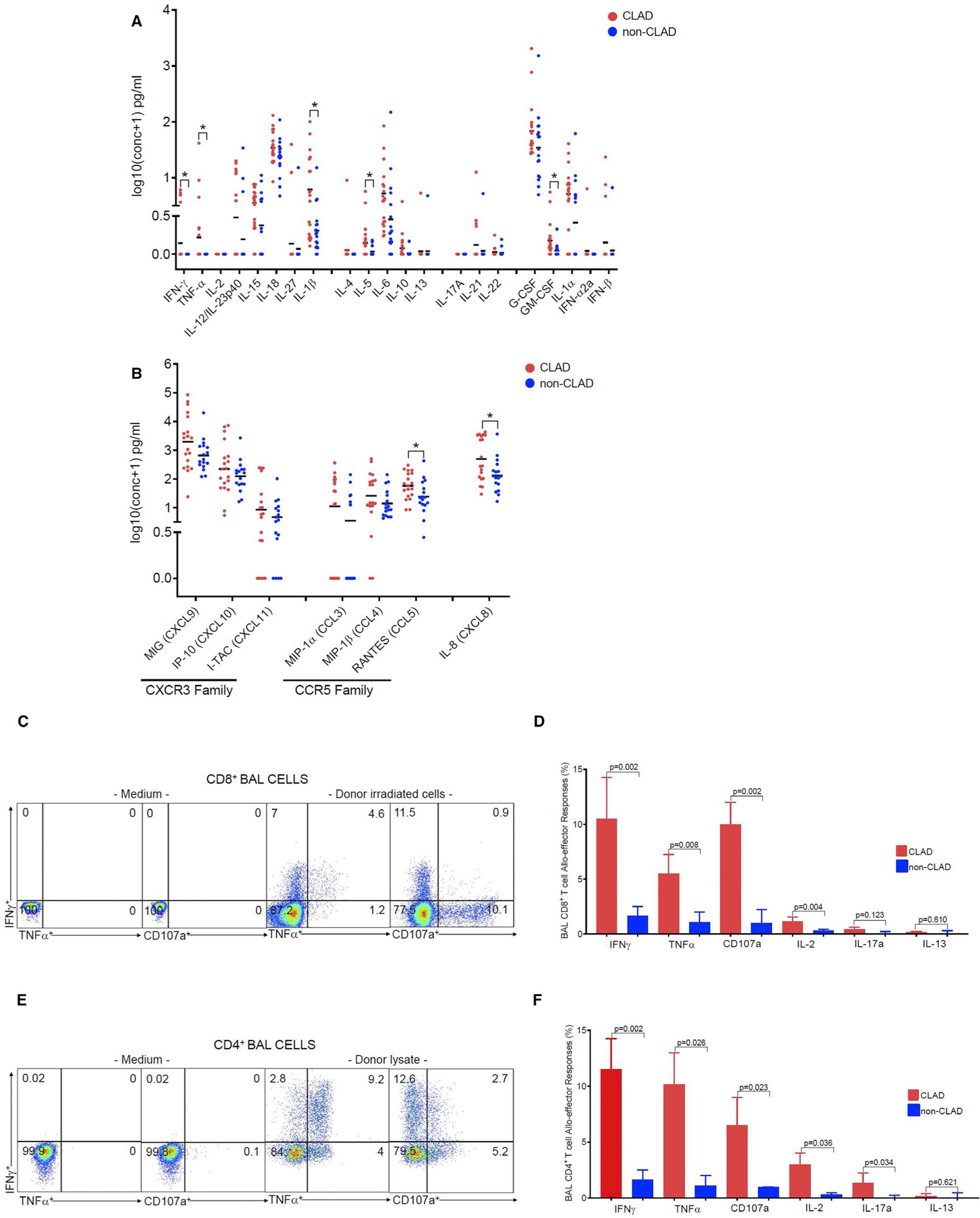
Using MSD-meso scale discovery assay system-U-PLEX Technology-multiplex immunoassay we show the poled data from a total of 36 LTRs with a subset of 19 CLAD and 17 non-CLAD from BAL cells supernatants for cytokines (A) and chemokines (B). (A) We analyzed multiplex assay for following cytokines: such as: IFNγ, TNFα, IL-2, IL-12/IL-23p40, IL-15, IL-18, IL-27, IL-1β (Type 1), IL-4, Il-5. IL-6. IL-10, IL-13 (Type 2), and IL-17A, IL-21, IL-22 (Type 17), and G-CSF, GM-CSF, IL1α, IFN-α2a, IFNβ. (B) We analyzed multiplex assay for following chemokines such as: CXCR3 family (MIG, IP-10, I-TAC) and CCR5 family (MIP-1α, MIP–1β, RANTES) and IL-8(CXCL8). Bars represent median values and P-values were calculated using the Mann-Whitney U test, with * representing statistical significance for P-value <.05. (C) Representative flow cytometric plots of CLAD LTRs showing donor allospecific CD8+ BAL T cell effector responses. Values represent frequencies data of intracellular cytokine staining following re-stimulation with donor irradiated PBMC (right 2 panels) or medium alone (left 2 panels), (unstimulated) of IFNγ+, TNFα+, and CD107a+, from a subset of 6 LTRs with CLAD. (D) Pooled data from the same subset of six LTRs, with CLAD (red bars) and six LTRs with non-CLAD (blue bars) showing the ex vivo BAL CD8+ T cell frequencies for IFNγγ+, TNFα+, CD107a+, IL-2+, IL-17a+, and IL-13+ from 12 LTRs. Bars represent median values and P-values were calculated using the Mann-Whitney U test. (E) Representative flow cytometric plots of CLAD LTRs showing donor allospecific CD4+ BAL T cell effector responses. Values represent frequencies data of intracellular cytokine staining following re-stimulation with PBMC donor lysate (right 2 panels) or medium alone (left 2 panels), (unstimulated) of IFNγ+, TNFα+, and CD107a+, from a subset of six LTRs with CLAD. (F) Pooled data from the same subset of six LTRs, with CLAD (red bars) and six LTRs with non-CLAD (blue bars) showing the ex vivo BAL CD4+ T cell frequencies for IFNγ+, TNFα+, CD107a+, IL-2+, IL-17a+, and IL-13+ from 12 LTRs total. Bars represent median values and P-values were calculated using the Mann-Whitney U test
TABLE 7.
Comparison of MSD and DGE results
| MSD |
RNAseq |
|||
|---|---|---|---|---|
| Cytokine/chemokine | P-value | Gene | Fold change | FDR P-value |
| IFN-γ | 0.062 | IFNG | 5.36 | 1.13E−04 |
| TNF–α | 0.038 | TNF | 2.03 | 0.1 |
| IL−2 | 0.089 | IL2 | 1.36 | 0.78 |
| IL−12 | 0.179 | IL12A | −1.15 | 0.02 |
| IL−15 | 0.143 | IL15 | 1.6 | 1 |
| IL−18 | 0.106 | IL18 | 1.2 | 0.2 |
| IL−27 | 0.539 | IL27 | 1.69 | 0.51 |
| IL−1β | 0.005 | IL1B | 3.16 | 3.72E−03 |
| IL−4 | 0.046 | IL4 | 2.27 | 0.26 |
| IL−5 | 0.026 | IL5 | 1.13 | 0.91 |
| IL−6 | 0.142 | IL6 | 3.47 | 0.01 |
| IL−10 | 0.063 | IL10 | 5.29 | 1.22E−03 |
| IL−13 | 0.786 | IL13 | 1.51 | 1 |
| GM-CSF | 0.016 | CSF2 | 2.31 | 0.25 |
| IL−17A | 0.11 | IL17A | 1.11 | 0.94 |
| IL−21 | 0.072 | IL21 | 8.48 | 0.03 |
| IL−22 | 0.661 | IL22 | NaN | NaN |
| G-CSF | 0.117 | CSF3 | 2.92 | 1.45E−03 |
| IL−1a | 0.058 | IL1A | 1.54 | 0.58 |
| IFN-a2α | 0.922 | IFNA2 | NaN | NaN |
| IFN-β | 0.227 | IFNB1 | −1.12 | 0.91 |
| MIG | 0.082 | CXCL9 | 9.65 | 2.11E−08 |
| IP−10 | 0.317 | CXCL10 | 5.97 | 6.98E−05 |
| I-TAC | 0.231 | CXCL11 | 6.98 | 2.86E−05 |
| MIP−1α | 0.074 | CCL3 | 4.53 | 8.61E−04 |
| MIP−1β | 0.139 | CCL4 | 3.97 | 1.35E−03 |
| RANTES | 0.033 | CCL5 | 1.6 | 0.11 |
| IL−8 | 0.016 | CXCL8 | 6.39 | 5.68E−08 |
To elucidate the cellular sources driving the Type-1 immune signature, we evaluated donor-specific alloimmune T cell effector responses in six patients with CLAD and six stable LTR controls, using BAL cells isolated at the time airway brushes were obtained. These cells were re-stimulated for 6 h in the presence or absence or either irradiated, CFSE-labeled donor PBMC (direct mixed lymphocyte reaction [MLR] to assess CD8+ T cells) or donor PBMC lysate (indirect MLR to assess CD4+ T cells). As shown in Figure 6C,D (gating strategy in Figure S4), the predominant allograft CD8+ alloeffector responses were IFN-γ, the cytotoxic degranulation marker, CD107a, and TNF-α with significantly increased frequencies of these responses in CLAD subjects. Similarly, the predominant responses from lung allograft CD4+ T cells were IFN-γ, TNF-α, CD107a, and IL-2 with either IL-17a or IL-13 responses only detected at very low levels. Together, these findings support lung allograft T cells from CLAD LTRs having increased frequencies of CD8+ and CD4+ T cells that produce hierarchically predominantly Type-1 effector cytokines in response to alloantigen.
4 |. DISCUSSION
Here, we report the first analysis of the distal airway transcriptome and a targeted BAL proteome in LTRs with CLAD. Our findings demonstrate a significant and differential Type-1 immune activation signature in the airway transcriptome and BAL proteome from CLAD LTRs, compared to stable LTR controls. The top DGE that we identified in our CLAD group was IDO1, with a 22-fold increased expression versus stable controls. Importantly, IDO1 is a known immune regulator that is potently induced by the signature Type-1 effector cytokine, IFN-γ and is produced by various immune cells including dendritic cells and macrophages, but also pulmonary and other epithelial cells.21,22 IDO1 is the rate-limiting oxidoreductase enzyme that catalyzes the degradation of the essential amino acid, tryptophan, to the kynurenine pathway and has been shown to suppress T cell responses.23 Notably, we detected marked upregulation of IDO1 along with increased airway transcripts for IFN-γ, as well as its downstream signaling molecules signal transducer and activator of transcription 1 (STAT1) and interferon-regulatory factor-1 (IRF-1), both previously shown to mediate IDO1 expression.24 Overall, our findings are consistent with IDO1 upregulation in response to a Type-1 alloimmune response in CLAD LTRs.25 Indeed, this appears consistent with previous studies that suggested IDO1 induction is protective for pulmonary epithelia during inflammation.26,27 Moreover, an earlier study in the rat orthotopic lung transplant model showed that overexpression of lentiviral IDO1, resulted in attenuation of ACR, supporting its role as an immune regulator in lung transplant.28 Interestingly, a recent study by Weigt et al. evaluated the BAL-cell transcriptome during ACR episodes using RNAseq analysis and reported a predominant cytotoxic T cell signature that included IFN-γ and other markers but not IDO1.7 As BAL cells are highly enriched for macrophages, the lack of IDO1 upregulation might suggest airway epithelial cells as a major cell source in our study. Taken together, our results showing substantial upregulation of IDO1 and suggest it as a potential useful biomarker for CLAD.
Another highly upregulated gene in the airway transcriptome during CLAD that we observed was tumor necrosis factor receptor superfamily 6b (TNFRSF6B), also known as decoy receptor 3 (DcR3).29 This molecule was the top upregulated TNFRSF member and, similar to IDO1, is an endogenous immune regulator that can neutralize other TNFRSF members. Notably, we found two of its three molecular targets, FasL and LIGHT were both upregulated in CLAD airways, whereas TNF-like molecule 1A (TL1A/TNFSF15) was not. These pro-inflammatory molecules are important mediators in apoptosis (FasL, LIGHT) and T cell co-stimulation (LIGHT) that promote Type-1 immune responses. Additionally, other costimulatory molecules including CD153 (CD30 ligand; TNFRSF8), TNFRSF9 (4–1BB), and TNF-related apoptosis-inducing ligand (TRAIL; TNFRSF10) were also significantly upregulated in the airway transcriptome supporting ongoing T cell activation in CLAD.30 Together, these costimulatory molecules, including FasL and LIGHT, have been implicated in the immune response during infection and autoimmunity, with TNFRSF6B/DcR3 interactions with FasL having been shown to confer resistant to apoptosis and regulate immune responses in binding LIGHT.31–33 Indeed, while TNFRSF6B/DR3 is not expressed in the mouse, a previous study using human recombinant DR3 demonstrated an attenuation of heart allograft and pancreatic islet rejection in mouse models.34,35 Furthermore, we found that IDO1 and TNFRSF6B expression were correlated across our cohort, with significantly increased levels in CLAD. Together, these data support the notion that Type-1 immune activation during CLAD leads to the co-expression of two endogenous immune regulators in the airway transcriptome, IDO1 and TNFRSF6B/DR3, that might be useful tandem biomarkers to differentiate CLAD.
Our targeted proteomic study of cytokines and chemokines in BAL fluid in our cohort demonstrated differential levels in CLAD versus controls that were complementary to our airway transcriptomic signatures. Here, we observed that increased levels of IL-1β mRNA and protein, a pro-inflammatory cytokine and canonical product of the inflammasome, significantly differentiated CLAD from non-CLAD LTRs. Our IPA analysis determined IL-1β as an important upstream regulator of the CLAD airway transcriptome, in addition to being downstream of the Type-1 cytokines TNF-α and IFN-γ. Furthermore, TNF-α protein was also differentially detected in CLAD and a major upstream regulator of our airway transcriptome using IPA analysis. Importantly, in our assessment of donor-specific alloeffector responses from BAL cells in CLAD, the Type-1 cytokines IFN-γ, TNF-α, and the cytotoxic degranulation marker CD107a were the major immune responses we detected from CD4+ and CD8+ T cells, compared to substantially lower Type-2 or Type-17 responses. These data support alloimmune T cells as an important contributing source to Type-1 immunity leading to downstream signaling signature in the airway transcriptome (eg, STAT1, IRF-1). Furthermore, increased levels of chemokines such as the IFN-γ-inducible chemokine CXCL9 (CXCR3 family), RANTES (CCL5; CCR5 family) and IL-8 (CXCL8) were detected at the mRNA and protein levels consistent with earlier reports implicating these chemokines in BOS.36–38 Together, these chemokines are important for the recruitment of T cells, monocytes (CXCL9, RANTES), neutrophils (IL-8) and other cells to the airways in CLAD. Thus, our targeted proteomic analysis of BAL fluid demonstrates an inflammatory milieu that complements the airway transcriptome signature in CLAD and provides evidence for IFN-γ, TNF-α, and IL-1β playing key roles.
In addition to a Type-1 immune signature in the airway transcriptome in CLAD, we also detected factors in the airway transcriptome supporting humoral immune activation and other immune responses. Several immunoglobulin genes were significantly upregulated including IGKC, which encodes for the constant region for immunoglobulin light chains and IGHA1 and IGHM that encode constant regions for the IgA and IgM heavy chains, respectively, supporting active antibody production in the airway. A role for Type-2 immune responses in CLAD can be inferred with significant upregulation of the lysophospholipase, Charcot-Leyden crystal galectin (produced by eosinophils and basophils), and increased detection of IL-5 protein in CLAD BAL. Importantly, eosinophils have been previously associated with acute lung allograft rejection.39 Interestingly, the Type-2 regulatory cytokine IL-10, was significantly increased in the airway transcriptome in CLAD but not detected at significant levels at the protein level in BAL fluid. Unexpectedly, we did not detect increased levels of IL-17, an effector cytokine previously implicated in BOS pathogenesis,40 in the airway transcriptome or BAL, nor were significant T cell frequencies detected in response to alloantigen re-stimulation. Together, our detection of these responses indicates a more broad and complex immune response in CLAD, nevertheless, our data support a hierarchical Type-1 immune predominance.
In addition to a Type-1 immune activation signature in the airway transcriptome, our analysis also demonstrated genes associated with tissue remodeling. The matrix metalloproteinases (MMP)-9 and MMP-2 are two matrix degrading enzymes that are critical in tissue repair and we found to be increased in CLAD airways, consistent with a recent report implicating MMP9 in CLAD.41 Furthermore, ADAM-like decysin-1, ADAMDEC1, is a disintegrin metalloproteinase that we observed was significantly upregulated in CLAD and previously reported to be a component of T cell–mediated rejection signature in renal transplant recipients, lung ACR, and pulmonary sarcoidosis.8,42,43 The serpin family member alpha-1 antichymotrypsin (SERPINA3) is protease that can be produced in response to metalloproteinases and has been implicated as a major differentially expressed gene in the idiopathic interstitial pneumonias.44 Together, these data suggest active tissue remodeling in CLAD in the setting of chronic inflammation.
There are several caveats to our study. Because our study was cross-sectional in design, we cannot definitively conclude that our airway transcriptome signature is causal for CLAD. Brushes were collected during bronchoscopies conducted in the clinical course of care, so the median time for collection of CLAD brushes was later than non-CLAD control brushes as they were collected during for-cause bronchoscopies beyond the typical surveillance period. From registry data, the rate of BOS increases by approximately 10% each year for the first 5 years posttransplant.45 Utilization of second year surveillance biopsies for the non-CLAD group allowed for time for patients to develop CLAD; however, CLAD would certainly become more prevalent over time among long-term survivors. There may be important differences in the degree of immunosuppression or other time-dependent factors that might impact airway gene expression and will require future investigation to fully characterize these influences. While our cohort was comprised predominantly of CLAD stage-1 LTRs, we did observe an increased intensity of the transcriptomic signature in patients with advanced CLAD (stages 2, 3), thus providing internal support within our cohort for our signature with CLAD progression. Moreover, our separate validation cohort provides external support for our airway transcriptome signature in CLAD and demonstrated a reasonable discriminatory value. Our study does raise the question of whether a Type-1 immune activation signature precedes incipient CLAD, which we plan to address in future studies. Furthermore, our cohort was comprised almost entirely of the BOS clinical endotype of CLAD, therefore it is possible that with a larger study cohort the airway transcriptome may differ between BOS versus RAS endotypes.
In summary, we report an airway transcriptome with a predominant Type-1 immune activation signature in a cross-sectional study of CLAD versus stable LTRs. A targeted analysis of the concomitant BAL proteome confirmed multiple cytokines and chemokines that were upregulated at both the mRNA and protein level, and mixed lymphocyte reaction analysis revealed T cells producing predominantly Type-1 effector cytokines in CLAD. Together, these data suggest persistent Type-1 alloimmune inflammation as an important driver in CLAD, with downstream immune activation detected in the airway transcriptome, comprised predominantly of airway epithelia and monocyte/macrophages. Assessment of the airway transcriptome may be useful for the molecular diagnosis of CLAD and provide new therapeutic targets and immune metrics to follow in future interventional studies in CLAD.
Supplementary Material
ACKNOWLEDGMENTS
Support: R01 HL133184/HL/NHLBI NIH HHS/United States (JFM), R01 HL151552/HL/NHLBI NIH HHS/United States (JRG), I01 CX002011/VA ORD CSR&D. Grant support from the Cystic Fibrosis Foundation (MCDYER17XX0 & GREENL16XX0). This research was also supported in part by the UPMC Immune Transplant and Therapy Center (JFM) and the University of Pittsburgh Center for Research Computing through the resources provided.
Abbreviations:
- ACR
Acute cellular rejection
- AUC
Area under the curve
- BAL
Bronchoalveolar lavage
- BOS
Bronchiolitis obliterans syndrome
- CLAD
Chronic lung allograft dysfunction
- DGE
Differential genes expressed
- FDR
False discovery rate
- IDO-1
Indoleamine 2, 3 dioxygenase 1
- ISHLT
International Society of Heart and Lung Transplant
- LTR
Lung transplant recipient
- MSD
Meso Scale Discovery
- qPCR
Quantitative polymerase chain reaction
- RAS
Restrictive allograft syndrome
- ROC
Receiver operator characteristics
- TNFRSF6B
Tumor necrosis factor receptor superfamily 6B
Footnotes
DISCLOSURE
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
DATA AVAILABILITY STATEMENT
Data available on request due to privacy/ethical restrictions.
REFERENCES
- 1.Meyer KC, Raghu G, Verleden GM, et al. An international ISHLT/ATS/ERS clinical practice guideline: diagnosis and management of bronchiolitis obliterans syndrome. Eur Respir J 2014;44:1479–1503. [DOI] [PubMed] [Google Scholar]
- 2.Verleden GM, Raghu G, Meyer KC, Glanville AR, Corris P. A new classification system for chronic lung allograft dysfunction. J Heart Lung Transplant: the official publication of the International Society for Heart Transplantation 2014;33:127–133. [DOI] [PubMed] [Google Scholar]
- 3.Sato M, Waddell TK, Wagnetz U, et al. Restrictive allograft syndrome (RAS): a novel form of chronic lung allograft dysfunction. J Heart Lung Transplant: the official publication of the International Society for Heart Transplantation 2011;30:735–742. [DOI] [PubMed] [Google Scholar]
- 4.Todd JL, Jain R, Pavlisko EN, et al. Impact of forced vital capacity loss on survival after the onset of chronic lung allograft dysfunction. Am J Respir Crit Care Med 2014;189:159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weigt S, Wallace W, Derhovanessian A, et al. Chronic allograft rejection: epidemiology, diagnosis, pathogenesis, and treatment. Seminars Respir Critical Care Med 2010;31:189–207. [DOI] [PubMed] [Google Scholar]
- 6.Gregson AL, Hoji A, Injean P, et al. Altered Exosomal RNA Profiles in Bronchoalveolar Lavage from Lung Transplants with Acute Rejection. Am J Respir Crit Care Med 2015;192:1490–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weigt SS, Wang X, Palchevskiy V, et al. Usefulness of gene expression profiling of bronchoalveolar lavage cells in acute lung allograft rejection. J Heart Lung Transplant 2019;38:845–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halloran KM, Parkes MD, Chang J, et al. Molecular assessment of rejection and injury in lung transplant biopsies. J Heart Lung Transplant: the official publication of the International Society for Heart Transplantation 2019;38:504–513. [DOI] [PubMed] [Google Scholar]
- 9.Sacreas A, Yang JYC, Vanaudenaerde BM, et al. The common rejection module in chronic rejection post lung transplantation. PLoS One 2018;13(10):e0205107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ray M, Horne W, McAleer JP, et al. RNA-seq in Pulmonary Medicine: How Much Is Enough? Am J Respir Crit Care Med 2015;192:389–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ling K-M, Garratt LW, Banerjee B, et al. Azithromycin partially mitigates dysregulated repair of lung allograft small airway epithelium. Transplantation 2020;104:1166–1176. [DOI] [PubMed] [Google Scholar]
- 12.Verleden GM, Glanville AR, Lease ED, et al. Chronic lung allograft dysfunction: Definition, diagnostic criteria, and approaches to treatment-A consensus report from the Pulmonary Council of the ISHLT. J Heart Lung Transplant: the official publication of the International Society for Heart Transplantation 2019;38:493–503. [DOI] [PubMed] [Google Scholar]
- 13.Balzar S, Fajt ML, Comhair SAA, et al. Mast cell phenotype, location, and activation in severe asthma. Data from the Severe Asthma Research Program. Am J Respir Crit Care Med 2011;183:299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Popescu I, Mannem H, Winters SA, et al. Impaired Cytomegalovirus Immunity in Idiopathic Pulmonary Fibrosis Lung Transplant Recipients with Short Telomeres. Am J Respir Crit Care Med. 2019;199:362–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Popescu I, Pipeling MR, Mannem H, et al. IL-12-Dependent Cytomegalovirus-Specific CD4+ T Cell Proliferation, T-bet Induction, and Effector Multifunction during Primary Infection Are Key Determinants for Early Immune Control. J Immunol 2016;196:877–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bradford E, Jacobson S, Varasteh J, et al. The value of blood cytokines and chemokines in assessing COPD. Respir Res 2017;18:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dabitao D, Margolick JB, Lopez J, Bream JH. Multiplex measurement of proinflammatory cytokines in human serum: comparison of the Meso Scale Discovery electrochemiluminescence assay and the Cytometric Bead Array. J Immunol Methods 2011;372:71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dalmas DA, Scicchitano MS, Mullins D, et al. Potential candidate genomic biomarkers of drug induced vascular injury in the rat. Toxicol Appl Pharmacol 2011;257:284–300. [DOI] [PubMed] [Google Scholar]
- 19.Halloran PF, Famulski KS, Reeve J. Molecular assessment of disease states in kidney transplant biopsy samples. Nat Rev Nephrol 2016;12. [DOI] [PubMed] [Google Scholar]
- 20.Todd JL, Vinisko R, Liu YI, et al. Circulating matrix metalloproteinases and tissue metalloproteinase inhibitors in patients with idiopathic pulmonary fibrosis in the multicenter IPF-PRO Registry cohort. BMC Pulmonary Medicine 2020;20:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol 2004;4:762–774. [DOI] [PubMed] [Google Scholar]
- 22.Moffett JR, Namboodiri MA. Tryptophan and the immune response. Immunol Cell Biol 2003;81:247–265. [DOI] [PubMed] [Google Scholar]
- 23.Lee GK, Park HJ, Macleod M, Chandler P, Munn DH, Mellor AL. Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology 2002;107:452–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du MX, Sotero-Esteva WD, Taylor MW. Analysis of transcription factors regulating induction of indoleamine 2,3-dioxygenase by IFN-gamma. J Interferon Cytokine Res. 2000;20:133–142. [DOI] [PubMed] [Google Scholar]
- 25.Liu X, Shin N, Koblish HK, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 2010;115:3520–3530. [DOI] [PubMed] [Google Scholar]
- 26.Lee S-M, Park HY, Suh Y-S, et al. Inhibition of acute lethal pulmonary inflammation by the IDO-AhR pathway. Proc Natl Acad Sci U S A 2017;114:E5881–E5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aldajani WA, Salazar F, Sewell HF, Knox A, Ghaemmaghami AM. Expression and regulation of immune-modulatory enzyme indoleamine 2,3-dioxygenase (IDO) by human airway epithelial cells and its effect on T cell activation. Oncotarget 2016;7:57606–57617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swanson KA, Zheng Y, Heidler KM, Mizobuchi T, Wilkes DS. CDllc+ cells modulate pulmonary immune responses by production of indoleamine 2,3-dioxygenase. Am J Respir Cell Mol Biol 2004;30:311–318. [DOI] [PubMed] [Google Scholar]
- 29.Lin WW, Hsieh SL. Decoy receptor 3: a pleiotropic immunomodulator and biomarker for inflammatory diseases, autoimmune diseases and cancer. Biochem Pharmacol 2011;81:838–847. [DOI] [PubMed] [Google Scholar]
- 30.Ward-Kavanagh LK, Lin WW, Sedy JR, Ware CF. The TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses. Immunity 2016;44:1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu KY, Kwon B, Ni J, Zhai Y, Ebner R, Kwon BS. A newly identified member of tumor necrosis factor receptor superfamily (TR6) suppresses LIGHT-mediated apoptosis. J Biol Chem 1999;274:13733–13736. [DOI] [PubMed] [Google Scholar]
- 32.Wan X, Zhang J, Luo H, et al. A TNF family member LIGHT transduces costimulatory signals into human T cells. J Immunol 2002;169:6813–6821. [DOI] [PubMed] [Google Scholar]
- 33.Shi G, Wu Y, Zhang J, Wu J. Death decoy receptor TR6/DcR3 inhibits T cell chemotaxis in vitro and in vivo. J Immunol 2003;171:3407–3414. [DOI] [PubMed] [Google Scholar]
- 34.Zhang J, Salcedo TW, Wan X, et al. Modulation of T-cell responses to alloantigens by TR6/DcR3. J Clin Invest 2001;107:1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu Y, Han B, Luo H, et al. DcR3/TR6 effectively prevents islet primary nonfunction after transplantation. Diabetes 2003;52:2279–2286. [DOI] [PubMed] [Google Scholar]
- 36.Belperio JA, Keane MP, Burdick MD, et al. Critical role for CXCR3 chemokine biology in the pathogenesis of bronchiolitis obliterans syndrome. J Immunol 2002;169:1037–1049. [DOI] [PubMed] [Google Scholar]
- 37.Weigt SS, Elashoff RM, Keane MP, et al. Altered levels of CC chemokines during pulmonary CMV predict BOS and mortality post-lung transplantation. Am J Transplant 2008;8:1512–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suwara MI, Vanaudenaerde BM, Verleden SE, et al. Mechanistic differences between phenotypes of chronic lung allograft dysfunction after lung transplantation. Transpl Int 2014;27:857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dosanjh AK, Elashoff D, Kawalek A, Moss RB, Esrig S. Activation of eosinophils in the airways of lung transplantation patients. Chest 1997;112:1180–1183. [DOI] [PubMed] [Google Scholar]
- 40.Burlingham WJ, Love RB, Jankowska-Gan E, et al. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. J Clin Invest 2007;117:3498–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pain M, Royer P-J, Loy J, et al. T Cells Promote Bronchial Epithelial Cell Secretion of Matrix Metalloproteinase-9 via a C-C Chemokine Receptor Type 2 Pathway: Implications for Chronic Lung Allograft Dysfunction. Am J Transplant 2017;17:1502–1514. [DOI] [PubMed] [Google Scholar]
- 42.Halloran PF, Venner JM, Madill-Thomsen KS, et al. Review: The transcripts associated with organ allograft rejection. Am J Transplant 2018;18:785–795. [DOI] [PubMed] [Google Scholar]
- 43.Crouser ED, Culver DA, Knox KS, et al. Gene expression profiling identifies MMP-12 and ADAMDEC1 as potential pathogenic mediators of pulmonary sarcoidosis. Am J Respir Crit Care Med 2009;179:929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steele MP, Luna LG, Coldren CD, et al. Relationship between gene expression and lung function in Idiopathic Interstitial Pneumonias. BMC Genom 2015;16:869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chambers DC, Cherikh WS, Harhay MO, et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Thirty-sixth adult lung and heart-lung transplantation Report-2019; Focus theme: Donor and recipient size match. J Heart Lung Transplant 2019;38(10):1042–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data available on request due to privacy/ethical restrictions.