

Abstract
Objective:
Electronic cigarette (e-cig) use has recently been implicated in promoting atherosclerosis. In this study we aimed to investigate the mechanism of e-cig exposure accelerated atherosclerotic lesion development.
Approach and Results:
Eight-week-old ApoE−/− mice fed normal laboratory diet were exposed to e-cig vapor (ECV) for 2 h/day, 5 days/week for 16 weeks. We found that ECV exposure significantly induced atherosclerotic lesions as examined by Oil Red O staining and greatly upregulated TLR9 expression in classical monocytes and in the atherosclerotic plaques, which the latter was corroborated by enhanced TLR9 expression in human femoral artery atherosclerotic plaques from e-cig smokers. Intriguingly, we found a significant increase of oxidative mitochondria DNA lesion in the plasma of ECV exposed mice. Administration of TLR9 antagonist prior to ECV exposure not only alleviated atherosclerosis and the upregulation of TLR9 in plaques, but also attenuated the increase of plasma levels of inflammatory cytokines, reduced the plaque accumulation of lipid and macrophages, and decreased the frequency of blood CCR2+ classical monocytes. Surprisingly, we found that cytoplasmic mtDNA isolated from ECV extract-treated macrophages can enhance TLR9 activation in reporter cells and the induction of inflammatory cytokine could be suppressed by TLR9 inhibitor in macrophages.
Conclusions:
E-cig increases level of damaged mtDNA in circulating blood and induces the expression of TLR9, which elevate the expression of proinflammatory cytokines in monocyte/macrophage and consequently lead to atherosclerosis. Our results raise the possibility that intervention of TLR9 activation is a potential pharmacologic target of ECV-related inflammation and cardiovascular diseases.
Keywords: Electronic cigarette, mtDNA damage, atherosclerotic lesion, TLR9
Graphical Abstract

Introduction
Atherosclerosis is a disease of large- and medium-sized arteries that is characterized by the formation of subendothelial plaques consisting of necrotic cores, accumulated modified lipids, extracellular matrix, smooth muscle cells, endothelial cells, leukocytes, and foam cells.1 Atherosclerosis is the pathological basis of a variety of cardiovascular diseases (CVDs), which are the leading causes of death worldwide.2 Cigarette smoking is a well-established major risk factor for CVDs, recognized as such for decades.3 A large body of evidence has indicated that smoking accelerates atherosclerosis and induces a hypercoagulable state.4–6 In the past 10 years, electronic cigarette (e-cig) use has increased rapidly in the United States7, 8 whereas the impact of e-cigs on CVDs has been reported only just recently. A randomized, double-blinded pilot study revealed that e-cigs worsen peripheral and central hemodynamics as well as arterial stiffness.9 Another study reported that e-cig aerosol increases arterial stiffness and oxidative stress, although to a lesser extent than conventional cigarette.10 Studies in animals have found that e-cigs have both rapid and long-term negative effects on arteries, inducing cardiac dysfunction and atherosclerosis.11, 12 However, despite the reported association of e-cigs with atherosclerosis, the underlying mechanisms of the pro-atherosclerotic effects of e-cigs remain unclear.
Toll-like receptors (TLRs) are a class of essential components of the innate immune system that recognize distinct kinds of pathogen-associated molecular patterns. Activation of TLRs triggers an intracellular signaling cascade mediated through MyD88 or TRIF, leading to the production of pro- and anti-inflammatory cytokines.13 TLR9 activation of murine and human macrophages has been shown to stimulate their inflammatory responses (TNF-α, IL-6) and accelerate their transformation into foam cells, an indication of plaque build-up.14, 15 Activation of TLR9 by relatively high dosage of oligonucleotide ODN1826 was found to increase plaque formation16 and TLR9 inhibition alleviated atherosclerotic progression in ApoE−/− mice fed a high-fat diet.17 Recently, evidence has accumulated to indicate that TLR9 plays a pivotal role in the development of vascular inflammation through proinflammatory activation of macrophages in angiotensin II-induced atherosclerosis.18 In addition, pharmacological blockade of TLR9 can attenuate atherosclerotic lesion formation, suggesting TLR9 may serve as a potential therapeutic target for atherosclerosis.18
TLR9 also recognizes endogenous self-DNA termed damage-associated molecular patterns (DAMPS) from injured mammalian cells.19 For example, mitochondrial DNA (mtDNA) is able to activate TLR9 and induces podocyte apoptosis.20 TLR9 activation by circulating mtDNA is reported to contribute to elevated arterial pressure and vascular dysfunction in spontaneously hypertensive rats.21 In fact, direct DNA damage is a critical mechanism underlying smoking-associated tumorigenesis (e.g. lung cancers).22–25 Smoke exposure is associated with increased mtDNA damage both in vitro and in vivo.26, 27 MtDNA damage is implicated in promoting adult atherogenesis by prenatal environmental tobacco smoke exposure in ApoE−/− mice.28 The detrimental effects of e-cigs on genomic DNA have also been recently identified. E-cig vapor (ECV)-exposed cells have significantly increased DNA strand breaks along with increased rates of apoptosis and necrosis.29 E-cig aerosols can suppress cellular antioxidant defenses and induce significant oxidative DNA damage.30 ECV damages DNA and reduces DNA-repair activity in mouse organs as well as in human cells.31 However, effect of e-cig on mtDNA damage, and the biological sensor(s) of the released mtDNA remains elusive. Importantly, no current study establishes the causal relationship between mtDNA damage-TLR9 activation during atherosclerotic process even in the setting of traditional cigarette smoking, let alone e-cig vaporing. In the present study, we examined the impact of ECV on DNA damage and the activation of TLR9 in ApoE−/− mice and investigated whether TLR9 blockade could alleviate e-cig induced development of atherosclerosis.
Materials and Methods
The authors declare that all supporting data are available within the article in the Data Supplement.
Animals
Eight-week-old mice (n = 5~10 per group) were subjected to whole-body exposure. The control mice were exposed to HEPA filtered room air. E-cig aerosol smoke was generated from Platinum V2 RED E-liquid (classic tobacco flavor containing 2.4% nicotine) by a Teague smoking machine (Model TE-2, Teague Enterprises, Woodland, CA). The aerosol was generated by heating 2 mL e-liquid as we previously described.32 Mice received 2 h exposures per day for 5 successive days per week, over a 16 week period. For the animal group with pharmacological TLR9 blockade, mice received intraperitoneal injections of TLR9 antagonist IRS86933 (5 mg/kg, 5′-TCCTGGAGGGGTTGT-3′) or Ctrl-ODN (5’-TCCTGCAGGTTAAGT-3; IDT, Coralville, IA) twice per week and 1 h prior to ECV exposure. The mice were euthanized with pentobarbital sodium (200 mg/kg) by intraperitoneal injection 24 h after the last exposure. Blood and tissues were collected and stored for further assays. Animal handling and experimentation were in accordance with the recommendation of the current NIH guidelines and were approved by the Temple University School of Medicine Institutional Animal Care and Use Committee.
Flow cytometry
Whole blood cells were stained and analyzed on a FACSCanto LSRII flow cytometer (BD Biosciences). Isotype controls were used to set appropriate gates. Detailed staining protocol and antibodies are described in supplementary materials. Data were analyzed acquired with FACSDiva (BD Biosciences) and analyzed with FlowJo 6.4.7v10.6 (Tree Star, Ashland, OR). For all samples, approximately 20,000 cells were analyzed to generate scatter plots. All events were collected from each sample to generate the scatter plots.
Multiplex ELISA for mouse cytokines
Aliquots of plasma from mice were analyzed using mouse cytokine 9-Plex ELISA kit from Boster Biological Technology (Pleasanton, CA) according to manufacturer’s instructions.
Histological and immunohistochemical studies
The formalin fixed heart and whole aorta were dehydrated in ethanol and embedded in paraffin, and 5-μm serial sections were stained with hematoxylin and eosin (Millipore-Sigma, Burlington, MA). Proximal aortic sections were obtained within the root. Sections were evaluated blindly to score for pathological changes. The expression of TLR9 (LS-B688, Lifespan, USA), adhesion molecule VCAM-1 (ab-134047, Abcam, USA) and macrophages (Mac2, LS-C62936, Lifespan) were examined by immunohistochemical staining with individual antibodies with appropriate controls suggested by manufactures. The stained tissue sections were scanned, positive areas outlined by 2 board-certified pathologists (blind for the animal treatment conditions), and positive area quantified by Imagescope software (Apeiro).
Atherosclerotic lesions
The atherosclerotic lesions were measured by both whole aorta en face oil Red O (ORO) stain and aortic root section ORO stain. To measure the en face lesion area of the aorta, mice were euthanized and then perfused with 1X phosphate-buffered saline with a constant pressure via the left ventricle. The whole aorta was immediately removed, fixed with 10% neutral buffered formalin for 5 min, then the aorta was washed with phosphate-buffered saline and stained with Sudan IV solution for 30 min. Excess stain was washed off with 70% ethanol for 30 min. The aorta was cut open longitudinally following carefully dissecting out adipose tissue in adventitia. The opened aorta was spread on a clean glass slide, followed by covering with another glass slide on the top, and then injecting water inside between two slides, followed by glue the edge of slides. The luminal side of the stained aorta was photographed. Adobe Photoshop and Image Pro plus 6.0 software (Media Cybernetics, Rockville, MD) were used to perform the image capture and analysis. The extent of atherosclerosis was expressed as the percent of surface area of the entire aorta covered by lesions.
To measure atherosclerotic lesion in aortic root, another subset of hearts and proximal aortas were dehydrated in 30% sucrose at 4°C overnight and embedded in OCT compound (Tissue-Tek, Elkhart, IN), frozen on dry ice, and then stored at −80°C until sectioning. Sequential 10-μm-thick cryosections throughout the aortic root, namely, from the origin of the aortic valves to the ascending aorta were cut and collected. Every other section was collected on poly-D-lysine-coated slides. The deposition of lipids was determined with ORO staining (Sigma-Aldrich, St. Louis, MO) and counterstained with hematoxylin and fast green for visualization of atherosclerotic lesions. Atherosclerotic lesions were quantified by taking an average of three cross sections from each aorta spaced 50 μm apart using Image Pro plus 6.0 software (Media Cybernetics, Rockville, MD). Briefly, the lesions were circled, and the area of each lesion for the quantified section on the slide was exported to a spreadsheet (in µm2). This was repeated for each of the slides, and the sum of the lesions area was calculated (µm2/section) for each mouse.
Measurement of cytoplasmic mtDNA lesion and plasma DNA damage
Murine monocyte/macrophage RAW 246.7 cells, maintained in DMEM (Thermo Scientific) plus 10% FBS and 1% antibiotics, were seeded in 60 mm cell-culture dish at density 2 × 106 cell/dish. After 24 h incubation, cells were treated with or without ECV extract (EVE; 0.5~2.5% v/v; equal to 2.5~12.5 cigarettes/day for conventional cigarettes normalized by nicotine intake which corresponds to the realistic situation of human daily exposure to cigarettes34) for 48 h and cytoplasmic mtDNA was extracted according to the protocol as previously described.35 Briefly, after treatment, 1% NP-40 was added and cells were scraped into a micro centrifuge tube, centrifuged at 13,000 rpm for 15 min at 4°C. The supernatant was collected into a new tube and cytoplasmic mtDNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen). Thapsigargin was used as a positive control in inducing mtDNA damage. As for plasma cell free DNA isolation, blood was gently drawn from mice using 16–18 gauge into a tube containing anticoagulant EDTA. The tube was gently inverted several times to mix and plasma was then immediately isolated from the blood by centrifugation at 1,600 x g for 10 min at 4°C. The cytoplasmic mtDNA lesion and plasma levels of DNA damage were measured using the DNA Damage Competitive ELISA (Thermo Scientific; Cat# EIADNAD) according to the manufacturer’s instruction. Both plasma DNA damage and cytoplasmic mtDNA lesion were expressed as the concentration of 8-OHdG.
Measurement of mtDNA/nDNA ratio in plasma cfDNA and cytoplasmic mtDNA
Mice plasma was first centrifuged at 16,000 × g for 10 min at 4°C and the supernatant was subjected to cell-free DNA (cfDNA) extraction using Apostle minimax cfDNA extraction kit (ApostleBio, CA, US). The mtDNA/nDNA ratio in plasma cfDNA and in the above cytoplasmic mtDNA were then measured by SYBR qPCR (Applied Biosystem, CA) with mtCO-1 and 18S rDNA primers (Table S1). The plasma mtDNA/nDNA ratio was calculated as the ratio of mtCO-1/18S rDNA. The relative fold-change of cytoplasmic mtDNA by EVE in macrophages was calculated using the 2−ΔΔCt method as compared to control-treated cells.”
In vitro analysis of TLR9 activation
To evaluate the TLR9 activation potency of the cytoplasmic DNA, we used engineered HEK293 cell lines (HEK-Blue) that stably express either mouse TLR9 (mTLR9) and an NF-κB reporter gene (Invivogen, San Diego, CA). In brief, HEK mTLR9 blue cells were maintained in selection medium (DMEM complete plus with Blasticidin 30 µg/mL and Zeocin 100 µg/mL) and 100 µL of cells in HEK blue detection medium were seeded in 96 well-plate (at density 2 × 104 cells/ well), followed by adding 20 µL of water or mtDNA (final treatment concentration 25 ng/µL). After 18–24 h incubation, the absorbance was measured at 620–655 nm.
TLR9 expression in human femoral artery atherosclerotic plaques
Femoral artery atherosclerotic lesions were identified and collected autopsy samples from non-smokers and e-cig smokers (>6 months) with the Institutional Review Board approval from Robert Wood Johnson Medical School, Rutgers (Pro2018001015). The expression of TLR9 was examined by immunohistochemical staining (TLR9 antibody: LS-B688, Lifespan, USA) using routine procedure. Demographic data of the human subjects were presented in supplementary Table S2. The procedures used in this study conform to the principles outlined in the Declaration of Helsinki.
Data analysis
Data are expressed as mean ± SEM. All in vitro experiments were technically repeated three times from 3 different cultures and the data values were scaled to controls where appropriate. GraphPad Prism 5.0 (GraphPad, La Jolla, CA) software was used for all statistical analyses. Data were tested for normality using the Shapiro-Wilks normality test (α=0.05). Comparison of 2 groups was accomplished using an unpaired Student’s t-test (two-sided) where appropriate. Statistical multiple comparisons between more than two groups were made by one-way analysis of variance (ANOVA) with Tukey’s post hoc analysis. For nonparametric analysis, Mann Whitney test was used to check statistical difference. P values of less than 0.05 were considered statistically significant.
Results
ECV exposure increased atherosclerosis development and TLR9 expression
We first compared atherosclerotic lesion progression in ECV-exposed ApoE−/− mice with air-exposed mice after 16 weeks. As shown in Fig. 1A, the results of en face aortic ORO staining demonstrated a significant increase of atherosclerosis burden in ECV-exposed ApoE−/− mice (P<0.001). ECV exposure also increased the intimal microscopic lesions in the cross-sectional aortic roots as evidenced by HE staining in Fig. 1B (P<0.001). Intriguingly, we observed that there was significantly higher expression of TLR9 in the plaque area of the ECV-exposed mice than that in the air-treated mice (Fig. 1C). More importantly, in the femoral arterial plaques from e-cig smokers (>6 months), there was higher TLR9 expression as compared to that in the plaques from non-smokers (P=0.0286; Fig. 1D). These results suggest an important role of TLR9 in e-cig smoking associated atherosclerosis.
Figure 1. E-cig vapor exposure increased atherosclerotic lesion and TLR9 expression in atherosclerotic plaques in normal laboratory diet-fed ApoE−/− mice, substantiated by the upregulation of TLR9 expression in femoral arterial plaques in e-cig smokers as compared to that in the plaques from nonsmokers.
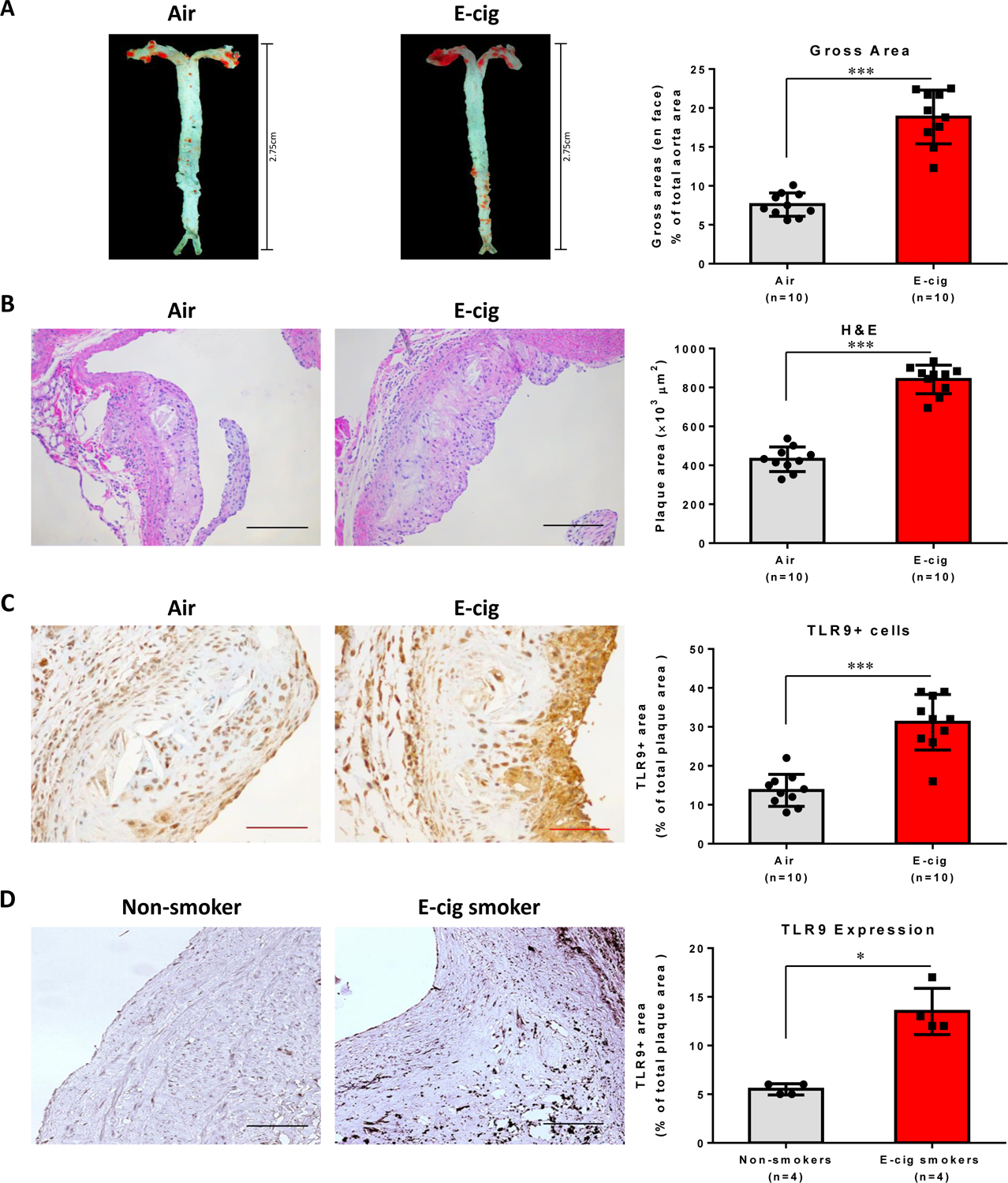
(A) Representative pictures of en face Oil Red O (ORO) staining of freshly dissected intact aorta and quantification analysis of atherosclerosis burden. 8-week-old mice were exposed to air or e-cig vapor for 2 h a day, 5 days per week for 16 weeks. (B) Hematoxylin-eosin (H&E) staining to measure the intimal microscopic lesions in the cross-sectional aortic roots. Scale bar: 100 µm. (C) TLR9 positive cells in atherosclerotic plaques in ApoE−/− mice. Cross-sectioned aortic roots were subjected to TLR9 immunohistochemistry staining. Scale bar: 50 µm. A-C: Data are shown as mean ± SEM (n=10 per group) and statistical analysis was performed using the Students’ t-test. ***P < 0.001. (D) TLR9 expression in the femoral arterial plaques from e-cig smokers (>6 months) as compared to in non-smoking-derived plaques in non-smokers. Scale bar: 100 µm. Data are shown as mean ± SEM (n=4 per group) and statistical analysis was performed using Mann-Whitney test. *P < 0.05.
TLR9 blockade attenuated ECV-exacerbated atherosclerosis in ApoE−/− mice
We next sought to investigate whether TLR9 blockade can inhibit ECV-induced atherosclerosis development. Mice in the ECV group were administrated with 5 mg/kg of IRS869 prior to ECV exposure. It was found that ECV significantly increased the lipid deposition (~2-fold; Fig. 2A) in the atherosclerotic lesion area and enhanced the intimal lesions (~1.9-fold) (Fig. 2B) in ApoE−/− mice after 16-week exposure as compared to control mice. In contrast, administration of mice with TLR9 antagonist IRS869 prior to ECV exposure significantly attenuated ECV-exacerbated atherosclerosis development (lipid deposition reduced by 50%; and intimal microscopic lesion decreased by 23%; all P<0.05) (Fig. 2A, B). ECV exposure had no significant effect on other metabolic parameters, such as total cholesterol, triglycerides, etc. (Fig. S1).
Figure 2. TLR9 inhibitor attenuated ECV-exacerbated atherosclerotic lesion development TLR9 inhibitor in ApoE−/− mice.
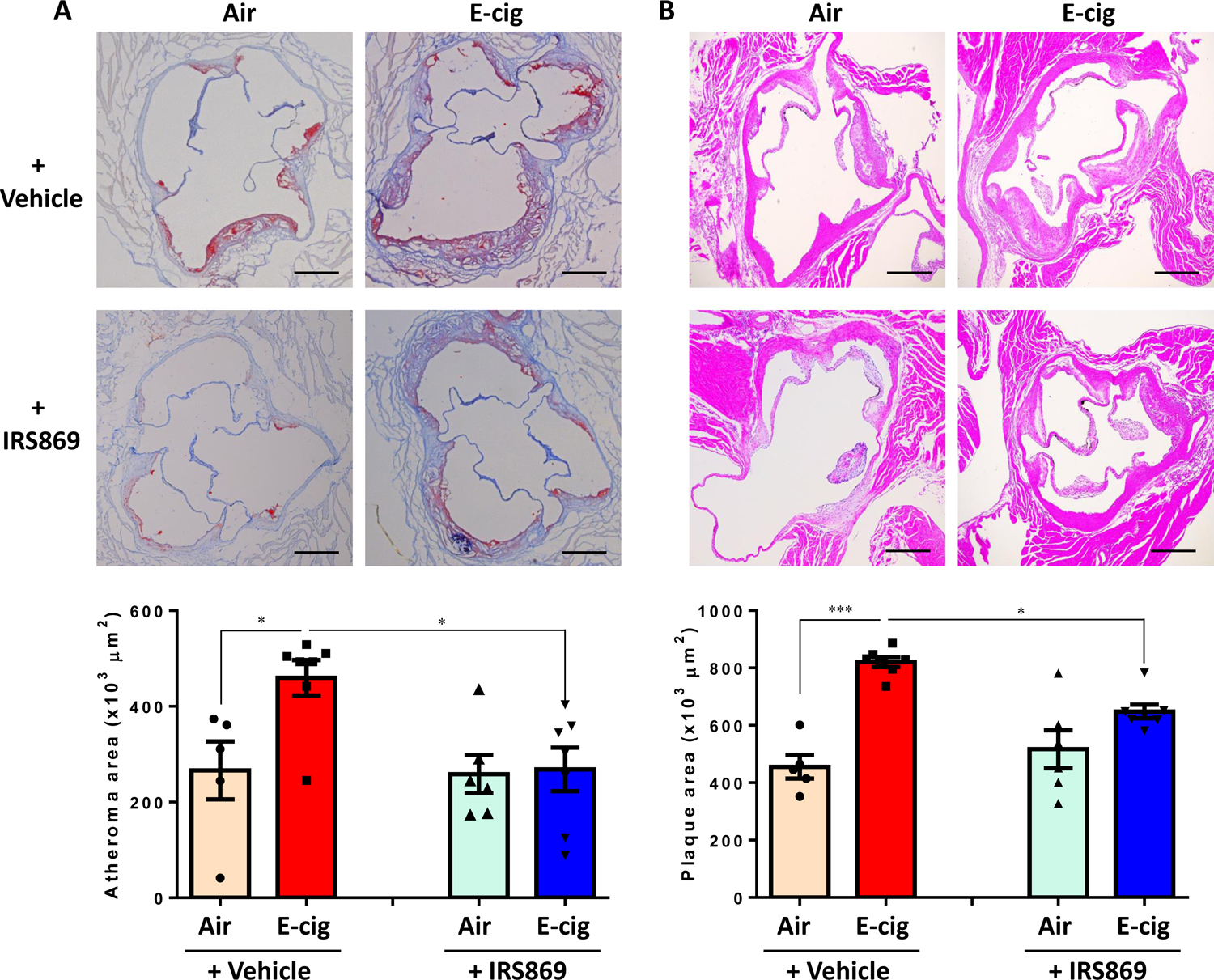
(A) Oil Red O (ORO) staining of the cross-sectioned aortic roots. Lipid deposition in plaques was quantified as ORO-positive lesion areas and expressed as percentage to the total area of the atheroma. (B). Cross-sectional hematoxylin-eosin staining to measure the intimal microscopic lesions in the aortic roots. Scale bar: 200 µm. Numbers of animals in each group: Air + Ctrl-ODN: n=5; E-cig + Ctrl-ODN: n=7; Air + IRS869: n=6; E-cig + IRS869: n=7. Data are shown as mean ± SEM and statistical multiple comparisons were made by one-way ANOVA with Tukey’s post hoc analysis. *P<0.05; ***P < 0.001.
ECV enhanced the expression of TLR9 in classical monocytes, which could be reduced by TLR9 inhibition
Smoking has been shown to activate human monocytes, leading to increased adhesion to the endothelium.36 We next determined whether monocyte activation was involved in e-cig-mediated atherosclerotic development by flow cytometry. The gating strategy was shown in Fig. S2. It was found that ECV increased the numbers of nonclassical monocytes in blood while had no effect on other two monocyte subsets (classical and intermediate monocytes; data not shown). Activation of TLR9 has been reported to stimulate inflammatory responses of monocytes/macrophages and accelerate their transformation into foam cells, an indication of atherosclerotic plaque build-up.14, 15, 37, 38 We thus further examined TLR9 expression in the different subsets of blood monocytes. There was high frequency TLR9 expression in all three subsets of monocytes at an average of 87.6% ± 13.3% in control mice, with the highest in classical monocytes (98.9% ± 1.2%) and the lowest in nonclassical monocytes (74.1% ± 134%; Fig. 3A). Interestingly, classical monocytes from e-cig-exposed mice expressed very significantly (P < 0.001) higher intensity of TLR9 (mean fluorescence intensity (MFI; 6598 ± 334) than cells from air-exposed mice (MFI 4416 ± 245; Fig. 3B). Pre-administration of mice with TLR9 antagonist IRS869 significantly reduced e-cig-upregulated TLR9 intensity (MFI 5220 ± 317, P<0.001) in classical monocytes. In contrast, there was lower frequency of TLR3 expression in all subsets of monocytes (27.4% ± 19.7%) and ECV exposure had no significant effect on TLR3 expression (Fig. S3).
Figure 3. TLR9 frequency in different subset of monocytes and the effect of ECV exposure on TLR9 expression classical monocytes of ApoE−/− mice.

(A) Representative histogram of TLR9 expression in different subsets of monocytes in air-exposed ApoE−/− mice. N=5. (B) Mean fluorescence intensity (MFI) of TLR9 expression in classical monocytes. Numbers of animals in each group: Air + Ctrl-ODN: n=5; E-cig + Ctrl-ODN: n=7; Air + IRS869: n=6; E-cig + IRS869: n=7. Data are shown as mean ± SEM and statistical multiple comparisons were made by one-way ANOVA with Tukey’s post hoc analysis. *P < 0.05; **P < 0.01.
TLR9 blockade decreased macrophage accumulation and the expression of adhesion molecule and TLR9 in atherosclerotic plaques
It has now been established that foam cells in atherosclerotic plaques are of bone marrow (BM) origin 39 although the mechanisms of monocyte recruitment into noninflamed aortas are not well defined. As shown in Fig. 4A, in addition to the upregulation of TLR9 expression in blood monocytes, ECV exposure significantly increased the expression of TLR9 in atherosclerotic plaques (P<0.001). Further double immunofluorescence staining revealed that macrophages are a major type of cells in the plaques that expressing high levels of TLR9 (Fig. S4). The expression of adhesion molecule VCAM-1 was also significantly increased after ECV exposure, both on the lesion surface and within the plaques (P<0.001, Fig. 4B). Pretreatment of mice with TLR9 inhibitor reduced the upregulation of both TLR9 and VCAM-1 in lesion sites (P<0.05). ECV exposure also remarkably enhanced the accumulation of Mac2+ macrophages in atherosclerotic plaques, which could be decreased by TLR9 inhibitor administration (Fig. 4C).
Figure 4. Pretreatment of mice with TLR9 inhibitor alleviated ECV exposure-induced macrophage accumulation and expression of adhesion molecule and TLR9 in atherosclerotic lesion.

(A) Representative pictures of immunostaining of cross-sectioned aortic root with TLR9 antibody (0.1 µg/mL) and quantification analysis between groups. (B) Representative pictures of immunostaining of cross-sectioned aorta with VCAM-1 antibody (1 µg/mL) and quantification analysis between groups. (C) Immunohistochemical staining of aortic cross-section with mouse macrophage marker Mac2 (0.2 µg/mL) and macrophage content was expressed as percentage of Mac2-positive cells. Scale bar: 100 µm. Numbers of animals in each group: Air + Ctrl-ODN: n=5; E-cig + Ctrl-ODN: n=7; Air + IRS869: n=6; E-cig + IRS869: n=7. Data are shown as mean ± SEM and statistical multiple comparisons were made by one-way ANOVA with Tukey’s post hoc analysis. *P<0.05; **P<0.01; ***P < 0.001.
ECV exposure increased CCR2+ cells in classical monocytes
Recent studies have suggested that Ly6Chigh inflammatory monocytes are more readily to migrate into atherosclerosis-prone arteries through CCR2 or CX3CR1 chemokine receptors and then differentiate into aortic macrophages.40 It was found that ECV exposure significantly increased the frequency of CCR2+ cells in classical monocytes (P<0.001) and pre-administration with TLR9 inhibitor could reduce the upregulation (P<0.05; Fig. 5A). On the contrary, the expression of CD62L in classical monocytes, which play an important role in lymphocyte-endothelial cell interactions was not affected either by e-cigs or the TLR9 inhibitor (P>0.05; Fig. 5B).
Figure 5. Effect of ECV exposure on the frequency of classical monocytes expressing CCR2 and CD62L in ApoE−/− mice.

(A) Representative histograms of CCR2 expression in classical monocytes in ApoE−/− mice exposed to air, e-cig, or e-cig with pre-administration of TLR9 inhibitor IRS869. (B) Expression of CD62L in classical monocytes. Numbers of animals in each group: Air + Ctrl-ODN: n=5; E-cig + Ctrl-ODN: n=7; Air + IRS869: n=6; E-cig + IRS869: n=7. Data are shown as mean ± SEM and statistical multiple comparisons were made by one-way ANOVA with Tukey’s post hoc analysis. *P < 0.05, ***P<0.001.
ECV induces inflammatory response which can be suppressed by TLR9 inhibitor
The production of pro-inflammatory cytokines plays an important role in smoking-associated CVDs risks. Fig. 6A–C show that ECV exposure significantly increased the plasma levels of a number of proinflammatory cytokines, including TNF-α, RANTES, and IL-6 in ApoE−/− mice (P<0.01). Pretreatment of the mice with TLR antagonist IRS869 significantly suppressed e-cig-induced upregulation of these cytokines/chemokines in the plasma (P<0.01). Further study showed that pretreatment of murine monocyte-macrophages ex vivo with IRS869 could suppress the induction of cytokines by plasma from ECV-exposed mice (P<0.05; Fig. 6D). Together, these results suggesting the TLR9 activation is involved in plasma-induced monocyte activation and inflammatory response from ECV-exposed mice.
Figure 6. Effect of ECV exposure on the plasma levels of inflammatory cytokines in ApoE−/− mice and impact of plasmas from different groups of mice on cytokine expression in RWA264.7 cells ex vivo.
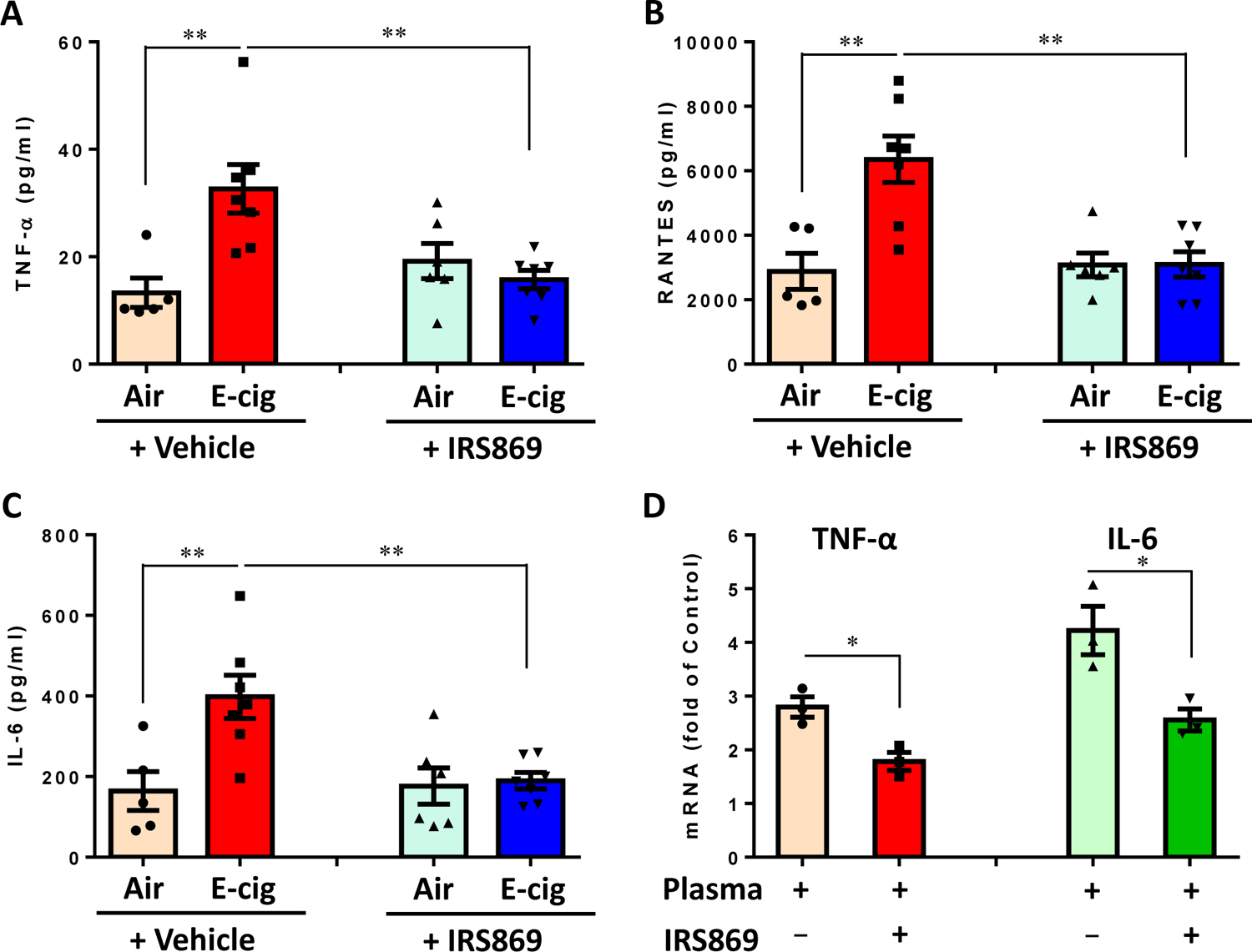
(A, B, C) Plasma were collected from different groups of ApoE−/− mice and subjected to immunoassay for a panel of inflammatory cytokines using the mouse Cytokine 9-Plex ELISA Kit. Images were scanned and the dot density was quantified by a parallel set of individual cytokine standards. N means the number of mice whose plasmas were used for cytokine measurement. Numbers of animals in each group: Air + Ctrl-ODN: n=5; E-cig + Ctrl-ODN: n=7; Air + IRS869: n=6; E-cig + IRS869: n=7. Data are shown as mean ± SEM and statistical multiple comparisons between different groups were made by one-way ANOVA with Tukey’s post hoc analysis. **P < 0.01. (D) Effect of plasma from ECV-exposed mice on proinflammatory cytokines in murine monocytes/macrophages. RAW264.7 cells were pretreated with or without IRS869 at 5 μM for 30 min, then with plasma from ECV-exposed ApoE−/− mice for 3 h. The expression of cytokines was examined by quantitative real time PCR. Data are shown as mean ± SEM from three independent experiments using plasma from 3 mice and statistical analysis was performed using Mann-Whitney test. *P < 0.05.
E-cig increased DNA damage of ApoE−/− mice
TLR9 is an intracellular immune sensor of dsDNA, particularly oxidized DNA and GC-rich DNA fragments.41 Recently, e-cig aerosol was reported to damage DNA and impair the cellular antioxidant defenses.30 We thus examined the DNA damage in e-cig exposed mice with or without TLR9 inhibitor pretreatment. 8-OHdG is one of the most representative products of oxidative modifications of DNA and has therefore been widely used as a biomarker for oxidative stress-related DNA damage. As shown in Fig. 7A, ECV exposure significantly increased the plasma levels of 8-hydroxy-2’-deoxyguanosine (8-OHdG) in ApoE−/− mice (P<0.01). ECV exposure also greatly increased the mtDNA/nDNA ratio (1.59 ± 0.63 to 4.27 ± 1.49) in the plasma, indicating damaged DNA in the plasma mainly attributed to the degradation of oxidatively damaged mtDNA (P<0.01; Fig. 7B). Intriguingly, unlike the ability to inhibit TLR9 activation and inflammatory response, pre-administration with TLR9 inhibitor could not reduce the DNA damage as compared to ECV exposure with Ctrl-ODN pretreatment.
Figure 7. Effect of e-cig on mitochondrial DNA damage.

(A) ECV exposure increased plasma levels of circulating damaged DNA in ApoE−/− mice. Plasma were collected from air or ECV-exposed ApoE−/− mice with or without IRS869 pre-administration. The plasma levels of circulating damaged DNA were measured by DNA damage ELISA kit and expressed by the plasma levels of 8-OHdG. Numbers of animals in each group: Air + Ctrl-ODN: n=5; E-cig + Ctrl-ODN: n=7; Air + IRS869: n=6; E-cig + IRS869: n=7. Data are shown as mean ± SEM and statistical multiple comparisons between different groups were made by one-way ANOVA with Tukey’s post hoc analysis. **P < 0.01. (B) The expression of mtCO-1 and 18S rDNA in the plasma circulating DNA was measured by SYBR qPCR and the ratio of mtDNA against nDNA was calculated. Numbers of animals in each group: Air + Ctrl-ODN: n=5; E-cig + Ctrl-ODN: n=7. Data are shown as mean ± SEM and statistical analysis was performed using the Students’ t-test. **P < 0.01. (C) EVE treatment upregulated cytoplasmic levels of mtDNA. RAW264.7 cells were treated with 0.5% of EVE for indicated period. The cytoplasmic mtDNA was extracted and the expression of mtCO-1 was measured by quantitative PCR. (D) HEK-Blue™ mTLR9 reporter cells were stimulated with 25 µg/ml of cytoplasmic mtDNA extracted from EVE-treated RAW264.7 cells after indicated time periods. After 24h incubation, NF-kB-induced SEAP activity was assessed using HEK-Blue™ Detection and reading the optical density (OD) at 655 nm. (E) The 8-OHdG levels in the cytoplasmic mtDNA from RAW264.7 cells treated with or without EVE for 48 h was measured by Competitive DNA Damage ELISA. (F) Pharmacological inactivation of TLR9 on lesioned mtDNA-induced inflammatory cytokine expression in macrophages. RAW264.7 cells were pretreated with Ctrl-ODN or IRS869 (10 µM) for 1 h and then exposed to cytoplasmic mtDNA from RAW264.7 cells treated with or without EVE. After 3 h, RNA was extracted and the mRNA expression of IL-6 was measured by quantitative PCR. Data are shown as mean ± SEM from three independent experiments and statistical analysis was performed using Mann-Whitney test. *P < 0.05 as compared with control treatment.
Cytoplasmic mtDNA induced by EVE could activate TLR9
We have now observed increased TLR9 MFI in classical monocytes and lesion site macrophages are a major type of cells expressing high level of TLR9. It has also been revealed that in vitro treatment of mouse macrophages with EVE could upregulate TLR9 expression (Fig. S6). Further, plasma from ECV-exposed mice was found to have the ability to activate TLR9 in HEK mTLR9 reporter cells, whereas there was no increase in the absorbance in HEK-Blue™ Null1 cells, demonstrating the specificity of the assay (Fig. S7). We next treated cultured macrophages with extracted the cytoplasmic mtDNA from EVE-treated macrophages and examined the functional significance of the damaged mtDNA in TLR9 activation. Fig. 7C shows that EVE treatment of RAW264.7 cells for 48 h increased the cytoplasmic mtDNA levels while there was no increase at 24 h and the levels diminished after 72 h. Correspondingly, cytoplasmic mtDNA from the 48 h EVE-treated macrophages significantly induced the activation of TLR9 in HEK mTLR9 reporter cells than that from control-treated RAW264.7 cells (Fig. 7D). To characterize the underlying mechanism of this difference, we examined the oxidative lesion in the cytoplasmic mtDNA from macrophages. As shown in Fig. 7E, 8-OHdG level was significantly higher in cytoplasmic mtDNA from EVE-treated RAW264.7 cells than that from control-treated cells, indicating that EVE treatment increased oxidative mtDNA lesion and the release of oxidized mtDNA into cytoplasma. Further, the cytoplasmic mtDNA from EVE-treated macrophages induced significantly higher expression of IL-6 in RAW264.7 cells in the presence of Ctrl-ODN (Fig. 7F). In contrast, pharmacological inactivation of TLR9 could attenuate the induction of IL-6 by cytoplasmic mtDNA from EVE-treated macrophages, suggesting TLR9 is the primary sensor for EVE-enhanced cytoplasmic mtDNA in RAW264.7 cells (Fig. 7F). In addition, siRNA knockdown the expression of TLR9 in macrophages alleviated the upregulation of IL-6 expression by cytoplasmic mtDNA from EVE-treated cells (Fig. S8).
Sex difference in response to e-cig treatment
Given the literature reports that females may respond to oxidative stress stimuli (such as those induced by chronic tobacco smoke exposure) differently than male,42–44 we also conducted sex specific analysis of our data, and results were illustrated in supplement table 3. In our first in vivo experiment, ECV exposure upregulated atherosclerotic lesion size and TLR9 expression in lesions of both male and female mice, both with statistical significance. In the followed-up experiment, similar trend of ECV-induced atherosclerosis, as well as lesion size reduction after TLR9 inhibitor treatment, was also observed in both sexes; however, the difference was only statistically significant in female mice. This is probably due to the limited male mouse number in one (Air + Ctrl-ODN) group (see supplemental table 3). Similarly, although both sexes showed trends of increased expression of TLR9, VCAM-1, Mac-2 in the lesions; increased expression of TLR9 MFI, CCR2 in classical monocytes; and increased plasma levels of RANTES, 8-OHdG, mtDNA/nDNA, the differences were only statistically significant in female mice. Given the same trends of changes for all the parameters in both sexes, we argue that the differences in statistical calculation is not likely biological based; rather it is more likely due to limited sample size.
Discussion
In the present study, our results show that ECV exposure induces atherosclerosis development, oxidative DNA damage and TLR9 activation in ApoE−/− mice, substantiated by enhanced TLR9 expression in e-cig smoking-associated atherosclerotic plaques from femoral artery in e-cig smoker. TLR9 signaling has recently been shown to promote macrophage activation and restoration of TLR9 in BM stimulates atherogenesis in Tlr9−/−Apoe−/− mice18, suggesting that hematopoietic cells contributes mainly to TLR9 activation-mediated atherosclerosis development. We found that peripheral classical monocytes exhibited increased TLR9 MFI and in the atherosclerotic plaque macrophages are a major type of cells that expressing TLR9. Pharmacological inactivation of TLR9 can attenuate lesioned mtDNA-triggered TLR9 activation in macrophages and alleviate atherosclerosis in vivo.
In the past 20 years, immune and inflammatory mechanisms have gained tremendous interest in studying atherosclerosis.1, 45, 46 It is now well established that foam cells in atherosclerotic plaques are mainly derived from bone marrow39 and inflammatory monocytes are a major cellular component infiltrating into atherosclerotic plaque.47–49 It has been shown that increase of inflammatory monocyte subset (Ly6Chigh monocytes) was associated with hypercholesterolemia in mice. As compared to Ly6Clow monocytes, Ly6Chigh monocytes are more readily in migrating to atherosclerosis-susceptible arteries.50 We here demonstrated that e-cig-mediated atherosclerosis was accompanied with no significant changes in numbers of classical and intermediate monocytes but increased numbers of nonclassical monocytes in blood. Given TLR9 activation has been reported to stimulate inflammatory responses of monocytes/macrophages and accelerate their transformation into foam cells,14, 15, 37, 38 we further examined TLR9 expression in the different subsets of blood monocytes and found there was high frequency of TLR9 in all three subset monocytes. In addition, ECV exposure increased the TLR9 MFI in classical monocytes and TLR9 expression atherosclerotic plaques, which could be suppressed by TLR9 inhibitor. Studies have suggested that classical monocytes are not only prone to enter developing atherosclerotic lesions, but also differentiate into aortic macrophages via chemokine receptors, such as CX3CR1, CCR2, and CCR5.40 Our study shows the increased frequency of CCR2+ classical monocytes by ECV exposure could be restored by TLR9 inhibition. On the contrary, the expression of CD62L – a cell adhesion molecule that play an important role in lymphocyte-endothelial cell interactions51, was affected neither by e-cigs nor TLR9 inhibition. Thus, it is likely that in ECV-mediated atherosclerosis development, classical monocytes utilized CCR2 to be recruited into inflamed arteries.
In addition to chemokine receptors that mediate monocyte trafficking, endothelial adhesion molecules (e.g. E- and P-selectin, and VCAM-1) also play important roles in the subendothelial accumulation of inflammatory monocytes.52 The upregulation of these molecules in lesion-prone areas is a hallmark of atherosclerosis.53 We here observed that e-cig-induced atherosclerosis was accompanied with upregulation of VCAM-1 expression in plaques. ECV exposure also upregulated plasma levels of RANTES (CCL5) and several other inflammatory cytokines (TNF-α, IL-6). Our results support a previous notion that combined inhibition of CCL2, CX3CR1, and CCR5 could abrogate BM monocytosis and almost abolish atherosclerosis in hypercholesterolemic, atherosclerosis-susceptible ApoE−/− mice.54
Induction of DNA damage is a critical mechanism underlying smoking-associated tumor development, particularly in the pathogenesis of lung cancers.22, 23, 25, 55 Subjects with coronary artery disease were found to have increased circulating nucleotide levels than healthy subjects.56 We observed that ECV exposure increased 8-OHdG levels in the plasma of ApoE−/− mice. This result is consistent with previous studies showing that e-cig aerosols could suppress cellular antioxidant defenses or DNA-repair activity and induce significant oxidative DNA damage both in vitro and in vivo.30, 31 Our earlier study also showed that e-cig aerosols could dysregulate the oxidative phosphorylation complexes, resulting in the disruption of the nuclear/mitochondrial stoichiometry and mitochondrial dysfunction.32 In the present study, we further characterized that the increased circulating DNA in ApoE−/− mice by ECV was enriched with mtDNA with oxidative DNA lesions compared to the genomic DNA. This novel results indicate that mtDNA is more susceptible to oxidative stress and consequently mtDNA is vulnerable to oxidation.41, 57
The correlation of mitochondrial oxidative stress with the progression of human atherosclerosis has been reported. Excessive production of mitochondrial oxidative stress was found in various cell types in atherosclerosis and with aging.58 Particularly, mitochondrial oxidative stress in lesioned myeloid cells was found to enhance atherosclerosis development in aged mice.59 Cigarette smoke-induced mtDNA damage was proposed to promote atherogenesis in ApoE−/− mice fed a standard laboratory diet, but underlying mechanism is not explored.28 Interestingly, in our study pre-administration with TLR9 inhibitor did not reduce plasma levels of 8-OHdG induced by e-cig, suggesting that inhibition of TLR9 activation/signaling but not ECV-induced DNA damage underlies the suppression of inflammatory response and atherosclerosis development. In addition, TLR9 inhibitor alone doesn’t appear to impact atherogenesis in ApoE−/− mice on a normal laboratory diet (Fig. S8), suggesting that the impact of TLR9 increases only in the setting of atherosclerosis and ECV. As compared to nonoxidized nuclear DNA fragments, oxidized DNA and GC-rich fragments of mtDNA are stronger TLR9-stimulating ligands.60 Our in vitro study showed that EVE could increase the cytoplasmic mtDNA in murine RAW264.7 macrophages. The aberrant release of mtDNA into the cytosol is known as mtDNA stress and was reported with the ability to prime antiviral innate immune response through engaging DNA sensor cGAS.61 We found that the EVE-treated macrophages also exhibited mtDNA stress as evidenced by the increased levels of cytoplasmic mtDNA as compared to control-treated cells. More importantly, similar to the induction of TLR9 activation by plasma from ECV-exposed mice, the cytoplasmic mtDNA from EVE-treated macrophages could more potently induce TLR9 activation than mtDNA from control cells. This TLR9 activation by e-cig-induced mtDNA stress is similar to a recent report showing that mitochondrial fission-induced mtDNA stress could activate TLR9 and initiate NF-κB signaling pathway, promoting tumor-associated macrophages recruitment and polarization.62 Macrophages are the major targets for damaged DNA to induce TLR9 signaling activation because circulating cfDNA can be more readily phagocytosed by macrophages and activate the endosomal receptor. Blockade of macrophage autophagy has been shown to ameliorate activated lymphocytes-derived DNA induced murine lupus, indicating autophagy is involved in the uptake of exogenous DNA.63 Recently, Gkirtzimanaki et al. showed that IFNα obstructs autophagic flux in systemic lupus erythematosus monocytes and leads to cytosolic mtDNA accumulation, suggesting autophagy plays an important in the clearance of damaged mitochondria.64 Efferocytosis can be another route for macrophages to intake the mito damage associated molecular pattern. Monocyte-derived macrophages from systemic lupus erythematosus patients were shown to have a reduced capacity to carry out efferocytosis,65 which could be associated with the high levels of auto-antibodies against cellular components such as DNA (anti-dsDNA or anti-ssDNA antibodies) in the serum.66 Nevertheless, the accumulation of ssDNA in the cytolasma of macrophages in atherosclerotic lesions obtained from Apoe−/− mice has been evidenced by immunogold staining, suggesting that macrophages are able to take up cfDNA in atherosclerotic lesions.18
Our further characterization of the mtDNA revealed that cytoplasmic mtDNA from EVE-treated cells had significantly higher levels of oxidative DNA marker 8-OHdG than that from control-treated cells. Due to the fact that oxidized DNA is stronger TLR9 stimulus than nonoxidized DNA,60 the higher potency to activate TLR9 by mtDNA from EVE-treated cells implicates that not only mtDNA stress but also oxidative mtDNA lesion underlie ECV-triggered TLR9 activation and atherosclerosis development in ApoE−/− mice.
The pivotal role of TLR9 has recently been reported in angiotensin II-induced atherosclerosis.18 There was increased circulating levels of cfDNA in angiotensin II –infused ApoE−/− mice whereas TLR9 blockade could suppress the proinflammatory activation of macrophages and vascular inflammation.67 In addition, the activation of TLR9 has been indicated by hepatocyte mtDNA in driving nonalcoholic steatohepatitis.68 More relevantly, spontaneously hypertensive rats were found to have elevated circulating mtDNA, which can activate TLR9 and contribute to vascular dysfunction.21 TLR9 activation of RAW264.7 cells was shown to stimulate the inflammatory responses and accelerate the transformation into foam cells.69 High Dose CpG ODN1826, an agonist ofTLR9, increases atherosclerotic plaque formation16 and pharmacological TLR9 inactivation can alleviate atherosclerotic progression17, suggesting a pro-atherosclerotic role of TLR9. Despite the above reports on the pro-atherosclerotic role of TLR9, an atheroprotective function of TLR9 is implicated by the conventional genetic deletion of TLR9 exacerbates atherosclerosis in ApoE−/− mice with high-fat diet by a 33% increase in lipid deposition and plaque size, and administration of TLR9 agonist reduced lesion severity.70 These controversial results implicate that the atherogenic effect of intracellular TLRs, including TLR9, might be context or cell type-dependent. In 2013, Lundberg et al. showed LDLR−/−:TLR3−/− BM chimeric mice were protected from atherosclerotic lesion formation when fed with high-fat diet. This result suggests that selectively TLR3-deficient in hematopoietic cells is atheroprotective rather than atheroprone in whole-body knockout situation.71 It also supports another report showing that selective deficiency of IFN-β in BM decreases atherosclerotic lesion formation.72 These results together implicate that hematopoietic intracellular TLR signaling is detrimental to atherosclerotic lesion development.73, 74 We here showed that e-cig significantly enhanced TLR9 intensity in circulating monocytes and TLR9 expression in atherosclerotic plaque, and administration of a specific TLR9 antagonist IRS869 prior to ECV exposure could significantly reduce the atherosclerotic lesions in ApoE−/− mice, demonstrating a pro-atherosclerotic role of TLR9 in this setting.
This study has some limitations which have to be pointed out. Firstly, we noticed sex differences in some experiments of our study, while the main findings in the experiments with 10 mice/group (5 in each sex) remain valid and had no sex differences in subgroup analyses. The noticed sex differences in the follow-up experiment in our view are likely due to the relatively smaller number of male mice, which should be addressed in future studies. Secondly, the induction of inflammatory cytokine IL-6 by cytoplasmic mtDNA from EVE-treated cells was significantly attenuated by TLR9 siRNA knockdown, however, it was still higher than that by cytoplasmic mtDNA from control-treated cells (Fig. S9). This could be attributed to the remaining TLR9 signaling as RNAi usually does not completely shut off the gene. Therefore, it warrants to use TLR9 deficiency animals for further study. On the other hand, it is also possible that besides TLR9, other DNA sensors (e.g. cGAS, AIM2) could also be involved in mediating the inflammatory response triggered by e-cig-induced mtDNA stress. Thirdly, the history of conventional cigarette smoking may confound the findings in smokers. Given that e-cig uses have been increasing in popularity in recent years, future studies to involve smokers using e-cig only or with a longer duration of e-cig use will more clearly characterize the role of TLR9 in vaporing-associated atherosclerosis.
In conclusion, our study shows that oxidative mtDNA damage and TLR9 activation of monocytes are critical mechanisms involved in e-cig-accelerated atherosclerosis development in ApoE−/− mice. Intervention of TLR9 activation is a potential treatment of e-cig aerosol-related inflammation and CVDs.
Supplementary Material
Highlights:
E-cig aerosols exacerbate atherosclerosis in ApoE−/− mice accompanied with TLR9 upregulation in atherosclerotic plaques.
E-cig aerosols cause the release of oxidatively lesioned mitochondrial DNA in plasma and induce monocytic TLR9 activation.
Pharmacological inactivation of TLR9 can attenuate lesioned mtDNA-triggered TLR9 activation in macrophages and alleviate atherosclerosis in vivo.
Acknowledgments
a) We acknowledge Dr. Michael Autieri from Temple University in providing the murine macrophage RAW264.7 cells. Dr. Gratian Salaru from the Flow Cytometry Core of Robert Wood Johnson Medical School of Rutgers University is acknowledged for flow cytometry data analyses.
b) Financial support from Bristol-Myers Squibb (New Jersey) in establishing the Temple Heart Transplantation Biorepository is also acknowledged. This study is partly supported by Cancer Center Support Grant P30CA072720.
c) Disclosure: none.
Abbreviations:
- CVDs
cardiovascular diseases
- TLR
Toll-like receptor
- ECV
e-cig vapor
- EVE
ECV extract
- MFI
mean fluorescence intensity
- ODN
oligodeoxynucleotides
- IRS
immunoregulatory DNA sequences
- e-cig
electronic cigarette
- mtDNA
mitochondrial DNA
- ORO
oil Red O
- cfDNA
cell-free DNA
- ANOVA
one-way analysis of variance
- 8-OHdG
8-hydroxy-2’-deoxyguanosine
- BM
bone marrow
References
- 1.Ross R Atherosclerosis--an inflammatory disease. The New England journal of medicine. 1999;340:115–126 [DOI] [PubMed] [Google Scholar]
- 2.Roth GA, Huffman MD, Moran AE, Feigin V, Mensah GA, Naghavi M, Murray CJ. Global and regional patterns in cardiovascular mortality from 1990 to 2013. Circulation. 2015;132:1667–1678 [DOI] [PubMed] [Google Scholar]
- 3.Health. NCfCDPaHPUOoSa UA. The health consequences of smoking—50 years of progress: A report of the surgeon general. 2014
- 4.Messner B, Bernhard D. Smoking and cardiovascular disease: Mechanisms of endothelial dysfunction and early atherogenesis. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:509–515 [DOI] [PubMed] [Google Scholar]
- 5.Gairola CG, Drawdy ML, Block AE, Daugherty A. Sidestream cigarette smoke accelerates atherogenesis in apolipoprotein e−/− mice. Atherosclerosis. 2001;156:49–55 [DOI] [PubMed] [Google Scholar]
- 6.Lee J, Cooke JP. The role of nicotine in the pathogenesis of atherosclerosis. Atherosclerosis. 2011;215:281–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McGraw D. Current and future trends in electronic cigarette use. International journal of psychiatry in medicine. 2015;48:325–332 [DOI] [PubMed] [Google Scholar]
- 8.McMillen RC, Gottlieb MA, Shaefer RM, Winickoff JP, Klein JD. Trends in electronic cigarette use among u.S. Adults: Use is increasing in both smokers and nonsmokers. Nicotine & tobacco research : official journal of the Society for Research on Nicotine and Tobacco. 2015;17:1195–1202 [DOI] [PubMed] [Google Scholar]
- 9.Franzen KF, Willig J, Cayo Talavera S, Meusel M, Sayk F, Reppel M, Dalhoff K, Mortensen K, Droemann D. E-cigarettes and cigarettes worsen peripheral and central hemodynamics as well as arterial stiffness: A randomized, double-blinded pilot study. Vascular medicine. 2018;23:419–425 [DOI] [PubMed] [Google Scholar]
- 10.Ikonomidis I, Vlastos D, Kourea K, Kostelli G, Varoudi M, Pavlidis G, Efentakis P, Triantafyllidi H, Parissis J, Andreadou I, Iliodromitis E, Lekakis J. Electronic cigarette smoking increases arterial stiffness and oxidative stress to a lesser extent than a single conventional cigarette: An acute and chronic study. Circulation. 2018;137:303–306 [DOI] [PubMed] [Google Scholar]
- 11.Olfert IM, DeVallance E, Hoskinson H, Branyan KW, Clayton S, Pitzer CR, Sullivan DP, Breit MJ, Wu Z, Klinkhachorn P, Mandler WK, Erdreich BH, Ducatman BS, Bryner RW, Dasgupta P, Chantler PD. Chronic exposure to electronic cigarettes results in impaired cardiovascular function in mice. Journal of applied physiology. 2018;124:573–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Espinoza-Derout J, Hasan KM, Shao XM, Jordan MC, Sims C, Lee DL, Sinha S, Simmons Z, Mtume N, Liu Y, Roos KP, Sinha-Hikim AP, Friedman TC. Chronic intermittent electronic cigarette exposure induces cardiac dysfunction and atherosclerosis in apolipoprotein-e knockout mice. American journal of physiology. Heart and circulatory physiology. 2019;317:H445–H459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Frontiers in immunology. 2014;5:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dushkin MI, Kovshik GG. Effect of toll-like receptor agonists on the formation of macrophage/foam cells upon acute peritonitis in mice. Bulletin of experimental biology and medicine. 2013;156:49–52 [DOI] [PubMed] [Google Scholar]
- 15.Karper JC, Ewing MM, Habets KL, de Vries MR, Peters EA, van Oeveren-Rietdijk AM, de Boer HC, Hamming JF, Kuiper J, Kandimalla ER, La Monica N, Jukema JW, Quax PH. Blocking toll-like receptors 7 and 9 reduces postinterventional remodeling via reduced macrophage activation, foam cell formation, and migration. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:e72–80 [DOI] [PubMed] [Google Scholar]
- 16.Krogmann AO, Lusebrink E, Steinmetz M, Asdonk T, Lahrmann C, Lutjohann D, Nickenig G, Zimmer S. Proinflammatory stimulation of toll-like receptor 9 with high dose cpg odn 1826 impairs endothelial regeneration and promotes atherosclerosis in mice. PloS one. 2016;11:e0146326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma C, Ouyang Q, Huang Z, Chen X, Lin Y, Hu W, Lin L. Toll-like receptor 9 inactivation alleviated atherosclerotic progression and inhibited macrophage polarized to m1 phenotype in apoe−/− mice. Disease markers. 2015;2015:909572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukuda D, Nishimoto S, Aini K, Tanaka A, Nishiguchi T, Kim-Kaneyama JR, Lei XF, Masuda K, Naruto T, Tanaka K, Higashikuni Y, Hirata Y, Yagi S, Kusunose K, Yamada H, Soeki T, Imoto I, Akasaka T, Shimabukuro M, Sata M. Toll-like receptor 9 plays a pivotal role in angiotensin ii-induced atherosclerosis. Journal of the American Heart Association. 2019;8:e010860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immune network. 2018;18:e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bao W, Xia H, Liang Y, Ye Y, Lu Y, Xu X, Duan A, He J, Chen Z, Wu Y, Wang X, Zheng C, Liu Z, Shi S. Toll-like receptor 9 can be activated by endogenous mitochondrial DNA to induce podocyte apoptosis. Scientific reports. 2016;6:22579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCarthy CG, Wenceslau CF, Goulopoulou S, Ogbi S, Baban B, Sullivan JC, Matsumoto T, Webb RC. Circulating mitochondrial DNA and toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovascular research. 2015;107:119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu Y, Morimoto K. Exposure level to cigarette tar or nicotine is associated with leukocyte DNA damage in male japanese smokers. Mutagenesis. 2008;23:451–455 [DOI] [PubMed] [Google Scholar]
- 23.Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, Campbell PJ, Vineis P, Phillips DH, Stratton MR. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354:618–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pryor WA, Stone K, Zang LY, Bermudez E. Fractionation of aqueous cigarette tar extracts: Fractions that contain the tar radical cause DNA damage. Chemical research in toxicology. 1998;11:441–448 [DOI] [PubMed] [Google Scholar]
- 25.Jorgensen ED, Zhao H, Traganos F, Albino AP, Darzynkiewicz Z. DNA damage response induced by exposure of human lung adenocarcinoma cells to smoke from tobacco- and nicotine-free cigarettes. Cell cycle. 2010;9:2170–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Z, Harrison CM, Chuang GC, Ballinger SW. The role of tobacco smoke induced mitochondrial damage in vascular dysfunction and atherosclerosis. Mutation research. 2007;621:61–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan D, Goerlitz DS, Dumitrescu RG, Han D, Seillier-Moiseiwitsch F, Spernak SM, Orden RA, Chen J, Goldman R, Shields PG. Associations between cigarette smoking and mitochondrial DNA abnormalities in buccal cells. Carcinogenesis. 2008;29:1170–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Z, Knight CA, Mamerow MM, Vickers K, Penn A, Postlethwait EM, Ballinger SW. Prenatal environmental tobacco smoke exposure promotes adult atherogenesis and mitochondrial damage in apolipoprotein e−/− mice fed a chow diet. Circulation. 2004;110:3715–3720 [DOI] [PubMed] [Google Scholar]
- 29.Yu V, Rahimy M, Korrapati A, Xuan Y, Zou AE, Krishnan AR, Tsui T, Aguilera JA, Advani S, Crotty Alexander LE, Brumund KT, Wang-Rodriguez J, Ongkeko WM. Electronic cigarettes induce DNA strand breaks and cell death independently of nicotine in cell lines. Oral oncology. 2016;52:58–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ganapathy V, Manyanga J, Brame L, McGuire D, Sadhasivam B, Floyd E, Rubenstein DA, Ramachandran I, Wagener T, Queimado L. Electronic cigarette aerosols suppress cellular antioxidant defenses and induce significant oxidative DNA damage. PloS one. 2017;12:e0177780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee HW, Park SH, Weng MW, Wang HT, Huang WC, Lepor H, Wu XR, Chen LC, Tang MS. E-cigarette smoke damages DNA and reduces repair activity in mouse lung, heart, and bladder as well as in human lung and bladder cells. Proceedings of the National Academy of Sciences of the United States of America. 2018;115:E1560–E1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bahmed K, Lin CR, Simborio H, Karim L, Aksoy M, Kelsen S, Tomar D, Madesh M, Elrod J, Messier E, Mason R, Unterwald EM, Eisenstein TK, Criner GJ, Kosmider B. The role of dj-1 in human primary alveolar type ii cell injury induced by e-cigarette aerosol. American journal of physiology. Lung cellular and molecular physiology. 2019;317:L475–L485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, Chang B, Duramad O, Coffman RL. Nucleic acids of mammalian origin can act as endogenous ligands for toll-like receptors and may promote systemic lupus erythematosus. The Journal of experimental medicine. 2005;202:1131–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kosmider B, Messier EM, Chu HW, Mason RJ. Human alveolar epithelial cell injury induced by cigarette smoke. PloS one. 2011;6:e26059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bronner DN, O’Riordan MX. Measurement of mitochondrial DNA release in response to er stress. Bio-protocol. 2016;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walters MJ, Paul-Clark MJ, McMaster SK, Ito K, Adcock IM, Mitchell JA. Cigarette smoke activates human monocytes by an oxidant-ap-1 signaling pathway: Implications for steroid resistance. Molecular pharmacology. 2005;68:1343–1353 [DOI] [PubMed] [Google Scholar]
- 37.Weaver LK, Chu N, Behrens EM. Tlr9-mediated inflammation drives a ccr2-independent peripheral monocytosis through enhanced extramedullary monocytopoiesis. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:10944–10949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu K, Chung YM, Endoh Y, Geczy CL. Tlr9 ligands induce s100a8 in macrophages via a stat3-dependent pathway which requires il-10 and pge2. PloS one. 2014;9:e103629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lessner SM, Prado HL, Waller EK, Galis ZS. Atherosclerotic lesions grow through recruitment and proliferation of circulating monocytes in a murine model. The American journal of pathology. 2002;160:2145–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. The Journal of clinical investigation. 2007;117:195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Konkova MS, Kaliyanov AA, Sergeeva VA, Abramova MS, Kostyuk SV. Oxidized cell-free DNA is a factor of stress signaling in radiation-induced bystander effects in different types of human cells. International journal of genomics. 2019;2019:9467029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaisar MA, Sivandzade F, Bhalerao A, Cucullo L. Conventional and electronic cigarettes dysregulate the expression of iron transporters and detoxifying enzymes at the brain vascular endothelium: In vivo evidence of a gender-specific cellular response to chronic cigarette smoke exposure. Neuroscience letters. 2018;682:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vina J, Gambini J, Lopez-Grueso R, Abdelaziz KM, Jove M, Borras C. Females live longer than males: Role of oxidative stress. Current pharmaceutical design. 2011;17:3959–3965 [DOI] [PubMed] [Google Scholar]
- 44.Borras C, Sastre J, Garcia-Sala D, Lloret A, Pallardo FV, Vina J. Mitochondria from females exhibit higher antioxidant gene expression and lower oxidative damage than males. Free radical biology & medicine. 2003;34:546–552 [DOI] [PubMed] [Google Scholar]
- 45.Hansson GK, Libby P. The immune response in atherosclerosis: A double-edged sword. Nature reviews. Immunology. 2006;6:508–519 [DOI] [PubMed] [Google Scholar]
- 46.Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: Lessons from mouse models. Nature reviews. Immunology. 2008;8:802–815 [DOI] [PubMed] [Google Scholar]
- 47.Zhang D, Fang P, Jiang X, Nelson J, Moore JK, Kruger WD, Berretta RM, Houser SR, Yang X, Wang H. Severe hyperhomocysteinemia promotes bone marrow-derived and resident inflammatory monocyte differentiation and atherosclerosis in ldlr/cbs-deficient mice. Circulation research. 2012;111:37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ghattas A, Griffiths HR, Devitt A, Lip GY, Shantsila E. Monocytes in coronary artery disease and atherosclerosis: Where are we now? Journal of the American College of Cardiology. 2013;62:1541–1551 [DOI] [PubMed] [Google Scholar]
- 49.Woollard KJ, Geissmann F. Monocytes in atherosclerosis: Subsets and functions. Nature reviews. Cardiology. 2010;7:77–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nicholson MW, Barclay AN, Singer MS, Rosen SD, van der Merwe PA. Affinity and kinetic analysis of l-selectin (cd62l) binding to glycosylation-dependent cell-adhesion molecule-1. The Journal of biological chemistry. 1998;273:763–770 [DOI] [PubMed] [Google Scholar]
- 52.Mestas J, Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends in cardiovascular medicine. 2008;18:228–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakai A, Kume N, Nishi E, Tanoue K, Miyasaka M, Kita T. P-selectin and vascular cell adhesion molecule-1 are focally expressed in aortas of hypercholesterolemic rabbits before intimal accumulation of macrophages and t lymphocytes. Arteriosclerosis, thrombosis, and vascular biology. 1997;17:310–316 [DOI] [PubMed] [Google Scholar]
- 54.Combadiere C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, Merval R, Proudfoot A, Tedgui A, Mallat Z. Combined inhibition of ccl2, cx3cr1, and ccr5 abrogates ly6c(hi) and ly6c(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649–1657 [DOI] [PubMed] [Google Scholar]
- 55.Tarik O, Zeynep O, Hasan D, Mustafa U, Ahmet Y, Mevlit I, Sahin A. Relationship between carbon monoxide intoxication and sister chromatid exchange in lymphocytes. Toxicology and industrial health. 2014;30:896–900 [DOI] [PubMed] [Google Scholar]
- 56.Sparks Daniel L., Heather Doelle CC Circulating nucleotides in health and disease. Receptor & Clinical Investigation. 2014;1:e344 [Google Scholar]
- 57.Cossarizza A, Pinti M, Nasi M, Gibellini L, Manzini S, Roat E, De Biasi S, Bertoncelli L, Montagna JP, Bisi L, Manzini L, Trenti T, Borghi V, Mussini C. Increased plasma levels of extracellular mitochondrial DNA during hiv infection: A new role for mitochondrial damage-associated molecular patterns during inflammation. Mitochondrion. 2011;11:750–755 [DOI] [PubMed] [Google Scholar]
- 58.Wang Y, Wang GZ, Rabinovitch PS, Tabas I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-kappab-mediated inflammation in macrophages. Circulation research. 2014;114:421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Y, Wang W, Wang N, Tall AR, Tabas I. Mitochondrial oxidative stress promotes atherosclerosis and neutrophil extracellular traps in aged mice. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:e99–e107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ermakov AV, Konkova MS, Kostyuk SV, Smirnova TD, Malinovskaya EM, Efremova LV, Veiko NN. An extracellular DNA mediated bystander effect produced from low dose irradiated endothelial cells. Mutation research. 2011;712:1–10 [DOI] [PubMed] [Google Scholar]
- 61.West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, Iwasaki A, Shadel GS. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bao D, Zhao J, Zhou X, Yang Q, Chen Y, Zhu J, Yuan P, Yang J, Qin T, Wan S, Xing J. Mitochondrial fission-induced mtdna stress promotes tumor-associated macrophage infiltration and hcc progression. Oncogene. 2019;38:5007–5020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li B, Yue Y, Dong C, Shi Y, Xiong S. Blockade of macrophage autophagy ameliorates activated lymphocytes-derived DNA induced murine lupus possibly via inhibition of proinflammatory cytokine production. Clinical and experimental rheumatology. 2014;32:705–714 [PubMed] [Google Scholar]
- 64.Gkirtzimanaki K, Kabrani E, Nikoleri D, Polyzos A, Blanas A, Sidiropoulos P, Makrigiannakis A, Bertsias G, Boumpas DT, Verginis P. Ifnalpha impairs autophagic degradation of mtdna promoting autoreactivity of sle monocytes in a sting-dependent fashion. Cell reports. 2018;25:921–933 e925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis and rheumatism. 1998;41:1241–1250 [DOI] [PubMed] [Google Scholar]
- 66.Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nature reviews. Rheumatology. 2010;6:280–289 [DOI] [PubMed] [Google Scholar]
- 67.Yasuda K, Yu P, Kirschning CJ, Schlatter B, Schmitz F, Heit A, Bauer S, Hochrein H, Wagner H. Endosomal translocation of vertebrate DNA activates dendritic cells via tlr9-dependent and -independent pathways. Journal of immunology. 2005;174:6129–6136 [DOI] [PubMed] [Google Scholar]
- 68.Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, Shlomchik MJ, Coffman RL, Candia A, Mehal WZ. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of tlr9. The Journal of clinical investigation. 2016;126:859–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sorrentino R, Morello S, Chen S, Bonavita E, Pinto A. The activation of liver x receptors inhibits toll-like receptor-9-induced foam cell formation. Journal of cellular physiology. 2010;223:158–167 [DOI] [PubMed] [Google Scholar]
- 70.Koulis C, Chen YC, Hausding C, Ahrens I, Kyaw TS, Tay C, Allen T, Jandeleit-Dahm K, Sweet MJ, Akira S, Bobik A, Peter K, Agrotis A. Protective role for toll-like receptor-9 in the development of atherosclerosis in apolipoprotein e-deficient mice. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:516–525 [DOI] [PubMed] [Google Scholar]
- 71.Lundberg AM, Ketelhuth DF, Johansson ME, Gerdes N, Liu S, Yamamoto M, Akira S, Hansson GK. Toll-like receptor 3 and 4 signalling through the trif and tram adaptors in haematopoietic cells promotes atherosclerosis. Cardiovascular research. 2013;99:364–373 [DOI] [PubMed] [Google Scholar]
- 72.Goossens P, Gijbels MJ, Zernecke A, Eijgelaar W, Vergouwe MN, van der Made I, Vanderlocht J, Beckers L, Buurman WA, Daemen MJ, Kalinke U, Weber C, Lutgens E, de Winther MP. Myeloid type i interferon signaling promotes atherosclerosis by stimulating macrophage recruitment to lesions. Cell metabolism. 2010;12:142–153 [DOI] [PubMed] [Google Scholar]
- 73.Cole JE, Kassiteridi C, Monaco C. Toll-like receptors in atherosclerosis: A ‘pandora’s box’ of advances and controversies. Trends in pharmacological sciences. 2013;34:629–636 [DOI] [PubMed] [Google Scholar]
- 74.Falck-Hansen M, Kassiteridi C, Monaco C. Toll-like receptors in atherosclerosis. International journal of molecular sciences. 2013;14:14008–14023 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.